population genetics, recombination histories & global pedigrees finding minimal recombination...
TRANSCRIPT

Population Genetics, Recombination Histories & Global Pedigrees
Finding Minimal Recombination Histories
1 2 3 4 1 2 3 4 1 234
Global Pedigrees
Fin
din
g
Co
mm
on
A
nc
es
tors
NOW
Population Genetics and Genealogies
NOW
Forw
ard in time
Bac
kwar
d in
tim
e

Wright-Fisher Model of Population Reproduction
Haploid Modeli. Individuals are made by sampling with replacement in the previous generation.
ii. The probability that 2 alleles have same ancestor in previous generation is 1/2N
Diploid Model
Individuals are made by sampling a chromosome from the female and one from the male previous generation with replacement
Assumptions
1. Constant population size
2. No geography
3. No Selection
4. No recombination

10 Alleles’ Ancestry for 15 generations
Generate new generations according to the haploid model
Sort generations forward in time, such that individual 1’s children in the next generation comes first, then the children of 2..
Pick individuals in the present and label edges to their ancestors back in time

Mean, E(X2) = 2N.
Ex.: 2N = 20.000, Generation time 30 years, E(X2) = 600000 years.
Waiting for most recent common ancestor - MRCA
P(X2 = j) = (1-(1/2N))j-1 (1/2N)
Distribution of time until 2 alleles had a common ancestor, X2?:
P(X2 > j) = (1-(1/2N))jP(X2 > 1) = (2N-1)/2N = 1-(1/2N)
1 2N
1
1 2N
1
2
j
1 2N
1
2
j

P(k):=P{k alleles had k distinct parents}
1 2N
1
(2N)k
k -> any2N *(2N-1) *..* (2N-(k-1)) =: (2N)[k]
k -> k
Ancestor choices:
k -> k-1
€
k
2
⎛
⎝ ⎜
⎞
⎠ ⎟(2N)[k−1]
€
P(k) =2N[k ]
(2N)k≈ (k 2 < 2N) 1−
k
2
⎛
⎝ ⎜
⎞
⎠ ⎟/2N ≈ e
−k
2
⎛
⎝ ⎜
⎞
⎠ ⎟/ 2N
For k << 2N:
k -> j
€
Sk, j (2N)[ j ]
Sk,j - the number of ways to group k labelled objects into j groups.(Stirling Numbers of second kind.

Expected Height and Total Branch Length
Expected Total height of tree: Hk= 2(1-1/k) +..+ 1/(k-1)) ca= 2*ln(k-1)
1
2
3
k
2
1
2/(k-1)
Branch Lengths
)1(
2
2/1
−=⎟⎟
⎠
⎞⎜⎜⎝
⎛kk
k
1/3
1
Time Epoch
i.Infinitely many alleles finds 1 allele in finite time.ii. In takes less than twice as long for k alleles to find 1 ancestors as it does for 2 alleles.
Expected Total branch length in tree, Lk: 2*(1 + 1/2 + 1/3 +..+ 1/(k-1)) ca= 2*ln(k-1)

6 Realisations with 25 leaves
Observations:
Variation great close to root.
Trees are unbalanced.

Sampling more sequences
The probability that the ancestor of the sample of size n is in a sub-sample of size k is
Letting n go to infinity gives (k-1)/(k+1), i.e. even for quite small samples it is quite large.
€
(n+1)(k−1)(n−1)(k+1)

Three Models of Alleles and Mutations.
i. Only identity, non-identity is determinable
ii. A mutation creates a new type.
i. Allele is represented by a line.
ii. A mutation always hits a new position.
i. Allele is represented by a sequence.
ii. A mutation changes nucleotide at chosen position.
Infinite Allele
Infinite Site
Finite Site
acgtgcttacgtgcgtacctgcattcctgcattcctgcat
acgtgcttacgtgcgtacctgcattcctggcttcctgcat

Final Aligned Data Set:
Infinite Site Model

Recombination-Coalescence Illustration Copied from Hudson 1991
Intensities
Coales. Recomb.
1 2
3 2
6 2
1 (1+b)
0
3 (2+b)
b
Bac
kwar
d in
tim
e

Local Inference of Recombinations
0000111
0001101
00
10
01
11
Four combinationsIncompatibility:
Myers-Griffiths (2002): Number of Recombinations in a sample, NR, number of types, NT, number of mutations, NM obeys:
€
NR ≥ NT − NM −1
0011
0101
T . . . GT . . . CA . . . GA . . . C
Recoding
•At most 1 mutation per column
•0 ancestral state, 1 derived state

”Observing” Recombinations: Hudson & Kaplan’s RM
If you equate RM with expected number of recombinations, this could be used as an estimator. Unfortunately, RM is a gross underestimate of the real number of recombinations.
0 0 0 0 0 1 0 0 0 0 0 1 1 1 1 1 1 1 1 1 0 0 0 0 0 0 0 00 0 1 0 0 0 0 0 0 0 0 1 1 1 1 1 1 1 1 1 0 0 0 0 0 0 0 00 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 00 0 0 0 0 0 0 0 0 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 00 0 0 1 1 1 1 1 1 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 10 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 1 0 1 0 1 1 10 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 0 11 1 1 1 1 1 1 1 1 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 0 11 1 1 1 1 1 0 0 1 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 0 1

Minimal Number of Recombinations
Last Local Tree Algorithm:
L21Data
2
n
i-1 i
1
Trees
The Kreitman data (1983): 11 sequences, 3200bp, 43(28) recoded, 9 different
How many neighbors?
€
(2n − 2)!
2n−1(n −1)!
€
n! (n −1)!
2n−1
14133 2 +− nn
€
~ n3
Bi-partitionsHow many local trees?
• Unrooted
• Coalescent

Metrics on Trees based on subtree transfers.
Pretending the easy problem (unrooted) is the real problem (age ordered), causes violation of the triangle inequality:
Tree topologies with age ordered internal nodes
Rooted tree topologies
Unrooted tree topologies
Trees including branch lengths

Observe that the size of the unit-neighbourhood of a tree does not grow nearly as fast as the number of trees
Allen & Steel (2001) Song (2003+)
Du
e to Yu
n S
ong
Tree Combinatorics and Neighborhoods
€
1
32n3 − 3n2 − 20n + 39( )
€
n! (n −1)!
2n−1
€
(2n − 3)!!=(2n − 2)!
2n−1(n −1)!
⎣ ⎦∑−
=
+−−2
12
2 )1(log2)2(4n
m
mn
14133 2 +− nn
€
2(n − 3)(2n − 7)

1
23
4
56
7
Due to Yun Song

minARGs: Recombination Events & Local Trees
True ARG
Reconstructed ARG
1 2 3 4 5
1 23 4 5
((1,2),(1,2,3))
((1,3),(1,2,3))
n=7, =10, =75
Minimal ARG
True ARG
Mutation information on only one side
Mutation information on both sides
0 4 Mb
Hudson-Kaplan
Myers-Griiths
Song-Hein
n=8, =40
n=8, =15
€
j−1
j=1
n−1
∑
€
E70(R) =132
€
E70(Rvisible ) = 46

Counting + Branch and Bound Algorithm
?
Exact len
gth
Lower bound
Up
per B
oun
d
0 31 912 13143 86184 304365 627946 789707 630498 324519 1046710 1727
289920
k-recom
bin
atination
n
eighb
orhood
k
Song, Y
.S., Lyngsø, R
.B. &
Hein, J. (2006) C
ounting all possible ancestral configurations of sample sequences in population genetics. IE
EE
/AC
M T
ransactions on C
omputational B
iology and Bioinform
atics, 3(3), 239–251 Lyngsø, R
.B., S
ong, Y.S
. & H
ein, J. (2008) Accurate com
putation of likelihoods in the coalescent w
ith recombination via parsim
ony. Lecture N
otes in Bioinform
atics: Proceedings of RE
CO
MB
2008 463–477
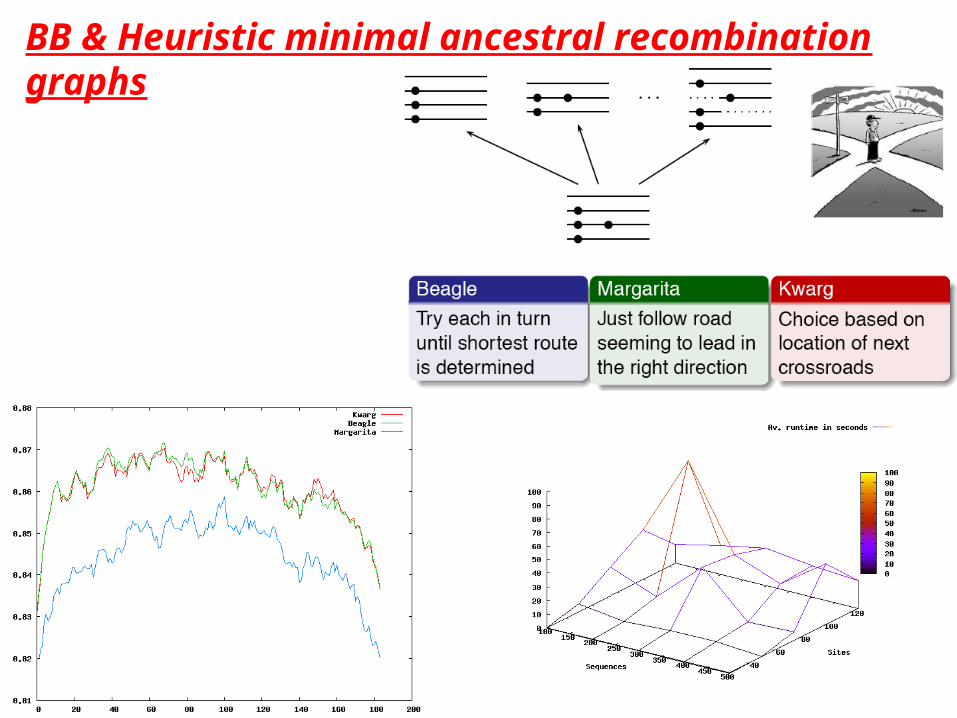
BB & Heuristic minimal ancestral recombination graphs

Time versus Spatial 1: Coalescent-Recombination (Griffiths, 1981; Hudson, 1983 - Wiuf & Hein, 1999)
Temporal Process Spatial Process
ii. The trees cannot be reduced to Topologies i. The process is non-Markovian
*
* =

Elston-Stewart (1971) -Temporal Peeling Algorithm:
Lander-Green (1987) - Genotype Scanning Algorithm:
Mother Father
Condition on parental states
Recombination and mutation are Markovian
Mother Father
Condition on paternal/maternal inheritance
Recombination and mutation are Markovian
Time versus Spatial 2: Pedigrees

Time versus Spatial 3: Phylogenetic Alignment
•Optimisation Algorithms
indels of length 1 (David Sankoff, 1973) Spatial
indels of length k (Bjarne Knudsen, 2003) Temporal
•Statistical Alignment
Spatial: Temporal:

- recombination 27 ACs
0
1
2
3
4
5
6
7
8
1
1
4
2
5
3
1
5
5
The Griffiths-Ethier-Tavare Recursions
No recombination: Infinite Site Assumption
Ancestral State Known
History Graph: Recursions Exists
No cycles
Possible Histories without Recombination for simple data example
+ recombination 9*108 ACs

1st
2nd
Ancestral configurations to 2 sequences with 2 segregating sites
mid-point heuristic
M
M
R
C
R
C
C

1st
2nd
0-ARG
5 states
1-ARG
+15 states
2-ARG
+10 states
, = 2, 0, = 1, 1, = 2, 2
0.1480.0370.032
0.1480.0820.074
0.1480.0900.085

Likelihood Calculations on the -ARG
010
010
101
101
110
Example:
0-ARG
2-ARG1-ARG

• For sites=7, sequences=6, then 10-7 th of states visited
• Cut-off -ARG, =2
-ARG likelihood calculation

Combining Ancestral Individuals and the Coalescent Wiuf & Hein, 2000.
Let T be the time, when somebody was everybody’s ancestor.
Fin
din
g C
om
mo
n A
nc
es
tors
.
NOW
Changs’ result: lim T*/log2(N) =1 prob. 1
Unify the two processes:
I. Sample more individuals II. Let each have 2 parents with probabilty p.
Result: A discontinuity at 1.
For p<1 change log2logp
Comment: Genetic Ancestors is a vanishing set within Genealogical Ancestors.

Reconstructing global pedigrees: SuperpedigreesSteel and Hein, 2006
The gender-labeled pedigrees for all pairs defines global pedigree
k
Gender-unlabeled pedigrees don’t!!
Ste
el, M
. & H
ein,
J. (
2006
) R
econ
stru
ctin
g pe
digr
ees:
a c
ombi
nato
rial
per
spec
tive
. Jou
rnal
of
The
oret
ical
Bio
logy
, 240
(3),
360
–367

•All embedded phylogenies are observable
•Do they determine the pedigree?
Genomes with and --> infinity recombination rate, mutation rate
Benevolent Mutation and Recombination Process
Counter example: Embedded phylogenies:

Infinite Sequences: From ARG to Pedigree
Given A/B/C/D/E above how much does that constrain set of pedigrees
How many pedigrees are compatible with A/B/C/D/E varying over data?
A. The ARG?
What can you observe from data (infinite sequences)?
C. Sequence of local trees?
D. Set of local trees? E. Set/Sequences of local unrooted tree topologies?
B. Sequence of neighbor pairs of local trees with recombination points
Going to neighbor triples, quadruples,.., be more restrictive than pairs?
F. Set/Sequences of local bipartitions? (neighbor pairs…

Infinite Sequences: From ARG to Pedigree
A. The ARG?
What can you observe from data (infinite sequences)?
C. Sequence of local trees?
D. Set of local trees? E. Set/Sequences of local unrooted tree topologies?
B. Sequence of pairs of local trees with recombination points
Going to triples, quadruples,.., be more restrictive than pairs?
1 2 3 4 1 2 3 4 1 234F. Set/Sequences of bipartitions