re: docket no. fda-2015-d-4848; human factors … · as the device hf final guidance notes, human...
TRANSCRIPT

701 Pennsylvania Avenue, Ste. 800
Washington, DC 20004–2654
Tel: 202 783 8700
Fax: 202 783 8750
www.AdvaMed.org
May 3, 2016
Division of Dockets Management (HFA-305)
Food and Drug Administration
5630 Fishers Lane, Room 1061
Rockville, MD 20852
RE: Docket No. FDA-2015-D-4848; Human Factors Studies and Related Clinical Study
Considerations in Combination Product Design and Development; Draft Guidance
for Industry and Food and Drug Administration Staff
Dear Sir or Madam:
AdvaMed represents manufacturers of medical devices, diagnostic products, and health
information systems that are transforming health care through earlier disease detection, less
invasive procedures, and more effective treatment. Our members range from the smallest to
the largest medical technology innovators.
AdvaMed appreciates the opportunity to comment on the draft guidance entitled “Human
Factors Studies and Related Clinical Study Considerations in Combination Product Design
and Development.” Our general comments are set forth below, and our specific comments
are in the attachment.
The relationship between this and other guidance documents, regulations, and standards:
This draft guidance is meant to complement the recently issued guidance for medical devices
(“Applying Human Factors and Usability Engineering to Medical Devices: Guidance for
Industry and FDA Staff,” issued 2/3/16), referred to hereafter as the “Device HF Final
Guidance.” Since June 2011, industry has relied on this guidance in understanding how to
best apply human factors and usability engineering to our combination products. While we
are pleased to see the collaboration between the Office of Combination Products and the
Centers in preparing this guidance for industry on the special considerations for human
factors information submitted in investigation and marketing applications, we would like to
better understand the relationship between these guidance documents and what additional
information this new guidance provides above and beyond the Device HF Final Guidance as
well as other guidance documents and standards related to human factors. The two FDA
human factors guidance documents are redundant for combination products that are regulated
as devices. It is overly burdensome, for example, to require combination products to conduct
two different risk analyses and submit two different sets of data to FDA.
For combination products, much of the same principles for HF studies apply as for devices,
such as the planning process (identifying intended users and use environments before critical
task identification), the use of commercially-equivalent product in the summative study, and
the simulation of an actual use environment, including training if applicable. It was not

FDA-2015-D-4848
May 3, 2016
Page 2 of 3
apparent why a combination product should be managed differently or why the review
process for these studies should be any different than for a device. Many of the comments in
the attached address some instances where terminology in this guidance is defined differently
than in other documents, or where information found elsewhere could not be found in this
draft guidance. Closer alignment between this guidance and existing documents may help
prevent increase in time or duplication of efforts by both applicants and FDA, for example,
by increasing the scope, formality, and time for investigational and marketing application
reviews.
Considering human factors studies to be a type of clinical study:
The title of this guidance is “Human Factors Studies and Related Clinical Study
Considerations…” (emphasis added). This title and many other instances throughout the
document (e.g., line 22, “the guidance describes how Human Factors studies relate to other
clinical studies” or the use of the adjective “major” before “clinical study” throughout the
document) imply that a human factors (HF) study is another type of, or minor, clinical study.
As the Device HF Final Guidance notes, human factors data can be collected as part of a
clinical study, but a human factors study is not the same as, nor a type of, clinical study.
There are distinct differences between the objectives and endpoints of human factors or
clinical studies. Whereas a clinical study assesses the safety and efficacy/effectiveness of a
drug product or a device for a proposed indication, a HF study of a drug-device combination
product assesses the safety and efficacy of the user interface of a combination product
regardless of the safety or efficacy of the drug product or device. Unless HF data are
collected as part of a clinical study, a HF study does not use the same type of study materials
as does a clinical study: A clinical study involves the clinical use of actual active drug
products or finished devices whereas a HF study uses samples without active drug product or
active drug product use assessed without delivery to a human (e.g., use of an injection
training pad, an aerosol/spray delivery to a controlled space or a device without its drug
constituent part). Most importantly, a HF study has predetermined participant demographics,
may involve a range of user training conditions, and relies on HF behavioral experts to
observe task deviations, user errors, near misses/close calls, and operational difficulties, and
to conduct knowledge probes about them. These study elements are typically not possible in
clinical studies or home use studies.
The regulatory implication of considering a HF Study to be a clinical study is that, for any
device modifications validated with a HF study, filing supplements may be considered to be
efficacy Prior Approval Supplements, with longer review times and submission costs for
combination products regulated as drugs. Additionally, as implied in footnote 7 in the draft
guidance, to consider a HF study to be a type of clinical study would require a case-by-case
decision about whether PDUFA IV User Fees apply. We recommend that the final guidance
address the type of drug or device supplement appropriate for a device change that is
supported with HF study data in a combination product regulated as a drug.

FDA-2015-D-4848
May 3, 2016
Page 3 of 3
Concerns about recommendations for combination products that are more prescriptive than
similar recommendations for devices, and which shift the accountability for design control,
risk assessment, and change control from the applicant to the FDA:
We appreciate that the intent of this guidance to answer questions specific to combination
products, and, as described in the Introduction and Scope, to promote timely review of
combination products. We disagree, however, that the FDA should assume the responsibility
for reviewing design changes, determining whether HF studies are necessary, and
determining appropriate tasks and study users. We would like to see more emphasis on the
use of formative study results. Per 21 CFR Part 4 and 21 CFR §820.30(g), applicants are
already expected to make risk-based decisions that are most appropriate for their products
and target users and validate that the user interface of a combination product is effective via
testing under actual or simulated use conditions. By transferring this accountability from
applicants to the FDA, the FDA takes on an increased burden of time and responsibility for
making decisions, translating to a longer application review than necessary or what is
currently expected for devices.
Thank you for the opportunity to submit these comments.
Sincerely,
/s/
Sharon A. Segal, Ph.D.
Vice President, Technology and Regulatory Affairs
Attachment
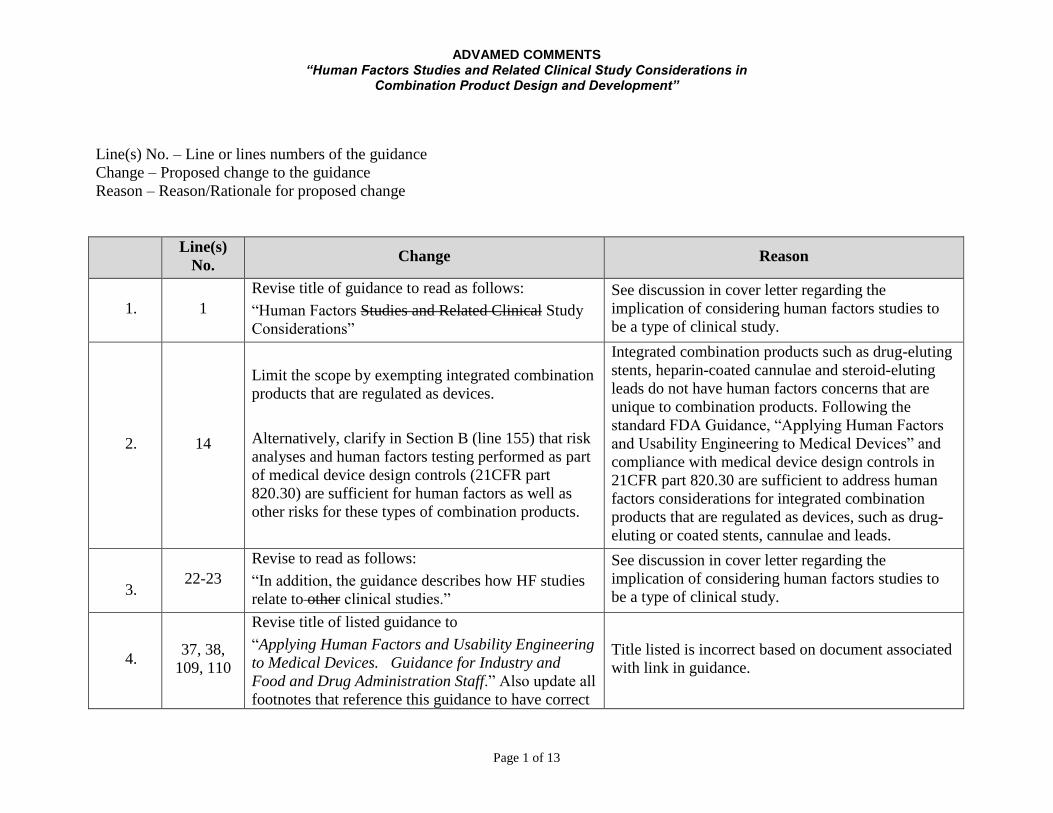
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 1 of 13
Line(s) No. – Line or lines numbers of the guidance
Change – Proposed change to the guidance
Reason – Reason/Rationale for proposed change
Line(s)
No. Change Reason
1. 1
Revise title of guidance to read as follows:
“Human Factors Studies and Related Clinical Study
Considerations”
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
2. 14
Limit the scope by exempting integrated combination
products that are regulated as devices.
Alternatively, clarify in Section B (line 155) that risk
analyses and human factors testing performed as part
of medical device design controls (21CFR part
820.30) are sufficient for human factors as well as
other risks for these types of combination products.
Integrated combination products such as drug-eluting
stents, heparin-coated cannulae and steroid-eluting
leads do not have human factors concerns that are
unique to combination products. Following the
standard FDA Guidance, “Applying Human Factors
and Usability Engineering to Medical Devices” and
compliance with medical device design controls in
21CFR part 820.30 are sufficient to address human
factors considerations for integrated combination
products that are regulated as devices, such as drug-
eluting or coated stents, cannulae and leads.
3. 22-23
Revise to read as follows:
“In addition, the guidance describes how HF studies
relate to other clinical studies.”
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
4. 37, 38,
109, 110
Revise title of listed guidance to
“Applying Human Factors and Usability Engineering
to Medical Devices. Guidance for Industry and
Food and Drug Administration Staff.” Also update all
footnotes that reference this guidance to have correct
Title listed is incorrect based on document associated
with link in guidance.

ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 2 of 13
Line(s)
No. Change Reason
title.
5. 65
Revise to read as follows:
“What is the role of HF studies as compared to other
types of clinical studies?”
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
6.
71,
footnote
7, last line
As applicable, FDA will determine whether a HF
study would meet these criteria.
Whether a HF study would meet these criteria
depends on whether the objectives, interventions, or
study materials are the same as those of a clinical
trial, for instance, if the study protocol involves
actual delivery (e.g., injection) of drug product or
placebo to humans.
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
7. 74
Revise to read as follows:
“maximizing the likelihood that of safe and effective
use of the device by the intended users will be safe
and effective for use by the intended users, for the
intended uses, and for the intended use environments.
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
8. 112
Revise to read as follows:
"1. Human Factors Study (or HF Study): A study of
human capabilities (physical, sensory, emotional, and
intellectual) and limitations to the design and
development of devices, systems, and environmentsA.
A HF study that assesses usability studies the
characteristics of the user interface that establish
effectiveness, efficiency, ease of user learning and
user satisfactionB. Other HF studies include heuristic
reviews, user inquiries, diary studies, and
ethnographic research. HF studies are conducted
The draft guidance’s definition of a Human Factors
(HF) study is narrower than the definition in the ISO
62366 standard (i.e., the ISO standard is inclusive of
more than just usability).
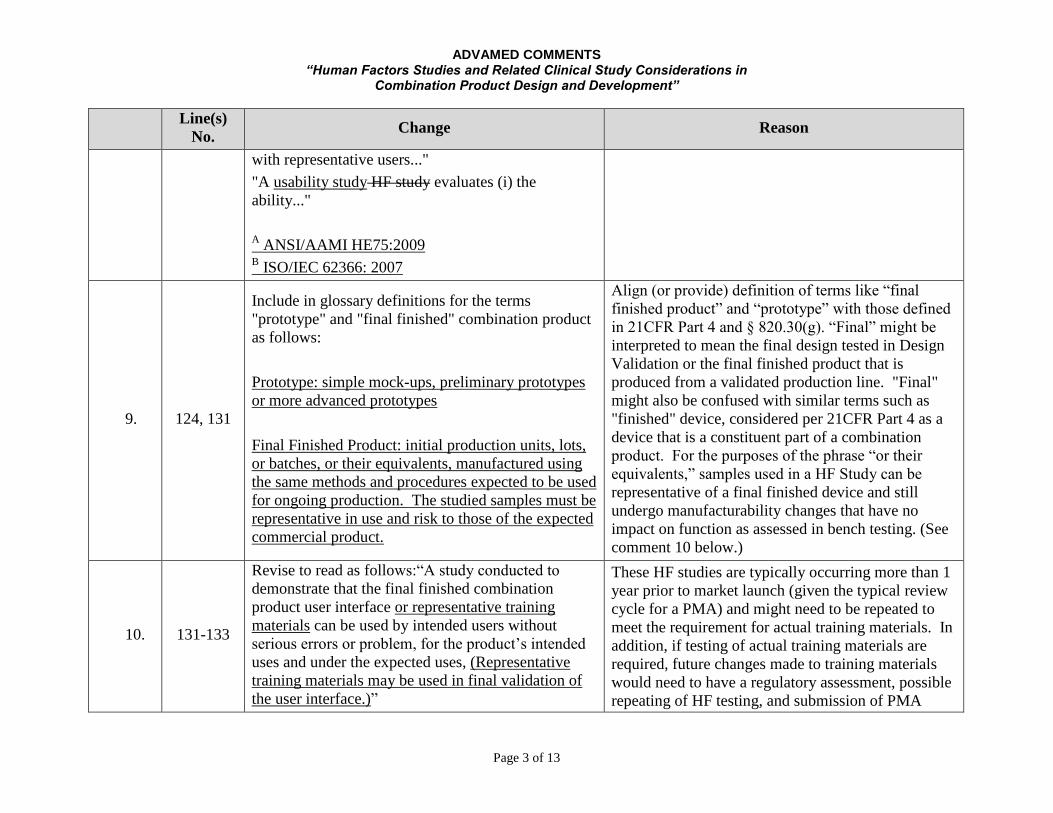
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 3 of 13
Line(s)
No. Change Reason
with representative users..."
"A usability study HF study evaluates (i) the
ability..."
A ANSI/AAMI HE75:2009
B ISO/IEC 62366: 2007
9. 124, 131
Include in glossary definitions for the terms
"prototype" and "final finished" combination product
as follows:
Prototype: simple mock-ups, preliminary prototypes
or more advanced prototypes
Final Finished Product: initial production units, lots,
or batches, or their equivalents, manufactured using
the same methods and procedures expected to be used
for ongoing production. The studied samples must be
representative in use and risk to those of the expected
commercial product.
Align (or provide) definition of terms like “final
finished product” and “prototype” with those defined
in 21CFR Part 4 and § 820.30(g). “Final” might be
interpreted to mean the final design tested in Design
Validation or the final finished product that is
produced from a validated production line. "Final"
might also be confused with similar terms such as
"finished" device, considered per 21CFR Part 4 as a
device that is a constituent part of a combination
product. For the purposes of the phrase “or their
equivalents,” samples used in a HF Study can be
representative of a final finished device and still
undergo manufacturability changes that have no
impact on function as assessed in bench testing. (See
comment 10 below.)
10. 131-133
Revise to read as follows:“A study conducted to
demonstrate that the final finished combination
product user interface or representative training
materials can be used by intended users without
serious errors or problem, for the product’s intended
uses and under the expected uses, (Representative
training materials may be used in final validation of
the user interface.)”
These HF studies are typically occurring more than 1
year prior to market launch (given the typical review
cycle for a PMA) and might need to be repeated to
meet the requirement for actual training materials. In
addition, if testing of actual training materials are
required, future changes made to training materials
would need to have a regulatory assessment, possible
repeating of HF testing, and submission of PMA
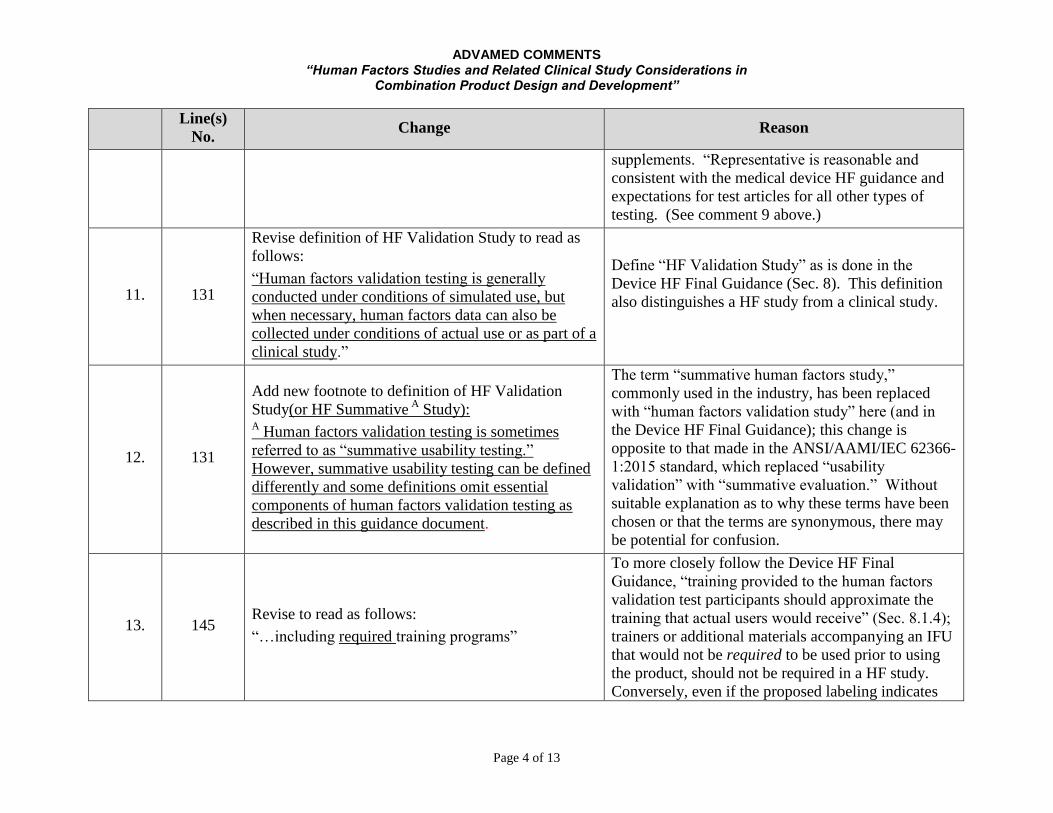
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 4 of 13
Line(s)
No. Change Reason
supplements. “Representative is reasonable and
consistent with the medical device HF guidance and
expectations for test articles for all other types of
testing. (See comment 9 above.)
11. 131
Revise definition of HF Validation Study to read as
follows:
“Human factors validation testing is generally
conducted under conditions of simulated use, but
when necessary, human factors data can also be
collected under conditions of actual use or as part of a
clinical study.”
Define “HF Validation Study” as is done in the
Device HF Final Guidance (Sec. 8). This definition
also distinguishes a HF study from a clinical study.
12. 131
Add new footnote to definition of HF Validation
Study(or HF Summative A
Study): A Human factors validation testing is sometimes
referred to as “summative usability testing.”
However, summative usability testing can be defined
differently and some definitions omit essential
components of human factors validation testing as
described in this guidance document.
The term “summative human factors study,”
commonly used in the industry, has been replaced
with “human factors validation study” here (and in
the Device HF Final Guidance); this change is
opposite to that made in the ANSI/AAMI/IEC 62366-
1:2015 standard, which replaced “usability
validation” with “summative evaluation.” Without
suitable explanation as to why these terms have been
chosen or that the terms are synonymous, there may
be potential for confusion.
13. 145
Revise to read as follows:
“…including required training programs”
To more closely follow the Device HF Final
Guidance, “training provided to the human factors
validation test participants should approximate the
training that actual users would receive” (Sec. 8.1.4);
trainers or additional materials accompanying an IFU
that would not be required to be used prior to using
the product, should not be required in a HF study.
Conversely, even if the proposed labeling indicates
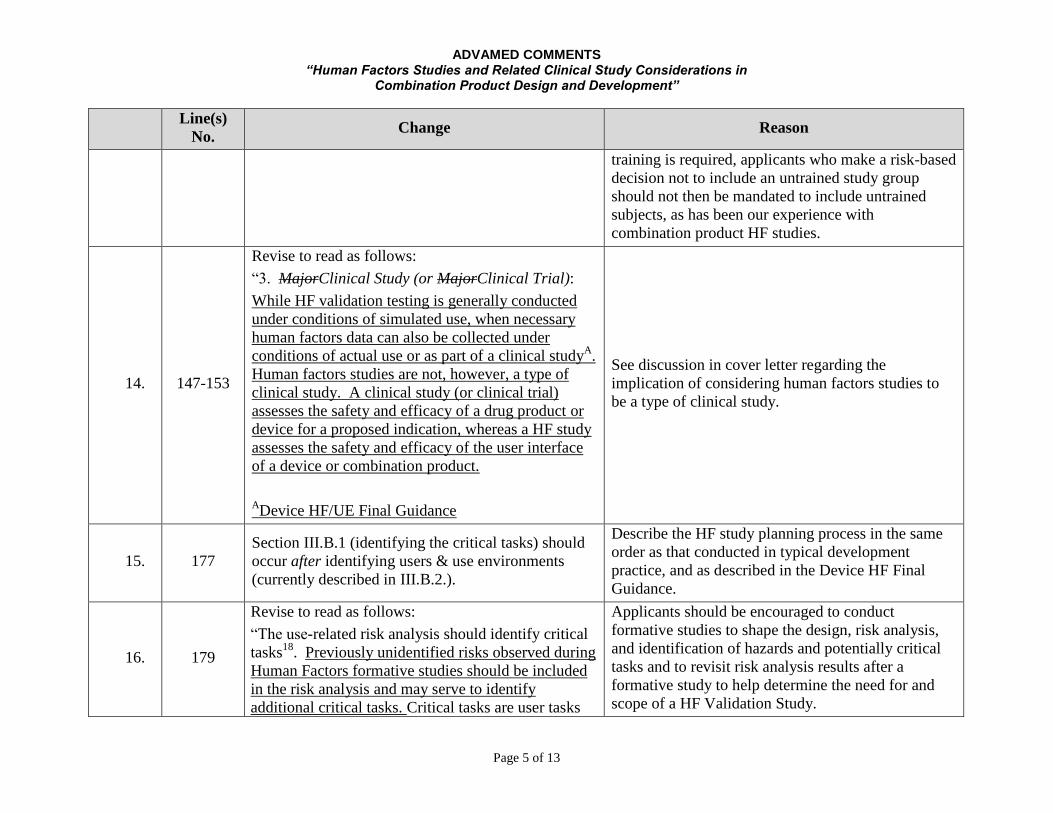
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 5 of 13
Line(s)
No. Change Reason
training is required, applicants who make a risk-based
decision not to include an untrained study group
should not then be mandated to include untrained
subjects, as has been our experience with
combination product HF studies.
14. 147-153
Revise to read as follows:
“3. MajorClinical Study (or MajorClinical Trial):
While HF validation testing is generally conducted
under conditions of simulated use, when necessary
human factors data can also be collected under
conditions of actual use or as part of a clinical studyA.
Human factors studies are not, however, a type of
clinical study. A clinical study (or clinical trial)
assesses the safety and efficacy of a drug product or
device for a proposed indication, whereas a HF study
assesses the safety and efficacy of the user interface
of a device or combination product.
ADevice HF/UE Final Guidance
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
15. 177
Section III.B.1 (identifying the critical tasks) should
occur after identifying users & use environments
(currently described in III.B.2.).
Describe the HF study planning process in the same
order as that conducted in typical development
practice, and as described in the Device HF Final
Guidance.
16. 179
Revise to read as follows:
“The use-related risk analysis should identify critical
tasks18
. Previously unidentified risks observed during
Human Factors formative studies should be included
in the risk analysis and may serve to identify
additional critical tasks. Critical tasks are user tasks
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
scope of a HF Validation Study.
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 6 of 13
Line(s)
No. Change Reason
that…. “
17. 194
Remove this example from the list and/or move to a
separate category: The user being able to safely
dispose of a used syringe. Failure to successfully
perform this task could result in needle sticks.
Applicants should use risk analysis to determine if
steps a user would follow independent of a particular
device (e.g. aseptic technique or standard disposal
practices) are considered critical.
18. 212
Add an additional bullet item
The user being able to appropriately work with a
software application that can affect the product
To include information related to the study of
combination products containing software
components
19. 221-222
Revise to read as follows:
“Intended users may be categorized into distinct user
groups by their different characteristics (e.g., use
responsibilities, tasks performed, age ranges, skills,
or experience levels)” or by different drug
indications, in the case of a drug-device combination
product.”
Similar to how the Device HF Final Guidance
identifies the dependence of usability on
"comorbidities (i.e., multiple conditions or diseases),"
we would like this guidance to point out that an
important characteristic of a user group, particularly
for a drug-delivery combination product, could be the
very disease state for which the product is indicated.
For example, if a hand-held device has been
approved for one indication but is going to treat a
new indication, a disease for which difficulty
grasping is a characteristic of users with that disease,
evaluating the validity of previous usability results
may be warranted.
20. 274
Add the following sentences after “…training.”19
” on
line 274:
”If you anticipate that most or all users would receive
minimal or no training, then the test participants in
the human factors validation test should not be
See comment on Line 145 above.
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 7 of 13
Line(s)
No. Change Reason
trained.” Untrained subjects are not required if it has
previously been determined that untrained users will
have increased difficulty using the product.”
21. 304
Add to Section C a statement that indicates, in the
case that a design problem is solved and the formative
study verifies that the solution has mitigated a
potential user risk, applicants may conclude, in the
formative study report, that the design issue has been
solved and the design is ready for a summative study
(perhaps with a greater or different population of
users), or that no summative study is necessary if
applicable.
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
scope of a HF Validation Study.
22. 312
Revise to read as follows:
“…clinical study, or after finalizing commercial
plans.). The study design should provide for the
identification of any unanticipated hazards or
unexpected use behaviors that were not previously
identified.”
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
scope of a HF Validation Study.
23. 318
Revise to read as follows:
“…the results of HF Formative studies inform the
design of the final finished combination product and
are inputs to the initial risk analysis and identification
of critical tasks.
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
scope of a HF Validation Study.
24. 319
Replace with: “Individual subjects who participate in
HF Formative studies may participate in the HF
Validation study but new participants should also be
recruited in order to avoid the potential for bias”
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 8 of 13
Line(s)
No. Change Reason
scope of a HF Validation Study.
25. 346
Move the following text to just before line 382:
“The study design should provide for the
identification of any unanticipated hazards or
unexpected use behaviors that were not previously
identified.”
This text is applicable to both types of Validation
studies
26. 362 Avoid use of “simulated” to define “actual-use”
study.
Using the term “simulated” to define “actual-use”
testing may be confusing in the attempt to
differentiate the terms “actual use,” “user study,”
(without the “HF” qualifier), and “HF Actual-Use
Validation Study.”
27. 369-380
Specify whether an actual drug would be used, and
whether it would be administered to a human.
Provide an example like that provided in the previous
paragraph.
Enhance description of an actual-use validation
study.
Helpful to avoid multiple interpretations by
applicants or the FDA as to how to interpret what
might be considered a “complex” product.
28. 369
Revise to read as follows:
“The other type of HF Actual-Use Validation study is
conducted in a real environment of use.”
Editorial -- Incomplete sentence, fragment, needs
clarification
29. 440-444
Revise to add footnote at the end of Line 444 as
follows:
“For the following two groups of combination
products, generally human factors data should be
submitted: (1) products for use outside the health care
environment or by laypersons (e.g., home-use
products, products for self-administration by patients
or lay-caregivers) and (2) combination products
Clarify which devices have a “constituent part”
requiring HF data. Suggest referencing the alternate
guidance document.

ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 9 of 13
Line(s)
No. Change Reason
having a device constituent part for which human
factors data should be submitted.[INSERT FOOTNOTE]
”
Footnote: “List of Highest Priority Devices for
Human Factors Review,” Draft Guidance for
Industry and Food and Drug Administration Staff
Feb 3, 2016.”
30. 441
Clarify the exclusion of OTC combination products
by adding a statement to the Introduction and Scope:
“This guidance does not apply to combination
products containing nonprescription drugs marketed
without an approved application (e.g., under a
monograph).”
The first group is combination products for use
outside the healthcare environment, such as at home.
Many types of OTC combination products which are
not reviewed in applications are used outside the
healthcare environment. Therefore, while the Scope
of this draft guidance does state that the focus is
combination products reviewed in an investigational
or marketing application, we recommend adding an
explicit exclusion.
31. 442
Provide further clarification on how an applicant
determines whether that device constituent requires
HFS data would be helpful, for example, by reference
to the Draft Guidance “List of Highest Priority
Devices for Human Factors Review.”
The second group is “combination products that
contain a device constituent requiring HFS data.”
Without this clarification, the second group appears
to overlap with the first group (line 441) since
combination products with a device constituent could
also be used outside the healthcare environment.
32. 440-447
Indicate (within the guidance or in the examples)
whether Design Validation data would be sufficient
for evaluating the usability of certain devices.
Clarify that a risk analysis should be completed for all
products (not just certain types).
For example, for products intended to be used by
medical professionals in a surgical setting (such as
surgeons, circulating/scrub nurses), Design
Validation may be sufficient.
Per QSRs, a risk analysis would be completed for all
combination products during development.
33. 444-445 Revise to read as follows:
“a risk analysis for the combination product should be
Applicants should be encouraged to conduct
formative studies to shape the design, risk analysis,
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 10 of 13
Line(s)
No. Change Reason
completed following formative HF studies and the
use-related risks reviewed to assess the need for HF
Validation (Summative) studies (see section III.B). If
the use-related risk analysis identifies the need for HF
Validation (Summative) studies, then a HF Validation
studies study should be conducted and the results
submitted for review.”
and identification of hazards and potentially critical
tasks and to revisit risk analysis results after a
formative study to help determine the need for and
scope of a HF Validation Study.
34. 476-477
Revise to read as follows:
“based on the use-related risk analysis, applicants
should determine whether differences are sufficiently
obvious to obviate the need for an HF Validation
study may be necessary”
Provide option (but not requirement) to applicants
that if, in the course of the design and development
process, an Applicant determines there is ambiguity
about whether a HF Validation Study is required,
then Applicants can (but are not required) to seek
advice from the FDA via the meeting process.
35. 503-505
Revise to read as follows:
“Applicants should determine, via their risk analysis
procedures, whether However, design changes made
after HF Validation that relate to identified critical
tasks or may result in new use-related errors or
hazards that could lead to harm should have new HF
Validation study assessments.”
Provide option (but not requirement) to applicants
that if, in the course of the design and development
process, an Applicant determines there is ambiguity
about whether a HF Validation Study is required,
then Applicants can (but are not required) to seek
advice from the FDA via the meeting process.
36. 516
Insert before line 516:
If applicants determine there is ambiguity about
whether additional HF testing is needed, they can
(but are not required) to seek advice from FDA.
Provide option (but not requirement) to applicants
that if, in the course of the design and development
process, an Applicant determines there is ambiguity
about whether a HF Validation Study is required,
then Applicants can (but are not required) to seek
advice from the FDA via the meeting process.
37. 522-525
Revise to read as follows:
“If after When making a design change to a
combination product, applicants are unclear what
Adding a time-consuming step of preparing briefing
documents and scheduling/conducting formal
meetings is not feasible when design changes are
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 11 of 13
Line(s)
No. Change Reason
FDA encourages applicants to expeditiously identify
the change plans and to discuss with the Agency the
types of HF and other clinical or non-clinical studies
that may be applicable, FDA encourages applicants to
review the changes with the Agency before the
applicant’s approval of the design changes are
validated.22
typical and frequent in the development process and
when there is already a process in place to manage
these changes. The request for a summary of changes
made to the device during the formative studies (i.e.
during device development) goes beyond the
information typically required by FDA in a device
submission. Development activities during the
iterative design process need not be reviewed as long
as the final device design is adequately supported by
verification and validation activities, including a HF
study. A comprehensive list and discussion of design
changes made in response to these historical studies
and design changes made for other reasons (such as
material or manufacturing changes), may be
burdensome and may not effectively explain why the
final design was chosen and studied in the HF
Validation Study.
38. 523-524
Revise to read as follows:
“HF, and other clinical studies, or non-clinical
studies”
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
39. 528-556
Remove or revise section IV.C
Or,
provide expected review periods more aligned with
IND review requirements in the event an applicant
determines an investigational application is the most
appropriate route for FDA review of a protocol.
Reconsider the expectation that the FDA review HF
information in an IND submission. In our
experience, FDA has routinely requested 3-4 months
for its reviews submitted in an IND information
amendment, and resolution of the FDA’s feedback
when received can and has taken additional months,
whereas IND applications with study protocols have
defined statutory FDA review periods of 30 days
wherein comments from FDA would be expected.
We would like to know if FDA is now planning to
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 12 of 13
Line(s)
No. Change Reason
review HFS protocols in 30 days.
40. 546 Remove requirement to include “intend to market”
labeling.
Per the Device HF Final Guidance, we agree the user
interface of the products tested in the summative
study should represent the final design. However, the
requirement to include "intend to market" labeling
may not be feasible. Regulatory submissions for
combination products regulated as drugs are often
prepared months or years before a product is
launched, and changes may be made in labeling
between the investigational stage and commercial
launch. Applicants should then be able to make the
determination whether the label change warrants any
additional usability testing. Final labeling may not be
in place at such an early investigational application
stage since studies may not have been conducted to
develop them; if labeling is to be included in a HF
study, it may likely be draft labeling. Carton labeling
is not finalized until a product is submitted for
approval, and text font and placement is often revised
by FDA in its final review.
41. 573-574
Provide expanded guidance regarding when a human
factors study data can be leveraged for different
products.
We would like to see guidance for users who wish to
leverage HF study data between:
- products which utilize the same device component
but deliver different drug products
- product that undergoes minor device design
changes that do not impact usability
42. 583
Revise to read as follows:
“HF studies of a combination product are conducted
as part of the product design controls development
21 CFR Part 820.30 does not specifically require a
human factors study.
ADVAMED COMMENTS “Human Factors Studies and Related Clinical Study Considerations in
Combination Product Design and Development”
Page 13 of 13
Line(s)
No. Change Reason
process”)”
43. 586-591
Revise to read as follows:
“However, the HF Validation study is not sufficient
to establish the safety and effectiveness use of the
combination product for the proposed indication.”
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
44. 593-599
Add a comment that HF study could be, but is not
required to be, conducted in parallel or after a clinical
study.
The results of one study would not impact the other
as HF studies and clinical studies have different
measures and outcomes.
45. 596
In cases where HF data could be collected from a
clinical study, provide further guidance (than what is
stated here) regarding what methods are
recommended for collecting these usability data.
See discussion in cover letter regarding the
implication of considering human factors studies to
be a type of clinical study.
46. 602
Revise to read:
.” .. combination product may be adequate without
the inclusion . . . “
Editorial -- Missing the word “be.”