regulatory affairs, qsite - hermon laboratories | home · · 2011-06-26regulatory affairs, qsite....
TRANSCRIPT

STERILIZATION VALIDATIONSTERILIZATION VALIDATION
Presented by:
Efrat Hartog-DavidRegulatory Affairs, Qsite

Introduction
SterilizationProcess used to transform product free from viable microorganisms
MicroorganismsBacteriaVirusesFungi

Introduction
Why sterilization is needed?y
Medical device is assembled in
t ll dcontrolled environment, but may contain ymicroorganisms

Introduction
Why sterilization is needed?y
Microorganisms may cause infectionInvasive devices enter normally sterile body tissue

Introduction
Sterilization purposep p
To inactivate the microorganisms
Transform the medical device into sterile

Introduction
Inactivation of microorganisms
D value:Time required to achieve inactivation ofachieve inactivation of 90% of a population of the microorganism
funder specific conditions

Sterility
The absence of microorganisms cannot be proven
A sterility assurance level (SAL) is used to define the sterility
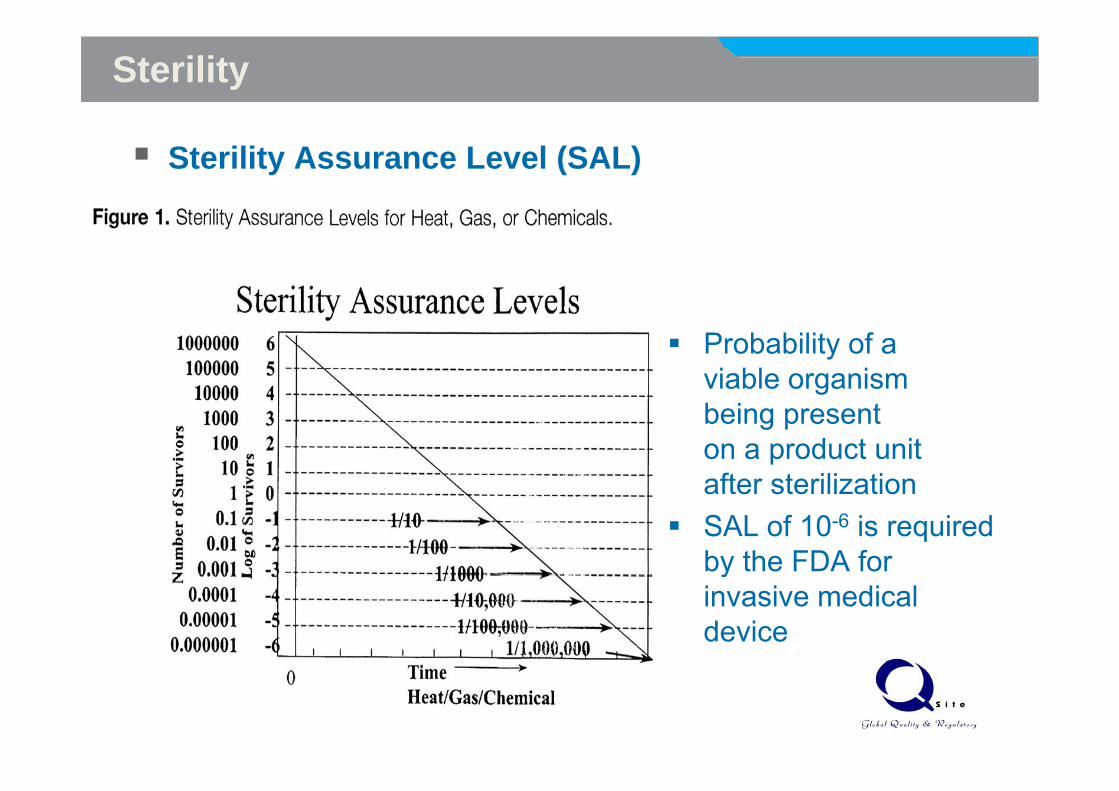
Sterility
Sterility Assurance Level (SAL)
Probability of a viable organism being present on a product unit after sterilizationafter sterilizationSAL of 10-6 is required by the FDA for yinvasive medical device

Sterilization methods
Moist heat
Ethylene oxide (ETO)Ethylene oxide (ETO)
Gamma radiation

ETO sterilization
Ethylene oxide (ETO)
Colorless flammable gas
Chemically reactivey
Irritating, carcinogenic, mutagenic gasg, g , g g
Alkylation reaction cause damage to DNA and y gproteins of microorganisms

ETO sterilization
ETO processing steps
Preconditioning/conditioningExposure to relative humidity and temperaturep y pEnsure uniformity of conditions
Sterilization cycleExposure to ETO gasg
AerationDissipation of remaining gases

ETO sterilization
Process parameters
Gas quantity>400mg/L
Temperaturep~45ºC
Relative humidity~35 – 80%
Exposure timeExposure time~90 – 360 minutes

ETO sterilization
AdvantagesMost product and packaging materials are compatibleRelatively low temperature process
DisadvantagesPenetration sometimes difficultResidualsLong process and release time

Gamma radiation sterilization
Gamma radiation
Electromagnetic radiation of short wavelength
High-energy photons are emitted from an isotope (Cobalt 60) so rce prod cing ioni ation thro gho t a(Cobalt 60) source producing ionization throughout a product
Cause damage to DNA and cellular structures of microorganismsmicroorganisms
Radiation dose is measured in kGy valuesRadiation dose is measured in kGy values

Gamma radiation sterilization
Gamma radiation effects on materials
BrittleColorOdorStiffnessSoftensToxicityChemical inertnessMelt temperature

Gamma radiation sterilization
AdvantagesDeep penetration powerNo residualsOnly one process parameter – timeLow temperature processp pRelease immediately after sterilization
DisadvantageNot all product and packaging materials are compatible

Sterility concerns

Sterilization validation
Documented procedure for obtaining, recording and p g, ginterpreting the results required to establish that a process will consistently yield product complying with predetermined specifications
SAL of 10-6 shall be demonstrated

Sterilization validation
Objectivesj
The sterilization process will consistently achieve sterility
The sterilization process will not have an adverse impact on the device or its packaging

ETO sterilization validation
Applicable standardspp
ANSI/AAMI/ISO 11135-1:2007 “Sterilization of health care products – Ethylene oxide -Part 1: Requirements for the development, validation and routine control of a sterilization process for medicalroutine control of a sterilization process for medical devices”
ANSI/AAMI/ISO 11135-2:2008 “Sterilization of health care products – Ethylene oxide -Part 2: Guidance on the application of ISO 11135-1”

ETO sterilization validation
ETO validation overview
Process and equipment characterizationProcess and equipment characterizationIQ (Installation Qualification)OQ (Operational Qualification)OQ (Operational Qualification)Product definitionPQ (Performance Qualification) – PhysicalPQ (Performance Qualification) PhysicalPQ (Performance Qualification) – MicrobiologicalDocumentationDocumentationRevalidation

ETO sterilization validation
IQ - Installation QualificationQ QIQ shall demonstrate that the sterilization equipment have been installed in accordance with their specification
OQ – Operational QualificationOQ shall demonstrate that the installed equipment is capable of delivering the specified process within defined tolerances

ETO sterilization validation
Product definition
Product familiesProduct configurationProduct and packaging materialsp g gDensityManufacturing environmentgBioburden
PCD (Product Challenge Device)

ETO sterilization validation
PQ - Performance Qualification
PQ shall use product or PCD to demonstrate that:
Equipment consistently operates in accordance with predetermined criteria
The process produces product that is sterile

ETO sterilization validation
PQ – PhysicalQ yPhysical PQ shall confirm the predetermined process parameters throughout the load, for the duration of the sterilization process
PQ - MicrobiologicalMicrobiological PQ shall confirm the effectiveness of the defined process in achieving the required SAL, for product/load combination

ETO sterilization validation
PQ - Physical
The physical PQ can be performed in parallel with the microbiological PQDuring sterilization process parameters are monitored:
Temperature Relative humidityPressure
Process parameters are within the required range

ETO sterilization validation
PQ – Microbiological
Determination of lethal rate of the sterilization process, according to one of the approaches:
Cycle calculation approachC l t th t d li i i ll 12 l d tiCycle parameters that deliver minimally 12 log reduction shall be calculated
Half cycle approach3 half cycles resulting in 6 log reduction shall be3 half cycles resulting in 6 log reduction shall be performed

ETO sterilization validation
PQ – Microbiological
Bioburden estimation
Validation cycles according to half cycle approachFractional cycle3 x Half cyclesFull cycle
Sterilized samples testingETO residual testing

ETO sterilization validation
Bioburden estimation
Population of viable microorganisms on or in the product
Test non-sterilized samples
Bioburden <100 CFU/product is considered relatively low
If bioburden > 1000 CFU/product the cleanness level of the manufacturing environment should be improvedimproved

ETO sterilization validation
Validation Cycles
Fractional cycle
3 x Half cyclesy
Full cycle

ETO sterilization validation
Biological Indicators (BI)
Test system containing 106 viable microorganisms to ensure SAL of 10-6
BI must be inserted in the most difficult location to sterilize in the product
Discs and Wires Paper StripsGl A lGlass Ampoule

Fractional cycleETO sterilization validation
Fractional cycle
Products inoculated with BIs Products
Product Sterility TestBI Sterility Test
BI product
X BIs show GROWTH If X>Y Y products show GROWTH
The BI is more resistant to the sterilization process, compared to the bioburden

ETO sterilization validationHalf cycle
Presents worst case scenario
Samples inoculated with BIs are subjected to half cycle
Following sterilization, BIs are removed from the samples and tested for BI sterility
All BIs should show NO GROWTH
ISO 11135: 3 half cycles must be run

ETO sterilization validation
Full cycley
One full cycle is requiredO e u cyc e s equ ed
Samples inoculated with BIs are subjected to full cycleSamples inoculated with BIs are subjected to full cycle
All BIs should show NO GROWTHAll BIs should show NO GROWTH
ETO residual testingETO residual testing

ETO sterilization validation
ETO residual testing
ETO is known to exhibit a number of biological effects:effects:
IrritationMutagenicityMutagenicityCarcinogenicity
ETO residual quantity is effected by productETO residual quantity is effected by product, packaging and process characteristicsThe ETO residuals should meet the acceptanceThe ETO residuals should meet the acceptance criteria according to ISO 10993-7Aeration may be prolongedAeration may be prolonged

ETO sterilization validation
Revalidation
Annually the status of the sterilization validation must be reviewed
Inspection of:BioburdenProduct design and packagingProcess equipment and parameters
Revalidation usually consist one half cycle and one full cycle

Gamma radiation sterilization validation
Applicable standards
ANSI/AAMI/ISO 11137-1:2006 “Sterilization of health care products – Radiation - Part 1: Requirements for development validation and routineRequirements for development, validation and routine control of a sterilization process for medical devices”
ANSI/AAMI/ISO 11137-2:2006 “Sterilization of health care products – Radiation - Part 2: Establishing the sterilization dose”
ANSI/AAMI/ISO 11137 3 2006ANSI/AAMI/ISO 11137-3:2006 Sterilization of health care products – Radiation - Part 3: Guidance on dosimetric aspectsp

Gamma radiation sterilization validation
Gamma radiation validation overview
Equipment characterizationEquipment characterizationIQ (Installation Qualification)OQ (Operational Qualification)OQ (Operational Qualification)Product definitionProcess definitionProcess definitionPQ (Performance Qualification)DocumentationDocumentationDose audit

Gamma radiation sterilization validation
P d t d fi itiProduct definition
P d t f iliProduct families
BioburdenSize of productNo. of componentsComplexity of productManufacturing environment

Gamma radiation sterilization validation
Process definition
Establishing the sterilization dose, according to one of the approaches:
The number of the bioburden is obtained and used t t th t ili ti dto set the sterilization dose
A t ili ti d i l t d d b t ti t dA sterilization dose is selected and substantiated

Gamma radiation sterilization validation
PQ
Dose mapping shall be carried out:Identify the sterilization dose accepted within each point inside the package
The dose mapping is dependent on:The dimensions and density of packaged productThe orientation of product within the package

Gamma radiation sterilization validation
VDMAX25 method
A bi b d h ll b 1000Average bioburden shall be ≤ 1000
Obtain samples of productSelect at least 10 products from each of 3 batches
Determine average bioburden
Obtain verification dose using Table 9 ( SO )(ISO 11137-2)

Gamma radiation sterilization validation
VDMAX25 method (cont.)
Perform verification dose experimentSelect 10 products from a single batchSelect 10 products from a single batchIrradiate at verification dose (±10%)Prod ct sterilit testProduct sterility test
I t t ti f ltInterpretation of results≤1 non-sterile – 25 kGy is substantiated
2 t il fi t i t=2 non-sterile – confirmatory experiment>2 non-sterile – sterilizaion dose shall be re establishedre-established

Gamma radiation sterilization validation
VDMAX25 method – confirmatory experimentMAX y p
Confirmatory verification dose experimentCo ato y e cat o dose e pe e tSelect 10 products from a single batchIrradiate at verification dose (±10%)Irradiate at verification dose (±10%)Product sterility test
Interpretation of resultsAll sterile – 25 kGy is substantiatedAll sterile 25 kGy is substantiated>1 non-sterile – sterilizaion dose shall be re-establishedre established

Gamma radiation sterilization validation
VDMAX25 method dose audit
Shall be performed quarterly
Obtain samples of productSelect at least 10 products from a single batchp g
Determine average bioburdeng
Perform verification dose experimentpSelect 10 products from a single batchIrradiate at verification dose (±10%)Irradiate at verification dose (±10%)Product sterility test

Gamma radiation sterilization validation
VDMAX25 method dose audit (cont.)
Interpretation of results
≤1 non-sterile – 25 kGy is substantiated=2 non-sterile – confirmatory experiment≥3, ≤6 non-sterile – augmentation of the sterilization dose≥7 non-sterile – sterilization dose shall be
bli h dre-established

Gamma radiation sterilization validation
VDMAX25 method dose audit – confirmatory experiment
Confirmatory verification dose experimentSelect 10 products from a single batchSelect 10 products from a single batchIrradiate at verification dose (±10%)Product sterility testProduct sterility test
Interpretation of resultsInterpretation of resultsAll sterile – 25 kGy is substantiated≥1 ≤4 non-sterile – augmentation of the sterilization≥1, ≤4 non-sterile augmentation of the sterilization dose≥5 non-sterile – sterilization dose shall be5 non sterile sterilization dose shall be re-established

Sterilization validation – additional tests
Additional tests for sterilized products
The sterilization process will not have an adverse impact on the device or its packaging:
Product functionality
Packaging integrityVisual inspectionVisual inspectionPeel testDye penetration test

Sterilization validation
M i t i i ff tiMaintaining process effectiveness
Routine monitoring of product bioburdenRoutine monitoring of product bioburden
Maintenance of the sterilization equipmentMaintenance of the sterilization equipment
Instrumentation used to monitor and control processInstrumentation used to monitor and control process parameters should be calibrated

Sterilization validation
A t f hAssessment of change
A h t i t d t k i l diA change to equipment, product, packaging, or loading pattern
Effect on effectiveness of the sterilization process
Effect on IQ, OQ or PQ validations
Documented rationale for decisions reached

Summary
Sterilization process is crucial for patient safety
Validation is required to establish that a process will consistently yield SAL of 10-6
process effectiveness must be maintained
Any change to product design or packaging shall be assessedassessed

Any questions?