revised manuscript m1:07442 doxorubicin induces
TRANSCRIPT

Revised Manuscript M1:07442
DOXORUBICIN INDUCES APOPTOSIS AND CD95 GENE EXPRESSION IN HUMAN PRIMARY ENDOTHELIAL CELLS
THROUGH A p53-DEPENDENT MECHANISM
Elisa Lorenzo1, Carmen Ruiz-Ruiz2, Antonio Jesús Quesada1, Gabriela Hernández1,
Antonio Rodríguez1, Abelardo López-Rivas2 and Juan Miguel Redondo1,*.
From the 1Centro de Biología Molecular “Severo Ochoa”, Consejo Superior de Investigaciones Científicas (CSIC), Universidad Autónoma de Madrid, Facultad de
Ciencias, E-28049, Madrid, Spain, and the 2Instituto de Parasitología y Biomedicina, calle Ventanilla 11, E-18001, Granada, Spain.
Running title: p53 regulates CD95 expression and apoptosis in HUVECs.
*Corresponding author. Mailing address: Centro de Biología Molecular “SeveroOchoa”, Consejo Superior de Investigaciones Científicas (CSIC)-UniversidadAutónoma de Madrid. Facultad de Ciencias. Cantoblanco. Madrid 28049, Spain.
1
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on January 4, 2002 as Manuscript M107442200 by guest on A
pril 6, 2018http://w
ww
.jbc.org/D
ownloaded from

Phone: 34-91-3978270Fax: 34-91-3978087E-mail: [email protected]
SUMMARY.
Regulation of the homeostasis of vascular endothelium is critical for the
processes of vascular remodeling and angiogenesis under physiological and
pathological conditions. Here we show that Doxorubicin (Dox), a drug used in
antitumor therapy, triggered a marked accumulation of p53, and induced CD95 gene
expression and apoptosis in proliferating human umbilical vein endothelial cells
(HUVECs). Transfection and site directed mutagenesis experiments using the CD95
promoter fused to an intronic enhancer indicated the requirement for a p53 site for Dox-
induced promoter activation. Furthermore, the p53 inhibitor PFT-α blocked both
promoter inducibility and protein upregulation of CD95 in response to Dox. Up-
regulated CD95 in Dox-treated cells was functional in eliciting apoptosis upon
incubation of the cells with an agonistic CD95 antibody. However, Dox-mediated
apoptosis was independent of CD95/CD95L interaction. The analysis of apoptosis in the
presence of PFT-α and z-VAD-FMK revealed that both p53 and caspase activation are
required for Dox-mediated apoptosis of HUVECs. Finally, Dox triggered Bcl-2
downregulation, cytochrome c release from mitochondria, and the activation of
caspases-9 and -3, suggesting the involvement of a mitochondrially-operated pathway
of apoptosis. These results highlight the role of p53 in the response of primary
endothelial cells to genotoxic drugs and may reveal a novel mechanism underlying the
antitumoral properties of Dox, related to its ability to induce apoptosis in proliferating
endothelial cells.
2
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

INTRODUCTION
The regulation of apoptosis in endothelial cells is critical for the integrity of
endothelium. Processes such as vascular remodeling and angiogenesis involve both
proliferation and apoptosis of vascular endothelial cells (1,2). In addition, endothelial
injury that results in apoptosis appears to play an important pathogenic role in the
progression of atherosclerotic lesions and many inflammatory disorders (3-8).
The CD95 (Fas/Apo-1) receptor, a member of the TNF/nerve growth factor
receptor family (9,10), triggers a potent apoptotic signal when bound to its natural
specific ligand CD95L (11). This apoptotic signal eventually results in the activation of
the caspase cascade (12). However, a number of stress agents may also elicit the
activation of downstream caspases through a different apoptotic pathway which
involves the release of cytochrome c (13).
Genotoxic stress by chemotherapeutic drugs such as Doxorubicin (Dox) has
been shown to activate apoptosis in a number of different cell types (14). Dox has been
used to treat a variety of cancers, and as in the case of other DNA-damaging-agents, it
induces the accumulation of the p53 tumor suppressor protein. This results in cell cycle
arrest and may lead to apoptosis (15,16); a mechanism that ensures the elimination of
these dangerous cells from the organism. Recently, p53 has been shown to regulate
CD95 gene expression through interactions with p53 binding motifs located within the
CD95 gene promoter and an enhancer situated within the first intron of the CD95 gene
(17). In several tumor cell lines the p53-mediated upregulation of CD95 has been
reported to be required for genotoxic drug-induced apoptosis (17,18).
3
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

CD95 and CD95L have been shown to be expressed on the surface of
endothelial cells (19-23). In the vascular endothelium, CD95L appears to negatively
regulate extravasation by inducing apoptosis of leukocytes as they cross the vessel wall
(24). Under homeostatic conditions, CD95 is expressed at lower levels in endothelial
cells that are resistant to CD95-mediated apoptotic cell death (19-22,24,25). Recently,
a number of cellular stresses including matrix detachment and exposure of cells to
oxidized LDL or hydrogen peroxide have been reported to upregulate endothelial cell
surface expression of CD95 (20,21,25), and to induce apoptosis through CD95/CD95L
interaction (20,25). In spite of this, very little is known about the mechanisms that
regulate CD95 gene expression in endothelial cells. To address this issue we have
searched for stimuli that induce CD95 expression and found that Dox promoted
apoptosis in human primary endothelial cells and was a potent inducer of CD95. In
these cells, Dox induced a sustained accumulation of p53 which regulated both CD95
gene expression and the apoptotic process. However, although Dox-induced CD95
protein was able to trigger apoptotic signals, it was not involved in the apoptosis
induced by the drug. Our findings show that, Dox induced apoptosis through a
mitochondrially operated/p53-dependent pathway, and it is noteworthy that significant
apoptosis was only seen in subconfluent endothelial cells These findings support a
major role for p53 in the regulation of CD95 and apoptosis of primary endothelial cells
exposed to genotoxic drugs.
The preferential effect of Dox inducing apoptosis on proliferating endothelial
cells may reveal a novel mechanism underlying the antitumoral activity of Dox. If Dox
is able to selectively trigger apoptosis in proliferating cells “in vivo”, this could result in
selective killing by the drug of endothelial cells involved in neovascularization,
4
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

including that required for the growth and dissemination of tumors. We also discuss the
potential advantages and disadvantages that would be derived from the use of p53
inhibitors for the prevention of the side effects of Dox in wound healing or ovulation.
5
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

EXPERIMENTAL PROCEDURES
Cell culture and reagents. HUVECs were isolated from umbilical veins as previously
described (26) and maintained in culture on 0.5% gelatin-coated tissue culture flasks in
medium 199 (Biowhittaker) containing 20% fetal calf serum (FCS), 50 µg/ml bovine
brain extract and 100 µg/ml heparin. The cells were used between passages 4 and 9. J-
HM1-2.2 Jurkat cells expressing the human muscarinic acetylcholine type 1 (HM1)
receptor were grown in RPMI 1640 medium (Gibco-BRL) supplemented with 10%
FCS. Doxorubicin (Dox), the calcium ionophore A23187, phorbol-12.13-dibutyrate,
6-diamidino-2-phenylindole (DAPI) and carbamylcholine chloride (Carbachol) were
purchased from the Sigma Chemical Co. The agonistic anti-CD95 CH11 mAB (IgM)
was purchased from Upstate Biotechnology (Lake Placid, NY). Antagonistic anti-CD95
DX2 mAb (IgG1), anti-CD95 ligand NOK-1 mAb (IgG1), and the matrix
metalloprotease inhibitor KB8301 were from Pharmingen. The generic caspase inhibitor
benzyloxycarbonyl-Val-Ala-DL-Asp-fluoromethylketone (z-VAD-FMK) was from
Bachem and the specific p53 inhibitor pifithrin-α (PFT-α) was from Calbiochem. TNF-
α (3.2 x 107 U/mg) was from Wichem.
Determination of apoptotic cells. Subconfluent HUVECs were plated in twenty four-
well tissue culture plates (4 x 104 cells/plate) precoated with 0,5% gelatin. Attached
cells were detached from the culture plates with a trypsin solution containing 3mM
EDTA (tryspin/EDTA). These cells were then collected together with floating cells,
washed once with cold phosphate-buffered saline (PBS), and incubated with 200 µl
staining buffer (0,1% sodium citrate, 0,02 mg/ml RNAase, 0,3% NP-40, 0,05 mg/ml
6
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

propidium iodide) for 30 min on ice. Then, 300 µl of staining buffer (without RNAase
and propidium iodide) were added and hypodiploid apoptotic cells were determined by
cytofluorometric analysis of DNA content in a FACScan cytofluorometer (Becton
Dickinson).
Flow cytometry. HUVECs were plated in six-well tissue culture plates (3 x 105
cells/plate) precoated with 0,5% gelatin. They were then treated as described in Results,
detached with 3mM EDTA, washed once with cold PBS and then incubated with one of
the following antibodies for 30 min on ice: anti-CD95 mouse monoclonal IgG antibody
DX2 (1 µg/ml), the anti-CD95L mAb NOK-1 (IgG1) (2 µg/ml), or the anti-ICAM-1
mAb RR1/1. After this incubation, cells were washed once with cold PBS and then
incubated with FITC-conjugated rabbit anti-mouse Ig (1/50; Dako) for 30 min on ice.
Cells were again washed with cold PBS and resuspended in PBS. Flow cytometry was
performed on a FACScan cytometer and analysed with Cell Quest software (Becton
Dickison).
mRNA analysis. Total RNA was isolated from attached HUVECs with the Ultraspect
system (Biotecx Laboratories, Inc), in accordance with the manufacturer’s instructions.
cDNAs were synthesized using an RNA PCR kit (Perkin Elmer) from 2-4 µg of total
RNA. PCR reactions were performed using the following primers: human CD95 sense,
5´-GGGTGAAGAGAAAGGAAGTACAG-3´, human CD95 antisense, 5´-
CCTTGGAGGCAGAATCATGA-3´; human CD95L sense, 5´-
CAGGACTGAGAAGAAGTAAAACCG-3´, human CD95L antisense, 5´-
CTCCAAAGATGATGCTGTG-3´; human β-actin sense 5´-
TGACGGGGTCACCCACACTGTGCCCATCTA-3´, and human β-actin antisense,
7
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5´-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3´. The PCR products obtained
were 615, 440 and 661 bp, respectively. Cycle conditions of the PCR reactions were 60s
at 95ºC, 60 s at 55ºC and 60 s at 72ºC for 35, 40 or 25 cycles for CD95, CD95L or β-
actin, respectively. Amplified cDNAs were separated by agarose gel electrophoresis and
bands visualized by ethidium bromide staining.
Plasmid constructs and transient-transfection assays. Genomic fragments of the CD95
upstream regulatory region spanning 391 bp from the ATG site of the CD95 gene was
amplified by PCR of human genomic DNA. These sequences were then cloned into the
BglII site of the pXP2 luciferase reporter plasmid to generate the pCD95 391 Luc
construct. This plasmid was used as the parental construct to generate the pI-CD95 391
Luc by cloning a 500 bp enhancer region from the first intron of the CD95 gene (17)
into the BamHI site of the pXP2 polylinker (upstream of the promoter region). The 500
bp fragment was also amplified from genomic cDNA using the primers ENH FAS5’:
cgggtccGTGAGCCCTCTCCTGCCCGGGT and ENHFAS3’:
cgggatcCCTGAAGGCTGCAGGCTCTCTCC. The pmI-CD95 391 Luc plasmid,
harboring mutations within the p53 sites of the intronic enhancer, was generated by site
directed mutagenesis of the pI-CD59 391 Luc with a kit from Quick Change,
Stratagene. Mutations were introduced using the following sense oligonucleotides: 5´-
AACTCCTGGAGGGGCCCTGACAAG-3´ and 5´-
CCCTGACAATAAAAGCCAAAGGT-3´ and their respective antisense oligonucleotides (mutated nucleo
in bold). The pBHA-941Luc plasmid including a 941 bp fragment of the upstream
regulatory region of the ICAM-1 promoter has been described previously (26). The
reporter plasmid pG13 Luc (p53 Luc), containing 13 tandem copies of the p53-
responsive element, was kindly donated by Dr. B. Vogelstein (Johns Hopkins
8
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

University School of Medicine, Baltimore, Maryland, US) (27). The pCDNA3-hbcl-2
plasmid, containing the cDNA for the human Bcl-2 human gene, was kindly provided
by Dr. Jacint Boix (University of Lleida, Spain). The pEGFP-spectrin expression vector
was kindly provided by Dr. Kalejta (Princeton University, New Jersey, USA) (28). The
pKBF-Luc construct includes a trimer of the NF-κB motif from the H-2Kb gene,
placed upstream of a herpes simplex virus thymidine kinase minimal promoter driving
the luciferase reporter gene (29).
For transient transfection experiments, HUVECs were plated in 100-mm tissue
culture dishes (1.5 x 106 cells/plate) the day before transfection. Cells were transfected
in 4ml of Dulbecco´s minimal essential medium containing 10% FCS, with 10 µg of the
indicated luciferase reporter plasmid by the calcium phosphate procedure as previously
described (30,31), with some modifications. Briefly, cells were incubated with
precipitated DNA until a mild cytopathic effect was observed (4,5 to 9h). Cells were
then washed twice with PBS, and detached with trypsin/EDTA from the culture dishes.
After centrifugation, cells were resuspended in OPTI-MEM (Life Technologies)
containing 0,5% FCS, and split among six well (35mm) tissue culture plates precoated
with 0,5% gelatin. After 24 h, transfected cells incubated with or without z-VAD-
FMK, were treated with different agents for an additional period of 14 h. Cells were
then detached with trypsin/EDTA, washed with PBS, and lysed. Luciferase activity was
measured according to the instructions of the Luciferase System Kit (Promega) in a
Sirius luminometer (Berthold, Germany). The expression of Renilla luciferase was used
as an internal control for the efficiency of transfection. A total of 0.1 µg Renilla
luciferase expression vector pRLCM (Promega) was used in cotransfection experiments.
9
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

In these experiments, 1/10 of cells cotransfected with both types of luciferase plasmid
were plated in 24-well tissue culture plates, and the cells treated in the same way as
those plated on 6-well tissue culture plates. After lysis with passive lysis buffer, Renilla
luciferase activity was measured with the Dual luciferase assay kit (Promega).
Subcellular fractionation and Western blot analysis. After treatment, subconfluent
attached HUVECs grown in 35-mm culture dishes were detached with trypsin/EDTA
and collected together with floating cells. They were then washed with cold PBS,
resuspended in 40µl Laemmli buffer and sonicated. For the detection of cytochrome c
release, cells were detached, washed with PBS, and the pellet was resuspended in 50 µl
cold lysis buffer (25 mM Tris-HCl pH 6,8, 250 mM sucrose, 1 mM EDTA, 0,005%
digitonin, 1mM DTT, 0,1mM PMSF, 1mM Benzamidin and 1µg per ml each of
aprotinin, leupeptin and pepstatin) for 30 s on ice. Lysates were centrifuged and the
supernatant containing the cytosolic fraction was removed and separated from
mitochondria. Cytosolic proteins were mixed with Laemmli buffer and sonicated. For
detection of p53 protein, 10 ¼g whole cell extract were resolved on 8% SDS PAGE
minigels. For all other proteins, 60 µg of extract (cytosolic or whole) were resolved on
12% SDS PAGE minigels. Proteins were transferred onto Immobilon membranes
(Millipore), and the blots were blocked with 5% w/v skimmed milk in PBS/0,05%
Tween 20 (PBST) for 1 h at room temperature. Blots were then washed three times for
10 min with PBST, and incubated in PSBT, 1% w/v milk at 4ºC overnight with the
indicated antibodies: anti-Bcl-2 (1:500, DAKO), anti-caspase-3 (1:1000, New
England, Biol.), anti-PARP (1:4000, Boehringer), anti-caspase-9 (1:250, New England
Biol.), anti-p53 (1:3000, Ab-7, Calbiochem) or anti-cytochrome c mAb (1µg/ml,
Pharmigen). Blots were again washed three times for 10 min with PBST and then
10
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

incubated with the corresponding HRP-coupled goat anti-rabbit or anti-mouse
secondary antibody (1:2000, DAKO). After three washes with PBST and once with
PBS, blots were visualized with the Amersham enhanced chemiluminescence (ECL)
detection reagents. Detection of α-tubulin (anti-α-tubulin: 1:40000, Sigma) was used
to control for loading of protein.
Nuclear extracts and EMSAs. For nuclear protein extraction, HUVECs grown in 150-
mm dishes were detached and then lysed with 400 µl of hypotonic buffer (10 mM
HEPES {pH 7,6}, 10 mM KCl, 0,1 mM EDTA, 0,1 mM EGTA, 1 mM DTT, 0,5 mM
phenylmethylsulfonyl fluoride (PMSF), 10 mM Na2Mo04, 2 µg per ml of pepstatin,
leupeptin and aprotinin, 0,75 mM spermidine, 0,15 mM spermine, and 1µg per ml of
pepstatin) containing 0,6% of NP40. Nuclei were then centrifugated and incubated in 50
µl of buffer C (20 mM HEPES {pH 7,6}, 0,4 mM KCl, 1 mM EDTA, 1 mM EGTA, 1
mM DTT, 0,5 Mm PMSF, 10 mM Na2Mo04, and 2 µg per ml of pepstatin, leupeptin
and aprotinin) for 30 min in a rocking platform. Nuclei were centrifuged at 15,000 x g
for 10 min, and the supernatants containing the nuclear extracts were immediately
stored at –80ºC. The protein concentration was quantified by the Bradford procedure.
Electrophoretic mobility shift assays (EMSAs) were performed by incubating
nuclear proteins (2 - 3 µg) with 1 µg poly (dI-dC) DNA carrier and 3 µl of 5 x DNA-
binding buffer (10% {wt/vol} polyvinylethanol, 12,5% {vol/vol} glycerol, 50 mM Tris
{pH 8}, 2,5 mM EDTA, 2,5 mM DTT) in a final volume of 15 µl on ice for 10 min.
Then 2 µl (1ng/µl) of 32P-labeled double-stranded oligonucleotide (108 cpm/µg) were
added to the reaction mixture, and it was incubated at room temperature for 30 min. For
competition experiments, a 30-fold molar excess of unlabeled oligonucleotide was
11
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

added before the addition of the probe. Where indicated, nuclear extracts were
incubated at room temperature for 10 min before addition of the probe with the
following antibodies: 2-4 µl of anti-p53 antiserum PAb421; 1 µl of antiserum 1226,
raised against the p65 NFκB subunit; or 1 µl of antiserum 1141, raised against the p50
NFκB subunit. DNA-protein complexes were resolved by electrophoresis on 4%
nondenaturing polyacrylamide gels. The sequences of the oligonucleotides (5´ to 3´)
used in these experiments were as follows:
ctagCTCCCCAACCCGGGCGTTCCCCAGCGAGG (human NFκB sequence –306 to
–278 of the 391 bp fragment of CD95 promoter, (32)) and
gatcCTCCTGGACAAGCCCTGACAAGCCAAGCCA (human p53 sequence located within of the introni
enhancer of the CD95 human gene (17)).
Inmunofluorescence experiments. To analyze the effect of Bcl-2 expression on the
viability of HUVECs treated with Dox, cells were cotransfected with 200 ng of the
pEGFP-spectrin expression plasmid together with 200 ng of either pCDNA3-hBcl-2
or the control pCMVβ-Galactosidase expression vector (33). The pGL3 Basic vector
(2,1 ¼g) was added as a DNA carrier in a total volume of 0.140 ml, and transfection
was performed by the calcium phosphate procedure in 35 mm tissue culture dishes.
After treatment, the cells were washed with PBS, fixed with 3,7% formaldehyde for 15
min, and washed for a further 10 min with 50 mM NH4Cl blocking solution in PBS.
Cells were then washed with PBS, permeabilized with a 0,1% triton-X100 for 10 min,
washed again with PBS, and stained with 1 ¼g/ml DAPI solution for 2 min. The cells
were examined under a fluorescence microscope, and GFP positive cells were scored
after counting a minimum of 1000 total cells for each condition. The efficiency of
12
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

transfection in Bcl-2– and β-Gal–expressing cells, determined in aliquots of
transfected cells just before the addition of Dox, was similar, (10-12%).
Data analyses. Data are presented as mean ± standard error (SE) of several
determinations. Differences between groups were tested for significance using Student´s
t test. Analysis of cell cycle by flow cytometry was performed by counting a total
population of at least 5000 cells. In these assays similar results were obtained in at least
three independent experiments, and the significance determined by the χ2 test.
13
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

RESULTS
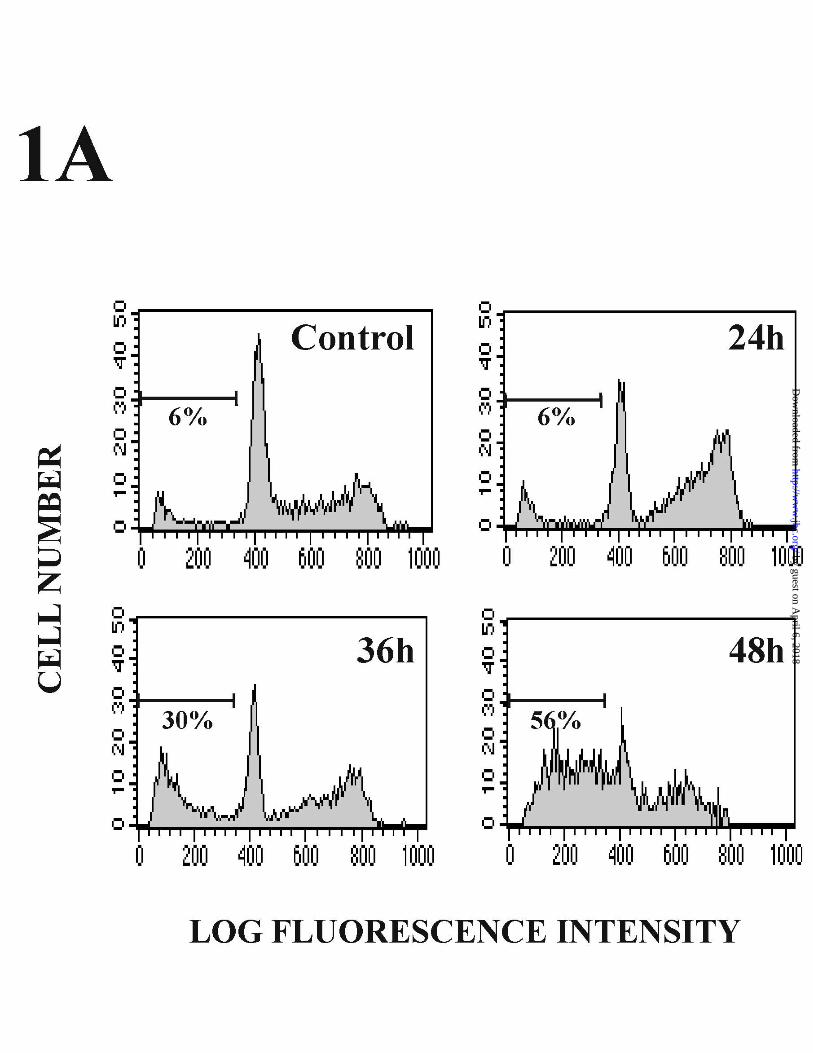
Doxorubicin induces apoptosis in subconfluent HUVECs.
Previous studies have shown that different genotoxic drugs induce apoptosis in
many types of cells (34-39). To analyze whether Dox affected the viability of primary
endothelial cells, we performed FACS analysis on propidium iodide-stained HUVECs
and determined the fraction of hypodiploid apoptotic cells following Dox treatment.
These experiments showed the presence of apoptotic cells after 36 h treatment, with a
further increase by 48 h. In addition, this cell cycle analysis demonstrated a marked
accumulation of Dox-treated cells in the G2-M phase after a 24 h treatment (Fig 1A).
Since we observed variability in the percentage of apoptotic cells induced by Dox in
different experiments, we analyzed whether cell confluence could influence the
sensitivity of HUVECs to Dox. As shown in Fig. 1 B, cell confluence significantly
affected the strength of Dox-mediated apoptosis in HUVECs. Dox failed to induce
apoptosis of confluent cells (100%) after a 30 h treatment, and only a slight increase in
the number of sub-G1 apoptotic cells (10-12%) was observed by 48 h under these
conditions. However, in cells plated at low cell density (40% confluence) apoptosis was
observed after 30 h, and an extensive number of sub-G1 apoptotic cells (up to 70%)
was detected after 48 h (Fig 1B).
Doxorubicin induces CD95 expression and apoptosis through a p53-dependent
mechanism in HUVECs.
Dox has been shown to up-regulate CD95 expression in a number of cells that
can become sensitized to death through CD95/CD95L interactions (34,35,40,41). In
some cases, the cellular response to DNA damage results in p53 accumulation, which is
14
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

required for the up-regulation of CD95 expression. To determine whether Dox could
induce CD95 protein expression at the cell surface of primary endothelial cells, we
analyzed CD95 expression levels by flow cytometry in Dox-treated HUVECs. Dox
induced a marked increase in the number of CD95-expressing HUVECs (Fig. 2A). This
increase took place in a dose-dependent manner (data not shown), and was consistent
with that detected in the CD95 mRNA levels by RT-PCR. In these experiments, the low
levels of CD95 mRNA found in unstimulated cells were clearly upregulated by Dox
after 10 h treatment, and further maintained for at least 24 h. As a control, the CD95
mRNA levels of Jurkat JHM1 T-lymphocytes were analyzed in parallel (Fig 2B). To
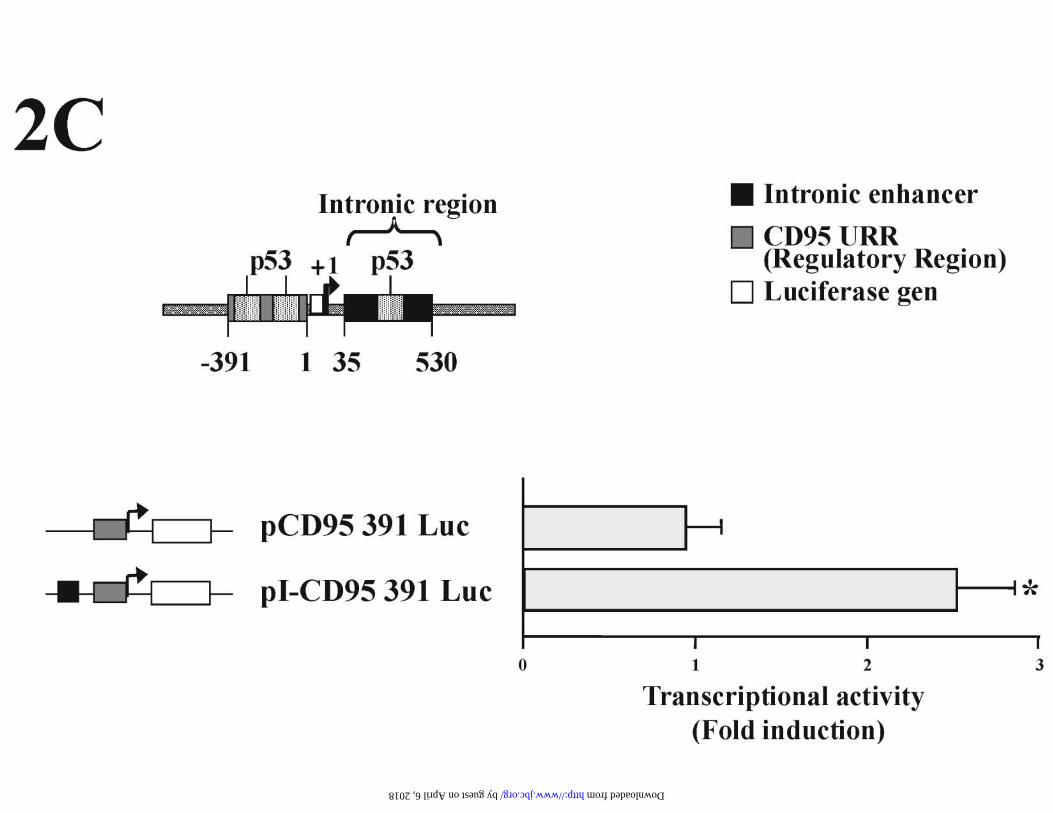
further investigate the mechanisms involved in the up-regulation of CD95 by Dox, we
analyzed the effect of the drug on CD95 promoter activity in HUVECs. We performed
transient transfection experiments to determine the transcriptional activity of a
luciferase reporter plasmid driven by a 391 bp fragment of the CD95 promoter (pCD95
391 Luc). This plasmid was further modified to contain a 500 bp p53-responsive
enhancer element from the first intron of the CD95 gene, inserted upstream of the
promoter fragment (pI-CD95 391 Luc). As shown in Fig 2C, Dox induced by two- to
threefold the transcriptional activity of CD95 reporter plasmid that harbored the p53
enhancer. The promoter fragment alone was not responsive to Dox, and the intronic
fragment containing the functional p53 binding site (17) was required to achieve a
significant promoter induction by Dox (Fig 2C). Together these results indicate that
Dox regulates CD95 expression in HUVECs, and suggest that p53 could be involved in
the CD95 up-regulation by Dox through transcriptional mechanisms.
To further analyze the role of p53 in CD95 expression we next tested whether
Dox regulated p53 protein expression in endothelial cells. To this end, we performed
15
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Western Blot experiments using whole cell extracts of HUVECs obtained at different
times after treatment with Dox. These assays showed that p53 protein, expressed at low
levels in unstimulated cells, was already induced at 4 h after Dox treatment, and reached
higher levels of expression by 12 h that were maintained for at least 36 h (Fig 3A).
In agreement with the results of the Western Blot experiments, EMSAs with
nuclear extracts of HUVECs and a probe including the p53 sequence of the CD95
intronic enhancer showed that p53 binding activity was significantly induced after Dox
treatment (Fig. 3B, left). The presence of p53 in the retarded complex was shown by the
addition of the PAb421 antibody, which completely shifted the specific complex. In
addition, p53 complex formation was efficiently competed by an excess of cold
homologous oligonucleotide (Fig. 3B, right).
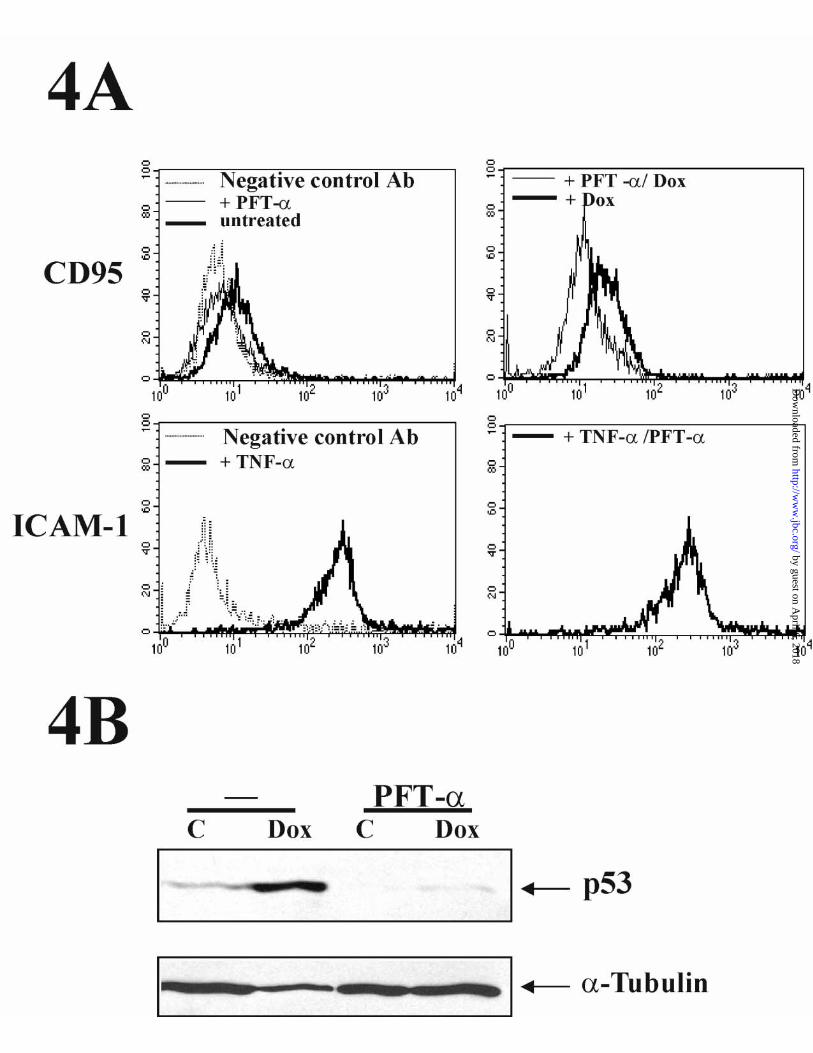
Noteworthily, flow cytometric analysis of CD95 expression in HUVECs
indicated that the specific p53 inhibitor PFT-α (42) blocked both constitutive and Dox-
induced expression of CD95 (Fig. 4A upper panels). By contrast, the TNF-α-induced
up-regulation of ICAM-1 cell surface expression (analyzed as a control in parallel
HUVEC cultures) was not affected by the treatment with PFT-α (Fig 4A, lower
panels). Therefore, the PFT-α-mediated inhibition of CD95 expression in HUVECs
was not due to a toxic or non-specific effect of the inhibitor. Control Western Blot
experiments, performed with extracts from aliquots of the same cells used for FACS,
showed that PFT-α efficiently inhibited both the basal and inducible levels of p53
protein (Fig. 4B).
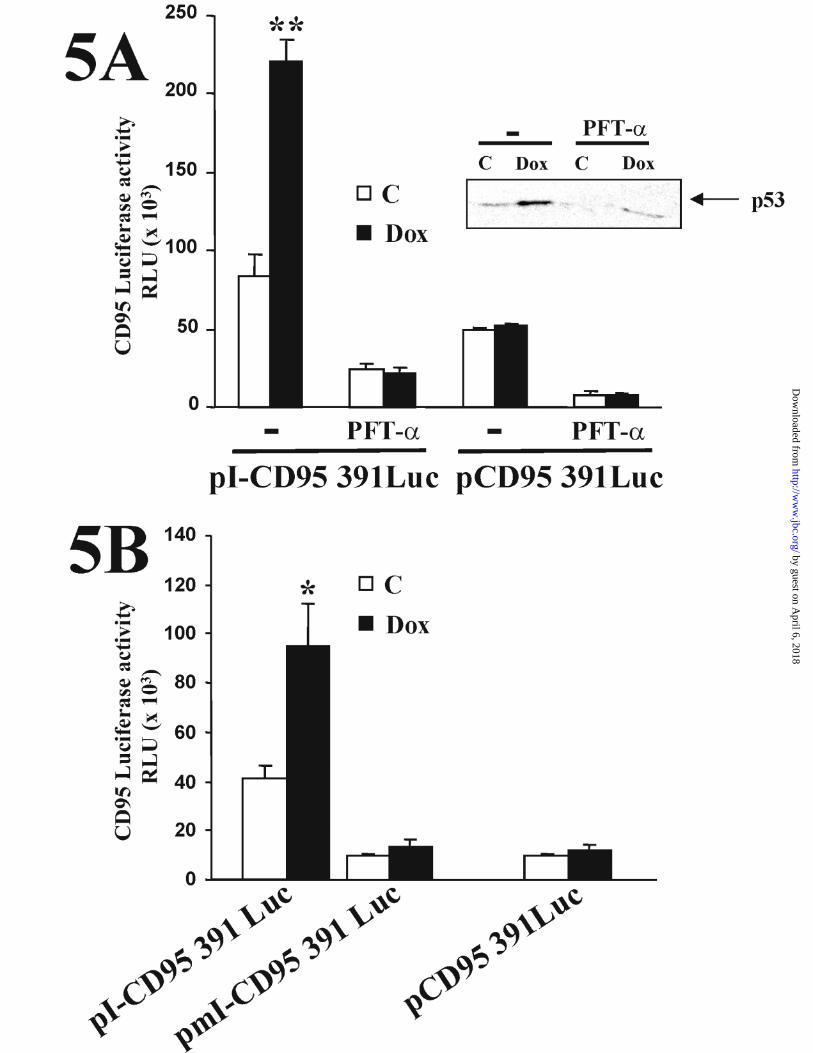
To evaluate the functional contribution of the intronic p53 site to the
transcriptional response of CD95 to Dox, we tested the effect of PFT-α in transient
transfection experiments with the pCD95 391 Luc and pI-CD95 391 Luc luciferase
16
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

reporter plasmids. As shown in Fig. 5 A, pre-treatment of cells with PFT-α completely
inhibited the induction of pI-CD95 391 Luc by Dox. Consistently with the FACS and
Western Blot experiments, PFT-α also inhibited the basal activity of the CD95
promoter constructs regardless of the presence of the intronic enhancer. Again, the
activity of PFT-α was controlled for by Western blot analysis of cell lysates from the
treated cells. (Fig. 5A, inset). As with the TNF-α↑mediated cell surface upregulation of
ICAM-1, the TNF-α-induced luciferase reporter activity of a 941 bp fragment of the
ICAM promoter was refractory to inhibition by PFT-α (data not shown). Evidence for
the functional involvement of the p53 site located within the CD95 intronic enhancer in
the Dox-induced activation of the CD95 promoter was provided by site directed
mutagenesis experiments. Mutation of this p53 site resulted in a reduction of the basal
and inducible transcriptional activity of the pmI-CD95 391 Luc construct that was
similar to that displayed by the enhancerless pCD95 391 Luc (Fig. 5B).
Dox has been shown to activate NFκB in a number of cell types (43-46). As
NFκB is also implicated in the transcriptional regulation of CD95 (32,47,48), we next
analyzed whether NFκB was also involved in the Dox-mediated activation of CD95 in
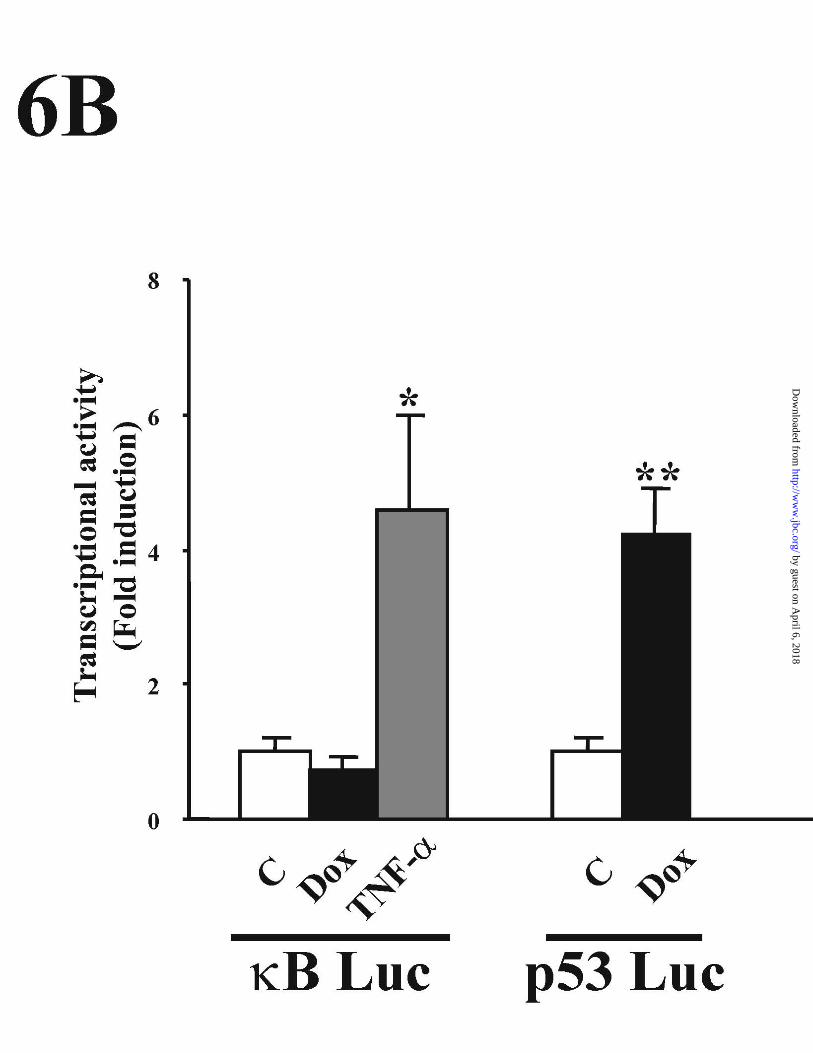
HUVECs. As shown in Fig 6A, Dox failed to induce NFκB binding activity to the -
306/-278 functional κB site of the CD95 promoter, whereas the binding of p65 and p50
NFκB subunits was induced by TNF-α. Similar results were obtained with a different
site; a κB motif of the IL-2 promoter that was also used as a probe in parallel EMSA
experiments. Furthermore, the activity of a NFκB reporter plasmid, that was activated
by TNF-α treatment of HUVECs, was not induced by Dox, whereas in parallel
experiments Dox efficiently induced the transcriptional activity of a p53-dependent
promoter (Fig. 6B).
17
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

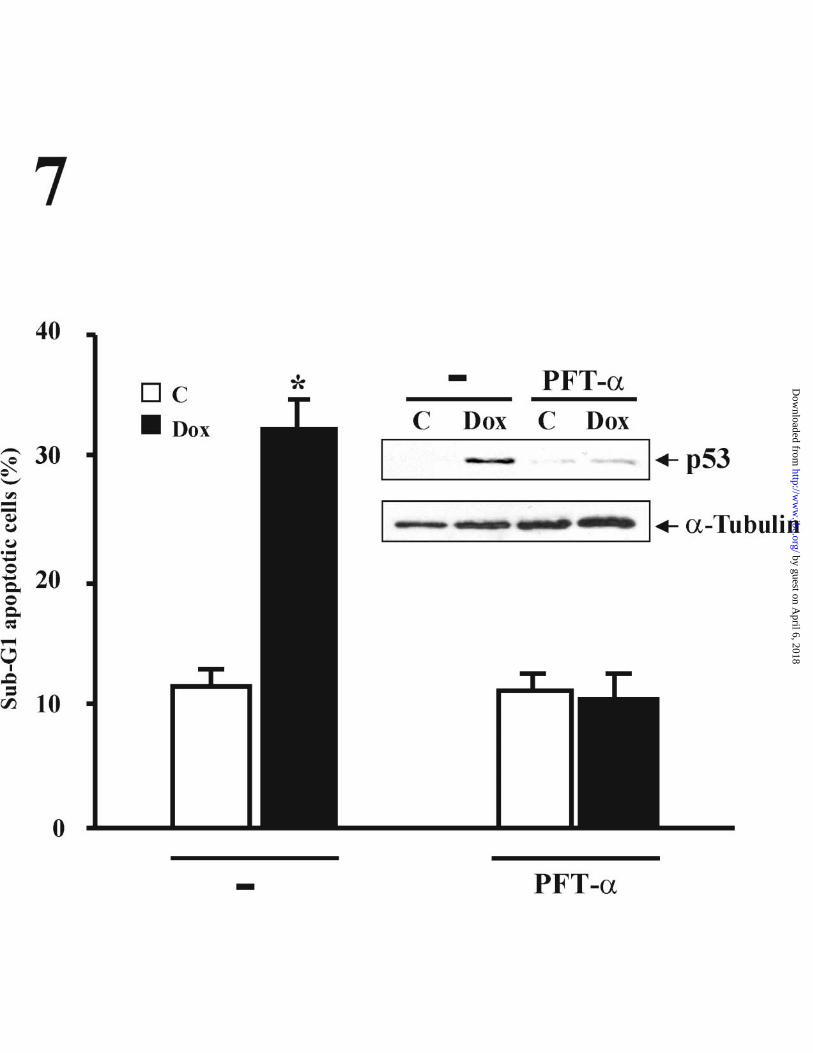
Given the involvement of p53 in the Dox-mediated apoptosis in many different
cell types (17,49-51), we next examined whether the induction of p53 expression was
related to the apoptosis triggered by the drug. For this purpose, we analyzed the effect
of PFT-α on the cell cycle of HUVECs exposed to Dox for 48 h. PFT-α treatment
completely prevented the appearance of sub-G1 apoptotic cells, indicating the
requirement for p53 in the Dox-induced apoptosis in HUVECs (Fig. 7). The efficient
inhibition of Dox-mediated p53 expression by PFT-α was confirmed by control
Western Blot experiments using whole cell extracts from the cells analyzed in these cell
cycle experiments (Fig. 7, inset).
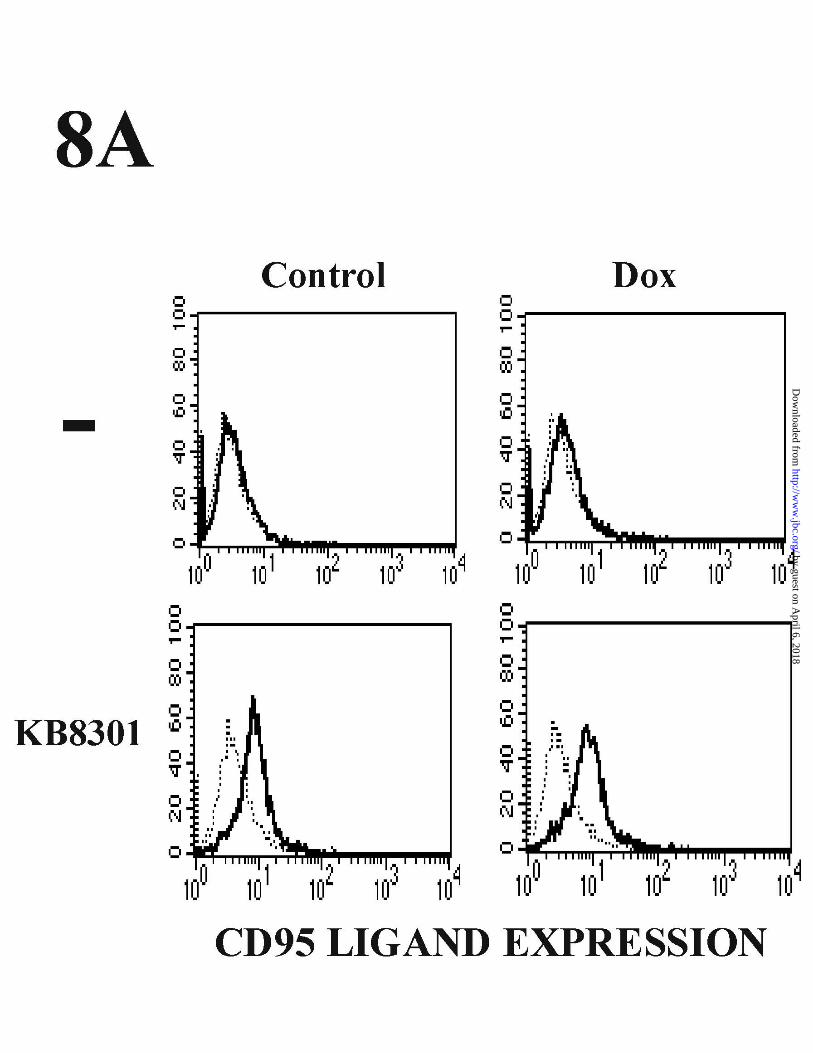
CD95/CD95L interaction does not mediate Dox-induced apoptosis in HUVEC.
As p53 was implicated in Dox-induced cell surface expression and gene
promoter activation of CD95, we tested whether signals delivered through CD95 were
involved in the apoptosis induced by Dox. We first evaluated whether Dox induced the
expression of CD95L, which could be involved in apoptosis through interaction with
CD95. As shown in Fig. 8 A CD95L was not detected in the surface of untreated
HUVECs by FACS. Low levels of CD95L were detected by HUVECs exposed to
KB8301, an inhibitor of matrix metalloproteinase previously shown to inhibit the
cleavage of membrane-bound CD95L (52). But Dox failed to induce the expression of
CD95L in the presence or absence of KB8301 (Fig. 8A). Moreover, Dox also failed to
induce the expression of CD95L mRNA in HUVECs, whereas in parallel experiments
CD95L mRNA expression was efficiently amplified in JHM1 Jurkat-derived cells
treated with Carbachol or PDBu plus ionophore (data not shown and (53)). Furthermore,
incubation of Dox-treated HUVECs with either the CD95 antagonistic antibody DX2 or
the CD95L blocking antibody NOK-1 failed to inhibit Dox-mediated apoptosis (Fig.
18
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8B). Control experiments using Carbachol and CD95 antibody in JHM1 cells
demonstrated the ability of NOK1 and DX2 antibodies to efficiently block apoptosis
triggered through CD95 activation (Fig. 8B). Nonetheless, the Dox-induced CD95
protein was able to trigger death signals as demonstrated by the marked induction of cell
death displayed by the CD95 agonistic antibody CH11 in Dox-treated HUVECs (Fig.
8B). Therefore, on the one hand these results indicate that Dox induces apoptosis in
HUVECs through a CD95/CD95L-independent mechanism, whereas on the other hand,
they also show that the CD95 receptor expressed after genotoxic drug treatment is able
to elicit functional apoptotic signaling in HUVECs.
Involvement of a p53-dependent caspase activation in the Doxorubicin-induced
apoptosis of HUVECs.
Since CD95/CD95L interaction was not involved in Dox-mediated apoptosis in
HUVECs we next analyzed the effect of Dox on the activation of the caspase cascade,
and the potential involvement of p53 in this process. We first determined the effect of
the general caspase inhibitor z-VAD-FMK on Dox-induced apoptosis. Cell cycle
analysis of HUVECs exposed to 100 µM z-VAD-FMK efficiently blocked apoptosis
induced by Dox (Fig. 9). Further confirmation of the involvement of caspases was
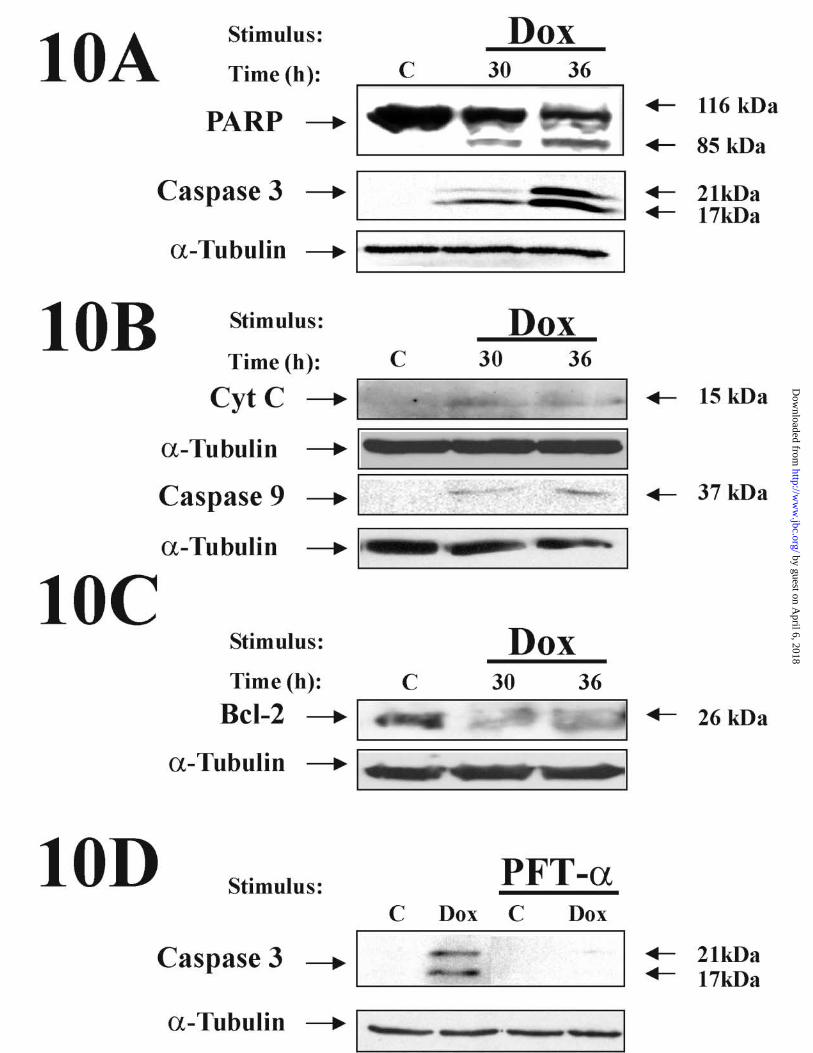
obtained by Western blot analysis of extracts of HUVECs treated with Dox for 30 to 36
h. As shown in Fig. 10 A, Dox led to the activation of executioner caspases, as revealed
by the proteolytic cleavage of the PARP nuclear substrate and the activation of caspase-
3. At earlier time points we failed to detect this activation (data not shown). In addition,
exposure of HUVECs to Dox resulted in release of cytochrome c release from the
mitochondria and caspase-9 activation (Fig. 10B). Moreover, Bcl-2 protein levels
19
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(reported to regulate caspase activation through the inhibition of cytochrome c release
(54,55)), were downregulated by Dox at times at which cytochrome c release was
detected (Fig. 10C, Fig. 10B). In addition, we performed experiments where GFP was
co-expressed with Bcl-2, or with β-Gal as a control, in HUVECs that were then treated
with Dox. When the cells were treated with Dox for 24 h, the number of viable, GFP-
expressing cells was reduced by 53-67% in two independent experiments (data not
shown) in cultures co-expressing the β-Gal control gene. In contrast, co-expression of
Bcl-2 completely prevented this loss of viability upon Dox treatment. This suggests that
down-regulation of Bcl-2 is a critical step in Dox-mediated apoptosis of HUVEC, and
together these data support the involvement of a mitochondrially-operated pathway of
apoptosis, triggered by Dox in endothelial cells. Although PARP and caspase-3
cleavage were already detected after 30 h of treatment, they reached higher levels by 36
h. However, maximal activation of caspase-9 was observed after a 30h treatment. This
probably reflects the activation of caspase-3 by caspase-9 (13).
Since the inhibition of p53 resulted in the blockade of apoptosis induced by Dox
(Fig. 7), we determined whether inhibition of p53 accumulation affected caspase
activation by Dox. As shown in Fig. 10 D, treatment of HUVECs with PFT-α
completely prevented activation of caspase-3 by Dox. This suggests that p53 could be
mediating Dox-induced apoptosis via activation of the caspase pathway in HUVECs.
Taken together, these findings support the hypothesis that Dox-induced
apoptosis in HUVECs is regulated by a p53-dependent mechanism, involving the
activation of downstream caspases, in a mitochondrially-operated apoptotic pathway.
20
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
Dox is a chemotherapeutic drug widely used in the treatment of a variety of
cancers including leukemias, sarcomas and breast cancer (36,37,56). Although Dox has
been shown to induce programmed cell death, the mechanisms by which Dox operates
appear to be different depending on the cell type analyzed. Thus, CD95/CD95L
interactions have been reported to mediate the drug-induced apoptosis in several tumor
cell lines (34,35,40) but not in others (36,37,56). Similarly, apoptosis by genotoxic
drugs appears to be dependent on p53 in hepatoma cells (18) but not in various breast
tumor xenografts (57).
Several recent reports have shown that treatment of endothelial cells with
different stimuli such as oxidized LDL, hydrogen peroxide or matrix detachment, result
in the upregulation of CD95 cell surface expression by endothelial cells (20,21,25).
However, the mechanisms that regulate CD95 gene expression in endothelial cells
remain poorly understood. In this study, we have looked for stimuli that induce CD95
expression and found that Dox induced both apoptosis and CD95 upregulation in human
primary endothelial cells through a p53-dependent mechanism. This upregulation of
CD95, however, was not involved in the Dox-induced activation of the caspase cascade
that led to the apoptosis of HUVECs.
We have found that Dox efficiently upregulates p53 protein expression and
DNA binding activity, and the transcriptional activity of the pG13 Luc p53-dependent
promoter in HUVECs. Since NFκB has been shown to transcriptionally regulate CD95,
we conducted parallel experiments to analyze the effect of Dox on the activation of
NFκB in HUVECs. However, in these experiments we did not detect any effect of Dox on the
binding or transcriptional activation by NFκB. Although the doses of Dox we used here
21
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

are lower than those shown to activate NFκB in some previous reports (44-46), other
authors have observed activation at lower doses (43). It is likely that the activation of
NFκB by Dox and the role of this in CD95 gene induction may depend on the cell type.
In this regard, Dox has been shown to activate NFκB and CD95 expression in hepatoma
cells but this expression was not transcriptionally mediated by NFκB (46).
The involvement of p53 in the regulation of CD95 gene expression in HUVECs
was demonstrated by transfection experiments, which showed that transcriptional
activity of the gene promoter was dependent on the presence of an intact p53 site within
the first intron of the CD95 enhancer. Previous studies have reported the importance of
the intronic region for transcriptional activation of the CD95 promoter in response to
p53 in hepatoma cells (17). We have shown that mutation of a critical p53 site within
this enhancer completely blocked Dox-mediated transcriptional activation of promoter
construct in HUVECs. Furthermore, the p53 inhibitor PFT-α blocked both the activity
of the promoter and the expression of CD95 at the cell surface induced by Dox, thus
suggesting that p53 regulates CD95 at the transcriptional level.
Although anti-CD95 or anti-CD95L blocking antibodies did not inhibit Dox-
mediated apoptosis, the CH11 agonistic antibody to CD95 was able to trigger apoptotic
signals in Dox-treated cells, showing that the cellular machinery required for CD95
apoptotic signaling, inactive in untreated HUVECs, was activated by Dox. Since
CD95L is expressed by activated circulating lymphocytes, the presence of functional
CD95 on the surface of endothelial cells might represent a potential risk for the
endothelium, and it is clear that tight regulatory mechanisms must operate to maintain
vascular integrity. In fact, despite the basal expression of CD95 found in endothelial
cells, these cells have been reported to be particularly resistant to CD95-mediated
22
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

apoptosis (19-22,24,25), and we have shown here that resting HUVECs fail to undergo
apoptosis after CD95 ligation. In view of these results, it will be very important to
investigate whether the sensitization to CD95 ligation and the apoptosis induced by Dox
that we have observed in HUVECs “in vitro” take place “in vivo”. It is important to
note that cardiotoxicity, impairment of wound healing, and renal and liver
complications are common side effects frequently found in patients treated with Dox
(58-62). As Dox was not able to induce significant apoptosis of confluent HUVECs, it
would also be of great interest to address whether the selective sensitivity of HUVECs
to Dox under subconfluent conditions is reflected in the proliferating cells of the
endothelium “in vivo”. If Dox is able to selectively trigger apoptosis of proliferating
(but not resting) endothelial cells “in vivo”, it is possible that part of its antitumoral
activity is mediated through this effect. In such a case, Dox could exert selective
apoptosis in endothelial cells involved in neovascularization, which would result in
disruption of blood vessel formation in growing tumors.
Our experiments clearly show that the caspase inhibitor z-VAD-FMK
prevented Dox-mediated apoptosis of HUVECs. A number of recent reports have
revealed a role for oxidative stress and the involvement of mitochondria in Dox-
mediated apoptosis in different cell systems including bovine aortic endothelial cells
(BAECs) and myocytes (38,58,63,64). We found that after 30-36 h treatment, Dox
induced cytochrome c release from mitochondria into the cytosol, caspase-9 activation,
and a concomitant downregulation of Bcl-2 protein levels. These results support the
involvement of a mitochondrially-operated apoptotic pathway induced by Dox in
endothelial cells. In this context, it is important to note that Bcl-2 has been shown to
prevent both the disruption of the inner mitochondrial membrane potential and the
23
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

release of cytochrome c from the mitochondria (54,55). Although, the precise molecular
mechanisms by which Bcl-2 prevents apoptosis are not completely clear, and appear to
be different depending of the cell type analyzed, Bcl-2 has been reported to act by an
antioxidant mechanism (65,66), and by interaction with different proapoptotic members
of the family (67,68). Since p53 has been reported to mediate Bcl-2 downregulation
(69,70), it is possible that the reported Dox-mediated activation of free radicals and
toxic metabolites (63,64) could initially trigger the activation of p53, leading to a p53-
dependent downregulation of Bcl-2, and activation of a mitochondrially-regulated
caspase cascade. This scenario would be consistent with our experiments showing that
Dox-mediated loss of cell viability is blocked in cells expressing Bcl-2. Although p53
may regulate others genes than Bcl-2 involved in apoptosis (69,71), it is possible that
the effect of PFT-α in preventing apoptosis of HUVECs is mediated, at least in part, by
its ability to prevent Dox-mediated downregulation of Bcl-2 levels.
The involvement of p53 in the regulated expression of CD95 and apoptosis by
Dox, and the prevention of apoptosis by PFT-α, point to the potential use of p53
inhibitors for the treatment of the impaired wound healing and ovulation that occur as
side effects of Dox in treated patients. However the inhibition of p53 may have
undesirable effects. Not only might this block the potential beneficial effect of Dox on
proliferating endothelial cells, it could also interfere with apoptosis of tumor cells that
sense DNA damage in response to genotoxic stress and trigger an apoptotic response
upon accumulation of p53. A careful analysis by evaluation of the effects of Dox and
p53 inhibitors “in vivo” will be required to elucidate these important issues.
24
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Gibbons, G. H., and Dzau, V. J. (1994) N. Engl. J. Med. 330(20), 1431-8.
2. Karsan, A., and Harlan, J. M. (1996) J. Atheroscler. Thromb. 3(2), 75-80
3. Bjorkerud, S., and Bjorkerud, B. (1996) Am. J. Pathol. 149(2), 367-80.
4. Han, D. K., Haudenschild, C. C., Hong, M. K., Tinkle, B. T., Leon, M. B., and
Liau, G. (1995) Am. J. Pathol. 147(2), 267-77.
5. Bochaton-Piallat, M. L., Gabbiani, F., Redard, M., Desmouliere, A., and
Gabbiani, G. (1995) Am. J. Pathol. 146(5), 1059-64.
6. Isner, J. M., Kearney, M., Bortman, S., and Passeri, J. (1995) Circulation 91(11),
2703-11.
7. Dimmeler, S., Hermann, C., and Zeiher, A. M. (1998) Eur. Cytokine Netw. 9(4),
697-8.
8. Dimmeler, S., and Zeiher, A. M. (2000) Circ. Res. 87(6), 434-9.
9. Oehm, A., Behrmann, I., Falk, W., Pawlita, M., Maier, G., Klas, C., Li-Weber,
M., Richards, S., Dhein, J., Trauth, B. C., and et al. (1992) J. Biol. Chem.
267(15), 10709-15.
10. Itoh, N., Yonehara, S., Ishii, A., Yonehara, M., Mizushima, S., Sameshima, M.,
Hase, A., Seto, Y., and Nagata, S. (1991) Cell 66(2), 233-43.
11. Suda, T., Takahashi, T., Golstein, P., and Nagata, S. (1993) Cell 75(6), 1169-78.
12. Muzio, M., Chinnaiyan, A. M., Kischkel, F. C., O’Rourke, K., Shevchenko, A.,
Ni, J., Scaffidi, C., Bretz, J. D., Zhang, M., Gentz, R., Mann, M., Krammer, P.
H., Peter, M. E., and Dixit, V. M. (1996) Cell 85(6), 817-27.
13. Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E.
S., and Wang, X. (1997) Cell 91(4), 479-89.
25
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14. Barry, M. A., Behnke, C. A., and Eastman, A. (1990) Biochem. Pharmacol.
40(10), 2353-62.
15. Clarke, A. R., Purdie, C. A., Harrison, D. J., Morris, R. G., Bird, C. C., Hooper,
M. L., and Wyllie, A. H. (1993) Nature 362(6423), 849-52.
16. Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K. W., and Vogelstein, B. (1997)
Nature 389(6648), 300-5.
17. Muller, M., Wilder, S., Bannasch, D., Israeli, D., Lehlbach, K., Li-Weber, M.,
Friedman, S. L., Galle, P. R., Stremmel, W., Oren, M., and Krammer, P. H.
(1998) J. Exp. Med. 188(11), 2033-45.
18. Muller, M., Strand, S., Hug, H., Heinemann, E. M., Walczak, H., Hofmann, W.
J., Stremmel, W., Krammer, P. H., and Galle, P. R. (1997) J. Clin. Invest. 99(3),
403-13.
19. Richardson, B. C., Lalwani, N. D., Johnson, K. J., and Marks, R. M. (1994) Eur.
J. Immunol. 24(11), 2640-5.
20. Sata, M., and Walsh, K. (1998) J. Clin. Invest. 102(9), 1682-9.
21. Suhara, T., Fukuo, K., Sugimoto, T., Morimoto, S., Nakahashi, T., Hata, S.,
Shimizu, M., and Ogihara, T. (1998) J. Immunol. 160(8), 4042-7.
22. Sata, M., Suhara, T., and Walsh, K. (2000) Arterioscler. Thromb. Vasc. Biol.
20(2), 309-16.
23. Cardier, J. E., Schulte, T., Kammer, H., Kwak, J., and Cardier, M. (1999) Faseb
J. 13(14), 1950-60.
24. Sata, M., and Walsh, K. (1998) Nat. Med. 4(4), 415-20.
25. Aoudjit, F., and Vuori, K. (2001) J. Cell. Biol. 152(3), 633-43.
26. Munoz, C., Castellanos, M. C., Alfranca, A., Vara, A., Esteban, M. A., Redondo,
26
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

J. M., and de Landazuri, M. O. (1996) J. Immunol. 157(8), 3587-97.
27. el-Deiry, W. S., Kern, S. E., Pietenpol, J. A., Kinzler, K. W., and Vogelstein, B.
(1992) Nat. Genet. 1(1), 45-9.
28. Kalejta, R. F., Shenk, T., and Beavis, A. J. (1997) Cytometry 29(4), 286-91.
29. Yano, O., Kanellopoulos, J., Kieran, M., Le Bail, O., Israel, A., and Kourilsky,
P. (1987) Embo J. 6(11), 3317-24.
30. Armesilla, A. L., Lorenzo, E., Gomez del Arco, P., Martinez-Martinez, S.,
Alfranca, A., and Redondo, J. M. (1999) Mol. Cell. Biol.19(3), 2032-43.
31. Hernandez, G. L., Volpert, O. V., Iniguez, M. A., Lorenzo, E., Martinez-
Martinez, S., Grau, R., Fresno, M., and Redondo, J. M. (2001) J. Exp. Med.
193(5), 607-20.
32. Chan, H., Bartos, D. P., and Owen-Schaub, L. B. (1999) Mol. Cell. Biol. 19(3),
2098-108.
33. Petrak, D., Memon, S. A., Birrer, M. J., Ashwell, J. D., and Zacharchuk, C. M.
(1994) J. Immunol. 153(5), 2046-51.
34. Fulda, S., Strauss, G., Meyer, E., and Debatin, K. M. (2000) Blood 95(1), 301-8.
35. Fulda, S., Sieverts, H., Friesen, C., Herr, I., and Debatin, K. M. (1997) Cancer
Res. 57(17), 3823-9.
36. Ruiz-Ruiz, M. C., and Lopez-Rivas, A. (1999) Cell Death Differ. 6(3), 271-80.
37. Petak, I., Tillman, D. M., Harwood, F. G., Mihalik, R., and Houghton, J. A.
(2000) Cancer Res. 60(10), 2643-50.
38. Kotamraju, S., Konorev, E. A., Joseph, J., and Kalyanaraman, B. (2000) J. Biol.
Chem. 275(43), 33585-92.
39. Gamen, S., Anel, A., Lasierra, P., Alava, M. A., Martinez-Lorenzo, M. J.,
27
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Pineiro, A., and Naval, J. (1997) FEBS Lett. 417(3), 360-4.
40. Friesen, C., Herr, I., Krammer, P. H., and Debatin, K. M. (1996) Nat. Med. 2(5),
574-7.
41. Nakamura, T., Ueda, Y., Juan, Y., Katsuda, S., Takahashi, H., and Koh, E.
(2000) Circulation 102(5), 572-8.
42. Komarov, P. G., Komarova, E. A., Kondratov, R. V., Christov-Tselkov, K.,
Coon, J. S., Chernov, M. V., and Gudkov, A. V. (1999) Science 285(5434),
1733-7.
43. Maestre, N., Tritton, T. R., Laurent, G., and Jaffrezou, J. P. (2001) Cancer Res.
61(6), 2558-61.
44. Das, K. C., and White, C. W. (1997) J. Biol. Chem. 272(23), 14914-20.
45. Gnad, R., Kaina, B., and Fritz, G. (2001) Exp. Cell. Res. 264(2), 244-9.
46. Tietze, M. K., Wuestefeld, T., Paul, Y., Zender, L., Trautwein, C., Manns, M. P.,
and Kubicka, S. (2000) Cancer Gene Ther. 7(10), 1315-23.
47. Zheng, Y., Ouaaz, F., Bruzzo, P., Singh, V., Gerondakis, S., and Beg, A. A.
(2001) J. Immunol. 166(8), 4949-57.
48. Kuhnel, F., Zender, L., Paul, Y., Tietze, M. K., Trautwein, C., Manns, M., and
Kubicka, S. (2000) J. Biol. Chem. 275(9), 6421-7.
49. Fuchs, E. J., McKenna, K. A., and Bedi, A. (1997) Cancer Res. 57(13), 2550-4.
50. Lowe, S. W., Ruley, H. E., Jacks, T., and Housman, D. E. (1993) Cell 74(6),
957-67.
51. Maecker, H. L., Koumenis, C., and Giaccia, A. J. (2000) Cancer Res. 60(16),
4638-44.
52. Suda, T., Hashimoto, H., Tanaka, M., Ochi, T., and Nagata, S. (1997) J. Exp.
28
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Med. 186(12), 2045-50.
53. Izquierdo, M., Ruiz-Ruiz, M. C., and Lopez-Rivas, A. (1996) J. Immunol.
157(1), 21-8.
54. Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer, D. D. (1997)
Science 275(5303), 1132-6.
55. Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., Peng, T. I.,
Jones, D. P., and Wang, X. (1997) Science 275(5303), 1129-32.
56. Villunger, A., Egle, A., Kos, M., Hartmann, B. L., Geley, S., Kofler, R., and
Greil, R. (1997) Cancer Res. 57(16), 3331-4.
57. Winthrop, M. D., DeNardo, S. J., Muenzer, J. T., Chi, S. G., and Gumerlock, P.
H. (1997) Cancer 80(12 Suppl), 2529-37.
58. Sarvazyan, N. (1996) Am. J. Physiol. 271(5 Pt 2), H2079-85.
59. Singal, P. K., and Iliskovic, N. (1998) N. Engl. J. Med. 339(13), 900-5.
60. Sasaki, T., Holeyfield, K. C., and Uitto, J. (1987) J. Clin. Invest. 80(6), 1735-41.
61. Llesuy, S. F., and Arnaiz, S. L. (1990) Toxicology 63(2), 187-98.
62. Okuda, S., Oh, Y., Tsuruda, H., Onoyama, K., Fujimi, S., and Fujishima, M.
(1986) Kidney Int. 29(2), 502-10.
63. Konorev, E. A., Kennedy, M. C., and Kalyanaraman, B. (1999) Arch. Biochem.
Biophys. 368(2), 421-8.
64. Sinha, B. K., Katki, A. G., Batist, G., Cowan, K. H., and Myers, C. E. (1987)
Biochem. Pharmacol. 36(6), 793-6.
65. Hockenbery, D. M., Oltvai, Z. N., Yin, X. M., Milliman, C. L., and Korsmeyer,
S. J. (1993) Cell 75(2), 241-51.
66. Kane, D. J., Sarafian, T. A., Anton, R., Hahn, H., Gralla, E. B., Valentine, J. S.,
29
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Ord, T., and Bredesen, D. E. (1993) Science 262(5137), 1274-7.
67. Oltvai, Z. N., Milliman, C. L., and Korsmeyer, S. J. (1993) Cell 74(4), 609-19.
68. Chao, D. T., and Korsmeyer, S. J. (1998) Annu. Rev. Immunol. 16, 395-419
69. Miyashita, T., Krajewski, S., Krajewska, M., Wang, H. G., Lin, H. K.,
Liebermann, D. A., Hoffman, B., and Reed, J. C. (1994) Oncogene 9(6), 1799-
805.
70. Wu, Y., Mehew, J. W., Heckman, C. A., Arcinas, M., and Boxer, L. M. (2001)
Oncogene 20(2), 240-51.
71. Raffo, A. J., Kim, A. L., and Fine, R. L. (2000) Oncogene 19(54), 6216-28.
30
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
Figure 1. Induction of apoptosis in Dox-treated subconfluent endothelial cells. (A)
HUVECs were incubated with 500 ng/ml Dox, and apoptosis was determined by FACS
analysis after propidium iodide-staining of nuclei at the indicated times after treatment.
(B) The effect of cell confluence on the sensitivity of HUVEC to Dox was analyzed by
cell cycle analysis as above, using cells plated at the indicated confluence and then
treated with 500 ng/ml Dox for 30 h or 48 h. Results are expressed as the percentage of
cells displaying a sub-G1 DNA content. The basal level of apoptosis (Control) was
monitored in parallel cultures of untreated HUVECs after 48 h. The data are
representative of three independent experiments, and the significance level (χ2) was
p < 0,01 in cells at either 60% or 40% confluence when comparing Dox-treated cells
for either 30 or 48 h versus non-treated cells at the same confluence and treatment
times, and in cells at 100% confluence when comparing Dox-treated cells for 48 h with
non treated cells.
Figure 2. Dox induces CD95 gene expression in HUVECs. (A) CD95 cell surface
expression was determined by flow cytometry. HUVECs were either incubated with
(solid black line) or without Dox (500 ng/ml) for 48 h. Cells were labeled with anti-
CD95 antibody and subjected to FACS analysis. The dotted line shows the baseline
levels displayed by the secondary antibody. (B) The levels of CD95 mRNA were
analyzed by RT-PCR using mRNA from HUVECs treated with 500 ng/ml Dox for the
times indicated. RT-PCR products of β-actin were used as control of RNA input, and
the CD95 RT-PCR product from resting Jurkat JHM1 cells was amplified as a positive
31
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

control. (C) HUVECs were transfected for 9 h with luciferase reporter plasmid driven
by a 391 bp fragment of the CD95 promoter (pCD95 391 Luc), or this construct
containing an intronic p53-reponsive enhancer element upstream of the promoter region
(pI-CD95 391 Luc). Twenty-four hours later, cells were treated with Dox for an
additional 14 h period. Luciferase activity is expressed as the fold induction over the
baseline levels of the transfected untreated control. Data are expressed as the mean and
± SE of three different experiments. *p < 0,01 (Student´s t test) when comparing pI-
CD95 391 Luc transfected cells with pCD95 391 Luc transfected cells after Dox
treatment.
Figure 3. Dox induces p53 protein expression and p53-DNA binding activity in
HUVECs. (A) Cells were treated either with or without Dox (500 ng/ml) for the
indicated times. Ten µg of total cell lysate were analyzed by Western Blot probed with
an anti-p53 antibody, or with an anti-α-tubulin antibody as a control for protein
loading. (B) Nuclear extracts from HUVECs stimulated for 1 h with 500 ng/ml Dox
were analyzed by EMSA with a probe containing the p53 site of the human CD95
intronic enhancer. EMSAs were performed in the presence or absence of the anti-p53
antiserum PAb421. The specific DNA-p53 complex is indicated by the arrow. A 30-
fold molar excess of unlabeled p53 oligonucleotide was added to the binding reaction in
order to determine the specificity of binding. A representative result of three
independent experiments is presented.
Figure 4. p53 is required for the Dox-mediated up-regulation of CD95 expression in
HUVECs. (A) HUVECs were pretreated with or without 30 µM PFT-α for 24 h, and
subsequently treated with or without 500 ng/ml Dox for an additional 48 h. Cell surface
expression of CD95 was determined by flow cytometry. As a control, the effect of the
32
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

same dose of inhibitor was tested on the cell-surface expression of I-CAM in HUVECs
stimulated with TNF-α (50 ng/ml) for 24 h. In all instances, fresh PFT-α was added
every 24 h. (B) The inhibitory effect of PFT-α was confirmed by Western Blot analysis
using anti-p53 and anti-α↑tubulin antibodies. Whole cell extracts from aliquots of cells
used in (A) were analyzed. Results representative of three independent experiments are
presented.
Figure 5. p53 is involved in the activation of CD95 gene promoter by Dox. (A)
HUVECs were transfected for 9 h with the pCD95 391 Luc or pI-CD95 391 Luc
reporter plasmids, (the latter plasmid containing an intronic p53-reponsive enhancer
element from intron 1 of the CD95 gene). Twenty four hours later, cells were either left
or pretreated with 30 µM PFT-α for 24 h, and left untreated or treated with 500 ng/ml
Dox for an additional 14 h period, after which the luciferase activity was determined.
The inhibitory effect of PFT-α was confirmed by Western Blot analysis using anti-p53
and whole cell extracts from aliquots of cells used in the transfection (A, inset). (B)
HUVECs were transfected for 9 h either with the pCD95 391 Luc or the pI-CD95 391
Luc reporter plasmids, or with the pmI-CD95 391 Luc plasmid containing the mutated
p53-intronic-binding site. Twenty four hours later, cells were left untreated or treated
with 500 ng/ml Dox for an additional 14 h, and then the luciferase activity was
determined. Data show one representative experiment of three performed, and are
expressed as the mean and ± SE of three different measurements. **p < 0,005
(Student´s t test) in A, and *p < 0,01 in B, for Dox-treated cells versus non-treated
cells.
Figure 6. Effects of Dox on NFκB–DNA binding activity and NFκB transactivation. (A)
Nuclear extracts from HUVECs, stimulated for 1 h with 500 ng/ml Dox or with 50
33
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ng/ml of TNF-α, were analyzed by EMSA with a probe containing the NFκB site of the
CD95 human promoter (nucleotides – 306 to - 278). Nuclear extracts from Dox- and
TNF-α-treated cells were incubated with the anti-p65 antiserum 1226 or with the anti-
p50 antiserum 1141 to detect NFκB subunits. The specific DNA-NFκB complex
containing p65 or p50 proteins is indicated by the arrow. (B) HUVECs were transfected
with the p53 reporter plasmid (pG13 Luc) or the NFκB luciferase reporter plasmid by
the calcium phosphate method. Twelve hours after transfection cells were stimulated
with TNF-α (50 ng/ml) or Dox (500 ng/ml) for an additional 12 h. The results are
expressed as the fold induction over the relative luciferase units (RLU) displayed by the
corresponding unstimulated transfected cells. Results of one representative experiment
of three performed are shown. Data are expressed as the mean ± SE of three
determinations. *p < 0.01 versus control experiment using kB Luc
promoter. **p < 0,001 versus control experiment using pG13 Luc promoter (Student´s t).
Figure 7. Inhibition of p53 accumulation prevents Dox-induced apoptosis in HUVECs.
Cells pretreated with or without PFT-α (30 µM) for 24 h were then incubated in the
presence or absence of 500 ng/ml Dox for an additional 48 h period. Apoptosis was
determined by cell cycle analysis of propidium iodide-stained nuclei. The results are
presented as the percentage of cells undergoing apoptotis (cells displaying a sub-G1
DNA content) The inhibition of p53 by PFT-α was confirmed by Western Blot analysis
using anti-p53 and anti- α↑ tubulin antibodies, and whole cell extracts from aliquots of
cells used for FACS analysis were analyzed. Data are expressed as the mean and ± SE
of three different experiments. *p < 0,01 (Student´s t test) when comparing Dox-
treated with non-treated cells.
Figure 8. Dox-induced apoptosis in HUVEC is not dependent on CD95/CD95L
34
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

interaction. (A) FACS analysis of CD95L expression in Dox-treated HUVECs. Cells
were preincubated with or without 10 µM of the matrix metalloprotease inhibitor
KB8301 for 30 min, and were then left untreated or treated with 500 ng/ml Dox for 36
h. To avoid the effects of potential KB8301 degradation, the inhibitor was re-added
every 12 h. Cells were labeled with anti-CD95L antibody and subjected to FACS
analysis. (B) Effects of CD95 and CD95L antibodies on Dox-mediated apoptosis.
HUVECs treated or untreated (-) with Dox (500 ng/ml) for 24 h were left untreated or
were reacted with 1 µg/ml of the DX2 antagonistic anti-CD95 mAb, 1µg/ml of the
NOK-1 antagonistic anti-CD95L mAb, or with 500 ng/ml of the CH11 agonistic anti-
CD95 antibody for an additional 24 h. Cell cycle analysis and determination of the Sub-
G1 apoptotic cells was monitored after 48 h of Dox treatment. The blocking activity of
the antagonist antibodies was confirmed in Jurkat JMH1 cells treated with Carbachol
(500 µM) or CH11 (20 ng/ml) for 10 h. Data are expressed as the mean and ± SE of
three different experiments. *p < 0,01 and **p < 0,001 (Student´s t test) when
comparing the corresponding situation with the indicated control.
Figure 9. Inhibition of Dox-induced apoptosis by z-VAD-FMK. HUVECs were
pretreated or with or without 100 µM of the caspase inhibitor z-VAD-FMK for 2 h,
and subsequently treated with 500 ng/ml Dox for 30 h. Apoptosis was measured by
FACS analysis of the cell cycle. Data are expressed as the mean and ± SE of three
different experiments.*p < 0,02 (Student´s t test) when comparing Dox-treated with
nontreated cells.
Figure 10. Dox induces activation of a mitochondrially -operated apoptotic pathway in
HUVECs. Cells were treated with Dox 500 ng/ml for 30 or 36 h and whole extracts
35
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(60µg) (A, C, D) or cytosolic extracts (B) were analyzed by Western blot with antibodies
raised against PARP, caspase-3, Bcl-2, cytochrome c, and caspase-9, as indicated. In
(D), HUVECs pretreated with 30 µM of PFT-α for 24 h were treated with Dox (500
ng/ml) for 36 additional hours, and 30 µM of fresh PFT-α was re-added every 12 h.
The proteolytic fragment of PARP (85 kDa), the intermediate cleaved forms of
caspase-9 (37 kDa), and caspase-3 (17 and 21 kDa) are indicated by arrows. Anti-α-
tubulin was probed to control for protein loading. A representative experiment of three
performed is shown.
.
36
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ACKNOWLEDGMENTS
We are very grateful to Dr. S. Bartlett for critical reading of the manuscript and editorial
assistance. We also thank to Drs Edmundo Fernández, Yolanda Rodríguez, Jesús
Vazquez, Miguel Campanero, Manuel Izquierdo and Emilia Mira for providing reagents
and advice. This work was supported by a shared Grant FEDER 1FD97-0514-CO2
from MEC-DGES and European Community (to JMR and ALR) and grant PM99-0116
from Ministerio de Educación y Cultura (MEC-DGES) of Spain to JMR. EL was
supported by a FPI fellowship from the MEC-DGES, and GH by Grant 8.3/24 1/2000
from the Comunidad Autónoma de Madrid. The Centro de Biología Molecular S.O. is
supported by a grant from the Fundación Ramón Areces.
37
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from







Rodríguez, Abelardo López-Rivas and Juan Miguel RedondoElisa Lorenzo, Carmen Ruiz-Ruiz, Antonio Jesús Quesada, Gabriela Hernández, Antonio
endothelial cells through a p53-dependent mechanismDoxorubicin induces apoptosis and CD95 gene expression in human primary
published online January 4, 2002J. Biol. Chem.
10.1074/jbc.M107442200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from