raymond steptoe senior research fellow diamantina institute for cancer, immunology and metabolic...
Post on 22-Dec-2015
215 views
TRANSCRIPT

Raymond SteptoeRaymond SteptoeSenior Research Fellow Senior Research Fellow
Diamantina Institute for Cancer, Immunology and Metabolic Diamantina Institute for Cancer, Immunology and Metabolic MedicineMedicine
University of Queensland, Princess Alexandra Hospital University of Queensland, Princess Alexandra Hospital
[email protected]@uq.edu.au
Tumour Tumour immunotherapyimmunotherapy

Overview
• History
• Review of immune effectors & immunity
• Basis for immunotherapy
• Passive (Adoptive) immunotherapy
• Active immunotherapy
• Overview of a clinical trial (DC therapy)

HistoryImmunotherapy: actively enhance immune response or passively
deliver immune effectors
1890’s Coley’s toxins ( streptococcus/staphylococcus)
Surgical intervention
Radiotherapy
Chemotherapy
1960’s – 70’s Understanding of role of immune system in animal models
1980’s Mixes of tumour cells and bacteria (e.g BCG) Cytokines: IFN-alpha, interleukin 2 Adoptive transfer of in vitro activated T cells
1990’s Peptide and recombinant antigen vaccines Gene-engineered tumour cell vaccines Dendritic cell vaccines

Why immunotherapy?Rationale is based on:
• evidence from mouse models -immune-compromised mice have increased incidence of cancers-immunisation induces tumour-specific immunity & reduces tumour mass/tumour growth
• clinical observations -spontaneous regressions-immunodeficiency increases some cancers-immune infiltrates –better prognosis-tumour specific T cells can be isolated
Use effectors of the immune system to kill tumours

Tumour antigens -immune targets Tumours are ‘altered self’
Tumour antigens are usually self-proteins selectively over expressed by a tumourcell type specific (e.g)
melanoma - MART-1/MelanA, tyrosinase , gp-100B cell lymphoma - idiotype, CD20AML - CD33Prostate cancer – PSA, prostatic acid phosphatase
shared (e.g.)MAGE-3Carcinoembryonic antigen (CEA)HER2/neu
Peptides defined from molecular approaches
Whole proteins defined by responses in tumour-bearing individuals
Non-selective methods (eluted peptides, fusions)

Effectors of the immune system
Modified from: Schuster et al., Biotechnology J. 2006. 1:138-
CellularCD4+ T cell-produces cytokines-helps for CD8+ T cells and B cells
CD8+ T cell (CTL)-direct lysis/killing of antigen-expressing cells
B cell -produces antibody (Ab)
Granulocyte- Ab-dependent cytotoxicity
Macrophage-cytokine-induced killing-Ab-dependent cytotoxicity
Natural killer cell-direct lysis of tumour cell target-Ab-dependent cytotoxicity
MolecularCytokine-direct tumour killing (e.g. TNF-)Antibody-coating of tumour cell – ADCC, CDC
Macrophage

Immune induction/effector pathways
Modified from Banchereau et al., Ann Rev Immunol. 2000
constitutive traffickingof dendritic cells
NAÏVE T CELLS
Priming and recirculation of effector T cells
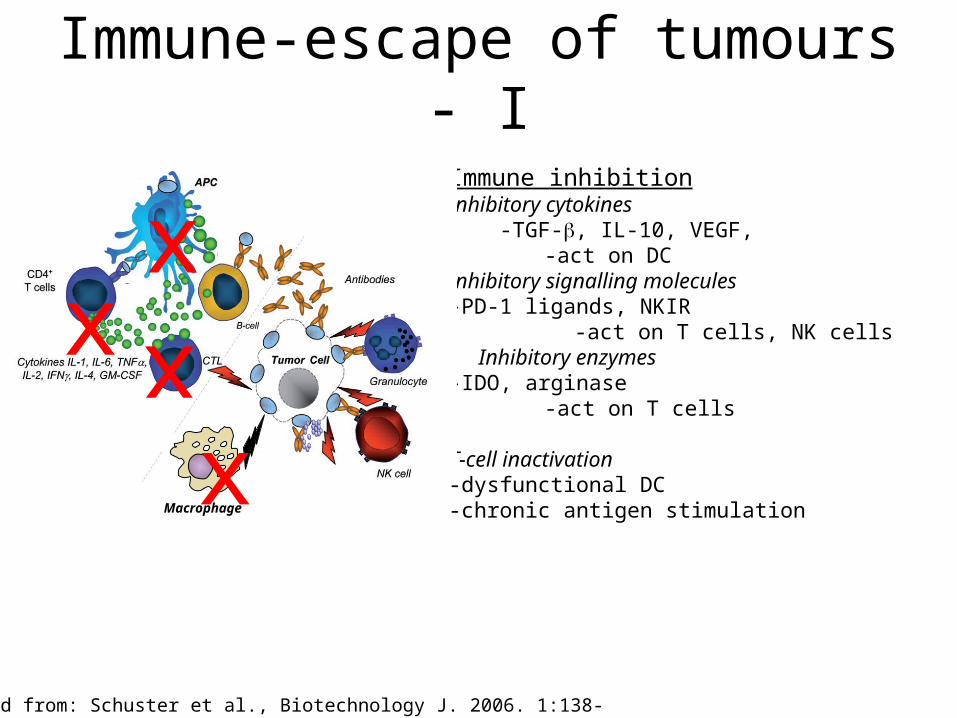
Immune-escape of tumours - I
Immune inhibitionInhibitory cytokines -TGF-, IL-10, VEGF, -act on DCInhibitory signalling molecules-PD-1 ligands, NKIR
-act on T cells, NK cellsInhibitory enzymes
-IDO, arginase-act on T cells
T-cell inactivation -dysfunctional DC-chronic antigen stimulation Macrophage
xxx
x
Modified from: Schuster et al., Biotechnology J. 2006. 1:138-

Macrophage
xx
xxx
Immune-escape of tumours - II
Antigenic loss variants
Loss/down regulation of antigen targets -Tumour Specific Antigens
-loss of CD4+ & CD8+ T-cell epitopes -CD20
-loss of antibody binding (Rituximab)
Loss of MHC class I / antigen processing -MHC class I expression -TAP etc. (for processing /loading)
- loss of CD8+ T cell recognition
Modified from: Schuster et al., Biotechnology J. 2006. 1:138-

Immunotherapy - purpose
• actively enhance immune response or passively deliver immune effectors
-boost impaired components
-replace missing elements

Passive (adoptive) immunotherapy
Transfer of efferent elements of the immune system
Effector T cells in-vitro activated T cells
Antibodies -surface antigens CD20 Non-Hodgkins lymphoma -growth factors / receptors HER2/neu breast cancer VEGF colorectal cancer
Macrophage
Effector T cellsAntibody

Passive (adoptive) immunotherapyAdoptive antibody therapy - targets surface molecules expressed or over-expressed by tumour cells
Antibody-dependent cytoxicity (ADCC), complement-dependent cytotoxicity (CDC) – cells are killed by these mechanismsCD20 Rituximab Non-Hodgkins lymphoma (NHL)CD33 Gemtuzumab Acute myelogenous leukemia (AML)CD52 Alemtuzumab Chronic lymphocytic leukemia (CLL)
Disruption of signalling through receptors or growth factors -prevents growth of cells
HER2/neu Herceptin Breast cancerVEGF Avastin Colorectal cancer (CRC)EGF-R Cetuximab Colorectal cancer (CRC)
-limitations-loss of antigen expression- large quantities required/expensive- surface molecules only – limits repertoire

Passive (adoptive) immunotherapyAdoptive cellular therapy (ACT) -provides an exogenous source of anti-tumour T cells
Patient’s own T cells are activated in vitro and retransferred -tumour specificity generated by:
-using defined tumour-specific antigen-tumour infiltrating lymphocytes
-most effective for highly immunogenic tumours-melanoma-EBV-induced post-transplant lymphoproliferative disorder-allogeneic HSCT for acute myelogenous leukemia
-may be boosted by concurrent immunisation etc.
-can target intracellular proteins, more diverse targets than antibody
-limitations-persistence of transferred cells
(overcome by lymphodepletion)-diverse specificities required-experimental procedure
Rosenberg et al. Nat Rev Cancer, 2008

Active immunotherapyAdjunctive therapy-promotes immune responsiveness
Immune-stimulatory cytokines-interleukin-2 (IL-2)
-boosts function of T cells, NK cells -interferon-2b (IFN-2b )
-mechanism unclear Limitations:
-side effects-limited effectiveness
Suppression of immune inhibitors-lymphodepletion (promotes expansion of antigen-specific T cells)-anti-CTLA4 (prevents inactivation of T cells)-anti-PD-L1 (prevents inactivation of T cells)
-limitations:
-experimental

Active immunotherapyVaccination (therapeutic) -boosts ‘ineffective’ T cell responses
Whole tumour vaccines
-tumour cells poorly immunogenic so immunogencity must be increased-addition of BCG-addition of adjuvants-use of allogeneic tumour cells -gene-engineering of tumour cells -cytokines-GM-CSF,
-costimulatory molecules B7
-evidence of T-cell priming often apparent in vitro, but with little clinical effect
-limitations-modest clinical effects-under development

Active immunotherapyVaccination (therapeutic) -boosts ‘ineffective’ T cell responses (and induces de-novo responses?)
Specific antigen vaccines -a range of tumour-specific antigens have now been defined (see Kim et al., - best prospects are those that are widely expressed in tumours
-synthetic peptide fragments-recombinant proteins -DNA/RNA
- delivery vectors -’conventional’ adjuvants- viral delivery-dendritic cells
-evidence of T-cell priming often apparent in vitro, but with little clinical effect
-limitations-modest clinical effects-under development

Active immunotherapyVaccination (prophylactic) -primes responses in a ‘naïve’ immune system and generates protective immunity
-limited applications
Gardasil (Merck) & Cervarix (GSK) – cervical cancer -exploits known features of the human papilloma virus life-cycle.
HPV-induced cervical cancer requires in infection with HPV (6,11,16,18 etc.)
-immunisation with virus-like particle containing HPV E6, E7 induces strong neutralising antibody responses and prevents HPV infection.
-limitations-not all pathogenic HPV serotypes targeted-cancer must be virally induced

OverviewApproved immunotherapies primarily passive strategies
Development of active immunotherapy has been slow-limited somewhat by stage of disease treated (ie late/advanced disease)
Immunotherapy (primarily) is considered an adjunct to ‘conventional’ therapies and particularly for clearing minimal residual disease of metastases
Noteable success have been very profound RituximabGardasil (Cervarix)

DC vaccines
Overview of a clinical trial

Considerations for DC vaccines
• Generation
• Antigen loading / maturation
• Administration
• Migration
• T-cell activation
• Monitoring: clinical markers/surrogate markers
• Quality control

Generation of DC for vaccine use• In-vitro conversion of monocytes
GM-CSF/IL-4 + maturation cocktail (IL-6, IL-1,TNF-, PGE2)
• Expansion from blood CD34+ stem cellsGM-CSF/TNF-
TNF- matures DC
labour intensive, expensive, QC issues
• Harvest from blood with / without growth factor-induced mobilisation
maturation procedure
limiting cell number obtained
DC are generated from each individual (because of MHC differences between individuals)
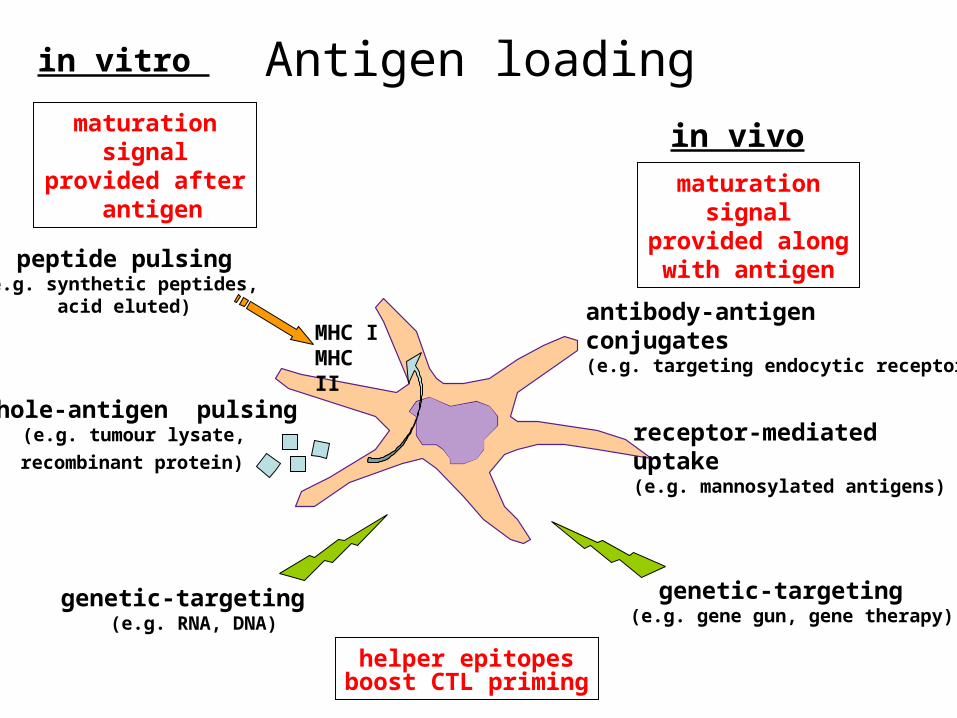
Antigen loadingin vitro
in vivo
peptide pulsing(e.g. synthetic peptides,
acid eluted)
whole-antigen pulsing(e.g. tumour lysate,
recombinant protein)
genetic-targeting (e.g. RNA, DNA)
genetic-targeting (e.g. gene gun, gene therapy)
antibody-antigen conjugates(e.g. targeting endocytic receptors)
receptor-mediated uptake(e.g. mannosylated antigens)
MHC IMHC II
helper epitopesboost CTL priming
maturation signal provided along
with antigen
maturation signal provided after
antigen

DC immunotherapy using autologous tumour cell antigens at PAH
Melanoma cell culture
DC culture
GM-CSF + IL-4
HBsAgPulse DC

Administration / Migration of DC vaccines
RoutesSubcutaneous:-requires correct coordinated
migration pattern
Intranodal -direct admin to lymph nodes
Dose?
Injection of DC vaccine induces a systemic anti-tumour response
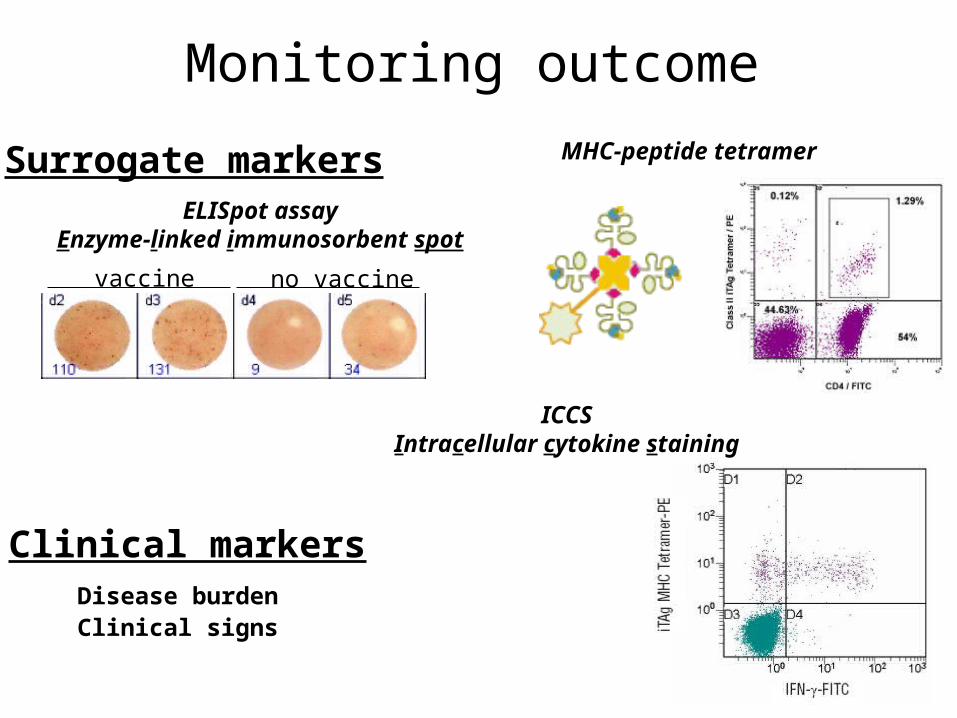
Monitoring outcome
no vaccinevaccine
ELISpot assayEnzyme-linked immunosorbent spot
MHC-peptide tetramerSurrogate markers
ICCSIntracellular cytokine staining
Clinical markersDisease burdenClinical signs

Clinical Response: +ve HBsAg response
Age/sex Site Dose (DCx106/kg)
Vaccines Resp Duration (months)
35F Liver, s.c. 0.05 5 PD - 67M skin 0.02 5 PD - 50M s.c., adrenal, LN 0.05 8 PR 3 40M s.c 0.07 8 CR 29+ 62M s.c., spleen, LN 0.09 8 PD - 75F Pelvis, lung 0.09 8 PR 8 48F s.c. 0.11 7 PD - 45F s.c. 0.07 8 PD - 60M Lung, s.c. 0.07 8 SD 2+ 39F Pelvis, LN, bowel 0.07 7 PD -
Resp: PD: patient died, PR: partial remission, CR: complete remission

Clinical Response: no HBsAg response
Age/sex Site Dose (DCx106/kg)
Vaccines Resp Duration (months)
41F LN 0.02 8 PD - 37F Abdomen, s.c.,
adrenal, spleen, lung 0.02 3 PD -
66M s.c. 0.02 3 PD - 43M s.c., brain 0.05 8 PD - 57F Liver, abdomen,
mediast., s.c., bone 0.07 4 PD -
52F Liver, lung 0.11 6 PD - 72M s.c. 0.07 5 PD - 44M Lung, spleen, s.c. 0.07 8 PD - 63M Adrenal, lung 0.07 8 PD - 71M Lung 0.07 5 PD - 31F LN, bowel 0.07 5 PD - 33M LN, lung 0.07 5 PD -
Resp: PD: patient died, PR: partial remission, CR: complete remission

Monitoring clinical outcome
Insert vitiligo
halo nevi
vitiligo hair depigmentation

Monitoring clinical outcome
ON ENTRY
4 MONTHS
Pelvis Chest

Overview- DC therapy
DC therapy :
Safety – very good
Outcome - not 100% effective (~ 20%) -disease and stage-dependent

Key points to remember
‘Approved’ immunotherapies primarily passive strategies
Development of active immunotherapy has been slow -still experimental
- large range of tumour-specific antigens defined for some tumours
-limited somewhat by stage of disease treated (ie late/advanced disease)
Immunotherapy (primarily) is considered an adjunct to ‘conventional’ therapies and particularly for clearing minimal residual disease of metastases
Noteable success have been very profound
Rituximab
Gardasil

Further reading
• Immunotherapy of melanoma:Fang et al., Journal of Investigative Dermatology, 128:2596- (2008).Kirkwood et al., Journal of Clinical Oncology, 26:3445- (2008).
• Adoptive T cell therapy:Rosenberg et al., Nature Reviews Cancer, 8:299-, (2008).
• DC therapy:Banchereau & Palucka, Nature Reviews Immunology, 5:296-, (2005).