rapamycin-induced endothelial cell death and...
TRANSCRIPT

Rapamycin-Induced Endothelial Cell Death and Tumor VesselThrombosis Potentiate Cytotoxic Therapy againstPancreatic Cancer
Christiane J. Bruns, Gudrun E. Koehl,Markus Guba, Maksim Yezhelyev,Markus Steinbauer, Hendrik Seeliger,Astrid Schwend, Anna Hoehn,Karl-Walter Jauch, and Edward K. GeisslerDepartment of Surgery, University of Regensburg, Regensburg,Germany
ABSTRACTPurpose: Despite current chemotherapies, pancreatic
cancer remains an uncontrollable, rapidly progressive dis-ease. Here, we tested an approach combining a recentlydescribed antiangiogenic drug, rapamycin, with standardgemcitabine cytotoxic therapy on human pancreatic tumorgrowth.
Experimental Design: Tumor growth was assessed inrapamycin and gemcitabine-treated nude mice orthotopi-cally injected with metastatic L3.6pl human pancreatic can-cer cells. H&E staining was performed on tumors, alongwith Ki67 staining for cell proliferation and immunohisto-chemical terminal deoxynucleotidyl transferase-mediatednick end labeling and CD31 analysis. Rapamycin-treatedtumor vessels were also directly examined in dorsal skin-foldchambers for blood flow after thrombosis induction. Celldeath in human umbilical vein endothelial cells was assessedby flow cytometry after annexin-V staining.
Results: Rapamycin therapy alone inhibited tumorgrowth and metastasis more than gemcitabine, with remark-able long-term tumor control when the drugs were com-bined. Mechanistically, H&E analysis revealed tumor vesselendothelium damage and thrombosis with rapamycin treat-ment. Indeed, dorsal skin-fold chamber analysis of rapamy-cin-treated tumors showed an increased susceptibility of
tumor-specific vessels to thrombosis. Furthermore, terminaldeoxynucleotidyl transferase-mediated nick end labeling/CD31 double staining of orthotopic tumors demonstratedapoptotic endothelial cells with rapamycin treatment, whichalso occurred with human umbilical vein endothelial cells invitro. In contrast, gemcitabine was not antiangiogenic and,despite its known cytotoxicity, did not reduce proliferationin orthotopic tumors; nevertheless, rapamycin did reducetumor proliferation.
Conclusions: Our data suggest a novel mechanismwhereby rapamycin targets pancreatic tumor endotheliumfor destruction and thrombosis. We propose that rapamy-cin-based vascular targeting not only reduces tumor vascu-larization, it decreases the number of proliferating tumorcells to be destroyed by gemcitabine, thus introducing a new,clinically feasible strategy against pancreatic cancer.
INTRODUCTIONPancreatic cancer remains a major unsolved health problem
with an estimated overall 5-year survival rate of only 1–4%,making it one of the leading causes of cancer-related mortality.Presently, over 80% of these patients have locally advanced ormetastatic disease at the time of diagnosis, which excludes eventhe possibility of curative surgery (1–3). Moreover, tumor con-trol in these cases is not normally successful with currentlyavailable systemic chemotherapy. In fact, a response rate ofone-quarter or less can be expected with standard chemotherapy,with a dismal median survival of �6 months (4, 5). With thisbackground, the question is what different approach, besidesstandard cytotoxic therapy, could be used to attack this aggres-sive, highly resistant form of cancer. One key to this questioncould lie in the emerging realization that pancreatic tumors maybe susceptible to antiangiogenic therapy (6–9).
Indeed, recent clinical studies suggest that pancreatic can-cer is highly angiogenesis dependent. More specifically, clinicalprognostic data indicate that expression of proangiogenic factorssuch as vascular endothelial growth factor (VEGF), epidermalgrowth factor, and thymidine phosphorylase positively corre-lates with a higher relapse rate and shorter patient survival (10,11). Furthermore, a high density of microvessels within pancre-atic tumors is a prognostic factor for early disease progression(10, 12, 13). Therefore, we hypothesized in the current studythat pancreatic cancer progression may be sensitive to antian-giogenic therapy, particularly when combined with a cytotoxicagent. With regard to antiangiogenic therapy, we chose to testwhether the mammalian target of rapamycin inhibitor rapamy-cin could be effective against metastasizing pancreatic cancer.This choice was based on our recent study showing that rapa-mycin is a potent antiangiogenic substance, working most ef-fectively at noncytotoxic, nanomolar concentrations (14). Theantiangiogenic activity of rapamycin is due, at least in part, to
Received 10/30/03; revised 12/11/03; accepted 12/16/03.Grant support: Grants from the Roche Organ Transplantation ResearchFoundation and the Deutsche Forschungsgemeinschaft (Grant BR 1614/3-1).The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.Note: C. Bruns and G. Koehl contributed equally to this work. Presentaddress for C. Bruns, M. Guba, M. Yezhelyev, H. Seeliger, and K. Jauchis the Department of Surgery, Ludwig-Maximilians University, Munich,Germany.Requests for reprints: Edward K. Geissler, Department of Surgery,University of Regensburg, Franz-Josef-Strauss-Allee 11, 93053 Regens-burg, Germany. Phone: 49-941-944-6964; Fax: 49-941-944-6886; E-mail: [email protected].
2109Vol. 10, 2109–2119, March 15, 2004 Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

inhibition of VEGF production and blockage of VEGF-medi-ated stimulation of endothelial cells. However, a clinically rel-evant corollary to this initial study was that nests of tumor cellsnot requiring angiogenesis continued to exist and eventuallyprogressed into larger masses once the rapamycin therapy wasdiscontinued. Therefore, in the present study, we tested thepossibility that the combination of cytotoxic chemotherapy withrapamycin could better control or reduce these nests of tumorcells over a long-term period. In the situation of pancreaticcarcinoma, our approach combines daily rapamycin treatmentwith repeated use of the best available cytotoxic drug for thisdisease, gemcitabine. Mechanistically, intracellular phosphoryl-ation of gemcitabine produces di- and triphosphate molecularforms capable of acting as a fraudulent base in DNA andinhibiting DNA synthesis-dependent ribonucleotide reductase(15), together producing a strong cytotoxic effect.
Using a model of metastatic human pancreatic cancer innude mice, our present study shows that antiangiogenic therapywith rapamycin alone has an antitumor effect exceeding that ofgemcitabine and that the combination of rapamycin and gem-citabine dramatically reduces long-term tumor growth and thedevelopment of metastases. Mechanistically, our data suggestthat rapamycin affects tumor vascularization and decreases thenumber of proliferating tumor cells, thereby enhancing the ef-fectiveness of gemcitabine’s cytotoxic activity against tumorgrowth. Moreover, this study provides the first evidence thattumor control achieved with rapamycin is associated with tumorvessel thrombosis related to the death of endothelial cells.Therefore, rapamycin promotion of thrombosis in new pancre-atic tumor vessels introduces a novel mechanism potentiallycontributing to its anticancer action.
MATERIALS AND METHODSPancreatic Cancer Model and Treatment Regimens.
The highly metastatic human pancreatic cancer cell line L3.6plwas maintained in cultures supplemented as described previ-ously (16). Using animal procedures approved by the localauthorities, 1 � 106 L3.6pl tumor cells were orthotopicallyimplanted in the subcapsular region of the pancreas of maleathymic 8–12-week-old nude mice (BALB/c nu/nu; CharlesRiver, Sulzfeld, Germany), as detailed previously (16). Afterimplantation, tumors were allowed to grow for 7 days beforetreatment initiation. At the start of treatment, the median tumorvolume in sacrificed mice is typically 18 mm3 (17). Tumor-bearing mice were randomized and subjected to the followingtreatment: (a) 1.5 mg/kg/day rapamycin (5 mg/ml stock solu-tion; Wyeth Pharma, Munster, Germany) by i.p. injection; (b)biweekly 50 or 100 mg/kg gemcitabine (Gemzar 1000 powderdissolved in 0.9% saline; Lilly, Giessen, Germany) by i.p.injection; (c) i.p. combination of 1.5 mg/kg/day rapamycin witheither 50 or 100 mg/kg gemcitabine biweekly; or (d) i.p. injec-tions of 0.9% saline control solution at corresponding timepoints (rapamycin and gemcitabine were diluted for injectionwith 0.9% saline).
Mice were sacrificed on day 28 after tumor cell injection inexperiments aimed at measuring tumor growth at a fixed point.Excised pancreatic tumors were weighed and measured. Thetumor volume was then calculated using the formula V �
�/6(a � b � c), where a, b, and c represent the length, width,and height of the mass. For H&E staining and immunohisto-chemical analysis, half of the primary tumor was fixed informalin for paraffin embedding, and the other half was pre-pared for frozen sectioning. Metastatic L3.6pl tumor growth wasalso evaluated. For metastases in the liver, macroscopicallyvisible tumor nodules (�1 mm) were noted on the liver surface.Furthermore, enlarged regional (celiac and para-aortic) lymphnodes were recorded. Liver and lymph node tissue were excisedand processed to confirm metastases by H&E staining.
In one experiment, all mice in the control group and 6 of 10mice from each treatment group were sacrificed as usual on day28 after orthotopic tumor cell injection. The pancreatic tumorand metastases were analyzed as described above. However, theremaining four mice in each treatment group were kept alive toobtain long-term data, and drug therapy was continued. Thosemice in good condition were kept alive until day 60; any miceshowing progressive tumor growth, signs of tumor burden, drugtoxicity (weight loss � 20%), or reduction in mobility to easilyaccess food and water were sacrificed. To monitor cancer pro-gression, the tumor mass was held between the fingers andmoved to the abdominal surface, where its size could be meas-ured using a caliper. Tumor volume was estimated by theformula V � �/6(a2 � b), where a is the width of the tumor, andb is the length of the tumor.
Immunohistochemical Staining for Ki67, Terminal De-oxynucleotidyl Transferase-Mediated Nick End Labeling(TUNEL), and CD31. Cell proliferation analysis was per-formed on paraffin-embedded tissues with standard Ki67 stain-ing techniques (18, 19). Briefly, a mouse antihuman Ki67 mono-clonal antibody (DAKO A/S, Glostrup, Denmark) was used inthe primary reaction. The DAKO EnVision System, containinga secondary horseradish peroxidase-conjugated antimouse anti-body complex, was used with 3,3�-diaminobenzidine to detectKi67. Sections were counterstained with Gill’s hematoxylin. Toquantify the amount of proliferation, all Ki67-positive and -neg-ative cells were counted in 10 random high-power fields (0.159mm2 at �100 magnification) per slide.
Colorimetric immunohistochemical staining for apoptoticcell death (TUNEL) was performed on paraffin-embedded tis-sue sections using the In Situ Cell Death Detection Kit (RocheDiagnostics, Mannheim, Germany) and the AEC substrate pack(Biogenex, Hamburg, Germany), according to the manufactur-ers’ instructions.
Analysis of apoptotic endothelial cells was performed onfrozen tissue sections using a previously described immunoflu-orescent CD31/TUNEL double-labeling technique (17). Briefly,sections were first incubated with a rat antimouse CD31/plate-let/endothelial cell adhesion molecule 1 monoclonal antibody(PharMingen, San Diego, CA), followed by staining with Texasred-conjugated goat antirat IgG (Jackson ImmunoResearch Lab-oratories, West Grove, CA). A TUNEL procedure was subse-quently performed using the Fluorescein Apoptosis DetectionSystem (Promega, Madison, WI).
Dorsal Skin-Fold Chamber (DSFC) Analysis. Tumorangiogenesis was analyzed in vivo via the transparent DSFCmodel, as described previously (20, 21). Chambers were inoc-ulated with 1 � 105 L3.6pl cells. The day after tumor inocula-tion, mice were treated i.p. with saline or 1.5 mg/kg/day rapa-
2110 Rapamycin-Induced Thrombosis Targets Pancreatic Cancer
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
mycin. On day 7, intravital microscopy (Zeiss Axiotech Variomicroscope; Gottingen, Germany) was performed on DSFCs toexamine tumor blood vessels. The entire tumor was examined,and these images (7–15 images/tumor) were recorded on videofor analysis (modified Sony 3CCD Color Video Camera; AVTHorn, Aalen, Germany). Vessel diameter was measured usingImage J software (from Wayne Rasband; Version 1.25s; NIH,Bethesda, MD) by generating horizontal grid lines every 50pixels. Tumor vessels crossing the grid lines were individuallymeasured, whereas vertically aligned vessels were not includedin the analysis.
Blood flow in tumor vessels in DSFCs was measureddirectly using a modified thrombosis induction technique (22).In principle, i.v. injected FITC-dextran (Mr 464,000; Sigma-Aldrich Chemicals, St. Louis, MO), when activated by pro-longed UV light irradiation, causes oxidative stress by free-radical production as well as activation of the thrombosiscascade (22). In our experiments, L3.6pl tumors were allowed togrow in DSFCs of nude mice for 7 days, with or withoutrapamycin treatment (1.5 mg/kg/day). Mice then received injec-tion via the tail vein with 0.5 ml of 5% FITC-dextran dissolvedin PBS. At the same time, mice also received i.v. injection with8 � 107 red blood cells that had been labeled with a redfluorescent stain (Red Fluorescent Cell Linker Kit; Sigma-Aldrich Chemicals). The fluorescent red blood cells could beeasily seen flowing through blood vessels in the tumors of theDSFCs by intravital microscopy. Phototoxic UV (Zeiss filter setEX BP 450–490, BSFT 510, EM BP 515–565) light wasdirectly applied to a vascular area of the tumor through a �20objective lens for 1 min, resulting in a dose of 1010 mW/cm2.Then, the vascular architecture was observed for 30 s undernormal bright-field light, followed by 30 s of RBC flow obser-vation under filtered light for red fluorescence (Zeiss filter setEX BP 546/12, BS FT 580, EM LP 590). The cycle of photo-toxic, bright-field, and red fluorescent light was repeated up toa maximum of 20 times. When all of the blood vessels withinthe area showed total occlusion (no blood flow), this time pointwas recorded, and light cycles were discontinued. In addition,normal vascular areas clearly outside the tumor region wereanalyzed in the same way.
In Vitro Cell Proliferation Assay. L3.6pl cells werecultured for 48 h in 96-well microtiter plates in medium with orwithout rapamycin or gemcitabine. Proliferation was assessedby adding bromodeoxyuridine (bromodeoxyuridine prolifera-tion kit; Roche Diagnostics GmbH, Mannheim, Germany) to
individual wells 4 h before completion of the 48-h incubationperiod and then measuring absorbance at 450 nm.
Fluorescence-Activated Cell-Sorting Analysis for CellDeath. Human umbilical vein endothelial cells (HUVECs)were cultured under normal conditions with endothelial cellbasal medium (PromoCell, Heidelberg, Germany) supplementedwith growth factors (PromoCell) and 2% fetal bovine serum, orthey were placed under minimal culture conditions, where cellswere deprived of fetal bovine serum and other supplements.Under supplement and serum-deprived conditions, recombinanthuman VEGF165 (R&D Systems, Wiesbaden, Germany) wasadded at a concentration of 50 ng/ml in the presence of increas-ing concentrations of rapamycin. After 8 h, HUVECs wereremoved from the culture dishes with gentle trypsinization,labeled with annexin V-FITC (R&D Systems), and analyzed byflow cytometry.
Statistical Analysis. Data are given as the mean � SEMin quantitative experiments. For statistical analysis of differ-ences between the groups, an unpaired Student’s t test wasperformed with InStat 3.0 Statistical Software (Graphpad Soft-ware, San Diego, CA).
RESULTSGrowth and Metastasis of Established Pancreatic Tu-
mors. To determine the potential for rapamycin treatment in apancreatic cancer situation, athymic nude mice received ortho-topic injection with metastatic human L3.6pl cancer cells. Pan-creatic tumors were allowed to become established for 7 daysbefore initiation of rapamycin or gemcitabine treatment. Stand-ard doses of rapamycin (1.5 mg/kg/day) and gemcitabine (100mg/kg, 2�/week) were used in the first group of experiments,and all animals were sacrificed 28 days after tumor cell injec-tion. All control mice and treated animals did develop primarypancreatic tumors, but the growth and extent of tumor progres-sion depended on the treatment regimen. Standard pancreaticcancer treatment with gemcitabine alone resulted in a significantreduction in the pancreatic tumor volume, compared with con-trol mice (Table 1). Interestingly, and unexpectedly, rapamycintreatment alone reduced tumor volume 2-fold more than stand-ard gemcitabine therapy. Furthermore, when rapamycin andgemcitabine treatment were combined, tumors were very small,growing to only 19% of the size observed with gemcitabinetreatment alone. Mice tolerated rapamycin well (0.8 � 1.6%weight gain), with animals treated with gemcitabine alone or
Table 1 Effect of rapamycin and gemcitabine on established human L3.6pl pancreatic tumors in nude miceAll values were obtained from mice sacrificed 28 days after orthotopic tumor cell injection.
Treatment(started 1 week after tumor cell injection)
Orthotopic pancreatic tumor Metastases (incidence)
Incidence Volume (mm3) Liver Lymph node
Saline (control) 8/8 1672 � 144 4/8 8/8Gemcitabine (100 mg/kg, 2�/week) 9/9 773 � 117a 2/9 9/9Rapamycin (1.5 mg/kg/day) 10/10 388 � 5a,b 2/10 5/10Rapamycin � gemcitabine 9/9 147 � 23a,b,c 1/9 0/9
a P � 0.00002 versus saline-injected controls.b P � 0.01 versus gemcitabine.c P � 0.001 comparing rapamycin � gemcitabine treatment versus rapamycin alone.
2111Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
rapamycin � gemcitabine experiencing some weight loss duringtherapy (7.7 � 1.6% and 12.7 � 1.9%, respectively).
Metastasis of pancreatic tumors was also affected by thedifferent treatment regimens. Lymph node metastases were re-duced by rapamycin treatment and completely eliminated bycombination therapy with rapamycin and gemcitabine, but treat-ment with gemcitabine alone did not reduce the incidence ofthese metastases (Table 1). On day 28, macroscopically visibleliver metastases were present in 50% of controls, and this tendedto be reduced in frequency by all three treatment regimens, withcombination therapy giving the lowest incidence.
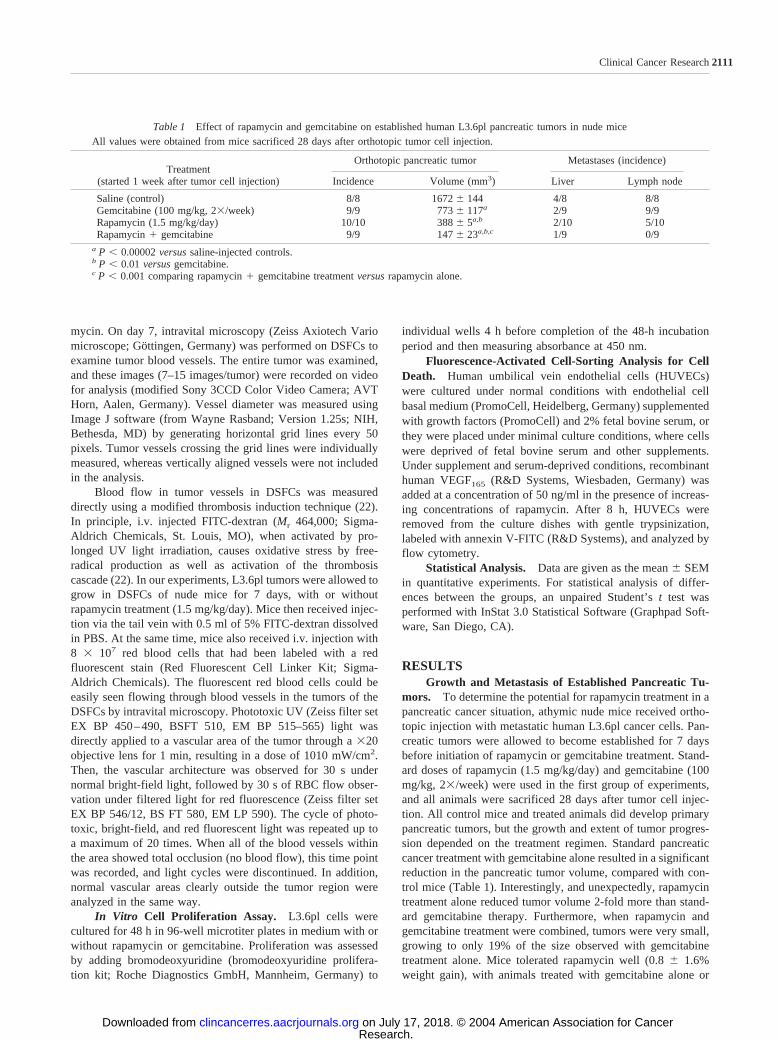
A second group of similar experiments was performed totest whether a lower dose of gemcitabine could be effective andsustained long-term. Results showed that in mice sacrificed at28 days, low-dose gemcitabine (50 mg/kg) inhibited tumorgrowth to the same degree as the higher dose (100 mg/kg; Fig.1A). However, combination therapy using high-dose gemcitab-ine combined with rapamycin did lead to a slightly greaterreduction in tumor volume, compared with the rapamycin com-bination with low-dose gemcitabine (P � 0.001). In these sameexperiments, all controls were sacrificed on day 28 because oftheir deteriorating condition, but 4 of 10 drug-treated mice werecontinued on therapy for as long as 60 days to determinelong-term effects (Fig. 1B). All mice on single-agent therapy orhigh-dose rapamycin � gemcitabine had to be sacrificed by day53 because of either tumor progression or therapy side effects.In contrast, all mice on low-dose gemcitabine � rapamycintherapy tolerated the treatment well and survived throughout theobservation period. Moreover, this treatment group showed anaverage total weight loss of �10% at day 60; between day 28and day 60, animal weight remained quite stable in this group(weight loss � 5%). Importantly, tumor growth estimationsmade by in vivo palpation measurements showed that the pan-creatic tumor volume remained stable in these mice between day40 (211 � 49 mm3) and day 60 (218 � 54 mm3), which is alsonearly identical to measurements made in sacrificed animalsfrom the same group on day 28 (Fig. 1A).
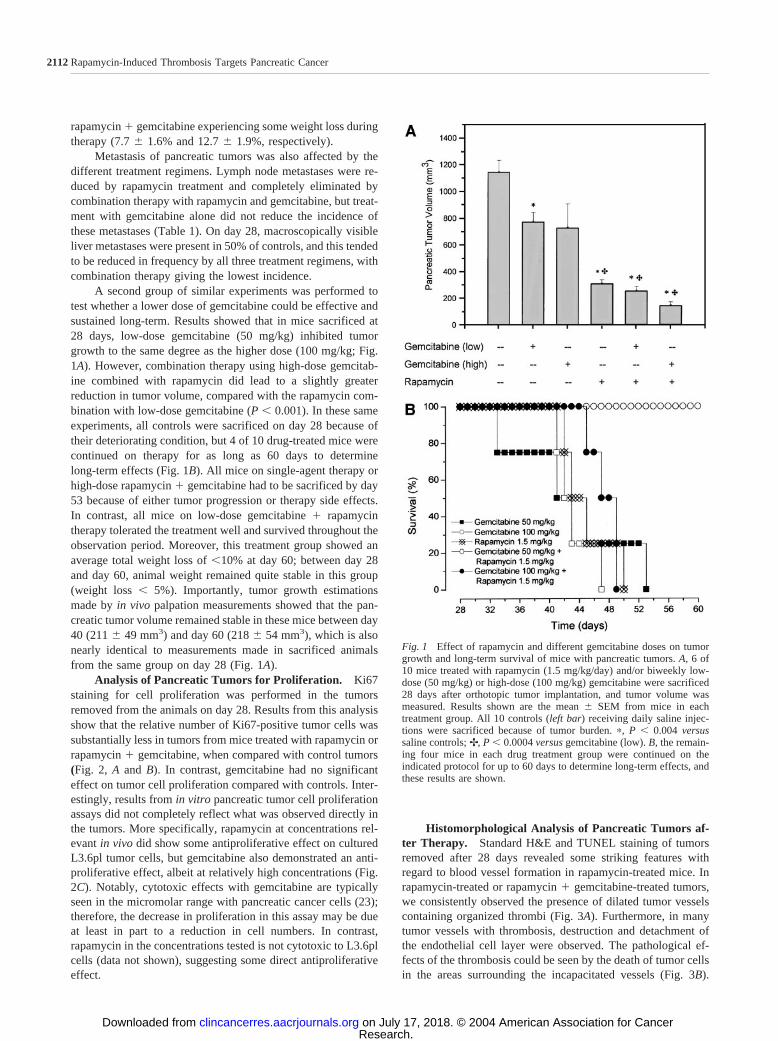
Analysis of Pancreatic Tumors for Proliferation. Ki67staining for cell proliferation was performed in the tumorsremoved from the animals on day 28. Results from this analysisshow that the relative number of Ki67-positive tumor cells wassubstantially less in tumors from mice treated with rapamycin orrapamycin � gemcitabine, when compared with control tumors(Fig. 2, A and B). In contrast, gemcitabine had no significanteffect on tumor cell proliferation compared with controls. Inter-estingly, results from in vitro pancreatic tumor cell proliferationassays did not completely reflect what was observed directly inthe tumors. More specifically, rapamycin at concentrations rel-evant in vivo did show some antiproliferative effect on culturedL3.6pl tumor cells, but gemcitabine also demonstrated an anti-proliferative effect, albeit at relatively high concentrations (Fig.2C). Notably, cytotoxic effects with gemcitabine are typicallyseen in the micromolar range with pancreatic cancer cells (23);therefore, the decrease in proliferation in this assay may be dueat least in part to a reduction in cell numbers. In contrast,rapamycin in the concentrations tested is not cytotoxic to L3.6plcells (data not shown), suggesting some direct antiproliferativeeffect.
Histomorphological Analysis of Pancreatic Tumors af-ter Therapy. Standard H&E and TUNEL staining of tumorsremoved after 28 days revealed some striking features withregard to blood vessel formation in rapamycin-treated mice. Inrapamycin-treated or rapamycin � gemcitabine-treated tumors,we consistently observed the presence of dilated tumor vesselscontaining organized thrombi (Fig. 3A). Furthermore, in manytumor vessels with thrombosis, destruction and detachment ofthe endothelial cell layer were observed. The pathological ef-fects of the thrombosis could be seen by the death of tumor cellsin the areas surrounding the incapacitated vessels (Fig. 3B).
Fig. 1 Effect of rapamycin and different gemcitabine doses on tumorgrowth and long-term survival of mice with pancreatic tumors. A, 6 of10 mice treated with rapamycin (1.5 mg/kg/day) and/or biweekly low-dose (50 mg/kg) or high-dose (100 mg/kg) gemcitabine were sacrificed28 days after orthotopic tumor implantation, and tumor volume wasmeasured. Results shown are the mean � SEM from mice in eachtreatment group. All 10 controls (left bar) receiving daily saline injec-tions were sacrificed because of tumor burden. �, P � 0.004 versussaline controls; ✢, P � 0.0004 versus gemcitabine (low). B, the remain-ing four mice in each drug treatment group were continued on theindicated protocol for up to 60 days to determine long-term effects, andthese results are shown.
2112 Rapamycin-Induced Thrombosis Targets Pancreatic Cancer
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
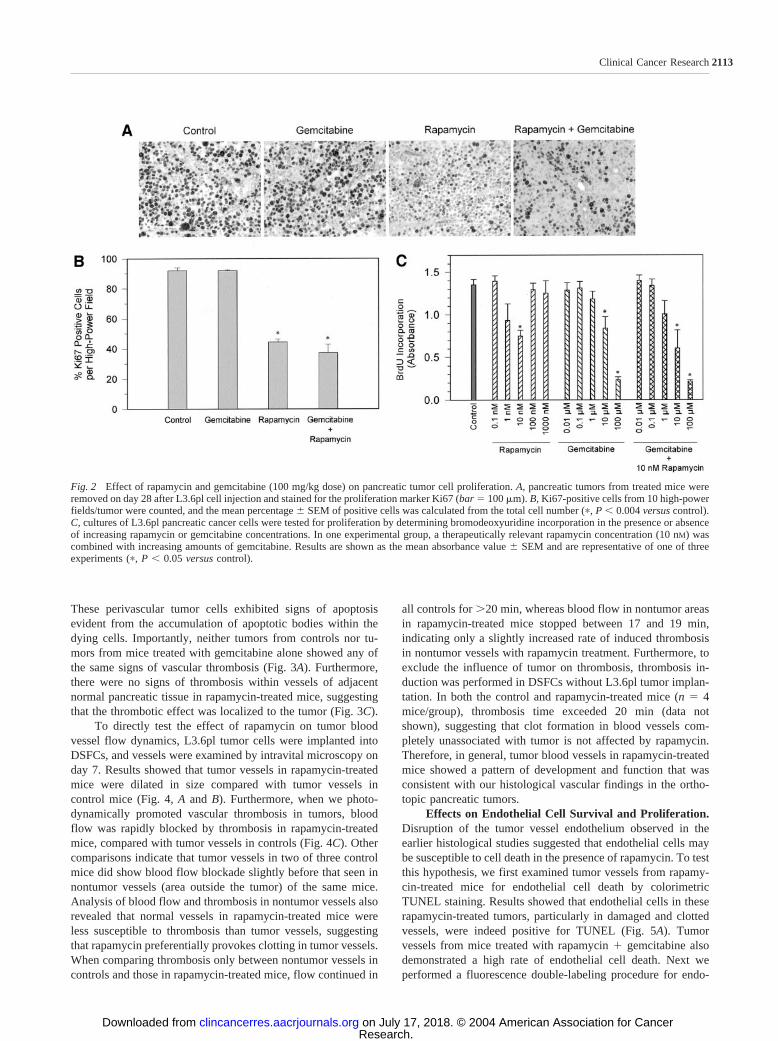
These perivascular tumor cells exhibited signs of apoptosisevident from the accumulation of apoptotic bodies within thedying cells. Importantly, neither tumors from controls nor tu-mors from mice treated with gemcitabine alone showed any ofthe same signs of vascular thrombosis (Fig. 3A). Furthermore,there were no signs of thrombosis within vessels of adjacentnormal pancreatic tissue in rapamycin-treated mice, suggestingthat the thrombotic effect was localized to the tumor (Fig. 3C).
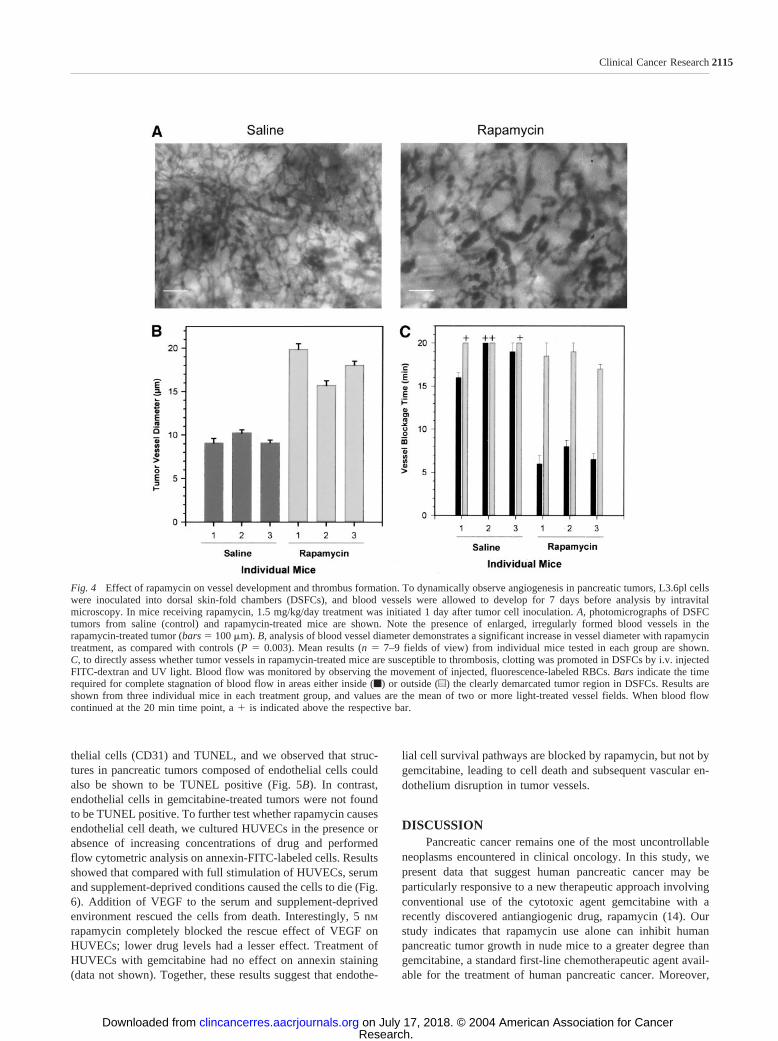
To directly test the effect of rapamycin on tumor bloodvessel flow dynamics, L3.6pl tumor cells were implanted intoDSFCs, and vessels were examined by intravital microscopy onday 7. Results showed that tumor vessels in rapamycin-treatedmice were dilated in size compared with tumor vessels incontrol mice (Fig. 4, A and B). Furthermore, when we photo-dynamically promoted vascular thrombosis in tumors, bloodflow was rapidly blocked by thrombosis in rapamycin-treatedmice, compared with tumor vessels in controls (Fig. 4C). Othercomparisons indicate that tumor vessels in two of three controlmice did show blood flow blockade slightly before that seen innontumor vessels (area outside the tumor) of the same mice.Analysis of blood flow and thrombosis in nontumor vessels alsorevealed that normal vessels in rapamycin-treated mice wereless susceptible to thrombosis than tumor vessels, suggestingthat rapamycin preferentially provokes clotting in tumor vessels.When comparing thrombosis only between nontumor vessels incontrols and those in rapamycin-treated mice, flow continued in
all controls for �20 min, whereas blood flow in nontumor areasin rapamycin-treated mice stopped between 17 and 19 min,indicating only a slightly increased rate of induced thrombosisin nontumor vessels with rapamycin treatment. Furthermore, toexclude the influence of tumor on thrombosis, thrombosis in-duction was performed in DSFCs without L3.6pl tumor implan-tation. In both the control and rapamycin-treated mice (n � 4mice/group), thrombosis time exceeded 20 min (data notshown), suggesting that clot formation in blood vessels com-pletely unassociated with tumor is not affected by rapamycin.Therefore, in general, tumor blood vessels in rapamycin-treatedmice showed a pattern of development and function that wasconsistent with our histological vascular findings in the ortho-topic pancreatic tumors.
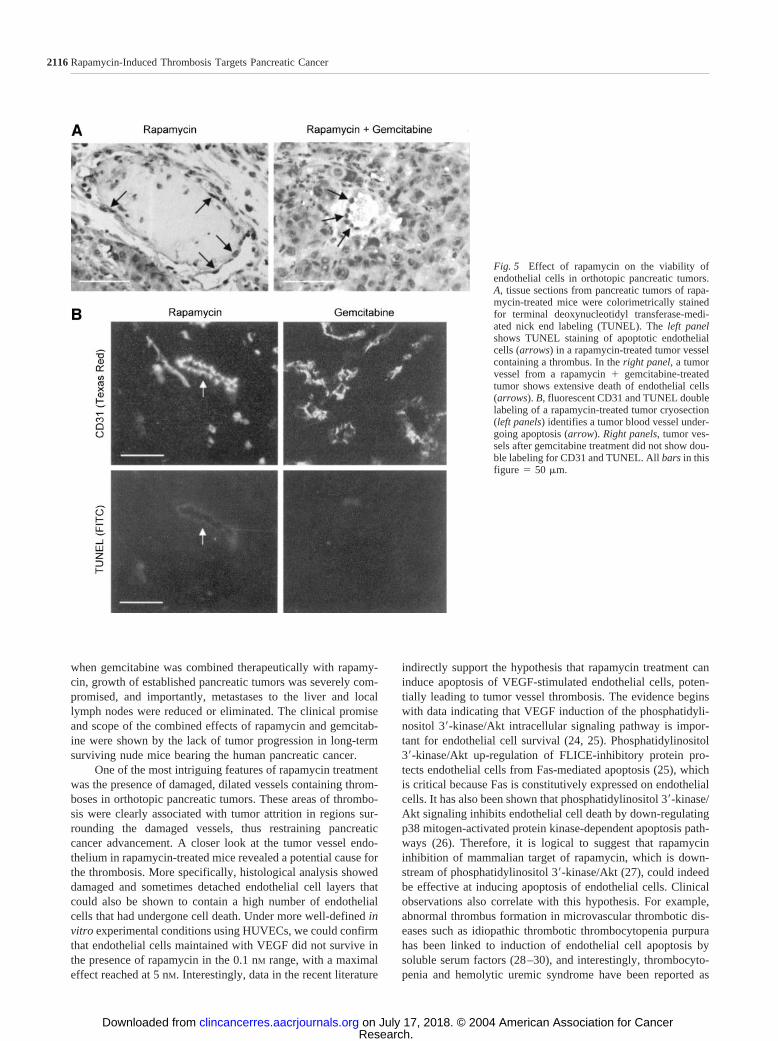
Effects on Endothelial Cell Survival and Proliferation.Disruption of the tumor vessel endothelium observed in theearlier histological studies suggested that endothelial cells maybe susceptible to cell death in the presence of rapamycin. To testthis hypothesis, we first examined tumor vessels from rapamy-cin-treated mice for endothelial cell death by colorimetricTUNEL staining. Results showed that endothelial cells in theserapamycin-treated tumors, particularly in damaged and clottedvessels, were indeed positive for TUNEL (Fig. 5A). Tumorvessels from mice treated with rapamycin � gemcitabine alsodemonstrated a high rate of endothelial cell death. Next weperformed a fluorescence double-labeling procedure for endo-
Fig. 2 Effect of rapamycin and gemcitabine (100 mg/kg dose) on pancreatic tumor cell proliferation. A, pancreatic tumors from treated mice wereremoved on day 28 after L3.6pl cell injection and stained for the proliferation marker Ki67 (bar � 100 m). B, Ki67-positive cells from 10 high-powerfields/tumor were counted, and the mean percentage � SEM of positive cells was calculated from the total cell number (�, P � 0.004 versus control).C, cultures of L3.6pl pancreatic cancer cells were tested for proliferation by determining bromodeoxyuridine incorporation in the presence or absenceof increasing rapamycin or gemcitabine concentrations. In one experimental group, a therapeutically relevant rapamycin concentration (10 nM) wascombined with increasing amounts of gemcitabine. Results are shown as the mean absorbance value � SEM and are representative of one of threeexperiments (�, P � 0.05 versus control).
2113Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Fig. 3 Rapamycin treatment results in the development of thrombi in pancreatic tumors. A, increasing magnification views of an area of pancreatictumor from control, rapamycin, gemcitabine, and combination treatment mice are shown. No thrombi were found in either control or gemcitabine-treated mice; however, the presence of thrombi was a predominant feature in tumors of mice treated with rapamycin alone or with rapamycin incombination with gemcitabine (�100, �200). At �400 magnification, normal blood vessels can be seen in control and gemcitabine-treated mousetumors, whereas clotted tumor vessels in rapamycin-treated mice show disruption of the endothelial layer (arrows). B, this photomicrograph showsa clotted vessel in a rapamycin � gemcitabine-treated tumor (arrow). Note the signs of tumor cell death (apoptotic bodies) in the area surroundingthe thrombosed vessel. C, areas of normal pancreatic tissue outside tumors treated with rapamycin (�gemcitabine) showed no evidence of vesselthrombosis (arrows). All bars in this figure � 50 m.
2114 Rapamycin-Induced Thrombosis Targets Pancreatic Cancer
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
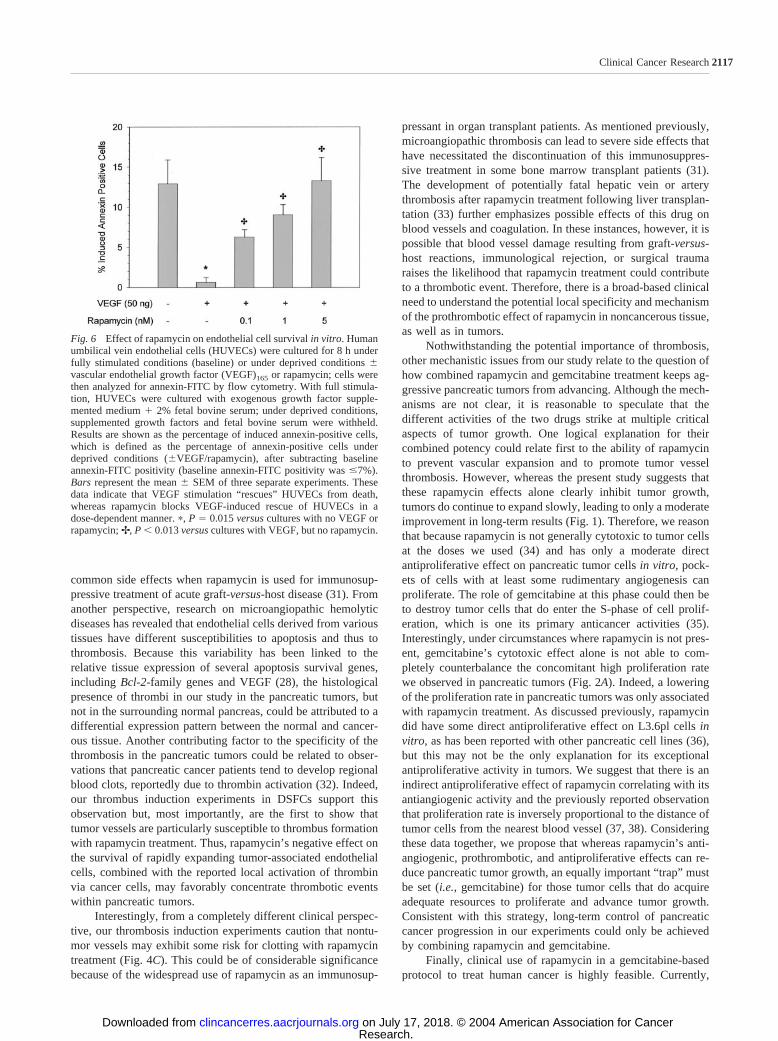
thelial cells (CD31) and TUNEL, and we observed that struc-tures in pancreatic tumors composed of endothelial cells couldalso be shown to be TUNEL positive (Fig. 5B). In contrast,endothelial cells in gemcitabine-treated tumors were not foundto be TUNEL positive. To further test whether rapamycin causesendothelial cell death, we cultured HUVECs in the presence orabsence of increasing concentrations of drug and performedflow cytometric analysis on annexin-FITC-labeled cells. Resultsshowed that compared with full stimulation of HUVECs, serumand supplement-deprived conditions caused the cells to die (Fig.6). Addition of VEGF to the serum and supplement-deprivedenvironment rescued the cells from death. Interestingly, 5 nM
rapamycin completely blocked the rescue effect of VEGF onHUVECs; lower drug levels had a lesser effect. Treatment ofHUVECs with gemcitabine had no effect on annexin staining(data not shown). Together, these results suggest that endothe-
lial cell survival pathways are blocked by rapamycin, but not bygemcitabine, leading to cell death and subsequent vascular en-dothelium disruption in tumor vessels.
DISCUSSIONPancreatic cancer remains one of the most uncontrollable
neoplasms encountered in clinical oncology. In this study, wepresent data that suggest human pancreatic cancer may beparticularly responsive to a new therapeutic approach involvingconventional use of the cytotoxic agent gemcitabine with arecently discovered antiangiogenic drug, rapamycin (14). Ourstudy indicates that rapamycin use alone can inhibit humanpancreatic tumor growth in nude mice to a greater degree thangemcitabine, a standard first-line chemotherapeutic agent avail-able for the treatment of human pancreatic cancer. Moreover,
Fig. 4 Effect of rapamycin on vessel development and thrombus formation. To dynamically observe angiogenesis in pancreatic tumors, L3.6pl cellswere inoculated into dorsal skin-fold chambers (DSFCs), and blood vessels were allowed to develop for 7 days before analysis by intravitalmicroscopy. In mice receiving rapamycin, 1.5 mg/kg/day treatment was initiated 1 day after tumor cell inoculation. A, photomicrographs of DSFCtumors from saline (control) and rapamycin-treated mice are shown. Note the presence of enlarged, irregularly formed blood vessels in therapamycin-treated tumor (bars � 100 m). B, analysis of blood vessel diameter demonstrates a significant increase in vessel diameter with rapamycintreatment, as compared with controls (P � 0.003). Mean results (n � 7–9 fields of view) from individual mice tested in each group are shown.C, to directly assess whether tumor vessels in rapamycin-treated mice are susceptible to thrombosis, clotting was promoted in DSFCs by i.v. injectedFITC-dextran and UV light. Blood flow was monitored by observing the movement of injected, fluorescence-labeled RBCs. Bars indicate the timerequired for complete stagnation of blood flow in areas either inside (f) or outside (u) the clearly demarcated tumor region in DSFCs. Results areshown from three individual mice in each treatment group, and values are the mean of two or more light-treated vessel fields. When blood flowcontinued at the 20 min time point, a � is indicated above the respective bar.
2115Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
when gemcitabine was combined therapeutically with rapamy-cin, growth of established pancreatic tumors was severely com-promised, and importantly, metastases to the liver and locallymph nodes were reduced or eliminated. The clinical promiseand scope of the combined effects of rapamycin and gemcitab-ine were shown by the lack of tumor progression in long-termsurviving nude mice bearing the human pancreatic cancer.
One of the most intriguing features of rapamycin treatmentwas the presence of damaged, dilated vessels containing throm-boses in orthotopic pancreatic tumors. These areas of thrombo-sis were clearly associated with tumor attrition in regions sur-rounding the damaged vessels, thus restraining pancreaticcancer advancement. A closer look at the tumor vessel endo-thelium in rapamycin-treated mice revealed a potential cause forthe thrombosis. More specifically, histological analysis showeddamaged and sometimes detached endothelial cell layers thatcould also be shown to contain a high number of endothelialcells that had undergone cell death. Under more well-defined invitro experimental conditions using HUVECs, we could confirmthat endothelial cells maintained with VEGF did not survive inthe presence of rapamycin in the 0.1 nM range, with a maximaleffect reached at 5 nM. Interestingly, data in the recent literature
indirectly support the hypothesis that rapamycin treatment caninduce apoptosis of VEGF-stimulated endothelial cells, poten-tially leading to tumor vessel thrombosis. The evidence beginswith data indicating that VEGF induction of the phosphatidyli-nositol 3�-kinase/Akt intracellular signaling pathway is impor-tant for endothelial cell survival (24, 25). Phosphatidylinositol3�-kinase/Akt up-regulation of FLICE-inhibitory protein pro-tects endothelial cells from Fas-mediated apoptosis (25), whichis critical because Fas is constitutively expressed on endothelialcells. It has also been shown that phosphatidylinositol 3�-kinase/Akt signaling inhibits endothelial cell death by down-regulatingp38 mitogen-activated protein kinase-dependent apoptosis path-ways (26). Therefore, it is logical to suggest that rapamycininhibition of mammalian target of rapamycin, which is down-stream of phosphatidylinositol 3�-kinase/Akt (27), could indeedbe effective at inducing apoptosis of endothelial cells. Clinicalobservations also correlate with this hypothesis. For example,abnormal thrombus formation in microvascular thrombotic dis-eases such as idiopathic thrombotic thrombocytopenia purpurahas been linked to induction of endothelial cell apoptosis bysoluble serum factors (28–30), and interestingly, thrombocyto-penia and hemolytic uremic syndrome have been reported as
Fig. 5 Effect of rapamycin on the viability ofendothelial cells in orthotopic pancreatic tumors.A, tissue sections from pancreatic tumors of rapa-mycin-treated mice were colorimetrically stainedfor terminal deoxynucleotidyl transferase-medi-ated nick end labeling (TUNEL). The left panelshows TUNEL staining of apoptotic endothelialcells (arrows) in a rapamycin-treated tumor vesselcontaining a thrombus. In the right panel, a tumorvessel from a rapamycin � gemcitabine-treatedtumor shows extensive death of endothelial cells(arrows). B, fluorescent CD31 and TUNEL doublelabeling of a rapamycin-treated tumor cryosection(left panels) identifies a tumor blood vessel under-going apoptosis (arrow). Right panels, tumor ves-sels after gemcitabine treatment did not show dou-ble labeling for CD31 and TUNEL. All bars in thisfigure � 50 m.
2116 Rapamycin-Induced Thrombosis Targets Pancreatic Cancer
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
common side effects when rapamycin is used for immunosup-pressive treatment of acute graft-versus-host disease (31). Fromanother perspective, research on microangiopathic hemolyticdiseases has revealed that endothelial cells derived from varioustissues have different susceptibilities to apoptosis and thus tothrombosis. Because this variability has been linked to therelative tissue expression of several apoptosis survival genes,including Bcl-2-family genes and VEGF (28), the histologicalpresence of thrombi in our study in the pancreatic tumors, butnot in the surrounding normal pancreas, could be attributed to adifferential expression pattern between the normal and cancer-ous tissue. Another contributing factor to the specificity of thethrombosis in the pancreatic tumors could be related to obser-vations that pancreatic cancer patients tend to develop regionalblood clots, reportedly due to thrombin activation (32). Indeed,our thrombus induction experiments in DSFCs support thisobservation but, most importantly, are the first to show thattumor vessels are particularly susceptible to thrombus formationwith rapamycin treatment. Thus, rapamycin’s negative effect onthe survival of rapidly expanding tumor-associated endothelialcells, combined with the reported local activation of thrombinvia cancer cells, may favorably concentrate thrombotic eventswithin pancreatic tumors.
Interestingly, from a completely different clinical perspec-tive, our thrombosis induction experiments caution that nontu-mor vessels may exhibit some risk for clotting with rapamycintreatment (Fig. 4C). This could be of considerable significancebecause of the widespread use of rapamycin as an immunosup-
pressant in organ transplant patients. As mentioned previously,microangiopathic thrombosis can lead to severe side effects thathave necessitated the discontinuation of this immunosuppres-sive treatment in some bone marrow transplant patients (31).The development of potentially fatal hepatic vein or arterythrombosis after rapamycin treatment following liver transplan-tation (33) further emphasizes possible effects of this drug onblood vessels and coagulation. In these instances, however, it ispossible that blood vessel damage resulting from graft-versus-host reactions, immunological rejection, or surgical traumaraises the likelihood that rapamycin treatment could contributeto a thrombotic event. Therefore, there is a broad-based clinicalneed to understand the potential local specificity and mechanismof the prothrombotic effect of rapamycin in noncancerous tissue,as well as in tumors.
Nothwithstanding the potential importance of thrombosis,other mechanistic issues from our study relate to the question ofhow combined rapamycin and gemcitabine treatment keeps ag-gressive pancreatic tumors from advancing. Although the mech-anisms are not clear, it is reasonable to speculate that thedifferent activities of the two drugs strike at multiple criticalaspects of tumor growth. One logical explanation for theircombined potency could relate first to the ability of rapamycinto prevent vascular expansion and to promote tumor vesselthrombosis. However, whereas the present study suggests thatthese rapamycin effects alone clearly inhibit tumor growth,tumors do continue to expand slowly, leading to only a moderateimprovement in long-term results (Fig. 1). Therefore, we reasonthat because rapamycin is not generally cytotoxic to tumor cellsat the doses we used (34) and has only a moderate directantiproliferative effect on pancreatic tumor cells in vitro, pock-ets of cells with at least some rudimentary angiogenesis canproliferate. The role of gemcitabine at this phase could then beto destroy tumor cells that do enter the S-phase of cell prolif-eration, which is one its primary anticancer activities (35).Interestingly, under circumstances where rapamycin is not pres-ent, gemcitabine’s cytotoxic effect alone is not able to com-pletely counterbalance the concomitant high proliferation ratewe observed in pancreatic tumors (Fig. 2A). Indeed, a loweringof the proliferation rate in pancreatic tumors was only associatedwith rapamycin treatment. As discussed previously, rapamycindid have some direct antiproliferative effect on L3.6pl cells invitro, as has been reported with other pancreatic cell lines (36),but this may not be the only explanation for its exceptionalantiproliferative activity in tumors. We suggest that there is anindirect antiproliferative effect of rapamycin correlating with itsantiangiogenic activity and the previously reported observationthat proliferation rate is inversely proportional to the distance oftumor cells from the nearest blood vessel (37, 38). Consideringthese data together, we propose that whereas rapamycin’s anti-angiogenic, prothrombotic, and antiproliferative effects can re-duce pancreatic tumor growth, an equally important “trap” mustbe set (i.e., gemcitabine) for those tumor cells that do acquireadequate resources to proliferate and advance tumor growth.Consistent with this strategy, long-term control of pancreaticcancer progression in our experiments could only be achievedby combining rapamycin and gemcitabine.
Finally, clinical use of rapamycin in a gemcitabine-basedprotocol to treat human cancer is highly feasible. Currently,
Fig. 6 Effect of rapamycin on endothelial cell survival in vitro. Humanumbilical vein endothelial cells (HUVECs) were cultured for 8 h underfully stimulated conditions (baseline) or under deprived conditions �vascular endothelial growth factor (VEGF)165 or rapamycin; cells werethen analyzed for annexin-FITC by flow cytometry. With full stimula-tion, HUVECs were cultured with exogenous growth factor supple-mented medium � 2% fetal bovine serum; under deprived conditions,supplemented growth factors and fetal bovine serum were withheld.Results are shown as the percentage of induced annexin-positive cells,which is defined as the percentage of annexin-positive cells underdeprived conditions (�VEGF/rapamycin), after subtracting baselineannexin-FITC positivity (baseline annexin-FITC positivity was �7%).Bars represent the mean � SEM of three separate experiments. Thesedata indicate that VEGF stimulation “rescues” HUVECs from death,whereas rapamycin blocks VEGF-induced rescue of HUVECs in adose-dependent manner. �, P � 0.015 versus cultures with no VEGF orrapamycin; ✢, P � 0.013 versus cultures with VEGF, but no rapamycin.
2117Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

rapamycin is approved for use in human organ transplantation asan immunosuppressive agent to prevent allograft rejection. Thedrug is maintained on a daily basis in patients for several yearsor indefinitely. An important corollary to this issue from ourstudy is that rapamycin exerts its most potent effect on endo-thelial cells near 5 nM, which coincides with serum drug levelstargeted in transplant patients. Therefore, it is reasonable tosuggest that long-term, continuous inhibition of tumor neoan-giogenesis is possible by incorporating these already thoroughlytested rapamycin treatment protocols into cancer treatment reg-imens. Another positive aspect of combining rapamycin withgemcitabine is that the latter agent can also be effectively andsafely administered over an extended period at a reduced dose(39), lending credibility to the potential of a clinical protocol forlong-term tumor control, as we were able to achieve in micewith low-dose gemcitabine � rapamycin treatment. Therefore,our study suggests that rapamycin and gemcitabine could offera novel, clinically feasible drug therapy to control pancreaticcancer disease progression, and the general strategy of combin-ing rapamycin with other cytotoxic drugs may also prove to beeffective for a broader range of cancers for which drug cyto-toxicity alone is not curative or does not provide tumor controlwith a favorable quality of life.
ACKNOWLEDGMENTSWe thank Dr. Hagen Blaszyk (Department of Pathology, Univer-
sity of Regensburg) for review of histological work presented in thispaper. We also thank Christine Wagner for excellent technical assist-ance on the project.
REFERENCES1. Brand RE, Tempero MA. Pancreatic cancer. Curr Opin Oncol 1998;10:362–6.2. Warshaw AL, Fernandes-del Castillo C. Pancreatic carcinoma.N Engl J Med 1992;326:455–65.3. Bramhall SR, Allum WH, Jones AG, et al. Treatment and survival in13,560 patients with pancreatic cancer, and incidence of the disease, inthe West Midlands: an epidemiological study. Br J Surg 1995;82:111–5.4. Rothenberg ML, Abbruzzese JL, Moore M, et al. A rationale forexpanding the endpoints for clinical trials in advanced pancreatic car-cinoma. Cancer 1996;78:627–32.5. Burris HA III, Moore MJ, Andersen J, et al. Improvements insurvival and clinical benefit with gemcitabine as first-line therapy forpatients with advanced pancreas cancer: a randomized trial. J Clin Oncol1997;15:2403–13.6. Kato H, Ishikura H, Kawarada Y, et al. Anti-angiogenic treatment forperitoneal dissemination of pancreas adenocarcinoma: a study usingTNP-470. Jpn J Cancer Res 2001;92:67–73.7. Ikeda N, Adachi M, Taki T, et al. Prognostic significance of angio-genesis in human pancreatic cancer. Br J Cancer 1999;79:1553–63.8. Niedergethmann M, Hildenbrand R, Wolf G, et al. Angiogenesis andcathepsin expression are prognostic factors in pancreatic adenocarci-noma after curative resection. Int J Pancreatol 2000;28:31–9.9. Seo Y, Baba H, Fukuda T, Takashima M, Sugimachi K. Highexpression of vascular endothelial growth factor is associated with livermetastasis and a poor prognosis for patients with ductal pancreaticadenocarcinoma. Cancer 2003;88:2239–45.10. Fujioka S, Yoshida K, Yanagisawa S, et al. Angiogenesis in pan-creatic carcinoma: thymidine phosphorylase expression in stromal cellsand intratumoral microvessel density as independent predictors of over-all and relapse-free survival. Cancer 2002;92:1788–97.11. Baker CH, Solorzano CC, Fidler IJ. Blockade of vascular endothe-lial growth factor receptor and epidermal growth factor receptor signal-
ing for therapy of metastatic human pancreatic cancer. Cancer Res2002;62:1996–2003.
12. Stipa F, Lucandri G, Limiti MR, et al. Angiogenesis as a prognosticindicator in pancreatic ductal adenocarcinoma. Anticancer Res 2002;22:445–9.
13. Kuehn R, Lelkes PI, Bloechle C, Niendorf A, Izbicki JR. Angio-genesis, angiogenic growth factors, and cell adhesion molecules areupregulated in chronic pancreatic diseases: angiogenesis in chronicpancreatitis and in pancreatic cancer. Pancreas 1999;18:96–103.
14. Guba M, von Breitenbuch P, Steinbauer M, et al. Rapamycininhibits primary and metastatic tumor growth by antiangiogenesis: in-volvement of vascular endothelial growth factor. Nat Med 2002;8:128–35.
15. Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of2�,2�-difluorodeoxycytidine on DNA synthesis. Cancer Res 1991;51:6110–7.
16. Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivoselection and characterization of metastatic variants from human pan-creatic adenocarcinoma by using orthotopic implantation in nude mice.Neoplasia 1999;1:50–62.
17. Bruns CJ, Harbison MT, Davis DW, et al. Epidermal growth factorreceptor blockade with C225 plus gemcitabine results in regression ofhuman pancreatic carcinoma growing orthotopically in nude mice byantiangiogenic mechanisms. Clin Cancer Res 2000;6:1936–48.
18. Torp SH. Diagnostic and prognostic role of Ki67 immunostaining inhuman astrocytomas using four different antibodies. Clin Neuropathol2002;21:252–7.
19. Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed,paraffin-embedded tissues: an enhancement method for immunohisto-chemical staining based on microwave oven heating of tissue sections.J Histochem Cytochem 1991;39:741–8.
20. Asaishi K, Endrich B, Gotz A, Messmer K. Quantitative analysis ofmicrovascular structure and function in the amelanotic melanoma A-Mel-3. Cancer Res 1981;41:1898–904.
21. Guba M, Cernaianu G, Koehl G, et al. A primary tumor promotesdormancy of solitary tumor cells before inhibiting angiogenesis. CancerRes 2001;61:5575–9.
22. Steinbauer M, Harris AG, Abels C, Messmer K. Characterizationand prevention of phototoxic effects in intravital fluorescence micros-copy in the hamster dorsal skinfold model. Langenbecks Arch Surg2000;385:290–8.
23. Bold RJ, Chandra J, McConkey DJ. Gemcitabine-induced pro-grammed cell death (apoptosis) of human pancreatic carcinoma is de-termined by Bcl-2 content. Ann Surg Oncol 1999;6:279–85.
24. Yu Y, Sato JD. MAP kinases, phosphatidylinositol 3-kinase, andp70 S6 kinase mediate the mitogenic response of human endothelialcells to vascular endothelial growth factor. J Cell Physiol 1999;178:235–46.
25. Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-medi-ated apoptosis via regulation of FLICE-inhibitory protein (FLIP). CircRes 2000;89:13–9.
26. Gratton JP, Morales-Ruiz M, Kureishi Y, et al. Akt down-regulationof p38 signaling provides a novel mechanism of vascular endothelialgrowth factor-mediated cytoprotection in endothelial cells. J Biol Chem2001;276:30359–65.
27. Inoki K, Li Y, Zhu J, Wu J, Guan KL. TSC2 is phosphorylated andinhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4:648–57.
28. Kim J, Wu H, Hawthorne L, Rafii S, Laurence J. Endothelial cellapoptotic genes associated with the pathogenesis of thrombotic microan-giopathies: an application of oligonucleotide genechip technology. Mi-crovasc Res 2001;62:83–93.
29. Dang CT, Magid MS, Weksler B, Chadburn A, Laurence J. En-hanced endothelial cell apoptosis in splenic tissues of patients withthrombotic thrombocytopenic purpura. Blood 1999;93:1264–70.
2118 Rapamycin-Induced Thrombosis Targets Pancreatic Cancer
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

30. Laurence J, Mitra D. Apoptosis of microvascular endothelial cellsin the pathophysiology of thrombotic thrombocytopenic purpura/sporadic hemolytic uremic syndrome. Semin Hematol 1997;34:98–105.31. Benito AI, Furlong T, Martin PJ, et al. Sirolimus (rapamycin) forthe treatment of steroid-refractory acute graft-versus-host disease.Transplantation 2001;72:1924–9.32. Wojtukiewicz MZ, Rucinska M, Zimnoch L, et al. Expression ofprothrombin fragment 1�2 in cancer tissue as an indicator of localactivation of blood coagulation. Thromb Res 2000;97:335–42.33. McAlister VC, Peltekian KM, Malatjalian DA, et al. Orthotopicliver transplantation using low-dose tacrolimus and sirolimus. LiverTranspl 2001;7:701–8.34. Eng CP, Sehgal SN, Vezina C. Activity of rapamycin (AY-22,989)against transplanted tumors. J Antibiot 1984;37:1231–7.
35. Auer H, Oehler R, Lindner R, et al. Characterisation of geno-toxic properties of 2�,2�-difluorodeoxycytidine. Mutat Res 1997;393:165–73.36. Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Reg-ulation of cell growth and cyclin D1 expression by the constitutivelyactive FRAP-p70s6K pathway in human pancreatic cancer cells. CancerRes 1999;59:3581–7.37. Takeda A, Stoeltzing O, Ahmad SA, et al. Role of angiogenesis inthe development and growth of liver metastasis. Ann Surg Oncol 2002;9:610–6.38. Folkman J. What is the evidence that tumors are angiogenesisdependent? J Natl Cancer Inst 1990;82:4–6.39. Heinemann V. Gemcitabine: progress in the treatment of pancreaticcancer. Oncology 2001;60:8–18.
2119Clinical Cancer Research
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

2004;10:2109-2119. Clin Cancer Res Christiane J. Bruns, Gudrun E. Koehl, Markus Guba, et al. Pancreatic CancerVessel Thrombosis Potentiate Cytotoxic Therapy against Rapamycin-Induced Endothelial Cell Death and Tumor
Updated version
http://clincancerres.aacrjournals.org/content/10/6/2109
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/10/6/2109.full#ref-list-1
This article cites 39 articles, 9 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/10/6/2109.full#related-urls
This article has been cited by 11 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/10/6/2109To request permission to re-use all or part of this article, use this link
Research. on July 17, 2018. © 2004 American Association for Cancerclincancerres.aacrjournals.org Downloaded from