poorly water-soluble drug nanoparticles via solvent evaporation in water-soluble porous polymers
TRANSCRIPT

P
Pw
AD
a
ARRAA
KPDAGPC
1
fowirmec(
cisedadesa
0h
International Journal of Pharmaceutics 447 (2013) 241– 250
Contents lists available at SciVerse ScienceDirect
International Journal of Pharmaceutics
jo ur nal homep a ge: www.elsev ier .com/ locate / i jpharm
harmaceutical nanotechnology
oorly water-soluble drug nanoparticles via solvent evaporation inater-soluble porous polymers
led D. Roberts, Haifei Zhang ∗
epartment of Chemistry, University of Liverpool, L69 7ZD, United Kingdom
r t i c l e i n f o
rticle history:eceived 7 January 2013eceived in revised form 15 February 2013ccepted 1 March 2013vailable online 13 March 2013
eywords:
a b s t r a c t
A generic method is described to form poorly water-soluble drug nanoparticles within water-solubleporous polymer by solvent evaporation. The simple dissolution of porous polymer with drug nanoparti-cles results in stable aqueous drug nanoparticle suspension under the optimized conditions. The porouspolymers were prepared by freeze-drying aqueous solutions of polyvinyl alcohol, polyethylene glycol,and a surfactant. They were then used as scaffolds for the formation of nanoparticles by initially soakingthem in an organic drug solution, followed with removing the solvent via evaporation under ambient
oorly water-soluble drugsrug nanocrystalsqueous nanosuspensionriseofulvinaclitaxelarbamazepine
conditions. This process was optimized for an antifungal drug griseofulvin, before being translated to anti-convulsant carbamazepine and antineoplastic paclitaxel via a similar procedure, with an aim to improvethe loading of drug nanoparticles. By varying certain process parameters a degree of control over theparticle size and surface charge could be attained, as well as the drug to stabilizer ratio (drug payload).Noticeably, aqueous paclitaxel nanoparticles (500 nm) were prepared which used the equivalent of 46%less stabilizer than the formulation Taxol®.
. Introduction
Overcoming poor aqueous solubility is a significant challengeaced by the pharmaceuticals industry at present, with over 40%f compounds in development pipelines classed as being poorlyater soluble (Kasim et al., 2003; Lipinski, 2002). This can lead to
ssues such as low bioavailability, erratic absorption profiles andeduced patient compliance from having to administer larger orore frequent doses (Rannard and Owen, 2009). The problem is
ven more acute in the field of oncology due to the high toxicity,ost and the side effects associated with many antineoplastic agentsShepherd, 2003).
There have been many attempts over the years to over-ome this issue through various formulation approaches. Thesenclude the application of emulsions (Masahiro, 2000), microemul-ions (Lawrence and Rees, 2000), solid dispersions (Vasconcelost al., 2007) and liposomes (Schwendener and Schott, 1996) asrug delivery systems. Design of polymer architectures (includingmphiphilic block copolymers, comb-shaped polymers, den-rimers, and hyperbranched polymers) has been extensively
mployed for delivery of hydrophobic drugs, by means of encap-ulation in micelles, nanoparticles, microspheres or capsules (Qiund Bae, 2006; Adams et al., 2003; Hoskins et al., 2012; Svenson,∗ Corresponding author. Tel.: +44 0151 7943545; fax: +44 0151 7943588.E-mail address: [email protected] (H. Zhang).
378-5173/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.ijpharm.2013.03.001
© 2013 Elsevier B.V. All rights reserved.
2009). These processes often experience problems such as poorphysical stability, difficulty in scale up and inability to achievehigh drug loading; issues which have so far prevented them frombeing widely adopted for wider use (Patravale et al., 2004). Anotherapproach to improving drug solubility is to form a nanosuspen-sion of the desired compound. A nanosuspension can be definedas a colloidal dispersion of pure drug nanoparticles (or nanocrys-tals) stabilized by appropriate dispersants (Liu et al., 2007, 2012).The principle advantage of employing a nanosuspension over otherdelivery methods is that the vehicle itself is composed of thepure drug, meaning a relatively high ratio of the active com-pound to stabilizing excipients can be achieved (Müller et al.,2011).
The standard methods employed to prepare nanosuspensionsare typically categorized as either “top-down” or “bottom-up”processes (Liu et al., 2012; Müller et al., 2011; Horn and Rieger,2001; Shegokar and Müller, 2010). Top-down methods typicallyform nanocrystals via the attrition or ablation of larger drug frag-ments, whereas bottom-up methods generally produce amorphousnanoparticles via controlled precipitation from solutions or emul-sions (Horn and Rieger, 2001). Although both categories have led tothe development of viable pharmaceutical products, the processeshave a number of limitations. For instance, top-down approaches
such as wet milling and high pressure homogenization are gen-erally not applicable to soft or temperature-sensitive compounds,and impurities may be introduced into the drug formulations byattrition of milling media or residual solvents.
242 A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250
−1), (b
oasn2mbIwwevOlFit
Psbsedaitbdtp
wt
Scheme 1. Chemical structures of (a) GF (MW 352.8 g mol
An emulsion freeze-drying approach was developed to formrganic or drug nanoparticles in situ within porous polymers; theddition of water to such materials causes them to rapidly dis-olve and release the nanoparticle payload to produce aqueousanoparticle dispersions (Zhang et al., 2008a; Grant and Zhang,011). This is an approach that may be applied without the aboveentioned limitations. For example, nanoparticle aggregation can
e avoided because they are supported within the porous scaffold.n this approach, oil-in-water (O/W) emulsions were formed first
ith the drugs dissolved in the oil phase. However, many poorlyater-soluble drugs are only soluble in polar organic solvents (e.g.,
thanol, acetone) particularly when the toxicity of the organic sol-ents is taken into consideration. It is extremely difficult to form/W emulsions from these polar organic solvents, thus seriously
imiting the application of the emulsion freeze-drying approach.urthermore, these organic solvents normally have very low melt-ng points (<−100 ◦C) and it is not economically effective to removehe solvents by freeze drying.
Recently, a 2-step procedure ‘Solvent Evaporation within Porousolymer’ (SEPP) was proposed to address the emulsion and organicolvent freeze-drying issue. Porous polymers were firstly preparedy freeze-drying. These materials were then soaked in organic drugolutions. After filtration, the organic solvent could be removed byvaporation rather than freeze drying (Qian et al., 2011). This proce-ure can be used for all types of organic solvents including ethanolnd acetone. The materials can be composed of various biocompat-ble polymers and surfactants, giving them the dual role of acting asemplates for the initial formation of nanoparticles as well as sta-ilizers upon dissolution. The materials may also be administeredirectly as a solid dose platform, which may be more convenienthan an aqueous nanosuspension under certain situations from a
harmacological perspective (Lee, 2003).In this study, the SEPP approach is applied to prepare poorlyater-soluble drug nanoparticles. The procedures are investigated
o produce porous polymers which are used to generate high ratio
) CBZ (MW 236.3 g mol−1) and (c) PTX (MW 853.9 g mol−1).
of drug nanoparticles to stabilizer in the formulations. The for-mulation and process parameters were initially optimized usinggriseofulvin (GF) as a model compound, before the process wastranslated to carbamazepine (CBZ) and paclitaxel (PTX); drugswhich differ in molecular weight and have little structural resem-blance (Scheme 1). No additional optimization was performed forthe preparation of CBZ and PTX nanosuspensions, suggesting thatthe formulation developed could be applicable to a wide range ofother hydrophobic drug compounds.
2. Materials and methods
2.1. Chemicals and reagents
Polyvinyl alcohol (PVA, 80% hydrolysed, MW 9000–10,000),polyethylene glycol (PEG, MW 20,000), sodium dodecyl sulfate(SDS), cetyltrimethylammonium bromide (CTAB), sodium deoxy-cholate (SDC) and dioctyl sodium sulfosuccinate (AOT) werepurchased from Sigma Aldrich and used as received. The drugsgriseofulvin (GF, >97% from Penecillium griseofulvum), carba-mazepine (CBZ) and paclitaxel (PTX, >95% from Taxus brevifolia)were also purchased from Sigma. Analytical grade acetone andmethanol were purchased from Fischer Scientific. Deionised waterwas used for the preparation of aqueous solutions.
2.2. Procedure to make porous polymer and drug nanoparticles
A freeze and freeze-drying approach was used to prepare a vari-ety of porous materials. Both O/W emulsions and aqueous solutionswere investigated, with the same freeze-drying procedure. It wasfound that the simpler preparation procedure with aqueous solu-
tions could produce porous materials for the formation of similarquality of drug particles. The optimized procedure for freeze-dryingaqueous solutions is described here. Typically, an aqueous solu-tion of PVA (1.5 wt%), PEG (0.5 wt%) and SDS (1 wt%) was firstly
urnal
pldra
s(wptptn
2
cascnglwd
M
wtalf
Y
to
2
cupca1ltdcD
sTbuoamwt
A.D. Roberts, H. Zhang / International Jo
repared. The solution was cryogenically frozen by submersion iniquid N2 before being freeze-dried in a Virtis AdVantage freezerier (duration: 36 h, shelf temperature: −2 ◦C). The resultant mate-ial was a dry white porous solid composed of 50% PVA, 16.6% PEGnd 33.3% SDS by mass.
To form drug nanoparticles, the porous material (c.a. 0.5 g) wasoaked in a solution of GF in acetone at the required concentrationtypically 3 mg cm−3) for approximately 20 min before the solventas drained, and any excess removed by gentle drying with tissueaper. The soaked material was then dried under vacuum at roomemperature for approximately 3 h, giving the dry, drug-loadedorous material. Upon the addition of water and gentle agitation,he material was dissolved to release the nanoparticles and form aanoparticle suspension.
.3. Drug payload and nanoparticle yield
The mass of drug to stabilizer (drug payload) and the % of drugonverted to nanoparticles as opposed to microparticles and othergglomerates (nanoparticle yield) were determined by UV–Vispectrophotometry (HP 8452A Diode array spectrophotometer). Afterentrifugation (3 min at 3000 rpm) to separate the suspendedanoparticles from the larger aggregates, each was dissolved toive a 1:1 methanol/water solution. These solutions were then ana-yzed by UV–Vis spectrophotometry and the drug concentrations
ere determined with reference to calibration curves prepared. Therug-payload was calculated using the following equation.
D = (Mp + Mnp)MT
(1)
here MD is the drug payload (mg drug per g of material), Mp ishe mass of drug as precipitates (mg), Mnp is the mass of the drugs nanoparticles (mg) and MT is the total mass of the initial drug-oaded material (g). The nanoparticle yield was calculated using theollowing equation.
ield = 100 × (Mnp)(Mp + Mnp)
(2)
The UV calibration curves (UV absorbance – drug concentra-ion) in 1:1 (v/v) for each investigated drug (GF, CBZ and PTX) werebtained and given in the supporting information (Fig. S1).
.4. Characterization
The hydrodynamic sizes and zeta-potentials of the nanoparti-le suspensions were measured by dynamic light scattering (DLS)sing a Malvern Zetasizer Nanoseries instrument at 25 ◦C. Theolydispersity index (PDI) of the nanoparticles was automaticallyalculated by the software and given as an index value between 0nd 1, where values closer to 0 indicated greater monodispersity.
cm3 of distilled water was added to a 20 mg portion of the drug-oaded porous materials followed by gentle agitation to dispersehe nanoparticles. Any microparticles or aggregates that formed onissolution of the composite material were removed by 3 min ofentrifugation at 3000 rpm (Eppendorf Centrifuge 5415 D) prior toLS measurements.
The morphology of the porous materials was characterized bycanning electron microscopy (SEM) using a Hitachi S-4800 SEM.he size and shape of the dry drug nanoparticles were assessedy the scanning transmission electron microscopy (STEM) detectorsing the same instrument. For SEM measurements, a small portionf the porous material was adhered to an aluminium SEM stud
nd sputter-coated with gold for 2 min at 25 mA prior to measure-ents being taken. For STEM, a drop of the dilute nanosuspensionas placed on a TEM grid with holy carbon film and allowedo dry before measurements were recorded. The macropore size
of Pharmaceutics 447 (2013) 241– 250 243
distribution of some porous polymers were determined by mer-cury intrusion porosimetry (MIP) using a Micrometrics AutoporeIV instrument. Measurements were performed on 20 mg portionsof the dry materials and the pressure range was between 0.1 and60,000 psi.
3. Results and discussion
3.1. Screening porous polymers for preparation of drugnanoparticles
This study investigated the SEPP method as a general approachfor the preparation of poorly water-soluble drug nanoparticles(Scheme 2). Water-soluble porous polymers were prepared byfreeze drying and then used as scaffolds to form drug nanopar-ticles within. The porous polymers were soaked in organic drugsolutions and then filtered. The organic solvent was then removedunder ambient conditions to form drug nanoparticles in the porouspolymers. With the freeze-drying approach, the porous polymerscould be produced from aqueous solutions or O/W emulsions (Qianet al., 2009). Due to the facile control on porosity, the freeze-dryingemulsion approach was initially used to produce porous polymers.Firstly, we investigated whether stable O/W emulsions could beformed. A range of hydrophilic polymers and surfactants wereevaluated (Table 1). These polymers are biocompatible with lowtoxicity and have been widely used as excipients particularly fororal formulation development (Ali and Kolter, 2012). The emulsionswere formed by dispersing cyclohexane into aqueous solutionscontaining known concentrations of polymer and surfactant. Thevolume ratio of cyclohexane to aqueous solution was 3:1. It can beseen that anionic surfactant SDS was better overall to form emul-sions, compared to non-ionic polymeric surfactant Pluronics F-127and cationic surfactant CTAB. The polymers that can be used for allthe surfactants to form emulsions include PVA 10K, PEG 1.5K, Dex-tran and HPMC. These emulsions can be freeze-dried to producehighly interconnected porous polymers.
In order to use these porous polymers as scaffolds, the stabil-ity of these scaffolds in a range of polar and non-polar organicsolvents was evaluated. If the polymers were dissolved/partiallydissolved/disintegrated in the solvents, they would be assumedunsuitable for further investigation. For the polymer/SDS compos-ites, it was found that PVA10K/SDS and HPMC/SDS were stable withmost of the solvents tested while PEG1.5K/SDS and Dextran/SDSwere dissolved in almost all of the solvents (Table S1). Fur-ther investigation with polymer/Pluronic F-127 and polymer/CTABshowed that HPMC/Pluronic F-127 and PVA 10K/CTAB were gen-erally stable towards selected solvents (Tables S2 and S3). Theseresults should be equally valid for the porous polymer/surfactantcomposites but prepared by freeze-drying of aqueous solutions.
GF was selected as the model drug to demonstrate the generalsuitability for the formation of nanoparticles. GF is a BCS class IIantifungal agent produced by the fungus P. griseofulvum, and thereis on-going research to prepare the drug as nanoparticles in orderto enhance the solubility (Kamiya et al., 2009; Trotta et al., 2003;Chattopadhyay and Gupta, 2001). For pharmaceutical applications,it is required to meet the residual solvent limits depending onorganic solvents used in the formulations (Dwivedi, 2002). Table 2gives the solubility of GF in various solvents and the recognizedresidual limits of these solvents (Dwivedi, 2002; Hu et al., 2010).GF-acetone solutions are selected for further investigation due tohigh solubility and high residual limit.
The initial evaluation was carried out with porous PVA 10K/SDSwhich was prepared by freeze-drying the O/W emulsions withcyclohexane dispersed in aqueous PVA solution (5 wt%) con-taining 5 wt% SDS. Following the SEPP procedure (Scheme 2),

244 A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250
Scheme 2. Schematic representation of the SEPP process. The porous monolith (a) is initially prepared by freezing and freeze-drying an aqueous polymer solution (or oil-in-water emulsion), before being soaked in an organic solution of the drug (b). The solvent is removed by evaporation under ambient condition, leaving the nanoparticles formedwithin the porous monolith (c). Dissolution of the material in water releases the nanoparticles which are simultaneously stabilized by the polymer/surfactant combination(d).
Table 1Screening results to determine the polymer/surfactant combinations in order to produce stable emulsions.
Polymer composition Surfactant (5 wt%)
Polymer Concentration (wt%) Pluronic F127 SDS CTAB
PVA (MW 10 K) 5 n/a + n/a1 + + +0.1 n/a + n/a
PVP (MW 40 K) 5 n/a − −1 − − −0.1 n/a + −
PVP (MW 10 K) 5 n/a + n/a1 + + n/a0.1 n/a + n/a
Chitosan 5 n/a − −1 − − −0.1 + − −
PEG (MW 1.5 K) 5 n/a + n/a1 + + +0.1 n/a + n/a
Dextran 5 n/a + +1 + + +0.1 + + +
HPMC 5 n/a − −1 + + +0.1
+, forms a stable emulsion, stable for 30 min without stirring; −, fails to form a stable em
Table 2GF solubility and residual solvent limits for the selected solvents.
Solvent GF solubility (mg cm−3) Residual solvent limit (ppm)
2-Propanol <1.5 High1-Butanol <0.5 HighCyclohexane <0.5 3880Acetone 33.6 HighToluene <4.5 890Pentane <0.5 High
n/a + +
ulsion; n/a, combinations not tested.
GF nanoparticle composites and nanoparticle dispersions wereprepared. Different concentrations of GF-acetone solutions weretested (Table S4). It was observed that higher concentrationscontributed to large number of aggregates after dissolution ofthe composites in water. This suggested what concentrationswould be suitable for formation of GF nanoparticles. Similar testswere also performed on porous PVA 10K/SDS composites, which
were simply prepared by freeze drying aqueous PVA solution(5 wt%) containing 5 wt% SDS. Other composite materials formedby freeze-drying of aqueous solutions and emulsions were alsoevaluated. All the observations (formation of stable dispersions)
A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250 245
F with dp rial (eb
spFfa
3
papddsiiststsaco
ig. 1. SEM micrographs showing the porous structures for the PVA–PEG materials
orous materials characterized by Hg intrusion porosimetry where (i) for the matey freeze-drying of 1.5 wt% PVA 10K and 0.5 wt% PEG 20K aqueous solutions.
uggested that the porous polymers from freeze-drying of aqueousolymer solutions generated similar or better results (Table S5).reeze-drying of aqueous polymer solution was thus employedor further investigation because this simplified the technique andvoided the use of organic solvent in making porous polymers.
.2. Optimization of porous polymers
A number of aqueous formulations were tested to produceorous polymers by freeze drying. The best candidate should beble to produce stable nanoparticle dispersions with the highestossible ratio of drug to polymer, by using high concentration ofrug solutions. It is likely that this optimized porous polymer can-idate may vary with different types of drugs and solvents. Drugolutions in acetone were investigated in this study. After test-ng with different porous polymers, a formulation composed of annitial solution of PVA 10K (1.5 wt%) and PEG 20K (0.5 wt%) waselected for further investigation. This formulation was selected forhe following reasons: (1) nanoparticle dispersions with a relativelymall size distribution and low nanoparticle PDI were formed fromhis combination; (2) PVA is a widely used and effective disper-
ant for the long-term stabilization of nanosuspensions; (3) PEG isbiocompatible polymer which has been shown to increase the cir-ulation time of nanoparticles in vivo due to suppression of surfacepsonisation (Moghimi and Szebeni, 2003); (4) porous polymer of
ifferent amounts of SDS (a) 2%, (b) 1%, (c) 0.5%, (d) 0.25%, (e) 0%, and (f) some of the), (ii) for (c) and (iii) for (a). All the PVA–PEG materials in discussion were prepared
this composition is stable in acetone. A wide range of drugs dissolvewell in acetone and it has a relatively high residual solvent limit forpharmaceutical applications (Dwivedi, 2002).
Surfactants were also incorporated into the polymers in orderto produce smaller nanoparticles and/or more stable aqueousnanoparticle dispersions. It was found that SDS was highly efficientin achieving these targets. Freeze-drying the aqueous polymer solu-tions produced layered porous materials, with no obvious effectsfrom the addition of SDS (Fig. 1) while freeze-drying emulsionswould form highly interconnected cellular porous materials (Qianet al., 2009). We do not expect the pore morphology would make anobvious difference for the soaking and removing solvent procedurein order to make drug nanoparticles. In addition to pore morphol-ogy, the porous polymers were also characterized by Hg intrusionporosimetery to show the porosity (or intrusion pore volume) andpore size distribution. The Hg intrusion data revealed a fairly broadbut similar pore size-distribution, with the majority of pores withinthe 50–200 �m range (Fig. 1f). No significant trends were foundbetween the porosity of the material and the nanoparticle size ordrug-payload.
3.3. GF nanoparticles and nanosuspensions
Using the selected porous PVA/PEG, with an initial GF-acetoneconcentration of 1 mg cm−3, nanoparticles were prepared which

246 A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250
Fig. 2. (a) Relationship of drug payload to GF-acetone concentration when thePVA–PEG materials were soaked in the solutions. (b) Shows the distributionovc
hatld(cyiio
nrdstttst
Fig. 3. GF nanoparticle sizes increase with soaking time when the PVA–PEG materialwas soaked in 1 mg cm−3 GF-acetone solution.
Fig. 4. (a) Change of nanoparticle yield as GF soaking concentration increases for
f nanoparticles and precipitates formed when GF-acetone concentrations werearied. (c) Relationship between nanoparticle yield and GF-acetone soaking con-entration for the PVA–PEG materials.
ad an average size of 413 nm, PDI of 0.31, payload of 3.6 mg g−1
nd a nanoparticle yield of 77.5%. It was found that by increasinghe concentration of the GF-acetone solution the total drug pay-oad increased linearly (Fig. 2a). The majority of the increase inrug mass was found to be the formation of precipitated particlesFig. 2b). Therefore, for the PVA/PEG scaffold, increasing the GF con-entration was not always beneficial as it led to a low nanoparticleield at high drug-soaking concentrations (Fig. 2c). The longer soak-ng time of PVA–PEG in GF-acetone solution (1 mg cm−3) was alsonvestigated. Unfavourably, this resulted in the consistent increasef GF nanoparticle size with the increase in soaking time (Fig. 3).
In order to improve payload while maintaining the druganoparticles below 500 nm, different surfactants were incorpo-ated into the material to provide additional stabilization uponissolution. Anionic surfactant SDS was firstly investigated. Fig. 4ahows how nanoparticle yield changes when GF-acetone concen-ration and SDS concentration are varied. It is clear that in general
he nanoparticle yield decreases with the increased GF concentra-ion. With the addition of SDS, the nanoparticle yield is improvedignificantly. At the concentration of 1 wt% SDS, ∼75% nanopar-icle yields are achieved for GF concentrations of 5 mg cm−3 andthe PVA–PEG material with various amounts of SDS. (b) The DLS plots by intensityshow the size distribution of GF nanoparticles in aqueous nanosuspensions withdifferent amounts of SDS. The GF-acetone concentration was 5 mg cm−3. (c) Rela-tionship between the average GF nanoparticle size and the SDS concentration forthe PVA–PEG material (the GF soaking concentration was 5 mg cm−3).

A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250 247
F ials as
7mcfm
aGonoisnGPgPtvd(ad
ipatwoTwnfyftaawd
ticles on the scale of <600 nm have been shown to accumulate attumour sites due to the enhanced permittivity and retention (EPR)mechanism (Yuan et al., 1995; Hiroshi, 2001; Cabral et al., 2011).
Table 3GF nanoparticles produced from the PVA–PEG material incorporated with differ-ent surfactants. GF-acetone soaking concentration was 5 mg cm−3. The surfactantconcentrations were 1 wt% of the initial solution. GF nanoparticle suspensions wereevaluated by DLS.
Material composition Particlesize (nm)
PDI Payload(mg g−1)
Nanoparticleyield (%)
PVA–PEG 595 0.18 20.5 19.3
ig. 5. SEM images of the precipitates formed from the PVA–PEG–surfactant mater
mg cm−3. The nanoparticle yield is not improved further whenore SDS (2 wt%) was added into the formulation. The best con-
entration of SDS in the initial solution (prior to freeze-drying) wasound to be 1 wt%; constituting 33.3 wt% of the resultant polymeric
onoliths.The effect of SDS on nanoparticle size was studied at the GF-
cetone concentration of 5 mg cm−3. Fig. 4b shows the DLS plots ofF nanoparticle suspensions with the presence of different amountf SDS. It can be seen that addition of SDS can lead to smaller GFanoparticles with narrow size distribution. And the average sizef the GF nanoparticles decreases consistently with the increas-ng amount of SDS in the formulation (Fig. 4c) while the particleize distributions (or PDI) do not change significantly. PDI of the GFanoparticles was studied for different concentrations of SDS andF-acetone solutions (Fig. S2). There was no obvious trend betweenDI and SDS/GF concentration. However, in most of the investi-ated samples, particularly for 0.5 and 1 wt% SDS formulations, theDI was less than 0.3, which indicated a reasonably narrow dis-ribution. It was also noticed that the porosity (or intrusion poreolume as measured by Hg porosimeter) of the polymer scaffoldsecreased with the increasing amount of SDS in the formulationFig. S3). However, under the same soaking conditions with GF-cetone solutions, there was no direct link observed between therug payload and the porosity of the materials (Fig. S4).
Other surfactants including AOT, SDC and CTAB were alsonvestigated. Like SDS, AOT has shown excellent stabilizationroperties. SDC was chosen due to its excellent biocompatibilitynd CTAB was investigated as it is a cationic surfactant unlikehe others. GF-acetone solutions with concentration of 5 mg cm−3
ere used to soak the above materials containing different typesf surfactants. The relevant results are summarized in Table 3.he use of different surfactants still produces drug nanoparticlesith low PDI values (in the range of 0.17–0.34). However, theanoparticle yields were significantly lower for the other sur-
actants particularly AOT formulation (25.7%), compared to theield of 74.8% for the SDS formulation. The payloads among theormulations are in the range of 29.7–46.5 mg g−1, much higherhan the PVA–PEG formulation. The solid precipitates were formed
fter dissolving drug nanoparticle–polymer composites in waternd followed by centrifuging at 3000 rpm for 3 min. SEM imagingas performed on the precipitates from the formulations withifferent surfactants as listed in Table 3. As shown in Fig. 5, thedetailed in Table 3. The surfactants are: (a) AOT, (b) CTAB, (c) SDC, and (d)SDS.
precipitates are formed from aggregation of small particles, usuallyat least one dimension of which are in the nanoparticle range.
3.4. Factors effecting particle size-distribution
Throughout the formulation optimization process for thePVA–PEG material it was noticed that varying certain parametershad an effect on the nanoparticle size-distribution. For exam-ple, Fig. 3 shows that under similar conditions increasing soakingtime resulted in larger GF particles. The particle size increased byabout 300 nm when the soaking duration increased from 10 min to120 min. It was also found that by increasing the concentration ofthe initial GF-acetone solution from 1 mg cm−3 to 5 mg cm−3, thenanoparticle size increased by about 100 nm (Fig. 6a). This trend canalso be easily observed from the DLS plots (Fig. 6b). The increaseof particle size with increased solution concentration is consis-tent with our previous study (Qian et al., 2011) and others (Aranazet al., 2009) where the nanoparticles were formed from solutionseither by solvent evaporation or freeze drying. Having a methodwhich is able to control nanoparticle size is desirable since this fac-tor can have a significant effect on the behaviour and fate of thenanoparticles in vivo (Waser et al., 1987). For example, relativelylarge particles give a slower and more prolonged drug release incomparison to smaller particles which release their payload morerapidly (Redhead et al., 2001). In cancer chemotherapy nanopar-
PVA–PEG–SDS 454 0.20 31.0 74.8PVA–PEG–AOT 657 0.17 46.5 25.7PVA–PEG–SDC 550 0.34 34.8 41.6PVA–PEG–CTAB 589 0.25 29.7 45.8

248 A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250
Fig. 6. The PVA–PEG materials were soaked in GF-acetone solutions of different con-centrations. Standard soaking time 20 min. (a) Plot of average particle size againstsw
3
pafictzc(
Ftb
Table 4Different drug nanoparticles (GF, CBZ and PTX) prepared using the PVA–PEG–SDSformulation. Drug-acetone soaking concentration was 3 mg cm−3. The drugnanoparticle suspensions were assessed by DLS.
Drug Particle size (nm) PDI Payload(mg g−1)
Nanoparticleyield (%)
oaking concentration and (b) GF nanoparticle size distribution measured by DLSith different concentrations of GF-acetone solutions.
.5. Effect of surfactant on zeta-potential of GF nanoparticles
By varying the type and concentration of surfactants used in theorous monolith, a degree of the zeta-potential control could bettained for the nanoparticles (Fig. 7). SDS, being an anionic sur-actant, gave the nanoparticles a negative zeta-potential, whichncreased linearly with increasing SDS concentration. CTAB, beingationic, showed an increasing positive charge as the concentra-ion was increased. Having a method which is able to control theeta-potential is important because the charge on the nanoparti-
les dictates their stability and their behaviour and toxicity in vivoZhang et al., 2008b).ig. 7. Change of zeta-potential for the aqueous GF nanoparticle suspensions whenhe concentrations of SDS (�) and CTAB (©) were varied. The concentrations wereased on aqueous PVA–PEG solutions.
GF 434 0.19 24.6 82.9CBZ 133 0.32 27.0 84.1PTX 500 0.41 21.2 94.2
3.6. Translation of the approach to CBZ and PTX
After optimizing the formulation for the preparation of GFnanoparticles, the approach was then translated to other poorlysoluble drugs. Using the optimized PVA–PEG–SDS materials (pre-pared by freeze drying the solutions containing 1.5 wt% PVA 10K,0.5 wt% PEG 20K, and 1 wt% SDS), nanosuspensions of the BCSclass II anticonvulsant CBZ and the BCS class IV antineoplasticPTX were prepared and characterized (Table 4). Even though thesecompounds have little structural resemblance (Scheme 1), similardrug-payloads (21.2–27.0 mg g−1), and nanoparticle yields (>80%,Fig. S5) were achieved. The mean particle sizes and PDI values didvary however, particularly in the case for CBZ where the averagenanoparticle size (133 nm) was significantly smaller in comparisonwith the other drugs (434 nm and 500 nm for GF and PTX respec-tively). The size of the drug nanoparticles in aqueous suspensionwas routinely measured by DLS throughout this study. Alterna-tively, dry drug nanoparticles could be imaged by STEM. Fig. 8shows the STEM images of drug nanoparticles as detailed in Table 4.TEM grids covered with holy carbon film were used for the imag-ing. The dark spots in Fig. 8 are nanoparticles. It is clear that allthe particles have sizes <500 nm. DLS measures the hydrodynamicsize of the nanoparticles in suspension so the sizes are always big-ger than those measured by STEM imaging. The drug nanoparticlesmeasured by STEM and DLS are comparable with each other. Onenoticeable feature is that thin nanowires grow from CBZ nanopar-ticles when the nanoparticles are on the TEM grid for a longer time.Fig. 8a–c is the TEM images from freshly prepared dry nanoparti-cles. When the dry nanoparticles are imaged again 3 months later,there is no change to GF and PTX nanoparticles but a large num-ber of nanowires are observed from CBZ nanoparticles (Fig. 8d). Itshould be noted that a small number of nanowires are present evenfor the freshly prepared CBZ TEM sample (Fig. 8c). These nanopar-ticles are likely formed during TEM sample preparation where adrop of nanoparticle suspension is placed on TEM grid and normallyallowed to dry overnight. It is thus clear that, for nanoparticles witha tendency to aggregate or re-crystallize, there is a potential advan-tage with the SEPP method because the nanoparticles are formedand stabilized within the polymer scaffolds. The composites can beeasily dissolved to produce aqueous suspensions. In the case of CBZ,due to its small average size of nanoparticles (133 nm, Table 4), ananoparticle suspension is formed instantly. However, for the otherformulations, due to the presence of large particles, additional stepssuch as centrifuging or nanofiltration may be necessary to generatesmall nanoparticle suspensions.
For CBZ, the nanoparticle yields only changed slightly whenthe drug concentrations were varied (Fig. 9a). A linear trend wasalso observed between the concentration of the initial CBZ–acetonesolution and the total payload of the drug (Fig. 9b). Due to the veryhigh costs, we did not carry out detailed study for PTX. Althoughthe highest payload possible is generally desirable from a phar-maceutical perspective, having a linear relationship such as this
should allow the payload to be controlled by simply varying theconcentration of the initial drug solution. The results indicate thatthe nature of the drug and drug-scaffold interaction have a signifi-cant influence on the resultant nanoparticles that are formed. Since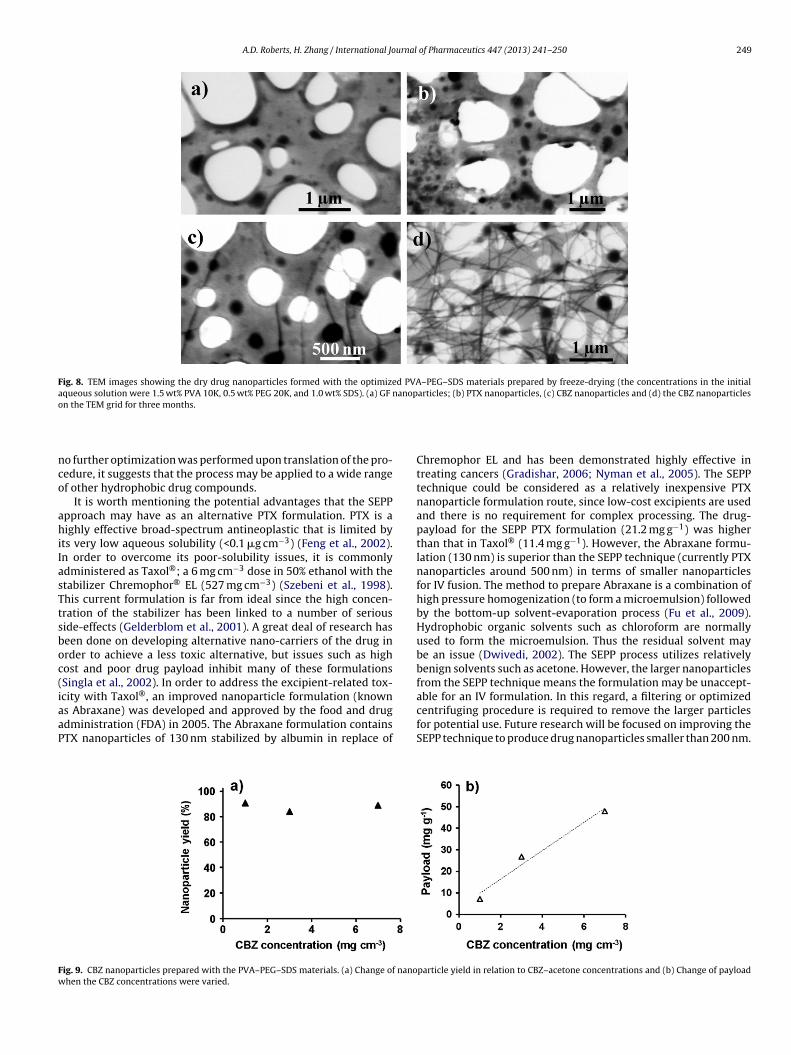
A.D. Roberts, H. Zhang / International Journal of Pharmaceutics 447 (2013) 241– 250 249
Fig. 8. TEM images showing the dry drug nanoparticles formed with the optimized PVA–PEG–SDS materials prepared by freeze-drying (the concentrations in the initialaqueous solution were 1.5 wt% PVA 10K, 0.5 wt% PEG 20K, and 1.0 wt% SDS). (a) GF nanoparticles; (b) PTX nanoparticles, (c) CBZ nanoparticles and (d) the CBZ nanoparticleso
nco
ahiIasTtsboc(iaaP
Fw
n the TEM grid for three months.
o further optimization was performed upon translation of the pro-edure, it suggests that the process may be applied to a wide rangef other hydrophobic drug compounds.
It is worth mentioning the potential advantages that the SEPPpproach may have as an alternative PTX formulation. PTX is aighly effective broad-spectrum antineoplastic that is limited by
ts very low aqueous solubility (<0.1 �g cm−3) (Feng et al., 2002).n order to overcome its poor-solubility issues, it is commonlydministered as Taxol®; a 6 mg cm−3 dose in 50% ethanol with thetabilizer Chremophor® EL (527 mg cm−3) (Szebeni et al., 1998).his current formulation is far from ideal since the high concen-ration of the stabilizer has been linked to a number of seriouside-effects (Gelderblom et al., 2001). A great deal of research haseen done on developing alternative nano-carriers of the drug inrder to achieve a less toxic alternative, but issues such as highost and poor drug payload inhibit many of these formulationsSingla et al., 2002). In order to address the excipient-related tox-city with Taxol®, an improved nanoparticle formulation (known
s Abraxane) was developed and approved by the food and drugdministration (FDA) in 2005. The Abraxane formulation containsTX nanoparticles of 130 nm stabilized by albumin in replace ofig. 9. CBZ nanoparticles prepared with the PVA–PEG–SDS materials. (a) Change of nanohen the CBZ concentrations were varied.
Chremophor EL and has been demonstrated highly effective intreating cancers (Gradishar, 2006; Nyman et al., 2005). The SEPPtechnique could be considered as a relatively inexpensive PTXnanoparticle formulation route, since low-cost excipients are usedand there is no requirement for complex processing. The drug-payload for the SEPP PTX formulation (21.2 mg g−1) was higherthan that in Taxol® (11.4 mg g−1). However, the Abraxane formu-lation (130 nm) is superior than the SEPP technique (currently PTXnanoparticles around 500 nm) in terms of smaller nanoparticlesfor IV fusion. The method to prepare Abraxane is a combination ofhigh pressure homogenization (to form a microemulsion) followedby the bottom-up solvent-evaporation process (Fu et al., 2009).Hydrophobic organic solvents such as chloroform are normallyused to form the microemulsion. Thus the residual solvent maybe an issue (Dwivedi, 2002). The SEPP process utilizes relativelybenign solvents such as acetone. However, the larger nanoparticlesfrom the SEPP technique means the formulation may be unaccept-able for an IV formulation. In this regard, a filtering or optimized
centrifuging procedure is required to remove the larger particlesfor potential use. Future research will be focused on improving theSEPP technique to produce drug nanoparticles smaller than 200 nm.particle yield in relation to CBZ–acetone concentrations and (b) Change of payload

2 urnal
4
fptfwpmhpsptotcipi
A
Utat
A
fp
R
A
A
A
C
C
D
F
F
G
G
G
H
H
H
50 A.D. Roberts, H. Zhang / International Jo
. Conclusions
The SEPM technique is a relatively simple and versatile methodor the preparation of poorly water-soluble drug nanoparticles inorous polymers and aqueous nanoparticle suspensions by dissolu-ion of the composites in water. The process was initially optimizedor the model drug GF before being translated to the other poorlyater-soluble drugs CBZ and PTX. No additional optimization waserformed upon translation, suggesting that the conditions useday be applied to prepare nanosuspensions of a wide range of other
ydrophobic drug compounds. A degree of nanoparticle size- andayload-control could be achieved by varying parameters such asurfactant composition and drug-soaking concentration. The zeta-otential of the nanoparticles could also be tuned by varying theype and concentration of the incorporated surfactants. The sizef the drug nanoparticles was found to vary with the nature ofhe drug, suggesting that the drug-scaffold interaction is signifi-ant in the formation of nanoparticles. More research, includingn vitro and in vivo characterization, is required to assess the fullotential of this technique, which may have broader applications
n pharmaceutics.
cknowledgements
The authors would like to thank the financial support from theniversity of Liverpool. Prof. Steve Rannard is thanked for use of
he DLS, and the CMD is acknowledged for access to state of thert facilities. Mr. Michael Barrow and Dr. Adham Ahmed are alsohanked for help with discussion and SEM measurements.
ppendix A. Supplementary data
Supplementary data associated with this article can beound, in the online version, at http://dx.doi.org/10.1016/j.lantsci.2004.08.011.
eferences
dams, M.L., Lavasanifar, A., Kwon, G.S., 2003. Amphiphilic block copolymers fordrug delivery. J. Pharm. Sci. 92, 1343–1355.
li, S., Kolter, K., 2012. Challenges and opportunities in oral formulation develop-ment. Am. Pharm. Rev. 15 (7).
ranaz, I., Gutierrez, M.C., Yuste, L., Rojo, F., Ferrer, M.L., del Monte, F., 2009.Controlled formation of the anhydrous polymorph of ciprofloxacin crystalsembedded within chitosan scaffolds: study of the kinetic release dependenceon crystal size. J. Mater. Chem. 19, 1576–1582.
abral, H., Matsumoto, Y., Mizuno, K., Chen, Q., Murakami, M., Kimura, M., Terada, Y.,Kano, M.R., Miyazono, K., Uesaka, M., Nishiyama, N., Kataoka, K., 2011. Accumu-lation of sub-100 nm polymeric micelles in poorly permeable tumours dependson size. Nat. Nanotechnol. 6, 815–823.
hattopadhyay, P., Gupta, R.B., 2001. Production of griseofulvin nanoparticles usingsupercritical CO2 antisolvent with enhanced mass transfer. Int. J. Pharm. 228,19–31.
wivedi, A.M., 2002. Residual solvent analysis in pharmaceuticals. Pharm. Technol.North Am. 26, 42–46.
eng, X., Yuan, Y.-J., Wu, J.-C., 2002. Synthesis and evaluation of water-soluble pacli-taxel prodrugs. Bioorg. Med. Chem. Lett. 12, 3301–3303.
u, Q., Sun, J., Zhang, W., Sui, X., Yan, Z., He, Z., 2009. Nanoparticle albumin – bound(NAB) technology is a promising method for anti-cancer drug delivery. RecentPat. Anticancer Drug Discov. 4, 262–272.
elderblom, H., Verweij, J., Nooter, K., Sparreboom, A., 2001. Cremophor EL: thedrawbacks and advantages of vehicle selection for drug formulation. Eur. J.Cancer 37, 1590–1598.
radishar, W.J., 2006. Albumin-bound paclitaxel: a next-generation taxane. ExpertOpin. Pharmacother. 7, 1041–1053.
rant, N., Zhang, H., 2011. Poorly water-soluble drug nanoparticles via an emulsion-freeze-drying approach. J. Colloid Interface Sci. 356, 573–578.
iroshi, M., 2001. The enhanced permeability and retention (EPR) effect in tumorvasculature: the key role of tumor-selective macromolecular drug targeting.
Adv. Enzyme Regul. 41, 189–207.orn, D., Rieger, J., 2001. Organic nanoparticles in the aqueous phase—theory, exper-iment, and use. Angew. Chem. Int. Ed. 40, 4330–4361.
oskins, C., Thoo-Lin, P.K., Cheng, W.P., 2012. A review on comb-shaped amphiphilicpolymers for hydrophobic drug solubilization. Ther. Deliv. 3, 59–79.
of Pharmaceutics 447 (2013) 241– 250
Hu, G., Li, H., Wang, X., Zhang, Y., 2010. Measurement and correlation of griseoful-vin solubility in different solvents at temperatures from (281.95 to 357.60) K. J.Chem. Eng. Data 55, 3969–3971.
Kamiya, S., Kurita, T., Miyagishima, A., Arakawa, M., 2009. Preparation of griseofulvinnanoparticle suspension by high-pressure homogenization and preservation ofthe suspension with saccharides and sugar alcohols. Drug Develop. Ind. Pharm.35, 1022–1028.
Kasim, N.A., Whitehouse, M., Ramachandran, C., Bermejo, M., Lennernäs, H., Hussain,A.S., Junginger, H.E., Stavchansky, S.A., Midha, K.K., Shah, V.P., Amidon, G.L., 2003.Molecular properties of WHO essential drugs and provisional biopharmaceuticalclassification. Mol. Pharm. 1, 85–96.
Lawrence, M.J., Rees, G.D., 2000. Microemulsion-based media as novel drug deliverysystems. Adv. Drug Delivery Rev. 45, 89–121.
Lee, J., 2003. Drug nano- and micro-particles processed into solid dosage forms:physical properties. J. Pharm. Sci. 92, 2057–2068.
Lipinski, C., 2002. Poor aqueous solubility – an industry wide problem in drug dis-covery. Am. Pharm. Rev. 5, 82–85.
Liu, Y., Miyoshi, H., Nakamura, M., 2007. Nanomedicine for drug delivery andimaging: a promising avenue for cancer therapy and diagnosis using targetedfunctional nanoparticles. Int. J. Cancer 120, 2527–2537.
Liu, Y., Xie, P., Zhang, D., Zhang, Q., 2012. A mini review of nanosuspensions devel-opment. J. Drug Target. 20, 209–223.
Masahiro, N., 2000. Places of emulsions in drug delivery. Adv. Drug Delivery Rev. 45,1–4.
Moghimi, S.M., Szebeni, J., 2003. Stealth liposomes and long circulating nanopar-ticles: critical issues in pharmacokinetics, opsonization and protein-bindingproperties. Prog. Lipid Res. 42, 463–478.
Müller, R.H., Gohla, S., Keck, C.M., 2011. State of the art of nanocrystals – specialfeatures, production, nanotoxicology aspects and intracellular delivery. Eur. J.Pharm. Biopharm. 78, 1–9.
Nyman, D.W., Campbell, K.J., Hersh, E., Long, K., Richardson, K., Trieu, V., Desai, N.,Hawkins, M.J., Von Hoff, D.D., 2005. Phase I and pharmacokinetics trial of ABI-007, a novel nanoparticle formulation of paclitaxel in patients with advancednonhematologic malignancies. J. Clin. Oncol. 23, 7785–7793.
Patravale, V.B., Date, A.A., Kulkarni, R.M., 2004. Nanosuspensions: a promising drugdelivery strategy. J. Pharm. Pharmacol. 56, 827–840.
Qian, L., Ahmed, A., Foster, A., Rannard, S.P., Cooper, A.I., Zhang, H., 2009. Systematictuning of pore morphologies and pore volumes in macroporous materials byfreezing. J. Mater. Chem. 19, 5212–5219.
Qian, L., Ahmed, A., Zhang, H., 2011. Formation of organic nanoparticles by sol-vent evaporation within porous polymeric materials. Chem. Commun. 47,10001–10003.
Qiu, L.Y., Bae, Y.H., 2006. Polymer architecture and drug delivery. Pharm. Res. 23,1–30.
Rannard, S., Owen, A., 2009. Nanomedicine: not a case of “One size fits all”. NanoToday 4, 382–384.
Redhead, H.M., Davis, S.S., Illum, L., 2001. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine908: in vitro characterisation and in vivo evaluation. J. Control. Release 70,353–363.
Schwendener, R.A., Schott, H., 1996. Lipophilic 1-�-d-arabinofuranosyl cytosinederivatives in liposomal formulations for oral and parenteral antileukemictherapy in the murine L1210 leukemia model. J. Cancer Res. Clin. Oncol. 122,723–726.
Shegokar, R., Müller, R.H., 2010. Nanocrystals: industrially feasible multifunctionalformulation technology for poorly soluble actives. Int. J. Pharm. 399, 129–139.
Shepherd, G., 2003. Hypersensitivity reactions to chemotherapeutic drugs. Clin. Rev.Allergy Immunol. 24, 253–262.
Singla, A.K., Garg, A., Aggarwal, D., 2002. Paclitaxel and its formulations. Int. J. Pharm.235, 179–192.
Svenson, S., 2009. Dendrimers as versatile platform in drug delivery applications.Eur. J. Pharm. Biopharm. 71, 445–462.
Szebeni, J., Alving, C.R., Muggia, F.M., 1998. Complement activation by cremophorEL as a possible contributor to hypersensitivity to paclitaxel: an in vitro study.J. Natl. Cancer Inst. 90, 300–306.
Trotta, M., Gallarate, M., Carlotti, M.E., Morel, S., 2003. Preparation of griseoful-vin nanoparticles from water-dilutable microemulsions. Int. J. Pharm. 254,235–242.
Vasconcelos, T., Sarmento, B., Costa, P., 2007. Solid dispersions as strategy toimprove oral bioavailability of poor water soluble drugs. Drug Discov. Today12, 1068–1075.
Waser, P.G., Müller, U., Kreuter, J., Berger, S., Munz, K., Kaiser, E., Pfluger, B., 1987.Localization of colloidal particles (liposomes, hexylcyanoacrylate nanoparticlesand albumin nanoparticles) by histology and autoradiography in mice. Int. J.Pharm. 39, 213–227.
Yuan, F., Dellian, M., Fukumura, D., Leunig, M., Berk, D.A., Torchilin, V.P., Jain,R.K., 1995. Vascular permeability in a human tumor xenograft: molecular sizedependence and cutoff size. Cancer Res. 55, 3752–3756.
Zhang, H., Wang, D., Butler, R., Campbell, N.L., Long, J., Tan, B., Duncalf, D.J., Foster, A.J.,Hopkinson, A., Taylor, D., Angus, D., Cooper, A.I., Rannard, S.P., 2008a. Formationand enhanced biocidal activity of water-dispersable organic nanoparticles. Nat.
Nanotechnol. 3, 506–511.Zhang, Y., Yang, M., Portney, N., Cui, D., Budak, G., Ozbay, E., Ozkan, M., Ozkan, C.,2008b. Zeta potential: a surface electrical characteristic to probe the interactionof nanoparticles with normal and cancer human breast epithelial cells. Biomed.Microdev. 10, 321–328.