regulatory framework for drug approvals v7 - sfda€¦ · each applicant will have a user id and a...
TRANSCRIPT

Page 1 of 96 v.7 (14/7/08)
Saudi Food & Drug Authority
Regulatory Framework for Drug Approvals
Pre-Market Assessment July 2008

DRAFT
Page 2 of 96 v.7 (14/7/08)
Table of Contents
Forward 3
Submission 4
Market Authorization Application (MAA) - Generics 7
MAA – New Chemical Entity 9
MAA – Biologicals 11
MAA – Orphan drugs 13
MAA – Veterinary drugs 15
Herbal Products 17
Renewal of MAA 20
Variation 21
Clinical Trials 24
Table of all Performance Targets 27
Appendix A: Glossary 28
Appendix B: Acronyms 30
Appendix C: Application Forms 31
Table of Figures
Figure 1: Flow chart of product assignment 5
Figure 2: Application form and product file submission process 6
Figure 3: MAA – Generics, NCE, Biologicals, Orphan, Veterinary and Herbal
19
Figure 4: Variation Type I & II 23
Figure 5: Clinical Trial 26

DRAFT
Page 3 of 96 v.7 (14/7/08)
Forward
The Drug Sector in the Saudi Food & Drug Authority (SFDA) has developed this document, “Regulatory Framework for Drug Approval”, to provide assistance for stakeholders on how to submit applications for various types of drug products. This document is an administrative instrument that outlines the requirements of various types of applications to be filled out and submitted to the SFDA.
Besides the Market Authorization Application (MAA) of various types of drug products, it also describes the renewal of MAA, variations and clinical trials applications. Various application forms and procedures for processing applications to marketing the product in Saudi Arabia are also included in this document.
It is important to note that the SFDA reserves the right to request information, material or defined conditions not specifically described in this document, in order to allow the administration to adequately assess the safety, efficacy and quality of drug products. The SFDA is committed to ensuring that such requests are justifiable and decisions are clearly documented.
It is expected that relevant guidances and standard operating procedures for the data requirement will follow. Therefore, this document should be read in conjunction with the other relevant and applicable guidance documents. A copy of this document as well as the SFDA's application forms can be found on our website:
http://www.sfda.gov.sa/En/Drug
The SFDA is fully committed to an orderly process for the review and authorization of pharmaceutical products, and we are working to develop procedures to implement those aspects of the initiative. We are also committed to assuring that stakeholders remain fully informed of our progress as we implement the initiative.

DRAFT
Page 4 of 96 v.7 (14/7/08)
All applicants should apply the Application Form ONLINE, then submit the Product File as hardcopy and electronically (CD or DVD).
A. Application Forms:
Steps in which each applicant have to follow in order to submit the application form.
1. Each applicant will have a user ID and a password
2. Go to the SFDA website
3. Log-in to apply
4. Choose the appropriate application form
5. Complete the application form:
• The application form can be saved partially as the applicant may complete
it in several steps, then submitting it once completed
6. The applicant then have to pay the right fee in order to book an appointment
through (SADAD Payment System)
• The applicant then can complete the application, submit it and schedule an
appointment
7. Schedule an appointment:
• The earliest appointment can be after 1 week up to 2 months. The
applicant can reschedule one week before the appointment. An automatic
reminder e-mail will be generated two weeks before the appointment and
sent to the applicant
• A reference number will be generated
8. On the appointed day, the applicant will handle the product file (hardcopy and
soft copy) and product samples with the reference number
9. The Director (Product Licensing Department) will assign a product manager according to the scheduling system to arrange the receiving of the product file (hardcopy and soft copy) and product samples for verification
10. The Drug Sector staff will verify the followings:
• The application form
• The product file (hardcopy and soft copy):

DRAFT
Page 5 of 96 v.7 (14/7/08)
i. If verified, generate a letter of acknowledgment with a
reference number
ii. If not, return with reason and write it down in the Drug Sector
database.
iii. The applicant will have a period of 60 days to complete the
requirements, otherwise the application will be automatically
deleted from the database
• The product samples
B. Product File (Submission Data):
The process of filing of the submission (dossier) is as follows:
1. Hardcopy of dossier, and
2. Electronic copy (CD or DVD)
• Both should be in the Common Technical Document (CTD) format
3. Product samples for assessment and testing
Figure 1: Flow chart of product assignment

DRAFT
Page 6 of 96 v.7 (14/7/08)
Figure 2: Flow chart of application form and product file submission process

DRAFT
Page 7 of 96 v.7 (14/7/08)
Market Authorization Application (MAA) – Generics:
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation/Validation:
1. The file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment:
1. The product file will be distributed to TWO groups: quality and safety. 2. Quality assessment: this will be performed by a review team. Once a review
is completed and a report is created, it will be forwarded to the product manager.
3. Safety assessment: this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
4. If a clarification of any group is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
5. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
6. Performance target: 120 days
C. Pricing
1. The Pricing Committee will decide the price according to the formula. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The final price will be written in a report and forwarded to the product
manager. 4. The company has the right to appeal in 30 days. 5. Performance target: 60 days
D. Testing
1. Samples received by SFDA headquarters will be sent to the Central lab.

DRAFT
Page 8 of 96 v.7 (14/7/08)
2. If more information or clarification is required, a letter is forwarded through the product manager to the company.
3. The central lab will generate a report either comply or not comply. 4. The results will be written in a report and forwarded to the product manager. 5. Performance target: 90 days.
Notes: • Non-complied products will be discussed with the company, and new
samples should be provided. Also, the performance target will stop. • The 1st batch imported after approval will be tested. Results will be in
30 days. The company should not distribute the product during this period. After 30 days, the company can start distributing but when the results are not complied with regulations, SFDA will ask the marketing authorization holder to recall the batch.
E. Product Licensing
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”.
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 day.
Total performance target = 165 days (≈ 24 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resumed after receiving the response.

DRAFT
Page 9 of 96 v.7 (14/7/08)
MAA – New Chemical Entity (NCE)
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation/Validation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and /or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment:
1. The product file will be distributed to THREE groups: quality, safety and efficacy
2. Quality group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
3. Safety group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
4. Efficacy group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
5. If a clarification of any group is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
6. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
7. Performance target: 245 days
C. Pricing:
1. The Pricing Committee will decide the price according to the formula. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The Pricing Committee will negotiate the decided price with the company. 4. The final price will be written in a report and forwarded to the product
manager. 5. Performance target: 60 days

DRAFT
Page 10 of 96 v.7 (14/7/08)
D. Testing:
1. Samples received by SFDA headquarters will be sent to the Central laboratory. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The central lab will generate a report either comply or not comply. 4. The results will be written in a report and forwarded to the product manager. 5. Performance target: 90 days Notes:
• Non-complied products will be discussed with the company, and new samples should be provided. Also, the performance target will stop.
• The 1st batch imported after approval will be tested. Results will be in 30 days. The company should not distribute the product during this period. After 30 days, the company can start distributing but when the results are not complied with regulations, SFDA will ask the marketing authorization holder to recall the batch.
E. Product Licensing
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”.
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 day.
Total performance target = 295 days (≈ 42 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.

DRAFT
Page 11 of 96 v.7 (14/7/08)
MAA – Biologicals:
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation/Validation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment
1. The product file will be distributed to THREE groups: quality, safety and efficacy
2. Quality group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
3. Safety group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
4. Efficacy group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
5. If a clarification of any group is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
6. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
7. Performance target: 245 days
C. Pricing:
1. The Pricing Committee will decide the price according to the formula. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The Pricing Committee will negotiate the decided price with the company. 4. The final price will be written in a report and forwarded to the product
manager. 5. Performance target: 60 days

DRAFT
Page 12 of 96 v.7 (14/7/08)
D. Testing
1. Samples received by SFDA headquarters will be sent to the Central laboratory. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The central lab will generate a report either comply or not comply. 4. The results will be written in a report and forwarded to the product manager. 5. Performance target: 90 days Notes:
• Non-complied products will be discussed with the company, and new samples should be provided. Also, the performance target will stop.
• The batch/lot release in accordance with the guidance/policy document
E. Product Licensing
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”.
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 day.
Total performance target = 295 days (≈ 42 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.

DRAFT
Page 13 of 96 v.7 (14/7/08)
MAA – Orphan drugs:
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation/Validation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 5 days
B. Assessment
1. The product file will be distributed to THREE groups: quality, safety and efficacy
2. Quality group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
3. Safety group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
4. Efficacy group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
5. If a clarification is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
6. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
7. Performance target: 60 days
C. Pricing:
1. The Pricing Committee will decide the price according to the formula. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The Pricing Committee will negotiate the decided price with the company. 4. The final price will be written in a report and forwarded to the product
manager. 5. Performance target: 30 days

DRAFT
Page 14 of 96 v.7 (14/7/08)
D. Testing
1. Samples received by SFDA headquarters will be sent to the Central laboratory. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The central lab will generate a report either comply or not comply. 4. The result will be written in a report and forwarded to the product manager. 5. Performance target: 60 days Notes:
• Non-complied products will be discussed with the company, and new samples should be provided. Also, the performance target will stop.
• The 1st batch imported after approval will be tested. Results will be in 30 days. The company should not distribute the product during this period. After 30 days, the company can start distributing but when the results are not complied with regulations, SFDA will ask the marketing authorization holder to recall the batch.
E. Product Licensing
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”:
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 days
Total performance target = 90 days (≈ 13 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.

DRAFT
Page 15 of 96 v.7 (14/7/08)
MAA – Veterinary drugs:
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment:
Important note:
o Vet generic will follow Human generic o Vet NCE will follow Human NCE
1. The product file will be distributed to THREE groups: quality, safety and efficacy (only NCE)
2. Quality group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
3. Safety group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
4. Efficacy group this will be performed by a review team. Once a review is completed and a report is created, it will be forwarded to the product manager.
5. If a clarification is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
6. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
7. Performance target: 150 days
C. Pricing:
1. The Pricing Committee will decide the price according to the formula. 2. If more information or clarification is required, a letter is forwarded through
the product manager to the company. 3. The final price will be written in a report and forwarded to the product
manager. 4. The company have the right to appeal in 30 days

DRAFT
Page 16 of 96 v.7 (14/7/08)
5. Performance target: 90 days
D. Testing:
1. Samples received by SFDA headquarters will be sent to the Central laboratory. 2. If more information or clarification is required, a letter is forwarded through
the administrator to the company. 3. The central lab will generate a report either comply or not comply. 4. The result will be written in a report and forwarded to the product manager. 5. Performance target: 90 days Notes:
• Non-complied products will be discussed with the company, and new samples should be provided. Also, the performance target will stop.
• The 1st batch imported after approval will be tested. Results will be in 30 days. The company should not distribute the product during this period. After 30 days, the company can start distributing but when the results are not complied with regulations, SFDA will ask the marketing authorization holder to recall the batch.
E. Product Licensing
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”:
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 days
Total performance target = 195 days (≈ 28 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response

DRAFT
Page 17 of 96 v.7 (14/7/08)
MAA – Herbal Products:
The Market Authorization Application will be subjected to the following processes:
A. Pre-evaluation/Validation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment:
1. The product file will be distributed to TWO groups: quality, and safety. 2. Quality group this will be performed by a review team. Once a review is
completed and a report is created, it will be forwarded to the product manager. 3. Safety group this will be performed by a review team. Once a review is
completed and a report is created, it will be forwarded to the product manager. 4. If a clarification is required, an “Assessment Inquiries” will group the
questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
5. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of the Product Licensing department.
6. Performance target: 110 days
C. Testing:
1. Samples received by SFDA headquarters will be sent to the Central laboratory. 2. If more information or clarification is required, a letter is forwarded through
the administrator to the company. 3. The central lab will generate a report either comply or not comply. 4. The results will be written in a report and forwarded to the product manager. 5. Performance target: 90 days Notes:
• Non-complied products will be discussed with the company, and new samples should be provided. Also, the performance target will stop.
• The 1st batch imported after approval will be tested. Results will be in 30 days. The company should not distribute the product during this period. After 30 days, the company can start distributing but when the

DRAFT
Page 18 of 96 v.7 (14/7/08)
results are not complied with regulations, SFDA will ask the marketing authorization holder to recall the batch.
D. Product Licensing:
1. Director will receive all the reports from the product manager (assessment, pricing and testing in addition to other requirements: GMP, inspection, company registration…etc) and forward them to the secretary of the “Registration Committee”:
a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings. c. The Registration Committee will either recommend approval, rejection
or further information are needed d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2. The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
a. The Director of Product Licensing department will issue the market authorization.
b. Performance target: 5 days
Total performance target = 155 working days (≈ 22 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.

Page 19 of 96 v.7 (14/7/08)
Figure 3: Schematic figure showing the different levels for getting a marketing authorization for
Generics, NCE, Biologicals, Orphan, Veterinary and Herbal products

Page 20 of 96 v.7 (14/7/08)
Renewal of Marketing Authorizations:
First renewal of Marketing Authorization with the SFDA, applicant has to submit a FULL product file.
Further renewal requirements will be determined in future publications.
The Renewal of Marketing Authorization will be subjected to the following processes:
1. The product manager will forward the renewal application to the Director. 2. The Director will review and forward the application to the Drug VP. 3. Drug VP will approve the renewal and forward it to the Director who will
issue the renewal authorization document. 4. Performance target: 30 days
Total performance target = 30 days (≈ 4 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.

Page 21 of 96 v.7 (14/7/08)
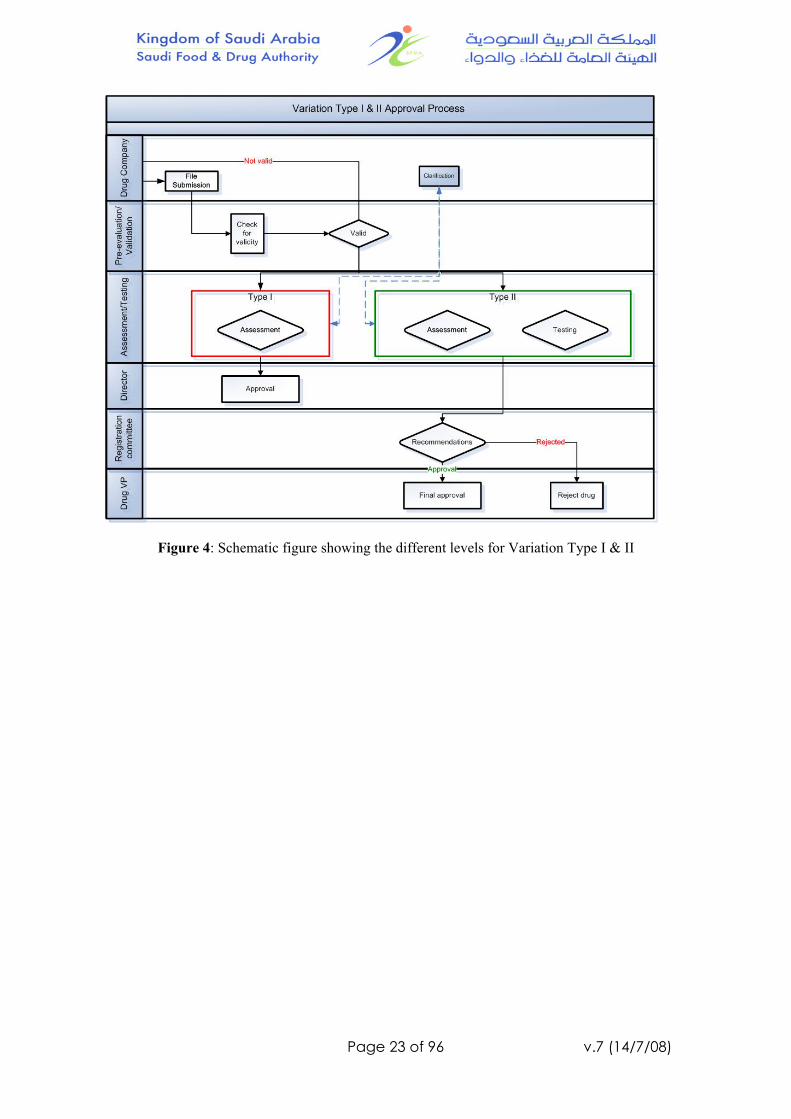
Variation Type I (Notifiable change) and Type II (Supplement to MA):
A. Pre-evaluation/Validation:
1. The product file will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The completed file with the product samples will go to the next step – assessment.
b. If any information is missing or incorrect, the company will be notified in writing. The company will be given an opportunity to complete the file within 60 days. If no response, the file will be properly destroyed and rejected.
2. Performance target: 10 days
B. Assessment
The product file will be distributed according to the variation type: 1. If type I (Notifiable change):
a. Product manager will receive the product file and assign a staff or more as needed.
b. The assigned staff will review/study the application/product file. c. Then, print the letter of acceptance or rejection and forward it to
the product manager, then the Director. d. Performance target: 90 days
2. If type II (Supplement to MA): a. Product manager will receive the product file and distribute it to
assessment and testing – as required. b. If more information or clarification is required, an “Assessment
Inquiries” will group the questions and will be forwarded to the company through the product manager.
c. The reports (recommendation for approval or rejection) collected by the product manager will be forwarded to the Director of Product Licensing department.
d. Performance target: 120 days
C. Product Licensing:
1. For type I (Notifiable change): a. The Director will approve the letter b. Notify the company. c. Performance target: 5 days
2. For type II (Supplement to MA): 1) Director will receive all the reports from the product manager and forward
them to the secretary of the “Registration Committee”: a. Secretary of the Registration Committee will set a meeting. b. The product manager will present the final findings.

DRAFT
Page 22 of 96 v.7 (14/7/08)
c. The Registration Committee will either recommend approval, rejection or further information are needed
d. The minutes goes to the Director of Product Licensing department e. Performance target: 30 days
2) The Director of Product Licensing department will forward the minutes to the Drug VP for approval.
A. The Director of Product Licensing department will issue the approval letter of changes.
B. Performance target: 5 days
Total performance target (Type I) = 105 days (15 weeks) Total performance target (Type II) = 165 days (≈ 24 weeks)
Note: the performance target in any step will STOP if a clarification or information is needed from the company, and resume after receiving the response.
Page 23 of 96 v.7 (14/7/08)
Figure 4: Schematic figure showing the different levels for Variation Type I & II

Page 24 of 96 v.7 (14/7/08)
Clinical Trials Approval:
The approvals of clinical trials have two sequential steps. The first step is protocol approval. The second step is the product assessment. However, in case if the submitted drug is already registered in KSA it will need only protocol approval.
A. Pre-evaluation/Validation:
1. The clinical trial protocol will be pre-evaluated/validated to ensure that all information provided are according to the requirements and/or guidelines:
a. The complete protocol file will go to the next step – assessment. b. If any information is missing or incorrect, the applicant will be
notified in writing. The applicant will be given an opportunity to complete the file within 60 days. If no response, the file will be rejected.
2. Performance target: 10 days
B. Assessment:
I. Clinical Trial Protocol
1. The clinical trial protocol will be assessed in the Clinical Trials Unit. 2. If a clarification is required, an “Assessment Inquiries” will group the
questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.
3. The recommendation for approval or rejection will be forwarded to the product manager.
4. Performance target: 25 days
II. Product assessment:
1. If the clinical trial protocol is approved, then the applicant has to submit the product file with samples – if the drug is not registered in KSA.
2. The product file will be distributed to quality and safety groups. 3. Quality group this will be performed by a review team. Once a review is
completed and a report is created, it will be forwarded to the product manager.
4. Safety group this will be performed by a review team. Once a review is completed, it will be forwarded to the product manager. The review includes the rational of the trial, protocol & safety of the subjects.
5. If a clarification is required, an “Assessment Inquiries” will group the questions and will be forwarded to the company through the product manager. The response of the “Assessment Inquiries” should be received within 30 days. If no response, the application will be rejected.

DRAFT
Page 25 of 96 v.7 (14/7/08)
6. The recommendation for approval or rejection will be forwarded to the product manager.
7. Performance target: 120 days
C. Testing
If required by SFDA, the product samples will be tested.
D. Product Licensing:
1. Director will receive all the reports from the product manager and forward them to the Drug VP.
2. Drug VP will approve the reports and forward it to the Director who will issue the clinical trial license.
a. Performance target: 10 days
Total performance target (protocol approval) = 45 days (≈ 7 weeks)
Total performance target (product assessment) = 140 days (20 weeks)

Page 26 of 96 v.7 (14/7/08)
Figure 6: Schematic figure showing the different levels for approving Clinical Trials

Page 27 of 96 v.7 (14/7/08)
Table of all Performance Targets
Process Total Performance Target
Marketing Authorization Application for Generics 165 days
Marketing Authorization Application for NCE 295 days
Marketing Authorization Application for Biologicals 295 days
Marketing Authorization Application for Orphan Product
90 days
Marketing Authorization Application for Veterinary drugs
195 days
Marketing Authorization Application for Herbal Products
155 days
Renewal of Marketing Authorization 120 days
Variation to a Marketing Authorization Type I: Notifiable Change
135 days
Variation to a Marketing Authorization Type II: Supplemental New Drug Application
165 days
Clinical Trial approval (protocol approval) 45 days
Clinical Trial approval (product assessment) 140 days

DRAFT
Page 28 of 96 v.7 (14/7/08)
Appendix A: Glossary
Biologicals A term used to group the biosimilars, biotech and vaccines.
Biosimilars
Therapeutic proteins produced by means of recombinant DNA technologies following the footsteps of an innovator product after the expiration of the innovator’s patent. They are complex and heterogeneous in their nature; hence they are not considered generics, but as closely similar to the innovator’s drug as possible.
Biotech
Products made via a set of biological techniques developed through basic research and now applied to research and product development. In particular, the industrial use of recombinant DNA, cell fusion and new bio-processing techniques.
Clinical trial The evaluation of a product in studies involving human subjects.
Common Technical Document
An international harmonized format for submissions for approval of pharmaceuticals for human use. The CTD provides a standardization of the presentation of the content.
Drug An article intended for use in the diagnosis, cure mitigation, treatment, or prevention of disease and which is intended to affect the structure or function of the body.
Generic drug Approved drugs that are no longer protected by patents and are approved for marketing by companies without the need for clinical trials; generic drugs are bioequivalent to the original approved drugs.
Herbal product A drug or preparation made from a plant or plants and used to prevent and treat diseases and ailments or to promote health and healing.
New Chemical Entity
A drug that contains a new active moiety that has been not approved by any regulatory authority.
Orphan drugs A drug that is prohibitively expensive and/or unprofitable to develop under normal circumstances.
Renewal of marketing authorization
A process of renewing the marketing authorization license every five years.
SADAD SADAD links between the commercial sector and the local banks; it offers the ability to collect its Customer payment electronically through all the banking channels in the kingdom around the clock.
Vaccines Preparations that contain antigenic substances capable of inducing a specific and active immunity against the infecting agent or the toxin or the antigen produced by it.
Validation The process of checking if documents satisfies a certain criterion

DRAFT
Page 29 of 96 v.7 (14/7/08)
Variation A process of informing the authority of any minor or major changes in the drug product.
Verification The process of checking that all required documents are existing in the file during the submission.
Veterinary drug
Any substance or mixture of substances manufactured, sold or represented for use in: • the diagnosis, treatment, mitigation or prevention of a disease,
disorder, abnormal physical state, or its symptoms, in animals, • restoring, correcting or modifying organic functions in
animals

DRAFT
Page 30 of 96 v.7 (14/7/08)
Appendix B: Acronyms
CTD Common Technical Document
Drug VP SFDA’s Drug Vice President
MAA Marketing Authorization Application
NCE New Chemical Entity
SFDA Saudi Food and Drug Authority

DRAFT
Page 31 of 96 v.7 (14/7/08)
Appendix C: Application Forms
Application form Page
Marketing Authorization Application of Medicinal Product for Human Use: New Drugs, Generic Drugs and Biologicals
32
Marketing Authorization Application of Orphan Product 46
Marketing Authorization Application of Medicinal Product for Veterinary Use
58
Marketing Authorization Application of Herbal Product 72
Application for Variation to a Marketing Authorization
Type I: Notifiable Change
80
Application for Variation to a Marketing Authorization
Type II: Supplemental New Drug Application
84
Clinical Trial Application Form 88

DRAFT
Page 32 of 96 v.7 (14/7/08)
אאא
א،א،אא Marketing Authorization Application of Medicinal Product
for Human Use
New Drugs, Generic Drugs and Biologicals
New Renewal
Registration No:
אאא،אא
אאאK
אK
The application form is to be used to apply for a marketing authorization of a medicinal product for human use submitted to Saudi Food & Drug Authority (SFDA).
A separate application form is needed for each strength and drug dosage form.
DRAFT
Page 33 of 96 v.7 (14/7/08)
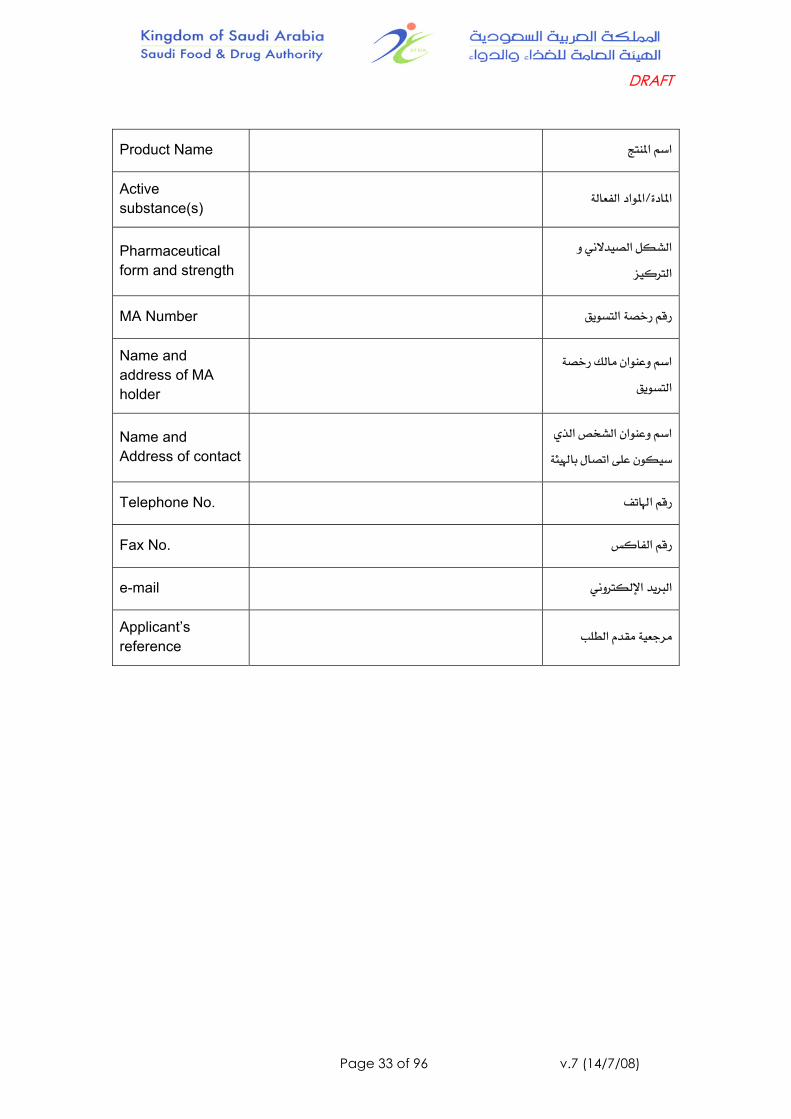
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אאא
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference א

DRAFT
Page 34 of 96 v.7 (14/7/08)
1. Type of Application
The following sections should be completed where appropriate.
1.1 This application concerns (Mandatory scope):
Annex (1) (New drugs including NCE)
Annex (2) (Biologicals including Biotech, blood and vaccines)
Annex (3) (Generics)
1.2 This application is submitted in accordance with the following:
1.2.1 New Drug Application (NDA)
New Drug
New Chemical Entity (NCE)
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
1.2.2 Generic Application
Generic Drug
Reference Product
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):

DRAFT
Page 35 of 96 v.7 (14/7/08)
1.2.3 Biologicals Application
Biotech
Biosimilars
Vaccines
Others:
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
1.2.4 Radiopharmaceuticals
Product name:
Product strength:
Product pharmaceutical form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
1.3 List of Countries where the product is authorized to market:
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
Attach letter of consent from the marketing authorization holder of the authorized product

DRAFT
Page 36 of 96 v.7 (14/7/08)
1.4 Consideration of this application:
Exceptional Circumstances
Priority Review
Regular Review
Date of approval by: (dd/mm/yyyy)
USA FDA
EMEA
Others
2. Marketing Authorization Application Particulars
2.1 Name(s) and ATC code:
2.1.1 Proposed (invented) name of the medicinal product:
2.1.2 Name of the active substance(s):
Note: ONLY one name should be given in the following order of priority: INN*, Ph.Eur., National Pharmacopoeia, common name, and scientific name.
* The active substance should be declared by its recommended INN, accompanied by its salt or hydrate form if relevant
2.1.3 Pharmacotherapeutic group: (Please use current ATC code)
ATC Code:
Group:
Note: If no ATC code has been assigned, please indicate if an application for ATC code has been made

DRAFT
Page 37 of 96 v.7 (14/7/08)
2.2 Pharmaceutical dosage form and Strength and reference pharmacopoeia:
2.2.1 Pharmaceutical dosage form:
Strength:
Reference Pharmacopoeia:
2.2.2 Route of administration:
2.2.3 Container, closure and administration device
For each type of pack give:
Package size(s):
Proposed shelf life:
Proposed storage conditions:
Samples/specimens:
2.3 Legal Status
2.3.1 Proposed dispensing/classification:
Prescription (None OTC)
Over the counter (OTC)
2.3.2 For products subject to medical prescription:
Product on special prescription*
Product on restricted prescription*
2.3.3 Supply for products NOT subject to medical prescription
Supply through pharmacies only
Supply through non-pharmacy outlets and pharmacies (if applicable)

DRAFT
Page 38 of 96 v.7 (14/7/08)
2.3.4 Promotion for products NOT subject to medical prescription
Promotion to health care professionals only
Promotion to the general public and health care professionals
2.4. Marketing Authorization Holder/Contact Person(s)/Company
2.4.1 Proposed marketing authorization holder/person legally responsible for placing the product on the market in the Kingdom of Saudi Arabia:
(Company) Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
2.4.2 Person/company authorized for communication on behalf of the applicant during the procedure in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, Attach letter of authorization (Annex )

DRAFT
Page 39 of 96 v.7 (14/7/08)
2.4.3 Person/Company authorized for communication between the marketing authorization holder and the SFDA after authorization, if different from 2.4.2 in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, attach letter of authorization
2.5 Manufacturers
Note: ALL manufacturing and control sites mentioned throughout the whole dossier MUST be in compliance and meet the GMP requirements.
Yes
No
2.6. Data requirements
New Drugs including NCE: o The data with respect to quality (chemistry and manufacturing)
should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o The data with respect to the clinical and non-clinical studies should be submitted in accordance with the CTD format.
o It is expected that if the product is approved by well established or recognized authorities (USA FDA, EMEA, Japan, TGA and Canada), a copy of the summary of approval should be submitted.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.

DRAFT
Page 40 of 96 v.7 (14/7/08)
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the marketing format with respect to the primary and secondary packaging.
Generics:
o The data with respect to quality (chemistry and manufacturing) should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o The data with respect to pharmaceutical equivalence with innovator's product used to establish the equivalency of the generic product.
o Evidence should be provided in support of the use of Saudi's reference product applied in comparative bio-studies.
o Information should be presented with respect to the active pharmaceutical ingredient (API) either in form of Drug Master File (DMF) or if the certificate of suitability is issued by EDQM a copy of such certificate should be included.
o It is expected that if the product is approved by well established or recognized authorities (US, Europe, Canada) a copy of the assessment may be provided in order to assist the evaluation process.
o The data with respect to bio-pharmaceutics (comparative bioavailability) should be submitted in accordance with the bioavailability guidelines. It should be noted that complete information should be provided with respect to the reference product used in comparative bio-studies.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the marketing format with respect to the primary and secondary packaging.
Biologicals:
o The data with respect to quality (manufacturing) should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o The data with respect to the clinical and non-clinical studies should be submitted in accordance with the CTD format.

DRAFT
Page 41 of 96 v.7 (14/7/08)
o It is expected that if the product is approved by well established or recognized authorities (US, Europe, Canada), a copy of the summary of approval should be submitted.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the marketing format with respect to the primary and secondary packaging.
o It should be noted that these products will be subjected to pre-approval inspection as well as lot release.
2.7 Certificate of Pharmaceutical Product (CPP)
• Please attach CPP in accordance with WHO guidelines. • If CPP is not available, please attach marketing authorization from the country
of origin (COR). Marketing authorization should include the following: 1. Product trade name in the COR. 2. Number and date of marketing authorization in the COR. 3. Name of active and inactive substances with their concentrations. 4. A statement that certifies the product is marketed in the COR. If this is
not possible, please specify the reasons and provide a marketing authorization showing that the product is marketed in one of the countries approved by SFDA.
5. Provide proof that the product has been marketed for no less than one year in the COR.
6. Pricing certificates in accordance with SFDA guidelines. 7. Provide Summary of Product Characteristics SPC. 8. Attach a product information insert and patient information.
2.8 Specify and list the materials of animal source contained in any component of the product?
Yes No
If yes, please describe in detail the animal source as well as the animal parts used in the processing of the material. Please note that any pork content has to be clearly specified. It should be noted that all material used must be free from BSE/TSE. If a certificate of suitability from EDQM is available, it should be attached.

DRAFT
Page 42 of 96 v.7 (14/7/08)
3. Scientific Advice 3.1 Was there formal scientific advice given by the SFDA for this medicinal
product?
Yes
No
If yes:
Date (dd/mm/yyyy):
Reference of the scientific letter:
Attach copy of the scientific letter (Annex )
3.2. Was there scientific recommendation(s) given by SFDA or Country of Origin for this medicinal product?
Yes
No
If yes,
Country(s):
Date (dd/mm/yyyy):
4. Pediatric Development Program
4.1. Is there a pediatric development program for this medicinal product?
Yes
No
If yes, please indicate the relevant section(s) in the dossier.

DRAFT
Page 43 of 96 v.7 (14/7/08)
5. Other Marketing Authorization Applications
Status of the application in other regulatory jurisdiction:
Name of the regulatory authority and status:
1.
Approved Rejected/Withdrawn Pending
2.
Approved Rejected/Withdrawn Pending
3.
Approved Rejected/Withdrawn Pending

DRAFT
Page 44 of 96 v.7 (14/7/08)
ANNEXED DOCUMENTS (WHERE APPROPRIATE)
Proof of payment
Letter of authorization on behalf of the applicant
Copy of the SFDA Manufacturing Authorization license (if available)
Copy of certificate(s) of suitability from European Department of Quality Medicine (EDQM) (if available)
Copy of Ph. Eur. Certificate(s) of suitability for TSE (if available)
Copy of approved Marketing Authorization(s) in other countries
List of Mock-ups and Samples/specimens sent with the application, as appropriate.

DRAFT
Page 45 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 46 of 96 v.7 (14/7/08)
א Marketing Authorization Application for Orphan Product
אאא،אא
אאאK
אK
The application form is to be used to apply for a marketing authorization of a medicinal product for human use submitted to Saudi Food & Drug Authority (SFDA).
A separate application form is needed for each strength and drug dosage form.

DRAFT
Page 47 of 96 v.7 (14/7/08)
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אאא
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference א

DRAFT
Page 48 of 96 v.7 (14/7/08)
1. Type of Application
The following sections should be completed where appropriate.
1.1 Orphan medicinal product information
Has Orphan designation been applied for this medicinal product?
Yes
No
If yes, Orphan Designation Procedure Number:
Pending
Orphan Designation Granted
Date (dd/mm/yyyy):
Country: USA EU GCC KSA Orphan Designation Refused
Date (dd/mm/yyyy):
Commission Decision Reference Number:
Orphan Designation Withdrawn
Date (dd/mm/yyyy):
1.2 Information relating to Orphan Market Exclusivity
Has any medicinal product been designated as an Orphan medicinal product in EU, FDA, GCC or SFDA for a condition relating to the indication proposed in this application?
Yes
No
If yes,
1. Please specify the drug authority Orphan Designation Number(s):

DRAFT
Page 49 of 96 v.7 (14/7/08)
2. Has any of the designated Orphan medicinal product(s) been granted a marketing authorization in any country?
Yes
No
Please specify:
Name:
Strength:
Pharmaceutical form:
Name of the marketing authorization holder:
Marketing authorization number(s):
Date of authorization:
If yes, is the medicinal product, subject of this application, considered as “similar” to any of the authorized Orphan Medicinal product(s)?
Yes
No
2. Marketing Authorization Application Particulars
2.1 Name(s) and ATC code:
2.1.1 Proposed (invented) name of the medicinal product:
2.1.2 Name of the active substance(s):
Note: ONLY one name should be given in the following order of priority: INN*, Ph.Eur., National Pharmacopoeia, common name, and scientific name.
* The active substance should be declared by its recommended INN, accompanied by its salt or hydrate form if relevant

DRAFT
Page 50 of 96 v.7 (14/7/08)
2.1.3 Pharmacotherapeutic group: (Please use current ATC code)
ATC Code:
Group:
Note: If no ATC code has been assigned, please indicate if an application for ATC code has been made
2.2 Pharmaceutical dosage form and Strength and reference pharmacopoeia:
2.2.1 Pharmaceutical dosage form:
Strength:
Reference pharmacopoeia:
2.2.2 Route of administration:
2.2.3 Container, closure and administration device
For each type of pack give:
Package size(s):
Proposed shelf life:
Proposed storage conditions:
Samples/specimens:
2.3 Legal Status
2.3.1 Proposed dispensing/classification:
Prescription (None OTC)
Over the counter (OTC)
2.3.2 For products subject to medical prescription:
Product on special prescription*
Product on restricted prescription*

DRAFT
Page 51 of 96 v.7 (14/7/08)
2.3.3 Supply for products NOT subject to medical prescription
Supply through pharmacies only
Supply through non-pharmacy outlets and pharmacies (if applicable)
2.3.4 Promotion for products NOT subject to medical prescription
Promotion to health care professionals only
Promotion to the general public and health care professionals
2.4. Marketing Authorization Holder/Contact Person(s)/Company
2.4.1 Proposed marketing authorization holder/person legally responsible for placing the product on the market in the Kingdom of Saudi Arabia:
(Company) Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
2.4.2 Person/company authorized for communication on behalf of the applicant during the procedure in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, Attach letter of authorization (Annex )

DRAFT
Page 52 of 96 v.7 (14/7/08)
2.4.3 Person/Company authorized for communication between the marketing authorization holder and the SFDA after authorization, if different from 2.4.2 in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, attach letter of authorization
2.5 Manufacturers
Note: ALL manufacturing and control sites mentioned throughout the whole dossier MUST be in compliance and meet the GMP requirements.
Yes
No
2.6. Data requirements
2.7 Certificate of Pharmaceutical Product (CPP)
• Please attach CPP in accordance with WHO guidelines. • If CPP is not available, please attach marketing authorization from the country
of origin (COR). Marketing authorization should include the following: 9. Product trade name in the COR. 10. Number and date of marketing authorization in the COR. 11. Name of active and inactive substances with their concentrations. 12. A statement that certifies the product is marketed in the COR. If this is
not possible, please specify the reasons and provide a marketing authorization showing that the product is marketed in one of the countries approved by SFDA.

DRAFT
Page 53 of 96 v.7 (14/7/08)
13. Provide proof that the product has been marketed for no less than one year in the COR.
14. Pricing certificates in accordance with SFDA guidelines. 15. Provide Summary of Product Characteristics SPC. 16. Attach a product information insert and patient information.
2.8 Specify and list the materials of animal source contained in any component of the product?
Yes
No
If yes, please describe in detail the animal source as well as the animal parts used in the processing of the material. Please note that any pork content has to be clearly specified. It should be noted that all material used must be free from BSE/TSE. If a certificate of suitability from EDQM is available, it should be attached.

DRAFT
Page 54 of 96 v.7 (14/7/08)
3. Scientific Advice 3.1 Was there formal scientific advice given by the SFDA for this medicinal
product?
Yes
No
If yes:
Date (dd/mm/yyyy):
Reference of the scientific letter:
Attach copy of the scientific letter (Annex )
3.2. Was there scientific recommendation(s) given by SFDA or Country of Origin for this medicinal product?
Yes
No
If yes,
Country(s):
Date (dd/mm/yyyy):
4. Pediatric Development Program
4.1. Is there a pediatric development program for this medicinal product?
Yes
No
If yes, please indicate the relevant section(s) in the dossier.

DRAFT
Page 55 of 96 v.7 (14/7/08)
5. Other Marketing Authorization Applications
Status of the application in other regulatory jurisdiction:
Name of the regulatory authority and status:
4.
Approved Rejected/Withdrawn Pending
5.
Approved Rejected/Withdrawn Pending
6.
Approved Rejected/Withdrawn Pending

DRAFT
Page 56 of 96 v.7 (14/7/08)
ANNEXED DOCUMENTS (WHERE APPROPRIATE)
Proof of payment
Letter of authorization on behalf of the applicant
Copy of the SFDA Manufacturing Authorization license (if available)
Copy of certificate(s) of suitability from European Department of Quality Medicine (EDQM) (if available)
Copy of Ph. Eur. Certificate(s) of suitability for TSE (if available)
Copy of approved Marketing Authorization(s) in other countries
List of Mock-ups and Samples/specimens sent with the application, as appropriate.

DRAFT
Page 57 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 58 of 96 v.7 (14/7/08)
אאא Marketing Authorization Application of Medicinal Product
for Veterinary Use
א،אאא
אאאאK
אאK
The application form is to be used to apply for a marketing authorization of a medicinal product for veterinary use submitted to Saudi Food & Drug Authority (SFDA).
A separate application form is needed for each strength and drug dosage form.
New Renewal
Registration No:

DRAFT
Page 59 of 96 v.7 (14/7/08)
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אאא
Species א
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference א

DRAFT
Page 60 of 96 v.7 (14/7/08)
1. Type of Application
The following sections should be completed where appropriate.
1.1 This application concerns (Mandatory scope):
Annex (1) (New drugs)
Annex (2) (Generics)
Annex (3) (Biologicals)
Annex (4) (Immunological veterinary medicinal product for the treatment of animal diseases subject to community prophylactic measures)
1.2 This application is submitted in accordance with the following:
1.2.1 New Drug Application
New Drug
New Chemical Entity (NCE)
Product name:
Product strength:
Pharmaceutical :
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
1.2.2 Generic Application
Generic Drug
Reference Product
Product name:
Product strength:
Dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):

DRAFT
Page 61 of 96 v.7 (14/7/08)
1.2.3 Biologicals Application
Biotech
Biosimilars
Vaccines
Others:
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
1.3 List of Countries where the product is authorized:
Product name:
Product strength:
Pharmaceutical dosage form:
Marketing Authorization holder:
Country of Origin Authorization:
Date (dd/mm/yyyy):
Attach letter of consent from the marketing authorization holder of the authorized product
1.4 Maximum Residual Limit (MRL) Status: (only for food producing species)
When the veterinary medicinal product is intended for use in food-producing animals, please provide the following information as available at the time of submission of the application (All substances contained in the product are subject to this requirement if they are pharmacologically active in the dose in which they are administered to the animal. Excipients not included in any of the Annexes of Council Regulation (EEC) No 2377/90 should also be listed and an appropriate justification given).

DRAFT
Page 62 of 96 v.7 (14/7/08)
Maximum Residue Limits (MRL) has been published in the Official Journal of the Communities:
Substance(s)
Annex
Species
Target tissue(s)
Remarks
OJ date of publication
Application for a Maximum Residue:
Substance(s)
Date of submission
Species
Remarks
1.5 Consideration of this application:
Exceptional Circumstances
Priority Review
Regular Review
Date of acceptance by CHMP (dd/mm/yyyy):

DRAFT
Page 63 of 96 v.7 (14/7/08)
2. Marketing Authorization Application Particulars
2.1 Name(s) and ATC code:
2.1.1 Proposed (invented) name of the medicinal product:
2.1.2 Name of the active substance(s):
Note: ONLY one name should be given in the following order of priority: INN*, Ph.Eur., National Pharmacopoeia, common name, and scientific name.
* The active substance should be declared by its recommended INN, accompanied by its salt or hydrate form if relevant
2.1.3 Pharmacotherapeutic group: (Please use current ATC code)
ATC Code:
Group:
Note: If no ATC code has been assigned, please indicate if an application for ATC code has been made
2.2 Pharmaceutical dosage form and Strength and standard (pharmacopoeia and professed and house):
2.2.1 Pharmaceutical dosage form:
Strength:
Standard:
2.2.2 Route of administration:
2.2.3 Container, closure and administration device
For each type of pack give:
Package size(s):
Proposed shelf life:
Proposed storage conditions:
Samples/specimens:

DRAFT
Page 64 of 96 v.7 (14/7/08)
2.3 Legal Status
2.3.1 Proposed administration:
ONLY by a Veterinary surgeon
By a Veterinary surgeon or under their direct responsibility
Others:
2.3.2 Proposed dispensing/classification:
Subject to medical prescription
NOT Subject to medical prescription
Subject to other controls:
2.3.2 For products subject to medical prescription:
Product on special prescription*
Product on restricted prescription*
2.3.3 For products subject to medical prescription:
Veterinary product on prescription which may be renewed (if applicable)
Veterinary product on prescription which may not be renewed (if applicable)
Veterinary product on special prescription
Veterinary product on restricted prescription
2.3.4 Supply for products not subject to medical prescription
Supply through pharmacies only
Supply through non-pharmacy outlets and pharmacies (if applicable)
Supply/administration by veterinary surgeons only

DRAFT
Page 65 of 96 v.7 (14/7/08)
Supply by pharmacies and/or veterinary surgeons for animals under their care
Supply through authorized distributor
General sale
2.3.5 Promotion for products not subject to medical prescription
Promotion to health care professionals only
Promotion to the general public and health care professionals
2.4. Marketing Authorization Holder/Contact Person(s)/Company
2.4.1 Proposed marketing authorization holder/person legally responsible for placing the product on the market in the Kingdom of Saudi Arabia:
(Company) Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
2.4.2 Person/company authorized for communication on behalf of the applicant during the procedure in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, Attach letter of authorization (Annex )

DRAFT
Page 66 of 96 v.7 (14/7/08)
2.4.3 Person/Company authorized for communication between the marketing authorization holder and the SFDA after authorization, if different from 2.4.2 in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, attach letter of authorization (Annex )
2.5 Manufacturers
Note: ALL manufacturing and control sites mentioned throughout the whole dossier MUST be in compliance and meet the GMP requirements.
Yes
No
2.6. Data requirements
New Drugs including NCE: o The data with respect to quality (chemistry and manufacturing)
should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o The data with respect to the clinical and non-clinical studies should be submitted in accordance with the CTD format.
o It is expected that if the product is approved by well established or recognized authorities (US, Europe, Canada), a copy of the summary of approval should be submitted.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the

DRAFT
Page 67 of 96 v.7 (14/7/08)
marketing format with respect to the primary and secondary packaging.
Generics:
o The data with respect to quality (chemistry and manufacturing) should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o Information should be presented with respect to the active pharmaceutical ingredient (API) either in form of Drug Master File (DMF) or if the certificate of suitability is issued by EDQM a copy of such certificate should be included.
o It is expected that if the product is approved by well established or recognized authorities (US, Europe, Canada) a copy of the assessment may be provided in order to assist the evaluation process.
o The data with respect to bio-pharmaceutics (comparative bioavailability) should be submitted in accordance with the bioavailability guidelines. It should be noted that complete information should be provided with respect to the reference product used in comparative bio-studies.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the marketing format with respect to the primary and secondary packaging.
Biologicals:
o The data with respect to quality (manufacturing) should be submitted in accordance with the quality guidelines. This information should be presented in CTD format.
o The data with respect to the clinical and non-clinical studies should be submitted in accordance with the CTD format.
o It is expected that if the product is approved by well established or recognized authorities (US, Europe, Canada), a copy of the summary of approval should be submitted.
o Complete information of the labels, package inserts or any associated promotional material used should be provided.
o A sufficient number of samples should be provided in order to perform complete testing. The samples must represent the

DRAFT
Page 68 of 96 v.7 (14/7/08)
marketing format with respect to the primary and secondary packaging.
o It should be noted that these products will be subjected to pre-approval inspection as well as lot release.
2.7 Certificate of Pharmaceutical Product (CPP)
• Please attach CPP in accordance with WHO guidelines. • If CPP is not available, please attach marketing authorization from the country
of origin (COR). Marketing authorization should include the following: 17. Product trade name in the COR. 18. Number and date of marketing authorization in the COR. 19. Name of active and inactive substances with their concentrations. 20. A statement that certifies the product is marketed in the COR. If this is
not possible, please specify the reasons and provide a marketing authorization showing that the product is marketed in one of the countries approved by SFDA.
21. Provide proof that the product has been marketed for no less than one year in the COR.
22. Pricing certificates in accordance with SFDA guidelines. 23. Provide Summary of Product Characteristics SPC. 24. Attach a product information insert and patient information.
2.8 Specify and list the materials of animal source contained in any component of the product?
Yes No
If yes, please describe in detail the animal source as well as the animal parts used in the processing of the material. Please note that any pork content has to be clearly specified. It should be noted that all material used must be free from BSE/TSE. If a certificate of suitability from EDQM is available, it should be attached.

DRAFT
Page 69 of 96 v.7 (14/7/08)
3. Scientific Advice 3.1 Was there formal scientific advice given by the SFDA for this medicinal
product?
Yes
No
If yes:
Date (dd/mm/yyyy):
Reference of the scientific letter:
Attach copy of the scientific letter (Annex )
3.2 Was there scientific recommendation(s) given by SFDA or Country of Origin for this medicinal product?
Yes
No
If yes,
Country(s):
Date (dd/mm/yyyy):
4. Other Marketing Authorization Applications
Status of the application in other regulatory jurisdiction:
Name of the regulatory authority and status:
1.
Approved Rejected/Withdrawn Pending
2.
Approved Rejected/Withdrawn Pending
3.
Approved Rejected/Withdrawn Pending

DRAFT
Page 70 of 96 v.7 (14/7/08)
ANNEXED DOCUMENTS (WHERE APPROPRIATE)
Proof of payment
Letter of authorization on behalf of the applicant
Copy of the SFDA Manufacturing Authorization license (if available)
Copy of certificate(s) of suitability from European Department of Quality Medicine (EDQM) (if available)
Copy of Ph. Eur. Certificate(s) of suitability for TSE (if available)
Copy of approved Marketing Authorization(s) in other countries
List of Mock-ups and Samples/specimens sent with the application, as appropriate.

DRAFT
Page 71 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 72 of 96 v.7 (14/7/08)
Marketing Authorization Application of Herbal Product
New Renewal
Registration No:
אאא،א
אאאאK
אK
The application form is to be used to apply for a marketing authorization of a herbal product for human use submitted to Saudi Food & Drug Authority (SFDA).
A separate application form is needed for each strength and drug dosage form.

DRAFT
Page 73 of 96 v.7 (14/7/08)
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אאא
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference א

DRAFT
Page 74 of 96 v.7 (14/7/08)
1. Initial Information
The following sections should be completed where appropriate.
Please tick any of the following that apply:
The medicinal product contains one or more vitamins or minerals from a new source. European Pharmacopoeia or USP or BP certificates of suitability cover all the vitamins or minerals.
The medicinal product contains one or more vitamins or minerals from a new source. European Pharmacopoeia certificates or USP or BP of suitability DO NOT cover all the vitamins or minerals.
The medicinal product contains one or more new excipients.
The medicinal product is a sterile medicinal product.
The medicinal product contains material from animal origin.
2. Marketing Authorization Application Particulars
2.1 Name(s) and ATC code:
2.1.1 Proposed (invented) name of the medicinal product:
2.1.2 Name of the active substance(s):
2.1.3 Pharmacotherapeutic group: (Please use current ATC code)
ATC Code:
Group:
Note: If no ATC code has been assigned, please indicate if an application for ATC code has been made
2.2 Pharmaceutical dosage form and Strength and reference pharmacopoeia:
2.2.1 Pharmaceutical dosage form:
Strength:
Reference pharmacopoeia:

DRAFT
Page 75 of 96 v.7 (14/7/08)
2.2.2 Route of administration:
2.2.3 Container, closure and administration device
For each type of pack give:
Package size(s):
Proposed shelf life:
Proposed storage conditions:
Samples/specimens:
2.3 Legal Status
Does this product have medical claims?
Yes
No
If yes, please specify:
2.4. Marketing Authorization Holder/Contact Person(s)/Company
2.4.1 Proposed marketing authorization holder/person legally responsible for placing the product on the market in the Kingdom of Saudi Arabia:
(Company) Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
2.4.2 Person/company authorized for communication on behalf of the applicant during the procedure in the Kingdom of Saudi Arabia:

DRAFT
Page 76 of 96 v.7 (14/7/08)
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, Attach letter of authorization (Annex )
2.4.3 Person/Company authorized for communication between the marketing authorization holder and the SFDA after authorization, if different from 2.4.2 in the Kingdom of Saudi Arabia:
Name:
Company Name:
Address:
Country:
Telephone:
Fax:
E-Mail:
Note: If different to 2.4.1 above, attach letter of authorization (Annex )
2.5 Manufacturers
Note: ALL manufacturing and control sites mentioned throughout the whole dossier MUST be in compliance and meet the GMP requirements.
Yes
No
2.6. Data requirements

DRAFT
Page 77 of 96 v.7 (14/7/08)
3. Scientific Advice 3.1 Was there formal scientific advice given by the SFDA for this medicinal
product?
Yes
No
If yes:
Date (dd/mm/yyyy):
Reference of the scientific letter:
Attach copy of the scientific letter (Annex )
3.2. Was there scientific recommendation(s) given by SFDA or Country of Origin for this medicinal product?
Yes
No
If yes,
Country(s):
Date (dd/mm/yyyy):
4. Other Marketing Authorization Applications
Status of the application in other regulatory jurisdiction:
Name of the regulatory authority and status:
4.
Approved Rejected/Withdrawn Pending
5.
Approved Rejected/Withdrawn Pending
6.
Approved Rejected/Withdrawn Pending

DRAFT
Page 78 of 96 v.7 (14/7/08)
ANNEXED DOCUMENTS (WHERE APPROPRIATE)
Proof of Payment
Proof of establishment of the applicant in the EEA
Letter of authorization for communication on behalf of the applicant/MAH
Curriculum Vitae of the Qualified Person for Pharmacovigilance
Justification for more than one manufacturer responsible for batch release
in the EEA
Flow-chart indicating the different sites involved in the manufacturing
process of the medicinal product (including sites involved in sampling and
testing for batch release of products manufactured in third countries)
Statement from the competent authority which carried out the inspection of
the manufacturing site(s)
Letter(s) of access to Drug Master File(s) or copy of Ph.Eur. Certificate(s) of
suitability
Copy of written confirmation from the manufacturer of the active substance
to inform the applicant in case of modification of the manufacturing process or
specifications
Ph.Eur.Certificate(s) of suitability for TSE
Written consent(s) of the competent authorities regarding GMO release in
the environment
List of Mock-ups or Samples/specimens sent with the application, as
appropriate

DRAFT
Page 79 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 80 of 96 v.7 (14/7/08)
א
Application for Variation to a Marketing Authorization
Type I: Notifiable Change
This application form will cover the following types of supplemental applications: (please, tick the appropriate box)
Generics New drugs Biologicals Herbals Veterinary drug
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אא
א
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference
א

DRAFT
Page 81 of 96 v.7 (14/7/08)
Instructions:
Tick the appropriate change and submit the details of changes with supportive data in CTD format.
All applicable sections of CTD format should be completed.
Change
Yes No
a. Administrative change(s)
1 Change in the name and/or address of the marketing
authorization holder
2 Change in the ownership of a marketing authorization
holder
3 Change in the name of the medicinal product
4 Change in the name and/or address of the manufacturer of
API
5 Change in the name and/or address of the manufacturer of
the finished product
6 Change in ATC Code:
a) Medicinal products for human use
b) Medicinal products for veterinary use
b. Drug Substance (API)
1 Change in the manufacturing site for the starting materials
or intermediates or the API
2 Change in the manufacturing process to improve the
quality and purity
3 Change in the analytical procedures
4 Change in the specifications
5 Change in the reference material

DRAFT
Page 82 of 96 v.7 (14/7/08)
Change
Yes No
6 Change in the Primary or Secondary packaging
7 Change in the re-testing date
c. Drug product
1 Change in the manufacturing site of simple dosage forms
such as immediate release tablets, oral solutions,
suspensions cream and ointments
2 Change in the composition if the product which has no
significant impact on the quality attributes
3 Changes in the equipments
4 Change in the manufacturing processes that has no
significant impact on the quality attributes of the product
5 Change in the specifications that improve the quality
attributes of the product
6 Change in the testing methods to improve the precision,
accuracy and suitability
7 Change in the excipients that will have no impact on the
quality or safety of the finished products
8 Change in the Primary or Secondary packaging that will not
impact on the quality attributes of the product
9 Changes in the expiration date of the product
d. Changes in the labeling and package insert
1 Change in the Product Characteristics, labeling and
package leaflet/insert
2 Change in the package size

DRAFT
Page 83 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 84 of 96 v.7 (14/7/08)
א
Application for Variation to a Marketing Authorization
Type II: Supplemental New Drug Application
This application form will cover the following types of supplemental applications: (please, tick the appropriate box)
Generics New drugs Biologicals Herbals Veterinary drugs
Product Name אא
Active substance(s) אLאאא
Pharmaceutical form and strength
אא
א
MA Number א
Name and address of MA holder
אא
א
Name and Address of contact
אאאאא
Telephone No. א
Fax No. א
e-mail אא
Applicant’s reference
א

DRAFT
Page 85 of 96 v.7 (14/7/08)
Instructions:
Tick the appropriate change and submit the details of changes with supportive data in CTD format.
All applicable sections of CTD format should be completed.
Change
Yes No
a. Administrative
1 Name of the medicinal product
2 Strength and/or Pharmaceutical dosage forms
3 Change in ATC Code:
a) Medicinal products for human use
b) Medicinal products for veterinary use
b. Drug Substance (API)
1 Change in the new supplier/source of the API
c. Drug product
1 Change in the manufacturing site of complex dosage forms
such as controlled released, sterile products, and inhalation
products
2 Change in the composition of the product which has a
significant impact on the quality, safety and efficacy of the
product
3 Change in the manufacturing processes that has a
significant impact on the quality, safety and efficacy of the
product
d. Clinical
1 New indication and/or contraindication due to the clinical
studies on the existing dosage form
2 Changes in the package inserts/leaflet due to safety issues

DRAFT
Page 86 of 96 v.7 (14/7/08)
Change
Yes No
e. Labeling and package insert
1 Product Characteristics, labeling and package leaflet/insert
2 Package size(s)

DRAFT
Page 87 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I hereby submit a notification for the above Marketing Authorization to be varied in
accordance with the proposals given above. I declare that (Please tick the appropriate declarations):
There are no other changes than those identified in this application;
The change(s) will not adversely affect the quality, efficacy or safety of the product
All conditions as set for the notification(s) concerned are fulfilled
The required documents as specified for the notification(s) concerned have been
submitted
Where applicable, SFDA fees have been paid
Fees paid (if applicable): ____________________________________________
Please specify fee category: _________________________________________
Orphan Drug fee exemption granted (attach fee Notification of the competent
authority)
I certify that the information submitted is true and accurate and changes will not be
made until they are approved by SFDA.
Company Director/CEO:
Signature:
Date:

DRAFT
Page 88 of 96 v.7 (14/7/08)
Clinical Trial Application Form
Please, tick the appropriate box:
Request for a Clinical Trial Authorization for an Investigational
Drug For Human Use
(complete sections A, B, C and D)
Request for a Clinical Trial Authorization
(complete sections A, B, C and D)
Request for a Clinical Trial Notification
(complete ONLY section E)

DRAFT
Page 89 of 96 v.7 (14/7/08)
Section A: Company Details
• Company Name:
• Company Registration No.:
• Contact person:
• Contact No. (24Hrs):
• Telephone:
• Fax:
• e-mail:
• Company address:
• Agent name:
Section B: Clinical Trial Details
• Title:
• Phase of trial:
Phase I Phase II Phase III Phase IV
• Therapeutic Area:
• Disease Area:
• Individuals:
Healthy Volunteer Patients Both
• Blindness:
Single Double Triple Open label
• Randomization:
Randomized Not Randomized
• Concurrent use of:
Placebo Comparator drug Concomitant drug

DRAFT
Page 90 of 96 v.7 (14/7/08)
• Protocol duration:
Starts (dd/mm/yyyy):
Ends (dd/mm/yyyy):
• Location of the trial:
Saudi Arabia ONLY
Saudi Arabia and GCC
Middle East
Specify countries:
• No. of centers in Saudi Arabia:
• Objectives of the trial:
1. ………………………………………………………….
2. ………………………………………………………….
3. ………………………………………………………….
4. ………………………………………………………….
5. ………………………………………………………….
• Ethical committee for hospitals (hospitals name):
1. ………………………………………………………….
2. ………………………………………………………….
3. ………………………………………………………….
4. ………………………………………………………….
• Is there any advertisement for recruitment?
Yes No

DRAFT
Page 91 of 96 v.7 (14/7/08)
Section C: Drug Details
C1 The Trial Drug details
• Drug Name (or code):
• Brand Name (if available):
• Pharmaceutical dosage form:
• Strength:
• Dose:
• Route of administration:
• Previous clinical trials:
C2 The Comparator drug details
• Drug Name (or code):
• Brand Name (if available):
• Pharmaceutical dosage form:
• Strength:
• Dose:
• Route of administration:
C3 The Concomitant drug(s) details
Drug name Brand Strength Dose Route

DRAFT
Page 92 of 96 v.7 (14/7/08)
Section D: Information of Local Centers Numbers of centers:
Center No. 1:
• Name:
• Investigator name:
• Specialty:
• Department:
• Telephone:
• Fax:
• e-mail:
Center No. 2:
• Name:
• Investigator name:
• Specialty:
• Department:
• Telephone:
• Fax:
• e-mail:
Center No. 3:
• Name:
• Investigator name:
• Specialty:
• Department:
• Telephone:

DRAFT
Page 93 of 96 v.7 (14/7/08)
• Fax:
• e-mail:
Center No. 4:
• Name:
• Investigator name:
• Specialty:
• Department:
• Telephone:
• Fax:
• e-mail:

DRAFT
Page 94 of 96 v.7 (14/7/08)
Section E: Clinical Trial Details of Notification
• Clinical Trial SFDA No.
• Title:
• Type of notification:
Add a new site
Delete a site
Add new patients
Extend trial time
Others
• Justification:
1. ………………………………………………………….
2. ………………………………………………………….
3. ………………………………………………………….
4. ………………………………………………………….
5. ………………………………………………………….
• Requester name:

DRAFT
Page 95 of 96 v.7 (14/7/08)
Required Documents:
Request for a Clinical Trial Authorization for an Investigational Drug for Human Use
• Study Protocol • Patient Information Sheet & Consent Form • Ethical Committee / IRB approval Letter • Investigator Brochure/CV • Investigator Financial Disclosure. • GMP certificate for manufacturer • Certificate of analysis for the investigational drug
Request for a Clinical Trial Authorization
• Study Protocol • Patient Information Sheet & Consent Form • Ethical Committee / IRB approval Letter • Investigator Financial Disclosure • Investigator Brochure/CV

DRAFT
Page 96 of 96 v.7 (14/7/08)
Declaration of the Applicant:
I confirmed that the information submitted in this application is true and accurate at the date of the submission.
I must inform the SFDA for any changes in the information submitted in this application.
I shall not initiate this trial until approvals are obtained from the ethical committees of the hospitals involved in this trial and the SFDA.
Applicant:
Signature:
Date:
Company Stamp: