proteins - koç hastanesihome.ku.edu.tr/okeskin/public_html/bolia_gerek_ozkan.pdf · proteins...
TRANSCRIPT

proteinsSTRUCTURE O FUNCTION O BIOINFORMATICS
The binding affinities of proteins interactingwith the PDZ domain of PICK1Ashini Bolia,1 Z. Nevin Gerek,1 Ozlem Keskin,2 Sefika Banu Ozkan,1* and Kumlesh K. Dev3*
1Department of Physics, Center for Biological Physics, Arizona State University Tempe, Arizona
2 Center for Computational Biology and Bioinformatics, Department of Chemical and Biological Engineering, Koc University,
Istanbul, Turkey
3Department of Physiology, Molecular Neuropharmacology, School of Medicine, Trinity College Institute of Neuroscience,
Trinity College Dublin, Ireland
INTRODUCTION
PDZ (PSD95/DlgA/Zo-1) domains are common to over 150 proteins
that are otherwise unrelated.1 These domains are �90 residues long and
consist of six b-strands (bA to bF) and two a-helices (aA and aB). Incanonical PDZ domains, the PDZ domain binds the C-terminus-located
PDZ motif of its interacting partner in an elongated groove between the
bB sheet and the aB helix of the PDZ domain that is termed the PDZ
binding groove.2 The amino acids in PDZ domains are numbered
according to their topographical location, for example, aB1 is the first
residue on the aB helix.3 The selectivity of a PDZ domain for its ligands
is dictated by the first residue of the a-helix B (aB1) of the PDZ
domain. The residue in the aB1 position of a Type I PDZ domain is
histidine, in Type II is generally hydrophobic and in Type III is usually
tyrosine.3 The amino acids of PDZ motifs (or ligands) are numbered in
relation to the residue at the extreme C-terminus, which is referred to as
position 0 (P0), with the subsequent residue from the C-terminus
occupying the -1 position (P-1). Type I PDZ domains bind PDZ motifs
consisting of X-Thr/Ser-X-hydrophobic residues, Type II recognize a
X-hydrophobic-X-hydrophobic residue motif, while Type III interact with
a X-Asp-Glu-X-hydrophobic residue motif; where X denotes any amino
acid.3 The relatively loose consensus of PDZ motifs allows PDZ domains
to bind to a range of ligands.
Protein interacting with C kinase (PICK1) is 416 residues in length con-
taining a single N-terminal PDZ domain that interacts with the PDZ motifs
of several proteins, which have roles in synaptic plasticity,4–12 neuronal cell
morphology,13 and mitochondrial-dependent apoptosis.14 The functions of
PICK1 are largely fulfilled by controlling the trafficking of its binding partners
and by facilitating their phosphorylation by recruiting protein kinase C-a(PKCa).15 The proteins interacting with PICK1 have roles in epilepsy,16,17
Abbreviations: PICK1, protein interacting with C kinase; PRS, perturbation response scanning; PDZ
(PSD95/DlgA/Zo-1)
Grant sponsors: Enterprise Ireland and Science Foundation Ireland, Ireland, National Science Foundation
through TeraGrid resources (NCSA)
*Correspondence to: Kumlesh K. Dev, Department of Physiology, Molecular Neuropharmacology, School of
Medicine, Trinity College Institute of Neuroscience, Trinity College Dublin, Ireland. E-mail: [email protected] and
Sefika Banu Ozkan, Department of Physics, Center for Biological Physics, Arizona State University Tempe,
Arizona. E-mail: [email protected].
Received 22 May 2011; Revised 30 December 2011; Accepted 3 January 2012
Published online 17 January 2012 in Wiley Online Library (wileyonlinelibrary.com).
DOI: 10.1002/prot.24034
ABSTRACT
Protein interacting with C kinase (PICK1)
is well conserved throughout evolution
and plays a critical role in synaptic plastic-
ity by regulating the trafficking and post-
translational modification of its interact-
ing proteins. PICK1 contains a single
PSD95/DlgA/Zo-1 (PDZ) protein–protein
interaction domain, which is promiscuous
and shown to interact with over 60 pro-
teins, most of which play roles in neuronal
function. Several reports have suggested
the role of PICK1 in disorders such as epi-
lepsy, pain, brain trauma and stroke, drug
abuse and dependence, schizophrenia and
psychosis. Importantly, lead compounds
that block PICK1 interactions are also
now becoming available. Here, a new
modeling approach was developed to
investigate binding affinities of PDZ inter-
actions. Using these methods, the binding
affinities of all major PICK1 interacting
proteins are reported and the effects of
PICK1 mutations on these interactions are
described. These modeling methods have
important implications in defining the
binding properties of proteins interacting
with PICK1 as well as the general struc-
tural requirements of PDZ interactions.
The study also provides modeling methods
to support in the drug design of ligands
for PDZ domains, which may further aid
in development of the family of PDZ
domains as a drug target.
Proteins 2012; 80:1393–1408.VVC 2012 Wiley Periodicals, Inc.
Key words: PICK1; molecular docking;
modeling; binding affinity.
VVC 2012 WILEY PERIODICALS, INC. PROTEINS 1393

pain,18,19 and also in brain trauma, stroke, excitotoxicity,
and cell death.20–22 Recent evidence from genetic associa-
tion studies and characterization of PICK1 knockout ani-
mals suggests a role for PICK1 in drug abuse and depend-
ence, schizophrenia, and psychosis.23–31 PICK1 has also
been reported to be expressed in insulin-producing pan-
creatic beta-cells where it may play a role in the neuroen-
docrine system.32
The PICK1 residues that interact with P-2 and P0 resi-
dues of PDZ motifs have been best studied. Interestingly,
for the ‘‘P-2 binding pocket,’’ the Lys83 residue (usually a
histidine in other PDZ domains) in PICK1 creates a
hydrogen bond with P-2 residues to preserve class I motif
binding and a hydrophobic interaction to satisfy Class II
motif binding.33–35 Additional residues involved in
interaction with P-2 residues of PDZ motifs include
Thr82, Val84, and Ala87 in the aB helix of PICK1.33,34
There are also a number of residues in PICK1 that create
a ‘‘P0 binding pocket’’ where P0 residues of PDZ motifs
interact. Firstly, the PDZ domain of PICK1 contains an
8–9 amino acid carboxylate (��COO��) binding domain
(CBD) containing the residues Lys27 and Asp28 (‘‘KD
motif ’’) commonly found in Type II PDZ domain inter-
acting with P0 residues.4,15 Second, the Ile37 residue in
the bB strand of PICK1 plays a critical role in interaction
with P-2 and P0 residues. Third, the carboxylate binding
motif comprises GLGF motif, which in PICK1 are resi-
dues Leu32-Ile33-Gly34-Ile35 (LIGI motif) in the bB sheet
play a role in PDZ motif interaction. Specifically, the res-
idues Ile33 and Ile35 of the LIGI motif and the Val86,
Ala87, Ile90 residues of the aB helix also interact with P0
residues of PDZ motifs.34 Residues at P0 and P-2 of
PDZ motifs also seem to influence each other, for exam-
ple a small residue at P0 (valine) would allow Ile37 in
PICK1 to adopt multiple conformations allowing versatil-
ity of the P-2 binding pocket.33
There are over 150 proteins that contain PDZ domains,
making this a large family of potential drug targets. How-
ever, to date, PDZ interactions represent a drug target that
still remains largely untapped.36 Recently, drugability of
PICK1 has been suggested by discovery of a lead com-
pound, FSC231, which inhibits PICK1 interactions.37,38
Recent studies have also helped characterize the structure-
activity relationship of the various domains found in
PICK1 giving insight into the binding properties of its
PDZ domain. Here, a new modeling approach was devel-
oped in order to further characterize the binding affinities
of all the major PICK1 interacting proteins. In this study,
the binding affinities of all PDZ motifs known to interact
with PICK1 are reported. These results further elucidate
the determinants of PICK1 binding and may aid in the de-
velopment of therapeutic agents aimed at regulating spe-
cific PICK1 interactions. The study also provides model-
ing methods to support in the drug design of ligands for
PDZ domains, which may further aid in development of
the family of PDZ domains as a drug target.
MATERIALS AND METHODS
Crystal structures applied
All PICK1 PDZ domain structures interacting with
peptides listed in Table I were analyzed in this study. The
atomic coordinates for the proteins used in this study
were obtained from RCSB Protein Data Bank
(www.rcsb.org), with PDB Ids 2GZV, 2PKU, and 1GZV.
The crystal structure of PICK1 protein bound with the
PKCa peptide (PDB code: 2GZV) was retrieved from the
PDB (http://www.rcsb.org/pdb).39 This structure was
used as a template and the following procedure was car-
ried out in our docking calculations: first, the original
PKCa peptide was redocked into a known PDZ domain
of the bound conformation (self-docking); then, the
computational point mutations were introduced into the
X-ray structure of the PKCa peptide via Swiss PDB
Viewer40 to obtain other peptides listed in Table I.
Generating multiple receptor conformations
An ensemble of the wild type (WT) and mutant ver-
sions (K27A, K27E, K83H, and K83V) of PICK1 were gen-
erated through Perturbation Response Scanning (PRS).
PRS couples elastic network models with linear response
theory.41 This PRS computes the fluctuation profile of a
protein upon an external force or a perturbation on a
single residue when a ligand approaches a receptor and
starts exerting external forces on the receptor structure
inducing conformational changes. To mimic this process,
a random force was applied in every direction to the a-carbon atom of each residue in the protein, the residue
displacement responses were recorded as a linear function
and were used to generate low-resolution receptor con-
formations (based on Ca atom). These responses upon
perturbation of each single residue capture the shift in
conformational change between the unbound and bound
conformations as previously shown.41 Therefore, the
multiple receptor conformation (MRC) ensemble gener-
ated from the perturbations include the conformations
that are sampled through the binding process. The
deformed/perturbed structures of the PICK1 crystal
structure were then clustered to discard the similar con-
formations generated from the perturbations on different
residues. After clustering, all atom minimization of the
clustered structures were performed to account for the
fluctuation in the side chains upon perturbation on the
backbone of the protein and to relieve strain in the sys-
tem. Thus, each perturbed structure was subjected to an
energy minimization of 50 steepest descent iterations fol-
lowed by 1000 conjugate gradient iterations using
AMBER force field along with a Generalized Born solva-
tion model.42 Finally, an ensemble docking for all these
different peptides using all the structures were generated
by PRS for the PICK1. The outline of this method is
depicted in Figure 1. For the mutant proteins, an initial
A. Bolia et al.
1394 PROTEINS

TableI
PICK1BindingAffinities
Interactor
Binding
site
PDZmotif
type
E bind
(kca
l/mol)
Family
mem
ber
Func
tion
Reference
Wild
type
PICK
1K27E
PICK
1K27A
PICK
1K83H
PICK
1K83V
PICK
1
Ephrin
B1
EphRe
ceptor
ligand
Torres
etal.,1998
YYKV
II2338.85
2320.45
2304.43
2301.76
2301.91
Parkin
E3ub
iquitin
ligase
Joch
etal.,2007
WFD
VII
2297.37
2285.42
2271.51
2272.62
2288.87
DAT
Dop
aminetran
sporter
Torres
etal.,2001
WLKV
II2291.93
2274.00
2301.13
2297.99
2292.42
JAM2A,B
,CJu
nctionad
hesion
molec
ule
Reym
ondet
al.,2007
SFVL
II2289.97
2278.34
2282.91
2283.15
2269.94
Nec
tin1a
,2a,2
d,3a
,3b,4
Cell-ad
hesion
molec
ules
Reym
ondet
al.,2007
EWYV
II2289.93
2270.72
2278.20
2285.37
2280.40
Syntenin
PDZprotein
Torres
etal.,1998
IPEV
II2280.73
2269.23
2269.87
2264.24
2265.12
NET
Norad
renalinetran
sporter
Torres
etal.,2001
WLA
III
2278.25
2267.13
2276.93
2285.33
2273.70
PrRP
Prolactin
releasingpe
ptiderece
ptor
Linet
al.,2001
SVVI
II2270.14
2234.65
2242.45
2255.71
2249.70
Anion
exch
ange
r21,22
Chlorid
e/bica
rbonateexch
ange
rsCo
wan
etal.,2000
AMPV
II2265.25
2259.05
2249.38
2246.14
2248.84
ARF1,ARF3
ADP2
ribosylationfactors
Takeya
etal.,2000
RNQK
X2262.11
2257.45
2230.85
2249.81
2230.45
ErbB
2/HER
2Ep
idermal
grow
thfactor
rece
ptor
Julin-Bastard
etal.,2001
DVP
VII
2260.81
2246.30
2260.07
2252.42
2256.86
Neu
roligin
1,2
Synaptic
cell-adhe
sion
molec
ules
Meyer
etal.,2004
TTRV
I2247.76
2263.31
2245.44
2252.19
2248.31
EphB
2,Ep
hA7
Ephrin
Rece
ptor
tyrosine
kinase
Torres
etal.,1998
SVEV
II2246.30
2214.34
2225.32
2227.38
2228.25
GLT1b
Glutamatetran
sporter
Bassanet
al.,2008
ETCI
I2246.09
2270.72
2258.22
2254.00
2258.20
MuS
KMusclespec
ifictyrosine
kina
seXiaet
al.,1999;Torreset
al.,1998
TVSV
II2242.86
2201.42
2227.86
2205.02
2215.32
Serin
erace
mace
Synthe
sisof
L-serin
eFujiiet
al.,2006
SVSV
II2242.25
2208.70
2217.03
2229.20
2229.92
Kalirin-7
Gua
nine
-nucleotide-exch
ange
Factor
Penzes
etal.,2001
STYV
I2240.14
2258.17
2242.46
2240.24
2256.37
GluR2,3
,4AMPA
rece
ptors
Dev
etal.,1999;X
iaet
al.,1999
SVKI
II2236.79
2213.58
2233.20
2222.56
2236.57
Aqu
aporin-1,2,9
Mem
branewater
chan
nels
Cowan
etal.,2000
SVIM
II2236.60
2205.88
2241.84
2230.91
2221.42
mGluR3,4
a,7,8
Metab
otropicglutam
aterece
ptors
Bou
dinet
al.,2000;D
evet
al.,2000;
ElFarOet
al.,2000;H
irbec
etal.,2002
NLVI
II2234.51
2230.76
2226.53
2222.23
2219.57
SERT
Serotonintran
sporter
Torres
etal.,2001
LNAV
II2233.98
2205.26
2232.99
2229.09
2211.48
ASIC1a,
2a,b
/BNaC
1Acid-sensingionch
anne
lHruska-Hag
eman
etal.,2002;
Dug
ganet
al.,2002
EIAC
II2221.09
2205.11
2219.56
2224.44
2217.72
CAR
CoxsackieB&
adenoviru
srece
ptor
Exco
ffonet
al.,2004
GSIV
I2217.38
2221.33
2220.35
2227.85
2228.99
PKCa
ProteinKina
seCa
Stau
ding
eret
al.,1995;1997
QSA
VI
2217.18
2232.90
2223.56
2220.36
2225.46
Calcineurin
Proteinph
osphatase(2B)
Iidaet
al.,2008
VDV
II2215.91
2202.02
2206.73
2196.11
2203.61
GluR5(2b,c),R
6Ka
inaterece
ptors(KAR)
Hirb
ecet
al.,2003
ETVA
I2215.78
2220.31
2201.31
2206.15
2207.75
LRP1B
LDLrece
ptor-related
protein
Shiro
shimaet
al.,2009
ETVA
I2215.78
2220.31
2201.31
2206.15
2207.75
UNC5H1
Netrin
rece
ptor
Williamset
al.,2003
EAEC
X2211.88
2200.28
2202.17
2196.33
2195.11
a7nA
ChRs
Nicotinic
Ace
tylcho
linerece
ptors
Bae
ret
al.,2007
c-term
atypical
TIS21
TPAIndu
cibleSe
quence
Linet
al.,2001
Internal
Arp2/3
Actin-Related
Proteins
Rocc
aet
al.,2008
c-term
acidic
NCS
-1NeuronalC
a21
sensor
(NCS
)Jo
etal.,2008
BAR
ICA69
IsletCe
llAutoa
ntigen
(diabetes)
Caoet
al.,2007
BAR
GRIP
PDZprotein
Luet
al.,2005
BAR
F-ac
tinMicrofilam
entG-actin
polymers
Rocc
aet
al.,2008
BAR
a-SNAP,b-SNAP
SolubleNSF
attachmen
tproteins
Han
leyet
al.,2002
BAR
CK2a
Catalytic
subu
nitproteinkina
se2
Xiao
etal.,2009
n.d.
GPO
CGolgi-assoc
iatedprotein
Xiao
etal.,2009
n.d.
KRIP6
KARinteractingproteinGluR6
Laezza
etal.,2008
n.d.
BindingaffinitiesforwildtypeandmutatedversionsofPICK1areshown.Higher
negativevalues
indicateahigher
bindingaffinityprediction.PDZinteractionsareindicated
withPDZbindingmotifresidues
andtypeofbinding
motifas
IandII.Alternatively,Xforatypical;BAR,BARdomaininteraction;n.d.notdetermined
areindicated
asinteractionsiteswithPICK1.
Modeling PICK1 Binding Affinities
PROTEINS 1395

receptor structure was generated by performing a point
mutation by Swiss PDB Viewer and following an all atom
minimization through AMBER package. Next, PRS was
applied to these initial structures in order to generate new
receptor ensemble for each mutant. Overall backbone
root-mean-square deviations (RMSDs) of the mutants
were observed, to some extent, different than WT PICK1.
Moreover, some side chain rotamers were significantly
changed in the mutant structures due to perturbation on
the backbone upon mutation through the PRS analysis,
which leads to changes in binding affinities.
ROSETTALIGAND docking analysis
All docking analysis for each of the structure in the en-
semble in this study was performed using ROSETTALI-
GAND43,44 protocol in the ROSETTA package. The
ligand flexibility was established by changing torsional
angles, and the backbone of the ligand and the whole
protein were held fixed throughout the docking simula-
tion. ROSETTALIGAND is a method specifically devel-
oped for docking ligands into protein binding sites. The
method uses a Monte Carlo minimization protocol to
optimize the rigid body position and orientation of the
ligand and the protein side-chain conformations. The
energy function includes van der Waals interactions, an
implicit solvation model, an electrostatics model, explicit
orientation hydrogen bonding potential, and empirically
derived torsional potentials. The ROSETTALIGAND pro-
tocol applied was substantially the same as described pre-
viously.44 The coordinates of the peptides were taken
from the crystallographic complexes and were treated as
a single residue. The peptide flexibility was introduced by
changing torsional angles and the backbone was held
fixed throughout the docking simulation. In this study,
the ligand position and orientation were perturbed ran-
domly with the translation of mean 0.1 A and rotations
of mean 38, respectively. Computation of 10,000 trajecto-
ries was performed to generate a comprehensive ensem-
ble of conformations of the receptor-ligand complex for
each peptide. The formation of a distinct binding funnel
in binding energy/RMSD plots was taken as an indication
of successful docking and the final docked conformations
were selected based on the lowest free energy pose in the
protein-binding site.
DrugScore binding affinity
After selecting the pose corresponding to the lowest
free energy of binding at the end of Rosetta docking, the
binding energy score of this complex using DrugScore45
was reassessed. Thus, the receptor domain of the docked
pose in PDB format and the ligand coordinates in mol2
format were submitted into DrugScore online (http://
pc1664.pharmazie.uni-marburg.de/drugscore/). Drug-
Score is a knowledge-based scoring function for protein–
ligand interactions that employs statistically derived pair
potentials using the distance-dependent occurrence fre-
quencies by which a particular ligand atom type is found
in contact with a protein atom type. Higher negative val-
ues indicate a higher binding affinity prediction. With
DrugScore, the binding selectivity preferences for various
peptides of PICK1 PDZ domain proteins were found to
be more significant than others.
Figure 1Flow chart of flexible docking method. In our flexible docking approach,
we generated an ensemble of receptor conformations through several
steps: (i) sequentially exerting random external force on each single-
residue, (ii) calculating the response fluctuation vector using Perturbation
Response Scanning (PRS) method, (iii) constructing the low resolution
deformed structures (i.e., backbone) using the response vectors after each
single residue perturbation, (iv) clustering the perturbed conformations
using k-clustering method, and (v) all-atom minimization of each
clustered conformation. Once the multiple receptor conformations
(MRCs) ensemble was completed, we performed a docking simulation
using the ROSETTALIGAND option in the Rosetta package for each
minimized structure in the ensemble. [Color figure can be viewed in the
online issue, which is available at wileyonlinelibrary.com.]
A. Bolia et al.
1396 PROTEINS

Figure 2Sequence analysis of PICK1. A: Alignment of PICK1 PDZ domain. Alignments were performed using Vector NTI software and the following
accession numbers were used: Human PICK1 Q9NRD5; Human PSD-95 P78352; NHERF O14745; Disks large homolog 1 Human Q1295; CSKP
O14936; Syntenin1 O00560; NOS1 P29475. Bovine PICK1 Q2T9M1; Macaque PICK1 Q4R7Q5; Murine PICK1 Q62083; Orangutan PICK1
Q5REH1; Rat PICK1 Q9EP80, Drosophila NP609582, C. Elegans NP502796. The residues important for interaction with PDZ motifs are
highlighted in bold. The residues underlined appear to be well conserved in PDZ domains Types I, II, and III. B: Domain structure of PICK1. ThePDZ and BAR domains, an N-terminal 18 residue acidic region, a 40 residue a-helical linker region, and the C-terminal acidic region are shown.
The residues in PICK1 that create the ‘‘P0 binding pocket’’ include (i) the carboxylate binding domain (CBD) residues Lys27 and Asp28 (‘‘KD
motif ’’), (ii) the Ile37 residue, and (iii) the GLGF-like motif (LIGI) residues Lue32, Ile33, Gly34, Ile35. The residues in PICK1 that create the ‘‘P-2
binding pocket’’ include (i) the Lys83 at position aB1 and (ii) the residues Thr82, Val84, and Ala87. Three lysine residues in the PICK1 BAR domain
are needed for PICK1 clustering are shown. Positively charged residues Arg76, Lys79, Lys81 (‘‘RKK motif ’’) and hydrophobic residues Cys44-Pro45-
Cys46 (CPC motif) essential for membrane, lipid, and zinc binding52,53 are also shown.
PROTEINS 1397
Modeling PICK1 Binding Affinities

Computational hotspots
The computational hotspots of the PICK1 complexes
were found using the HotPoint Server (http://
prism.ccbb.ku.edu.tr/hotpoint). This model uses an intui-
tive efficient method to determine computational hot
spots based on conservation, solvent accessibility and sta-
tistical pairwise residue potentials (PP) of the interface
residues as we have recently described.46,47 The PICK1-
peptide complexes in Figure 3 are modeled using PRISM
as described previously48,49 and Chimera.50
RESULTS
Categorization of PICK1 residues intobinding pockets
To categorize PICK1 residues into binding pockets for
P-4 to P0 residues of PDZ motifs, sequence alignments of
several PDZ domains were performed (Fig. 2) and all
PDZ motifs that interact with PICK1 were analyzed (Table
I). For the ‘‘P-2 binding pocket,’’ alignment data showed
Lys83 is uniquely found in PICK1 compared to histidine
and is well-conserved suggesting the importance of
PICK1’s diverse binding ability throughout evolution (Fig.
2). For the other three residues suggested to play a role in
P-2 residue binding, namely Thr82, Val84, and Ala87 (Ref.33), the alignment data showed that Thr82 (or Ser) is well
conserved in Types I, II, and III PDZ domains, Val84 dis-
played little homology, and the small sized Ala87 (normally
Val in Type I PDZ domains) occurred in PICK1 that may
allow additional ‘‘space’’ to accept a diverse set of P-2 resi-
dues (Fig. 2). For the ‘‘P0 binding pocket,’’ alignment data
showed the residues Lys27 and Asp28 (‘‘KD motif ’’) were
commonly found in Type II PDZ domains, that the Ile37
is reasonably well conserved between PDZ domains and
that PICK1 contains and GLGF-like motif (LIGI) (Fig. 1).
In support of a preference for valine at P0,33–35 this resi-
due was found in a majority of PICK1 interacting Type II
PDZ motifs (Table I). Taken together, we classify (i) the
‘‘P-2 binding pocket’’ as containing Lys83 together with
Thr82, Val84, and Ala87; and (ii) ‘‘P0 binding pocket’’ as
comprising the ‘‘KD motif ’’ (Lys27 and Asp28), the Ile37
residue, and the LIGI motif (Ile33, Ile35). Notably, neither
the ‘‘RKK motif ’’ (Arg76, Lys79, Lys81) nor the CPC motif
(Cys44-Pro45-Cys46), which are both important for mem-
brane binding51–53 were found to be conserved in PDZ
domains, indicating a unique role for PICK1’s PDZ do-
main in membrane association (Fig. 2).
Modeling of PICK1 interaction residues intohotspots
Next, four of the best-studied PDZ motifs of PICK1
namely, GluR2 (-SVKI), PKCa (-QSAV), DAT (-WLKV),
and Ephrin B1 (-YYKV) were modeled. The data showed
PICK1 residues Lys83 and Ala87 (P-2 binding pocket) and
the ‘‘KD motif ’’ (Lys27, Asp28) and the Ile37 residue (P0
binding pocket) interacted with the GluR2 PDZ motif (-
SVKI). In addition, the PICK1 residues Gly39 and Val84
made contact with Ephrin B1 (-YYKV) and PKCa (-
QSAV) peptides. For the PKCa peptide (-QSAC), the
Lys83 of PICK1 also made contact with P-3 (Gln) (Fig. 3).
To investigate the binding hot spots54 of the four PICK1
complexes, alanine mutations in PICK1 were analyzed and
the change in binding affinity with the HotPoint Server47
was examined. In the GluR2 peptide (-SVKI) the residues
P-2 (Val) and P0 (Ile) made the major binding contribu-
tions, as did the residues P-2 (Ser) and P0 (Val) for the
PKCa peptide (-QSAV). In the PICK1-GluR2 complex
(PDB ID: 2pku) the residues Lys83 and Ala87 of the P-2
binding pocket and the KD motif (Lys27), the Ile37 residue,
the LIGI motif (Ile33, Ile35), as well as the Ile90 residue of
the P0 binding pocket were all found to represent hot
spots. The Ala87 residue of PICK1 made hydrophobic
interactions with P-2 (Val) of GluR2 (-SVKI) making a
‘‘P-2 binding pocket’’ as expected. In the PICK1-PKCacomplex (modeled structure), the residue Lys83 of the P-2
binding pocket, and the Ile37 residue, the LIGI motif
(Leu32, Ile33, Ile35), as well as Ile90 of the P0 binding
pocket represented hot spots. Interestingly, the side chain
atoms of Ser36 of PICK1 also made strong H-bonds with
the backbone atoms of P-2 (Ser) of PKCa (-QSAV) and
were involved in creating a ‘‘P-2 binding pocket’’ (Fig. 3).
These results are in agreement with previous studies.33–35
Binding affinities of PICK1 interactingproteins
To date almost 60 proteins have been shown to inter-
act with PICK1, which were sorted according to their
PDZ binding affinities (Table I). Using a flexible docking
approach (see Fig. 1 and methods for details), the bind-
ing affinities of all the PDZ motifs known to interact
with PICK1 were computed (Fig. 4). This data displays
the poses of PICK1 complexes corresponding to the low-
est binding energies and their rescored binding affinity
values. Importantly, the predicted binding energy scores
agree qualitatively with experiments where data is avail-
able. For example, the dopamine transporter (DAT) pep-
tide (-WLKV) binds to the PDZ domain of PICK1 with
higher affinity than the PKCa peptide (-QSAV).34 In
agreement the predicted binding affinity of DAT
(2291.93 kcal/mol) was found to be higher than PKCa(2217.18 kcal/mol). Furthermore, experimental analysis
indicates that the affinity of GLT1b (-ETCI) for PICK1 is
significantly better than that of PKCa55 and slightly bet-
ter than that of GluR2,55 on the other hand it is slightly
lower than that of the DAT.34 Likewise, our predicted af-
finity value of GLT1b (2246.09 kcal/mol) is in between
the affinities of DAT (2291.93 kcal/mol) and GluR2
(2236.79 kcal/mol) peptides. On the other hand, binding
energy scores obtained by rigid docking did not correlate
A. Bolia et al.
1398 PROTEINS
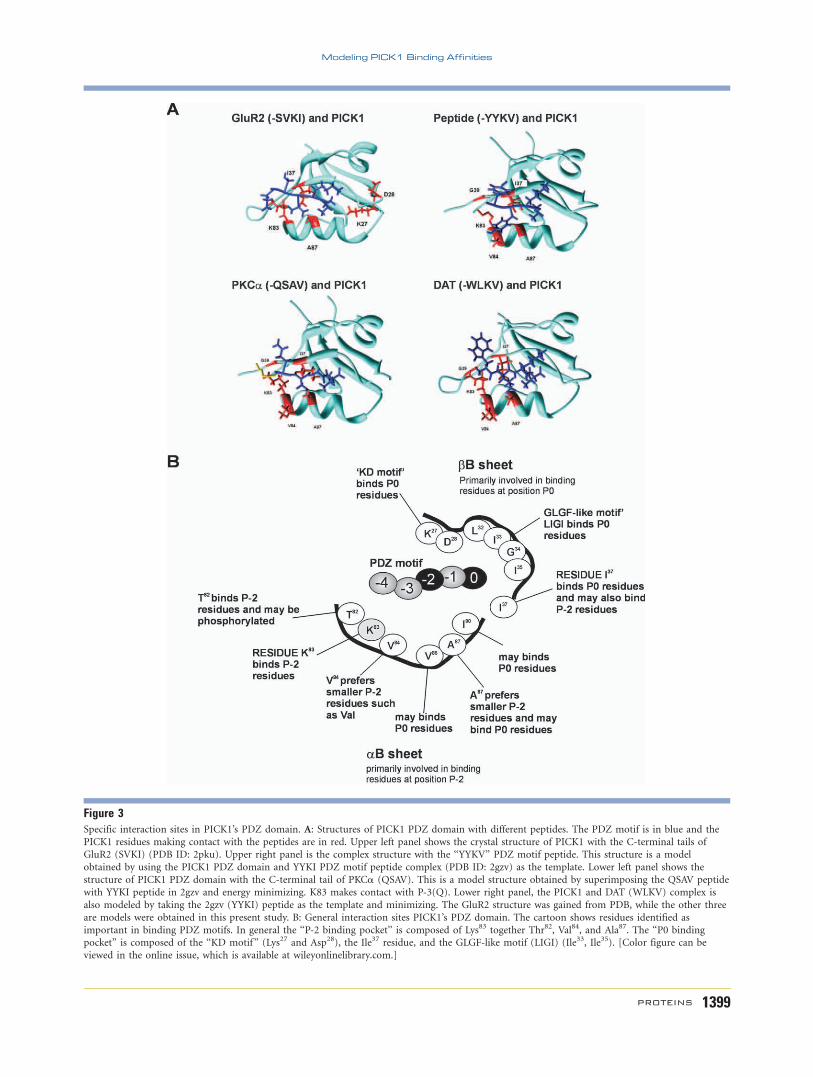
Figure 3Specific interaction sites in PICK1’s PDZ domain. A: Structures of PICK1 PDZ domain with different peptides. The PDZ motif is in blue and the
PICK1 residues making contact with the peptides are in red. Upper left panel shows the crystal structure of PICK1 with the C-terminal tails of
GluR2 (SVKI) (PDB ID: 2pku). Upper right panel is the complex structure with the ‘‘YYKV’’ PDZ motif peptide. This structure is a model
obtained by using the PICK1 PDZ domain and YYKI PDZ motif peptide complex (PDB ID: 2gzv) as the template. Lower left panel shows the
structure of PICK1 PDZ domain with the C-terminal tail of PKCa (QSAV). This is a model structure obtained by superimposing the QSAV peptide
with YYKI peptide in 2gzv and energy minimizing. K83 makes contact with P-3(Q). Lower right panel, the PICK1 and DAT (WLKV) complex is
also modeled by taking the 2gzv (YYKI) peptide as the template and minimizing. The GluR2 structure was gained from PDB, while the other three
are models were obtained in this present study. B: General interaction sites PICK1’s PDZ domain. The cartoon shows residues identified as
important in binding PDZ motifs. In general the ‘‘P-2 binding pocket’’ is composed of Lys83 together Thr82, Val84, and Ala87. The ‘‘P0 bindingpocket’’ is composed of the ‘‘KD motif ’’ (Lys27 and Asp28), the Ile37 residue, and the GLGF-like motif (LIGI) (Ile33, Ile35). [Color figure can be
viewed in the online issue, which is available at wileyonlinelibrary.com.]
Modeling PICK1 Binding Affinities
PROTEINS 1399

with the order of experimental binding affinities. Specifi-
cally, the rigid docking predicted that GluR2 and DAT
peptides have the same affinity and higher than PKCa.
The K27 and K83 binding hotspots in PICK1
Previous studies show that mutation of K27A (in the
‘‘KD motif ’’) in PICK1 prevents binding to both Class I
Figure 4Binding affinities of PDZ motifs interacting with PICK1. The sorted binding affinity values of C-terminal peptides interacting with the PDZ
domain of PICK1 and the ribbon diagrams of the complex structure with the best score. Higher negative values indicate a higher binding affinity
prediction. The key residues near the binding pocket making side interactions with the peptide are also shown.
A. Bolia et al.
1400 PROTEINS

(PKCa, -QSAV) and Class II (GluR2, -SVKI)
motifs.4,11,15 In contrast, a K27E mutation in PICK1
reduces binding to P0 residues and disrupts interactions
with Class II motifs (GluR2, -SVKI) while maintaining
binding with Class I motifs (PKCa, -QSAV).33,36 This
may explain why the ‘‘KD motif ’’ is conserved in Type II
PDZ domains but not found in Type I PDZ domains
(Fig. 2). Importantly, the replacement of Lys83 with His83
(K83H) in PICK1 enhances the affinity for Class I motifs
(PKCa, -QSAV), and decreases the affinity for Class II
motifs (DAT, -WLKV).34 In contrast, mutation of Lys83
to Val83 (K83V) in PICK1 enhances affinities for both
class I (PKCa, -QSAV) and Class II (DAT, -WLKV) PDZ
motifs.33,34,52 On the basis of these experimental stud-
ies, four mutations K27E, K27A, K83H, and K83V of
PICK1 were investigated (Table I). In agreement with ex-
perimental observations36 the binding energy score of
GluR2 (-SVKI) was found to significantly decrease,
whereas that of PKCa (-QSAV) was enhanced with K27E
PICK1 (Table I). Moreover, the data showed a disruption
in binding affinity of mGluR7 (-NLVI) upon K27E muta-
tion as observed experimentally.4 In most cases, the K27E
mutation and the K27A mutation increased the binding
affinity for Class I peptides whereas it decreased those
for Class II peptides (Table I). However, there were out-
liers; for example, binding energy of GluR5-2b (Class I
peptide) decreased, while binding of DAT and NET
(Class II peptides) increased upon K27A mutation. Bind-
ing energy of PKCa (-QSAV), but not DAT (-WLKV)
was also higher with K83H PICK1, as previously
reported.34 In general, mutations at position Lys83 (K83V
and K83H) did not perturb the Class I type interactions,
and in some cases caused an increase in binding energies,
while they disturbed the interactions with Class II pep-
tides (Table I). As in the case of K27A, the DAT, and
NET (Class II) peptides were not affected from K83V and
K83H mutations. All mutations also disturbed the bind-
ing of PICK1 to atypical peptides. In order to compare
the accuracy of flexible docking in predicting binding
affinities, the binding scores without incorporating the
flexibility of PICK1 was also computed. Specifically, resi-
dues were mutated in the wild type crystal structure for
cases where experimental data is available and an energy
minimization was performed to obtain a reasonable mu-
tant structure. These energy minimized mutant structures
were used for ROSETTA docking. The flexible docking
results with PRS showed better agreement with the ex-
perimental affinity data15,34,36 compared with the rigid
docking results (Table II). For PKCa, the flexible docking
method captured the increase in binding affinity upon
K83V and K83H mutants that was comparable with the
experimental affinity results,4,15,34,36 yet rigid docking
failed to predict these. Moreover, rigid docking was not
able to predict the change in binding affinity of DAT in
K83H or K83V mutation. Overall, these results suggest
that the conformational dynamics of PICK1 is critical in
binding affinities, and besides affecting critical interac-
tions, mutations lead to change in conformational dy-
namics, which affects binding affinities.61,62,63
GluR2 interaction with wild typeand mutant PICK1
In GluR2, the carboxyl group of P0 Ile (-ESVKI) forms
a hydrogen bond in the LIGI motif of PICK1, whereas
the side chain of P-1 Lys (-ESVKI) makes minimal con-
tact with PICK1.52 In addition, the P-2 Val (-ESVKI)
interacts with the side chain of Lys83 and the methyl
group of Ala87 of PICK1. The negatively charged P-3
phosphorylated Ser (-ESVKI) also interacts with the posi-
tively charged Lys83 in PICK1. Finally, a stabilizing
hydrogen bond and side chain interactions are also found
between the amino group of Lys83 on PICK1 and the car-
bonyl of P-4 Glu (-ESVKI).52 In agreement with these
observations, the docking method used in this study
indicated that P-3 Ser (-SVKI) interacts with the Lys83 at
the aB1 position. Furthermore, it was observed that P-2
Val (-SVKI) forms a hydrophobic association with the
aliphatic side chain of Lys83 of PICK1 and also interacts
with the Ile37 of PICK1. This strong hydrophobic interac-
tion appeared to increase the binding affinity (-236.79
kcal/mol) of GluR2 for WT PICK1 as proposed earlier.34
Table IIComparison of Rigid and Flexible Docking with PRS
Peptide Mutation
Rigid docking Flexible docking with PRS
Experimental affinityDDE# Agreement with experiments DDE# Agreement with experiments
GluR2 (-SVKI) K27E 230 Yes 223.20 Yes DecreasePKCa (-QSAV) K27A 0 Yes 6.38 Yes Still interacts
K27E 1 Yes 15.72 Yes Still interactsK83H 25 No 3.18 Yes IncreaseK83V 26 No 8.28 Yes Increase
DAT (-WLKV) K83H 0 No 6.06 No DecreaseK83V 23 No 0.47 Yes Increase
Flexible docking results show better agreement with the experimental affinity data as compared to the rigid docking results. #DDE 5 DEWT 2 DEMut.
Modeling PICK1 Binding Affinities
PROTEINS 1401

The K27E PICK1 interaction with GluR2 showed destabi-
lization between P-3 Ser and Ile37 and a loss of interac-
tion between P-2 Val and Lys83, which could have
accounted for the decreased binding affinity (2213.58
kcal/mol). The K27A PICK1 also showed a similar loss of
interaction between P-2 Val and Lys83 and decreased
binding (2233.20 kcal/mol). The data showed that the
interaction between P-3 Ser and Lys83 were lost in K83H
PICK1. Furthermore, the phosphorylated P-3 Ser did not
interact with the His83 in PICK1, which explains the
decrease in binding affinity of K83H PICK1 (2222.56
kcal/mol). For K83V PICK1, the interactions between P-3
Ser and Val83, and P-2 Val and Ala87, which were not
observed between WT PICK1 and GluR2, might have
compensated for the loss of interaction between P-3 Ser
and Lys83 and explain the similar binding affinities of
K83V PICK1 (2236.57 kcal/mol) and WT PICK1.
PKCa interaction with wild type andmutant PICK1
Further analysis of the PKCa-PICK1 interactionshowed that P-2 Ser and P0 Val of the PKCa motif(-QSAV) interacts with Ala87 (P-2 binding pocket) ofPICK1. It was also observed that P-2 Ser interacts withside chains of Ile37 (P-2 binding pocket) and P0 Valinteracts with Ile33, Ile35, and Ile90 (P0 binding pocket)of PICK1, in agreement with previous observations33,34
Figure 5Ribbon diagrams of PKCa and DAT with PICK1. Interactions shown between PKCa (-QSAV) for (A) WT-PICK1 and (B) mutant K83H PICK. Also
shown are interactions between DAT (-WLKV) for (C) WT-PICK1 and (D) mutant K83V-PICK1. PKCa and DAT peptides are show with side-chains. The critical residues interacting with PKCa and DAT peptide are shown and labelled. The mutated residues Lys83, Val83, and His83 are
shown in dark grey. The residues whose interactions with PKCa are lost after K83H mutation are Ser36 and Ile35. [Color figure can be viewed in
the online issue, which is available at wileyonlinelibrary.com.]
A. Bolia et al.
1402 PROTEINS

[Fig. 5(A)]. Analysis on the lowest energy docked pose ofK27E PICK1 with PKCa showed an interaction betweenP-2 Ser and Lys83 in PICK1. As proposed earlier, thisinteraction increases the binding affinity of PKCa forK27E PICK1 (2232.90 kcal/mol) as compared with WT
PICK1 (2217.18 kcal/mol). The data showed that the
interactions of P0, P-1, P-2, and P-3 residues of PKCa(-QSAV) were more stable with K27A PICK1 (2223.56
kcal/mol). Interestingly, the P-2 Ser interacts with His83
in the PDZ domain of K83H PICK1 [Fig. 5(B)], which is
not observed at the P-2 residue in Class II peptides. As
proposed in previous studies,33–36 the K83H mutation
allowed a hydrogen bond between P-2 Ser in PKCa(-QSAV) and His83 in PICK1, which might have compen-
sated for the loss of interaction between P-2 Ser and
Lys83 in PICK1. Thus, the interaction between P-2 Ser
and His83 might account for the slight increase in the
binding affinity of PKCa for K83H PICK1 (2220.36 kcal/
mol). Likewise, interaction between P-2 Ser and Val83
might have compensated for the loss of interaction
between P-2 Ser and Lys83 in K83V PICK1. This might
explain the increase in binding affinity for K83V PICK1
(-225.46 kcal/mol).
DAT interaction with wild type andmutant PICK1
The data generated also showed that P-2 Leu in DAT
(-WLKV) interacts with Val84 and Ala87 (P-2 binding
pocket) in PICK1 and also with Ile37 of the bB strand of
PICK1 [Fig. 5(C)]. These interactions might contribute
towards the high binding affinity of DAT for PICK1
(2291.93 kcal/mol). Further analysis indicated that a
hydrophobic interaction between aliphatic side chain of
P-1 Lys in DAT (-WLKV) and Phe53 of PICK1 is stabi-
lized significantly in K27A PICK1. In contrast, the inter-
action between P-2 Leu of DAT (-WLKV) and Ser36 of
PICK1 is destabilized in K27A PICK1, whereas it is stabi-
lized in K27E PICK1. Therefore, we propose that the P-2
Leu and Ser36 might form an unfavourable interaction
and the destabilization of this interaction along with the
stabilization of a hydrophobic interaction of the aliphatic
side chain of P-1 Lys of DAT and Phe53 in K27A PICK1
could increase the binding affinity (2301.13 kcal/mol)
compared to K27E PICK1 (2274.00 kcal/mol). Analysis
also showed that interactions between P-2 Leu of DAT (-
WLKV) with His83, Val84, and Ile37 in PICK1 became
more stabilized in K83H PICK1. Thus, these strong inter-
actions between P-2 Leu of DAT (-WLKV) and the cru-
cial residues of PICK1 increases the binding affinity of
DAT for K83H PICK1 (2297.99 kcal/mol). Likewise, it
was observed that the interactions between P-2 Leu with
Val83 and Val84 became more stabilized in K83V PICK1
[Fig. 5(D)]. Although data showed increased binding
energies for K83V and K83H PICK1, the experimental
analysis suggests an increase in affinity of DAT for K83V
PICK1, while a decrease in binding affinity for K83H
PICK1. To investigate this further, we found that some
hydrogen bond interactions were lost between DAT pep-
tide and K83H PICK1 mutant suggesting a possible rea-
son for decrease in binding affinity for K83H PICK1 as
seen in experimental results. However, our scoring func-
tion was not sensitive enough to capture this effect in its
binding score. Overall, the analysis suggests that the K83V
mutation is not perturbed due to a more stabilized inter-
action of P-2 Leu of DAT (-WLKV) with the mutant res-
idue at Val83 as proposed earlier,34 yet it is not sensitive
to small changes in binding affinity detected by experi-
mental methods.
Computational docking analysis of thePICK1 compound FSC231
The first small-molecule inhibitor (FSC231) of the
PDZ domain in PICK1 has been identified among
�44,000 compounds with a fluorescent polarization
assay.37,38 The fluorescent polarization binding experi-
ments showed that the FSC231 compound has higher af-
finity for the K83H mutant than for the wild type of
PICK1.37 The affinity for PICK1 compound FSC231 was
also analyzed and predicted using the docking
approaches explained above. The chemical structure of
the compound was generated with the PRODGR server
(http://davac1.bioch.dundee.ac.uk/programs/prodrg/
prodrg.html) and then used a geometric-based conforma-
tional sampling technique called FRODA (Framework Ri-
gidity Optimized Dynamics Algorithm)56 to generate a
variety of different topological structures of the com-
pound. These structures were docked into the wild type
and K83H mutant of PICK1 structures. The predicted
binding energy values agree with fluorescent polarization
binding experiments showing a higher affinity of FSC231
for the K83H mutant (299.50 kcal/mol) than for the
wild type (295.69 kcal/mol) (Fig. 6).
Correlation of flexible versus rigid dockingwith experimental data
Previous studies have shown that incorporating back-
bone flexibility of PDZ domains increases the prediction
accuracy in determining their peptide binding specific-
ity57–63 and therefore in their binding selectivity.55 As
indicated above, in this study, a PDZ flexibility
approach was introduced into the docking simulations
through the generation of an ensemble of multiple re-
ceptor conformations (MRCs) together with a newly
developed coarse grain approach, where an elastic net-
work method (ENM) was coupled with linear response
theory (LRT).64 The LRT states that the response behav-
ior is related to the equilibrium fluctuations in the
unperturbed state (i.e., unbound state). Therefore, after
constructing the structural information of the unbound
form using ENM (i.e., a purely mechanical model that
Modeling PICK1 Binding Affinities
PROTEINS 1403

views a protein structure as a network of harmonic resi-
due–residue interactions), the LRT was used to formu-
late the response of the protein structure to ligand bind-
ing and to obtain structural changes of the given struc-
ture. These computed structural changes, upon binding,
were integrated into the docking simulations through
generation of multiple conformations of the receptor
(PDZ domain). To further determine if this flexible
modeling approach increased the prediction accuracy
compared with rigid modeling (Table II), both flexible
and rigid modeled binding values were compared with
experimental data and correlation plots were generated.
In these comparisons the binding energy scores were
compared with binding energy of experimental results
using Ki values. Specifically, a quantitative comparison
was made between the predicted and experimental affin-
ities34 of wild-type and single point mutations of DAT
and PKCa peptides bound to wild-type and mutated
PICK1 (K83H). The binding energy was computed for
all cases with both rigid and flexible docking by PRS
(Table III). Simultaneously, the experimental binding
free energy (DG) values were also calculated from the
experimental Ki values. The data showed some disagree-
ment in both flexible and rigid values compared to ex-
perimental data, likely due to the stringent test of pre-
dicting the change of affinity per single mutations.
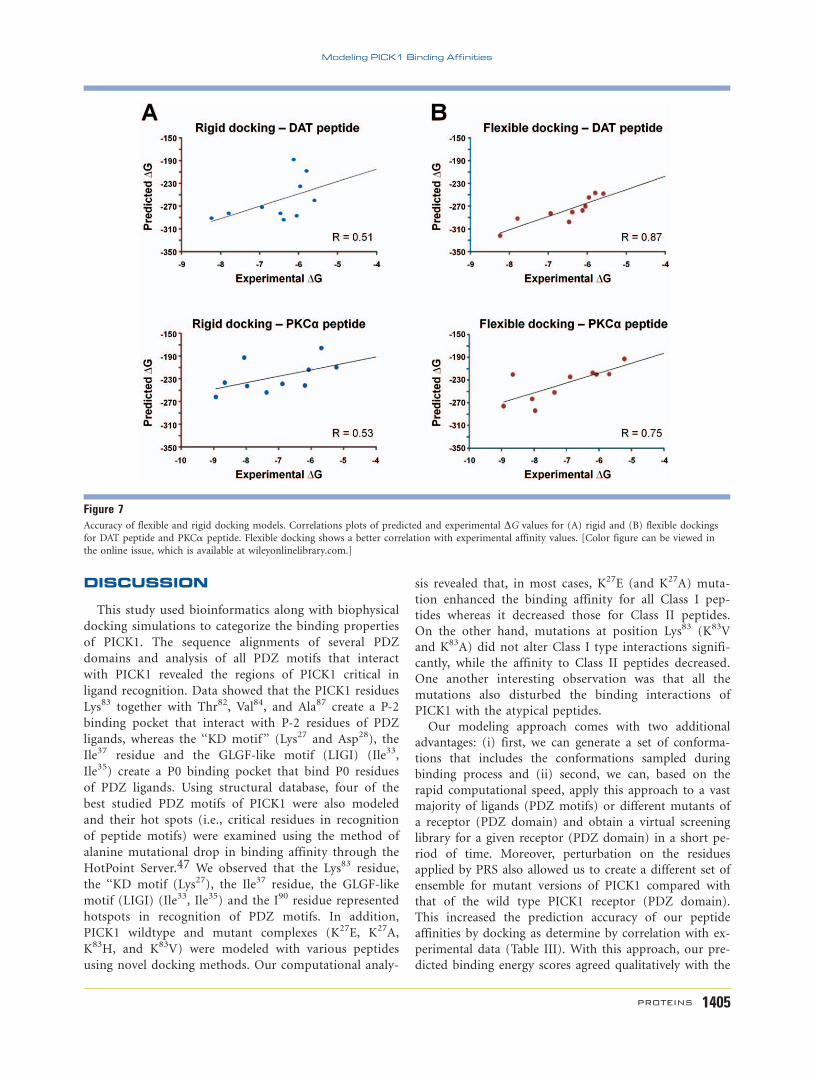
Nevertheless, the correlation between the experimental
affinity and predicted affinities showed a value of 0.87
for flexible docking as compared with 0.51 for rigid
docking for the DAT peptide (Fig. 7). Similarly for the
PKCa peptide, a correlation of 0.75 for flexible docking
and 0.53 for rigid docking was obtained (Fig. 7). This
analysis demonstrated further that the accuracy and pre-
diction of the flexible PRS docking approach provided
better correlation with the experimental data compared
with rigid docking methods.
Table IIICorrelation of Docking Models with Experimental Data
PDZ Motif Experimental Theoretical
PICK1 DAT Ki DG DG Rigiddocking
DG FlexibleDocking
WT 2LKV 2.3 27.790 2282.62 2291.932 SKV 42 26.047 2287.04 2270.262 LKI 9.5 26.939 2272.03 2282.622 LKL 37 26.123 2188.12 2277.322 LKA 49 25.954 2235.15 2254.80
K83H 2LKV 21 26.463 2282.63 2297.992 SKV 1.1 28.232 2291.24 2321.712 LKI 24 26.382 2294.11 2280.202 LKL 64 25.794 2207.99 2246.632 LKA 90 25.590 2260.10 2247.96
Correlation 0.51 0.87
PICK1 PKCa Ki DGDG Rigiddocking
DG flexibledocking
WT 2SAV 33 26.191 2241.74 2217.182LAV 1.7 27.971 2242.70 2283.652SAI 77 25.683 2175.64 2219.652SAL 166 25.222 2209.55 2192.982SAA 40 26.076 2213.93 2220.22
K83H 2SAV 0.54 28.659 2236.75 2220.362LAV 10.4 26.884 2238.89 2224.602SAI 1.46 28.062 2192.79 2263.462SAL 4.6 27.374 2253.94 2251.652SAA 0.34 28.937 2261.94 2275.84
Correlation 0.53 0.75
Docking results show comparison between experimental binding free energy (DG)with rigid and flexible docking methods.
Figure 6Binding affinity of FSC231. The lowest binding energy scores and their corresponding structures of docking FSC231 compound to (A) the wild type
and (B) the mutant (K83H) of PICK1s are displayed as ribbon diagrams. The key residues of the PICK1 domain making side interactions with
FSC231 are also shown. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
A. Bolia et al.
1404 PROTEINS
DISCUSSION
This study used bioinformatics along with biophysical
docking simulations to categorize the binding properties
of PICK1. The sequence alignments of several PDZ
domains and analysis of all PDZ motifs that interact
with PICK1 revealed the regions of PICK1 critical in
ligand recognition. Data showed that the PICK1 residues
Lys83 together with Thr82, Val84, and Ala87 create a P-2
binding pocket that interact with P-2 residues of PDZ
ligands, whereas the ‘‘KD motif ’’ (Lys27 and Asp28), the
Ile37 residue and the GLGF-like motif (LIGI) (Ile33,
Ile35) create a P0 binding pocket that bind P0 residues
of PDZ ligands. Using structural database, four of the
best studied PDZ motifs of PICK1 were also modeled
and their hot spots (i.e., critical residues in recognition
of peptide motifs) were examined using the method of
alanine mutational drop in binding affinity through the
HotPoint Server.47 We observed that the Lys83 residue,
the ‘‘KD motif (Lys27), the Ile37 residue, the GLGF-like
motif (LIGI) (Ile33, Ile35) and the I90 residue represented
hotspots in recognition of PDZ motifs. In addition,
PICK1 wildtype and mutant complexes (K27E, K27A,
K83H, and K83V) were modeled with various peptides
using novel docking methods. Our computational analy-
sis revealed that, in most cases, K27E (and K27A) muta-
tion enhanced the binding affinity for all Class I pep-
tides whereas it decreased those for Class II peptides.
On the other hand, mutations at position Lys83 (K83V
and K83A) did not alter Class I type interactions signifi-
cantly, while the affinity to Class II peptides decreased.
One another interesting observation was that all the
mutations also disturbed the binding interactions of
PICK1 with the atypical peptides.
Our modeling approach comes with two additional
advantages: (i) first, we can generate a set of conforma-
tions that includes the conformations sampled during
binding process and (ii) second, we can, based on the
rapid computational speed, apply this approach to a vast
majority of ligands (PDZ motifs) or different mutants of
a receptor (PDZ domain) and obtain a virtual screening
library for a given receptor (PDZ domain) in a short pe-
riod of time. Moreover, perturbation on the residues
applied by PRS also allowed us to create a different set of
ensemble for mutant versions of PICK1 compared with
that of the wild type PICK1 receptor (PDZ domain).
This increased the prediction accuracy of our peptide
affinities by docking as determine by correlation with ex-
perimental data (Table III). With this approach, our pre-
dicted binding energy scores agreed qualitatively with the
Figure 7Accuracy of flexible and rigid docking models. Correlations plots of predicted and experimental DG values for (A) rigid and (B) flexible dockings
for DAT peptide and PKCa peptide. Flexible docking shows a better correlation with experimental affinity values. [Color figure can be viewed in
the online issue, which is available at wileyonlinelibrary.com.]
Modeling PICK1 Binding Affinities
PROTEINS 1405

available experimental data. For example, our analysis
shows that Ephrin B1, Parkin and DAT (Type II sequen-
ces) have strong affinities while the atypical peptide of
UNCH5 has the weakest affinity. With the same
approach, we also examined the two crucial binding hot-
spots of PICK1, namely Lys83 and Lys27. Using the single
mutation of K27E in our flexible docking, we observed
that the binding energy score of GluR2 significantly
decreased, whereas that of PKCa enhanced with this mu-
tant version of PICK1 (Table I). Likewise, we found
increased binding energy of PKCa, but not of DAT, whenwe mutated Lys83 to His, which is also observed in muta-
tional binding analysis.34 Moreover, our docking analysis
showed a disruption in binding affinity of GluR7 upon
K27E mutation as observed experimentally.4 Notably,
these binding affinity changes were unlikely to be
obtained if a single crystal structure of PICK1 had been
used in our computational analysis. Thus, the accuracy
in our flexible docking method lies in incorporating the
binding-induced conformational changes through PRS. It
is known that when a ligand (PDZ motif) approaches a
receptor (PDZ domain), it exerts force on the binding
site. Likewise, by exerting small random forces on the
binding residue site, we computed the displacement of
each residue and also incorporated those displacement
results into our docking analysis. This enabled us to inte-
grate both backbone and side chain conformational
changes of the receptor (PDZ domain) in the docking
analysis. These small conformational changes in the back-
bone can lead to a significant change in the side chain,
thus simulating the changes of how a ligand (PDZ motif)
interacts with the receptor (PDZ domain).
In closing, we suggest these new computational analy-
ses methods will advance our understanding of the struc-
ture and binding relationships of PICK1. We have previ-
ously highlighted the value of PDZ domains as novel
drug targets for disease.65 The study provides modeling
methods to aid in the drug design of ligands for PDZ
domains, which may further aid in development of the
family of PDZ domains as a drug target.
ACKNOWLEDGMENTS
The authors thanks the Fulton High Performance
Computing Initiative at ASU for computer time. The
authors would like to thank Dr. Graham Sheridan,
Yvonne Kuma, Ciara Twomey and Shane Lyons for help
with the manuscript.
REFERENCES
1. Ponting CP. Evidence for PDZ domains in bacteria, yeast, and
plants. Protein Sci 1997;6:464–468.
2. Jemth P, Gianni S. PDZ domains: folding and binding. Biochemis-
try 2007;46:8701–8708.
3. Sheng M, Sala C. PDZ domains and the organization of supramo-
lecular complexes. Annu Rev Neurosci 2001;24:1–29.
4. Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Naka-
nishi S. PICK1 interacts with and regulates PKC phosphorylation of
mGLUR7. J Neurosci 2000;20:7252–7257.
5. Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir
RL. Calcium-permeable AMPA receptor plasticity is mediated by
subunit-specific interactions with PICK1 and NSF. Neuron
2005;45:903–915.
6. Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-
induced AMPA receptor trafficking. EMBO J 2005;24:3266–3278.
7. Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S,
Zhang M, Xia J. Lipid binding regulates synaptic targeting of
PICK1, AMPA receptor trafficking, and synaptic plasticity. J Neuro-
sci 2006;26:2380–2390.
8. Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the
AMPA receptor subunit GluR2/3 with PDZ domains regulates hip-
pocampal long-term depression. Proc Natl Acad Sci USA
2001;98:11725–11730.
9. Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate
receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recy-
cling after NMDA receptor-induced internalization. J Neurosci
2007;27:13903–13908.
10. Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge
GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA
receptor-dependent bidirectional synaptic plasticity. Neuron
2008;57:872–882.
11. Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA
receptors by the synaptic PDZ domain-containing protein PICK1.
Neuron 1999;22:179–187.
12. Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar
long-term depression requires PKC-regulated interactions between
GluR2/3 and PDZ domain-containing proteins. Neuron 2000;28:
499–510.
13. Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-
mediated actin polymerization by PICK1 regulates neuronal mor-
phology and AMPA receptor endocytosis. Nat Cell Biol
2008;10:259–271.
14. Wang WL, Yeh SF, Huang EY, Lu YL, Wang CF, Huang CY, Lin WJ.
Mitochondrial anchoring of PKCa by PICK1 confers resistance to
etoposide-induced apoptosis. Apoptosis 2007;12:1857–1871.
15. Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ do-
main protein PICK1 with the COOH terminus of protein kinase C-
alpha. J Biol Chem 1997;272:32019–32024.
16. Bertaso F, Zhang C, Scheschonka A, de Bock F, Fontanaud P, Marin
P, Huganir RL, Betz H, Bockaert J, Fagni L, Lerner-Natoli M.
PICK1 uncoupling from mGluR7a causes absence-like seizures. Nat
Neurosci 2008;11:940–948.
17. Zhang CS, Bertaso F, Eulenburg V, Lerner-Natoli M, Herin GA,
Bauer L, Bockaert J, Fagni L, Betz H, Scheschonka A. Knock-in
mice lacking the PDZ-ligand motif of mGluR7a show impaired
PKC-dependent autoinhibition of glutamate release, spatial working
memory deficits, and increased susceptibility to pentylenetetrazol. J
Neurosci 2008;28:8604–8614.
18. Baron A, Deval E, Salinas M, Lingueglia E, Voilley N, Lazdunski M.
Protein kinase C stimulates the acid-sensing ion channel ASIC2a
via the PDZ domain-containing protein PICK1. J Biol Chem
2002;277:50463–50468.
19. Garry EM, Moss A, Rosie R, Delaney A, Mitchell R, Fleetwood-
Walker SM. Specific involvement in neuropathic pain of AMPA
receptors and adapter proteins for the GluR2 subunit. Mol Cell
Neurosci 2003;24:10–22.
20. Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis
contributes to cellular injury after neuronal trauma. Cell Death Dif-
fer 2009;16:1665–1680.
21. Dixon RM, Mellor JR, Hanley JG. PICK1-mediated glutamate
receptor subunit 2 (GluR2) trafficking contributes to cell death in
oxygen/glucose-deprived hippocampal neurons. J Biol Chem
2009;284:14230–14235.
A. Bolia et al.
1406 PROTEINS

22. Joch M, Ase AR, Chen CX, MacDonald PA, Kontogiannea M, Cor-
era AT, Brice A, Seguela P, Fon EA. Parkin-mediated monoubiquiti-
nation of the PDZ protein PICK1 regulates the activity of acid-
sensing ion channels. Mol Biol Cell 2007;18:3105–3118.
23. Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of
AMPA receptor trafficking and signaling molecule transcripts in the
prefrontal cortex in schizophrenia. Synapse 2006;60:585–598.
24. Bousman CA, Glatt SJ, Everall IP, Tsuang MT. Genetic association
studies of methamphetamine use disorders: a systematic review and
synthesis. Am J Med Genet B Neuropsychiatr 2009;150:1025–1049.
25. Dev KK, Henley JM. The schizophrenic faces of PICK1. Trends
Pharmacol Sci 2006;27:574–579.
26. Dracheva S, McGurk SR, Haroutunian V. mRNA expression of
AMPA receptors and AMPA receptor binding proteins in the cere-
bral cortex of elderly schizophrenics. J Neurosci Res 2005;79:868–
878.
27. Fujii K, Maeda K, Hikida T, Mustafa AK, Balkissoon R, Xia J,
Yamada T, Ozeki Y, Kawahara R, Okawa M, Huganir RL, Ujike H,
Snyder SH, Sawa A. Serine racemase binds to PICK1: potential rele-
vance to schizophrenia. Mol Psychiatry 2006:11:150–157.
28. Ghasemzadeh MB, Vasudevan P, Mueller C, Seubert C, Mantsch JR.
Region specific alterations in glutamate receptor expression and
subcellular distribution following extinction of cocaine self-adminis-
tration. Brain Res 2009;1267:89–102.
29. Hong CJ, Liao DL, Shih HL, Tsai SJ. Association study of PICK1
rs3952 polymorphism and schizophrenia. Neuroreport 2004;15:
1965–1967.
30. Ishiguro H, Koga M, Horiuchi Y, Inada T, Iwata N, Ozaki N, Ujike
H, Muratake T, Someya T, Arinami T. PICK1 is not a susceptibil-
ity gene for schizophrenia in a Japanese population: association
study in a large case-control population. Neurosci Res
2007;58:145–148.
31. Matsuzawa D, Hashimoto K, Miyatake R, Shirayama Y, Shimizu E,
Maeda K, Suzuki Y, Mashimo Y, Sekine Y, Inada T, Ozaki N, Iwata
N, Harano M, Komiyama T, Yamada M, Sora I, Ujike H, Hata A,
Sawa A, Iyo M. Identification of functional polymorphisms in the
promoter region of the human PICK1 gene and their association
with methamphetamine psychosis. Am J Psychiatry 2007;164:1105–
1114.
32. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J,
Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C.
STRING 8—a global view on proteins and their functional interac-
tions in 630 organisms. Nucleic Acids Res 2009;37:D412–D416.
33. Elkins JM, Papagrigoriou E, Berridge G, Yang X, Phillips C, Gileadi
C, Savitsky P, Doyle DA. Structure of PICK1 and other PDZ
domains obtained with the help of self-binding C-terminal exten-
sions. Protein Sci. 2007;16:683–694.
34. Madsen KL, Beuming T, Niv MY, Chang CW, Dev KK, Weinstein
H, Gether U. Molecular determinants for the complex binding
specificity of the PDZ domain in PICK1. J Biol Chem
2005;280:20539–20548.
35. Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. PICK1
targets activated protein kinase Calpha to AMPA receptor clusters
in spines of hippocampal neurons and reduces surface levels of the
AMPA-type glutamate receptor subunit 2. J Neurosci 2001;21:5417–
5428.
36. Dev KK, Nakanishi S, Henley JM. The PDZ domain of PICK1 dif-
ferentially accepts protein kinase C-alpha and GluR2 as interacting
ligands. J Biol Chem 2004;279:41393–41397.
37. Thorsen TS, Madsen KL, Rebola N, Rathje M, Anggono V, Bach A,
Moreira IS, Stuhr-Hansen N, Dyhring T, Peters D, Beuming T,
Huganir R, Weinstein H, Mulle C, Stromgaard K, Ronn ,LC, Gether
U. Identification of a small-molecule inhibitor of the PICK1 PDZ
domain that inhibits hippocampal LTP and LTD. Proc Natl Acad
Sci USA 2010;107:413–418.
38. Bach A, Stuhr-Hansen N, Thorsen TS, Bork N, Moreira IS, Fryden-
vang K, Padrah S, Christensen SB, Madsen KL, Weinstein H, Gether
U, Strømgaard K. Structure-activity relationships of a small-mole-
cule inhibitor of the PDZ domain of PICK1. Org Biomol Chem
2010;8:4281–4288.
39. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig
H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic
Acids Res 2000;28:235–242.
40. Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an
environment for comparative protein modeling. Electrophoresis
1997;18:2714–2723.
41. Atilgan C, Gerek ZN, Ozkan SB, Atilgan AR. Manipulation of con-
formational change in proteins by single-residue perturbations. Bio-
phys J 2010;99:933–943.
42. Tsui V, Case DA. Molecular dynamics simulations of nucleic acids
with a generalized born solvation model. J Am Chem Soc
2000;122:2489–2498.
43. Davis IW, Baker D. RosettaLigand docking with full ligand and re-
ceptor flexibility. J Mol Biol 2009;385:381–392.
44. Meiler J, Baker D. ROSETTALIGAND: protein-small molecule
docking with full side-chain flexibility. Proteins 2006;65:538–548.
45. Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function
to predict protein-ligand interactions. J Mol Biol 2000;29:337–356.
46. Tuncbag N, Gursoy A, Keskin O. Identification of computational
hot spots in protein interfaces: combining solvent accessibility and
inter-residue potentials improves the accuracy. Bioinformatics
2009;25:1513–1520.
47. Tuncbag N, Keskin O, Gursoy A. HotPoint: hot spot prediction
server for protein interfaces. Nucleic Acids Res 2010;38:W402–
W406.
48. Aytuna AS, Gursoy A, Keskin O. Prediction of protein-protein
interactions by combining structure and sequence conservation in
protein interfaces. Bioinformatics 2005;21:2850–2855.
49. Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A. PRISM:
protein interactions by structural matching. Nucleic Acids Res
2005;33:W331–W336.
50. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM,
Meng EC, Ferrin TE. UCSF Chimera—a visualization system for ex-
ploratory research and analysis. J Comput Chem 2004;25:1605–
1612.
51. Madsen KL, Eriksen J, Milan-Lobo L, Han DS, Niv MY,
Ammendrup-Johnsen I, Henriksen U, Bhatia VK, Stamou D, Sitte
HH, McMahon HT, Weinstein H, Gether U. Membrane localization
is critical for activation of the PICK1 BAR domain. Traffic
2008;9:1327–1343.
52. Pan L, Wu H, Shen C, Shi Y, Jin W, Xia J, Zhang M. Clustering
and synaptic targeting of PICK1 requires direct interaction between
the PDZ domain and lipid membranes. EMBO J 2007;26:4576–
4587.
53. Shi Y, Zhang L, Yuan J, Xiao H, Yang X, Niu L. Zinc binding site
in PICK1 is dominantly located at the CPC motif of its PDZ do-
main. J Neurochem 2008;106:1027–1034.
54. Clackson T, Wells JA. A hot spot of binding energy in a hormone-
receptor interface, Science 1995;267:383–386.
55. Bassan M, Liu H, Madsen KL, Armsen W, Zhou J, Desilva T, Chen
W, Paradise A, Brasch MA, Staudinger J, Gether U, Irwin N, Rosen-
berg PA. Interaction between the glutamate transporter GLT1b and
the synaptic PDZ domain protein PICK1. Eur J Neurosci
2008;27:66–82.
56. Wells S, Menor S, Hespenheide B, Thorpe MF. Constrained geomet-
ric simulation of diffusive motion in proteins. Phys Biol
2005;2:S127–S136.
57. Lockless SW, Ranganathan R. Evolutionarily conserved pathways of
energetic connectivity in protein families. Science 1999;286:295–
299.
58. Dhulesia A, Gsponer J, Vendruscolo M. Mapping of two networks
of residues that exhibit structural and dynamical changes upon
binding in a PDZ domain protein. J Am Chem Soc 2008;130:8931–
8939.
Modeling PICK1 Binding Affinities
PROTEINS 1407

59. Ho BK, Agard DA. Conserved tertiary couplings stabilize elements
in the PDZ fold, leading to characteristic patterns of domain con-
formational flexibility. Protein Sci 2010;19:398–411.
60. Kong Y, Karplus M. Signaling pathways of PDZ2 domain: a molec-
ular dynamics interaction correlation analysis. Proteins 2009;74:
145–154.
61. Gerek ZN, Keskin O, Ozkan SB. Identification of specificity and
promiscuity of PDZ domain interactions through their dynamic
behavior. Proteins 2009;77:796–811.
62. Gerek ZN, Ozkan SB. A flexible docking scheme to explore the
binding selectivity of PDZ domains. Protein Sci 2010;19:914–928.
63. Smith CA, Kortemme T. Structure-based prediction of the peptide
sequence space recognized by natural and synthetic PDZ domains. J
Mol Biol 2010;402:460–474.
64. Ikeguchi M, Ueno J, Sato M, Kidera A. Protein structural change upon
ligand binding: linear response theory. Phys Rev Lett 2005;94:078102.
65. Dev KK. Making protein interactions druggable: targeting PDZ
domains. Nat Rev Drug Dis 2004;3:1047–1056.
1408 PROTEINS
A. Bolia et al.