reaction of transition metal complexes via silicon
TRANSCRIPT

叩何て
Reaction of Transition Metal Complexes via Silicon
ー
Hypervalent Structure
z
(ケイ素超原子価構造を経由する遷移金属錯体の反応)
、
絡
会、ー弘司ser
、ae--4-'IHTwp'titd:
可台
理学研究科
物質分子系専攻
2009年 3..月
福本晃造
(Kozo F叫ωrnoto)
‘

Contents
List of Abbreviations and Complexes
Chapter 1 Generallntroduction I
1-1 Prologue 2
1-2 Prepαγα!ion ofPentαcoordinαte Silicon Compounds 3
1-3 N→Si Coordination to an Organosilane 4
1-4 0→Si Coordination toαn Orgω10silαne 5
1-5 References 7
Chapter 2 GeometricalIsomerization ofMolybdenum Complexes with
Phosphites Cαtalyzed by Neutral or Cationic Silicon Compound
11
2-1 Introduction 12
12 2-1・1 Silicon Catalyst in Chemical Reactions
2-1・2 facィnerlsomerization of Phosphite Molybdenum Complexes 15
• 2-2 fiαc-mer Isomer匂αtionCαtαIyzed by Me3SiCl 18
2・2・1 fac-mer Isomerization of MO(CO)3 {P(OR)3} 3 with Me3SiCl 18
2・2・2 Possibility offac-mer Isomerization with Adventitious HCl 21
2-2-3 Plausible Mechanism for fac-mer Isomerization Promoted
by Me3SiCl
2-2-4 Observation of Pentacoordinate Silyl Complexes as Intermediates 23
22
2・2・5 Supporting Results to the Reaction~Mechanism 25
2-2-6 Reaction offac-Mo(CO)3(phosphite)3 with Group 14 Substrates 26
• 2・3 Isomerization Cata/yzed by a Lewis Acid
2ふ 1 fac-mer Isornerization of MO(CO)3 {P(OR)3h with a Lewis Acid
2-3-2 Structure of mer-2.4
。。。O
A
U
フu
ペ,L
4
3
...
、
IV

Reactloll ofTransltlOl1 MetaI Con中lexesVla SillCOl1 Hypen1alel1t Structure
2-3-3 Two Types of Reactions of a Phosphlte Complex with a Lewis Acid 33
2ふ 4 Plausible Isomerization Mechanism offac-mer Isomeriztion by 34
Me3SiOTf
2-3-5 cis-trans Isomerization of MO(CO)4(phosphite)2 by Me3SiOTf
2-3-6 Reaction offac-Mo(CO)3(phosphite)3 and
cis-Mo(CO)4(phosphite)2 with BF3. OEh
,
37
38
2-4 Conclusion
2世 5 Experimental Section
2 -6 References
のY
A
U
K
U
2
J
J
ザ
d-'
Chapter3 N-CN Bond Cleavage ofCyanamide bya Transition Metal
CompI.α Bearing a Si.か1Ligand
3-1 Introduction
49
50
50 3-1-1 Character of Cyanamides (R2CN)
3-1-2 C-CN Bond Cleavage of Organonitriles witb an lron Complex 52
3-2 N-CN Bond Cleavage with a Silyl-Iron Complex 53
3-2-1 N-CN Bond Cleavage ofMasked Cyanamides 53
3-2-2 N四 CNBond Cleavage ofNon-Masked cyanamides 55
3-2-3 Optimization of Solvent and Reaction Time in the N-CN 56
Bond Cleavage ofMe2NCN
3・2・4 Synthesis, Isolation, and Characterization ofN-Silylated
112-Amidino lron Complex
58
3-3 Catalytic N-CN Bond Cleavage
3-4 Unsaturated Bond Cleavage by an lron Complex
3-4・1 C=S Bond Cleavage ofMe2NCHS
3・4-2 C=S Bond Cleavage ofMeN=C=S
ぅ
3
戸、J
戸、JA
ツ
バυ
K
U
ζ
U
ζ
り
11

、‘
、3骨 5 Conclusion
3-6 Experimentα1 Section
3・7 Refe;γences
2
J
d
“,
AU
7/
ヴ/
00
4
•
h
Acknowledgement 83
References for the Thesis 84
.
,
•
、
‘. ..
111 、

22h'
ReactlO71 ofTrmlSttiol1 Metal Complexes Vla Silrcoll Hypervalel1t Structure
List 0/ Ahbreviations
TrimethyIsilyl, Me3Si TMS
diahF事Z433も
31
・
...,.
丁目fluoromethanesulfonate,OS02CF3 OTf
Ben勾,1,C6HSCH2 Bn
か hexy1, C6H J 3 n-Hex
cyclo-hexyl, C6H J J cyclo-Hex
~
? 2,5・norbomadienenbd
¥ Cyclopentadienyl,ポーCsHsCp 会民
'a・
b
咽.
PentamethylcyclopentadienyI,ポーCsMes
fac-2.1
L
CH3CN
List 0/ Conlplexes
Pyridine
No. of Compounds
Cp*
py
P(OMe)3 fac-or mer-2.2
P(OEt)3 fac骨 ormer-2.3
• P(OPh)3 fac-or mer-2.4
L
OC4fhL Ocv| 可 L
C o
fac
4
J-a-w
v,
P(NMeCH2CH2NMe )(OMe)
No.ofCompounds L
fac-or mer-2.5
cis-2.6
y
Oc,,..!、doc'10、Cハ
C '-/ 。
P(OMe)3 Clト ortrans・2.7
P(OPh)3 ClS-or trans-2.8
L
OCII ,~L, ,\\co oc'1、Co
L
PPh2(OMe) ClS-or trans・2.9
lV
L
oct,JaHdL OC〆1、Co
L mer
tγαns
L
oc,J-川 L
oc'γ、CハC '-/
O ClS

普
ES--s'
g詰P
Fe‘
ぷ、、VSiEb
。3.1
R氏
名~':> No. of Compounds R,R'
3.3
3.5
3.7
R=H,R'=Et
R = Me, R') = Me2Ph
RzH,R'=Ph
RJ:: g説。
_Fe 1"/ I、
O¥ J メてZN¥γSiR'~
Me2N ~
No. of Compounds R, R'
3.4
3.6
R=H,R'=Et
R = Me, R'3 = Me2Ph
No.ofCompounds [M] Rs
3.8 Fe Mes
L45 3.9 Fe fuMe
3.2 Fe Hs id Ml • 。cEC¥CH3 3.10 Fe fuI 。
3.11 Mo(CO) Mes
3.12 Mo(CO) Hs
宅設....Feぶ H
ハc' ¥、C'..
v _S よーR3
SI/ 問問e2
AU
一n一u一o-
nr一
m一日
M
o
-
-
U
1
U
C
一33
FI
一
O
一
O
一N一 一E
m
pA
一一一-一
R
R
z ピ:下u‘ } No.ofCompounds R
.. 3.15 R=Et •
-・L
3.16 R=Ph R3Si
v •

ReactlOn ofTransltlO17 Metal Complexes via Silzcoll Hypervale11.t Structure
.,.
-am引
azze--Z4省、.p'''b,‘.. ,,、z
,‘h
,,.,,ae・d
・
-h
手可、
容 4~

‘' ‘ 、
』
•
穐
4・•
Chapter 1. Generallnlroduchon
Chapter 1.
Geoeral Introduction ~
~
、
、

ReactlOll ofTrmlslt101l Metal Complexes Vla SzllCOll Hypervalenf Structure
Fd, ぷ,
n‘t fV
....

d
Gent'TaJ Ifltrvductlltn
(/1 〈
官、九f晶. ..
Otaptf'r 1
Cbapter 1.
General lotroduction
SF
,,s'崎
ha
,
‘
•

Reactzon 0/ TransitlOn Metalαmplexes vza Silicon Hypervalent Structure
1・1Prologue
Silicon situating just below carbon in the periodic table has pronounced c~acity
for the enlargement of the coordination sphere. Compounds of silicon with
coordination number greater than four have been known since the beginning of the 19th
cenωry, when Gay-Lussac 1・1and J. Davyt 2 first observed, independently, the formation
of the [SiF 6f・ ionand of the adduct of SiF 4 with ammonia. The formation and
structure of hypercoordinate siIicon compounds continue to be an area of lively
interest,l 3・15 which has been reviewed.1 6・114 ln recent year, interest in
hypercoordinate silicon compounds has grown considerably, because of their
unexpected reactivity. lnterest in this field started with studies of nucleophilic
activation at silicon. lt was shown that some reactions, such as racemization of
optically active silicon compounds, hydrolysis and alcholysis of tetracoordinate
chlorosilanes, are highly accelerated by nucleophiles (Scheme ト1).1 15 These studies
suggested that a pentacoordinate intermediate reacts with nucleophiles faster than does
the starting te甘acoordinatesilane. The enhanced reactivity of pentacoordinate si1icon
compounds in nucleophilic substitutions was demonstrated experimental1yl 16 and was
supported by calculations.1 17-18
ln contrast to the well喧 developed organosilicon chemistry concerning
H、'O-H
RL,!.,、川CI
R2寸l、R3
Nu
Hydrolysis .. R1R2R3SiOH
+ HCI
(陀tention)
-H20
R1R2R3SiCI + Nu .:;;司
e
n
a
--------e-o
r
o
---a hM
e
s--
陣
0
.
l
l
S
H1ぺ
--a
r
L
S
1
v
d
•••••
+
m
AU
Bl内
φ
c
h
k
が
旬
、
一
」
u
uぃ、
u
u
-
u
n
N11釘llN4LWNll釘llNi
九,
h-,m山
112
1
,2
a
R
R
R
R
d
白』
4E--a
n
---a AU
F-o
o
p』ra b』n
r
ve
弘M
SE
o
n
o
&t a
.u m
e
p』a
R
司且
噌且品』m
e
hu n--
QU
CI
R1". I ~ + Nu
R2~1i-R.j竜一一ー1 ・NuNu
+ Nu 、¥-Nu
racemiza討on
2

coordination ln of sllicon
Generallntroducttol1
of hypervalency role
Chapter 1
the strucれ汀es,hypervalent silicon
ln this thesis, 1 describe the chemis甘yof transition metals is much less investigated.
first example of trialkylhalosilane catalyst for geometncal isomerization of octahedral
0今Slwith compound sihcon hypervalent a where complexes metal 甘ansltlon
1 also describe unprecedented N・CNbond cleavage of interaction plays a crucial role.
ae弓Snw刷、友、J
・46veee£f
、h角叫・
、• 、
.・、
where the formation of a hypervalent silicon compound with cyanamide (R2N・CN)、
働、h
N今Siinteraction in a coordmation sphere of a transition metal is a trigger of the N・CN
, ‘ .
、、
bond activation.
lt might be pe口Inentto take a cursory look at examples of N今Siand 0今Si
,
、
interaction to afford pentacoordinate silicon compounds and summarize briefly findings J‘・
following the according to
about pentacoordinate sihcon compounds.
1-2 Preparation 0/ Pentacoordinate Silicon Compounds
prepared may be specles silicon Pentacoordinate ー、
general methods 1 12:
a or to ny
--、‘,J'EE-a
---A eq. ( e.g.) organosilane an to anlon of an addition By 、‘,J
---E-
JSE
‘、
、‘,f-EEA
噌
E'arzt
、
spirosilane (e.g.) eq. 1・2)119-120 to give an anionic pentacoordinate silicon complex.
PhnSI(OMe )4-n + MeO・[K.18・crown-6t(n = 1・3)
•
(1・2)
By inter-or in甘amolecularcoordination of a neutral donor to sihcon, giving
Et3NH+
[PhnSI(OMe)s・nnK,18・crown-6t
RO-SI 担・
"
+ MeOH + Et3N 2
s;to 0
(2)
a neutral or a cationic pentacoordlnate silicon complex, depending on the nature of the
3
substituents (e.g., Figure 1-1).1 21・126
司.
a・.

Reachon ofTranSlbon Metal Compleχes Vla SZlrC071 Hypervalent Structure
F__ F -... , Me2Nー"'Si-F
F__ F 、三,、N-Si-F
N
Me2N ""$iXYZ
.-
F: F
o S,-F
Arλ0)
kv
fsα
Figure 1-1. Oragenosilicon compounds dooated by a oeutral donor.
(3) By substitution of an organosilane: (a) by a bidentate ligand to give an
anloruc or a cationic pentacoordinate complex according to the nature of the bidentate
ligands (e.g., eq. 1-3)) 19 or (b) by triethanolamine or another trialkanolamine to give
sila仕anes,or by tris(2-aminoethyl)amine to give triazasilatranes (e.g., eq.ト4).120,127
Si(OR)4 + 2 "IRO-SiナO。 BunNH3+ + 3ROH (1-3)
郎 i問+ 3R'OH (1-4) ー・ー
R
1・3N→SiCoordination toαn Organosilane
Numerous neutral pentacoordinate siIico n compounds have been preparated. F or
example,intramolecular chelation of a donor atom to the silicon atom can be facilitated (Figure. 1-2).1 28
〉Si← NMe2 :::~i←NMe2
a b
Figure 1・2・Chelateligands 00 N→Si intramolecular coordination .
Variable tempera印reI H NMR studies 1 29 of monofunctional and bifunctional
4
、F

General J1ltroductt0l1
derivatives c, d and e showed two signals for diastereotopic N-methyl groups at low
Chapter 1.
The bond energies temperature出usindicating intramolecular coordination of N→Si.
were calculated to lie in the range 38.4-72 kJ mor1, the stability of the chelated form
19F NMR studies of depends on X in the order: 0 R く H< F, SF < OAc, Cl, Br.
ss'a'IJea--'
Resonance f1uorosilanes f established the apicophilicity of X related to fluorine.1 30
due to axial f1uorine is found at relatively low field, and an upfield shi合 indicates
The experiments showed fluorine to be more occupanon of an equatoria1 site.1 31
• 』
However, fluonne appears to be apicophi1ic tban H, OR and NR2, but less than C1.1 30
more apicophilic than chlorine in compound g. ) 32 司、
F CI u ヅN-ーSトーF
/ N
g
Me F -:".f
Me2Nー--SトーX
f
Me X -:"_f
Me2Nー--Sト-X
e
X = OR, H, F, SF, OAc, CI, Br
Me
Me2Nー-Si-X
d
Me H -:".f
Me2Nー--Si-X
c
, d
Figure 1・3.Pentacoordioation silane stabilized by N atom.
In 1970, Boer and van Remoortere determined, by X-ray crystallography, the
1・40→SiCoordination to an Organosilane
•
They found that one silicon atom is pentacoordinated structure of compound h.1 33
with a distorted trigonal bipyramidalal geome訂y,the axial sites being occupied by the
The coordinative 0→Si bond is longer (2.613 A) than the covalent two oxygen atoms.
0・Sibond (1.670 A).
Numerous compounds with Si-halogen bonds and 0→Si coordination have been
the that showed of i determination structure crystal X岬 rayThe described. I 34
Si(l )-Cl(1) bond at pentacoordinate silicon is 15 % longer (2.35 A) than the Si(2)-Cl(2)
The geometIγaround the silicon bond at tetracoordinate silicon (2.05 A) (Figure 1-4).
aωm is a slightly deformed trigonal bipyramidal with an 0→Si distance of 1.913 A, . •
which is much shorter than the O-+Si coordinative bond in h.
5

kv
‘a
・叩.‘‘‘
sep
験
Reactwn ofTransitioll Metalαmplexes Vla Sillcon Hypervalent Structure
1 749ベザe
c~J ふ5A,.
胸
Fm刊¥仰
向吋」
い
od
k
23引でpo_j込2A
Aハ
り
A
5
eq~リ日
目J;KME
¥-O夕、、Nr¥SIC
A
J
、〈∞
引
胸
p'
20¥fNS
M
F「
「
¥M
:srhsf
十戸川J
H
/胸
A
{
p
円。
h
Figure 1-4. Pentacoordination silane stabilized by 0 atom.
The fact that in these complexes the length of the coordinative bond depends on
the other equatorial ligand is particularly well il1ustrated for compounds j, k and 1
In j of the白rsttype Three types of compounds can be distinguished. (Figure 1 -4). 4官
compound, the coordinative O-Si distance is 2.395人whereasthe length of出eSi-F
bond is 1.652 A, which is only 0.1 A longer than an SトFbond for tetrahedral Si.I.35・
I36
The geometry around the silicon atom is that of a tetrahedron monocapped by the
In this case, the geome仕y
In the chloro compound k (second type), the 0→Si coordinative bond
is shorter than that in the firstザpecompound(e.g.,j).135
oxygen atom.
+tr‘24弘
《
LY--‘z‘拓t-E1ze、‘,aZ47
In the third type, around the silicon corresponds to a slightly distorted bipyramidal.
including a compound 1, the O-Si dl~tance (1.749 A) is much more shorter than that in k,
whereas the opposite X-Si bonds are very long (3.734 A for the Si-I distance in 1).135-1 37
The geome仕yaround the silicon atom in the derivative 1 is that of inverted (compared to
The changes in the O-Si the first type) tetrahedron monocapped by iodide ions.
lengths and in the g~ometries around the silicon atoms on going from j to k and 1 mimic
the course of SN2 substitution at silicon occurring with inversion of configuration. j
represents the initial attack of the oxygen at the silicon, k the transition state, and 1 the
final attack close to the “reaction product. ,,1 38
6

Generallntroduchon
Gay心 lssac、1.L.;Thenard, L. J. Memores des Physlque et de Chlmie de la Societe
Chapter 1.
1-5 References
-E・
E
・-
.•••
‘.
車事,守
aez'eet
味. 司、
、、
d'Arcueil1809、102,317.
Davy, J. Phil. Trans. Roy. Soc. London 1812, 102, 352. 1.2
(a) Corriu, R. J. P.; Guerin, C.~ Moreau, J. J. E. Top. Stereochem. 1984,15,43. (b) 1.3
Comu, R. J. P.; Guenn, C.~ Moreau, J. J. E. The Chemistry o/Organic Silicon
Compounds. Patai, S.釦 dRappoport, Z.; Eds. (John Wiley, Chichester, 1989), 、
Yamamura, M.; Kano, N.; Kawashima, T.1. Org. Chem. 2008, 73, 8244.
Chapter 4.
1.4
Dorsey, C. L.; Gabbai, F. P. Organometallics 2008, 27, 3065. 1.5
Tandura, S. N.; Voronkov, M. G.; Alekseev, N. V. Main Group Met. Chem. 1987,
10, 209.
Shk1over, V. E.; Struchkov, Y. T.; Voronkov, M. G. Russ. Chem. Rev. 1989, 58, 211.
1.6
1.7
Lukevics, E.; Pudova, 0.; Sturkovich, R. Molecu/ar Structure olOrganosilicon 1.8 P司
Compounds (Ellis Horwood, Chichester, 1989). ‘
Coπiu, R. J. P.; Young, J. C.刀leChemisnアザOrganlcSilicon Compounds, Patai, 1.9 • 、
S. And Rappoport, Z.; Eds (John Wiley, Chichester, 1989), Chapter 20.
1.10 Gel'mbol'dt, V. 0.; Ennan, A. A. Russ. Chem Reν. 1989, 58, 371.
1.11 Holmes, R. R. Chem. Reν. 1990, 90, 17.
1.12 Chuit, C.; Corriu, R. J. P.; Reye, C.; Young, 1. C. Chem. Rev. 1993,93,1371.
•
1.13 Chult, C.; Comu, R. 1. P.; Reye, C. Chemisl1ァザめ'pervalentCompounds, Akiba,
-同
K.; Eds (Wiley-VCH, 1999), 81.
1.14 K.ira, M.; Zhang, L. C. Chemist,アザHypervalentCompounds, Akiba, K.; Eds
(Wiley-VCH, 1999), 147.
1.15 (a) Corriu, R. J. P.; DabosI, G; Martineau, M. J. Organomet. Chem. 1978, 150,27.
(b) Corriu, R. J. P.; Dabosi, G.; Martineau, M. J. Organomel. Chem. 1978, 154, 33.
(c) Corriu, R. J. P.; Dabosi, G.; Martineau, M.J. Organomet. Chem. 1980,186,25.
1.16 Corriu, R. 1. P. J. Organomet. Chem. 1990,400,81.
1.17 Deiters, J. A.; Holmes, R. R. J. Am. Chem. Soc. 1990,112,7197.
1.18 Gordon, M. S.; Caπoll, M. T.; Davis, L. P.; Burggraf, • W. J. Phys. Chem. 1990, 94,
, 7
8125 .
. ‘
4・

Reaction ofTransttton MetaI Complexes vza SlLZCOl'l Hypervalen.t Structure
1.19 Holmes, R. R. Chem. Rev. 1996, 96, 927.
1.20 Frye, C. L. J Am. Chem. Soc. 1970, 92, 1205.
1.21 Corriu; R. 1. P.; Mazhar, M.; Poirier, M.; Royo, G J Organomet. Chem. 19お,306, @〉‘,.vh
‘,,ぜhU426企R1av-EEazg&V鍋F
、,‘‘dh'nya
『,,旬、屯aョ.,.
C5.
1.22 Klebe, G.; Hensen, K.; Fuess, H. Chem. Ber. 1983, 116, 3125.
1.23 (a) Corriu, R. J. P.: Royo, G.: de Saxe, A. J. Chem. Soc. Chem. Commun. 1980, 892.
(b) Boyer, J.; Breiere, C.; Ca凶, F.; Corriu, R. 1. P.; Kpoton, A.; PO凶er,M.;
Royo, G.; Young, 1. C. J Chem. Soc. Dalton Trans. 1989, 43.
1.24 Voronkov, M. G.; Frolov, Yu. L.; D'yakov, V M.; Chipanina, N. N.; Gubanova, L.
1.; Gavrilova, G. A.; Klyba, L. V; Aksamentova, T. N. J. Organomet. Chem. 1980,
201, 165.
1.25 Onan, K. D.; McPhail, A. T.; YoderョC.H.; Hil1yard, R. W. J Chem. Soc. Chem.
Commun. 1978,209.
1.26 Macharashvili, A. A.; Shk1over, V. E.; Struchkov, Yu. T.; Oleneva, G 1.; K.ramarova,
E. P.; Shipov, A. G.; Baukov, Y 1. J Chem. Soc. Chem. Commun. 1988, 683.
1.27 Verkade, 1. G. Coord Chem. Rev. 1994, 13ス233.
1.28 Jastrzebski, 1. T. B.H.; van Koten, G・Adv.Organomet. Chem. 1993, 35, 241.
1.29 Corriu, R. 1. P.; Royo, G.; de Saxce, A. J Chem. Soc. Chem. Commun. 1980, 892.
1.30 Corriu, R. 1. P.; Poirer, M., Royo, G. J Organomet・Chem.1982,233, 165.
1.31 Breliere, C.; Carre, F.; Corriu, R. 1. P.; de Saxce, A.; Poirier, M.; Royo, G. J.
Organomet. Chem. 1981, 205, Cl.
1.32 Klebe, G.; Hensen, K. J Chem. Soc. Dalton Trans. 1985, 5.
133Boer,F.p.;van Remoortere,F.p.l dm.Chem.Soc-1970392,801.
1.34 Onan, K. D.; McPhail, A. T.; Yoder, C. H.; Hillyard, R. W. J Chem. Soc. Chem.
Commun. 1978, 209.
1.35 Macharashvili, A. A.; Shklover, V E.; Struchkov, Y. T.; Oleneva, G. 1.: Kramarova_
E. P.; Sh帆 A G;BauKMJ l chem S0C Chm Commm:198368J- 「
1.36 Macha凶 li,A. A.; 畑町 V.E.;ωrnnik川 N.Y.;吋m,M 主
Struchkov, Y・T.;Baukov, Y・1.;Oleneva, G・1.;Kramarova, E. P.; Shipov, A・G・l
Organomet. Chem. 1989, 359, 13.
1.37 Macharashvili, A. A.; Shklover, V. E.; Struchkov, Y・T.;Baukov, Y 1.; Kramarova,
E PJ OIeneva,G.1.l OF2Gnomet.Chem.1987,327F167.
、.. ‘,",‘, .. e電E.
.
.
,.,
,tep--、aFFa,'tR4
8

)¥1
Ch伊ter1 Generallnlroduchon
h川J
伽
m一-,
1.38 Voro凶(ov,M. G.; Pestunovich, V. A.; Baukov, Y. 1. 0,宮anomel.Chem. USSR 1991,
4, 593.
aHU
,,,.aEa-
-
-
伽間町
内
、
19簡
を司
.,
~
f育、
t乱

Reacttoll ofTral1sitiol1 Metal Complexes VlQ Silico7t Hyperoale71t Structure
,.

Chapter 2. Geometncal IsomenzatlOn 01 Molybdenum CompLexes wlth Phosphltes Catalyzed by Neulral or Ulhonic Slllcon Compound
Chapter 2.
Geometrical Isomer包ationof Molybdenum Complexes with Phosphites Catalyzed
by Neutral or Cationic Silicon Compound

ReactlO71 ofTral151t1on Metal Complexes VLa StlrcOll HyperoaJent Strucfure
2-1 lntroduction
2-1-1 Silicon Catalyst in Chemical Reactions ,...・
Organosilicon reagents are widely used, especial1y for organic syntheslS, because
of unique reactivity making highly efficient and selective organic reactlons possibIe,
ready availability, and relatJvely low toxicity.2 )・22 1n particular, they have extensively
been used for carbon-carbon bond formation in the past three decades. And, several
synthetically valuable named reactions have been created such as出eMukaiyama aldoI
reaction wIth silyl enolates (Scheme 2・1/3-2 4, the HosomトSakuraireaction with
allylsilanes (Scheme 2_2)25・26,and the Hiyama coupling wlth alkenyl-, alkynyl-, and
arylsilanes (Scheme 2_3).27 1n these reactions, silicon compounds play important roles
with the aid of a Lewis acid or a transltion-metal complex, and are finaIly converted into
other si1icon compounds. 1n other words, these reactions require a stoI'chiometric
amount of silicon compounds.
9SiMe3 1) LewlS Aαd OH 。。
zムR3R1人
H+
R1'γw 2) desilylation R2 R2
desiJylation E
r R3 ...LA LA、O 5sh Mゆ ...... 0
,LA 0" i 、0。っ R~ 。R2
I -LA R1JGH MV
'ーH R1 R3 R1' y"R3 LA = Lewis Acid R2 R2
Scheme 2-1. Mukaiyama aldol reaction.
9Lewis Acid ~Jl +グ~SiMe3 こ
Rγ 、Rど' イハ
LA、、O
quench
R1 R1R2
Scheme 2-2. Hosomi-Sakurai reaction.
12
'
回
_1 -一_. -,
・・R ・一一・・ν., J司
一一--・-• J山守1:...・.一,
‘9・・一,_・
・ ~ I..... 一一_. E ・-" 一-.&WI!・
一・--.一一_ ... 一一一一-_.
"" ・・・・・・_.. 6司迦司
』動羽目
』

唱・.
‘
Chapter 2. Geometncal Isomenzahon 01 MoかbdenumComplexes wltl1 Phosphztes Catalyzed by Neutral or Cah011lc Szllcon Compound
A Pdo,ligand 4 内
R1-X + Rn~Si-R2 一 一一-..R'-RL 。 F-or base
R"3Si = (RO)3Si, R3・nFnSi,etc.
ヘマ 重R1-X
R1 F寸。 F 。
,
¥ __.-し2Pd、
/ R2SiY3
+
oxidative / X R2SiY 3 t E addition 3
~ t ra nsmet al ati on ‘
L2Pd ハa
‘
|R1什 ,R1/¥fw~ 巧圃眠
redu ctive ....__
L2Pd、2XSiY3elimination R
Scheme 2-3. Hiyama Coupling.
R2 H20
、ORHO'"
...._,...、... ( 0
Bn02CHN、、、て".........R2
R1 R1
45・90%, 72-88 % de
HO
Bn02CHN、、
R1 .R1
、ORO OR
Me3SIOTf Me3SiO
NHC02Bn
--司舟HV
Bn02CHN R1 .R1
Me3SiO
QSiMe3
h RVAR2
NHC02Bn ん•
Scheme 2・4.Me3SiOTf-catalyzed ring-opening.
13

JS、Avtra'J'aEleSEEL-L6ta‘tsF
、SEa-V4E4f-3eeJ‘avs!‘t='r,bb也、,ba--F
圃
R3SiB(OTf)4,
Reaction ofTral1srtLOI1 Metal Complexes Vla SillCOI1 Hypervalent Structure
R3SiOTf, reported: been have catalysts silicon-based Several
These catalysts can be described as R3SiB(OTf)3Cl, R3Si(S02F)2, and R3SiN(Tf)2・22
F or example, Kobayashi et al. containing R3St and are considered to be a Lewis acid.
benzyl of nng -openlng TMSOTf-catalyzed the reported
1,2・synhigh with enolate silyl a with (3圃 oxyte仕ahydropyran-2-yl)carbamates
They proposed the reaction mechanism involving diastereoselectivity (Scheme 2_4)28.
the coordination of Me3Si+ to the ring申 oxygenfollowed by ring-opening activation to
of a addition nucleophilic After the in termediate. 10n lmlDlum acyclic an form
仕imethylsilylatednucIeophile to the iminium proton, TMSOTf is regenerated by the
仕if1ateanion attack onto the trimethylsilyl group of the nucleophile.
silicon four由 coordinateneutral (a silane described report no contrast, ln
I t has been speculated that a compound) serving as a catalyst instead of a reagent.
In this chapter, 1 report the frrst silane is not a catalyst any more than an alkane is.
and related silane Cl, Br, 1) (X = example of a silane-catalyzed reactlon. Me3SiX
[MO(CO)3 {P(OR)3} 3] of lsomenzatlon facサnercatalyze compounds room at
temperan江e.
14
。叩pter2. Geomemcallsomenzatwn of Molybdenum Complexes wlth Phosphl加 Catalyzedby Neulral or Cahomc Srllcon Compound
、、、-JJ
、2-1-2 fac-mer Isomer包aitonof Phosphite Molybdenum Complexes
手、、、、.・
‘、
、...... .. ",,-
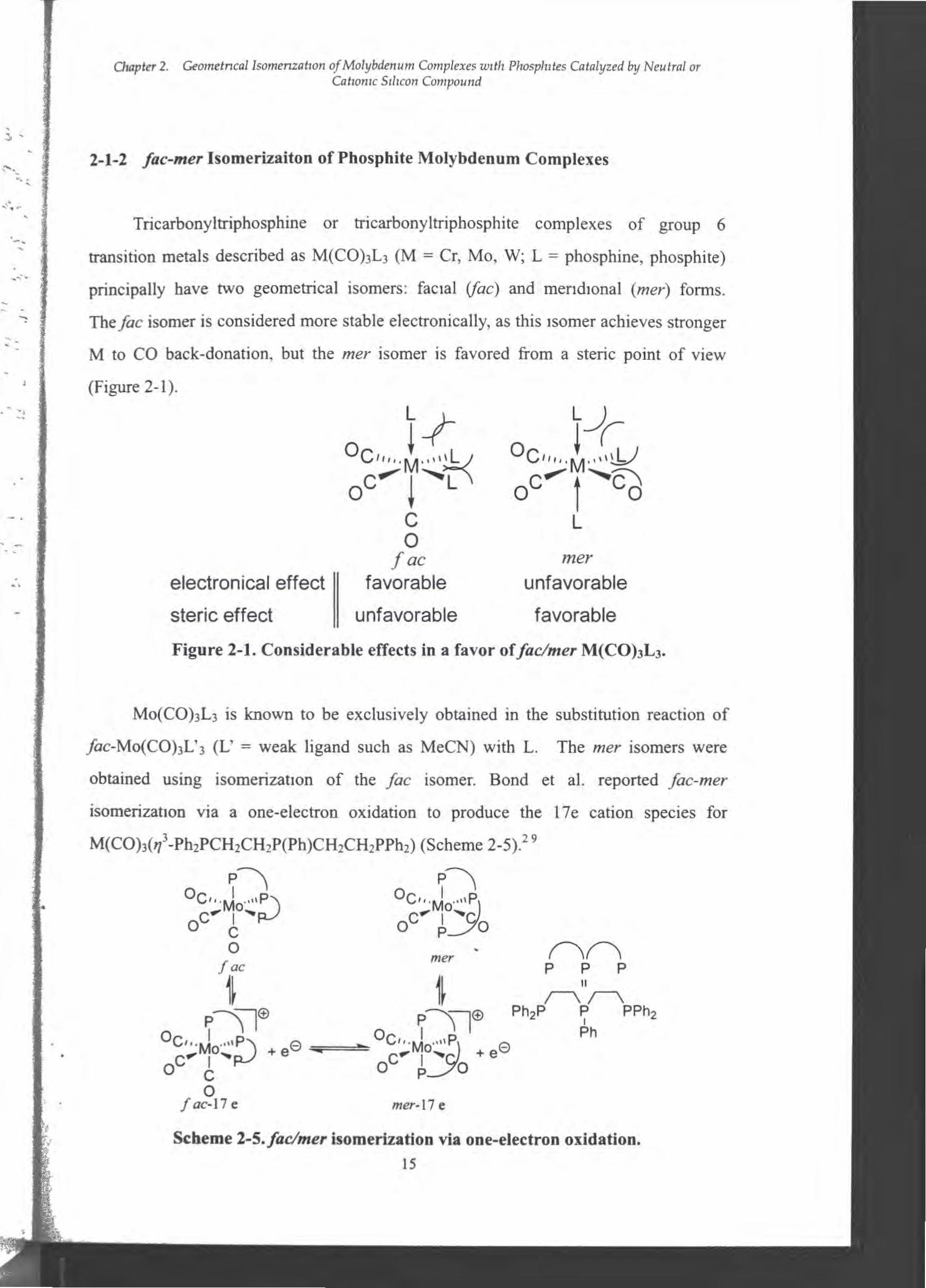
‘、Tricarbonyl仕iphosphine or tricarbonyltriphosphite complexes of group 6
transition metals described as M(CO)3L3 (M = Cr, Mo, W; L = phosphine, phosphite)
principal1y have two geometrical isomers: faclal (戸c)組 dmendlonal (mer) forms.
The fac isomer is considered more stable electronically, as this lsomer achieves stronger
M to CO back-donation, but the mer isomer is favored合oma steric point of view
、目 、崎、
、、・、、
‘』司、
、、、
司除
』 (Figure 2・1). -、、.‘. 、.
op iI V ユ:M
Ocv↓ c o
fac electronical effect 11 favorable
meγ
unfavorable
favorable
〆hGL ー •
、司F . 、
-a‘、
steric effect unfavorable
Fi伊 re2・1.Considerable effects in a favor offac/mer M(CO)3L3・
Mo(C0)3L3 is known to be excIusively obtained in the substitution reaction of
fac-Mo(CO)3L'3 (L' = weak ligand such as MeCN) with L. The mer isomers were
obtained using isomerizatlon of the fac isomer. Bond et al. reported fac-mer
isomerizatlon via a one-electron oxidation to produce the 1 7 e cation species for
M( CO )3(.,,3 -Ph2PCH2CH2P(Ph )CH2CH2PPh2) (Scheme 2・5).29
•
。c~"ïc o
fac
-て--,oP ¥ I
Or. I . ,~イ A'v' .二Moご・・ ) ...ρ匂
ocvl可 pノ ー
c o
fac・17e mer-17 e
c' O
• (¥(¥ P P P
mer
pp・-、
01'" .
コム問。ネコ +e8 0'"'
fー¥rー"¥f pph2
Ph
•
Scbeme 2...5. fac/mer isomerization via one-electron oxidation.
15

-句‘・1
、aE--va
,‘.‘.-'eVt・-,‘‘ete‘eza‘z‘‘4geh--t.,‘・・1・,町aJEhWEE-,‘z'u,守会
E1e
Reachol1 ojTrn71Srtl01l Metal Complexcs t'ra SIltcon Hyperva1cnt Structure
for JsOmenzatlon related the a1. et Nakazawa
bpy W; Mo, Cr, (M 一一
The reaction of the fac isomer with BF 3・OJit2gives
cOlllplex fac-M(bpy){P(NMeCH2CH2NMe)} -1, which tben
2,2' -bipyridine) (Scheme 2・6).2J 0
the cationic phosphenium
It does so because the phosphenium ligand shows s甘ongπisomerizes to the mer fo口n.
one-electron complete (not center metal electron置 deficientan to leading acidity,
/
¥
/
N4/
o-v
cpiN'
A
、
/
ociMlCO
h
九〆、1Nwd
寸+o 、 c,NI,,人‘けcO'"N〆丁、Cハ, P.、 〉--N N-
し_j
f ac-phosphenium complex
寸+
-ー
oxidation, but partial oxidation).
』'
司,
ι
F」o
qu
r「ロu
reported
fac-M(CO)3(bpy) {P(NMeCH2CH2NMe )(OMe)}
。、 cNI,,人川cO
"'N〆丁、Cハ~ _p, '-'
'Nぐ, 'OMe 仁,./N,
mer-phosphenium complex
Scheme 2-6・fac/merisomerization of a phosphenium complex.
been has lsomenzatJon fαc-mer Thermal for
fac-Mo( CO)3( '72 -dppe){ P( O'Pr)3 } 2 11 and by Howe 11 et al. for fac-M( CO)3 {P( 0 R)3 h (M
a1. et Rousche by reported
Howel1 et a1. performed also computatJonal calculation using the = Cr, Mo, W) 2 12.
lsomenzatlon fac・merthe proposed and program MM2 the
The fac-mer isomerization can be accomplished
for mechanism M(CO)3{P(OR)3}3 (Scheme 2-7).
conslsteng planes tongonal two of rotatJon by either and P(2)-P(3)ーC(l)of
or those consisting of P(2)ーC(l)ーC(3)and P(1)-C(2)-P(3)・P(l)ーC(2)-C(3)
these faces are rendered non-equivalent due to the oItentation ofthe P(OR)31igmds,the h
σb
u
0
・nT
rotatlon-energy profile is independent of the choice of face pairs.
16

F
, -
J‘h 遺
句、.‘.、『
‘h、
働、
『
. 、.
‘ -.
'‘ ‘・'‘
' ,-
-
αUlpter 2. Geometncal Isomenzahon 01 M,仰bdenumComplexes wlth Ph叩 hltesCatalyzed by Neutral or 白 tzomc5ilrco1'1 Compound
P(OR)3
OCI '.:.; ~:.:..' ¥ P(ORh 。c〆|、P(OR)3
C 。111
(1 )
;:l'EM・(2)1'(3)
fac
P(必か3)
cωつ1¥(2)C(1 )
octahedral S佐ucture
P(OR)3
OCIハムリP(OR)3c〆|、c。 。
P(OR)3
111
(1 )
(3) "M・け (3) (2)〆 1#(1)
mer
P .. (1 )
P(3)一一一C(1) ¥て MY
C(3).マゥーC(2)P(2)
octahedral structure
Scheme 2・7.fac/mer isomerization via a trigonalprism structure .
•
•
17

RcactlOl1 ofTra"sttwl1 Mctal ωmplexes Vla 5伽 071Hypc7"'1.mli!lIf Slructurc
2・2 fac・InerIsolller;zat;on Catalyzed by Me]SiCI
2・2..1 fac-n,er Isomerization of MO(CO)3{P(ORhh with Me)SiCI ....
The reaction of fac-[Mo(CO)J {P(OMe)3} 3] (jac-2.2) WJth 1 equiv of Me3SiCl in
CH2Ch at room temperature was examined. The reaction was monitored by measuring
the 31p{lH} NMR spec凶 m. A singlet at 164.8 ppm because of fac-2.2 decreased with
time, 1nstead a triplet at 167.1 ppm (JpP = 44.3 Hz) and a doublet at 175.3 ppm (Jpp =
41. 7 Hz) appeared. The two new slgnals could be a町ibuted to
mer-Mo(CO)3 {P(OMe)3} ~ (mer-2.3) 合om the couphng pattems. A食er 3 h,
equilibrium between fac・2.2and mer-2.2 was estabhshed wlth the ratlO of 1 : 3.4. No
fac-mer isomerization occurred at room temperature 1n CH2Cb wlthout Me3SiCl. N.ext,
the reactions with 0.5 equlv and 0.1 equlv of Me3SiCl were examlned. Table 2-1
(entries 1-3) shows that the final equihbrium positIon was tndependent of l.be amount
of Me3SiCl used貴 althoughit takes a longer time to reach the equilibrium when the
amount of Me3SiCl is reduced The equilibnum ratto resembles that obtained in
thermolysis by Howell et a1. (1 : J / 12. These results show that Me3S1CI actsぉ a
catalyst for fac-mer isomerization.
The corresponding isomerization catalyzed by Me3SiCl was observed for
triethylphosphite complex (fac-2.3), tnphenylphosphite complex ifac-2.4), and
diamino-substi削除dphosphite complex (fac-2.5) (entries 4-6)・ ln aIl cases, the 31 P
NMR spec仕aof the reaction mixture suggested the formation of the corresponding mer
isomers; fac・2.3:164.8 ppm (~); mer-2.3: 167.1 ppm (t, Jpp = 44.3 Hz, cis-PP), 175.3
ppm (d, Jpp = 41.7 Hz, trans-PP);fac・2.4:145.4 ppm (s); mer-2・4:148.8 ppm (しみ'p-
52.1 Hz, cおーPP),155・7ppm (d, Jpp = 52.1 Hz, trans-PP); fac・2.5:146.1 ppm (s);
mer-2.5: 143.9 ppm (d, Jpp = 46.9 Hz, cis-PP), 180.9 ppm (t, Jpp = 46.9 Hz, trans-PP)・
The isomerization ofβc-2・4to Iner-2・4was much slower (more than 72 h)出組出atof
fac-2・2to mer-2.2 (3 h), which might be attributed to a ste行ccause. The reaction of
fac・2.5is noteworthy. We found that the reaction of fac・2.5with Me3SiOTf involves
abs仕action of one OMe as an anion企om the phosphorus to give a cationic
18
"
、

Chapter 2. Geometncal Isomerizahon 01 Molybdenum Complexes wltlt Phosphztes Cαtalyzed by Neu tral or CatlOntC Szl1Con Compound
Table 2・1.Isomerization offac-Mo(CO)3{P(ORhh by Me3SiCl in CH2Ch at •
room temperature.
P(OR)3
Oc 'I'I..! ,1¥¥¥ P(ORh __ MO __
OC-- I、P(ORh
c o
P(ORh
0cthJ 川 P(ORhハfIIII"'"''';''、、ハ
0"'" I vO P(ORh
-、、
、(~
Me3SiCI ... 句
、 ~. ‘、
P(ORh ‘ ー、
、、. fac・2.2 P(OMeh ・mer-2.2
fac・2.3 P(OEth mer-2.3
fac・2.4 P(OPh h mer-2.4
fac・2.5P(内MeCH2CH2NMe)(OMe)mer-2.5
entry complex Me3SiCI (eq) time (h) fac: mer
fac・2.2 1 .0 3 1 : 3.4 .
2 fac・2.2 0.5 5 1 : 3.4 』
3 fac・2.2 0.1 8 1 : 3.4 ••
4 fac-2.3 1 .0 2 1 : 2.2 . . 量4 回
fac・2.4 1 : 1.3a 5 1.0 72
‘、. 竃 6 fac・2.5 1 .0 12 1 : 0.5
a Ihe i~o.meriza~~o ~s v~ry ~lo\y, a~9 the ~quiJiþriu!D is~n9t ?ttaj!)ed ~fter ].2 h .Th~ f~,!l ratio obtamed by usin.g Me3SiOTf is 1 : 30
-・ .. (see Chaptぽ 2-3-1).
~
-"
3
, ,
ausd
¥MM'
¥fNOovs
ぬ
/NKPいい5
α'~川、ィ
2
/川
lpi愉lco釦
ハいN(
九グ
f
〆
q
o
T1
0
時l
i
e
2
一
¥MM'
JQん
Jwkd
iIN~
川、.4.
{Vpi肋lco
j
N
'
/
ハ
い
σ'
O
C
LP
Me3SiOTf
、
-、
. ¥'
.. b h • Scheme 2・8.Generation of a cationic pbosphenium complex in tbe reaction with
Me3SiOTf. う
19

Reaction ofTransitioll Metal Complexes via SillCOn Hypervalent Structure
phosphenium complex, fac-[Mo(CO)3 {P(NMeCH2CH2NMe )(OMe)} 2 {P(NMeCH2)2} ]+
(Scheme 2_8),213 In con仕ast,based on 31p NMR monitoring, the reaction of fac・2.5
with Me3SiCl generated no cationic phosphenium complex, which suggests that the
pathway of the isomerization promoted by Me3SiC1 differs from that involving a
cationic phosphenium complex produced by Me3SiOTf,
20
, a
• ,
e
.. •
戸
F
L
,
"
v 、、
, a
‘

f:ィ
1jms 、、、lC・La、、
'‘ ~1 、‘、.
α1apter 2. GeometncallsomenzatlOn 01 Molybdenum Complexes wtth Phosphltes Catalyzed by Neutral or CatlOnlC Slllcon Compound
2・2・2 Possibility of fac-mer Isomer包ationwith Adventitious HCl
1n our experiments合yCH2Ch distilled from CaH2 was used. However, there is
a possibility that adventitious HCl合omMe3SiCl hydrolysis (eq. 2-1) could be
responsible for the observed isomerization. Thus, the 29Si NMR of Me3S1Cl in CH2Ch
was measured. N 0 signal other than a singlet at甘ibutableto Me3SiCl was observed,
indicating no silicon compound derived from Me3SiCl hydrolysis exist (less than 5%, if
釦 y).We observed出atthe isomerization of戸c・2.2is completed within 8 h in the
presence of 10 mol% of Me3SiC1. If 10% of the Me3SiCl produces adventitious HCl,
出enthe amount of HCl would be 1 mol% based on fac・2.2in the above conditions.
The CH2Ch solution of fac・2.2containing 1 mol% of HCl was prepared and monitored
by the 31 P NMR measurement and no isomerization was observed after 10 h. These
results clearly indicate that the fac-mer isomerization is catalyzed by Me3SiCl but not
by HCl.
Me3SiCI + H20 Me3SiOH + HCI (2・1)
•
21

React馴 o!TrnnsitlO'1Metal ω刈
Plausible Mechanism for fac・",erIsomerization Promoted by Me3SiCI
lsomenzatton the _fac-n1er for mechanJsm plausible a 2・9presen岱Scheme
2..2..3
The sihcon atom in Me3SiCl interacts with one oxygen in promoted by Me3SiCl.
of mteractJon An (α). structure hypervalent Si an fOffi1 to ligands P(OR)3
p}ausible because may be phosphJte lD oxygen an wlth silicon te廿acoordinated
formation of pentacoordlnated StliCOD compounds by coordinatJon of a carbonyl oxygen
of coordination the weakens 1.4). The interaction Sectton reported (see has been
P(OR)3.Me3S1Cl toward the central metal and makes the ligand bulky, thereby makes
the tngonalprism structure in Scheme 2・7possible leadlng to decrease the isomenzarion
The foIlowing dissociation of the energy barrier, which gives its mer isomer (P).
•
-Fea--.46
P(ORb
o CIIt..! ..¥¥¥P(ORh
0cvl、。P(OR)J
+Me3SiCI
P
斤ler
P(OR)J
0ch,j川側3
OC"'-γ、cI -0
Me ハ,/P(OR)2
Me‘三〆¥/、J¥R
ClMe
-Me3SiCf
Me3SiCl gives the mer isomer reproducing企eeMe1SlCL
Isomenzation
__Me Me¥¥いSi--
M~〆\CI
+Me3SiCI
P(ORb
OCI1,.. j川附3
o C""i。、P附 3
c o
P(ORh
OCI1,.. L.¥¥¥p附 3
c~'ï~、.. I "'~(ORh
8 む-R
fac
-Me3SiCI
α
Scheme 2-9・Proposedfac-mer isomerizaoon mechanism catalyzed by Me3SiCI・
22

Geometncal Isomenzation of Molybdenum Complexes wtth Phosp}utes Catalyzed by Neutral or ωtzomc SZfrCOl1 Compound
The 31p and 29Si NMR measurements of a CH2Cb solution contaJnJngfac-2.2 and
Observation of Pentacoordinate Silyl Complexes as Intermediates
Olapter 2,
2個2-4
t$tFeS22』句
Z
‘‘S5.
,
To obtaln some Me3SiCl showed no slgnals assignable to eitherαor P in Scheme 2・9.
evidence of a and Il, we dissolved fac-2.2 in Me3SiCl and measured the 31p and 29Si
In additlon to a The 31 P NMR is shown in Fig. 2・2,NMR spec甘aof the solution.
singlet at 166.7 ppm attributable to fac・2.2and a doublet at 176.2 ppm and a tnplet at
168,8 with Jpp = 41.7 Hz attributable to mer-2.2, five signals were observed clearly.
Among them, a triplet at 181.3 ppm (assignable toαP(3) from the molecular symmetry)
10
岡崎町。
;;j m叫
h川可
ぬ‘,、、‘,
ー
(lq拘
崎 01
~è~
and a doublet at 173.9 ppm (assignable toαP(l), (2)) wlth Jpp = 41,7 Hz are attnbutable to '7JllOG
The remaining three signals at 184.8 ppm (dd, JpP = 41.7 and 229.3 Hz, assignable α. 目白
to PP(3)), 174.3 ppm (dd, Jpp = 44.3 and 229.3 Hz, assignable to PP(I)), and 166.2 ppm (t,
The product ratio of aJll Jpp = 44.3 Hz, assignable to IlP(2)) are attributable to p.
estimated from signal intensity (1 : 6) was different合omthat of fac-2.2/mer-2.2 (1 :
NMR 29Si The is thermodynamically determined. that the latter indlcating 3.4),
showed two singlets at 6.62 ppm and 6.53 ppm in addltion to a strong slnglet at 29.8
Coordination of nitrogen or oxygen to sihcon ppm attributable to the so]vent Me3S1C1.
to fonn a pentacoordinate bond has been shown to produce a s汀ongshielding effect of
Kummer et a1. reported the 29Si NMR chemical shift at the silicon chemical shin-214
11.1 ppm in CD2Ch for an Si hypervalent compound with Cl and N in apical positions
The 13C and two Me groups and one CH2 group in equatorial positions (Chart 2・1).215
Two lumps was also measured. NMR of the Me3SiCl solution containing fac・2.2
consisting of several signals were observed at 213.9-214.1 ppm and 218.0-218.2 ppm.
These signals are attributable to tenninal carbonyl ligands, indicating that Me3SiCl does
Therefore, not have an interaction with an oxygen atom in tenninal carbonyl ligand.
E4,eaaiwav
t
d
守‘,生eq.
4、te-e予
our observations suggest the formation of hypervalent Si compounds.
23
J E
叶
L'
,

ー,
μVA守谷主密集出L-
• ,
Reaction ofTransztion Metal Complexes Vla SiliCOl1 Hyperoalent Structure
(;,) L~_
怜, Chart 2-1. 29Si NMR spectrum of pentacoordinate silicom compound. 下'
‘
s ,
"',
F
個、.、財
• •
•
,d『
.
J
,4444S之、,
K2)
示。)
JU~い」ん???小九J叫仰げ vf ¥¥'-If恥いw
18.0 lふ 18bo l弘 l完olJ;5 1Jい・・1L'沿m
P(1 )
OC111,a J 川 向
。C"~O、cI ""0
Me ハ/P(3)
Me‘三〆¥,/ー1,R
CI' 'Me P
fac
ふ
rm2)
点
斤7erA
e
M
R
/
k
α
mpャ、少
η
川、
Maw
P11附llco
cグ。
o
A3)
α
Figure 2・2・31PNMR spectrum of a Me3SiCl solution at room temperature in
Which fac-2.2 is dissolved.
24

,
Chapter 2. Geometncallsomerizahol1 of Molybdenum Complexes wlth Phosphztes Catalyzed by Neutral or Caho7UC Szllcon Compound
2・2・5 Supporting Results to the Reaction Mechanism
ln the previous section, 1 proposed that an interaction of Si 1n Me3SiCl with 0 in
P(OR)3 causes 出efac-mer isomerization of Mo(C0)3(phosphite)3・ Next,the
isomerization of MO(CO)3(Phosphine)3 (phosphine = PEt3, BUn3) having no OR group
on a coordinating phosphorus was examined. No fac-mer isomerization was
con白nned(Scheme 2-10) suggesting that a phosphite oxygen plays a crucial role in
3
・eed‘.. 、,b
,,,‘,,ra‘
3
3
R
R
p
p
u
R
川
、
品
位
plIM--CO回目
u
h
'
=
a
'
F
.
,
M
C
C
R
m
o
o
o
r
e
m
p」z
fId
Me3SiCI no reaction
Scheme 2・10.Treatment of Mo(CO)J(phosphine)3 with Me3SiCI.
To elucidate whether fac-mer isomerization involves a phosphite dissociation
process or not, a crossover experiment was performed. Addition of Me3SiCl to a
CH2Ch solution containing both fac・2.2andfac・2.3yielded mer-2.2 and mer-2.3, but
form neither phosphite products exchange such as fac-
事 mer-[Mo(CO)3 {P(OMe)3}2 {P(OEt)3}], nor OR exchange products such as fac-or
mer-[Mo(CO)3{P(OMe)3}2{P(OMe)2(OEt)}] (Scheme 2-11). These results show
intramolecular isomerization, which is consistent with the mechanism shown in Scheme
2-9. 司邑
• observed
f~c・2.2 mer-2.2 今
J
司
d
F』広』
0
0
ゐ
p町
ぽ川、
μ
町・
lル肌
tlCOル
h
d
F
f
cc
。。+ し川、
n
e
e
M
M
h
ρρ
e
p
p
川
川
、
口
町・
l愉
lcop
h
,
れ
cc
o
o
Me3SICI -fac・2.3 mer-2.3
)
3
芯
日
制
,
d
O
M
vmρ
o
h
e
p
c
e
釧
川
、
側
,, .. 、
‘,d
,,,.、
plIM--P
h
山〆
FU
、J
o
d
e
d
-
M
ゆ一
h
ρ
0
1
門一
e
p
C決
問
一
側
川
、
何
-EME
・E
・-,,EE
‘、
、冒dF
a
,,.‘、
川U
一plMlp
φt-
,
O-''
n-cy
o
d
Scbeme 2・11.Reaction of fac・2.2and fac-2.3 with Me3SiCI.
2S
or

drゆ内、,
AeL-f
.、,A
『"、
for
RCQct701l ofTrrmsltrotl Metal Complexe5 VIQ S,hC011 Hypcruale71t Structure
Reaction of fac・MO(CO)3(phosphite)3with group 14 substrates
lsomenzatlon promote fac-mer can that Me3SiCl shown it has been
2-2-6
As ,
for fac-2.2. exammed was silanes other of actlVlty fac-[Mo(CO)3(phosphite )3],
Me2SiCb, MeSiCh, and SiC14 promoted the isomerizatton, but some by-products were
di chlorophosphite, monochlorophosphi旬、that considered formed. also or
仕ichlorophosphitecomplexes were generated in the ligand exchange between OR and
Such ligand exchange was already reported.2 16 CI via hypervalent silicon compounds.
For example, C引 ucleophilesreact with acetals in the presence of Me3SiX with the
ln the reactlon mechanism, the pentacoordinate replacement of the alkoxy group.
R1, /OR3
R2〆 "'+Nu-M
X
Scheme 2-12・ReactioDof aD acetal with a nucleopbile in the presence of Me3SiX.
ee--,.‘,‘,
-F'
•
R1 X "' + )-0
R2K3
R30SiMe3 :;¥oシ〆
ー:;¥073
silicon compound induces the ligand exchange (Scheme 2-12).
R'、 ノOR3
〆グーγX SIMe3
¥
,園町・・
i
q
e
,P円
J
M
O+013
〉
X
R
R
-MX
Me3SIX
Nu-M 祖国岡
R1.... ノ
OR3
R2~ "OR3
.,晶B
・・・,‘,
In contrast, SiMe4, with no chloride substituent, did Dot promote isomerization at Therefore, among the MenSiC4・n(n = 0-4) series, Me3SiCl is出ebest promoter,
al1.
合omits superior hypervalent might stem which
elec仕onegative同 osubstituents (Cl and 0 from phosphite) in apical positions and three elec仕onreleasing
arrangement:
groups (three Me groups) in equatotiaI positions.
Si, Ge, Sn). Table 2-2 shows the activity of R3EX (E = Bromosllane and
The
‘=
iodosilane (entries 2, 3) show better activity than that of chlor・osilane(entry 1)・
26

Geometrical Isomenzation of Molybdenum Complexes wzth Phosphztes Catalyzed by NeutraJ or ωt10mc Sil1con Compoul1d
order is parallel to elec仕ondensity on the central Si reflected by 29Si NMR chemical
Ozapter 2.
The Ph3SiCl (entry shift (Me3SiC1: 35.5 ppm, Me3SiBr: 26.5 ppm, Me3SiI: 18.5 ppm).
of the bulky substituents. 4) does not promote isomerization, presumably because -‘ .
Slow isomerization was observed in the reaction with (EtO)3SiC1 (entry 5), which might 込J、‘・・
be attributable to the weak electron releasing ability of an OEt group compared to that J
j
、‘eミ
A similar fac-mer ratio was obtained for entries 1, 2, 3, and 5, of the Me group.
meaning白紙 theratio is determined thermodynamically and is unaffected by the added
till
-h
Chlorogermane and chlorotin (entries 6 and 7) shows worse activity than that silane. 、‘
order of the 14 group the electrophilic order is parallel to ηle of chlorosilane. !
!
、、、
Table 2-2. Isomerization offac・2.2with halides.8
elements.2 ) 7 -‘、
fac: mer time (h) R3SiX entry :lj
1 : 3.4
1 : 3.6
3
く 0.1
Me3SiCI
Me3SiBr 2
1 : 3.7 < 0.1 Me3Sil 3
no reaction
1 : 3.4
25
25
Ph3SiCI
(EtO)3SiCI
4
5
1 : 3.4 25 Me3GeCI 6
1 : 3.4 3 days Me3SnCI 7
afac・2.2was treated with 1 equiv of R3EX (E = Si, Ge, Sn) in CH2C12 at room temperature.
‘・
27
JEise--i
i

94号室毒要港義H守也事
二
Reacho77 01 Trmls, tlCJ11 Mefalαmplcxes vra 5,I,c071 Hypervalcn f Structurc
Isomerization Catalyzed by Q Lewis Acids 2-3
fac-Iner Isomerization of Mo(CO)) {P(ORh}J with a Lewis Acid 2-3・1
neutral
...
by tbe of Mo(CO)3 {P(OR)3}3 1 discussed the fac-n1er isomerization J
川
-Tyh何
@
M
V
,.Slmilar isomerizations were silicOD compound, e.g. Me)SiCl, in the previous chapters.
Complexfac・2.2was also examined by a cationic silicon compound, e.g. Me3SiOTf. 尚子、p
dequimolar amount of Me3SiOTf was added an 組 dCH2Ch m dissolved
f
‘.4主g-dee--
room
Three
at
temperature and the reactloD was mODJtored by the 31p NMR measurement.
The observed. me,..・.2.2and fac・2.2to attributed were which signals were
isomerization reached at an equibrium after 1.5 h and the the jおか2.2: mer.骨 2.2ratio was s'
・‘
The reactions Next question is whether Me3SiOTf works as a catalyst or not. 1 : 3.4.
offac・2.2with 0.5 and 0.1 equivaJent of Me3S10Tf revealed that the final equilibrium
. position did not depend 011 the amount of Me3SiOTf used although it took a longer time
to reach the equilibrium when the amount of Me3SiOTfwas reduced.
Reactions ofj必 2.3andfac・2.4with Me3SJOTf were a1so examined in CH2Ch at
1n the 31 P NlV1R spec汀a,new t¥νo slgnals asslgnable to mer-2.3 room temperature.
were observed in the reaction of fac・2.3,and new sImilar signals attributable to mer,岨 2.4
equilibrium .角c-merratios The of fac・2.4.reactlon the observed in were were
independent of the amount of Me3SiOTf used, showing that Me3SiOTf serves as a
The results together with those forβc・2.2are shown in Table 2-3. catalyst. ηle
equilibriurn fac-mer ratios are quite dependent on the kind of the phosphite ligand. It
should be noted that these values are equal to those for the isomerization promoted by
Me3SiX (X = CI, Br, 1) (Section 2-2・5),and the value for fac-2.2 is similar to白紙
Therefore, it can be said that the values are derived企omthe repo此edby Howelf 12.
thermodynamic stability between the fac and mer i mers, not from the stability of the
intermediates created from a Mo complex and a catalyst (presumably Me3St, vide in合a).
catalyzed by MeOTf as The corresponding isomerization acid was
Table 2-4 shows that MeOTf can work as a catalyst
in the jbc-mu isomerization,although it takes a longer time to reach the equiblimn-
Lewis a
28
examined for fac-2.2 and fac-2.4.

、.
. 険
-、、、
‘
e
, .
•
αlIlPtl許 2. Ge仰側1符e的 Cω'011ω叩mηlen:::1
Cαtzo】HηnrηtκCS111C01η1 Coηmpound
The isomerization of fac-2.4 by MeOTf demands a longer reaction period than
Me)SiOTf and Me3SiCl. The results may be rationalized by the lower oxophilicity for
Mぜthanfor Me1Si-+.
Table 2・3.Isomerization of Mo(CO)J{P(OR)Jh by Me3SiOTf.a, b
P(OR)3 P(ORb
OCtll,. !川¥¥¥P(OR)3
O C"了、I .....P(ORb
TMSOTf a
ー咽司ーー
OCtll,. L..¥¥¥¥¥P側 3
OC引-、CO
P(ORb c o
P(ORb fac:庁1erc
P{OMe)3 fac・2.2 mer-2.2 1:3.4
P(OEtb fac・2.3 mer-2.3 1 : 2.2
P(OPhh fac・2.4 mer・2.4 1 : 30
a 1.0,0.5 and 0.1 equvalents based on the fac complex were used. b Reactlon conditions: room temperature, 0.1 M CH2CI2 solution.
C f ac:mer equilibnum ratio after completion of the isomerization.
Table 2-4. lsomerization of Mo(CO)J{P(OR)3h by R'OTf(R' = Me, Me3Si).a. b
P(OR)3 P(OR)3
I . _,__, 0,... . I OCI11,.! _.川町ORh LewM~d OCIII , ~ln. ,,\\\\P(ORh C"γ、十 C"了、cI .....P(OR)3 O¥.,"" I ~CO
C P(ORb 。∞mplex Lewis aCld reaction time fac: mer
fac・2.2 0.5 eq. Me3SiOTf th 1 : 3.4
fac・2.2 0.5 eq. MeOTf 4 days 1 : 3.4
fac・2.4 1.0 eq. Me3SiOTf 0.5 h 1 : 30
fac・2.4 1.0 eq. MeOTf 4 days 1 : 0.06b
a Reaction conditions: room temperature, 0.1 M CH2CI2 solution.
b The isomerization is very slow, and the equilibrium is not attained d蛤r4 days.
29

;bFvsd腸,JSE一台J,〆
Reactiol1 ofTransltion Metal Compleχes Vla Sillcon Hypervalent Structure
Structure of mer圃 2.42-3-2
Complex mer-2.4 could be isolated from the reaction mixture of fac-2.4 and
A食era treatment of fac・2.4with TMSOTf in CH2Ch, a white〆powderTMSOTf.
with washed was 30) mer-2.4 and of fac・2.4mlxture a IS which
hexane/CH2Ch/benzene (100/1/ 1) solution many times to obtain the pure complex
several Although yield. 53% ln mer-2.4・0.5CH2Ch・0.5C6H6as formulated
mer-M(CO)3L3 (M = Cr, Mo, W) type complexes have been reported, this is the釘rst
The structure was confirmed by preparation and isolation of mer-M(CO)3 {P(OPh)3}3・
The ORTEP drawing is depicted in Figure 2-3 and the crystal data the X -ray analysis.
among the first example IS This X-ray s仕uctureare summarized in Table 2-5.
The X-ray structure of mer-M(CO)3(tertiary phosphorus compound)3 type complexes.
beDNeen fac-and The structural comparison fac・2.4was reported previously2 18.
The bond distance of mer-Mo(CO)3 {P(OPh)3}3 revealed some interesting points.
-e
Mo-C2 for mer-2.4 (2.017 A) is clearly shorter than those of Mo-Cl (2.041 A) and
As C202 ligand is trans to P(OPh)3, the CO ligand can Mo・C3(2.041 A) (Table 2-6).
get more 1t-back donation企omthe central metal than C 1 01 and C303 ligands which
The mean Mo-C bond distance for fac・2.4(1.986 A) is shorter are mutually trans.
than that for mer-2.4 (2.033 A), reasonably understood by greaterル backdonation form
Another the Mo to the CO Iigands for fac・2.4because of the trans P(OPh)3 ligand.
[目lnteresting point is that the mean Mo-P bond distance for fac・2.4(2.435 A) is longer
The difference may stem企omthe steric repulsion than that for mer-2.4 (2.417 A).
f{IJ
約HUU
《EE
、
t-b,‘
•• {
¥
。
30
beween P(OPh)3 ligands infac-2.4.

~
4
4
αapter 2. Geometrtcallsomenzahon of Molybdenum Complexes wrth Phosphztes Catalyzed by Neutral or Catzonzc Sllzcon Compound
、.、帳、3
、,,a,,
aphn
Figure 2・3.ORTEP drawing of mer-2.4・0.5CH2Ch・0.5C6H6(50010 probability
el1ipsoids) showing the numbering system.
.、一、‘4f(~
~' ~ 、e n8
:ご~\
、h
‘J ( 14
-KJ
(I}
tプき』
-a、‘ー、‘
, ~
『、
~ ~.
-.
. 』、 ~ }o
, 01 ¥1
{ ~耳}
~, ι
l ~
'寵 Table 2-5. Crystal data for mer-2.4. •
, Empirical formula Cω 50~9C岱!fOÜ I2P3 lv,A3 5525.4(4) a
-唱 • F ormula weight 1192.30 Z 4
Crystal system monocliruc μ,cml 4.36
Crystal size(mm3) 0.45 x 0.35 x 0.15 Dωω, g!cm3 1.433
Temperature CC) -70.0 No. of unique ret1ections 41908
Space group P2dn (No. 14) No. of llsed ret1ections 12498
αA 13.7319(5) I No. of variables 712
b, A 18.2658(7) IR 0.051 J
c. A 22.1401(9) Rw 0.111
βdeg 95.744(2) GOF 1.06
31

a
,、,‘、gmF
pbv芸
er--
~
ReactlOl1 ofTrmlsltlOIl Metal Complexes VUl 5'[lCO'1 Hypervalent Structure
Table 2-6. Selected bond distance (A) and angJes (deg) for mer-2.4.
2.042(1 ) Mol-Cl
Bond Distances
2.4320(4) Mol-PI
2.017(2) Mol-C2 2.3792(4) 恥10トP2
2.041(1)
91.50(4)
Mol-C3
P2-Mol-C3
BondAngles
2.4390(4)
174.25(1)
Mol骨 P3
PI-Mol-P2
92.30(4) P3-Mol-Cl 89.56(1) PI-Mol-P3
175.91(4) P3-Mol-C2 83.08(4) PI-Mol由 Cl
92.38(4) P3・Mol-C394.53(4) PI-Mol目 C2
CI-Mol-C2 、88.43(6)
174.58(6) CI-MoI同 C3
94.23(4)
89.76(1 )
Pl-恥101-C3
P2-Mol-P3 ' h
87.09(6) C2-Mol-C3 91.24(4) P2-Mol-Cl
e
P2-Mol-C2
32
86.21(4)

chap信T2. Geometncallsomer印刷ofMoゆ伽lI1nComplexes wltlt Phosp111郎 CatalyzedbyNeutral or Catt01JlC Sl1zcoll Compound
2・3・3 Two Types of Reactions of a Pbosphite Complex with a Lewis Acid
...... ・・
In Section 2・3・1.1 described fac-mer isomerization for molybdenunl complex
bearing phosphlte (P(OMe)3) in the reaction with Me1SiOTf. It has been reported that
another type react10n takes place for transition metal complexes bearing
diamino-substttuted phosphite(s): fonnatton of a cationic phosphenium complex by OR
組 ionabs甘actionas ShOWD io eq. 2-2, Scheme 2-6, and Scheme 2・8. Transition metal
complexes with a phosphenium hgand have a町 actedconsiderable attention because a
cationic phospheniurn (PR2) is isolobal with a singlet carbene, silylene, and the heavier
~ 19-~ 13 congenem.
Phosphenium complex fonnation has been reported for many transltion metal 4
bearing a diamlno-substltuted phosphite; complexes
224-230 M(bpy)(CO)} {PσωleCH2CH2NMe)(OR)}
M(dppe)(CO)3 {P(NMeCH2C民自Me)(OR)}215,
M(bpy)(C0)2{P(NMeCH2CH2NMe)(OR)}22 26 229・23O,
M(CO)3 {P(NMeCH:!CHAMe)(OR))3213,
CpM(COh(ER)) {P(NMeCH2CH2NMe)(OR)} (M
M(CO)4 {P(NMeCH2CH2内Me)(OR)}/I1,
W)23ト234, and Cr, Mo,
CpM(CO)(ER3){P(NMeCH2CH2NMe)(OR)} (M = Fe, Ru) (ER3 = CH3, SiMe3, GeMe3,
SnMe3i 13. 228, 235-240 for reactlons Systematlc researches
Mo(bpy)(CO)J{PXY(OR)} with BF3'OEt2 revealed the effect ofthe substituents (X, Y)
00 the stability of cationic phosphenium cornplexes; the stabihty increases with
increasing the number of amlno substituents on the phosphenium phosphorus.2 27 ... ・
Y
。
¥ I +
N
(2・2)Lewis acid ーt
-OR-
33
,
of

Reactwl1 ofTrallsztion Metal C0111plexes via Siltcon Hyperoalent S伽 C仰向
2-3-4 Plausible Isomer包ationMechanism of fac-mer Isomel包ationby Me3SiOTf
Regarding isomerization of MO(CO)3{P(OR)3}3 promoted by Me3SiOTf, two
mechanisms are conceivable: mechanisms via a phosphenium complex and via a TMS+
adduct. The mechanism via a phosphenium complex is shown in Scheme 2-13. As
transition-metal complexes bearing a diamino-substituted phosphite have been reported
to react with a Lewis acid to give cationic phosphenium complexes by OR-abstraction
as shown in eq 2・2,a similar OR-abstraction may take place in the reaction of
MO(CO)3 {P(OR)3}3 with TMSOTf to produce a cationic phosphenium complex ifac' in
Scheme 2-14). Then, isomerization企omfac'to mer' is expected to take place. The
similar isomerization has been reported previously (Scheme 2-6), where the driving
force of the fac-mer isomerization is thought ωbe the gain of more 1トbackdonation for
白ephosphenium ligand. The reaction of mer' with Me3SiOR formed would give
mer-Mo(CO)3 {P(OR)3}3 with regenerabon of TMSOTf. However, this catalytic cycle
seems not plausible based on the following observations. (i) A complex having a
phosphenium ligand was not detected in the reaction of MO(CO)3 {P(OR)3}3 with
Me3SiOTf. (ii) After the仕eatmentof faト 2.4with 1 equiv of Me3SiOTf in the
presence of 1 equiv of Me3SiOMe in CH2Ch, the 31p NMR spec仕aof tbe reaction
mixture were measured and fac-2.4 and mer-2.4 were detected but fac-and
mer-Mo(CO)3{P(OPh)3}2{P(OPh)2(OMe)} were not detected at all. This indicates that
the reaction of mer' with Me3SiOR to give nler in Scheme 2・13does not proceed.
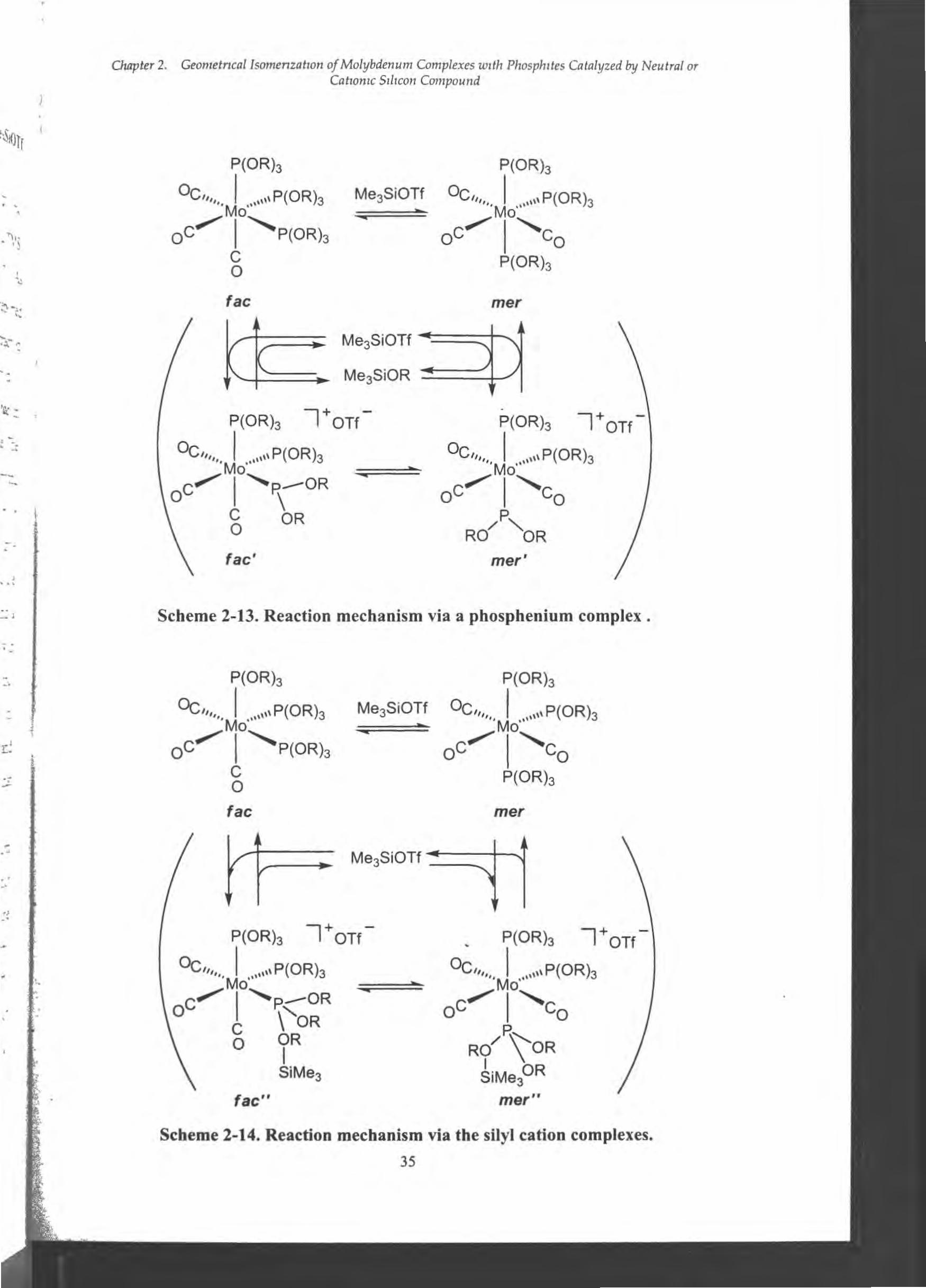
The other isomerization mechanism is shown in Scheme 2・14. τhesilicon atom
in Me3St interacts with one oxygen in P(OR)3 ligands to form fac", but does not
abstract the OR group as an anion. The interaction weakens the coordination of the
P(OR)3(SiMe3) ligand toward the central metal and makes the ligand bulky, thereby
decreasing the isomerization energy barrier to give its mer isomer (mer").
Dissociation of TMS+企omlner" gives mer-Mo(CO)3 {P(OR)3} 3 with regeneration of
Me3SiOTf. This isomerization mechanism is similar to that by Me3SiX (X = Cl, Br, 1)
(Section 2-2-3).
34
,
•
• ぜ
Chapter 2. Geonzetrzcal Isomenzatlon 01 Molybdenum Complexes wlth Phosphltes Catalyzed by Neutral or CatJOnlC Srllcon Compound
、
吻4
P(ORh
0c,hj川 P(ORh
F〆〆]o、、OC- I ""P(ORh
c o
、• 、
d、
子"、• I、‘'
“ 』、
~引、、
司‘、4‘_.... 門
、
a
-・
s定こ 宅
fac
P(ORh
Me3SiOTf OChhJ け川 P(ORh
OC〆)。¥co
P(ORh
mer
W
H
R
O
ゆ
a
s'
内
O
a
F
4
3
J
F
Q
M
M
M
T8
0
+
コ
、
寸
mho
-
orv庁
、npp¥、〈
保
川
、
円ーー肋llco
、引〆q
d
』, • a 、 .、
-・.、-h 、
主、、、
fac'
T
O
+
寸
町ρ
o
h
p
c
t
阿
川
¥
¥
O
町||附||久
、ヘ〆
w
q
c
。mer'
Scheme 2・13.Reaction mechanism via a phosphe凶umcomplex.
3
しM
れ
V
R
αρ
3
町、
P
m
J
¥
c
町||九州
llco向
句〆
T'
O
+
ミ寸
O
O
R
句
αJ、OR
M
1n
町
恥
1101s
侃
川
、
円ーl附
|iCO
句〆
ー‘ i
ー. •
-1
、L~
-ー. . _.
F d .
-a
,降、
.; ‘・
-‘' -、
、、、
fac"
ρ
o
h
p
c
h
阿
川
¥
保
,,EE1
、J
J
・-1
PIM--P
札
/c
o
c
o
mer
Me3SiOTf
~ P(ORh 寸+OTf-
OC,ヘj 川、P(ORh
戸〆〆]o、、《0""' I しO
ザ、OR
~:.._ OR SiMe3
mer'f
Scbeme 2-14. Reaction mechanism via the silyl catioD complexes.
35

Reachol'l o!TrallSrtlOn Metal Complexes Vla 511ic01l Hypervalel1.t Structure
There is a possibility that dissociation of one of the phosphites in
MO(CO)3 {P(OR)3} 3 induces the isomerization and Me3SiOTf promotes the dissociation.
To check the possibility, a crossover experiment was cooducted. Both fac.-2.2 and
fac-2.3 were dissolved in CH2Ch, Me3SiOTf was added, and the products were
estimated合om白e31 P NMR spectra of the resulting CH2Ch solution. Signals
assignable to mer-2.2 and n,er-2.3, in addition to fac・2.2andfac・2.3were observed, but
those due to phosphite exchange products such as fac- or
mer-Mo(CO)3 {P(OMe)3}2 {P(OEt)3} and OR exchange products such as fac- or
mer-Mo(CO)3 {P(OMe)3}2 {P(OMe)2(OEt)} were not detected. These results strongly
suggest that neither phosphite dissociation nor OR-abstraction shown in Scheme 2-13 is
involved in the fac-mer isomerization. Therefore, we proposed白eisomerization
mechanism shown in Scheme 2-14, though the intermediates have oot been observed.
36
•
、
守‘
..
‘・
司v
内
肉、
.... 、A

、、、f
‘ 、 ‘
、
“C 、
、、
、向
、、、.
、.
晴、
αapteT 2. Geo側m隠附et的T門rcωalIωS却omen氾za仙tr削OCatれ10m口tκC5111κCω01ηI CoωJ川1ηlpOUω4υyηldf
2・3-5 cis-trans Isomerization of MO(CO)4(phosphite)2 by Me3SiOTf
ln additioD to the isomerization of fac-Mo(CO)3 {P(OR)3 h promoted by
Me3SiOTf, the isomerization of ci~トMO(CO)4{P(OR)3 } 2 was also lnvestigated. The
results are shown in Table 2・7. The cis-trans isomerization occurred in the presence of
Me3SiOTf and did not in the absence of Me3SiOTf for cis・2.7,and cis・2.9,and
Me3SiOTf worked as a catalyst. The 31p NMR signals of trans・2.7and trans-2.9 were
observed at 173.4 and 155.1 ppm, respectively. In con廿ast,cis・2.8did not isomerize
to trans-2.8 even in the presence of Me3SiOTf. F or the cis-trans isomerization, the
reaction pathway similar to出atfor the fac・merisomerization of Mo(CO)3 {P(OR)3}3
shown in Scheme 2-14 is proposed. Interaction of Me3St with an oxygen in the
P(OR)3 ligands may initiate the cis-trans isomenzation. The basicity of the phosphite
oxygen in cts・MO(CO)4{P(OR)3}2 IS considered to be less than that in
fac-Mo(CO)3{P(ORhh because the former complex has more CO ligands in number
being a s甘ongπ-accepterligand. Among cis圃 2.7,cis・2.8,and cis・2.8,cis・2.8has least
oxygen basicity because of tbe substituents (Ph vs Me). Therefore, cis曹 2.8may not have
enough basicity on the oxygen to form an interaction with Me3Si+.
Table 2-7. Isomenzation of MO(CO)4{P(OR)3}2 by M~SiOTf in CH2Ch at room
tempera旬 re.a.b
P(ORh
OC111..! -'川CO
OC"γ、I ~P(ORh c o
P(ORh
P(OMe)3 cis・2.7
P(OPhh cis...2.8
PPh2(OMe) cis・2.9
P(ORh
TMSOTf a
『司電回目・・
ÛCI11 •• !川¥¥¥CO
o c'lv、CoP(OR)3
cis: trans C
‘
trans・2.7 1 : 0.9
trans・2.8 no reaction
trans・2.9 1 : 2.3
a 1.0, 0.5 and 0.1 equvalents based on the cis complex were used.
b Reaction conditions: room temperature, 0.1 M CH2CI2 solution
C cis:lrans equilibrium ratio after completion of the isomerization.
37

Reactiol1 ofTransitwn Metal Compleχes杭 aSilicon Hypervalent Structure
2-3-6 ReactioD of fac・MO(CO)3(phosphite)3and cis・MO(CO)4(Pbosphite)2with
BF3・OEt2".
Me3SiOTf and BF3・OEh are effective Lewis acids to obtain a cationic
phosphenium complex by an OR anioD. abstraction from a diamino帽 substituted
phosphite ligand in a transition metal complex. In contrast, Me匂3S凱iOTfdoes no飢t
aめbs鈎甘a泌ct 叩 OR anion from a P(ρOR'め)3ligand 0ぱffacひ-Mo州(CO)3{伊P(ρOR町)3汁}3and
Ci釘s
T刊he的r印eば的fおor爪e久, reactions 0ぱf j戸bCひ-MO(CO)3{仔P(ρOMeめ)3}3 (仰(acひ-2.2勾) and
cis-Mo(CO)4 {P(OMeの)3汁}2(やcis-2.7)with BF3・OEt2were examined and it was found that
BF3・OEt2causes some complicated reactions in addition to isomerization.
The 31p NMR spectrum of the reaction mixture of fac・2.2and an equimolar
amount of BF3・OEt2in CH2Ch showed several unidentified signals in addition to a
signals assignable to mer-2.2. These signals increased in intensity with time but the
singlet due to the starting complex (jac-2.2) stiH remained after several hours.
The reaction of cis・2.7with an equimolar amount of BF3・oEt2 in CH2Ch at room
temperature was fo1'lowed by the 31 P NMR measurement. After 4 h, in addition to a
S廿ongsinglet due to cis・2.7,a doublet of doublet at 164.5 ppm (d,ゐF= 1157.1, and Jpp
= 46.9 Hz) and a doublet at 163.4 ppm (d, Jpp = 46.9 Hz) were observed. The large
coupling constant (1157.1 Hz) suggests the existence of a P-F bond and the small
coupling constant (46.9 Hz) indicates that two phosphorus ligands訂 ecis.1O each 0白er.
Therefore, the formation of cis骨 MO(CO)4{P(OMe)3}{P(OMe)2F} was proposed. The
similar ORlF substitution reaction has been reported? 16, 224, 227, 229 The 31p NMR
spectrum a食er24 h, signals due to cis-Mo(CO)4{P(OMe)3} {P(OMe)2F} increased in
intensity and a new singlet at 173.4 ppm attributable to trans・2.7was observed. ln
addition, several unidentified signals were observed. Therefore, it was found in the
reaction of cis-2.7 with BF3・OEt2,relatively fast OMe/F substitution reaction and
relatively slow cis to trans isomerization and some unidentified reactions take place.
38
,
,.
ρ 1・r
♂ F
令
』・b
し、
~~ .,.,.
tJJ
P'rt'
伶
a.,は唱
仇引Et
・
.‘

Chapter 2. Geometncal Isomenzahon 01 Molybdenum Complexes wztll PllOsphltes Catalyzed by Neulral or CatlOntC Stlzcon Compound
、 2・4 Conclusion
-属、!・
ln conclusion, this study described, for the first time, a halosilane-catalyzed
reaction. Results showed that an interaction of a silane compound with one phosphite
oxygen to form a hypervalent silicon structure reduces the fac・merisomerization energy
barrier of MO(CO)3(phosphite)3・ Thehypervalent silicon species was detectable
spectroscopically. This geome甘icalisomerizations were aIso observed in the presence
of a catalytic amount of Me3SiOTf. Crossover experiments suggest出ata ligand
、
、、.“
、:f、
.、
‘、t
、、、
ご 1 dissociation IS削 involvedin the isomerization. The ca凶yticcyc1e involving an
! 附則ionof the山 on伽 nin Me3St with one叩 enln P(OR)3 liga時 hasbeen
proposed. The first isolation and the X-ray s甘ucωre組 alysiswere attained for
I mer-Mo(CO)3 {P(OPh)3} 3 which was obtained by the Me3SiOTf-assisted isomenzation
offaひMo(COh{P(OPh)3} 3・ Similarcis-trans isomerizatlon of MO(CO)4(phosphite)2
proceeded by Me3SiOTι
, •
、
39

-・・E
Reactlo1J ofTranslt1oH Mefal Complexes t'la SllrCC1J Hyperval(!I1t Structure
2・5Experilnental Sect;on
General Remarks. All reactions were carried out under an atmosphere ....of dry
nitrogen by using standard Schlenk tube technlques. All solvents were purified by
distillation: THF and benzene were distilled合ornsodiumlbenzophenone, hexane and
n-pentane were distilled from sodium metal, CH3CN and CH2Ch were dlstilled企om
CaH2・ Allsolvents were stored under nitrogen atmosphere. TMSOTf, MeOTf and
BF 30Eh were distilled and were stored under a nl仕ogenatmosphere. Other reagents
employed in this research were used as received. Column chromatography was done
quickly in the air.
IR spectra were recorded on a Perkln-Elmer Spectrum One spec甘orneter. A JEO L
丹~M-AL400 spec甘ometerwas used to obtain JH, J3C、29旬、 31pNMR spec町a. lH, 13C
and 29Si NMR data were referenced to Me4S1. 3 J P NMR data were referenced to 85 %
H3P04・
Preparation of fac・Mo(CO)J(NCMe)J(fac-2.1). Fac・2.1was prepared according to
literature methods.241
MO(CO)6 (17.7 g, 6.70 mmoI) in CH3CN (120 mL) was
introduced into a 100 mL Schle叫〈旬beequipped wlth a magnetic st廿barand a reflux
condenser connected to an oil bubble. The solution was beated to reflux under
dinitrogen for 5 h to result in a dark-yellow solution, at which point the IR spec甘um
indicated that MO(CO)6 was completely transformed to fac・2.1. After filtration to
remove insoluble materials, volatile materials were removed under reduced pressure.
The resulting precipitate was ~'ashed with n-pentane and dried in vacuo to give a gray
powder of fac・2.1(16.6 g, 5.48 mmol, 81.7 %).
Preparation of fac・MO(CO)3{P(OMeh}J ifac・2.2). Fac・2.2 was synthesized
accordjng to the literature methods.2 12 P(OMe)3 (0.65 mL, 5.51 mmol) was added ωa
solution of Mo(CO)3(NCMe)3 (fac・2.1)(0.55 g, 1.81 mmol) io THF (20 mL) in a
Schlenkぬbe,and the reaction mixture was stirred for 4 h at room temperature. After
volatile materials were removed under reduced pressure, the resulting precipitate was
40
同d
、,
、,
附叩
円 円唱 l
、、
、,、
‘'、‘
l
,
,、ー
•
¥ '¥
、、
•
-Lτ 12
P、t可、
t(

'
. "' .
.
• ‘ 、
、
. • L 1.
、、崎町‘
‘判
、もL
干、
・.、ー
231
、
•
2
.
毎J
』
J
¥
•
Oulpter 2. Geomemcallsomenzahorl of Moかbd,印刷 Complexeswlth Phosphz的 CatalyzedmJ Neutral or caれomcSllzcon Compound
washed with hexane a few times and dried in vacuo to give a white powder offac-2.2
(0.92 g, 1.67 mmol, 92 %). lH NMR (8, in CDCb): 3.61 (d, 27H, 3JpH = 10.8 Hz,
OCH3). 13C{IH} NMR (8, in CDCh): 51.4 (m, OCH3), 215.9 (m, CO). 31peH}
NMR (8, m CDCh): 168.0 (s). IR (cm-I, in CDCh): v (CO) 1880,1967 .
Preparation of fac・Mo(CO)J{P(OEt)J}3怖か2.3).2必 Fac-2.3 was prepared in a
manner similar to出atfor fac・2.2. P(OEt)3 (0.67 rnL, 3.90 mmol) was added to a
solution of MO(CO)3(NCMe)J (jac・2.1)(0.39 g, 1.30 mmol) in THF (20 mL) in a
Schlenk tube, and the reaction mixture was stlηed for 4 li at room temperature. After
volatile materials were removed under reduced pressure, the resulting precipitate was
I washed with hexane a few times at・78oC and dried in vacuo to give a white powder of
fac-2.3 (0.61 g, 1.2 mmol, 92 0/0). lH NMR (8, in CDCh): 1.22 (m, 27H, OCH2CH3),
3.96 (m, 18H, OCH2CH3). 13CCH} NMR (8, in CDCb): 16.4 (m, OCH2CH3), 59.7 (s,
OCH2α-h), 216.4 (s, CO). 31p{IH} NMR (8, in CDCh): 161.2 (s). IR (cm-J, in
CDCh): v (CO) 1860, 1963 .
PreparatioD of fac-Mo(CO)3{P(OPh)3}J (jac・2.4).2・42,2・43 Fac・2.4was prepared in a
m釦 nersimilar to that for fac・2.2. P(OPh)3 (1.36 mL, 5.19 mmol) was added to a
solution of Mo(CO)J(NCMe)3 (jaひ2.1)(0.52 g, 1.72 rnmol) in THF (20 mL) in a
Schlenk tube, and the reaction mixture was stirred for 4 h at roorn temperature. After
volatile matenals were removed under reduced pressure, the resulting precipitate was
wasbed witb bexane a few times and dried in vacuo to give a white powder of fac・2.4
(1.57 g, 1.41 mmol, 82 %). lH NMR (8, in CDCh): 6.98・7.18(m, 45H, Ph). 13CCH}
N恥iR(o, in CDCh): 121.9 (s,p-Ph), 124.3 (s, m-Ph), 129.4 (s, o-Ph), 152.1 (s, ipso・Ph),
212.2 (m, CO). 3Ip{'H} NMR (8, in CDCI3): 145.Q (s). IR (cm-1, in CDCh): v (CO)
1917, 1992 .
Prepara“00 of fac-Mo(CO)3{P(NMeCH2CH2NMe)(OMe)}J ifac・2.5). Fac・2.5was
synthesized according to白elitεrature meぬOdS.2.13P(NMeCH2CH2NMe)(OMe) (0.68
mL, 4.7 mmol) was added ωa solution of Mo(C0)3(NCMe)3 (向c・2.1)(0.44 g, 1.50
41

Reactioll ofわ'ansltionMetal Complexes Vla Silicon Hypervalent Structure
mmol) in THF (20 mL) in a Schlenk tube, and the reaction mixture was stirred for 4 h at
room temperature. After volatile materials were removed under reduced pressure, the
resulting precipitate was washed with hexane a few times and dried in vacuo tG-_give a
white powder offac-2.5 (0.66 g, 1.1 mmol, 71 %)・ lHNMR (0, in CDCb): 2.80 (m,
18H, NCH3), 3.14 (m, 6H, NCH2), 3.17 (m, 9H, OCH3), 3.27 (m, 6H, NCH2)・ 13CeH}
NMR (0, in CDCh): 33.59 (m, NCH3), 50.20 (m, OCH3), 52.25 (m, NCH2), 218.28 (m,
CO)・ 31p{IH}NMR (0, in CDCh): 149.36 (s). IR (cm-¥ in CDCh): v (CO)
1841,1944.
Isolation of mer-Mo(CO)3{P(OPh)3h (mer-2.4). A CH2Ch solution (20 mL)
containing fac・2.4(2.3 g, 2.07 mmol) and TMSOTf (0.38 mL, 2.08 mmol) was stirred
for 1.5 h at room tempera旬re. After volatile materials were removed under reduced
pressure, the resulting precipitate was washed with hexane (3 mL, 5 times) at・780C
and dried in vacuo to give a white po,vder of a mixture offac-2.4 and mer-2.4 (1 : 30).
To obtain pure Iner-2.4, the powder was washed with hexane/CH2Ch = 100/1 solution
(5 mL, 20 times) at room temperature, and the powder was dried in vacuo (1.22 g, 1.10
mmol, 53 %). lH NMR (0, in CDCh): 6.83-7.25 (m, 45H, Ph). 13CeH} NMR (0, in
CDCh): 115.5 (s, Ph), 121.8 (s, Ph), 124.4 (s, Ph), 129.5 (s, Ph), 129.7 (s, Ph), 129.9 (s,
Ph), 152.1 (s, Ph), 155.7 (s, Ph), 208.0 (m, CO), 213.0 (m, CO). 31p{JH} NMR (0, in
CDCh): 148.6 (t, 2Jpp = 46.9 Hz, equatorial-P), 155.4 (d, 2Jppニ 46.9Hz, apical-P)・ IR
(cm-1, in CDCh): v (CO) 1931,1813.
Preparation of MO(CO)4(nbd) (nbd = 2,5-norbornadiene) (2.6)・ Complex2.6 was
prepared according to literature methodsμ4 A hexane solution (30 mL) of MO(CO)6
(4.1 g, 15.4 mmol) and 2ふnorbomadiene(4.7 mL, 46.2 mmol) was introduced into a
100 mL Schlenk tube equipped with a magnetic stir bar and a reflux condenser
connected to an oil bubble. The mixture was heated to reflux under diniなogenfor 2
days to result in a dark-brown solution, at which point the IR spec甘umindicated
MO(CO)6 was completely transformed to 2.6. After filtration to remove insoluble
materials, volatile materials were removed under reduced pressure. The resulting
42
, ν
dnrF --f
グt
4
‘uv
dEav ぬ‘‘.
nuI
伽my
C::P
fI rr,
コpe:
「、旬、
-t
,,
'hH1.
・1
《九
uw,,-l
l
.、
.,
-VEU
l
A
“MW 1
Pltj
\\~
lJj2
ー‘Ha--
aHH
am樋h
弘川町""刊
-h
i -内iz-LV
恥
HH''
h
n
.
n
m
h
u
w
M
H川
V
U
A
'仏
v
z
d
n
P町
自国1
(I~
泊r
M
洲可
bM、
Lm叫
h陀
h
w

αapter 2. Geometrical Isomenzation 0/ Mo旬bdenumComplexesωlth PhosphJtes Catalyzed btJ Neutral or Catlomc SIUcon Compoul1d
、.
“
、‘、‘、、、、precipitate was washed with hexane at・78oC and dried in vacuo to give a dark-orange
powder of2.6 (3.5 g, 10.6 rnmol, 69 %). 、・v、、
、、
Preparation of cis-Mo(CO)4{P(OMe)3h (cis・2.7).2・45 P(OMe)3 (0.52 mL, 4.42
mmol) was added to a solution of MO(CO)4(nbd) (2.6) (0.73 g, 2.21 mmol) in CH2Ch
( 1 0 mL) in a Schlenk tube, and the reaction mixture was stirred for 4 h at room
temperature. After volatile materials were removed under reduced pressure, the
resulting precipitate was washed with hexane a few times at・78oC and dried in vacuo
to give a white powder of cis・2.7(0.97 g, 2.13 mmol, 96 %). lH NMR (o, in CDCh):
3.62 (d, 3JpH = 5.6 Hz, 9H, OCH3). 13C{'H} NMR (o, in CDCb): 51.4 (s, OCH3),
二 I 207.9(t, 2JpC= 14.1 HZ,cis且 CO),212.1 (t, 2JpC = 13.3 Hz, trans-CO). 3lpeH} NMR
、4
、-ー“・
、
"'" ,: (o, in CDCh): 165.6 (s). IR(cm-J, in CDCb): v (CO) 2036, 1921.
,
Preparation of cIs-MO(CO)4{P(OPh)3h (cis・2.8).2・46 P(OPh)3 (0.83 mL, 4.92 mmoI)
was added to a solution ofMo(CO)4(nbd) (2.6) (0.81 g, 2.42 mmol) in CH2Ch (10 mL)
in a Schlenk tube, and the reaction mixture was stirred for 4 h at room temperature.
After volatile materials were removed under reduced pressure, the resulting precipitate
was washed with hexane a few times at・78oC and dried in vacuo to give a white
powder of cis・2.8(l.87 g, 2.26 mmol, 93 %). 'H NMR (o, in CDCh): 7.18骨 7.36(m,
30H, Ph). I3CeH} NMR (o, in CDCh): 121.6 (s, p-Ph), 124.9 (s, m-Ph), 129.8 (s,
0・Ph),151.4 (t, 2Jpc = 4.2 Hz, ipso-Ph), 205.5 (t, 2JpC = 13.3 Hz, cis-CO), 212.1 (t,2Jpc
= 18.2 Hz, trans-CO). 31p{'H} NMR (o, in CDCh): 165.6 (s). IR (cm-¥ in CDCb): v
(CO) 2036, 1921.
PreparatioD of cis・MO(CO)4{PPh2(OMe)}2(cis・2-.9).2.47 PPh2(OMe) (0.76 tnL, 3.82
mmol) was added to a solution of MO(CO)4(nbd) (2.6) (0.63 g, 1.91 mmol) in CH2Ch
(10 mL) in a Schlenk tube, and the reaction mixture was stirred for 3 h at room
館mperature. After volatile materials were removed under reduced pressure, the
resulting precipitate was washed with hexane a few times atヴ8oC and dried in vacuo
to give a white powder of cis-2.9 (1.17 g, 1.82 mmol, 96 %). 'H NMR (o, in CDCh):
43

-・・r
ReactlO7I ojTransltlol1 Mefal Complexes vza Srtlco71 Hypeπmleu f Structure
3川, 3JpH = 4.9 Hz, 3H, 0 ω, 7.40-7.53 (rn, 10H、問 13C{ IH} NMR仇 in
CDCh):引い, OCH3), 128.1 (げPh),130.1 (s, m-乎判Ph),
139.ρO(いt'2勺}pてc= 1η5.8 Hz, ipso-Ph), 209.5 (t, :. }pc = 10.3 Hz, cis-CO), 214.6 (t, ~)Pc = 9.5
Hz, trans-CO)・ 31pCH}NMR (8, in CDCh): 144.9 (s)・ lR(cm-I, in CDCh): v (CO)
2026, 1912.
Treatment of complexes with Me3SiC1, T九iSOT久島teOTf and BF30Et2・
fac・MO(CO)3(phosphite)3and cis・MO(CO)4(phosphite):!(0.06 mmol) were treated with
an appropriate amount of Me3SiCl, TMSOT仁MeOTfandBF30Et2 in CH2Ch (0.6 mL)・
Then, the solutions were subjected to the 3 J P NMR measurements at room temperature
at appropriate intervals.
Detection of Intermediates in the isomerization of jおひ2.2. fac・2.2(100 mg, 0.18
mmol) was dissolved in Me3SiC1 (0.6 mL) and the solution was subjected to the 31p and
29Si NMR measurements at room tempera同re.
X-ray Crystal Structure Determination of Iner-2.4. Crystals of mer-2.4 suitable for
an X-ray di飴action study were obtained through crystallization 企om
CH2Chlhexane/benzene for a fe¥v days. The single crystal was mounted in a glass
capillary. Data for mer-2.4 were collected at・70oC to a maximum 20 value of 55.0 0 on
RigakulMSC Mercury CCD area噌 detectordiffractometer equipped with monochromated
MoKαradiation (λ=0.71070 A). Cell constants and an orientation matrix for data
collection corresponded to a primitive monoclinic cell with dimensions of a =
11.9900(3)人b= 20.7000(4) A, c = 24.1000(1)入, α=67.630(4) 0, s = 77.640(6) 0, y =
87.590(6) 0, Z = 4 and V = 53980(3) A3. P-1 (No. 2) was selected as the space group,
which led to successful refinements. All calculations for mer-2.4 were performed with
the teXan crystallographic software package of Molecular Structure Corporation、 H
atoms were refined using a riding model, with C-H = 0.95 A, and fixed individual
displacement parameters [U150 (H) =凡 (C)]. The cηstal was formulated as mer-2.4・
0.5CH2Ch・0.5C6H6・TheCH2Ch molecule was disordered in two positions with 50:50
44
レ令

戸、
J・
‘、
~
‘ 司,.‘
郵
4・
例p
'‘
、a
、.
、
'
。即断2. Gωme加・ω1Isomenzanon 01 Molybdenum Complexes wrth Ph05戸1ltesCatalyzed by Neutral OT
Ca{lol1lc SI1lcon Compound
probability in the unit cell.
‘
4S

Reaction ofTra71Siれ011九1etalComplexes via 51l1C071 Hypervalel1 t 5tructure
2-6 References
2.1 Larson, G. L. In The Chemistry 01 Organic Silicon Compounds; Patai, S.,
Rappoport, Z., Eds.; Wiley: London 1989; p763.
2.2 Miura, K.; Hosomi, A. In Main Group Meωls in Organic 砂nthesisYamamoto, H.,
Oshima, K., Eds.;Wiley-VCH: Weinheim, Germany 2004; p 409.
2.3 Mukaiyama, T. In Organic Reactions; Wiley, New York 2000.
2.4 Gennari, C. ln Comprehensive Organic Synthesis; Trost, B. M., Fleming, 1., Eds.;
Pergamon Press, Oxford 1991, 2, Chap. 2.4, p 629.
2.5 Hosomi, A. Acc. Chem. Res. 1988,21,200.
2.6 Fleming, 1. In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.;
Pergamon Press, Oxford 1991, 2, Chap. 2.2, p 563.
2.7 (a) Hiyama, T. In Metal-Catalyzed Cross Coupling Reactions; Wiley, Weinheim
1998, Chap. 10, p 421. (b) Hatanaka, Y.; Hiyama, T. Synlett 1991, 845.
2.8 Sugiura, M.; Hagio, H.; Hirabayashi, R.; Kobayashi, S. J. Am. Chem. Soc. 2001,
123,12510.
2.9 Bond, A. M.; Colton, R.; Feldberg, S. W.; Mahon, P. J.; Whyte, T. Organometallics
1991, 10, 3320.
2.10 Nakazawa, H.; Yama思lchi,Y.; Miyoshi, K. J. Organomet. Chem. 1994, 465, 193.
2.11 Rousche, 1. -C.; Dobson, G. R. Inorg・Chim.Acta 1978, 28, L139.
2.12 Howell, J. A. S.; Yates, P. C.; Ashford, • F.; Dixon, D. T.; Warren, R. J. Chem. Soc.
Dalton Trans. 1996, 20, 3959.
2.13 Nakazawa, H.; Miyoshi, Y.; Katayama, T.; Mizuta, T.; Miyoshi, K.; Tsuchida, N.;
Ono, A.; Takano, K. Organometallics 2006,25, 5913.
2.14 Williams, E. A. ln The Chemisttアザ OrganicSilicon Compounゐ;Patai, S.,
Rappoport, Z., Eds.; John Wiley & Sons Ltd. 1989; p 511.
2.15 Kummer, D.; Halim, S. H. A.; K吐lS,W.; Mattem, G J. Organomet. Chem. 1993,
446, 51.
2.16 Dilma, A. D.; loffe, S. L. Chem. Reν~ 2003, 103, 733.
2.17 Giju, K. T.; Proft, F. D.; Geerlings, P. J. Phys. Chem. A 2005, 109, 2925.
46
,
, ,
, ,
,、k .*
、、..-E
,
、a司
.-'‘. ・...~
• • .-
,、
、偽・. .
‘z-伺‘
•.
am
・
AM司、,A
・、•.
1 1
、可、
.
. ・、
、、1
、、.、、、、.、
.‘.v‘‘事、‘,‘、、
:ぉ

!,'
a、
-・H
。即位T2. Geometncallsomenzahorl 01 Molybdemwz Complexes uf1th Phospllltes Catalyzed by Neutral or Cano1!lC SZIICOU Compound
2.18 Dobson, G R.; Rettenmaier, A. 1.1norg. Chim. Acta 1972, 6,433.
2.19 Cowley‘ A. H.; Kemp, R. A. Chem. Rev. 1985,85, 367.
2.20 (a) Sanchez, M.: Mazieres‘M. R.; Lamande, L.; Wolf, R. Multiple Bonds and Low
Coordination in Phosphonls Chemistry, Regitz, M., Scherer, O. ]., Eds., Thieme,
New York 1990: Chapter D 1. (b) Schmidpeter, A. Multiple Bonds and Low
Coordination in Phosphorns Chemistη, Regitz, M., Scherer, O. 1., Eds., Thieme,
New York 1990; Chapter D2.
2.21 Gudat、D.Coord. Chem. Reν.1997,163,71.
2.22 Nakazawa. H. J. Organomet. Chem. 2000,61人349.
2.23 Nakazawa, H. Aνd. Organomet. Chem. 2004, 50,107.
2.24 Nakazawa, H.: Ohta, M.; Miyoshi, K.; Yoneda, H. Organometallics 1989,8, 638.
2.25 Nakazawa‘H.; Yamaguchi, Y.; Miyoshi, K. 1. Organomet. Chem. 1994, 465, 193.
2.26 Nakazawa, H.; Yamaguchi, Y.; Mizuta, T.; Miyoshi, K. Organometallics 1995, 14,
4137.
2.27 Yama思lchi,Y.: Nakazawa, H.; Itoh, T.; Miyosht, K. Bull. Chem. Soc. Jpn. 1996, 69,
983.
2.28 Nakazawa, H.; Yamaguchi, Y.; Miyoshi, K. Phosphorus Su抑rSilicon 1996, 109,
129.
2.29 Nakazawa, H.; Yamaguchi雪 Y.;Mtyoshi, K.; Nagasawa, A. Organometallics 1996,
15,2517.
2.30 Takano, K.; Tsumura, H.; Nakazawa, H.; Murakata, M.; Hirano, T.
Organometallics 2000, 19, 3323.
2.3l Nakazawa, H.; Kishishita, M.; Yoshinaga, S.; Yamaguchi, Y.; Mizuta, T.; Miyoshi,
K.1. Organomet. Chem. 1997, 529,423.
2.32 Nakazawa, H.; Kishishita, M.; Miyoshi, K. Phosphorus Sulfur Silicon 1999, 144,
45.
2.33 Nakazawa, H.; Kishishita, M.; lshiyama, T.; Mizuta, T.; Miyoshi, K. J. Organomet.
Chem. 2001, 617-618, 453.
2.34 Nakazawa, H.; Yamashita, Y.; Miyoshi, K. Phosphorus Sulfur Silicon 2002, 177,
1533.
47
、

-・・「氏側t"州 .. '{ lf"tf呉川'州 A1t-tdlい糊,,1(,1作 r'", !'J1i h 1句}J-IIlPf'f"C fd1州t~tt\U1"w
2.35 Naka油、,'8. H.; Yamagudll. Y: Ml/uta. T‘ lc:hunura. S.; MJ)'Iω加. K.
OlJ:tlIIυmetel'"' 1 1995. 14. 4(),l~
2.36 Nakals¥¥8, H,; Yamagudu. Y.. 1¥flYωhl. Kυ'1:cJN01tte( ull 1<.¥ 1996. J 5, 13..17
2.37 Naw.aw8, H.; Yanlagu('hl~ Y . K州抑制‘』4.MtFwin-KOrronowfuJluwE"?a
J6t 4626.
2.38 Ka¥¥'anlUf3. K.: Naku.Jl¥¥ a. H ‘t\~)\o~h l. Kυ'1!.OI1υ,nefulhハ 3φ99.lN. J 517.
2.39 Kawamura, K. :λ‘akaza、~a. H . ~1. ¥ (l!¥ln. Kυ'''Cω10ffl('fall,c.. ¥ 1999. /I( 47M5
2.40 Naknl..aw3‘H . Kblu~h刷、 \1 . 1"a~1ot('. 1 . !\.i瓜anUtu - N . iahwanu‘ T. ~ MI)'o鈎t.
K. Clrent. Lt'lf 2000. .:!JO
2.41 Lt. ('.-1.: 、'eh.¥¥', -Y. Jleng. S・¥1..Lc~. <i.H .1 ピカヌuf10nl('f (~hem . 2001. 620"
106・112.
2.42 Podblanc, P. R.; BIgorgne. ~i 8,,1/ So{ ('111m 1, 1 cJn( e J 962. Jヌ01
2.43 Alyea. E. C'.:トer事Uぉ00.G. Song. S・(),.f( fu C" ¥f J 99夕、('5人213N
.2 44 Cho¥¥二T.J . ¥Vu‘Mぺ.L.u. L -K J (),"RlJl1付f11c'1 Chem 1985..."1¥ ,.ぐねぐ37.
2.45 Ogilvle. F. B ; Jenklns. J. M.. Verkadc. J (. .J Am (h~m ル'}( 1970. 92. 1916.
2.46 Dobson. G R . Rcttenn1alc:r.ヘJ111(11叉 (111m A( Icl 1972.的.F07
2.47 Gary. M. G: Krruhan7d. C ~ .J ()t1!onomel Chpm 1983、:4/、201
48

1
、•
、i句・
•
時
‘ 、、.
αapter3. N-CN Bond Cleavage of Cyanamrde句aTransitron Metal Complα Beanl1g a Si旬1Lrgand
Chapter 3.
N-CN Bond Cleavage of Cyanamide by a Transition Metal Complex Bearing a
Silyl Ligand
‘
49
ー‘-

React101l of TranSlhol1 Metal Complexes Vla Silicon Hypervalen t Structure
3-1 Introduction
3-1-1 Character of Cyanamides (R2NCN) "
ln the last two decades, considerable efforts have been devoted to cleavage of
carbon-hydrogen,3 1 carbon-carbon/2 carbon-nitrogen33
and carbon-oxygen.3 4 Direct
cleavage of these bonds provides several advantages in organic syntheses, including
atom efficiency, low environmentalload, and the potential for unusual chemoselectivity.
Here 1 focus atlention on the N-CN bond cleavage of cyanamide (R2N-CN) by a
住ansitionmetal complex.
H2N-CN, the simplest cyanamide, consists of an amino group and a cyano group
and is synthesized from chlorocyane and ammonia at first time in 1838 (von Braun
reaction)? 5 Nowadays, most cyanamides including H2N圃 CNhave been prepared企om
calcium cyanamide (CaNCN), which has been obtained from calucium carbide (CaC)
and dinitrogen (N2).3 6
H2N-CN and HN=C-NH (carbodiimide) are referred to as tautomers. This
phenomenon indicates that N-CN bond has a double bond character (eq. 3-1).
M川一一一c
-Mパ
H
aH門
H N=C二N
H ー一一萌ー (3-1)
Cyanamide Carbodiimide
Other cyanamides have similar double bond character in the N-CN bond. Clmningham
et al. reported that the N-CN bond length in Me(p-C6~Cl)N-CN is 1.331 A, which lies
just between those ofa normal N-C single bond (1.47 A) and an N=C double bond (1.27
A) (Figure 3_1).37
As mentioned above, the N四 CNbond in cyanamide is s甘ongand di伍cultto
cleavage. The von Braun reaction is the only reaction known to date to cleave the
R2N -CN bond.3 8 However, it requires harsh reaction conditions (s仕ongacid or base
50
向Eb
h
N刈
h吋
自、
4い・
liu司、u
C\~

ヘ:
、、、
ヘ、、‘ν
・・2、h
-、
-・
』 . ‘・‘
.... . 姐句、.
、.‘『
.,.. 0
、-•
J
•
αapter3. N-CN Bond αeavage ofCyanamzde by a Transitlon Metal Complex Beanng a Szlyl Llgand
General Bond Distance N ' CN
N-C
N=C
N三C
1.47 A 1.27 A
1.16 A CI
Figure 3-1. N-CN bond length of Me(p-C~4CI)N岨CN.
R
••
R'
Figure 3・2.Cyanamide molecular model.
conditions).
Another general character of cyanamide is that the molecule has two nitrogen
atoms, which are both electrophilic sites (Figure 3-2). For example, a Lewis acid BH3
attacks the N atom of the amine合agmentselectively in the reaction of Me2NCN with
BH3. The fact demonstrates that the N atom of the amine fragment has higher
nucleophilicity than白紙 ofthe cyano group. The民 bythe selective reaction for the
cyano group is difficult.
51
‘

Reaction ofTransltwn Metal Compleχes Vla 51li∞n Hypervalent 5tructure
3-1・2 C圃 CNbond Cleavage of Organonitriles with an Iron Complex
Nakazawa et al. recently reported reactions of C-CN bond cleavage of
organonitriles promoted by a silyl-iron complex.32
(d) The essence of the reaction
mechanism for Me-CN bond cleavage is depicted in eq. 3-2. Acetonitrile coordinates
to a 16e silyl-iron complex Cp(CO)Fe(SiEt3) produced from Cp(CO)2Fe(SiEt3) (3.1) in
a photoreaction to give anηI-Meαcomplex, which is converted into anポ弛CN
complex. Then, silyl migration企omFe to the nitrile nitrogen atom occurs to form
η,2 -iminoacyl complex. The transition state of this step has a pentacoordinate silicon
structure. The theoretical calculation revealed that the activation energy is only 4.8
kcal/mol. Following C-C bond cleavage on the coordination sphere gives a
methylsilylisocyanide complex. The rl-coodination of acetonitrile through the C=N
7トbondinduces silyl migration, which then causes C-CN bond cleavage. The reaction
sequences stimulate me to examine the possibility of R2N-CN bond cleavage by a
silyl-iron complex because the replacement of the R group in RCN by an NR2 group
yields cyanamide. Herein, 1 describe the first R2N-CN bond cleavage reaction by a
transition-metal complex, isolation of an intermediate, and establishment of a catalytic
cycle involving R2N-CN bond cleavage.
~iEt3 ~iEt3 I I ~ !日]-~三C-Me ー. [民]111111
C
Me η2圃MeC~
complex
[Fe] = CpFe(CO)
SiEt'l
I ,~ I ~ ~
~[Fe]~ 11 、C
Me pentacoordinate silicon structre
ゲーMeCN
complex (3-2)
/SiEt3 ιN ,、/
/- .... 唾
[Fe]
'Me methylsilylcyan ide complex
SiEt'l / V
[Fe]くド¥
_ Me '1ιiminoacyl
complex
52
j ,
・1'• 1
qL-
hて
rtH、
パVH
-A山い
1
、
:
:
・
4』
tvbnM肌
」F
、付札出,に
M'
,ーへ、、、nuauH
3Jil':
~'Î: v:e,
4
+-
羽句A凶
te碍
n日
Ract
SIl!n'
もl~
~iä

~
、、
、、、
叫〈島、
-..
場、
、
Chapter3. N-CN Bond Cleavage ofCyanaJmde by a TrmlSlhonル1etalComplex Beanng a 511yl Llgand
3・2 N・CNBond Cleavage with a Silyl圃.Jron C omplex
3-2・1 N-CN Bond Cleavage of Masked Cyanamides
The amino D1町ogenatom in R2NCN has higher nucleophilicity than the cyano
ni甘ogeoatom. Coordlnation of cyanamide to the 16e Fe species, Cp(CO)Fe(SiEt3),
through the amino oitrogeo, may reduce the acttvity of出elron complex toward
R2N-CN bond cleavage (Figure 3・3). Derivation of cyanamide into the borane adduct
at the amino nl甘ogen‘R2N(BX))CN(X = H, F)39, mIght engender more effective
R~N-CN bond cleavage because of masking of the lone pair electrons 00 the amino
m甘ogen.
加 nddeavage 屯司面M
」子一 Fe 骨ー 。c、 R、 SiEt3N
C
NR2
R2N-C三N
cyano N coord matlon
ミ2Fe
oC/¥SiEt3
R2N-C三N
ammo N
∞ordinatlon
志~ん stop
OC、、
f¥SlEtret
R/~ 、CNW
R
Figure 3・3.Expected paths in the treatment of 16e Fe complex with Me2NCN.
A THF solurion containing Cp(C0)2Fe(StEt3) (3.1) and R2NCN bearing a Lewis
acid was irradiated with a 400 W medium pressure mercury arc lamp at room
temperature for 24 h and Et3SiCN formed was detennined by gas chromatography.
The results are listed in Table 3・1. A small amount of Et3SiCN was produced in the
reaction of BH) adduct of Me2NCN, indicating the N-CN bonds was cleaved (entη1 ).
Similar results were obtalned for BH3 adducts of other cyanamides (entries 2 to 4). lt
should be noted that be目erN-CN bond cleavage without BH3 (vide in企a). No N・CN
bond cleavage was attained for cyanamide was obset:Ved for the BF 3 and AICb adducuts
(entries 5 and 6).
53

RcnctJo7t 01 Trans,ho" Mctal (c>1t1l'lex何 ptnSJ/tαm Hypr'やoklltStru(t fJ r~
,
Table 3・1.Photoreaction of 3.1 with cyanamides bearing with Lewis acid.
Cp(C0)2(SIEt3) (3.1) + substrate hv for 24H ェ Et'lSiCNIn THF -
entry substrate ,守E-O
N
du、,
eu引
Y
一3c日
G
GC Yleld of Et3SfCN entry substrate
川川
bpuf3ω3ω
ur6rkH-H
一
EMlhB10810
pH3 O-CN
pF3 Me-N-CN
Me
AICI3
Me-N-CN
Me
0010
140/0 4 19 0/0
180/0 5 00
/0 2
3 8 0/0 6
•
54
‘' ,
、
旬,
>
旬、-
乞

、¥-、 -、‘.>
‘ 'ー、、.、
CJurpter3. N-CN BOl1d Cleavage ofCyanamzde b:ヲaTransltlOn Metal CompleχBeanng a 5匂JLlgand
3・2・2 N・CNBond Cleavage of Non-Masked Cyanamides
The reaction of 3.1 with cyanamides without a Lewis acid was examined. The
results are presented in Table 3・2. Although the yields of Et3SiCN are less than 50%,
these N-CN bonds are cleaved. The reaction of H2NCN is noteworthy (entry 6). The
H2N-CN bond has a double bond character because H2N-CN (cyanamide) lS a tautomer
of HN =C= NH (carbodiimide) (Chapter 3・1・1). Therefore, H2N-CN bond is stronger
than other R2N-CN. The first H2N-CN bond cleavage is attainable in our reaction
conditions, although the efficiency remalllS insufficient.
Table 3・2.Photoreaction of cyanamides with 3.1.
Cp(COb(SiEt3) (3.1) + substrate Et3SiCN in THF, hv, 24 h
entry substrate GC Yield of entry substrate
Et3SiCN
¥ 51 0/0
O 「一¥N-CN N-CN 4 / ¥_ーj
n-Hex
CN-CN 、
2 N-CN 30 %) 5 /
n斗~ex
。一CN
H 、3 41 010 6 N-CN ,
H
avield of Et3SiCN obtained by GC. bln 1,2・dichloroethane.
町、
55
•
GC Yield of Et3SiCNa
32 Of<)
260/0
200lob

Reactzol1 ofTranslhon Metal αmplexes vωSihcoll Hypervalent Structu
3-2-3 Optimization of Solvent and Reaction Time in the N-CN Bond Cleavage of
Me2NCN ....
Optimization of a reaction solvent and time in the reaction of Me2NCN with a
silyl-iron complex 3.1 under photo-irradiatlon was performed. The attempted solvents
were listed in Table 3・3. Halogen-containing solvents (entries 1 and 2) and toluene
(en仕Y3) showed relatively good results. N岨 coordinativesolvents except NEt3,
however, showed worse results (entries 4, 6-8). Other solvents showed unfavorable
results except DMF (entries 9・13). Consequently, 1,2-dichloroethane, toluene and
DMF seem to be good solvents.
Table 3・3.Photoreaction of Me2NCN with 3.1 in various solvents.8
entry Solvents
Me + N-CN
Me'
GC Yield of Et3SiCN
entry Solvents ,Tf o
・
-N一
出氾一
%%%νmνmνm
mH持一辺
O
氾
V
U
u
r
LHh、-
門
」
『
』
4
E
E
r
H
』
C
E一
G
~ Et3SiCN hv, 24 h
3.1
N
l 、~DMSO
DMF O
nBu人H
EtOH
H20/ NEt3
= 1 /9
43 010 8 chloroform
2 1,2-dichloroethane 52 0/0
、,n
U
4
1
4.‘ ..
4
1
E
a
-
-
,
3
4
5
6
toluene 500/0
27010
43 0/0
10 0/0
刊
阻
(
バ
)12
13
7 260/0
a Complex 3.1 was treated with 1 equiv of Me2NCN at room temperature under photo田 irradiationfor 24 h.
•
Figure 3-4 showes the time course chart of the yield of Et3SiCN in the reaction of
Cp(CO)2Fe(SiEt3) with Me2NCN under photo-irradiation. The chart indicates that出e
stoichiometric cleavage was completed around 11 hours.
56

宅~ n
,、
、
r'".
~ 、
‘
、
Chaple1'3 N-CN Bc".,dαeatlQgt ofCyanamlde uy a Transltron Metal Complex Beanng a 5t1yl Llga7ld
50
ヌ 40
‘-Z 30 。、一ω 4回F,3
W 20 ‘ 、。由
:2 10 @
〉 。。
_ -.A__ __‘---
5 10 Time I h
目 o_ _・ 同局四-噌"“・・---・・4時 ...,. - 帥・ ,
1 5 20 25
Figure 3-4. Time course of the yield of Et3SiCN in the reaction of 3.1 witb
MelNCN回目derpboto-irradiation (entηr 4 in Table 4-4).
‘・
57

-・r
ReactlOll ofTranSl tl011 ^仰lαmplexestlla S仰
み2・4 Synthesis, Isolation, and Characterization of N...SiJylatedイ帽AmidinoIron
Complex ,
Reaωωaction∞ns判e句quenω悶 nb凶li即I
wit白hcyanamide (いeq.3-3引3り). We attempted to isolate N-silylated rJ九amidinoiron
SiEt3 宇iEt3I I f'J
内一倍C-NR2→州Jf'JRっ
η2.R2NCN
∞mplex
SiEt'l
/〉N..... [Fe]ミII
、C
NR2 pen匂coordinatesilicon structure (3申 3)
[Fe] = CpFe(CO)
ηT圃 R2NCN
complex
司
J
…M
4
/ Aγ
同
C
N
/
]
¥
e
「「
, SiEh / ..,
[Fe]く「¥ NR2
N-silytated
rf-amidino
complex
complex as one intermediate, but it was unsuccessful in the reaction of 3.1 with
Me2NCN. However, reactions with Me2NCN of (CSR5)Fe(CO)(py)(SiR'R九)(py =
pyridine), considered as a synton of a 16e complex (CsRs)Fe(CO)(SiR'R"2), led to
isolation of N-silylatedη,2 -amidino iron complexes (Scheme 3-1). Heating a solution
containing 3.3 and Me2NCN in benzene at 500C for 10 h yielded 3.4 quantitatively
according to the NMR measurements, but the isolation as a solid was failed. In
contrast, a reaction of 3.5 with Me2NCN yielded 3.6, which can be isolated as dark-red
powders in 85% yield. The unprecedentedポ田amidinocomplex was confirmed using
X-ray analysis (Figure 3・5,Tables 3・4,3-5). The iron takes a distorted three-legged
piano-stool struc旬rewith anη,2 -amidino fragment. The bond distance of Nl・C2
(1.327 A) is shorter than that of a typical N-C single bond (e.g., C子Nl= 1.455 A,
C4-Nl = 1.458 A),組 dis rather similar to that of an N=C double bond (e.g., N2-C2 =
1.303 A). The sum of angles around Nl is 359.90• These structural characters are
58
, 、
-F ..
p
aF
N_ ~
~
~-

‘h
i a
j
、
h--e-司
、‘
.、
•
『
, ..
‘' ・8
-" 、-・・r
.・f
-J.
' 、.
, . ,
•
'" J色、
‘ ,
Chapter3. N-CN Bond Cleavage 01 Cyanamide by a TranstNoll Metal Complex Bearmg a Szlyl Llgand
consistent with Sp2 hybridization of Nl. The C3-NトC2-N2fragment is nearly planar
with a torsion angle of 2.6(4)0. Both IH and 13C NMR spectra show that the structure
in a solid state is maintained in solution. Two NCH3 resonances were observed at
room temperature m 'H and J3C NMR (2.37 ppm (s) and 3.05 ppm (s) in 'H NMR, 39.6
ppm (s) and 42.4 ppm (s) in 13C NMR), reflecting that the C2-N 1 bond does not rotate
企eelyat room temperature.
pu
N
M
R
5
R
dl小hv
o
N、J
W
百H
H
¥
n
v
tlt
司
4
h
'
+
m
R
α
R
c
一二
1
n
5
¥
〈、
m
R
'
F
N
.
J
1
肖
a
e、ヘ
/
/
・
町
三-iFie-J同
庁
、
い
J
r
m
司
ハ
ν
e
a
d
MフゲA
U e
a
v' nb
M川
d 500C
in C6D6t 10 h
R = H; R',R" = Et 3.3 R = Me; R' = Ph; R" = Me 3.5
R = H; R',R" = Et 3.4 R = Me; R' = Ph; R" = Me 3.6
Scheme 3-1. Syntbesis of N -silylated 112 -amidino iron complexes.
•
‘・
59
、
Reacholl ofTra1lsttJol1 Metal Complexes vta SzlZCOI1 Hyperoalent Structure
Figure 3δ.ORTEP drawing of 3.6 showing the number system.
C19
C18 C13 (_軍C14C15
Table 3-4. Crystal data for 3.6.
Empirical formula C22H32FeN20S1 I V, A司 2251.0(4)
Formula welght 424.44 Iz 4
Crystal system monoclinic !仏cm-I 7.358
Crystal size(mm3) O. 18 x O. 1 0 x 0.06 ID伺 Icdラダcm3 1.252
Temperature CC) -70.0 I No. of unlque ref1ections 16617
Space group P21/n (No. 14) I No. of used ref1ections 5062 。A 12.2300( 12) I No. ofvariables 373
b, A 9.0822(8) 0.0680
c入 20.872(2) 0.1077
βdeg 103.847(5) I GOF 1.116
60
....
--・・・・・E
量
ー...
冨:
-璽軍医
,
""""
, .・‘
k
¥
、
t
.
1 、
、:
、、

•
-•
,
Chapter3. N-CN Bond Cleavage 01 Cyanamide by a Transltion Metal CcmplαBeanng a Sllyl Llgand
Fel-N2
Fel-C2
SiトC5
Sil-C7
NI-C2
NI-C4
N2-Fe1-Cl
CI-Fel-C2
N2-Sil-C6
C5-Sil-C6
C6-Sil-C7
C2-NI-C4
Fel-N2-Sil
Sil骨 N2-C2
Fel-C2-Nl
NI-C2-N2
、、
Table 3・5.Selected bond distance (A) and angles (deg) for 3.6.
Bond Distances
2.043(2) Fel-Cl l.732(2)
1.859(2) Sil-N2 l.7181 (18) .
1.866(3) Sil-C6 1.847(3)
1.880(2) Ol-Cl 1.167(3)
1.327(3) N トC3 1.455(3)
1.458(3) N2-C2 1.303(2)
BondAngles
100.34(10) N2-FeトC2 38.65(8)
94.65(12) N2-Sil-C5 110.25(12)
107.96(13) N2-Sil-C7 107.79(10)
110.58(17) C5-Sil-C7 110.05(14)
110.15(15) C2-NI-C3 123.4(2)
119.5(2) C3-NI-C4 117.0(2)
134.04(12) Fel-N2-C2 63.04(13)
141.24(19) FeトCI-01 175.0(2)
149.40(17) Fel-C2-N2 78.31 (15)
132.3(2)
..
61

-園町一
ReactlOll ofTran.sihol1 Metal Complexes VIQ $;l,C011 Hypervale111 Structure
Complexes 3.4 and 3.6 were subjected to a thermal reaction. Although 3.6
produced a small amount of PhMe2SiCN on heating in toluene at 1100
C for 24 h, 3.4
gave Et3SiCN in 62% yield on heating in toluene at 700
C for 24 h (Schem~3-2)・ The
results sぬho仰wclearly tぬha幻ta如nN孔Jιふ-sil
N恒 CNbond cleavage of cyanamide.
話BFe
oC¥、iJN¥'/ SiEt~
Me2N -
3.4 Scheme 3-2. Thermal reaction of 3.4.
~ 70 oc ー Et~SiCN
in toluene, 24 h V
620/0
62
必炉'
EL
'1d
-LF'
、~I
川中併、d
o
‘
-LV
H ~)ぺ
、,a' h
f
au,d
,,at
三;nて4
•
'r-‘
huw
/Q
-
Ma
--'--E・
tf
J
hMwe--a
,aj
a
z
z-
,,
nhwM
ハ4HV
1
hw
AHUV
偽‘恥,、
r(、l旬、55k
hhf;
a
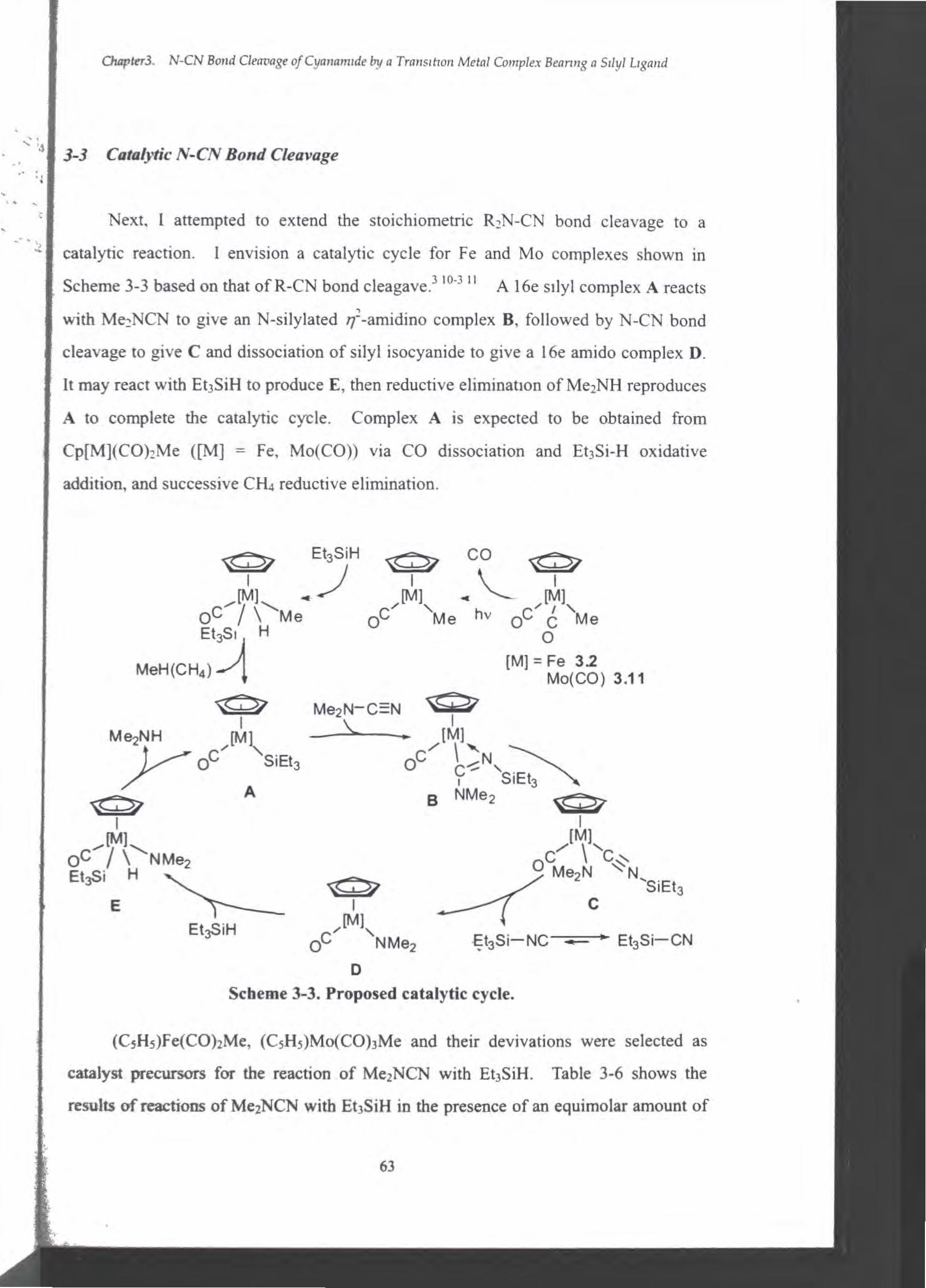
‘
3-3 CQtaかがcN-CN Bond Cleavage
Chapter3. N-CN Bond Clea-vage ofCyanamlde by a TranSlhon Me旬1Complex Benrzng a Sllyl Llgalld
、.、w、、
••..
,
、.
• 、
、.、‘、、
. 、Next、1attempted to extend the stoichiometric R2N-CN bond cleavage to a
catalytic reaction. 1 envision a catalytic cycle for F e and Mo complexes shown in
Scheme 3-3 based on that of R-CN bond cleagave.3 10-3 11 A 16e s11yl complex A reacts
with Me~NCN to give釦 N-silylatedr/ -amidino complex 8, followed by N-CN bond
c1eavage to give C and dissociation of silyl isocyanide to give a 16e amido complex D.
It may react with Et3SiH to produce E, then reductive eliminat10n of Me2NH reproduces
A to complete the catalytic cycle. Complex A is expected to be obtained from
Cp[M](CO)::!Me ([M] = Fe, Mo(CO)) via CO dissociation and Et3Si-H oxidative
-、"、‘, ~、十、、、
addition, and successive Cfu reductive elimination.
ゼヂ戸、ヲ Et3SiH
~~)~ _J OC. / ¥ 'Me
MeH(CH4)
話診 cO 宅診I ~ I
,仰1 ~ .......... 、ー jM]
。,C' "Me hv OC'己¥Me
0
[M] = Fe 3.2 Mo(CO) 3.11
ミ~
r-.)M~_._ 0- ~IヒI
協 2N-C三N 中キ Jt01oC L2N¥
γSiEt3 B NMe2
ーム
A g詰F
Jf¥:11 0C'l-Cふ
M&JN "'N_ ー、SiEt3c
~Ç'>>
.JM]、
。C~I ¥メNMe2目指 H
E ミ主?7
a仰}。C' 、NMe2
D
Scheme 3-3. Proposed catalytic cycle.
-・ーーー
Et~jH Et3Si-CN 与t3Si-NC
(CsHs)Fe(CO)2Me, (CsHs)Mo(CO)3Me and their devivations were selected as
catalyst preαE鈎 rsfor the reaction of Me2NCN with Et3SiH. Table 3・6shows the
results of reactions of Me2NCN witb Et)SiH in the presence of an equimolar amount of
63
e

Table 3圃 7.Catalytic cleavage under photolysis or heating.
Me2NCN + Et3SIH cat. 3.2 or 3.12
Et3SiCN in toluene 、.、
entry cat. [M]:[N]:[Sir condition temp/OC time/h TONb
rr~
3.2 1:10:10 hv 25 24 0.4 司・・・E め-2 3.12 1 : 10 : 10 hv 25 24 1.4 冒・・・・・h l¥1'l 冒圃E
3 3.2 1 : 1 : 1 A 80 12 。ITl!n
ー-.4 3.12 1 : 1 : 1 d 100 12 0.52
5C 3.12 1: 10: 1000 d 100 48 7.9
6C 3.12 1: 1000:5000 A 100 120 32.3
。Molarratio of a transition metal complex, Me2NCN, and Et3SiH. bCalculated from the isolated
Et3SiCN. The values are based on the concentratlOD of a仕ansitionmetal complex. cIn企eesolvent.
React1011 ofTrm似ttOl1Metal αmplexes via Siliωn Hypervalel1t Structure
a transition-metal methyl complex. An iron complex and a molybdenum complex with
a C5Hs ligand (3.2 and 3.11) showed better reactivity than complexes with a modified
....
Cp ligand.
Table 3-6. Yield of Et3SiCN in the cleavage with metbyl complexes.
Me complex+Et3SiH+Me2NCN hv~ Et3SiCN in toluene
淫三yl
Fe‘ 。c、、、MeC 0
3.10
手rMeQ Fe“ Fe、
oC1、 k¥MeoCοh、Me
も も
話ニジ
Mo_ _Mo、
ハ.C/j¥'MeハC'_:' ¥、Mev C C 'OJ C C
O~ b 0 0
3.11 3.12 3.8 3.9 3.2
一%
%
一nU今
L
一内
ζnJ』
e一
m一
パ一h
h
q
一2μ
pu
一
免u-e
一
Yield of Et3SiCN 51 % 170
/0
52 % 19 % 44% 49%
38%
41 % 10 % 28%
Next, the catalytic activity of 3.2 and 3.12 was examined. The results are shown
in Table 3-7. En廿Y1 shows that the Fe complex does not work as a catalyst, whereas
the Mo complex does under photo-irradiation conditions (entry 2). The Mo complex
shows catalytic activity even under thernlal conditions (entries 4-6).
64
,け
μ'
ψl
h在、『
C¥ar,d
¥¥,
和・1
tS:h.
... f •
..!.1.
、.

1 .、、-之、.叫、、ι
、、、
弘
ー
『
.‘
、ー、
Otapteβ. N-CN Bond Cleavage of Cyanamlde by a Transltz071 Metal Complex Beanng a Silyl LIgand
3-4 Unsaturated Bond Cleavage by an Iron Complex
3-4・1 C=S Bond Cleavage of Me2NCHS
In Section 3ふえ 1described the synthesis of N-silyla削r1-amidlnocomplex
which corresponds to an intennediate in the N-CN bond c1eavage sequences of
cyanamide. The pyridine complex 3.3 was treated in toluene with
N,N-dimethylthioformamide, Me2NCHS containing a C=S unsaturated bond in toluene
for 3 hours at 50 oC to generate quantitatively a Fischer-type carbene complex 3.13
(Scheme 3・4). Complex 3.14 was also obtained as a dark-red powder in 90 % yield in
the reaction of the pyndine complex 3.7 with Me2NCHS.
淫三~
(C/;?¥SiRq uo-R = Et 3.3
R = Ph 3.7
+ 'N人H
淫ζ診ジd. 50 oC
/Feく、:::-_" H i川n川附tωolu附馴e町n眠1
V NMeつR3Si 辺
R=Et 3.13 100%eHNMRYield)a
R = Ph 3.14 90 % (Isolated Yield) a Complex 3.13 was not isolated as a solid.
Scheme 3-4. Reaction of iron complex bearing pyridine with Me2NCHS.
The structure of 3.14 was confrrmed by X-ray crystal s廿uctureanalysis (Figure
3-7, Tables 子8,3-9). Complex 3.14 adopts a three-legged piano幽 stoolgeome甘y:the
iron center possesses one Cp, one carbonyl書 onethiosiloxy and one amino-substituted
carbene企agment. Tbe bond distance of F e 1・C3(1.8891 A) is shorter than that of a
typical Fe-C bond in a Fischeトtypecarbene (e.g・ヲ Dillen et al. reported 1,974 A in a
iron complex) (Figure 3-6),3 12 The bond length between the nitrogen atom Nl and the
、1
Ph Ph CO....S 、 1/-"
C二 Fe一一一;Fe(CO)3,l¥/J Me2N T 60 "s
¥ -- Ph
1.974 A Figure 3..6. Fe=C bond length in a Fischer-type carbene complex.
65

Reachon ofTransiれ011Metal Complexes Vla St1icol1 Hypel・valentStructure
C27
C21
Table 3-8. Crystal data for 3.14.
Empirica1 formula C27H27FeNOSSj lV,A3
F ormula weight 497.51 Z
Crystal system monoclinic μ, cm
Crystal size(mm3) 0.45 x 0.30 x 0.30 Dcalcd, g/cm3
Tempera制reCC) -70.0 No. ofunique reflections
Space group P2t1n (No. 14) No. ofused ref1ections
a. A 16.037(2) I No. of variables
b, A 9.3525(11) IR
c. A 16.402(3) Rw
βdeg 101.572(3) GOF
,
66
E
...
2410.0(5)
4
7.813
1.371
23293
5411
373
0.0328
0.1051
1.007
-、
週間
墨田
戸困回
fel.
fel・
w
~I.I
S¥-
o 51.
SI-'
C¥.
τーー、E ・h

Chapter3. N-CN Bond Cleavage ofCyal1amlde by n Transltwn Metal Complex Beanng a Sllyl Llgand
Table 3-9. Selected bond distance (A) and angles (deg) for 3.14.
Bond Distances
Fel-Sl 2.3371(3) Fel-C3 1.8891(15)
FeトC4 1.7434(17) S I-Si 1 2.1016(5) .
NI-Cl 1.466(2) 0トC4 1.153(2)
NトC3 1.306(2) NI-C2 1.473(2)
Bond Angles
S トFel-C3 96.42(5) SI-Fel-C4 87.39(5)
C3-Fel-C4 97.34(7) Fel-S1-Sil 115.482( 19)
Sl-Sil-C5 112.96(4) Sl-Si l-Cl1 113.45(5)
Sl-Sil-C17 107.24(5) C5-Sil-Cl1 106.44(7)
C5-Si l-C 17 107.72(7) C ll-SiトC17 108.86(7)
CI-I-C2 113.75(13) CI-NI-C3 123.56(13)
C2-Nl・C3 122.54(14) Fe l-C3-Nl 135.96(12)
Fe トC4-01 174.95(16)
..
67
、

.--
RenctlOl1 ofTransltlOH Metal Complexes l'la Sillcol1 Hyperva/eul Structure
carbene carbon atom C3 is also shorter than that of aザpicalN-C single bond (1.47 A).
This tendency is very similar to that for the N-silylated r/ -amidino complex 3.6. The
1止:
sun1 of angles around N 1 is 359.85 o.
hybridization ofN 1.
Proposed reaction mechanism is shown in Scheme 3・5. A pyridine in the
These characters are consistent with Sp2
I
IJU ‘
ー
、.• 1
precursor is released to give CpFe(CO)(SiR3). The C=S bond coordinates to the
16・electronspecies in an r/ -fashion, followed by silyl migration to the sulfer atom to
give a and successive C-S bond cleavage in the coordination sphere, thereby yielding a
Fischer-ザpecarbene iron complex. Comparison of eq. 3-3 with Scheme 3-5 is
interesting. The N-silylated rr-amidino complex exhabits N-CN bond cleavage,
whereas a exhibits C働 Sbond cleavage instead of C-N bond cleavage. The reaction
co汀espondsto the convertion ofC=S double bond into C=Fe double bond.
S
' N.....C、H
Lふ
t.
‘.
‘
1 1
謡 言
。c/;¥SiR3ο
淫三~
寸+(C/?~ N 、
υ16 e complex
-ー
話ミp
_Fe‘
ハC' 工、SiR-:t
Me2N/:二 S
silyl mlgratlon r • .-
g三ジ_Fe..._ 叫
ん .C"""U、S
Me〆、 SiR3
話三ジ_Fe. ~
ハc,..... ¥ :8、。;ロv _r.' ¥",)11、3Me2N' -H
ミミ詔
ぷ"""""/e\~SiR3ML/C-s eぅN I
ιH
、It4
、d
zz ‘
f 、
ー
a pentacoordinate silicon complex
Scbeme 3-5. Proposed reaction mechanism in the cleavage of C=S bond.
、‘
.跡内、、
‘,
w 、
』
68

、
Ch叩ter3. N-CN Bond Cleavage of Cyanamlde by a TranSlllon Metal Complex Beanng a 51旬1Llgand
百}
.、、‘.‘
3-4-2 C=S Bond Cleavage of MeN出 C=s
、司
‘
町、a 、可、
The仕eatmentof 3.3 in toluene wlth methyl isothiocyanate MeNCS containing a
C=N unsaturated bond in addition to a C=S bond for 2.5 hours at 50 oC generated an
isocyanide complex 3.15 quantitatively (Scheme 3・6). Complex 3.16 could be also
obtained as a dark-red powder in 80 % yield in the reaction ofthe pyndine complex 3.7
with MeNCS.
戸‘
、、、
、‘
淫三2_Fe‘
ハC/JE¥SiRquo-R = Et 3.3 R = Ph 3.7
、・.
淫B,Fel¥
in toluene, 2.5 h ハc/¥"'C、υS、NMe
R3Si'
R = Et 3.15 100010 CH NMR Yield)a
R = Ph 3.16 80 % (Isolated Yield)
a Complex 3.15 was not isolated as a solid.
~ ・・・. d 50 oC
、+ N=C=S
Me{
Scheme 3・6.Reaction of the pyridine complex with MeNCS.
The structure of the isocyanide complex 3.16 was confirmed by X-ray analysis
(Figure 3・9,Tables 3・10,3-11). The iron takes a distored three-legged piano田 stool
structure with a Cp, a carbonyl, a thiosiloxy and an isocyanide Iigand. The bond
distances of F eトC2(1.843 A) and N1・C2(1.152 A) are similar to those of other
isocyanide complexes (e.g., Adams et al. reported 1.805 A assignable to Fe=C bond and
1.158 A assignable to C N bond )3 13 (Fi♂Jre 3・8). The bond angles of Fe 1・C2-Nl
and CトN1・C2are 176.5 0 and 177.6 0 respectively. The angle of Fe-S-Si in a
literature complex is 129.01 0.3
14
...
0
1.805 A'--:'、¥r//C¥rf〆COe一一一十e
Cp
OC/7ks OC ¥.....1
129.01 0 Si(t-Buh .N' 0
f-Bu'
Figure 4・8.Bond distances and bond angle.
69

ReactlOn ofTransitlOl1 Metal Conψlexes Vla SlhCOll Hyperoalent Structure
Figure 3-9.0RTEP drawing of 3.16 showing the Dumber system.
('26 C25 ('22
....
C2 Nl
,〆
p
‘、,』'ak・
rbBa
F
、, 、
rbEh
iへ
¥ '.
A7
・、• .
、代、
、1
¥ ¥
~ I入
Table 3・10.Crystal data for 3.16.
Empirical formula C26H23FeNOSSi I y, deg 74.018(16) L
F ormula weight 481.47 I V, A3 1127.7(5) ョー-...
Crystal system triclinic Iz 2
Crystal size(mm3) 0.40 x 0.40 x 0.40 lμ,cdl 8.323
Temperature CC) -70.0 Dcalぬ glcm3 1.418
Space group P-1 (No. 2) No. ofunique reflections 11012
a. A 8.639(2) No. ofused reflections 5007
b, A 8.698(2) I No. of variables 372
c, A 16.057(4) IR 0.0491
α:, deg 84.537(19) Rw 0.1753
βdeg 76.620(16) GOF 1.003
70
、、
-、

ch呼,ter3. N-CN Bond Cleavage of Cyanamide by a TransztlOn Metal CompI釘 Beannga 5zIyI Llgand
Table 3-11. Selected bond distance (A) and angles (deg) for 3.16.
Bond Distances
Fel-Sl 2.3344(7) Fel-C2 1.843(2)
Fel-C3 1.759(2) S I-Si 1 2.0936(7) .
Sil-C4 1.881(2) Sil-CIO 1.884(3)
Sil-C16 1.891 (2) 01-C3 1.155(3)
NI-Cl 1.432(3) NトC2 1.152(3)
Bond Angles
SI-Fel-C2 94.84(8) SI-Fel-C3 88.49(8)
C2-FeトC3 95.04(11 ) Fel-S1-Sil 114.39(3)
SI-Sil-C4 105.59(8) SI-Sil-CI0 113.37(7)
SI-Sil-CI6 115.68(7) C4-Sil-CI0 107.08(12)
C4置 Sil-C16 104.10(11 ) CI0-Sil-CI6 110.12(11)
CI-NI-C2 177.6(2) Fel-C2-Nl 176.5(2)
Fel-C3-01 179.4(2)
71
•

" ,
ReactlOn ofTransitzol1 Metal Contplexes Vla SilzcOl1 HypervaLent Structure
The reaction exhibited C=S double bond cleavage in isothiosyanate, which has
not been reported to date. The reaction is expected to proceed in the similar
mechanism shown in Scheme 3・5,but silyl migration企omF e to S in isothiocyanate.
l
,r・,,,,
‘,‘EJ'
harsh
e¥slfl
,捌
1] .使
由5i
1
4
• 、、、
即5
•
,
72

、
~
• 、
ν
、
OuzpteT3 N-CN Bcmd Cleat)age of Cyanmnrde byαTransltwn九lfetalComplex Bean1tg a 511yl Llgand
3-5 Conclusion
The R2N・CNbond is known to be s仕ongand not broken readlly. Only under
harsh conditions the bond is cleaved (von Braun reaction). 1 could exhiblt the first
example of R2N-CN bQ.Dd cleavage under mild conditions by using a transition metal
catalyst. In the reaction, silyl tlUgration合oma甘ansitionmetal to a nitrogen atom of
'11-coordinated cyanamide is a trigger of the successive N-CN bond activation. The
migration may be owing to出eprope口yof silicon to take readily a hypervalent s汀ucture.
N-silylated ,,2 -arnidino iron complex coπesponding to the complex right after the silyl
migranon could be isolated and characterized by X-ray analysis.
73
電

Reachon of TrallSi tiOll Metalωmplexes via Silicon Hypervalent 5削 C仰向
3・6Experimental Section
General Remarks. All reactions were carried out under an atmosphere of dry
nitrogen by using standard Schlenk tube techniques. All solvents were purified by
distillation: ether and toluene were distilled合omsodiumlbenzophenone, pentane and
hexane were distilled合omsodium rnetal, and CH2Ch was distilled企omCaH2・ Al1
solvents were stored under nitrogen atmosphere. Most chemicals were commercialIy
available except diーかhexylcyanamide,N-cyanopiperidine, N-cyanomorpholine, and
N-cyanopyrrolidine. Column chromatography was done quick1y with non-anhydrous
pentane in the air. Photo-irradiation was performed with a 400 W medium-pressure
mercury arc lamp at room temperature under nitrogen atmosphere.
IR spec廿awere recorded on a Perkin-Elmer Spectrum One spec仕ometer. A JEO L
丹~M-AL400 spectrometer was used to obtain IH, 13Cラ 29SiNMR spec町a. All NMR
data were referenced to Me4Si.
Preparation of cyanamides. Di-n-hexylcyanamide, N-cyanopiperidine,
N-cyanomorpholine, and N-cyanopyrrolidine were synthesized according to 白e
literature methods.315 Secondary amine (10.0 mmol) was added to 20 mL of
anhydrous ether at room temperature with stirring and the mixture was cooled to 0 oC.
To the solution was added dropwise over 30 min 20 mL of anhydrous ether containjng
cyanogen bromide (5.0 mmol, 0.530 g). Then, the reaction mixture was allowed to
waロnto room temperaωre, and was stirred for 2 h. Precipitates of ammonium chloride
formed were removed by filtratlon, and the filtrate was concentrated under reduced
pressure to a small volume, which was distilled to give the desired cyanamide. 百le
spec甘oscopicdata eH and I3C NMR and IR data) were identical with those in the
literatures.
Photoreaction of cyanamides with Cp(CO)2Fe(SiEt3) (3.1). ln a typical reaction, a
solution of dimethylcyanamide (0.0787 mmol, 6.57μL) and Cp(CO)2Fe(SiEt3) (3.1)
(0.0787 mmol, 23.0 mg) in toluene (0.31 mL, 0.25 M solution) was photo-irradiated.
74
. ‘-
f
3rw
抑!
a'aav--
1Cl'lv
.. ‘ ザtt
''bl
同HH
nvt
例
γ
・・・
令
rt,
,a・向日
νE
llll~'
‘、 d
V“:
.~ r .‘
, r
l .....
,¥
i
,Eeh
nHFI
p"e
、1弘、
1, ...:
l t
l
~l
t~.
hw ト¥

、.
、
e、
、
。凶'pter3.N-CN Bond αeavage ofCyanamlde by a Tra71Slhon Metal Complex Beanng 0 Sllyl Llgolld
After removal of volatile materials at 3000 Pa, the residue was dissolved in a smal1
amount ofpent釦 eand loaded on a silicagel column, and eluted with pentane to isolate
triethylsilyl cyanide.
Preparation of Cp *(CO)(py)Fe(SiMe2Ph) (py = pyridine) (3.5)・ Comp1ex3.5 was
prep紅 edin a manner similar to that for Cp(CO)(py)Fe(SiEt3).3 10 A solution of
Cp.(C0)2Fe(SiMe2Ph) (2.21 mmol, 844 mg) and pyridine (11.0 mmol, 0.893 mL) in
to)uene (10 mL) was subjected to photo-irradiation for several hours. Removal of
volatile materials under reduced pressure led to the formation of a dark-red solid, which
was washed three times with hexane at -78 oC. Pure crystals of 3.5 were obtained by
recrystallization with hexane at -20 oC (1.66 mmol, 721 mg, 75 %). 1H NMR (o, in
C6D6): 0.72 (s, 3H, SiCH3), 0.63 (s, 3H, SiCH3), 1.42 (s, 15H, CsMes), 5.96 (s, lH,
p-NCsHs), 6.46 (s, 2H, m-NCsHs), 7.23・7.80(m, 5H, Ph), 8.31 (s, 2H, 0・NCsHs).
J3C {IH} NMR (o, in C6D6): 2.7 (s, SiCH3), 3.6 (s, SiCH3), 10.0 (s, CsMes), 90.4 (s,
CsMes), 123.2 (s, p-NCsHs), 126.7 (s, Ph), 127.1 (s, Ph), 133.2 (s, m喧 NCsHs),134.1 (s,
Ph), 150.5 (s, o-NCsHs), 156.6 (s, Ph), 223.4 (s, CO). 29Si NMR (O, ln C6D6): 34.5 (s).
IR (cm-1, in C6D6): v (CO): 1874.
Preparation of Cp(CO)(py)Fe(SiPb3) (PY = pyridine) (3.η. Comp1ex 3.7 was
prepared in a manner similar to that for Cp(CO)(py)Fe(SiEt3).310 A solutioD
containing Cp(C0)2Fe(SiPh3) (0.740 mmol, 323 mg) and pyridine (13.1 mmol, 1.06
mL) in toluene (2.5 mL) was subjected to photo-irradiation for 1 day. CO was
degassed合omtbe solution in the first 1 hour, 3 hours and 12 hours. Removal of
volatile materials under reduced pressure led to the formation of a dark-red solid, which
was washed three times with ether at・78oC. Pure crystals of 3.7 were obtained by
recrysta1Iization with ether at・20oC (0.518 mmol, 252 mg, 70 %). 1H NMR (o, in
CDCh): 4.37 (s, 5H, CsHs), 6.64 (s, 2H, m岬 NCsHs),7.20・7.28(m, 15H, Ph), 7.63 (s, lH,
p-NC掛け, 8.43(s, 2H, o-NCsHs). 13CeH} NMR (o, in CDCh): 82.7 (s, CsHs), 123.1
(s, p岬 NCsHs),126.9 (s, Ph), 127.9 (s, Ph), 134.4 (s, m-NCsHs), 135.4 (s, Ph), 144.3 (s,
75

品提j
d J Bt詮i
・・・・・・・・・圃圃・・・・・圃・・・・・圃・圃圃・・・・・・・・・・・・・・・園田・・・・・・・圃・・・・・・・・圃圃・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・
ReactlOl'l ofTransltion Metal Complexes via 5111coI1 Hypervalent 5tructure
0・NCsHs),158.0 (s, Ph), 222・6(s, CO)・ 29SiNMR (0, in CDCh): 39.5 (s)・ IR(cm-1,
in CH2Ch): v (CO): 1896. Anal. Calc. for C29H2SFeNOSi: C, 71.46; H, 5.17; N, 2.87.
Found: C, 71.7とH,5.22; N, 2.87.
Syntbeses of N-silylated
Cp(CO)Fe{ぷ(C,l¥チC(NMe2)=N(SiEt3)}
η2-amidino complexes
and (3.4)
Cp* (CO)Fe{}(2( C,l¥トC(NMe2)=N(SiMe2Pb)}(3.6)・ Complex3.3 (1.37 mmol, 592
mg) was treated with Me2NCN (1.37 mmol, 0.114 mL) in toluene at 50 oC for 10 h.
Removal of volatile materials under reduced pressure led to the formation of the
corresponding N-silylated 112-amidino complex 3.4 as a dark-red oil in 100 % lH NMR
yield. ln a similar way, the N-silylated 172 -amidino complex 3.6 was obtained as
orange powders which were purified by washing with pentane at -78 oC ( 1.16 mmol,
492 mg, 85 %)・ 3.4;lH NMR (0, in C6D6): 0.64 (q, 6H, SiCH2CH3, 3 JHH = 75.9 Hz),
0.95 (t, 9H, SiCH2CH3, 3 JHH = 75.9 Hz), 2.49 (s, 3H, NCH3), 3.20 (s, 3H, NCH3), 4.36
(s, 5H, Cp). 13C{lH} NMR (8, in C6D6): 7.1 (s, Siα12CH3), 7.7 (s, SiCH2α13), 39.1
(s, NCH3), 42.8 (s, NCH3), 79.5 (s, Cp), 194.8 (s, NCN), 219.5 (s, CO). 29Si NMR (0,
in C6D6): 7.1 (s). IR (cm-¥ in toluene): v (CO): 1902. 3.6; lH NMR (0, in C6D6):
0.32 (s, 3H, SiCH3), 0.47 (s, 3H, SiCH3) 1.72 (s, 15H, CsMes), 2.37 (s, 3H, NCH3), 3.05
(s, 3H, NCH3) 7.22-7.60 (m, 5H, Ph) 13C{lH} NMR (0, in C6D6): 0.9 (s, SiCH3), 3.1
(s, SiCH3), 10.9 (s, CsMes), 39.6 (NCH3), 42.4 (s, NCH3), 89.9 (s, CsMes), 128.6 (s, Ph),
129.2 (s, Ph), 133.8 (s, Ph), 141.1 (s, Ph), 204.0 (s, NCN), 220.6 (s, CO). 29Si NMR (0,
in C6D6): 35.4 (s). IR (cm-1 , in toluene): v (CO): 1884.
Photoreaction of dimetbylcyanamide with trietbylsilane in tbe presence of methyl
complex. A solution containing methyl complex 3.2, 3.8, 3.9, 3.10, 3.11 or 3.12
(0.0150 mmol), dimethylcyanamide (0.150 mmol, 12.5μL) and位iethylsilane(0.150
mmol, 24.0μL) in toluene (3.0 mL) was photo-irradiated. After removal of volatile
materials at 3000 Pa, the residue was dissolved in a small amount of pentane aad was
charged on a silicagel column to isolate triethylsilyl cyanide.
76
ー3・
....' ・ー
理 -・・咽圃圃圃圃圃圃圃圃・・圃園田園田圃圃置圃圃圃園周咽』岨唱団・
同一一一
UUEERI--E-
.・
、、...,
~
通J~
d
w
M
m
u
aV-J--
伶
刊
州
山
F
ー
‘asaw a
、ι‘E・
-W1
・mv
1
、‘
l
『i
e¥J[f,
i i
明
H1
ha
、、
A-1
-‘‘、
.‘ 、.
':34
司 e
u..J
''JM' e,.
1 ,、 g
¥. ) 1
-、,1 .. ~
自けu
・1 1 FL
i
f
、
-旬
。、Fare
聞週
La
pt、
、、‘、
可I1
Icm
、uuu叩
1
l
‘.
nL
、u
、am、
』 圃聞
、‘4・,、、、“‘,,、,J
、
,

、
、
1
守
, ,
• ,
Chnpter3. N-CN Bond Cleavage ofCyanarmde by a TransltlOn Metal Complex Beanng a SzlyI Llga71d
Thermal reaction of dimethylcyanamide with triethylsilane in the presence of
metbyl complex. ln stoichiometric reactions, a solution containing methyl complex
3.2 or 3.12 (0.0150 mmol), dimethylcyanamlde ( 0.0150 mmol, 12.5μL) and
triethylsilane (0.0150 II}IIlol, 24.0μL) ln toluene ( 3.0 mL) was heated at 80 oC or
100 oC under ni住ogenatmosphere. Then, triethylsilylcyanide formed was isolated in
the similar manner described above. The catalytic activities of 3.2 and 3.12 were also
examined.
Syntheses of carben complexes Cp(CO)Fe(=CHNMe2)(SSiR3) (R = Et, 3.13; R = Ph,
3.14). Complex 3.7 (0.343 mmol, 167 mg) was仕eatedwith Me2NCHS (0.3 70 mmol,
31.1μL) in toluene (6 mL) at 50 oC for 3 h. Removal of volatile materials under
reduced pressure led to the formation of the corresponding carbene complex 3.14 as
dark-red powders which were puri白edby washing with ether at・60oC ( 0.300 mmol,
149 mg, 88 %). ln a simtlar way, the carbene complex 3.13 was obtained as a dark-red
oil in 100 % IH NMR yield. 3.13; IH NMR (0, in C6D6): 0.92 (m, 6H, SiCH2CH3),
1.24 (m, 9H, SiCH2CH3), 2.42 (s, 3H, NCH3), 3.20 (s, 3H, NCH3) 4.49 (s, 5H, CsHs),
11.99 (s, lH" Fe=CH). 13CeH} NMR (0, in C6D6): 8.74 (s, SiCH2CH3), 9.59 (s,
SiCH2CH3), 44.4 (s, NCH3), 53.6 (s, NCH3), 86.0 (s, CsHs), 223.77 (s, CO), 256.0 (s,
Fe=C). 29S1 NMR (0, in C6D6): 22.6 (s). IR (cm-t, in toluene): v (CO): 1932. 3.14;
IH NMR (0, in C6D6): 2.17 (s, 3H, NCH3), 3.05 (s, 3H, NCH3) 4.19 (s, 5H, CsHs),
7.21・8.03(m, 15H, Ph), 11.74 (s, lH, Fe=CH). 13CeH} NMR (0, in C6D6): 44.4 (s,
NCH3), 53.4 (s, NCH3), 85.7 (s事 CsHs),127.62 (s, Ph), 128.8 (s, Ph), 136.5 (s, Ph),
140.8 (s, Ph), 222.9 (s, CO), 255.8 (s, Fe=C). 29Si NMR (0, in C6D6): 1.8 (s). IR
(cm.l, in toluene): v (CO): 1937. Anal. Calc. for C27H27FeNOSSi: C, 65.18; H, 5.47; N,
2.82. Found: C, 65.04; H, 5.41; N, 2.75.
Synthesis of carben complexes Cp(CO)Fe(C=NMe)(SSiR)) (R = Et, 3.15; R = Ph,
3.16). Complex 3.7 (0.401 mmol, 196 mg) was treated with MeNCS (0.428 mmol,
29.3μ,L) in toluene (5 mL) at SO oC for 2.5 h. Removal of volatile materials under
77

ReactlOn ofTransltlOll Metal Complexes vza SillCOn. Hypervalent Structure
reduced pressure led to the formation of the corresponding carbene complex 3.16 as
dark-red powders which were purified by washing with ether at -60 oC ( 0.311 mmol,
150 mg, 78 %)・ lna similar way, the carbene complex 3.15 was obtained as a datk-red
oil in 100 % IH NMR yield. 3.15; IH NMR (3, in C6D6): 0.98 (m, 6H, SiCH2CH3),
1.29 (m, 9H, SiCH2CH3), 2.16 (s, 3H, NCH3), 4.32 (s, 5H, CsHs)・ 13C{JH}NMR (3,
in C6D6): 8.75 (s, Siα12CH3), 9.61 (s, SiCH2α13), 29.78 (s, NCH3), 83.3 (s, C5H5),
162.6 (s, FeCN), 219.9 (s, CO)・ 29SiNMR (3, in C6D6): 16.1 (s)・ IR(cm-1, in
toluene): v (CO): 1962, v (C=N): 2172. 3.16; IH NMR (3, in C6D6): 1.86 (s, 3H,
NCH3), 4.03 (s, 5H, CsHs), 7.19・8.07(m, 15H, Ph)・ 13C{lH}NMR (3, in C6D6): 29.74
(s, NCH3), 83.4 (s, CSH5), 127.7 (s, Ph), 128.8 (s, Ph), 136.3 (s, Ph), 140.4 (s, Ph), 161.3
(s, FeCN), 218.4 (s, CO)・ 29SiNMR (3, in C6D6): 1.9 (s)・ IR(cm-t, in toluene): v
(CO): 1973, v (C N): 2180. Anal. Calc. for C26H23FeNSSi: C, 64.86; H, 4.82; N, 2.91.
Found: C, 64.82; H, 4.77; N, 2.83.
X-ray Crystal structure determination of 3ふ Dark曲 redcrystals of 3.6 suitable for
an X-ray diffraction study were obtained through crystallization企ompentane. The
single crystal was mounted in a glass capillary. Data for 3.6 were collected at -70 oC
on Rigaku/MSC Mercury CCD area-detector diffractometer equipped with
monochromated MoKαradiation (λ=0.71070 A). Calculations for 3.6 were performed
with the teXane crystallographic software package of Molecular Structure Corporation.
Crystal Data: C22H32FeN20Si, M = 424.44, orange plate, 0.18 x 0.10 x 0.06 mm,
monoclinic, space group P2dc (No. 14), a = 12.2300(12) A, b = 9.0822(8) A, c =
20.872(2) A,β= 103.847(5)0, V = 2251.0(4) A3, Z = 4,μ(MoKα) = 7.358 cm-1, Dωc-
1.252 g/cm3, 16617 ref1ections col1ected, 5062 (1) 3σ乃uniqueref1ections were used in
all calculations, number ofvariables = 373, R = 0.0680, Rw = 0.1077, and goodness of
白t=1.116.
X-ray Crystal structure determination of 3.14 Dark-red crystals of 3.14 suitable for
組 X-raydi飴actionstudy were obtained through crystallization企omCH2Ch/hexane.
The single crystal was mounted in a glass capillary. Data for 3.14 were collected at 20
78
• ,
fi}~l
4ド
ds
,W
M
H
μ
4
1
伺山「
p
打い
hu
,,=
qMM
AU
一
-hμ!
叫
U
T
Fi
n
n
-hy
pWPL ¥¥t
'ri~
mu
nt
、、u
l~
ゐ
J白
~~
、、
、民

ミ
、h-、
、
"
Chap館r3.N-CN Bond Cleavage 01 Cyanamlde by a Transltlonん1etalComplex Bearing a 511yl Llgand
。C on RigakulMSC Mercury CCD area-detector dif企actometer equipped with
monochromated MoKαradiation (λ=0.71070 A). Calculations for 3.14 were
performed with the teXane crystallographic sofu入rarepackage of Molecular Structure
Corporation. Crystal Data: C27H27Fel、~OSSi, M = 497.51, red block, 0.45 x 0.30 x
0.30 mm, monoclinic, space group P21/c (No. 14), a = 16.037(2) A, b = 9.3525(11)人c
= 16.402(3) A, P = 101.572(3)0, V= 2410.0(5) A乙Z= 4, Jl(MoKα) = 7.813 cm-¥ Dωc
= 1.371 g/cm七16617reflections collected, 5411 (1 > 3σ乃uniquereflections were used
in all calculations, number of vanables = 373, R = 0.0328, Rw = 0.1051, and goodness
of fit = 1.007.
X岨 rayCrystal structure determination of 3.16. Dark-red crystals of 3.16 suitable
for an X-ray di飴action study were obtained through crystallization 合om
CH2Chlhexane. The single crystal was mounted in a glass capillary. Data for 3.16
were col1ected at 20 oC on RigakulMSC Mercury CCD area-detector di飴actometer
equipped with monochromated MoKαradiation (入=0.71070A). Calculations for 3.16
were perfoロnedwith the teXane crystallographic software package of Molecular
S甘uctureCorporation. Crystal Data: C26H23FeNOSSi, M = 481.47, red block, 0.40 x
0.40 x 0.34 mm, triclinic, space group P-1 (N o. 2), a = 8.639(2)人b= 8.698(2)人c=
16.057(4) A,α= 84.537(10) o,p = 101.572(3)0, y = 74.018(16) 0, V= 1127.7(5) A3, Z =
2,μ(MoKα) = 8.323 cm-t, Dcalc = 1.418 g1cm3, 11012 reflections collected, 5007σ 〉
3σl) unique reflections were used in all calculations, number of variables = 372, R =
0.0491, Rw = 0.1753, and goodness offit = 1.003.
79
、

圃「
Reactloll ofTralls1tio71 MetaJ Complexes VJa SlliCOIl HypervaJent Structure
3-7 References
3.1 See for example: (a) Ritleng, V.; Sirlin, C.; Pfeffer, M. Chem. Rev. 2002, 1 02~ 1731.
(b) Dyker, G. Angew. Chem., lnt. Ed. 1999,38, 1698. (c) Naota, T.; Takaya, H.;
Murabashi, S. 1. Chem. Rev. 1998, 98, 2599. (d) Zhang, Y.; Li, C.-J.; Angew. Chem.,
lnf. Ed. 2006, 45, 1949. (e) Ma, K.; Piers, W. E.; Parvez, M. 1. Am. Chem. Soc.
2006, 128,3303. (ηKakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Murai, S. 1. Am.
Chem. Soc. 2003, 125, 1698. (g) Goosen, L. J. Angew. Chem., lnt. Ed. 2002,41,
3775. (h) Arndtsen, B. A.; Bergman, R. G.; Mobley, T. A.; Peterson, T. H. Acc.
Chem. Res. 1995, 28, 154. (i) Crabtree, R. H. 1. Organolnet. Chem. 2004, 689,
4083.
3.2 See for example: (a) Jun, C.-H. Chem. Soc. Rev. 2004, 33, 610. (b) Rybtchinski, B;
Milstein, D. Angew. Chem., lnt. Ed. 1999, 38, 870. (c) Storsberg, J.; Yao, M.-L.;
Ocal, N.; De Meijere, A.; Adam, A. E. W.; Kaufmann, D. E. Chem. Commun. 2005,
5665. (d) Nakazawa, H.; Kawasaki, T.; Miyoshi, K.; Suresh, C. H.; Koga, N.
Organometallics 2004, 23, 117. (e) Chianese, A. R.; Zeglis, B. M.; Crabtree, R. H.
Chem. Commun. 2004, 2176. (f) Terao, Y.; Wakui, H.; Satoh, T.; Miura, M.;
Nomura, M. J. Am. Chem. Soc. 2001, 123, 10407.
3.3 (a) Lei, Y.; Wrobleskl, A. D.; Golden, J. E.; Powell, D. R.; Aube, 1. 1. Am. Chem.
Soc. 2005, 127,4552. (b) Fran, L.; Yang, L.; Guo, C.; Foxman, B. M.; Ozerov, O.
V. Organometallics 2004, 23, 4778. (c) Yao, M.-L.; Adiwidjaja, G.; Kanfmann, D.
E. Ang飢え Chem.,Int. Ed. 2002, 41, 3375. (d) Niklas, N.; Heinemann, F. W.;
Hampel, F.; Alsfasser, R. Angew. Chem., lnt. Ed. 2002,41, 3386.
3.4 See for example: (a) Maercker, A. Angew. Chem., Int. Ed. 1987, 2瓜 972.(b)
Kabalka, G. W; Yao, M.ーし;Borel1a, S.; Goins, L. K. Organometallics 2007, 26,
4112. (c) Szu, P.・h.;He, X.; Zhao, L.; Liu, H.-w. Angew. Chem., Int. Ed. 2005, 44,
6742. (d) Casado, F.; Pisano, L.; Farriol, M.; Gallardo, 1.; Marquet, J.; Melloni, G.
J. Org. Chem. 2000, 65,322.
3.5 Bineau, A. Ann. Chim. Phys. 1838, s2ν67, 225.
3.6 Frank, A.; Caro, N. Ger. Patent 1895, 88363.
80
* ,
、
LV
l
,
.. 、
-F
• 曲、,
,E 、、
-v
・、
t
r、
huv
.‘
次,.n
、A 1
・、
L 1
a . . 4・-
'‘ ~
、.
内
( I、
、、

ぺ
、
、予
、
• ‘
Chapter3. N-CN Bond Cleavage 01 Cyanamlde旬。TransltlOnMetalωmplex Beanng a 5z1yl Lzgalld
3.7 Cl1nningham, 1. D.; Light, M. E.; Hursthouse, M. B. Acat Cη1St. Sect. C} Cryst.
Struct. Commun. 1999,55,1833.
3.8 E. B. Vliet, Organic秒ntheses,Coll. Vol. 1941, 1, 201.
3.9 Syntheses of boran adduct of cyanamide and confirmation of the adduct at the
amino nitrogen hav,e been repo口ed:Henneike, H. F.; Drago, R. S. lnorg. Chem.
1968, 7, 1908・1915.
3.10 Nakazawa, H.; ltazaki, M.; Kamata, K.; Ueda, K. Chem. Asian. 1. 2007, 2,
882-888.
3.11 Nakazawa, H.; Kamata, K.; Itazaki, M. Chem. Commun. 2005, 4004.
3.12 Dillen, 1. L. M.; van Dyk, M. M.; Lo包,S. J Chem. Soc., Dalton Trans. 1989,
2199.
3.13 Adams, R. D.; Cotton, F. A.; Troup, 1. M. Inorg. Chem. 1974, 13, 257.
3.14 Kuckman, T. 1.; Doπ由aus,F.; Bolte, M.; Lerner, H.-W.; Holthausen, M. C.;
Wagner, M. Eur. J. Inorg. Chem. 2007, 1989.
3.15 Overman, L. E.; Tsuboi, S.; Roos, J. P.; Taylor, C. F. J Am. Chem. Soc. 1980,102,
747.
81
' ‘ . ,

Reaction ofTransztlO1'l Metal Complexes vza SillC01'l Hypervalent Structure
,.
J
82 、、
e
•
mr MMr
-FE
‘
saaal,
川伊
,,‘.
LHH
ptaLV
r・18Anuv
ぜB
-
h胤
v
t
似
-A1ul'
O
却
い
ド
ー
1、l品
i
l
・
2
u
?
1
4
M
ば
ALl-
本
町
t
4
K
A
W
阿-u
w
K
2
4
1
¥
o
t
hu nu
rvl
COR
W
M
h

Acknowledgement
The thesis for a doctorate is completed by summarizing the research results which
were carried out from 2004 to 2009 in Coordination Chemistry Laboratory, Department
ofChemis甘y,Graduate School of Science, Osaka City University. At first, 1 would like ,
to express my deep gratitude to Professor Hiroshi Nakazawa. Because of his direction
and help, 1 completed this dissertation and lived a happy life in the laboratory. Secound
1 would like to thank Dr. Masumi Itazaki whose comments contributed to my work.
1 owe a very impo口antdebt to Professor Akio lchimura and Professor Hiroshi
Tsukube at Osaka City University for their kind advice and reviewing this出esis-
1 am deeply indebted to a11 members of Professor Nakazawa 's laboratory,
particularly Mr. Koji Kamata, Mr. Masaharu Ohba, Mr. Ryohei Nakamura, Ms. Seijo Ou,
Ms. Rie Ujihara, Ms. Kumiko Nito, Mr. Tsukuru Oya , Ms. Michiho Kasa and Mr. Yuta
Nakai for the enjoyable discussions and expenmental assistance. My special thanks are
due to Mr. Akina Nigi, Mr. Jun Okamoto, Mr. Ken-Ichiro Okuma, Mr. Kensuke Ueda,
Mr. Kunihisa Noichi, Mr. Masahiro Kamitani, Ms. Makiko Minakata, Mr. Wataru
Na.kamura, Ms. Yuka Sigesato, Mr. Yasuhiro Hashimoto, Dr. Naumov Roman, and a11
other co11eagues in Coordination Chemistry Laboratory for their enormous
collaborations, kind help, and企iendship.
1 also gratefully appreciate the financial support of Nakato Scholarship
Foundation and Japan Securities Scholarship Foundation that made it possible to
complete my thesis.
Finally, 1 would like to express my heartfelt acknowledgment to my parents,
Nobuaki and Ju凶ωFukumoto,and my sister, Rie Fukumoto, for their patient assistance,
kind help and continuous encouragement. 1 also wish to thank my創ends,especially Ms.
Akiyo Miyajima, for their heartfelt encouragement.
Kozo Fukumoto
Department of Chemisttγ,
Graduate School of Science,
Osaka City University
March 2009

J
•
Reactio1l ofTra11sitioll Metal Complexes via Silicon Hypervalent Sか明C仰向
,
References for the Thesis
(1) Silane瞳 CatalyzedReaction:戸c-merlsomerization of [MO(CO)3(Phosphite)3}-
Kozo Fukumoto, Hiroshi Nakazawa
Organolnetallics 2007, 26, 6505・6507.
(2) Geometrical isomerization offac/mer-Mo(CO)3(Phosphiteh and
cis/trans-Mo(CO)4(phosphite)2 catalyzed by Me3SiOS02CF3
Kozo Fukumoto, Hiroshi Nakazawa
Journal olOrganometalfic Chemisf7)ノ2008,693,1968-1974.
(3) N-CN Bond Cleavage ofCyanamides by a Transition-metal Complex
Kozo Fukumoto, Tsukuru Oya, Masumi Itazaki, Hiroshi Nakazawa
Journal 01 American Chemical Socie砂2009,131, 38-39.
• 1
t
h
旬、札、川向切に、吋ヘ川、同.mu油
h
¥
伽川町ぬ吋むい川、恥
九
川
叫
同
恥
MmLhr¥勺
84 、、
• d・

• cJ I ~
毒 aヤ
:J_jJ.Jj記J
Volume 26, Number 26, December 1ス2007。Copyflght2007
Ameflcan Chemlcal Soclety
•
Communications
Silane-CataJyzed Reaction: fac一merIsomerization of [Mo( CO))(phosphite ))]
Kozo Fukumoto and HlrOsht Nakazawa*
Depanment (ずChemlSln..Gradtlate SchρυI of S( lencど.OsaJ...a CIハUml'erSlハ.SU17ll¥osh,-Au. OsaJ..a 5 )8-8585, Japan
Re(ell'ed Septt>mTer 3、z∞7
swnman M~ SIX (X = CI. Br 1) and reLated sdane compounds “抱れこ~fac-m~rωmi'nこω<>nofIMoICOJI{P(ORJJ}1/ 01 mom k~ralur~ A h¥perva/enr s,hcon strucωre has been shm~ n 釦 pltna cruCtaJ role m the catahlrc reactlOn
Because ot山引ruruque. efficlem. and切 lectJw陀 acU、lty.tn
addmon to lhetr陀 adyavaJlabt Uty and low tO;(ICJt)'. organosl h・
con compounds have ~en ~,del) u切 d.es肘C1allyfor organtL s}nu配sest..= Extensl've studIes o¥<er the past three decades havc e,annned slhcon comlX>UOOs and have created many useful reacb何 S. FnOSl of wtnch ,"¥<olve converslOn ot a sJhcon com卯田叫 mlOanother slhcon compound 10 addttJOn. several 日bc仰のωed catalysts have 恥en de、e)oped R~S10Tf.
RlS.B(σTf4. R,S.8fOTfhCl. R,SI!'(SU2Fh. and R1S,N(Tfh 2 τ"hese ca'aly~lS c釦 bede拡 nbedas contammg R ~SJ + and ar定C明 lStdeTedLewIS aclds. 10 contrasL 00 陀p側 descnbe~ sllane U 舵UU'alfour-c侃)f,由nateslhcon compound)犯 rvlngas a catalYSI Inslead of a reagent. lt has beeo s戸culated白紙 a引lane15 f削 acatalySl any n即 reth留1an a.lk.ar柁 ISThlS IS the firSI 陀戸川 ofa St凶le-<:誠alァzedreactJ∞ MelS1X (X = CI. 8r. 1) g凶陀凶剖臼加Je∞m肘undscatatyze fac-mer IsomenzatJon of[~似COhfP(ORh }JJ;輔氏)()m tempera仰陀
Reg宿命ngM(COhL, (M = Cr. Mo. W: L == phosphme. PtI04T抑制.pFFf1C1pall) two gωmetncallso問 "eXlstfOCla1 ifac)
• 10 w蜘Jm concspc:期金encc ~bould bc 3d伽鈍ed E-fTUl11 _az:at時.~J 伺晶a-ωatJP
(J) Lar領m、ol ln Th~ 0""'1$1" nf ~anlc S,lw伺 Compmmd$.陶創.
S.R勾ゆop:机 Z.,E品.Wdcy 1.0酬釦s.1989. p763 (2)輔Nn.Jt.肱l'SOfflI.A ln Mallf Gmtlf1 MttaJ3 m OrRcm,( Svnthun.
Y .. 刷 3師、 H.O晴間車 K.Bcb .Wdey-VCH Wem.helm. Omnany. 2慌)4.,409
and mendJOnal (mer) fonns Thefac Isomer IS consldered mo陀
stable electro01cally. as thls J~omer achleves stronger M to CO back-donatJon. bUI the mer Jsomer IS fa'vored from a stenc pomt ot Vlev.羽leM(CO),L, complexes p陀 parでdto dale from their 汀an"JtJOn-meta1prec山'SOrsand L have excluslvely fac geometry R陀 mer I somers we陀 obtameduSlOg IsomenzatJon of the fac Jsomer, 80nd et al陀 portedfac-merJsomenzatton vla a one-eleclrOn O;(ldatIOn to produce the 17e catlon specles for M(COh(ゲ-PhzkH2CHzP伊h)CH2CH2PPh~)‘We repo巾 d出erelated Jsomenzation for fac-[M(COh(bpy){P(NMeCH2h (0Mc)}] (bpy = 2.2'-bJpyndme. P(NMeCH~h(OMe) = P( NMeCH~CH2NMe )(OMe)).ηle react10n of the fac Isomer 州 th8F,・OEt:;:glves the calloruc phosphemum compJex fac・
[Mo(bpy )(COh {P(NMeCH2h} Jへwhlchthen Isomenzes to山emer form. It d促 S鈎 becausethe phospheOlum hgand shows strong汀 aCJ也ty.leadjng to an electron-deficienl metal cente~ (not complete one-e)ectron OXldat1on. bul partlal oXJdation)プThermalfac-mer Isomenzatlon has been陀 portedby Rousche et al. for fac・fMo(COh('12-dppe) {P(O'Prh} }、 andby Howell et a) for fac-[M(COh{P(ORh} ,] 6
We exammed the reactlOn offac-[Mo(CO),{P(OMehb1 ifac-1) with 1 equlv of MelSiCJ m CH2CI1 at roof!l_ te~peraturc. The陀 actJoowas monitored by measunng山e¥'P{ 'H} NMR
{司)BOD<t A M. Colton. R . Feldberg. S W . M凶lQn.P J • Whyte. T OrllOltomtfalf,cs 1991. 10. 3320
(4) Nakazawa. H . Yamagucht. Y . Mtyoshl. K J Or8an()m~t Clttm W剣.465.19ヲ
(ラ)Rousche. J .C . Dobson. G R lnorll Ch"". ACla 1978. 28. L Il9 (6) Howell, J A S. Yales. P C . A命ford.N F, Ot~on. 0 T . Wru胃 n.
R J Chtm S何伽/rrmTrart$ 19鮒 .20.'959
10.1ωI/om70088幻 ccc.$37.∞ <02∞7 Am何回nChemlcaJ S∞iety Poblka幻 ∞ onWeb 11/1012∞7

句
、
.
f
,、
hrV3
・セホiさY、re.LJr'h寸.
•• a ゐ.‘
3に台、
事
t
P(ORh
o c",.l ...,P(OR)a
OC〆T、COP(ORb
P(OR)J
OC" I ."p問 3_MO_
OC.... I、P(ORh
8
P(ORb Oc,, JJ(OR}3 OC〆 γ、c。
P(ORb
庁'l8r
+MssSICIミ
150円lenzallon
‘
b
e
M
h
b
R
/1¥α
R
R
一
ls
po:〉L句
H/t
h
np¥(
釦
E
L
批
…
山
川
、
M
h
門
l蜘
118
川〆
cc
o
o
~
官昆
18501825180017751750172517001675 ppm a
Figure 1.引PNMR spectrum of a Me3S1Cl solution at temperatu陀 10whlchfac・1is dlSS0)ved
(OMe)}2{P(NMeCH2)2}]+.9 In con仕おし on the baslS of 31 NMR morutoring, the reactlOn of fac・4Wl出 Me3S1CIgenerate no catiomc phospheruum complex. which suggests that pathway of the lsomerizatlon promoted by Me3S1Cl di仔e回that involving a catlonic phosphenium complex produced Me3SiOTf.
Scheme 1 presents a plausible mechanism for the fac-isomerization promoted by Me3S1Cl. The slhcon滋omMe3SiCI mteracts with one oxygen m P(OR)3 ligands to a SI hypervalent s町ucωre(a). The mteraction weak:ens coordination of P(ORh・Me3SiCltoward the central meta1 makes the ligand bulky. thereby decreasing the i energy banier to give its mer isomer (b). Tbe dissociation of the Me3SiCl g1Ves出emer isomer, 合'eeMe3SiCl. The 31 P and 29Si NMR measuremen岱 ofa CH2Ch
contaimng fac・1and Me3SiCI showed no signals assignable either a or b in Scheme 1. To obtain some evidence of a and we dissolvedfac-l in Me3SiCI and measured the 11p and NMR spec凶 ofthesol凶 on.The 11p N加IRis shown in 1. ln addition to a smglet at 166.7 ppm attributable to and a doublet at 176.2 ppm and a triplet at 168.8 with Jpp
spectrum. A tnplet at 167.1 ppm (Jpp = 44.3 Hz) and a doublet at 175.3 ppm (Jpp = 41.7 Hz) attnbutable to mer-l appeared at the expense of an mtensIty of a smglel at 164.8 pprn because offac・1.After 3 h. eqUllJbrium betweenfac-l and mer-l was estabhshed wlth the ratlO of 1.3 4 I Next the react10ns with 0.5 and 0.1 eqmv of Me3S1Cl were examined Table 1 (entnes 1-3) shows that the final eqUlhbrium posltlon was found not to depend on the amount of Me3S1Cl used. al出oughit takes a longer tIme to reach the eqUllibnum when the amount of Me3SiCl IS reduced. The equilibnum ratio resembles that obtamed m thermolysls by Howell et al. (1 3). These results sbow that Me3SJCl acts as a catalyst for !ac-mer】somenzatton.
In our expenments命yCH2Ch dlst1lled from CaH2 was llsed. However. there is a possibllity出atadventltlous HCI from Me3S1Cl hydrolysls could be responslble for the observed 1 somerizatlOn. Thus, the 29S1 NMR of Me3SiCl 10 CH2Ch was measured. No signal other th加 asmglet attributable to Me)SiCl was observed, indicatmg no sllJcon compound denved from Me3SiCl hydrolYSlS eXJsts ()ess出an5%. lf any). We observed that the isomenzation of fac・1is completed within 8 h m the p陀senceof 10 mol % of Me3SiCl. lf 10% of出eMe3SiCl produces adventltious HCl, then the amount of HCl would be 1 rnol % based onfac-l under the above condluons. The CH2Ch solution offac・1cont出mng1 mol % of HCl was prepared and monitored by the .)且PNMR measurement. and no Isomenzation was observed after 10 h. These results clearly mdlcate that the fac-mer isomenzatlOn is catalyzed by Me3SiCl but not by HCl.
The coπespondmg lsomerizatlOn catalyzed by Me3SiCI was observed for the tnethyl phosphite complexfac・2,出e出phenylphosphite complexfac-3, and the diammo-substltuted phosphite complexfac・4.In all cases. the 3Jp NMR spec町aof the reacnon rruxture suggested出eforrnation of出ecorrespon也ngmer Jsomers.8 The isomerizatlOn offac・3to mer-3 was much slower (more than 72 h) than that offac・1to mer-l (3 h), which might be attributed to a steric cause. The reactlOn of fac・4IS noteworthy. We found that the reaction offac-4 with Me3S10Tf abstracts one OMe as an anion from the phosphorus to give a cationic phosphenium complex, fac-[Mo(C0)1{P(NMeCH2h
(9) Nakazawa, H • Mlyoshl, Y ,Katayama、T.Mlzuta, T.; Mlyoshi, Ts帥 .1da,N., Ono; A., Taka肌 KOrganOFmlites m6.25.F913.
、、
. '
M舵 ba1. Proposed fac-mer 150m町izationCatalyzed by Me~jCI
Scbeme lsomerlzation by Me3SiCl in CH1Clz at Room Temperature
11
...
-MeaSiCI
P(ORh
OC". .! ..,P(OR)J
OC勺。、Co
Me r'\/P(OR~ e r'¥
Mh5F L CI' Me
fo( mer
134 134 1 34 122 1 1 30
105
ume (h)
43ε300
ヲ-つ-つ-
吋J
t』
a 。TheIsomenzatlOn IS veηslow, and lhe eqUlhbnum IS not altamed after 72 h The finaJ ratIo obtamed by uSJIlg Me,SIOTf IS 1 30 (uDpubhshed resu1ls)
b
fac
r十1
(7) In the absence of Me,SlCl no fac-mer lsomenzauon occurred at roorn t~rnperature m CH2Ch
(8) HP{IH} NMR (161 7 MHz.. CH2Ch, 25 oc. reference 85% H,PU4、d, ppm) fac・1.1648 (s), mer-l. 167 1 (t, }pp = 44 3 Hz. CIS-PP), 1753 (d. Jr・7・=417Hz. rrans-PP),fac・2,1454 (s), mer-2, 1488 (t,}pp = 52.1 Hz, CIS-PP). 1557 (d, JpP = 521Hz, rrans-PP). fac・3,146 ) (s), mer-3, 143 9 (d. }pp = 46 9 Hz, CIS-PP), 1809 (t、Jpp= 469Hz.. rrans-PP)
Organometallics~ Vol 26, No 26, 2007 6506
Table 1.
Me3SICI P(ORb
Oc f.. .! ...' P(ORb OC〆1。、P(OR)3
8
fac
+Me3SICt --------ZEe--,.,.
c
cu ‘‘, e
M
mer・1mer-2 mer.・3mer-4
P(ORb
P(OMe),3 P(OEth P(OPh)J
P(NMeCH2h(OMe}
fac・1fsc・2fac・3fsc-4
ωnt of Me,SICJ (eqUlv)
O『
J100o
'IAUAU't'tEa
comp1ex
fac・1fac・1fac・1fac・2
fac・3fac-4
entry
'ーヲ申弓‘J
必且守
主
J
ro
aAb〈ハ
〈
r1'1
わ

.> 、、
•• 、,
• 、.. ' t 、>")
• Iぜ、& 耳、
.~
.'ミ、-、
,
I内.‘:-l ,、
、、
司を
:-
. 句. 〈
十
JHall-ugUHF-
,.内iraH
"、"'、,F ・-・-
J ~
,停・44島ー
_, 、.'
,
‘' l'
41.7 Hz attributable to mer-l,自veslgnals we陀 observedclearly. Among them, a tnplet at 181.3 ppm and a doub1et al 173 9 mm wi山 JpP= 41.7 Hz can be asslgned to a. The triplel IS assignable to P(OMeh. Me)SICl because of the mo1ecular 9mrnetry.百時間mamingthree slgnals at 1848 ppm (dd. JpP i;: 41.7 and 229.3 Hz)、 174.3ppm (dd. Jpp = 44 3 and 229.3 習が,and 166.2 ppm (t‘Jpp = 443Hz) can be reasonably .lSsigned respectlvely to P(OMeh・Me"¥SICland trans and ClS .f(OMeh in P(OMeh. Me3S1Cl m b The ・"SINMR showed 側 osinglets at 6.62 and 6.53 ppm m addlllon to a strong stnglet が29.8ppm attnbutable to the solvent Me"¥SlCl. CoOrdtnallon
nitrogen or oxygen to s111con to fonn a pentacoordtnate bond been shown to produce a strong shleldmg effect ot the
chemlcal shlft. 10 Kummer et al repoロedthe :!C/S1 NMR ICaJ Shlft at 11 1 ppm m CD2Cb for an SI hyperva1ent
Wllh C1 and N m aplcal posltlons and two Me groups one CH2 group m equatonal posttlons 11 The J1C NMR of MeiSICl solullOn contammgfac・1was also measured Two
conS1stmg of severaJ Slgr】剖swe陀 observedat 213 9-214 1 2180-218.2 ppm. These slgna1s are annbulable to tenrunal
1 hgands. mdlcating山atMe3SICl does not have an Wllh an oxygen atom m the tennmal carbonyl hgand
fo陀 ,our observal1ons suggest山eforrnatton of hyperva1ent compounds No fac-mer Isomenzatton was confirmed m reactlons
Me)SlCl and fac-[Mo(CO))(phosphIne)"¥l (phosphme PEt3・BUn3)havmg no OR group on a coordmatmg phos-
s, suggesung that a phosphite oxygen plays a cruclal role fac-mer lS0menzatlon To elucldate whether fac-mer lsomerizatJon mvolves a
russoClatlon process or not、acrossover expenment wぉ perforrned AddttlOn of Me3SICl to a CH2Ch solutlon cont剖mngbothfac・1andfac・2Ylelded mer-l and mer-2 but formed nenher phosprute exchange products such as fac・ and
r-[Mo(COh{P(OMehh{P(OEt)3}] nor OR exchange prod・such as fac-and mer-[Mo(COh{P(OMehh{P(OMeh I}]. These results show rntramolecular lsomerizatJon. whlch
is conSlstent with the mechamsm shown tn Scheme J 百leresults showed出atMe)SiCl can promote fac-mer
for fac-[Mo(COh(phosphlte)J], the actlvJty of other s11anes was exarruned for fac-l Me2SICh, MeSICl), and
(10) Wl1hams. E A In The ChemlStry olOrgam( SILlcon Compolmdf. S . Rappopon. Z . Eds . Wlley London. 1989、P511
(11) Kummer. D • Hahm. S H A, Kuhs, W , Mattem. G J Organomet Chem. 1993,446. 51
Orgαnometal!tcs Vol 26. No 26. 2ω7 6507
Table 2. lsomeril.8tion or lac・1Promoted b~ R、SiX.entry R‘SIX tHnc Ch, Jac mer
Me 、S.CI 1 1 ~ 4 'ヲ Me,S.Br <0 J 136 1 Me,StI <0 J 137 4 Ph,C).CI 25 00 rea(..o 5 ( f::.tOhS.CI 2号 J '¥ ~
IJ lac・1w白山c:ltedwuh I equ.¥ o(民、s.Xtn CH~CI 、 al fl∞m temperature
SIC1" promoted lhe l ~omenzatlOn. but somc byproducts were also formed 1n contrast, SIMe~. wJth no chlondc ~ubstltuent. did not promole Isomenzauon at all Thcrefore. among the MeIlSIC1"-n (n = 0-4) senes. Me,SICI IS the best promoter. WhlCh mlght stem from llS supenor hype円 alentaπangement two electronegatlve substJtuenl!> (CI and 0 from phosphJte) m aplcal posltlOns and three electron -releasmg groups (three Me groups) m equatonal poslllons TabJe 2 shows the R1SIX aCllvlty Bromostlane and lodo~í lane (entnes 2 and 3) show actlvtty better than that of ch10rosJlane PhlSICI (entry 4) does not promote IsomenzatlOn. presumably becausc of the bulky substltuents Slow lsomenzatlOn was obsened m the reactlon wlth (EtOhSJCl (entry 5), WhlCh mlghl be attnbutable to the weak elec汀on-releasmgablltty of an OEt group compared to that of the Me group A slmtlar fac-mer ratlo wぉ obtamedfor entnes 1 -3 and 5. meamng出atthe ratJo IS determmed thermodynamlcally and IS unaffected by the added stlane 1n conclustOn、thJSpaper descnbes, for the first llme. a
halosllane-catalyzed reactlOn The results showed that an lOteracllon of sllane wnh one phosphlle oxygen to form a hypervalent slbcon s凶 cturereduces the fac-mer IsomenzatJon energy bamer of [Mo(CO),(phosphJteh). The hypervalent s111con specles was detectable spectroscoplcally ThJS findmg IS beheved to be a sta口mgpomt for expJonng the potenllaI catalyllc actl vlty of halosllanes.
Acknowledgment. ThlS work was suppo口edby a Grant-m-Ald for SClence Research on Pnonty Area (No 18033044‘
Chemlstry of CoordmatlOn Space. and No. 19027047. Synerglstlc Effect of EJements)台omthe Mlmstry of Educa-1Ion, Culture, Sports. SClence and Technology
Supporting InformatioD A vaHable: Detalied expenmental procedures and the charactenzauon of products This matenal 15 avrulable free of charge vla the Intemet at http'lIpubs acs org
OM7008827

Available online at www sciencedlrect.com _-"
‘ーン ScienceDirect
ELSEVIER Journal of Organometdlhc Chemlstry 693 (2008) 1968-1974 www e)sevJer
".
Geometrical isomerization of fac/mer圃 MO(CO)3(phosphite)3and cis/ trans-Mo( CO)4(phosphite)2 catalyzed byお1e3SiOS02CF3
Kozo Fukumoto, Hiroshi Nakazawa事
Depmtm仰 t01 Chen11Scn Graduate 8ι1100/ 01 SCfence Osαka C/[l' Vlllversfll. SUlIln oslu-ku. Osaka 558・8585,Japan
Recelved 16 January 2008、recelvedm revlsed form 18 Februaf) 2008. accepted 21 February 2008 Avallable onhne 4 March 2008
Abstract
Geometrical IsomerizatlOn of fac-Mo(COhL3 (L = P(OPh)3、 P(OMe)3, P(OEt)J) to the mer fom1 and that of cls-Mo(CO)4L2 (L = P(OPhh, P(OMe)3, PPh2(OMe)) to the lrallS form were observed m CH2Ch at room temperature in the presence of a catalytic amount of Me3SiOS02CF3 (TMSOTf). Crossover ex.periments suggest that a hgand dlssoclation 15 not involved in the lsomerization. A cataJytlc cyc1e mvolving an mteractlOn of the slhcon atom m Me~Sl"" wlth one oxygen 10 P(OR), hgands has been proposed. The first isolatlOn and the X-ray structure anaJysls were attained for mer-Mo(CO)J{P(OPhhb through the TSMOTf-asSlSted lsomerization of fac・Mo(C0)3{P(OPhh}3・
@ 2008 Elsevler B V AII nghts reserved
KeYlI'ords、GeometncalISOmenZatlon, Catalysls; Mo complex, Phospl勺te
1. lntroductioD metal complexes; M(bpy)(C0)3{P(NMeCH2h(OR)} [6-12], M(dppe)(COh{P(NMeCH2h(OR)} [7], M(bpy)・(COh{P(NMeCH2h(OR)h [8,11,12], M(CO)3{P(NMe-CH2h(OR)}3 [13], M(CO)4{P(NMeCH2)2(OR)h [13], CpM(C0)2(ER3){P(NMeCH2h(OR)} (M = Cr, Mo, W) [14-17], and CpM(CO)(ER3HP(NMeCH2h(OR)} (M =
Fe, Ru) (ER3 = CH3, SiMe3, GeMe3, SnMe3) [10,18-23]. Systematic researches for reactlOns of Mo(bpy)・(COh{PXY(OR)} with BF3・ OEt2revealed the effect of the substituents (X, Y) on白estab出tyof cationic phosphe-nium complexes; the stability increases with increasing the number of amino substituents on the phosphenium phos-phorus [9]. TMSOTf has been demonstrated to be an appropriate Lewis acid for fac-Mo(CO)J{P(NMe-CH2h(OR)}J and cis-Mo(CO)4{P(NMeCH2h(OR)h [13].
During the course of investigation of suitability of a Lewis acid to yield a cationic phosphenium complex by OR anion abstractlOn, we obtained unexpected results in the reaction with TMSOTf of fac-Mo(CO)3{P(ORhb and cis-MO(CO)4{P(ORhh having no amino-substituent on也ecoordinating phosphites; TMSOTf does not abstract叩
Transition metal complexes with a phosphenium ligand (+PR2) have attracted considerable attention because a cationic phosphenium is isolobal with a smglet carbene, silylene, and the heavier congeners [1-5]. One of the best methods fo1' preparation of cationic phospheni-um complexes of transition metals IS OR anion abstrac-tion by a Lewis acid such as BF3・OEt2or Tl'vlS0Tf (Me3SiOS02CF3) from coordinatmg dlamino-substituted phosphite P(NMeCH2h(OR) as shown in Eq. (1) [4,5]. This method is applicable for many types of transltIon
ヘ
ーN
R
JN
ご
μ¥O
M
¥ 1+
Mー Pくコ 、、,,,,
••••• ,,,‘、
Lewis acid
-OR-
-Correspondmg author E-mαtI address ndkazawa@osaka-cu ac JP (H Nakazawa)
0022-328X1$・seefront matter @ 2008 Elsevler B V. AII nghts reserved dOl 10 10 16/J.Jorganchem 2008 02 024
, 、、
, v
~~
"eDlぜ::,.... ¥111. r e凶 ,)1,
,がL;「{け $11h
'.Y.t p~\" L tiki
uム:,:,,~.z.. 1 .•
ィピ
ptかJ
tZ3
1f (1[
ω';
¥1e S¥ ¥
1 its也,
T :[も恥
u
r
i
t
叶-h
一、目lurド
-s
i
・
e
-
1
.-V同
.hklhk
tuj'ぺ
d42m1
』
1t
LfRJLhw時
-ha
民
らゆ
Aれ
.、、、JV
nuF
、n守、
。

、κFuJ...-umoto. H Naka::a¥wlJournal olOrganomelalhc Chenllsln 693 (2008) 1968-1974 1969 、
雪 辱 )RanlOn to give a catioruc phosphenium complex、butpro・~otes 也e geometrical isomerizatlon of出ecomplexes (fac-
、 警rJterand CIS-lrans lsomerizatlOn). and lt works as a catalyst
、ま TbeMo(CO)司L司complexes(M = Cr、Mo,W; L = phos-‘phosphite) have been syntheslZed in the faclal form
M(CO)~L; (L' = weekly coordinating 2e-donor d) and L. The mer isomers were obtamed by using
tlon of the correspondmg fac lsomers. Bond
al. reported出ata one electron-oXldatlOn promotes
r isomenzatlOn for M(CO)1{Ph2PCH2CH:?・P(Ph)CH:!CH:!P Ph:!-K3 P} [14]. We reported that the reac-
tion of (ac-M(COh(bpy){P(NMeCH2h(OMe)} with
BF3 -OEt:! yields a catiomc phosphem山ncomplex、fac-
[M(COh(bpy){P(NMeCH2h}了、WhlChthen lsomenzes to 也emer form [7-9]. The strongπ-acldlty of a phosphemum
ligand causes an electron deficlent metal center WhlCh may
induce thefac一merlsome氾 atlOnThermalfac-mer lsomer-
ization has been reported by Rousche et a1. for fac-Mo(COh(η:!-dppe){ P(OIPrh} (25] and by Howell et al.
for fac-M(COh{P(ORhh [26] We recently found that
Me3StX (X = Cl, Br. 1) promotes the fac-mer Isomenza-
t10n for fac-Mo(COh{P(ORhb [27].
2. Resulお anddiscussion
2 J. PreparatlOn of fac・Mo(COh{P(ORh}3and
cls-Mo( CO )4{P( OR) j} 2
Tbe fac・Mo(CO)JtP(ORhh complexes were obtamed
in good yteld by modIf}'ing the hterature methods [28] Mo(CO)J(NCMe)J was treated wlth P(OR)l m 1'3 molar
ratJo m THF at room temperature (Eq (2)) A preparative
method for cls-Mo(CO)4{P(ORhb was bnefty descnbed m
the lJterature. where MO(CO)4(NMe2(CH2hNMe2) was used as an Mo source [29] We modlfied the startmg com-plex. MO(CO)4(nbd) (nbd = norbornadJene) and P(OR)3 were treated m 1:2 molar ratlO in CH2Ch at room temper-
ature to give ClS・MO(CO)4{P(ORhbm excellent yield (Eq (3)).
Me C
L N
~",. _I 1.,NCMe mTHF Oc",. I 川 LMo_ + 3L Mo
at r t 。c〆|、L(2) CMe c
o c o
L = P(OPhh y,eld 82% P(OMe)3 92% P(OEth 92%
。 。c C
cb,,, 1..1 In CH2Cl2 uc"" I ....'¥ L
〆10l+ 2L Mo:・
at r.t (3)
c o
L = P(OPh)3 Yleld 92% P(OMe)3 96% P円12(OMe) 96%
2 2. ReactlOn of fac・Mo(CO hL3 lVlth TMSOTf
The fac-Mo(CO)3{P(OPhhbゆか1)was dlsso1ved m CH2C12 and an eqUlmolar amount of TMSOTf was added at room temperature and the reaction was momtored by the、
:JI P NMR measurement A tnplet at 148.2 ppm (t、ソpp= 47.0 Hz) and a doublet at 155.2 ppm (d, 2Jpp = 47.0 Hz) were newly observed at the expense of a smg1et at 144.4 ppm attnbuted to fac・1.The new slgnals were rea-sonably assigned to mer-l based on thelr coupling pattern and the couplmg constant. The isomenzation reached at an eqUllIbnum after 1.5 h and the fac-l.mer-l ratlO was 1 30.
We confirmed that fac・1dld not IsomerlZe to mel'-l in CH2C12 m the absence of TMSOTf at room temperature and even at the reftux temperature, showmg that TMSOTf promotes its isomerrzatlOn. Next questlOn IS whether TMSOTf works as a catalyst or not The reactions of fac・1wlth 0.5 and 0 1 eqUlva]ent of TMSOTf revealed that the final eqUlhbnum posltlOn dld not depend on the amount of TMSOTf used although it took a longer tlme to reach the eq凶 libnumwhen the amount of TMSOTf was red uced.
The mer-l could be ls01ated from the reactlOn mJxture of fac-l and TMSOTf After a treatment of fac・1wlth TMSOTf in CH2Ch, a whlte powder WhlCh IS a mixture of fac-l and mω汁 (130) was washed wlth hexane/ CH2Cl:Jbenzene (100/1/1) solutlOn many times to obtam the pure complex formulated as mer-l. 0 5CH2C12・
0.5C6H6 in 530/0 yleld Although several mer-M(C0)3L3 (M = Cr, Mo, W) type complexes have been reported, this IS the first preparatlOn and lsolation of mer-
M(CO)3{P(OPh)司h・Thestructure was confirmed by the X-ray analysls. The ORTEP drawmg 15 deplcted in Fig. 1
and the crystal data are summarIzed m Table 1. Tlus
X-ray structure IS the first example among mer-
M(CO)3(tertlary phosphorus compound)J type complexes.
The X-ray structure of fac-l was repo口edprevlOusly [29]
The structural companson between fac- and mer-
Mo(CO)3{P(OPh)3)3revealed some Interestpg points.
The bond dlstance of Mo-C2for mer-I (2L018A)1s clearly
shorter than those of Mo-Cl (2.041 A) and Mo-C3
(2.041 A). As C202 ligand is trans to P(OPh)J, the CO
hgand can get more n:-back donation from the central metal than C101 and C303 ligands whJch are mutualJy
lrans. The mean Mo-C bond dlstance for fac・1(1.986 A)
is shorter than that for mer-l (2.033 A), reasonably under-
stood by greater n:-back donatlOn from the Mo to the CO
ligands for fac・1because of the Irans P(OPhh ligand.
Another interesting POill_t IS that the mean Mo-P bond dlS-
tance for fac・1(2.435 A) is longer than that for mer-l
(2.417 A). The difference may stem from the steric repul-
sion between P(OPh)J hgands in fac・1.The mer-l did not isomerize to fac-l in CH2Ch at room
temperature, but it did by the additlOn of an equimolar
amount or even a catalytic amount of TMSOTf. After seY-
eral hours. the fac・ l :mer~l ratlo was 1:30. The results men~
tioned above show that the equilibrium ratio is detenmned

1970 κFukumOlO. H NakaごawalJollrnalofOrganometalll(, Chemlslry 693 (2008) /968-/974
ロー
FJg. 1 ORTEP drawmg of mer-l 05CH2C12 0 5C6H6 (50% probabtlity elhpsotds) showmg the numbermg systemAII hydrogen atoms and the solvated CH2CI2 and C6H6 molecules are omttted for clanty Selected bond dtstances (λ) and bond angles (0) Mol-PJ, 23796(7), Mol-P2、24319(7): Mol-P3, 2.4390(7), Mol-Cl, 2041(3), Mol-C2、2.018(3),Mol-C3, 2041(3), PドMol-P2.17424(2), PI-Mol-P3, 8976(2)、P2-Mol-P3, 8957(2); PI-MoJ-Cl, 91 21(8), PI-Mo-C2, 86 15(8), PI-Mo-C3, 91 51(8)
Table 1 Crystal data for me,.-l 05CH2Ch 05C6H6
Empmcal formula Formula wetght Crystal system Crystal slze (mm3) Temperature (0C) SPFe goup a (A)
b (A) c (A) s (0)
V(λ3)
Z μ(cm-I
)
D国)c(g/cm3)
Number of umque reflecttons Number of used reftect10ns Number of vanables R
RIY Goodness-of-F t t
CωSoH49CIMo012P3 1192.30 Monochnlc 045 x 0.35 x 0 15 -700 ntfn (No 14) 137319(5) 18.2658(7) 22.1401(9) 95.744(2) 5525.4(4) 4
436 1433 41908 12498 712 0.051 o 111 1.06
thermodynamically and TMSOTf serves as a catalyst for the fac-mer lsomerization.
Reactions 0ぱffac-羽恥M“州O叫(CO)3{P町(OMe仏hh(仰舟cは-2)川an凶dfacひ. Mo叫(C∞Oh{伊例P町(ρOE臥町t)3山}3(仰fac-めwithTMSωOT甘fwer印7右ealso exam-ined in CH2Cαha似troom t旬empe釘ratur印e.ln the 31p NMR spectra, a triplet at 166.9 ppm (t, 2 Jpp = 41.7 Hz) and a doublet at 175.1 ppm (d, 2Jpp = 41.7 Hz) assignable to mer-2 were observed in the reaction of fac・2,and a triplet at 162.7 ppm (t, 2 Jpp = 41.7 Hz) and a doublet at
170.5 ppm (d, 2 Jpp = 41.7 Hz) attributable to mer-3 observed in the reaction of fac・3.The equilibrium fi mer ratios were independent of the amount of used, showing that TMSOTf serves as a catalyst. results together with those for fac・1are shown in Ta 2. The eqUllibrium fac-mer ratios are quite"'dependent the kind of the phosphite ligand. lt should be noted t these values are equal to those for the isomerization moted by Me3SiX (X = Cl, Br, 1) [27], and the value ~ fac・2is similar to that reported by Howell [26]. Theref~ it can be said that the values are derived from the dynamic stability between the fac and mer isomers, from the stability of the intermediates created from Mo complex and a catalyst (presumably TMS+, vide infra)
2.3. Isomerizαtion mechanism
Regarding isomerizatIon of Mo(CO)3{P(ORhh pro-moted by TMSOT仁 twomechanisms are conceivable: mechanisms Vla a phosphenium complex and via a TMS+ adduct.
A mechanism Vla a phospheniwn complex is shown in Scheme l. As transition-metal complexes bearmg a dia-mino-substituted phosphite have been reported to react with a Lewis acid to give cationic phosphenium complexes by OR -abstraction as shown in Eq. (1), a similar OR-abstraction may take place in the reaction of Mo(COh {P(OR)3} 3 wlth TMSOTf to produce a cationic phospheni-um complex (fac' in Scherne 1). Then, isomerization from fac' to meY is expected to take place. The similar isomeri-zation has been reported previously (Eq. (4)), where the driving force of the fac-mer isomerization is thought to be the gain of more 1t-back donation for the phosphenium ligand. The reaction of me,.' wIth TMSOR formed would give mer-Mo(CO)3{P(ORhh with regeneration of TMSOTf. However, this catalytic cycle seems not plausible based on the following observations. (i) A complex having a phosphenium ligand was not detected in the reaction of Mo(COh {P(ORhh with TMSOTf. (ii) After the treatment
Table 2 Isomertzatton of Mo(COh{P(ORhh by TMSOTf 10 CH2C12 at room temperature
P(ORh
0c,,JHoumR)3
oc〆下、、I '_P(OR)3 c o
P(ORh
0仏 JJ附 3
Oc〆I、c。P(ORb
TMSOTfa
『司句ーー
3しμ
3
司
3-
、‘,J
SM
Lrh
利一
mMB
O
一ooo
pppp
fac.merb
1 :30 1 :3.4 1 :2.2
fac-l fac・1
fac-3
meωr-me官1'-2mer-3
01 1.0, 0.5 and 0.1 eq叩uv刊刈a剖i怜entωsbased 。∞nth恥1潟efacc∞。mp凶)e飢xwer陀e凶 e吋d.bj角ac:m昭erequtl山h巾bm可lU叩』江m町mr悶atJoafter c∞。mplet10nof the i“some町r官 a剖hωonl
, 、
, J
?ザl、
2
.‘
h匹
信Jν
吹け
u'
1依
E
h
s・
川
,HE
O
-
ir
k
-
mJ陶仰い
配
刈
-ta
U凶
,川団川目・
Uれ
市
町
同
制
恥
配
は
¥
ヘ‘1
・k
fa,lstk
いいーハV
vi川町、地・
{
AU
川
町
11Rirt-ihmt』山
嶋
崎
-uhhhL刊ぬ畑町
hhqh油
輸hm一
声
、
命
、
内
Hvhw杭加
叫
滋
け
刊
h山叫
mmuhn‘nvAm判
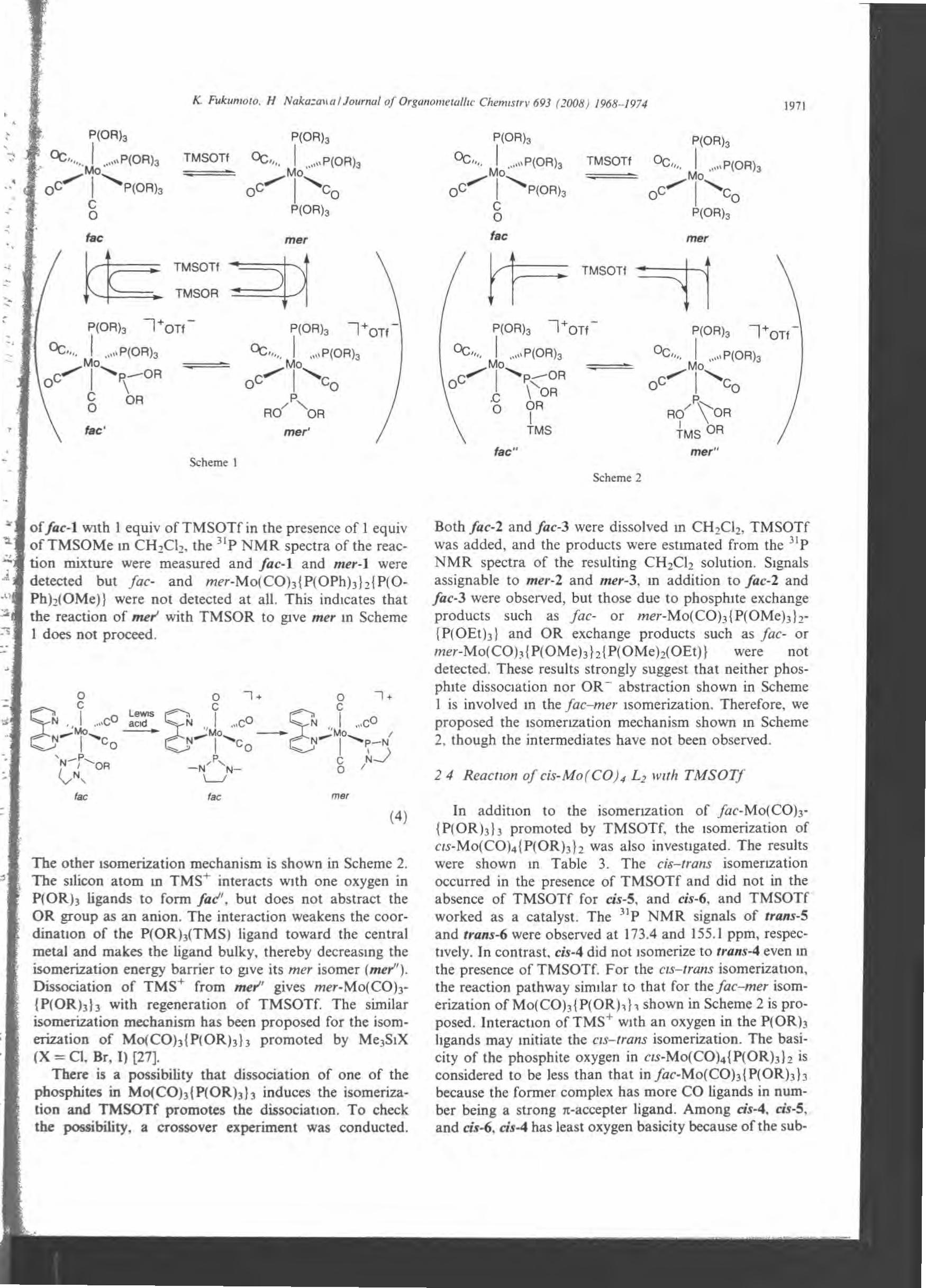
K Fukum010. H Naka:alla!Journal ofOrganometalllC Chemlslrv 693 (2008) 1968-1974 1971 、
,、
、、
-、剛 、、.
‘ 、、-ゐ
.‘ B・
品、.
、、
、主
‘島、ー、
、、、a‘
, 、
、‘e
-、』
P(ORh
h , jJρRb
p〆rlo、、。C- I -P(ORb
c o
抱c
P(ORb
TMSOTf UC",..1川 ¥¥P(ORb
。c/10¥coP(ORb
P(OR)a
cbh j川 P{OR)3
p〆/io、、。じ I ""P(OR)a c o fac
P{ORb
TMSOTf U(で|VLJ,,,, j 川、P(OR)a
0C/ 1
。¥co
P(OR)a
mer mer
TMSOTf TMSOTf
TMSOR 二二〉
P(ORh 寸+OTf-
UC.... . J ..¥¥¥ P(ORb
。c"",• -,。、ヤOR
8bR
P(OR)a寸+OTf-
Oc"" "1 ..¥¥¥ P(OR)a
。c"", "1。、
PぐOR
よ¥"'OR
る ?RTMS
~(ORh 寸 +OTf
cb,,,JJ(OR)3
。c/10¥coP、
RO/ "OR
P(OR)3 寸+OTf-
OC"',.1 .,¥¥¥ P(ORh
。c/)。¥CO
RO/、OR
~uC" OR TMS ‘, 勉c・ mer' 、 危c" mer"
Scheme 1 -、
Scheme 2
-、
offac-l則 th1 equiv of TMSOTf in the presence of 1 equiv ofTMSOMe m CH1Ch, the .1lp NMR spectra of the reac-tion mixture were measured and fac-l and mer-j were detected but fac- and mer-Mo(COh{P(OPhhh{P(O・
Phh(OMe)} were not detected at alL This indlcates that the reaction of ~r' with TMSOR to gtve mer m Scheme 1 does not proceed.
Both fac・2and fac・3were dissolved m CH2Ch, TMSOTf was added, and the products were estrrnated from the 31p NMR spectra of the resulting CH2Ch solution. Slgnals assignable to mer-2 and mer-3, m addition to fac・2and fac・3were observed, but those due tωop凶hos叩ph加llteexchange p戸roωducはts such aωS facひ- 0町r mer
{伊P(ωOEt)川3汁}and OR exchange products such aおsfacひ- or mer-Mo叫(CO)3{P町(OMe吋h汁b孔{P町(0恥Me吋)2(OEt)} we釘re not detected. These results strongly suggest that neither phos-phlte dissoClation nor OR -abstraction shown in Scheme 1 is involved m the fac-mel・lsomerization.Therefore, we proposed the lsomenzation mechanism shown m Scheme 2, though the intermediates have not been observed.
寸
♂
0
.
0C4
0
00-
d
川ぴ
。 i+
q?"co f W〆;~O""""'P_Nも Jb んJ
o / 2 4 Reactwn of cis・MO(CO)4L2 wlth TMSOTf
fac fac 庁lsr
(4) In additlOn to the isomenzation of fac-Mo(CO)r {P(ORhb promoted by TMSOT仁thelsomerization of cls-Mo(CO)4{P(ORhh was also invest1gated. The results were shown m Table 3. The ci8-lrans isomenzation occurred in the presence of TMSOTf and did not in the absence of TMSOTf for cis・5,and cis・6,and TMSOTf worked as a catalyst. The 31 P NMR signals of trans-5 and trans・6were observed at 173.4 and 155.1 ppm, respec-tIvely. 1n contrast. cis-4 did not lsomerize to trans-4 even m the presence of TMSOTf. F or the C1S-trans isomerizatlOn,
the reaction pathway sinular to that for the fac-mer isom-erization of Mo(CO)3{P(OR)汁'¥shown in Scheme 2 is pro・posed. InteractlOn of TMS+ wlth an oxygen in the P(OR)J hgands may mitiate the Cls-Irans isomerization. The basi-city of the phosphite oxygen in clS-Mo(CO)4{P(ORhb is considered to be less than that in fac-Mo(COh{P(ORhb because the former complex has more CO ligands in num-ber being a st10ngπ-accepter ligand. Among ds・4.cis・1and cis・6.cis-4 has least oxygen basicity because of the sub-
The other lsomerization mechanism is shown in Scheme 2. The sllicon atom m TMS+ interacts wlth one oxygen in P(ORh ligands to form J註どにbut does not abstract the OR group as an anion. The interaction weakens the coor-dinatlon of the P(ORh(TMS) ligand toward the central metal and makes the ligand buLky, thereby decreasmg the 也om釘包ationenergy barrier to gtve its mer isomer (mer"). Dissociation of TMS+ from me〆I gives mer-Mo(CO)J-{P(ORh}J with regeneration of TMSOTf. The similar isomerization mechanism has been proposed for the isom-erization of Mo(COh{P(ORhb promoted by Me3S1X (X=α. Br, 1) (27].
There is a possibility that dissociation of one of the pbosphites in Mo(COh{P(ORhh induces the isomeriza-tion and TM以)Tfpromotes白edissociatton. To check the戸溺ibility,a cros:ωver experiment was conducted.

1972 κFukrmlOto. H Naka;awa I JOllrIJa/ oj Organomewllrc Clrem/Hr)・693(2008) 1968.-/974
Table 3 ISOmenZd110n of Mo(CO).dP(ORhb by TMSOTf m CH2CI2 al room
temperature
P(ORh
0cJ Jo p〆γ、、
OC'" I ~P(ORb c o
P(OR)3
oC/I,..l川 cO
oC引~"'COP(ORb
TMSOTf a
ー司司r-ー
P(ORh (I~ Irα/lST
P(OPhh ds-4 franl-4 no redcuon
P(OMeh cIs-5 trans・5 109 PPh2{OMe) cis・6 trallト6 J 2 3
i¥ 1 O. 0 5 and 0.1 equvalents based on the (./s complex were ul>ed b CIS"lrans eqwhbnum ratlo afler complet1on of the IsomenzatJon
stituents (Ph vs. Me). Therefore、cis・4may not have enough basicity on the oxygen to form an mteractlOn wlth TMS"'.
2.5. Reaction offac-Mo(CO)3L3 and cis・Mo(CO)-IL]wlth BF3・OE12
TMSOTf and BF 3・OEt2are effective Lewis acids to
obtam a catlOnic phosphemum complex by an OR amon
abstractlOn from a dlammo-substltuted phosphite ligand in a transition metal complex. In contrast, TMSOTf does
not abstract an OR anlOn from a P(ORh hgand of
fac-M o( CO h {P( 0 R h} 3加 dcis-Mo(CO)4{P(ORhh, but promotes the fac-mer and Cls-trans isomerizatlOn There-
fore, reactions of fac-Mo(COh{P(OMehb (角c・2)and cis-M o( CO)4 {P( 0 Me) 3 } 2 ( cis・5) with BF3・OEt2 were
examined and it was found that BF 3・OEt2causes some cornplicated reactions in additlOn to isornerizahon.
The 31p NMR spectrum of the reaction mixture of fac・2
and an equimolar amount of BF 3・OEt2in CH2Ch showed several unidentified slgnals in addition to signals assignable to mer-2. These signals increased in intensity wlth time but the singlet due to the starting cornplex (fac・2)st111 remained after several hours.
The reaction of cis・5with an equimolar amount of
BF3・OEt2in CH2Ch at room ternperature was foUowed by the 3 J P NMR rneasurernent. After 4 h, in addition to a strong singlet due to cis・5,a doublet of doublet at 164.5pprn (d, J]PF= 1157.1, and 2Jpp=46.9Hz) and a doublet at 163.4 ppm (d, ~Jpp = 46.9 Hz) were observed. The large coupling constant (1157.1 Hz) suggests the exis-tence of a P-F bond and the small coupling constant (46.9 Hz) indicates that two phosphorus ligands are cis to each other. Therefore, the formation of cis-Mo(CO)4{P(O・Meh} {P(OMe)2F} was proposed. The similar OR/F sub-stitutlOll reaction has been reported [6,9,11]. The 31 P NMR spectrum after 24 h, signals due to cis・MO(CO)4・{P(OMeh} {P(OMe)2F} increased in intensity and a new singlet at 173.4 ppm attributable to trans・5was observed. In addition, several unidentified signals were observed.
Therefore, it was found in the reaction of cis・5BF3 .OEt2, relatively fast OMe/F substitution and relativeJy slow cis to trans isomerization and ullldenttfied reactions take place.
. (~れ
3. Experimental -事 f,
3 1 General remarks
Al1 reactlOns were carried out under an atmosphere
dry nitrogen by usmg Schlenk tube techniques. CH2Ch was distilled from CaH2, and hexane and THF were dis-
tilled from sodmm metal. These were stored under ru
atmosphere. Mo(CO)3(NCMe)3 [30] and MO(CO)4(nbd)
[31] were prepared accordmg to the literature methods.
Fac-Mo(CO)3(L)3 and cis・MO(CO)4(Lh (L = phosphite) were prepared by the modification of the published proce・
dures (?8.29]. IR spectra were recorded on a Perkin-Elmer
Spectrum One spectrometer. A JEOL JNM-AL400 spec-trometer was used to obtain 1 H, 13C, and 31 P NMR spec-tra. IH and 1 ~C NMR data were referenced to Me4Sl. 31p
NMR data were referenced to 85% H3P04・
1〔時
~tl ~ ~・'
説、~..
ρi、¥i:ゆl
fg,ddl
F~\ 'j1 ~
,Ii ¥. 1, f. Pi
'4fll
,
,
以""',‘t
3 2. PreparaTlOl1 of fac-Mo( CO) 3{P( OPhh}J (fac・1)
A THF solutlOn (20 rnL) contaimng Mo(CO)3(NCMe)3 (0.52 g, 1.72 rnmol) and P(OPhh (1.36 rnL, 5.19 mmol) was
stlrred for 4 h at room temperature. After volatile matenals were removed under reduced pressure、theresulting precip-
itate was washed with hexane a few times and dned in
vacuo to give a wlute powder of fac・1(1.57 g, 1.41 rnmol,
-820/0). 1 H NMR (ム in CDC13): 6.98-7.18 (m, Ph). 13C{ 1 H} NMR (<5, in CDCh): 121.9 (s, p-Ph), 124.3 (s, m・P旬、 129.4 (s, 0・Ph),152.1 (s, 自フso・Ph),212.2 (m, CO). 31peH} NMR (ム inCDCI3): 145.0 (s). IR (cm-1, m
CHCh): ,, (CO) 1917, 1992.
a--
-LF
hst、肉量、
•• ,
a40
-F JHHV
,
,
•••
‘、‘s ,
.‘ 司
p
・a
・-
•• 島ド、
‘.,
3 3. Preparαtion of fac・MO(CO)3{P(OMe)3}3(fac.・2)
A THF solution (20 mL) containmg Mo(CO)3(NCMe)3 (0.55 g、1.81mrnol) and P(OMeh (0.65 mL, 5.51 mmol)
was stirred for 4 h at roorn temperature. After volatile
materials were removed under reduced pressure, the resulting precipitate was washed with hexane a few times
and dried in vacuo to give a white powder of fac・2
(0.92 g, 1.67 rnmol, 920/0). lH NMR (ム inCDCb): 3.61
(d, 3 JpH = 10.8 Hz、OCH3).13C{ IH} NMR (ム inCDCh):
51.4 (m, OCH3), 215.9 (rn, CO). 31p{ lH} NMR (ム in
CDCI3): 168.0 (s). IR (cm-1, in CDCh):ν(CO) 1880, 1967.
、
,
‘ー
‘
=
、『、
h
!と
th
l
r
t
a
L
H
U円
z
l
h
J
‘1euc
p寸、
6、
民
lp住1A
1w-mU川崎、
4
b
ぃ・hw同札
rkh
m
34. PreparaTion of fiαC-MO(CO)3{P(OEt)3h (fac・3)
A THF solution (20 rnL) containing Mo(COh(NCMe)J (0.39 g, 1.30 mmol) and P(OEt)3 (0.67 mL, 3.90 mmol) was
stirred for 4 h at roorn temperature. After volatile materials
were rernoved under reduced pressure, the resulting precip-itate was washed with hexane a few times at -78 oc組 d
? d l!.Q/c
d

K Fukumoro. H Naka::awa / Journal o/Organomeralli( Chenllstrr 693 (2008) 1968-1974
-t in vacuo to give a wlute powder of fac・3(0.61 g,
.2 mmol, 920/0). 1 H NMR (d, in CDCh): 1.22 (m, 3H, 2CH3)、3.96(m、2H,OCH1CH~). 13C{ lH} NMR (ム
in CDCh): 16.4 (m, OCH1CH3). 59.7 (s, OCl-hCH分、16.4 (m, CO). 3lpeH} N恥1R(d, in CDCl~): 161.2 (s). (cm-I. in CHCh)・ν(CO)1860, 1963.
、守
3.5 Preparation 0/cis-Mo(CO)4{P(OPhh}2 (cis・4)
# • A CH1C12 solution (10 mL)∞ntainmg MO(CO)4(nbd) (0.81 g, 2.46 mmoりandP(OPh)3 (0.83 mL, 4.92 mmol) was stlrred for 4 h at room temperature. After volatile mate-nals were removed under reduced pressure. the resultmg preclpltate was washed with hexane a few tlmes at -78 oC and dned m vacuo to give a whlte powder of cis・4(1.87 g、2.68 mrnol, 92%). 1H NMR (d , in CDCh): 7.18-7.36 (m, Ph). 13C{IH} NMR (<5, in CDCI3): 121.6 (s, p-Ph), 1249 (s, m-Ph), 129.8 (s、0・Ph),151 4 (t, 2 J PC = 42Hz, lpSO-Ph), 205.5 (t, 2JpC = 13.3 Hz, cis-CO), 209.5 (t, 2JpC = 17.4 Hz、trans-CO) 31p{IH} NMR (<5, m CDC13): 151.4 (s). IR (cm-1, in CHCl司)・ v(CO) 1940,2046,
-w
・4
36 Preparatlon 01 C1S・MO(CO)4{P(OMe)J}2(cis-5)
A CH2Ch solutlon (10 mL) contammg MO(CO)4(nbd) (0.73 g、2.21mmol) and P(OMe)J (0.52 mL, 4.42 mmol) was stirred for 4 h at room temperature After volatile materials were removed under reduced pressure, the resu1t-ing precipitate was washed wlth hexane a few tlmes at -78 oC and dned in vacuo to glve a orange stlcky powder ofcis・5(0.97 g, 2.13 mrnol, 96%). I H NMR (ム inCDCI3). 3.62 (s、OCH3).I3C{ 1H} NMR (ム inCDC13): 51 4 (s,
OCH3), 207.9 (t, 2JpC = 14.1 Hz, ClS・CO),212.1 (t, 2 Jpc = 13.3 Hz, trans-CO). 31peH} NMR (d, in CDCh): 165.6 (s) IR (cm-I, m CHCh):ν(CO) 2036, 1921.
3 7 Preparation 01 crs-Mo( COノパPPh2(OMe)h(cis・6)
A CH2Ch solution (10 mL) contammg MO(CO)4(nbd) (0.63 g, 1.91 mmol) and PPh2(OMe) (0.76 mL, 3 82 mmol) was stirred for 3 h at room temperature After volatlle materials were removed under reduced pressure, the result-ing precipitate was washed with hexane a few times at -78 oC and dried m vacuo to give a whlte powder of cis-6 (1.17 g, 1.82 mmol, 960/0). IH NMR (d, m CDC13): 3.27 (s, 3 H, OCH3), 7.40-7.53 (m, 10H, Ph). 13C{IH} NMR (d, m CDC13): 53.6 (s, OCH3), 128.1 (s, p-Ph),
130.1 (s, m-Ph), 131.2 (t, 2JpC = 6.6 Hz, 0・Ph),139.0 (t, 2 Jpc = 15.8 Hz, ipso・Ph),209.5 (t, 2JpC = 10.3 Hz, cis・CO),214.6 (t, 2JpC = 9.5 Hz, trans-CO). 31p{ IH} NMR(ムinCDCI3): 144.9 (s). IR (cm-I, in CHCI3): v (CO) 2026, 1912.
3.8. Isolalion 01 mer-Mo( CO )J{P( OPh) 3}J (mer-l)
A solution of fac・1(2.31 g, 2.08 mmol) and TMSOTf (0.38 mL, 2.08 mmol)凶 CH2Ch (20 mL) was stirred for
1973
1 5 h at room temperature. After volatile materials were removed under reduced pressure. the resultmg preclpltate was washed wlth hexane (3 mL, 5 times) at -780C and dried m vacuo to give a whlte powder of a mlxture of fac・1and mer-l (1:30). To obtain pure mer-J, the powder was washed with hexane/CH2Ch = 100/1 solutlOn (5 mL. 20 times) at room temperature, and the powder was dned in vacuo (1.22 g, 1 I_Q mmol, 53%). 'H NMR (d, in CDCI3) 683-7.25 (m, Ph).リ C{'H} NMR (c5, m CDCh). 121 6 (s句
p-Ph), 124.2 (s、m-Ph),129.4 (s, 0・Ph),151.9 (s, lpso-Ph),
208.0 (m, CO), 2)30 (m, CO). 31p{ IH} NMR (仇 inCDC13): 148.6 (t, 2 Jpp = 46.9 Hz‘equatonal-P), 155.4 (d. 1. Jpp = 469Hz, apical-P). IR (cm -1, m CHCI3) ¥' (CO) 1931,1813
39 X-rα)' cryslα1 Slructure determmαtlOn 01 mer-}
Crystals of mer-l sUltable for an X-ray dl仔'ractionstudy were obtamed through crystallizatlon from CH2CIJhex-ane/benzene for a few days. The smgle crystal was mounted in a glass capillary. Data for mer-I were collected at -70oC on Rigaku/MSC Mercury CCD area-detector dlf-fractometer eqUlpped wlth monochromated Mo K~ radla-tlOn. CalculatlOns for mer-l were performed wlth the teXsan crystallographlc software package of Molecular Structure Corporation. H atoms werc refined using a ridmg model, wlth C-H = 0.95 A, and tixed indlvldual dlsplace-ment parameters [U,so (H) = Ueq(C)]. The crystal was for-mulated as mer-l 0.5CH2Ch・0.5C6H6・ The CH2C12
molecule was dlsordered m two posltlOns wlth 50:50 prob-ablhty m the unit cell.
Acknowledgements
This work was suppo口edby a Grant-m-Aid for Science Research on Prionty Area (No. 18033044、Chemistryof CoordinatIon Space, and No. 19027047, Synerglstic Effect of Elements) from the Ministry of Educatlon, Culture, Sports, SClence and Technology.
Appendix A. Supplementary material
X-ray crystallographlc data m CIF format for mer-l. This material IS available free of charge Vla the Intemet at http://pubs.acs.org. Supplementary data assoclated with thls article can be found、in the online version、atdoi.l0.1 016/j.]organchem.2008.02.024.
References
[1] A H Cowley、RA Kemp. Chem Rev 85 (1985) 367. (2) (a) M Sanchez. M R Mazleres. L Lamande、RWolf. m M Regt旬、
0.1 Scherer (Eds). Multtp1e Bonds and Low Coord1Oatlon m Phosphoru~ Chemlstry. Thleme. New York, 1990 (Chapter 0 1),
(b) A Schmldpeter、 10 M Regl旬、oJ. Scherer (Eds). Multlple Bonds and Low Coordmat1on m Phosphorus Chemlstry. Thleme、New York, 19卯 (Chapter02).
(3) O. Gudat. Coord Chem Rev. 163 (1997) 71

『穣司欝
1974 K FUl..lUfWW. 11λ'OAd=41" tllJU-limol C)( OrKcJ'Wm(Tullu Chf!III/S1rl 693 (~OO8) 196ι1914 l
l'
s
、』
BB.
,,
aa--z
、
l
i
・d"旬m
刊・Z
Et引t
・1z‘‘,
i
s
,.
例“ Na.ka.lllwa.J Orpnon剖Chem61 I (1仮泊)349 {51 H Nakau¥¥A, A小 .Org .. no~l Chem 50 (2ω41 )07 {6) H Nllk.a7~協益。 M Ohta. K M,>osh1. H Yoneda. Organon、euuhcぉ8
1198916'¥8 (7] H N,dauωa、YYamaguchl. K M 1) 0山 .JOrE・!nomt'tChem -U>5
(19941 193 ~8) H N..k低温愉a.Y Y.unaguじtu.T Mllut.t. K Ml'ω1¥1. 0,容dnom~.
1'1lJhC') 14 tl995J 4137 191 y Yum.t~uchl‘ 11 N.u..ll..t鳩.t.T Itoh. K MI)O::.hl. Bul1 Chem So,
Jpn 69 {19%J 98'¥ {IO] H Nak<tLl¥o¥a、Y Yama@uchl. K MηOShl. Pho、phoru!oSulfur SlIlcon
Rclul 日em109 (1996け 29fll] H Naka1A1偽札 y y弘mBgu~hl. K MI\ O~hl. A NagJSd鳩d、Organo・
mel.tlh~ 15 ( t抑 制 1511{l2) K T ..~.mo. JI T刈 munt.1i N..tkaz油W札 M KUf.tk.ttJ. T Hlfdno. 。rB.組"のhlC'talhω19110001H:!J fD) 11 N..lstlA鴨池.Y Ml)O)hl. T Koil的 dnl.t.T Ml1Ut.t. K M ,~ 0ぬ1.N
T >uch,dd. A Ono. K T .sk..nl). Or~..nomelalh山 2~ (封切6159)" [14} lf N..k;u.oll桃.1. M K I~lu~h,t... S YO::.hllld~"‘ y y dma~u~hl. T
MI/Ul.t. K M1)~h,. J Org.tno~t 仁 hem 529 (l9'n1 ~1' {151 11 N..l," A鳩.t.M KJ~hl~htrd . K Mη。、hl.Pho品川Hlru..~ulfur Slhcon
RdJ¥ [Icm 1斜(lml.s ~
{16) H N..ka.la鳩ぬ.M K,!>ht~t・I1 t .l T hhl~ .lm.t . 1 MII'¥Jt.t. K MI¥m.hl. J Orgunotnel Chem 617 -618 ,~()(t ll 45'¥
{l7} 11 Nal.ua~d. 、、.1m品hlt4 K M 1¥ O~hl . Pho~phoru, Sulfur. 511ILon Rcl31 Elem J 77 (20021 I 513
[18) H Ndk..za" 8. Y Yamaguchl. T Ml却 ¥8.S. lchullur~ K. Organomellllhc~ 14 (1995) 4ゐ35
Na~aLCI
(1996) 13'7 [20J H N山Ut".l.Y Y cun.1guchl. K. Kawamufd. K Miyoslu.Or:伊場
metalho J(l (199714616 夜
[21 J K K釧 川伊'Jmu山r以、
( 1999引11“51げ7 ;F{ロ22勾]K Kd 鳩仇恥..1‘.lml汎川u刷u“ . H N 祖laz却awaム‘ K MJ砂yoω畠d山ht】lt. Or暗gB創nom、刊elall加h白
( 1999) 4785 123] H Nd)...lZaW.l. M Klshl~hJt.t . T Nakamo¥o. N Mak.mu問、
lsh l~ ama. K Mlyo~llJ . Chem ~tl (2000, 230 (24) A M Bond. R ('011側、 SW Fdd~rg. P.J Mahon. T.
Orgdnom~tdlhC':, 10 ( 1991) 3320
[25] J・C R()u~che. G R Dobson‘ lnol g Chlm Acta 28 Ll "'9
p叫JA S Ho、),ell.P C Yate~. N F Ashford. D T Dlxon. R Wan-en. C'hem So<:. Ddlton Trans 20 (1996) 3959
[27] K Fukumoto. H Nd~azaw3. Organomelalhc:, 26 (2∞7) (.505 [28] E C AI~もS‘ G F CI gul>on. S -Q Song. Acla Cry!ltaUogr (' 5 J (1
2238 [29] G R Dob、on.A J RettcnOlQler宅 lnorg Chlln ACl.. 6
507 [30] D P T dlC. W R Kmpple. J M Augl. Inor{l. Chem )
413 [~ 1 ) M A scnncll. L Pratt. G Wllkm~on . J Chcm Soc (196112037
au偶MRド
川町刷同い
s 、‘• 、、
•

es-sta菖昆
f'
号
事
)t川tlrl1‘1川11凶川<1刈11ルiトトfοB仏4Il(陀削(~ωmち河川刊111凶】吋iω i凶So閃閃ω》陀陀刈CCd'訓i(e什州州、寸引前h竹、Yv
• Subscnberaccess by OSAKA CITY UNIV
N#CN Bond Cleavage of Cyanamides by a Transition-Metal Complex Kozo Fukumoto, Tsukuru Oya, Masumi Itazaki, and Hiroshi Nakazawa
J. Am. Chem. Soc., 2009,131 (1),38・39・001:10.1 021/Ja807896b・PublicationOate (Web): 10 Oecember 2008
Downloaded from http://pubs.acs.org on January 9, 2009
命 @I , 宅診
。C13¥S向
,/"
ミ~
0漁
About This Article
Additlonal resources and features assoclated with thls article are available withln the HTML verslon:
• Supporting Informatlon • Access to high resolutlon figures • Lmks to artlcles and content related to thls artlcle • Copyright permisslon to reproduce figures and/or text from this article
l v削 theFull Text HTML
ACS Publications High quality. High impact. Joumrll 01 lt1り Am~ r,( ~'I Cnerrll('cll S(><.IPly 1ちjlul¥h、h>edby ttlもんOt'rtc. ltl (' h心fnHcll
SoClely 11日 S川内nlh"tret=>1 l"I W W可:;111(1Ulllllf)C /0036

/一
一ea
F
b
,〆
/,Hf〆
/
!AV--&
人
'
/
が
札FJhvfJ
Jco~広jsPubUshed on Web 12ハ0/2008
N-CN Bond Cleavage of Cyanamides by a Transition-Metal Complex ". 〆、
少智正、
,.. Kozo 印刷moto,Tsukuru Oya, Masumi Itazaki, and Hiroshi Nakazawゲ
Departmenl 0/ Chenuslη" Graduate SchooL 0/ SClellce, Osaka Clty Unwerslt)', SunUVOShl-ku, Osaka 558・8585,Japan
Q Recelved October 6, 2008, E-mall nakazawa@scl osaka-cu ac lP
(-
.‘r-r
are cleaved (entnes 2-6) The reaction
noteworthy (entry 6). The H2N-CN bond has
character because H2N-CN (cyananllde) 18 a tautomer
HN=C=NH (carbodllmlde) Therefore H2N-CN bond JS
than other R2N -CN The first H2N-CN bond cleavage is
m our reactlOn conditions, although the efficJency rem創nsinsuf守
ficlenl
of H2NCN
a double bonds In the last two decades, considerable e仔'ortshave been devoted
to cleavage of unreactlve bonds such as carbonーhydrogen,1
carbon-carbon,2 carbon-nitrogen,3 and carbon-oxygen 4 DJrect
cleavage of these bonds prov1des sever剖 adv加 lagesIs org釦 lC
syntheses、includmgatom efficien句、 lowenvlConmenlalload, and
由epotent1al for unusual chemoselectl vity
" .. ,..‘
I ~ 1
Table 1.. PhotoreactJon of Cyanarmdes Wlth Cp(CO)2Fe(SIE~)Ø
hν alrl R2N-CN + Cp(COhFe(SiEt1)一一一→E句SlCN
In tolucoe
Scheme 1.. Reactlon Pathway of Me-CN Bond Cleavage
一/SIEt3e日
C /
[Fe) 'Me
Me
~IEt3 I N i佐e)'"Iー+
C
Me
?lEt3
[Fe)-N!!!C-Me -ー
5] 30 41
32 26 2(/
14 0 18
銀瞬 ・yleld (%)a ttme (h)
民,,~
222222444
・l'l
・t・-'l'i「44444
e5
陀
mm
げ
北
山
N
吋L山山
NNc
mmmrhにmw
N
Hリ郎
防
口
川
町
m
N
C布
3
l
MJ…・川
問
問
~ω附附洲
町
cqqqNhhH
MU日制
作
M仲件H陀
.MMG,
substrates enlry
121456789
庁2-lm1nωcy
∞mplex ポ-MeCN∞mplex
'11-MeCN complex
[Fe) = CpFe(CO)
,
9
an
u.,t
.~・凶
•• • 1
• ‘、、
)FM¥引いし川、川
rhhhwh匂U
九州旬、処
hAMJmu、
。Yteldof Et,StCN obtarned by GC b In 1,2-dlch1oroe由加e
Cyanamide has a lone palr of electrons on the arruno nitrogen
10 addluon to the cyano rutrogen. Coordination of cy釦 amideto
the 16e Fe spec1es. Cp(CO)Fe(SIEt~), through出earn100 nitrogen,
may reduce the actlvity of the lron complex toward R1N-CN bond
cleavage DerivatlOn of cyanamide mto出eborane adduct at the
ammo mtrogen, R2N(BX3)CN (X = H, F),1 rrught engender more
effectlve R2N-CN bond cleavage because of masking of the lone
p担relectrons on the ammo rutrogen The results (fable 1,印刷出
7-9) showed that出eintroduct1on of borane into cyanamtde dld
not faclhtate R2N-CN bond cleavage, instead, it reduced the
activlty, presumably because of stenc hmdrance. React10n sequences resernbling those in Scheme 1 are expected
for the reacuon of 1 wi出 cyanamide.We attempted to isolate the
N-sllylatedη2-amidino 1fon intermediate, but出erωcllonof 1 wi出
Me2NCN w部 unsuccessfulHowever, reactlOns WJ出 Me2NCNof (CsRs)Fe(CO)(py)(SiR'R" 2) (py = pyridine), considered as a
syn出onof a 16e complex (C'iRs)Fe(CQ)(SiR'R九),8led to isolation of N-sllylatedη2-amidino iron complexes (Scheme 2). Heating a
solutlOn containing 2 and Me2NCN in benzene at 50 oC for 10 h yielded 3 quantitatively ac-cording to the NMR measurements、but
the isolation as a solid fru.led 1n contrast. a reaction of 4 wi出Me2NCN yielded 5, WhlCh can be isolated as a dark-red powder in 85% yield. The unprecedented η2・amidinocomplex was confinned using X-ray analysis (Scheme 2).8 The iron takes a distorted three-legged piano-stool struc同時 withanη2_釦せdinofragment. The bond
distance of N l-C2 (1.327 A) is shorter than血atof a typical N-C 8ingle bond (e.g., C3-Nl = 1.455人C4-Nl= 1458 A) and is
We recently reported reacUons of C-CN bond cleavage o[ organonitnles promo也dusmg a silyl-lron complex 2d The e~~ence of the reactlOn mechanlsm for Me-CN bond c1eavage IS deplcted in Scheme 1 Aαtorutnle coordmates to a 16e sllyl-1ron complex
Cp(CQ)Fe(SlEt3) produced from Cp(CO)2Fe(SIEt3) (1) m a pho・
tor凶 cttonto gt ve anη'-MeCN complex, WhlCh IS converted into
anη2-MeCN complex Then, silyl mlgratlOn from Fe to the rutnle nitrogen atom occurs to form 1J2-lrrunoacyl complex, followed by
C-C bond cleavage on the coordmatlOn sphere to give a methyl-
sIlyhsocyanide complex The η2-coordmation of acetorutnle through
C=N 7r-bond induces sIlyl mJgratlon, whIch then causes仁一CN
bond cleavage The react10n sequences stlmulate us to examtne the pOSSJbility ofR2N-CN (cyananude) bond cleavage by a sllyl-lron
complex because the replacement of the R group in RCN b)加
NR2 group YJelds cyanarrude The R2N-CN bond lS known to be
strong and not broken readJly For example, the N-CN bond length in Me(p-C6l4Cl)N-CN 1S reported to be 1 331 A, whIch ltes Just
between伽 tof a normal N一Csmgle bond (1.47 A)釦 dthat of an N=C double bond (1.27 A) 5 The von Braun reactlon is the only
reaction known to date to cleave R2N-CN bond 6 However, lt
requires harsh reactJOn conditions (strong acid or base condltions) Herein, we describe the first R2N-CN bond cleavage reaction by
transition-metal complex, isolatlOn of an mtermedlate, and establishment of a catalytic cycle involving R2N-CN bond
cleavage. A solution of an eqUlmolar amount of dlmethylcyanamide and
出esllyl-iron complex 1 in toluene was irradiated with a 400-W
medium pressure mercury arc lamp (pyrex filtered) at room temperature for 12 h The lH NMR spec佐umand the GC analysis
of the reaction mixture showed formation of Et3SiCN.百leyleld was 51 % (entry 1 10 Table 1), showing that出eMe2N-CN bond
cleavage could be attained at room temperature using a silyl-iron
complex. Results of reactions wi出 othercyanarnides are presented in Table
1 Although the yields of Et3SiCN are less than 50%, these N -CN
a
@2∞9An鳩山enChe刑囚IS田 tety10.1021/ja807896b CCC: $40.75 38・J.AM. CHEM. SOC. 2009, 131,38-39

COMMUNICATIONS
Catalytlc Cleavagea Under PhotolYSIS or Heatlng Tsble 2.
C31 6 or 7
Me.,NCN + El"¥StH一一一一一El"¥StCNJn loJucnι
condl!Jons
。NoMe~N -CN bond cJeavage took place 10 the absence of 6 or 7 1) Molar ratlo of a transltlon metal complex Me,NCN.釦 dEt、StH'Ca1culated from the Isolated EhS1CN The vaJues are based on the concentratlOn of a tr加lsIt10nmetal complex tlln frec solvent
TOW
04 1 4
0 052 79 323
tlmeJh
24 24 12 12 48 120
lemp/~C
おお初∞∞∞
uhaaaa
[M) (N) [SI)T
1 10 10 1 10 10 1 1 1 1 1 1
1 10 1000 1 10005000
伺 1
正O『,
正
O
『,
『,
司I
enlry
I
I
't「ノ』勾‘,
a且『
CJ〆O
smular to白紙 ofan N-C double bond (e.g・.N2-C2 =し303
). The sum of angJes around N 1 15 359 90. These 5tructural
包'e cons1stent with Sp2 hybndization of N J The
-NI-C2-N2 fragment IS near)y pl釦 arWl出 atorslon ang)e of 2.6(4)0. 80th lH and I3C NMR spectra show that the structure 10
~ solid state IS ma.tntained in solution Two NCH, resonances were
observed at room temperarure 10 t H and I3C NMR、retlectJOgthat
the C2-N 1 bond does not rotate freely at room temperature
Scheme 2. Synthesls of N・Sllylated,,2-Amldlno Iron Complexes
N υ
~ 宅2妥fe
F、,、0"" .!.......N.
ア 'SIRR"', Me
2N •
A5CPC -ルh・2NCN
eeDe10t、
ぜ
lhp
Complexes 3 and 5 were su句ected(Q a thermal reaCUon
Al出ough5 produced a small amount of PhMe2S1CN on heat10g 10 toluene at 1 IO oC for 24 h. 3 gave EtlSiCN 10 62~ yleld on heatlOg
in benzene at 70 oC for 24 h The results show that an N-stlylated
ηl-amJdmo complex is釦 lntermedlate10 the N-CN bond cleavage
of cyan釘rude.
mtermedlate 10 the reactton pathway Cata)yttc N-CN bond
cleavage reacuon was a1so attamed usmg a methyJ molybdenum complex Studles are underway to deslgn translllon-metal catalysts
to improve acttvJty and to eluc1date the reaction mechan1sm
ORTEP drao川 崎of5
N'$相81剖,向mlOl間州、∞m凶ex
R=HR'R"=EI3 R=Me R =Ph R"=Me 5
R" H R・R".Et 2 R=胸R''" pt¥. R" ::胸 4
Acknowledgment. ThlS work was supported by a Grant-lO-AId
for SClence Research on Pnonty Area (No 20036043. SynerglStlC
Effect of Elements) from the Mimstry of Educatlon. CuJture‘Sports、
SC1ence and Technology
¥$ ; ?7 Et3S:判 定~I J I
OC"件ィι ぷ〆[附問M問}
Eγ 宅::?;-矛
拘苦拘N州H J叫,...",--0.... 'SIEt3
宅::?7
dプ~~NMezEゆ H '-...,、--ー
Supporting Information A vailable: Detalled expenmental proce-dures and the charactenzatlon of products ThlS matenal IS ava11able free of charge Vla the Intemet at http //pubs acs org
C? 屯~¥ I
ーニ::-..1M) hv oCゴと¥蜘。
Scheme 3. Proposed CatalytJc Cycle
(1) S民 forexample (a) Rnleng V Slrhn. C Pf~ffer M Chem Reu 2002 /02. 1731-1770 (b) Dyker. G Angt'l¥ Chem 1111 Ed. 1999 38 1698-1712 (c) Naota. T Takaya H. Murabasru. S 1 Chem Reu 1998.98. 2599-2660 (d) zh叩 gY Ll. C J Angt'¥1 Chem. 1m Ed訓)6.451949-1952 (e) Ma K. Plers. W E Parvez M J Am Chem Soc 2刷)6128 3301-3312 (ηKaklUCru F Kan S. 181. K . ChataJll N Mural. S J Am Chem Soc 2003 125. 1698-1699 (g) Goosen L J Ange}¥ Chem. 1m Ed 2002. 41 3775-3778 (h) AmdlSen B A Bergman. R G Mobley. T A Peterson T H Acc Chem Res 1995 28 154-162 (1) Crabtree. R H J Organomet Clzem 2制ト~. 689 4083-4091
(2) See for example (a) Jun C -H Chem 50c Rez- 2ω4. 33. 6ゆる18(b) Rybtduoskl a Mtlstem D Ange¥1 Chem 1m Ed 1999 38. 87ι883 (C) Storsberg. J Yao. M -L.伐 alN. De MelJere. A . Adam、AE W. i<.aufmann. b E Chem Commun 2005 5665-5666 (d) Nakazawa. H . Kawasakl. T . Mlyoshl K. Suresh C H. Kogn N ()rganoml!!al~l_cs ~刷)4.23. 117-126 (e)"Cruanese. A R. Zeghs. B-M. Crabtree. R _!i Chem CommWl 2ω4 2176ー引77(町 Terao~ Y WakU1. H . Satoh. T . M!Ura M. Nomura. M J Am Chem Soc 2001. 123.ゆ407-10408
(3)(a)Lm.Y.WrobleSKI-A D.Golden.J E,Powell-D R.Aube-J J Am Chem Soc 2ω5 J 27, 4552-4553 (b) Fran. L Yang L . Guo. C . Foxman B M. Ozerov. 0 V OrgallomeraJllct 2制胤 23.4778-4787(c) Yao. M L, AdlWldJ司Ja.G. Kanfmann. D E Ange~ Chem .!nt f!i: 2002 _41.3375-3378 (d) Nildas N. Hetnemann. F W. Hampe1. F Alsfasser. R Angt')¥
Chem. /111 Ed 2002,41,3386-3388 (4) See for exarnple (a) Maercker. A Ange"-Chem In_~ E<!: Engl 1987. 2"~.
972-989 (b)' Kabalka. G W Yao:' M -L. BoreUa S 90IOs.. L _ K OrganQmetaIllcs 2007.26.4112-4114 (c) Szu:Y -_h ~ ~e..:.. X ~ao. し L則、H・wAngew Chem.. 1m Ed 2ωS. +4. 6742-6746 (d) Casado. F . Ptsano. L. FamoJ. M包 Gallardo1. Marquet. J • Melloru. G J Org Chem 2000. 65.322-331
(5) Cunmngham, ( D. L1ght. M E. Hurslho_u_se. ~ _ B Acar Cnsrallogr. Sect C Cr¥'ft 5truct Comnllln 1999.55. 1833-1835
(6) Vhet. E B Org Svnrh 1941 1.201-202 (7) Syn山esesof bOran adducl of cyananude and con自nnauonof the adducl al
the anuno s1町ogenhave been repo代edHennelke. H F. Drago. R S lnorg Chem 1968. 7.円08-1915
(8) Selected bond lengths (A)佃 dangles (deg) Ç2-~ l. l_!27(~).. Ç~-~2, 1303(2). C1-Nl, 1455(3). C4-NI. 1 458(3). C~:-NI.-C3_12? ~i2)._C~NI-C4, 1195(2), C司-NI-C4. 1170(2). Fel-C2-N 1. 14940( 17). Fe1-C2-N2. 7811(15). N 1-C2-N2. 1323(1). ,=el-N2-Sd.日404(12).Fe1-N2-C2. 6304(13): StI-N2-C2. 14124(19)
(9)(a)Nakazawa,H,Kamata.K.Itaab.M ChentCommm2805.4004-4∞6 (b) Nakazawa. H . It低成1.M . Kamatn. K.. U吋a.K Chem -Aslan J. 1J.附7.2. 882-888
JA807896B
References 仰1'"Fe 6 Mo(CO) 7
宅三;?;>
ν12N心NM句 ミ~
OC/旬、h---y叩 "'N、SIEt_,C
Et,5r-NC-=→ EI;sSI-CN
Next. we anempted to extend the stOtChlOmetnc R2N -CN bond
cleavage to a catalync reactton We envlsion a catalytic cycle for
Fe and Mo compJexes shown 10 Scheme 3 based on that of R -CN
bond cleavage.9 A 16e siJyl complex A reacts with Me.2NCN to give an N-silylatedη2・amldinocomplex 8. followed by N-CN
bond cleavage to glve C. and dtSSOClatJon of sIlyl tsocyanlde to
gJve a 1仇 amidocompJex D. It may reac( with EhSiH (0 produce
E. then reductive elimination of Me2NH regenerates A to compJele
the ca凶 yticcycle. To produce 16e intermediate A. CpMo(CO)'¥-Me (7) or CpFe(CO)2Me (6) seem to be good precursors because
succむ SlveCO dissoclation. Et)Si-H oxjdatlve additlon. and CH4
reductive ehminallon will gJve A. ResuJts of reactions of Me2NCN and EhSiH m the presence of
a catalytic amount of Fe comp)ex 6 or Mo complex 7 are p陀 sented
in Table 2. Entry I there shows that the Fe compJex does not work 錨 acatalyst, whereas the Mo complex does. under photoiπadlation
conditions (entry 2).百leMo comp}ex shows catalytic actlvity even
under thennal conditions (entries 4-6). In ωnclusion. this paper describes山efirst example of inert
N-CNbondclωvage by a釘arlSitionmetaJ ∞mpJex. An N-silylated η2-amidino complex c釦 beisolated. It was shown to be an
B
Me宣N-C三Nミ』
h
Q
ーハD
一A
C~
E
39 J. AM. CHEM. SOC.・VOL131. NO 1. 2009