reaktionstechnische untersuchungen zur selektiven ... · pdf filedoktor-ingenieur vorgelegt...
TRANSCRIPT

Reaktionstechnische Untersuchungen
zur selektiven Desorption durch Mikrowellenstrahlung
bei heterogen katalysierten Reaktionen am Beispiel der
Hydroxylierung von Benzen mit N2O
Der Technischen Fakultät der
Universität Erlangen-Nürnberg
zur Erlangung des Grades
Doktor-Ingenieur
vorgelegt von
Jörg Münch
Erlangen 2007

Als Dissertation genehmigt von der Technischen Fakultät der Universität Erlangen-Nürnberg
Tag der Einreichung: 25.04.2007
Tag der Promotion: 14.11.2007
Dekan: Prof. Dr.-Ing. habil. J. Huber
Berichterstatter: Prof. Dr. W. Schwieger
Prof. Dr. M. Willert-Porada

Die vorliegende Arbeit entstand im Zeitraum Januar 2001 bis März 2005 am Lehrstuhl für
Technische Chemie I (heute: Lehrstuhl für Chemische Reaktionstechnik) der Friedrich-
Alexander-Universität Erlangen-Nürnberg.
An erster Stelle möchte ich meinem Doktorvater Herrn Prof. Dr. W. Schwieger für das mir stets
entgegengebrachte Vertrauen und die Unterstützung während meiner Tätigkeit am Lehrstuhl
danken. Der von ihm gewährte wissenschaftliche Freiraum bei der Durchführung und Gestal-
tung der Arbeit, sowie seine zahlreichen Tipps und Anregungen haben zum Gelingen der Arbeit
entscheidend beigetragen. Nicht zuletzt war auch das hervoragende Arbeitsklima in der
„Zeolith“-Gruppe sein Verdienst. Weiterhin möchte ich mich auch bei Herrn Prof. G. Emig und
Herrn Prof. P. Wasserscheid für die freundliche Unterstützung und Aufnahme am Institut
bedanken.
Frau Prof. Dr. M. Willert-Porada danke ich für die Übernahme des Koreferats.
Einen wesentlichen Beitrag zum Erfolg der Arbeit leisteten auch auch eine Reihe von Dipolm-
und Studienarbeitern, bei denen ich mich an dieser Stelle ebenfalls herzlich bedanken möchte.
In zeitlicher Reihenfolge waren dies Herr S. Pfadler, Herr N. Olong , Frau A. Scholz und Herr
S. Gopalakrishnan.
Ein besonderer Dank gilt Herrn Dr. R. Herrmann für seine Betreuung und die zahlreichen Dis-
kussionen, Impulse und Ratschläge zum Thema Mikrowelle.
Darüber hinaus möchte ich allen Mitgliedern der Forschungsgruppe um Prof. Schwieger dan-
ken, die eine angenehme und erfolgreiche Arbeit möglich gemacht haben. Während meiner Zeit
am Lehrstuhl waren dies: Dr. F. Scheffler, Dr. T. Selvam, Dr. M. Rauscher, Dr. O. Gravenhorst,
O. Scharf, V. Herzog, Dr. A. Unger, M. Pröll, H. Baser, Dr. A. Zampierie, Dr. G. Mabande, A.
Avhale, J. Bauer und S. Gopalakrishnan.
Natürlich danke ich auch allen Kollegen aus anderen Arbeitsgruppen am Lehrstuhl, die diese
Promotion zu einem wissenschaftlichen aber auch perönlichen Gewinn gemacht haben. An die-
ser Stelle sei besonders mein Bürokollege Dr. U. Hiemer erwähnt, der nicht nur mit seinem
Fachwissen auf dem Gebiet der Hydroxylierung eine große Hilfe war. Weiterhin möchte ich
Herrn Dr. M. Köstner, Herrn Dr. H. Schäfer, Herrn Dr. H. Freund, Herrn Dr. F. Kießlich und
Herrn Ch. Schneider danken.
Dank gebührt auch allen weiteren Mitarbeitern des Lehrstuhls, wie Frau R. Müller, Herrn P.
Widlok und Herrn S. Smolny für die Charakterisierung zahlreicher Proben, Herrn H. Gerhard,
der immer ein offenes Ohr für alle technischen Probleme hatte, der mechanischen Werkstatt mit
Herrn M. Schmacks und Herrn A. Manke, Herrn G. Dommer in der Elektronikwerkstatt und
Herrn W. Fischer für alle Rechnerfragen. Dem Sekretariat mit Frau M. Menuet, Frau H. Hajas
und Frau P. Singer danke ich für die stets freundliche Hilfe bei der Bewältigung der
verwaltungs- und organisatorischen Arbeiten.
Schließlich gilt mein ganz besonderer Dank meiner Frau Andrea und Tochter Leonie sowie
meinen Eltern für ihr Verständnis und ihre Unterstüzung.


In Dankbarkeit meiner Familie
Es ist nicht genug, zu wissen,
man muß auch anwenden;
es ist nicht genug, zu wollen,
man muß auch tun.
Johann Wolfgang von Goethe


Inhaltsverzeichnis
1 Einleitung ..................................................................................................................... 1
2 Mikrowellen als Energieträger in der Reaktionstechnik......................................... 6
2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie................................................................... 6
2.1.1 Mechanismen der dielektrischen Erwärmung............................................................................. 9 2.1.1.1 Intramolekulare Effekte ....................................................................................................... 13
2.1.2 Dielektrische Erwärmung - mathematische Beschreibung ....................................................... 14 2.1.2.1 Frequenz und Temperaturabhängigkeit der Dielektrizitätskonstante ................................... 16 2.1.2.2 Wirkleistung......................................................................................................................... 21
2.1.3 An Oberflächen adsorbierte Spezies ......................................................................................... 23
2.2 Mikrowellengeräte............................................................................................................................ 25
2.2.1 Multimode-Mikrowellengeräte ................................................................................................. 25 2.2.2 Monomode-Mikrowellengeräte ................................................................................................ 27
2.3 Einsatzgebiete - Stand der Technik ................................................................................................ 28
2.3.1 Organische Synthesen............................................................................................................... 28 2.3.1.1 Synthesen in der Flüssigphase ............................................................................................. 29 2.3.1.2 Synthesen, die Feststoffe als Support verwenden ................................................................ 30 2.3.1.3 Synthesen unter Verwendung von Feststoffkatalysatoren ................................................... 31 2.3.1.4 Diskussion der beobachteten Mikrowelleneffekte ............................................................... 32
2.3.2 Heterogene Gasphasenkatalyse im Mikrowellenfeld ................................................................ 34 2.3.2.1 Methanzersetzung und oxidierende Methankupplung.......................................................... 36 2.3.2.2 Oxidationsreaktionen ........................................................................................................... 38 2.3.2.3 NOx und SOx Reduktion....................................................................................................... 43 2.3.2.4 Andere Reaktionen............................................................................................................... 47 2.3.2.5 Diskussion der beobachteten Mikrowelleneffekte ............................................................... 50
2.3.3 Einfluss von Mikrowellen auf Sorptionsvorgänge.................................................................... 58 2.3.3.1 Trocknung ............................................................................................................................ 58 2.3.3.2 Regeneration von Adsorbentien ........................................................................................... 59 2.3.3.3 Konkurrenzadsorption.......................................................................................................... 61
3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren .......................... 66
3.1 Verwendung von Phenol .................................................................................................................. 66
3.2 Industrielle Produktionsverfahren für Phenol .............................................................................. 67
3.3 Wirtschaftliche Gesichtspunkte - Marktentwicklung Phenol....................................................... 69
3.4 Hydroxylierung von Benzen mit N2O ............................................................................................. 73
3.4.1 ZSM-5 Zeolithe......................................................................................................................... 74 3.4.1.1 Historisches.......................................................................................................................... 74 3.4.1.2 Struktur ................................................................................................................................ 75 3.4.1.3 Synthese und postsynthetische Modifizierung ..................................................................... 77 3.4.1.4 Katalytische Eigenschaften .................................................................................................. 79 3.4.1.5 Sorptionseigenschaften ........................................................................................................ 81 3.4.1.6 Dielektrische Eigenschaften................................................................................................. 84

II INHALTSVERZEICHNIS
3.4.2 Reaktionsmechanismus .............................................................................................................89 3.4.3 Desaktivierung ..........................................................................................................................92
4 Zielsetzung der Arbeit...............................................................................................98
5 Eingesetzte Katalysatoren ......................................................................................100
5.1 Auswahlkriterien ............................................................................................................................100
5.2 Klassische Katalysatorcharakterisierung.....................................................................................101
5.2.1 Elementaranalyse ....................................................................................................................101 5.2.2 Strukturanalyse........................................................................................................................101 5.2.3 Adsorptionseigenschaften .......................................................................................................102 5.2.4 Säureeigenschaften..................................................................................................................103 5.2.5 Ergebnisse ...............................................................................................................................104
5.3 Charakterisierung der dielektrischen Eigenschaften ..................................................................108
5.3.1 Versuchsdurchführung ............................................................................................................108 5.3.2 Unbeladene, dehydratisierte Katalysatorproben......................................................................109
5.3.2.1 Modulabhängigkeit.............................................................................................................110 5.3.3 Mit Phenol beladene, dehydratisierte Katalysatorproben........................................................111
6 Entwicklung der Mikrowellenanlage.....................................................................114
6.1 Konzeption ......................................................................................................................................115
6.2 Komponenten der Anlage ..............................................................................................................118
6.2.1 Dosierung ................................................................................................................................118 6.2.2 Reaktor ....................................................................................................................................120 6.2.3 Temperaturmessung und Regelung .........................................................................................122 6.2.4 Mikrowellensystem.................................................................................................................124 6.2.5 Analysensystem ......................................................................................................................126 6.2.6 Anlagensteuerung....................................................................................................................127 6.2.7 Modifikation des Versuchsaufbaus für die Untersuchung der N2O-Zersetzung .....................128
6.3 Untersuchungen zur Idealität des eingesetzten Reaktors............................................................128
6.3.1 Druckverlust............................................................................................................................129 6.3.2 Axiale Rückvermischung ........................................................................................................129 6.3.3 Radiale Gradienten in der Strömungsgeschwindigkeit ...........................................................129 6.3.4 Isothermie / Temperaturgradienten .........................................................................................130
6.4 Für Sorptionsuntersuchungen eingesetzte Apparaturen und Analytik .....................................132
7 Reaktionstechnische Untersuchungen...................................................................134
7.1 N2O-Zersetzung ..............................................................................................................................134
7.1.1 Versuchsdurchführung ............................................................................................................135 7.1.2 H-ZSM-5 (I) ............................................................................................................................135 7.1.3 Fe-ZSM-5 (I)...........................................................................................................................136 7.1.4 Fe-ZSM-5 (II) .........................................................................................................................137 7.1.5 Co-ZSM-5 ...............................................................................................................................138 7.1.6 Zusammenfassung und Diskussion der Ergebnisse.................................................................139

INHALTSVERZEICHNIS III
7.2 Hydroxylierung von Benzen mit N2O ........................................................................................... 140
7.2.1 Versuchsdurchführung............................................................................................................ 140 7.2.2 H-ZSM-5 (I)............................................................................................................................ 141
7.2.2.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C.......................................... 141 7.2.2.2 Variation der Reaktionstemperatur .................................................................................... 142
7.2.3 Fe-ZSM-5 (I) .......................................................................................................................... 143 7.2.3.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C.......................................... 144 7.2.3.2 Variation der Reaktionstemperatur .................................................................................... 144 7.2.3.3 Variation der Mikrowellenleistung .................................................................................... 146
7.2.4 Fe-ZSM-5 (II) ......................................................................................................................... 147 7.2.4.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C.......................................... 147 7.2.4.2 Variation der Reaktionstemperatur .................................................................................... 148 7.2.4.3 Variation der Mikrowellenleistung .................................................................................... 149 7.2.4.4 Variation der Verweilzeit ................................................................................................... 150 7.2.4.5 Überstöchiometrische Benzenzugabe ................................................................................ 156
7.2.5 Co-ZSM-5............................................................................................................................... 159 7.2.5.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C.......................................... 159 7.2.5.2 Variation der Reaktionstemperatur .................................................................................... 160 7.2.5.3 Variation der Mikrowellenleistung .................................................................................... 161 7.2.5.4 Variation der Verweilzeit ................................................................................................... 161 7.2.5.5 Wasserzugabe zum Feed .................................................................................................... 168
7.2.6 Diskussion der Ergebnisse ...................................................................................................... 172 7.2.6.1 Vergleich der Katalysatoren bei einer Reaktionstemperatur von 400 °C........................... 172 7.2.6.2 Einfluss der Reaktionstemperatur ...................................................................................... 174 7.2.6.3 Einfluss der Verweilzeit ..................................................................................................... 176 7.2.6.4 Überstöchiometrische Benzenzugabe ................................................................................ 176 7.2.6.5 Wasserzugabe zum Feed .................................................................................................... 176 7.2.6.6 Auswirkungen der Mikrowelleneinstrahlung..................................................................... 177
7.2.7 Zusammenfassung .................................................................................................................. 179
8 Charakterisierung des Desorptionsverhaltens ..................................................... 181
8.1 Versuchsdurchführung .................................................................................................................. 181
8.2 In situ beladene Proben (aus Einsatz in der Reaktion) ............................................................... 182
8.2.1 H-ZSM-5 (I)............................................................................................................................ 183 8.2.2 Fe-ZSM-5 (II) ......................................................................................................................... 185
8.3 Ex situ beladene Proben................................................................................................................. 187
8.3.1 H-ZSM-5 (I)............................................................................................................................ 187 8.3.2 Fe-ZSM-5 (II) ......................................................................................................................... 191
8.4 Zusammenfassung und Diskussion der Ergebnisse..................................................................... 196
9 Zusammenfassung und Ausblick ........................................................................... 200
10 Summary and Outlook............................................................................................ 208
Literaturverzeichnis ....................................................................................................... 215
Symbolverzeichnis .......................................................................................................... 225

IV INHALTSVERZEICHNIS
Appendix..........................................................................................................................230
A t-Plots der N2-Sorptionsmessungen ...............................................................................................230
B Kurvenfits der NH3-TPD-Profile...................................................................................................232
C Berechnung Na-Anteil in Na-ZSM-5.............................................................................................234
D Temperaturprofil des verwendeten Reaktors ..............................................................................235
E Konstruktionszeichnung des verwendeten Reaktors...................................................................236
F Bestimmung der axialen Bodenstein-Zahl über empirische Korrelationsbeziehungen............237
G Verweilzeitverteilung des verwendeten Reaktors ........................................................................238
H Ergänzende Messungen in der Hydroxylierung von Benzen mit N2O .......................................239

1 Einleitung
Mikrowellentechniken wurden erstmals in den 40’er Jahren des letzten Jahrhunderts in
größerem Umfang verwendet. Während des zweiten Weltkriegs beschränkte sich der Ein-
satz jedoch ausschließlich auf Radaranwendungen für militärische Zwecke [1, 2]. Mehr
durch Zufall wurde entdeckt, dass die emittierte Strahlung auch zum Erwärmen von Was-
ser geeignet ist. 10 Jahre später waren dann erste Mikrowellenöfen auf dem freien Markt
erhältlich. Der große Durchbruch im Bereich des Haushaltssektors kam aber erst in den
70’er Jahren aufgrund der durch Massenproduktion fallenden Preise für die benötigten
Magnetrone [3-5]. Heute findet man kaum mehr eine moderne Küche ohne solch einen
Mikrowellenofen. Das gleiche könnte schon bald auch für chemische Labors zutreffen.
Wie auch im Haushaltsbereich ist einer der größten Vorteile der Mikrowellen, dass sich
Reaktionslösungen deutlich schneller aufheizen lassen als mit konventionellen Methoden
[6]. Seit Mitte der 70’er Jahre experimentieren Chemiker mit Mikrowellen als Energieträ-
ger für chemische Reaktionen [7]. Bei den ersten Versuchen wurden gewöhnliche Haus-
haltsmikrowellengeräte verwendet. Führte man eine Reaktion in der Mikrowelle anstatt
mit einer konventionellen Heizung durch, zeigten sich meist sehr erstaunliche Ergebnisse
– in der Regel um vielfach höhere Umsatzgrade bzw. deutlich verkürzte Reaktionszeiten.
Weiterhin wurden veränderte Selektivitäten oder auch chemo-, regio-, und stereoselektive
Effekte beschrieben [8]. Das Problem bei vielen dieser Experimente war jedoch, dass im
Mikrowellenfeld zum damaligen Zeitpunkt weder eine Temperatur- noch Druckmessung
des Reaktionsgefäßes möglich war. Die Reproduzierbarkeit der Ergebnisse lies außerdem
meist sehr zu wünschen übrig [6]. Da sich viele dieser beobachteten Phänomene zu die-
sem Zeitpunkt nicht eindeutig erklären ließen, kam es zu einer Unterscheidung in so
genannte „nichtthermische“ und „thermische“ Mikrowelleneffekte [9]. „Thermische“
Effekte sind auf die dielektrische Heizwirkung der Mikrowellen zurückzuführen, welche
im Vergleich zu anderen Heizmethoden ein abweichendes Temperaturregime hervorrufen
kann. „Nichtthermische“ Effekte können hingegen nur durch den Einfluss elektromagneti-
scher Strahlung hervorgerufen werden und beeinflussen das thermische Regime nicht
[10]. Ein Beispiel für solch einen „nichtthermischen“ Mikrowelleneffekt ist die Herabset-
zung der Gibbs’schen Aktivierungsenergie durch die Strahlungsenergie der Mikrowellen
[11], während die selektive Aufheizung bestimmter an der Reaktion beteiligter Spezies
einen „thermischen“ Effekt darstellt [12].

2 1 Einleitung
Heute ist man sich zumindest darüber einig, dass Mikrowellen eine direkte, gleichmäßi-
gere und schnellere Aufheizung einer Reaktionslösung bei inversen Temperaturprofilen
ermöglichen. Weiterhin wird davon ausgegangen, dass Mikrowellen ein selektives Heizen
bestimmter Phasen/Stoffgruppen ermöglichen. Der Mechanismus, der diesen Effekten zu
Grunde liegt, ist jedoch bis heute nicht vollständig verstanden. Außerdem ist nicht klar,
wie sich diese Effekte auf eine chemische Reaktion auswirken können [13]. In den letzten
Jahren hat sich jedoch immer deutlicher gezeigt, dass die in der Literatur beschriebenen
„nichtthermischen“ Effekte nicht erklärbar sind, bzw. sich auf „thermische“ Effekte
zurückführen lassen [14]. Darüber hinaus haben theoretische Berechnungen bestätigt, dass
Mikrowellenstrahlung bei den für Heizzwecke eingesetzten Frequenzen von 2,45 GHz
bzw. 900 MHz keinen Einfluss auf die für chemische Reaktionen verantwortliche Valenz-
elektronen haben, und somit auch keine molekularen Veränderungen hervorrufen kann
[15]. Die Diskussion über die Wirkungsweise von Mikrowellen und wie sich diese Effekte
für chemische Reaktionen sinnvoll einsetzen lassen, wird in den letzten Jahren immer
intensiver geführt. Es werden viele Stimmen laut, welche ein systematischeres Vorgehen
bei der Erforschung mikrowellenunterstützter Reaktionen und damit auch eine bessere
Aufklärung der zugrunde liegenden Effekte fordern [10].
Ein Problem hierbei ist schon teilweise gelöst. Auf dem Markt gibt es mittlerweile kom-
merziell erhältliche Mikrowellensysteme für organische Synthesen, welche eine exakte
Prozesskontrolle (Temperatur, Druck, etc.) in der Flüssigphase ermöglichen. Fehler (z. B.
durch inakkurate Temperaturmessung), wie sie bei den ersten Experimenten in Haus-
haltsmikrowellen auftraten, lassen sich somit vermeiden [6]. Für heterogen katalysierte
Reaktionen, die ebenfalls schon seit Anfang der 80’er Jahre [16] im Mikrowellenfeld
untersucht werden, ist dies (noch) nicht der Fall. Seit 1995 ist die Zahl der Veröffent-
lichungen, welche sich mit der heterogenen Katalyse im Mikrowellenfeld beschäftigen,
deutlich gestiegen. Zahlreiche heterogene Reaktionssysteme wurden untersucht, meist mit
dem Ergebnis, dass die Mikrowellenstrahlung dazu führt, dass die Reaktion schon bei
deutlich niedrigeren Temperaturen mit vergleichbaren Umsatzgraden abläuft bzw. dass es
zu einer Veränderung der Produktzusammensetzung kam. Die Ursachen dieser in der
heterogenen Katalyse mit Mikrowellen beobachteten Effekte sind bisher jedoch noch
nicht befriedigend aufgeklärt [4]. Will man die Effekte von Mikrowellen auf heterogen
katalysierte Reaktionen untersuchen, sind zuverlässige Vergleichsmessungen zwischen
konventionellen und mit Mikrowellen geheizten Experimenten unabdingbar. Vorrausset-
zung hierfür ist natürlich eine exakte Temperaturmessung und Prozesskontrolle. Dies ist
innerhalb starker elektromagnetischer Wechselfelder (Mikrowelle) jedoch nicht unprob-
lematisch [10].

1 Einleitung 3
Um eine systematische Aufklärung der Mikrowelleneffekte zu ermöglichen, ist es nötig,
sich auf bereits gut erforschte Reaktionssysteme zu konzentrieren, bei denen nur einer der
in der Literatur diskutierten Mikrowelleneffekte auftreten kann (siehe Kapitel 2.3).
Aus der Literatur ist bekannt, dass eine selektive Aufheizung bestimmter Phasen bzw.
Stoffgruppen durch Mikrowellenstrahlung möglich ist [6]. Der technische Nutzen solch
einer selektiven Heizung liegt auf der Hand, stellt man sich vor, dass es zahlreiche Reak-
tionen gibt, bei denen eine „Temperaturdifferenz“ zwischen verschiedenen an der Reak-
tion beteiligten Stoffen wünschenswert ist. Beispielsweise könnte man ein Edukt selektiv
aufheizen, während das temperaturempfindliche Produkt nicht durch die Mikrowellen
beeinflusst wird. Die „makroskopische“ Reaktionstemperatur ließe sich somit deutlich
verringern, da die benötigte Aktivierungsenergie direkt den Reaktanden zugeführt und
gegebenenfalls sofort verbraucht wird, während Bereiche, in denen keine Edukte vorhan-
den sind, nicht geheizt werden. Dies ist mit konventionellen Heizmethoden undenkbar.
Einige Forschergruppen beschäftigen sich auch mit dem Einfluss von Mikrowellenstrah-
lung auf Sorptionsvorgänge an Oberflächen [17, 18]. Diese Untersuchungen zeigen, dass
die Mikrowellen eine gänzlich andere Wirkungsweise als konventionelle Heizmethoden
auf diese aus Adsorbens und Adsorptiven bestehenden Mehrstoffsysteme haben. Im
Arbeitskreis um BATHEN bzw. SCHMIDT-TRAUB sind zahlreiche Publikationen ent-
standen [17, 19, 20], welche sich mit der Regenerierung von Festbett-Adsorbern durch
Mikrowellenstrahlung beschäftigen. Bei diesen Untersuchungen hatte sich gezeigt, dass es
mit Hilfe der Mikrowellenstrahlung möglich ist, selektiv polare Spezies vom Adsorbens
zu desorbieren, während unpolare Moleküle beinahe unbeeinflusst auf der Oberfläche
verbleiben. Dieser Einfluss der Mikrowellen auf Ad- und Desorptionsvorgänge könnte
auch während einer Reaktion eine entscheidende Rolle spielen.
Ausgehend von diesen Erkenntnissen des Arbeitskreises um SCHMIDT-TRAUB über den
Einfluss von Mikrowellenstrahlung auf Desorptionsvorgänge an Oberflächen war es ein
Ziel der vorliegenden Arbeit, die Auswirkungen von Mikrowellenstrahlung auf Sorptions-
vorgänge während einer chemischen Reaktion zu untersuchen. Als Reaktionssystem
wurde die Hydroxylierung von Benzen zu Phenol mit Distickstoffmonoxid (N2O) als
Oxidationsmittel an Zeolithkatalysatoren gewählt.
Diese Reaktion ist von großem wirtschaftlichem Interesse. Seit den 90’er Jahren ist man
auf der Suche nach einem Alternativprozess zum Hock-Verfahren, welches heute mit
einem Marktanteil von ca. 90 % die wichtigste Route der Phenolproduktion darstellt.
Problematisch erweist sich bei diesem Verfahren die Koppelproduktion von Aceton. Die
Wirtschaftlichkeit dieses Prozesses ist somit direkt an den Marktwert des Acetons gebun-
den. Zwar wächst der Markt für Phenol seit 2001 jährlich um ca. 4 %, der Markt für
Aceton weist dagegen nur eine Steigerungsrate von ca. 3 % auf, was auf lange Sicht zu

4 1 Einleitung
einer Überproduktion an Aceton führen wird [21]. Weiterhin handelt es sich beim Hock-
Verfahren um einen dreistufigen Prozess mit entsprechend großem anlagentechnischen
Aufwand. Auch hier bietet die Hydroxylierung von Benzen mit N2O deutliche Vorteile
gegenüber dem Stand der Technik. Seit 1996 untersucht die Firma SOLUTIA diesen Pro-
zess in einer Pilotanlage [22]. Als eines der größten Probleme bei diesem Verfahren hat
sich allerdings die sehr kurze Standzeit des Katalysators erwiesen. Es wird davon ausge-
gangen, dass während der Reaktion gebildeter hard coke für die schnelle Desaktivierung
verantwortlich ist. Der Mechanismus der Desaktivierung wurde bisher jedoch noch nicht
vollständig aufgeklärt. Bisher gibt es nur Hinweise darauf, dass das während der Reaktion
gebildete Phenol aufgrund seiner starken Adsorption am Katalysator als so genannter
„Koksprecursor“ fungiert und für den Aktivitätsrückgang verantwortlich sein soll [23, 24].
Diese Arbeit steht im Zusammenhang mit einer Reihe von Arbeiten am Lehrstuhl für
Chemische Reaktionstechnik der Universität Erlangen-Nürnberg, welche ein umfassende-
res Verständnis der Vorgänge bei der Hydroxylierung von Benzen mit N2O zum Ziel hat-
ten, um eine wissenschaftliche Grundlage für die technische Realisierbarkeit dieses
Prozesses zu erhalten. Dabei wurden sowohl die materialseitige Optimierung der Kataly-
satoren bzw. die Aufklärung der Struktur-Wirkbeziehungen [25], sowie reaktionstechni-
sche Aspekte (Erstellung einer detaillierten Reaktionskinetik) [24, 26] verfolgt. Der
Einsatz von Mikrowellen als Heizmedium kann wiederum als Ansatz einer prozessseiti-
gen Optimierung dieser Reaktion, aber auch als gezielter Eingriff in den Reaktionsmecha-
nismus verstanden werden und ergänzt bzw. erweitert die anderen Arbeiten.
Weiterhin erfüllt diese Reaktion die Voraussetzungen, um die selektive Aufheizung von
an einer Reaktion beteiligter Spezies durch Mikrowellen untersuchen zu können:
a.) Das Reaktionssystem wurde in den letzten 20 Jahren intensiv erforscht [24, 26-
28]. Der Reaktionsmechanismus ist weitestgehend aufgeklärt, ebenso die Bildung
von Nebenprodukten und der Einfluss der verschiedenen Reaktionsparameter
(Temperatur, Eduktzusammensetzung, Verweilzeit) auf die Reaktion (vgl. Kapitel
3.4).
b.) Während der Reaktion kommt es zu einer erheblichen Desaktivierung durch
Kohlenstoffablagerungen. Als Ursache wird die starke Adsorption des Phenol an
den aktiven Zentren des Katalysators und die daraus resultierende Weiterreaktion
zu soft coke in der Literatur genannt (vgl. Kapitel 3.4.3).
c.) Es existieren Katalysatoren, welche in der Hydroxylierung aktiv sind und keine
Mikrowellenstrahlung adsorbieren (vgl. Kapitel 3.4.1.6). Dies ist nach BATHEN
[19] eine Voraussetzung für eine selektive Desorption adsorbierter Spezies durch
Mikrowellen.

1 Einleitung 5
d.) Während der Reaktion liegen sowohl unpolare (Benzen) als auch polare Stoffe
(Phenol) an der Katalysatoroberfläche adsorbiert vor.
Wird die Desaktivierung durch die starke Adsorption von Phenol an den aktiven Zentren
des Katalysators hervorgerufen, könnte der Einsatz von Mikrowellen längere Katalysa-
torstandzeiten ermöglichen.
Es ist zu erwarten das Phenol durch die Mikrowellenstrahlung selektiv aufgeheizt wird.
Dadurch kann eine Desorption von der Katalysatoroberfläche erreicht und die Weiterre-
aktion zu Kohlenstoffablagerungen verhindert bzw. verringert werden (siehe Abbildung
1.1). Eine Desaktivierung findet in diesem Fall nicht, oder zumindest stark verlangsamt
statt.
Sind für die Desaktivierung die selben aktiven Zentren verantwortlich wie für die Hydro-
xylierung des Benzens werden durch die beschleunigte Desorption des Phenol zusätzliche
aktive Zentren frei. Dies hat wiederum höhere Umsatzgrade zur Folge.
Abbildung 1.1: Einsatz von Mikrowellen zur selektiven Desorption des Phenols von der
Katalysatoroberfläche während der Hydroxylierung von Benzen mit N2O
Diese Reaktion ist somit aus wissenschaftlicher Sicht hervorragend geeignet, um den Ein-
fluss von Mikrowellen auf Sorptionsvorgänge während einer heterogen katalysierten Gas-
phasenreaktion zu untersuchen. Außerdem ist auch ein beträchtlicher wirtschaftlicher
Anreiz gegeben, da der Einsatz von Mikrowellen ein Weg sein könnte, um die Desaktivie-
rung des Katalysators während dieser Reaktion zu verhindern.

2 Mikrowellen als Energieträger in der Reaktionstechnik
Im folgenden Kapitel werden die physikalischen Grundlagen der Mikrowellenstrahlung
und der Vorgänge bei der Wechselwirkung dieser Strahlung mit Materie behandelt. Die
Mechanismen, die zu einer Erwärmung eines Körpers durch elektromagnetische Wellen
beitragen, werden aufgezeigt (Kapitel 2.1). In Kapitel 2.2 werden die üblicherweise im
Labor eingesetzten Mikrowellengeräte erläutert. Anschließend wir ein Überblick über den
Stand der Literatur zum Einsatz von Mikrowellenstrahlung zur Förderung chemischer
Reaktionen und zur Beeinflussung von Sorptionsvorgängen an porösen Feststoffen gege-
ben (Kapitel 2.3).
2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie
Als Mikrowellen werden elektromagnetische Wellen in einem Frequenzbereich zwischen
300 MHz und 30 GHz bezeichnet [29]. In Abbildung 2.1 wird die Einordnung ins elektro-
magnetische Spektrum veranschaulicht. Im niederen Frequenzbereich grenzen sie an die
Radiowellen, im hochfrequenten Bereich an die Infrarotstrahlung. Die korrespondierenden
Wellenlängen bewegen sich demnach zwischen 1 m und 30 cm.
Abbildung 2.1: Elektromagnetisches Spektrum
Mikrowellen werden aufgrund ihres vergleichsweise geringen Energieinhalts den so
genannten nicht-ionisierenden Strahlungen zugeordnet. Anders als beispielsweise bei der
UV- oder Röntgenstrahlung ist der Energieinhalt der Mikrowellenstrahlung zu gering, um
eine Bindungsspaltung oder ein Entfernen von Elektronen aus den Orbitalen eines Atoms
zu bewirken [30, 31]. Eine Wasserstoffbrückenbindung besitzt eine Bindungsenergie von
ca. 4 - 42 kJ/mol, während die Energie von Mikrowellenphotonen bei einer Frequenz von
2,45 GHz in etwa 0,001 kJ/mol beträgt [4].

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 7
Durchlaufen Mikrowellen ein Medium so kann die Strahlung von diesem absorbiert
werden. Dies kann durch so genannte Resonanz (Molekül wird durch Aufnahme eines
Quants in einen definiert höheren Energiezustand versetzt) oder durch Umwandlung in
thermische Energie geschehen. Moleküle oder Molekülgruppen werden in Rotation oder
Schwingung versetzt. Diesen Resonanz-Effekt macht man sich bei der Mikrowellenspekt-
roskopie zunutze. Es kann sowohl das ganze Molekül um die eigene(n) Achse(n) rotieren
oder einzelne Atome eines Moleküls schwingen um ihre Gleichgewichtslage.
Schwingungs und Rotationsanregung sind gekoppelt, so dass ein breites Spektrum von
Energiezuständen resultiert, während bei klassischen Atomspektren nur durch Elektro-
nenübergänge Energie adsorbiert oder emittiert wird. Änderungen des Schwingungszu-
standes treten aber vorrangig im Bereich der Infrarotstrahlung, bzw. bei infrarotnahen
Mikrowellenfrequenzen (fernes Infrarot) auf. Wird das Molekül mit ausreichend Energie
(Frequenz) angeregt kann es aus dem Grundzustand in den ersten angeregten
Schwingngszustand gehoben werden. In der Regel wird nun bei verschiedenen Frequen-
zen die Absorption gemessen. Um exakte Adsorptionsspektren zu erhalten, muss die
Wechselwirkung der Moleküle untereinander minimiert werden. Deshalb wird wie meist
mit verdünnten (Vakuum) Gasen gearbeitet. Die Adsorptionslinien ergeben sich aus der
Energiedifferenz quantenmechnisch festgelegter Energieniveus von Molekülrotationen
(Rotationsübergang) oder Molekülschwingungen (Schwingungsübergang). Im Bereich der
fernen infraroten Strahlung kommt es vorrangig zur Rotation ganzer Moleküle, während
im mittleren oder normalen Infrarot Atome oder Atomgruppen an ihren Bindungen zur
Schwingung angeregt werden. Die IR-Spektroskopie wird deshalb häufig zur Strukturauf-
klärung unbekannter Substanzen verwendet, während durch Mikrowellenspektroskopie
beispielsweise die Bindungslänge einfacher Moleküle bestimmt wird [32-34].
Mikrowellen der für Heizzwecke eingesetzten Wellenlängen rufen jedoch vorrangig eine
Umwandlung der Strahlung in thermische Energie hervor, z.B. durch Rotation von Dipo-
len um ihren Ladungsschwerpunkt (vgl. Relaxation S. 11), magnetische Verluste oder
Leitungsverluste [35]. Auf diese Effekte wird in Kapitel 2.1.1 und 2.1.2 noch näher einge-
gangen.
Wie alle elektromagnetischen Strahlungen können Mikrowellen beim Auftreffen auf einen
Körper in Abhängigkeit seiner Materialeigenschaften reflektiert (z. B. Metalle) oder
absorbiert (z. B. Wasser) werden bzw. ein Material ohne Wechselwirkung
(z. B. Quarzglas) durchdringen [29]. Die Erwärmung von Stoffen durch Mikrowellen birgt
einige entscheidende Vorteile gegenüber anderen Verfahren. Bei herkömmlichen
Methoden wird die Wärme entweder durch Wärmeleitung, Konvektion oder
Wärmestrahlung an das zu erwärmende Gut übertragen. Die Energieabgabe erfolgt somit
an die Oberfläche des Stoffes und wird anschließend durch Wärmeleitung ins Innere wei-
tergeleitet. Die Folge ist ein von Außen nach Innen abnehmendes Temperaturgefälle im
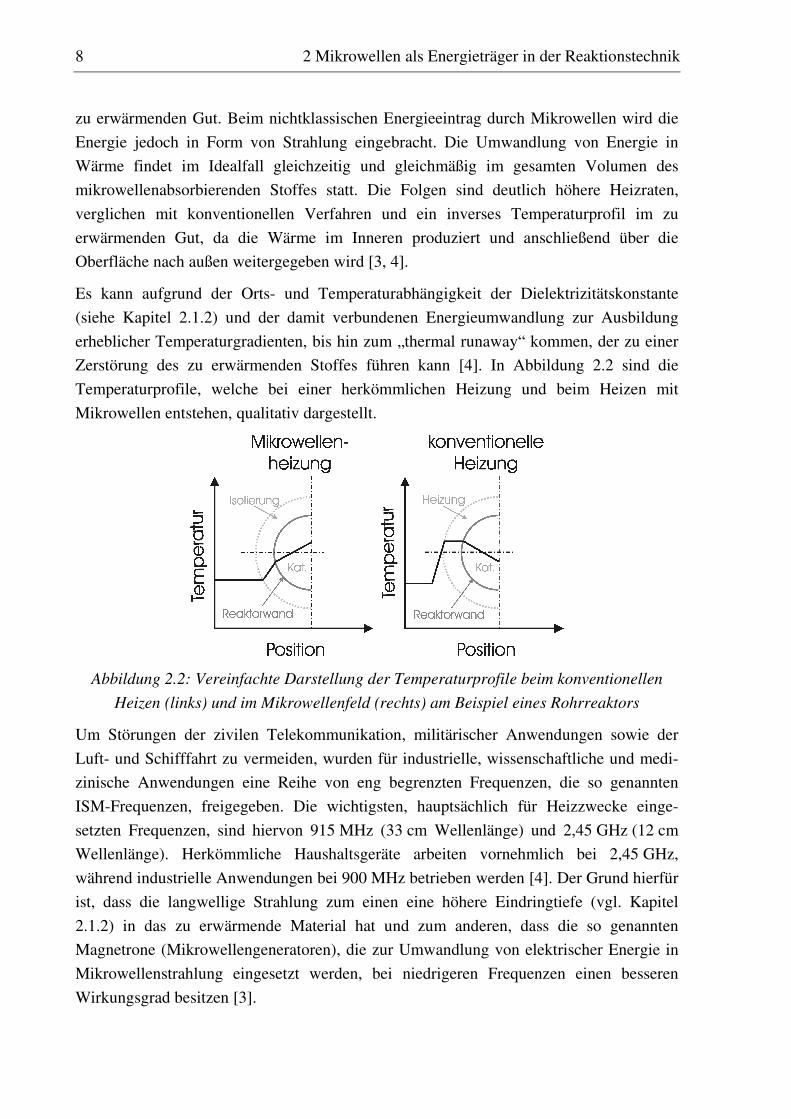
8 2 Mikrowellen als Energieträger in der Reaktionstechnik
zu erwärmenden Gut. Beim nichtklassischen Energieeintrag durch Mikrowellen wird die
Energie jedoch in Form von Strahlung eingebracht. Die Umwandlung von Energie in
Wärme findet im Idealfall gleichzeitig und gleichmäßig im gesamten Volumen des
mikrowellenabsorbierenden Stoffes statt. Die Folgen sind deutlich höhere Heizraten,
verglichen mit konventionellen Verfahren und ein inverses Temperaturprofil im zu
erwärmenden Gut, da die Wärme im Inneren produziert und anschließend über die
Oberfläche nach außen weitergegeben wird [3, 4].
Es kann aufgrund der Orts- und Temperaturabhängigkeit der Dielektrizitätskonstante
(siehe Kapitel 2.1.2) und der damit verbundenen Energieumwandlung zur Ausbildung
erheblicher Temperaturgradienten, bis hin zum „thermal runaway“ kommen, der zu einer
Zerstörung des zu erwärmenden Stoffes führen kann [4]. In Abbildung 2.2 sind die
Temperaturprofile, welche bei einer herkömmlichen Heizung und beim Heizen mit
Mikrowellen entstehen, qualitativ dargestellt.
Abbildung 2.2: Vereinfachte Darstellung der Temperaturprofile beim konventionellen
Heizen (links) und im Mikrowellenfeld (rechts) am Beispiel eines Rohrreaktors
Um Störungen der zivilen Telekommunikation, militärischer Anwendungen sowie der
Luft- und Schifffahrt zu vermeiden, wurden für industrielle, wissenschaftliche und medi-
zinische Anwendungen eine Reihe von eng begrenzten Frequenzen, die so genannten
ISM-Frequenzen, freigegeben. Die wichtigsten, hauptsächlich für Heizzwecke einge-
setzten Frequenzen, sind hiervon 915 MHz (33 cm Wellenlänge) und 2,45 GHz (12 cm
Wellenlänge). Herkömmliche Haushaltsgeräte arbeiten vornehmlich bei 2,45 GHz,
während industrielle Anwendungen bei 900 MHz betrieben werden [4]. Der Grund hierfür
ist, dass die langwellige Strahlung zum einen eine höhere Eindringtiefe (vgl. Kapitel
2.1.2) in das zu erwärmende Material hat und zum anderen, dass die so genannten
Magnetrone (Mikrowellengeneratoren), die zur Umwandlung von elektrischer Energie in
Mikrowellenstrahlung eingesetzt werden, bei niedrigeren Frequenzen einen besseren
Wirkungsgrad besitzen [3].

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 9
2.1.1 Mechanismen der dielektrischen Erwärmung
Die Grundlagen für das theoretische Verständnis der Wechselwirkung von Mikrowellen
mit Materie wurden in den frühen 50’er Jahren des letzten Jahrhunderts von ARTHUR
VON HIPPEL und seinen Mitarbeitern gelegt. Unter seiner Leitung entstand auch eine der
ersten Datenbanken von dielektrischen Eigenschaften verschiedenster chemischer Sub-
stanzen, Materialien und Nahrungsmitteln [36]. Die Wärmeerzeugung durch Mikrowellen
wird fast ausschließlich durch die elektrische Komponente des Wechselfelds hervorgeru-
fen. Bringt man eine Flüssigkeit oder einen Feststoff in das elektrische Feld, wird eine
Kraft auf polare oder geladene Partikel ausgeübt. Können sich diese frei in ihrer Umge-
bung bewegen, wird durch das Feld ein Strom in dem Stoff induziert, welcher durch
ohmsche Verluste zu einer Erwärmung führt [37]. Bringt man so genannte Dielektrika
(Isolatoren, Nichtleiter) [38] in ein elektrisches Feld, wird im Inneren ein Gegenfeld
aufgebaut, das das äußere Feld jedoch nicht kompensiert. Durch Ausrichtung elektrischer
Dipole werden die Ladungsschwerpunkte getrennt und dabei eine Polarisation innerhalb
des Stoffes erzeugt [39]. Durch diese Polarisation wird Energie des elektrischen Feldes
gespeichert.
Man unterscheidet im Allgemeinen fünf Polarisationsmechanismen, die im Folgenden
genauer betrachtet werden sollen [40, 41]. Eine Übersicht gibt Abbildung 2.3.
1. Elektronenpolarisation:
Das elektrische Feld E erzwingt bei unpolaren Molekülen eine Polarisation auf
molekularer oder atomarerer Ebene. Die Elektronenwolken der Atome werden so
verschoben, dass im zeitlichen Mittel die Ladungsschwerpunkte von Atomkern und
Atomhülle nicht mehr zusammenfallen (↔ Resonanz).
2. Atompolarisation:
Bei Molekülen mit unterschiedlichen Atomen werden die bindenden Elektronen von
den beiden Atomen unterschiedlich stark angezogen. Fallen die Ladungsschwerpunkte
beider Atome zusammen, ist das Molekül nach außen hin zwar neutral, es existieren
aber gegensätzliche Partialladungen an beiden Atomen. Durch die elektromagnetische
Strahlung wird dieser Gleichgewichtszustand gestört, da die Ladungen sich in
Richtung des elektrischen Feldes anordnen. Es kommt durch diese so genannte
atomare Polarisation zur Ausbildung eines Dipols (↔ Resonanz).
3. Ionenpolarisation:
Ist ein Material aus gegensätzlich geladenen Ladungsträgern aufgebaut, kommt es
durch Anlegen eines Feldes zu einer Verschiebung der Ladungsträger im Gitter
entsprechend ihrer Ladung. Der Stoff wird durch das elektrische Feld polarisiert (↔
Resonanz).

10 2 Mikrowellen als Energieträger in der Reaktionstechnik
4. Orientierungspolarisation:
Stoffe, welche von Natur aus permanente Dipole besitzen (z. B. Wasser) unterliegen
im Mikrowellenfeld wie bereits oben beschrieben der so genannte Orientierungspola-
risation, bei der sich die Dipole unter dem Einfluss des elektrischen Feldes in Richtung
der Feldlinien ausrichten (↔ Relaxation).
5. Grenzflächenpolarisation (Maxwell-Wagner Polarisation):
Bei elektrisch heterogenen Stoffen kann es durch Anlegen eines elektrischen Feldes zu
einer Ungleichverteilung von Ladungen entlang von Grenzflächen kommen. Sind
beispielsweise leitende Partikel in eine nicht leitende Matrix eingebettet, wandern die
gut beweglichen Ladungsträger an die Grenzflächen zwischen den Partikeln und dem
umgebenden Medium. Dieser Effekt führt zu einer höheren Polarisation als sie im
homogenen Material auftreten würde (↔ Relaxation).
Abbildung 2.3: Übersicht Polarisationsmechanismen nach [40, 41]
In Abhängigkeit von der Frequenz und den stoffspezifischen Eigenschaften des mit
Mikrowellen bestrahlten Materials überlagern sich die beschriebenen Effekte in unter-
schiedlichem Ausmaß.
Befinden sich N Moleküle pro Raumeinheit in einem elektrischen Feld, so setzt sich die
Gesamtpolarisation P folgendermaßen zusammen [40]:
(Gl. 2.1)
Wobei m das durchschnittliche Gesamtmoment pro Molekül darstellt.
P N m= ⋅

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 11
Die Gesamtpolarisation P kann als Moment pro Raumeinheit oder als induzierte Oberflä-
chendichte der Ladung betrachtet werden. Für m gilt:
(Gl. 2.2)
E ist hierbei die durchschnittliche auf ein Molekül wirkende Feldstärke.
Die Gesamtpolarisierbarkeit tα setzt sich frequenzabhängig anteilsmäßig aus den fünf
oben beschriebenen Mechanismen zusammen [37, 40, 42]:
(Gl. 2.3)
eα = Polarisierbarkeit durch Elektronenpolarisation
aα = Polarisierbarkeit durch Atompolarisation
iα = Polarisierbarkeit durch Ionenpolarisation
oα = Polarisierbarkeit durch Orientierungspolarisation
gα = Polarisierbarkeit durch Grenzflächenpolarisation
Die Frequenz der Mikrowellenstrahlung hat einen Einfluss auf die Polarisierung und den
Wärmeeintrag. Für die beim Heizen mit Mikrowellen eingesetzten Frequenzen von
900 MHz und 2,45 GHz können die Therme αe und αa in Gleichung 2.3 beispielsweise
vernachlässigt werden, da die zur Polarisation über diese Effekte benötigten Zeitskalen
viel kleiner sind als die eingesetzten Mikrowellenfrequenzen [43]. Diese Effekte sind
vorwiegend im Bereich der ultravioletten Strahlung zu beobachten. Bei einer Anregung
mit der geigneten Frequenz kommt es zu Resonanz [44].
Der Anteil der Ionenpolarisation im Bereich von Mikrowellenfrequenzen ist ebenfalls sehr
gering und wird im Allgemeinen vernachlässigt. Ionenpolarisation tritt hauptsächlich im
Frequenzbereich von 1011-1013 Hz auf, d.h. im Gebiet zwischen Mikrowellen und
Infrarotstrahlung es entstehen definierte Schwingungen/Rotationen (Resonanz) [44].
Die so genannte Orientierungspolarisation ist der dominierende Effekt bei den üblicher-
weise für Heizzwecke eingesezten Mikrowellenfrequenzen. Durch diesen Effekt
erwärmen sich viele Flüssigkeiten im Mikrowellenfeld. Permanente Dipole richten sich
parallel zur Feldrichtung aus. Wird das Feld abgeschaltet nehmen die Moleküle wieder
eine zufällige Orientierung ein. Dieser Vorgang wird als Relaxation des Systems
bezeichnet. Da die Moleküle (Dipole) eine Masse/Trägheit besitzen erfolgt eine
Relaxation nicht beliebig schnell. Die Heizwirkung der Mikrowellen beruht nun darauf,
dass es sich um ein elektrisches Wechselfeld hoher Frequenz handelt. Die Dipole richten
tm Eα= ⋅
t e a i o gα α α α α α= + + + +

12 2 Mikrowellen als Energieträger in der Reaktionstechnik
sich in einem Dielektrikum analog dem oszillierenden elektrischen Feld aus. Können die
Dipole den Feldwechseln folgen wird analog zum statischen Feld Energie gespeichert. Ist
die Frequenz so hoch, dass die Moleküle (aufgrund ihrer Trägheit) dem Feld nichtmehr
folgen können kommt es zu einer Phasenverschiebung zwischem dem Feld und der
Polarisation wodurch sich die Substanz erwärmt. Elektrische Energie wird in Wärme
umgewandelt („dielektrischer Verlust“) [37]. Wird die Frequenz noch weiter erhöht sinkt
der Energieeintrag wieder, da die Moleküle zu träge sind, um von den Feldwechseln
beeinflusst zu werden. Die Polarisierbarkeit ändert sich nicht schlagartig, sondern über ein
breites Frequenzband, das Dispersionsgebiet genannt wird. DEBYE hat die dielektrische
Erwärmung so interpretiert, dass es zwischen benachbarten Molekülen zu Reibung
kommt, wodurch sich Wärme entwickelt. Durch das Wechselfeld eingebrachte kinetische
Energie wird in innere Energie des Mediums (Wärme) umgewandelt [45].
Beim Prozess der dielektrischen Erwärmung spielt auch die Dichte des Mediums eine
Rolle. Mit abnehmender Atom- bzw. Molekülzahl sinkt die Wahrscheinlichkeit für Stöße
bzw. einer Wechselwirkung der Partikel untereinander. Die Umwandlung von Strahlung
in thermische Energie ist in der Gasphase ist nach DEBYE folglich sehr gering, während
die Wärmeentwicklung in der Flüssigphase groß ist. Nichtsdestotrotz kann es zu einer
Rotationsanregung der Moleküle durch die Mikrowellen in der Gasphase kommen (vgl. S.
7). Auch die Viskosität eines Stoffes hat einen Einfluss auf den Energieeintrag durch
Mikrowellen. Ist die Beweglichkeit der Moleküle in einer Phase eingeschränkt, verringert
sich auch die Heizwirkung durch die Strahlung. Beispielsweise lässt sich Wasser gut
durch Mikrowellen erwärmen, während Eis kaum einen Energieeintrag durch Mikrowel-
len erfährt, da das Kristallgitter die Bewegung der Wassermoleküle stark einschränkt [46-
48].
Der Effekt der Grenzflächenpolarisation tritt zwar im Bereich der für Heizzwecke einge-
setzten Frequenzen auf, der Energieeintrag über diesen Effekt ist jedoch meist stark limi-
tiert [42]. Dieser Mechanismus dominiert bei Frequenzen um 50 MHz [49]. Für den
Energieeintrag ist die Relaxation der Ladungsträger verantwortlich.
In Abbildung 2.4 wird die Frequenzabhängigkeit der Polarisationseffekte veranschaulicht.
Aufgetragen sind die Dielektrizitätskonstante und der Verlustfaktor (vgl. Kapitel 2.1.2)
über der Frequenz (Strahlungsart).

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 13
Abbildung 2.4: Frequenzabhängigkeit der Polarisationsmechanismen, schematisch nach
[44, 49, 50]
Der Heizeffekt der Mikrowellenstrahlung hängt somit sowohl von der Frequenz als auch
von der eingestrahlten Leistung ab. Der maßgeblich für die Erwärmung eines Stoffes im
Bereich der Mikrowellenfrequenzen verantwortliche Effekt ist die Orientierungspolarisa-
tion.
2.1.1.1 Intramolekulare Effekte
Obwohl der Energieeintrag zumeist auf den makroskopischen Effekt der Orientierungs-
polarisation zurückgeführt wird, bei dem ganze Moleküle zur Rotation angeregt werden,
können bei einigen Spezialfällen auch lokalisierte Rotationsphänomene beobachtet wer-
den. Diese Effekte treten bei sehr großen, starren Molekülen auf, die polare Gruppen
(z. B. -OH, -NH2) besitzen. Diese Gruppen verhalten sich wie auf einer Oberfläche veran-
kerte Dipole und können durch die Mikrowellenstrahlung in Rotation versetzt werden
[47]. Von DAVIES und MEAKINS wurden beispielsweise die dielektrischen Eigenschaf-
ten von 3-tert-Butylphenol in Decalin (Decahydronaphthalin) untersucht [42]. Dabei fan-
den sie heraus, dass bei niedrigen Mikrowellenfrequenzen (<109 Hz) eine molekulare
Rotation zu beobachten ist, während bei höheren Frequenzen (>1010 Hz) die Hydro-
xylgruppe angeregt wird. Ein weiterer Nachweis lokalisierter Rotationsphänomene gelang
beim Triphenylhydroxymethan, welches sich in seinem dielektrischen Verhalten deutlich
vom Triphenylchloromethan unterscheidet. Dies kann nur durch eine Rotation der Hydro-
xylgruppe erklärt werden [47]. Ob diese selektive Anregung polarer Gruppen zu veränder-
ten chemischen Eigenschaften des Moleküls führt, ist jedoch noch nicht untersucht

14 2 Mikrowellen als Energieträger in der Reaktionstechnik
worden. Es kann davon ausgegangen werden, dass dieser Effekt vorrangig bei höheren
Frequenzen auftritt. Bei den für Heizzwecke eingesetzten Frequenzen von ca. 109 Hz
dürfte dieser Effekt nur in speziellen Einzelfällen eine Rolle spielen.
2.1.2 Dielektrische Erwärmung - mathematische Beschreibung
Als Maß für die Energieeinkopplung (Umwandlung von elektromagnetischer Energie in
Wärme) durch Mikrowellen wird der Tangens des Verlustwinkels δ, oft auch als Dissipa-
tionsfaktor D bezeichnet, herangezogen [4, 5, 29, 44]. Er beschreibt die Phasenverschie-
bung zwischen dem elektrischen Feld und der Polarisation des Materials.
(Gl. 2.4)
Er ist auf die absolute Dielektrizitätskonstante ε (Permittivität) bezogen, welche die
dielektrischen Eigenschaften eines Materials beschreibt. Sie ist ein Produkt der stoffspezi-
fischen relativen Dielektrizitätskonstante εr (Permittivitätszahl) und der elektrischen
Feldkonstante ε0.
(Gl. 2.5)
Die relative Dielektrizitätskonstante εr charakterisiert die Stärke der Ladungsträgerver-
schiebung des Dielektrikums und lässt sich nach DEBYE [45] als komplexe Zahl darstel-
len.
(Gl. 2.6)
Der Realteil εr’ wird auch (ideale) Dielektrizitätskonstante genannt und ist ein Maß für die
Polarisierbarkeit des Stoffes, welche wiederum die Energiespeicherfähigkeit charakteri-
siert. Der Imaginärteil εr’’ wird auch als Verlustwert oder dielektrischer Verlustfaktor
bezeichnet und charakterisiert die Fähigkeit des Materials, elektromagnetische Energie in
Wärme umzuwandeln. Er setzt sich aus den Faktoren der verschiedenen Polarisationsme-
chanismen und Leitungsverlusten zusammen [4].
(Gl. 2.7)
Hierbei steht σ für die Leitfähigkeit des Materials und ω für die Winkelgeschwindigkeit
(Kreisfrequenz) der Strahlung. Dieser Term ist in der Regel stark Frequenz- und Tempe-
raturabhängig (vgl. Kapitel 2.1.2.1).
tanDε
δε
′′= =
′
0rε ε ε= ⋅
r r riε ε ε′ ′′= +
0
( ) ( ) ( ) ( ) ( )r e a o i
σε ω ε ω ε ω ε ω ε ω
ε ω′′ ′′ ′′ ′′ ′′= + + + +
2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 15
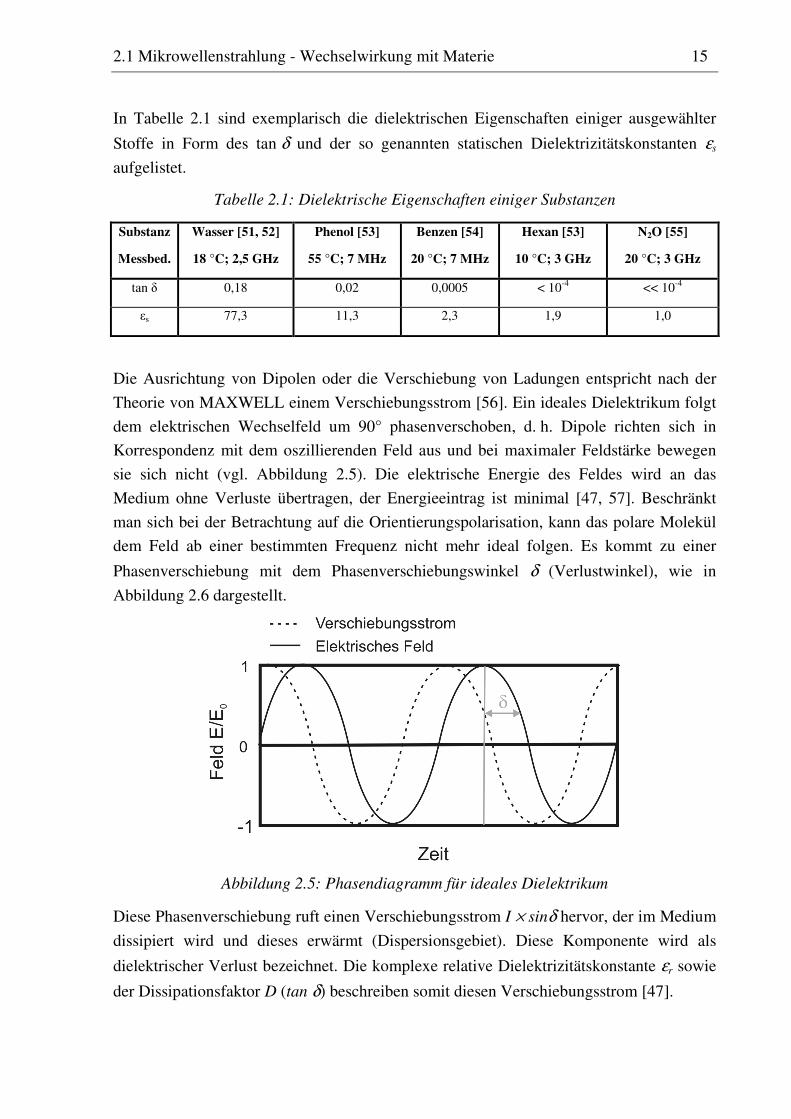
In Tabelle 2.1 sind exemplarisch die dielektrischen Eigenschaften einiger ausgewählter
Stoffe in Form des tan δ und der so genannten statischen Dielektrizitätskonstanten εs
aufgelistet.
Tabelle 2.1: Dielektrische Eigenschaften einiger Substanzen
Substanz
Messbed.
Wasser [51, 52]
18 °C; 2,5 GHz
Phenol [53]
55 °C; 7 MHz
Benzen [54]
20 °C; 7 MHz
Hexan [53]
10 °C; 3 GHz
N2O [55]
20 °C; 3 GHz
tan δ 0,18 0,02 0,0005 < 10-4 << 10-4
εs 77,3 11,3 2,3 1,9 1,0
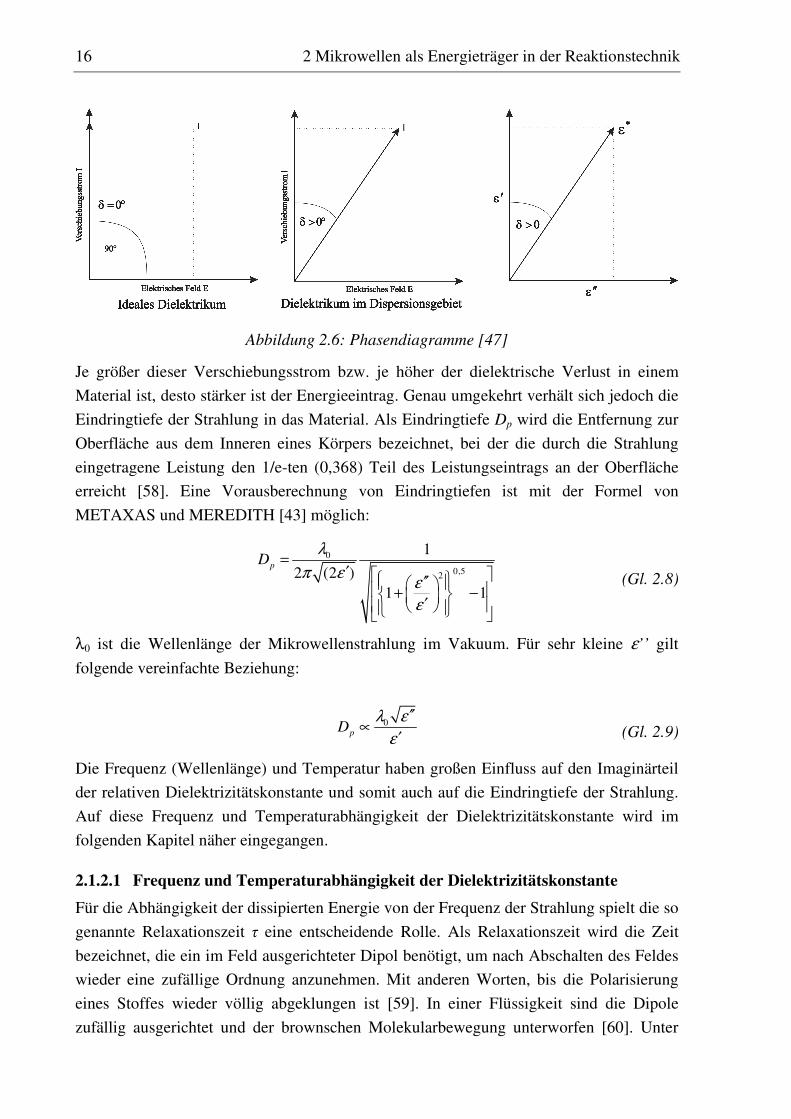
Die Ausrichtung von Dipolen oder die Verschiebung von Ladungen entspricht nach der
Theorie von MAXWELL einem Verschiebungsstrom [56]. Ein ideales Dielektrikum folgt
dem elektrischen Wechselfeld um 90° phasenverschoben, d. h. Dipole richten sich in
Korrespondenz mit dem oszillierenden Feld aus und bei maximaler Feldstärke bewegen
sie sich nicht (vgl. Abbildung 2.5). Die elektrische Energie des Feldes wird an das
Medium ohne Verluste übertragen, der Energieeintrag ist minimal [47, 57]. Beschränkt
man sich bei der Betrachtung auf die Orientierungspolarisation, kann das polare Molekül
dem Feld ab einer bestimmten Frequenz nicht mehr ideal folgen. Es kommt zu einer
Phasenverschiebung mit dem Phasenverschiebungswinkel δ (Verlustwinkel), wie in
Abbildung 2.6 dargestellt.
Abbildung 2.5: Phasendiagramm für ideales Dielektrikum
Diese Phasenverschiebung ruft einen Verschiebungsstrom I × sinδ hervor, der im Medium
dissipiert wird und dieses erwärmt (Dispersionsgebiet). Diese Komponente wird als
dielektrischer Verlust bezeichnet. Die komplexe relative Dielektrizitätskonstante εr sowie
der Dissipationsfaktor D (tan δ) beschreiben somit diesen Verschiebungsstrom [47].
16 2 Mikrowellen als Energieträger in der Reaktionstechnik
Abbildung 2.6: Phasendiagramme [47]
Je größer dieser Verschiebungsstrom bzw. je höher der dielektrische Verlust in einem
Material ist, desto stärker ist der Energieeintrag. Genau umgekehrt verhält sich jedoch die
Eindringtiefe der Strahlung in das Material. Als Eindringtiefe Dp wird die Entfernung zur
Oberfläche aus dem Inneren eines Körpers bezeichnet, bei der die durch die Strahlung
eingetragene Leistung den 1/e-ten (0,368) Teil des Leistungseintrags an der Oberfläche
erreicht [58]. Eine Vorausberechnung von Eindringtiefen ist mit der Formel von
METAXAS und MEREDITH [43] möglich:
(Gl. 2.8)
λ0 ist die Wellenlänge der Mikrowellenstrahlung im Vakuum. Für sehr kleine ε’’ gilt
folgende vereinfachte Beziehung:
(Gl. 2.9)
Die Frequenz (Wellenlänge) und Temperatur haben großen Einfluss auf den Imaginärteil
der relativen Dielektrizitätskonstante und somit auch auf die Eindringtiefe der Strahlung.
Auf diese Frequenz und Temperaturabhängigkeit der Dielektrizitätskonstante wird im
folgenden Kapitel näher eingegangen.
2.1.2.1 Frequenz und Temperaturabhängigkeit der Dielektrizitätskonstante
Für die Abhängigkeit der dissipierten Energie von der Frequenz der Strahlung spielt die so
genannte Relaxationszeit τ eine entscheidende Rolle. Als Relaxationszeit wird die Zeit
bezeichnet, die ein im Feld ausgerichteter Dipol benötigt, um nach Abschalten des Feldes
wieder eine zufällige Ordnung anzunehmen. Mit anderen Worten, bis die Polarisierung
eines Stoffes wieder völlig abgeklungen ist [59]. In einer Flüssigkeit sind die Dipole
zufällig ausgerichtet und der brownschen Molekularbewegung unterworfen [60]. Unter
0pD
λ εε
′′∝
′
0
0,52
1
2 (2 )1 1
pDλ
π εεε
=′ ′′ + − ′

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 17
dieser Vorraussetzung und Berücksichtigung des Stoke-Theorems beschreibt DEBYE die
Relaxationszeit eines kugelförmigen Moleküls in einem viskosen Medium mit folgender
Formel [45]:
(Gl. 2.10)
Wobei η die dynamische Viskosität des Mediums, r der Radius des Dipols, T die absolute
Temperatur und k die Boltzmann-Konstante ist. DEBYE beschreibt die Relaxationszeit
also in Form von Widerstandskräften im Medium. Für Feststoffe ist dieser Ansatz jedoch
nicht gültig. Mit Hilfe der Boltzmann Statistik lässt sich jedoch für einen idealen
Feststoff, in dem die Dipole durch die Potentialbarriere Ua getrennt sind, folgender
Zusammenhang aufstellen [37]:
(Gl. 2.11)
Hierbei stellt εs die statische Dielektrizitätskonstante bei sehr niedrigen Frequenzen dar
und ε∞ ist die Hochfrequenz-Dielektrizitätskonstante. Beide sind als Grenzwerte reelle
Zahlen [51]. 1/ν ist die Zeit, die für eine einzelne Schwingung benötigt wird [29].
Es hat sich gezeigt, dass für viele Feststoffe und Flüssigkeiten die Beschreibung der
Relaxationszeit mit Hilfe der Onsager-Gleichung [61] zulässig ist:
(Gl. 2.12)
Bei dieser Gleichung repräsentiert N die Anzahl der Moleküle; m ist die Masse eines
Moleküls.
Für eine polare Flüssigkeit, in der alle Moleküle die gleiche Relaxationszeit besitzen, stellt
DEBYE folgenden Zusammenhang zwischen der Relaxationszeit und der relativen
Dielektrizitätskonstante her [45, 51]:
(Gl. 2.13)
Die Variable ω ist die Winkelgeschwindigkeit, für die gilt [30]:
(Gl. 2.14)
Wobei f die Frequenz der Strahlung repräsentiert.
34 r
kT
π ητ =
/ ( 2)
( 2)
aU kTse ε
τν ε∞
+=
+
24 ( )
9 ( )s s
s
Nm
kT
π ε ε ετ
ε ε∞
∞
+ +=
+
1s
ri
ε εε ε
ωτ∞
∞
−= +
+
2 fω π=
18 2 Mikrowellen als Energieträger in der Reaktionstechnik
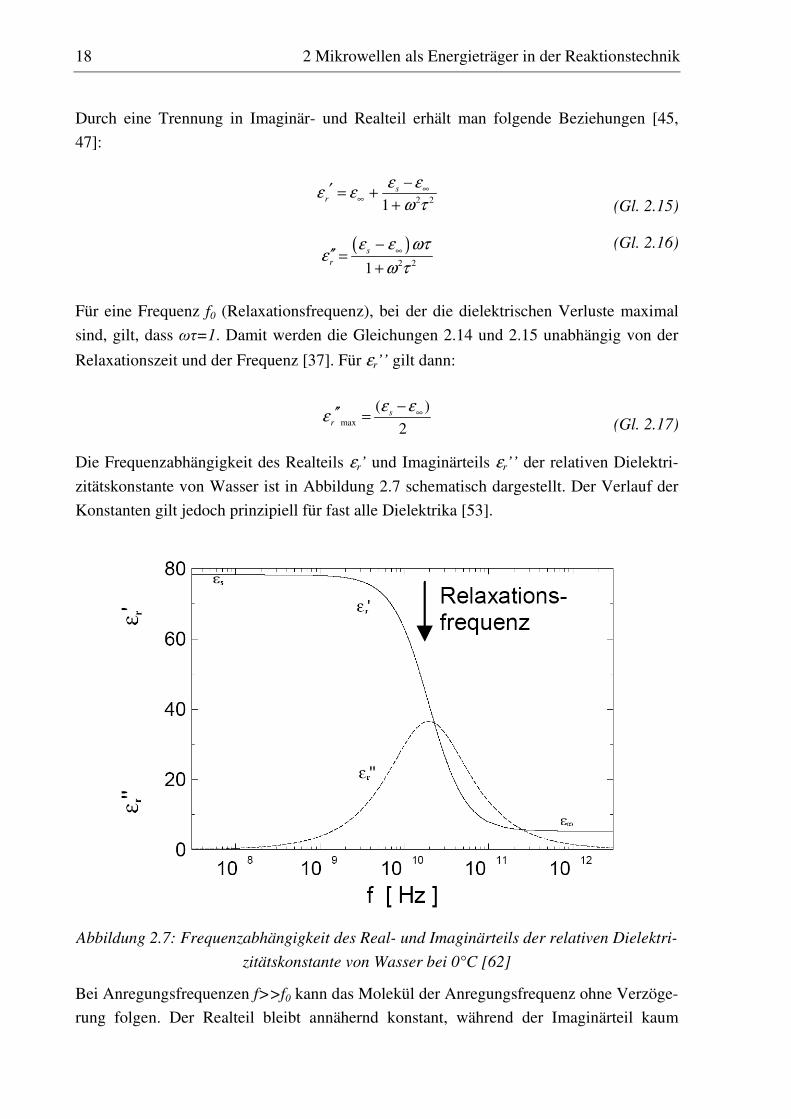
Durch eine Trennung in Imaginär- und Realteil erhält man folgende Beziehungen [45,
47]:
(Gl. 2.15)
(Gl. 2.16)
Für eine Frequenz f0 (Relaxationsfrequenz), bei der die dielektrischen Verluste maximal
sind, gilt, dass ωτ=1. Damit werden die Gleichungen 2.14 und 2.15 unabhängig von der
Relaxationszeit und der Frequenz [37]. Für εr’’ gilt dann:
(Gl. 2.17)
Die Frequenzabhängigkeit des Realteils εr’ und Imaginärteils εr’’ der relativen Dielektri-
zitätskonstante von Wasser ist in Abbildung 2.7 schematisch dargestellt. Der Verlauf der
Konstanten gilt jedoch prinzipiell für fast alle Dielektrika [53].
Abbildung 2.7: Frequenzabhängigkeit des Real- und Imaginärteils der relativen Dielektri-
zitätskonstante von Wasser bei 0°C [62]
Bei Anregungsfrequenzen f>>f0 kann das Molekül der Anregungsfrequenz ohne Verzöge-
rung folgen. Der Realteil bleibt annähernd konstant, während der Imaginärteil kaum
2 21s
r
ε εε ε
ω τ∞
∞
−′ = ++
( )2 21
sr
ε ε ωτε
ω τ∞−
′′ =+
max
( )
2s
r
ε εε ∞−′′ =

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 19
messbare Werte annimmt. Im Bereich von Mikrowellenfrequenzen befindet man sich im
so genannten Dispersionsgebiet, d. h. es kommt zu einer Phasenverschiebung. Der Real-
teil der relativen Dielektrizitätskonstante nimmt mit weiter steigender Frequenz ab, wäh-
rend der Imaginärteil εr’’ bei der Relaxationsfrequenz f0 sein Maximum durchläuft. Bei
dieser Frequenz ist der Energieeintrag durch die Mikrowellen maximal. Bei höheren Fre-
quenzen können die Moleküle dem Feld immer schlechter folgen, der Imaginärteil geht
gegen Null und der Realteil erreicht einen konstant niedrigen Wert.
DEBYE hat seine Betrachtungen auf den im Mikrowellenfrequenzbereich bedeutendsten
Effekt, die Orientierungspolarisation, beschränkt. Wie bereits erwähnt, können jedoch
auch die Grenzflächenpolarisation und Leitungseffekte zur Erwärmung eines Stoffes bei-
tragen.
Für den Fall einer Grenzflächenpolarisation hat WAGNER [41] zusammen mit seinem
Kollegen MAXWELL ein einfaches Modell entwickelt. Sie betrachten ein nicht leitendes
Medium, in dem kugelförmige, leitende Partikel verteilt sind. An Inhomogenitäten bzw.
Grenzflächen innerhalb der Matrix kommt es durch Anlegen des Feldes zum Ladungsauf-
bau. Für den dielektrischen Verlustfaktor eines Volumenelements v durch Grenzflächen-
polarisation ergibt sich damit folgender Zusammenhang:
(Gl. 2.18)
Wobei σ die Leitfähigkeit der leitenden Phase und ε’ deren Dielektrizitätskonstante ist.
Dieses Modell konnte durch Messungen an sphärischen Cu-Phatlocyanin Partikeln, wel-
che in Paraffin eingebettet waren, bestätigt werden [63].
Zusätzlich zu den bereits beschriebenen Effekten kann es in vielen Materialien auch zu
Leitungsverlusten kommen. Unter Berücksichtigung eines speziellen Leitungsterms lässt
sich Gleichung 2.12 dann folgendermaßen schreiben [37]:
(Gl. 2.19)
Diese Leitungsverluste treten in einer Vielzahl von Systemen auf. Der Energieeintrag
durch Mikrowellen in Salzlösungen und vielen Feststoffen wird von diesem Effekt domi-
niert [37].
max10 2 2
9
1,8 10 10i
fνε ωτε
σ ω τ′ ′′ = ⋅ +
0
( )
(1 )s
r
j
j
ε ε σε ε
ωτ ωε∞
∞
−= + −
+
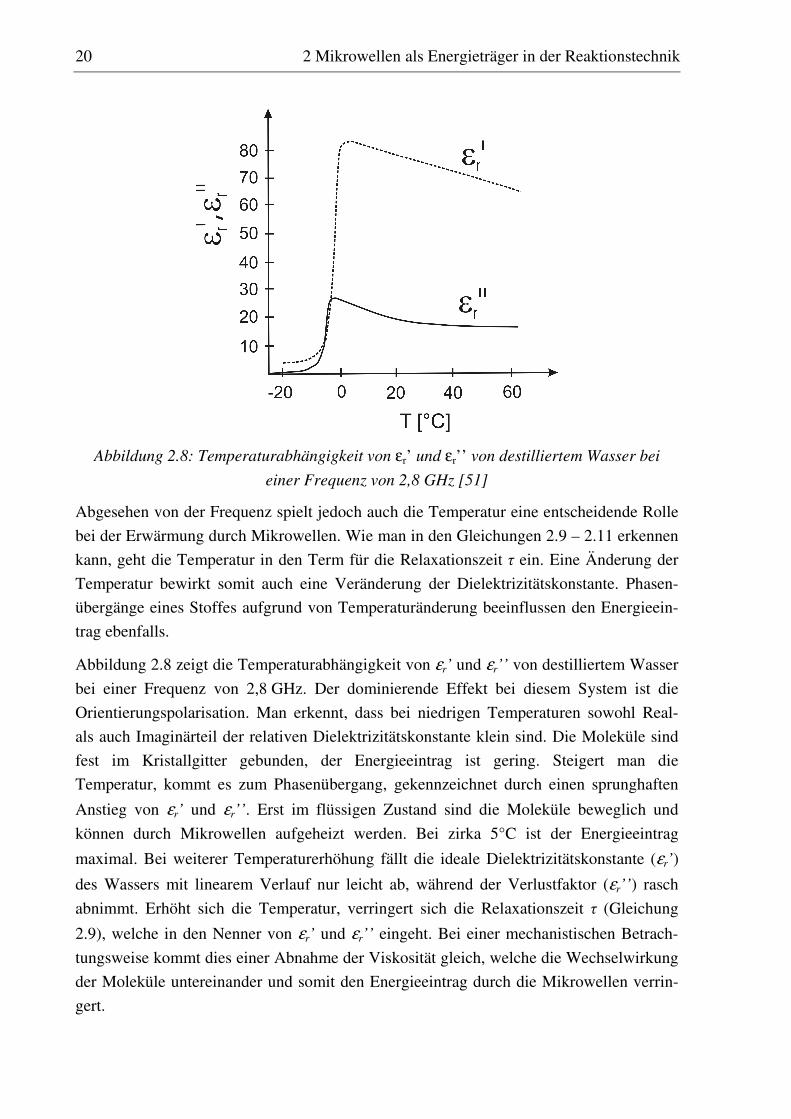
20 2 Mikrowellen als Energieträger in der Reaktionstechnik
Abbildung 2.8: Temperaturabhängigkeit von εr’ und εr’’ von destilliertem Wasser bei
einer Frequenz von 2,8 GHz [51]
Abgesehen von der Frequenz spielt jedoch auch die Temperatur eine entscheidende Rolle
bei der Erwärmung durch Mikrowellen. Wie man in den Gleichungen 2.9 – 2.11 erkennen
kann, geht die Temperatur in den Term für die Relaxationszeit τ ein. Eine Änderung der
Temperatur bewirkt somit auch eine Veränderung der Dielektrizitätskonstante. Phasen-
übergänge eines Stoffes aufgrund von Temperaturänderung beeinflussen den Energieein-
trag ebenfalls.
Abbildung 2.8 zeigt die Temperaturabhängigkeit von εr’ und εr’’ von destilliertem Wasser
bei einer Frequenz von 2,8 GHz. Der dominierende Effekt bei diesem System ist die
Orientierungspolarisation. Man erkennt, dass bei niedrigen Temperaturen sowohl Real-
als auch Imaginärteil der relativen Dielektrizitätskonstante klein sind. Die Moleküle sind
fest im Kristallgitter gebunden, der Energieeintrag ist gering. Steigert man die
Temperatur, kommt es zum Phasenübergang, gekennzeichnet durch einen sprunghaften
Anstieg von εr’ und εr’’. Erst im flüssigen Zustand sind die Moleküle beweglich und
können durch Mikrowellen aufgeheizt werden. Bei zirka 5°C ist der Energieeintrag
maximal. Bei weiterer Temperaturerhöhung fällt die ideale Dielektrizitätskonstante (εr’)
des Wassers mit linearem Verlauf nur leicht ab, während der Verlustfaktor (εr’’) rasch
abnimmt. Erhöht sich die Temperatur, verringert sich die Relaxationszeit τ (Gleichung
2.9), welche in den Nenner von εr’ und εr’’ eingeht. Bei einer mechanistischen Betrach-
tungsweise kommt dies einer Abnahme der Viskosität gleich, welche die Wechselwirkung
der Moleküle untereinander und somit den Energieeintrag durch die Mikrowellen verrin-
gert.
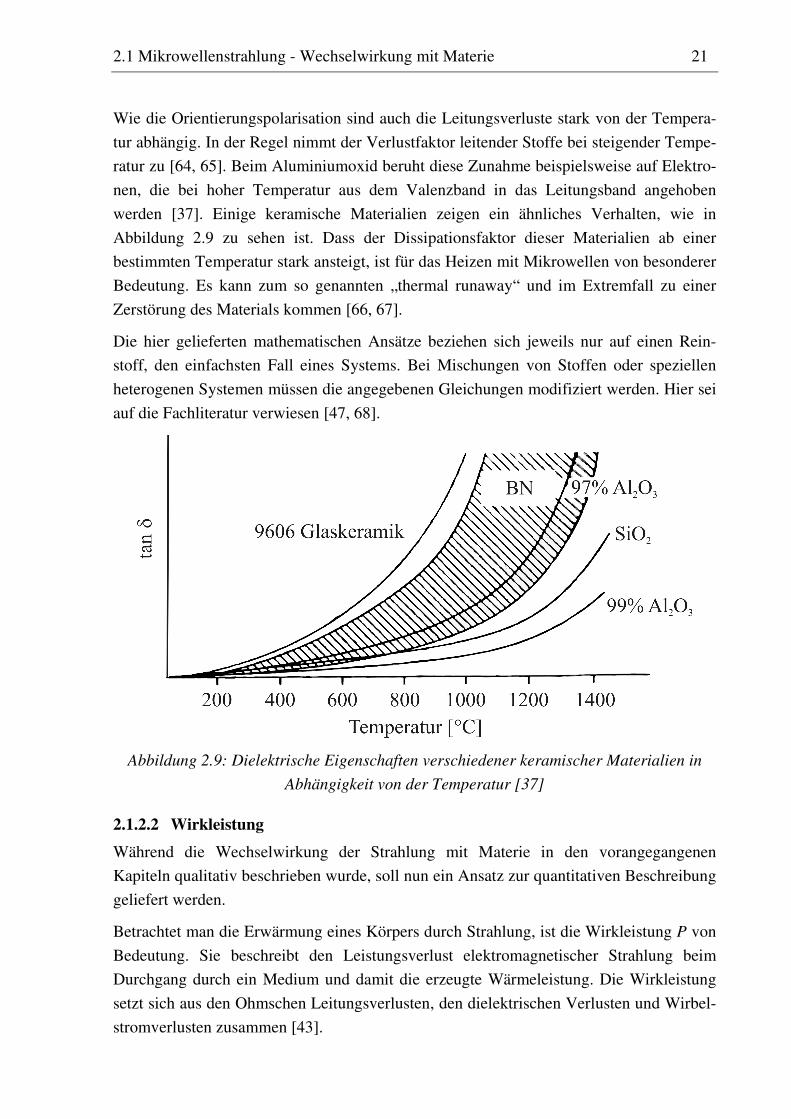
2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 21
Wie die Orientierungspolarisation sind auch die Leitungsverluste stark von der Tempera-
tur abhängig. In der Regel nimmt der Verlustfaktor leitender Stoffe bei steigender Tempe-
ratur zu [64, 65]. Beim Aluminiumoxid beruht diese Zunahme beispielsweise auf Elektro-
nen, die bei hoher Temperatur aus dem Valenzband in das Leitungsband angehoben
werden [37]. Einige keramische Materialien zeigen ein ähnliches Verhalten, wie in
Abbildung 2.9 zu sehen ist. Dass der Dissipationsfaktor dieser Materialien ab einer
bestimmten Temperatur stark ansteigt, ist für das Heizen mit Mikrowellen von besonderer
Bedeutung. Es kann zum so genannten „thermal runaway“ und im Extremfall zu einer
Zerstörung des Materials kommen [66, 67].
Die hier gelieferten mathematischen Ansätze beziehen sich jeweils nur auf einen Rein-
stoff, den einfachsten Fall eines Systems. Bei Mischungen von Stoffen oder speziellen
heterogenen Systemen müssen die angegebenen Gleichungen modifiziert werden. Hier sei
auf die Fachliteratur verwiesen [47, 68].
Abbildung 2.9: Dielektrische Eigenschaften verschiedener keramischer Materialien in
Abhängigkeit von der Temperatur [37]
2.1.2.2 Wirkleistung
Während die Wechselwirkung der Strahlung mit Materie in den vorangegangenen
Kapiteln qualitativ beschrieben wurde, soll nun ein Ansatz zur quantitativen Beschreibung
geliefert werden.
Betrachtet man die Erwärmung eines Körpers durch Strahlung, ist die Wirkleistung P von
Bedeutung. Sie beschreibt den Leistungsverlust elektromagnetischer Strahlung beim
Durchgang durch ein Medium und damit die erzeugte Wärmeleistung. Die Wirkleistung
setzt sich aus den Ohmschen Leitungsverlusten, den dielektrischen Verlusten und Wirbel-
stromverlusten zusammen [43].

22 2 Mikrowellen als Energieträger in der Reaktionstechnik
Mit Hilfe der Maxwell’schen Gleichungen lässt sich für ein Volumenelement V die Leis-
tungsbilanz folgendermaßen herleiten [17, 43, 69]:
(Gl. 2.20)
Der linke Term der Gleichung beschreibt die Gesamtstrahlungsleistung des Systems. Das
erste Integral auf der rechten Seite ist die durch Leitungsverluste eingebrachte Energie, zu
der sich der Energieeintrag durch dielektrische und magnetische (Wirbelstrom) Verluste
hinzuaddiert. Der Imaginärteil der Gleichung beschreibt die im Volumenelement gespei-
cherte elektromagnetische Energie. Diese ist bei harmonischer Erregung konstant und
liefert keinen Beitrag zur Erwärmung. Hr
ist die magnetische Feldstärke. Der Vektor
S E H= ×r r r
wird auch Poynting’scher Vektor genannt. Sein Betrag entspricht der
Strahlungsleistung pro Flächeneinheit, während seine Richtung mit der Feldrichtung über-
einstimmt [17, 70].
Die durchschnittliche absorbierte Leistung in einem Volumen V lässt sich somit wie folgt
schreiben [43]:
(Gl. 2.21)
Vernachlässigt man Diffusion und Wärmeverluste, erhält man für den Temperaturanstieg
∆T im Zeitintervall t [43]:
(Gl. 2.22)
mit der Dichte ρ und der Wärmekapazität C des Materials.
Die Vorausberechnung der eingebrachten Leistung und des Temperaturanstiegs anhand
dieser Formeln erweist sich in der Praxis jedoch ebenso wie die Bestimmung des auf Seite
14 beschriebenen Dissipationsfaktors als schwierig, da eine genaue Kenntnis des dielektri-
schen Verlustfaktors εr’’ und der lokalen elektrischen Feldstärke erforderlich ist. εr’’ ist
beispielsweise von der Temperatur und Frequenz abhängig und nur mit großem
experimentellen Aufwand zu bestimmen [4], während die Ermittlung der lokalen elektri-
schen Feldstärke bei realen Mehrkomponentensystemen ebenfalls kaum möglich ist.
Mathematische Modelle zu Berechnung oder Abschätzung von Dielektrizitätskonstanten
sind derzeit nicht verfügbar.
( ) ( )2 2 2 2 20 0 0 0
V
v v v
SdV
E dV E H dV j H E dVσ ωε ε µ µ ω µ µ ε ε
∇ =
′′ ′′ ′ ′− − + − +
∫
∫ ∫ ∫
r
r r r r r
2 2. . . 0 0 tanleit dielekt mag r rP P P P E V E Vωε ε ωε ε δ′′ ′= + + = =
r r
20 tanr ET
t C
ωε ε δρ
′∆=
r

2.1 Mikrowellenstrahlung - Wechselwirkung mit Materie 23
2.1.3 An Oberflächen adsorbierte Spezies
Prinzipiell sind an Oberflächen adsorbierte Moleküle im Mikrowellenfeld den gleichen
Effekten unterworfen wie freie Moleküle. Bei den üblicherweise für Heizzwecke einge-
setzten Frequenzen sind dies vor allem die Orientierungspolarisation und gegebenenfalls
Grenzflächeneffekte. Wie bereits erläutert, spielen aber auch der Aggregatszustand,
Dichte, Viskosität und Temperatur des zu erwärmenden Stoffes eine entscheidende Rolle.
Die Dielektrizitätskonstanten und Verlustfaktoren gebundener Moleküle sind also nicht
direkt mit denen freier Moleküle vergleichbar. Ausgehend von Debye’s Modell [45]
verschiebt sich die Relaxationsfrequenz f0 eines Stoffes mit zunehmender Viskosität zu
niedrigeren Frequenzen. Für das Beispiel Wasser und einer Mikrowellenfrequenz von
2,45 GHz, bedeutet dies, dass der Verlustfaktor bei zunehmender Viskosität bzw.
abnehmender Temperatur zunimmt, da die Relaxationsfrequenz von freiem Wasser bei
0 °C mit ca. 20 GHz (vgl. Abbildung 2.7) deutlich größer ist. Nach dem Phasenübergang
zu Eis ist die Beweglichkeit der Moleküle so weit vermindert, dass der Verlustfaktor
gegen Null geht. Die Relaxationsfrequenz von Eis liegt viel niedriger, bei ca. 10 kHz [71].
Bei der einfachsten Betrachtungsweise geht man davon aus, dass die Relaxationsfrequenz
für gebundenes Wasser zwischen der von Eis und freiem Wasser liegt. Die Moleküle auf
der Oberfläche verhalten sich wie Eis, die darüber liegenden Schichten nähern sich immer
mehr den dielektrischen Eigenschaften von freiem Wasser an [72, 73].
Von BOCKRIS ET AL. [74] wurde ein Modell entwickelt, welches die dielektrische
Relaxation von zwei adsorbierten Lagen Wasser beschreibt. Er unterscheidet hierbei zwei
Mechanismen, wie in Abbildung 2.10 dargestellt ist.
Abbildung 2.10: Mechanismen der Relaxation gebundenem Wassers [74]
Zum einen wird die Rotation von Molekülen in der ersten Schicht (A) berücksichtigt und
zum anderen die „desorptive Relaxation“, bei der ein Molekül aus einer der unteren Lagen

24 2 Mikrowellen als Energieträger in der Reaktionstechnik
in eine darüber liegende springt (B). Auf dieser Grundlage wurde ein Modell entwickelt,
mit dem sich die Relaxationszeit des gebundenen Wassers berechnen lässt [74, 75]. Nach
diesem Modell sind somit auch eine Anregung der ersten Moleküllage und eine Desorp-
tion der adsorbierten Moleküle möglich.
Thermodielektrische Messungen von OR ET AL. [76] und SERBIN [77] an mit Wasser
beladenen Erden haben dieses Modell bestätigt. Bei ihren Untersuchungen zeigte sich,
dass der Verlustfaktor dieser Stoffsysteme bei abnehmenden Beladungsgraden sogar
ansteigt. Ähnliche Ergebnisse wurden auch von BOYARSKII ET AL. veröffentlicht [78].
Mit Hilfe der Deby’schen Gleichungen wurde ein Modell entwickelt, mit dem sich die
Dielektrizitätskonstanten von an Tonen gebundenem Wasser berechnen lassen (vgl.
Abbildung 2.11).
Abbildung 2.11: Dielektrizitätskonstante von mit Wasser beladenen Tonen nach [78]
Von weiteren Arbeitsgruppen wurden die dielektrischen Eigenschaften von an mikrokri-
stalliner Cellulose gebundenem Wasser [79] und mit Wasserhüllen umschlossenen
Makromolekülen [80] untersucht.
Für andere Stoffsysteme wie beispielsweise an Zeolithen gebundenes Wasser oder organi-
sche Moleküle gibt es bisher noch keine Ansätze zur mathematischen Beschreibung der
Dielektrizitätskonstanten.

2.2 Mikrowellengeräte 25
2.2 Mikrowellengeräte
Die derzeit im Labormaßstab eingesetzten Mikrowellengeräte lassen sich in zwei Gruppen
einteilen. Zum einen werden so genannte Multimode-Mikrowellengeräte verwendet, zum
anderen Monomode-Geräte. Die Multimode-Geräte entsprechen in ihrer Bauweise einer
herkömmlichen Haushaltsmikrowelle und sind damit relativ kostengünstig. Die meisten
kommerziell erhältlichen Labormikrowellensysteme arbeiten nach diesem Prinzip.
2.2.1 Multimode-Mikrowellengeräte
Als Mikrowellengeneratoren für Erwärmungszwecke werden meist Magnetrone [3, 37]
verwendet. Sie arbeiten üblicherweise im 2,45 GHz Band. Die erzeugten Leistungen
können von einigen 100 W bis zu mehreren Kilowatt reichen [70]. Die Energie wird über
einen so genannten Applikator an das zu erwärmende Gut gebracht. Üblicherweise einge-
setzte Applikatoren sind Hohlraumresonatoren und Feiraumapplikatoren, welche im Prin-
zip wie eine Antenne arbeiten [70].
Abbildung 2.12: Aufbau eines Multimode-Geräts für Laboranwendungen [4]
In Abbildung 2.12 ist der Aufbau eines Multimode-Mikrowellengeräts für den Einsatz in
der heterogenen Katalyse schematisch dargestellt. Ein Magnetron ist über eine Hohllei-
tung (Wellenleiter) mit dem als Hohlraumresonator fungierenden Mikrowellenraum
(Cavity) verbunden. Damit die Energie möglichst gleichmäßig in der Cavity verteilt wird,
befindet sich in der Nähe der Einkopplung ein metallischer Reflektorflügel
(Modenrührer), an dem die Mikrowellen reflektiert und ständig in verschiedene Richtun-
gen umgelenkt werden. An den Wänden der Cavity werden die Wellen ebenfalls reflek-
tiert. Die Mikrowellenenergie wird so auf die verschiedenen Schwingungsmoden des
Hohlraumresonators verteilt [70]. Durch geeignete Wahl der Geometrie der Cavity, des

26 2 Mikrowellen als Energieträger in der Reaktionstechnik
Hohlleiters und des Modenrührers lässt sich eine relativ homogene Feldverteilung im
Mikrowellenraum erreichen.
Eine Leistungsregelung einfacher Geräte ist aufgrund der verwendeten LC-Netzteile nur
nach dem Puls-Pause Prinzip möglich. Eine Reduzierung der Leistung erfolgt über Takten
der Maximalleistung (z. B. 4 s ein, 6 s aus). Nach diesem Verfahren ist eine Temperatur-
kontrolle des zu erwärmenden Gutes nicht möglich, periodische Schwankungen der Tem-
peratur sind die Folge. Labormikrowellengeräte (z.B. der Firmen MLS und CEM) ver-
wenden deshalb häufig spezielle Schaltnetzteile, welche den Anodenstrom des
Magnetrons regeln [4]. Mit diesen Geräten ist eine exaktere Temperaturregelung möglich.
Im Gegensatz zu Haushaltsmikrowellen wird im Labor häufig ständiger Zugang zum zu
erwärmenden Material, beispielsweise für Temperatur oder Druckmessung oder auch Pro-
benentnahme oder Zufuhr benötigt. Zu diesem Zweck kann die Wand des Mikrowellen-
raums durchbohrt und mit einem Metallstutzen versehen werden, der das Austreten von
Mikrowellenstrahlung verhindert. Dieser Stutzen muss leitend mit der Cavity verbunden
werden und fungiert als zylindrischer Wellenleiter. Die minimale Wellenlänge λc, die in
solch einem Wellenleiter „transportiert“ werden kann, ergibt sich zu [37]:
(Gl. 2.23)
wobei a der Durchmesser des Wellenleiters ist. Bei einer Mikrowellenfrequenz von
2,45 GHz muss die Öffnung somit kleiner als 3,5 cm sein, damit es nicht zu Leckage-
strahlung kommt. Bei größeren Durchmessern kommt es im Zylinder zu einer Wellenlei-
tung, die jedoch mit einer Schwächung der Strahlung einhergeht. Je größer der Durchmes-
ser, desto länger muss auch der Stutzen ausgelegt werden, damit keine Strahlung austritt.
Als Faustformel gilt, dass bei einer Frequenz von 2,45 GHz und einem Durchmesser von
6 cm der Stutzen mindestens 12 cm, d.h. eine Wellenlänge lang sein muss, um Leckagen
zu vermeiden [37]. Es gibt jedoch auch die nötigen Berechnungsgrundlagen, um für
verschiedene Durchmesser und Stutzenmaterialien die benötigte Länge zu berechnen [37].
Die Temperaturmessung im Mikrowellenfeld erweist sich meist als problematisch.
Herkömmliche Quecksilber- oder Alkohol-Thermometer sind nicht geeignet, da sie sich
aufheizen würden. Herkömmliche Thermoelemente sind ebenfalls nur bedingt einsetzbar,
da durch das elektromagnetische Wechselfeld Ströme induziert werden, die eine Tempe-
raturmessung verfälschen. Deshalb kommen meist Xylen-Thermometer, Gasdruck-Ther-
mometer, faseroptische Sensoren oder Infrarot-Pyrometer zum Einsatz [4, 37].
3,412 ,c aλ =

2.2 Mikrowellengeräte 27
2.2.2 Monomode-Mikrowellengeräte
Teurer als die Multimode-Geräte und mit höherem experimentellem Aufwand verbunden
sind so genannte Monomode-Mikrowellengeräte [81]. In Abbildung 2.13 ist der Aufbau
eines Monomode-Mikrowellensystems schematisch dargestellt.
Abbildung 2.13: Aufbau eines Monomode-Mikrowellenreaktors nach [4, 82]
Die Probe wird bei diesen Systemen direkt in einen rechteckigen Wellenleiter eingebaut,
in dem eine stehende Welle einer bestimmten Feldkonfiguration (Mode) angeregt wird.
Mit Hilfe des Kurzschlussschiebers lässt sich die Resonanzfrequenz des Systems
einstellen. Über den Richtkoppler wird die hin- und rücklaufende Leistung gemessen und
der Zirkulator stellt sicher, dass keine Strahlung zum Magnetron zurückreflektiert wird.
Der 3-Stift-Tuner ermöglicht eine Abstimmung der MW-Leistung am Katalysator [81-83].
Der Vorteil solcher Systeme liegt im Allgemeinen darin, dass die Feldstärken deutlich
höher sind als in Multimode-Geräten. Die Feldkonfiguration lässt sich so einstellen, dass
der Energieeintrag an der Probe maximal wird. Weiterhin ist eine präzisere Einstellung
der Feldstärke bzw. der eingestrahlten Leistung als in Multimode-Geräten möglich. Die
Feldverteilung am Ort der Probe kann weitestgehend homogen sein. Außerdem können
diese Systeme zur Bestimmung dielektrischer Eigenschaften von Proben verwendet wer-
den, da sich eine Verschiebung der Resonanzfrequenz durch Einbringen einer Probe in
den Hohlleiter messen lässt.
Als nachteilig erweist sich jedoch, dass der Probenraum durch die Größe des Hohlleiters
begrenzt ist. Solche Hohlleiter haben üblicherweise Abmessungen von einigen Millime-
tern bis zu wenigen Zentimetern (siehe Gl. 2.23). In der heterogenen Gasphasenkatalyse
wurden bisher überwiegend solche Systeme eingesetzt [4, 81].
Für detailliertere Informationen über Aufbau und Funktionsweise von Mikrowellengerä-
ten sei auf die einschlägige Fachliteratur verwiesen [3, 43, 51, 58, 70].

28 2 Mikrowellen als Energieträger in der Reaktionstechnik
2.3 Einsatzgebiete - Stand der Technik
Mikrowellen werden im industriellen Maßstab zur Trocknung [84-87], Vulkanisation und
Devulkanisation von Elastomeren [88, 89], Polymerisation [90], chemischen Gasphasen-
abscheidung [91], sintern von Keramiken und Stählen [92, 93], in der
Lebensmittelindustrie [94] bis hin zu Spezialanwendungen wie der Wiederaufbereitung
von Atommüll [95] eingesetzt. Eine Übersicht der Einsatzgebiete von Mikrowellen findet
sich bei THUÉRY [96].
Den Einzug in die chemischen Labors hielten die Mikrowellengeräte bereits Mitte der
70’er Jahre des letzten Jahrhunderts. Dort kamen sie zunächst als schnelles Heizmedium
für nasschemische Aufschlüsse zum Einsatz [7, 97]. Seit damals wurden die Techniken
immer weiter verbessert und heute ist die Probenvorbereitung mit Hilfe von Mikrowellen
gängige Praxis in fast jedem chemischen Labor [98]. Erst rund 10 Jahre später folgten
dann erste Veröffentlichungen, die sich mit der Verwendung von Mikrowellen als Ener-
gieträger für organische Synthesen beschäftigen [99, 100]. Seitdem ist die Zahl der
Publikationen zu diesem Themenkomplex auf weit über 700 angestiegen. Aber auch im
Bereich der anorganischen Synthese wurden in den letzten Jahren Mikrowellen immer
häufiger eingesetzt, beispielsweise bei der Herstellung von Metallboriden [101] oder der
Zeolithsynthese [102, 103]. Im Vergleich zu dem enormen Interesse, das der
Mikrowelleneinsatz bei den organischen Chemikern hervorrief, verhielten sich die
Forscher, die sich mit heterogener Katalyse beschäftigten, eher konservativ. Zwar wurden
die Mikrowellen bereits Anfang der 80’er Jahre von WAN [16] in der heterogenen
Katalyse eingesetzt, die Zahl der Arbeitsgruppen, die sich mit diesem Themenkomplex
beschäftigen, stieg aber erst seit Mitte der 90’er Jahre auf heute ca. 20 an [4].
2.3.1 Organische Synthesen
Traditioneller Weise erfolgt die Energiezufuhr bei organischen Synthesen über Ölbäder,
Sandbäder und Heizmanschetten. Das verdampfende Lösungsmittel wird über Rückfluss-
kühler rückgeführt. Nachteilig bei diesen Heizmethoden ist, dass sehr lange Aufheizzeiten
nötig sind und meist Temperaturgradienten im Reaktionsgefäß entstehen. Lokale Über-
temperaturen an der Gefäßwand können zu Zersetzung der an der Reaktion beteiligten
Stoffe oder zu Nebenreaktionen führen. Im Gegensatz dazu erlauben die Mikrowellen
einen direkten Energieeintrag in das Reaktionsmedium ohne die Gefäßwand zu erwärmen.
Ist der Versuchsaufbau gut ausgelegt, kann eine einheitliche Temperaturverteilung im
gesamten Reaktionsvolumen erreicht werden. Die Aufheiz- und Abkühlzeiten sind
drastisch verringert und unerwünschte Nebenreaktionen aufgrund lokaler Temperatur-
überhöhungen können in einigen Fällen vermieden werden [5]. Ein weiterer Grund für die
weite Verbreitung der Mikrowellenöfen in der organischen Chemie ist, dass normale
Haushaltsmikrowellengeräte mittlerweile extrem billig auf dem Markt verfügbar sind.

2.3 Einsatzgebiete - Stand der Technik 29
Eine Vielzahl der Publikationen auf diesem Gebiet beschreibt den Einsatz solcher
„Küchengeräte“. Dies bringt jedoch auch erhebliche Probleme mit sich, worauf im
folgenden Kapitel näher eingegangen wird. Da es sich bei diesem Themenkomplex um ein
extrem breit gestreutes Gebiet handelt, wird in dieser Arbeit lediglich versucht, einen
Überblick über die untersuchten Reaktionstypen mit wenigen ausgewählten Beispielen zu
geben. Eine umfangreichere Darstellung findet sich in den Übersichtsartikeln von
LINDSTRÖM ET AL. [5], CADDICK [104], WHITTAKER ET AL. [105], MAJETICH
ET AL. [106], ABRAMOVITCH [107] und STRAUSS [108]. In den folgenden Kapiteln
soll eine Einteilung in Reaktionen, die unter Einsatz von Lösungsmitteln ablaufen und
lösungsmittelfreie Synthesen erfolgen.
2.3.1.1 Synthesen in der Flüssigphase
Die in diesem Kapitel vorgestellten mikrowellenunterstützten Synthesen haben alle die
Verwendung eines polaren Lösungsmittels oder Reaktanden gemeinsam. Im Allgemeinen
wird mit Wasser, Säuren, Laugen, Alkoholen, Ketonen, Glykolen oder Furanen gearbeitet,
welche einen hohen dielektrischen Verlustfaktor aufweisen und sich durch die Mikrowel-
len sehr gut aufheizen lassen. Ein Unterschied muss jedoch klar hervorgehoben werden.
Einerseits gibt es in der Literatur mikrowellengestützte Untersuchungen, die in Rückfluss-
systemen unter Umgebungsdruck durchgeführt werden (Kapitel 2.3.1.1.2), andererseits
werden geschlossene Druckgefäße verwendet (Kapitel 2.3.1.1.1). Als Vergleich wird in
der Literatur jedoch immer die konventionelle Synthese bei Umgebungsdruck unter Rück-
flussbedingungen herangezogen.
2.3.1.1.1 Synthesen unter erhöhtem Druck
Eine der ersten Reaktionen, die unter Verwendung einer Mikrowellenheizung durchge-
führt wurde, war eine Veresterung. Mitte der 80’er Jahre untersuchte die Arbeitsgruppe
von GEDYE ET AL. [109] die Fischer-Veresterung von Benzoesäure mit Methanol. Zum
Einsatz kam dabei eine handelsübliche Haushaltsmikrowelle mit einem verschlossenem
Reaktionsgefäß aus Teflon. Dabei zeigte es sich, dass nach nur 5 min Reaktionsdauer eine
Ausbeute von 76 % erreicht werden konnte. Beim konventionell beheizten Vergleichsex-
periment wurde nach 80 min lediglich 90 % Ausbeute erzielt. Spätere Untersuchungen
[110] in einem Mikrowellensystem mit Druckkontrolle bei 3,8 bar zeigten sogar 92 %
Ausbeute nach nur 1 min.
Von der gleichen Arbeitsgruppe [99] wurde auch die Reaktion von Benzylchlorid mit
Paracynophenoxyd-Ionen beschrieben. Unter Mikrowellenbedingungen konnten über
70 % Ausbeute in 3 min erreicht werden, während unter klassischen Bedingungen mehr
als 12 h benötigt werden, um vergleichbare Ausbeuten zu realisieren.

30 2 Mikrowellen als Energieträger in der Reaktionstechnik
Auch zahlreiche Dehydrierungsreaktionen wurden unter Mikrowellenbedingungen unter-
sucht. GUERDIA [111] beschäftigte sich beispielsweise mit der säurekatalysierten
Hydrolyse von Diazepam zu 2-Methylamino-5-Chlorobenzophenon. Unter Mikrowellen-
bedingungen konnten die Umsatzgrade um den Faktor 12 erhöht werden.
Weitere Beispiele für Mikrowellensynthesen unter erhöhtem Druck sind Diels-Alder
Reaktionen, Adler-Bong Reaktionen, ortho-Claisen Umlagerungen, Substitutionen,
Oxidationen, Reduktionen und viele mehr [106].
All diesen Untersuchungen ist jedoch gemeinsam, dass unter Verwendung von Mikro-
wellen in Verbindung mit geschlossenen Reaktionsgefäßen deutlich verkürzte Synthese-
zeiten bzw. um ein Vielfaches erhöhte Reaktionsgeschwindigkeiten beobachtet werden
konnten.
2.3.1.1.2 Synthesen unter Umgebungsdruck
Neben den Reaktionen, die unter erhöhtem Druck durchgeführt werden, gibt es jedoch
auch zahlreiche Untersuchungen, die Mikrowellensynthesen in offenen Gefäßen zum
Gegenstand haben. Diese Synthesen werden unter Umgebungsdruck mit Rückflusskühlern
durchgeführt. Die Versuchsdurchführung ist somit mit den konventionellen Syntheseme-
thoden vergleichbar. Der Unterschied besteht allein darin, dass die Heizung der Reakti-
onslösung über Mikrowellen erfolgt.
Von BOSE ET AL. [112] wurde die Diels-Alder Reaktion von Maleinsäureanhydrid mit
Anthracen in Diglyme als Lösungsmittel untersucht. Um eine Ausbeute von 90 % zu
erreichen, wurden unter Mikrowellenbedingungen eine Minute, unter konventionellen
Bedingungen mit Benzen als Lösungsmittel jedoch 90 min benötigt. GUPTA ET AL.
[113] untersuchten die Synthese von Chalkonen mittels Aldol-Kondensation und fanden
heraus, dass sich die Reaktionszeiten auf Minuten, verglichen mit mehreren Stunden bei
der konventionellen Methode, verkürzen ließen.
Ähnliche Steigerungen der Reaktionsgeschwindigkeiten wurden auch bei zahlreichen
anderen organischen Synthesen, die in der Mikrowelle unter Umgebungsdruck durchge-
führt wurden, beobachtet [114-117].
Bei vielen dieser Untersuchungen kamen auch homogene Katalysatoren wie Säuren,
Basen oder Metallkatalysatoren zum Einsatz [118, 119]. Wie auch bei den nicht
katalysierten Reaktionen zeigten sich beim Einsatz von Mikrowellen deutliche Steigerun-
gen der Reaktionsgeschwindigkeiten.
2.3.1.2 Synthesen, die Feststoffe als Support verwenden
Anstatt eine Synthese in der Flüssigkeit durchzuführen, laufen zahlreiche Reaktionen auch
dann ab, wenn die Edukte auf einem Feststoff adsorbiert vorliegen. Diese Methode ist aus

2.3 Einsatzgebiete - Stand der Technik 31
umwelttechnischer und industrieller Sicht von großem Interesse, da sich Lösungsmittelab-
fälle minimieren lassen. In zahlreichen Veröffentlichungen wird vom Einsatz von Mikro-
wellen bei diesen so genannten „dry media“ Reaktionen berichtet. Das Vorgehen ist dabei
üblicherweise, dass die Reaktanden in einer kleinen Menge CH2Cl2 gelöst werden, wel-
ches anschließend von einem Feststoff mit großer Oberfläche adsorbiert wird. Solche
„Supports“ können beispielsweise Montmorillonit-Tone, Aluminiumoxide (Tonerde), mit
Alkalimetallfluoriden dotiertes Aluminiumoxid und Silikate sein. Dabei kommen einige
Millimol Reaktanden auf ein oder zwei Gramm Support. Nach erfolgter Reaktion werden
die Produkte dann mit einem geeigneten Lösungsmittel wieder vom Support entfernt. Mit
Hilfe der Mikrowellen wird bei dieser Methode meistens der Feststoff aufgeheizt, wäh-
rend die Verlustfaktoren der beteiligten Reaktanden vergleichsweise gering sind [37, 105].
Von BRAM ET AL. [120] wurde die Kondensationsreaktion von 1-Brom-Oktan mit
Kaliumacetat auf Aluminiumoxid untersucht. Bei der herkömmlichen Methode im Ölbad
bei 75 °C dauert diese Reaktion über 40 h, unter Mikrowelleneinstrahlung nur wenige
Minuten. Im Arbeitskreis um VARMA sind zahlreiche Reaktionen auf „Solid Support“
Basis untersucht worden. Darunter die Synthese von Thioketonen, verschiedene
Oxidations- und Reduktionsreaktionen oder die Umwandlung von Aldehyden zu Nitrilen
[121]. BEN ALLOUM ET AL. berichten, dass bei der Umsetzung verschiedener Alkohole
an Montmorillonit unter Mikrowellenbestrahlung eine andere Produktzusammensetzung
bei deutlich höheren Umsatzgraden als bei Verwendung eines konventionellen Heizmedi-
ums gefunden wurde [122].
Zusammenfassend lässt sich sagen, dass bei einer Vielzahl von Reaktionen, die unter Ver-
wendung eines Feststoffes als Support unter Mikrowellenbestrahlung durchgeführt
wurden, deutlich verkürzte Reaktionszeiten beziehungsweise höhere Umsatzgrade
beobachtet wurden als bei Verwendung einer herkömmlichen Heizmethode.
2.3.1.3 Synthesen unter Verwendung von Feststoffkatalysatoren
Analog zu den Untersuchungen in Flüssigphasensystemen (vgl. Kapitel 2.3.1.1) wurden
auch Reaktionen im Mikrowellenfeld getestet, bei denen der Reaktionslösung ein Fest-
stoffkatalysator zugemischt wird.
Beispielsweise beschäftigten sich RADOIU ET AL. mit der Oxidation von Benzen mit
Wasserstoffperoxid an Zeolithen als Katalysator [123]. Bei dieser heterogenen katalysier-
ten Flüssigphasenreaktion kam ein Titansilikalith zum Einsatz. Der Reaktor wurde unter
Rückfluss (Umgebungsdruck) betrieben und die Temperaturmessung erfolgte außerhalb
der verwendeten Haushaltsmikrowelle zwischen dem Reaktionsgefäß und dem Rückfluss-
kühler. Dabei zeigte es sich, dass das Aufheizverhalten einen entscheidenden Einfluss auf
die Wasserstoffperoxidzersetzung und Phenolproduktion hatte. Ein sehr schnelles Aufhei-
zen führte zwar zu einem hohen Umsatz des H2O2, die Phenolausbeute war jedoch sehr

32 2 Mikrowellen als Energieträger in der Reaktionstechnik
gering. Die beste Phenolausbeute ließ sich durch ein langsames, gleichmäßiges Aufheizen
in der Mikrowelle realisieren. Dabei schienen jedoch nicht das Heizmedium, sondern die
Temperaturverteilung und die Aufheizgeschwindigkeit entscheidend zu sein.
Von anderen Autoren wurde die Flüssigphasenoxidation von n-Hexan mit TS-1 (Titansili-
kalith) im Mikrowellenfeld untersucht [124]. Es wird von deutlich gesteigerten
Reaktionsraten bei gleicher Temperatur unter Mikrowellenbedingungen im Vergleich zu
einer konventionellen Heizung berichtet.
Ein ähnlich großer Einfluss der Mikrowellen auf heterogen katalysierte Reaktionen in der
Flüssigphase wurde auch von HÁJEK und RADOIU bei der Umsetzung von
tert-Butylphenolen an Montmorillonit (KSF) beobachtet [125]. Dabei wurden sowohl
höhere Umsatzgrade als auch veränderte Selektivitäten unter Mikrowellenstrahlung fest-
gestellt. Der Autor geht davon aus, dass es durch die Mikrowellen zu einer selektiven
Aufheizung der am Katalysator absorbierten polaren Stoffe kommt.
PIPUŠ ET AL. untersuchten die Veresterung von Benzoesäure mit 2-Ethylhexanol [126-
128]. Dabei wurde sowohl ein speziell entwickelter kontinuierlich betriebener Strömungs-
rohrreaktor (Multimode) als auch ein Rührkesselreaktor (Monomode) getestet. Besonde-
res Augenmerk wurde auf eine exakte Temperaturmessung (faseroptischer Sensor) und
die Berechnung der Temperaturverteilung sowie des Leistungseintrags in den Reaktoren
gelegt. Verschiedene homogene und heterogene Katalysatoren wurden in der Veresterung
getestet. Bei den kinetischen Untersuchungen im Mikrowellenfeld zeigten sich deutlich
verkürzte Reaktionszeiten. Unter Berücksichtigung der Erkenntnisse bezüglich Leistungs-
eintrag und Temperaturverteilung beim Mikrowellenheizen in den verwendeten Reaktoren
und der bekannten Kinetik der Reaktion, ließ sich dieser Effekt exakt vorausberechnen.
Die Kinetik der Veresterung beim konventionellen Heizen entsprach somit der beim
Mikrowellenheizen. Für die verkürzten Reaktionszeiten waren eine veränderte Tempera-
turverteilung im Reaktor und verkürzte Aufheizzeiten durch die Mikrowellen verantwort-
lich. Andere Autoren konnten dieses Ergebnis bestätigen [129].
Neben Reaktionen in der Flüssigphase und Synthesen, bei denen der Support lediglich als
Energiewandler für die Reaktion dient, gibt es auch „dry media“ Untersuchungen, bei
denen ein katalytisch aktiver Support verwendet wird. Die experimentelle Vorgehens-
weise entspricht der in Kapitel 2.3.1.2 beschriebenen. Beispiele für solche Reaktionen
finden sich bei BALALAIE ET AL. [130, 131], BADGAR ET AL. [132] und HÁJEK ET
AL. [125].
2.3.1.4 Diskussion der beobachteten Mikrowelleneffekte
Die teilweise immensen Steigerungen der Reaktionsgeschwindigkeiten, über die in der
Literatur berichtet wurde, führten schnell zu einer Diskussion über einen „spezifischen

2.3 Einsatzgebiete - Stand der Technik 33
Mikrowelleneffekt“ [122, 133] oder auch „nichtthermischen“ [11, 134] Mikrowelleneffekt
als Ursache für die beobachteten Phänomene.
Bei den Reaktionen, die in geschlossenen Druckgefäßen durchgeführt wurden, liegt die
Erklärung für die höheren Reaktionsraten auf der Hand. Zum einen werden Drücke bis
40 bar im Reaktionsgefäß aufgebaut, zum anderen steigt damit der Siedepunkt des ver-
wendeten Lösungsmittels, wodurch Temperaturen erreicht werden können, die bis zu
100 °C über denen der konventionellen Synthese liegen können [37]. Man kann somit
folgern, dass es sich vorrangig um thermische Mikrowelleneffekte handelt.
2.3.1.4.1 Super heating
Anders sieht das bei den Reaktionen aus, die unter Umgebungsdruck und Rückfluss, also
analog der konventionellen Synthese, durchgeführt wurden. Es kann hier weder mit einer
Temperatur- oder Drucküberhöhung gerechnet werden. Trotzdem zeigen sich gesteigerte
Reaktionsgeschwindigkeiten, wenn auch nicht so extrem wie bei den in geschlossenen
Gefäßen durchgeführten Reaktionen. Eine mögliche Erklärung wurde Anfang der 90’er
Jahre von BOND ET AL. [135] und BAGHURST ET AL. [136] geliefert. Sie fanden
heraus, dass viele organische Lösungsmittel beim Aufheizen durch Mikrowellenstrahlung
ihren Siedepunkt erst bei Temperaturen erreichen, die bis zu 20 °C höher sind als unter
konventionellen Bedingungen. Diese Lösungsmittel besitzen im Allgemeinen einen hohen
dielektrischen Verlustfaktor, der bei steigender Temperatur noch zunimmt. Es wird nun
davon ausgegangen, dass für den Beginn des Siedens die Bildung von Bläßchen („Siede-
keime“) an der Gefäßwand notwendig ist. Da durch die Mikrowellen jedoch ein, vergli-
chen mit konventionellen Heizmethoden, inverses Temperaturprofil erzeugt wird, ist die
Wand des Gefäßes beim Heizen mit Mikrowellen der kälteste Punkt der Reaktionsmi-
schung. Bis es nun zur Bildung von Bläßchen an der Gefäßwand kommt, wird das Innere
der Reaktionsmischung überheizt. Dieser Effekt wird auch als „super heating“ bezeichnet.
Man geht davon aus, dass dieses Phänomen in der Regel für die gesteigerten Reaktionsge-
schwindigkeiten in offenen Systemen verantwortlich ist [137].
2.3.1.4.2 Hot spots und Temperaturgradienten
Der bereits beschriebene Effekt des super heating kann nicht für alle Beobachtungen ver-
antwortlich gemacht werden. In den letzten Jahren hat sich gezeigt, dass viele der
„nichtthermischen“ Effekte einfach auf eine unzureichende Versuchsdurchführung und
Prozesskontrolle zurückgeführt werden können [137]. Eine Ursache hierfür ist zumeist die
Verwendung von handelsüblichen Haushaltsmikrowellen, die eine schlechte Feldvertei-
lung und Leistungssteuerung besitzen. Dadurch kommt es zur Ausbildung von lokalen
„hot spots“, die deutlich höhere Temperaturen besitzen als die gemessene Bulk-Tempe-
ratur. Zudem ist eine exakte Temperaturmessung im Mikrowellenfeld schwierig und teuer
(vgl. Kapitel 2.2). Dies hat dazu geführt, dass sich viele veröffentlichte Ergebnisse von

34 2 Mikrowellen als Energieträger in der Reaktionstechnik
anderen Arbeitsgruppen nicht reproduzieren ließen. Beispielsweise berichtete SUN ET
AL. [114], dass die Hydrolyse von ATP bei gleicher Reaktionstemperatur 25-fach schnel-
ler mit Mikrowellenheizung abläuft als bei konventioneller Heizung. Nachdem die Expe-
rimente später in einer Versuchsapparatur mit verbesserter Temperaturkontrolle durchge-
führt wurden, zeigten sich keinerlei Unterschiede mehr zum konventionellen Versuch
[138]. Bei den von GUPTA ET AL. [113] veröffentlichten Ergebnissen über die Synthese
von Chalkonen im Mikrowellenfeld (Reaktionszeiten von Stunden auf Minuten verkürzt)
konnten die beobachteten Effekte auf einen Fehler in der Versuchsdurchführung zurück-
geführt werden. Beim Mikrowellenversuch wurde mehr Lösungsmittel verdampft als beim
konventionellen Experiment, wodurch es zu einer Aufkonzentration der Reaktionslösung
kam. Die Versuche wurden später in einem Mikrowellensystem durchgeführt, das mit
einem Rückflusskühler ausgestattet war und es konnte nur noch eine sehr kleine Steige-
rung der Reaktionsgeschwindigkeit beobachtet werden. Diese wurde auf ein Überheizen
der Reaktionslösung bzw. „hot spots“ zurückführt [11].
Bei Reaktionen, welche auf einem Feststoff als Support ablaufen, wird davon ausgegan-
gen, dass entweder die Reaktanden oder der Support einen hohen dielektrischen Verlust-
faktor aufweisen müssen, um die Unterschiede der konventionellen Versuche zu den
Mikrowellenversuchen erklären zu können. Bei der in Kapitel 2.3.1.2 beschriebenen
Reaktion von 1-Brom-Oktan mit Kaliumacetat lässt sich das mit Kaliumacetat beladene
Aluminiumoxid hervorragend mit Mikrowellen aufheizen. Innerhalb weniger Minuten
können bei Leistungen von ca. 500 W Temperaturen über 500 °C erreicht werden [120].
Man geht davon aus, dass es bei diesem Prozess zur Bildung lokaler Überhitzungen im
Bereich des Kaliumacetats kommt, wodurch die Reaktion beschleunigt wird. Die gemes-
sene Bulk-Temperatur ist somit niedriger als die am Kaliumacetat. Eine selektive Aufhei-
zung von Feststoffbereichen oder adsorbierten Reaktanden wird ebenfalls von einigen
Autoren vermutet [125]. Bei Feststoffen, welche gute Mikrowellenabsorber sind, ist es
zudem wahrscheinlich, dass es zur Bildung von lokalen „hot spots“ aufgrund einer
ungleichmäßigen Feldverteilung kommt [139]. Auch hier herrschen mikroskopisch
Temperaturen, die deutlich höher sind als die messbaren Bulk-Temperaturen.
Man geht heute davon aus, dass fast immer thermische Effekte für die beobachteten
Phänomene verantwortlich sind. Ein „spezifischer Mikrowelleneffekt“ ist unwahrschein-
lich und dürfte weniger Einfluss auf den Ablauf von Reaktionen haben, als in den ersten
Publikationen zu diesem Themenkomplex vermutet wurde [5].
2.3.2 Heterogene Gasphasenkatalyse im Mikrowellenfeld
Die Idee, Mikrowellen als Energiequelle bei heterogen katalysierten Reaktionen einzuset-
zen, wurde von WAN Anfang der 80’er Jahre entwickelt [140]. Ein erstes Patent geht auf
das Jahr 1982 zurück, in dem der Abbau von chlorierten Kohlenwasserstoffen an fein

2.3 Einsatzgebiete - Stand der Technik 35
verteiltem para- oder ferromagnetischen Material beschrieben wird. An Eisenpulver rea-
gieren Chlorkohlenwasserstoffe unter Mikrowelleneinstrahlung zunächst zu Eisenchlorid
und anschließend unter Sauerstoffeinwirkung zu Kohlendioxid und Wasser ab [16].
Ausgehend von der Tatsache, dass viele Reagenzien Mikrowellen nur ungenügend absor-
bieren, wurde von WAN eine Methode entwickelt, bei der dem Reaktionsgemisch ein
Mikrowellenabsorber zugegeben wird, der sich durch die Mikrowellen gut aufheizen lässt
und der Reaktion die benötigte Energie liefert. Eine Erweiterung dieser Methode bestand
darin, als Mikrowellenabsorber einen Katalysator zu verwenden. Dieses Konzept wurde
unter dem Namen „Mikrowellenkatalyse“ bekannt. Vorwiegend wurden metallische
Katalysatoren (z. B. Drahtnetze) verwendet, die zum Teil auch in eine Mikrowellentrans-
parente Matrix (z. B. Al2O3) eingebettet waren. Bei den verwendeten Mikrowellengeräten
handelte es sich ausschließlich um Monomode-Systeme. Aufgrund der großen Leistungs-
dichten dieser Mikrowellengeräte kann der Katalysator sehr schnell auf hohe Temperatu-
ren gebracht werden. Eine „Temperaturregelung“ wird von WAN über Pulsen der Mikro-
wellenstrahlung im Bereich von Millisekunden realisiert. Diese Vorgehensweise hat den
Vorteil, dass überschüssige Reaktionswärme in den Pulspausen an die umgebende Matrix
weitergegeben wird. Die Pulsdauer wird nun so gewählt, dass die benötigte Aktivierungs-
energie für die Reaktion am Katalysator bereitgestellt wird und die Reaktion startet. Wäh-
rend der Heizpausen wird die überschüssige Wärme vom Katalysator an die umgebende
Matrix abgegeben, wodurch sich Folgereaktionen vermeiden lassen. Katalysatorbett und
Gasphase weisen niedrigere Temperaturen auf als die Katalysatorpartikel. Durch Einstel-
len der Pulsdauer und Totzeiten lässt sich somit die Selektivität der Reaktion steuern
[140].
Diese Methode wurde an einer Reihe von unterschiedlichen Reaktionstypen getestet. Bis
Mitte der 90’er Jahre erschienen ca. 15 Veröffentlichungen aus diesem Arbeitskreis.
Ansonsten war das Interesse an der „Mikrowellenkatalyse“ verhalten. Erst später kam es
zu einem langsamen Anstieg der Publikationen zu diesem Themenkomplex bis heute auf
ca. 70. Ein gemeinsames Merkmal beinahe aller Veröffentlichungen ist, dass stark
Mikrowellen absorbierende Katalysatoren und Monomode-Mikrowellengeräte verwendet
werden. Im Folgenden soll eine Einteilung der Publikationen anhand der untersuchten
Reaktionssysteme erfolgen.
Abschließend sei noch erwähnt, dass es mit Hilfe von Mikrowellenstrahlung möglich ist,
an Metallen ein Mikrowellenplasma zu zünden, in dem katalytische Reaktionen ablaufen
können [141-143]. Auf diese Technik soll im Folgenden jedoch nicht näher eingegangen
werden.

36 2 Mikrowellen als Energieträger in der Reaktionstechnik
2.3.2.1 Methanzersetzung und oxidierende Methankupplung
Die wohl am intensivsten untersuchte Reaktion unter Mikrowellenstrahlung ist die
Umsetzung von Methan zu höheren gesättigten und ungesättigten Kohlenwasserstoffen.
Diese Reaktion ist von besonderem wirtschaftlichem Interesse, da in Anbetracht der stetig
abnehmenden Erdölreserven auf lange Sicht eine alternative Rohstoffquelle für die chemi-
sche Industrie gefunden werden muss. Weiterhin sind die natürlichen Gasvorkommen,
welche zum Großteil aus Methan bestehen, umfangreicher als die Erdölvorkommen und
zudem ist Methan deutlich preiswerter als Erdöl auf dem Weltmarkt verfügbar. Deshalb
wurde in den vergangenen Jahren intensiv nach einem einstufigen Verfahren gesucht,
welches eine Nutzung dieser natürlichen Ressource ermöglicht.
Im Laufe der 80’er Jahre wurden mehrere Verfahren zur Umsetzung des Methans entwi-
ckelt, unter anderem auch die oxidierende Kupplung [144] und die Hochtemperatur-Pyro-
lyse [145] zu C2-Kohlenwasserstoffen (Ethan, Ethylen). Bereits Anfang der 90’er Jahre
erkannten WAN ET AL., dass seine Methode der Mikrowellenkatalyse bei dieser Reak-
tion deutliche Vorteile bringen sollte [146]. Bei der konventionellen Pyrolyse werden
hohe Temperaturen und Drücke für die Methanumsetzung benötigt. Dadurch kommt es
zur Bildung unerwünschter Nebenprodukte und teilweiser Rückreaktion. Erste Untersu-
chungen [147, 148] in einem ungepulsten Mikrowellenreaktor brachten nicht den ge-
wünschten Erfolg. Die erreichten Umsatzgrade waren, verglichen mit konventioneller
Heizung, niedriger und die Bildung von C4-Kohlenwasserstoffen wurde gefördert. Später
wurden Si-geträgerte Ni-Katalysatoren und gepulste Mikrowellenstrahlung eingesetzt, um
die Bulk-Temperatur gegenüber der Katalysatortemperatur niedrig zu halten. Mit dieser
Methode konnte ein Methanumsatz von ca. 15 %, mit Produktselektivitäten zu C2 zwi-
schen 97 und 85 % und zwischen 3 – 15 % für C3 erreicht werden. Über eine Variation der
Puls-Pause-Zeiten ließ sich die Selektivität zu den zwei Gruppen einstellen. Die einge-
setzte Mikrowellenleistung betrug durchschnittlich 2 kW (Maximal 12 kW) bei Pulszeiten
von ca. 0,3 s und Pulspausen von ca. 5 s. Die Reaktortemperaturen (Bulk) lagen dabei nur
knapp über Umgebungstemperatur und Umgebungsdruck [146, 148]. In einem weiteren
Artikel wurde die katalytische Methan- und 1-Penten-Kopplung an Ni-Pulver beschrieben
[149].
Weit häufiger als die Methankopplung wird jedoch die oxidierende Dehydrierung von
Methan untersucht (oxidative Methankupplung). Ein vereinfachtes Reaktionsnetzwerk ist
in Abbildung 2.14 dargestellt.

2.3 Einsatzgebiete - Stand der Technik 37
CH4
C2H
6C
2H
4
COx
Abbildung 2.14: Reaktionsschema der oxidativen Methankupplung nach [4]
Unter klassischen Bedingungen lassen sich bei diesem Verfahren derzeit lediglich
Methanumsätze von 10-15 % bei Selektivitäten zu C2-Kohlenwasserstoffen von 80-85 %
erreichen. Ein Grund hierfür ist die geringe Stabilität der Produkte bei den für die Kupp-
lung benötigten Reaktionstemperaturen. Es kommt zur Totaloxidation [4].
Erste Untersuchungen dieser Reaktion mit Mikrowellenheizung wurden von BOND ET
AL. in einem Monomode-Mikrowellensystem bei konstanter Leistung durchgeführt.
Dabei kam ein Na-Al-Mischkatalysator zum Einsatz. Bei Temperaturen zwischen 300 und
600 °C konnten in den Mikrowellenversuchen Selektivitäten zu C2 von 50-60 % und 40-
50 % CO2 erzielt werden. Unter konventionellen Bedingungen war der Katalysator in die-
sem Temperaturbereich jedoch inaktiv. Vergleichbare Selektivitäten ließen sich erst bei
höheren Temperaturen um 800 °C erreichen [150].
Später wurde von BOND und MOYES ein basisches Oxid (1 % Sr, 15 % La auf MgO)
getestet. Auch mit diesem Katalysator ließen sich Selektivitäten zu C2 bis zu 80 % in
einem Temperaturbereich unter 600 °C erreichen. Ähnliche Selektivitäten konnten bei
Einsatz einer konventionellen Heizung erst bei 300 - 400 °C höheren Reaktionstemperatu-
ren erzielt werden [12].
ROUSSY ET AL. führten vergleichbare Versuche mit einer Reihe von Mischoxiden
durch. Als Katalysatoren wurden geträgertes und reines SmLiO2 [151] La2O3, La2ZrO7
[152], Li/MgO und BaBiO3-x [153] getestet. Alle Mischoxide bis auf La2ZrO7 zeigten
unter Mikrowellenheizung bei niedrigen Methanumsatzgraden (geringe Temperaturen)
deutlich höhere Selektivitäten zu C2 als bei konventioneller Heizung. Bei höheren Umsät-
zen konnte kein Unterschied zwischen Mikrowellen- und konventioneller Heizung gefun-
den werden. Beispielsweise ergab sich unter Mikrowellenheizung für den LiMgO-Kataly-
sator bei einer Reaktionstemperatur von 600 °C ein Methanumsatz von 10 % bei einer
Selektivität zu C2 von 60 %, während bei klassischer Heizung nur Umsätze von 2 % mit
Selektivitäten von 10 % zu C2 gemessen wurden. Lediglich an dem La2ZrO7-Katalysator
konnten keine Unterschiede zwischen den beiden Heizmethoden festgestellt werden.
Von HONG, CHEN und DAI wurde der Einfluss von Mikrowellenstrahlung auf die oxi-
dative Methankupplung an Perowskitkatalysatoren und anderen Mischoxiden untersucht
[154, 155]. Dabei zeigte es sich, dass bei den mit Mikrowellen geheizten Experimenten
schon bei 200 °C (Perowskite) bzw. 300 °C (andere Mischoxide) niedrigeren Reaktions-
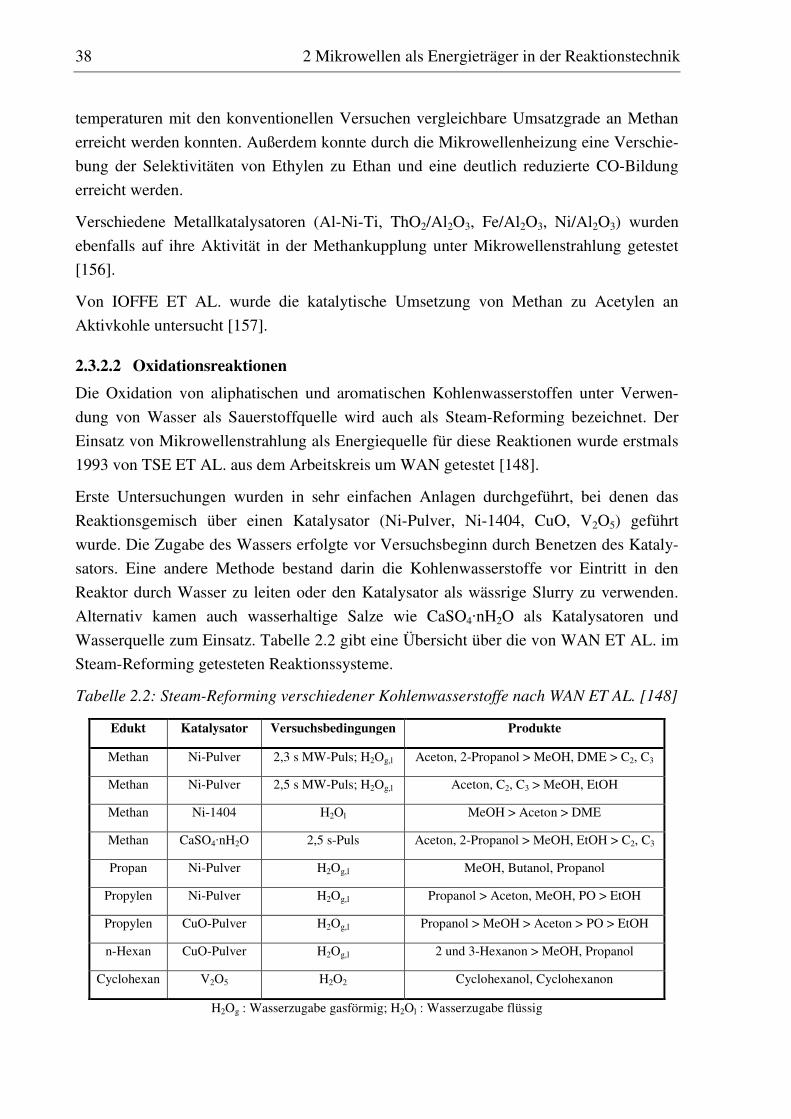
38 2 Mikrowellen als Energieträger in der Reaktionstechnik
temperaturen mit den konventionellen Versuchen vergleichbare Umsatzgrade an Methan
erreicht werden konnten. Außerdem konnte durch die Mikrowellenheizung eine Verschie-
bung der Selektivitäten von Ethylen zu Ethan und eine deutlich reduzierte CO-Bildung
erreicht werden.
Verschiedene Metallkatalysatoren (Al-Ni-Ti, ThO2/Al2O3, Fe/Al2O3, Ni/Al2O3) wurden
ebenfalls auf ihre Aktivität in der Methankupplung unter Mikrowellenstrahlung getestet
[156].
Von IOFFE ET AL. wurde die katalytische Umsetzung von Methan zu Acetylen an
Aktivkohle untersucht [157].
2.3.2.2 Oxidationsreaktionen
Die Oxidation von aliphatischen und aromatischen Kohlenwasserstoffen unter Verwen-
dung von Wasser als Sauerstoffquelle wird auch als Steam-Reforming bezeichnet. Der
Einsatz von Mikrowellenstrahlung als Energiequelle für diese Reaktionen wurde erstmals
1993 von TSE ET AL. aus dem Arbeitskreis um WAN getestet [148].
Erste Untersuchungen wurden in sehr einfachen Anlagen durchgeführt, bei denen das
Reaktionsgemisch über einen Katalysator (Ni-Pulver, Ni-1404, CuO, V2O5) geführt
wurde. Die Zugabe des Wassers erfolgte vor Versuchsbeginn durch Benetzen des Kataly-
sators. Eine andere Methode bestand darin die Kohlenwasserstoffe vor Eintritt in den
Reaktor durch Wasser zu leiten oder den Katalysator als wässrige Slurry zu verwenden.
Alternativ kamen auch wasserhaltige Salze wie CaSO4·nH2O als Katalysatoren und
Wasserquelle zum Einsatz. Tabelle 2.2 gibt eine Übersicht über die von WAN ET AL. im
Steam-Reforming getesteten Reaktionssysteme.
Tabelle 2.2: Steam-Reforming verschiedener Kohlenwasserstoffe nach WAN ET AL. [148]
Edukt Katalysator Versuchsbedingungen Produkte
Methan Ni-Pulver 2,3 s MW-Puls; H2Og,l Aceton, 2-Propanol > MeOH, DME > C2, C3
Methan Ni-Pulver 2,5 s MW-Puls; H2Og,l Aceton, C2, C3 > MeOH, EtOH
Methan Ni-1404 H2Ol MeOH > Aceton > DME
Methan CaSO4·nH2O 2,5 s-Puls Aceton, 2-Propanol > MeOH, EtOH > C2, C3
Propan Ni-Pulver H2Og,l MeOH, Butanol, Propanol
Propylen Ni-Pulver H2Og,l Propanol > Aceton, MeOH, PO > EtOH
Propylen CuO-Pulver H2Og,l Propanol > MeOH > Aceton > PO > EtOH
n-Hexan CuO-Pulver H2Og,l 2 und 3-Hexanon > MeOH, Propanol
Cyclohexan V2O5 H2O2 Cyclohexanol, Cyclohexanon
H2Og : Wasserzugabe gasförmig; H2Ol : Wasserzugabe flüssig

2.3 Einsatzgebiete - Stand der Technik 39
Bei der Oxidation von Methan in einem gepulsten Monomode-System konnten bei mode-
raten Temperaturen (kurze Pulse) gute Ausbeuten der Oxidationsprodukte Aceton und 2-
Propanol erreicht werden. Bei höheren Temperaturen (lange Pulse) wurden verstärkt C2-
und C3-Kohlenwasserstoffe gebildet. Große Umsätze konnten auch bei Propylen
beobachtet werden, mit hoher Selektivität zu Propanol. Bei der Oxidation von Cyclohexan
wurde H2O2 anstatt Wasser als Oxidationsmittel verwendet, da sich nur dann höhere
Umsätze von Cyclohexan erzielen ließen.
In weiteren Veröffentlichungen von WAN ET AL. wird die Umsetzung von Kohlendioxid
oder Kohle mit Wasser an Ni-Katalysatoren im Mikrowellenfeld beschrieben [140, 158,
159].
Genauere Angaben über die Versuchsbedingungen, vor allem die Katalysatorbetttempe-
ratur, sind in den Artikeln jedoch nicht zu finden, genauso wenig ein Vergleich mit der
Katalysatoraktivität unter konventionellen Bedingungen.
Weitere Untersuchungen zum Steam-Reforming von Methan, Ethan, Propan, und Butan
im Mikrowellenfeld wurden von COONEY ET AL. durchgeführt. Dabei kam SiC,
welches Mikrowellenstrahlung hervorragend absorbiert, als Katalysator zum Einsatz [160,
161].
In weiteren Veröffentlichungen zur Partialoxidation von Methan kamen Mischoxidkataly-
satoren (10 % Co/ZrO2, 10 % Ni/La2O3, 10 % Co/La2O3) zum Einsatz [155, 162]. Beim
Vergleich mit dem konventionell geheizten Experiment zeigte sich, dass vergleichbare
Methanumsätze unter Mikrowellebedingungen bereits bei deutlich niedrigeren Tempera-
turen erreicht werden konnten. Weiterhin führte die Verwendung von Mikrowellen als
Heizmedium zu einer Steigerung der CO-Selektivität (vgl. Tabelle 2.3).
Tabelle 2.3: Partialoxidation von Methan an Mischoxidkatalysatoren [155, 162]
Katalysator T [°C] Umsatz CH4 [%] Selektivität [%]
CO CO2
Konventionelle Heizung
10 % Co/ZrO2 800 94 93 7
10 % Ni/La2O3 800 91 92 8
Mikrowellen Heizung
10 % Co/ZrO2 800 100 99 1
10 % Ni/La2O3 700 100 100 <1

40 2 Mikrowellen als Energieträger in der Reaktionstechnik
Die Untersuchungen der bisher vorgestellten Publikationen wurden immer in Monomode-
Mikrowellengeräten durchgeführt. Lediglich zwei Forschungsgruppen setzen derzeit
Multimode-Geräte in der heterogenen Katalyse ein.
Der Arbeitskreis um ONDRUSCHKA beschäftigt sich seit circa drei Jahren mit der
Propanoxidation mit Luft an Katalysatoren mit Perowskitstruktur im Mikrowellenfeld
[163, 164]. Ausgehend von ersten Untersuchungen in einer herkömmlichen Haushaltsmik-
rowelle, bei der die Temperaturmessung über ein Thermoelement erfolgte, wurde ein Re-
aktorsystem entwickelt, bei dem die Temperaturmessung über ein im Katalysatorbett
liegendes Thermoelement und ein IR-Pyrometer erfolgt. Die ursprünglich eingesetzte
Haushaltsmikrowelle wurde durch einen modifizierten (spezielle Schaltnetzteile und zwei
über Drehantennen einkoppelnde Magnetrone) Panasonic Mikrowellenofen ersetzt. Mit
dieser Apparatur ließen sich Temperaturschwankungen während des Mikrowellenheizens
auf wenige Kelvin reduzieren. Unter anderem wurden verschiedene phasenreine, bis
1200 °C thermostabile Katalysatoren (LaCoO3, LaMnO3, La0,5Sr0,5MnO3) im Mikrowel-
lenfeld untersucht. In Tabelle 2.4 sind die Propanumsatzgrade, die beim konventionellen
und Mikrowellenheizen erreicht wurden, für einen LaMnO3-Katalysator gegenüberge-
stellt. Die bei diesen Versuchen im Mikrowellenfeld gemessenen Temperaturdifferenzen
zwischen Thermoelement und IR-Pyrometer betrugen etwa 20 K.
Tabelle 2.4: Propanoxidation mit Luft an LaMnO3-Katalysator (5 g Kat.; 22 l/h; 0,4 vol%
Propan in Luft) [163]
Propanumsatz [%] Temperatur [°C]
Konventionell Mikrowelle
400 44 45
500 91 89
Man erkennt, dass bei gleicher Katalysatorbetttemperatur kaum Unterschiede im Propan-
umsatzgrad bei den verschiedenen Heizmethoden gefunden wurden.
In weiteren Untersuchungen kamen unter anderem Mischungen eines La0,6Sr0,4CoO3 mit
einem LaMnO3 Katalysator zum Einsatz. In Tabelle 2.5 sind die Propanumsatzgrade die-
ser Katalysatormischung für verschiedene Reaktionstemperaturen und die unterschiedli-
chen Heizmethoden dargestellt.
Vergleichbare Umsatzgrade konnten beim Mikrowellenheizen bereits bei ca. 50 °C nied-
rigeren Reaktionstemperaturen als beim konventionellen Heizen erreicht werden. Bei der
Untersuchung der zwei Einzelkatalysatoren zeigte es sich, dass das LaMnO3 besser an die
Mikrowellenstrahlung ankoppelt. Die Temperaturunterschiede bei vergleichbaren Um-
satzgraden waren größer (≈ 70 °C bei 10 % Propanumsatz) als bei der Mischung. Das

2.3 Einsatzgebiete - Stand der Technik 41
La0,6Sr0,4CoO3 ließ sich jedoch nur schwer durch die Mikrowellen aufheizen und es
konnten nur geringe Unterschiede zwischen konventionell und mikrowellenbeheizten Ex-
perimenten beobachtet werden. Allerdings wurde vom Autor als Problem angeführt, dass
im verwendeten Reaktor „deutliche“ axiale und radiale Temperaturgradienten beim Mik-
rowellenheizen auftraten.
Tabelle 2.5: Katalysatormischung aus La0,6Sr0,4CoO3 und LaMnO3 (3 g Kat.; 10,5 l/h; 0,4
vol% Propan in Luft)
Propanumsatz [%] Temperatur [°C]
Konventionell Mikrowelle
400 10 30
450 30 50
500 50 70
Ein Arbeitskreis aus China setzt ebenfalls Multimode-Mikrowellengeräte in der
heterogenen Katalyse ein. Veröffentlichungen von LIU ET AL. beschreiben die Oxidation
von Toluol zu Benzoesäure [165] und die Herstellung von Polyakrylnitril (PAN) aus o-
Xylol [166] an V2O5-Katalysatoren. Bei diesen Untersuchungen kam eine handelsübliche
Haushaltsmikrowelle zum Einsatz. Die Temperaturmessung erfolgte mittels eines Queck-
silberthermometers. Als Reaktor wurde ein Quarzglas-Kolben verwendet, in den von oben
das Feed-Gas einströmte. In diesen Kolben wurde die Katalysatorschüttung eingebracht.
Im unteren Bereich der Katalysatorschüttung war eine Kapillare angebracht, über die das
Produktgas aus dem Reaktor ausströmen kann (kein plug-flow!). Die Produkte wurden in
Aceton absorbiert und nach der Reaktion analysiert.
Bei der Oxidation von Toluol mit einem Sauerstoff-Stickstoffgemisch wurde ein
V2O5/TiO2-Katalysator eingesetzt, dem bei einigen Versuchen noch gut mikrowellenab-
sorbierende Komponenten wie MoO3, WO3, Nb2O5, Ta2O5 oder schlechte Mikrowellenab-
sorber wie CeO2, Tl2O, K2O, P2O5 zugemischt wurden [165]. Tabelle 2.6 zeigt die
Umsatzgrade von Toluol und die Selektivitäten zu Benzoesäure und Benzaldehyd bei ver-
schiedenen Temperaturen und Heizmethoden.
Durch den Einsatz von Mikrowellen konnten im niedrigen Temperaturbereich deutlich
höhere Toluolumsatzgrade bei gleicher Temperatur und höhere Selektivitäten zur Benzoe-
säure erzielt werden. Bei vergleichbaren Umsatzgraden bzw. hohen Reaktionstemperatu-
ren waren die Selektivitäten zu Benzoesäure jedoch deutlich geringer als beim konventio-
nellen Heizen. Als Ursache wurde eine Totaloxidation des Toluols zu COx vermutet.

42 2 Mikrowellen als Energieträger in der Reaktionstechnik
Tabelle 2.6: Selektivoxidation von Toluol (1,6 g Kat. 5% V2O5/TiO2,
Toluol:O2:N2=1:9:35, GHSV=5000 h-1, Integraler Umsatz über 2 h)
Selektivität [%] T [°C] Umsatz Toluol [%]
Benzoesäure Benzaldehyd
Konventionelle Heizung
500 18 2 5
550 42 19 15
600 75 55 10
Mikrowellen Heizung
500 80 15 9
550 94 10 5
600 99 4 2
Durch das Zumischen von Mikrowellenabsorbern ließen sich die Selektivitäten bezüglich
Benzoesäure im Mikrowellenexperiment bei 500 °C noch steigern, während das Zumi-
schen von schlechten Mikrowellenabsorbern zu einer Verringerung der Selektivität
bezüglich Benzoesäure führte.
Die Oxidation von o-Xylol zu Polyakrylnitril wurde an V2O5/SiO2 Katalysatoren getestet.
Dabei zeigten sich ähnliche Ergebnisse wie bei der bereits diskutierten Toluoloxidation.
Die Umsatzgrade an o-Xylol waren im Mikrowellenfeld bei gleicher Temperatur immer
deutlich größer als beim konventionell geheizten Vergleichsexperiment. Die Selektivität
zu PAN war beim Mikrowellenexperiment in einem Temperaturbereich bis 600 °C ver-
gleichbar der des konventionellen Versuchs, bei höheren Temperaturen zeigte sich jedoch
ein starker Rückgang, während bei der herkömmlichen Heizmethode die Selektivität
annähernd konstant blieb.
Von PERRY ET AL. wurde die CO Oxidation mit Sauerstoff an Platin oder Palladium auf
γ-Al2O3 untersucht [167]. Diese Reaktion wurde als eine Art Testreaktion verwendet, um
die Temperatur der Metallpartikel auf dem Träger zu bestimmen. Ziel dieser Arbeit war es
zu zeigen, ob eine lokale Temperaturüberhöhung an den Metallpartikeln durch die
Mikrowellenheizung möglich ist. Eine ausführlichere Diskussion der Ergebnisse findet
sich in Kapitel 2.3.2.5.1.1.
Der Arbeitskreis um MA in China beschäftigte sich Ende der 90’er Jahre mit der katalyti-
schen Verbrennung von Dieselruß zur Abgasreinigung [168]. Als Katalysatoren wurden
verschiedene Metalloxide (Fe2O3, CuO) bzw. Metallpartikel auf diversen Trägermateria-
lien (SiO2, TiO2, ZrO2) getestet. Es wurde ein Monomode-Mikrowellensystem und ein

2.3 Einsatzgebiete - Stand der Technik 43
IR-Pyrometer zur Temperaturmessung verwendet. Beim Vergleich der mit Mikrowellen
geheizten katalytischen Verbrennung mit der konventionell beheizten Reaktion zeigte
sich, dass beim Einsatz von Mikrowellen die Zündtemperaturen für die Verbrennung um
bis zu 300 °C tiefer liegen als bei Verwendung einer elektrischen Heizung. Dieser große
Unterschied wurde von den Autoren auf einen „nichtthermischen“ Mikrowelleneffekt
zurückgeführt. Bei einigen Katalysatoren konnte auch eine Steigerung der Selektivität zu
CO beim Heizen mit Mikrowellen beobachtet werden.
Ein anderes Problem in der Abgastechnik stellt die Vergiftung von Dreiwegekatalysatoren
durch SO2 dar. Von TURNER ET. Al. [169] wurden deshalb Untersuchungen an kommer-
ziell erhältlichen Automobil-Dreiwegekatalysatoren im Mikrowellenfeld durchgeführt.
Zum Einsatz kam ein Monomode-Mikrowellensystem, die Temperaturmessung erfolgte
jedoch nicht direkt im Katalysatorbett. Stattdessen wurde die Abgastemperatur mit einem
Thermoelement bestimmt und auf die Reaktionstemperatur extrapoliert. Als Feed wurde
eine Mischung von CO, Propan und Sauerstoff in Helium verwendet. Dabei zeigte sich,
dass die CO- und Propanumsätze bei Mikrowellenheizung in einem Temperaturbereich
von 200 - 230 °C deutlich höher waren als im konventionell beheizten Vergleichsexperi-
ment. Teilweise wurden auch kleinere Mengen von SO2 zugemischt, um die Desaktivie-
rung des Katalysators zu untersuchen. Die im Abgasbereich wichtige „Anspringtempera-
tur“ (50 % des maximal erreichbaren Umsatzes) der Reaktion wurde durch die Mikro-
wellenheizung deutlich verringert. Anschließend wurden die Versuche mit Spuren des
Katalysatorgiftes SO2 im Feed wiederholt. Bei konventioneller Heizung verschlechterten
sich die Umsatzgrade und die ursprüngliche Aktivität des Katalysators konnte nicht mehr
erreicht werden. Beim Mikrowellenexperiment waren die Umsatzgrade jedoch auch mit
SO2 im Feed noch höher als im konventionellen Experiment ohne SO2.
2.3.2.3 NOx und SOx Reduktion
Eine relativ neue und ökologisch sehr nützliche Anwendung der Mikrowellenkatalyse ist
die Entfernung der Luftschadstoffe SOx und NOx aus Abgasen. Das Ziel ist es hierbei,
SO2 zu elementarem Schwefel und die Stickoxide zu Sauerstoff und Stickstoff zu reduzie-
ren. Forschungsbedarf besteht auf diesem Sektor vor allem im Bereich mobiler Anwen-
dungen. Für den Kraftwerks- und Nutzfahrzeugsektor steht bereits eine effiziente und
technisch ausgereifte Methode zur Entstickung von Abgasen zur Verfügung: die SCR-
DeNOx-Technologie. Ein Nachteil an diesem Verfahren ist jedoch die Verwendung von
NH3 oder Harnstoff als Reduktionsmittel, welches den Einsatz für Personenkraftfahrzeuge
erschwert.
In einem Patent von 1985 beschäftigt sich WAN ET AL. bereits mit der Entschweflung
beim Hydrockracken von Pech [170]. Die ersten Untersuchungen zur Abgasreinigung un-
ter Verwendung von Mikrowellen wurden wiederum im Arbeitskreis um WAN 1990

44 2 Mikrowellen als Energieträger in der Reaktionstechnik
durchgeführt [148]. Dabei kam die für den Arbeitskreis typische Vergehensweise (ge-
pulste Monomode-Mikrowelle, Ni/Cu/Fe-Katalysatoren) zum Einsatz. Als Eduktgemisch
wurden entweder 5 % SO2 in Luft oder 25 % NO in N2 oder He aufgegeben. Mit einem
Ni-Katalysator gelang es, mehr als 99 % des SO2 über einen längeren Zeitraum (5 h) zu
Schwefel abzubauen. Eine Regenerierung des Katalysators war durch Mikrowellenhei-
zung unter H2-Atmosphäre möglich. Der gleiche Katalysator wurde auch in der
NO-Reduktion getestet. Es konnten Umsatzgrade über 90 % für einen Zeitraum von 6 h
erreicht werden. Zwar war eine deutliche Alterung an den Katalysatoren zu beobachten,
eine Regenerierung ließ sich jedoch ohne Probleme durchführen.
Ein anderes Verfahren zur Abgasreinigung unter Einsatz von Mikrowellen wurde von
CHA entwickelt [171-174]. Bei dieser Technik werden die Schadstoffe zunächst an Kohle
adsorbiert und anschließend im Mikrowellenfeld reduziert. Streng genommen handelt es
sich hierbei jedoch nicht um Katalyse, weil die Kohle während der Reduktion verbraucht
wird.
Weitere Veröffentlichungen beschäftigen sich mit der Mikrowellenheizung von in der
Automobilindustrie eingesetzten Dreiwegekatalysatoren. Bei der Abgasreinigung von
Automobilkatalysatoren stellt das Kaltstartverhalten ein großes Problem dar. Während
dieser Anlaufphase ist eine Entfernung der Schadstoffe aufgrund der geringen Katalysa-
tortemperaturen nicht möglich. Dieses Problem ließe sich durch ein Vorheizen des Kata-
lysators beseitigen. Der Einsatz einer elektrischen Heizung ist jedoch aufgrund der hohen
benötigten Ströme und kurzen Zeitdauer nur schwer realisierbar. Deshalb schlagen einige
Autoren die Verwendung von Mikrowellenstrahlung zur Vorheizung des Katalysators vor.
MA und ZHANG untersuchten die katalytische Abgasreinigung mit Mikrowellen an einen
Wabenkatalysator mit Platin, Palladium und Ruthenium [175, 176]. Dabei zeigte es sich,
dass die technisch bedeutsamen „Anspringtemperaturen“ für die NO-und Kohlenwasser-
stoffumsetzung im Abgas bei Mikrowellenheizung deutlich niedriger lagen als bei Einsatz
einer konventionellen elektrischen Heizung. Andere Autoren setzten statt der üblicher-
weise verwendeten Wabenkatalysatoren eine spezielle Schaumkeramik ein, welche
Mikrowellenstrahlung sehr gut absorbiert [177].
Eine weitere Möglichkeit der Stickoxidentfernung aus Abgasen besteht darin, Kohlenwas-
serstoffe als Reduktionsmittel zu verwenden. Bei der Reduktion von NO mit Methan und
Sauerstoff haben sich Zeolithe als aktive Katalysatoren erwiesen [178]. Der erste Autor,
der die Verwendung von Mikrowellen als Heizmedium bei dieser Reaktion beschreibt,
war CHANG ET AL. im Jahr 1999 [179]. Die verwendeten Katalysatoren wurden über
Ionenaustausch eines Na-ZSM-5 mit einem Si/Al-Verhältnis von 56 hergestellt. Eine
Übersicht der eingesetzten Zeolithe gibt Tabelle 2.7.
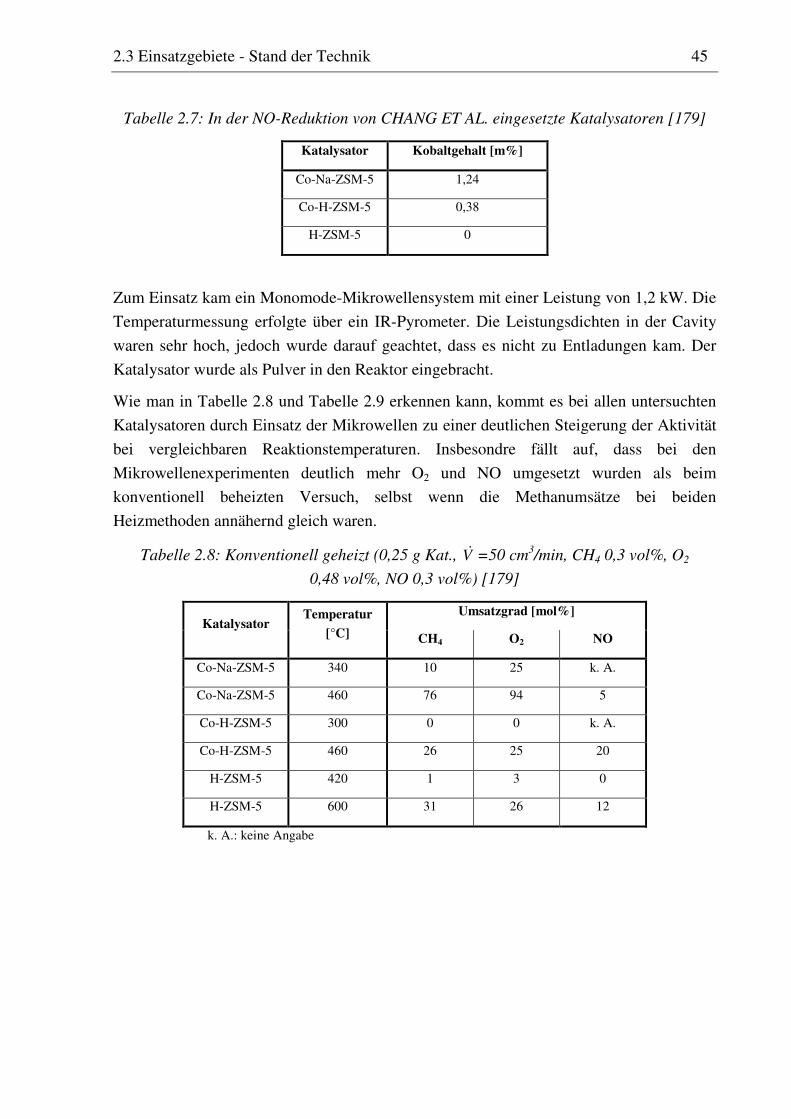
2.3 Einsatzgebiete - Stand der Technik 45
Tabelle 2.7: In der NO-Reduktion von CHANG ET AL. eingesetzte Katalysatoren [179]
Katalysator Kobaltgehalt [m%]
Co-Na-ZSM-5 1,24
Co-H-ZSM-5 0,38
H-ZSM-5 0
Zum Einsatz kam ein Monomode-Mikrowellensystem mit einer Leistung von 1,2 kW. Die
Temperaturmessung erfolgte über ein IR-Pyrometer. Die Leistungsdichten in der Cavity
waren sehr hoch, jedoch wurde darauf geachtet, dass es nicht zu Entladungen kam. Der
Katalysator wurde als Pulver in den Reaktor eingebracht.
Wie man in Tabelle 2.8 und Tabelle 2.9 erkennen kann, kommt es bei allen untersuchten
Katalysatoren durch Einsatz der Mikrowellen zu einer deutlichen Steigerung der Aktivität
bei vergleichbaren Reaktionstemperaturen. Insbesondre fällt auf, dass bei den
Mikrowellenexperimenten deutlich mehr O2 und NO umgesetzt wurden als beim
konventionell beheizten Versuch, selbst wenn die Methanumsätze bei beiden
Heizmethoden annähernd gleich waren.
Tabelle 2.8: Konventionell geheizt (0,25 g Kat., V& =50 cm3/min, CH4 0,3 vol%, O2
0,48 vol%, NO 0,3 vol%) [179]
Umsatzgrad [mol%] Katalysator
Temperatur
[°C] CH4 O2 NO
Co-Na-ZSM-5 340 10 25 k. A.
Co-Na-ZSM-5 460 76 94 5
Co-H-ZSM-5 300 0 0 k. A.
Co-H-ZSM-5 460 26 25 20
H-ZSM-5 420 1 3 0
H-ZSM-5 600 31 26 12
k. A.: keine Angabe

46 2 Mikrowellen als Energieträger in der Reaktionstechnik
Tabelle 2.9: Mikrowellenheizung (0,5 g Kat., V& =100 cm3/min, CH4 0,2 vol%, O2
0,4 vol%, NO 0,4 vol%; Co-Na-ZSM5: CH4 0,25 vol%, O2 0,4 vol%, NO 0,3 vol% ) [179]
Umsatzgrad [mol%] Katalysator Temperatur [°C]
CH4 O2 NO
Co-Na-ZSM-5 338 100 70 72
Co-Na-ZSM-5 402 100 76 75
Co-H-ZSM-5 293 37 91 77
Co-H-ZSM-5 395 33 94 85
H-ZSM-5 370 25 100 35
H-ZSM-5 402 26 100 35
Vergleichbare Untersuchungen im Mikrowellenfeld wurden auch von anderen Autoren
durchgeführt [180]. Dabei wurden ein In-ZSM-5 (2,1 m% Indium) und ein
In-Fe2O3-ZSM-5 (2,1 m% Indium, 16,8 m% Fe2O3) getestet, welche durch Imprägnierung
hergestellt wurden. Die Ergebnisse dieser Untersuchungen finden sich in Abbildung 2.15.
Abbildung 2.15: NO-Umsätze an einem In-Fe2O3-ZSM5 (1:8:20) in Abhängigkeit von der
Reaktionstemperatur bei verschiedenen Heizmethoden und bei Zugabe von Wasser als
Störkomponente [180]
Dabei zeigte sich, dass beim Heizen mit Mikrowelle am In-Fe2O3-Katalysator bereits bei
beinahe 200 °C niedrigeren Reaktionstemperaturen vergleichbare NO-Umsätze erreicht
werden konnten. Der In-ZSM-5 wiederum zeigte unter konventionellen Bedingungen
annähernd die gleiche Aktivität, ließ sich durch Mikrowellen jedoch nicht aufheizen, so
dass keine Umsätze erzielt werden konnten. Diese Versuche wurden dann mit

2.3 Einsatzgebiete - Stand der Technik 47
Wasserdampf im Eduktgemisch wiederholt. Als Folge konnte ein deutlicher
Aktivitätsverlust beobachtet werden, der bei Verwendung von Mikrowellen als
Heizmedium jedoch deutlich geringer ausfiel. Dies wurde vom Autor auf eine Desorption
des Wassers von den aktiven Zentren des Katalysators durch die Mikrowellenstrahlung
zurückgeführt.
Weitere Untersuchungen wurden von TANG ET AL. an Co-H-ZSM-5 und Ni-H-ZSM-5
durchgeführt. Hierbei wurden jedoch bewusst elektrische Entladungen am Katalysator
durch die Mikrowellen herbeigeführt [181].
Der Arbeitskreis von MINGOS untersuchte die Reduktion von SO2 mit Methan an
MoS2-Katalysatoren [182]. Auch hier wurden wieder im Vergleich zum konventionellen
Experiment deutlich höhere Umsatzgrade bei niedrigeren Reaktionstemperaturen durch
den Einsatz der Mikrowellen als Heizmethode erreicht. Da diese Untersuchungen auch für
die Diskussion der Mikrowelleneffekte eine große Bedeutung haben, wird auf diese
Arbeiten in Kapitel 2.3.2.5.1.1 noch näher eingegangen.
2.3.2.4 Andere Reaktionen
In diesem Kapitel sollen die mikrowellenunterstützten Reaktionen vorgestellt werden,
welche sich nicht in die bereits behandelten Kategorien einordnen lassen.
Zunächst sei hier auf die weiteren Arbeiten im Umfeld von WAN hingewiesen, in denen
unter anderem die Zersetzung organischer Halogenverbindungen [183], die Synthese von
Blausäure aus Methan und Ammoniak [184], die Herstellung von Acetylen durch Cracken
von Benzen [185] und die mikrowellenunterstützte Zersetzung von Alberta Öl-Sanden
und Bitumen [186] beschrieben wird. Bei all diesen Untersuchungen kam die für WAN
typische Vorgehensweise zum Einsatz (vgl. S. 34).
Die 2-Propanol-Zersetzung an mit Alkalimetallen (Li, Na, K) dotierter Kokosnuss-Aktiv-
kohle und Graphit wurde von BOND ET Al. untersucht [187]. Es kam eine Monomode-
Mikrowelle mit einer maximalen Leistung von 750 W zum Einsatz. Die Reaktions-Tem-
peratur wurde mit einem faseroptischen Sensor gemessen. Dabei zeigte sich, dass beim
Einsatz der Mikrowelle als Heizmethode ein vergleichbarer Umsatzgrad an 2-Propanol
bereits bei deutlich geringeren Reaktionstemperaturen erreicht werden konnte. Die Tem-
peraturunterschiede bei einem Umsatzgrad von 50 % zwischen Mikrowellenheizung und
konventioneller Heizung am Graphit-Katalysator betrugen teilweise mehr als 150 °C, bei
der Aktivkohle mehr als 50 °C. Neben der Aktivitätssteigerung konnte jedoch auch ein
Einfluss der Heizmethode auf die Selektivität zum Reaktionsprodukt Aceton festgestellt
werden. Während die Mikrowellenheizung am Graphit zu keiner Änderung der Aceton-
Selektivität führte, waren die Selektivitäten an der Aktivkohle deutlich niedriger als unter
konventionellen Bedingungen. Auf die vermuteten Ursachen wird im folgenden Kapitel
noch näher eingegangen.

48 2 Mikrowellen als Energieträger in der Reaktionstechnik
Weitere Autoren berichten von der Isomerisierung von Kohlenwasserstoffen unter
Mikrowelleneinwirkung. SEYFRIED ET AL. beschäftigten sich mit der 2-Methylpentan-
Isomerisierung an einem Pt/Al2O3-Katalysator (0,2 m% Pt) [188, 189].
Das verwendete Monomode-Mikrowellensystem hatte eine maximale Leistung von
200 W. Die Temperaturmessung erfolgte über Thermoelemente, welche an der Reaktorau-
ßenseite befestigt waren. Bei diesen Untersuchungen wurde sowohl eine Vorbehandlung
der Katalysatoren (Reduktion in H2) im Mikrowellenfeld getestet als auch die Reaktion
unter Mikrowelleneinstrahlung. Die Ergebnisse sind in Tabelle 2.10 zusammengefasst. In
der Veröffentlichung wird nur die Selektivität der gebildeten Hexanisomere betrachtet, da
die Umsatzgrade des 2-Methylpentans immer deutlich kleiner als 10 % waren.
Bei den Untersuchungen zeigte sich, dass sowohl die Heizmethode während der Kataly-
satorvorbehandlung als auch während der Reaktion einen entscheidenden Einfluss auf die
Isomerisierung hatten. Durch Verwendung von Mikrowellen als Heizmedium während der
Reaktion ließ sich die Selektivität zu den Isomeren beinahe verdoppeln. Der gleiche
Effekt zeigte sich jedoch auch, wenn der Katalysator während der Reaktion konventionell
geheizt wurde und nur die Vorbehandlung mit Mikrowellen erfolgte. Dies legte den
Schluss nahe, dass es durch die Mikrowellen zu einer Veränderung des Katalysators
kommt. Bestätigt wurde dies auch durch die Beobachtung, dass sich die Isomer-Selekti-
vität bei einem bereits mit Mikrowellen vorbehandelten Katalysator durch den Einsatz
von Mikrowellen während der Reaktion nicht mehr steigern ließ.
Tabelle 2.10: Isomer-Selektivität der 2-Methylpentan-Isomerisierung an einem
Pt/Al2O3-Katalysator bei verschiedenen Heizmethoden in der Katalysatorvorbehandlung
(Reduktion in H2 bei 350 °C) und während der Reaktion (260 °C) [188]
Heizmethode Experiment Nr.
Vorbehandlung Reaktion Isomer-Selektivität [%]
1 Konv. Konv. 41
2 Konv. MW 79
3 Katalysator aus Experiment 2 Konv. 48
4 MW Konv. 81
5 MW MW 79
Ebenfalls mit einer Isomerisierungsreaktion beschäftigt sich eine Publikation von
ZHOLOBENKO ET AL. [190]. Untersucht wurde die Isomerisierung von 1-Buten an
Zeolithkatalysatoren. Ziel dieser Arbeiten war es, den Einfluss der Mikrowellenheizung
auf die Aktivität eines Ferrierit (Si/Al = 9,8), dessen Struktur und das Deaktivierungsver-
halten während der Reaktion zu bestimmen. Bei der eingesetzten Monomode-Mikrowelle
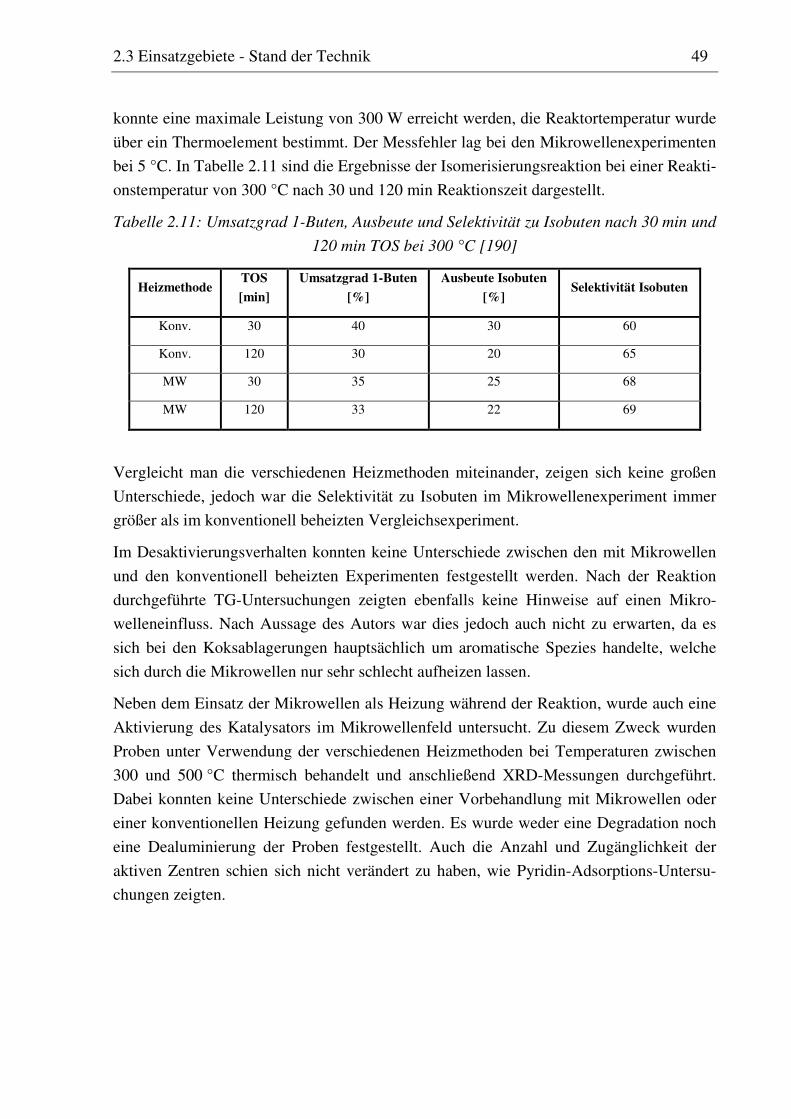
2.3 Einsatzgebiete - Stand der Technik 49
konnte eine maximale Leistung von 300 W erreicht werden, die Reaktortemperatur wurde
über ein Thermoelement bestimmt. Der Messfehler lag bei den Mikrowellenexperimenten
bei 5 °C. In Tabelle 2.11 sind die Ergebnisse der Isomerisierungsreaktion bei einer Reakti-
onstemperatur von 300 °C nach 30 und 120 min Reaktionszeit dargestellt.
Tabelle 2.11: Umsatzgrad 1-Buten, Ausbeute und Selektivität zu Isobuten nach 30 min und
120 min TOS bei 300 °C [190]
Heizmethode TOS
[min]
Umsatzgrad 1-Buten
[%]
Ausbeute Isobuten
[%] Selektivität Isobuten
Konv. 30 40 30 60
Konv. 120 30 20 65
MW 30 35 25 68
MW 120 33 22 69
Vergleicht man die verschiedenen Heizmethoden miteinander, zeigen sich keine großen
Unterschiede, jedoch war die Selektivität zu Isobuten im Mikrowellenexperiment immer
größer als im konventionell beheizten Vergleichsexperiment.
Im Desaktivierungsverhalten konnten keine Unterschiede zwischen den mit Mikrowellen
und den konventionell beheizten Experimenten festgestellt werden. Nach der Reaktion
durchgeführte TG-Untersuchungen zeigten ebenfalls keine Hinweise auf einen Mikro-
welleneinfluss. Nach Aussage des Autors war dies jedoch auch nicht zu erwarten, da es
sich bei den Koksablagerungen hauptsächlich um aromatische Spezies handelte, welche
sich durch die Mikrowellen nur sehr schlecht aufheizen lassen.
Neben dem Einsatz der Mikrowellen als Heizung während der Reaktion, wurde auch eine
Aktivierung des Katalysators im Mikrowellenfeld untersucht. Zu diesem Zweck wurden
Proben unter Verwendung der verschiedenen Heizmethoden bei Temperaturen zwischen
300 und 500 °C thermisch behandelt und anschließend XRD-Messungen durchgeführt.
Dabei konnten keine Unterschiede zwischen einer Vorbehandlung mit Mikrowellen oder
einer konventionellen Heizung gefunden werden. Es wurde weder eine Degradation noch
eine Dealuminierung der Proben festgestellt. Auch die Anzahl und Zugänglichkeit der
aktiven Zentren schien sich nicht verändert zu haben, wie Pyridin-Adsorptions-Untersu-
chungen zeigten.

50 2 Mikrowellen als Energieträger in der Reaktionstechnik
2.3.2.5 Diskussion der beobachteten Mikrowelleneffekte
Wie auch bei den organischen Synthesen (vgl. Kapitel 2.3.1.4) kam es im Bereich der
heterogenen Gasphasenkatalyse im Mikrowellenfeld zu einer heftigen Diskussion darüber,
wie sich die experimentellen Ergebnisse theoretisch erklären lassen. Eine Einteilung in
„nichtthermische“ und „thermische“ Mikrowelleneffekte erschien auch hier sinnvoll.
2.3.2.5.1 Thermische Mikrowelleneffekte
2.3.2.5.1.1 Lokalisierte Übertemperaturen – „hot spots“
Fast alle Untersuchungen der heterogenen Gasphasenkatalyse im Mikrowellenfeld weisen
zwei Gemeinsamkeiten auf:
1. Der Katalysator wird mit Hilfe der Mikrowellenstrahlung auf die gewünschte
Reaktionstemperatur gebracht.
2. Das Reaktionsverhalten deutet darauf hin, dass die Reaktionstemperatur höher ist
als die tatsächlich gemessene Katalysatorbetttemperatur.
Ein Großteil der Autoren erklären ihre Ergebnisse deshalb mit der Bildung so genannter
„hot spots“ im Katalysatorbett. Als problematisch erweist sich jedoch, dass diese Tempe-
raturüberhöhungen aufgrund ihrer geringen Größe meist nicht direkt messtechnisch nach-
gewiesen werden können. Es werden Größenordnungen im Nanometerbereich (mikrosko-
pische Inhomogenitäten; z. B. Sauerstofffehlstellen in Perowskiten) [4] bis hin zu 1 mm
(makroskopische Inhomogenitäten) diskutiert [139], also weit unter der Auflösung gängi-
ger Temperaturmessverfahren. Weiterhin ist umstritten, ob sich diese lokalen Tempera-
turüberhöhungen während der Mikrowellenstrahlung dauerhaft aufrechterhalten lassen,
oder ob die Temperaturabgabe an die Umgebung so schnell erfolgt, dass es innerhalb
kürzester Zeit zu einem Temperaturausgleich kommt.
Bei der Ausbildung dieser „hot spots“ kann man theoretisch zwei Fälle unterschieden.
1. Mikroskopische Bereiche des Katalysators (z.B. Metallpartikel bei geträgerten
Katalysatoren) können aufgrund der stofflichen Zusammensetzung mit den
Mikrowellen besser wechselwirken und werden stärker aufgeheizt. Besitzt das
Material eine relativ schlechte Wärmeleitfähigkeit, kommt es zur Ausbildung
von lokalen Überhitzungen im Katalysator.
2. Aufgrund einer inhomogenen Feldverteilung (technische Ursachen vgl. Kapitel
2.2) oder makroskopischer Inhomogenitäten des Katalysators werden
bestimmte Bereiche des Katalysatorbetts stärker erwärmt.
Diese beiden Fälle werden im Folgenden ausführlich diskutiert.

2.3 Einsatzgebiete - Stand der Technik 51
1.) Bei einem geträgerten Metallkatalysator erscheint es zunächst einleuchtend, dass die
Metallpartikel eine höhere Temperatur annehmen als der verwendete Support. Das Kata-
lysatorbett und die umgebende Gasphase sind kälter als die katalytisch aktiven Bereiche.
Dadurch lassen sich unerwünschte Nebenreaktionen zurückdrängen und höhere Selekti-
vitäten zum Zielprodukt realisieren [191].
Von PERRY ET AL., die sich mit der CO-Oxidation an Pd/Pt-γ-Al2O3-Katalysatoren
beschäftigte, wurden Berechnungen durchgeführt, um die durch Mikrowellenheizung
entstehenden Übertemperaturen an Metallpartikeln theoretisch zu erfassen [167, 191]. Das
Ergebnis war, dass lokale Übertemperaturen durch Mikrowellenheizung an den Metall-
partikeln (1-100 µm) theoretisch nicht auftreten dürften. Nichtsdestotrotz zeigte es sich,
dass die Katalysatoren im Mikrowellenfeld deutlich aktiver waren als unter konventio-
nellen Bedingungen. Die Autoren führen diese Beobachtung auf Temperaturgradienten im
Katalysatorbett bzw. eine fehlerhafte Temperaturmessung im Mikrowellenfeld zurück.
Untersuchungen mit einer Wärmebildkamera zeigten, dass es während des Mikrowellen-
heizens zu axialen Temperaturgradienten (20 °C) im Reaktor kam. Außerdem war das
Thermoelement während der reaktionstechnischen Untersuchungen an der kältesten Stelle
des Reaktors platziert. Um diese Fehlerquellen auszuschließen, wurde in weiteren Experi-
menten nur eine kleine Menge des Katalysators verwendet, die als dünne Schicht
zwischen zwei Bereiche Inertmaterial (reines α-Al2O3) in den Reaktor eingebaut wurde.
Die Temperaturmessung erfolgte nun in dieser Zone des Katalysators. Bei den Untersu-
chungen der CO-Oxidation zeigte sich nun, dass unter Mikrowellenbedingungen immer
noch eine höhere Katalysatoraktivität zu verzeichnen war als unter Verwendung einer
konventionellen Heizmethode. Die Differenzen fielen jedoch deutlich geringer aus als bei
den ersten Experimenten, bei denen der Katalysator nicht nach der „sandwich“ Methode
in den Reaktor eingebaut wurde. In einer dritten Versuchsreihe wurde schließlich das
Inertmaterial α-Al2O3 gegen γ-Al2O3 ausgetauscht. Das gesamte Katalysatorbett bestand
damit aus dem gleichen Material und nur in einem schmalen Streifen in der Bettmitte war
katalytisch aktives Metall enthalten. Temperaturgradienten sollten somit weitestgehend
vermieden werden. Bei dieser Versuchsanordnung ließen sich keine Unterschiede zwi-
schen den zwei Heizmethoden in der CO-Oxidation feststellen. Die Autoren werten dies
als klaren Beweis, dass es durch die Mikrowellenheizung nicht zu einer selektiven
Aufheizung der aktiven Zentren (Metallpartikel) kommt. Für die zwischen den beiden
Heizmethoden beobachteten Unterschiede in der CO-Oxidation seien lediglich Tempera-
turgradienten im Katalysatorbett und eine fehlerhafte Temperaturmessung verantwortlich.
Von THOMAS wurde der Einfluss von Mikrowellenstrahlung auf geträgerte Metallparti-
kel theoretisch untersucht [192]. Bei diesen Berechnungen wurde die Frequenz der Strah-
lung berücksichtigt. Das Ergebnis war, dass eine Temperaturüberhöhung wahrscheinlicher
ist für große Partikel (>60 µm) und hohe Frequenzen. Bei den herkömmlich verwendeten

52 2 Mikrowellen als Energieträger in der Reaktionstechnik
Frequenzen kleiner 3 GHz ist die zu erwartende Temperaturüberhöhung an dispers ver-
teilten Metallpartikeln jedoch vernachlässigbar klein.
2.) Von ZHANG und MINGOS, die sich mit der katalytischen Zersetzung von H2S [10,
139, 182] an MoS2-Katalysatoren im Mikrowellenfeld beschäftigten, wurde ein indirekter
Beweis für die Bildung von hot spots aufgrund makroskopischer Inhomogenitäten des
Katalysators gefunden. Bei dieser stark endothermen Reaktion wird H2S zu H2 und S2 zer-
setzt. Es kamen zwei verschiedene Katalysatoren mit dem gleichen MoS2-Anteil zum Ein-
satz. Ein Katalysator wurde mittels Imprägnierung des Trägers (γ-Al2O3) mit MoS2, der
andere durch Mischen der zwei Komponenten und anschließendes Verpressen hergestellt.
Bei den katalytischen Messungen zeigte sich, dass die H2S-Umsätze unter konventionellen
Heizbedingungen annähernd den zu erwartenden Gleichgewichtsumsätzen der Reaktion
bei den eingestellten Temperaturen entsprachen. Unter Mikrowellenbedingungen lagen
die Umsätze jedoch deutlich höher. Dabei war der durch Mischen hergestellte Katalysator
aktiver als der mit MoS2 imprägnierte. Um einen Umsatzgrad von 4 % zu erreichen, wa-
ren unter konventionellen Bedingungen ca. 800 °C nötig, unter Mikrowellenbedingungen
jedoch nur 650 °C am imprägnierten Katalysator bzw. 500 °C am verpressten Katalysator.
Bei XRD-Untersuchungen der getesteten Katalysatoren konnte nur festgestellt werden,
dass sich Teile des Trägers von γ- zu α-Al2O3 umgewandelt hatten. Diese Umwandlung
findet jedoch erst bei Temperaturen statt, die mindestens 200 °C über der maximal einge-
stellten Reaktionstemperatur lag. Die Größe dieser Bereiche betrug ca. 90 µm. Es konnte
festgestellt werden, dass sich das im frischen Katalysator dispers verteilte MoS2 zu hexa-
gonalen Kristallen zusammengeschlossen hatte. Der Schmelzpunkt von MoS2 liegt jedoch
mindestens 400 °C höher als die maximal gemessene Reaktionstemperatur. An zusätzlich
durchgeführten elektronenmikroskopischen Aufnahmen zeigten sich kugelförmige Parti-
kel aus MoS2 und Al2O3 mit einer Größe von bis zu 1000 µm. Für die Autoren ein Beweis
für die Existenz von lokalisierten hot spots. Die höhere Aktivität des durch Mischen
hergestellten Katalysators wurde mit Hilfe von Dielektrizitätsmessungen erklärt. Dieser
Katalysator ließ sich durch die Mikrowellen deutlich besser aufheizen und war deutlich
inhomogener, was zu einer verstärkten Bildung von hot spots bzw. noch größeren
Übertemperaturen führte. Diese Unterschiede zwischen den zwei Katalysatoren deuteten
jedoch darauf hin, dass es nicht zu lokalen Überhitzungen an einzelnen MoS2-Partikeln
kommt. Wären lediglich die MoS2-Partikel heißer als der Träger, müssten sich beide
Katalysatortypen in der Reaktion gleich verhalten. Viel wahrscheinlicher schien es, dass
Inhomogenitäten im Katalysator oder der elektrischen Feldverteilung für die Bildung der
hot spots verantwortlich waren. Deshalb wurden theoretische Berechnungen durchgeführt,
um die Übertemperatur an einem MoS2-Partikel auf γ-Al2O3 als Träger abzuschätzen.
Dabei zeigte sich, dass MoS2-Partikel in der Größenordnung von Millimetern vorliegen
müssten, um eine signifikante Temperaturüberhöhung zu bewirken. Während des

2.3 Einsatzgebiete - Stand der Technik 53
Mikrowellenheizens kam es also wahrscheinlich nicht zu einer „mikroskopischen“
Aufheizung des MoS2, sondern zu makroskopischen Übertemperaturen im Katalysator,
welche mit einer erheblichen Reorganisation des Katalysators einhergingen. Diese Ergeb-
nisse konnten an vergleichbaren Katalysatoren auch in der hydrierenden Entschweflung
von Thiophen [10] und in der Reduktion von SO2 mit Methan [182] bestätigt werden.
Neben den Untersuchungen an metallischen Katalysatoren finden sich jedoch auch bei
anderen Autoren Hinweise auf die Ausbildung von „makroskopischen“ hot spots. Bei
Untersuchungen zur 2-Propanolzersetzung an Aktivkohle oder Graphit fanden BOND ET
AL. eindeutige Hinweise für lokale Temperaturüberhöhungen im Katalysatorbett [187].
Wurde bei diesen Experimenten das Trägergas (Stickstoff) abgeschaltet, waren kleine,
glühende Bereiche im Reaktor zu erkennen. Dieses Phänomen schien zufällig in verschie-
denen Bereichen des Katalysatorbettes aufzutauchen. Der Autor führt dies auf die lokali-
sierte Ausbildung von Plasma zurück. Auch wenn es während der Reaktion nicht zur
Zündung eines Plasmas kam, sind doch lokale Übertemperaturen im Katalysatorbett zu
vermuten. Als Ursache dieses Phänomens kommen temporär auftretende Feldinhomoge-
nitäten in Frage.
Auch von STUERGA und GAILLARD wird vermutet, dass sich viele Mikrowelleneffekte
auf ein nicht isothermes Katalysatorbett zurückführen lassen [15].
2.3.2.5.1.2 Selektive Mikrowellenheizung von Oberflächenspezies
Neben den hot spots werden jedoch auch noch andere thermische Effekte diskutiert.
Untersuchungen von BOND ET Al. an Zeolithen zeigten, dass sich während einer Reak-
tion gebildeter Koks mit Hilfe von Mikrowellenstrahlung selektiv aufheizen und abbren-
nen lässt [12]. Im Unterschied zu den bisher diskutierten Effekten wird also nicht der
Katalysator selbst, sondern eine Oberflächenspezies selektiv aufgeheizt.
Ein vergleichbarer Effekt, der ebenfalls in der Literatur diskutiert wird, ist die selektive
Aufheizung von Oberflächen-Hydroxylgruppen des Katalysators durch Mikrowellen. Bei
den Untersuchungen zur 2-Propanolzersetzung an Aktivkohle zeigte sich unter Mikro-
wellenbestrahlung eine deutlich verschlechterte Selektivität zum Aceton [187]. Die Auto-
ren führen dies darauf zurück, dass Hydroxylgruppen am Katalysator durch die Mikro-
wellenstrahlung zerstört werden. Das bei der Reaktion freiwerdende Wasser wird nun zum
Wiederaufbau der Hydroxylgruppen verwendet, wodurch die Dehydrogenierung unter
Wasserfreisetzung bevorzugt abläuft.
ZHOLOBENKO ET Al. vermuten bei der Isomerisierung von 1-Buten an Zeolithen eben-
falls einen Einfluss der Mikrowellenstrahlung auf Hydroxylgruppen [190]. Da diese Grup-
pen eine maßgebliche Rolle bei dieser Reaktion spielen und sich aufgrund ihrer polaren
Natur eventuell gut durch die Mikrowellenstrahlung aufheizen lassen, könnte man so den
Mikrowelleneinfluss auf die Isomerisierung erklären. Bei FTIR-Untersuchungen der

54 2 Mikrowellen als Energieträger in der Reaktionstechnik
Hydroxylgruppen konnten jedoch keine Unterschiede zwischen einem konventionell oder
mit Mikrowellen behandelten Katalysator festgestellt werden.
Auch von anderen Autoren wird vermutet, dass die Mikrowellen einen Einfluss auf adsor-
bierte Oberflächenspezies ausüben, bisher konnte jedoch noch kein Beweis für diese
Theorie geliefert werden [165, 169, 175].
2.3.2.5.2 Nichtthermische Mikrowelleneffekte
Neben den bereits beschriebenen thermischen Mikrowelleneffekten vermuten jedoch
einige Autoren, dass es unter bestimmten Bedingungen auch zu nicht thermischen
Effekten durch die Mikrowellenstrahlung kommen kann [153, 189, 193]. Von KLIMOV
ET AL. wurde beispielsweise die Ethylen-Epoxidierung an Ag/α-Al2O3-Katalysaoren
getestet [194]. Bei diesen Untersuchungen wurde der Katalysator gleichzeitig
konventionell durch Einblasen heißer Luft und mit Mikrowellenstrahlung erwärmt. Bis
auf einen einzigen Katalysator konnten keine Unterschiede zwischen rein konventioneller
oder kombinierter Heizmethode gefunden werden. Da der an diesem Katalysator gefun-
dene Effekt nur bei reduzierenden Bedingungen auftrat, vermuten die Autoren, dass die
Ausbildung von Defekten auf der Silberoberfläche im Mikrowellenfeld gehemmt abläuft.
Zum gegenwärtigen Zeitpunkt geht man davon aus, dass es sich bei den meisten Fällen
um rein thermische Mikrowelleneffekte handelt. Die häufigsten Ursachen für unter-
schiedliche Aktivitäten oder Selektivitäten bei heterogen katalysierten Reaktionen im
Mikrowellenfeld sind das Auftreten von „makroskopischen“ hot spots, Temperaturgra-
dienten im Katalysatorbett, höhere Katalysatortemperaturen im Vergleich zur Gaspha-
sentemperatur oder einfach fehlerhafte Temperaturmessungen [4].
Tabelle 2.12 gibt eine Zusammenfassung der in Kapitel 2.3.2 zitierten Literatur.

2.3 Einsatzgebiete - Stand der Technik 55
Tabelle 2.12: Literaturübersicht Kapitel 2.3.2
Reaktion Katalysator MW-System T [°C] / p [kPa] „MW-Effekt“ Lit.
Abbau von
chlorierten
Kohlenwasserstoffen
Fe; para- oder
ferromagnetische
Metalle
Monomode;
gepulst k. A. / k. A.
Anregung freier
Elektronen der
Katalysatoroberfläche
[16,
140]
Methanzersetzung Ni Monomode,
gepulst >1400 / 100
Katalysatoroberfläche
ist wärmer als Bett- oder
Gastemperatur
[146]
Methanzersetzung Ni Monomode,
gepulst k. A. / 100
Selektive Aufheizung
ferromagentischer
Zentren der
Katalysatoroberfläche
[147,
148]
Methan- und 1-
Penten-Kopplung Ni
Monomode,
gepulst k. A. / 2-50 k. A. [149]
Oxidierende
Methandehydrierung Na-Aluminat
Monomode;
kontinuierlich 200-900 / U Hot spots [150]
Oxidierende
Methandehydrierung La, Sr/MgO
Monomode;
kontinuierlich 240-550 / U
Hot Spots; Katalysator
wärmer als Bett- oder
Gastemperatur
[12]
Oxidierende
Methandehydrierung SmLiO2
Monomode;
kontinuierlich 200-500 / U
Hot Spots; Katalysator
wärmer als Bett- oder
Gastemperatur
[151]
Oxidierende
Methandehydrierung La2O3; La2ZrO7
Monomode;
kontinuierlich 200-500 / U
Hot Spots; Katalysator
wärmer als Bett- oder
Gastemperatur
[152]
Oxidierende
Methandehydrierung
Li/MgO;
BaBiO3-x
Monomode;
kontinuierlich 550-800 / U
Katalysator wärmer als
Gasphase [153]
Oxidierende
Methandehydrierung
Perowskite;
Mischoxide
Monomode;
kontinuierlich 400-600 / U Hot Spots [154]
Oxidierende
Methandehydrierung
Ni/ZrO2;
Ni/La2O3;
CoZrO2;
Co/La2O3
Monomode;
kontinuierlich 400-800 / U
Hot Spots; von konv.
Heizung abweichender
Reaktionsmechanismus
[155]
Oxidierende
Methandehydrierung
Al-Ni-Ti;
ThO2/Al2O3;
Fe/Al2O3;
Ni/Al2O3
Monomode;
kontinuierlich k. A. / 100-120
Aufheizung aktiver
Zentren;
Oberflächenentladungen
[156]
Oxidierende
Methandehydrierung Aktivkohle
Monomode;
gepulst k. A. / U
Oberfläche wärmer als
Bett- oder
Gastemperatur
[157]
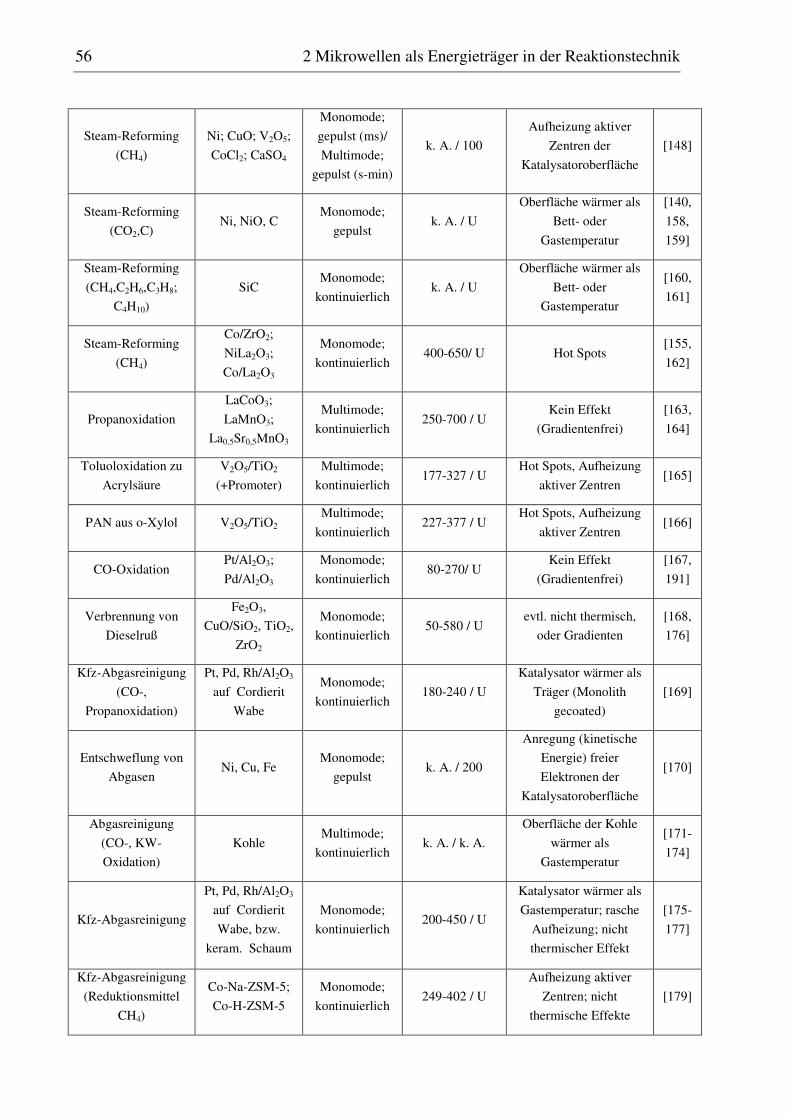
56 2 Mikrowellen als Energieträger in der Reaktionstechnik
Steam-Reforming
(CH4)
Ni; CuO; V2O5;
CoCl2; CaSO4
Monomode;
gepulst (ms)/
Multimode;
gepulst (s-min)
k. A. / 100
Aufheizung aktiver
Zentren der
Katalysatoroberfläche
[148]
Steam-Reforming
(CO2,C) Ni, NiO, C
Monomode;
gepulst k. A. / U
Oberfläche wärmer als
Bett- oder
Gastemperatur
[140,
158,
159]
Steam-Reforming
(CH4,C2H6,C3H8;
C4H10)
SiC Monomode;
kontinuierlich k. A. / U
Oberfläche wärmer als
Bett- oder
Gastemperatur
[160,
161]
Steam-Reforming
(CH4)
Co/ZrO2;
NiLa2O3;
Co/La2O3
Monomode;
kontinuierlich 400-650/ U Hot Spots
[155,
162]
Propanoxidation
LaCoO3;
LaMnO3;
La0,5Sr0,5MnO3
Multimode;
kontinuierlich 250-700 / U
Kein Effekt
(Gradientenfrei)
[163,
164]
Toluoloxidation zu
Acrylsäure
V2O5/TiO2
(+Promoter)
Multimode;
kontinuierlich 177-327 / U
Hot Spots, Aufheizung
aktiver Zentren [165]
PAN aus o-Xylol V2O5/TiO2 Multimode;
kontinuierlich 227-377 / U
Hot Spots, Aufheizung
aktiver Zentren [166]
CO-Oxidation Pt/Al2O3;
Pd/Al2O3
Monomode;
kontinuierlich 80-270/ U
Kein Effekt
(Gradientenfrei)
[167,
191]
Verbrennung von
Dieselruß
Fe2O3,
CuO/SiO2, TiO2,
ZrO2
Monomode;
kontinuierlich 50-580 / U
evtl. nicht thermisch,
oder Gradienten
[168,
176]
Kfz-Abgasreinigung
(CO-,
Propanoxidation)
Pt, Pd, Rh/Al2O3
auf Cordierit
Wabe
Monomode;
kontinuierlich 180-240 / U
Katalysator wärmer als
Träger (Monolith
gecoated)
[169]
Entschweflung von
Abgasen Ni, Cu, Fe
Monomode;
gepulst k. A. / 200
Anregung (kinetische
Energie) freier
Elektronen der
Katalysatoroberfläche
[170]
Abgasreinigung
(CO-, KW-
Oxidation)
Kohle Multimode;
kontinuierlich k. A. / k. A.
Oberfläche der Kohle
wärmer als
Gastemperatur
[171-
174]
Kfz-Abgasreinigung
Pt, Pd, Rh/Al2O3
auf Cordierit
Wabe, bzw.
keram. Schaum
Monomode;
kontinuierlich 200-450 / U
Katalysator wärmer als
Gastemperatur; rasche
Aufheizung; nicht
thermischer Effekt
[175-
177]
Kfz-Abgasreinigung
(Reduktionsmittel
CH4)
Co-Na-ZSM-5;
Co-H-ZSM-5
Monomode;
kontinuierlich 249-402 / U
Aufheizung aktiver
Zentren; nicht
thermische Effekte
[179]

2.3 Einsatzgebiete - Stand der Technik 57
Kfz-Abgasreinigung
(Reduktionsmittel
CH4)
In-ZSM-5;
In-Fe2O3-ZSM-5
Monomode;
kontinuierlich 200-650 / U Nicht thermischer Effekt [180]
NO-Reduktion
(Reduktionsmittel
CH4)
Co-H-ZSM-5;
Ni-H-ZSM-5
Monomode;
kontinuierlich;
discharge
150-680 / U Nicht thermischer Effekt [181]
Reduktion von SO2
mit CH4 MoS2
Multimode;
kontinulierlich 450-800 / U Hot Spots [182]
Zersetzung org.
Halogenverbindunge
n
Fe-Oxid/Al2O3 Monomode,
gepulst k. A. / U
Aufheizung aktiver
Zentren [183]
Synthese von
Blausäure aus CH4
und NH3
Ru, W, Pt, Co,
Ni, Pb, Ag,
Cu/α,γ-Al2O3
Monomode,
gepulst k. A. / U
Aufheizung aktiver
Zentren [184]
Synthese von
Acetylen durch
cracken von Benzen
Ni; H-Y; Al-
SR115; Si-
SP115; Al-
SR115-Ni; Si-
SP115-Ni
Monomode,
gepulst k. A. / U
Aufheizung aktiver
Zentren; Oberfläche
wärmer als Bett- oder
Gastemperatur
[185]
Zersetzung von
Alberta Öl-Sanden
und Bitumen
Ni; Cu; Ni/Al;
Cu/Al
Multimode,
gepulst (s) k. A. / 100
Katalysatoroberfläche
wärmer als Bett- oder
Gastemperatur
[186]
2-Propanol-
Zersetzung
Li, Na,
K/Aktivkohle
Monomode;
kontinuierlich 250-530 / U
Selektive Aufheizung
von Oberflächen-
Hydroxylgruppen; Hot
Spots
[187]
2-Methylpentan-
Isomerisierung Pt/γ-Al2O3
Monomode;
kontinuierlich 260-340 / U
Selektive Aufheizung
von „adsorbiertem“ H2O
und des Pt
[188,
189,
193]
1-Buten-
Isomerisierung Ferrierit
Monomode;
kontinuierlich 300 / U
Selektive Aufheizung
polarer Spezies [190]
Zersetzung von H2S MoS2/γ-Al2O3 Monomode;
kontinuierlich 377-800 / U Hot Spots
[10,
139]
Hydrierende
Entschwefelung von
Thiophen
MoS2/γ-Al2O3 Monomode;
kontinuierlich 180-600 / U Hot Spots [10]
Modellierung Metall/Support - - Hot Spots [192]
Ethylen-
Epoxidierung Ag/α-Al2O3
Multimode;
kontinuierlich 215 / U Nicht thermischer Effekt [194]
* k. A.: keine Angabe *U: Umgebungsdruck

58 2 Mikrowellen als Energieträger in der Reaktionstechnik
2.3.3 Einfluss von Mikrowellen auf Sorptionsvorgänge
Wie im letzten Kapitel beschrieben wurde, vermuten einige Autoren, dass der Einfluss der
Mikrowellenstrahlung auf eine selektive Wechselwirkung mit den am Katalysator adsor-
bierten Spezies zurückzuführen ist. Bisher sind jedoch noch keine Publikationen erschie-
nen, welche sich speziell mit dieser Thematik auseinandersetzen. Allerdings gibt es zahl-
reiche Untersuchungen zur Trocknung von porösen Schüttgütern und zur Regeneration
von Adsorbentien durch Mikrowellenenergie.
2.3.3.1 Trocknung
Wie bereits erwähnt, zählt die Trocknung zu den am intensivsten untersuchten und indus-
triell sehr häufig eingesetzten Mikrowellenanwendungen. Die Mikrowellentrocknung
bietet gegenüber herkömmlichen Verfahren verschiedene Vorteile. Insbesondre ist es
möglich, feuchte Gutsanteile selektiv zu trocknen, wenn der Feststoff im Vergleich zur
Flüssigkeit eine kleine Dielektrizitätskonstante besitzt. Der Energieeintrag erfolgt direkt
am gebundenen oder freien Wasser. Dadurch verläuft der Trocknungsvorgang schonender
und der Energieaufwand wird minimiert [195, 196]. Trotz des selektiven Energieeintrags
wird von vielen Autoren davon ausgegangen, dass sich die einzelnen Gutsbereiche in
einem thermodynamischen Äquilibrium befinden. Das heißt, der Feststoff, die Flüssigkeit
und die Gasphase in einem Kontrollvolumen besitzen zu jeder Zeit die gleiche Temperatur
[197-199]. Die Trocknung von Zeolithen vom Typ 13X wurde von RUOSSY ET AL.
[200] und BENCHANAA ET AL. [201] untersucht. Dabei zeigte es sich, dass die Trock-
nung zunächst sehr rasch erfolgt. Es kommt zu einem Temperaturanstieg, der mit einer
Desorption des gebundenen Wassers einhergeht. Dabei wird jedoch vorrangig das Wasser
und nicht der Zeolith selbst aufgeheizt. Schon nach kurzer Zeit erfolgt dann eine Stabili-
sierung der Temperatur. Die maximal erreichbare Temperatur hängt von der eingestrahl-
ten Mikrowellenleistung ab. Einhergehend mit der fortschreitenden Trocknung kommt es
zu einer Abnahme der pro Zeiteinheit desorbierten Wassermenge und anschließend zu
einem Rückgang der Temperatur. Die Trocknung schreitet nur noch sehr langsam weiter
voran. Analog zur desorbierten Wassermenge ändern sich auch die dielektrischen Eigen-
schaften des Zeolith. Imaginär- und Realteil der Dielektrizitätskonstante nehmen im Ver-
lauf der Trocknung immer weiter ab, bis sich schließlich ein konstant niedriger Wert ein-
stellt. Je höher die eingestrahlte Mikrowellenleistung, umso schneller erfolgt auch der
Rückgang der Dielektrizitätskonstanten. Die Anfangsbeladungen des Zeolith mit Wasser
waren bei diesen Untersuchungen sehr hoch (ca. 27 %). Aus diesen Ergebnissen lässt sich
der Schluss ziehen, dass eine Trocknung durch Mikrowellen umso effektiver möglich ist,
je höher die Anfangsbeladung des Zeolith mit Wasser ist.

2.3 Einsatzgebiete - Stand der Technik 59
2.3.3.2 Regeneration von Adsorbentien
In der Industrie werden Adsorber unter anderem für die Entfernung organischer
Lösungsmittel aus Abgasströmen eingesetzt. Nachdem die Aufnahmekapazität der Adsor-
bentien überschritten ist, muss einer Regenerierung oder Entsorgung erfolgen. Seit Mitte
der 80’er Jahre wird auch der Einsatz von Mikrowellen als Energieträger für die Regene-
rierung solcher beladener Adsorbentien untersucht [202]. Im Gegensatz zur Trocknung
sollen in diesem Fall nicht Wasser, sondern meist organische Stoffe desorbiert werden.
Verschiedene Publikationen beschäftigen sich mit dieser Thematik, insbesondre aus den
Arbeitskreisen um SCHMIDT [203-205] und BATHEN [17, 20, 206-208] sind zahlreiche
Veröffentlichungen erschienen. Im Folgenden sollen einige dieser Ergebnisse der Gruppe
um BATHEN exemplarisch vorgestellt werden.
Bei allen Versuchen kam eine Glaskolonne zum Einsatz, welche in einen speziell kon-
struierten Applikatorraum eingebaut werden konnte. Die Mikrowellen (2,45 GHz) wurden
an mehreren Stellen des Applikators über Koaxialkabel eingespeist. Es handelte sich
somit um ein Multimode-System. Die Temperatur des Festbettes wurde mit faseroptischen
Sensoren aufgenommen. Die Beladung des Adsorbers erfolgte aus der Gasphase in der
Anlage. Als Trägergas für die Ad- und Desorption wurde Stickstoff verwendet. Am
Ausgang der Kolonne konnte die Konzentration der desorbierten Komponenten im
Trägergas mit einem Fourier-Transformations-Infrarot-Spektrometer bestimmt werden.
Bei diesen Untersuchungen wurden zwei Adsorbentien verwendet, welche sich in ihren
dielektrischen Eigenschaften stark unterscheiden. Zum einen wurde Envisorb B+, welches
auf einer Silikagelmatrix basiert, in die fein verteilte Aktivkohle eingebunden ist, verwen-
det. Zum anderen kam ein hydrophober, dealuminierter Zeolith (DAY) zum Einsatz. Wäh-
ren das Material mit Aktivkohle sehr gut Mikrowellen absorbiert, ist der Zeolith nahezu
transparent für Mikrowellenstrahlung und kann somit kaum aufgeheizt werden.
Die beiden Adsorbentien wurden mit Mischungen polarer und unpolarer Stoffe bei 23 °C
beladen und anschließend durch Mikrowellenstrahlung unterschiedlicher Intensität rege-
neriert.
In Abbildung 2.16 ist der Desorptionsverlauf eines mit polaren Komponenten
(Ethanol/Aceton) beladenen, für Mikrowellen transparenten Adsorbens gezeigt. Man
erkennt, dass es sehr rasch zu einer Desorption beider Spezies kommt. Die Desorption
geht mit einem Temperaturanstieg auf ca. 70 °C einher. Nach 25 min gehen die Desorbat-
konzentrationen rasch zurück, der Großteil der adsorbierten Moleküle hat das Festbett
verlassen. Die Temperatur im Bett sinkt auf einen konstanten Wert (55 °C). Nach 40 min
ist die Desorption vollständig abgeschlossen. Die Restbeladung des Bettes beträgt ca. 5 %
der Anfangskonzentrationen beider Komponenten.

60 2 Mikrowellen als Energieträger in der Reaktionstechnik
Abbildung 2.16: Mikrowellendesorption eines mikrowellentransparenten Adsorbens;
Beladung mit zwei polaren Adsorptiven nach [20]
Ein gänzlich anderer Verlauf ergibt sich bei einer Beladung mit einer Mischung aus pola-
ren (Ethanol) und unpolaren (Toluol) Adsorptiven (vgl. Abbildung 2.17).
Abbildung 2.17: Mikrowellendesorption eines mikrowellentransparenten Adsorbens;
Beladung mit einem polaren (Ethanol) und einem unpolaren (Toluol) Adsorptiv nach [20]
Der polare Stoff wird sehr schnell desorbiert, während die Desorbatkonzentration des
unpolaren Stoffes zunächst leicht ansteigt und sehr schnell auf einen konstant niedrigen
Wert abfällt. Der Temperaturverlauf ist mit dem zweier polarer Adsorptive vergleichbar.
Während der Desorption des Ethanol steigt die Temperatur auf ca. 65 °C an und fällt
wieder ab, nachdem das Desorptionsmaximum durchlaufen wurde. Nach 2 h verbleibt
kaum noch Ethanol auf dem Adsorbens, während die Restbeladung an Toluol noch 25-
30 % der Ausgangsbeladung beträgt. Es kommt zu einer selektiven Aufheizung und an-
schließender Desorption des polaren Stoffes durch die Mikrowellen.

2.3 Einsatzgebiete - Stand der Technik 61
Ein komplett anderes Verhalten zeigt das Adsorbens Envisorb B+. Es erfolgt keine selek-
tive Aufheizung der adsorbierten Spezies, sondern eine Erwärmung des gesamten Fest-
bettes durch die Mikrowellen. Beim Desorptionsverlauf zeigen sich kaum Unterschiede
zwischen Mischungen aus polaren Stoffen oder unpolaren mit polaren Adsorptiven. Ein
Beispiel des Desorptionsverlaufs ist in Abbildung 2.18 gezeigt. Die Regeneration verläuft
deutlich langsamer als beim mikrowellentransparenten Adsorbens und es ist keine selek-
tive Desorption polarer Spezies möglich. Sowohl Toluol als auch Ethanol werden
aufgrund der Temperaturerhöhung des Festbettes desorbiert. Entsprechend zeigt sich auch
ein unterschiedlicher Temperaturverlauf während der Desorption. Es kommt zu einer
langsamen Aufheizung auf eine Temperatur über 100 °C, bis die Desorption vollständig
abgeschlossen ist. Anschließend bleibt die Temperatur konstant. Ein Absinken der Tem-
peratur nach beendeter Desorption wie beim transparenten Adsorbens ist nicht zu beo-
bachten. Die Adsorptive haben keinen Einfluss auf den Temperaturverlauf während der
Desorption.
Abbildung 2.18: Mikrowellendesorption eines nicht mikrowellentransparenten Adsorbens;
Beladung mit zwei polaren Adsorptiven nach [20]
Diese Untersuchungen zeigen, dass eine selektive Aufheizung von an einem Feststoff ad-
sorbierten polaren Spezies durch Mikrowellenstrahlung möglich ist. Eine Vorraussetzung
hierfür muss jedoch sein, dass die adsorbierte Spezies eine deutlich höhere Dielektrizi-
tätskonstante als der Feststoff aufweist.
2.3.3.3 Konkurrenzadsorption
Von KOBAYASHI ET AL. wurde die konkurrierende Adsorption von Chlorkohlenwas-
serstoffen und Wasser an NaY-Zeolithen untersucht [209]. Unter konventionellen
Bedingungen wird bevorzugt Wasser adsorbiert, da dieser Zeolith stark hydrophil ist.
Durch den Einsatz von Mikrowellenstrahlung ließ sich das Sorptionsgleichgewicht jedoch

62 2 Mikrowellen als Energieträger in der Reaktionstechnik
zu den Chlorkohlenwasserstoffen hin verschieben. Der Autor erklärt dies durch eine
selektive Aufheizung, bzw. Desorption des Wassers durch die Mikrowellen, wodurch
mehr freie Sorptionsplätze für die Kohlenwasserstoffe zur Verfügung stehen.
Ein Artikel von TURNER ET AL. [18] befasst sich ebenfalls mit Sorptionsvorgängen im
Mikrowellenfeld. Bei diesen Arbeiten wurde sowohl die Desorption als auch die konkur-
rierende Adsorption von Cyclohexan und Methanol an Zeolithen (Silikalith und DAY)
untersucht. Dabei kam ein Monomode-Mikrowellensystem mit einer maximalen Leistung
von 300 W, das bei einer Frequenz 2,45 GHz arbeitet, zum Einsatz. Die Temperaturmes-
sung erfolgte über faseroptische Sensoren. Zunächst wurde das Aufheizverhalten der un-
beladenen Zeolithe untersucht. Zu diesem Zweck wurden die Zeolithe zunächst in der
Mikrowelle bei 150 °C für 2 h getrocknet und anschließend auf Raumtemperatur abge-
kühlt. Dann wurden die Proben bei verschiedenen Mikrowellenintensitäten bestrahlt, bis
eine konstante Temperatur erreicht wurde. Abbildung 2.19 zeigt den Temperaturanstieg
der getesteten Zeolithe in Abhängigkeit von der eingestrahlten Mikrowellenleistung und
von den adsorbierten Stoffen.
Abbildung 2.19: Temperaturerhöhung des Zeolithbettes in Abhängigkeit von der
eingestrahlten Mikrowellenleistung und vom Adsorptiv nach [18]
Man erkennt, dass sich der Silikalith durch die Mikrowellen deutlich besser aufheizen
lässt. Die Autoren führen dies auf eine höhere Konzentration von Silanolgruppen auf der
Zeolithoberfläche und auf die höhere Dichte des Zeoliths zurück. Die Silanolgruppenkon-
zentrationen der zwei Proben wurden jedoch nicht experimentell bestimmt. Neben der
Temperatur des Bettes wurde bei diesen Experimenten auch die Gasphasentemperatur
gemessen. Dabei zeigte es sich, dass die Temperatur des Gases immer deutlich kleiner
war als die Temperatur des Zeoliths. Die Energieaufnahme durch die Silanolgruppen
musste somit deutlich höher sein als der Wärmeabtransport an die umgebende Gasphase.

2.3 Einsatzgebiete - Stand der Technik 63
Da Zeolithe sehr schlechte Wärmeleiter sind, ziehen die Autoren den Schluss, dass die
Silanolgruppen nochmals eine höhere Temperatur aufweisen als das gesamte Katalysator-
bett. Die Energieaufnahme erfolgt an der Oberfläche der Zeolithe und der Wärmeabtrans-
port ist zu langsam um ein thermisches Gleichgewicht zu erreichen. Anschließend wurden
die Zeolithe mit Methanol oder Cyclohexan beladen und eine Desorption durch Mikro-
wellenstrahlung durchgeführt. Die Anfangsbeladungen der Zeolithe finden sich in Tabelle
2.13.
Tabelle 2.13: Anfangsbeladungen der Desorptionsuntersuchungen [18]
System Beladung [Mol Adsorbat/kg Adsorbens]
Cyclohexan / DAY 2,1
Cyclohexan / Silikalith 0,9
Methanol / DAY 4,5
Methanol / Silikalith 1,7
Dabei wurde zunächst eine Leistung von 40 W eingestellt. Nachdem sich ein Gleichge-
wicht eingestellt hatte, wurde die Leistung zunächst auf 80 und danach auf 120 W erhöht.
Bei den mit Methanol beladenen Proben kam es zu einer raschen Desorption, welche mit
einem Temperaturanstieg des Katalysatorbettes einherging (vgl. Abbildung 2.19). Bei
einer eingestrahlten Leistung von 40 W betrug der Temperaturanstieg am Zeolith DAY
beispielsweise 25 °C, während am unbeladenen Zeolith nur 5 °C gemessen wurden. Die
gleiche Beobachtung wurde auch beim Silikalith gemacht. Hier betrug die Temperaturer-
höhung bei 40 W mit Methanol 37 °C, am unbeladenen Zeolith jedoch nur 9 °C. Die Stei-
gerung der eingebrachten Mikrowellenleistung hatte eine weitere Erhöhung der Betttem-
peraturen zur Folge. Diese war am Silikalith jedoch deutlich größer als am DAY. Ein ganz
anderes Verhalten zeigte sich, wenn mit Cyclohexan beladene Proben untersucht wurden
(Abbildung 2.19). Die Temperaturüberhöhungen am DAY entsprachen exakt denen des
unbeladenen Zeoliths. Beim Silikalith konnte eine minimal höhere Temperatur festgestellt
werden (maximal 5 °C bei 120 W) als bei der unbeladenen Probe. Eine höhere Mikro-
wellenleistung führte zu einer Steigerung der Temperaturüberhöhung im Bett. Bei
Betrachtung der Desorptionsrate fiel auf, dass sowohl bei einer Beladung mit Methanol
als auch mit Cyclohexan die Desorption zu Beginn des Desorptionsvorganges schneller
ablief. Trotz Steigerung der eingebrachten Mikrowellenleistung wurde immer weniger
desorbiert. Am Zeolith DAY wurden nach Steigerung der Leistung auf 120 W insgesamt
12 % des Methanols desorbiert, am Silikalith 21 %. Bei den Experimenten mit
Cyclohexan konnten am DAY beinahe 50 %, am Silikalith 44 % der ursprünglich
vorhandenen Beladung entfernt werden. Die deutliche Temperaturerhöhung bei den mit
Methanol beladenen Proben führt der Autor auf eine selektive Aufheizung und Desorption

64 2 Mikrowellen als Energieträger in der Reaktionstechnik
des adsorbierten Alkohols zurück. Bei den mit Cyclohexan beladenen Proben kam es zu
keiner Temperaturerhöhung, da dieser Stoff nur eine vernachlässigbar kleine
Dielektrizitätskonstante besitzt. Die Desorption war vorwiegend auf die
Temperaturerhöhung des Adsorbens zurückzuführen.
Neben der Mikrowellendesorption der Einzelkomponenten wurde jedoch auch der Ein-
fluss der Mikrowellenstrahlung auf die konkurrierende Adsorption von Methanol und
Cyclohexan untersucht. Zu diesem Zweck wurden die Zeolithe zunächst bei Raumtempe-
ratur mit beiden Komponenten aus der Gasphase beladen, bis sich ein Gleichgewicht
eingestellt hatte. Anschließend wurde lediglich die Mikrowelle mit einer Leistung von 40
/ 80 / 120 W zugeschaltet, bis sich wieder ein Gleichgewicht einstellen konnte. Das Bett
wurde während des gesamten Versuchs mit einer Mischung aus Methanol und
Cyclohexan überströmt. Nach Einschalten der Mikrowelle kam es wieder zu einem An-
stieg der Betttemperatur, wie sie auch schon bei der Desorption der mit Methanol belade-
nen Proben beobachtet wurde. Im Desorbat konnte für kurze Zeit nach dem Einschalten
bzw. einer Steigerung der Mikrowellenleistung eine Erhöhung der Methanolkonzentration
und eine Erniedrigung der Cyclohexankonzentration beobachtet werden. Schon nach
wenigen Minuten erreichten die Desorbatkonzentrationen der beiden Stoffe jedoch wieder
den Ausgangswert. Ein neues Gleichgewicht hatte sich eingestellt. Nachdem sich das
Equilibrium bei einer Leistung von 120 W eingestellt hatte, wurde die Leistung wieder auf
80 W abgesenkt, danach auf 40 W und schließlich wurde die Mikrowelle wieder abge-
schaltet. Im Desorbat konnte nach jedem Absenken der Leistung und damit fallender
Betttemperatur diesmal ein Anstieg der Cyclohexan- und ein Rückgang der Methanolkon-
zentration beobachtet werden. Durch die Mikrowellen ließ sich das Sorptionsgleich-
gewicht auf der Zeolithoberfläche in Richtung des unpolaren Stoffes Cyclohexan ver-
schieben. Da allerdings jeder Leistungswechsel auch mit einer Temperaturänderung
verbunden war, musste das Sorptionsgleichgewicht der beiden Stoffe bei den effektiven
Betttemperaturen berücksichtigt werden. Deshalb wurden die theoretisch zu erwartenden
Sorptionsverhältnisse mit den bei den Experimenten gemessenen verglichen. In
Abbildung 2.20 sind die Adsorptionsverhältnisse von Cyclohexan und Methanol in
Abhängigkeit von der Temperatur an den beiden getesteten Zeolithen aufgetragen. Man
erkennt deutlich, dass theoretisch bei einer steigenden Temperatur ein Rückgang der
Cyclohexanbeladung zu erwarten ist. Durch die Mikrowelleneinstrahlung wurde zwar die
Betttemperatur erhöht, jedoch die Cyclohexanbeladung vergrößert. Der Autor führt dies
auf eine selektive Desorption des Methanols zurück, wodurch bevorzugt Cyclohexan
adsorbieren kann.

2.3 Einsatzgebiete - Stand der Technik 65
Abbildung 2.20: Errechnete und gemessene Adsorptionsverhältnisse bei konkurrierender
Adsorption von Cyclohexan und Methanol an Zeolithen unter Mikrowelleneinstrahlung in
Abhängigkeit von der Temperatur nach [18]
Die Untersuchungen von Turner zeigen, dass es möglich ist, durch die Mikrowellen
selektiv an einer Oberfläche adsorbierte Spezies aufzuheizen, sofern diese polar sind. Es
kommt zu einer lokalen Übertemperatur auf der Feststoffoberfläche. Bei einer konkurrie-
renden Mehrkomponentenadsorption wird das Sorptionsgleichgewicht zu den unpolaren
Spezies hin verschoben, da es zu einer selektiven Desorption der polaren Adsorptive
kommt.
Neben experimentellen Untersuchungen gibt es jedoch auch Ansätze, die Wechselwirkung
von Mikrowellenstrahlung mit Zeolithen und auf deren Oberfläche adsorbierten
Molekülen mit Hilfe von Molecular Modelling zu beschreiben. Von BLANCO ET AL.
wurde die Mikrowellenheizung verschiedener Zeolithe (Y, Silikalith) in Abhängigkeit von
der angelegten Feldstärke simuliert [210]. Dabei zeigte es sich, dass im Zeolith enthaltene
Kationen bei Mikrowellenbestrahlung höhere Temperaturen annehmen als im thermischen
Equilibrium. Berechnungen von mit Benzen und Methanol beladenen Zeolithen zeigten
weiterhin, dass folgender Zusammenhang gilt:
ZeolithBenzenMethanol TTT >>> (Gl. 2.24)
Bei Mikrowellenheizung kommt es also nicht zur Ausbildung eines thermischen Gleich-
gewichts auf molekularer Ebene, sondern es entstehen lokale Übertemperaturen an ge-
bundenen Spezies, welche umso größer sind, je höher die Polaritätsunterschiede der ad-
sorbierten Spezies sind.

3 Hydroxylierung von Benzen mit N2O an ZSM-5
Katalysatoren
Die folgenden Kapitel geben einen Überblick über die Verwendung von Phenol (Kapitel
3.1), die bisher eingesetzten Methoden zu seiner Produktion (Kapitel 3.2) und die
wirtschaftliche Notwendigkeit ein neues Verfahren dafür zu entwickeln (Kapitel 3.3). An-
schließend wird ein Alternativverfahren, die Hydroxylierung von Benzen mit N2O vor-
gestellt und dessen Vorteile im Vergleich zu gängigen Verfahren aufgezeigt (Kapitel 3.4).
Auf die für diese Reaktion geeigneten Katalysatoren wird ebenso näher eingegangen wie
auf den zugrunde liegenden Reaktionsmechanismus.
3.1 Verwendung von Phenol
Mit einer Produktion von über 7 Millionen Tonnen im Jahr 2004 [21] gehört Phenol zu
den wichtigsten chemischen Zwischenprodukten. Abbildung 3.1 zeigt eine Übersicht der
Märkte, die Phenol verbrauchen.
Abbildung 3.1: Weltweite Verwendung von Phenol 2002 [211]
Der größte Teil des Phenols wird für die Herstellung von Bisphenol-A verwendet. Es
dient als Ausgangsstoff für die Herstellung von Epoxydharzen und Polycarbonaten, wel-
che vor allem in der Produktion optischer Speichermedien (CD, DVD) eingesetzt werden.

3.2 Industrielle Produktionsverfahren für Phenol 67
Dieser Bereich zeigte in den letzten Jahren auch die größten Wachstumsraten. Wurden
1995 nur 32 % der Phenolproduktion für die Herstellung von Bisphenol-A eingesetzt, wa-
ren es 2002 bereits 41 %. Der Bereich der Phenolharze, welcher der zweitgrößte
Abnehmer für Phenol ist, zeigte in den letzten Jahren dagegen eine stark rückläufige Ten-
denz. Wurden 1995 37 % der Weltphenolproduktion für die Herstellung dieser Harze
verwendet, waren es 2002 nur noch 28 %. Die Phenolharze werden vor allem für die Her-
stellung von Lacken, Leimen, Pressmassen und Schaumstoffen verwendet. Ein weiterer
großer Teil des Phenols wird zu Xylenolen (Adipinsäure) und Caprolactam weiterverar-
beitet, welche ihrerseits der Kunstfaserherstellung (Nylon) dienen. Kleinere Mengen
werden in der Produktion von Alkylphenolen eingesetzt, die zur Herstellung von
Antioxidantien, Pflanzenschutzmitteln, Alterungsschutzmitteln und Zusätzen in der
Kunststoff- und Gummiindustrie verwendet werden. Die Pharmaindustrie verbraucht
ebenfalls größere Mengen an Phenol. Anders als beim Bisphenol-A bzw. den
Phenolharzen, sind die Anteile dieser Bereiche am Gesamtphenolverbrauch über die
letzten Jahre jedoch relativ konstant. Beim Caprolactam war von 1995 bis heute ein
leichter Rückgang um 2 % zu verzeichnen, während der Anteil der Xylenole um 2 %
zunahm. Insgesamt befinden sich jedoch all diese Märkte seit den 90’er Jahren im
Wachstum, was wiederum zu einem erhöhten Phenolbedarf auf dem Weltmarkt führte
(vgl. Kapitel 3.3) [211-213].
3.2 Industrielle Produktionsverfahren für Phenol
Die erste Quelle für Phenol war die Gewinnung aus Steinkohleteer (Carbolöl) [38]. Da
diese Ressource jedoch nur sehr begrenzt zur Verfügung steht und aus 1 t Steinkohle nur
ca. 250 g Phenol gewonnen werden [38], wurde bereits im neunzehnten Jahrhundert ein
Verfahren für die synthetische Herstellung entwickelt: Das so genannte Benzensulfonat-
verfahren. Heute sind sechs technische Herstellungsmethoden für Phenol bekannt [214]:
• Benzensulfonatverfahren
• Raschig-Hooker-Prozess
• Alkalische Hydrolyse von Chlorobenzen
• Dehydrierung von Cyclohexanol oder Cyclohexanon
• Toluoloxidation
• Cumolverfahren (Hock-Verfahren)
Von diesen Verfahren kommt heute aus wirtschaftlichen Gründen fast nur noch das
Cumolverfahren zum Einsatz. In den USA wurden im Jahr 2002 98 % des Phenols auf
diesem Syntheseweg hergestellt [211]. Auch Weltweit ist dieses Verfahren am weitesten
verbreitet. Allein in Japan werden noch größere Mengen Phenol durch Toluoloxidation

68 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
und über die Dehydrierung von Cyclohexanol/-hexanon produziert [213]. Deshalb soll im
Folgenden nur auf das Cumolverfahren näher eingegangen werden.
Der Prozess beruht auf den Forschungsergebnissen von HOCK und LANG, die in den
40’er Jahren des letzten Jahrhunderts entdeckten, dass man Cumolhydroperoxid zu Phenol
und Aceton aufspalten kann. Das Verfahren lässt sich in drei Teilschritte untergliedern,
die in Abbildung 3.2 dargestellt sind.
Abbildung 3.2: Cumolverfahren [212, 214]
Im ersten Schritt des Cumolverfahrens wird Benzen mit Propen zu Cumol alkyliert. Dies
kann entweder nach dem SOLUTIA/KELLOG-Flüssigphasenprozess mit AlCl3 als Kata-
lysator erfolgen oder in der Gasphase. Beim Gasphasenprozess wird Benzen mit einem
Gemisch aus Propan und Propen an Phosphorsäure auf Kieselgur als Katalysator umge-
setzt [213]. Bei neueren Verfahren werden jedoch vor allem Zeolithe als Katalysatoren
eingesetzt. MOBIL/RATHEON verwendet beispielsweise Zeolithe vom Typ MCM und
ENICHEM den Typ Beta. Andere Zeolithtypen (Mordenite, MgAPSO) sind jedoch auch
geeignet. Der Großteil (> 90 %) der heute betriebenen Anlagen arbeitet nach dem Gaspha-
senverfahren mit Zeolithen als Katalysator [215]. Die Cumolherstellung ist meist an
Raffinerien gekoppelt, da der Ausgangsstoff Benzen entweder aus Reformatbenzin (USA)
oder der Pyrolysefraktion von Steamcrackern (Westeuropa, Japan) stammt. Propen wird
CH3
CH2
CH3
CH3
CH3
CH3 Na
2CO
3
CH3
CH3
OOH
O2
CH3
CH3
OOH H2SO
4OH
CH3
CH3
O
I. Benzenalkylierung zu Cumol
+
Friedel-Crafts-Alkylierung
[KAT]
Benzen Propen Cumol
II. Oxidation von Cumol zu Cumolhydroperoxid
90-130 °C8-10 bar
Cumol
- Emulsion
Cumolhydroperoxid
+
III. Säurekatalytische Spaltung des Cumolhydroperoxid
Cumolhydroperoxid
ca. 50 °C
Phenol
+
Aceton

3.3 Wirtschaftliche Gesichtspunkte - Marktentwicklung Phenol 69
ebenfalls im Steamcracker oder in FCC-Anlagen produziert. Das Cumol wird wiederum
fast ausschließlich im Hock-Prozess zu Phenol weiterverarbeitet [212].
Im zweiten Schritt des Cumolverfahrens erfolgt dann eine Oxidation des Cumol mit
Sauerstoff zu Cumolhydroperoxid in der Flüssigphase [212, 213, 216]. BP/HERKULES
und KELLOGG haben ein Verfahren entwickelt, bei dem das Cumol in einer Na2CO3-
Emulsion aufgegeben wird. Bei einem Alternativprozess von HERKULES und
PHENOLCHEMIE wird unverdünnt in homogener Phase gefahren. Als kritisch bei die-
sem Schritt erweist sich die Nebenproduktbildung ausgehend von Cumolhydroperoxid
und die Explosionsgefahr. Deshalb wird nur bei niedrigen Sauerstoffkonzentrationen
(< 8 %) und einer Cumolhydroperoxidkonzentration von 20 - 30 % am Ende der Reaktion
gearbeitet. Außerdem verläuft die Reaktion autokatalytisch und ist stark exotherm
(800 kJ/kg Cumolhydroperoxid), weshalb die freigesetzte Wärme durch Kühlschlangen
abgeführt werden muss, in denen Cumol/Wasser verdampft wird. Bei diesem Schritt kön-
nen Selektivitäten von 95 – 98 % zu Cumolhydroperoxid erreicht werden. Anschließend
wird das Oxidat auf 75 - 85 % Cumolhydroperoxid aufkonzentriert und in den nächsten
Prozessschritt übergeben.
Im dritten Schritt des Verfahrens erfolgt eine säurekatalytische Spaltung des Cumol-
hydroperoxid zu Phenol und Aceton [212, 213, 216]. Dabei kann entweder in einer
homogenen Lösung von verdünnter H2SO4 mit Aceton oder Phenol (BP, HERKULES),
oder heterogen mit 40 %-iger H2SO4 (PHENOLCHEMIE) gearbeitet werden. Auch diese
Reaktion ist stark exotherm (1680 kJ/kg Cumolhydroperoxid). Bei vollständigem Umsatz
des Cumolhydroperoxid können Selektivitäten von 95 % zu Phenol/Aceton erreicht wer-
den. Pro t Phenol werden 0,62 t Aceton produziert. Als Nebenprodukte fallen α-Methyl-
styrol, Mesityloxid und höhersiedende Komponenten an. Nach der Abtrennung des
Katalysators oder Neutralisation des Reaktionsgemisches erfolgt eine mehrstufige
destillative Trennung von Aceton, Cumol, α-Methylstyrol und Phenol. Es können
Phenolreinheiten von bis zu 99,99% und 99,75 % reines Aceton erreicht werden. Das
α-Methylstyrol wird katalytisch zu Cumol hydriert und in den Prozess zurückgeführt.
3.3 Wirtschaftliche Gesichtspunkte - Marktentwicklung Phenol
Seit Beginn der 90’er Jahre stieg der Weltverbrauch an Phenol auf 5 Mio. t im Jahr 1995
an [217]. Danach konnte ein konstant hoher Zuwachs von ca. 4,6 % bis zum Jahr 2000
verzeichnet werden. In Abbildung 3.3 sieht man deutlich, dass die verfügbaren Kapazitä-
ten der Hersteller deutlich über der Nachfrage auf dem Weltmarkt lagen. Allerdings muss
davon ausgegangen werden, dass diese Kapazitäten in der Realität nicht auf Dauer
erreichbar sind. Deshalb wird auch manchmal eine effektive Kapazität eingeführt, welche
die nutzbaren Kapazitäten über einen längeren Zeitraum veranschaulicht. Die in
Abbildung 3.3 aufgetragene „nutzbare Kapazität“ beruht auf einer Schätzung der Firma

70 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
SHELL und stellt etwa 92 % der angegebenen Kapazitäten dar. Man erkennt, dass sich
diese nutzbaren Kapazitäten bis Ende der 90’er Jahre sehr nahe an der Nachfrage auf den
Märkten bewegten. Deshalb wurden viele bestehende Anlagen verbessert oder neue An-
lagen geplant und gebaut, so dass ab dem Jahr 2000 ein deutlicher Anstieg der verfügba-
ren Kapazität zu verzeichnen war. Beispielsweise wurde von der Firma INEOS PHENOL
(PHENOLCHEMIE), einem der größten Phenolhersteller weltweit, 1999 eine neue
Anlage in Mobile (USA) errichtet, welche ab dem Jahr 2000 mit einer Kapazität von
400000 t/Jahr die Produktion aufnahm [52]. Kleinere Produktionsanlagen der Firmen
SHELL SUN, ENICHEM (POLIMERI EUROPA), ERTISA, MITSUI, FORMOSA
CHEMICALS und weiterer Firmen folgten bis zum Jahr 2002 [21]. Diese Investitionen
schienen Ende der 90’er Jahre dringend notwendig, um den Bedarf des Marktes abdecken
zu können. Im Jahr 2001 kam es jedoch zu einem drastischen Einbruch des Phenolmarktes
aufgrund einer weltweiten Rezession. Der Markt wuchs in den nächsten Jahren zwar wie-
der kontinuierlich, jedoch nur mit einer Rate von 4,3 %. Dieser Unterschied wirkt im Ver-
gleich zu den erwarteten 4,6 % zunächst klein, hat jedoch dazu geführt, dass bis heute
(2004) Überkapazitäten vorhanden sind. Der Zuwachs der letzten beiden Jahre ist vor
allem auf die verstärkte Nachfrage nach Polycarbonaten und die rasch wachsenden
Märkte in Asien zurückzuführen [21].
Abbildung 3.3: Weltweite Phenolnachfrage, Produktionskapazitäten und die nutzbaren
Kapazitäten (ca. 92 % der maximalen Kapazität) für den Zeitraum 1996 – 2004 [21]

3.3 Wirtschaftliche Gesichtspunkte - Marktentwicklung Phenol 71
Bei Prognosen für die Marktentwicklung nach 2004 ist es interessant, die drei größten
Märkte Nordamerika, Westeuropa und Asien getrennt zu betrachten. Für Westeuropa wird
beispielsweise in den nächsten Jahren nur ein sehr geringer Zuwachs des Phenolmarktes
von ca. 2,6 % erwartet, da kaum neue Polycarbonat-Anlagen in Planung sind. Außerdem
ist die Phenolproduktion in Europa sehr kostspielig, da schon heute nicht genügend
Cumol auf dem Markt zur Verfügung steht. Außerdem stiegen die Importkosten für Ben-
zen in den letzten Jahren stark an und die Investitionskosten für neue Benzenanlagen sind
in Europa sehr hoch. Deshalb sind bis zum Jahr 2012 auch kaum neue Phenolkapazitäten
geplant. Einige kleine Kapazitätserweiterungen von NOVAPEX (2004), BOREALIS
(2007), POLIMERI EUROPA (2007) und ERTISA (2008) von insgesamt ca.
300000 t/Jahr könnten ausreichen, um den Markt für die nächsten 8 Jahre zu sättigen [21].
Ähnlich sieht die Entwicklung in Nordamerika aus. Es wird mit einem Wachstum von ca.
3 % gerechnet. Ursache für dieses Wachstum ist ein steigender Phenolbedarf in den USA
und zunehmende Exporte nach Europa. Da in Nordamerika jedoch seit 2001 erhebliche
Überkapazitäten bestehen, sind keine größeren Kapazitätsausweitungen geplant [21].
Ganz anders ist die Lage in Asien. Dort wird mit einem Wachstum des Phenolmarktes
zwischen 6 und 7 % gerechnet. In den nächsten 4 Jahren sollen dort neue Produktionsan-
lagen mit einer Kapazität von über 1 Mill. t/Jahr entstehen. Der Schwerpunkt der Investi-
tionen wird in China liegen wo CHANG CHUN, FCFC, LG CHEM, KHUOMO, GAO
QIAO, SHELL und weitere Firmen zusätzliche Kapazitäten schaffen wollen. Bis zum Jahr
2009 wird es dadurch zu einem immensen Kapazitätsüberhang in China kommen [21].
Da das Hock-Verfahren die Phenolherstellung dominiert, muss jedoch auch die Marktent-
wicklung für Aceton betrachtet werden, da pro t Phenol 0,6 t Aceton anfallen. Bis zum
Jahr 1985 reichte die Acetonproduktion aus dem Cumolverfahren noch nicht aus, um den
Weltbedarf zu decken [21]. Danach konnte der Acetonmarkt vollständig durch die
Acetonproduktion der Phenolhersteller abgedeckt werden. Da der Acetonverbrauch seit
Mitte der 90’er Jahre deutlich langsamer (ca. 3 %/Jahr) anwuchs als der des Phenols (ca.
4,6 %/Jahr), kam es bereits im Jahr 1998 zu einer Überproduktion von Aceton (vgl.
Abbildung 3.4), welche bis zum Jahr 2000 immer weiter anstieg. Dies führte zu einer
Schwächung des Acetonmarktes und zu höheren Produktionskosten für Phenol. Erst die
globale Rezession und die damit verbundenen Rückgänge der Phenolproduktion bewirk-
ten wieder einen ausgeglichenen Acetonmarkt bis heute. Aceton wird vor allem für die
Produktion von Methylmethacrylat (31 %), Methylisobutylketon (9 %), Bisphenol-A
(16 %) und als Lösemittel (28 %) verwendet. Diese Zwischenprodukte werden wiederum
zu Kunststoffen, Farben, Lacken und Harzen weiterverarbeitet [21, 212, 213]. Die stärks-
ten Zuwachsraten ließen sich hier beim Methymethacrylat und Bisphenol-A verzeichnen.
Mittlerweile wurde jedoch ein neuer umweltschonenderer Prozess entwickelt, bei dem
Methylmetacrylat aus Isobuten und Ethylen hergestellt wird. Dieser hat etwa 30 % niedri-
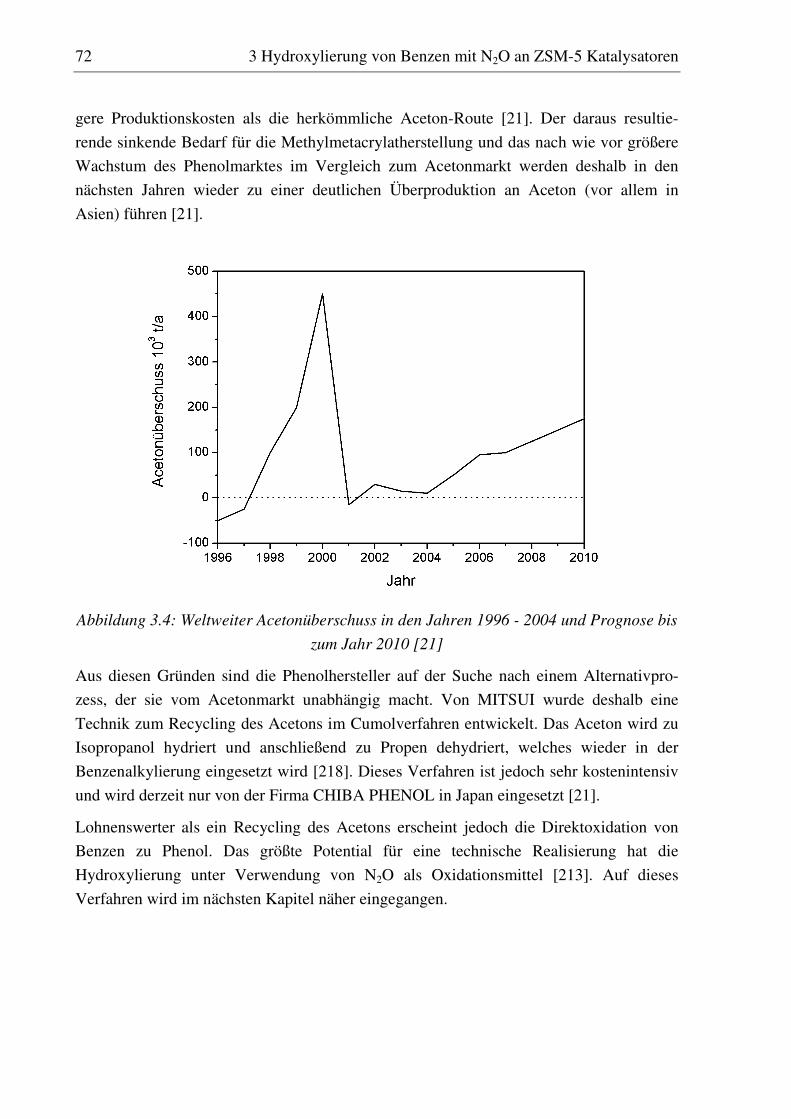
72 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
gere Produktionskosten als die herkömmliche Aceton-Route [21]. Der daraus resultie-
rende sinkende Bedarf für die Methylmetacrylatherstellung und das nach wie vor größere
Wachstum des Phenolmarktes im Vergleich zum Acetonmarkt werden deshalb in den
nächsten Jahren wieder zu einer deutlichen Überproduktion an Aceton (vor allem in
Asien) führen [21].
Abbildung 3.4: Weltweiter Acetonüberschuss in den Jahren 1996 - 2004 und Prognose bis
zum Jahr 2010 [21]
Aus diesen Gründen sind die Phenolhersteller auf der Suche nach einem Alternativpro-
zess, der sie vom Acetonmarkt unabhängig macht. Von MITSUI wurde deshalb eine
Technik zum Recycling des Acetons im Cumolverfahren entwickelt. Das Aceton wird zu
Isopropanol hydriert und anschließend zu Propen dehydriert, welches wieder in der
Benzenalkylierung eingesetzt wird [218]. Dieses Verfahren ist jedoch sehr kostenintensiv
und wird derzeit nur von der Firma CHIBA PHENOL in Japan eingesetzt [21].
Lohnenswerter als ein Recycling des Acetons erscheint jedoch die Direktoxidation von
Benzen zu Phenol. Das größte Potential für eine technische Realisierung hat die
Hydroxylierung unter Verwendung von N2O als Oxidationsmittel [213]. Auf dieses
Verfahren wird im nächsten Kapitel näher eingegangen.

3.4 Hydroxylierung von Benzen mit N2O 73
3.4 Hydroxylierung von Benzen mit N2O
Erste Untersuchungen zur Benzenhydroxylierung mit N2O als Oxidationsmittel wurden
von IWAMOTO ET AL. 1983 veröffentlicht. Er verwendete V2O5- oder MoO3-Katalysa-
toren, an denen Raum-Zeit-Ausbeuten von ca. 0,1 gPhenol/(gKat.·h) erreicht werden konnten,
bei einer maximalen Selektivität zu Phenol von ca. 71 % [219]. Zeolithe vom Typ ZSM-5
wurden erstmals von ONO ET AL. in dieser Reaktion getestet [220]. An diesen
Katalysatoren konnten deutlich höhere Raum-Zeit-Ausbeuten und Selektivitäten zu
Phenol erreicht werden. Dieser Katalysatortyp gilt für die Benzenhydroxylierung mit N2O
bis heute als am besten geeignet [24]. Allerdings finden sich in der Literatur Hinweise,
dass sich auch andere Zeolithe für diese Reaktion eignen. Beispiele sind Zeolithe vom
Typ Y [221], Beta [222], MCM-22 [222] und ZSM-11 [223].
Die Grundlage für eine technische Realisierung dieser Reaktion wurde schließlich Anfang
der 90’er Jahre in der Forschergruppe um PANOV am Boreskov Institut (BIC) geschaffen
[224]. Sie verwendeten Zeolithe des Typs Fe-ZSM-5 und führten die katalytische Aktivi-
tät auf das eingebrachte Eisen zurück [225]. Seitdem sind zahlreiche Patente zu diesem
Thema angemeldet worden. Eine Übersicht der Patentliteratur findet sich bei
REITZMANN [24]. Die Reaktionsgleichung und die von PANOV aufgestellten
Reaktionsbedingungen der Benzenhydroxylierung sind in Abbildung 3.5 dargestellt.
N2O
OH
N2+ +
300-450 °C, 1 bar
Kat.
Benzen Phenol
Abbildung 3.5: Direktoxidation von Benzen mit N2O [28, 226]
Die Rechte an sämtlichen Patenten dieser Forschungsgruppe wurden von
MONSANTO/SOLUTIA erworben, die 1996 ankündigte, bis zum Jahr 2000 eine Anlage
mit einer Phenolkapazität von 150000 t/Jahr in Pensacola (Florida, USA) nach diesem
Verfahren betreiben zu wollen [22, 227]. Die Direktoxidation sollte in den firmeneigenen
Herstellungsprozess für Adipinsäure, welches zu Nylon 6,6 weiterverarbeitet wird,
integriert werden [228]. Die Adipinsäureherstellung geht üblicherweise von Cyclohexan
aus. Alternativ existiert jedoch auch ein Verfahren, welches Phenol als Rohstoff verwen-
det [212]. Beide Varianten setzen große Mengen an N2O frei, das als Treibhausgas gilt
und die Ozonschicht schädigt [229]. Während der Direktoxidation von Benzen ließe sich
dieses N2O für die Phenolherstellung im Gesamtprozess umweltschonend recyceln [22].
Als Vorteile der Hydroxylierung von Benzen mit N2O gegenüber dem Cumolverfahren
werden angeführt [226, 228, 230]:

74 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
• hohe Phenolausbeuten
• hohe Prozesssicherheit
• geringere Investitionskosten
• keine Koppelprodukte
• das umweltschädliche Abfallprodukt N2O aus der Adipinsäureherstellung kann für
die Benzenhydroxylierung genutzt werden.
Im Jahr 2001 gaben SOLUTIA und ihre Partnerfirma JLM jedoch bekannt, dass die ge-
plante Anlage in Pensacola nicht gebaut wird. Als Gründe wurden die Überkapazitäten
auf dem Phenolmarkt angeführt, welche durch die in den Jahren 2000/2001 neu geschaf-
fenen Kapazitäten anderer Phenolhersteller verursacht wurden (vgl. Kapitel 3.3) [227].
3.4.1 ZSM-5 Zeolithe
3.4.1.1 Historisches
Zeolithe wurden im Jahre 1756 von dem Schwedischen Chemiker AXEL FRIEDRICH
CRONSTEDT entdeckt. Der Name rührt von ihrer Eigenschaft her, dass beim Erhitzen
stetig und ohne Änderung der Kristallstruktur Wasser abgegeben wird. Er setzt sich aus
den griechischen Begriffen „zeo“ (sieden) und „lithos“ (Stein) zusammen [231]. Nach
einer moderneren Definition von BRECK handelt es sich bei den Zeolithen um
„kristalline, hydratisierte Alumosilikate, synthetisiert oder natürlich vorkommend, mit
Gerüststruktur, die Alkali- bzw. Erdalkalikationen enthalten“ [232]. Heute sind über 100
Zeolithstrukturen bekannt, von denen etwa 40 in der Natur vorkommen [233, 234]. Für
wichtige industrielle Prozesse werden heute ausschließlich synthetische Zeolithe
verwendet. Sie besitzen den Vorteil, dass sie schnell in den benötigten Mengen hergestellt
und für die entsprechende Anwendung mit den gewünschten Eigenschaften
maßgeschneidert werden können. Vom Einsatz der Zeolithe in der Katalyse wurde
erstmals 1960 berichtet [235]. Heute kommen Zeolithe in petrochemischen Bulk-
Prozessen ebenso zum Einsatz [236, 237] wie bei der Herstellung chemischer
Zwischenprodukte und Feinchemikalien [238, 239]. Eine komplette Übersicht der
verschiedenen Zeolithtypen sowie ihrer chemischen Eigenschaften und technischen
Einsatzgebiete würde den Rahmen dieser Arbeit sprengen. Eine Zusammenstellung der
verschiedenen Zeolithtypen findet sich in [234]. Im Folgenden wird nur der ZSM-5
Zeolith ausführlich besprochen. Dieser Zeolithtyp wurde von Mobile für den Einsatz in
der Petrochemie entwickelt, von der sich auch der Name Zeolite Socony Mobile No. 5
(ZSM-5) herleitet [240]. Die industriellen Einsatzgebiete sind beispielsweise das FCC
(fluid catalytic cracking) und das Hydrocracking [241], die Oligomerisierung von
Olefinen [242], die Xylolisomerisierung [243] und die Alkylierung von Aromaten [244].
Die folgenden Kapitel geben einen Überblick über die Struktur, Herstellungsweise und die
Eigenschaften dieses Zeolithtyps.

3.4 Hydroxylierung von Benzen mit N2O 75
3.4.1.2 Struktur
Zeolithe sind aus so genannten primären Baueinheiten zusammengesetzt. Dabei handelt es
sich um Tetraeder, in deren Zentrum sich entweder ein Silizium- oder Aluminiumatom
(T-Atome) befindet, welches von 4 Sauerstoffatomen umgeben ist. Diese Tetraeder bilden
sekundäre Baueinheiten (SBU), indem zwei Zentralatome wiederum über Sauerstoffatome
verknüpft werden. Aus diesen sekundären Baueinheiten setzen sich Polyeder und schließ-
lich das Zeolithgerüst als dreidimensionale Struktur zusammen. Anhand der sekundären
Baueinheiten erfolgt die Einteilung der Zeolithe in verschiedene Klassen [232, 245]. In
Abbildung 3.6 wird der Aufbau des ZSM-5 Zeolithen nach diesem Schema veranschau-
licht.
Abbildung 3.6: Schematischer Aufbau des Zeoliths ZSM-5 aus Baueinheiten nach [246]
Die sekundäre Baueinheit des ZSM-5 bildet 5-Ringe (SBU: 5+1-Ring), weshalb dieser
Zeolith den Pentasilen zugeordnet wird. Diese gruppieren sich zu Polyedern, die wie-
derum Ketten bilden, aus denen die pseudodreidimensionale Gerüststruktur aufgebaut ist
[247]. Abbildung 3.7 zeigt die Porenstruktur des ZSM-5 mit den charakteristischen Kanal-
abmessungen.
Abbildung 3.7: Dreidimensionale Porenstruktur des ZSM-5 nach [23]
Das Porengefüge des ZSM-5 besteht aus parallel angeordneten Kanälen mit elliptischen
Öffnungen, welche aus 10 Sauerstoffatomen gebildet werden und eine Größe von

76 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
5,3 x 5,6 Å besitzen. Senkrecht dazu verlaufen sinoidale Kanäle, mit den Abmessungen
5,1 x 5,6 Å., welche ebenfalls von 10 Sauerstoffatomen umgeben sind [245]. An den
Kanalkreuzungspunkten ergeben sich Hohlräume mit einer Größe von ca. 9 Å [241]. Die
Porenöffnungen liegen somit in der Größenordnung des kinetischen Durchmessers von
Benzen [232].
Alle Alumosilikate lassen sich durch die allgemeine Strukturformel
/ 2 2 2( ) ( )x n x yM AlO SiO mH O⋅ ⋅ ⋅ (Gl. 3.1)
beschreiben. M ist ein Kation mit der Ladung n, welches die durch das Aluminium entste-
hende negative Ladung ausgleicht. Der Anteil x an Aluminium im Gitter bestimmt somit
die Anzahl der Kationen, welche frei beweglich sind und gegen andere Kationen ausge-
tauscht werden können [232]. Die kleinste sich wiederholende Einheit eines Zeolithgitters
wird auch Elementarzelle genannt und lässt sich für den ZSM-5 folgendermaßen
schreiben [234]:
/ 96 192 216x n x xM Al Si O H O− ⋅ (Gl. 3.2)
Eine Zelle wird demnach aus 96 TO4-Tetraedern aufgebaut und besitzt im Allgemeinen
eine orthorhombische Symmetrie. Für ein Si/Al-Verhältnis von 86 ergeben sich folgende
Abmessungen [247]:
a ≈ 20,07 ± 0,01 Å, b ≈ 19,92 ± 0,01 Å, c ≈ 13,42 ± 0,01 Å
mit einem Einheitsvolumen von 5365 Å3.
Als Kenngröße für den Zeolith dient das Verhältnis von Silizium zu Aluminium (Si/Al-
Verhältnis), welches einen entscheidenden Einfluss auf die katalytischen Eigenschaften
des Zeolithen besitzt. Häufig wird auch das molare Verhältnis der Oxide angegeben, wel-
ches als Modul M bezeichnet wird:
2
2 3
2SiO Si
Al O Al
n nModul M
n n= = (Gl. 3.3)
Nach der Regel von LÖWENSTEIN [248] können zwei Aluminiumatome aufgrund ihrer
Ladung nicht direkt über ein Sauerstoffatom miteinander verbunden werden. Deshalb gibt
es einen Mindestwert für das Si/Al-Verhältnis, jedoch kein Maximum [248]. Für den
ZSM-5 können Si/Al-Verhältnisse von 10 - ∞ (Silikalith) erreicht werden [249].
Neben dem eigentlichen ZSM-5 gibt es jedoch auch metallosilikatanaloge Zeolithe, wel-
che die gleiche Struktur besitzen, jedoch andere T-Atome. Dies können 2 bis 6-wertige
Kationen wie beispielsweise Eisen sein [238]. Für die Benzenhydroxylierung spielen
solche Ferrisilikate mit ZSM-5-Struktur eine wichtige Rolle [24].

3.4 Hydroxylierung von Benzen mit N2O 77
3.4.1.3 Synthese und postsynthetische Modifizierung
Die klassische Synthese des ZSM-5 erfolgt hydrothermal aus einer Reaktionsmischung,
welche üblicherweise folgende Komponenten enthält [250, 251]:
• Kieselsol oder Wasserglas als SiO2-Quelle
• Natriumaluminat, Aluminiumnitrat oder andere Aluminiumsalze als Al2O3-Quelle
• Mineralisierungsagens (OH-)
• Templat
Schematisch kann die Synthese in folgende Teilschritte untergliedert werden [251]:
• Gelpräparation
• Übersättigung
• Keimbildung
• Kristallwachstum
Im ersten Schritt wird durch geeignetes Zusammenmischen der Ausgangssuspensionen
oder Sole ein amorphes Gel gebildet. Dieses dient als Reservoir für die Reaktanden. Über
das Mineralisierungsagens wird die Löslichkeit des silikatischen Gels und somit die Kon-
zentration der Reaktanden gesteuert [251]. Der entscheidende Bestandteil ist jedoch das
Templat, welches als eine Art Schablone mit strukturdirigiernden Eigenschaften dient.
Nach SCHWIEGER können folgende Templatklassen unterschieden werden: organische
Kationen, organische Moleküle und organische Anionen [252]. Eines der am häufigsten
verwendeten Template ist das Tetrapropylammonium Kation (TPA+), welches meist als
Hydroxid [253] oder kostengünstigeres Bromid [254] der Reaktionslösung zugegeben
wird.
Die Kristallisation erfolgt anschließend hydrothermal in Autoklaven unter erhöhtem
Druck, üblicherweise bei Temperaturen zwischen 150 und 200 °C. Die Kristallisations-
zeiten können dabei von wenigen Stunden bis zu mehreren Tagen reichen [254]. Bei die-
sem Vorgang kommt es zunächst zu einer Übersättigung mit anschließender Keimbil-
dungsphase, auf die eine Kristallwachstumsphase folgt [251, 255]. Da es sich bei
Zeolithen um thermodynamisch metastabile Phasen handelt, haben die Reaktionsparame-
ter eine entscheidenden Einfluss auf das Gelingen der Synthese [255].
Neben der üblicherweise eingesetzten templatgestützten Kristallisation besteht jedoch
auch die Möglichkeit, die ZSM-5-Synthese templatfrei unter Verwendung bestimmter
Pufferlösungen durchzuführen. Diese Methode hat jedoch den Nachteil, dass nur
Si/Al-Verhältnisse kleiner 50 erreicht werden können, und dass die Kristallisationszeiten
meist deutlich länger sind als bei der Synthese mit Templaten [252].

78 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
Da der Eisenanteil des ZSM-5 eine entscheidende Rolle bei der Hydroxylierung von
Benzen mit N2O spielt (vgl. Kapitel 3.4.2), muss an dieser Stelle noch erwähnt werden,
dass beinahe alle synthetisierten ZSM-5 Zeolithe einen geringen Eisenanteil enthalten
können, welcher aus Verunreinigungen in den Ausgangsstoffen (Kieselsol, Wasserglas)
oder dem Reaktionsgefäß selbst (Edelstahlautoklav) herrührt.
Nach der Synthese liegt der Zeolith in der so genannten „as-synthesized“-Form vor. Der
Zeolith enthält noch das Templat, Wasser und Natrium-Ionen auf den Extragitterplätzen.
Da die Eigenschaften dieser Ausgangsform für einen Einsatz in der Katalyse nicht optimal
sind, schließen sich deshalb meist mehrere Nachbehandlungsschritte an. Zunächst werden
das Templat und Wasser thermisch entfernt (Kalzinieren). Man erhält die so genannte
Na-Form des Zeoliths, welche in der Regel nicht katalytisch aktiv ist. Deshalb wird meist
ein Ionentausch der Extragitterkationen in flüssiger Phase durchgeführt. Um den Kataly-
sator in die H-Form zu überführen, werden die Na-Kationen zunächst gegen NH4+-Ionen
ausgetauscht. Anschließend erfolgt eine thermische Behandlung, bei der Ammoniak
abgespalten wird und ein Proton an den Kationenplätzen zurückbleibt (vgl. Abbildung
3.8) [256].
Abbildung 3.8: Ionenaustausch in Zeolithen nach [256]
Neben einwertigen Kationen können jedoch auch mehrwertige wie Fe, Co, Cu oder Pd
eingetauscht werden. Dabei kann ein dreiwertiges Kation (z. B. Fe3+) die negativen
Ladungen von drei Gitteraluminiumatomen kompensieren (siehe Abbildung 3.9). Der
Abstand des Kations von den Aluminiumatomen wird durch das Si/Al-Verhältnis des
Zeolithen bestimmt. Einhergehend mit einem größeren Abstand der Kationen vom Gitter
nimmt die elektrische Feldstärke im Gitter zu und die Dichte des Zeoliths ab [232].
Neben dem Ionenaustausch in der Flüssigphase besteht jedoch auch die Möglichkeit,
Kationen durch Festkörperionentausch oder Sublimation aus der Gasphase einzubringen.
Bei diesem Verfahren wird jedoch bereits die Ammonium- oder H-Form des Zeoliths als
Ausgangsmaterial verwendet [257].

3.4 Hydroxylierung von Benzen mit N2O 79
Abbildung 3.9: : Schematische Darstellung der Koordination eines dreiwertigen Kations
(Fe3+) bei einem Zeolithen mit niedrigem und hohem Modul nach [232]
Nach dem Ionentausch können sich weitere Nachbehandlungsschritte anschließen. Oft ist
es vorteilhaft, den Zeolith bei einer hohen Temperatur thermisch zu behandeln, wodurch
es zu einer Dehydroxylierung und Dealuminierung kommt (vgl. Kapitel 3.4.1.4), oder eine
Silanierung durchzuführen.
3.4.1.4 Katalytische Eigenschaften
Grundlegende Eigenschaften, die ZSM-5-Zeolithe für den Einsatz in der Katalyse qualifi-
zieren, sind vor allem ihre regelmäßige Porengeometrie, welche sich in der Größenord-
nung einfacher aromatischer Moleküle bewegt, ihre hohe Temperaturbeständigkeit und
ihre Eigenschaft als Festkörpersäure zu wirken [238].
Die Azidität von Zeolithen hängt in erster Linie von ihrer chemischen Zusammensetzung
ab. An den Stellen im Gitter, an denen vierwertige Siliziumatome durch dreiwertige Alu-
miniumatome ersetzt sind, entstehen negative Ladungen, welche durch die Extragitter-
kationen kompensiert werden. Tauscht man (wie in Kapitel 3.4.1.3 beschrieben)
Natriumkationen eines Zeolithen gegen Protonen aus, entstehen verbrückte Hydro-
xylgruppen (Si-OH-Al), welche als Brønsted-Säure-Zentren fungieren (siehe Abbildung
3.10). Die Anzahl und Stärke dieser Zentren haben einen entscheidenden Einfluss auf die
Aktivität des Katalysators. Je kleiner das Modul des Zeolithen, desto höher ist sein
Aluminiumanteil und damit die Zahl der Brønsted-Säure-Zentren [245]. Genau umgekehrt
verhält es sich jedoch mit der Stärke der einzelnen Zentren. Nach SANDERSON lässt sich
die Elektronegativität einer chemischen Verbindung mit unterschiedlichen Atomtypen
berechnen. Für Zeolithe ergibt sich, dass die Stärke eines einzelnen Zentrums und damit
die Säurestärke mit steigendem Modul des Zeolithen zunimmt. Berechnungen von
MORTIER haben gezeigt, dass die Säurestärke von Zeolithen jedoch ab einem Modul von
16 nicht weiter ansteigt, sondern einen Sättigungswert erreicht [258].

80 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
O Si
O
O O
OO
O
O
Si
Si
Si Si
Si
Si
O
O
O
O
O
Si
O
O
Al
O
OO
H
Abbildung 3.10: Brønsted-Säure-Zentrum im Zeolithgitter nach [259]
Seit langem wird ein experimenteller Nachweis der Säurestärke der Brønsted-Zentren in
Zeolithen angestrebt. Hierfür kommen verschiedene Methoden in Frage, wie infrarot-
spektroskopische Untersuchungen, NH3-TPD, die Adsorption starker Basen, Testreak-
tionen oder 1H-NMR. Viele dieser Untersuchungen bestätigten die Berechnungen von
MORTIER, führten jedoch auch zu nicht übereinstimmenden Ergebnissen, so dass dieses
Thema bis heute intensiv diskutiert wird [24, 260].
Neben den bereits beschriebenen verbrückten Hydroxylgruppen tragen jedoch auch so
genannte terminale (endständige) OH-Gruppen zur Azidität des Zeolithen bei. Man geht
davon aus, dass sie direkt an ein Siliziumatom gebunden sind, also ohne Aluminium in
unmittelbarer Umgebung [261]. Dies kann auf der äußeren Oberfläche der Zeolithkristalle
oder an Fehlstellen und silikatischen Verunreinigungen im Inneren des Zeoliths der Fall
sein [262, 263]. Man hat festgestellt, dass ZSM-5-Zeolithe mit hohem Modul mehr dieser
Silanolgruppen aufweisen, als solche mit niedrigem [264]. Außerdem ist die Säurestärke
dieser Gruppen unabhängig vom Modul und im Allgemeinen sehr gering [261].
Neben den Brønsted-Säure-Zentren gibt es aber auch Lewis-Säure-Zentren in Zeolithen.
Sie entstehen nach einer Dehydroxylierung oder Dealuminierung der H-Form der
Zeolithe. Bei der Dehydroxylierung wird aus zwei Brønsted-aziden Hydroxylgruppen
Wasser abgespalten und es bleibt ein dreifach koordiniertes Aluminium- und ein positiv
geladenes Siliziumatom im Gitter zurück, welches als Lewis-Säure wirkt (siehe
Abbildung 3.11) [265].

3.4 Hydroxylierung von Benzen mit N2O 81
Abbildung 3.11: Bildung eines Lewis-Säure-Zentrums aus zwei Brønsted-Säure-Zentren
nach [259]
Einige Autoren sind jedoch der Meinung, dass sich der Dehydroxylierung immer eine
Dealuminierung anschließen muss [266]. Nach der Dehydroxylierung kommt es zur
Bildung einer Extragitter [AlO]+-Spezies, welche auch als „true Lewis-acid-site“ bezeich-
net wird. Andere Autoren gehen jedoch davon aus, dass die Kalzinierungstemperatur für
die Ausbildung der beiden möglichen Al-Spezies verantwortlich ist. Niedrige Temperatu-
ren führen zu einer Dehydroxylierung, hohe zu einer Dealuminierung [267].
Bis heute ist die Entstehung und Stabilität der Gitter-Lewis-Zentren in ZSM-5-Zeolithen
umstritten [268].
Neben der Säurewirkung der Zeolithe spielt aber auch die so genannte Formselektivität
eine große Rolle für die Katalyse. Man unterscheidet dabei drei Fälle. Im ersten Fall, der
„reactant shape selectivity“, können nur solche Moleküle umgesetzt werden, die kleiner
sind als der Porendurchmesser des Zeoliths. Sind an einer Reaktion mehrere Spezies
beteiligt, welche kleiner als der Porendurchmesser sind, wird die Diffusion der großen
Moleküle behindert und kleinere werden bevorzugt umgesetzt. Bei der „product shape
selectivity“ kann ein Produkt nicht entstehen, wenn der Durchmesser größer ist als der
Porendurchmesser, oder die Diffusion der größeren Moleküle behindert wird. Im Falle
einer „restricted transition state shape selectivity“ läuft eine Reaktion nicht ab, weil ein
Übergangszustand während der Reaktion größer als die Porenabmessungen des Katalysa-
tors ist [238, 245]. Diese Formselektivität der Zeolithkatalysatoren wird bei zahlreichen
großtechnischen Prozessen ausgenutzt [269].
3.4.1.5 Sorptionseigenschaften
Die Adsorption der an der Reaktion beteiligten Spezies am Katalysator spielt eine
entscheidende Rolle bei heterogen katalysierten Reaktionen [270]. Zeolithe können in
Abhängigkeit des Si/Al-Verhältnisses hydrophil oder hydrophob sein. Ein hoher Alumini-
umanteil im Zeolith bewirkt eine größere Polarität und bedeutet eine größere Anzahl an
lewis-aziden Kationen, welche als Adsorptionsplätze für polare Moleküle dienen. Alu-
miniumreiche Zeolithe (z. B. Zeolith X) sind deshalb hydrophil, während siliziumreiche
Zeolithe (Silikalith) als hydrophob eingestuft werden [23, 236, 271]. Während chemi-
schen Reaktionen werden die Reaktanden am Katalysator adsorbiert und dadurch aufkon-
zentriert, weshalb die Reaktion quasi unter erhöhtem Druck abläuft [271]. Kommt es zu

82 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
einer Kapillarkondensation der Reaktanden in den Poren des Zeoliths, liegt sogar ein
quasiflüssiger Zustand vor, auch wenn die Edukte gasförmig aufgegeben wurden [272].
Das unterschiedliche Sorptionsverhalten der an einer Reaktion beteiligter Spezies hat
somit einen entscheidenden Einfluss auf den Ablauf der Reaktion. Idealerweise erfolgt
eine schnelle Adsorption der Edukte und eine schnelle Desorption (schwache Adsorption)
der Produkte. Sind die Produkte zu stark adsorbiert, treten vermehrt Folgereaktionen und
eine Produktinhibierung der Reaktion auf [271].
In der vorliegenden Arbeit sind die Adsorption und Desorption von Benzen und Phenol in
ZSM-5 Zeolithen von besonderer Bedeutung.
In der Literatur gibt es Berichte über eine Reihe von Untersuchungen zum Sorptions-
verhalten von Benzen an ZSM-5 Zeolithen, jedoch nicht für das Phenol. Da es aufgrund
der kleinen Beladungen messtechnisch schwierig ist, Sorptionsuntersuchungen bei
höheren Temperaturen durchzuführen, sind die Literaturwerte meist nur bis zu Tempe-
ratur von 250 °C gültig. Benötigt werden jedoch Sorptionsmessungen unter Reaktionsbe-
dingungen, welche bei der Benzenhydroxylierung mit N2O im Allgemeinen jedoch über
300 °C liegen [273]. Bei REITZMANN [24] findet sich ein Überblick der Literaturwerte
für die Sorptionsenthalpien von Benzen in ZSM-5 Zeolithen. Die Werte reichen je nach
eingesetztem Zeolith und eingestellter Benzenbeladung von 35 - 92 kJ/mol. Hier sollen
jedoch nur einige allgemeine Trends, welche aus diesen Untersuchungen abzuleiten sind,
wiedergegeben werden.
• Der Aluminiumanteil im ZSM-5 Zeolithen und damit die Polarität des Gitters hat
nur geringen Einfluss auf die Sorption von Benzen.
• Punktladungen im Gitter (z. B. Natriumkationen) bewirken eine stärkere
Adsorption von Benzen.
• Die Sorptionsenthalpien von Benzen sind beladungsabhängig. Bei hohen Benzen-
beladungen sind die Sorptionsenthalpien kleiner als bei niedrigen.
Neben den experimentellen Untersuchungen gibt es jedoch auch Simulationsrechnungen
von KLEMM ET AL. [274], welche die Konkurrenzadsorption von Benzen und Phenol in
ZSM-5 Zeolithen beschreiben.
In Tabelle 3.1 sind die errechneten isosteren Sorptionsenthalpien für Benzen und Phenol
an drei ZSM-5-Modifikationen zusammengefasst.

3.4 Hydroxylierung von Benzen mit N2O 83
Tabelle 3.1: Isostere Sorptionsenthalpien für Benzen und Phenol [274]
Isostere Adsorptionsenthalpie [kJ/mol] Zeolith
Benzen Phenol
Silikalith 58,1 61,7
H-ZSM-5 (Si/Al = 95) 58,0 62,8
Na-ZSM-5 (Si/Al = 95) 83,3 105,2
Der Vergleich der errechneten isosteren Sorptionsenthalpien für Benzen mit Literatur-
werten zeigte eine sehr gute Übereinstimmung [274]. Weiterhin zeigen diese Berechnun-
gen, dass das Phenol am Na-ZSM-5-Katalysator deutlich stärker adsorbiert wird als das
Benzen. Neben den Sorptionsenthalpien wurden an diesen Modellkatalysatoren auch die
Adsorptionsisothermen von Benzen und Phenol und die Henry-Konstanten bei verschie-
denen Temperaturen simuliert. In Abbildung 3.12 ist das Verhältnis der errechneten
Henry-Konstanten für Benzen und Phenol in Abhängigkeit von der Temperatur
aufgetragen.
Abbildung 3.12: Verhältnis der Henry-Konstanten zwischen Benzen und Phenol in
Abhängigkeit von der Temperatur [274]
Man erkennt in Abbildung 3.12, dass die KP/KB-Werte für alle Katalysatoren mit
steigender Temperatur abnehmen. Das Verhältnis der Konstanten bei Silikalith und
H-AlZSM-5 zeigt nur geringe Unterschiede und nähert sich mit steigender Temperatur 1
an. Auch bei Raumtemperatur sind die Werte nur geringfügig größer als 1. Ein deutlich
anderes Verhalten zeigt der Na-ZSM-5, der aufgrund der Punktladung am Natrium deut-
lich polarer als die anderen Katalysatoren ist. Gerade bei niedrigen Temperaturen, aber

84 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
auch im Bereich der Reaktionstemperaturen der Benzenhydroxylierung, wird das Phenol
deutlich stärker adsorbiert als das Benzen.
Der Autor schließt aus diesen Untersuchungen, dass sich ein geringer Natriumgehalt des
Zeolithen positiv auf die Reaktion auswirken sollte, da sich die Adsorption des Phenols
verringert und somit eine Produktinhibierung bzw. unerwünschte Nebenreaktionen ver-
mieden werden.
Auf einen großen Einfluss der Adsorption des Phenols auf die Hydroxylierung von
Benzen mit N2O deuten auch die Untersuchungen von HÄFELE und REITZMANN hin
[233, 275, 276]. Bei diesen Untersuchungen zeigte es sich, dass die Fahrweise mit
Benzenüberschuss zu einer höheren Phenolselektivität und einer verringerten Desaktivie-
rung des Katalysators führt. Als Ursache wird von den Autoren vermutet, dass das Phenol
aufgrund seiner starken Adsorption zur Bildung von Nebenprodukten und einer Verko-
kung des Katalysators führt. Durch den Benzenüberschuss wird das Phenol von der Ober-
fläche verdrängt und so dieses Problem verringert. Von PERATHONER [277] und SELLI
[278] wird dem Phenol aufgrund seiner starken Adsorption am Katalysator ebenfalls eine
zentrale Rolle bei der Desaktivierung während der Hydroxylierung von Benzen mit N2O
zugeschrieben. Die Arbeiten von VENUTO [23, 239] und PARTON [279] weisen bei der
Phenolalkylierung an Zeolithen auf Probleme durch die starke Adsorption von Phenol hin.
Auf den Zusammenhang zwischen Phenoladsorption und Desaktivierung während der
Hydroxylierung von Benzen mit N2O wird in Kapitel 3.4.3 noch unter dem Gesichtspunkt
reaktionstechnischer Aspekte eingegangen.
3.4.1.6 Dielektrische Eigenschaften
Über die dielektrischen Eigenschaften von Zeolithen ist relativ wenig bekannt. Das Ver-
halten von Zeolithen im Mikrowellenfeld gleicht im Grunde dem keramischer Materialien
(vgl. Kapitel 2.1.2.1). Die Dielektrizitätskonstanten von Zeolithen sind von ihrer chemi-
schen Zusammensetzung und Struktur, der eingestrahlten Mikrowellenfrequenz, der Tem-
peratur und vor allem dem Wassergehalt abhängig [232, 280]. Einzig für den stark
hydrophilen Zeolith A sind ausführliche Untersuchungen in der Literatur zu finden.
Deshalb soll zunächst dieser Zeolithtyp exemplarisch behandelt werden. Informationen zu
Struktur und Zusammensetzung dieses Zeolith Typs finden sich bei BRECK [232] und im
Zeolith-Atlas [234].
In Abbildung 3.13 ist der dielektrische Verlustfaktor von Zeolith A Pulver in Abhängig-
keit der eingestrahlten Frequenz und dem Wassergehalt des Zeolithen aufgetragen. Wie in
Kapitel 2.1 beschrieben wird Strahlung einer Frequenz von etwa 300 MHz – 30 GHz
(3⋅108 – 3⋅1010 Hz) als Mikrowelle bezeichnet, d. h. für eine Mikrowellenerwärmung sind
nur die hohen in der Grafik dargestellten Frequenzen relevant. Für einen mit Wasser
beladenen Zeolithen erkennt man zwei deutliche Absorptionsmaxima (Typ I, II) deren

3.4 Hydroxylierung von Benzen mit N2O 85
Ausprägung mit zunehmendem Wassergehalt zunimmt und deshalb auf die Relaxation
von gebundenem Wasser zurückgeführt wird. Der Typ I bei niedrigerer Frequenz wird auf
Wassermoleküle in den großen α-Käfigen des Zeoliths zurückgeführt und der Typ II auf
die verbleibenden Adsorptionsplätze. Betrachtet man den dehydratisierten Zeolith zeigt
sich ein gänzlich anderer Verlauf. Die Absorptionsmaxima des Typs I und II sind nicht
vorhanden, stattdessen erkennt man einen anderen Relaxationsprozess bei einer höheren
Frequenz. Diese Absorption vom Typ III kann nur auf die Na-Ionen im Zeolith
zurückgeführt werden. Da dieses Maximum jedoch bei Beladung des Zeoliths mit Wasser
verschwindet, geht man davon aus, dass der Relaxationsprozess des Typs II auf eine
Wechselwirkung dieser Ionen mit den Wassermolekülen zurückzuführen ist. Außerdem
steigt der Verlustfaktor ε’’ beim trockenen Zeolith im niedrigen Frequenzbereich
(< 104 Hz) deutlich stärker an als beim mit Wasser beladenen Zeolith [232].
Abbildung 3.13: Dielektrischer Verlustfaktor von Zeolith A Pulver in Abhängigkeit der
eingestrahlten Frequenz [232]
Diese Untersuchungen zeigen, dass die Mikrowellenfrequenz eine entscheidende Rolle bei
der dielektrischen Erwärmung von Zeolithen spielt. Beinahe alle in der Literatur verfügba-
ren Daten über das Aufheizverhalten von Zeolithen im Mikrowellenfeld wurden jedoch
bei der technisch relevanten Frequenz von 2,45 GHz ermittelt.
Erste Untersuchungen von KOMARNENI ET AL. [281] zeigten bereits, dass sich viele
Zeolithe durch Mikrowellen einer Frequenz von 2,45 GHz bis zum Schmelzpunkt erhitzen
lassen. Der Autor führt die Wechselwirkung mit dem Mikrowellenfeld auf eine Anregung
von Ionen im Zeolith zurück. Von OHGUSHI ET AL. [280] wurde das Aufheizverhalten
verschiedener Zeolithe vom Typ A (3A, 4A, 5A) untersucht. Es wurde der Wassergehalt
(2-20 %) der Proben, die Starttemperatur (100-500 °C) und das Probenvolumen (4,0 cm3,

86 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
13,4 cm3) variiert. Zum Einsatz kam eine Haushaltsmikrowelle mit einer Frequenz von
2,45 GHz bei 850 oder 900 W Leistung. Die Proben wurden in einen Probenhal-
ter/Isolierung aus hochreinem Aluminiumoxid eingebaut. Die Temperaturmessung
erfolgte über ein Thermoelement, welches von einer mit dem Gehäuse leitend verbunde-
nen Metallhülle vor dem Feld abgeschirmt wurde. Bei der Betrachtung der angegebenen
Probentemperaturen muss jedoch berücksichtigt werden, dass sich das Probengefäß ohne
Zeolith während der Mikrowellenbestrahlung auf ca. 150 °C aufheizte. Die Erwärmung
der Probe erfolgte somit nicht ausschließlich über den Zeolith. Im Folgenden sollen die
Ergebnisse dieser Untersuchungen zusammengefasst werden:
• Der Wassergehalt der Proben spielte bei einer Erwärmung der Zeolithe ausgehend
von Raumtemperatur eine entscheidende Rolle. Je höher der Wassergehalt war,
desto schneller erfolgte die Aufheizung. Während sich die dehydrierten Zeolithe
nur bis zu einer Temperatur von ca. 200 °C im Mikrowellenfeld aufheizen ließen,
konnten die feuchten Zeolithe 3A und 4A auf Temperaturen über 800 °C erwärmt
werden. Beim Zeolith des Typs 5A konnten jedoch auch bei hohem Wassergehalt
nur Temperaturen um 200 °C erzielt werden.
• Wurde eine Desorption des Wassers verhindert, verlief die Aufheizung der
Zeolithe schneller.
• Große Proben (13,4 cm3) ließen sich durch die Mikrowellen deutlich besser
aufheizen als kleine (4,0 cm3). Bei großen Probenvolumen (Probenmassen) kam es
beim hydratisierten Zeolith 3A zu einem thermal runaway, während kleine Proben
des selben Materials nur Temperaturen von 200 °C erreichten. Der Autor spricht
von einem „Masseneffekt“ und führt ihn auf die unterschiedlichen
Wärmeabgabeflächen zur Umgebung zurück. Die Heizwirkung der Mikrowellen
erfolgt im Inneren der Probe, die Wärmeabgabe über die Oberfläche. Wird diese
im Verhältnis zum Volumen verringert, kommt es zu einer stärkeren Aufheizung.
• Die Ausgangstemperatur der Proben hatte einen entscheidenden Einfluss auf die
Erwärmung. Wurden dehydrierte Proben der Zeolithe 3A und 4A auf Temperatu-
ren über 200 °C vorgeheizt, kam es auch bei diesen Proben zu einem Temperatur-
anstieg auf über 800°C.
• Die chemische Zusammensetzung und Struktur der Zeolithe dominierte das
Aufheizverhalten bei höheren Temperaturen. Während sich vorgeheizte Proben
(> 200 °C) der Zeolithe 3A und 4A sehr gut durch Mikrowellen aufheizen ließen,
wurde beim Typ 5A kein weiterer Temperaturanstieg im Mikrowellenfeld beo-
bachtet. Der Autor führt dies auf die unterschiedliche Kationenverteilung und
Kationentypen in den Zeolithen zurück. Zeolithe des Typs 4A enthalten aus-
schließlich Na-Ionen, 3A Na- und K-Ionen und 5A Na- und Ca2+-Ionen.

3.4 Hydroxylierung von Benzen mit N2O 87
Der Einfluss der Kationen auf die dielektrischen Eigenschaften von 4A und 5A Zeolithen
wurde von OHGUSHI ET AL. in zwei Publikationen genauer beschrieben [282, 283].
Während die Typen 4A und 3A Kationen an drei verschiedenen Gitterplätzen (8-
Sauerstoff-Ringe, 6-Sauerstoff-Ringe und 4-Sauerstoff-Ringe) besitzen, ist beim Zeolith
5A nur einer dieser Kationenplätze besetzt. Die Kationen befinden sich im Zentrum eines
6-Rings aus Sauerstoffatomen. Für die Anregung dieser Kationen durch Mikrowellen ist
jedoch eine sehr hohe Temperatur des Zeolithen (> 800 °C) nötig. Außerdem spielt auch
die Art der Kationen eine Rolle bei der Anregung durch Mikrowellen. Na- lassen sich
besser als K- und diese wiederum besser als Ca2+-Ionen durch Mikrowellen aufheizen. Für
die Aufheizbarkeit durch Mikrowellen ergibt sich somit folgender Zusammenhang:
4A > 3A >> 5A
Die Erwärmung der Zeolithe über Mikrowellen erfolgt somit nach zwei Mechanismen.
Bei niedrigen Temperaturen erfolgt der Energieeintrag hauptsächlich über gebundenes
Wasser, bei hohen Temperaturen ausschließlich über die Kationen des Zeolithen.
Diese Ergebnisse decken sich auch mit den Untersuchungen von WHITTINGTON und
MILESTONE [284], der die Aufheizung verschiedener Zeolithtypen durch Mikrowellen-
strahlung testete. Bei diesen Untersuchungen kam eine Haushaltsmikrowelle (2,45 GHz)
bei einer Leistung von 700 W zum Einsatz. Die Temperatur wurde mit einem Thermo-
element nach Abschalten der Mikrowelle gemessen.
Die Untersuchungen zielten ursprünglich darauf ab, das Aufheizverhalten reiner
H-Formen von Zeolithen zu bestimmen. Es stellte sich jedoch heraus, dass sich diese
praktisch nicht durch Mikrowellen erwärmen ließen. Deshalb wurden verschiedene
Zeolithtypen sowie Parameter, wie der Na- und Wassergehalt variiert. In Tabelle 3.2 ist
die Temperatur einiger der getesteten Zeolithe nach 15 Minuten Mikrowellenbestrahlung
aufgetragen. Es zeigte sich, dass der Wassergehalt eine entscheidende Rolle bei der
Aufheizung der Zeolithe durch Mikrowellenstrahlung spielt. Höhere Wassergehalte
bewirken eine raschere Aufheizung bis zu einem Temperaturbereich von ca. 200 °C. Der
Na-Gehalt des Zeoliths ist vor allem für das Erreichen höherer Temperaturen bzw. bei der
Aufheizung trockener Zeolithe ausschlaggebend. Werden Na-Ionen gegen Protonen
getauscht, verschlechtert sich der Energieeintrag durch Mikrowellenstrahlung mit steigen-
dem Austauschgrad. Die Art der Ionen hat ebenfalls einen Einfluss. Tauscht man Na-
gegen K-Ionen, lässt sich der Zeolith schlechter durch Mikrowellen erwärmen. Je mehr
Kationen ein Zeolith enthält, desto besser absorbiert er Mikrowellenstrahlung.. Weiterhin
wird von den Autoren vermutet, dass die Position der Kationen im Zeolith eine Rolle
spielt. Eine eindeutige Zuordnung ist jedoch schwierig, weil die Verteilung der Kationen
auf unterschiedliche Extragitterplätze nicht immer bekannt ist und weil eine Migration der
Kationen bei Temperaturerhöhung auftreten kann. Es wird jedoch vermutet, dass die

88 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
Ionen, welche sich in größeren Hohlräumen des Zeoliths befinden, bevorzugt durch
Mikrowellen angeregt werden. Hierfür spricht die Beobachtung, dass Faujasite, welche
große Hohlräume (Superkäfige) besitzen, Mikrowellen besser absorbieren als Zeolithe
vom Typ ZSM-5.
Tabelle 3.2: Aufheizverhalten verschiedener Zeolithtypen; Temperaturen nach 15 min bei
einer Leistung von 700 W [284]
Zeolith (Si/Al) Na+ gegen H+
getauscht [%]
Wassergehalt
[%]
Temperatur
[°C]
0 0 130
0 29 >1000
2 0 120
59 0 70
NaY (2,39)
59 30 200
KY (90 % K, 10 % Na) 0 0 100
0 0 590 NaA (1,02)
13 0 180
0 0 90 KA (75 % K, 25 % Na)
0 23 220
Na-ZSM-5 (20) 0 0 160
0 0 100 Na-ZSM-5 (31)
0 7 150
Von besonderer Bedeutung für die vorliegende Arbeit ist, dass sich ZSM-5-Zeolithe, wie
sie in der Benzenhydroxylierung eingesetzt werden, nur sehr schlecht durch Mikrowellen
erwärmen lassen. Die reine H-Form dieses Zeolithtyps ist annähernd transparent für Mik-
rowellenstrahlung. Über den Einfluss anderer für die Katalyse relevanter Kationen (Fe,
Co, Cu) auf die dielektrischen Eigenschaften von Zeolithen ist in der Literatur nichts
bekannt.

3.4 Hydroxylierung von Benzen mit N2O 89
3.4.2 Reaktionsmechanismus
Obwohl die Hydroxylierung von Benzen mit N2O an ZSM-5-Katalysatoren bereits seit
langem intensiv erforscht wird, herrscht über den Reaktionsmechanismus und die in der
Hydroxylierung aktiven Zentren noch Unklarheit. In der Literatur werden vier Mechanis-
men diskutiert, welche jeweils an einem unterschiedlichen Zentrum des Katalysators
ablaufen. Von ONO ET AL. [220] wurde ein Mechanismus über Brønsted-Säure-Zentren
vorgeschlagen. PANOV ET AL. [225] gehen davon aus, dass Extragittereisenspezies die
zentrale Rolle bei der Benzenhydroxylierung spielen und KUSTOV ET AL. [285] postu-
lierte einen Mechanismus über Gitter-Lewis-Säure-Zentren. Die Gruppe um
HÖLDERICH [286] vermutet wiederum, dass Extragitteraluminiumspezies die Reaktion
katalysieren. In den Arbeiten von KLEMM [287] und REITZMANN [24] wurden die
postulierten Mechanismen und der Reaktionsablauf der Benzenhydroxylierung bereits
detailliert beschrieben.
Von MELONI ET AL. [278, 288] wurden verschiedene Fe-ZSM-5 Zeolithe in der
Benzenhydroxylierung getestet. Dabei wurden das Si/Al-Verhältnis und der Eisenanteil
der Zeolithe variiert. Es zeigte sich, dass die Azidität keinen Einfluss auf die Hauptreak-
tion hat, jedoch maßgeblich an der Desaktivierung des Katalysators beteiligt ist (vgl.
Kapitel 3.4.3). Als aktives Zentrum werden binukleare Extragittereisenspezies vorgeschla-
gen, da ein eisenfreier H-ZSM-5 (<< 0,005 m% Fe2O3) keine katalytische Aktivität zeigte.
Bereits geringste Mengen an Eisen (0,07 m% Fe2O3) genügten jedoch, um einen in der
Hydroxylierung aktiven Katalysator zu erhalten.
HENSEN ET AL. [289] synthetisierten ebenfalls einen eisenfreien, aluminiumreichen
ZSM-5 (< 0,001 m% Fe), der in der Hydroxylierung keine Aktivität zeigte.
Zu einem vergleichbaren Ergebnis führten auch die Untersuchungen von KUBÁNEK ET
AL. [290] an H-ZSM-5 Katalysatoren mit geringem Eisenanteil. Es konnte kein
Zusammenhang zwischen der katalytischen Aktivität in der Benzenhydroxylierung und
der Anzahl an Brønsted- oder Lewis-Säure-Zentren gefunden werden. Dagegen wurde ein
beinahe linearer Zusammenhang zwischen der Phenolbildung und dem Eisengehalt der
Zeolithe festgestellt.
Auch der Mechanismus über Extragitteraluminiumspezies wird mittlerweile angezweifelt,
obwohl ein höherer Aluminiumanteil des ZSM-5 im Allgemeinen zu einer höheren Akti-
vität des Katalysators in der Benzenhydroxylierung führt. Stattdessen gehen einige
Autoren davon aus, dass das Aluminium den Ausbau von Eisen aus dem Gitter fördert
[290] oder eine Aktivierung des Eisens bewirkt [225, 291]. Andere Autoren nehmen an,
dass das Aluminium zwar eine am aktiven Zentrum beteiligte Spezies darstellt, jedoch nur
in Verbindung mit Eisen [289].

90 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
Viele weitere Untersuchungen deuten darauf hin, dass Eisen die zentrale Rolle in der Ben-
zenhydroxylierung mit N2O spielt [289, 291-297]. Auch deutet einiges darauf hin, dass
andere in den Zeolith eingebrachte Metalle, wie z. B. Ru, Co, V, Cr und Ga zwar einen
positiven Einfluss auf die katalytische Aktivität der ZSM-5-Zeolithe haben können,
beispielsweise durch eine Stabilisierung der Eisenzentren oder einen Ausbau von Eisen
aus dem Gitter bewirken, jedoch nicht direkt am Reaktionsmechanismus beteiligt sind
[291, 295].
Deshalb soll im Folgenden der Mechanismus über Extragittereisenspezies genauer be-
trachtet werden.
PANOV ET AL. unterscheiden zwei Teilschritte bei der Reaktion von Benzen mit N2O,
die in Abbildung 3.14 dargestellt sind. Im ersten Schritt bildet sich durch Zersetzung von
N2O eine elektrophile, atomare, ungeladene Oberflächensauerstoffspezies, welche als
α-Sauerstoff bezeichnet wird. Dieser aktivierte Sauerstoff reagiert anschließend mit
Benzen zu Phenol [225]. DUBKOV ET AL. vermuten, dass in diesem zweiten Teilschritt
die CH-Bindung nicht gespalten, sondern ein Arenoxid als Übergangszustand gebildet
wird [298]. Diese These wurde von KACHUROVSKAYA ET AL. durch DFT-
Simulationen bestätigt [293]. Die Zentren, an denen sich α-Sauerstoff bildet, identifizierte
PANOV als Extragittereisenspezies im Inneren des ZSM-5-Zeoliths. Eisen in
tetraedrischen Gitterpositionen kann nach seinen Untersuchungen als aktive Spezies
ausgeschlossen werden. Über die Struktur und den Aktivitätszustand der von PANOV
identifizierten Eisenspezies herrscht jedoch noch Unklarheit [225].
N2
OH
N2O Z
Z O
Z O +
+ + Z
Abbildung 3.14: Bildung und Reaktion von α-Sauerstoff mit Benzen zu Phenol [225]
Die Gruppe um PANOV geht davon aus, dass es sich bei der aktiven Eisenspezies um
isolierte binukleare, sauerstoffverbrückte Eisencluster handelt, welche sich mit einer
hohen Dispersion im Inneren der Zeolithkanäle befinden (vgl. Abbildung 3.15). Vor der
Beladung mit N2O liegt ein gemischter Oxidationszustand (Fe(II)/Fe(III)) vor, danach
stellt sich ein volloxidierter Zustand (Fe(III)/Fe(III)) ein. Gitteraluminium stabilisiert die
Eisencluster und begünstigt zudem den Ausbau von Eisen aus dem Gitter [225, 291].
Diese These wird auch von KUBÁNEK ET AL. [290], PÉREZ-RAMÍREZ ET AL. [296]
und OVERWEG ET AL. [299] vertreten.

3.4 Hydroxylierung von Benzen mit N2O 91
Fe
OH
OH OH
OH
Fe
OH
OH
Fe
O
OH OH
Fe
OH
OH
III III IIIII
Abbildung 3.15: Schematische Darstellung von binuklearen Extragittereisenclustern im
ZSM-5 nach [300]
Als problematisch in der Theorie, dass der Reaktionsmechanismus über Extragittereisen
verläuft, erweist sich jedoch, dass zwar bei niedrigen Eisengehalten der Katalysatoren
eine Korrelation zwischen dem Eisengehalt und dem Benzenumsatzgrad besteht, bei
höheren Eisenanteilen jedoch ein Plateau erreicht wird und keine weitere Aktivitätssteige-
rung beobachtet werden kann [225, 290]. KHARITONOV ET AL. führen dies auf eine
Produktinhibierung durch Phenol zurück [301]. JIA ET AL. [297] untersuchten ebenfalls
den Einfluss des Eisengehalts in der Benzenhydroxylierung. Bei einem steigenden Eisen-
gehalt im Zeolithen ergab sich eine sinkende Turnoverfrequenz des Eisens. Aus diesen
Ergebnissen und Untersuchungen in der Reduktion von NOx wurde gefolgert, dass es drei
verschiedene Eisenspezies im Zeolith gibt, welche unterschiedliche Reaktionen katalysie-
ren. Monomolekulare Eisenionen sind für die Hydroxylierung von Benzen zu Phenol
verantwortlich, bimolekulare katalysieren die Reduktion von NOx und größere
Eisenoxidcluster führen zu einer Totaloxidation. Weiterhin wird vermutet, dass die mono-
und bimolekularen Eisenzentren durch azide Zentren vor einem Zusammenwachsen zu
großen Eisenoxidclustern geschützt werden.
Zum gegenwärtigen Zeitpunkt ist es am wahrscheinlichsten, dass Eisenspezies die
zentrale Rolle bei der Benzenhydroxylierung mit N2O spielen. Klärungsbedarf besteht
jedoch noch in der Frage, welche Eisenspezies die Reaktion katalysiert und wie sich wei-
tere Eigenschaften des Zeolithen, wie Brønsted- und Lewis-Azidität oder die Anwesenheit
anderer Metalle im Zeolith, auf diese Zentren auswirken.

92 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
3.4.3 Desaktivierung
Eines der Hauptprobleme für die technische Umsetzung der Hydroxylierung von Benzen
mit N2O ist die schnelle Desaktivierung der ZSM-5 Katalysatoren durch Kohlenstoffabla-
gerungen. Tatsächlich kann ein beinahe vollständiger Aktivitätsverlust in einem Zeitraum
von wenigen Stunden auftreten [24, 275, 277]. Neben der Erforschung des
Reaktionsmechanismus, mit der sich die meisten Publikationen beschäftigen, ist es des-
halb notwendig, die für eine Desaktivierung verantwortlichen Faktoren zu identifizieren
und eine Verbesserung der Standzeiten über reaktionstechnische Maßnahmen oder Modi-
fikationen am Katalysator herbeizuführen.
Von BURCH und HOWITT [302] wurde die Regenerierung von in der
Benzenhydroxylierung eingesetzten Katalysatoren untersucht. Durch eine Behandlung mit
Stickstoff bei einer Temperatur von 500 °C konnte nur eine teilweise Reaktivierung des
Katalysators festgestellt werden, bei Verwendung von Sauerstoff wurde die ursprüngliche
Aktivität jedoch wieder vollständig hergestellt. Die Autoren unterscheiden deshalb zwei
Koksspezies. Einerseits wird während der Benzenhydroxylierung soft coke gebildet, der
zu einer Blockierung der Zeolithkanäle führt, sich jedoch bei höheren Temperaturen
(> 500 °C) im Stickstoffstrom zersetzt oder desorbieren lässt. Andererseits entsteht an den
sauren Zentren des Zeoliths auch hard coke, welcher nur durch einen Abbrand mit Sauer-
stoff entfernt werden kann. Der Anteil des soft coke ist, gemessen an der gesamten Koks-
menge, jedoch gering. Der untersuchte Katalysator vom Typ ZSM-5 wies nach einer
Reaktionsdauer von 120 min eine Kohlenstoffbeladung von 22,35 mgC/gKat. auf, bei
einem Anteil an soft coke von 16,3 %. Durch Stickstoffsorptionsmessungen konnte ge-
zeigt werden, dass bereits kleine Koksmengen zu einem erheblichen Rückgang der mess-
baren Katalysatoroberfläche führen (vgl. Tabelle 3.3). Es kommt zu einer Verblockung
der Zeolithporen und nicht zu einer homogenen Belegung mit Kohlenstoff [302].
Tabelle 3.3: Rückgang des Porenvolumens und Zunahme des C-Volumens nach Einsatz
eines ZSM-5 Zeolithen in der Hydroxylierung von Benzen mit N2O für verschiedene
Reaktionszeiten nach [302]
Reaktionsdauer Rückgang der Porenvolumens* Anteil C am Gesamtporenvolumen*
[min] [%] [%]
30 42 7
120 47 9
1440 55 11
* bezogen auf frischen Katalysator; durch N2-Sorption ermittelte Werte
Auch andere Autoren berichten, dass unter Einsatz von Sauerstoff/Luft eine vollständige
Regenerierung des Katalysators erfolgen kann [24, 303]. Aus wirtschaftlicher Sicht ist es
jedoch nötig, eine möglichst lange Standzeit des Katalysators bei möglichst hoher
Aktivität zu gewährleisten.

3.4 Hydroxylierung von Benzen mit N2O 93
Will man dieses Problem beseitigen, stellt sich die Frage, nach welchem Mechanismus die
Desaktivierung der Benzenhydroxylierung abläuft. Einige neue Untersuchungen deuten
darauf hin, dass die Desaktivierung nicht vom Edukt Benzen ausgeht, sondern vom
Phenol, das als so genannter „Koksprecursor“ fungiert [24, 277, 278, 297].
Für ein umfassendes Verständnis des Desaktivierungsmechanismus müssen deshalb vor
allem die Nebenreaktionen, ausgehend von Phenol, betrachtet werden. Bei der Entstehung
der Nebenprodukte sind zwei Reaktionspfade zu unterscheiden. Einerseits kann es zu
einer weiteren Hydroxylierung des Phenols kommen, andererseits können auch Polyphe-
nole und andere Kondensationsprodukte (mehrkernige Aromaten) durch eine Weiterreak-
tion des Phenols mit einem Phenol- oder Benzenmolekül entstehen (vgl. Abbildung 3.16).
Sowohl das Phenol selbst als auch die mehrfach hydroxylierten Spezies und die
Polyaromaten können zur Bildung von hard coke beitragen [277, 288].
Abbildung 3.16: Reaktionsnetzwerk der Benzenhydroxylierung [277, 288]
MELONI ET AL. vermuten, dass der soft coke aufgrund der Größe der Moleküle an
Kanalkreuzungspunkten, den Porenöffnungen und der äußeren Oberfläche der
Zeolithkristalle, gebildet wird. Moleküle sind teilweise dort gefangen, reagieren weiter zu
hard coke und blockieren ganze Bereiche der Zeolithstruktur für die Reaktanden. Nach
dem Starten der Reaktion wird demnach zunächst soft coke gebildet, dessen Anteil an der
gesamten Koksmenge jedoch mit Fortschreiten der Reaktion immer geringer wird, bis

94 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
schließlich nur noch hard coke vorhanden ist. Dies wurde durch Koksanalysen nach ver-
schiedenen Reaktionszeiten bestätigt [288].
PERATHONER ET AL. gehen weiterhin davon aus, dass nicht die mehrfach hydroxy-
lierten Nebenprodukte maßgeblich für die Desaktivierung verantwortlich sind, sondern
dass der dominante Desaktivierungsmechanismus über die Koppelprodukte des Phenols
verläuft (vgl. Abbildung 3.16) [277].
Die These, dass die Desaktivierung über mehrkernige Aromaten abläuft, deckt sich auch
mit den Erkenntnissen, dass die Standzeiten an Zeolithen mit größeren Poren/Hohlräumen
wie dem BEA-Zeolith oder Faujasit verglichen mit ZSM-5-Zeolithen deutlich geringer
sind, da die Bildung von größeren Molekülen dort leichter stattfinden kann [24, 225].
Untersuchungen von KHARITONOV ET AL. [301] zeigten, dass 5 - 15 % des in der
Benzenhydroxylierung umgesetzten Benzens zu Koks weiterreagiert und eine rasche
Desaktivierung des Katalysators bewirkt. Es konnte jedoch keine Korrelation zu der Akti-
vität des Katalysators in der Hydroxylierung gefunden werden. Stattdessen wurde eine
Korrelation zwischen der Anzahl an Lewis-Säure-Zentren (bestimmt über IR-
Spektroskopie) und der Desaktivierungsgeschwindigkeit festgestellt.
Neuere Ergebnisse von MELONI ET AL. [278, 288] zeigen ebenfalls einen Zusammen-
hang zwischen der Art und Stärke der Säure-Zentren des Katalysators und der
Deaktivierung bei der Benzenhydroxylierung. Bei diesen Untersuchungen zeigte es sich,
dass der beste getestete Katalysator zwar die geringste Anzahl starker Säure-Zentren
besaß, jedoch eine hohe Aktivität in der Hauptreaktion und die beste Langzeitstabilität.
Andere Katalysatoren mit einer größeren Anzahl starker Säure-Zentren zeigten zwar die
gleiche Anfangsaktivität in der Benzenhydroxylierung, erfuhren jedoch auch eine deutlich
schnellere Desaktivierung. Der Autor folgert aus seinen Untersuchungen, dass die
Säurezentren maßgeblich für die Desaktivierung verantwortlich sind, nicht jedoch für die
Aktivität in der Benzenhydroxylierung. Außerdem besteht ein direkter Zusammenhang
zwischen der Stärke der Säurezentren und der Geschwindigkeit der Koksbildung.
Als Ursache für die von Phenol ausgehenden Nebenreaktionen und der daraus resultieren-
den Verkokung des Katalysators wird die starke Wechselwirkung des Phenols mit den
Säure-Zentren des Katalysators und die langsame Rückdiffusion aus den Zeolithkanälen
angeführt (siehe Kapitel 3.4.1.5) [24, 277, 278]. Das Phenol ist im Vergleich zum Benzen
deutlich reaktiver, was darauf zurückgeführt werden kann, dass Phenol ein, verglichen mit
Benzen, niedriges Ionisationspotential besitzt. Die Folge ist, dass an Brønsted-Säure-Zent-
ren Carbenium-Ionen gebildet werden, welche zur Entstehung von höhermolekularen
Verbindungen und schließlich Kohlenstoffablagerungen führen [297].
Aus der Sicht der Katalysatorentwicklung ist es somit nach dem derzeitigen Stand der
Erkenntnisse notwendig, Katalysatoren zu entwickeln, welche insgesamt nur eine geringe

3.4 Hydroxylierung von Benzen mit N2O 95
Anzahl an Brønsted- und Lewis-Säure-Zentren aufweisen, jedoch viele in der Hydroxylie-
rung aktive Eisenzentren.
Neben einer Modifikation der Katalysatoren gibt es jedoch auch reaktionstechnische
Maßnahmen, um das Standzeitverhalten in der Benzenhydroxylierung zu verbessern.
Die einfachste Methode, die Desaktivierung während der Benzenhydroxylierung zu ver-
langsamen und eine weitere Hydroxylierung des Phenols zu Nebenprodukten zu verhin-
dern, ist die überstöchiometrische Zugabe von Benzen im Verhältnis zu N2O. Diese Fahr-
weise mit Benzenüberschuss wird bereits im ersten Patent des Boreskov Instituts
angewendet [224]. Aber auch andere Autoren berichten von einer deutlichen
Verlangsamung der Desaktivierung, sofern Benzen im Überschuss vorliegt [24]. Als
Ursache für das verbesserte Standzeitverhalten wird die Verdrängung des Phenols von der
Katalysatoroberfläche angeführt. Freiwerdende Zentren werden sofort von einem Ben-
zenmolekül besetzt, Phenolmoleküle können somit nicht adsorbiert werden und eine
Weiterreaktion wird verhindert. Die Vorteile dieser überstöchiometrischen Fahrweise
liegen in einer verlangsamten Desaktivierung bei gleichzeitiger Erhöhung der Phenol-
selektivität bezüglich N2O, da Nebenreaktionen unterdrückt werden. Als nachteilig
erweist sich der geringe Benzenumsatzgrad. Bei einer technischen Realisierung dieses
Prozesses muss nicht umgesetztes Benzen aus dem Produktgemisch abgetrennt und dem
Eduktgemisch wieder zugeführt werden. Dies verursacht zusätzliche Kosten [24].
Eine weitere Methode, die Desaktivierung bei technischen Verfahren zu verlangsamen
und die Aktivität des Katalysators zu steigern, ist das so genannte „steaming“. Dabei ist
grundsätzlich eine Vorbehandlung des Katalysators mit Wasserdampf oder die Zugabe
während der Reaktion zu unterscheiden [304, 305].
JIA ET AL. [297] berichten von einer Aktivitätssteigerung eines getesteten Fe-ZSM-5 um
das Dreifache, wird der Katalysator mit Wasserdampf bei einer Temperatur von 650 °C
für 2 h vorbehandelt. Die Phenolausbeute des unbehandelten Katalysators lag bei einer
Reaktionstemperatur von 400 °C bei ca. 20 %, während sich durch das steaming Ausbeu-
ten über 60 % realisieren ließen. Zudem konnte trotz der deutlich höheren Phenolausbeu-
ten über einen Zeitraum von 3 h nur eine geringe Desaktivierung beobachtet werden. Der
Autor führt die Aktivitätssteigerung auf einen Ausbau von Aluminium und Eisen aus dem
Gitter durch die hydrothermale Vorbehandlung zurück, so dass es zu einer Ausbildung
zusätzlicher aktiver Zentren auf Extragitterpositionen kommt.
Vergleichbare Ergebnisse wurden auch von und PILLAI ET AL. [306] veröffentlicht.
Durch eine Wasserzugabe zum Feed konnten die Benzenumsatzgrade und Phenolausbeu-
ten deutlich gesteigert werden. Die Desaktivierung ging signifikant zurück. Eine Dealu-
minierung war unter den Versuchsbedingungen unwahrscheinlich. Deshalb führen die

96 3 Hydroxylierung von Benzen mit N2O an ZSM-5 Katalysatoren
Autoren diese Effekte auf eine Verdrängung des Phenols von den aktiven Zentren des
Katalysators zurück.
Auch andere Autoren berichten von einer deutlichen Aktivitätssteigerung der Fe-ZSM-5
Katalysatoren durch solch eine hydrothermale Vorbehandlung bei hoher Temperatur
(> 500 °C) [277, 288, 296, 306].
MELONI ET AL. [288] untersuchten neben einer Vorbehandlung der Katalysatoren auch
die Zugabe von Wasserdampf während der Reaktion. Durch eine Vorbehandlung eines
Katalysators mit Wasserdampf (30 - 50 % Wasserdampf in Luft) bei einer Temperatur
von 600 °C für 5 h konnte eine deutliche Aktivitätssteigerung bei gleichzeitiger Verringe-
rung der Desaktivierung beobachtet werden. Im Vergleich zum „trocken“ bei 800 °C kal-
zinierten Katalysator ließ sich der anfängliche Benzenumsatzgrad von ca. 5 % auf beinahe
20 % steigern. An einem weiteren Katalysator wurde die Zugabe von Wasserdampf wäh-
rend der Reaktion untersucht. Zunächst wurde der Katalysator unter Zugabe von trocke-
nem feed getestet. Anschließend wurde der Versuch unter zusätzlicher Wasserzugabe, bei
Beibehaltung der Partialdrücke von Benzen und Phenol wiederholt. Dabei zeigte es sich,
dass sowohl die Aktivität als auch die Selektivität zu Phenol gesteigert werden konnten.
Die Desaktivierung ließ sich durch die Wasserzugabe zum Feed verringern. Dabei fiel die
Aktivitätssteigerung durch die Wasserzugabe zum Feed jedoch geringer aus wie nach der
oben beschriebenen hydrothermalen Vorbehandlung. Der Benzenumsatzgrad zu Beginn
der Reaktion ließ sich lediglich von ca. 5 auf 6 % steigern. Deutlicher war der Unterschied
beim Desaktivierungsverhalten. Während die Aktivität des Katalysators ohne Wasserzu-
gabe innerhalb von 6 h um 35 % zurückging, war bei zusätzlicher Wasserzugabe nur ein
Rückgang um 10 % zu beobachten. Bei der Untersuchung der Katalysatoren nach 6 h
Reaktion zeigte es sich, dass am Wasserdampf beaufschlagten Katalysator deutlich weni-
ger Koksablagerungen vorhanden waren als am Katalysator aus der Reaktion ohne Wasser
im feed. Die Autoren führen dies auf eine „Reinigung“ der Katalysatoroberfläche durch
das Wasser während der Reaktion zurück.
Auch bei den Untersuchungen von MOTZ Et AL. [286] konnte eine deutliche
Aktivitätssteigerung in der Benzenhydroxylierung durch eine hydrothermale Behandlung
der Katalysatoren festgestellt werden.
Von HIEMER [26] wurde ebenfalls eine Wasserzugabe zum Feed während der Benzen-
hydroxylierung untersucht. Dabei zeigte es sich, dass zwar keine Aktivitätssteigerung des
Katalysators beobachtet werden konnte, die Selektivität zum Zielprodukt Phenol jedoch
deutlich abnahm. Der Autor führt dies auf unerwünschte Folgereaktionen des Phenols mit
dem Wasser zurück.
Aus diesen Untersuchungen lässt sich der Schluss ziehen, dass durch die Wasserzugabe
zwei verschiedene Effekte resultieren. Zum einen wird durch die Behandlung der Kataly-

3.4 Hydroxylierung von Benzen mit N2O 97
satoren mit Wasser bei hoher Temperatur Aluminium und Eisen aus dem Gitter ausge-
baut, was zu einer dauerhaften Verbesserung der Katalysatoraktivität führt. Zum anderen
kann durch eine Wasserzugabe während der Reaktion die Aktivität gesteigert und die
Desaktivierung deutlich herabgesetzt werden, da vergleichbar mit der Fahrweise mit Ben-
zenüberschuss, das Phenol von den für die Desaktivierung verantwortlichen Zentren des
Katalysators verdrängt wird. Allerdings kann die Wasserzugabe während der Reaktion
auch zu unerwünschten Folge/Nebenreaktionen führen, welche die Selektivität zum Ziel-
produkt Phenol erniedrigen [26].

4 Zielsetzung der Arbeit
Zielsetzung der Arbeit war es, die Wirkung von Mikrowellenstrahlung auf die Hydroxy-
lierung von Benzen mit N2O an Zeolithkatalysatoren des Typs ZSM-5 zu untersuchen.
Diese heterogen katalysierte Reaktion kann als typisches Modell einer Reaktion angese-
hen werden, bei der polare und unpolare Spezies beteiligt sind, welche sich unterschied-
lich stark durch Mikrowellen erwärmen lassen.
Dabei galt es folgende wissenschaftliche Fragestellungen zu klären:
1. Wie wirken sich bestimmte Katalysatoreigenschaften und Reaktionsparameter
auf die Hydroxylierung von Benzen mit N2O aus und über welche Mechanis-
men läuft die Desaktivierung ab?
2. Welchen Einfluss hat Mikrowellenstrahlung auf die Hydroxylierung von
Benzen mit N2O an ausgewählten ZSM-5 Katalysatoren, insbesondre auf deren
Desaktivierungsverhalten und ist eine prozessseitige Optimierung der Reaktion
auf diesem Weg möglich?
3. Kann eine selektive Aufheizung des Phenols beziehungsweise eine selektive
Desorption von der Katalysatoroberfläche durch Mikrowellenstrahlung erreicht
werden?
Um eine Bewertung der Ergebnisse der reaktionstechnischen Untersuchungen zu ermögli-
chen, war es nötig eine Reihe von Voruntersuchungen durchzuführen. Dabei waren
sowohl die katalytisch relevanten Parameter aber auch die dielektrischen Eigenschaften
von besonderem Interesse. Diese Arbeiten umfassten eine
• klassische Charakterisierung der verwendeten Katalysatoren hinsichtlich ihrer
physikalisch-chemischen Eigenschaften,
• Charakterisierung der eingesetzten Katalysatoren hinsichtlich ihres Aufheizverhal-
tens im Mikrowellenfeld, und
• Experimente zur Aufheizung von am Katalysator adsorbiertem Phenol durch
Mikrowellenstrahlung.

4 Zielsetzung der Arbeit 99
Da kommerziell keine Anlagen verfügbar waren, welche für die Durchführung der reakti-
onstechnischen Untersuchungen geeignet erschienen, musste zunächst eine Reihe
technischer Themen bearbeitet werden:
• Konzeption und Aufbau einer teilautomatisierten Laboranlage
• Entwicklung eines Reaktorkonzepts, welches zuverlässige Vergleichsmessungen
zwischen konventionell und mit zusätlicher Mikrowellenstrahlung geheizten
Experimenten ermöglicht. Voraussetzungen hierfür sind wiederum eine exakte
Temperaturmessung und die Vermeidung von Temperaturgradienten im Reaktor,
welche durch die Mikrowellenheizung hervorgerufen werden können.
Nach Abschluss dieser Arbeiten konnte mit den der reaktionstechnischen Untersuchungen
begonnen werden, welche
• Messungen zum Einfluss der Mikrowellenstrahlung auf die N2O-Zersetzung,
• Vergleiche von konventionell und mit zusätzlicher Mikrowellenstrahlung geheiz-
ten Experimenten in der Hydroxylierung von Benzen mit N2O, und
• Variationen der Reaktionsparameter der Hydroxylierungsreaktion wie:
Temperatur, Verweilzeit, Feedzusammensetzung und eingestrahlte Mikrowellen-
leistung
umfassten.
Weiterhin wurden die während der Reaktion am Katalysator vorliegende Spezies Benzen,
Phenol und Koks hinsichtlich ihres Desorptionsverhaltens vom Katalysator untersucht. Im
Einzelnen wurden nachstehende Analysen durchgeführt:
• Ermitteln der Temperaturabhängigkeit der Desorption von Benzen und Phenol
vom Katalysator und der Koksbeladung nach dem Einsatz der Katalysatoren in der
Hydroxylierungsreaktion
• Experimente zur Desorption von Phenol, Benzen und deren Mischungen vom
Katalysator durch Mikrowellenstrahlung

5 Eingesetzte Katalysatoren
5.1 Auswahlkriterien
Aus der Literatur ist bekannt, dass sich verschiedene Katalysatortypen für die Hydroxylie-
rung von Benzen mit N2O als Oxidationsmittel eignen. Die besten Ergebnisse wurden mit
Zeolithen des Typs ZSM-5 erzielt (vgl. Kapitel 3.4). Dieser Katalysatortyp lässt sich
zudem im dehydratisierten Zustand nur geringfügig durch Mikrowellen erwärmen (vgl.
Kapitel 3.4.1.6) und erfüllt somit die Anforderungen, welche für eine selektive Desorption
adsorbierter Spezies durch Mikrowellenstrahlung benötigt werden (vgl. Kapitel 2.3.3).
Im Rahmen dieser Arbeit wurden deshalb vier Katalysatoren des Typs ZSM-5 ausgewählt,
welche sich aufgrund ihrer Eigenschaften deutlich hinsichtlich ihres Verhaltens in der
Reaktion unterscheiden sollten.
1) Ein Katalysator des Typs H-ZSM-5.
Im Folgenden wird dieser Katalysator als H-ZSM-5 (I) bezeichnet.
2) Ein Katalysator vom Typ Fe-ZSM-5 mit einem geringen Fe-Gehalt und kleinen
Kristalliten. Der Vorteil dieses Katalysators liegt in den kurzen Diffusionswegen,
was sich beispielsweise in der Hydroxylierung von Toluol mit N2O zu Kresol als
vorteilhaft erwiesen hat [307]. Mit Hilfe von SEM-Aufnahmen wurden
Kristalldurchmesser dieses Katalysators < 50 nm ermittelt. Diese Messungen
wurden im Rahmen der Arbeit von HIEMER [26] durchgeführt. Der Katalysator
wurde 2 h bei einer Temperatur von 900 °C im Stickstoffstrom kalziniert.
Im Folgenden wird dieser Katalysator als Fe-ZSM-5 (I) bezeichnet.
3) Ein Katalysator vom Typ Fe-ZSM-5 mit einem hohen Fe-Gehalt, welcher sich
durch viel Extragittereisen auszeichnet. Da in der Literatur häufig ein
Mechanismus über Extragittereisen postuliert wird (vgl. Kapitel 3.4.2) sollte dieser
Katalysator Vorteile bei der Hydroxylierung bringen.
Im Folgenden wird dieser Katalysator als Fe-ZSM-5 (II) bezeichnet.
4) Ein Katalysator vom Typ Co-ZSM-5. Dieser Katalysator wurde ausgewählt, da
Co-Zeolithe für ihre hohe Aktivität in der N2O-Zersetzung bekannt sind. Wie in
Kapitel 3.4.2 beschrieben, stellt die N2O-Zersetzung einen entscheidenden Schritt
bei der Hydroxylierung von Benzen mit N2O dar. Der Katalysator wurde bei einer

5.2 Klassische Katalysatorcharakterisierung 101
Temperatur von 900 °C für 2 h im Stickstoffstrom kalziniert.
Im Folgenden wird dieser Katalysator als Co-ZSM-5 bezeichnet.
Neben diesen Katalysatoren, welche in der Hydroxylierung von Benzen mit N2O getestet
wurden, kamen jedoch noch weitere Zeolithe bei der Untersuchung der dielektrischen
Eigenschaften zum Einsatz. Unter anderem wurde ein Na-ZSM-5 als Referenzkatalysator
für die Aufheizexperimente verwendet. In der Literatur wird berichtet, dass sich dieser
Katalysatortyp gut durch Mikrowelleneinstrahlung erwärmen lässt (vgl. Kapitel 3.4.1.6).
Darüber hinaus wurden noch weitere Katalysatoren des Typs H-ZSM-5 hinsichtlich ihrer
Aufheizung durch Mikrowellen untersucht (H-ZSM-5 (II-VII)). Auf eine klassische
Katalysatorcharakterisierung (vgl. Kapitel 5.2) wurde bei diesen Katalysatoren jedoch
weitestgehend verzichtet.
5.2 Klassische Katalysatorcharakterisierung
Üblicherweise werden verschiedene ex situ Charakterisierungsmethoden angewendet, um
die katalytisch relevanten Eigenschaften eines Katalysators zu bestimmen. Da der
Schwerpunkt dieser Arbeit jedoch nicht im Bereich der Katalysatorentwicklung
beziehungsweise Synthese lag, werden die Ergebnisse dieser Charakterisierungsmethoden
nur kurz vorgestellt. Für detailliertere Informationen über die verschiedenen Methoden
und deren Hintergründe soll hier nur auf die Spezialliteratur verwiesen werden [260, 308-
310]. Stattdessen wird besonderes Augenmerk auf die dielektrischen Eigenschaften und
damit das Aufheizverhalten der Katalysatoren im Mikrowellenfeld gelegt.
5.2.1 Elementaranalyse
Die chemische Zusammensetzung der Feststoffkatalysatoren wurde mit Hilfe der Induk-
tiv-gekoppelten-Plasma-Emmissionsspektroskopie (ICP) ermittelt. Der Aufschluss der
Proben erfolgte in einer Labormikrowelle des Typs CEM MDS-2000 im geschlossenen
Druckbehälter (40 min bei 520 W; Aufschlussgemisch: HF:HCl:HNO3 = 4:1:1) [311,
312]. Dabei ist vor allem die Bestimmung des Modul M (vergleiche S. 76) sowie des
Gehalts an Natrium bzw. katalytisch aktiven Ionen wie Eisen oder Cobalt von Bedeutung.
Die Messungen wurden mit einem Gerät der Firma Perkin Elmer vom Typ Plasma 400
durchgeführt.
5.2.2 Strukturanalyse
Neben der Zusammensetzung ist jedoch auch die Struktur des Katalysators von besonde-
rem Interesse. Zeolithe können Fremdphasen enthalten, welche die Qualität des Kataly-
sators beeinträchtigen. Die Strukturaufklärung erfolgte mit Hilfe der Röntgenpulver-
diffraktometrie (XRD). Bei dieser Methode werden Röntgenbeugungsmuster der Kataly-
satoren aufgenommen und mit Werten aus der Literatur oder spezieller Datenbanken [234,

5 Eingesetzte Katalysatoren 102
313] verglichen, um die Phasenreinheit zu bestimmen. Will man die Kristallinitäten
quantitativ erfassen, wird die Peakintensität oder die Peakfläche eines oder mehrerer
Peaks des Beugungsspektrums mit der eines kristallinen Standards verglichen. Von
SCHWIEGER [255] wurde eine Methode entwickelt, welche mit α-Al2O3 als externem
Standard arbeitet. Dabei wird der so genannte QAl-Wert als Richtwert für die Kristallinität
einer Probe herangezogen. Es gilt:
2 3
( -5)
( - )
ZSM
Al
Al O
IQ
I α
= mit: 2 3
( -5) (2 23,1)
( - ) 2 35,2 2 43,30,5 ( )ZSM
Al O
I I
I I Iα
Θ=
Θ= Θ=
=
= ⋅ + (Gl. 5.1)
QAl-Werte um 1 entsprechen einer Kristallinität von 100 %. Mit dieser Methode lassen
sich somit verschiedene Katalysatoren anhand eines einzigen Wertes hinsichtlich ihrer
Qualität vergleichen. Die Messungen erfolgten mit einem Gerät der Firma Phillips Typ
X’pert pro. Ein Grund für niedrige QAl-Werte kann jedoch auch eine geringe Kristal-
litgröße der Zeolithe sein. Kleine Kristallite führen zu einer Verringerung des QAl-Wertes,
was jedoch nicht als Strukturverlust zu werten ist [24].
5.2.3 Adsorptionseigenschaften
Zur Bestimmung der Oberfläche von mikro- oder mesoporösen Festkörpern führt man
häufig Physisorptionsmessungen mit N2 bei 77,3 K durch. Als charakteristische Kennzahl
werden in der Regel die BET-Oberflächen angegeben. Dies macht bei Zeolithen wenig
Sinn, da die BET-Theorie von einer Mehrschichtbelegung der Oberfläche mit den
N2-Molekülen ausgeht. Die Porendurchmesser der Zeolithe bewegen sich jedoch in der
Größenordnung des Durchmessers der N2-Moleküle, so dass es nicht zu einer Belegung
der Oberfläche, sondern zu einer Füllung der Poren kommt. Deshalb wird bei Zeolithen
häufig das Mikroporenvolumen als charakteristischer Texturparameter angegeben. Die
N2-Sorptionsisotherme von ZSM-5 Zeolithen entspricht im Idealfall einer Typ I-Isotherme
(IUPAC). Ist dies der Fall, lässt sich das Mikroporenvolumen grafisch durch den Schnitt-
punkt einer Tangente an das Plateau der Sorptionsisotherme mit der y-Achse des Isother-
men-Plots bestimmen [309]. Eine weitere Methode zur Ermittlung des Mikroporenvolu-
mens ist die Auswertung des so genannten t-Plots. Dabei wird das adsorbierte Stickstoff-
volumen nicht wie beim Isothermen-Plot über den Relativdruck P/P0 aufgetragen, sondern
über eine aus P/P0 berechnete statistische Filmdicke. Die Berechnung dieser Filmdicke
kann über verschiedene Gleichungen erfolgen.
In dieser Arbeit wurden die Mikroporenvolumina durch die Auswertung des t-Plots
ermittelt. Dabei kam die Beziehung nach Harkins & Jura zum Einsatz. Die Ermittlung der
Mikroporenvolumina erfolgte durch Anlegen einer Tangente an den t-Plot im Bereich
zwischen 0,5 und 0,7 nm. Für detailliertere Informationen zu dieser Auswertemethode sei
auf die Spezialliteratur verwiesen [310]. Die Bestimmung der Sorptionsisothermen

5.2 Klassische Katalysatorcharakterisierung 103
erfolgte mit einem Gerät der Firma Micromeritics vom Typ ASAP 2000. Vor Beginn der
Messung wurden die Proben 2 Stunden bei einer Temperatur von 200 °C im Vakuum
ausgeheizt.
5.2.4 Säureeigenschaften
Die Säureeigenschaften der verwendeten Zeolithe wurden durch die Temperaturprogram-
mierte-Desorption (TPD) von Ammoniak ermittelt. Bei dieser Methode wird an einer
Katalysatorprobe bei einer Temperatur von 80 – 100 °C Ammoniak adsorbiert. Die beim
Aufheizen der Probe desorbierte Ammoniakmenge wird als Funktion der Temperatur
aufgezeichnet. Man erhält einen charakteristischen Kurvenverlauf mit zwei Peaks. Das
erste Desorptionsmaximum bei niedriger Temperatur kann nach KAPUSTIN [314] und
BAGNASCO [315] vor allem den an Lewis-Zentren gebundenen Ammoniak, der
Hochtemperaturpeak hingegen den an Brønsted-Zentren adsorbierten Ammoniak
zugeordnet werden. Aus der Peaklage kann die Stärke der Säurezentren abgeleitet werden.
Je höher die Temperatur der Peakmaxima, desto stärker sind die Säurezentren. Unter der
Annahme, dass die Ammoniakmoleküle stöchiometrisch an den Brønsted-Zentren
adsorbiert vorliegen, lässt sich durch geeignete Dekonvolutierung des TPD-Plots und
anschließende Integration der Peaks die Anzahl der Zentren bzw. die Menge der
dreiwertigen Gitteratome bestimmen.
Eine detaillierte Beschreibung der eingesetzten Versapparatur und Vorgehensweise bei
der Auswertung der Messdaten findet sich bei SEITZ und REITZMANN [24, 316].

5 Eingesetzte Katalysatoren 104
5.2.5 Ergebnisse
Tabelle 5.1 gibt einen Überblick der Charakterisierungsdaten, welche für die in der
Hydroxylierung von Benzen mit N2O eingesetzten Katalysatoren ermittelt wurden.
Tabelle 5.1: Charakterisierungsdaten der in der Hydroxylierung von Benzen mit N2O
eingesetzten ZSM-5-Zeolithe
Hersteller SCM SCM SCM A. Unger [23]
Messmethode Parameter H-ZSM-5 (I) Fe-ZSM-5 (I) Fe-ZSM-5 (II) Co-ZSM-5
Modul M [-] 58 79 92 86
Na [m%] 0 0 0,06 0,01
Fe [m%] 0,04 0,19 1,16 0,05
Co [m%] n.b. n.b. n.b. 0,89
XRD Q Al [-]* 0,80 0,56 0,62 0,59
N2-Sorption V MP [cm³/g] 0,138 0,119 0,132 0,112
Fläche 1 [-] 920 234 559 620
Fläche 2 [-] 1471 839 1144 866
T 1 [°C] 203 190 197 168
T 2 [°C] 357 351 386 418
ICP
Katalysator
NH
3-T
PD
n.b.: Parameter nicht bestimmt * Auswertung der Peakflächen SCM: Süd-Chemie AG
Die ICP-Untersuchungen zeigen, dass sich die Module der eingesetzten Zeolithe im
Bereich zwischen 58 (H-ZSM-5 (I) und 92 (Fe-ZSM-5 (II)) bewegen. Die Katalysatoren
Fe-ZSM-5 (II) und Co-ZSM-5 weisen geringe Gehalte an Natrium auf, welche auf eine
nicht vollständige Überführung der Natriumform in die H-Form, bzw. Verunreinigungen
zurückzuführen sind.
Eisen konnte bei allen getesteten Zeolithen detektiert werden, d.h. auch beim H-ZSM-5 (I)
und dem Co-ZSM-5. Diese Eisenanteile sind auf Verunreinigungen der bei der
Zeolithsynthese eingesetzten Ausgangsstoffe zurückzuführen. Die beiden Katalysatoren
Fe-ZSM-5 (I) und Fe-ZSM-5 (II) weisen deutlich höhere Eisengehalte auf, wobei der
Fe-ZSM-5 (II) nochmals die sechsfache Menge an Eisen enthält wie der Fe-ZSM-5 (I).
Der Fe-ZSM-5 (II) zeigt aufgrund des hohen EisenIII-Anteils eine deutliche Braunfärbung,
während der Fe-ZSM-5 (I) weiß ist.
Die Abbildung 5.1 zeigt die Röntgenbeugungsmuster der eingesetzten Zeolithe. Beim
Vergleich mit der Literatur [234] zeigt sich, dass alle Katalysatoren die ZSM-5-typischen
Reflexe und keine erkennbaren Fremdphasen aufweisen.

5.2 Klassische Katalysatorcharakterisierung 105
Abbildung 5.1: Röntgenbeugungsmuster der in der Hydroxylierung von Benzen mit N2O
eingesetzten Katalysatoren
Bei der Betrachtung der berechneten QAl-Werte (siehe Tabelle 5.1) zeigt sich jedoch, dass
sich Werte zwischen 0,56 und 0,8 ergeben. Für die Berechnung der QAl-Werte wurden die
Peakflächen, und nicht die Intensitäten der Peaks ausgewertet. Lediglich der H-ZSM-5 (I)
weist eine hohe Kristallinität (QAl = 0,8) auf. Alle anderen Katalysatoren besitzen QAl-
Werte um 0,6. Dieser Verlust an Kristallinität kann darauf zurückzuführen sein, dass die
vermessenen Katalysatorproben bereits häufig in der Hydroxylierung von Benzen mit
N2O eingesetzt wurden. Es handelt sich nicht um frisch synthetisierte Zeolithe. Der
Fe-ZSM-5 (I) und Co-ZSM-5 wurden zudem bei einer Temperatur von 900 °C kalziniert,
was ebenfalls zu einer Beeinträchtigung der Zeolithstruktur führen kann.
Zur Ermittlung der Mikroporenvolumina der Zeolithe wurden N2-Sorptionsisothermen
aufgezeichnet. Die Ergebnisse sind in Abbildung 5.2 dargestellt. Auffällig ist zunächst,
dass alle Sorptionsisothermen einen Knick im Bereich des Relativdrucks p/p0 ≈ 0,2
aufweisen. Die Form entspricht nicht einer reinen TypI-Isotherme. Eine Ursache für
dieses Verhalten kann eine sekundäres Porensystem sein, welches nach der Synthese oder
dem Kalziniervorgang entsteht, wobei allerdings auch der Aluminiumgehalt eine Rolle
spielt [317]. Dieses Verhalten wird bei REITZMANN [24] eingehend diskutiert, weshalb
in dieser Arbeit auf weitere Ausführungen verzichtet wird. Es zeigt sich bei den
untersuchten Proben kein ausgeprägten Sättigungswert. Die Isothermen ähneln somit ehr

5 Eingesetzte Katalysatoren 106
einer Typ IV-Isothermen mikro-meso-poröser Materialien. Da es aber auch bei Typ IV-
Isothermen einen klar abgegrenzten linearen Bereich gibt, ist eine Ermittlung der BET-
Oberfläche und Porenvolumina auch in diesem Fall möglich [318].
Die N2-Sorptionskapazitäten der Zeolithe Fe-ZSM-5 (I), Fe-ZSM-5 (II) und Co-ZSM-5
sind nahezu identisch, der H-ZSM-5 (I) weist dagegen eine höhere N2-Sorptionskapazität
auf.
Abbildung 5.2: N2-Sorptionsisothermen der in der Hydroxylierung von Benzen mit N2O
eingesetzten Katalysatoren
Wie bereits in Kapitel 5.2.3 erläutert erfolgte die Bestimmung der Mikroporenvolumina
anhand der t-Plots nach Harkins & Jura. Diese sind in Appendix A dargestellt. Die
ermittelten Mikroporenvolumina finden sich in Tabelle 5.1.
Es ergibt sich somit folgende Einteilung der eingesetzten Zeolithe nach der Höhe des
Mikroporenvolumens:
H-ZSM-5 (I) > Fe-ZSM-5 (II) > Fe-ZSM-5 (I) > Co-ZSM-5
Die Säureeigenschaften der Zeolithe wurden mit Hilfe der Temperaturprogrammierten
Desorption von Ammoniak bestimmt (vgl. Kapitel 5.2.4). Die ermittelten NH3-TPD-
Profile sind in Abbildung 5.3 dargestellt. Auffällig ist zunächst, dass sich bei keiner der
untersuchten Proben ein klar abgegrenzter Hochtemperaturpeak ausmachen lässt. Der
Verlauf zeigt eine Schulter bei hohen Temperaturen. Beim Fe-ZSM-5 (I) ist zudem auch
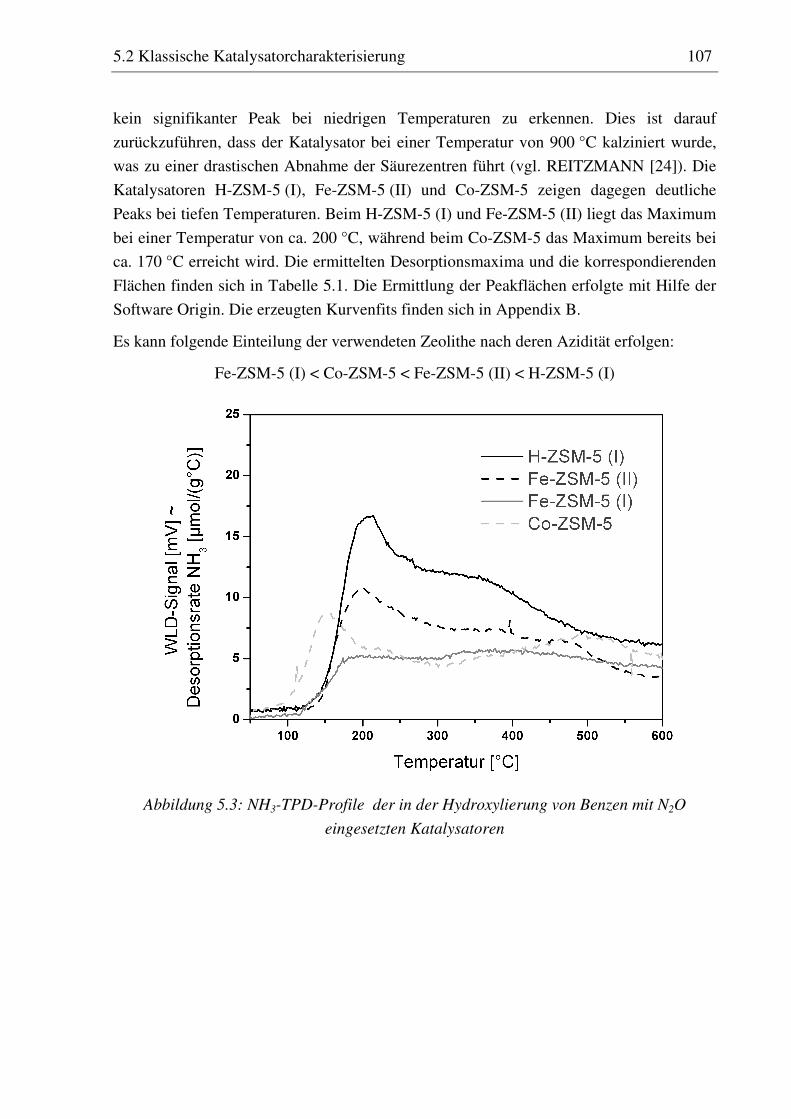
5.2 Klassische Katalysatorcharakterisierung 107
kein signifikanter Peak bei niedrigen Temperaturen zu erkennen. Dies ist darauf
zurückzuführen, dass der Katalysator bei einer Temperatur von 900 °C kalziniert wurde,
was zu einer drastischen Abnahme der Säurezentren führt (vgl. REITZMANN [24]). Die
Katalysatoren H-ZSM-5 (I), Fe-ZSM-5 (II) und Co-ZSM-5 zeigen dagegen deutliche
Peaks bei tiefen Temperaturen. Beim H-ZSM-5 (I) und Fe-ZSM-5 (II) liegt das Maximum
bei einer Temperatur von ca. 200 °C, während beim Co-ZSM-5 das Maximum bereits bei
ca. 170 °C erreicht wird. Die ermittelten Desorptionsmaxima und die korrespondierenden
Flächen finden sich in Tabelle 5.1. Die Ermittlung der Peakflächen erfolgte mit Hilfe der
Software Origin. Die erzeugten Kurvenfits finden sich in Appendix B.
Es kann folgende Einteilung der verwendeten Zeolithe nach deren Azidität erfolgen:
Fe-ZSM-5 (I) < Co-ZSM-5 < Fe-ZSM-5 (II) < H-ZSM-5 (I)
Abbildung 5.3: NH3-TPD-Profile der in der Hydroxylierung von Benzen mit N2O
eingesetzten Katalysatoren

5 Eingesetzte Katalysatoren 108
5.3 Charakterisierung der dielektrischen Eigenschaften
Für die Untersuchung des Mikrowelleneinflusses auf heterogen katalysierte Reaktionen
sind die dielektrischen Eigenschaften eines Katalysators von besonderer Bedeutung. Wie
die Ergebnisse von REUß ET AL. [20] (vgl. Kapitel 2.3.3.2) zeigen, ist ein für Mikro-
wellenstrahlung transparenter Katalysator die Grundvoraussetzung für eine selektive
Desorption adsorbierter Moleküle durch Mikrowellenstrahlung. Über die dielektrischen
Eigenschaften der Zeolithe ist in der Literatur jedoch wenig bekannt. Zwar gibt es
Hinweise, dass sich H-ZSM-5 Zeolithe nur sehr schlecht durch Mikrowellen erwärmen
lassen und dass Metallkationen (Natrium) dabei eine entscheidende Rolle spielen.
Gesicherte Aussagen, wie sich ein Zeolith im Mikrowellenfeld verhalten wird, sind aus
der Literatur jedoch nicht direkt ableitbar (vgl. Kapitel 3.4.1.6).
Deshalb erfolgte zunächst die Charakterisierung einer Auswahl von Zeolithen des Typs
ZSM-5 hinsichtlich ihres Aufheizverhaltens im Mikrowellenfeld.
Die chemische Zusammensetzung der getesteten Katalysatoren findet sich in Tabelle 5.2.
Tabelle 5.2: Chemische Zusammensetzung der hinsichtlich ihres Aufheizverhaltens im
Mikrowellenfeld untersuchten Zeolithe (ICP)
Zeolith Na-ZSM-5 H-ZSM-5 (I) H-ZSM-5 (II) H-ZSM-5 (III) H-ZSM-5 (IV)Modul M [-] 40 58 86 45 98
Na [m%] 1,7* 0,00 0,01 0,00 0,00
Zeolith H-ZSM-5 (V) H-ZSM-5 (VI) H-ZSM-5 (VII) Fe-ZSM-5 (I) Fe-ZSM-5 (II) Co-ZSM-5Modul M [-] 101 162 217 79 92 86
Na [m%] 0,00 0,00 0,00 0,00 0,06 0,01
*: errechneter Wert (Berechnung siehe Appendix C)
5.3.1 Versuchsdurchführung
Für die Aufheizversuche wurden die pulverförmigen Katalysatoren zunächst für
2 Stunden bei 400 °C im Stickstoffstrom dehydratisiert. Anschließend wurde 1 g pulver-
förmiger Katalysator in 7 g getrocknetes Hexan (Fluka 99,5 %) dispergiert und auf ca.
25 °C temperiert. Die Bestrahlung dieser Mischung aus Hexan und Zeolith erfolgte für
240 s mit einer Leistung von 120 W in einer Labormikrowelle (CEM MDS-2000). Eine
längere Aufheizdauer wurde nicht untersucht, da bei einer zu großen Temperaturerhöhung
das Lösungsmittel Hexan verdampfen würde und dadurch die Messergebnisse verfälscht
werden. Für die Temperaturmessung kam der in der Labormikrowelle integrierte
faseroptische Sensor zum Einsatz, dessen Messgenauigkeit mit 0,01 °C angegeben ist.
Die Aufheizversuche wurden, um Messfehler zu minimieren, für jeden Katalysator
dreimal wiederholt und der Mittelwert gebildet.
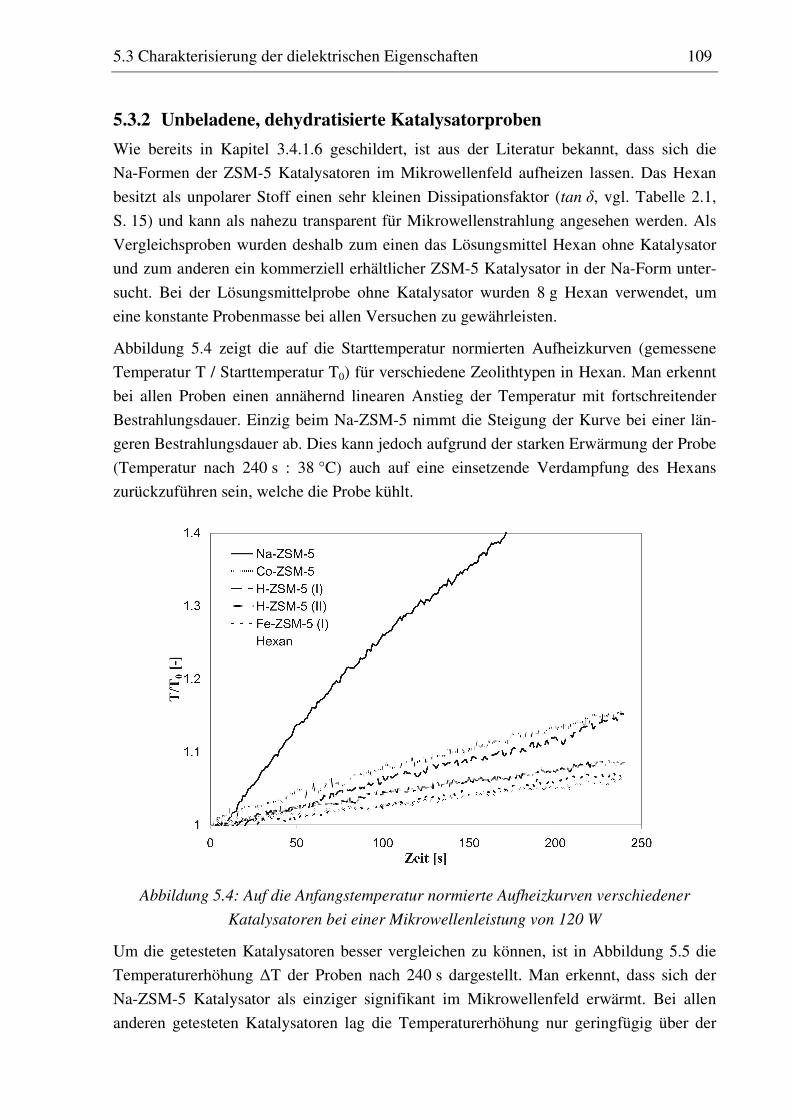
5.3 Charakterisierung der dielektrischen Eigenschaften 109
5.3.2 Unbeladene, dehydratisierte Katalysatorproben
Wie bereits in Kapitel 3.4.1.6 geschildert, ist aus der Literatur bekannt, dass sich die
Na-Formen der ZSM-5 Katalysatoren im Mikrowellenfeld aufheizen lassen. Das Hexan
besitzt als unpolarer Stoff einen sehr kleinen Dissipationsfaktor (tan δ, vgl. Tabelle 2.1,
S. 15) und kann als nahezu transparent für Mikrowellenstrahlung angesehen werden. Als
Vergleichsproben wurden deshalb zum einen das Lösungsmittel Hexan ohne Katalysator
und zum anderen ein kommerziell erhältlicher ZSM-5 Katalysator in der Na-Form unter-
sucht. Bei der Lösungsmittelprobe ohne Katalysator wurden 8 g Hexan verwendet, um
eine konstante Probenmasse bei allen Versuchen zu gewährleisten.
Abbildung 5.4 zeigt die auf die Starttemperatur normierten Aufheizkurven (gemessene
Temperatur T / Starttemperatur T0) für verschiedene Zeolithtypen in Hexan. Man erkennt
bei allen Proben einen annähernd linearen Anstieg der Temperatur mit fortschreitender
Bestrahlungsdauer. Einzig beim Na-ZSM-5 nimmt die Steigung der Kurve bei einer län-
geren Bestrahlungsdauer ab. Dies kann jedoch aufgrund der starken Erwärmung der Probe
(Temperatur nach 240 s : 38 °C) auch auf eine einsetzende Verdampfung des Hexans
zurückzuführen sein, welche die Probe kühlt.
Abbildung 5.4: Auf die Anfangstemperatur normierte Aufheizkurven verschiedener
Katalysatoren bei einer Mikrowellenleistung von 120 W
Um die getesteten Katalysatoren besser vergleichen zu können, ist in Abbildung 5.5 die
Temperaturerhöhung ∆T der Proben nach 240 s dargestellt. Man erkennt, dass sich der
Na-ZSM-5 Katalysator als einziger signifikant im Mikrowellenfeld erwärmt. Bei allen
anderen getesteten Katalysatoren lag die Temperaturerhöhung nur geringfügig über der

5 Eingesetzte Katalysatoren 110
des reinen Lösungsmittels Hexan. Sie können damit als annähernd transparent für Mikro-
wellenstrahlung angesehen werden. Auch die getesteten eisenhaltigen Zeolithe zeigen
keine stärkere Erwärmung als die H-Formen.
0
2
4
6
8
10
12
14
Na-
ZSM-5
Co-
ZSM
-5
H-Z
SM-5
(I)
H-Z
SM-5
(II)
Fe-ZSM
-5 (I
)
Fe-ZSM
-5 (I
I)
Hex
an
∆∆ ∆∆T
[°C
]
Abbildung 5.5: Temperaturerhöhung der Zeolithpulver nach 240 s
Mikrowelleneinstrahlung (120 W)
Auffällig ist jedoch, dass sich bei den Katalysatoren Co-ZSM-5 und H-ZSM-5 (I) ein grö-
ßerer Temperaturanstieg zeigte als bei den restlichen Proben (ausgenommen Na-ZSM-5).
Dieses Verhalten lässt sich beim Co-Katalysator eventuell auf die vorhandenen Metall-
kationen zurückführen, welche durch die Mikrowellenstrahlung angeregt werden.
Die Unterschiede im Aufheizverhalten der getesteten Zeolithe können anhand dieser
Untersuchungen nicht eindeutig erklärt werden. Deshalb wurde zusätzlich der Einfluss des
Moduls auf das Aufheizverhalten im Mikrowellenfeld untersucht (siehe nächstes Kapitel).
5.3.2.1 Modulabhängigkeit
Um den Einfluss des Si/Al-Verhältnisses der Zeolithkatalysatoren auf die Aufheizung im
Mikrowellenfeld zu ermitteln, wurde eine Reihe von Katalysatoren der H-Form, welche
sich in ihrem Modul unterscheiden, untersucht.
Wie man in Abbildung 5.6 erkennen kann, ließ sich jedoch keine Abhängigkeit des
Aufheizverhaltens bzw. der dielektrischen Eigenschaften dieser Katalysatoren von Modul
feststellen. Alle Proben zeigten einen beinahe identischen Temperaturanstieg, welcher
geringer als beim H-ZSM-5 (I) war. Weiterhin unterscheiden sich alle getesteten Kataly-
satoren nur geringfügig in ihrem Na-Gehalt, so dass dieser Einflussfaktor ausgeschlossen
werden kann. Der Unterschied im Aufheizverhalten des H-ZSM-5 (I) muss deshalb nicht

5.3 Charakterisierung der dielektrischen Eigenschaften 111
auf der chemischen Zusammensetzung, sondern auf anderen Parametern wie beispiels-
weise der Korngrößenverteilung im Katalysatorpulver oder dessen Dichte beruhen.
0
1
2
3
4
5
H-Z
SM-5
(III)
H-Z
SM-5
(I)
H-Z
SM-5
(II)
H-Z
SM-5
(IV)
H-Z
SM-5
(V)
H-Z
SM-5
(VI)
H-Z
SM-5
(VII)
∆∆ ∆∆T
[°C
]
0
50
100
150
200
250
Mod
ul M
[-]
∆∆∆∆T [°C]
Modul M [-]
Abbildung 5.6: Temperaturerhöhung nach 240 s Mikrowelleneinstrahlung (120 W) in
Abhängigkeit vom Modul M des Katalysators
5.3.3 Mit Phenol beladene, dehydratisierte Katalysatorproben
Neben den unbeladenen Katalysatoren wurden auch mit Phenol beladene Proben hinsicht-
lich ihres Aufheizverhaltens im Mikrowellenfeld untersucht. Zu diesem Zweck wurden
zwei dehydratisierte Katalysatoren aus der Gasphase mit Phenol beladen. Die
Beschreibung der verwendeten Apparaturen und die Vorgehensweise zur Beladung der
Katalysatoren findet sich in Kapitel 6.4.
Abbildung 5.7 zeigt zunächst die normierte Aufheizung der Katalysatorproben mit fort-
schreitender Bestrahlungsdauer bei einer Mikrowellenleistung von 120 W. Verglichen
werden jeweils eine unbeladene und eine mit Phenol beladene Katalysatorprobe des
H-ZSM-5 (I) und Fe-ZSM-5 (I). Nach einer kurzen Einlaufphase zeigt sich auch bei
diesen Proben ein annähernd linearer Verlauf des Temperaturanstiegs mit fortschreitender
Bestrahlungsdauer. Betrachtet man zunächst die unbeladenen Katalysatorproben, fällt die
deutlich stärkere Erwärmung der H-ZSM-5 (I) im Vergleich zum Fe-ZSM-5 (I) auf. Die
mit Phenol beladenen Katalysatoren zeigen eine deutlich stärkere Erwärmung als die
unbeladenen Proben. Zudem heizt sich der Fe-ZSM-5 (I) im beladenen Zustand sogar
stärker auf als der H-ZSM-5 (I) mit Phenol.

5 Eingesetzte Katalysatoren 112
Abbildung 5.7: Auf die Starttemperatur normierte Aufheizkurven von dehydratisierten und
mit Phenol beladenen Katalysatoren bei einer Mikrowelleneinleistung von 120 W
Phenolbeladung: H-ZSM-5 (I) 7,3 m%; Fe-ZSM-5 (I) 9,7 m%
Für eine einfachere Diskussion der Ergebnisse soll im Folgenden wiederum die Tempe-
raturüberhöhung nach 240 s betrachtet werden (vgl. Abbildung 5.8).
Wie man in Abbildung 5.8 erkennen kann, ist der Temperaturanstieg bei den mit Phenol
beladenen Proben, wie bereits diskutiert, höher als bei den unbeladenen Katalysatoren.
Ursache hierfür ist, dass Phenol als polares Molekül einen hohen Dissipationsfaktor
(tan δ: 0,02) besitzt und sich durch die Mikrowellen gut erwärmen lässt. Das sich die
beladenen Proben nach 240 s nur ca. 1 °C stärker aufheizen als die unbeladenen, liegt zum
einen daran, dass der Anteil des Phenols an der gesamten Probenmasse gering ist (Bela-
dung des Katalysators weniger als 10 m%) und dass gegebenenfalls ein Teil der einge-
brachten Energie nicht für die Erwärmung der Probe, sondern für die Desorption des
Phenols von der Katalysatoroberfläche verbraucht wird. Offensichtlich ist jedoch, dass
sich der Energieeintrag in die Probe durch das an der Oberfläche des Katalysators adsor-
bierte Phenol erhöht und zu einer verstärkten Aufheizung der Probe führt.
Es fällt auf, dass sich der Phenol beladene Fe-ZSM-5 (I) verhältnismäßig stärker erwärmt
als die beladene Probe des H-ZSM-5 (I). Die Ursache hierfür ist die mit 9,7 m% deutlich
höhere Phenolbeladung der Fe-Katalysators im Vergleich zu 7,3 m% der H-Form. Man
erkennt somit sehr gut, dass eine höhere Beladung auch zu einer stärkeren Erwärmung der
Probe führt. Es sei hier der Vollständigkeit halber noch erwähnt, dass beide Proben bei

5.3 Charakterisierung der dielektrischen Eigenschaften 113
identischen Bedingungen (Temperatur: 200 °C; Phenol-Partialdruck: 24000 Pa) beladen
wurden.
0
1
2
3
4
5
H-ZSM-5 (I) Fe-ZSM-5 (I)
∆∆ ∆∆T
[°C
]
unbeladen
Phenol beladen
Abbildung 5.8: Temperaturerhöhung von unbeladenen und mit Phenol beladenen
Katalysatoren nach 240 s Mikrowelleneinstrahlung (120 W);
Phenolbeladung: H-ZSM-5 (I) 7,3 m%; Fe-ZSM-5 (I) 9,7 m%
Abschließend muss darauf hingewiesen werden, dass mittels dieser Versuche nur qualita-
tive Aussagen über die dielektrischen Eigenschaften der Katalysatoren getroffen werden
können. Es ist beispielsweise nicht möglich, Dielektrizitätskonstanten zu bestimmen.
Auch ist der Vergleich mit Aufheizversuchen aus der Literatur schwierig. Wie in Kapitel
3.4.1.6 diskutiert wird, spielt der Versuchsaufbau (Probenträger, Isolierung, etc.) bzw. die
Probenmasse eine entscheidende Rolle bei der Erwärmung von Katalysatoren im
Mikrowellenfeld. Weiterhin muss berücksichtigt werden, dass die dielektrischen Eigen-
schaften auch von der Probentemperatur abhängen. Bei höheren Temperaturen wird der
Dissipationsfaktor und damit der Energieeintrag gegebenenfalls größer. Diese Versuche
zeigen jedoch deutlich, dass sich trockene Zeolithe des Typs ZSM-5 nur sehr schlecht
durch Mikrowellenstrahlung aufheizen lassen. Dabei spielt scheinbar der Na-Gehalt des
Katalysators eine entscheidende Rolle. Als einziger Katalysator zeigte der Na-ZSM-5
einen deutlichen Temperaturanstieg. Alle anderen getesteten Katalysatoren waren annä-
hernd transparent für die Mikrowellenstrahlung und ließen sich praktisch nicht aufheizen.
Dieses Ergebnis bestätigt die Untersuchungen von WHITTINGTON ET AL. [284], wel-
cher das Aufheizverhalten verschiedener Zeolith-Katalysatoren untersucht hat (vgl.
Kapitel 3.4.1.6).

6 Entwicklung der Mikrowellenanlage
Die Herausforderung bei der Planung einer Anlage zur Untersuchung des Mikrowellen-
einflusses auf heterogen katalysierte Gasphasenreaktionen besteht darin, ein Anlagen-
bzw. Reaktorkonzept zu entwickeln, welches die üblicherweise bei Einsatz einer Mikro-
wellenheizung auftretenden Phänomene wie die Ausbildung von „hot spots“ oder Tempe-
raturgradienten (vgl. Kapitel 2.2), vermeidet. Ziel muss es sein, identische Reaktions-
bedingungen (Temperatur, Druck, …) unter Mikrowelleneinstrahlung und beim konventi-
onell beheizten „Referenzexperiment“ zu erreichen, um einen möglichen Einfluss der
Mikrowellenstrahlung auf das Reaktionssystem erkennen zu können. Sind der verwendete
Katalysator und die Edukte, wie im Fall der Hydroxylierung von Benzen mit N2O, zudem
transparent für Mikrowellenstrahlung, kann die gewünschte Reaktionstemperatur nicht
durch eine alleinige Mikrowellenheizung erreicht werden. Die Bereitstellung der für die
Reaktion benötigten Aktivierungsenergie muss immer über eine konventionelle Heizung
(z.B. elektrische Widerstandsheizung) erfolgen.
Somit ergeben sich folgende Anforderungen an das Reaktorsystem, um eine bestmögliche
Vergleichbarkeit eines konventionell beheizten Experiments und eines Mikrowellenexpe-
riments zu gewährleisten:
• Mikrowellen- und Referenzexperiment sind in der gleichen Anlage und Reaktor
ohne Umbaumaßnahmen durchführbar
• Simultane konventionelle und Mikrowellenheizung des Katalysatorbetts (Reaktors)
sind möglich
• Exakte Temperaturmessung auch unter Mikrowelleneinstrahlung
• Vermeidung von axialen und radialen Temperaturgradienten
Unter Einhaltung dieser Anforderungen wurde ein neuartiges Reaktorkonzept entwickelt,
welches in Kapitel 6.2.2 ausführlich beschrieben wird. Das Kapitel 6.1 gibt zunächst einen
Überblick über die Anlage, in der die Benzenhydroxylierung und die Untersuchungen zur
N2O-Zersetzung durchgeführt wurden. Auf die Hauptkomponenten der Laboranlage wird
in Kapitel 6.2 gesondert eingegangen.

6.1 Konzeption 115
6.1 Konzeption
Abbildung 6.1 zeigt ein Fließbild der Anlage, in der die Hydroxylierung von Benzen mit
N2O und die Experimente zur N2O-Zersetzung durchgeführt wurden. Die Anlage lässt
sich in 5 Haupteinheiten untergliedern:
• Dosierung
• Reaktor
• Mikrowellensystem
• Analytik
• Anlagensteuerung und Messdatenerfassung
Das Kernstück der Anlage bildet der Reaktor (vgl. Kapitel 6.2.2) und das
Mikrowellensystem (vgl. Kapitel 6.2.4).
Der Reaktor kann über einen Bypass umgangen werden, um mit Hilfe der Analytik die
Dosiergenauigkeit des Verdampfers zu überprüfen. Mit Hilfe von Dreiwegehähnen kann
parallel zur Messung des Bypasses eine Regenerierung oder N2-Spülung des Katalysators
vorgenommen werden, wodurch eine Zeitersparnis bei der Versuchsdurchführung ermög-
licht wird. Das bei der Regenerierung (Spülen) entstehende Abgas wird direkt in den
Abzug geleitet. Das Eduktgemisch (während der Bypassmessung) oder das Reaktionsge-
misch nach Austritt aus dem Reaktor wird über eine Mischzelle geleitet, an deren Eingang
Methan als interner Standard für den Gaschromatographen (vgl. Kapitel 6.2.5) zudosiert
wird. Der Gasstrom wird anschließend geteilt, wobei ein Teilgasstrom durch die Proben-
schleife des Gaschromatographen geführt wird. Der verbleibende Abgasstrom und das
Abgas aus der Probenschleife werden jeweils durch mit N-Methyl-Pyrrolidon (NMP)
gefüllte Waschflaschen geleitet, um die organischen Bestandteile (Aromaten) im Abgas
auszukondensieren. Das eingesetzte Lösungsmittel NMP wurde wegen seines hohen Sie-
depunktes von 202 °C ausgewählt.
In der Bypass-Leitung sowie vor und nach dem Reaktor sind Druckaufnehmer angebracht,
die eine Überwachung des Drucks in der Anlage und eine Bestimmung des Druck-
verlustes im Reaktor ermöglichen. Außerdem können eventuell in den Leitungen auftre-
tende Verstopfungen, die durch ein Auskondensieren von Reaktionsprodukten hervorge-
rufen werden können, leicht erkannt werden. Über ein in der Abgasleitung installiertes
Nadelventil (NV) lässt sich der Hauptstrom, von dem der Analysenstrom abgeteilt wird,
so regulieren, dass kein signifikanter Druckanstieg auftritt. Ein Überdruckventil vor dem
Eingang des Reaktors soll zudem verhindern, dass bei einem eventuell auftretenden
raschen Druckanstieg der Reaktor beschädigt wird.
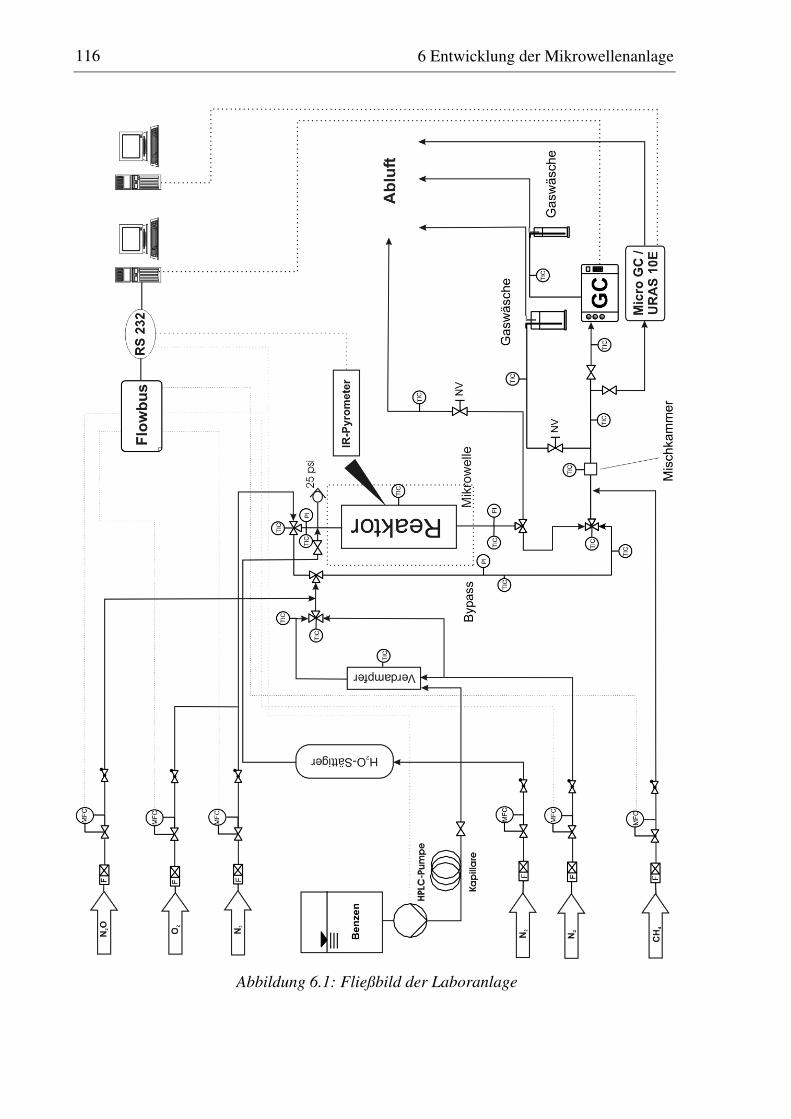
6 Entwicklung der Mikrowellenanlage 116
Abbildung 6.1: Fließbild der Laboranlage

6.1 Konzeption 117
Alle Verbindungsleitungen sind aus Edelstahl gefertigt und werden mit Hilfe von Ther-
moelementen, elektrischen Heizbändern und Temperaturreglern auf einer Temperatur von
mindestens 200 °C gehalten, um ein Auskondensieren von hochsiedenden Stoffen zu
verhindern. Bis auf die Zuleitung zum Gaschromatographen haben alle Rohrleitungen
einen Außendurchmesser von 6 mm.
Die gesamte Anlage befindet sich aus Sicherheitsgründen in einem Abzug, damit im Fall
einer Leckage giftige bzw. karzinogene Edukte und Produkte sofort abgesaugt werden.
Die Dosierung der gasförmigen Komponenten erfolgt mit Hilfe handelsüblicher Massen-
durchflussregler der Firma Bronkhorst-Hitec. Neben Methan (interner Standard für
Gaschromatograph) können Stickstoff als Trägergas für die Verdampfung der Flüssigkei-
ten sowie N2O zudosiert werden. Für eine Regenerierung des Katalysators nach dem Ein-
satz in der Benzenhydroxylierung stehen Massendurchflussregler für Stickstoff und
Sauerstoff zur Verfügung.
Die Dosierung des bei Raumtemperatur flüssigen Benzens erfolgt über eine HPLC-
Pumpe, welche in einen Edelstahlverdampfer fördert. Wasser wird über einen Sättiger
dosiert. Da die Analytik nicht für eine Bestimmung von Wasser ausgelegt ist, erfolgt die
Einspeisung in die Zuleitung zum Reaktor. Eine Bypassmessung ist nicht möglich. Die
detaillierte Beschreibung der Gas- und Flüssigdosierung ist in Kapitel 6.2.1 zu finden.
Für die Konzentrationsbestimmung von Benzen, Phenol und Benzochinon wird ein
Gaschromatograph der Firma Varian eingesetzt. Die Detektion von N2O ist mit diesem
Gerät nicht möglich. Für Untersuchungen in der N2O-Zersetzung kommt ein NDIR-Pho-
tometer oder ein Mikrogaschromatograph zum Einsatz. Während der Untersuchungen in
der Benzenhydroxylierung werden diese Geräte über einen Hahn vom Rest der Anlage
abgetrennt, da sie bei niedrigen Temperaturen arbeiten und eine Kondensation von
Aromaten in den Geräten stattfinden würde. Nähere Informationen zu den Analysengerä-
ten finden sich in Kapitel 6.2.5.
Die Steuerung der Massendurchflussregler, HPLC-Pumpe und Reaktortemperatur erfolgt
über den Anlagenrechner, welcher auch die Messdatenerfassung des Gaschromatographen
übernimmt. Der Massendurchflussregler für die Dosierung des Sättiger-Trägergases sowie
die Temperaturregler der Rohrbegleitheizungen und des Verdampfers können nur manuell
bedient werden. Ein zweiter Rechner ist für die Steuerung und Messdatenerfassung des
Mikrogaschromatographen verantwortlich. Weitere Informationen zur Anlagensteuerung
und Messdatenerfassung finden sich in Kapitel 6.2.5 und 6.2.6.

6 Entwicklung der Mikrowellenanlage 118
6.2 Komponenten der Anlage
6.2.1 Dosierung
Die Gase Stickstoff (Inert- und Trägergas für
die Verdampfung), Sauerstoff (zur Regene-
ration des Katalysators) und Methan (interner
Standard des Gaschromatographen) werden
der zentralen Gasversorgung des Instituts
entnommen. Distickstoffmonoxid wird in
einer separaten Flasche bereitgestellt. Nach
der Druckminderung werden die Gase über
thermische Massendurchflussregler [319] der
Firma Bronkhorst-Hitec dosiert. Technische
Daten der verwendeten Massendurchfluss-
regler finden sich in [320].
Das flüssige Benzen wird über eine HPLC-
Pumpe der Firma Knauer (Typ K120) in den
Verdampfer gefördert. Abbildung 6.2 zeigt
den Aufbau des verwendeten Edelstahlver-
dampfers (1.4571). Das Benzen strömt durch
eine dünne Kapillare zur Fritte. Um eine vor-
zeitige Verdampfung des Benzens in der
Kapillare und Pulsationen zu vermeiden,
wird das Benzen vor Eintritt in den
Verdampfer auf eine Temperatur von 16 °C
gebracht. Zu diesem Zweck wird der Pum-
penkopf mit Hilfe eines Kryostaten gekühlt.
Außerdem befindet sich die Kapillare bis zu
einer Entfernung von 1 cm vor der Fritte in
der Trägergaszuleitung, was die Gefahr einer
vorzeitigen Verdampfung ebenfalls verrin-
gert. Auf diese Weise wird eine gleichmäßige
Verdampfung des Benzens an der Metallfritte
erreicht, welche auf eine Temperatur von ca.
130 °C temperiert ist. Oberhalb der Fritte
befindet sich eine Glaskugelschüttung, wel-
che zusätzlich Pulsationen dämpft.
Abbildung 6.2: Verdampfer für Benzen

6.2 Komponenten der Anlage 119
Mit diesem Verdampferkonzept wird eine Dosiergenauigkeit von ± 1 % des jeweiligen
Normvolumenstroms an Benzen im gesamten Betriebsbereich erreicht.
Die Zugabe von Wasser als zusätzliche Komponente bei der Benzenhydroxylierung
erfolgt über einen Sättiger (Abbildung 6.3). Die Glasapparatur besteht aus zwei Teilen. Im
großen Behälter wird Stickstoff über eine Glasfritte durch Wasser geleitet und es bildet
sich ein Stickstoff-Wasser-Gemisch in der darüber liegenden Gasphase. Dieses Gemisch
strömt anschließend in den zweiten (kleineren) Behälter. Beide Gefäße besitzen einen
äußeren Mantel, durch den ein Heizmedium geleitet werden kann. Zuerst wird das Heiz-
medium in den großen Behälter eingespeist und anschließend über einen Schlauch zum
kleinen Behälter weitergeleitet. Dadurch ist die Temperatur im zweiten Gefäß etwas nied-
riger. Überschüssiges Wasser kondensiert und sammelt sich am Boden. Auf diese Weise
wird immer eine konstante Sättigung erreicht, unabhängig vom eingestellten Volumen-
strom des Trägergases. Die Überleitung für das Wasser-Gas-Gemisch zwischen den zwei
Gefäßen ist gut isoliert, die Leitung für das Heizmedium nicht. Über zwei im Gasraum der
Behälter angebrachte Thermoelemente kann die Temperatur der Gasmischung bestimmt
werden.
Die Sättigertemperatur (Temperatur des Heizmediums) wird über einen Badthermostaten
mit äußerem Umlauf eingestellt.
Die verdampfte Wassermenge pro Zeiteinheit wird rechnerisch in Abhängigkeit von der
Gasphasentemperatur im zweiten (kleinen) Behälter ermittelt (siehe S. 168).
Abbildung 6.3: Sättiger für Wasser

6 Entwicklung der Mikrowellenanlage 120
6.2.2 Reaktor
Als problematisch bei der Konstruktion des Reaktors erwies sich, dass eine kombinierte
Elektrische- und Mikrowellenheizung möglich sein sollte. Die üblicherweise für Labor-
Reaktoren verwendeten Heizmanschetten können aufgrund ihrer Wechselwirkung mit
dem Mikrowellenfeld nicht eingesetzt werden. Bei Reaktoren für Mikrowellen-
anwendungen wird deshalb häufig der Edukt-Gasstrom auf die Reaktionstemperatur (oder
höher) vorgeheizt. Eine Regelung der Katalysatorbett-Temperatur erweist sich mit diesem
Verfahren jedoch als schwierig. Weiterhin kommt es zur Ausbildung axialer Temperatur-
gradienten, da sich das Gas nach Eintritt in den Reaktor immer weiter abkühlt.
Um diese Probleme zu umgehen, wurde ein Reaktor entwickelt, welcher durch eine elekt-
rische Heizpatrone im Kern des Reaktors beheizt wird. Abbildung 6.4 zeigt eine schemati-
sche Darstellung des verwendeten Reaktors.
Abbildung 6.4: Schematische Darstellung des Reaktors
Der Reaktor besteht aus einem inneren Quarzglas-Rohr (Ø 2,1 cm; Länge 56 cm) und
einem äußeren Rohr (Ø 3,5 cm), welches sich in der Mitte erweitert (Ø 4 cm). Die zwei
Rohre sind an ihren Enden verbunden und bilden einen äußeren Mantel, in dem der Kata-
lysator auf einer Quarzglasfritte platziert werden kann. Dieses Design erlaubt einen
kleinen Durchmesser an den Durchführungen des Mikrowellengehäuses und aufgrund der

6.2 Komponenten der Anlage 121
Erweiterung in der Mitte des Reaktors genügend Raum, um eine größere Katalysator-
menge einzubauen.
Über eine zweite Quarzglasfritte ist das Katalysatorbett vom oberen Teil des Reaktors
abgetrennt. Zwischen den beiden Fritten befindet sich ein Stutzen, über den der
Katalysator eingefüllt werden kann. Sowohl der obere als auch untere Teil des Reaktor-
mantels ist mit Quarzglasbruch (Ø 0,8 - 1 mm) befüllt, um eine Rückvermischung der
Edukte/Produkte zu vermeiden bzw. ein plug-flow Verhalten des Reaktors zu erreichen.
Auch der Katalysator wurde mit Quarzglasbruch verdünnt, um hot spots aufgrund der
stark exothermen Reaktion zu vermeiden und eine gleichmäßige Durchströmung des
Katalysatorbetts zu ermöglichen.
Als Werkstoff für den Reaktor und Inertmaterial wurde Quarzglas gewählt, weil es einer-
seits keine Mikrowellenstrahlung absorbiert und andererseits eine Temperaturmessung
direkt am Katalysator mittels eines Infrarot-Pyrometers (vgl. Kapitel 6.2.3) erlaubt.
Der äußere Mantel des Reaktors ist mit den Rohrleitungen der Anlage über Quarzglas-
rohre (Ø 6 mm) und Swagelock VCO-Verschraubungen verbunden und wird von oben
nach unten durchströmt (vgl. Abbildung 6.4).
Im inneren Rohr des Reaktors ist eine Heizpatrone eingebaut. Die verwendete Heizpat-
rone (Hersteller Ihne&Tesch) ist aus Edelstahl gefertigt (Ø 1 cm; Passung H7; Länge
45 cm). Der obere Teil (15 cm) wird nicht aktiv beheizt, sondern allein über Wärmelei-
tung erwärmt. Im unteren Teil des Reaktors ist eine zweite Heizpatrone (Hersteller
Ihne&Tesch) eingebaut (Ø 1 cm; Passung H7; Länge 11 cm). Der Einsatz der metalli-
schen Heizpatronen im Mikrowellenfeld ist deshalb möglich, weil sie sehr gut leitend mit
dem Mikrowellengehäuse verbunden sind (siehe Kapitel 6.2.4). Im Inneren der Heizpatro-
nen sind Thermoelemente eingebaut, welche eine Messung der Kerntemperatur des
Reaktors erlauben. Diese Thermoelemente werden nicht durch das Mikrowellenfeld
beeinflusst, da sie durch die Hülle der Heizpatronen vor dem Feld abgeschirmt sind. Das
Thermoelement der oberen Heizpatrone befindet sich auf Höhe der Mitte der Katalysator-
schüttung. Beide Heizpatronen werden von einem auf Passung (H7) gefertigten Keramik-
rohr (Haldenwanger ALSINT 99,7) umgeben, um eine gleichmäßige Wärmeabgabe an das
innere Quarzglasrohr des Reaktors zu ermöglichen. Außerdem bestünde die Gefahr einer
Überhitzung (Zerstörung) der Heizpatronen, wenn diese ohne einen passgenau gefertigten
Wärmeüberträger verwendet werden. Die eingesetzte Keramik ist weitestgehend transpa-
rent für Mikrowellenstrahlung.
Diese Anordnung der Heizpatronen erzeugt eine isotherme Zone im mittleren Bereich des
Reaktors (Länge 6 cm), in dem der Katalysator eingebaut ist. Das Temperaturprofil des
Reaktors findet sich in Appendix D. Die maximale Reaktionstemperatur liegt bei 600 °C.
Im oberen Bereich des Reaktors ist die Temperatur deutlich niedriger als die am

6 Entwicklung der Mikrowellenanlage 122
Katalysatorbett (bei 300 °C ca. 150 °C). Im unteren Teil kann die Temperatur über die
zweite Heizpatrone frei eingestellt werden. Dies hat den Vorteil, dass die Durchführungen
am Mikrowellengehäuse auch bei hohen Reaktionstemperaturen nur geringen thermischen
Belastungen ausgesetzt sind.
Von außen ist der Reaktor mit einer für Mikrowellenstrahlung transparenten Keramik-
isolierung umgeben (Hersteller: MLS).
Durch dieses Design werden zum einen axiale Temperaturgradienten im Katalysatorbett
vermieden, da die Wärme unabhängig vom Mikrowellenfeld durch die Heizpatrone
eingebracht wird. Das Katalysatorbett bildet einen Kreisring mit kleinem Durchmesser,
wodurch radiale Temperaturgradienten minimiert werden. Weiterhin ist eine exakte
Temperaturmessung (Regelung) der Katalysatortemperatur möglich (siehe Kapitel 6.2.3).
Eine Konstruktionszeichnung des Reaktors findet sich in Appendix E.
6.2.3 Temperaturmessung und Regelung
Als problematisch bei der Temperaturmessung im Mikrowellenfeld erweist sich, dass her-
kömmliche Thermoelemente aus Metall nicht ohne Einschränkungen eingesetzt werden
können (vgl. Kapitel 2.2.1).
Die Messung der Reaktortemperatur erfolgt deshalb mit einem Infrarot-Pyrometer
(Hersteller: Impac; Typ IP-120). Dieses Gerät besitzt den Vorteil, dass es in einem
Wellenlängenbereich (2 -2,8 µm) misst, welcher nicht von Quarzglas emittiert wird [321].
Die Größe des Messflecks des Pyrometers kann zwischen 0,25 mm und 1 cm eingestellt
werden. Bei den in dieser Arbeit diskutierten Experimenten wurde eine Messfleckgröße
von ca. 2 mm verwendet. Durch einen im Gerät integrierten Laserpointer wird die
Fokussierung ermöglicht. Die Temperaturmessung erfolgt in etwa in der Mitte der
Katalysatorschüttung (axial), wie in Abbildung 6.4 (S. 120) skizziert.
Da dieses Gerät die Temperatur der Reaktorwand aus Quarzglas nicht detektiert, wird die
Temperatur der im Strahlengang liegenden Katalysatorpartikel gemessen. Dies hat den
Vorteil, dass eine Temperaturänderung am Katalysator direkt und schnell (keine Trägheit
wie bei herkömmlichen Thermoelementen) erkannt werden kann.
Um die Temperatur eines Materials mit einem IR-Pyrometer exakt bestimmen zu können,
muss zunächst der Emissionsfaktor dieses Materials ermittelt werden [321]. Zu diesem
Zweck werden die Katalysatoren vor dem Einsatz in der Reaktion in einem Ofen auf
200 - 300 °C erwärmt. Die Probentemperatur wird mit einem herkömmlichen Thermo-
element bestimmt. Anschließend erfolgt die Temperaturmessung mit Hilfe des IR-
Pyrometers, wobei der verwendete Emissionsfaktor des Geräts über die Gerätesoftware
(InfraWin) angepasst wird, bis eine zum Thermoelement identische Temperatur angezeigt
wird. Die Emissionsfaktoren der getesteten Zeolithe liegen im Bereich von ca. 0,8.

6.2 Komponenten der Anlage 123
Weiterführende Informationen zur Funktionsweise von IR-Pyrometern finden sich in
[321].
Neben der Temperaturmessung mittels des IR-Pyrometers ist auch die Bestimmung der
Temperatur der Heizpatronen über die integrierten Thermoelemente möglich. Das Ther-
moelement der oberen Heizpatrone liegt auf Höhe der Mitte der Katalysatorschüttung
(vgl. Abbildung 6.4, S. 120) und liefert die Temperatur der Heizpatrone im Zentrum des
Reaktors bzw. im Mittelpunkt des Kreisrings der Katalysatorschüttung.
Abbildung 6.5 zeigt exemplarisch die gemessenen Temperaturen während eines
Experiments in der Hydroxylierung von Benzen mit N2O bei einer Mikrowellenleistung
von 200 W.
Man erkennt, dass die vom IR-Pyrometer gemessene Temperatur praktisch keine Abwei-
chung vom voreingestellten Wert (400 °C) zeigt. Die Temperatur der Heizpatrone (innen
liegendes Thermoelement) schwankt um ca. ± 1 %. Ursache hierfür ist der verzögerte
Wärmeübergang von der Heizpatrone an das Katalysatorbett aufgrund der Keramikum-
mantelung der Patrone, welche eine hohe Wärmekapazität besitzt.
Der Temperaturverlauf bei einem konventionell beheizten Experiment ist dem in
Abbildung 6.5 gezeigten vergleichbar.
Abbildung 6.5: Temperaturmessung während eines Mikrowellenexperiments; T = 400 °C;
Mikrowellenleistung 200 W; Stellgröße: Signal des IR-Pyrometers

6 Entwicklung der Mikrowellenanlage 124
Die Regelung der Reaktortemperatur erfolgte in dieser Arbeit immer anhand des Mess-
Signal des IR-Pyrometers. Das in der Heizpatrone eingebaute Thermoelement wurde nur
zu Kontrollzwecken verwendet. Über ein externes Temperaturmessgerät lässt sich die
Temperatur der Heizpatrone protokollieren. Prinzipiell kann auch das Mess-Signal des
Thermoelements für die Regelung verwendet werden.
Als Regeleinheit wird ein Eurotherm-Regler (Typ 2416) benutzt, welcher die Temperatur
des IR-Pyrometers als Stellgröße verwendet. Die Leistungsabgabe an die Heizpatrone
erfolgt stufenlos zwischen 0 und 500 W (die maximale Leistungsaufnahme der Heiz-
patrone beträgt 1000 W). Wird durch die Mikrowelle zusätzlich Wärme in das System
eingebracht, verringert sich die Leistungsabgabe an die Heizpatrone. Ein Schwingen der
Temperatur bei Zu- oder Abschalten der Mikrowelle kann auf diese Weise minimiert
werden und das Regelverhalten ist besser als bei den üblicherweise eingesetzten digitalen
Leistungsstellern. Diese arbeiten bei einer festgelegten Leistung, wobei die Temperatur
vom Regler über Puls-Pause-Zyklen konstant gehalten wird.
6.2.4 Mikrowellensystem
Bei der verwendeten Mikrowellenausstattung handelt es sich um eine umgebaute Labor-
mikrowelle der Firma Milestone (Typ Ethos 1600), die in Abbildung 6.6 zu sehen ist. Die
maximale Leistungsabgabe beträgt 1000 W. Das Multimode-Mikrowellengerät arbeitet
bei einer Frequenz von 2,45 GHz und wurde hinsichtlich einer konstanten Leistungsab-
gabe und homogenen Feldverteilung konzipiert [322].
Der Mikrowellenofen besitzt an der Ober- und Unterseite Stutzen (Ø 3,5 cm), die als
Durchführung für den Reaktor dienen (vgl. Abbildung 6.7). Am Ende der Heizpatronen
sind Kappen aus Stahl befestigt, welche sich exakt über diese Stutzen schieben lassen und
ein Austreten von Mikrowellenstrahlung verhindern. Auf diese Weise sind die Heizpatro-
nen leitend mit dem Mikrowellengehäuse verbunden. Sie fungieren als „Wand“ des
Mikrowellenraums und reflektieren die Strahlung. Beide Kappen besitzen eine Öffnung
(Ø 6,2 mm) für die Quarzglasrohre, welche als Zu- und Ausgang des Reaktors dienen. Der
obere Stutzen besitzt einen Flansch (Ø 12 cm), welcher durch Schrauben mit dem Mikro-
wellengehäuse verbunden wird. Dadurch steht eine genügend große Öffnung zur Verfü-
gung, durch die der Reaktor in den Mikrowellenraum eingebaut werden kann. An der
Seitenwand der Mikrowelle ist ein weiterer Stutzen angebracht, welcher als optischer
Zugang für das IR-Pyrometer dient.

6.2 Komponenten der Anlage 125
Abbildung 6.6: Mikrowellenapparat und Teile des Laboraufbaus
Die Stutzen verhindern, dass Mikrowellen-
strahlung austreten kann, auch wenn keine
Heizpatronen (Verschlusskappen) installiert
sind (vgl. Kapitel 2.2.1).
Die Zuführungen für den Reaktor sind von
außen isoliert.
Bei jedem Versuch wird zur zusätzlichen
Sicherheit das Mikrowellengehäuse mit
einem Mikrowellen-Leck-Tester auf austre-
tende Strahlung hin untersucht.
Nach dem Einbau des Reaktors wird dieser
mit einem keramischen Isolationsmaterial
ummantelt (Hersteller MWS), welches für
Mikrowellen durchlässig ist, sehr gute Isola-
tionseigenschaften besitzt und sich für Tem-
peraturen bis maximal 1000 °C eignet (siehe
Abbildung 6.6).
Die Steuerung der Labormikrowelle wird von
dem zum System gehörenden Laborrechner
(LabTerminal) übernommen. Mit Hilfe der
Software (ContWAVE) lässt sich die gewünschte Leistung frei einstellen und der Wert
der aktuellen Leistungsabgabe der Magnetrone auslesen. Weiterhin ist eine zeitabhängige
Abbildung 6.7: Schematische Darstellung der
Labormikrowelle mit eingebautem Reaktor

6 Entwicklung der Mikrowellenanlage 126
Leistungssteuerung sowie ein An- oder Ausschalten der Mikrowellenabgabe nach einer
bestimmten Zeit möglich. Außerdem lässt sich eine temperaturabhängige
Leistungsregelung über die Software realisieren. Zu diesem Zweck sind Eingänge für
Thermoelemente an der Labormikrowelle vorhanden. An diese kann das innen liegende
Thermoelement der Heizpatrone angeschlossen werden. Die in der Software eingestellte
Temperatur wird durch eine Variation der Mikrowellenleistung konstant gehalten. Diese
Temperaturregelung über die Leistung der Mikrowelle macht nur Sinn, wenn sich ein
Material (Katalysator) im Reaktor befindet, das sich sehr gut durch die Mikrowellen
aufheizen lässt. Die Heizpatrone muss in diesem Fall bei einer konstanten Leistung
betrieben werden. Der Eurotherm-Regler hat keine Regelfunktion und das IR-Pyrometer
dient nur der Temperaturkontrolle.
In dieser Arbeit wird jedoch immer bei einer konstanten Leistungsabgabe der Mikrowelle
gearbeitet. Die Temperaturregelung erfolgt durch den Eurotherm-Regler, die vom IR-
Pyrometer gelieferte Temperatur dient als Regelgröße.
6.2.5 Analysensystem
Für die Analysen der Edukt- und Produktzusammensetzung bei der Hydroxylierung von
Benzen mit N2O wird ein Gaschromatograph der Firma Varian (Typ 3400) verwendet. Die
Probenahme erfolgt über ein 6-Port-Ventil mit einer Probenschleife (Volumen 250 µl).
Das Gerät ist mit einer HP-5 Kapillarsäule (Länge 25 m; Ø 0,53 mm, Filmdicke 0,52 µm)
zur Trennung der Kohlenwasserstoffe ausgerüstet. Die Analyse der einzelnen Kompo-
nenten erfolgt über einen Flammenionisationsdetektor (FID). Diese Anordnung erlaubt die
Analyse der Aromaten, die restlichen Gase (z. B. N2O) können nicht detektiert werden.
Die Zykluszeit für eine Messung beträgt inklusive der benötigten Abkühlungszeit 8 min.
Näheres zur Funktionsweise von Gaschromatographen findet sich bei GOTTWALD
[323].
Die Steuerung des Gaschromatographen, der Messdatenerfassung und der Auswertung
(Integration der Chromatogramme) erfolgt über den Anlagenrechner mit Hilfe des
Softwarepakets VarianStar.
Zur Quantifizierung der einzelnen Stoffe wird die Methode des internen Standards einge-
setzt [324]. Dem zu analysierenden Gemisch wird eine definierte Menge an Methan
zugegeben. Temperatur- und Druckschwankungen in der Probenschleife, welche die
analysierte Probenmenge (Probenschleifenvolumen 250 µl) beeinflussen, können auf
diese Weise herausgerechnet werden, da sich auch die bekannte Menge an Methan verän-
dert. Die exakten Stoffmengenströme der zu analysierenden Substanzen lassen sich über
den bekannten Stoffmengenstrom des Methans anhand vorher bestimmter RMR-Werte
(relative molar response) errechnen.

6.2 Komponenten der Anlage 127
Eine detaillierte Beschreibung der Vorgehensweise zur Bestimmung der RMR-Werte und
zur Auswertung der Analysen findet sich in [325]. Retentionszeiten, RMR-Werte,
Betriebs-Parameter und detailliertere Informationen zum verwendeten Gaschromato-
graphen können in [320, 325] nachgeschlagen werden.
6.2.6 Anlagensteuerung
Die Steuerung der Anlage erfolgt über ein im Rahmen dieser Arbeit konzipiertes und
programmiertes VisualBasic Programm für Windows. Es bietet die Möglichkeit, die
Massendurchflussregler, HPLC-Pumpe, Temperaturregler für den Reaktor und das IR-
Pyrometer anzusprechen. Die Bedienung ist leicht verständlich. Das Hauptfenster des
Programms zeigt ein Fließbild der Anlage mit Anzeigen der aktuell vorliegenden Mess-
parameter der Massendurchflussregler und der Reaktortemperatur. Über ein Eingabefeld
können die Sollwerte für die Massendurchflussregler (in Nml/min des entsprechenden
Gases) festgelegt und an die Geräte übergeben werden. Unter-Menüs erlauben eine
Ansteuerung der HPLC-Pumpe und der Temperaturregler des Reaktors. Weiterhin können
Zeitprogramme für die Durchflüsse und Reaktortemperatur definiert werden. Eine
Protokollierung der Anlagenparameter (nach vorgegebenen Zeitintervallen) in eine Text-
datei ist ebenfalls möglich.
Die Ansteuerung der Massendurchflussregler erfolgt über ein Serverprogramm
(Flow DDE32) der Firma Bronkhorst-Hitec, welches über die serielle Schnittstelle des
Rechners (RS232) mit dem Bus-System der Massendurchflussreglers verbunden ist. Das
Programm zur Anlagensteuerung kommuniziert mit diesem Server, nicht direkt mit den
Geräten.
Die HPLC-Pumpe und die Eurotherm-Regler werden direkt über die serielle Schnittstelle
des Rechners (RS232) angesteuert. Auf diesem Weg werden auch die Messwerte des IR-
Pyrometers ausgelesen.
Die Kommunikation des Rechners mit dem Gaschromatographen läuft über eine spezielle
Schnittstellenkarte (AD-Wandler) der Firma Varian bzw. der Software VarianStar (vgl. S.
126).
Eine Einstellung der Geräteparameter (z. B. Emissionsfaktor) des IR-Pyrometers kann nur
über die zum Gerät gehörende Software (InfraWin) erfolgen, nicht durch das Anlagen-
steuerungsprogramm.
Weitere Informationen zur verwendeten Anlagensteuerungssoftware finden sich bei
EWANE und GOPALAKRISHNAN [320, 325].

6 Entwicklung der Mikrowellenanlage 128
6.2.7 Modifikation des Versuchsaufbaus für die Untersuchung der N2O-
Zersetzung
Für die Untersuchungen zur N2O-Zersetzung im Mikrowellenfeld war eine Modifikation
der Laboranlage nötig, welche im Folgenden erläutert wird.
Mit der in Kapitel 6.2.5 beschriebenen Analytik (GC) ist keine Analyse von N2O möglich.
Deshalb kam ein N2O-Detektor der Firma Hartmann & Braun vom Typ Uras 10E zum
Einsatz. Dieses Gerät arbeitet nach dem Prinzip eines NDIR-Photometers und erlaubt
Konzentrationsmessungen von N2O im Bereich zwischen 0 und 1000 ppm. Das Mess-
prinzip basiert auf der Vergleichsmessung der Adsorption von Kontinuumsstrahlung im
Infrarotbereich (NDIR), die durch eine Probenküvette und eine Referenzküvette läuft
[326]. Aufgrund der unterschiedlichen Extinktion kann nach dem Lambert-Beer’schen
Gesetz [327] die Konzentration des zu messenden Gases bestimmt werden. Die Mess-
werte werden am Display des Geräts abgelesen, eine Messdatenerfassung über Soft-
ware/Rechner ist nicht möglich.
Da dieses Messgerät jedoch nicht über den gesamten Versuchszeitraum zur Verfügung
stand, wurde bei den Untersuchungen am Katalysator Co-ZSM-5 ein MicroGC der Firma
Chrompack (Typ: CP-2002) zur Konzentrationsbestimmung von N2O verwendet. Im
Unterschied zu herkömmlichen Gaschromatographen zeichnet sich dieses Gerät vor allem
durch seine kompakte Bauweise und sehr kurze Analysenzeiten aus. Die Steuerung erfolgt
über einen separaten Computer mit der Software Maestro. Detaillierte Informationen zur
Funktion, Bauweise Säulen- und Betriebsparametern dieses Geräts finden sich bei
HIEMER [26]. Ein interner Standard wird bei der Bestimmung der N2O-Konentration
nicht verwendet.
Als Feed wurde ein Prüfgas mit 950 ppm N2O in Helium (Linde, Prüfgas Kl. 1)
eingesetzt. Die Dosierung erfolgte über den Anlagen-MFC für N2O, welcher entsprechend
dieses Gasgemisches neu kalibriert wurde.
6.3 Untersuchungen zur Idealität des eingesetzten Reaktors
Die Modellierung eines Reaktors ist vor allem für die kinetische Untersuchung einer
Reaktion von Bedeutung. Mit Hilfe eines geeigneten Reaktormodells lässt sich die Kinetik
der Reaktion vom Reaktor entkoppeln. Aber auch für ein Katalysatorscreening ist die
Kenntnis des Modells nötig [328]. Üblicherweise strebt man bei einem Strömungsrohr-
reaktor ideales plug-flow Verhalten an. Ist dies nicht gewährleistet, kommt es zur
Ausbildung axialer und radialer Temperatur- und Konzentrationsgradienten, welche die
Aktivitäts- und Selektivitätsmessungen durch Nichtlinearitäten verfälschen [329].
Aufgrund des neuartigen Reaktordesigns war es nötig zu überprüfen, ob axiale und radiale
Konzentrations- und Temperaturgradienten vernachlässigt werden können.

6.3 Untersuchungen zur Idealität des eingesetzten Reaktors 129
Um ein pseudohomogenes, eindimensionales plug-flow Modell für einen Rohrreaktor zu
rechtfertigen, müssen verschiedene Parameter wie Druckverlust, axiale Rückvermischung,
radiale Gradienten in der Strömungsgeschwindigkeit und Temperaturgradienten überprüft
werden.
6.3.1 Druckverlust
Nach RASE [328] soll der Druckverlust über die Schüttung bei Strömungsrohrreaktoren
weniger als 10 % des Gesamtdrucks betragen, um Impulsbilanzen bei der Modellierung
des Reaktors vernachlässigen zu können. Der Druckverlust lässt sich über die Ergun
Gleichung berechnen [328] oder über Druckaufnehmer vor und nach dem Reaktor experi-
mentell bestimmen. Für den verwendeten Reaktor zeigten Differenzdruckmessungen, dass
selbst bei der höchsten in der Reaktion eingestellten Temperatur (450 °C) und Durchfluss
(390 Nml/min) ein relativer Druckverlust von 1,5 %, bezogen auf atmosphärischen
Betriebsdruck, nicht überschritten wurde.
6.3.2 Axiale Rückvermischung
Die axiale Rückvermischung der Moleküle in einem Reaktor lässt sich in der Verweilzeit-
verteilung erkennen. Eine idealer Strömungsrohrreaktor kann durch das so genannte
Dispersionsmodell beschrieben werden. Die bestimmende Kennzahl ist in diesem Fall die
Bodenstein-Zahl als Maß für das Verhältnis von erzwungener Konvektion zur Dispersion.
Überschreitet diese Kennzahl einen bestimmten Wert, kann axiale Dispersion
vernachlässigt werden. Nach BAERNS ET AL. ist dies für Bodenstein-Zahlen größer 100
der Fall [330]. In der Arbeit von PFADLER [331] konnte gezeigt werden, dass für die in
dieser Arbeit vorliegenden Randbedingungen und den verwendeten Reaktor Bodenstein-
Zahlen von 100 weit überschritten werden. Des Weiteren wurden Sprungmarkierungs-
experimente durchgeführt, welche die Annahme eines plug-flow-Models für den verwen-
deten Reaktor rechtfertigen [331]. Die Berechnung der Bodenstein-Zahlen kann in
Appendix F nachgeschlagen werden. Die Ergebnisse der Markierungsexperimente zur
Bestimmung des Verweilzeitverhaltens finden sich in Appendix G.
6.3.3 Radiale Gradienten in der Strömungsgeschwindigkeit
Ein Problem bei Rohrreaktoren ist das Auftreten von radialen Strömungsgeschwindig-
keitsgradienten. In Rohrreaktoren wird in diesem Zusammenhang häufig von der Rand-
gängigkeit gesprochen. Zwischen der Katalysatorschüttung und der Reaktorwand bilden
sich Kanäle mit niedrigerem Strömungswiderstand aus, wodurch in diesen Bereichen
höhere Strömungsgeschwindigkeiten resultieren. In diesem Fall ergeben sich in den unter-
schiedlichen Bereichen des Festbetts auch unterschiedliche Katalysatorbelastungen, was
sich wiederum auf die Produktivität des Reaktors auswirkt [328]. Modelle für eine
zuverlässige Abschätzung von radialen Gradienten in der Strömungsgeschwindigkeit sind

6 Entwicklung der Mikrowellenanlage 130
jedoch selten und nicht universal anwendbar, da viele Einflussfaktoren (z. B. Aufbau- und
Art der Schüttung, Partikelform) eine Rolle spielen [332]. Deshalb werden auch oft unter-
schiedliche geometrische Kriterien vorgeschlagen [328, 333].
RASE [328] gibt folgendes Abschätzkriterium an, um radiale Gradienten in der
Strömungsgeschwindigkeit auszuschließen:
10R
P
d
d> (Gl. 6.1)
Dieses Kriterium kann allerdings nicht direkt auf den eingesetzten Reaktor angewendet
werden, da es sich nicht um einen Kreisförmigen, sondern einen Kreisringquerschnitt
handelt. Setzt man für eine Abschätzung in Gleichung 6.1 den Durchmesser des Kreis-
rings ein, erreicht man mit dem verwendeten Quarzglasbruch Werte zwischen 8 und 10.
Rechnet man mit dem Außendurchmesser des Reaktors, erhält man jedoch um ein vielfa-
ches höhere Werte. Die im Rahmen dieser Arbeit durchgeführten Stufenmarkierungsexpe-
rimente (siehe Appendix G) zeigen jedoch keine Hinweise auf Randgängigkeit.
6.3.4 Isothermie / Temperaturgradienten
Besonders kritisch bei der Bestimmung reaktionstechnischer Größen erweist sich das
Auftreten von Temperaturgradienten im Reaktor [329]. Diese verfälschen die experimen-
tell bestimmten Werte stark, weil die eingestellten Betriebsbedingungen nicht eindeutig
einer Temperatur zuzuordnen sind.
In der vorliegenden Arbeit muss jedoch zwischen zwei verschiedenen Ursachen unter-
schieden werden, welche Temperaturgradienten hervorrufen können:
1. Reaktionen mit einer starken Wärmetönung, wie die Benzenhydroxylierung, sind
problematisch. Die während der Reaktion freigesetzte Wärme muss kontinuierlich
aus dem Reaktor abgeführt werden, da sonst hot spots entstehen oder es zum
runaway kommt. Dabei stellt vor allem die übliche Bauweise von Labor-Strö-
mungsrohrreaktoren ein Problem dar. Die Wärme wird dem Rohr-Reaktor von
Außen über eine Heizmanschette zugeführt. Wird nach dem Starten der Reaktion
im Reaktor Wärme frei, kann diese nur schlecht nach Außen abgeführt werden, da
die Heizungen in der Regel eine sehr große Wärmekapazität besitzen. Aus diesem
Grund ist auch die Temperaturregelung bei diesen Systemen sehr träge. Es kommt
zu Ausbildung von axialen und radialen Temperaturgradienten, die umso stärker
ausgeprägt sind, je größer der Reaktordurchmesser gewählt wird. Weiterhin kann
es auch zu einer axialen Wärmedispersion kommen, welche wiederum axiale
Temperaturgradienten zur Folge hat.
2. Durch den Einsatz von Mikrowellen als Heizmedium kann es ebenfalls zur Ausbil-
dung von Temperaturgradienten kommen. Ursache hierfür ist die Wirkungsweise

6.3 Untersuchungen zur Idealität des eingesetzten Reaktors 131
der Mikrowellen, welche einen Körper von innen heraus erwärmen (vgl. S. 8). Ist
die Wärmeabfuhr nach Außen behindert, entstehen radiale Temperaturgradienten
im Katalysatorbett. Außerdem erweist sich eine inhomogene Feldverteilung im
Mikrowellenraum als problematisch. Die Feldstärken sind in verschiedenen Berei-
chen des Katalysatorbettes unterschiedlich und damit auch die Temperatur. Dies ist
die Ursache für die häufig in der Mikrowellenliteratur diskutierten hot spots (vgl.
Kapitel 2.3.1.4).
Zu 1.: Um Temperaturgradienten aufgrund der Wärmetönung einer Reaktion zu
vermeiden, gibt es verschiedene Möglichkeiten. Wird eine Betriebsweise bei hoher
Verdünnung (wie in dieser Arbeit mit Stickstoff als Inertgas) gewählt, kann die Wärme
gut abgeführt werden und es kommt nicht zur Ausbildung von Temperaturgradienten.
Durch eine Verdünnung des Katalysatorbetts mit Quarzglasbruch kann die Gefahr von
Temperaturgradienten noch weiter reduziert werden, weil sich die Reaktionsgeschwindig-
keit pro Volumenelement verringert [329]. Entscheidend zur Vermeidung von radialen
Temperaturgradienten trägt jedoch auch das spezielle Reaktordesign mit Kreisringquer-
schnitt bei. Der Durchmesser des Katalysatorbetts ist klein (Ø 0,8 cm) und es steht eine
große Wärmeabgabefläche zur Verfügung. Die maximale Temperaturüberhöhung, die
nach dem Starten der Benzenhydroxylierung im Katalysatorbett gemessen werden konnte,
betrug 3 °C. In diesem Fall wurde die eingestellte Betttemperatur jedoch bereits nach ca.
3 min Reaktionsdauer wieder erreicht. Eine axiale Wärmedispersion kann zudem auf-
grund der im Verhältnis zum Reaktordurchmesser großen Länge der Katalysatorschüttung
(ca. 5 cm) vernachlässigt werden [329, 330].
Zu 2.: Aufgrund dieses speziellen Reaktordesigns können auch radiale Temperaturgra-
dienten durch das Mikrowellenfeld vernachlässigt werden. Der geringe Durchmesser der
Schüttung (Ø 0,8 cm) gewährleistet eine gute Ableitung der Wärme nach außen. Axiale
Temperaturgradienten und hot spots lassen sich durch eine homogene Feldverteilung ver-
meiden (siehe Kapitel 6.2.4). Ist der Wärmeeintrag in das Katalysatorbett durch die
Mikrowellen zudem gering, besteht keine Gefahr, dass sich axiale Gradienten im
Katalysatorbett ausbilden, da das Temperaturprofil des Reaktors über die Heizpatrone
aufgeprägt wird. Dies ist bei den in dieser Arbeit beschriebenen Experimenten der Fall.
Die verwendeten Katalysatoren sind nahezu transparent für Mikrowellen und die während
der Reaktion im Reaktor vorhandene Masse an Phenol ist so gering, dass keine
signifikante Aufheizung des gesamten Katalysatorbetts (große Masse) zu erwarten ist. Am
Fe-ZSM-5 (I) werden z. B. bei den Versuchseinstellungen T = 400 °C, N2O:Benzen = 1:1,
τmod = 90 (g·min)/mol zu Beginn der Reaktion 31 mg/min Phenol gebildet.
Wird der Katalysator zusätzlich mit Quarzglas verdünnt, verringert sich die Gefahr von
Temperaturgradienten noch weiter, da weniger Mikrowellen absorbierendes Material pro
Volumenelement vorliegt.

6 Entwicklung der Mikrowellenanlage 132
6.4 Für Sorptionsuntersuchungen eingesetzte Apparaturen und
Analytik
Die Beladung der Katalysatorproben mit Benzen, Phenol bzw. deren Mischungen erfolgte
in einer speziellen Beladungsapparatur, welche in Abbildung 6.8 dargestellt ist und sich
aus 3 Teilen zusammensetzt. Zunächst wird der Katalysator in einen Probenträger (Pos 1)
eingebaut, welcher an der Seite eine Öffnung besitzt und dessen Boden aus einer Glas-
fritte besteht. Der Probenträger wird anschließend in das Gefäß 2 eingebaut, welches mit
Stickstoff gefüllt ist und bereits das Adsorptiv enthält. Über einen Schliff wird der Pro-
benträger mit Gefäß 2 luftdicht verschlossen und mit einer Klammer fixiert. Anschließend
erfolgt die Beladung der Probe mit dem Adsorptiv, indem das Gefäß für eine bestimmte
Dauer auf die gewünschte Temperatur in einem Trockenschrank gebracht wird. Nach
Abschluss der Adsorption wird der Probenträger in ein weiteres mit Stickstoff gefülltes
Gefäß (Pos 3) eingebaut und auf Raumtemperatur abgekühlt. So lässt sich verhindern,
dass während des Abkühlvorgangs überschüssiges Adsorptiv auf dem Probenträger und
Katalysator kondensiert.
Bei dieser Vorgehensweise erfolgt somit keine Adsorption bei Umgebungsdruck, da das
Gefäß luftdicht verschlossen ist, während es aufgeheizt wird. Außerdem erhöht sich der
Systemdruck zusätzlich durch das verdampfende Adsorptiv. Dies muss bei einer Berech-
nung der herrschenden Partialdrücke berücksichtigt werden.
Die Desorption erfolgte in einen Probenträger aus Quarzglas, welcher in einer Labor-
mikrowelle der Firma CEM (Typ: Mars 5) eingebaut und mit Stickstoff gespült werden
konnte. Der Probenträger wurde bei den im Rahmen dieser Arbeit durchgeführten Unter-
suchungen nicht mit einer Isolierung ummantelt.
Abbildung 6.8: Beladungsapparatur
(1: Probenhalter; 2: Beladungsgefäß; 3: Abkühlgefäß)

6.4 Für Sorptionsuntersuchungen eingesetzte Apparaturen und Analytik 133
Für die thermogravimetrischen Messungen kam ein Gerät der Firma TA Instruments vom
Typ SDT 2960 zum Einsatz. Bei der Thermogravimetrie wird der Gewichtsverlust einer
Probe als Funktion der Temperatur gemessen, während die Probe einem geregelten
Temperaturprogramm unterworfen ist. Die Probe/Waage wird bei der Messung mit einem
konstanten Gasstrom gespült. Im Rahmen dieser Arbeit wurden Stickstoff und syntheti-
sche Luft als Spülgase verwendet.
Das aus der Thermowaage austretende Abgas wurde mit einem Quadrupol-Mas-
senspektrometer der Firma Balzers (Typ: ThermoStar 200) analysiert, welches die Detek-
tion relativer Atommassen von 1 bis 100 u erlaubt. Für detailliertere Informationen über
diese Messmethoden und die physikalischen Grundlagen sei auf die Fachliteratur
verwiesen [334-336].

7 Reaktionstechnische Untersuchungen
In diesem Kapitel werden die Ergebnisse der reaktionstechnischen Untersuchungen zur
Hydroxylierung von Benzen mit N2O diskutiert. Hierbei soll zunächst der Einfluss von
Mikrowellenstrahlung auf die N2O-Zersetzung gesondert betrachtet werden, weil diesem
Teilschritt in der Literatur eine zentrale Rolle im Reaktionsmechanismus zugeschrieben
wird (vgl. Kapitel 3.4.2). Die Präsentation der Messergebnisse aus der Benzenhydroxylie-
rung unter Mikrowelleneinstrahlung erfolgt im zweiten Teil dieses Kapitels.
7.1 N2O-Zersetzung
Nach der Theorie von PANOV ET AL. [225] muss für die heterogen katalysierte
Hydroxylierung von Benzen mit N2O zu Phenol zunächst eine aktive Oberflächensauer-
stoffspezies (α-Sauerstoff) an den Fe-Zentren des Katalysators gebildet werden, welche
mit dem Benzen weiterreagiert. Die Zersetzung des N2O bildet somit den ersten Teilschritt
in der Hydroxylierung und nimmt eine zentrale Rolle beim Gesamtprozess ein. Deshalb
wird die N2O-Zersetzung auch häufig getrennt von der Hydroxylierung untersucht [300,
337-341]. Da N2O als Treibhausgas fungiert, besteht jedoch auch aus umwelttechnischen
Gesichtspunkten ein steigendes Interesse an der Erforschung dieser Reaktion [342].
Ziel dieser Untersuchungen war es einerseits festzustellen, ob Mikrowelleneinstrahlung
einen Einfluss auf diese Reaktion hat und andererseits die Katalysatoren hinsichtlich ihrer
Aktivität in der N2O-Zersetzung zu charakterisieren.
Aus theoretischer Sicht ist eigentlich keine Wechselwirkung der Mikrowellen mit dem
N2O zu erwarten, da der Dissipationsfaktor wie bei allen Gasen sehr klein ist (vgl. Seite
15). Ein Energieeintrag der Mikrowellen könnte somit nur am Katalysator selbst erfolgen.
Als problematisch erweist sich jedoch, dass die Dielektrizitätskonstanten (bzw. Dissipati-
onsfaktoren) am Reinstoff gemessen werden. Während der Reaktion liegt das N2O jedoch
an der Oberfläche gebunden vor. Wie in Kapitel 2.1 beschrieben, hat dies eine Änderung
der dielektrischen Eigenschaften zur Folge. Außerdem treten bei adsorbierten Stoffen
Grenz- und Oberflächeneffekte auf, welche die Dielektrizitätskonstante verändern. Des-
halb war es nötig, den Einfluss der Mikrowellenstrahlung auf die N2O-Zersetzung
detailliert zu untersuchen.

7.1 N2O-Zersetzung 135
7.1.1 Versuchsdurchführung
Die verwendete Versuchapparatur und Analytik sind in Kapitel 6 ausführlich beschrieben.
Für die Untersuchungen wurde jeweils 1 g Katalysator (Partikelgröße 1,0 – 1,2 mm) ein-
gesetzt. Um eine gleichmäßige Durchströmung des Katalysatorbetts zu gewährleisten
bzw. die Ausbildung von axialen Temperaturgradienten aufgrund der exothermen Reak-
tion zu vermeiden, wurde der Katalysator mit 20 ml Quarzglasbruch (Partikelgröße 0,8 –
1,0 mm) vermischt.
Vor jedem Versuch erfolgte eine Trocknung der Katalysatoren für zwei Stunden im N2-
Strom (200 Nml/min) bei 400 °C. Anschließend wurde die gewünschte Reaktionstempe-
ratur, Verweilzeit (Gasstrom) und Mikrowellenleistung eingestellt und die Reaktion ge-
startet. Um eine Verfälschung der Messergebnisse durch gegebenenfalls auftretende
Einlaufeffekte nach dem Starten der Reaktion zu vermeiden, wurden die Messwerte nach
30 min Reaktionsdauer („Time-On-Stream“; TOS) ausgewertet.
Bei den Katalysatoren H-ZSM-5 (I), Fe-ZSM5- (II) und Co-ZSM-5 wurde der Mikrowel-
leneinfluss auf die N2O-Zersetzung in einem Temperaturbereich zwischen 300 und 500 °C
bestimmt. Zu diesem Zweck wurde jeweils ein konventionell beheizter Versuch und ein
Experiment, bei dem zusätzlich Mikrowellen einer Leistung von 120 W eingestrahlt
wurde, durchgeführt. Die Messungen erfolgten bei zwei Verweilzeiten, welche auch in der
Benzenhydroxylierung eingestellt wurden (vgl. Kapitel 7.2). Die Angabe der Verweilzeit
erfolgt in dieser Arbeit als so genannte modifizierte Verweilzeit τmod, in der die eingesetzte
Katalysatormasse eingeht. Es gilt:
mod W/F Katalysator
ges
m
nτ = = (Gl. 7.1)
7.1.2 H-ZSM-5 (I)
Die N2O-Umsatzgrade des Katalysators H-ZSM-5 (I) in Abhängigkeit von der
Reaktionstemperatur, Verweilzeit und Mikrowelleneinstrahlung sind in Abbildung 7.1
dargestellt. Betrachtet man zunächst die Kurve für τmod = 90 (g·min)/mol ohne Mikrowel-
leneinstrahlung (●), so erkennt man, dass der Katalysator bei Temperaturen unter 400 °C
kaum aktiv ist. Erst ab einer Temperatur von 430 °C steigen die Umsatzgrade mit zuneh-
mender Temperatur rasch an.
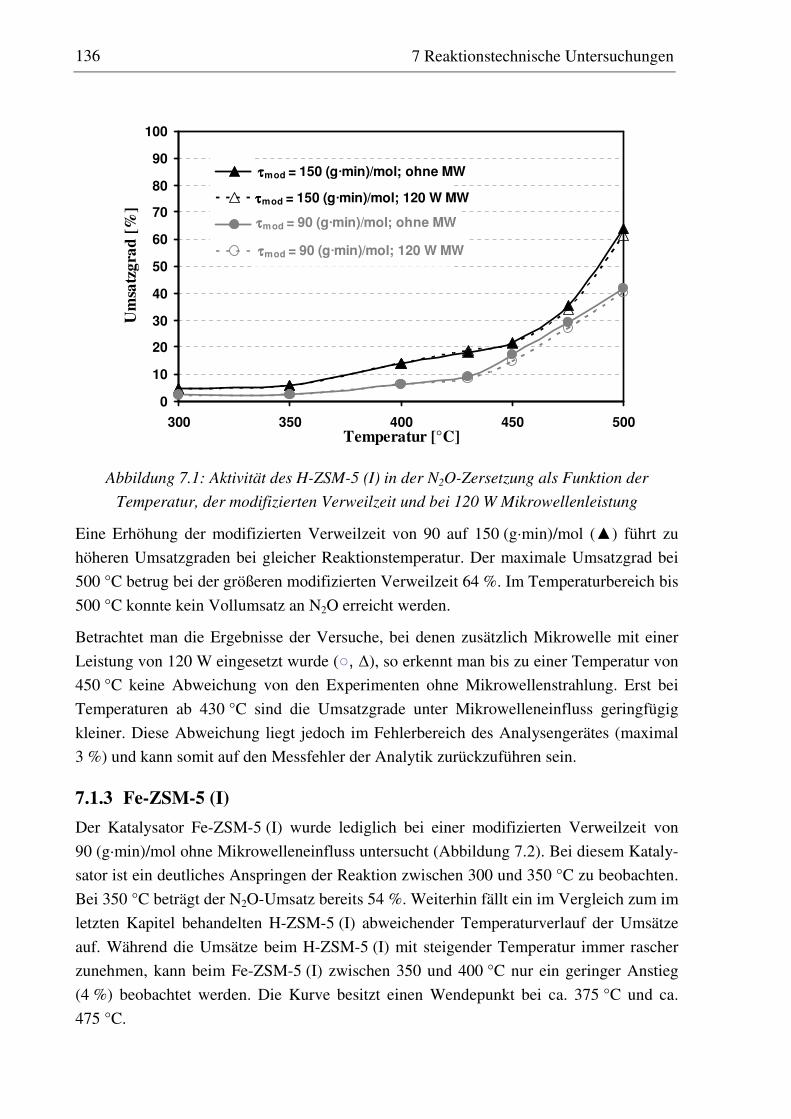
7 Reaktionstechnische Untersuchungen 136
0
10
20
30
40
50
60
70
80
90
100
300 350 400 450 500Temperatur [°C]
Um
satz
grad
[%
]
ττττmod = 150 (g·min)/mol; ohne MW
ττττmod = 150 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; ohne MW
Abbildung 7.1: Aktivität des H-ZSM-5 (I) in der N2O-Zersetzung als Funktion der
Temperatur, der modifizierten Verweilzeit und bei 120 W Mikrowellenleistung
Eine Erhöhung der modifizierten Verweilzeit von 90 auf 150 (g·min)/mol (▲) führt zu
höheren Umsatzgraden bei gleicher Reaktionstemperatur. Der maximale Umsatzgrad bei
500 °C betrug bei der größeren modifizierten Verweilzeit 64 %. Im Temperaturbereich bis
500 °C konnte kein Vollumsatz an N2O erreicht werden.
Betrachtet man die Ergebnisse der Versuche, bei denen zusätzlich Mikrowelle mit einer
Leistung von 120 W eingesetzt wurde (○, ∆), so erkennt man bis zu einer Temperatur von
450 °C keine Abweichung von den Experimenten ohne Mikrowellenstrahlung. Erst bei
Temperaturen ab 430 °C sind die Umsatzgrade unter Mikrowelleneinfluss geringfügig
kleiner. Diese Abweichung liegt jedoch im Fehlerbereich des Analysengerätes (maximal
3 %) und kann somit auf den Messfehler der Analytik zurückzuführen sein.
7.1.3 Fe-ZSM-5 (I)
Der Katalysator Fe-ZSM-5 (I) wurde lediglich bei einer modifizierten Verweilzeit von
90 (g·min)/mol ohne Mikrowelleneinfluss untersucht (Abbildung 7.2). Bei diesem Kataly-
sator ist ein deutliches Anspringen der Reaktion zwischen 300 und 350 °C zu beobachten.
Bei 350 °C beträgt der N2O-Umsatz bereits 54 %. Weiterhin fällt ein im Vergleich zum im
letzten Kapitel behandelten H-ZSM-5 (I) abweichender Temperaturverlauf der Umsätze
auf. Während die Umsätze beim H-ZSM-5 (I) mit steigender Temperatur immer rascher
zunehmen, kann beim Fe-ZSM-5 (I) zwischen 350 und 400 °C nur ein geringer Anstieg
(4 %) beobachtet werden. Die Kurve besitzt einen Wendepunkt bei ca. 375 °C und ca.
475 °C.

7.1 N2O-Zersetzung 137
0
10
20
30
40
50
60
70
80
90
100
300 350 400 450 500Temperatur [°C]
Um
satz
grad
[%
]
ττττmod = 90 [(g·min)/mol] ohne MW
Abbildung 7.2: Aktivität des Fe-ZSM-5 (I) in der N2O-Zersetzung als Funktion der
Temperatur
7.1.4 Fe-ZSM-5 (II)
Analog zum H-ZSM-5 (I) wurde auch der Katalysator Fe-ZSM-5 (II) getestet (Abbildung
7.3). Im Vergleich zu der bereits diskutierten H-Form fällt sofort auf, dass der Katalysator
mit Eisen bei identischen Temperaturen deutlich aktiver ist. Bis zur Temperatur von
430 °C sind die Umsatzgrade mehr als doppelt so hoch wie beim H-ZSM-5 (I). Bei einer
Temperatur von 350 °C und einer modifizierten Verweilzeit von 90 (g·min)/mol beträgt
der N2O-Umsatzgrad am H-ZSM-5 (I) beispielsweise 2,4 %, während am Fe-ZSM-5 (II)
bereits 7 % umgesetzt werden. Noch deutlicher ist der Unterschied zum Fe-ZSM-5 (I), der
unter entsprechenden Bedingungen bereits einen Umsatzgrad von 54 % aufweist.
Im Temperaturbereich über 400 °C nimmt die Aktivität rasch zu. Bei 500 °C wird
Vollumsatz an N2O erreicht. Eine Steigerung der Verweilzeit von 90 auf 150 (g·min)/mol
führt bei Temperaturen unter 450 °C zu einer Steigerung des N2O-Umsatzgrades um ca.
10 %. Bei 475 und 500 °C hat die Erhöhung der Verweilzeit kaum einen Einfluss, da
beinahe das gesamte N2O umgesetzt wird.
Betrachtet man die Versuche, bei denen zusätzlich Mikrowelle mit einer Leistung von
120 W eingestrahlt wurde (○, ∆), zeigt sich das gleiche Bild wie beim H-ZSM-5 (I). Es ist
im Rahmen des Messfehlers kein Einfluss der Mikrowellenstrahlung auf die N2O-
Zersetzung feststellbar.

7 Reaktionstechnische Untersuchungen 138
0
10
20
30
40
50
60
70
80
90
100
300 350 400 450 500Temperatur [°C]
Um
satz
grad
[%
]
ττττmod = 150 (g·min)/mol; ohne MW
ττττmod = 150 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; ohne MW
Abbildung 7.3: Aktivität des Fe-ZSM-5 (II) in der N2O-Zersetzung als Funktion der
Temperatur, der modifizierten Verweilzeit und bei 120 W Mikrowellenleistung
7.1.5 Co-ZSM-5
Als dritter Katalysator wurde der Co-ZSM-5 in der N2O-Zersetzung getestet (Abbildung
7.4). Im niedrigen Temperaturbereich um 300 °C entsprechen die Umsatzgrade dieses
Katalysators annähernd denen des Fe-ZSM-5 (II) bei entsprechenden modifizierten Ver-
weilzeiten. Die Umsatzgrade steigen dann jedoch sehr schnell an, so dass bei 430 °C
beinahe Vollumsatz des N2O erreicht wird. Eine Steigerung der modifizierten Verweilzeit
von 90 auf 150 (g·min)/mol hat identische Auswirkungen wie beim Fe-ZSM-5 (II). Bei
Umsatzgraden kleiner 90 % führt die Erhöhung der modifizierten Verweilzeit zu einer
Zunahme des Umsatzgrades von ca. 10 %, bei sehr hohen Umsätzen ist kaum noch ein
Effekt durch eine Änderung der Verweilzeit zu beobachten.
Im Temperaturbereich ab 350 °C zeigte der Co-ZSM-5 somit von allen untersuchten
Katalysatoren die höchste Aktivität.
Auch bei diesem Katalysator konnte kein signifikanter Einfluss der Mikrowelleneinstrah-
lung auf die Aktivität in der N2O-Zersetzung festgestellt werden. Die geringen Abwei-
chungen dürften auf der Messungenauigkeit der verwendeten Analytik beruhen.
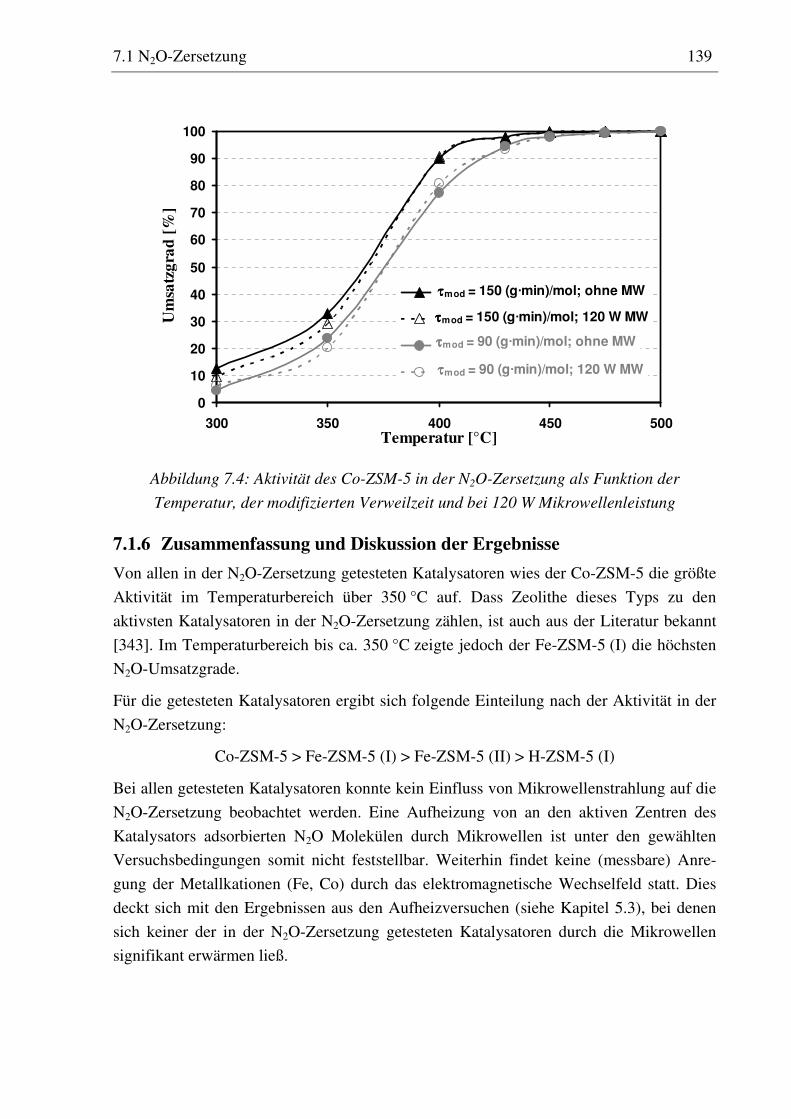
7.1 N2O-Zersetzung 139
0
10
20
30
40
50
60
70
80
90
100
300 350 400 450 500Temperatur [°C]
Um
satz
grad
[%
]
ττττmod = 150 (g·min)/mol; ohne MW
ττττmod = 150 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; 120 W MW
ττττmod = 90 (g·min)/mol; ohne MW
Abbildung 7.4: Aktivität des Co-ZSM-5 in der N2O-Zersetzung als Funktion der
Temperatur, der modifizierten Verweilzeit und bei 120 W Mikrowellenleistung
7.1.6 Zusammenfassung und Diskussion der Ergebnisse
Von allen in der N2O-Zersetzung getesteten Katalysatoren wies der Co-ZSM-5 die größte
Aktivität im Temperaturbereich über 350 °C auf. Dass Zeolithe dieses Typs zu den
aktivsten Katalysatoren in der N2O-Zersetzung zählen, ist auch aus der Literatur bekannt
[343]. Im Temperaturbereich bis ca. 350 °C zeigte jedoch der Fe-ZSM-5 (I) die höchsten
N2O-Umsatzgrade.
Für die getesteten Katalysatoren ergibt sich folgende Einteilung nach der Aktivität in der
N2O-Zersetzung:
Co-ZSM-5 > Fe-ZSM-5 (I) > Fe-ZSM-5 (II) > H-ZSM-5 (I)
Bei allen getesteten Katalysatoren konnte kein Einfluss von Mikrowellenstrahlung auf die
N2O-Zersetzung beobachtet werden. Eine Aufheizung von an den aktiven Zentren des
Katalysators adsorbierten N2O Molekülen durch Mikrowellen ist unter den gewählten
Versuchsbedingungen somit nicht feststellbar. Weiterhin findet keine (messbare) Anre-
gung der Metallkationen (Fe, Co) durch das elektromagnetische Wechselfeld statt. Dies
deckt sich mit den Ergebnissen aus den Aufheizversuchen (siehe Kapitel 5.3), bei denen
sich keiner der in der N2O-Zersetzung getesteten Katalysatoren durch die Mikrowellen
signifikant erwärmen ließ.

7 Reaktionstechnische Untersuchungen 140
7.2 Hydroxylierung von Benzen mit N2O
Die Katalysatoren H-ZSM-5 (I), Fe-ZSM5 (I), Fe-ZSM-5 (II) und Co-ZSM-5 wurden in
der Hydroxylierung von Benzen mit N2O untersucht. Damit wurde eine Reihe von Kataly-
satoren ausgewählt, welche sich in ihren chemischen und katalytischen Eigenschaften
deutlich unterscheiden (vgl. Kapitel 5.1). Das Hauptaugenmerk der Experimente lag
jedoch nicht darin, die Eignung dieser Katalysatoren für die Benzenhydroxylierung zu
untersuchen, sondern zu ermitteln, ob ein Einfluss von Mikrowelleneinstrahlung auf die
Reaktion feststellbar ist. Zu diesem Zweck wurde jeder Katalysator bei identischen Reak-
tionsbedingungen (Temperatur, Verweilzeit, Feedzusammensetzung) sowohl unter kom-
binierter Heizung (konventionell + Mikrowelle) sowie unter ausschließlicher Verwen-
dung der konventionellen Heizung (d. h. ohne Mikrowelle) getestet. Die Reaktionstempe-
ratur, eingestrahlte Mikrowellenleistung und Verweilzeit wurden variiert. Niedrigere
Reaktionstemperaturen sollten nach REITZMANN [24] die Desorption des Phenols
erschweren und damit günstigere Voraussetzungen für den Nachweis einer selektiven
Desorption durch Mikrowellen darstellen. An den Katalysatoren Fe-ZSM-5 (II) und
Co-ZSM-5 erfolgte zudem eine Variation der modifizierten Verweilzeit. Aus den Arbeiten
von HÄFELE [233] und REITZMANN [24] geht hervor, dass sich eine Fahrweise mit
überstöchiometrischer Benzenzugabe positiv auf das Desaktivierungsverhalten der ZSM-5
Zeolithe auswirkt. Deshalb wurden am Katalysator Fe-ZSM-5 (II) Versuche mit Benze-
nüberschuss im Eduktgemisch durchgeführt. Am Katalysator Co-ZSM-5 wurde außerdem
der Einfluss von Wasser auf das Reaktionssystem untersucht. Im Folgenden sollen die
Ergebnisse für jeden Katalysator zunächst gesondert betrachtet werden. Abschließend
folgt eine vergleichende Diskussion und Zusammenfassung der Ergebnisse.
7.2.1 Versuchsdurchführung
Laborversuchsanlage und Analytik werden in Kapitel 6 ausführlich beschrieben. Die bei
den Untersuchungen verwendete Katalysatormenge betrug jeweils 1 g (Partikelgröße 1,0 –
1,2 mm). Der Katalysator wurde mit 20 ml Quarzglasbruch (Partikelgröße 0,8 – 1,0 mm)
verdünnt.
Der Versuchsablauf war folgender:
• Spülen des Katalysators mit Stickstoff (200 Nml/min) für eine Stunde
• Einstellen der Reaktionstemperatur, Verweilzeit (N2-Ströme des Verdampfers und
Sättigers), Benzen- und N2O-Molenstrom
• Bypassmessung (Überprüfung der Dosiergenauigkeit des Verdampfers)
• Gegebenenfalls Starten des Sättigers (der Katalysator wurde aus anlagentechni-
schen Gründen bereits während der Bypassmessung mit der Stickstoff-Wasser-
Mischung überströmt)

7.2 Hydroxylierung von Benzen mit N2O 141
• Starten der Reaktion
• Gegebenenfalls Starten der Mikrowelle
• Starten der GC-Messungen nach 5 min TOS
Nach jedem Experiment wurde der Katalysator für eine Stunde bei einer Temperatur von
530 °C mit einer Mischung aus Stickstoff (140 Nml/min) und Sauerstoff (60 Nml/min)
regeneriert. Auf diese Weise ließ sich die ursprüngliche Aktivität der Katalysatoren
wieder herstellen. Genauere Hinweise zur Versuchsdurchführung und zur Auswertung der
Messungen finden sich bei GOPALAKRISHNAN [320].
7.2.2 H-ZSM-5 (I)
7.2.2.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C
In diesem Kapitel werden die Messungen am Katalysator H-ZSM-5 (I) behandelt.
Abbildung 7.5 zeigt das Standzeitverhalten dieses Katalysators bei 400 °C ohne zusätzli-
chen Einsatz von Mikrowellen. Anhand des Diagramms sollen charakteristische Merk-
male aufgezeigt werden, welche unabhängig von den Reaktionsparametern für alle
Messungen an diesem Katalysator gelten. Betrachtet man den Umsatzgrad an Benzen (X
Benzen), so fällt auf, dass der stärkste Rückgang der Aktivität zwischen 5 und 13 min
TOS zu verzeichnen ist. Der Benzenumsatzgrad fällt innerhalb der ersten 8 min von 25
auf 19,7 %. Dies entspricht einem relativen Umsatzrückgang bzw. einer relativen Desak-
tivierung von ca. 23 % bezogen auf die Anfangsaktivität (nach 5 min TOS). Mit fort-
schreitender Reaktionsdauer bzw. fallenden Umsatzgraden nimmt die Desaktivierungs-
geschwindigkeit immer weiter ab. Um einen Vergleich der Desaktivierung an den ver-
schiedenen Katalysatoren unabhängig vom Benzenumsatzgrad zu ermöglichen, wird im
Folgenden immer der relative Rückgang des Umsatzgrades von Benzen zwischen 5 und
61 min TOS betrachtet. Im Beispiel aus Abbildung 7.5 entspricht dies einer relativen
Desaktivierung von 41 % nach 61 min TOS.
Die Ausbeute an Phenol (Y Phenol) verhält sich ähnlich wie der Benzenumsatzgrad. Der
stärkste Rückgang ist zu Beginn der Reaktion zu verzeichnen.
Betrachtet man nun die auf Benzen bezogene Selektivität zu Phenol (S Phenol), so fällt
auf, dass mit fortschreitender Reaktion ein Anstieg zu verzeichnen ist. Nach 5 min TOS
beträgt die Selektivität zu Phenol lediglich 64 %. Bereits nach 61 min wird ein Wert von
70 % überschritten. Nach 109 min wird eine auf Benzen bezogene Phenolselektivität von
76 % erreicht. Zu Beginn der Reaktion ist die Desaktivierungsgeschwindigkeit am
größten, d.h. es wird wahrscheinlich nicht Phenol, sondern Koks gebildet, was sich in
einer niedrigen Selektivität zu Phenol widerspiegelt. Wird die Desaktivierung im Lauf der
Reaktion langsamer, steigt wiederum die Selektivität zu Phenol an.

7 Reaktionstechnische Untersuchungen 142
Als einziges Nebenprodukt wurde Benzochinon detektiert, welches während der gesamten
Reaktionsdauer mit einer konstanten auf Benzen bezogenen Selektivität (S Benzochinon)
von 2,9 % gebildet wurde.
0
10
20
30
0 20 40 60 80 100 120
TOS [min]
X B
en
ze
n, Y
Ph
en
ol [%
]
0
20
40
60
80
100
S P
he
no
l [%
]
X Benzen Y Phenol S Phenol
rel. Desaktivierung = 41 %
Abbildung 7.5: Standzeitverhalten H-ZSM-5 (I)
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol)
7.2.2.2 Variation der Reaktionstemperatur
Die Temperaturabhängigkeit der Hydroxylierung wurde in einem Temperaturbereich
zwischen 300 und 400 °C untersucht. Bei einer Reaktionstemperatur von 300 °C war der
Katalysator gänzlich inaktiv. Abbildung 7.6 zeigt das katalytische Verhalten nach 13 min
TOS bei einer Temperatur von 350 und 400 °C. Es sind jeweils ein Experiment ohne Mik-
rowellenstrahlung und ein Vergleichsexperiment, bei dem Mikrowellen mit einer Leistung
von 200 W eingestrahlt wurden, gegenübergestellt.
Betrachtet man zunächst die Experimente, welche ohne Mikrowelle durchgeführt wurden,
so fällt auf, dass der Benzenumsatzgrad von 6,5 % bei 350 °C auf 19,7 % bei 400 °C
ansteigt. Die korrespondierenden Ausbeuten nehmen ebenfalls zu. Die Selektivität zu
Phenol ist bei höherer Temperatur jedoch um 10 % niedriger. Weiterhin verlangsamt sich
die relative Desaktivierung (∆X) zwischen 5 und 61 min von 47 % bei 350 °C auf 41 %
bei 400 °C. Dies lässt sich nur durch eine beginnende Totaloxidation (CO2) der Reaktan-
den und des Kokses durch N2O erklären. Dieses Verhalten wurde bereits in den Arbeiten
von REITZMANN [24] und HIEMER [26] beschrieben.
Benzochinon wurde erst ab bei einer Reaktionstemperatur von 400 °C detektiert.

7.2 Hydroxylierung von Benzen mit N2O 143
Abbildung 7.6: Einfluss von Temperatur und Mikrowelleneinstrahlung auf das
katalytische Verhalten des H-ZSM-5 (I)
(N2O:Benzen = 1:1, τmod = 90 (g·min)/mol), TOS = 13 min)
Ein Vergleich der Experimente ohne Mikrowelleneinstrahlung mit den Messungen bei
einer Mikrowellenleistung von 200 W zeigt, dass durch die Mikrowellen keine signifi-
kanten Änderungen des Reaktionsverhaltens bewirkt wurden. Bei einer Temperatur von
350 °C sind die Benzenumsatzgrade, Phenolausbeuten und Selektivitäten von Phenol
nahezu identisch, unabhängig davon, ob der Versuch mit Mikrowellen durchgeführt
wurde. Lediglich die Desaktivierung zwischen 5 und 61 min TOS ist um 1,5 % verringert.
Diese Abweichung bewegt sich jedoch im Rahmen des Messfehlers bzw. der Dosierge-
nauigkeit. Bei einer Reaktionstemperatur von 400 °C zeigen sich ebenfalls keine signifi-
kanten Unterschiede zwischen den Experimenten mit und ohne Mikrowelle. Durch die
Mikrowellen ist eine minimale Erhöhung der Phenolselektivität (von 69 auf 71 %) und der
Desaktivierung (Zunahme 0,6 %) zu beobachten. Die Selektivität zum Folgeprodukt
Benzochinon wurde dagegen von 2,9 auf 2,3 % erniedrigt. Das Standzeitverhalten der
Mikrowellenexperimente entsprach dem der Messungen ohne zusätzliche Mikrowellen-
einstrahlung. Es lässt sich somit zusammenfassen, dass am Katalysator H-ZSM-5 (I) kein
signifikanter Einfluss von Mikrowellenstrahlung auf das Reaktionssystem festgestellt
werden konnte.
7.2.3 Fe-ZSM-5 (I)
Als zweiter Katalysator wurde der Fe-ZSM-5 (I) in der Benzenhydroxylierung unter
Mikrowelleneinstrahlung eingesetzt. Das im Katalysator enthaltene Eisen fungiert als
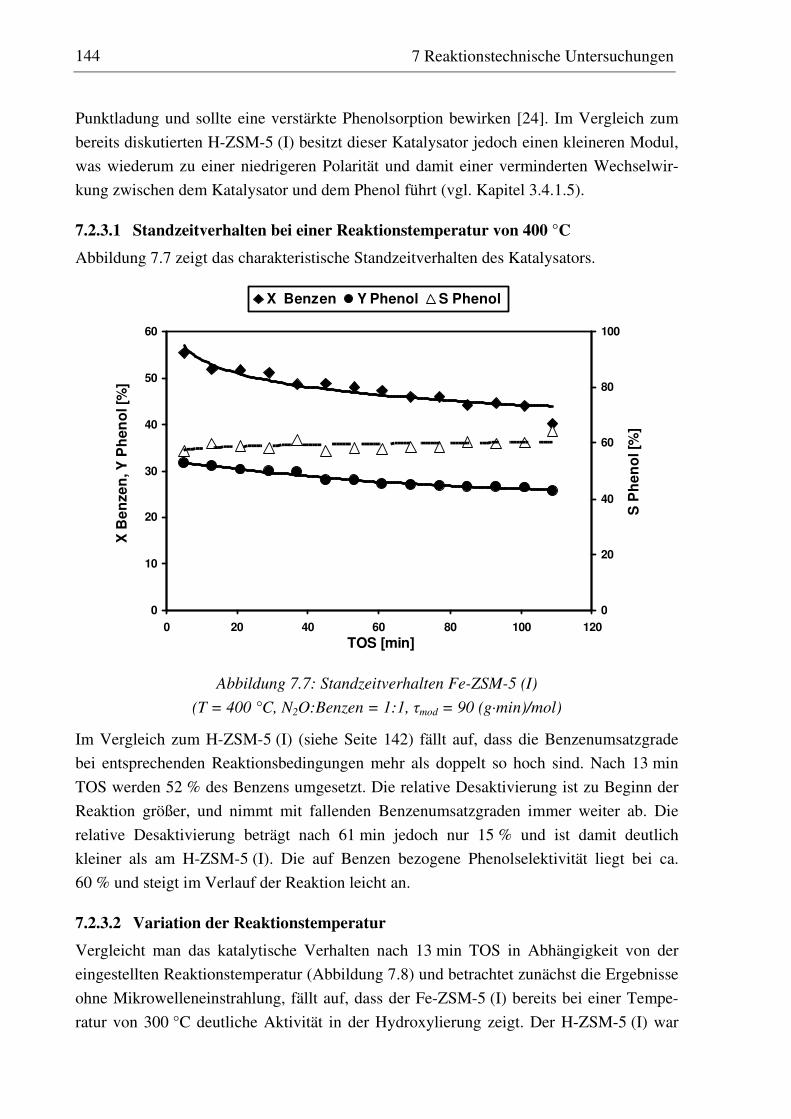
7 Reaktionstechnische Untersuchungen 144
Punktladung und sollte eine verstärkte Phenolsorption bewirken [24]. Im Vergleich zum
bereits diskutierten H-ZSM-5 (I) besitzt dieser Katalysator jedoch einen kleineren Modul,
was wiederum zu einer niedrigeren Polarität und damit einer verminderten Wechselwir-
kung zwischen dem Katalysator und dem Phenol führt (vgl. Kapitel 3.4.1.5).
7.2.3.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C
Abbildung 7.7 zeigt das charakteristische Standzeitverhalten des Katalysators.
0
10
20
30
40
50
60
0 20 40 60 80 100 120
TOS [min]
X B
en
ze
n, Y
Ph
en
ol [%
]
0
20
40
60
80
100
S P
he
no
l [%
]
X Benzen Y Phenol S Phenol
Abbildung 7.7: Standzeitverhalten Fe-ZSM-5 (I)
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol)
Im Vergleich zum H-ZSM-5 (I) (siehe Seite 142) fällt auf, dass die Benzenumsatzgrade
bei entsprechenden Reaktionsbedingungen mehr als doppelt so hoch sind. Nach 13 min
TOS werden 52 % des Benzens umgesetzt. Die relative Desaktivierung ist zu Beginn der
Reaktion größer, und nimmt mit fallenden Benzenumsatzgraden immer weiter ab. Die
relative Desaktivierung beträgt nach 61 min jedoch nur 15 % und ist damit deutlich
kleiner als am H-ZSM-5 (I). Die auf Benzen bezogene Phenolselektivität liegt bei ca.
60 % und steigt im Verlauf der Reaktion leicht an.
7.2.3.2 Variation der Reaktionstemperatur
Vergleicht man das katalytische Verhalten nach 13 min TOS in Abhängigkeit von der
eingestellten Reaktionstemperatur (Abbildung 7.8) und betrachtet zunächst die Ergebnisse
ohne Mikrowelleneinstrahlung, fällt auf, dass der Fe-ZSM-5 (I) bereits bei einer Tempe-
ratur von 300 °C deutliche Aktivität in der Hydroxylierung zeigt. Der H-ZSM-5 (I) war
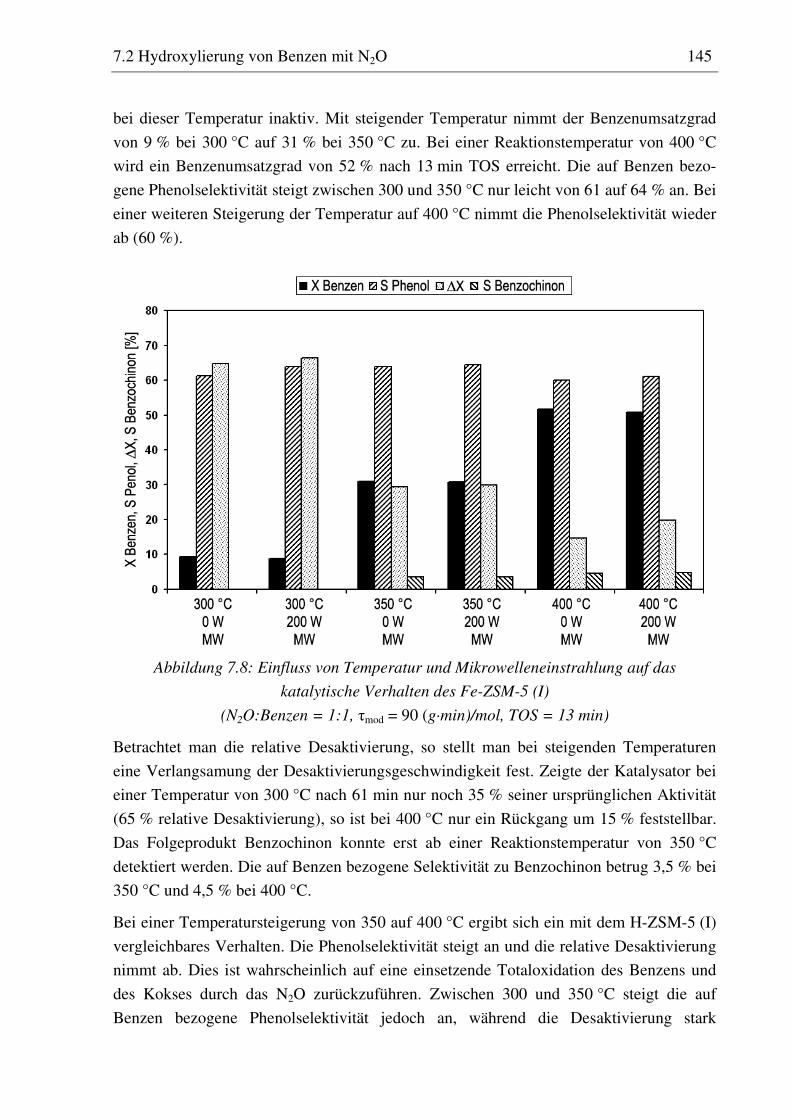
7.2 Hydroxylierung von Benzen mit N2O 145
bei dieser Temperatur inaktiv. Mit steigender Temperatur nimmt der Benzenumsatzgrad
von 9 % bei 300 °C auf 31 % bei 350 °C zu. Bei einer Reaktionstemperatur von 400 °C
wird ein Benzenumsatzgrad von 52 % nach 13 min TOS erreicht. Die auf Benzen bezo-
gene Phenolselektivität steigt zwischen 300 und 350 °C nur leicht von 61 auf 64 % an. Bei
einer weiteren Steigerung der Temperatur auf 400 °C nimmt die Phenolselektivität wieder
ab (60 %).
Abbildung 7.8: Einfluss von Temperatur und Mikrowelleneinstrahlung auf das
katalytische Verhalten des Fe-ZSM-5 (I)
(N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
Betrachtet man die relative Desaktivierung, so stellt man bei steigenden Temperaturen
eine Verlangsamung der Desaktivierungsgeschwindigkeit fest. Zeigte der Katalysator bei
einer Temperatur von 300 °C nach 61 min nur noch 35 % seiner ursprünglichen Aktivität
(65 % relative Desaktivierung), so ist bei 400 °C nur ein Rückgang um 15 % feststellbar.
Das Folgeprodukt Benzochinon konnte erst ab einer Reaktionstemperatur von 350 °C
detektiert werden. Die auf Benzen bezogene Selektivität zu Benzochinon betrug 3,5 % bei
350 °C und 4,5 % bei 400 °C.
Bei einer Temperatursteigerung von 350 auf 400 °C ergibt sich ein mit dem H-ZSM-5 (I)
vergleichbares Verhalten. Die Phenolselektivität steigt an und die relative Desaktivierung
nimmt ab. Dies ist wahrscheinlich auf eine einsetzende Totaloxidation des Benzens und
des Kokses durch das N2O zurückzuführen. Zwischen 300 und 350 °C steigt die auf
Benzen bezogene Phenolselektivität jedoch an, während die Desaktivierung stark

7 Reaktionstechnische Untersuchungen 146
zurückgeht. Dies könnte auf eine behinderte Desorption des Phenols bei niedrigen Tempe-
raturen zurückzuführen sein, welche zu einer beschleunigten Desaktivierung führt.
Betrachtet man die Experimente bei einer Mikrowelleneinstrahlung von 200 W und
vergleicht diese mit den Referenzversuchen ohne Mikrowelle, so sind nur geringe Unter-
schiede feststellbar. Allerdings zeichnen sich die gleichen Trends ab, wie sie bereits am
H-ZSM-5 (I) bei einer Reaktionstemperatur von 400 °C beobachtet wurden. Durch die
Mikrowellenstrahlung erniedrigen sich die Benzenumsatzgrade, während die Phenolse-
lektivität leicht ansteigt. Die relative Desaktivierung nimmt durch die Mikrowellen zu.
Die Unterschiede sind jedoch gering. Bei einer Reaktionstemperatur von 300 °C ging der
Umsatzgrad durch die Mikrowelleneinstrahlung um 0,5 % zurück, während die Phenolse-
lektivität um 2,6 % und die relative Desaktivierung um 1,8 % anstiegen. Diese Abwei-
chungen bewegen sich im Fehlerbereich der Analytik bzw. der Dosierung. Aus theoreti-
scher Sicht müsste die Wirkung der Mikrowellen bei niedrigen Reaktionstemperaturen am
größten sein, da in diesem Temperaturbereich (300 °C) eine behinderte Phenoldesorption
auftreten könnte. Aufgrund der geringen Unterschiede zwischen den Experimenten mit
und ohne Mikrowelleneinstrahlung lässt sich diese Annahme jedoch nicht bestätigen.
7.2.3.3 Variation der Mikrowellenleistung
Am Beispiel der Reaktionstemperatur 350 °C soll exemplarisch ein Einfluss der einge-
strahlten Mikrowellenleistung auf das Reaktionssystem diskutiert werden. Wie man in
Abbildung 7.9 erkennen kann, hat eine Steigerung der Mikrowellenleistung wiederum nur
geringe Auswirkungen auf die Reaktion.
Allerdings bestätigt sich der Trend, dass die Mikrowelleneinstrahlung zu einer Steigerung
der Phenolselektivität bei kleineren Umsatzgraden und leicht erhöhter relativer
Desaktivierung zwischen 5 und 61 min TOS führt. Die Unterschiede sind jedoch gering,
der maximale Rückgang des Umsatzgrades betrug 1,4 % (bei 400 W), die größte Steige-
rung der Phenolselektivität 6 % (bei 300 W) und die relative Desaktivierung nahm maxi-
mal 2,3 % (bei 300 W) zu. Damit liegen die Abweichungen im Bereich des Messfehlers,
allerdings zeigt sich dieser Trend bei allen Experimenten mit Mikrowelleneinstrahlung.
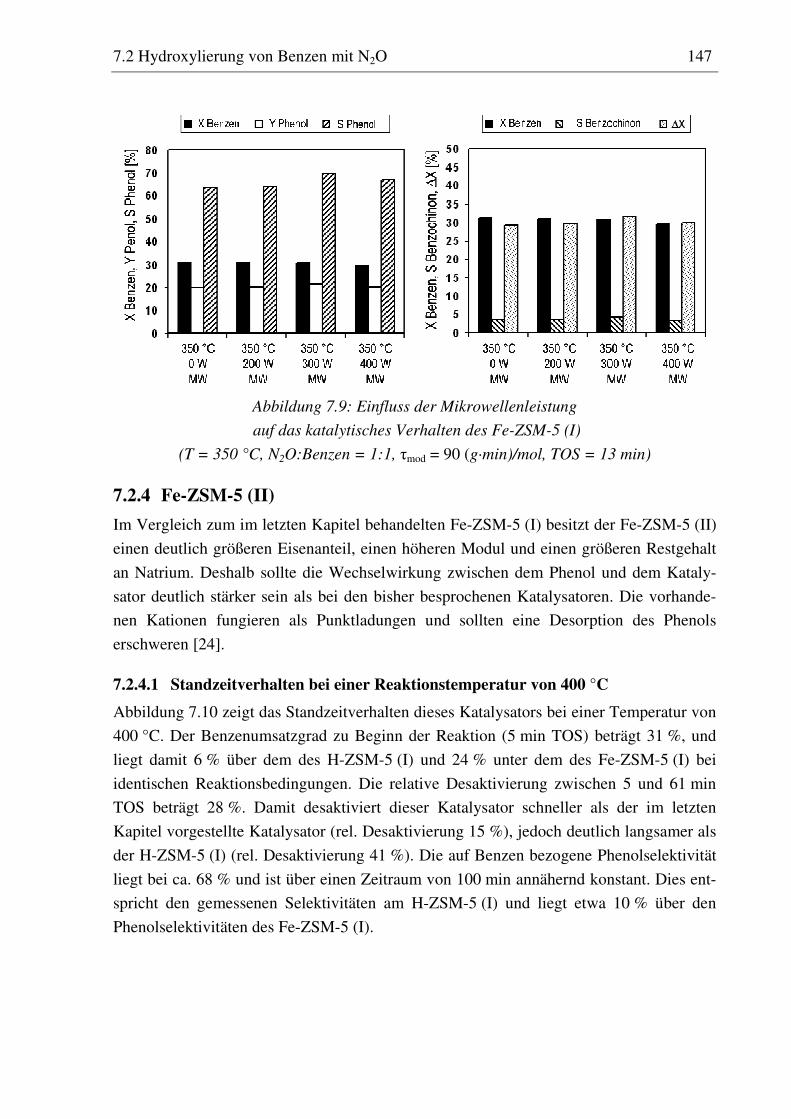
7.2 Hydroxylierung von Benzen mit N2O 147
Abbildung 7.9: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Fe-ZSM-5 (I)
(T = 350 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
7.2.4 Fe-ZSM-5 (II)
Im Vergleich zum im letzten Kapitel behandelten Fe-ZSM-5 (I) besitzt der Fe-ZSM-5 (II)
einen deutlich größeren Eisenanteil, einen höheren Modul und einen größeren Restgehalt
an Natrium. Deshalb sollte die Wechselwirkung zwischen dem Phenol und dem Kataly-
sator deutlich stärker sein als bei den bisher besprochenen Katalysatoren. Die vorhande-
nen Kationen fungieren als Punktladungen und sollten eine Desorption des Phenols
erschweren [24].
7.2.4.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C
Abbildung 7.10 zeigt das Standzeitverhalten dieses Katalysators bei einer Temperatur von
400 °C. Der Benzenumsatzgrad zu Beginn der Reaktion (5 min TOS) beträgt 31 %, und
liegt damit 6 % über dem des H-ZSM-5 (I) und 24 % unter dem des Fe-ZSM-5 (I) bei
identischen Reaktionsbedingungen. Die relative Desaktivierung zwischen 5 und 61 min
TOS beträgt 28 %. Damit desaktiviert dieser Katalysator schneller als der im letzten
Kapitel vorgestellte Katalysator (rel. Desaktivierung 15 %), jedoch deutlich langsamer als
der H-ZSM-5 (I) (rel. Desaktivierung 41 %). Die auf Benzen bezogene Phenolselektivität
liegt bei ca. 68 % und ist über einen Zeitraum von 100 min annähernd konstant. Dies ent-
spricht den gemessenen Selektivitäten am H-ZSM-5 (I) und liegt etwa 10 % über den
Phenolselektivitäten des Fe-ZSM-5 (I).

7 Reaktionstechnische Untersuchungen 148
0
10
20
30
40
0 20 40 60 80 100 120
TOS [min]
X B
en
ze
n, Y
Ph
en
ol [%
]
0
20
40
60
80
100
S P
he
no
l [%
]
X Benzen Y Phenol S Phenol
Abbildung 7.10: Standzeitverhalten Fe-ZSM-5 (II)
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol)
7.2.4.2 Variation der Reaktionstemperatur
Das katalytische Verhalten in Abhängigkeit von der Reaktionstemperatur nach 13 min
TOS (Abbildung 7.11) ähnelt dem des Fe-ZSM-5 (I) (Abbildung 7.8, S. 145). Bei einer
Steigerung der Reaktionstemperatur von 300 auf 350 °C steigt der Benzenumsatzgrad von
7 auf 16 % an. Bei einer Reaktionstemperatur von 400 °C wird ein Benzenumsatzgrad von
28 % erreicht. Im niedrigen Temperaturbereich ändert sich die Phenolselektivität kaum
und liegt bei ca. 74 % nach 13 min TOS. Bei 400 °C zeigt sich jedoch ein deutlicher
Rückgang der Selektivität auf 67 %. Betrachtet man die relative Desaktivierungs-
geschwindigkeit, so fällt auf, dass bei einer Temperatur von 350 °C ein Maximum erreicht
wird. Dies deutet darauf hin, dass die Desaktivierung nicht durch eine erschwerte Desorp-
tion des Phenols hervorgerufen wird. Wäre dies der Fall, sollte die relative Desaktivierung
bei niedrigen Temperaturen stärker ausgeprägt sein. Dieses Verhalten konnte am Fe-
ZSM-5 (I) beobachtet werden. Das Nebenprodukt Benzochinon wurde erst ab einer Reak-
tionstemperatur von 350 °C detektiert. Die auf Benzen bezogene Benzochinonselektivität
betrug bei 350 °C 4 % und bei 400 °C 5 %.
Führt man einen Vergleich der Experimente ohne Mikrowelleneinstrahlung mit den Mik-
rowellenexperimenten bei entsprechender Temperatur durch, so zeigen sich die gleichen
Tendenzen wie am Fe-ZSM-5 (I). Die zusätzliche Mikrowelleneinstrahlung führt zu ge-

7.2 Hydroxylierung von Benzen mit N2O 149
ringfügig höheren Selektivitäten, kleineren Benzenumsatzgraden und einer schnelleren
Desaktivierung.
Diese Trends sind am Fe-ZSM-5 (II) stärker ausgeprägt, wie man in Abbildung 7.11
erkennen kann. Bei einer Reaktionstemperatur von 300 °C lässt sich die Selektivität von
74 auf 77 % steigern. Der Benzenumsatzgrad geht von 7 auf 6 % zurück. Im Gegenzug
nimmt die relative Desaktivierung um 10 % zu. Der Mikrowelleneinfluss ist bei niedrige-
ren Temperaturen stärker ausgeprägt. Bei einer Temperatur von 400 °C sind nur minimale
Unterschiede feststellbar. Die Phenolselektivität steigt durch die Mikrowellen um 1,8 %
an, die Desaktivierung um 1,7 %. Die auf Benzen bezogene Benzochinonselektivität
nimmt durch die Mikrowelleneinstrahlung bei 350 °C von 3,7 auf 2,2 % ab. Bei 400 °C ist
ein Anstieg von 5,2 auf 5,7 % zu beobachten.
Abbildung 7.11: Einfluss von Temperatur und Mikrowelleneinstrahlung auf das
katalytische Verhalten des Fe-ZSM-5 (II)
(N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
7.2.4.3 Variation der Mikrowellenleistung
Anhand von Abbildung 7.12 soll der Einfluss der Mikrowellenleistung diskutiert werden.
Wie sich im letzten Kapitel gezeigt hat, führt eine Steigerung der Mikrowellenleistung am
Fe-ZSM-5 (I) nur zu einer geringfügigen Verstärkung des Mikrowelleneinflusses auf die
Reaktion. Dies bestätigt sich auch bei den Untersuchungen an diesem Katalysator.
Betrachtet man die Phenolselektivität bei steigender Mikrowellenleistung, ist eine
Tendenz zu steigenden Selektivitäten bei höherer Leistung erkennbar. Bei 200 W ist der

7 Reaktionstechnische Untersuchungen 150
Effekt jedoch weniger stark ausgeprägt als bei 120 W. Problematisch ist, dass sich die
Messungen nur geringfügig unterscheiden und somit Messfehler in Betracht gezogen wer-
den müssen. Bei den Benzenumsatzgraden ist lediglich eine Mittelwertabweichung von
0,6 % feststellbar. Auch die relative Desaktivierung nimmt zwischen 100 und 120 W nur
um 1 % zu. Bei einer Mikrowellenleistung von 200 W ist jedoch eine um 10 % stärkere
Desaktivierung zu beobachten. Dies macht deutlich, dass sich die Unterschiede der ein-
zelnen Versuche an der Grenze der Versuchsgenauigkeit (Versuchsdurchführung und
Analytik) bewegen. Der Vergleich absoluter Werte ist demnach nur eingeschränkt mög-
lich. Es zeigen sich jedoch Trends, welche in gleicher Form bei verschiedenen Reaktions-
bedingungen und Katalysatoren auftreten.
Abbildung 7.12: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Fe-ZSM-5 (II)
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
7.2.4.4 Variation der Verweilzeit
Alle bisher vorgestellten Messungen wurden bei einer modifizierten Verweilzeit von
90 (g·min)/mol durchgeführt. Eine Steigerung der Verweilzeit sollte dazu führen, dass
durch die Mikrowellen hervorgerufene Effekte auf das Reaktionssystem deutlicher zum
Vorschein kommen, da alle Reaktionsprodukte und insbesondre das Phenol länger mit den
Mikrowellen und dem Katalysator in Kontakt kommen. Deshalb wurde der Katalysator
auch bei einer modifizierten Verweilzeit von 120 und 155 (g·min)/mol getestet.
7.2.4.4.1 Reaktionstemperatur: 300 °C
Abbildung 7.13 zeigt das katalytische Verhalten in Abhängigkeit von der modifizierten
Verweilzeit und Mikrowelleneinstrahlung bei einer Temperatur von 300 °C. Betrachtet
man zunächst die Messungen ohne Mikrowelleneinstrahlung, erkennt man einen Anstieg
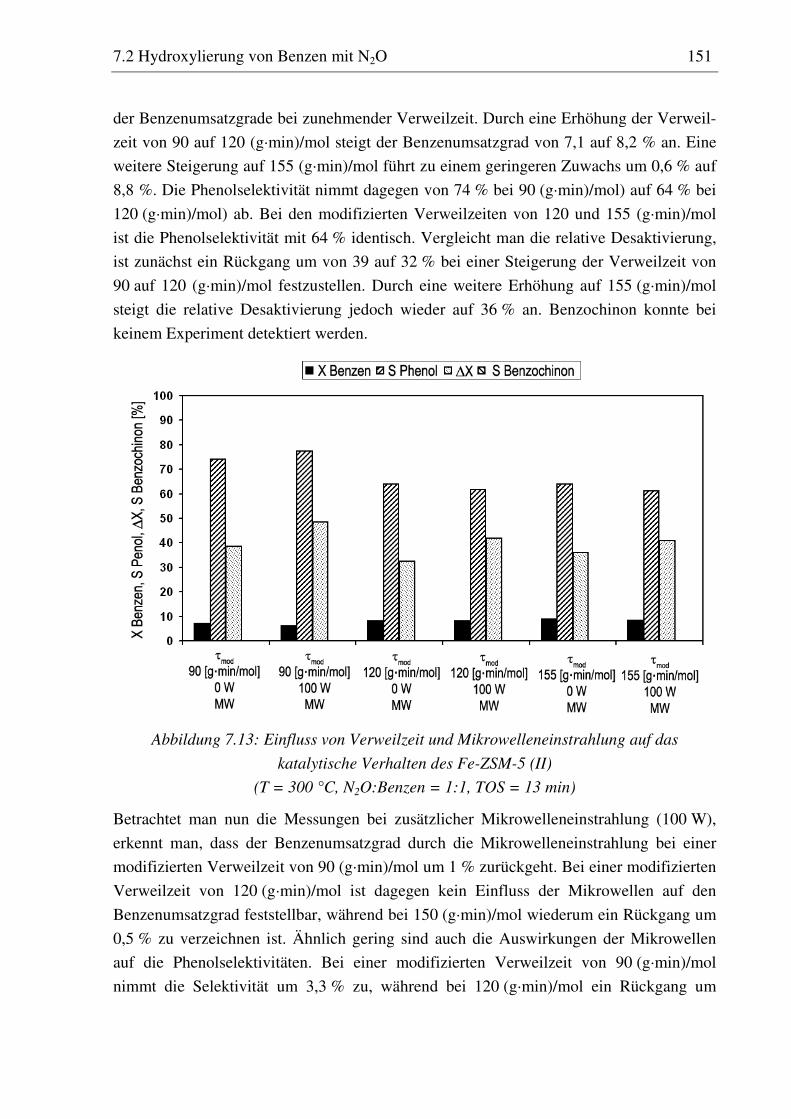
7.2 Hydroxylierung von Benzen mit N2O 151
der Benzenumsatzgrade bei zunehmender Verweilzeit. Durch eine Erhöhung der Verweil-
zeit von 90 auf 120 (g·min)/mol steigt der Benzenumsatzgrad von 7,1 auf 8,2 % an. Eine
weitere Steigerung auf 155 (g·min)/mol führt zu einem geringeren Zuwachs um 0,6 % auf
8,8 %. Die Phenolselektivität nimmt dagegen von 74 % bei 90 (g·min)/mol) auf 64 % bei
120 (g·min)/mol) ab. Bei den modifizierten Verweilzeiten von 120 und 155 (g·min)/mol
ist die Phenolselektivität mit 64 % identisch. Vergleicht man die relative Desaktivierung,
ist zunächst ein Rückgang um von 39 auf 32 % bei einer Steigerung der Verweilzeit von
90 auf 120 (g·min)/mol festzustellen. Durch eine weitere Erhöhung auf 155 (g·min)/mol
steigt die relative Desaktivierung jedoch wieder auf 36 % an. Benzochinon konnte bei
keinem Experiment detektiert werden.
Abbildung 7.13: Einfluss von Verweilzeit und Mikrowelleneinstrahlung auf das
katalytische Verhalten des Fe-ZSM-5 (II)
(T = 300 °C, N2O:Benzen = 1:1, TOS = 13 min)
Betrachtet man nun die Messungen bei zusätzlicher Mikrowelleneinstrahlung (100 W),
erkennt man, dass der Benzenumsatzgrad durch die Mikrowelleneinstrahlung bei einer
modifizierten Verweilzeit von 90 (g·min)/mol um 1 % zurückgeht. Bei einer modifizierten
Verweilzeit von 120 (g·min)/mol ist dagegen kein Einfluss der Mikrowellen auf den
Benzenumsatzgrad feststellbar, während bei 150 (g·min)/mol wiederum ein Rückgang um
0,5 % zu verzeichnen ist. Ähnlich gering sind auch die Auswirkungen der Mikrowellen
auf die Phenolselektivitäten. Bei einer modifizierten Verweilzeit von 90 (g·min)/mol
nimmt die Selektivität um 3,3 % zu, während bei 120 (g·min)/mol ein Rückgang um

7 Reaktionstechnische Untersuchungen 152
2,4 % bzw. bei 155 (g·min)/mol 2,6 % zu beobachten ist. Diese Abweichungen bewegen
sich jedoch im Bereich des Messfehlers.
Ein klarer Trend ist dagegen bei den relativen Desaktivierungen ∆X zu erkennen. Bei
allen getesteten Verweilzeiten ist ein deutlicher Anstieg durch die Mikrowelleneinstrah-
lung zu verzeichnen, der bei niedrigen Verweilzeiten stärker ausgeprägt ist. Der Anstieg
beträgt 9,8 % bei 90 (g·min)/mol, während sich bei 120 (g·min)/mol nur eine Zunahme um
9,4 %, bzw. 4,9 % bei 155 (g·min)/mol zeigt.
Um den Einfluss der Mikrowellenleistung zu verifizieren, wurde bei einer modifizierten
Verweilzeit von 155 (g·min)/mol die Leistung von 100 auf 200 W gesteigert (siehe
Abbildung 7.14 ). Man erkennt, dass die Verdopplung der Leistung zu einer Verstärkung
der bereits diskutierten Effekte führt. Der Benzenumsatzgrad ändert sich nicht, während
die Phenolselektivität im Vergleich zum Experiment mit 100 W Mikrowellenleistung
nochmals um 1,3 % zurückgeht. Am deutlichsten macht sich der Einfluss bei der relativen
Desaktivierung bemerkbar. Diese nimmt um 4,5 % von 41 auf 45,5 % zu. Eine deutlich
höhere Leistung verstärkt den Einfluss der Mikrowellen auf das Reaktionssystem nur
geringfügig.
Abbildung 7.14: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Fe-ZSM-5 (II)
(T = 300 °C, N2O:Benzen = 1:1, τmod = 155 (g·min)/mol, TOS = 13 min)
Das entsprechende Diagramm zur Leistungsvariation bei einer modifizierten Verweilzeit
von 120 (g·min)/mol findet sich in Appendix H, Abbildung H.1.

7.2 Hydroxylierung von Benzen mit N2O 153
7.2.4.4.2 Reaktionstemperatur: 350 °C
Abbildung 7.15 zeigt das katalytische Verhalten in Abhängigkeit von der modifizierten
Verweilzeit und Mikrowelleneinstrahlung bei einer Temperatur von 350 °C.
Ein Vergleich der Experimente ohne Mikrowelleneinstrahlung bei verschiedenen modifi-
zierten Verweilzeiten zeigt, dass der Benzenumsatzgrad bei einer Erhöhung von 90 auf
120 (g·min)/mol von 16,2 auf 12,4 % abnimmt. Bei einer Verweilzeit von
155 (g·min)/mol liegt der Benzenumsatzgrad dagegen bei 19,4 %. Ein ähnliches Bild zeigt
sich auch bei den Phenolselektivitäten. Im Vergleich zur modifizierten Verweilzeit
90 (g·min)/mol (S Phenol: 74 %) ist die Selektivität bei 120 (g·min)/mol um 6 % niedriger
(S Phenol: 68 %), bei 155 (g·min)/mol dagegen um 3 % höher (S Phenol: 77 %). Auch die
relative Desaktivierung nimmt zwischen 90 und 120 (g·min)/mol von 46 auf 37 % ab. Bei
einer weiteren Steigerung der Verweilzeit auf 155 (g·min)/mol ist eine relative
Desaktivierung von 38 % zu verzeichnen. Die Benzochinonselektivität beträgt 3,7 % bei
90 (g·min)/mol, 2,0 % bei 120 (g·min)/mol und 4,1 % bei 155 (g·min)/mol.
Abbildung 7.15: Einfluss von Verweilzeit und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Fe-ZSM-5 (II)
(T = 350 °C, N2O:Benzen = 1:1, TOS = 13 min)
Der Einfluss der Mikrowellen (100 W) ist je nach eingestellter Verweilzeit sehr unter-
schiedlich. Bei einer modifizierten Verweilzeit von 90 (g·min)/mol geht der Benzenum-
satzgrad von 16,1 auf 11 % zurück, während bei höheren Verweilzeiten ein geringfügiger
Anstieg von 0,6 % (120 (g·min)/mol), bzw. 1,0 % (155 (g·min)/mol) zu beobachten ist.

7 Reaktionstechnische Untersuchungen 154
Das Entgegengesetzte Verhalten zeigen die Phenolselektivitäten. Bei niedrigen Verweil-
zeiten ist kaum ein Einfluss der Mikrowellen feststellbar (Rückgang um 1 % bei
90 (g·min)/mol; Anstieg um 2 % bei 120 (g·min)/mol) während bei 155 (g·min)/mol ein
Selektivitätsrückgang von 77,4 auf 67,1 % zu verzeichnen ist. Weiterhin bewirkt die Mik-
rowelle wieder einen Anstieg der relativen Desaktivierung. Dieser ist jedoch nur bei
120 (g·min)/mol) deutlich ausgeprägt. Bei dieser Verweilzeit erhöht sich die relative
Desaktivierung von 37,4 auf 55,5 %. Bei 90 (g·min)/mol) beträgt der Anstieg von ∆X
lediglich 2,4 %, bei 155 (g·min)/mol) 0,23 %. Die Benzochinonselektivität geht bei
90 (g·min)/mol) von 3,7 auf 2,2 % durch die Mikrowellen zurück. Bei 120 (g·min)/mol)
ist dagegen ein Anstieg von 2,0 auf 2,3 % und bei 155 (g·min)/mol) ein Rückgang von 4,1
auf 3,4 % durch die Mikrowelleneinstrahlung zu beobachten.
In Abbildung 7.16 ist die Variation der Mikrowellenleistung bei einer modifizierten
Verweilzeit von 120 (g·min)/mol) dargestellt. Man erkennt, dass sich bei einer höheren
Mikrowellenleistung der Trend zu höheren Phenolselektivitäten und schnellerer relativer
Desaktivierung verstärkt. Bei einer Steigerung der Leistung von 100 auf 200 W ändert
sich der Benzenumsatzgrad nur gering (Rückgang von 13 auf 12 %), während die Phenol-
selektivität von 70 auf 74 % ansteigt. Entsprechend nimmt die relative Desaktivierung ∆X
von 55 auf 58 % zu. Die Benzochinonselektivität steigt bei einer Erhöhung der Leistung
von 2,3 auf 2,5 % an.
Abbildung 7.16: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Fe-ZSM-5 (II)
(T = 350 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)
Das entsprechende Diagramm zur Leistungsvariation bei einer modifizierten Verweilzeit
von 155 (g·min)/mol findet sich Appendix H, Abbildung H.2.

7.2 Hydroxylierung von Benzen mit N2O 155
7.2.4.4.3 Reaktionstemperatur: 400 °C
Abbildung 7.17 zeigt das katalytische Verhalten in Abhängigkeit von der modifizierten
Verweilzeit und Mikrowelleneinstrahlung bei einer Temperatur von 400 °C.
Zunächst sollen wiederum die Ergebnisse ohne Mikrowelleneinstrahlung verglichen wer-
den. Bei einer Steigerung der modifizierten Verweilzeit 90 und 120 (g·min)/mol) nimmt
der Benzenumsatzgrad von 28 auf 29 % zu. Bei einer weiteren Erhöhung der Verweilzeit
auf 155 (g·min)/mol) nimmt der Umsatz deutlich stärker auf 39 % zu.
Die Phenolselektivität bleibt unabhängig von der eingestellten Verweilzeit annähernd
konstant (66,9 % bei 90 (g·min)/mol), 65,9 % bei 120 (g·min)/mol), 66,6 % bei
155 (g·min)/mol)). Auch die relative Desaktivierung verändert sich nur geringfügig (28 %
bei 90 (g·min)/mol), 30 % bei 120 (g·min)/mol), 27 % bei 155 (g·min)/mol)). Die Benzo-
chinonselektivität steigt mit zunehmender Verweilzeit tendenziell an. Bei einer Erhöhung
von 90 (g·min)/mol) auf 120 (g·min)/mol) nimmt die Benzochinonselektivität von 5,2 auf
5,5 %, bzw. auf 5,8 % bei 155 (g·min)/mol) zu.
Abbildung 7.17: Einfluss von Verweilzeit und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Fe-ZSM-5 (II)
(T = 400 °C, N2O:Benzen = 1:1, TOS = 13 min)
Auch der Einfluss der Mikrowellenstrahlung auf die Reaktion ist bei 400 °C gering. Der
Benzenumsatzgrad nimmt durch die Mikrowellen bei 90 (g·min)/mol) lediglich um 0,2 %
zu, bei 120 (g·min)/mol) um 0,3 % ab und bei 155 (g·min)/mol) um 1,4 % zu. Im Rahmen
der Messgenauigkeit kann somit keine Auswirkung der Mikrowellen auf den Benzenum-
satzgrad gefolgert werden. Auch die Auswirkung auf die Phenolselektivitäten ist gering.

7 Reaktionstechnische Untersuchungen 156
Bei 90 (g·min)/mol) steigt die Phenolselektivität um 1,8 %, bei 120 (g·min)/mol) um
4,2 %, und bei 155 (g·min)/mol) ist ein Selektivitätsrückgang um 2,6 % durch die Mikro-
wellen zu verzeichnen. Betrachtet man die relative Desaktivierung, so zeigt sich eine
Tendenz zu schnellerer Desaktivierung durch Mikrowellen, welche bei höheren Verweil-
zeiten deutlicher ausgeprägt ist. Bei 90 (g·min)/mol) nimmt ∆X um 1,7 %, bei
120 (g·min)/mol) um 7,4, und bei 155 (g·min)/mol) um 8,8 % zu. Die Benzochinonselek-
tivität steigt bei einer Verweilzeit von 90 (g·min)/mol) von 5,2 auf 5,7 % und bei
120 (g·min)/mol) von 5,5 auf 5,8 %. Bei der Verweilzeit 155 (g·min)/mol) ist dagegen ein
Rückgang von 5,8 auf 5,2 % durch die Mikrowellen zu beobachten.
Durch eine Steigerung der eingesetzten Mikrowellenleistung lassen sich die beschriebe-
nen Effekte nur noch geringfügig steigern.
In Abbildung 7.18 kann man erkennen, dass bei einer Verweilzeit von 120 (g·min)/mol)
bei einer Leistung von 200 W keine Änderung des Benzenumsatzgrades im Vergleich
zum Experiment bei 100 W Leistung festzustellen ist. Die Phenol- und Benzochinonse-
lektivitäten bleiben ebenfalls nahezu konstant (S Phenol: Anstieg um 1 %, S Benzochi-
non: Abnahme um 0,2 %)). Die relative Desaktivierung nimmt dagegen von 37 auf 40 %
zu. Dies sind somit 10 % mehr als beim konventionell beheizten Vergleichsexperiment.
Abbildung 7.18: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Fe-ZSM-5 (II)
(T = 400 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)
7.2.4.5 Überstöchiometrische Benzenzugabe
In der Literatur wird berichtet, dass sich die Desaktivierung des Katalysators während der
Hydroxylierung von Benzen mit N2O durch eine Fahrweise mit Benzenüberschuss
verlangsamen lässt [233]. Deshalb wurden einige Versuche bei einem Eduktverhältnis

7.2 Hydroxylierung von Benzen mit N2O 157
N2O:Benzen = 1:4 durchgeführt. Zu diesem Zweck wurden der Benzenmolenstrom und
die modifizierte Verweilzeit konstant gehalten, während die N2O-Menge im Feed redu-
ziert wurde.
Ein Vergleich der Messungen bei stöchiometrischer N2O-Zugabe mit den Ergebnissen bei
Benzenüberschuss (Abbildung 7.19) ohne Mikrowelleneinstrahlung zeigt einen deutlichen
Rückgang der Benzenumsatzgrade über den gesamten Temperaturbereich, da bei einer
Fahrweise mit vierfachen Benzenüberschuss maximal ein Benzenumsatzgrad von 25 %
erreichbar ist. Bei 350 °C wird ein Benzenumsatzgrad von 6,5 % erreicht (zum Vergleich:
bei stöchiometrischer N2O-Zugabe 16 %), bei 400 °C ein Umsatz von 21 % (bei stöchio-
metrischer N2O-Zugabe 28 %). Dies macht deutlich, dass gerade bei hohen Reaktionstem-
peraturen eine Fahrweise mit Benzenüberschuss sinn macht, da beinahe das gesamte N2O
zu Phenol umgesetzt wird. Die auf N2O bezogenen Phenolselektivitäten sind damit sehr
hoch. Dies ist aus wirtschaftlicher Sicht von Interesse, da zum gegenwärtigen Zeitpunkt
N2O eine größere Wertschöpfung besitzt als Benzen. Zudem lässt sich eine Rückführung
des nicht umgesetzten Benzens aus anlagentechnischer Sicht leicht realisieren [26].
Betrachtet man die auf Benzen bezogenen Phenolselektivitäten, zeigt sich eine Verbesse-
rung gegenüber einer stöchiometrischen Feedzusammensetzung. Bei 350 °C nimmt die
Selektivität von 73 auf 77 % zu, bei 400 °C von 67 auf 69 %. Die relative Desaktivierung
nimmt bei beiden Temperaturen um ca. 3 % ab.
Abbildung 7.19: Einfluss von Feedzusammensetzung und Temperatur
auf das katalytische Verhalten des Fe-ZSM-5 (II)
( τmod = 90 (g·min)/mol, TOS = 13 min)

7 Reaktionstechnische Untersuchungen 158
Vergleicht man die Versuche bei Benzenüberschuss mit den entsprechenden Mikro-
wellenexperimenten (Abbildung 7.20) zeigen sich identische Auswirkungen wie bei
stöchiometrischer Feedzusammensetzung. Die Benzenumsatzgrade nehmen mit steigender
Mikrowellenleistung ab, die Phenolselektivitäten und Desaktivierungsgeschwindigkeiten
erhöhen sich. Dieser Effekt ist bei Fahrweise mit Benzenüberschuss sogar noch deutlicher
ausgeprägt. Bei einer Reaktionstemperatur von 350 °C und einer Mikrowellenleistung von
200 W beträgt die Phenolselektivität ca. 85 % und liegt damit 8 % über der des Experi-
ments ohne Mikrowelle. Allerdings steigert sich die relative Desaktivierung von 42 auf
73 %. Bei einer Temperatur von 400 °C sind die Unterschiede geringer, die Phenolselek-
tivitäten bei 200 W Mikrowelle sind aber noch 5 % größer als beim Referenzversuch ohne
Mikrowelle. Die relative Desaktivierung nimmt von 25 auf 29 % zu.
Abbildung 7.20: Einfluss von Temperatur und Mikrowelleneinstrahlung auf das
katalytische Verhalten des Fe-ZSM-5 (II) bei überstöchiometrischer Benzenzugabe
(N2O:Benzen = 1:4, τmod = 90 (g·min)/mol, TOS = 13 min)
Weitere Messungen mit überstöchiometrischer Benzenzugabe, die bei einer modifizierten
Verweilzeit von 120 (g·min)/mol durchgeführt wurden, finden sich Appendix H,
Abbildung H.3.
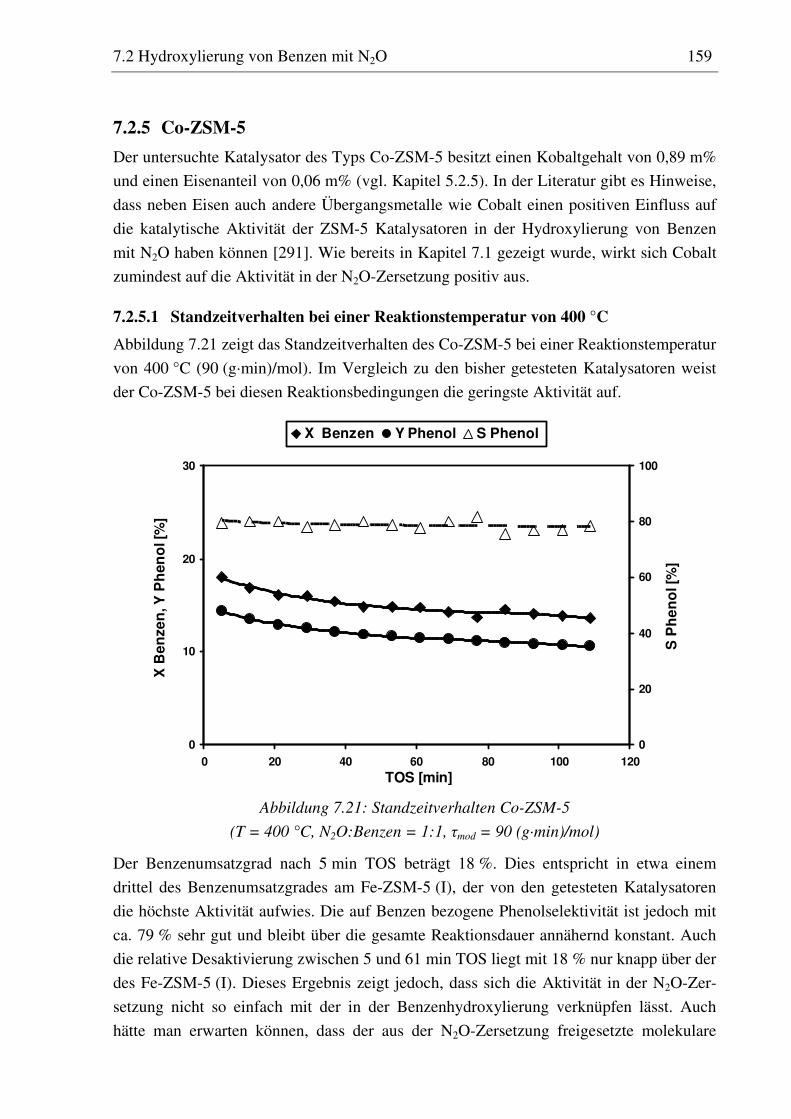
7.2 Hydroxylierung von Benzen mit N2O 159
7.2.5 Co-ZSM-5
Der untersuchte Katalysator des Typs Co-ZSM-5 besitzt einen Kobaltgehalt von 0,89 m%
und einen Eisenanteil von 0,06 m% (vgl. Kapitel 5.2.5). In der Literatur gibt es Hinweise,
dass neben Eisen auch andere Übergangsmetalle wie Cobalt einen positiven Einfluss auf
die katalytische Aktivität der ZSM-5 Katalysatoren in der Hydroxylierung von Benzen
mit N2O haben können [291]. Wie bereits in Kapitel 7.1 gezeigt wurde, wirkt sich Cobalt
zumindest auf die Aktivität in der N2O-Zersetzung positiv aus.
7.2.5.1 Standzeitverhalten bei einer Reaktionstemperatur von 400 °C
Abbildung 7.21 zeigt das Standzeitverhalten des Co-ZSM-5 bei einer Reaktionstemperatur
von 400 °C (90 (g·min)/mol). Im Vergleich zu den bisher getesteten Katalysatoren weist
der Co-ZSM-5 bei diesen Reaktionsbedingungen die geringste Aktivität auf.
0
10
20
30
0 20 40 60 80 100 120
TOS [min]
X B
en
ze
n, Y
Ph
en
ol [%
]
0
20
40
60
80
100
S P
he
no
l [%
]
X Benzen Y Phenol S Phenol
Abbildung 7.21: Standzeitverhalten Co-ZSM-5
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol)
Der Benzenumsatzgrad nach 5 min TOS beträgt 18 %. Dies entspricht in etwa einem
drittel des Benzenumsatzgrades am Fe-ZSM-5 (I), der von den getesteten Katalysatoren
die höchste Aktivität aufwies. Die auf Benzen bezogene Phenolselektivität ist jedoch mit
ca. 79 % sehr gut und bleibt über die gesamte Reaktionsdauer annähernd konstant. Auch
die relative Desaktivierung zwischen 5 und 61 min TOS liegt mit 18 % nur knapp über der
des Fe-ZSM-5 (I). Dieses Ergebnis zeigt jedoch, dass sich die Aktivität in der N2O-Zer-
setzung nicht so einfach mit der in der Benzenhydroxylierung verknüpfen lässt. Auch
hätte man erwarten können, dass der aus der N2O-Zersetzung freigesetzte molekulare

7 Reaktionstechnische Untersuchungen 160
Sauerstoff die Totaloxidation des Benzens fördert. Die gemessene hohe Phenolselektivität
widerspricht dem jedoch. Zu entsprechenden Erkenntnissen gelangte auch REITZMANN
[24].
7.2.5.2 Variation der Reaktionstemperatur
Betrachtet man das katalytische Verhalten dieses Katalysators nach 13 min TOS in
Abhängigkeit von der Reaktionstemperatur (Abbildung 7.22), fällt zunächst auf, dass die
Phenolselektivität bei 300 °C mit 40 % sehr gering ist. Bei einer Temperatursteigerung auf
350 °C verdoppelt sich die Selektivität auf 80 %. Eine weitere Steigerung der Reaktions-
temperatur auf 400 °C wirkt sich jedoch nicht weiter auf die Phenolselektivität aus. Bei
allen anderen getesteten Katalysatoren waren die Phenolselektivitäten bei 300 und 350 °C
annähernd identisch bzw. war bei einer Erhöhung der Temperatur auf 400 °C ein deutli-
cher Rückgang zu beobachten.
Abbildung 7.22: Einfluss von Temperatur und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Co-ZSM-5
(N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
Der Benzenumsatzgrad bei 300 °C beträgt nur 2,6 %. Eine Desaktivierung war über den
gesamten Reaktionszeitraum nicht zu beobachten. Erst bei einer Temperatur von 350 °C
(Benzenumsatzgrad: 5 %) tritt eine deutliche Desaktivierung (15 %) auf. Bei einer Steige-
rung der Temperatur auf 400 °C ist nur ein leichter Anstieg der relativen Desaktivierung
um 3 % zu verzeichnen. Benzochinon konnte erst ab einer Temperatur von 400 °C detek-
tiert werden (S Benzochinon: 2,7 %).

7.2 Hydroxylierung von Benzen mit N2O 161
Vergleicht man die Mikrowellenexperimente mit den Referenzversuchen ohne Mikro-
wellen bei identischen Reaktionsbedingungen, so zeigt sich, wie auch bei allen anderen
getesteten Katalysatoren, kaum ein Einfluss auf die Benzenumsatzgrade, während die
Phenolselektivität und Desaktivierung zunehmen. Bei 300 °C geht der Benzenumsatzgrad
beispielsweise um 0,5 % zurück, die Phenolselektivität steigt um 4 % an. Bei 350 und
400 °C nimmt die Phenolselektivität durch die Mikrowelleneinstrahlung um jeweils 6 %
zu. Bei 350 °C steigt die Desaktivierung um 15 %, bei 400 °C um 20 % durch die
Mikrowellen an. Die bereits bei den anderen Katalysatoren beobachteten Trends zeichnen
sich somit wieder ab, am deutlichsten bei der Desaktivierungsgeschwindigkeit.
7.2.5.3 Variation der Mikrowellenleistung
Abbildung 7.23 zeigt exemplarisch den Einfluss der Mikrowellenleistung bei einer
Reaktionstemperatur von 350 °C. Der Trend zu abnehmenden Umsatzgraden bestätigt
sich zwar, jedoch ist der stärkste Rückgang des Benzenumsatzgrades bei einer Leistung
von 200 W gerade einmal 0,25 %. Deutlicher sind der maximale Anstieg der Phenolse-
lektivität von 6 % und die Verdopplung der relativen Desaktivierung zu erkennen.
Abbildung 7.23 Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Co-ZSM-5
(T = 350 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
Die entsprechenden Diagramme zur Variation der Mikrowellenleistung bei 300 und
400 °C finden sich Appendix H, Abbildung H.4 – H.5.
7.2.5.4 Variation der Verweilzeit
Wie der Katalysator Fe-ZSM-5 (II) wurde auch der Katalysator vom Typ Co-ZSM-5 bei
einer modifizierten Verweilzeit von 90, 120 und 155 (g·min)/mol getestet.
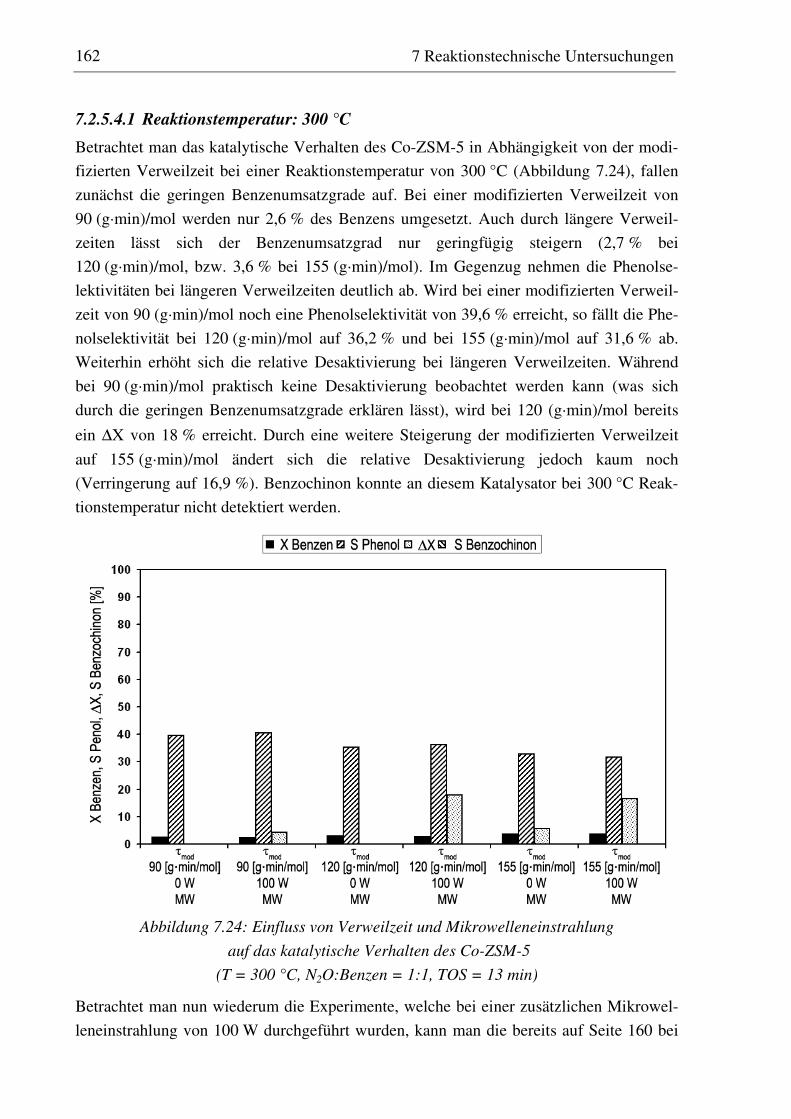
7 Reaktionstechnische Untersuchungen 162
7.2.5.4.1 Reaktionstemperatur: 300 °C
Betrachtet man das katalytische Verhalten des Co-ZSM-5 in Abhängigkeit von der modi-
fizierten Verweilzeit bei einer Reaktionstemperatur von 300 °C (Abbildung 7.24), fallen
zunächst die geringen Benzenumsatzgrade auf. Bei einer modifizierten Verweilzeit von
90 (g·min)/mol werden nur 2,6 % des Benzens umgesetzt. Auch durch längere Verweil-
zeiten lässt sich der Benzenumsatzgrad nur geringfügig steigern (2,7 % bei
120 (g·min)/mol, bzw. 3,6 % bei 155 (g·min)/mol). Im Gegenzug nehmen die Phenolse-
lektivitäten bei längeren Verweilzeiten deutlich ab. Wird bei einer modifizierten Verweil-
zeit von 90 (g·min)/mol noch eine Phenolselektivität von 39,6 % erreicht, so fällt die Phe-
nolselektivität bei 120 (g·min)/mol auf 36,2 % und bei 155 (g·min)/mol auf 31,6 % ab.
Weiterhin erhöht sich die relative Desaktivierung bei längeren Verweilzeiten. Während
bei 90 (g·min)/mol praktisch keine Desaktivierung beobachtet werden kann (was sich
durch die geringen Benzenumsatzgrade erklären lässt), wird bei 120 (g·min)/mol bereits
ein ∆X von 18 % erreicht. Durch eine weitere Steigerung der modifizierten Verweilzeit
auf 155 (g·min)/mol ändert sich die relative Desaktivierung jedoch kaum noch
(Verringerung auf 16,9 %). Benzochinon konnte an diesem Katalysator bei 300 °C Reak-
tionstemperatur nicht detektiert werden.
Abbildung 7.24: Einfluss von Verweilzeit und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Co-ZSM-5
(T = 300 °C, N2O:Benzen = 1:1, TOS = 13 min)
Betrachtet man nun wiederum die Experimente, welche bei einer zusätzlichen Mikrowel-
leneinstrahlung von 100 W durchgeführt wurden, kann man die bereits auf Seite 160 bei
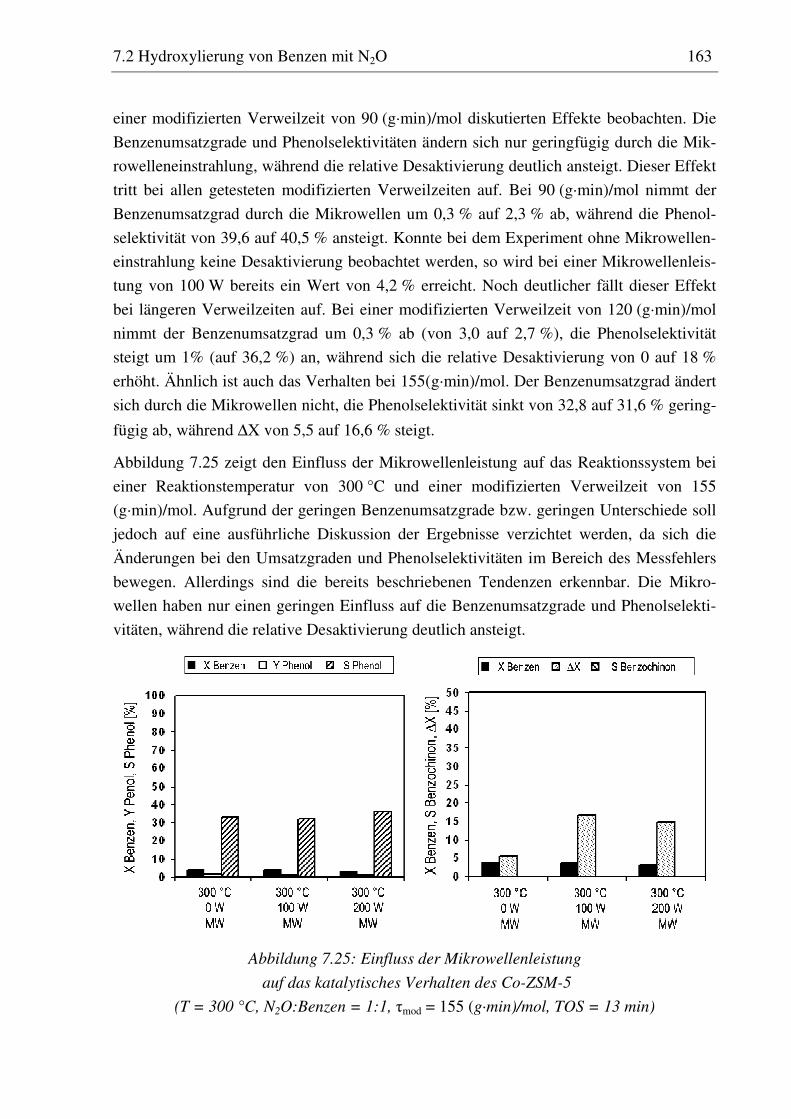
7.2 Hydroxylierung von Benzen mit N2O 163
einer modifizierten Verweilzeit von 90 (g·min)/mol diskutierten Effekte beobachten. Die
Benzenumsatzgrade und Phenolselektivitäten ändern sich nur geringfügig durch die Mik-
rowelleneinstrahlung, während die relative Desaktivierung deutlich ansteigt. Dieser Effekt
tritt bei allen getesteten modifizierten Verweilzeiten auf. Bei 90 (g·min)/mol nimmt der
Benzenumsatzgrad durch die Mikrowellen um 0,3 % auf 2,3 % ab, während die Phenol-
selektivität von 39,6 auf 40,5 % ansteigt. Konnte bei dem Experiment ohne Mikrowellen-
einstrahlung keine Desaktivierung beobachtet werden, so wird bei einer Mikrowellenleis-
tung von 100 W bereits ein Wert von 4,2 % erreicht. Noch deutlicher fällt dieser Effekt
bei längeren Verweilzeiten auf. Bei einer modifizierten Verweilzeit von 120 (g·min)/mol
nimmt der Benzenumsatzgrad um 0,3 % ab (von 3,0 auf 2,7 %), die Phenolselektivität
steigt um 1% (auf 36,2 %) an, während sich die relative Desaktivierung von 0 auf 18 %
erhöht. Ähnlich ist auch das Verhalten bei 155(g·min)/mol. Der Benzenumsatzgrad ändert
sich durch die Mikrowellen nicht, die Phenolselektivität sinkt von 32,8 auf 31,6 % gering-
fügig ab, während ∆X von 5,5 auf 16,6 % steigt.
Abbildung 7.25 zeigt den Einfluss der Mikrowellenleistung auf das Reaktionssystem bei
einer Reaktionstemperatur von 300 °C und einer modifizierten Verweilzeit von 155
(g·min)/mol. Aufgrund der geringen Benzenumsatzgrade bzw. geringen Unterschiede soll
jedoch auf eine ausführliche Diskussion der Ergebnisse verzichtet werden, da sich die
Änderungen bei den Umsatzgraden und Phenolselektivitäten im Bereich des Messfehlers
bewegen. Allerdings sind die bereits beschriebenen Tendenzen erkennbar. Die Mikro-
wellen haben nur einen geringen Einfluss auf die Benzenumsatzgrade und Phenolselekti-
vitäten, während die relative Desaktivierung deutlich ansteigt.
Abbildung 7.25: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Co-ZSM-5
(T = 300 °C, N2O:Benzen = 1:1, τmod = 155 (g·min)/mol, TOS = 13 min)

7 Reaktionstechnische Untersuchungen 164
Die Ergebnisse der Untersuchungen zur Variation der Mikrowellenleistung bei 90 und
120 (g·min)/mol finden sich Appendix H, Abbildung H.4 und Abbildung H.6.
7.2.5.4.2 Reaktionstemperatur: 350 °C
Abbildung 7.26 zeigt das Katalytische Verhalten des Co-ZSM-5 in Abhängigkeit von der
modifizierten Verweilzeit und Mikrowelleneinstrahlung bei einer Reaktionstemperatur
von 350 °C. Betrachtet man zunächst wieder die konventionell beheizten Experimente,
zeigt sich ein ähnliches Bild wie bei 300 °C (vgl. Kapitel 7.2.5.4.1). Die Benzenum-
satzgrade steigen bei längeren Verweilzeiten an, während die Phenolselektivitäten sinken.
Der Benzenumsatzgrad beträgt bei 90 (g·min)/mol 5,4 % bei 120 (g·min)/mol 7,4 % und
bei 155 (g·min)/mol 9,0 %. Im Gegenzug fallen die Phenolselektivitäten von 79,7
(90 (g·min)/mol) auf 72,7 (120 (g·min)/mol) bzw. 65,3 % bei einer modifizierten Ver-
weilzeit von 155 (g·min)/mol ab. Im Unterschied zu den bereits diskutierten Ergebnissen
bei einer Reaktionstemperatur von 300 °C nimmt die relative Desaktivierung jedoch nur
zwischen 90 und 120 (g·min)/mol von 14,8 auf 24,2 % zu, während bei 155(g·min)/mol
wieder ein Rückgang von ∆X auf 14,1 % zu verzeichnen ist. Auch bei einer Reaktions-
temperatur von 350 °C konnte an diesem Katalysator unter den verwendeten Reaktions-
bedingungen kein Benzochinon detektiert werden.
Abbildung 7.26: Einfluss von Verweilzeit und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Co-ZSM-5
(T = 350 °C, N2O:Benzen = 1:1, TOS = 13 min)
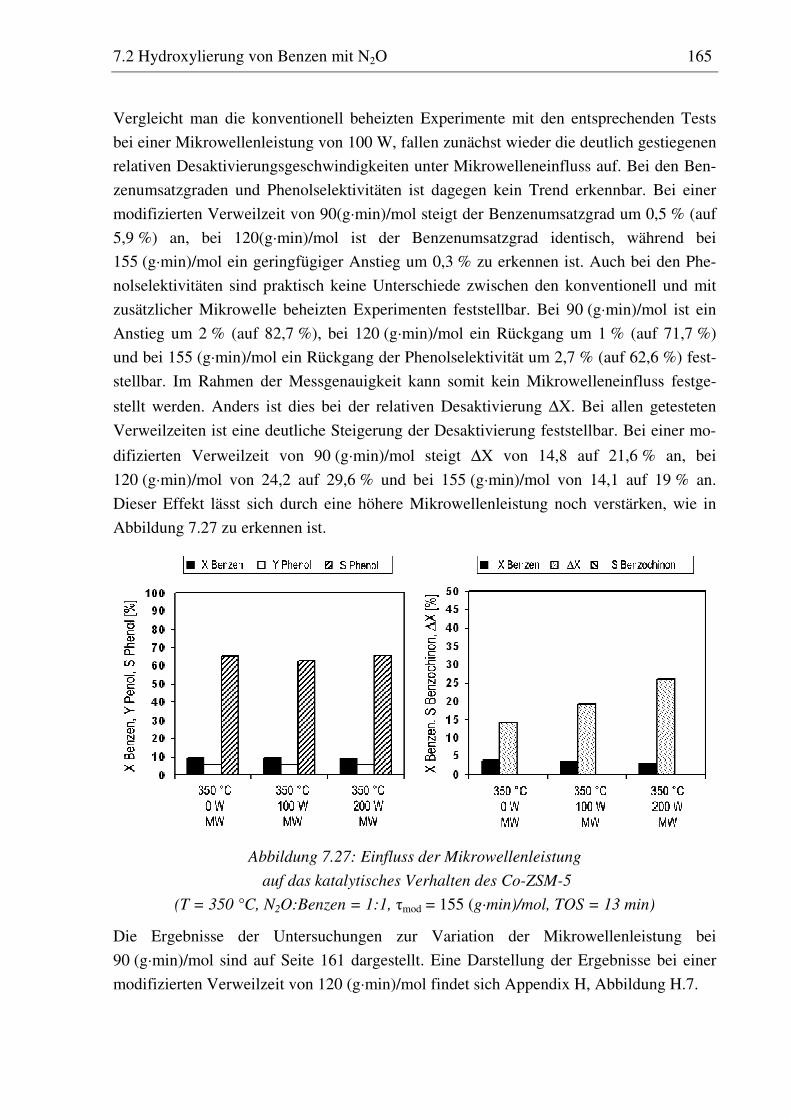
7.2 Hydroxylierung von Benzen mit N2O 165
Vergleicht man die konventionell beheizten Experimente mit den entsprechenden Tests
bei einer Mikrowellenleistung von 100 W, fallen zunächst wieder die deutlich gestiegenen
relativen Desaktivierungsgeschwindigkeiten unter Mikrowelleneinfluss auf. Bei den Ben-
zenumsatzgraden und Phenolselektivitäten ist dagegen kein Trend erkennbar. Bei einer
modifizierten Verweilzeit von 90(g·min)/mol steigt der Benzenumsatzgrad um 0,5 % (auf
5,9 %) an, bei 120(g·min)/mol ist der Benzenumsatzgrad identisch, während bei
155 (g·min)/mol ein geringfügiger Anstieg um 0,3 % zu erkennen ist. Auch bei den Phe-
nolselektivitäten sind praktisch keine Unterschiede zwischen den konventionell und mit
zusätzlicher Mikrowelle beheizten Experimenten feststellbar. Bei 90 (g·min)/mol ist ein
Anstieg um 2 % (auf 82,7 %), bei 120 (g·min)/mol ein Rückgang um 1 % (auf 71,7 %)
und bei 155 (g·min)/mol ein Rückgang der Phenolselektivität um 2,7 % (auf 62,6 %) fest-
stellbar. Im Rahmen der Messgenauigkeit kann somit kein Mikrowelleneinfluss festge-
stellt werden. Anders ist dies bei der relativen Desaktivierung ∆X. Bei allen getesteten
Verweilzeiten ist eine deutliche Steigerung der Desaktivierung feststellbar. Bei einer mo-
difizierten Verweilzeit von 90 (g·min)/mol steigt ∆X von 14,8 auf 21,6 % an, bei
120 (g·min)/mol von 24,2 auf 29,6 % und bei 155 (g·min)/mol von 14,1 auf 19 % an.
Dieser Effekt lässt sich durch eine höhere Mikrowellenleistung noch verstärken, wie in
Abbildung 7.27 zu erkennen ist.
Abbildung 7.27: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Co-ZSM-5
(T = 350 °C, N2O:Benzen = 1:1, τmod = 155 (g·min)/mol, TOS = 13 min)
Die Ergebnisse der Untersuchungen zur Variation der Mikrowellenleistung bei
90 (g·min)/mol sind auf Seite 161 dargestellt. Eine Darstellung der Ergebnisse bei einer
modifizierten Verweilzeit von 120 (g·min)/mol findet sich Appendix H, Abbildung H.7.

7 Reaktionstechnische Untersuchungen 166
7.2.5.4.3 Reaktionstemperatur: 400 °C
Analog zu den letzten Kapiteln wurde auch eine Reaktionstemperatur von 400 °C
hinsichtlich der Verweilzeiteinflusses am Co-ZSM-5 untersucht (siehe Abbildung 7.28).
Man erkennt die gleichen Tendenzen wie im letzten Kapitel bei einer Reaktionstemperatur
von 350 °C beschrieben. Durch eine Steigerung der modifizierten Verweilzeit steigen die
Benzenumsatzgrade an, während die Phenolselektivitäten sinken. Bei Verwendung der
konventionellen Heizung ohne Mikrowelle wird bei 90 (g·min)/mol ein Benzenum-
satzgrad von 16,9 %, bei 120 (g·min)/mol 20,6 % und bei 155 (g·min)/mol 23 % beo-
bachtet. Die korrespondierenden Phenolselektivitäten fallen von 80,2 % (bei
90 (g·min)/mol) auf 76,1% bzw. 70,3 % bei 155 (g·min)/mol. Analog zu den Ergebnissen
bei 350 °C steigt die relative Desaktivierung zwischen 90 und 120 (g·min)/mol von 18,3
auf 33,6 % an, während bei 155 (g·min)/mol wieder ein Rückgang auf 32,2 % zu erkennen
ist.
Benzochinon konnte nur bei einer modifizierten Verweilzeit von 155 (g·min)/mol bei
einer Selektivität von 2,7 % detektiert werden.
Abbildung 7.28: Einfluss von Verweilzeit und Mikrowelleneinstrahlung
auf das katalytische Verhalten des Co-ZSM-5
(T = 400 °C, N2O:Benzen = 1:1, TOS = 13 min)
Vergleicht man die konventionell beheizten Experimente mit den Tests bei einer zusätzli-
chen Mikrowellenleistung von 100 W, erkennt man wiederum nur geringe Unterscheide
bei den Benzenumsatzgraden und Phenolselektivitäten. Allerdings zeichnet sich auch bei
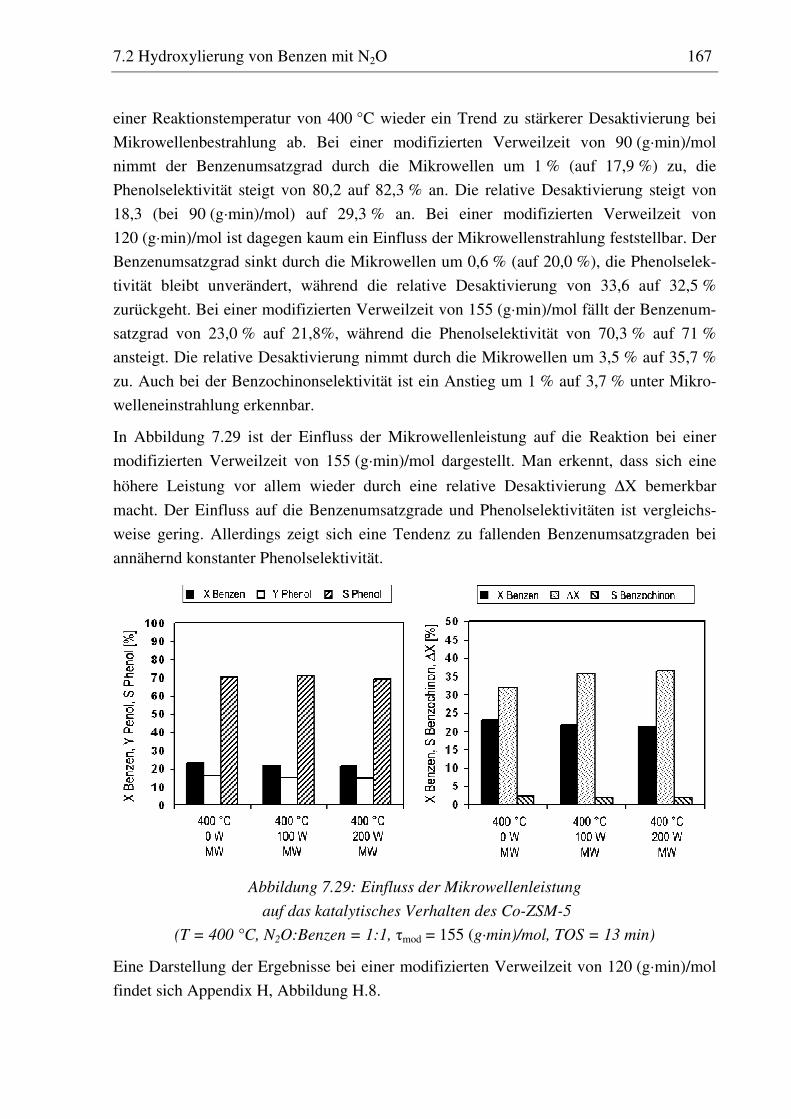
7.2 Hydroxylierung von Benzen mit N2O 167
einer Reaktionstemperatur von 400 °C wieder ein Trend zu stärkerer Desaktivierung bei
Mikrowellenbestrahlung ab. Bei einer modifizierten Verweilzeit von 90 (g·min)/mol
nimmt der Benzenumsatzgrad durch die Mikrowellen um 1 % (auf 17,9 %) zu, die
Phenolselektivität steigt von 80,2 auf 82,3 % an. Die relative Desaktivierung steigt von
18,3 (bei 90 (g·min)/mol) auf 29,3 % an. Bei einer modifizierten Verweilzeit von
120 (g·min)/mol ist dagegen kaum ein Einfluss der Mikrowellenstrahlung feststellbar. Der
Benzenumsatzgrad sinkt durch die Mikrowellen um 0,6 % (auf 20,0 %), die Phenolselek-
tivität bleibt unverändert, während die relative Desaktivierung von 33,6 auf 32,5 %
zurückgeht. Bei einer modifizierten Verweilzeit von 155 (g·min)/mol fällt der Benzenum-
satzgrad von 23,0 % auf 21,8%, während die Phenolselektivität von 70,3 % auf 71 %
ansteigt. Die relative Desaktivierung nimmt durch die Mikrowellen um 3,5 % auf 35,7 %
zu. Auch bei der Benzochinonselektivität ist ein Anstieg um 1 % auf 3,7 % unter Mikro-
welleneinstrahlung erkennbar.
In Abbildung 7.29 ist der Einfluss der Mikrowellenleistung auf die Reaktion bei einer
modifizierten Verweilzeit von 155 (g·min)/mol dargestellt. Man erkennt, dass sich eine
höhere Leistung vor allem wieder durch eine relative Desaktivierung ∆X bemerkbar
macht. Der Einfluss auf die Benzenumsatzgrade und Phenolselektivitäten ist vergleichs-
weise gering. Allerdings zeigt sich eine Tendenz zu fallenden Benzenumsatzgraden bei
annähernd konstanter Phenolselektivität.
Abbildung 7.29: Einfluss der Mikrowellenleistung
auf das katalytisches Verhalten des Co-ZSM-5
(T = 400 °C, N2O:Benzen = 1:1, τmod = 155 (g·min)/mol, TOS = 13 min)
Eine Darstellung der Ergebnisse bei einer modifizierten Verweilzeit von 120 (g·min)/mol
findet sich Appendix H, Abbildung H.8.

7 Reaktionstechnische Untersuchungen 168
7.2.5.5 Wasserzugabe zum Feed
Anhand des Co-ZSM-5 wurde der Einfluss von Wasser auf das Reaktionssystem unter-
sucht. Aus der Literatur ist bekannt, dass sich die Wasserzugabe zum Feed positiv auf die
Reaktion auswirken kann (vgl. Kapitel 3.4.3).
7.2.5.5.1 Versuchsdurchführung
Bei den Experimenten mit Wasserzugabe zum Feed wurde das Wasser über einen Sättiger
(vgl. Kapitel 6.2) dosiert. Die Sättigertemperatur betrug 85 °C. Als Trägergas kam Stick-
stoff mit einem Volumenstrom von 100 Nml/min zum Einsatz. Die Molenströme der
Edukte und des Stickstoffs wurden gegenüber den Experimenten ohne Wasserzugabe
nicht verändert. Der Sättigungsdampfdruck von Wasser bei einer Temperatur von 85 °C
beträgt 578 mbar [344]. Damit ergibt sich eine Wasserbeladung des Trägergases von
Y = 0,8808. Dies entspricht einem Wasservolumenstrom von 137 Nml/min. Der Gesamt-
volumenstrom der Experimente mit Wasserzugabe vergrößert sich somit um diesen Betrag
gegenüber den Experimenten ohne Wasserzugabe. Die modifizierte Verweilzeit verringert
sich auf 58 (g·min)/mol. Bei den Untersuchungen ohne zusätzliche Wasserzugabe beträgt
die modifizierte Verweilzeit dagegen 90 (g·min)/mol.
7.2.5.5.2 Ergebnisse
Abbildung 7.30 zeigt das katalytische Verhalten bei einer Reaktionstemperatur von
350 °C in Abhängigkeit von der Wasserzugabe und Mikrowelleneinstrahlung.
Durch die zusätzliche Wasserzugabe verdoppelt sich der Benzenumsatzgrad von ca. 5 auf
10 %, trotz der kürzeren Verweilzeit. Allerdings wurden viele Nebenprodukte detektiert,
so dass die auf Benzen bezogene Phenolselektivität von ca. 80 % ohne Wasserzugabe auf
42 % mit Wasser zurückgeht. Dies bedeutet, dass der Benzenumsatzgrad durch die
Wasserzugabe zwar ansteigt, die Phenolausbeuten jedoch beinahe identisch sind. Die
relative Desaktivierung fällt von ca. 15 auf 9 %. Benzochinon wurde nicht detektiert.
Offensichtlich kommt es zu einer Aktivitätssteigerung des Katalysators, allerdings
verschlechtert sich die Phenolselektivität aufgrund unerwünschter Folgeprodukte des
Phenols. In den GC-Chromatogrammen wurden zahlreiche Peaks detektiert, welche bei
den Experimenten ohne Wasserzugabe nicht beobachtet werden konnten. Eine
Identifizierung bzw. Quantifizierung dieser Stoffe wurde nicht durchgeführt.
Im Gegensatz zu den Untersuchungen ohne Wasserzugabe zeigt sich ein signifikanter
Einfluss der Mikrowellen auf die Reaktion, sobald Wasser zum Feed zugegeben wird.
Bei einer Mikrowellenleistung von 500 W fällt der Benzenumsatzgrad um 2 % und die
Phenolselektivität erhöht sich auf 75 %. Gleichzeitig geht die relative Desaktivierung auf
12 % zurück. Es muss erläutert werden, dass die Mikrowellen bei dieser hohen Leistung
zu einer Aufheizung des Mikrowellensystems führen, da zwar viel Leistung eingestrahlt

7.2 Hydroxylierung von Benzen mit N2O 169
wird, jedoch nur ein vergleichsweise geringer Betrag über die Aufheizung der Reaktanden
abgeführt wird. Um eine Beschädigung des Mikrowellenofens zu vermeiden, wurde die
Mikrowelleneinstrahlung gepulst. Jeweils 3 min vor Beginn einer GC-Messung wurde die
Mikrowelle für 4 min gestartet, dann wieder abgeschaltet. Es ergibt sich somit ein Puls-
Pause Zyklus von 4 min, da alle 8 min eine Probe gezogen wurde (vgl. Kapitel 6.2.5).
Abbildung 7.30: Katalytisches Verhalten des Co-ZSM-5 in Abhängigkeit von
Wasserzugabe und Mikrowelleneinstrahlung
(T = 350 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, τmod = 58 (g·min)/mol (mit
Wasser), TOS = 13 min)
Bei einer Reaktionstemperatur von 400 °C führt die Wasserzugabe zu einem Anstieg des
Benzenumsatzgrades von ca. 17 auf 22 % (vgl. Abbildung 7.31).
Die Phenolselektivität geht von 80 auf 59 % zurück. Das Verhalten entspricht den
Beobachtungen bei 350 °C. Die relative Desaktivierung fällt jedoch auf ca. 3,5 % ab, bei
350 °C war dagegen ein Anstieg zu verzeichnen. Die auf Benzen bezogene
Benzochinonselektivität geht durch die Wasserzugabe um 1 % zurück. Dies kann jedoch
auch auf die kürzere Verweilzeit bei zusätzlicher Wasserzugabe zurückzuführen sein.
Das Referenzexperiment mit Mikrowellen wurde bei einer konstanten Mikrowellenleis-
tung von 50 W durchgeführt. Es kommt zu einem Rückgang des Benzenumsatzgrades um
3 % gegenüber dem Experiment ohne Mikrowelleneinstrahlung. Die Phenolselektivität
steigt um 5,5 % an. Dieses Verhalten entspricht den Beobachtungen, die bei 350 °C
gemacht wurden. Allerdings ist ein Anstieg der relativen Desaktivierung zwischen 5 und
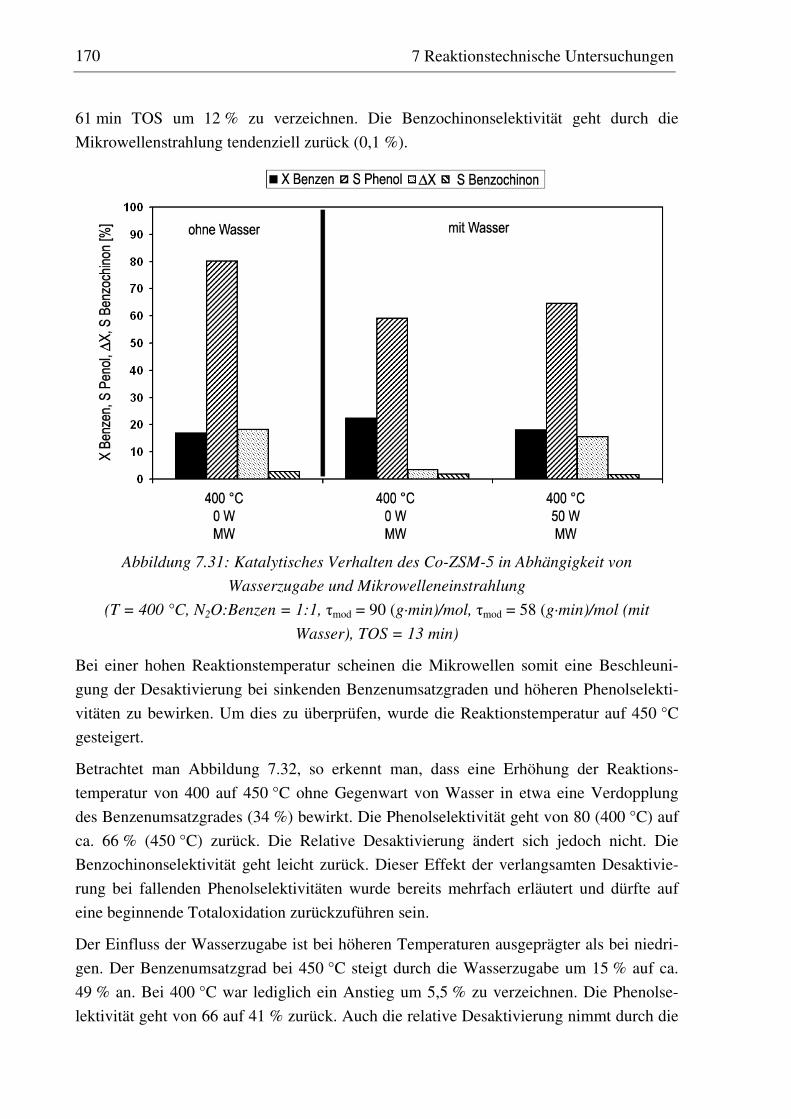
7 Reaktionstechnische Untersuchungen 170
61 min TOS um 12 % zu verzeichnen. Die Benzochinonselektivität geht durch die
Mikrowellenstrahlung tendenziell zurück (0,1 %).
Abbildung 7.31: Katalytisches Verhalten des Co-ZSM-5 in Abhängigkeit von
Wasserzugabe und Mikrowelleneinstrahlung
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, τmod = 58 (g·min)/mol (mit
Wasser), TOS = 13 min)
Bei einer hohen Reaktionstemperatur scheinen die Mikrowellen somit eine Beschleuni-
gung der Desaktivierung bei sinkenden Benzenumsatzgraden und höheren Phenolselekti-
vitäten zu bewirken. Um dies zu überprüfen, wurde die Reaktionstemperatur auf 450 °C
gesteigert.
Betrachtet man Abbildung 7.32, so erkennt man, dass eine Erhöhung der Reaktions-
temperatur von 400 auf 450 °C ohne Gegenwart von Wasser in etwa eine Verdopplung
des Benzenumsatzgrades (34 %) bewirkt. Die Phenolselektivität geht von 80 (400 °C) auf
ca. 66 % (450 °C) zurück. Die Relative Desaktivierung ändert sich jedoch nicht. Die
Benzochinonselektivität geht leicht zurück. Dieser Effekt der verlangsamten Desaktivie-
rung bei fallenden Phenolselektivitäten wurde bereits mehrfach erläutert und dürfte auf
eine beginnende Totaloxidation zurückzuführen sein.
Der Einfluss der Wasserzugabe ist bei höheren Temperaturen ausgeprägter als bei niedri-
gen. Der Benzenumsatzgrad bei 450 °C steigt durch die Wasserzugabe um 15 % auf ca.
49 % an. Bei 400 °C war lediglich ein Anstieg um 5,5 % zu verzeichnen. Die Phenolse-
lektivität geht von 66 auf 41 % zurück. Auch die relative Desaktivierung nimmt durch die

7.2 Hydroxylierung von Benzen mit N2O 171
Wasserzugabe deutlich ab (5 %). Die Benzochinonselektivität ist bei Zugabe von Wasser
zum Reaktionssystem wiederum leicht rückläufig (Rückgang: 0,7 %).
Abbildung 7.32: Katalytisches Verhalten des Co-ZSM-5 in Abhängigkeit von
Wasserzugabe und Mikrowelleneinstrahlung
(T = 450 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, τmod = 58 (g·min)/mol (mit
Wasser), TOS = 13 min)
Bei einer Temperatur von 450 °C wurden Experimente mit einer konstanten Mikrowel-
lenleistung von 50 W und bei gepulster Mikrowellenzugabe von 500 W durchgeführt
(4 min 500 W, dann 4 min Pause). Der Benzenumsatzgrad geht durch eine konstante
Mikrowelleneinstrahlung von 50 W um ca. 13 % zurück. Im Gegenzug steigt jedoch die
Phenolselektivität auf ca. 62 % an und erreicht somit beinahe den Wert ohne Wasserzu-
gabe. Die relative Desaktivierung steigt durch die Mikrowelleneinstrahlung um 12 % an.
Die Benzochinonselektivität fällt um 0,7 %. Eine Erhöhung der Mikrowellenleistung auf
500 W (gepulst) bringt nur noch geringfügige Änderungen gegenüber dem Experiment bei
50 W mit sich. Der Benzenumsatzgrad steigt um 2 % an, die Phenolselektivität geht leicht
zurück (1 %) und die relative Desaktivierung nimmt um 3 % ab. Bei der
Benzochinonselektivität nach 13 min TOS ist kein Einfluss der Mikrowellen feststellbar.
Diese Veränderungen sind wahrscheinlich auf das Pulsen der Strahlung zurückzuführen,
da bei 50 % der gesamten Reaktionsdauer keine Mikrowelleneinstrahlung eingebracht
wurde. Gegenüber dem Experiment bei einer konstanten Mikrowellenleistung von 50 W
(ohne pulsen!) sind höhere Benzenumsatzgrade, abnehmende Phenolselektivitäten und
eine geringere Desaktivierung die Folge. Dies bestätigt die in den letzten Kapiteln
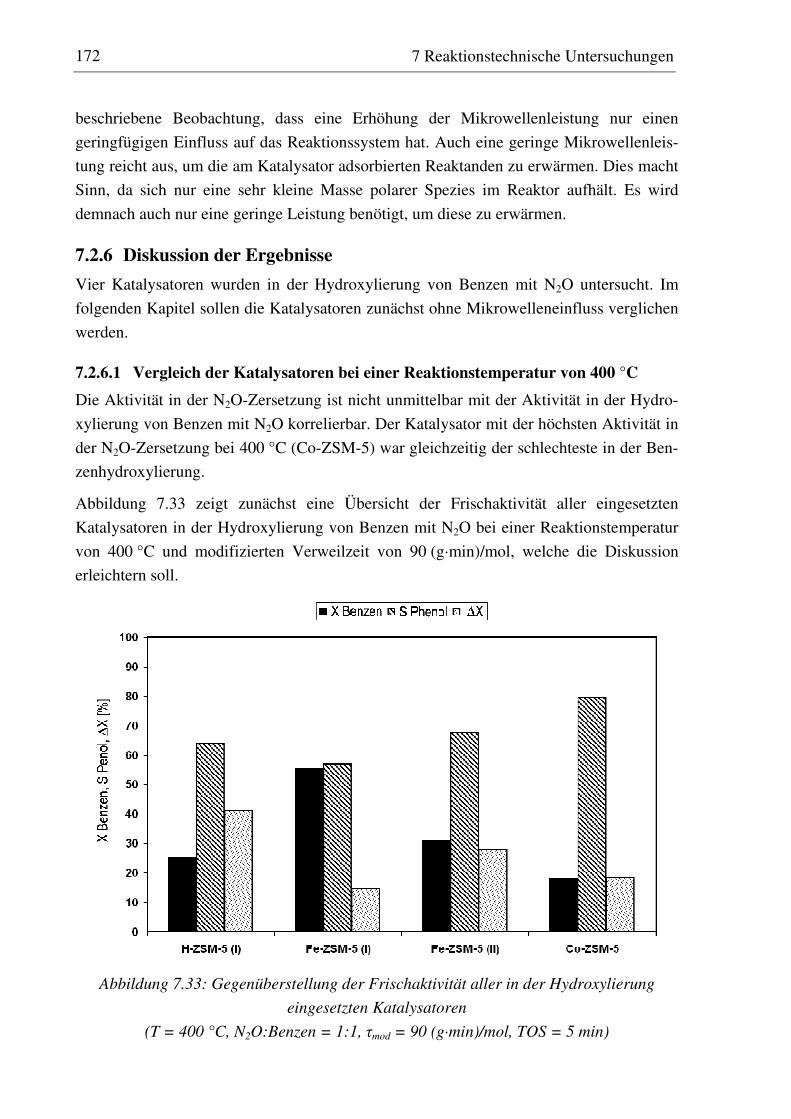
7 Reaktionstechnische Untersuchungen 172
beschriebene Beobachtung, dass eine Erhöhung der Mikrowellenleistung nur einen
geringfügigen Einfluss auf das Reaktionssystem hat. Auch eine geringe Mikrowellenleis-
tung reicht aus, um die am Katalysator adsorbierten Reaktanden zu erwärmen. Dies macht
Sinn, da sich nur eine sehr kleine Masse polarer Spezies im Reaktor aufhält. Es wird
demnach auch nur eine geringe Leistung benötigt, um diese zu erwärmen.
7.2.6 Diskussion der Ergebnisse
Vier Katalysatoren wurden in der Hydroxylierung von Benzen mit N2O untersucht. Im
folgenden Kapitel sollen die Katalysatoren zunächst ohne Mikrowelleneinfluss verglichen
werden.
7.2.6.1 Vergleich der Katalysatoren bei einer Reaktionstemperatur von 400 °C
Die Aktivität in der N2O-Zersetzung ist nicht unmittelbar mit der Aktivität in der Hydro-
xylierung von Benzen mit N2O korrelierbar. Der Katalysator mit der höchsten Aktivität in
der N2O-Zersetzung bei 400 °C (Co-ZSM-5) war gleichzeitig der schlechteste in der Ben-
zenhydroxylierung.
Abbildung 7.33 zeigt zunächst eine Übersicht der Frischaktivität aller eingesetzten
Katalysatoren in der Hydroxylierung von Benzen mit N2O bei einer Reaktionstemperatur
von 400 °C und modifizierten Verweilzeit von 90 (g·min)/mol, welche die Diskussion
erleichtern soll.
Abbildung 7.33: Gegenüberstellung der Frischaktivität aller in der Hydroxylierung
eingesetzten Katalysatoren
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 5 min)

7.2 Hydroxylierung von Benzen mit N2O 173
Es fällt auf, dass der Fe-ZSM-5 (I) die höchste Aktivität (Benzenumsatzgrad 55 % nach
5 min TOS) in der Hydroxylierung besitzt. Die relative Desaktivierung ∆X zwischen 5
und 61 min TOS ist mit 15 % zudem die geringste aller getesteten Katalysatoren. Die auf
Benzen bezogene Phenolselektivität ist jedoch mit ca. 60 % ebenfalls die niedrigste. Der
Fe-ZSM-5 (II) zeigte die zweithöchste Anfangsaktivität (Benzenumsatzgrad 31 % nach
5 min TOS) bei einer relativen Desaktivierung von 28 %. Die Phenolselektivität ist jedoch
mit 68 % höher als am Fe-ZSM-5 (I). Der Katalysator der H-Form (H-ZSM-5 (I)) zeigt
einen Umsatzgrad von 25 % zu Beginn der Reaktion (5 min TOS) bei einer
Phenolselektivität von ca. 70 %. Die geringste Aktivität in der Benzenhydroxylierung
wies der Co-ZSM-5 auf, der nach 5 min TOS lediglich einen Benzenumsatzgrad von 18 %
erreichte. Die Phenolselektivität war mit 80 % allerdings die höchste aller getesteten
Katalysatoren. Die relative Desaktivierung zwischen 5 und 61 min TOS war mit 18 %
gering. Diese Ergebnisse zeigen, dass die relative Desaktivierung nicht vom
Benzenumsatzgrad abhängt, da der Katalysator mit der höchsten Anfangsaktivität auch
gleichzeitig die geringste relative Desaktivierung zwischen 5 und 61 min TOS aufweist.
Die bei allen Katalysatoren niedrigen Phenolselektivitäten lassen sich durch die starke
Koksbildung und eine Totaloxidation zu CO2 erklären, da keine signifikanten Mengen
aromatischer Nebenprodukte detektiert wurden.
Weiterhin zeigt sich, dass die Phenolselektivitäten bei geringen Umsatzgraden an Benzen
tendenziell höher sind.
Interessant ist, dass alle Katalysatoren außer dem Co-ZSM-5, eine leicht ansteigende
Selektivität zum Zielprodukt mit fortschreitender Reaktionsdauer zeigen.
Zum einen kann dies durch eine behinderte Diffusion oder Desorption des Phenols im
Zeolith erklärt werden. Zu Beginn der Reaktion gebildetes Phenol benötigt sehr lange um
den Katalysator zu verlassen, und geht erst zu einem späteren Zeitpunkt wieder in die Bi-
lanzen ein.
Eine weitere Erklärung für dieses Phänomen ist, dass eine Koksbildung ausgehend von
Phenol mit fortschreitender Reaktionsdauer langsamer abläuft. Mit fortschreitender TOS
wird ein immer kleinerer Anteil des gebildeten Phenols zu Koks umgesetzt. Dies lässt sich
nur so erklären, dass verschiedene Zentren für die Bildung des Phenols und die Weiterre-
aktion des Phenols zu Koks verantwortlich sind. Vor allem diese für die Desaktivierung
verantwortlichen Zentren werden durch den gebildeten Koks blockiert. Die in der Hydro-
xylierung aktiven Zentren werden damit nur indirekt über eine Blockierung der Kanäle
des Zeoliths für die Reaktion „ausgeblendet“. Mit Hilfe dieser Theorie kann auch erklärt
werden, warum die Aktivität in der Hydroxylierung von Benzen mit N2O nicht mit der
relativen Desaktivierungsgeschwindigkeit korreliert. Abbildung 7.34 zeigt die relative
Desaktivierung ∆X zwischen 5 und 61 min und den Benzenumsatzgrad nach 13 min TOS

7 Reaktionstechnische Untersuchungen 174
in Abhängigkeit von der Fläche des bei der NH3-TPD ermittelten Hochtemperaturpeak bei
einer Reaktionstemperatur von 400 °C.
Die Fläche des Hochtemperaturpeak stellt ein Maß für die Anzahl starker Säurezentren
dar (vgl. Kapitel 5.2.4). Man erkennt einen linearen Zusammenhang zwischen der relati-
ven Desaktivierung ∆X und der Zahl dieser Zentren, jedoch keine Korrelation mit der
Aktivität in der Benzenhydroxylierung. Dieses Ergebnis deckt sich auch mit
Beobachtungen aus der Literatur [278, 288, 301] am identischen Katalysatortyp bzw.
Reaktionssystem. Somit liegt der Schluss nahe, dass die Desaktivierung unabhängig von
der gewünschten Hauptreaktion über unterschiedliche Mechanismen (Zentren) abläuft.
Ein Ziel bei der Katalysatorentwicklung für die Benzenhydroxylierung muss es somit
sein, die Zahl der Säurezentren möglichst gering zu halten, während die Zahl der in der
Hauptreaktion aktiven Zentren (Eisen?) maximiert werden muss.
Abbildung 7.34: Abhängigkeit des Benzenumsatzgrades und der relativen
Desaktivierungsgeschwindigkeit ∆X zwischen 5 und 61 min TOS von Fläche des mittels
NH3-TPD ermittelten Hochtemperaturpeak
(T = 400 °C, τmod = 90 (g·min)/mol, Benzen:N2O = 1:1, TOS = 5 min)
7.2.6.2 Einfluss der Reaktionstemperatur
Bei allen Katalysatoren wurde die Reaktion bei einer Temperatur von 300, 350 und
400 °C und einer modifizierten Verweilzeit von 90 (g·min)/mol untersucht.
Der Katalysator H-ZSM-5 (I) zeigte bei 300 °C keine Aktivität. Eine Steigerung der Tem-
peratur von 350 auf 400 °C führte zu einer verringerten Phenolselektivität bei abnehmen-
der Desaktivierung.

7.2 Hydroxylierung von Benzen mit N2O 175
Beim Katalysator Fe-ZSM-5 (I) ist die Phenolselektivität zwischen 300 und 350 °C bei-
nahe identisch, bei 400 °C ist jedoch ein deutlicher Rückgang zu verzeichnen. Die relative
Desaktivierung ∆X nimmt mit steigender Temperatur ab. Dies kann auf eine starke
Adsorption oder behinderte Diffusion des Phenols zurückzuführen sein, welches lange im
Katalysator verweilt und zu Koks weiterreagiert.
Der Katalysator Fe-ZSM-5 (II) zeigt ein anderes Verhalten. Eine Steigerung der Reakti-
onstemperatur von 300 auf 350 °C vermindert die Phenolselektivität gering, während die
relative Desaktivierung deutlich ansteigt. Eine weitere Temperaturerhöhung auf 400 °C
führt zu einem Rückgang der Phenolselektivität bei geringerer Desaktivierung. Dieses
Ergebnis widerspricht der Annahme, dass stark adsorbiertes Phenol für die Desaktivierung
verantwortlich ist. Eine Erklärung könnte wiederum sein, dass die Desaktivierung von
einer Weiterreaktion des Phenols zu Koks ausgeht. Diese Reaktion läuft bei höheren
Temperaturen schneller ab. Ein Koksabbrand durch N2O ist bei 350 °C dagegen noch
nicht zu erwarten.
Beim Co-ZSM-5 ist ebenfalls ein Anstieg der relativen Desaktivierung ∆X zwischen 300
und 350 °C zu beobachten, während die auf Benzen bezogene Phenolselektivität stark
zunimmt. Allerdings sind die Benzenumsatzgrade am Co-ZSM-5 bei 300 °C mit 2,6 %
sehr gering, was eine exakte Bestimmung der Phenolselektivitäten erschwert. Der Mess-
fehler der Analytik bzw. Dosierschwankungen wirken sich bei diesen geringen Umsätzen
stark aus. Deshalb soll auf eine eingehende Diskussion verzichtet werden. Eine weitere
Steigerung auf 400 °C hat kaum noch eine Auswirkung. Die relative Desaktivierung steigt
geringfügig an, die auf Benzen bezogene Phenolselektivität geht leicht zurück.
Das unterschiedliche Verhalten der vier getesteten Katalysatoren bei einer Erhöhung der
Reaktionstemperatur kann nur durch sich überlagernde Effekte erklärt werden, welche in
unterschiedlichen Temperaturbereichen dominieren. Nimmt die Phenolselektivität durch
eine Erhöhung der Temperatur ab, während die relative Desaktivierung ansteigt, kann dies
durch eine verstärkte Weiterreaktion von Phenol zu Koks erklärt werden. Verschlechtern
sich bei einer Temperaturerhöhung die Phenolselektivitäten bei einem gleichzeitigen
Rückgang der relativen Desaktivierung ∆X, deutet dies auf eine einsetzende Totaloxida-
tion des Kokses und eventuell des Phenols hin. Dieser Effekt tritt vor allem bei höheren
Reaktionstemperaturen (400 °C) auf.

7 Reaktionstechnische Untersuchungen 176
7.2.6.3 Einfluss der Verweilzeit
An den Katalysatoren Fe-ZSM-5 (II) und Co-ZSM-5 wurde die Reaktion zusätzlich bei
modifizierten Verweilzeiten von 120 und 155 (g·min)/mol untersucht. Eine Erhöhung der
Verweilzeit hat am Fe-ZSM-5 (II) folgende Auswirkungen:
• 300 °C: XBenzen ↑; SPhenol ↓; ∆X →
• 350 °C: XBenzen ↑; SPhenol →; ∆X ↓
• 400 °C: XBenzen ↑; SPhenol →; ∆X →
Am Co-ZSM-5 bewirkt eine Steigerung der Verweilzeit:
• 300 °C: XBenzen ↑; SPhenol ↓; ∆X ↑
• 350 °C: XBenzen ↑; SPhenol ↓; ∆X ↑
• 400 °C: XBenzen ↑; SPhenol ↓; ∆X ↑
(Zeichenerklärung: ↑ Zunahme; ↓ Rückgang; → keine Änderung)
Durch eine Erhöhung der modifizierten Verweilzeit wird mehr Benzen umgesetzt, aller-
dings reagiert auch Phenol in Abhängigkeit des eingesetzten Katalysators verstärkt zu
Folgeprodukten und Koks weiter, was sich durch abnehmende Phenolselektivitäten
bemerkbar macht. Die relative Desaktivierung nimmt durch eine Erhöhung der Verweil-
zeit am Co-ZSM-5 tendenziell zu, beim Fe-ZSM-5 (II) ist dagegen kein eindeutiger Trend
zu erkennen.
7.2.6.4 Überstöchiometrische Benzenzugabe
Am Katalysator Fe-ZSM-5 (II) wurden Experimente mit überstöchiometrischer Benzen-
zugabe (Benzen:N2O = 4:1) durchgeführt. Im Vergleich zu stöchiometrischer Fahrweise
verringern sich die Benzenumsatzgrade deutlich, die auf Benzen bezogene Phenolselek-
tivität steigt an und die relative Desaktivierung nimmt geringfügig ab. Bei einer Reakti-
onstemperatur von 400 °C wird beinahe das gesamte N2O zu Phenol umgesetzt. Dieses
Verhalten wird auch in der Literatur beschrieben [26, 233] und auf eine „Reinigung“ der
Katalysatoroberfläche durch das überschüssige Benzen zurückgeführt. Am Katalysator
adsorbiertes Phenol wird durch Benzen verdrängt und dadurch eine Weiterreaktion von
Phenol zu Nebenprodukten und Koks verhindert.
7.2.6.5 Wasserzugabe zum Feed
Am Co-ZSM-5 wurde zusätzlich die Wasserzugabe zum Feed bei 350, 400 und 450 °C
getestet. Die Auswirkungen waren bei allen getesteten Reaktionstemperaturen folgende:
• Anstieg der Benzenumsatzgrade
• Abnahme der auf Benzen bezogenen Phenolselektivitäten

7.2 Hydroxylierung von Benzen mit N2O 177
• Rückgang der relativen Desaktivierung ∆X
Dieses Verhalten lässt sich nach PILLAI ET AL. [306] durch die Verdrängung des
Phenols von den aktiven Zentren des Katalysators durch das Wasser erklären. Die Weiter-
reaktion des Phenols zu Koks wird verhindert, was zu höheren Benzenumsatzgraden bei
langsamerer Desaktivierung führt. Ein ähnlicher Effekt tritt auch bei einer Fahrweise mit
Benzenüberschuss auf (vgl. Kapitel 7.2.4.5).
Allerdings kommt es durch das Wasser auch zu unerwünschten Nebenreaktionen oder
Folgereaktionen. In den GC-Chromatogrammen konnten bei den Experimenten mit
Wasserzugabe zahlreiche Peaks beobachtet werden, welche im Vergleichsexperiment
ohne Wasserzugabe nicht vorhanden waren. Eine Identifizierung bzw. Quantifizierung
dieser Peaks wurde im Rahmen dieser Arbeit jedoch nicht durchgeführt. Aus diesem
Grund sinken wiederum die auf Benzen bezogenen Phenolselektivitäten. Zu vergleichba-
ren Ergebnissen kam auch HIEMER [26].
7.2.6.6 Auswirkungen der Mikrowelleneinstrahlung
Alle vier Katalysatoren wurden hinsichtlich ihres Verhaltens unter Mikrowelleneinstrah-
lung bei verschiedenen Reaktionsbedingungen getestet. Ein Vergleich der Experimente
ohne Mikrowelleneinstrahlung mit den entsprechenden Mikrowellenexperimenten zeigt
nur geringe Unterschiede. Die Benzenumsatzgrade und Phenolausbeuten differieren im
Durchschnitt um weniger als 1 %. Diese Abweichungen liegen im Bereich der Dosierge-
nauigkeit des Verdampfers und könnten auch durch Ungenauigkeiten in der Versuchs-
durchführung oder Messfehler verursacht sein. Allerdings zeigen sich Tendenzen, welche
bei allen getesteten Katalysatoren und Reaktionsbedingungen durch die Mikrowellenein-
strahlung zu beobachten waren:
• Die relative Desaktivierung ∆X erhöht sich
• Die Benzenumsatzgrade verringern sich leicht nach 13 min TOS
• Bei kurzer Verweilzeit Tendenz zu höheren auf Benzen bezogenen Phenol-
selektivitäten durch Mikrowelleneinstrahlung
• Bei hoher Verweilzeit Tendenz zu erniedrigten Phenolselektivitäten durch die
Mikrowelleneinstrahlung
• Eine Erhöhung der Mikrowellenleistung zeigt nur eine geringe Wirkung, allerdings
verstärken sich die beobachteten Tendenzen
Der am deutlichsten ausgeprägte Effekt ist die Beschleunigung der Desaktivierung durch
die Mikrowellen. Dies deutet darauf hin, dass es nicht zu einer selektiven Desorption des
Phenols kommt. Stattdessen heizen sich die adsorbierten Phenolmoleküle durch die Mik-
rowellen auf, werden jedoch nicht vorrangig desorbiert, sondern reagieren beschleunigt zu

7 Reaktionstechnische Untersuchungen 178
Koks weiter. Dies erklärt auch die sinkenden Benzenumsatzgrade nach 13 min TOS, da
die Desaktivierung beschleunigt abläuft und der Aktivitätsrückgang nach dieser Zeit
schon weiter fortgeschritten ist.
Weiterhin ist es denkbar, dass Folgeprodukte des Phenols (siehe Abbildung 3.16; S. 93)
durch die Mikrowellen selektiv aufgeheizt werden, was wiederum die Bildung von Koks
(hard coke) beschleunigt.
Die verringerten Phenolselektivitäten bei hoher Verweilzeit lassen sich ebenfalls durch
eine Aufheizung des Phenols erklären. Die Weiterreaktion zu Koks wird gefördert. Dem
widerspricht die Beobachtung, dass die Phenolselektivitäten bei niedrigen modifizierten
Verweilzeiten durch die Mikrowelleneinstrahlung ansteigen.
Dies kann durch zwei sich überlagernde Effekte erklärt werden. Zum einen fördert die
Mikrowellenstrahlung die Desorption des Phenols von den aktiven Zentren, was eine
Erhöhung der Phenolselektivität zur Folge hat. Eventuell kann auch die Diffusion des
Phenols durch die Mikrowellenheizung beschleunigt werden. Zum anderen wird aber auch
die Weiterreaktion des Phenols zu Koks gefördert, was sich in einer rascheren
Desaktivierung bemerkbar macht.
Die eingestrahlte Mikrowellenleistung hat kaum einen Einfluss auf das Reaktionsverhal-
ten. Allerdings verstärken sich die beobachteten Tendenzen bei höheren Leistungen. Da
die Masse an polaren Stoffen, welche die Mikrowellenstrahlung absorbieren sehr gering
ist, liegt die Erklärung auf der Hand. Auch bei geringen Leistungen wird bereits genügend
Energie eingestrahlt, um die Stoffe optimal aufzuheizen. Am Fe-ZSM-5 (I) werden bei-
spielsweise bei den Versuchseinstellungen T = 400 °C, N2O:Benzen = 1:1, τmod = 90
(g·min)/mol zu Beginn der Reaktion (5 min TOS) lediglich 31 mg/min Phenol gebildet.
Ein höherer Energieeintrag bewirkt lediglich, dass mehr ungenutzte Energie im System
vorhanden ist, welche sich in einer Aufheizung der Wände des Mikrowellenofens (und der
in den Ofen integrierten „Grundlast“) bemerkbar macht. Dies ist ein klassisches Problem
der Mikrowellentechnik und kann bei billigen Geräten sogar zur Zerstörung der
Magnetrone führen.
Warum keine Verbesserung des Desaktivierungsverhaltens durch die Mikrowellen
bewirkt werden konnte, soll anhand der TG-MS-Untersuchungen im folgenden Kapitel
näher erläutert werden.
Der Einfluss von Wasser auf das Reaktionssystem wurde am Katalysator Co-ZSM-5
untersucht. Wie bereits in Kapitel 7.2.5.5 beschrieben, führt die Anwesenheit von Wasser
im Reaktionssystem zu einer Steigerung der Benzenumsatzgrade. Da sich viele Folgepro-
dukte des Phenols bilden, nimmt die auf Benzen bezogene Phenolselektivität jedoch ab.
Die relative Desaktivierung geht durch die Wasserzugabe zurück.

7.2 Hydroxylierung von Benzen mit N2O 179
Betrachtet man nun die Experimente mit zusätzlicher Mikrowelleneinstrahlung, so
verschiebt sich das Reaktionsverhalten wieder in Richtung der Fahrweise ohne Wasserzu-
gabe. Der Benzenumsatzgrad verringert sich, während die auf Benzen bezogenen Phenol-
selektivität und die Desaktivierungsgeschwindigkeit deutlich ansteigen. Es kommt zu
einer Aufheizung des adsorbierten Wassers und einer Verdrängung von der Katalysator-
oberfläche durch die Mikrowellen. Dies bestätigt die Beobachtungen in der Literatur, dass
durch Mikrowelleneinstrahlung eine Verschiebung des Sorptionsgleichgewichts hin zu
unpolaren Stoffen möglich ist (siehe Kapitel 2.3.3).
7.2.7 Zusammenfassung
Es konnte keine Korrelation zwischen der Aktivität der Katalysatoren in der N2O-Zerset-
zung und der Aktivität in der Hydroxylierung von Benzen mit N2O festgestellt werden.
Ein Vergleich der Katalysatoren bei einer Reaktionstemperatur von 400 °C zeigte, dass
die relative Desaktivierung nicht von der Frischaktivität der Katalysatoren abhängt.
Es wurden leicht ansteigende Phenolselektivitäten bei fortschreitender Reaktionsdauer
bzw. Desaktivierung beobachtet.
Ein Vergleich der durch NH3-TPD ermittelten Anzahl starker Säurezentren mit der
Frischaktivität der Katalysatoren und der relativen Desaktivierung nach 5 min TOS zeigte
eine Korrelation mit der relativen Desaktivierung der Katalysatoren, nicht jedoch mit der
Aktivität in der Hydroxylierung.
Eine Erklärung für dieses Verhalten kann sein, dass diese Säurezentren für die Desaktivie-
rung verantwortlich sind, jedoch nicht für die Aktivität in der Hydroxylierung. Die Wei-
terreaktion des Phenols zu Koks läuft nicht an den Zentren ab, an denen das Phenol gebil-
det wird.
Bei allen Katalysatoren wurde die Reaktion bei einer Temperatur von 300, 350 und
400 °C und einer modifizierten Verweilzeit von 90 (g·min)/mol untersucht. Ist eine starke
Adsorption oder behinderte Diffusion des Phenols an den aktiven Zentren des
Katalysators für die Desaktivierung verantwortlich, sollte die Desaktivierung mit
steigender Reaktionstemperatur zurückgehen und die Phenolselektivität ansteigen. Dies
konnte lediglich beim Fe-ZSM-5 (I) bei einer Temperatursteigerung von 300 auf 350 °C
beobachtet werden. Die Ergebnisse deuten darauf hin, dass es noch zwei weitere, sich
überlagernde Effekte gibt. Zum einen kommt es durch Steigerung der
Reaktionstemperatur zu einer rascheren Desaktivierung des Katalysators durch eine
beschleunigte Weiterreaktion des Phenols zu Koks, zum anderen setzt bei höheren
Temperaturen (400 °C) eine Totaloxidation des Phenols und Kokses ein, was zu einer
langsameren relativen Desaktivierung führt.

7 Reaktionstechnische Untersuchungen 180
Eine Steigerung der Verweilzeit führte zu höheren Benzenumsatzgraden, wirkte sich
jedoch negativ auf die auf Benzen bezogenen Phenolselektivitäten und die Desaktivierung
aus.
Eine Fahrweise mit überstöchiometrischer Benzenzugabe steigerte die auf Benzen bezo-
genen Phenolselektivitäten und verringerte die Desaktivierung.
Durch Wasserzugabe zum Feed lies sich die Aktivität erhöhen und die Desaktivierung
verringern, jedoch nahm die Phenolselektivität aufgrund von Nebenreaktionen ab.
Der Einfluss der Mikrowellenstrahlung auf die Reaktion war gering, allerdings zeigten
sich Tendenzen, welche bei allen Katalysatoren zu beobachten waren. Bei bestimmten
Reaktionsbedingungen konnten gesteigerte Phenolselektivitäten beobachtet werden, der
stärker ausgeprägte Effekt war jedoch eine beschleunigte Desaktivierung. Es kommt zu
einer selektiven Aufheizung des Phenols durch die Mikrowellen, was eine Desorption von
den aktiven Zentren bewirken kann, jedoch auch eine beschleunigte Weiterreaktion des
Phenols oder dessen Folgeprodukten zu Koks.
Wurde die Reaktion mit Wasserzugabe zum Feed unter Mikrowelleneinstrahlung durch-
geführt, verschob sich das Reaktionsverhalten in Richtung der Fahrweise ohne Wasserzu-
gabe. Das adsorbierte Wasser wird durch die Mikrowellen aufgeheizt und von der
Katalysatoroberfläche verdrängt.
Es konnte somit gezeigt werden, dass ein gezielter Eingriff in den Reaktionsablauf und
das Sorptionsverhalten bei heterogen katalysierten Reaktionen durch eine selektive Auf-
heizung an der Reaktion beteiligter Spezies durch Mikrowellen möglich ist.

8 Charakterisierung des Desorptionsverhaltens
Nach Auswertung der Ergebnisse in der Hydroxylierung von Benzen mit N2O galt es, fol-
gende Fragestellungen zu klären:
• Wie wirken sich Mikrowellen auf die adsorbierten Stoffe aus?
• Ist eine selektive Desorption des Phenols von der Katalysatoroberfläche durch
Mikrowellen möglich, wenn keine Reaktion abläuft?
• Ist die Desorption des Phenols für die Desaktivierung des Katalysators bei der
Benzenhydroxylierung ausschlaggebend?
Zu diesem Zweck wurden zunächst Katalysatorproben nach dem Einsatz in der Hydroxy-
lierung von Benzen mit N2O einer TG-MS-Analyse (kombinierte Thermogravimetrie /
Massenspektrometrie) unterzogen. Weiterhin wurden frische Katalysatorproben mit Ben-
zen, Phenol sowie Mischungen aus beiden Stoffen beladen und anschließend mit Mikro-
wellen bestrahlt. An den so präparierten Proben wurden ebenfalls TG-MS-Untersuchun-
gen durchgeführt.
8.1 Versuchsdurchführung
Zunächst wurde der getrocknete Katalysator (0,3 g; bei 400 °C 2 h mit Stickstoff gespült)
in der in Kapitel 6.4 beschriebenen Apparatur mit den gewünschten Substanzen beladen.
In der vorliegenden Arbeit wurde die Adsorption immer bei 200 °C für eine Dauer von 2 h
durchgeführt.
Der auf diese Weise beladene Katalysator wurde anschließend in 3 Teilproben aufgeteilt.
Ein Drittel wurde mit Mikrowellen bestrahlt, um eine Desorption der adsorbierten Stoffe
zu untersuchen. Zu diesem Zweck wurde der beladene Katalysator für 2 h bei einer
Leistung von 400 W behandelt. Der eingesetzte Probenträger wurde mit 100 Nml/min
Stickstoff gespült, um den Abtransport der desorbierten Substanzen sicherzustellen.
Das zweite Drittel wurde ohne Mikrowellen für 2 h bei Raumtemperatur mit Stickstoff
gespült (100 Nml/min).
Sowohl die frisch beladene als auch die auf verschiedene Weise nachbehandelten Proben
wurden anschließend einer TG-MS-Analyse unterzogen.

182 8 Charakterisierung des Desorptionsverhaltens
Während der TG-Messung wurden die Proben (Probenmasse ca. 20 mg) von Raumtempe-
ratur auf 700 °C mit einer Rampe von 10 °C/min aufgeheizt. Als Trägergas kam Stickstoff
(40 Nml/min) zum Einsatz. Anschließend wurde das Trägergas auf synthetische Luft um-
gestellt (40 Nml/min) und die Temperatur (700 °C) für weitere 30 min konstant gehalten.
Mit Hilfe des Massenspektrometers konnten die desorbierten Spezies während der TG-
Messung identifiziert werden. Ausgewertet wurden die Atommassen 78,1 für Benzen und
94,1 für Phenol in den MS-Spektren. Diese Vorgehensweise erlaubt es, sowohl die Menge
und Art der adsorbierten Spezies zu bestimmen, die sich im Stickstoffstrom desorbieren
lassen (soft coke), als auch die Masse an hard coke, welche nur durch Abbrand mit Luft
ermittelt werden kann.
Diese Vorgehensweise ist in Abbildung 8.1 schematisch dargestellt.
Abbildung 8.1: Schema der Probenvorbereitung
8.2 In situ beladene Proben (aus Einsatz in der Reaktion)
Proben der Katalysatoren H-ZSM-5 (I) und Fe-ZSM-5 (II) wurden nach ihrem Einsatz in
der Hydroxylierung von Benzen mit N2O einer TG-MS-Analyse unterzogen, um Informa-
tionen über die Kokszusammensetzung und Koksmenge (soft bzw. hard coke) zu gewin-
nen. Als soft coke werden im Folgenden diejenigen Spezies bezeichnet, welche sich
während der TG-Messung bis zu einer Temperatur von 700 °C im Stickstoffstrom (bei
einer Aufheizrate der TG von 10 °C/min) desorbieren lassen. Als hard coke die Stoffe,
welche sich erst nach dem Umschalten auf Luft als TG-Trägergas bei einer Temperatur
von 700 °C abbrennen lassen. Dies ist formal nicht ganz korrekt (vgl. S. 189), hilft jedoch
bei der Diskussion der Ergebnisse.
Von besonderem Interesse war bei diesen Untersuchungen, ob sich Unterschiede zwi-
schen den Experimenten ohne Mikrowellen und den bei identischen Reaktionsbedingun-
gen mit zusätzlicher Mikrowelleneinstrahlung durchgeführten Experimenten zeigen.

8.2 In situ beladene Proben (aus Einsatz in der Reaktion) 183
Die Reaktion wurde jeweils über einen Zeitraum von 2 h durchgeführt. Anschließend
wurde der Katalysator im Reaktor für 3 min mit Stickstoff (200 Nml/min) gespült, um das
Reaktionsgemisch aus dem Reaktor zu entfernen.
8.2.1 H-ZSM-5 (I)
Abbildung 8.2 zeigt den Masseverlust von zwei Katalysatorproben des H-ZSM-5 (I) nach
der Benzenhydroxylierung während der TG-Messung.
Abbildung 8.2: Vergleich von zwei Proben des Katalysators H-ZSM-5 (I) aus der
Benzenhydroxylierung; T = 400 °C, τmod = 90 (g·min)/mol, Benzen:N2O = 1:1 nach 2 h
TOS; Reaktion ohne Mikrowelleneinstrahlung (0 W) und bei 200 W
Die X-Achse gibt die Zeit während der TG-MS-Messung an. Auf der linken Y-Achse ist
die Masse der Probe (bezogen auf die Einwaage) aufgetragen. Auf diese Achse sind die
schwarzen Kurven bezogen, anhand derer die Änderung der Probenmasse während der
TG-Messung abgelesen werden. Auf der rechten Seite sind zwei Y-Achsen aufgetragen.
Zum einen kann an der äußeren Skala die Probentemperatur (hellgraue Kurve) zu einem
bestimmten Zeitpunkt während der Messung abgelesen werden. Im linken Bereich des
Diagramms (mit „Stickstoff“ gekennzeichnet) erfolgt die Aufheizung der Probe auf
700 °C mit einer Rampe von 10 °C/min unter Stickstoffstrom, im rechten Bereich des
Diagramms (mit „Luft“ gekennzeichnet) wird die Temperatur konstant auf 700 °C
gehalten und die Probe mit Luft überströmt. Zum anderen ist auf der inneren Skala der
linken Y-Achse (dunkelgraue Kurven) das während der Messung detektierte MS-Signal
für Benzen und Phenol aufgetragen. Der Übersichtlichkeit halber und da sich die

184 8 Charakterisierung des Desorptionsverhaltens
Kurvenverläufe zwischen den Proben aus Experimenten mit oder ohne Mikrowelle nicht
unterscheiden, wird im Folgenden immer nur das MS-Signal für eine Probe aufgetragen.
Die mit „0 W“ bezeichnete Probe stammt aus einem Experiment ohne Mikrowellen-
einstrahlung, die „200 W“ Probe aus der Reaktion bei einer Mikrowellenleistung von
200 W. Die Reaktionsbedingungen waren bis auf die Mikrowelleneinstrahlung bei beiden
Experimenten identisch. Die Ergebnisse der reaktionstechnischen Untersuchungen finden
sich in Kapitel 7.2.2.
Es fällt zunächst auf, dass die beiden TG-Kurven nach 10 min bei einer Temperatur von
ca. 90 °C auseinander laufen. Betrachtet man die MS-Signale (graue Kurven, rechte Y-
Achse, Skala innen) erkennt man, dass dieser Unterschied auf die Desorption von Benzen
zurückzuführen ist, welches in einem Temperaturbereich von 80 bis 250 °C desorbiert
wird. Das Desorptionsmaximum wird bei etwa 160 °C erreicht. Die MS-Signale der mit
Mikrowellen behandelten Probe sind nicht in der Grafik enthalten, entsprechen in ihrem
Verlauf jedoch denen aus der konventionell beheizten Reaktion. Am Katalysator aus dem
MW-Experiment war somit mehr Benzen adsorbiert als an dem aus der nur konventionell
beheizten Reaktion. Dies dürfte jedoch auf den Spülvorgang nach Beendigung der Reak-
tion zurückzuführen sein, da Benzen nur schwach adsorbiert vorliegt. Vermutlich wurde
die Probe aus der Reaktion unter Mikrowelleneinstrahlung geringfügig länger mit Stick-
stoff gespült. Der weitere Verlauf der TG-Kurven ist beinahe identisch. Betrachtet man
das MS-Signal für Phenol, erkennt man, dass eine Phenoldesorption in einem Tempera-
turbereich zwischen 200 und 350 °C stattfindet. Das Maximum wird bei ca. 250 °C
erreicht. In einem Temperaturbereich von 400 bis 700 °C kommt es dann zu einem
steileren Abfall der TG-Kurven und zu einem Anstieg des MS-Signals für Benzen mit
einem Maximum bei ca. 600 °C.
Diese Benzendesorption bei sehr hohen Temperaturen ist überraschend, zumal Phenol
schon bei deutlich niedrigeren Temperaturen desorbiert. Es ist daher wahrscheinlich, dass
der „Benzenpeak“ bei hohen Temperaturen nicht auf Benzen zurückzuführen ist. In die-
sem Zusammenhang ist es von Bedeutung, dass große Moleküle in einem Mas-
senspektrometer „fragmentieren“, d.h. zu kleineren Bruchstücken zerfallen. Dieser Hoch-
temperaturpeak ist wahrscheinlich auf Fragmente höhermolekularer Nebenprodukte oder
Koksspezies zurückzuführen, welche die identische Atommasse besitzen wie Benzen.
Dies beweisen die Untersuchungen zur separaten Benzenbeladung, bei denen kein solcher
Hochtemperaturpeak beobachtet werden konnte (vgl. Kapitel 8.3.1).
Die ermittelte Menge an soft coke war 3,1 % bei der Probe aus der Reaktion unter Mikro-
welleneinstrahlung und 3,6 % bei der Probe aus dem konventionell beheizten Vergleichs-
experiment. Dies ist auf eine unterschiedliche Menge an adsorbiertem Benzen zurückzu-
führen.
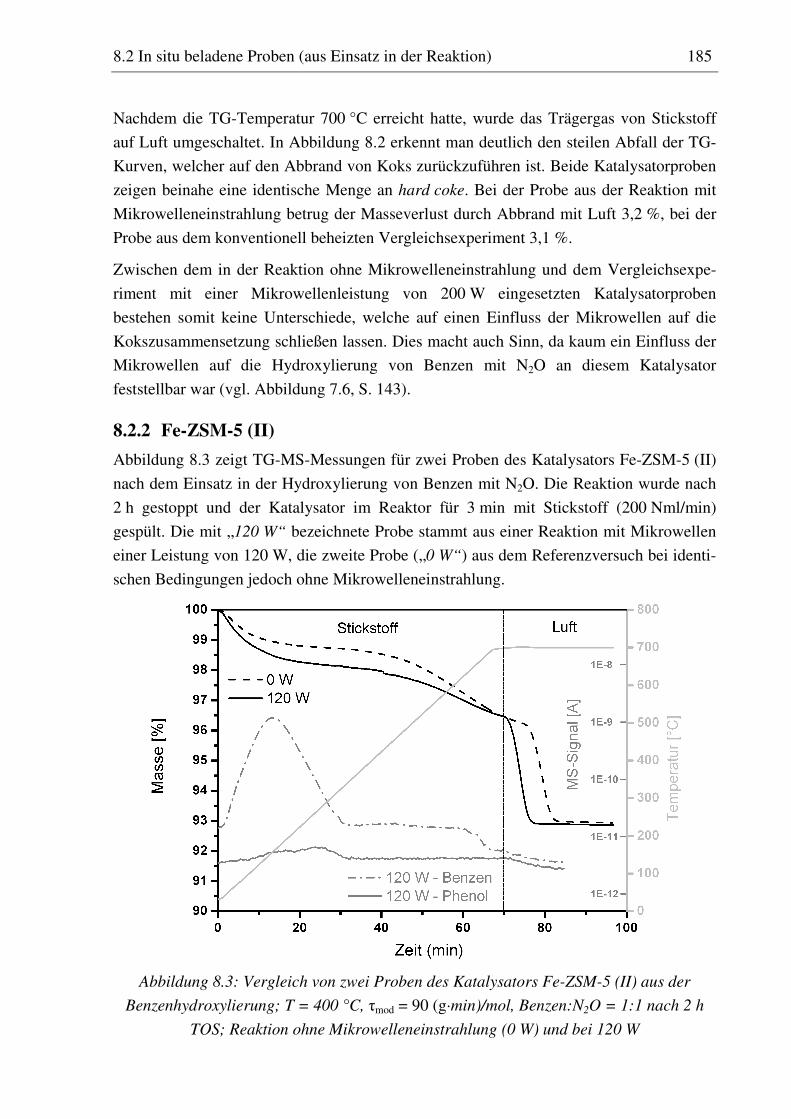
8.2 In situ beladene Proben (aus Einsatz in der Reaktion) 185
Nachdem die TG-Temperatur 700 °C erreicht hatte, wurde das Trägergas von Stickstoff
auf Luft umgeschaltet. In Abbildung 8.2 erkennt man deutlich den steilen Abfall der TG-
Kurven, welcher auf den Abbrand von Koks zurückzuführen ist. Beide Katalysatorproben
zeigen beinahe eine identische Menge an hard coke. Bei der Probe aus der Reaktion mit
Mikrowelleneinstrahlung betrug der Masseverlust durch Abbrand mit Luft 3,2 %, bei der
Probe aus dem konventionell beheizten Vergleichsexperiment 3,1 %.
Zwischen dem in der Reaktion ohne Mikrowelleneinstrahlung und dem Vergleichsexpe-
riment mit einer Mikrowellenleistung von 200 W eingesetzten Katalysatorproben
bestehen somit keine Unterschiede, welche auf einen Einfluss der Mikrowellen auf die
Kokszusammensetzung schließen lassen. Dies macht auch Sinn, da kaum ein Einfluss der
Mikrowellen auf die Hydroxylierung von Benzen mit N2O an diesem Katalysator
feststellbar war (vgl. Abbildung 7.6, S. 143).
8.2.2 Fe-ZSM-5 (II)
Abbildung 8.3 zeigt TG-MS-Messungen für zwei Proben des Katalysators Fe-ZSM-5 (II)
nach dem Einsatz in der Hydroxylierung von Benzen mit N2O. Die Reaktion wurde nach
2 h gestoppt und der Katalysator im Reaktor für 3 min mit Stickstoff (200 Nml/min)
gespült. Die mit „120 W“ bezeichnete Probe stammt aus einer Reaktion mit Mikrowellen
einer Leistung von 120 W, die zweite Probe („0 W“) aus dem Referenzversuch bei identi-
schen Bedingungen jedoch ohne Mikrowelleneinstrahlung.
Abbildung 8.3: Vergleich von zwei Proben des Katalysators Fe-ZSM-5 (II) aus der
Benzenhydroxylierung; T = 400 °C, τmod = 90 (g·min)/mol, Benzen:N2O = 1:1 nach 2 h
TOS; Reaktion ohne Mikrowelleneinstrahlung (0 W) und bei 120 W

186 8 Charakterisierung des Desorptionsverhaltens
Die Ergebnisse der reaktionstechnischen Untersuchungen finden sich in Kapitel 7.2.4.
Vergleicht man die TG-Kurven der beiden Katalysatorproben, so fällt zunächst auf, dass
der Katalysator aus der Reaktion ohne Mikrowellen im niedrigen Temperaturbereich (bis
180 °C) einen geringeren Masseverlust aufweist als die Probe aus der Reaktion unter
Mikrowelleneinstrahlung. Wie auch beim H-ZSM-5 (I) wird im Temperaturbereich zwi-
schen 80 und 250 °C vor allem Benzen desorbiert (Maximum bei 158 °C). Phenol wird im
Massenspektrometer zwischen 180 und 350 °C detektiert. Die größte Phenolmenge wird
bei 266 °C desorbiert. In diesem Temperaturbereich sind jedoch keine Unterschiede der
TG-Kurven zu erkennen. Beide Proben enthalten etwa gleich viel Phenol. Erst ab einer
Temperatur von ca. 450 °C zeigt die Probe aus der Reaktion ohne Mikrowellen eine stär-
kere Masseabnahme als der Katalysator aus der Reaktion bei 120 W Mikrowellenleistung.
Ein Hochtemperaturpeak mit der Atommasse von Benzen, wie er am H-ZSM-5 (I) beo-
bachtet wurde, ist nicht zu erkennen.
Bei einer Temperatur von 700 °C weisen beide Proben einen identischen Masseverlust
von ca. 3,5 % auf (soft coke). Der abweichende Verlauf der TG-Kurven bei Temperaturen
zwischen 450 und 700 °C deutet jedoch auf Unterschiede in der Kokszusammensetzung
hin. Der Katalysator aus der Reaktion ohne Mikrowelleneinstrahlung enthält mehr Koks
(bzw. Nebenprodukte), welcher sich durch Ausheizen bei hoher Temperatur entfernen
lässt.
Die Menge an hard coke, welche sich durch den Abbrand mit Luft bei 700 °C bestimmen
lässt, ist bei beiden Proben mit 3,6 % identisch.
Es zeigt sich somit, dass die Mikrowellen nur einen geringen Einfluss auf die Kokszu-
sammensetzung und die Menge der adsorbierten Spezies haben. Allerdings erkennt man
am Fe-ZSM-5 (II) Unterschiede in der Menge der Desorbierten Spezies zwischen 450 und
700 °C, die auf einen Einfluss der Mikrowelleneinstrahlung hindeuten. Interessant ist,
dass Phenol bei beiden Katalysatoren vorwiegend im Temperaturbereich unter 300 °C
desorbiert wurde. Dies widerspricht der Argumentation, dass Phenol aufgrund seiner star-
ken Adsorption am Katalysator zu einer Desaktivierung während der Benzenhydroxylie-
rung führt.
Außerdem weist der Fe-ZSM-5 (II) im Vergleich zum H-ZSM-5 (I) eine größere Menge
an Koks (Adsorbat) auf, obwohl er in der Hydroxylierung von Benzen mit N2O eine
höhere Aktivität bei geringerer Desaktivierung zeigte (vgl. Kapitel 7.2). Vergleicht man
die Experimente ohne Mikrowelle bei identischen Reaktionsbedingungen, so weist der
H-ZSM-5 (I) eine Beladung von 6,7 % auf, während der Fe-ZSM-5 (II) 7,1 % Massever-
lust zeigt. Die Menge an hard coke ist beim Fe-ZSM-5 (II) mit 3,6 % ebenfalls größer als
am Katalysator in der H-Form (3,1 %). Die relative Desaktivierungsgeschwindigkeit in
der Benzenhydroxylierung ist somit nicht direkt an die gebildete Koksmenge gekoppelt.

8.3 Ex situ beladene Proben 187
8.3 Ex situ beladene Proben
Die Beladung der Katalysatorproben erfolgte nach der in Kapitel 8.1 geschilderten Vorge-
hensweise. Die Beschreibung der verwendeten Apparaturen findet sich in Kapitel 6.4.
8.3.1 H-ZSM-5 (I)
Der Katalysator wurde mit Benzen, Phenol oder simultan mit beiden Stoffen beladen. Im
Folgenden sollen jeweils die bei Raumtemperatur im Stickstoffstrom gespülten Proben
mit den unter zusätzlicher Mikrowelleneinstrahlung behandelten verglichen werden. Es
wird nur das MS-Signal der nicht mit Mikrowellen bestrahlten Probe betrachtet, da sich
die MS-Signale der unterschiedlich vorbehandelten Proben lediglich in ihrer Intensität,
jedoch nicht im Verlauf unterscheiden.
Abbildung 8.4 zeigt TG-MS-Messungen einer Katalysatorprobe, welche mit Benzen bela-
den wurde.
Abbildung 8.4: Mit Benzen beladene Probe des Katalysators H-ZSM-5 (I); Partialdruck
Benzen = 13250 Pa; 0 W: N2 gespült; 400 W: N2 gespült bei 400 W
Mikrowelleneinstrahlung
Ein Vergleich der TG-Kurven zeigt, dass die Mikrowelleneinstrahlung nur einen geringen
Einfluss auf die Benzendesorption hat. Die mit Mikrowellen behandelte Probe weist im
niedrigen Temperaturbereich einen etwas geringeren Masseverlust auf (weniger Benzen
wird desorbiert), allerdings gleichen sich die Kurven rasch an, bis schließlich eine nahezu
identische Masseabnahme von ca. 3,9 % bei einer Temperatur von 700 °C erreicht wird.
Bei einer Temperatur von ca. 400 °C weisen die Kurven einen Wendepunkt auf, welcher
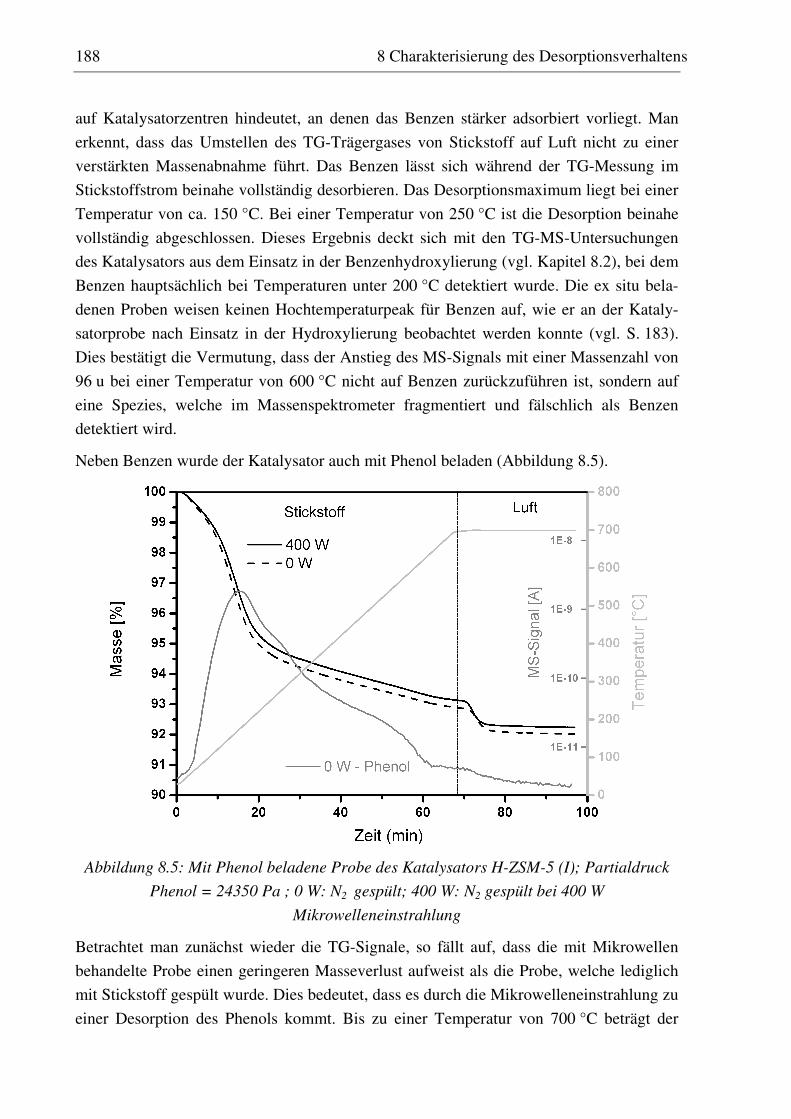
188 8 Charakterisierung des Desorptionsverhaltens
auf Katalysatorzentren hindeutet, an denen das Benzen stärker adsorbiert vorliegt. Man
erkennt, dass das Umstellen des TG-Trägergases von Stickstoff auf Luft nicht zu einer
verstärkten Massenabnahme führt. Das Benzen lässt sich während der TG-Messung im
Stickstoffstrom beinahe vollständig desorbieren. Das Desorptionsmaximum liegt bei einer
Temperatur von ca. 150 °C. Bei einer Temperatur von 250 °C ist die Desorption beinahe
vollständig abgeschlossen. Dieses Ergebnis deckt sich mit den TG-MS-Untersuchungen
des Katalysators aus dem Einsatz in der Benzenhydroxylierung (vgl. Kapitel 8.2), bei dem
Benzen hauptsächlich bei Temperaturen unter 200 °C detektiert wurde. Die ex situ bela-
denen Proben weisen keinen Hochtemperaturpeak für Benzen auf, wie er an der Kataly-
satorprobe nach Einsatz in der Hydroxylierung beobachtet werden konnte (vgl. S. 183).
Dies bestätigt die Vermutung, dass der Anstieg des MS-Signals mit einer Massenzahl von
96 u bei einer Temperatur von 600 °C nicht auf Benzen zurückzuführen ist, sondern auf
eine Spezies, welche im Massenspektrometer fragmentiert und fälschlich als Benzen
detektiert wird.
Neben Benzen wurde der Katalysator auch mit Phenol beladen (Abbildung 8.5).
Abbildung 8.5: Mit Phenol beladene Probe des Katalysators H-ZSM-5 (I); Partialdruck
Phenol = 24350 Pa ; 0 W: N2 gespült; 400 W: N2 gespült bei 400 W
Mikrowelleneinstrahlung
Betrachtet man zunächst wieder die TG-Signale, so fällt auf, dass die mit Mikrowellen
behandelte Probe einen geringeren Masseverlust aufweist als die Probe, welche lediglich
mit Stickstoff gespült wurde. Dies bedeutet, dass es durch die Mikrowelleneinstrahlung zu
einer Desorption des Phenols kommt. Bis zu einer Temperatur von 700 °C beträgt der

8.3 Ex situ beladene Proben 189
Masseverlust der nicht mit Mikrowellen behandelten Probe 7,1 %, während die bestrahlte
Probe lediglich einen Masseverlust von 6,9 % aufweist. Der absolute Unter-schied ist
gering, allerdings muss berücksichtigt werden, dass die Desorption bei Raumtemperatur
durchgeführt wurde. In diesem Temperaturbereich ist eine Desorption aufgrund des
polaren Charakters des Phenols bzw. der starken Wechselwirkung mit dem Zeolith nur
schwer möglich.
Nach dem Umschalten des TG-Trägergases von Stickstoff auf Luft ist ein steiler Abfall
der Massenkurven zu beobachten. Es kommt zu einem Abbrand von adsorbierten Spezies
(bzw. Koks), welche sich nicht unter Stickstoffatmosphäre im Temperaturbereich unter
700 °C desorbieren ließen. Der Anteil dieses hard coke war bei beiden Proben nahezu
identisch (0 W: 0,82 %; 400 W: 0,86 %). Wahrscheinlich kommt es schon während der
Beladung bei einer Temperatur von 200 °C zur Bildung von Koks. Dies wird auch durch
die Beobachtung bestätigt, dass die ursprünglich weiße Katalysatorprobe nach dem Bela-
dungsvorgang eine Graufärbung aufwies. Natürlich ist es auch vorstellbar, dass ein Anteil
des Phenols während der TG-MS-Messung im Stickstoffstrom aufgrund der langsamen
Diffusion nicht desorbiert wird. Weiterhin hat sich gezeigt, dass Phenol während der TG-
MS-Messung zu Koks weiterreagiert. Bricht man die TG-MS-Messung vor dem
Umschalten des Trägergases auf Luft ab, sind die mit Phenol beladenen Proben intensiver
gau-schwarz gefärbt als nach dem Beladungsvorgang. Es scheint somit einleuchtend, dass
auch die Desaktivierung während der Benzenhydroxylierung mit N2O ausgehend von
Phenol abläuft.
Betrachtet man das MS-Signal, so zeigt sich, dass in einem Temperaturbereich bis 600 °C
Phenol desorbiert wird. Das Maximum liegt jedoch bei einer Temperatur von ca. 180 °C.
Der Großteil des Phenols wird bei einer Temperatur unter 300 °C desorbiert (0 W: 5,7%),
während zwischen 300 und 700 °C nur noch eine kontinuierliche Massenabnahme um
insgesamt ca. 1,4 % (0 W) festzustellen ist. Die Temperatur, bei der die maximale
Phenolmenge desorbiert wurde liegt somit ca. 30 °C über der Temperatur, bei der das
Maximum der Benzendesorption beobachtet wurde.
Abbildung 8.6 zeigt die TG-MS-Messungen einer Katalysatorprobe, welche simultan mit
Benzen und Phenol beladen und anschließend mit Stickstoff gespült (0 W) bzw. mit Mik-
rowellenstrahlung nachbehandelt wurde (400 W). Betrachtet man die TG-Kurven, so
erkennt man einen Verlauf, wie er sich bereits bei der mit Phenol beladenen Probe
(Abbildung 8.5) zeigte. Die mit Mikrowellen behandelte Katalysatorprobe weist einen
geringeren Masseverlust (6,3 %) im Temperaturbereich bis 700 °C auf als die Probe, wel-
che lediglich mit Stickstoff gespült wurde (6,7 %). Es kam also zu einer Desorption
während des Nachbehandlungsschritts mit Mikrowellen. Unter Lufteinwirkung bei 700 °C
ist wiederum deutlich ein Abbrand von Koks zu erkennen. Der Masseverlust durch
Abbrand des hard coke war wiederum bei beiden Proben beinahe identisch (0 W: 0,78 %;
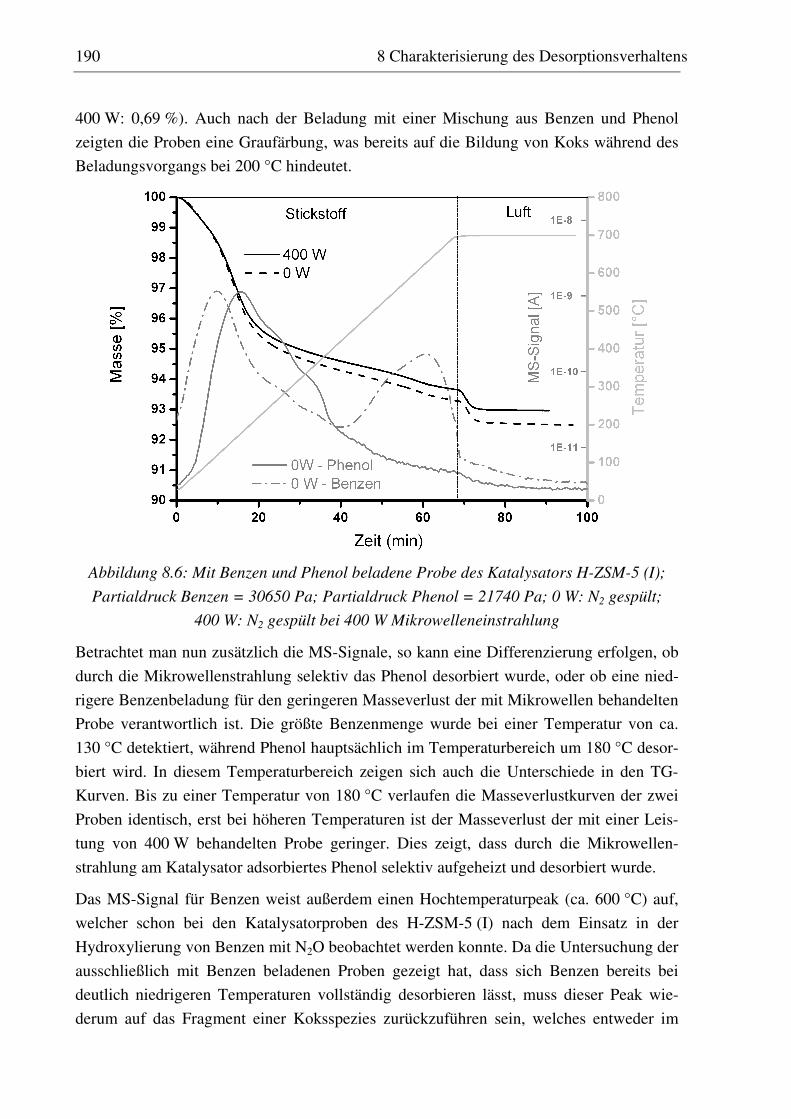
190 8 Charakterisierung des Desorptionsverhaltens
400 W: 0,69 %). Auch nach der Beladung mit einer Mischung aus Benzen und Phenol
zeigten die Proben eine Graufärbung, was bereits auf die Bildung von Koks während des
Beladungsvorgangs bei 200 °C hindeutet.
Abbildung 8.6: Mit Benzen und Phenol beladene Probe des Katalysators H-ZSM-5 (I);
Partialdruck Benzen = 30650 Pa; Partialdruck Phenol = 21740 Pa; 0 W: N2 gespült;
400 W: N2 gespült bei 400 W Mikrowelleneinstrahlung
Betrachtet man nun zusätzlich die MS-Signale, so kann eine Differenzierung erfolgen, ob
durch die Mikrowellenstrahlung selektiv das Phenol desorbiert wurde, oder ob eine nied-
rigere Benzenbeladung für den geringeren Masseverlust der mit Mikrowellen behandelten
Probe verantwortlich ist. Die größte Benzenmenge wurde bei einer Temperatur von ca.
130 °C detektiert, während Phenol hauptsächlich im Temperaturbereich um 180 °C desor-
biert wird. In diesem Temperaturbereich zeigen sich auch die Unterschiede in den TG-
Kurven. Bis zu einer Temperatur von 180 °C verlaufen die Masseverlustkurven der zwei
Proben identisch, erst bei höheren Temperaturen ist der Masseverlust der mit einer Leis-
tung von 400 W behandelten Probe geringer. Dies zeigt, dass durch die Mikrowellen-
strahlung am Katalysator adsorbiertes Phenol selektiv aufgeheizt und desorbiert wurde.
Das MS-Signal für Benzen weist außerdem einen Hochtemperaturpeak (ca. 600 °C) auf,
welcher schon bei den Katalysatorproben des H-ZSM-5 (I) nach dem Einsatz in der
Hydroxylierung von Benzen mit N2O beobachtet werden konnte. Da die Untersuchung der
ausschließlich mit Benzen beladenen Proben gezeigt hat, dass sich Benzen bereits bei
deutlich niedrigeren Temperaturen vollständig desorbieren lässt, muss dieser Peak wie-
derum auf das Fragment einer Koksspezies zurückzuführen sein, welches entweder im

8.3 Ex situ beladene Proben 191
Massenspektrometer entsteht oder durch die Zersetzung von Koksspezies während der
TG-MS-Messung bei hohen Temperaturen. Die Analyse der Katalysatorprobe, welche nur
mit Phenol beladen wurde, zeigte ebenfalls einen Peak der Atommasse 96 bei 600 °C,
obwohl diese Probe niemals mit Benzen in Berührung kam. Auf eine Darstellung des MS-
Signals für Benzen wurde in der entsprechenden Grafik (Abbildung 8.5) jedoch
verzichtet.
8.3.2 Fe-ZSM-5 (II)
Neben dem H-ZSM-5 (I) wurde auch der Katalysator Fe-ZSM-5 (II) mit Benzen, Phenol
und simultan mit beiden Substanzen beladen.
Abbildung 8.7 zeigt die TG-MS-Messungen einer Katalysatorprobe, welche mit Benzen
beladen und anschließend entweder mit Stickstoff gespült (0 W) oder mit Mikrowellen
(400 W) behandelt wurde. Auf ein Umschalten des TG-Trägergases von Stickstoff auf
Luft wurde bei dieser Messung verzichtet, da sich bei den Untersuchungen am
H-ZSM-5 (I) bereits herausgestellt hatte, dass es nicht zur Bildung von hard coke während
der Benzenbeladung kommt.
Abbildung 8.7: Mit Benzen beladene Probe des Katalysators Fe-ZSM-5 (II); Partialdruck
Benzen = 6450 Pa; 0 W: N2 gespült; 400 W: N2 gespült bei 400 W
Mikrowelleneinstrahlung
Betrachtet man zunächst wieder die TG-Kurven, so zeigt sich ein Verlauf, welcher mit
dem des Benzen beladenen H-ZSM-5 (I) (Abbildung 8.4) vergleichbar ist. Die mit
Mikrowellen behandelte Probe weist einen geringeren Masseverlust im niedrigen
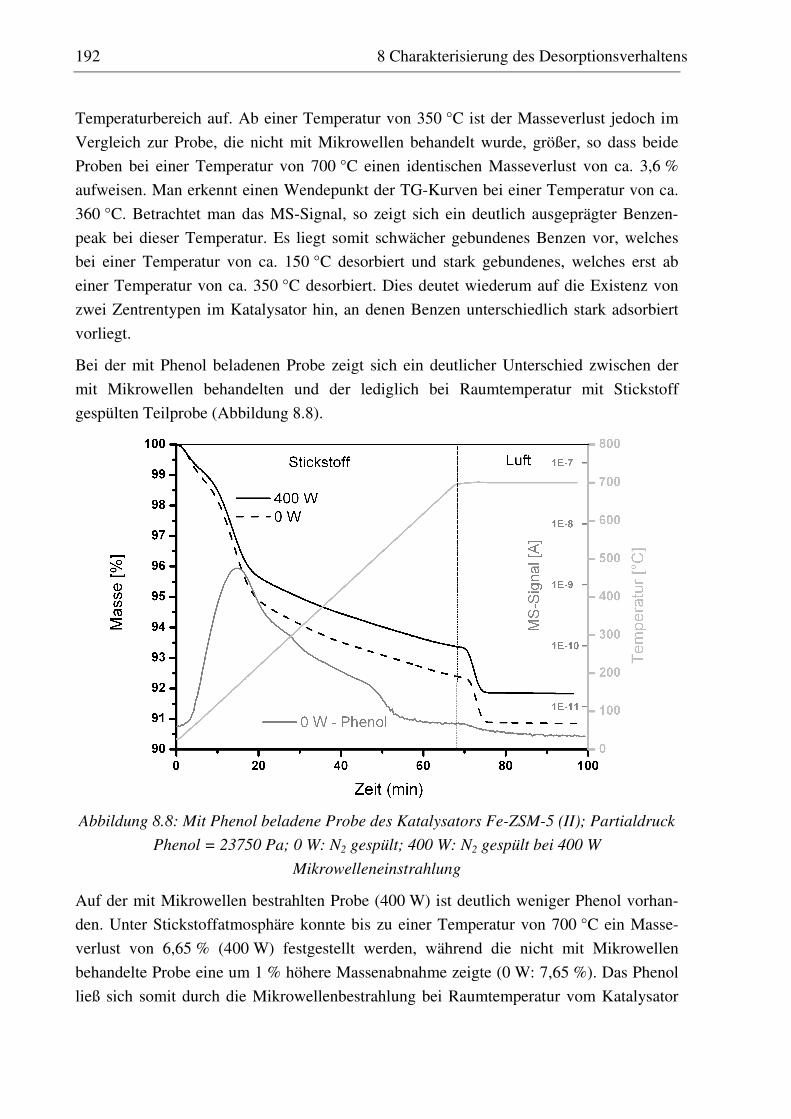
192 8 Charakterisierung des Desorptionsverhaltens
Temperaturbereich auf. Ab einer Temperatur von 350 °C ist der Masseverlust jedoch im
Vergleich zur Probe, die nicht mit Mikrowellen behandelt wurde, größer, so dass beide
Proben bei einer Temperatur von 700 °C einen identischen Masseverlust von ca. 3,6 %
aufweisen. Man erkennt einen Wendepunkt der TG-Kurven bei einer Temperatur von ca.
360 °C. Betrachtet man das MS-Signal, so zeigt sich ein deutlich ausgeprägter Benzen-
peak bei dieser Temperatur. Es liegt somit schwächer gebundenes Benzen vor, welches
bei einer Temperatur von ca. 150 °C desorbiert und stark gebundenes, welches erst ab
einer Temperatur von ca. 350 °C desorbiert. Dies deutet wiederum auf die Existenz von
zwei Zentrentypen im Katalysator hin, an denen Benzen unterschiedlich stark adsorbiert
vorliegt.
Bei der mit Phenol beladenen Probe zeigt sich ein deutlicher Unterschied zwischen der
mit Mikrowellen behandelten und der lediglich bei Raumtemperatur mit Stickstoff
gespülten Teilprobe (Abbildung 8.8).
Abbildung 8.8: Mit Phenol beladene Probe des Katalysators Fe-ZSM-5 (II); Partialdruck
Phenol = 23750 Pa; 0 W: N2 gespült; 400 W: N2 gespült bei 400 W
Mikrowelleneinstrahlung
Auf der mit Mikrowellen bestrahlten Probe (400 W) ist deutlich weniger Phenol vorhan-
den. Unter Stickstoffatmosphäre konnte bis zu einer Temperatur von 700 °C ein Masse-
verlust von 6,65 % (400 W) festgestellt werden, während die nicht mit Mikrowellen
behandelte Probe eine um 1 % höhere Massenabnahme zeigte (0 W: 7,65 %). Das Phenol
ließ sich somit durch die Mikrowellenbestrahlung bei Raumtemperatur vom Katalysator

8.3 Ex situ beladene Proben 193
desorbieren. Über das MS-Signal kann das Maximum der Phenoldesorption bei einer
Temperatur von etwa 175 °C bestimmt werden.
Phenol ließ sich durch das Massenspektrometer in einem Temperaturbereich von
80 - 550 °C nachweisen. Die TG-Kurven der unterschiedlich vorbehandelten Proben
zeigen jedoch im niedrigen Temperaturbereich bis ca. 280 °C die stärksten Abweichun-
gen.
Nach dem Umschalten des TG-Trägergases von Stickstoff auf Luft bei einer Temperatur
von 700 °C kann wiederum ein steiler Abfall der Massenkurven beobachtet werden, wel-
cher aus dem Abbrand von hard coke resultiert. Beide Proben weisen einen beinahe
identischen Anteil von hard coke auf (0 W: 1,50 %; 400 W: 1,52 %). Die Proben des
Fe-ZSM-5 (II) zeigten, wie auch die des H-ZSM-5 (I), bereits nach dem Beladungsvor-
gang mit Phenol eine Graufärbung.
Um den Einfluss der Aufheizgeschwindigkeit während der TG-MS-Messung zu verifizie-
ren, wurde eine weitere Probe mit Phenol beladen und anschließend mit Stickstoff gespült.
An dieser Probe wurde eine TG-MS-Messung bei einer Aufheizrate von 10 °C/min und
1 °C/min durchgeführt. Die Messung bei einer Aufheizrate von 10 °C/min ist in
Abbildung 8.9 dargestellt. Wie zu erwarten entspricht der Verlauf dem in Abbildung 8.8,
allerdings ist der Anteil an soft coke der zweiten Messung um 0,65 % höher, der hard coke
Anteil ist hingegen identisch. Dies ist evtl. auf eine Abweichung während des
Beladungsvorgangs zurückzuführen oder auf veränderte TG-MS-Parameter, da die zweite
Messung mit 1 Jahr Abstand durchgeführt wurde. Betrachtet man das MS-Signal zeigt
sich wiederum bei einer Temperatur von 175 °C ein Desorptionsmaximum von Phenol.
Phenol wurde bei einer Temperatur von ca. 80 - 500 °C detektiert. Ab 420 °C ist die
detektierte Konzentration jedoch nahe an der Nachweisgrenze des Massenspektrometers.
Generell ist die MS-Signalintensität bei der in Abbildung 8.9 gezeigten Messung im
Vergleich zu Abbildung 8.8 deutlich geringer. Dies ist auf Reparaturarbeiten am
Massenspektrometer zurückzuführen, welche zwischenzeitlich durchgeführt wurden.
Vergleicht man nur die TG-Kurven der Messung, welche mit einer Aufheizrate von
10 °C/min (Abbildung 8.9) durchgeführt wurde und die Messung mit einer Aufheizrate
von 1 °C/min (Abbildung 8.10), so fällt zunächst auf, dass der Masseverlust bis zu einer
Temperatur von 700 °C mit 8,92 % bei der Aufheizrate von 1 °C/min um 0,63 % höher
ist, als bei der Vergleichsmessung mit der schnelleren Aufheizrate (Abbildung 8.9). Der
Anteil an hard coke ist mit 0,82 % dagegen deutlich niedriger. Durch das langsame Auf-
heizen konnte bis zu einer Temperatur von 700 °C im Stickstoffstrom mehr adsorbierte
Spezies desorbiert werden.
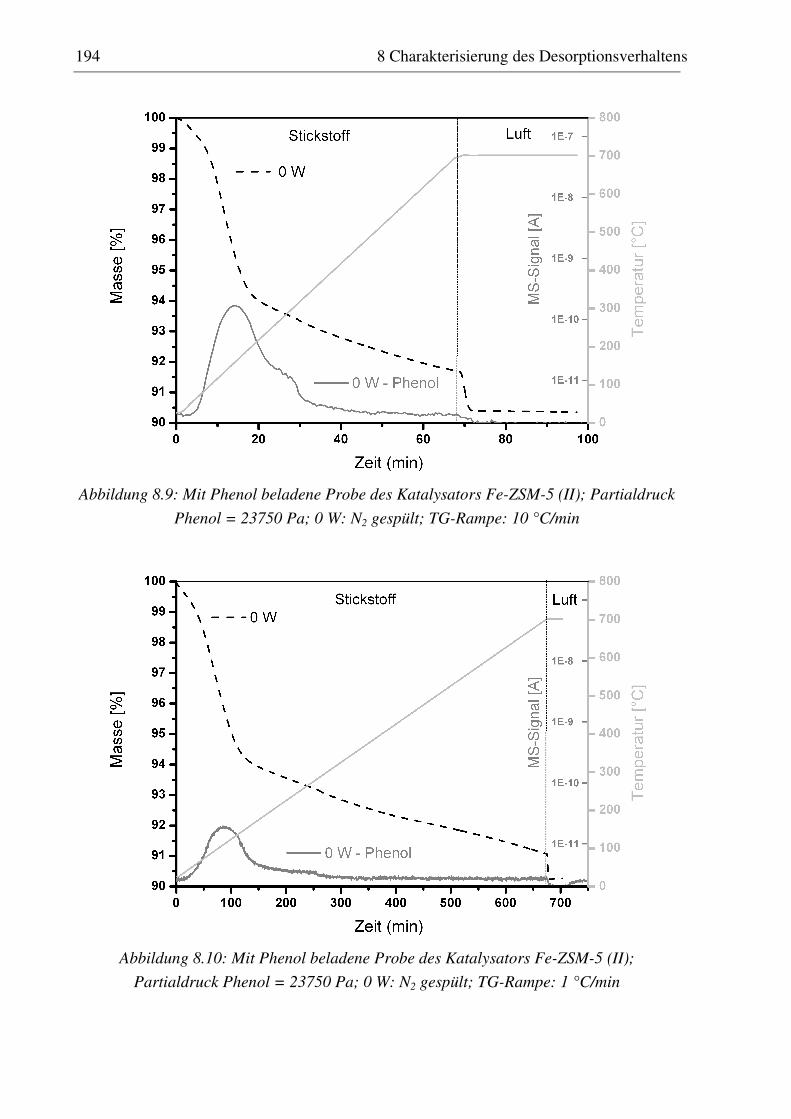
194 8 Charakterisierung des Desorptionsverhaltens
Abbildung 8.9: Mit Phenol beladene Probe des Katalysators Fe-ZSM-5 (II); Partialdruck
Phenol = 23750 Pa; 0 W: N2 gespült; TG-Rampe: 10 °C/min
Abbildung 8.10: Mit Phenol beladene Probe des Katalysators Fe-ZSM-5 (II);
Partialdruck Phenol = 23750 Pa; 0 W: N2 gespült; TG-Rampe: 1 °C/min

8.3 Ex situ beladene Proben 195
Vergleicht man die MS-Signale der beiden Messungen (Abbildung 8.9, Abbildung 8.10)
fällt zunächst auf, dass das MS-Signal bei der Messung mit der langsamen Aufheizrate
deutlich schwächer ist. Dies ist darauf zurückzuführen, dass bei einer langsameren Auf-
heizrate pro Zeiteinheit weniger Phenol desorbiert wird. Weiterhin verschiebt sich das
Desorptionsmaximum für Phenol zu niedrigeren Temperaturen (110 °C). Ab einer Tempe-
ratur von 325 °C wurde vom Massenspektrometer bei der Messung mit 1 °C/min kein
Phenol mehr detektiert, während bei einer Rampe von 10 °C/min erst bei etwa 500 °C die
Nachweisgrenze des Massenspektrometers erreicht wurde.
Diese Untersuchungen machen deutlich, dass die Menge des desorbierten Phenols und
auch die Temperatur bei der Phenol vom Katalysator desorbiert wird maßgeblich von der
Aufheizrampe und damit der Zeit bestimmt wird. Der Abtransport aus dem Katalysator
erfolgt offensichtlich extrem langsam, was wiederum die Bildung von Koksspezies för-
dert. Somit ist die in dieser Arbeit vorgenommene Einteilung in soft und hard coke
eigentlich nicht korrekt, da die exakte Menge an soft coke nur durch eine Messung bei
einer sehr langsamen Aufheizrate bzw. sehr hoher Messdauer bestimmt werden kann.
Eine weitere Katalysatorprobe wurde simultan mit Phenol und Benzen beladen
(Abbildung 8.11). Die TG-Massenkurven der unterschiedlich behandelten Proben weisen
bis zu einer Temperatur von ca. 180 °C keinen Unterschied auf. Bei höheren Temperatu-
ren fällt die Masse der mit Stickstoff gespülten Probe stärker ab, als die der mit Mikro-
wellen behandelten. Das MS-Signal zeigt, dass bei dieser Temperatur vornehmlich Phenol
desorbiert wurde (Maximum der Phenoldesorption: ca. 180 °C). Das Maximum der Ben-
zendesorption lag dagegen bei etwa 130 °C. Dies zeigt wiederum deutlich, dass durch die
Mikrowellenbestrahlung vornehmlich Phenol desorbiert wurde. Das Benzen blieb unbe-
einflusst. Unter Stickstoffatmosphäre (bis 700 °C) ergab sich der Masseverlust der nicht
mit Mikrowellen vorbehandelten Probe zu 6,33 %, während die bei 400 W behandelte
Probe nur eine Beladung von 5,74 % aufweist. Nach dem Umstellen des TG-Trägergases
von Stickstoff auf Luft ist wiederum ein rascher Masseverlust durch den Abbrand von
Koks zu beobachten. Der Anteil von hard coke betrug 1,36 % bei der mit Mikrowellen
behandelten Probe (400 W), während die Vergleichsprobe (0 W) nur einen Koksanteil von
1,12 % aufwies. Dieses Ergebnis deutet darauf hin, dass schon während der Behandlung
mit Mikrowellen nicht nur Phenol desorbiert, sondern auch Koks gebildet wurde.
Bei der Betrachtung der MS-Signale fällt wiederum ein Anstieg des Benzensignals bei
Temperaturen über 600 °C auf, welcher eventuell von einer Koksspezies herrührt, die erst
bei hohen Temperaturen desorbiert wird oder sich bei diesen Temperaturen zersetzt (vgl.
S. 190).
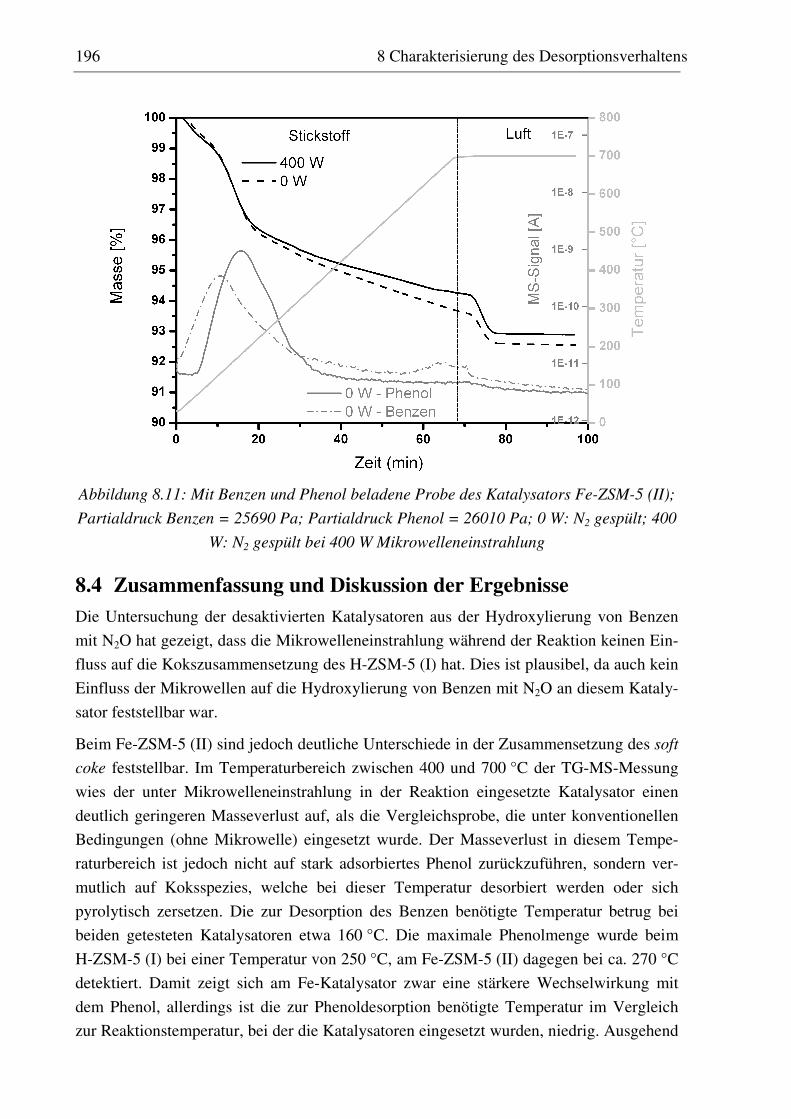
196 8 Charakterisierung des Desorptionsverhaltens
Abbildung 8.11: Mit Benzen und Phenol beladene Probe des Katalysators Fe-ZSM-5 (II);
Partialdruck Benzen = 25690 Pa; Partialdruck Phenol = 26010 Pa; 0 W: N2 gespült; 400
W: N2 gespült bei 400 W Mikrowelleneinstrahlung
8.4 Zusammenfassung und Diskussion der Ergebnisse
Die Untersuchung der desaktivierten Katalysatoren aus der Hydroxylierung von Benzen
mit N2O hat gezeigt, dass die Mikrowelleneinstrahlung während der Reaktion keinen Ein-
fluss auf die Kokszusammensetzung des H-ZSM-5 (I) hat. Dies ist plausibel, da auch kein
Einfluss der Mikrowellen auf die Hydroxylierung von Benzen mit N2O an diesem Kataly-
sator feststellbar war.
Beim Fe-ZSM-5 (II) sind jedoch deutliche Unterschiede in der Zusammensetzung des soft
coke feststellbar. Im Temperaturbereich zwischen 400 und 700 °C der TG-MS-Messung
wies der unter Mikrowelleneinstrahlung in der Reaktion eingesetzte Katalysator einen
deutlich geringeren Masseverlust auf, als die Vergleichsprobe, die unter konventionellen
Bedingungen (ohne Mikrowelle) eingesetzt wurde. Der Masseverlust in diesem Tempe-
raturbereich ist jedoch nicht auf stark adsorbiertes Phenol zurückzuführen, sondern ver-
mutlich auf Koksspezies, welche bei dieser Temperatur desorbiert werden oder sich
pyrolytisch zersetzen. Die zur Desorption des Benzen benötigte Temperatur betrug bei
beiden getesteten Katalysatoren etwa 160 °C. Die maximale Phenolmenge wurde beim
H-ZSM-5 (I) bei einer Temperatur von 250 °C, am Fe-ZSM-5 (II) dagegen bei ca. 270 °C
detektiert. Damit zeigt sich am Fe-Katalysator zwar eine stärkere Wechselwirkung mit
dem Phenol, allerdings ist die zur Phenoldesorption benötigte Temperatur im Vergleich
zur Reaktionstemperatur, bei der die Katalysatoren eingesetzt wurden, niedrig. Ausgehend

8.4 Zusammenfassung und Diskussion der Ergebnisse 197
von der Annahme, dass es zu einer Behinderung der Reaktion durch stark adsorbiertes
Phenol kommt (vgl. Kapitel 3.4.3), sollte ein Großteil des Phenols erst bei deutlich höhe-
ren Temperaturen (> 400 °C) desorbiert werden. Dies ist konnte nicht beobachtet werden.
Es hat sich gezeigt, dass der Fe-ZSM-5 (II) eine deutlich höhere Koksbeladung aufwies
als der H-ZSM-5 (I), obwohl die relative Desaktivierung des Fe-Katalysator während der
Reaktion deutlich geringer war. Die relative Desaktivierungsgeschwindigkeit in der
Benzenhydroxylierung ist somit nicht direkt an die gebildete Koksmenge gekoppelt.
Die TG-MS-Untersuchungen der ex situ beladenen Proben machen deutlich, dass eine
Nachbehandlung mit Mikrowellen keinen Einfluss auf die Benzendesorption hat. Das
Peakmaximum für Benzen (MS-Signal) wurde während der TG-Messung bei einer Tem-
peratur von ca. 150 °C detektiert. Der Fe-ZSM-5 (II) zeigte einen zweiten Benzenpeak bei
einer Temperatur von etwa 350 °C. Dies deutet auf verschiedene Zentren im Katalysator
hin, an denen das Benzen unterschiedlich stark adsorbiert vorliegt. Bei den Untersuchun-
gen der mit Benzen beladenen Proben konnte kein hard coke detektiert werden. Das
Benzen lässt sich unter N2-Atmosphäre vollständig vom Katalysator desorbieren.
Die ex situ (bei 200 °C) mit Phenol beladenen Proben zeigen nach dem Beladungsvorgang
eine Graufärbung, welche schon auf die Bildung von Koks hindeutet. Während der TG-
MS-Messung wird nicht das gesamte Phenol desorbiert, sondern es kommt zu einer weite-
ren Koksbildung. Dies konnte durch die TG-MS-Untersuchungen bestätigt werden, die
einen deutlichen Anteil von hard coke nach dem Aufheizen auf 700 °C nachweisen. Diese
Ergebnisse deuten darauf hin, dass das Phenol als Koksprecursor fungiert. Während der
TG-Messung wurde das Peakmaximum für Phenol (MS-Signal) im Temperaturbereich um
180 °C erreicht, bei der untersuchten hohen Beladung konnte Phenol jedoch bis zu einer
Temperatur von ca. 550 °C mit dem Massenspektrometer detektiert werden. Eine
Untersuchung bei der eine langsamere Aufheizrate während der TG-MS-Messung gewählt
wurde, zeigte jedoch, dass der Abtransport des Phenols aus dem Katalysator sehr langsam
erfolgt. Durch die verlangsamte Aufheizung wurde das Desorptionsmaximum für Phenol
von 180 auf 110 °C verschoben. Weiterhin konnte ab einer Temperatur von 325°C bereits
kein Phenol mehr detektiert werden.
Eine Behandlung der Proben mit Mikrowellen bei Raumtemperatur führte zu einer
Desorption des Phenols.
Die TG-MS-Untersuchungen der simultan mit Benzen und Phenol beladenen Katalysator-
proben beweisen, dass eine selektive Desorption des Phenols durch die Bestrahlung mit
Mikrowellen möglich ist. Das Benzen bleibt unbeeinflusst, während Phenol desorbiert
wird. Abweichungen in den TG-Kurven der unterschiedlich vorbehandelten Proben (ohne
Mikrowelleneinstrahlung bzw. bei einer Leistung von 400 W) zeigen sich nur in dem
Bereich, in dem Phenol desorbiert wird (ca. 180 °C). Bei den mit Mikrowellen vorbehan-

198 8 Charakterisierung des Desorptionsverhaltens
delten Proben ist der Masseverlust in diesem Temperaturbereich geringer als bei den nicht
mit Mikrowellen behandelten Proben. Außerdem konnte eine geringfügige Koksbildung
durch die Vorbehandlung mit Mikrowellen beobachtet werden.
Greift man die zu Beginn dieses Kapitels (S. 181) formulierten Fragen auf, so lassen sich
aus den TG-MS-Untersuchungen folgende Schlüsse ziehen:
• Eine Wechselwirkung der Mikrowellenstrahlung mit adsorbierten Benzen ist nicht
feststellbar. Phenol lässt sich hingegen durch die Mikrowelleneinstrahlung aufhei-
zen und vom Katalysator desorbieren. Unter den gegebenen Versuchsbedingungen
ließ sich lediglich ein geringer Anteil des adsorbierten Phenols durch die Mikro-
wellen desorbieren. Dies kann so erklärt werden, dass es zwar zu einer Tempera-
turerhöhung des Phenols durch die Mikrowellen kommt, die erreichbare Tempe-
raturdifferenz ist jedoch relativ gering.
• Nur Phenol fungiert als Koksprecursor. Bereits während der Beladung der Proben
bei einer Temperatur von 200 °C kommt es zu Koksablagerungen am Katalysator,
welche sich nur durch den Abbrand in Luft beseitigen lassen. Die zur Desorption
des Phenols benötigten Temperaturen sind kleiner als 300 °C. Der Abtransport des
Phenols aus dem Katalysator erfolgt jedoch sehr langsam (Diffusionslimitierung).
Da die Hydroxylierung von Benzen mit N2O üblicherweise bei deutlich höheren
Temperaturen durchgeführt wird, ist bei den getesteten Katalysatoren nicht mit
einer Produktinhibierung durch stark adsorbiertes Phenol zu rechnen. Problema-
tisch ist vielmehr der sehr langsame Abtransport des Phenols aus dem Katalysator.
Damit ist die Verweilzeit des Phenols im Katalysator hoch und somit auch die
Wahrscheinlichkeit einer Weiterreaktion zu Koks.
• Liegen sowohl Benzen als auch Phenol auf der Katalysatoroberfläche adsorbiert
vor, wird Phenol durch die Mikrowellen selektiv aufgeheizt und desorbiert.
Mit Hilfe dieser Ergebnisse lassen sich die Beobachtungen, welche während der Benzen-
hydroxylierung unter Mikrowelleneinstrahlung gemacht wurden (vgl. Kapitel 7.2) folgen-
dermaßen interpretieren.
Durch die Mikrowellen kommt es während der Reaktion zwar zu einer selektiven Aufhei-
zung des Phenols, die TG-MS-Untersuchungen beweisen jedoch, dass die Desorption des
Phenols nicht nur durch die Temperatur limitiert ist. Der Großteil des Phenols kann bereits
bei Temperaturen kleiner 300 °C desorbiert werden. Problematisch ist vielmehr der lang-
same Abtransport des Phenols aus dem Katalysator (Diffusionslimitierung). Eine zusätzli-
che Aufheizung des Phenols führt zu einer beschleunigten Desaktivierung, da Phenol
deutlich reaktiver ist als Benzen. Bereits bei Temperaturen, welche unter den für die
Hydroxylierung benötigten liegen, kommt es zu Koksablagerungen, wie die mit Phenol

8.4 Zusammenfassung und Diskussion der Ergebnisse 199
beladenen Proben gezeigt haben. Wird Phenol während der Reaktion durch die Mikro-
wellen zusätzlich aufgeheizt, beschleunigt sich auch die Weiterreaktion zu Koks.

9 Zusammenfassung und Ausblick
In der Literatur wird berichtet, dass der Einsatz von Mikrowellen als Heizmedium für
heterogen katalysierte Reaktionen Vorteile gegenüber konventionellen Heizmethoden hat.
Die Ursachen der in der heterogenen Katalyse mit Mikrowellen beobachteten Effekte sind
bisher jedoch noch nicht befriedigend geklärt. Eine mögliche Wirkungsweise der Mikro-
wellen ist es, bestimmte Phasen oder Stoffgruppen selektiv aufzuheizen (Kapitel 2.1).
Desorptionsvorgänge an Oberflächen lassen sich auf diese Weise gezielt beeinflussen. Ob
dies auch im Verlauf einer Reaktion möglich ist wurde bisher noch nicht gezielt unter-
sucht (Kapitel 2.3).
Ziel der Arbeit war es, die Wirkung von Mikrowellenstrahlung am Beispiel der Hydroxy-
lierung von Benzen mit N2O an Zeolithkatalysatoren des Typs ZSM-5 zu untersuchen.
Diese Reaktion ist von großem wirtschaftlichem Interesse. Ein Problem dieses Verfahrens
stellt jedoch die rasche Desaktivierung des Katalysators dar. Ausgangspunkt war, dass
während der Reaktion gebildetes Phenol aufgrund der starken Adsorption am Katalysator
für den Aktivitätsrückgang verantwortlich ist (Kapitel 3). Weiterhin sind sowohl polare
(Phenol) als auch unpolare Stoffe (Benzen) an der Reaktion beteiligt. Diese heterogen
katalysierte Reaktion kann somit als typisches Modell einer Reaktion angesehen werden,
bei der die selektive Heizwirkung der Mikrowelle ausgenutzt werden könnte.
Die zu klärenden Fragestellungen dieser Arbeit waren,
1. wie sich bestimmte Eigenschaften des Katalysators auf die Reaktion auswirken
und über welche Mechanismen die Desaktivierung abläuft,
2. welchen Einfluss Mikrowellenstrahlung auf die Hydroxylierung von Benzen mit
N2O an ausgewählten ZSM-5 Katalysatoren, insbesondre auf das Desaktivierungs-
verhalten hat und
3. ob eine selektive Aufheizung des Phenols beziehungsweise eine selektive Desorp-
tion von der Katalysatoroberfläche durch Mikrowellen prinzipiell möglich ist.
Im Folgenden werden die Ergebnisse der im Rahmen dieser Arbeit durchgeführten Unter-
suchungen zusammengefasst.

9 Zusammenfassung und Ausblick 201
Voruntersuchungen
In der Hydroxylierung von Benzen mit N2O kamen vier ausgewählte Katalysatoren des
Typs ZSM-5 zum Einsatz, welche sich deutlich hinsichtlich ihres Verhaltens in der Reak-
tion unterscheiden. Da in der Literatur wenig über die dielektrischen Eigenschaften dieser
Katalysatoren bekannt ist (Kapitel 3.4.1.6) erfolgte neben der klassischen Charakterisie-
rung (Kapitel 5.2) auch eine Charakterisierung hinsichtlich des Verhaltens im Mikro-
wellenfeld (Kapitel 5.3).
Durch Aufheizexperimente im Mikrowellenfeld konnte gezeigt werden, dass sich alle in
der Benzenhydroxylierung eingesetzten dehydratisierten Katalysatoren kaum durch
Mikrowellen erwärmen lassen. Auch in den Katalysatoren eingebundene Eisen- oder
Kobalt-Spezies führen nicht zu einer stärkeren Wechselwirkung mit dem Feld. Dies ist
eine Voraussetzung, um die selektive Desorption von am Katalysator adsorbierten Stoffen
durch Mikrowelleneinstrahlung untersuchen zu können (Kapitel 2.3.3). Anhand eines
Vergleichskatalysators in der Na-Form wurde gezeigt, dass im Katalysator enthaltene Na-
Ionen maßgeblich für die Erwärmung dieses Katalysatortyps durch Mikrowellen verant-
wortlich sind. Weiterhin wurde gezeigt, dass das Modul des ZSM-5 keinen Einfluss auf
die Aufheizbarkeit des Katalysators durch Mikrowellen hat.
Mit Phenol beladene Proben der eingesetzten Katalysatoren zeigten demgegenüber eine
stärkere Aufheizung durch die Mikrowellen. Am Katalysator adsorbiertes Phenol kann
also mit den Mikrowellen wechselwirken und führt zu einem Temperaturanstieg.
Diese Ergebnisse belegen, dass die ausgewählten Katalysatoren und das Reaktionssystem
auf Basis der in der Literatur beschriebenen Voraussetzungen geeignet sind, um eine
selektive Aufheizung bzw. selektive Desorption adsorbierter Spezies während einer
Reaktion zu ermöglichen.
Technische Aufgaben
Zur Durchführung der reaktionstechnischen Experimente wurde eine teilautomatisierte
Laboranlage konzipiert und gebaut, welche für die Untersuchung verschiedener heterogen
katalysierter Reaktionen geeignet ist (Kapitel 6).
Ein geeignetes Reaktorkonzept wurde entwickelt, welches zuverlässige Vergleichsmes-
sungen zwischen konventionell und mit Mikrowellen beheizten Experimenten ermöglicht
(Kapitel 6.2.2). Dieser Reaktor erlaubt eine kombinierte Heizung mittels Mikrowellen und
elektrischer Widerstandsheizung. Mikrowellen- und Referenzexperiment sind im selben
Reaktor, bzw. in der selben Laboranlage ohne Umbaumaßnahmen durchführbar. Es ist
eine exakte Temperaturmessung und Regelung der Katalysatorbetttemperatur im Mikro-
wellenfeld möglich.

202 9 Zusammenfassung und Ausblick
Es wurde gezeigt, dass sich der Reaktor wie ein ideales Strömungsrohr verhält und wei-
testgehend frei von Temperaturgradienten ist, wie sie häufig bei der Verwendung von
Mikrowellen als Heizmedium beobachtet werden.
Erst durch die Entwicklung dieses Reaktors war ein zuverlässiger Vergleich zwischen
konventionell und mit zusätzlicher Mikrowelleneinstrahlung beheizten Reaktionen mög-
lich.
Reaktionstechnische Untersuchungen
Voraussetzung für die Bewertung der Ergebnisse in der Hydroxylierungsreaktion war die
Untersuchung der N2O-Zersetzung, weil diesem Teilschritt in der Literatur eine zentrale
Rolle im Reaktionsmechanismus der Benzenhydroxylierung zugeschrieben wird (Kapi-
tel 7.1).
• Es wurde gezeigt, dass sich die untersuchten Katalysatoren deutlich in ihrer Aktivität
hinsichtlich der N2O-Zersetzung unterscheiden.
• Bei keinem Katalysator konnte ein Einfluss der Mikrowellenstrahlung auf die N2O-
Zersetzung festgestellt werden.
Das Hauptaugenmerk dieser Arbeit lag jedoch darin, den Einfluss der Mikrowellen auf die
Hydroxylierung von Benzen mit N2O zu untersuchen, insbesondre ob eine selektive Auf-
heizung des Phenols unter Reaktionsbedingungen durch Mikrowellen möglich ist. Zu
diesem Zweck wurde jeder Katalysator unter konventionellen Bedingungen und unter
Mikrowelleneinstrahlung getestet (Kapitel 7.2). Die Reaktionstemperatur und einge-
strahlte Mikrowellenleistung wurden variiert. An den Katalysatoren Fe-ZSM-5 (II) (hoher
Eisenanteil) und Co-ZSM-5 erfolgte zudem eine Variation der modifizierten Verweilzeit
(Kapitel 7.2.4.4 und 7.2.5.4). Weiterhin wurden am Katalysator Fe-ZSM-5 (II) Versuche
mit Benzenüberschuss im Eduktgemisch durchgeführt (Kapitel 7.2.4.5). Am Katalysator
Co-ZSM-5 wurde außerdem der Einfluss von Wasser auf das Reaktionssystem untersucht
(Kapitel 7.2.5.5).
• Anhand eines Vergleichs der vier eingesetzten Katalysatoren bei einer Reaktions-
temperatur von 400 °C unter konventionellen Bedingungen konnte gezeigt werden,
dass die relative Desaktivierung nicht von der Frischaktivität der Katalysatoren
abhängt. Die Phenolselektivitäten stiegen mit fortschreitender Reaktionsdauer leicht
an.
• Es wurde eine kein Zusammenhang zwischen der durch NH3-TPD ermittelten
Anzahl an Säurezentren und der Frischaktivität der Katalysatoren in der Hydroxylie-
rung von Benzen mit N2O gefunden. Allerdings ergab sich eine annähernd lineare
Korrelation zwischen der Anzahl starker Säurezentren und der relativen Desaktivie-
rung nach 61 min TOS.

9 Zusammenfassung und Ausblick 203
Je größer die Zahl dieser Zentren, desto schneller verlief auch die Desaktivierung.
Dies ist ein Hinweis darauf, dass die Phenolbildung und die Desaktivierung unter-
schiedlichen Mechanismen folgen, bzw. dass verschiedene aktive Zentren an den
diesen Mechanismen beteiligt sind.
• Die Aktivität in der N2O-Zersetzung war nicht mit der Aktivität in der Hydroxylie-
rung von Benzen mit N2O korrelierbar. Eine hohe Aktivität in der N2O-Zersetzung
wirkte sich nicht zwangsläufig positiv auf die Hydroxylierung von Benzen mit N2O
aus.
• Eine Variation der Reaktionstemperatur zwischen 300 und 400 °C bei einer modifi-
zierten Verweilzeit von 90 (g·min)/mol zeigte, dass nur am Fe-ZSM-5 (I) (kleine
Kristallite) bei einer Temperatursteigerung von 300 auf 350 °C ein Anstieg der
Phenolselektivität bei gleichzeitig verringerter relativer Desaktivierung beobachtet
werden konnte. Dies kann als eine starke Adsorption bzw. behinderte Diffusion des
Phenols bei niedrigen Reaktionstemperaturen an diesem Katalysator gedeutet wer-
den. Am Fe-ZSM-5 (II) und Co-ZSM-5 wurde dagegen eine Zunahme der relativen
Desaktivierung zwischen 300 und 350 °C verzeichnet. Eine Temperatursteigerung
auf 400 °C führte bei allen Katalysatoren zu einer Verringerung der auf Benzen
bezogenen Phenolselektivitäten und relativen Desaktivierung. Dies kann so erklärt
werden, dass es mehrere sich überlagernde Effekte gibt, welche in bestimmten Tem-
peraturbereichen dominieren. Zum einen kommt es bei mittleren (350 °C) Tempe-
raturen am Fe-ZSM-5 (II) und Co-ZSM-5 zu einer beschleunigten Weiterreaktion
des Phenols zu Koks und somit rascherer Desaktivierung, zum anderen setzt bei
höheren Temperaturen (400 °C) eine Totaloxidation des Phenols und Kokses ein,
was mit einer langsameren Desaktivierung einhergeht.
• Eine Steigerung der Verweilzeit führte zu höheren Benzenumsatzgraden, wirkte sich
jedoch negativ auf die auf Benzen bezogenen Phenolselektivitäten und die Desakti-
vierung aus.
• Durch eine überstöchiometrische Benzenzugabe konnten die auf Benzen bezogenen
Phenolselektivitäten gesteigert und die Desaktivierung verringert werden. Dieses
Verhalten wird in der Literatur durch eine Verdrängung des Phenols von der Kataly-
satoroberfläche durch Benzen erklärt, wodurch sich eine Weiterreaktion des Phenols
zu Nebenprodukten und Koks vermeiden lässt.
Am Katalysator Co-ZSM-5 wurden die Auswirkungen einer Wasserzugabe zum Feed
untersucht.
• Im Vergleich zu den Experimenten ohne Wasserzugabe konnten gesteigerte
Benzenumsatzgrade bei abnehmenden auf Benzen bezogenen Phenolselektivitäten
und verringerter relativer Desaktivierung erreicht werden. Dieses Verhalten lässt

204 9 Zusammenfassung und Ausblick
sich ähnlich wie bei der Fahrweise mit Benzenüberschuss durch eine Verdrängung
des Phenols von den aktiven Zentren des Katalysators erklären. Die Folge ist eine
verringerte relative Desaktivierung. Allerdings kam es zu unerwünschten Nebenre-
aktionen mit dem Wasser, welche sich in verringerten Phenolselektivitäten wider-
spiegelten.
Die Katalysatoren wurden unter den oben beschriebenen Reaktionsbedingungen ebenfalls
unter zusätzlicher Mikrowelleneinstrahlung getestet. Der Einfluss der Mikrowellenstrah-
lung auf die Reaktion war gering. Allerdings zeigten sich Tendenzen, welche bei allen
Katalysatoren zu beobachten waren.
• Bei kurzen Verweilzeiten konnten gesteigerte Phenolselektivitäten beobachtet
werden, während bei hoher Verweilzeit die Phenolselektivität durch die Mikrowel-
lenstrahlung zurückging.
• Entgegen der zu Beginn der Arbeit aufgestellten Hypothese, dass Phenol durch die
Mikrowellen selektiv aufgeheizt und desorbiert wird, was zu einer verringerten
Desaktivierung führen sollte, wurde eine beschleunigte Desaktivierung beobachtet.
Diese Ergebnisse geben wiederum einen Hinweis darauf, dass zwei konkurrierende
Mechanismen durch die Mikrowellen beeinflusst werden. Zum einen kommt es zu einer
selektiven Aufheizung und Desorption des Phenols, was sich in gesteigerten Phenolselek-
tivitäten bei kleinen Verweilzeiten widerspiegelt. Zum anderen führt die Aufheizung des
Phenols oder dessen Folgeprodukten durch die Mikrowellen jedoch auch zu einer
beschleunigten Weiterreaktion zu Koks.
Weitere Ergebnisse der Mikrowellenexperimente waren:
• Eine Steigerung der eingestrahlten Mikrowellenleistung führte nur zu einer schwa-
chen Verstärkung der beobachteten Effekte, da die Masse an polaren Stoffen, welche
sich im Reaktor befindet und die Mikrowellenstrahlung absorbiert sehr gering ist. Es
wurde bereits bei geringen Leistungen genügend Energie eingestrahlt, um die Stoffe
optimal aufzuheizen.
• Bei den Experimenten mit zusätzlicher Wasserzugabe zum Feed ließ sich das
Reaktionsverhalten durch die Mikrowelleneinstrahlung in Richtung des Experiments
ohne Wasser verschieben. Das adsorbierte Wasser wird durch die Mikrowellen
aufgeheizt und von der Katalysatoroberfläche verdrängt.
Es konnte gezeigt werden, dass ein gezielter Eingriff in den Reaktionsablauf der Hydro-
xylierung von Benzen mit N2O durch eine selektive Aufheizung des Phenols möglich ist.
Die Experimente mit Wasser im Feed belegen zudem, dass eine Verschiebung von Sorpti-
onsgleichgewichten am Katalysator durch die Mikrowelleneinstrahlung erreicht werden
kann.

9 Zusammenfassung und Ausblick 205
Charakterisierung des Desorptionsverhaltens
Um Informationen über die Temperaturabhängigkeit der Desorption von Benzen und
Phenol vom Katalysator und die Koksbeladung nach dem Einsatz der Katalysatoren in der
Hydroxylierung zu erhalten, wurden Proben des H-ZSM-5 (I) und Fe-ZSM-5 (II) einer
TG-MS-Messung nach der Reaktion unterzogen (Kapitel 8.2).
Es stellte sich heraus, dass der H-ZSM-5 (I) eine deutlich geringere Koksbeladung auf-
wies als der Fe-ZSM-5 (II), obwohl die relative Desaktivierung des Fe-Katalysators deut-
lich geringer war. Die relative Desaktivierung war somit nicht direkt mit der gebildeten
Koksmenge korrelierbar. Ein Vergleich von Proben des H-ZSM-5 (I) aus der Reaktion mit
und ohne Mikrowelleneinstrahlung zeigte keinen Einfluss auf die Kokszusammensetzung.
Beim Fe-ZSM-5 (II) wiesen die Proben hingegen deutliche Unterschiede in der Kokszu-
sammensetzung auf. Sowohl am H-ZSM-5 (I) als auch am Fe-ZSM-5 (II) wurde die
maximale Benzenmenge bei einer Temperatur von 160 °C desorbiert. Das Maximum der
Phenoldesorption lag beim Katalysator der H-Form bei 250 °C und beim Fe-ZSM-5 (II)
bei 270 °C.
Anschließend wurden TG-MS-Untersuchungen von ex situ mit Benzen, Phenol und deren
Mischung beladenen Proben der Katalysatoren durchgeführt (Kapitel 8.3). Eine Ver-
gleichsprobe wurde jeweils mit Mikrowellen behandelt, um den Einfluss der Bestrahlung
auf die adsorbierten Spezies zu untersuchen. Dabei konnten folgende Beobachtungen
gemacht werden:
• Benzen ließ sich während der TG-MS-Messung im N2-Strom vollständig vom
Katalysator desorbieren. Das Maximum der Benzendesorption lag bei beiden
Katalysatoren bei 150 °C. Am Fe-ZSM-5 (II) wurde ein zusätzlicher Benzenpeak
mit einem Maximum bei 350 °C beobachtet. Beim Aufheizen auf 700 °C wurde
kein hard coke gebildet. Nach einer Vorbehandlung der beladenen Proben mit
Mikrowellenstrahlung bei Raumtemperatur zeigte sich keine Veränderung der TG-
MS-Profile.
• Alle untersuchten Katalysatorproben wiesen bereits nach dem Beladungsvorgang
mit Phenol eine Graufärbung auf. Nach dem Aufheizen der Proben bei der TG-
MS-Messung auf 700 °C in N2-Strom konnte ein deutlicher Anteil an hard coke
nachgewiesen werden, der sich nur durch den Abbrand mit Luft beseitigen ließ. Es
kam also auch ohne Einsatz in der Hydroxylierung von Benzen zu einer Verko-
kung des Katalysators. Das Desorptionsmaximum für Phenol lag bei Temperaturen
unter 200 °C. Phenol fungierte als Koksprecursor. Der Abtransport des Phenols aus
dem Katalysator erfolgte sehr langsam. Durch eine Vorbehandlung der Proben mit
Mikrowellen bei Raumtemperatur konnte Phenol desorbiert werden.

206 9 Zusammenfassung und Ausblick
• Anhand der TG-MS-Untersuchungen der mit Benzen und Phenol beladenen
Proben konnte gezeigt werden, dass eine selektive Desorption des Phenols durch
Mikrowelleneinstrahlung möglich war, während das Benzen unbeeinflusst blieb.
Unter den angewandten Versuchsbedingungen ließ sich jedoch nur ein kleiner
Anteil des Phenols desorbieren. Auch bei einer Mischbeladung konnte hard coke
am Ende der TG-MS-Messung detektiert werden.
Diese Untersuchungen unterstützen die Aussage der Literatur, dass Phenol als Kokspre-
cursor fungiert. Selbst nach dem Beladen der Katalysatorproben mit Phenol bei 200 °C,
also einer Temperatur die deutlich unter der für die Hydroxylierungsreaktion benötigten
liegt, konnte eine Verkokung der Proben beobachtet werden.
Für eine thermische Desorption des Phenols genügen jedoch bereits Temperaturen um
200 °C, was der Theorie widerspricht, dass stark adsorbiertes Phenol für die Desaktivie-
rung verantwortlich ist. Vielmehr können die Ergebnisse so gedeutet werden, dass zwar
die langsame Diffusion des Phenols problematisch ist, da eine Weiterreaktion des Phenols
zu Koks bereits bei niedrigen Temperaturen erfolgt und sich deshalb die lange Verweilzeit
des Phenols im Katalysator als nachteilig erweist, jedoch nicht vorrangig eine zu starke
Adsorption.
Weiterhin konnte gezeigt werden, dass eine selektive Aufheizung von am Katalysator
adsorbiertem Phenol und dadurch eine selektive Desorption vom Katalysator durch
Mikrowelleneinstrahlung möglich ist.
Ausblick
Auf Basis der Ergebnisse dieser Arbeit scheint es so, dass das Problem der Desaktivierung
bei der Hydroxylierung von Benzen mit N2O, an den heute verfügbaren Katalysatoren des
Typs ZSM-5, nicht mit Prozessseitigen Maßnahmen zu lösen ist. Deshalb sollte in
Zukunft daran gearbeitet werden für die Hydroxylierung von Benzen mit N2O maßge-
schneiderte Katalysatoren zu entwickeln. Diese dürfen nur möglichst wenig der für die
Desaktivierung verantwortlichen Zentren enthalten und müssen eine raschere Diffusion
des Phenols erlauben. Auf diese Weise ließe sich das Problem der raschen Desaktivierung
ohne Verlust an Frischaktivität verringern.
Da es mit Hilfe der Mikrowellen möglich war gezielt in den Ablauf einer Reaktion ein-
zugreifen, sollten weitere Reaktionen untersucht werden, bei denen der Einsatz der
Mikrowellen von Nutzen sein könnte. Gerade die starke Wechselwirkung der Mikrowel-
len mit am Katalysator adsorbiertem Wasser kann bei bestimmten Reaktionen vorteilhaft
sein. Beispielsweise stellt Wasser bei der SCR-Reaktion von Stickoxiden mit NH3 als Re-
duktionsmittel an vanadiumhaltigen Katalysatoren ein Problem dar [345]. Eine Verdrän-
gung des Wassers von der Katalysatoroberfläche durch den Einsatz von Mikrowellen-
strahlung könnte diese Probleme eventuell beseitigen.

9 Zusammenfassung und Ausblick 207
Wie diese Arbeit jedoch gezeigt hat, kann es durch die Mikrowellen durchaus auch zu
unerwünschten Nebenreaktionen kommen. Der Einsatz von Mikrowellen zur Unterstüt-
zung heterogen katalysierter Reaktionen muss deshalb für die jeweilige Reaktion im
Detail geprüft werden.

10 Summary and Outlook
It is reported in literature that deploying microwaves as a heating method for
heterogeneously catalysed reactions has advantages over conventional heating methods.
The reasons for the effects observed have yet to be satisfactorily explained. One possible
effect could be the microwaves selectively heating specific phases or substance classes
(Chap. 2.1). Desorption processes on surfaces can be affected in this manner. Whether this
effect can occur during a heterogeneously catalysed reaction has not yet been investigated
(Chap 2.3).
The objective of the thesis was to investigate the effect of microwave radiation using the
example of hydroxylation of benzene with N2O on zeolite catalysts of the type ZSM-5.
This reaction is of major economic interest. However, one problem of this process is the
fast deactivation of the catalyst. The starting point was that strongly adsorbed phenol
produced by the reaction is responsible for the decline in activity (Chap. 3). Moreover,
polar (phenol) as well as nonpolar (benzene) substances are involved in the reaction. This
heterogeneously catalyzed reaction can be considered as a typical model of a reaction that
could benefit from the selective heating of the microwave.
Questions to be clarified by this work were:
1. How do specific properties of the catalyst affect the reaction and which mechanism
is responsible for the deactivation?
2. Which influence does microwave radiation have on the hydroxylation of benzene
with N2O on selected ZSM-5 catalysts, and in particular on the deactivation?
3. Is a selective heating of the phenol, respectively a selective desorption from the
catalyst surface possible using microwave?
The results of the investigations carried out within this work are summarized below.
Preliminary investigations
For the investigation of the hydroxylation of benzene with N2O, four catalysts of the type
ZSM-5 were chosen which clearly differ in behavior in the reaction. Since there is not
much information available in literature concerning the dielectric properties of these

10 Summary and Outlook 209
catalysts (Chap. 3.4.1.6) a characterization of the behavior in the microwave field was
carried out alongside classical characterization (Chap. 5.2-5.3).
Via heating-up experiments in the microwave-field, it was shown that the dehydrated
catalysts used for the hydroxylation of benzene can hardly be heated up by microwave.
Also iron- or cobalt-species incorporated in the catalyst do not lead to an enforced
interaction with the electromagnetic field. This is prerequisite to being able to investigate
the selective desorption of adsorbed species on the catalyst surface by microwave (Chap.
2.3.3). By means of a reference catalyst in the sodium-form it was shown that sodium-ions
contained in the catalyst are primarily responsible for the warm-up of the catalyst by
microwave. Furthermore, it was identified that the module of the ZSM-5 does not have
any influence on the ability to heat up the catalyst by microwave.
On the other hand, samples of catalysts loaded with phenol showed increased heat-up by
microwave. In other words phenol adsorbed on the catalyst surface is capable of
interacting with the microwave and causes a temperature increase.
These results prove that the selected catalysts and the reaction system, based on the
prerequisites described in literature, are suitable for facilitating selective heating, or a
selective desorption of adsorbed species respectively, during a reaction.
Technical tasks
To perform the experiments in reaction engineering, a partly automated laboratory facility
was designed and built, suitable for the investigation of different heterogeneously
A reactor concept was developed that makes a reliable comparison between
conventionally and microwave heated experiments possible (Chap. 6.2.2). This reactor
enables a combined microwave and electrical resistance heating. Microwave- and
conventional experiments can be conducted in the same reactor and facility without
modifications being necessary. An exact temperature measurement and control of the
reactor bed are possible in the microwave field.
It was shown that the reactor behaves like an ideal plug-flow reactor and is (as far as
possible) free from temperature-gradients which are frequently observed using
microwaves for heating.
Only through the development of this reactor did a reliable comparison between reactions
heated conventionally and those with additional microwave radiation become possible.
Investigations in reaction engineering
Prerequisite for evaluating the results of hydroxylation was the investigation of the N2O-
desomposition, since this individual step plays an important role in the reaction-
mechanism of the benzene hydroxylation (Chap. 7.1).

210 10 Summary and Outlook
• It was shown that the catalysts investigated differ significantly in their activity for
N2O-decomposition.
• An influence of the microwave radiation on the N2O-decomposition could not be
determined with any catalyst.
Focus of this work was to investigate the influence of microwave on the hydroxylation of
benzene with N2O, especially whether a selective heating of the phenol is possible by
microwave or not. For this purpose, each catalyst was tested under conventional
conditions and under microwave irradiation (Chap. 7.2). The reaction temperature and
microwave power emitted were varied. For the catalysts Fe-ZSM-5 (II) (high Fe-content)
and Co-ZSM-5 the modified residence time was varied (Chap. 7.2.4.4 and 7.2.5.4).
Furthermore tests with an excess of benzene in the feed were conducted using the catalyst
Fe-ZSM-5 (II) (Chap. 7.2.4.5). In addition to this, the influence of water on the reaction
system was tested using the catalyst Co-ZSM-5 (Chap. 7.2.5.5).
• Using a comparison of the four catalysts at a reaction temperature of 400 °C under
conventional conditions, it was shown that the relative deactivation does not depend
on the fresh activity of the catalyst. The selectivity towards phenol increases slightly
with progressing time on stream.
No relationship was found between the quantity of acid sites determined by NH3-
TPD and the fresh activity of the catalysts in the hydroxylation of Benzene with
N2O. However, there was an approximately linear correlation between the quantity
of strong acid sites and the relative deactivation after 61 minutes time on stream.
The larger the number of acid sites, the faster the deactivation was. This is an
indication that phenol formation and relative deactivation result follow different
mechanisms, or that different sites are involved in these mechanisms.
• The activity in the N2O-decompositin reaction did not correlate with the activity in
the hydroxylation of benzene with N2O. A high activity in N2O-decomposition does
not necessarily have a positive effect on the hydroxylation of benzene with N2O.
• A variation in reaction temperature between 300 and 400 °C at a modified residence
time of 90 (g·min)/mol showed, that an increase of the selectivity towards phenol
with simultaneously reduced relative deactivation could just be observed over Fe-
ZSM-5 (I) (small crystallites) by increasing the temperature from 300 to 350 °C.
This can be interpreted as a strong adsorption of the phenol or restricted diffusion in
this catalyst at low reaction temperatures. On the other hand, for the Fe-ZSM-5 (II)
and Co-ZSM-5, an increase of the relative deactivation was recorded between 300
and 350 °C. A temperature increase to 400 °C led to a decrease of the selectivity
towards phenol (referred to benzene) and relative deactivation with all catalysts. This

10 Summary and Outlook 211
can be explained through superimposed effects, which dominate in defined
temperature ranges. Over Fe-ZSM-5 (II) and Co-ZSM-5 the reaction of phenol
towards coke is accelerated at medium temperatures (350°C) leading to faster
deactivation. On the other hand total oxidation of phenol and coke occurs at higher
temperatures (400 °C) which decreases deactivation.
• An increase in residence time led to higher benzene conversions, but had a negative
effect on the selectivity towards phenol (referred to benzene) and deactivation
behavior.
• By means of a leaner-than-stoichiometric addition of benzene the selectivity towards
phenol (referred to benzene) was increased and deactivation was decreased. This
behavior is explained in literature as a displacement of the phenol from the catalyst
surface by benzene. One can avoid a further reaction of the phenol towards coke and
byproducts.
The influence of additional water in the feed (steaming) was tested using the catalyst Co-
ZSM-5.
• In comparison to the experiments without steaming, an increased benzene
conversion, a lowered selectivity towards phenol (referred to benzene) and a
decreased deactivation was achieved. This can be explained by a displacement of the
phenol from the active sites of the catalyst, similar to the effect of benzene in excess.
The consequence is lowered relative deactivation. However, undesired side reactions
involving water occurred, which led to a decreased selectivity towards phenol.
The catalysts were also tested under additional microwave radiation within the above
described reaction conditions. The influence of the microwave on the reaction was low.
However, tendencies appeared which were observed with all catalysts.
• Increased selectivity towards phenol was observed at short residence time, while
selectivity to phenol was decreased by microwave at high residence time.
• Contrary to the hypothesis proposed at the beginning of the work, that phenol will be
heated selectively and desorbed by microwave, which should lead to a reduced
deactivation, an accelerated deactivation was observed.
Once more these results indicate that two competing mechanisms are influenced by the
microwaves. On the one hand, phenol is selectively heated up and desorbed by
microwave, leading to an increased selectivity towards phenol at low residence time. On
the other, heating of phenol or its derivates by microwave causes an accelerated coke
formation.
Further results of the microwave experiments were:

212 10 Summary and Outlook
• An increase of the microwave power applied just led to weak reinforcement of the
effects observed, since the mass of polar substances absorbing microwave resident in
the reactor is very low. Even at low power sufficient energy was supplied to heat up
the substances optimally.
• Applying microwaves to a reaction with water in the feed led to a shift of the
reaction behavior towards the experiment without water. The adsorbed water is
heated up and displaced from the catalyst surface by the microwave.
It was shown that specific manipulation of the reaction kinetics of the hydroxylation of
benzene with N2O is possible by heating phenol selectively. The experiments with water
in the feed furthermore prove that a shift of sorption equilibrium can be achieved by
microwave radiation.
Characterization of the deactivation behavior
To get information about the temperature dependency of the desorption of benzene and
phenol from the catalyst surface and coke loading, catalysts H-ZSM-5 (I) and Fe-ZSM-
5 (II) used in the hydroxylation reaction were subject to a TG-MS-analysis (Chap. 8.2).
It turned out that the H-ZSM-5 (I) had a clearly lower coke loading than the Fe-ZSM-5
(II), although that the relative deactivation of the Fe-catalyst was significantly lower.
Therefore the relative deactivation could not be correlated with the amount of coke
generated. A comparison of samples of the H-ZSM-5 (I) from reactions with and without
microwave radiation did not demonstrate any influence on the coke composition. As far as
the Fe-ZSM-5 (II) was concerned, however, the tests showed clear differences in the coke
composition. The maximum amount of benzene was desorbed at a temperature of 160 °C
for both the H-ZSM-5 (I) and the Fe-ZSM-5 (II). The maximum of the phenol desorption
was at 250 °C for the H-form and at 270 °C for Fe-ZSM-5 (II).
TG-MS-analysis was carried out afterwards with catalyst samples loaded ex situ with
benzene, phenol and a mixture of both (Chap. 8.3). A reference sample was treated with
microwave in each case in order to examine the influence of the irradiation on the
adsorbed species. The following observations were made:
• Benzene could be desorbed during TG-MS-analysis with N2-flow completely. The
maximum of benzene desorption was at 150 °C for both catalysts. The Fe-ZSM-
5 (II) showed a second maximum at 350 °C. While heating up to 700 °C no hard
coke was built. Subsequent to a pretreatment with microwave at room temperature
no change of the TG-MS-profiles was observed.
• All catalysts that were investigated showed grey coloring directly after loading
with phenol. After heating up to 700 °C during TG-MS-analysis with N2-flow a
significant amount of hard coke was detected, which could only be burned off by

10 Summary and Outlook 213
combustion with air. A coking of the catalyst therefore occurred without the use in
the hydroxylation of benzene. The maximum of phenol desorption was observed at
temperatures lower than 200 °C. Phenol acted as a coke precursor. The removal of
the phenol from the catalyst took place extremely slowly. It was possible to desorb
phenol by pretreating the samples with microwave at room temperature.
• On the basis of the TG-MS-analysis of samples loaded with benzene and phenol it
could be pointed out that a selective desorption of phenol was possible while
benzene remained unaffected by microwave. However just a small fraction of the
phenol could be desorbed within the applied test conditions. Also for samples
loaded with a mixture of benzene and phenol hard coke was detected after TG-MS-
analysis.
These investigations support the statement in literature that phenol acts as a coke
precursor. Even after loading the catalyst samples with phenol at 200 °C, in other words a
temperature which is clearly lower than required for the hydroxylation, a coking of the
samples could be observed.
However temperatures of about 200 °C are already sufficient for a thermal desorption of
the phenol which disagrees with the theory that strongly adsorbed phenol is responsible
for the deactivation. More than this, the results can be interpreted in such a way that the
slow diffusion of phenol is problematic, since a further reaction of phenol to coke takes
place already at low temperatures, and therefore the long residence time of phenol in the
catalyst proves to be disadvantageous, but not primarily a too strong adsorption.
Furthermore, it could be shown that a selective heating of phenol adsorbed on the catalyst
and thereby a selective desorption from the catalyst by microwave radiation, is possible.
Outlook
It seems on basis of the results of this work that the problem of deactivation during the
hydroxylation of benzene with N2O with the ZSM-5-type catalysts available today cannot
be solved by process measures. Therefore, future work should be to develop customized
catalysts for the hydroxylation of Benzene with N2O. These have to contain as few sites
responsible for deactivation as possible, and have to facilitate faster diffusion of the
phenol. By this means the problem of a fast deactivation could be reduced without
lowering the fresh activity of the catalyst.
Since it was possible to interfere with the course of a reaction by microwave irradiation,
further reactions which might benefit from the use of microwave ought to be examined.
Indeed, the strong interaction of the microwave with water adsorbed on the catalyst could
be advantageous for certain reactions. For example water is problematic in the SCR-
reaction of nitrogen oxides with NH3 as reductive using catalysts containing vanadia

214 10 Summary and Outlook
[340]. Displacement of the water from the catalyst surface by the use of microwave
radiation could possibly solve these problems.
However, this work showed that microwave can certainly cause undesired side reactions,
too. The use of microwave to support heterogeneously catalyzed reactions must therefore
be examined in detail for the respective reaction.

Literaturverzeichnis
[1] A.J. Bahr, Microwave nondestructive testing Methods, Gordon and Breach Science Publishers, New York (1982)
[2] E. Nyfors, Industrial microwave Sensors, Artech House, Norwood (1989) [3] E. Pehl, Mikrowellentechnik, Vol. 2, Dr. Alfred Hüthig Verlag, Heidelberg (1989) [4] H. Will, P. Scholz, et al., Chem. Ing. Tech. 74/8 (2002) S. 1057 [5] P. Lindström, J. Tierney, et al., Tetrahedron 57 (2001) S. 9225 [6] D. Adam, Nature 421 (2003) S. 571 [7] A. Abu-Samara, J.S. Morris, et al., Anal. Chem. 47 (1975) S. 1475 [8] M. Hájek, Microwaves in Organic Synthesis: Microwave catalysis in organic synthesis
Ed. A. Loupy, Wiley-VCH, Weinheim (2002) [9] C. Shibata, T. Kashima, et al., Jpn. J. Appl. Phys. 35 (1996) S. 316 [10] X. Zhang, D. Hayward, et al., Catal. Lett. 88/1-2 (2003) S. 33 [11] R. Laurent, A. Laporterie, et al., J. Org. Chem. 57 (1992) S. 6231 [12] G. Bond and R. Moyes, Applications of Microwaves in Catalytic Chemistry, in
Microwave-Enhanced Chemistry, Ed. H.M. Kingston and S.J. Haswell, ACS, Washington D.C. (1997)
[13] M. Radoiu and M. Hájek, J. Mol. Cat. A: Chem. 186 (2002) S. 121 [14] M. Hájek, Czech. Chem. Commun. 62/3 (1997) S. 347 [15] D.A. Stuerga, K. Gonon, et al., Tetrahedron 49/28 (1993) S. 6229 [16] J.K.S. Wan, Method for disposal of chemical waste Patent, US4345983 (1982) [17] D. Bathen, VDI Reihe 3, Fortschritt-Berichte: Untersuchungen zur Desorption durch
Mikrowellen, VDI-Verlag, Düsseldorf (1993) [18] M. Turner, R. Laurence, et al., AIChE J. 46/4 (2000) S. 758 [19] D. Bathen, Chem. Ing. Tech. 71/1-2 (1999) S. 89 [20] J. Reuß, D. Bathen, et al., Chem. Eng. Technol. 25/4 (2002) S. 381 [21] K. Takeno. Phenol & Acetone Market Outlook And Investment Update in Asia in
Centre for Management Technology Forum, Centre for Management Technology Forum, Bangkok, Thailand (2004)
[22] M. Mccoy, Chemical Market Reporter 250/27 (1996) S. 21 [23] P. Venuto, Microporous Mesoporous Mater. 2/5 (1994) S. 297 [24] A. Reitzmann, Direktoxidation von Benzol zu Phenol mit Distickstoffmonoxid an
modifizierten Zeolithkatalysatoren, Dissertation, Universität Erlangen-Nürnberg (2001)
[25] A. Unger, Der Zusammenhang von Katalysatorstruktur und Katalyse bei der N2O-Zersetzung und der selektiven Benzoloxidation mit N2O an Zeolithkatalysatoren des Typs ZSM-5, Dissertation, Universität Erlangen-Nürnberg (2006)
[26] U. Hiemer, Reaktionstechnische Untersuchungen zur Benzolhydroxylierung, Dissertation, Universität Erlangen-Nürnberg (2004)
[27] G.I. Panov, A.K. Uriate, et al., Catal. Today 41/4 (1988) S. 365 [28] G.I. Panov, Cattech 4/1 (2000) S. 18

216 Literaturverzeichnis
[29] H. Kingston, Microwave enhanced Chemistry. Fundamentals, sample preparation and Applications, American Chemical Society, Washington (1997)
[30] P.A. Tipler, Physik, Spektrum akademischer Verlag, Heidelberg (1995) [31] E. Hering, R. Martin, et al., Physik für Ingenieure, 4. Auflage, VDI-Verlag,
Düsseldorf (1992) [32] M. Hesse, H. Meier, et al., Spektroskopische Methoden in der organischen Chemie, 6.
Auflage, Thieme Verlag, Stuttgart (2002) [33] H. Günzler and H.-U. Gremlich, IR-Spektroskopie: Eine Einführung, Wiley-VCH,
Weinheim (2003) [34] C.H. Townes and A.L. Schawlow, Microwave Spectroscopy, Dover Publications, New
York (1975) [35] M. Grünberg, Untersuchungen und Modellbildung zur Mikrowellenerwärmung von
Lebensmitteln im Mikrowellenherd, Dissertation, Universität Karlsruhe (1993) [36] A.V. Hippel, Dielectric and Waves, Ed. M.I.O. Technology, MIT Press, Cambridge,
Massachusetts USA (1954) [37] D. Michael, D. Mingos, et al., Microwave Dielectric Heating Effects in Chemical
Synthesis, in Chemistry Under Extreme or Non-Classical Conditions, Ed. R.V. Eldik and C.D. Hubbard, Wiley-VCH, New York, Weinheim (1996)
[38] Römpp Lexikon Chemie, 10. Auflage, Georg Thieme Verlag, Stuttgart (1997) [39] N. Hill, W. Vaughan, et al., Dielectric Properties and Molecular behaviour, Van
Nostard, New York (1969) [40] A.V. Hippel, Dielectric Materials and Applications, Artech House, New York (1954) [41] K. Wagner, Arch. Elektrotech. 2 (1914) S. 371 [42] M. Davies and R. Meakins, J. Chem. Phys. 26 (1957) S. 1584 [43] A. Metaxas and R. Meredith, Industrial Microwave heating, Peter Perigrinus, London
(1983) [44] E. Döring, Werkstoffkunde der Elektrotechnik, 2. Auflage, Friedrich Vieweg,
Braunschweig, Wiesbaden (1988) [45] P. Debye, Polar Molecules, Dover Publications, New York (1945) [46] M. Davies, Some Electrical and optical aspects of Molecular behaviour, Pergamon
Press, Oxford (1965) [47] C. Gabriel, S. Gabriel, et al., Chem. Soc. Rev. 27/3 (1998) S. 213 [48] E.H. Grant, R.J. Sheppard, et al., Dielectric Behaviour of Biological Molecules in
Solution, Calderon Press, Oxford (1978) [49] Basic Science, www.ibmm-microtech.co.uk/pages/science/basic.htm (2004) [50] Kapitel 8: Dielektrische Keramiken, http://www.uni-
saarland.de/fak8/fuwe/fuwe_de/Lehre/downl.html (2006) [51] G. Nimtz, Mikrowellen Einführung in Theorie und Anwendung, BI-Wiss. Verl.,
Mannheim (1990) [52] Hydrocarbon Process. 79/2 (2000) S. 45 [53] F. Öhme, Erdöl Kohle Erdgas P. 12/8 (1959) S. 623 [54] D. Craig, Dielectric Analysis of Pharmaceutical Systems, Taylor and Francis, London
(1995) [55] A. Maryott and F. Buckley, Circular: Table of Dielectric Constants and Electric
Dipole Moments of Substances in Gaseous State, Vol. 537, National Bureau of Standards (1953)
[56] Advances in heat Transfer: Advances in heat Transfer, Ed. J. Hartnett, T. Irvine, et al., Vol. 33, Academic Press, New York (1999)
[57] Water - dielectric and Microwave radiation, www.martin.chaplin.btinternet.co.uk/microwave.html (2004)

Literaturverzeichnis 217
[58] R. Meredith, IEE Power Series: Engineers' Handbook of Industrial Microwave Heating, Ed. A.T. Johns and D.F. Warne, Vol. 25, The Institution of Electrical Engineers, London (1998)
[59] Dipole relaxation, www.tf.uni-kiel.de/matwis/amat/elmat_en/kap_3/backbone/r3_3_2.html (2004)
[60] H. Fröhlich, Theory of Dielectrics, 2. Auflage, Oxford University Press, Oxford (1958)
[61] C. Böttcher, Theory of Electric Polarisation, Elsevier, Amsterdam (1952) [62] W. Leschnik. Feuchtemessung an Baustoffen - Zwischen Klassik und Moderne in
Feuchtetag 99, Vol. BB 69-CD, DGZfP-Berichtsband, Berlin (1999) [63] B. Hamon, Aust. J. Phys. 6 (1953) S. 304 [64] R.J. Meakins, Trans. Faraday Soc. 51 (1955) S. 953 [65] C. Smyth, Dielectric Behaviour and Structure, McGraw-Hill, New York (1955) [66] G. Roussy, A. Mercier, et al., J. Microwave Power EE. 20 (1985) S. 47 [67] V.M. Kenkre, L. Skala, et al., J. Mater. Sci. 26/9 (1991) S. 2483 [68] C. Persch, Messung von Dielektrizitätskonstanten im Bereich von 0,2 bis 6 GHz und
deren Bedeutung für die Mikrowellenerwärmung von Lebensmitteln, Dissertation, Technische Universität Darmstadt (1997)
[69] D.C. Dibben and A.C. Metaxas, J. Microwave Power EE. 29 (1994) S. 242 [70] E. Pehl, Mikrowellen in der Anwendung, Hüthig, Heidelberg (1993) [71] P. Hoekstra and W.T. Doyle, J. Colloid Interface Sci. 36 (1971) S. 513 [72] V.I. Kvlividze, A.V. Krasnushkin, et al., Properties of surface films and stratums of
Water, in Surface Water Films in Dispersed Media, MSU, Moskau (1984) [73] M.L. Belaya and V.G. Levadnyi, New in Life, Science, Engineering, Series - Physics
11 (1987) S. 3 [74] J. Bockris, E. Gileadi, et al., J. Chem. Phys. 44 (1966) S. 1445 [75] J.B. Hasted, Aqueous Dielectrics, Chapman and Hall, London (1973) [76] D. Or and J.M. Wraith, Water Resour. Res. 35/2 (1999) S. 371 [77] G. Serbin, Microwave Thermodielectric Behaviour of Soil-Water Mixtures and Their
Potential Effects on Microwave Remote Sensing, Master Thesis, Ben Gurion University of Negev (2001)
[78] D.A. Boyarskii, V.V. Tikhonov, et al., Prog. Electron. Res. 35 (2002) S. 251 [79] M. Kent and W. Meyer, J. Phys. D: Appl. Phys. 16 (1983) S. 915 [80] S. Pal, S. Balasubramanian, et al., J. Chem. Phys. 120/4 (2004) S. 1912 [81] D. Michael, P. Mingos, et al., Applications of microwave Dielectric Heating Effects to
Synthetic Problems in Chemistry, in Microwave-Enhanced Chemistry, Ed. H.M. Kingston and S.J. Haswell, American Chemical Society, Washington, DC (1997)
[82] M. Buryan, Entwicklung von Auslegungsprinzipien für mikrowellen- und gasbeheizte Hybrid-Sinteröfen, Dissertation, TU Freiberg (2004)
[83] M. Teffal and A. Gourdenne, Eur. Polym. J. 19 (1981) S. 543 [84] G. Roussy, J. Thiebaut, et al., J. Microwave Power EE. 19/4 (1984) S. 243 [85] D. Goerz and J. Jolly, J. Microwave Power EE. 2/3 (1967) S. 87 [86] Microwave conditioning of leather, The Electricity Council, Case History, Report, EC
3608/12/76 (1976) S. 1 [87] H. Chen, Z. Shen, et al., J. Microwave Power EE. 17/1 (1982) S. 11 [88] Microwave rubber curing, case history, The Electricity Council, Report, EC 3349/1/76
(1976) S. 1 [89] S. Israel, J. Chabert, et al., Club Microondes (1981) S. 66 [90] A. Gourdenne, A. Maassarant, et al. Quelles aspects de la polymérisation par
chauffage diélectique micro-ondes. in Comptes-rendus du Symposium international

218 Literaturverzeichnis
sur les applications énergétiques des micro-ondes, Vol. 14, IMPI-CFE, Monaco (1979)
[91] H. Linn, K. Rebstock, et al., Method for manufacturing coated short fibers, Patent, US6143376 (2000)
[92] K. Rödiger, K. Dreyer, et al. Microwave Processing of Cemented Carbides in 8th International Conference on Microwave and High Frequency Heating, University of Bayreuth, Chair of Materials Processing, Bayreuth (2001)
[93] C. Siligardi, C. Leonelli, et al., J. Eur. Ceram. Soc. 20 (2000) S. 177 [94] H. Schubert and M. Regier. Novel and Traditional Microwave Applications in the
Food Industry in 8th International Conference on Microwave and High Frequency Heating, University of Bayreuth, Chair of Materials Processing, Bayreuth (2001)
[95] E. Bonek, K. Knotik, et al. Microwave hardening of free-falling radioactive droplets in 12th European Microwave Conference, Helsinki (1982)
[96] J. Thuéry, Microwaves: Industrial, Scientific, and Medical Applications, Artech House, London (1992)
[97] A. Abu-Samara, J.S. Morris, et al., Trace. Subst. Environ. Health 9 (1975) S. 297 [98] J. Walter, S. Chalk, et al., Overwiew of Microwave-Assisted Sample Preparation, in
Microwav-Enhanced Chemistry, Ed. H. Kingston and S. Haswell, American Chemical Society, Washington (1997)
[99] R. Gedye, F. Smith, et al., Tetrahedron Lett. 27 (1986) S. 279 [100] R. Guigerre, T. Bray, et al., Tetrahedron Lett. 27 (1986) S. 4945 [101] D. Mingos, A. Whittaker, et al., Br. Ceram. Trans. 91 (1992) S. 124 [102] D. Mingos, A. Whittaker, et al., Chem. Rev. 20 (1991) S. 1 [103] A. Arafat, J.C. Jansen, et al., Zeolites 13 (1993) S. 162 [104] S. Caddick, Tetrahedron 51 (1995) S. 10403 [105] A.G. Whittaker and D.M.P. Mingos, J. Microwave Power EE. 29/4 (1994) S. 195 [106] G. Majetich and K. Wheless, Microwave Heating in Organic Chemistry - An Update,
in Microwave-Enhanced Chemistry, Ed. H.M. Kingston and S.J. Haswell, American Chemical Society, Washington (1997)
[107] R.A. Abramovitch, Org. Prep. Proced. Int. 23 (1991) S. 683 [108] C.R. Strauss, Aust. J. Chem. 52 (1999) S. 83 [109] R.N. Gedye, F.E. Smith, et al., Can. J. Chem. 66 (1988) S. 17 [110] G. Majetich and R. Hicks, J. Microwave Power EE. 30 (1988) S. 27 [111] M. Guerdia, Anal. Chim. Acta 224 (1989) S. 123 [112] A. Bose, M. Manhas, et al., J. Org. Chem. 56 (1991) S. 6968 [113] R. Gupta and S. Paul, Indian J. Chem., Sect B 34 (1995) S. 61 [114] W.C. Sun, P.M. Guy, et al., J. Org. Chem. 53 (1988) S. 4414 [115] K. Dema-Khalaf and A. Morales-Rubio, Anal. Chim. Acta 281 (1993) S. 249 [116] M.P. Vasquez-Tato, Synth. Lett. 7 (1993) S. 506 [117] A.K. Bose, B.K. Banik, et al., Synth. Lett. 8 (1993) S. 575 [118] M. Radoiu, I. Calinescu, et al., J. Microwave Power EE. 34/3 (1999) S. 144 [119] A. Breccia and B. Esposito, J. Microwave Power EE. 34/1 (1999) S. 3 [120] G. Bram, A. Loupy, et al., Tetrahedron 46 (1990) S. 5167 [121] R.S. Varma, Pure Appl. Chem. 73/1 (2001) S. 193 [122] B. Alloum, B. Labiad, et al., J. Chem. Soc., Chem. Commun. (1989) S. 386 [123] M. Radoiu, I. Calinescu, et al., J. Microwave Power EE. 35/2 (2000) S. 86 [124] P. Kooyman, G. Luijks, et al., J. Mol. Catal. A: Chem. 111 (1996) S. 167 [125] M. Hájek and M. Radoiu, J. Mol. Catal. A: Chem. 160 (2000) S. 383 [126] I. Plazl, G. Pipuš, et al., AIChE J. 43/3 (1997) S. 754 [127] G. Pipuš, Ind. Eng. Chem. Res. 41 (2002) S. 1129

Literaturverzeichnis 219
[128] G. Pipuš, I. Plazl, et al., Chem. Eng. J. 76 (2000) S. 239 [129] K. Kabza, B. Chapados, et al., J. Org. Chem. 65 (2000) S. 1210 [130] S. Balalaie and A. Arabanian, Green Chem. 2 (2000) S. 274 [131] S. Balalaie, A. Arabanian, et al., Monatsh. Chem. 131 (2000) S. 945 [132] B. Badgar and S. Kasture, Green Chem. 2 (2000) S. 154 [133] J. Berlan, P. Giboreau, et al., Tetrahedron Lett. 32 (1991) S. 2363 [134] K.D. Raner, C.R. Strauss, et al., J. Org. Chem. 58 (1993) S. 950 [135] G. Bond, R. Moyes, et al., Chem. Ind. (1991) S. 686 [136] D.R. Baghurst and D.M.P. Mingos, J. Chem. Soc., Chem. Commun. (1992) S. 674 [137] R.N. Gedye and J.B. Wei, Can. J. Chem. 76 (1998) S. 525 [138] E.G.E. Jahngen, R.R. Lentz, et al., J. Org. Chem. 55 (1990) S. 3406 [139] X. Zhang, D. Hayward, et al., Chem. Commun. 9 (1999) S. 975 [140] J.K.S. Wan, Res. Chem. Intermed. 19/2 (1993) S. 147 [141] S. Suib and R. Zerger, J. Catal. 139 (1993) S. 383 [142] C. Cundy, Collect. Czech. Chem. Commun. 63 (1998) S. 1699 [143] Y.S. Mok and D.J. Koh, Ind. Eng. Chem. Res. 42 (2003) S. 2960 [144] G.E. Keller and M.M. Bahasin, J. Catal. 73 (1982) S. 9 [145] L.F. Albright, B.L. Crynes, et al., Pyrolysis: Theory and industrial practice.,
Academic Press, New York (1983) [146] J.K.S. Wan, M. Tse, et al., J. Microwave Power EE. 25/1 (1990) S. 32 [147] J.K.S. Wan, Conversion of methane to ethylene and Hydrogen, Patent, US 4574038
(1968) [148] M.Y. Tse, M. Depew, et al., Res. Chem. Intermed. 13 (1990) S. 221 [149] K.L. Cameron, M.C. Depew, et al., Res. Chem. Intermed. 16 (1991) S. 57 [150] G. Bond, R.B. Moyes, et al., Catal. Today 17 (1993) S. 427 [151] G. Roussy, J. Thiebaut, et al. Quality Enhancements Using Microwaves in 28th
Microwave Symposium Industrial Microwave Power Institute, Clifton, VA (1993) [152] Foundations and Industrial Application of microwave and Radio Frequency Fields, in
Foundations and Industrial Application of microwave and Radio Frequency Fields, Ed. G. Roussy and J.A. Pearce, Wiley&Sons, New York (1995)
[153] G. Roussy, E. Marchal, et al., Fuel Process. Technol. 50 (1997) S. 261 [154] C. Chen, P. Hong, et al., React. Kinet. Catal. Lett. 61 (1997) S. 175 [155] X. Bi, P. Hong, et al., React. Kinet. Catal. Lett. 66 (1999) S. 381 [156] Y. Tanashev, V. Fedoseev, et al., Catal. Today 42 (1998) S. 333 [157] M. Ioffe, S. Pollington, et al., J. Catal. 151 (1995) S. 349 [158] J.K.S. Wan, G. Bamwenda, et al., Res. Chem. Intermed. 16 (1991) S. 241 [159] G. Bamwenda, E. Moore, et al., Res. Chem. Intermed. 17 (1992) S. 243 [160] D.O. Cooney and Z. Xi, Fuel Sci. Technol. Int. 14 (1996) S. 1111 [161] D.O. Cooney and Z. Xi, Fuel Sci. Technol. Int. 14 (1996) S. 1315 [162] X. Bi, P. Hong, et al., Chin. Chem. Lett. 9 (1998) S. 975 [163] H. Will, P. Scholz, et al., Chem. Ing. Tech. 74/9 (2002) S. 1258 [164] H. Will, P. Scholz, et al., Chem. Ing. Tech. 76/6 (2004) S. 700 [165] Y. Liu, Y. Lu, et al., Appl. Catal., A. 170 (1998) S. 207 [166] Y. Liu, Y. Lu, et al., Catal. Today 51 (1999) S. 147 [167] W. Perry, J. Katz, et al., J. Catal. 171 (1997) S. 431 [168] J. Ma, M. Fang, et al., Appl. Catal., A. 159 (1997) S. 211 [169] M. Turner, R. Laurence, et al., Catal. Lett. 71/3-4 (2001) S. 133 [170] J.K.S. Wan and J.F. Kriz, Hydrodesulphurization of hydrocracked pitch Patent, US
4545879 (1985)

220 Literaturverzeichnis
[171] Cha, Process and reactor for char-gas oxide reactions by radiofrequency catalysis, Patent, US 5269892 (1993)
[172] Cha, Process for selected gas oxide removal by radiofrequency catalysts, Patent, US 5246554 (1993)
[173] Cha, Process for oxide reactions by radiofrequency-char catalysis, Patent, US 5256265 (1993)
[174] Y. Kong and C. Cha, Energy Fuels 9 (1995) S. 971 [175] J. Tang, T. Zhang, et al., J. Catal. 211 (2002) S. 560 [176] Ma, Emission Control, Patent, US 5180559 (1993) [177] Y. Zhenming, Z. Jinsong, et al., Appl. Catal., B 34 (2001) S. 129 [178] Y. Li and J. Armor, Appl. Catal., B 1 (1992) S. 1 [179] Y. Chang, A. Sanjurjo, et al., Catal. Lett. 57/4 (1999) S. 187 [180] X. Wang and T. Zhang, Chem. Commun. (2000) S. 279 [181] J. Tang and T. Zhang, Catal. Lett. 73/2-4 (2001) S. 193 [182] X. Zhang, D. Hayward, et al., Appl. Catal., B 33 (2001) S. 137 [183] T. Dinesen, M. Tse, et al., Res. Chem. Intermed. 15 (1991) S. 113 [184] J.K.S. Wan, Res. Chem. Intermed. 20/1 (1994) S. 29 [185] G. Bamwenda, M. Depew, et al., Res. Chem. Intermed. 19/6 (1993) S. 553 [186] M. Depew, S. Lem, et al., Res. Chem. Intermed. 16 (1991) S. 213 [187] G. Bond, R.B. Moyes, et al., Top. Catal. 1 (1994) S. 177 [188] L. Seyfried, F. Garin, et al., J. Catal. 148 (1994) S. 281 [189] G. Roussy and J. Pearce, Foundations and industrial applications of microwaves and
radio frequency fields: physical and chemical processes, Wiley&Sons, Chinchester (1995)
[190] V. Zholobenko and E. House, Catal. Lett. 89/1-2 (2003) S. 35 [191] W. Perry, D. Cooke, et al., Catal. Lett. 47/1 (1997) S. 1 [192] J. Thomas, Catal. Lett. 49/3-4 (1997) S. 137 [193] J. Thiebaut, G. Roussy, et al., Catal. Lett. 21/1-2 (1993) S. 133 [194] A. Klimov, B. Balzhinimaev, et al., Kinet. Catal. 39 (1998) S. 511 [195] N. Ahr, E. Brandauer, et al., Chem. Tech. 46/4 (1994) S. 216 [196] I.W. Turner and K. Bremhorst, Dry. Technol. (1992) S. 635 [197] I.W. Turner and P.G. Jolly, Dry. Technol. 9 (1991) S. 1209 [198] H. Feng, J. Tang, et al., AIChE J. 47/7 (2001) S. 1499 [199] T. Constant, C. Mpyne, et al., AIChE J. 42/2 (1996) S. 359 [200] G. Roussy, A. Zoulalian, et al., J. Phys. Chem. 88 (1984) S. 5702 [201] M. Benchanaa, M. Lallemant, et al., Thermochim. Acta 152 (1989) S. 43 [202] H. Burkholder and R. Demarco, Ind. Eng. Chem. Fundam. 25/3 (1986) S. 414 [203] A. Weissenberger and P. Schmidt. Microwave-Enhanced Regeneration of Adsorbents
in Mat. res. Soc. Symp., Vol. 347, San Francisco (1994) [204] D. Price and P. Schmidt, J. Microwave Power EE. 32/3 (1997) S. 145 [205] D. Price and P. Schmidt, JAPCA J. Air Waste Ma. 48 (1998) S. 1146 [206] J. Reuß, D. Bathen, et al., Berichte aus der Verfahrenstechnik: Parameterstudie zur
Mehrkomponentendesorption durch Mikrowellen, Ed. H. Schmidt-Traub, Shaker, Aachen (2000)
[207] D. Bathen and H. Schmidt-Traub, Chem. Ing. Tech. 68/4 (1996) S. 434 [208] D. Bathen, C. Hummelt, et al., Chem. Ing. Tech. 71/1-2 (1999) S. 89 [209] S. Kobayashi, Y. Kim, et al., Chem. Lett. 25/9 (1996) S. 769 [210] C. Balnco and S. Auerbach, J. Am. Chem. Soc. 124 (2002) S. 6250 [211] Phenol, www.the-innovation-group.com/ChemProfiles/Phenol.htm (2002)

Literaturverzeichnis 221
[212] K. Weissermel and H.-J. Arpe, Industrielle Organische Chemie, 5. Auflage, Wiley-VCH, Weinheim (1998)
[213] Phenol/Acetone/Cumene, Report, 96/97-2 (1997) [214] U. Onken and A. Behr, Chemische Prozeßkunde, Thieme Verlag, Stuttgart (1996) [215] G.R. Meima, M.J.M.V.D. Aalst, et al. Catalysis on Solid Acids and Bases in DGMK
Conference, Vol. 9601, DGMK, Berlin (1996) [216] Phenol, in Ullmann's Encyclopedia of Industrial Chemistry, VCH, Weinheim (1999) [217] A. Budzinski, Chemische Industrie 5 (1996) S. 13 [218] H. Fukuhara and F. Matsunaga, Phenol preparation process and propylene recovery
therefrom, Patent, US 5017729 (1991) [219] M. Iwamoto, J. Hirata, et al., J. Phys. Chem. 87 (1983) S. 903 [220] Y. Ono, K. Tohmori, et al., Stud. Surf. Sci. Catal. 41 (1988) S. 75 [221] R. Burch and C. Howitt, Appl. Catal., A 103 (1993) S. 135 [222] G. Jutta and R. Lobo, Stud. Surf. Sci. Catal. 135 (2001) S. 4392 [223] C.R. Buechler, J.R. Ebner, et al., Process For The Hydroxylatio Of Benzene, Patent,
WO001998015514A1 (1996) [224] A.S. Kharitonov, G.I. Panov, et al., Preparation of phenol or phenol derivatives
Patent, US 5110995 (1992) [225] G.I. Panov, A.S. Kharitonov, et al., Appl. Catal., A 98 (1993) S. 1 [226] A.K. Uriarte, M.A. Rodkin, et al., Stud. Surf. Sci. Catal. 110 (1997) S. 857 [227] Chemical Market Reporter 8 (2001) S. 7 [228] Chem. Eng. Prog. (1997) S. 16 [229] B.W. Riley and J.R. Richmond, Catal. Today 17 (1993) S. 277 [230] Eur. Chem. News (2000) S. 59 [231] A.F. Cronstedt, Der Königl. Schwedischen Akademie der Wissenschaften neue
Abhandlungen aus der Naturlehre, Haushaltskunst und Mechanik 18: Von einer unbekannten Bergart, Zeolithes genannt, Vol. 111, Königl. Schwedischen Akademie der Wissenschaften, Stockholm (1757)
[232] D.W. Breck, Zeolite Molecular Sieves, Vol. 2, Wiley, New York (1974) [233] M. Häfele, Gasphasenhydroxylierung von Benzol mit Distickstoffmonoxid an
Katalysatoren vom Pentasil-Typ, Dissertation, Universität Erlangen-Nürnberg (1997) [234] Internetsiete der Intl. Zeolite Assoc. - Structure Commission, Datenbanken,
http://www.iza-structure.org/databases/ (2004) [235] P.B. Weisz and V.J. Frilette, J. Phys. Chem. 64 (1960) S. 382 [236] W. Hölderich and E. Gallei, Chem. Ing. Tech. 56/12 (1984) S. 908 [237] U. Graeser, W. Keim, et al., Erdöl Kohle Erdgas P. 5 (1995) S. 208 [238] W. Hölderich, M. Hesse, et al., Angew. Chem. 100 (1988) S. 232 [239] P.B. Venuto, Stud. Surf. Sci. Catal. 105 (1997) S. 811 [240] E. Roland, Zeolites as Catalysts, Sobents and Detergent Builders, Ed. J. Weitkamp,
Elsevier, Amsterdam (1989) [241] J. Weitkamp, S. Ernst, et al., Chem. Ing. Tech. 58/8 (1986) S. 623 [242] W.E. Garwood, Am. Soc. Symp. Ser. 218 (1983) S. 371 [243] W.O. Haag. in 6th Int. Zeol. Conf., Butterworth, Reno (1983) [244] W.W. Kaeding, L.B. Young, et al., J. Catal. 89 (1984) S. 264 [245] J. Weitkamp, Solid State Ionics 131 (2000) S. 175 [246] L. Puppe, Chem. unserer Zeit 4 (1986) S. 117 [247] D.H. Ohlson, G.T. Kokotailo, et al., J. Phys. Chem. 85 (1981) S. 2238 [248] W. Löwenstein, Am. Mineral. 39 (1954) S. 92 [249] S. Szostak, Handbook of Molecular Sieves, van Nostrand, New York (1992)

222 Literaturverzeichnis
[250] J.-L. Guth and K. Kessler, Synthesis of Alumosilicate Zeolites and Related Silica-Based Materials, in Catalysis and Zeolites, Ed. J. Weitkamp and L. Puppe, Springer-Verlag, Berlin (1999)
[251] R.A. Racoczy, Hydrothermalsynthese ausgewählter Zeolithe und ihre Charakterisierung durch Adsorption, Dissertation, Universität Stuttgart (2004)
[252] K.H. Bergk, W. Schwieger, et al., Stud. Surf. Sci. Catal. 65 (1991) S. 185 [253] R.J. Argauer and G.R. Landolt, Crystalline Zeolite ZSM-5 And Method Of Preparing
The Same Patent, US 3702886 (1970) [254] P.A. Jacobs and J.A. Martens, Stud. Surf. Sci. Catal. 33 (1987) S. 500 [255] W. Schwieger, Die templatfreie Pentasilkristallisation - Der Einfluß von
Synthesebedingungen auf Eigenschafts-Anwendungs-Beziehungen, Habilitationsschrift, Universität Halle-Wittenberg (1994)
[256] R.M. Barrer, Hydrothermal Chemistry of Zeolites, Academic Press, London (1982) [257] H.G. Karge, Stud. Surf. Sci. Catal. 105 (1997) S. 1901 [258] W.J. Mortier, J. Catal. 55 (1978) S. 138 [259] D. Barthomeuf, Acidic Catalysis with Zeolites, in Zeolites: Science and Technology,
Ed. F. Ribeiro, A. Rodrigues, et al., Martinus Nijhoff Publishers, The Hague (1984) [260] J. Weitkamp and L. Puppe, Catalysis and Zeolites-Fundamentals and Applications,
Springer Verlag, Berlin (1999) [261] J.C. Vedrine, A. Auroux, et al., J. Catal. 59 (1979) S. 248 [262] J. Datka and Z. Piwowarska, Zeolites 8 (1988) S. 30 [263] B. Kraushaar-Czarnetzki, Characterization and modification of zeolites and related
materials, Dissertation, Technische Universität Eidhoven (1989) [264] G.O. Brunner, Zeolites 7 (1987) S. 9 [265] J.B. Uytterhoeven, L.G. Christner, et al., J. Phys. Chem. 72 (1968) S. 1768 [266] G.H. Kühl, J. Phys. Chem. Solids 38 (1970) S. 1259 [267] T. Chen, A. Men, et al., Catal. Today 30 (1996) S. 189 [268] S.M. Campbell, D.M. Bibby, et al., J. Catal. 161 (1996) S. 338 [269] N.Y. Chen, W.E. Garwood, et al., Shape Selective Catalysis in Industrial Applications,
Marcel Decker, New York (1989) [270] N.Y. Chen, T.F. Degnan, et al., Molecular Transport and Reaction in Zeolites -
Design and Application of Shape Selective Catalysts, VCH, New York (1994) [271] W. Hölderich, Stud. Surf. Sci. Catal. 58 (1991) S. 631 [272] P.S. Landis and P.B. Venuto, Adv. Catal. 31 (1963) S. 259 [273] T. Masuda, Y. Fujikata, et al., Microporous Mesoporous Mater. 23 (1998) S. 157 [274] E. Klemm, J. Wang, et al., Microporous Mesoporous Mater. 26 (1998) S. 11 [275] M. Häfele, A.Reitzmann, et al., Appl. Catal., A 150 (1997) S. 153 [276] A. Reitzmann, E. Klemm, et al., Chem. Eng. J. 90 (2002) S. 149 [277] S. Perathoner, F. Pino, et al., Top. Catal. 23/1-4 (2003) S. 125 [278] E. Selli, I. Rossetti, et al., Appl. Catal., A. 262 (2004) S. 131 [279] R.F. Parton, J.M. Jacobs, et al., Stud. Surf. Sci. Catal. 46 (1989) S. 162 [280] T. Ohgushi, S. Komarneni, et al., J. Porous Mater. 8 (2001) S. 23 [281] S. Komarneni and R. Roy, Mater. Lett. 4 (1986) S. 107 [282] T. Oghushi, K. Nomaka, et al., Bull. Chem. Soc. Jpn. 62 (1989) S. 2998 [283] T. Oghushi and S. Sato, J. Solid State Chem. 87 (1990) S. 95 [284] B.I. Whittington and N.B. Milestone, Zeolites 12 (1992) S. 815 [285] L.M. Kustov, A.L. Tarasov, et al., Catal. Today 61 (2000) S. 123 [286] J.L. Motz, H. Heinichen, et al., J. Mol. Catal. A: Chem. 136 (1998) S. 175 [287] E. Klemm, Direktsynthese von Phenol und Kresol, Habilitationsschrift, Universität
Erlangen-Nürnberg (2000)

Literaturverzeichnis 223
[288] D. Meloni, R. Monaci, et al., J. Catal. 214 (2003) S. 169 [289] E.J.M. Hensen, Q. Zhu, et al., J. Catal. 220 (2003) S. 260 [290] P. Kubánek, B.Wichterlova, et al., J. Catal. 211 (2002) S. 109 [291] L.V. Pirutko, V.S. Chernyavsky, et al., Appl. Catal., A 227 (2002) S. 143 [292] V.I. Sobolev, K.A. Dubkov, et al., Appl. Catal., A 141 (1996) S. 185 [293] N.A. Kachurovskaya, G.M. Zhidomirov, et al., Catal. Lett. 86/1-3 (2003) S. 25 [294] K. Yoshizawa, Y. Shiota, et al., J. Phys. Chem. B 107/41 (2003) S. 11404 [295] L.V. Pirutko, A.K. Uriarte, et al., Microporous Mesoporous Mater. 48 (2001) S. 345 [296] J. Pérez-Ramírez, F. Kapteijn, et al., J. Catal. 214/1 (2003) S. 33 [297] J. Jia, K.S. Pillai, et al., J. Catal. 221 (2004) S. 119 [298] K.A. Dubkov, V.I. Sobolev, et al., J. Mol. Catal. A: Chem. 123 (1997) S. 155 [299] A.R. Overweg, M.W.J. Craje, et al., J. Catal. 223 (2004) S. 262 [300] V.I. Sobolev, G.I. Panov, et al., J. Catal. 139 (1993) S. 435 [301] A.S. Kharitonov, G.A. Sheveleva, et al., Appl. Catal., A 98 (1993) S. 33 [302] R. Burch and C. Howitt, Appl. Catal., A. 106 (1993) S. 167 [303] R. Leanza, I. Rosetti, et al., Appl. Catal., A. 205 (2001) S. 93 [304] J.R. Rostrup-Nielsen, Catal. Today 37 (1997) S. 225 [305] J. Zhu and S.L.T. Andersson, Appl. Catal., A 53 (1998) S. 251 [306] K.S. Pillai, J. Jia, et al., Appl. Catal., A 264 (2004) S. 133 [307] B. Vogel, Reaktionstechnische Untersuchungen zur Direktsynthese von Kresolen aus
Toluol und Distickstoffmonoxid an Zeolithkatalysatoren vom MFI-Typ, Dissertation, Universität Erlangen-Nürnberg (2005)
[308] B. Imelik and J.C. Vedrine, Catalyst characterisation - Physical Techniques for Solid Materials, Plenum Press, New York (1994)
[309] Handbook of Heterogeneous Catalysis, Ed. G. Erltl, H. Knözinger, et al., Vol. 2, 1. Auflage, VCH, Weinheim (1997)
[310] P.A. Webb and C. Orr, Analytical Methods in Fine Particle Technology, Micromeritics Instrument Corp., Norcross, USA (1997)
[311] Innovative methods for inorganic sample preparation, Report, ANL/ACL-92/1 (1992) S. 32
[312] Publikationen zum Thema Mikrowellenaufschluß Systeme (MDS 2000, STAR 2 & STAR 6 , MARS 5 sowie SpectroPrep), http://www.cem.de/index.html?http://www.cem.de/documents/publikationen/mikrowellenauf.htm (2006)
[313] The International Centre for Diffraction Data®, http://www.icdd.com/ (2006) [314] G.I. Kapustin, T.R. Brueva, et al., Appl. Catal., A 42 (1988) S. 239 [315] G. Bagnasco, J. Catal. 159 (1996) S. 249 [316] M. Seitz, Gezielte Modifizierung von Zeolithkatalysatoren mit Silan, Dissertation,
Universität Erlangen-Nürnberg (2000) [317] P. Houdec, A. Smieskova, et al., Collect. Czech. Chem. Commun. 63 (1998) S. 141 [318] R.H. Müller and W. Mehnert, Particle and Surface Characterisation Methods,
medpharm Scientific Publishers, Stuttgart (1997) [319] Massendurchflussmesser- und Regler für Gase, http://www.wagner-
msr.de/prod_fra.htm (2005) [320] S. Gopalakrishnan, Effects of microwave heating on heterogeneously catalysed
reactions, Master Thesis, Universität Erlangen-Nürnberg (2004) [321] G. Impac, Pyrometer Handbuch, IMPAC GmbH, Frankfurt a. Main (2004) [322] Ethos Micro Synth Tech. , http://www.milestonesci.com/synth-tech.php (2003) [323] W. Gottwald, GC für Anwender, VCH Verlagsgesellschaft, Weinheim (1995) [324] G. Schomburg, Gaschromatographie, 2. Auflage, VCH-Verlag, Weinheim (1987)

224 Literaturverzeichnis
[325] O.N. Ewane, The Influence of Microwave Radiation on the Direct Hydroxylation of Benzene to Phenol with Nitrous Oxide, Master Thesis, Universität Erlangen-Nürnberg (2003)
[326] H.J. Hediger, Quantitative Photometrie im ultravioletten und infraroten Spektralbereich, Hüthig, Heidelberg (1985)
[327] P.W. Atkins, Pysikalische Chemie, Verlag Chemie, Weinheim (1990) [328] H.F. Rase, Fixed-Bed Reactor Design and Diagnostics, Butterworths, Boston (1990) [329] J.A. Moulijn and F. Kapteijn, Handbook of Heterogeneous Catalysis, Ed. G. Ertl, H.
Knözinger, et al., Vol. 3, VCH, Weinheim (1998) [330] M. Baerns, H. Hofmann, et al., Chemische Reaktionstechnik, 2. Auflage, Georg
Thieme Verlag, Stuttgart (1992) [331] S. Pfadler, Untersuchungen zum Sorptionsverhalten von Benzol und Phenol bei
konventioneller thermischer und mikrowellenunterstützter Desorption, Studienarbeit, Universität Erlangen-Nürnberg (2002)
[332] D. Vortmeyer and J. Schuster, Chem. Eng. Sci. 38 (1983) S. 1691 [333] J.A. Moulijn, A. Tarfaoui, et al., Catal. Today 11 (1991) S. 1 [334] M.E. Brown, Introduction to Thermal Analysis, Chapman and Hall, London (1988) [335] C. Brunee and H. Voshage, Massenspektrometrie: Physikalische und Apparative
Grundlagen, Vol. 1, Thiemig Verlag, München (1964) [336] C. Brunee and H. Voshage, Massenspektrometrie: Anwendungen, Vol. 2, Thiemig
Verlag, München (1964) [337] K.A. Dubkov, N.S. Ovanesyan, et al., J. Catal. 207 (2002) S. 341 [338] B.R. Wood, J.A. Reimer, et al., J. Catal. 209 (2002) S. 151 [339] J.A.Z. Piterse, S. Booneveld, et al., Appl. Catal., B 51 (2004) S. 215 [340] J. Pérez-Ramírez, F. Kapteijn, et al., Catal. Commun. 3 (2002) S. 19 [341] A. Heyden, B. Peters, et al., J. Phys. Chem. B 109 (2005) S. 1857 [342] M. Kögel, B.M. Abu-Zied, et al., Catal. Commun. 2 (2001) S. 273 [343] F. Kapteijn, G. Marban, et al., J. Catal. 167 (1997) S. 256 [344] N. Schmitz, SF Dampfdruck für Windows. 2005, Software-Factory: Schifferstadt [345] C. Walz, NOx-Minderung nach dem SCR-Verfahren: Untersuchungen zum Einfluß des
NO2-Anteils, Dissertation, Universität Karlsruhe (2000) [346] R.C. Reid, J.M. Prausnitz, et al., The properties of gases and liquids, McGraw-Hill,
New York (1987)

Symbolverzeichnis
Abkürzungen
BET Spezifische Oberfläche nach Brunauer-Emmett-Teller [m2/g]
F Filter
FCC fluid catalytic cracking
FID Flammenionisationsdetektor
FTIR Furrier Trnasformations Infrarot Spektroskopie
GC Gaschromatograph
GHSV gas hourly space velocity [h-1]
HPLC high pressure liquid chromatography
I Relative Intensität
ICP Induktiv-gekoppelte-Plasma-Emmissionsspektroskopie
IR Infrarot
k. A. keine Angabe
konv. konventionell
M Modul des Zeolithen (nSiO2/nAl2O3)
m% Anteil auf Gesamtmasse bezogen [%]
MFC Mass-Flow-Controler
MFI Strukturtyp ZSM-5
MS Massenspektrometer
MW Mikrowelle
NDIR Nichtdispersive Infrarotabsorption
Nml Milliliter unter Normbedingungen [Nml]
NMP n-Methyl-Pyrrolidon
NMR nuclear magnetic resonance

226 Symbolverzeichnis
NV Nadelventil
PI Druckanzeige (Pressure Indicator)
PID Proportional, Integral, Derivate - Regler
RMR relative molar response
SBU secondary building unit
SCR selective catalytic reduction
TG Thermogravimetrie
TG-MS kombinierte Thermogravimetrie-Massenspektroskopie
TIC Temperaturregelung (temperature indication control)
TOS Time-on-stream [min]
TPD Temperaturprogrammierte-Desorption
UHF ultra high frequency
UV ultraviolett
WLD Wärmeleitfähigkeitsdetektor
XRD Röntgenpulverdiffraktometrie (X-Ray Diffraction)
ZSM-5 Zeolite Socony Mobile Nr. 5
↑ Zunahme
→ keine Änderung
↓ Rückgang
Lateinische Symbole
a Durchmesser des Wellenleiters [m]
C Wärmekapazität [J/K]
dP Durchmesser Partikel [m]
dR Durchmesser Reaktor [m]
D Dissipationsfaktor [-]
Dp Eindringtiefe [m]
E Elektrisches Feld (Feldstärke) [V/m]
Er
Elektrisches Feld (Vektor) [V/m]

Symbolverzeichnis 227
f Frequenz [Hz]
F Gesamtvolumenstrom (Feed) [mol/min]
f0 Relaxationsfrequenz [Hz]
Hr
magnetische Feldstärke [A/m]
I Strom [A]
k Boltzmann Konstante [J/K]
KB Henry-Konstante Benzen [-]
KP Henry-Konstante Phenol [-]
M Metallkation
M Modul [-]
m Masse [kg]
m durchschnittliches Gesamtmoment pro Molekül [Nm]
N Molekülanzahl [-]
n Elementarladung [-]
n Stoffmenge [mol]
P Gesamtpolarisation [N/m2]
P Leistung [W]
p Druck, Partialdruck [Pa]
Pdielekt Polarisation durch Dielektrizität [N/m2]
Pelekt Polarisation durch Leitfähigkeit [N/m2]
Pmag Polarisation durch Magnetismus [N/m2]
QAl Kristallinitätsmaß [-]
r Radius [m]
t Zeit [min]
T Temperatur [K] [°C]
S Benzochinon Selektivität zu Benzochinon [%]
S Phenol Selektivität zu Phenol [%]
Sr
Poynting’scher Vektor [W/m2]
Ua Potentialbarriere [-]

228 Symbolverzeichnis
V& Volumenstrom [m3/h]
V Volumen [m3]
W Katalysatormasse [g]
X Benzen Benzenumsatzgrad [%]
Y Beladung (Wasser) [-]
Y Phenol Phenolausbeute [%]
griechische Symbole
αa Polarisierbarkeit durch Atompolarisation [N⋅m2/V]
αe Polarisierbarkeit durch Elektronenpolarisation [N⋅m2/V]
αg Polarisierbarkeit durch Grenzflächenpolarisation [N⋅m2/V]
αi Polarisierbarkeit durch Ionenpolarisation [N⋅m2/V]
αo Polarisierbarkeit durch Orientierungspolarisation [N⋅m2/V]
αt Gesamtpolarisierbarkeit [N⋅m2/V]
δ Verlustwinkel [°]
∆X relative Desaktivierung zwischen 5 und 61 min TOS [%]
∆T Temperaturanstieg [°C]
λ0 Wellenlänge von Mikrowellen im Vakuum [m]
λc minimale für Wellenleiter benötigte Wellenlänge [m]
ε absolute Dielektrizitätskonstante (Permittivität) [A⋅s/V⋅m]
ε0 elektrische Feldkonstante (Permittivitätszahl) [A⋅s/V⋅m]
ε' ideale Dielektrizitätskonstante Realteil [-]
ε" ideale Dielektrizitätskonstante Imaginärteil [-]
εa" Dielektrizitätskonstante für Atompolarisation Imaginärteil [-]
εe" Dielektrizitätskonstante für Elektronenpolarisation Imaginärteil [-]
εi" Dielektrizitätskonstante für Ionenpolarisation Imaginärteil [-]
εo" Dielektrizitätskonstante für Orientierungspolarisation
Imaginärteil
[-]
εr relative Dielektrizitätskonstante [-]

Symbolverzeichnis 229
εr' relative Dielektrizitätskonstante Realteil
(ideale Dielektrizitätskonstante)
[-]
εr" relative Dielektrizitätskonstante Imaginärteil (Verlustwert) [-]
εs statische Dielektrizitätskonstante [-]
ε ∞ Hochfrequenz-Dielektrizitätskonstante [-]
η dynamische Viskosität [kg/m⋅s]
ν Verlustfaktor pro Volumenelement [-]
ρ Dichte [kg/m3]
σ Leitfähigkeit [S/m]
τ Relaxationszeit [s]
τmod modifizierte Verweilzeit [g⋅min/mol]
θ Glanzwinkel [°]
ω Winkelgeschwindigkeit [s-1]
1/ν Zeit Pro Schwingung [s]

Appendix
A t-Plots der N2-Sorptionsmessungen
Abbildung A.1: t-Plot nach Harkins & Jura für den Katalysator H-ZSM-5 (I)
Abbildung A.2: t-Plot nach Harkins & Jura für den Katalysator Fe-ZSM-5 (I)
Appendix 231
Abbildung A.3: t-Plot nach Harkins & Jura für den Katalysator Fe-ZSM-5 (II)
Abbildung A.4: t-Plot nach Harkins & Jura für den Katalysator Co-ZSM-5
232 Appendix
B Kurvenfits der NH3-TPD-Profile
Abbildung B.1: Fit des NH3-TPD-Signals des Katalysators H-ZSM-5 (I)
Abbildung B.2: Fit des NH3-TPD-Signals des Katalysators Fe-ZSM-5 (I)
Appendix 233
Abbildung B.3: Fit des NH3-TPD-Signals des Katalysators Fe-ZSM-5 (II)
Abbildung B.4: Fit des NH3-TPD-Signals des Katalysators Co-ZSM-5

234 Appendix
C Berechnung Na-Anteil in Na-ZSM-5
Der Na-Gehalt des Katalysators Na-ZSM-5 wurde nicht durch ICP bestimmt, sondern
errechnet.
Es wurde angenommen, dass gilt:
nAl nNa= .
Mit der für den Typ ZSM-5 geltenden Summenformel für eine Elementarzelle (vgl.
S. 76):
/ 96 192 216x n x xM Al Si O H O− ⋅ und dem Modul M = 40 (nSi = 20·nAl), lässt sich x wie folgt
berechnen:
9620 4,57
xx
x
−= → = .
Somit ergibt sich für den Na-ZSM-5 folgende Summenformel:
4,57 4,57 91,43 192 216Na Al Si O H O⋅ .
Über die Molmassen der beteiligten Elemente und die errechneten stöchiometrischen
Faktoren ergibt sich die Molmasse einer Elementarzelle zu:
6155,4 g/mol
Die Molmasse des in der Elementarzelle enthaltenen Natriums beträgt 105,1 g/mol.
Dies entspricht einem Anteil an der Elementarzelle von 1,7 m%.

Appendix 235
D Temperaturprofil des verwendeten Reaktors
Abbildung D.1: Abweichung von der Solltemperatur entlang der Reaktorachse; Die
Ordinate stellt das Zentrum der Katalysatorschüttung dar
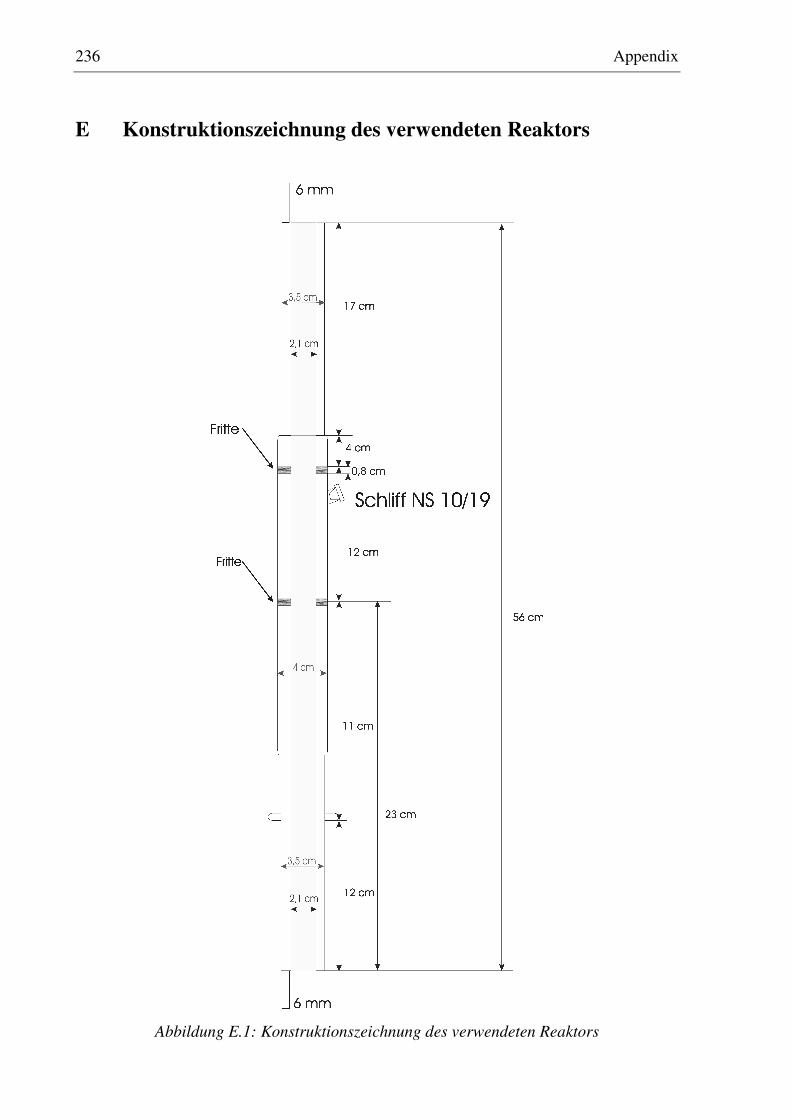
236 Appendix
E Konstruktionszeichnung des verwendeten Reaktors
Abbildung E.1: Konstruktionszeichnung des verwendeten Reaktors

Appendix 237
F Bestimmung der axialen Bodenstein-Zahl über empirische
Korrelationsbeziehungen
Es gilt:
(Gl. F.1)
(Gl. F.2)
Durch Umformen erhält man:
(Gl. F.3)
Einfluss von Füllkörpern auf das Dispersionsverhalten von Gasströmungen:
(Gl. F.4)
Für die vorherrschenden Normvolumenströme von 200 bzw. 100 Nml/min, was bei einer
Temperatur von 473 K und der vorherrschenden Reaktorgeometrie einer Strömungsge-
schwindigkeit von 0,0510, bzw. 0,0255 m/s entspricht, ergeben sich mit Hilfe des in [346]
angegebenen Diffusionskoeffizienten DBenzen-Stickstoff, der bei 473 K 0,2 cm²/s beträgt, eine
axiale Peclet-Zahl von 3,1 bzw. 2,6. Hierfür wurde mit dem hydraulischen Durchmesser
für einen Kreisring gearbeitet.
Setzt man die Peclet-Zahl in Gleichung F.3 ein, so ergeben sich bei einer Länge von
34 cm vom Reaktoreingang bis zur unteren Fritte für die verwendete Kornfraktion Boden-
stein-Zahlen von 734 bis 1319.
ax
rax
D
LuBo
⋅= 0
ax
pax
D
duPe
⋅= 0
p
raxax
d
LPeBo ⋅=
)/(Re8,315,0
Re3,01
ScScPe ppax ⋅++
⋅=

238 Appendix
G Verweilzeitverteilung des verwendeten Reaktors
Zur Bestimmung des Verweilzeitverhaltens des verwendeten Reaktors wurden Stufen-
markierungsexperimente mit Sauerstoff durchgeführt. Die Anlage wurde mit Stickstoff
gespült (200 Nml/min) und zu einem definierten Zeitpunkt wurde bei konstantem Volu-
menstrom auf ein Gemisch aus 90 vol% N2 und 10 vol% O2 umgestellt. Mit Hilfe des in
Kapitel 6.8 beschriebenen Massenspektrometers wurde das Sauerstoffsignal während des
Experiment überwacht. Diese Messungen bestätigen das bei einem mit Quarzglasbruch
gefüllten Reaktor die Annahme eines Pfropfenströmungsmodells (plug-flow) gerechtfer-
tigt ist.
Abbildung G.1: Stufenmarkierungsexperiment am mit Quarzglasbruch gefüllten Reaktor
und am leeren Reaktor

Appendix 239
H Ergänzende Messungen in der Hydroxylierung von Benzen
mit N2O
Abbildung H.1: Katalytisches Verhalten des Fe-ZSM-5 (II) als Funktion der
Mikrowellenleistung
(T = 300 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)
Abbildung H.2: Katalytisches Verhalten des Fe-ZSM-5 (II) als Funktion der
Mikrowellenleistung
(T = 350 °C, N2O:Benzen = 1:1, τmod = 155 (g·min)/mol, TOS = 13 min)

240 Appendix
Abbildung H.3: Katalytisches Verhalten des Fe-ZSM-5 (II) als Funktion der Temperatur
und Mikrowellenleistung (N2O:Benzen = 1:4, τmod = 120 (g·min)/mol, TOS = 13 min)
Abbildung H.4: Katalytisches Verhalten des Co-ZSM-5 als Funktion der
Mikrowellenleistung
(T = 300 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)

Appendix 241
Abbildung H.5: Katalytisches Verhalten des Co-ZSM-5 als Funktion der
Mikrowellenleistung
(T = 400 °C, N2O:Benzen = 1:1, τmod = 90 (g·min)/mol, TOS = 13 min)
Abbildung H.6: Katalytisches Verhalten des Co-ZSM-5 als Funktion der
Mikrowellenleistung
(T = 300 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)
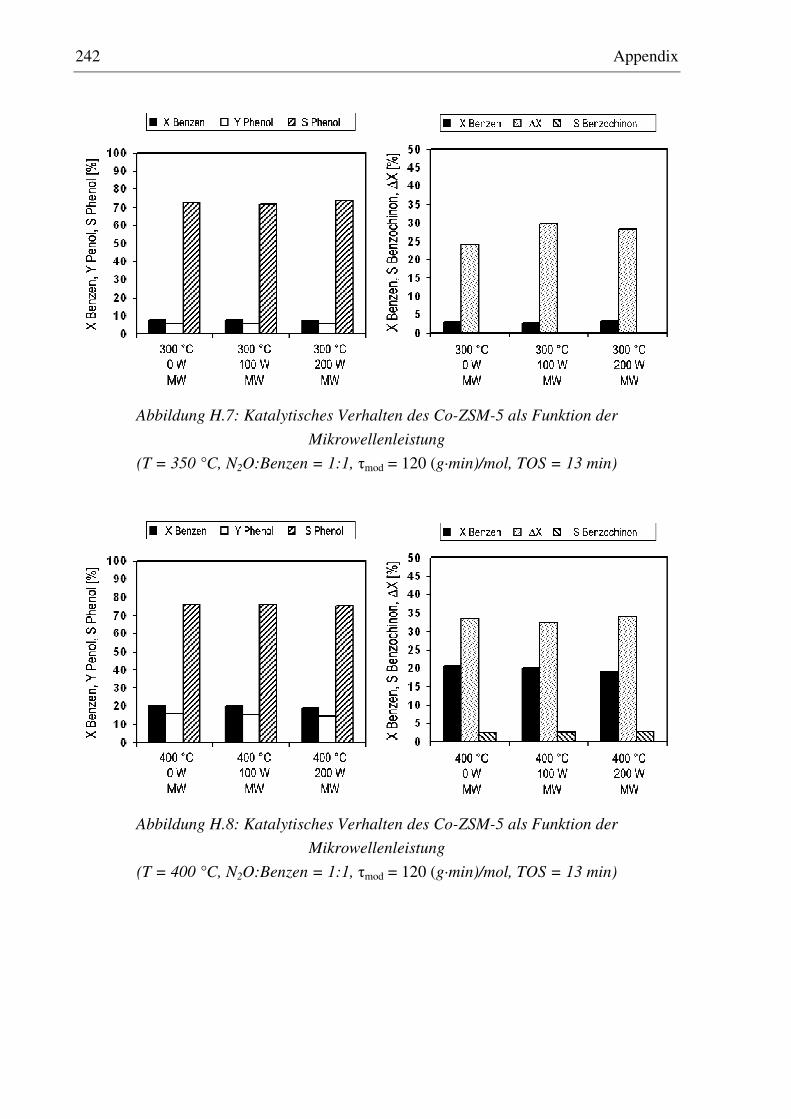
242 Appendix
Abbildung H.7: Katalytisches Verhalten des Co-ZSM-5 als Funktion der
Mikrowellenleistung
(T = 350 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)
Abbildung H.8: Katalytisches Verhalten des Co-ZSM-5 als Funktion der
Mikrowellenleistung
(T = 400 °C, N2O:Benzen = 1:1, τmod = 120 (g·min)/mol, TOS = 13 min)