poly(meth)acrylates obtained by cascade reaction
TRANSCRIPT

Feature Article
Poly(meth)acrylates Obtained by CascadeReaction
Dragos Popescu, Helmut Keul,* Martin Moeller*
Preparation, purification, and stabilization of functional (meth)acrylates with a high dipolemoment are complex, laborious, and expensive processes. In order to avoid purification andstabilization of the highly reactive functional monomers, a concept of cascade reactions wasdeveloped comprising enzymatic monomer synthesis and radical polymerization. Transacylationofmethyl acrylate (MA) andmethylmethacrylate (MMA)with different functional alcohols, diols,and triols (1,2,6-hexanetriol and glycerol) in the presence ofNovozyme 435 led to functional (meth)acrylates. After theremoval of the enzyme by means of filtration, removal ofexcess (meth)acrylate and/or addition of a new monomer,e.g., 2-hydroxyethyl (meth)acrylate the (co)polymerizationvia free radical (FRP) or nitroxide mediated radicalpolymerization (NMP) resulted in poly[(meth)acrylate]swith predefined functionalities. Hydrophilic, hydrophobicas well as ionic repeating units were assembled within thecopolymer. The transacylation of MA and MMA with diolsand triols carried out under mild conditions is an easy andrapid process and is suitable for the preparation of sensitivemonomers.
Introduction
Polymeric materials are indispensable in modern society.
They are widely used as commodity materials in everyday’s
life and as highly advanced materials in electronics,
machinery, communication, transportation, pharmacy,
medicine, etc. Today, a society without polymeric materials
is hard to imagine.
Polymers which are prepared fully or partially from
acrylates or methacrylates are called poly(meth)acrylates.
Numerous fields of application of poly(meth)acrylates
are known, such as fiber protection agents in laundry
H. Keul, M. Moeller, D. PopescuInstitute of Technical and Macromolecular Chemistry, RWTHAachen and DWI an der RWTH Aachen e.V., Pauwelsstr. 8,D-52056 Aachen, GermanyE-mail: [email protected]; [email protected]
Macromol. Rapid Commun. 2011, 32, 559–572
� 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim wileyonlin
detergents, corrosion protection, or as complexing agents.
The end use of poly(meth)acrylates depends on the
monomer composition or the modification of the polymers
after their synthesis. Polymers with pendant functional
groups have gained much attention in the last few years[1]
and are of interest as starting materials for complex
polymer architectures or for polymers with customized
properties for different applications.[2]
The development of new polymeric materials is closely
linked to the polymerization procedures. Radical polymer-
ization is a widely used technique in industry for the
preparation of polymers, like poly(styrene) and poly(-
methyl methacrylate). Although radical chain reactions are
known since the twenties of the last century, it was not
until the mid 1950 s that the free-radical polymerization
was introduced into technical applications. Nowadays,
free-radical polymerization is used for the synthesis of
many important classes of polymers such as polymetha-
elibrary.com DOI: 10.1002/marc.201000725 559

560
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
crylates, polystyrene, polychloroprene, polyacrylonitrile,
polyethylene, and a large number of copolymers. Many
reviews on this subject are available.[3–5] The major
advantage of radical polymerization is that it is carried
out under relatively undemanding conditions: water or
other impurities are well tolerated; the reactions occur in a
broad temperature range from 0 to 100 8C;[6] high molecular
weight polymers are obtained without removal of stabi-
lizers present in commercial monomers and are also
produced in the presence of trace amounts of oxygen. It
is for these reasons that radical polymerization has been
adopted for many industrial polymer syntheses.
The major drawbacks of conventional radical polymer-
izations are related to the lack of control over the polymer
microstructure which is particularly important for the
preparation of more complex polymer structures such as
block copolymers or gradient copolymers. The design of
complex macromolecular architectures has become a
particular focus in polymer science. Some of these
architectures possess unique properties, which make them
interesting candidates for specialty applications in nanos-
tructured and biomedical materials. Such precise macro-
molecular syntheses employ concepts of ‘‘living’’ polymer-
ization. The concept of living polymerization was
introduced by Szwarc for the anionic polymerization of
Helmut Keul studied chemistry at the University of TeUniversity of Karlsruhe, Germany. Post-doctoral workUniversity ofMichigan (Ann Arbor), USA, was followedAachen in 1986. His current interests regard the controstructure-property relationships. At the timeDr. Keul isTechnical Chemistry and Macromolecular Chemistry (IT200 scientific publications and 20 patents.
Martin Moller was born in 1951. He received his Ph.D.Lynen-Research Fellow of the Alexander von Humboldtof Massachusetts, Amherst (USA), he returned to Freilecturer on Macromolecular Chemistry from the UniDepartment of Chemical Technology, University of TwUlm and Head of the Department of Organic ChemistrTextile Chemistry and Macromolecular Chemistry of RWinstitute DWI. He ismember of Deutsche Akademie derthe State of North-RhineWestphalia. In 2003, he was awas elected scientific expert of Deutsche Forschungsgfur Angelegenheiten der Sonderforschungsbereiche’ oscientific journals. He has published about 450 articlefunctional nanostructures, synthesis and structure pblockcopolymers, surface modification, self organizati
Dragos Popescuwas born in 1980 in Bucharest (Romanhe received the M.Sc. degree in therapeutical chemistresearch scholarship at DWI an der RWTHAachen e.V. (Gobtained the Ph.D. degree under the supervision of ProChemistry, RWTH Aachen and DWI an der RWTH Aachmultifunctional and reactive poly(meth)acrylates. SinComposite Resins in the Chemistry and Synthesis gro
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
styrene,[7,8] later the concept was transferred to cationic
vinyl polymerization[9,10] and to ionic (anionic and cationic)
ring opening polymerizations (ROPs).[11–14] Much later, at
the end of 20th century, controlled radical polymerization
(CRP) was introduced: atom transfer radical polymerization
(ATRP),[15–20] nitroxide mediated radical polymerization
(NMP),[21,22] and reversible addition fragmentation chain
transfer polymerization (RAFT).[23,24] All these CRP methods
are based on a fast equilibrium between dormant and active
species. The equilibrium constants are low, keeping the
concentration of the active species very low (ca. 10�7
mol � L�1). As a result, termination and transfer reactions are
minimized, and CRP can be achieved under appropriate
polymerization conditions.
Although the CRP processes—ATRP, NMP, and RAFT—
were developed within the last decade, these techniques
already find applications in the commercial production of
new materials.[25] Block copolymers based on acrylates find
applications as thermoplastic elastomers,[26] adhesives,
lubricants, gels, and coatings. However, they can also be
used for more sophisticated applications such as specialized
chromatographic packing[27] or controlled drug-delivery in
cardiovascular stents.[28]
The CRP techniques allow the straightforward synthesis
of well-defined polymers and copolymers with desired
chnology, Bucharest, Romania, and received his PhD in 1973 at theat the University of Karlsruhe and visiting research scientist at thebymoves to the University of Bayreuth in 1984 and the University oflled synthesis of multifunctional and reactive polymers as well asleading a research group of fifteen Ph.D. students at the Institute ofMC) of the RWTH Aachen University. He is an author of more than
from the University of Freiburg in 1981. After working as a Feodor-Foundation at the Department of Polymer Science of the Universityburg in 1982. In 1988, he received his qualification as a universityversity of Freiburg. From 1989 till 1993, he was professor at theente (NL). From 1993 to 2002, he was professor at the University ofy III / Macromolecular Chemistry. In 2002, he became professor forTH Aachen University and since 2003, he is also the director of theTechnikwissenschaften (acatech) and of the Academy of Sciences ofwarded the Korber European Science Award. From 1999 to 2005, heemeinschaft (DFG). Since 2006, he is member of ‘Senatsausschussf the DFG. He is member of the editorial board of several polymers. His main research interests are macromolecular chemistry androperty relationships in branched and hyper-branched polymers,on of polymers in the bulk and in thin films.
ia). In 2003 he obtained his B.Sc. degree in biochemistry and in 2005ry at the University of Bucharest (Romania). After a three monthermany), he started in October 2005with his Ph.D. work. In 2010 hefessor Martin Moller at Institute of Technical and Macromolecularen e.V. His research focused on the chemoenzymatic synthesis ofce August 2010 he is working as a project coordinator for DSMup.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
molecular weight, molecular weight distribution, composi-
tion and microstructure, architecture and functionality.
These parameters strongly influence the response to
external stimuli as for example temperature in stimuli-
responsive polymers.[29] Depending on the structure,
polymers exhibit a phase transition from soluble to
insoluble upon heating in aqueous solution. Such polymers
show a lower critical solution temperature (LCST) which is
based on the existence of hydrogen bonding between the
solvent molecules and the polymer chain. Thus, in aqueous
solutions, below the LCST, the polymer chains are soluble
and exist in a random coil conformation. Weakening of
the hydrogen bonds at higher temperatures causes entropy-
driven phase transition of the polymer to a hydrophobic
collapsed state.[30] Polymers with an LCST show a sudden
and mostly reversible change from hydrophilic to hydro-
phobic that makes them attractive for switchable materials
in biotechnological applications including drug delivery
systems,[31] tissue engineering,[32] and biomolecule separa-
tion[33,34] but are also of interest for industrial applications
in catalysis,[35] coatings, and even in textile materials.
Synthesis of Functional Polyacrylates
The target of macromolecular engineering is to design and
to control the structural parameters of multifunctional
polymers such as chain length, polydispersity, functional
composition, and microstructure, in order to adjust their
macroscopic properties.[36] Homo- and copolymers with
functional groups as lateral substituents are of increasing
interest in polymer science. Several strategies were
developed for the preparation of functional polymers such
as (i) (co)polymerization of functional monomers and
(ii) polymer analogous reactions using polymers with
reactive groups. Polymerization of functional monomers to
yield functional polymers sometimes is not the method of
choice due to the difficulty of both the preparation of the
monomers and their polymerization.[37] The second con-
cept for the preparation of functional polymers consists of
the modification of the pending groups of a polymer.
Acrylates are an important class of monomers for
technical applications that are found in a variety of
consumer products. Based on their reactivity and function-
ality they are used for the preparation of polymers of
different architecture and resins for surface coatings.
Acrylate chemistry is by far the most commonly used
technology in UV-cured coatings. It involves the introduc-
tion of cross-linking functionalities in the form of acrylate
groups to create binders for the use in surface formulations.
Hydroxy functional acrylates and methacrylates are
interesting precursors for hydrophilic and water-soluble
polymers which are promising functional polymers for
biotechnological applications,[38,39] including biomedical
and pharmaceutical products such as contact lenses, dental
www.MaterialsViews.com
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
materials, encapsulated cells, carriers for controlled drug
delivery as well as hydrogels.[31,40,41] Hydroxy functional
acrylates and methacrylates are versatile and ideal co-
monomers for cross-linking via the pendant hydroxy group;
they can be used as precursors for further chemical
modification leading to novel building blocks.
For the preparation of functional acrylates or methacry-
lates the chemical synthesis is based for example on the
reaction of acrylic and methacrylic acid with cyclic ethers
like ethylene oxide and propylene oxide.[42] When cyclic
ethers are not available or not reactive enough, the reaction
of rather expensive acid chlorides or active esters of
(meth)acrylic acids[43–45] with alcohols or amines is the
method of choice. Chemical synthesis with nonactivated
(meth)acrylic alkyl esters requires high temperatures,
pressure, acidic catalysts, polymerization inhibitors, and
complex purification procedures to isolate the mono-
(meth)acrylates from the mixture of mono- and multi-
functional monomers, leading to low overall yields. In the
case of base-catalyzed transesterification the products are
often complex mixtures, occasionally colored. In order to
remove the coloration and the unconverted reactants it is
necessary to work up the product mixtures by means of
costly and inconvenient washing procedures or by distilla-
tion. However, the monomers often are not stable under
distillation conditions and polymerization may occur.
Further development of syntheses of functional acrylates
and methacrylates as well as of multifunctional poly-
(meth)acrylates is an area of major interest because of their
high performance potential.
Enzyme Catalyzed Transacylation
An alternative to conventional chemical syntheses for the
production of fine chemicals is the enzyme catalyzed
transformation. In contrast to ‘‘chemical’’ metal catalysts,
which might be toxic, enzymes are natural (‘‘green’’)
biocatalysts. In their natural environment enzymes
are efficient catalysts and show high chemoselectivity,
regioselectivity, and enantioselectivity. More than
hundred years ago, it was shown that enzymes can be
used in organic solvents.[46]. Their high selectivity and
activity in organic solvents make them ideal catalysts for
chemical conversions. In vivo, enzymatic catalyses obey
two fundamental characteristics: the first is the ‘‘key and
lock’’ principle proposed by Emil Fischer in 1894 which
points out the relationship between an enzyme and a
substrate and the second suggested by Linus Pauling
explaining the decrease of the activation energy by the
formation of an enzyme-substrate complex (transition-
state). However, the interest for biocatalysis did not boost
until the 1980 s with the general acceptance that enzymes
can catalyze unnatural reactions efficiently in organic
solvents.[47]
. 2011, 32, 559–572
H & Co. KGaA, Weinheim561
562
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
In recent years, the employment of triacylglycerol lipases
as biocatalysts for transacylation reactions (transesterifica-
tion) has emerged as a potential route to replace conven-
tional chemical methods. The main reason for this is the
hope for more efficient processes with higher chemo-, regio-,
and stereoselectivity. In addition, the use of lipases as
catalysts is a non-toxic and environmentally friendly
technology and requires mild operating conditions com-
pared to many chemical procedures.[48,49] Besides, the
availability of lipases from different microbial sources
possessing specificity of action and the fact that their
catalytic activity can be easily regulated are some of the
highlights that most chemical catalysts do not possess. So
far, the most widely used enzyme for esterification and
transacylation reactions is lipase B fromCandidaantarctica
(CALB), immobilized by adsorption on a macroporous
acrylic resins, [Lewatit VP OC 1600, Bayer, poly(methyl
methacrylate-co-divinylbenzene)] called Novozyme 435,
which has exceptionally high activity and versatility.[50–53]
The yeast C. antarctica was originally isolated in Antarctica
and was found to produce two lipase variants (CALA and
CALB).[54] CALB belongs to the a/b-hydrolase-fold super-
family which contains enzymes that evolved from a
Asp
O
O
NNH
His
H,CH3
O
OCH3
H O
Ser
Asp
O
O
NNH
His
H O
Ser
O OR
H,CH3
k-4k4 H,CH3
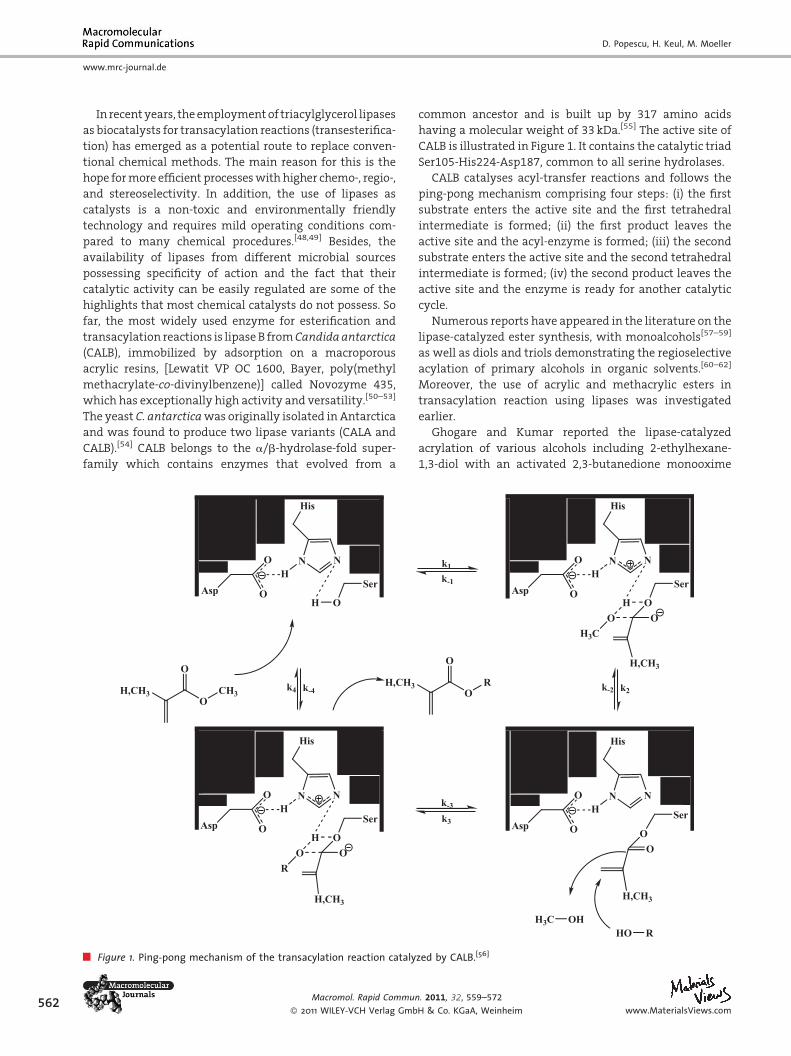
Figure 1. Ping-pong mechanism of the transacylation reaction cataly
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
common ancestor and is built up by 317 amino acids
having a molecular weight of 33 kDa.[55] The active site of
CALB is illustrated in Figure 1. It contains the catalytic triad
Ser105-His224-Asp187, common to all serine hydrolases.
CALB catalyses acyl-transfer reactions and follows the
ping-pong mechanism comprising four steps: (i) the first
substrate enters the active site and the first tetrahedral
intermediate is formed; (ii) the first product leaves the
active site and the acyl-enzyme is formed; (iii) the second
substrate enters the active site and the second tetrahedral
intermediate is formed; (iv) the second product leaves the
active site and the enzyme is ready for another catalytic
cycle.
Numerous reports have appeared in the literature on the
lipase-catalyzed ester synthesis, with monoalcohols[57–59]
as well as diols and triols demonstrating the regioselective
acylation of primary alcohols in organic solvents.[60–62]
Moreover, the use of acrylic and methacrylic esters in
transacylation reaction using lipases was investigated
earlier.
Ghogare and Kumar reported the lipase-catalyzed
acrylation of various alcohols including 2-ethylhexane-
1,3-diol with an activated 2,3-butanedione monooxime
Asp
O
O
NNH
His
k1k-1
H O
Ser
O OH3C
H,CH3
Asp
O
O
NNH
His
Ser
OO
H,CH3
H3C OHHO R
k2k-2
k-3k3
O
OR
zed by CALB.[56]
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
acrylate,[63] while enzymatic transacylation of vinyl
acrylate with alcohols using immobilized lipase from
C. cylindracea was reported by Ikeda et al.[64] A frequent
disadvantage of these reactions is the need to use activated
acrylates, such as the oximes or vinyl esters, which are
expensive and difficult to obtain. Hajjar et al. described
the enzymatic transacylation of cyclic and open-chain
alkane diols with ethyl acrylate using a lipase from
Chromobacterium viscosum. The reaction proceeds with
an excess of alkyl acrylate with respect to the diol in a
system without solvent producing a mixture of mono- and
bisacrylates.[65]
A highly stabilized, immobilized pig liver esterase was
found to be effective in catalyzing both hydrolysis and
transesterification reactions of methyl and ethyl esters of
acrylic and methacrylic acids. The enzyme was successfully
employed for the preparation of hydroxy and dihydroxy
alkyl acrylates and methacrylates without formation of bis-
or tris(meth)acrylates by using an alcohol excess of 50–70%
v/v.[66] The lipase PS-30 Pseudomonas species-catalyzed
preparation of carbamoyloxyethyl methacrylate by trans-
acylation of 2-hydroxyethyl carbamate with vinyl metha-
crylate in a solvent mixture of toluene/THF (3:1) was
reported by Derango et al.[67] Complete conversion is
achieved with the specific vinyl methacrylate reactant,
since vinyl alcohol liberated is removed from the reaction
equilibrium in the form of acetaldehyde.
In another study, Warwel et al. used Novozyme 435 as
biocatalyst for the transacylation of methyl acrylate (MA)
and methyl methacrylate (MMA) with unsaturated fatty
alcohols,[68] while Athawale et al. performed a comprehen-
sive study of the reaction parameters governing the
enzymatic synthesis of geranyl methacrylate using porcine
pancreatic lipase and 2,3-butanedione mono-oxime metha-
crylate as acyl donor in diisopropyl ether as the solvent.[69] A
comparative study of the enzymatic synthesis of acrylic
acid esters of different alcohols using 2,3-butanedione
mono-oxime acrylate and vinyl acrylate as acylating agents
and the immobilized lipase from C. cylindracea (CCL) was
reported.[70] The rate of conversion was fastest when the
oxime acrylate was used. Effect of solvents on the rate of
conversion was studied and diisopropyl ether was proved
to be a better solvent than CHCl3 and THF. Various alcohols
were used to study the effect of the structure of the alcohol
on the rate of conversion. Among the linear alcohols
studied, ease of conversion was found to be of the order
n-octanol>n-hexanol>n-butanol. In the case of cyclo-
hexyl methanol, highest conversion (80%) was achieved.
Epoxy-containing (meth)acrylic esters can also be
obtained by lipase-catalyzed reaction. Thus, Xin et al.
described for the first time the synthesis of glycidyl acrylate
from glycidol and vinyl acrylate studying the influence of
three different lipases, four solvents, and polymeric
additives on the conversion.[71] Quantitative conversions
www.MaterialsViews.com
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
were not obtained since all the reactions were terminated at
a maximum conversion of 75% after 4 h. However, due to
their high preparation costs, such acrylic acid derivates are
not of technical interest.
This drawback was overcome by one of the BASF patents
where epoxy-containing (meth)acrylic esters were
obtained by transacylation of (meth)acrylic esters available
on industrial scale and alcohols comprising epoxy groups in
the presence of lipase B from C. antarctica.[72] Numerous
other patents on the lipase catalyzed transacylation have
appeared in the literature showing the industrial relevance
of this research.[73–78]
Chemoenzymatic Approach toward PolymericMaterials
An exhaustive study on combining chemical and enzyma-
tical steps was performed by the groups of Palmans and
Heise in Eindhoven.[79] Thus, the first combination of an
enzymatic polymerization with a chemical polymerization
technique: lipase-catalyzed ROP of e-caprolactone and ATRP
of styrene was reported.[80,81] This approach was extended
by using methyl-substituted e-caprolactones as a substrate
for the enzymatical step and MMA for ATRP.[82] A novel
chemoenzymatic approach toward polymeric materials
by integration of enzymatic ROP with nitroxide mediated
polymerization was described as well.[83] Peeters et al.
reported for the first time the synthesis of branched
polymers following the alternative strategy of self con-
densing vinyl polymerization (SCVP) of fully enzymatically
generated macroinimers by radical polymerization.[84] Yu
and Lowe reported the enzymatic reaction between an
amino alcohol and vinyl methacrylate followed by RAFT
polymerization of the newly synthesized monomer.[85] The
Institute of Technical and Macromolecular Chemistry,
RWTH Aachen, for the first time reported that chloroper-
oxidase (CPO) is stable in scCO2/H2O biphasic media. This
allows the cascading of chemical and enzymatic reactions
for the synthesis of optically enriched (R)-sulfoxide.[86]
The chemo-enzymatic methodology opens new oppor-
tunities for the efficient integration of enzymes and
chemical catalysts toward cascade reactions. A broad
literature presents the chemo-enzymatic synthesis of
polyacrylates containing sucrose functionalities based on
the cascade reaction methodology.[87–92]
This paper will point out the features and the advantages
of using the concept of cascade reactions in the synthesis of
different multifunctional poly(meth)acrylates. The synth-
eses of functional acrylate and methacrylate monomers
and their polymerization leading to multifunctional and
reactive poly(meth)acrylates will be discussed. Transacyla-
tion of MA and MMA as a substrate with different
functional alcohols in the presence of Novozyme 435 leads
to a mixture of functional monomers which subsequently
. 2011, 32, 559–572
H & Co. KGaA, Weinheim563

564
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
are ‘‘in situ’’ copolymerized—after removal of Novozyme
435 by filtration—via free radical polymerization or
nitroxide mediated polymerization, resulting in poly-
(meth)acrylates with predefined functionalities designed
for a wide range of applications. Depending on the
application, the excess of M(M)A is removed by evaporation
and, or another functional monomer like HE(M)A is added to
the monomer mixture, which later is converted into a
reactive repeating unit with phenyl chloroformate obtain-
ing a functional and reactive copolymer suitable for further
polymer analogous reactions with primary amines.[93–95]
Chemoenzymatic Synthesis of(Meth)acrylates and Poly(meth)acrylates
The synthesis of highly functional and reactive poly-
(meth)acrylates via a chemical route is very demanding and
requires highly pure starting materials as well as inhibitors
in order to prevent the undesired polymerization. It was our
goal to develop a straightforward, mild, and rapid way for
the preparation of functional and reactive poly(meth)acry-
lates. The concept of our work is illustrated in Figure 2.
Starting with a technical monomer MMA or MA and a
H,CH3
OO + HO
H2C X
Enzymatictransacylation
H,CH3
OO
X
O
H,CH3
OO
H,CH3
OO
OH
OH
O
OHHO
O
HO
Free radicalpolymerization
Nitroxide mediatedpolymerization
Thermorespons
G(M)A
NP
DHH(M)A 2H
Figure 2. Schematic representation of the synthesis of functional an
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
functional alcohol, diol, or triol in an enzymatically
catalyzed step which is a transacylation catalyzed by
Novozyme 435, a mixture of monomers is obtained at
equilibrium. After the removal of Novozyme by filtration
the mixture of monomers can be polymerized via free
radical polymerization or CRP. As a CRP we have chosen
nitroxide mediated polymerization due to the polarity of
the hydroxy acrylates which were copolymerized for the
synthesis of novel poly(hydroxy acrylates)s. In some cases,
excess of MMA or MA was removed prior to polymerization
and another functional monomer, HE(M)A was added to the
mixture. The copolymers thus obtain are subjected to a
polymer analogous reactions using phenyl chloroformate
in order to prepare highly reactive poly(meth)acrylates
bearing phenyl carbonate groups which are known to react
fast and without side reaction with nucleophiles like for
example amines.
Enzymatic Synthesis of Functional (Meth)acrylates
In order to prepare functional acrylates or methacrylates
we investigated the transacylation of MA and MMA as
substrates and different functional monoalcohols as
reagents with Novozyme 435 as catalyst. Under certain
O
X
H,CH3
OO
H,CH3
OO
H,CH3
OO
N
O
O
H,CH3
O
OH
H,CH3
O
Bacteriostatic polymers
H,CH3
OO
N
Peptide/Protein-Polymer Conjugates
H,CH3
OO
O
O
O
ive polymers
G(M)A
PCE(M)A
P(M)A
DEGE(M)A D(M)A
DMAP(M)A
d reactive polymethacrylates via a cascade reaction.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
conditions this transacylation occurs with the result of a
mixture of two (meth)acrylates and two alcohols: the
starting materials, the functional (meth)acrylate, and the
methanol formed. It is worth mentioning that all transa-
cylation reactions were performed in bulk and no side
reactions occurred.[96]
All functional mono-alcohols used—dodecanol (D-ol), 3-
dimethylamino-1-propanol (DMAP), diethylene glycol
monomethyl ether (DEGME), and diethylene glycol ethyl
ether (DEGEE)—were successfully transacylated both, with
MA and MMA.
When two different alcohols were used simultaneously
for the transacylation reaction of the same substrate, the
following molar ratio of the components was chosen:
M(M)A/R1OH/R2OH¼ 1:0.5:0.5.
Optimization of the Transacylation of MethylAcrylate (MA) and Methyl Methacrylate (MMA) withFunctional Alcohols
In order to optimize the enzyme-catalyzed transacylation
reactions, the influence of different parameters on the
conversion of M(M)A was studied: (i) the influence of the
weight ratio substrate/enzyme, (ii) the conversion as a
fuction of time (time necessary to reach the equilibrium),
(iii) the influence of temperature, and (iv) the influence of
the molar ratio substrate/alcohol.
(i) T
www
he first remark with respect to the influence of the
enzyme concentration is the following: in the absence
of the enzyme no conversion or a conversion as low as
2 mol-% for DMAPMA and 5 mol-% for DMAPA is
observed. With increasing concentration of Novozyme
435 (1, 5, and 10 wt.-% with respect to M(M)A) the
reaction rate increases. For all enzyme concentrations
the conversion of MA is higher than that of MMA. One
reason for this result might be the higher sterical
demand of MMA as compared to MA in the enzymatic
transacylation reaction. In addition, MMA is a less
efficient acyl donor than MA.[97,98]
(ii) S
tudying the time necessary to reach the equilibriumthe same result—acrylates react faster than metha-
crylates—was obtained. The nature of the alcohol
used defines the reaction rate. The highest product
concentrations are found for D-ol: at equilibrium
49 mol-% DA is formed while DMA did not reach the
equilibrium: 36.5 mol-% after 144 h and 42 mol-% after
264 h. For both DMAP esters a concentration of 46 mol-%
is reached, however, at different times (for DMAPA
within 24 h, for DMAPMA within 96 h). The DEGME
and DEGEE esters result in the lowest concentration
with 34 mol-%.
(iii) T
he effect of temperature on the transacylation ofM(M)A was also studied for a molar ratio of M(M)A/
.MaterialsViews.com
Macromol. Rapid Commun. 201
� 2011 WILEY-VCH Verlag GmbH & C
functional alcohol 1:1 in bulk with an enzyme
concentration of 10 wt.-% with respect to M(M)A at
r.t. and at 70 8C. Under the conditions applied higher
conversion of the products are achieved at 70 8C than
at r.t. after 24 h. For acrylates the differences in
conversion are small and therefore the reaction at r.t.
should be preferred because of the milder reaction
conditions. For methacrylates a higher temperature is
needed to obtain an acceptable conversion within
24 h.
(iv) F
inally the influence of the molar ratio M(M)A/alcoholon the transacylation efficiency at 70 8C and 24 h in
bulk with 10 wt.-% concentration of Novozyme 435
with respect to M(M)A was studied by decreasing the
ratio M(M)A/functional alcohol from 1:1 to 1:5 in
order to shift the equilibrium toward product forma-
tion. For the reaction of D-ol with MMA, an increase of
the alcohol concentration leads to a slight decrease of
DMA from 35 to 28 mol-%. For DMAP, an increase of
the alcohol concentration has no significant effect on
the conversion of MMA (33 mol-% vs. 30 mol-%). The
most significant change in the efficiency of the
transacylation by increasing the concentration of
the alcohol is found in reaction of MMA with DEGME.
When DEGME is used in an equimolar ratio with
respect to the substrate, 30 mol-% of DEGMMA are
formed, while, when a fivefold excess of DEGME is
used only 10 mol-% DEGMMA are obtained. An
explanation for these results might the inhibition
effect of the alcohol on the lipase. In the case of MA
as the substrate, with increasing alcohol concentra-
tion the absolute conversion of MA increases except
in the case of DEGME supporting our theory that
the DEGMEE inhibits the enzyme. In enzymatic
transacylation, the highest conversions are observed
for D-ol while the lowest are observed for DEGME.
Using an equimolar ratio the following conversions
were achieved: 45 mol-% DA, 40 mol-% DMAPA,
and 35 mol-% DEGMA, while using a molar
ratio substrate/alcohol of 1:5 the conversions were
76 mol-% for DA, 43 mol-% for DMAPA, and 30 mol-%
for DEGMA.
These results suggest that for an enzymatic transacyla-
tion the increase of the concentration of one component
does not have the expected result due to the fact that the
reaction medium is changed and consequently the enzyme
activity is changed. Based on these results we concluded
that the optimum conditions for the enzyme catalyzed
transacylation of M(M)A with different functional mono-
alcohols are: M(M)A/ROH¼ 1:1 mole/mole; t¼ 24 h;
T¼ 70 8C and an enzyme concentration of 10 wt.-% with
respect to M(M)A.
1, 32, 559–572
o. KGaA, Weinheim565

Table 1. Enzymatic transacylation of M(M)A with functional alco-hols (R–OH): molar composition of (M(M)A) and R(M)A.a)
M(M)A R(M)A
mol-% mol-%
MMA (86) DMA (14)
MA (55) DA (45)
MMA (73) DMAPMA (27)
MA (56) DMAPA (44)
MMA (79) DEGMMA (21)
MA (69) DEGMA (31)
MMA (81.5) DEGEMA (18.5)
MA (70) DEGEA (30)
a)All reactions were performed at r.t. for 24 h using 10 wt.-%
Novozyme 435 with respect to M(M)A. All the concentrations
were determined using 1H NMR spectroscopy.
566
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
The molar ratios of the functional monomers obtained
R(M)A and M(M)A are summarized in Table 1.
Enzymatic Synthesis of Hydroxyl Functional(Meth)acrylates
This approach for the preparation of functional acrylate
monomers was expanded to the more demanding synth-
eses of hydroxy functional acrylates and methacrylates via
an enzymatic transacylation of MA and MMA with
different diols and triols.[99,100] In this way, mono- and
dihydroxy functional (meth)acrylate monomers were
obtained which are difficult to synthesize via conventional
methods and are difficult to purify and stabilize. To
investigate the effect of the alcohol structure used as
substrate on the formation of hydroxy functional mono(-
meth)acrylate monomers, symmetrical diols (ethylene
glycol; 1,3-propane diol; 1,4-butane diol; 1,5-pentane diol;
1,6-hexane diol), substituted symmetrical diols (2-methyl-
1,3-propane diol; neopentyl glycol), an asymmetrical diol
(1,2-propane diol), a symmetrical triol (glycerol) as well as
an asymmetrical triol (1,2,6-hexane triol) were selected.
Since the enzymatic transacylation is reversible mono-, di-,
and tri-functional (meth)acrylates can be obtained. None-
theless, sterical hindrance at the active site of the enzyme is
expected to suppress multiple acylation as well as acylation
of secondary alcohol groups. In addition the enantio-
selectivity during transacylation of the asymmetrical
racemic diol (1,2-propane diol) and triol (1,2,6-hexane triol)
was determined. Furthermore, it should be noted that the
reaction rate and product distribution depend on the
alcohol/M(M)A ratio,[43–45] the specificity of Novozyme 435
and the water activity.[101]
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
To obtain a better understanding of the mechanism of
the lipase-catalyzed transacylation we conducted kinetic
studies using 1,3Pdiol, 1,4Bdiol, 1,2Pdiol as well as glycerol
and MA and MMA as acyl donors. These alcohols were used
in order to synthesize hydroxy functional acrylates and
methacrylates that may be suitable for a wide variety of
applications.[31,38–40,102,103] 2-Methyl-2-butanol was used
as the solvent because of its polarity allowing the
dissolution of the alcohols while the enzyme retains its
catalytically active conformation.[104,105] The reaction
temperature was set to 50 8C, which is within the optimum
temperature range for the enzyme stability.[106] In order to
decrease the formation of bis(meth)acrylates, the molar
ratio acyl donor/alcohol was set to 1/number of OH groups
in the alcohol according to a theoretical model presented in
the literature.[43–45] The results show that acrylates react
faster than methacrylates. Further, the final conversions of
glycerol and 1,2Pdiol are lower than those of 1,3Pdiol and
1,4Bdiol which is explained by the higher polarity of the
reaction medium, by the higher sterical demand of these
substrates, and by the increase in viscosity of the medium
which makes diffusion to the active center of the lipase
difficult.[107] To these arguments, the lower reactivity of
secondary alcohol groups in glycerol and 1,2Pdiol should be
added. As a consequence, the unsubstituted a,v-diols
(1,3Pdiol and 1,4Bdiol) reach higher conversions than
a-substituted diols (glycerol and 1,2Pdiol).
Selectivity in Transacylation of Diols
The effect of the structure of the diols on the enzymatic
transacylation was also studied. For all cases except
1,3Pdiol, the final conversion of MA is higher than that of
MMA being in the range of 59–78 and 50–73 mol-%,
respectively. The conversion of both esters with EG is a
special case due to the hydrophilicity of EG, which may trap
the tightly-bound water necessary for retaining the tertiary
structure of the enzyme and thereby causing a rapid
deactivation of the lipase with the consequence of low
conversion.[101]
In the case of symmetrical diols (1,3Pdiol, 1,4Bdiol,
1,5Pdiol, and 1,6Hdiol) the product distribution mostly
follows the statistical rule as described in literature[43,44] for
chemical transacylation reactions, i.e., the enzyme does not
have any selectivity for the formation of mono(meth)acry-
late over bis(meth)acrylates. Nonetheless, in the case of the
reaction between MA or MMA and 1,3Pdiol the concentra-
tion of monosubstituted product is slightly higher than in
the case of 1,4 Bdiol, 1,5 Pdiol, and 1,6 Hdiol and also higher
than expected for the statistical product distribution. This is
most likely due to the chain length of the diol; the
monosubstituted product formed in the reaction with 1,3
Pdiol is more sterically hindered than longer diols. As a
result the formation of the disubstituted compounds is
suppressed. As expected, this effect is slightly stronger in
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
the reaction with MMA. The effect of steric hindrance was
further evaluated by the use of substituted symmetrical
diols 2 Me1,3 Pdiol and NPG. Not surprisingly the highest
concentration of mono(meth)acrylates and the lowest of
bis(meth)acrylates were obtained with these diols. Again
the sterical hindrance of the monosubstituted compound
which could react with the acyl donor to result bis(meth)a-
crylates plays an important role. Iglesias et al.[43,44] found
that 73� 3 mol-% monoacrylate and 80 mol-% mono-
methacrylate are formed in the reaction of NPG and acid
chloride with a molar ratio NPG/acid chloride of 7:3, while
the calculated statistical amount of mono(meth)acrylate is
88 mol-%. Comparing these results with results obtained by
enzymatic transacylation having a similar molar ratio NPG/
M(M)A of 2:1, one can notice that higher amounts of the
monosubstituted (meth)acrylates (95 mol-% for the acry-
late and 92 mol-% for the methacrylate) are formed proving
the advantage of using Novozyme 435. One more important
observation was made when evaluating the product ratio
with time. In fact, the conversion of monoacrylate
formed in this reaction increases from 39 mol-% (t¼ 24 h)
to 56 mol-% (t¼ 120 h) with respect to the acyl donor MA,
while the concentration of bisacrylate remains constant at
3 mol-% up to 120 h. The same behavior was observed for
2Me1,3Pdiol where the concentration of the monoacrylate
increases from 60 to 67 mol-% and the concentration of
bisacrylates remained constant. This result is a conse-
quence of the sterical hindrance of the monoacrylates
which can not further react with the MA to form the
bisacrylates. A possible explanation for the higher con-
centration of 2-methyl hydroxypropyl acrylate (2MHPA)
than of 3-hydroxy neopentyl acrylate (HNPGA) is again
attributed to the sterical hindrance, NPG is more severely
sterically hindered than 2Me1,3Pdiol due to the second
methyl group in C2 position. Moreover, the higher sterical
demand of the transition state in enzymatic transacylation
leads to the formation of lower amounts of disubstituted
products in enzymatic transacylation than in chemical
transacylation.
When 1,2Pdiol, an asymmetrical diol comprising both a
primary and a secondary hydroxyl group, was used a very
small amount of bisacrylates (2 mol-%) was observed while
the formation of bismethacrylates was not detected at all.
This suppression of bis(meth)acrylate formation is due to
the lower reactivity of the secondary hydroxy group as well
as the increased sterical hindrance caused by the vicinity of
the two hydroxyl groups. However, the molar ratio of
monosubstituted (meth)acrylates is 4:1 (77:21 and 80:20)
which is similar to the ratio observed for the chemical
synthesis of 2-hydroxypropyl acrylate from propylene
oxide and acrylic acid where 25 mol-% of the minor isomer
(2-hydroxy isopropylacrylate, 2HIPA) is formed.[42,108]
Nonetheless, the chemical route is more demanding and
requires high purity reagents, CuCl as inhibitor, pyridine as
www.MaterialsViews.com
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
solvent as well as high temperature[108] in comparison to
the enzymatic catalyzed reaction.
Being a racemic diol the stereoselectivity of the lipase
was also determined. It was found that both enantiomers
reacted at the primary hydroxyl group to give 37 mol-% of
one isomer and 34 mol-% of its enantiomer. In contrast to
this result, it was previously reported for benzyl alcohol
derivatives that Novozyme 435 discriminates between
R and S enantiomers, since the enantioselectivity of
Novozyme 435 is very high for the secondary benzylic
alcohols but much less for the aliphatic alcohols.[109,110] In
addition, this difference might be an effect of the substrate
and the polarity of the solvent used (2Me2BuOH) which
apparently suppresses the selectivity of Novozyme 435. In
addition, the system may contain traces of water which also
has an effect on the enantioselectivity ofC. antarctica lipase
B (CAL-B) catalyzed reactions, since water might
be simultaneously a competitive and enantioselective
inhibitor and a competitive substrate.[111,112]
Selectivity in Transacylation of Triols
In the case of glycerol, a symmetrical triol, two equivalent
primary hydroxyl groups and a secondary hydroxyl group
of lower reactivity are involved in the reaction with the
acyl donor. Garcia et al. studied the direct synthesis of
monomers derived from glycerol and unsaturated acid
chlorides in a stoichiometrical ratio obtaining five products:
two mono-, two di-, and one trisubstituted monomer.[45]
Using Novozyme 435, and a molar ratio (meth)acrylate/
glycerol of 1:3 only two mono(meth)acrylates are formed
and traces of bis(meth)acrylates. The major product
obtained is the 1-glyceryl (meth)acrylate in a concentration
of 92 mol-% (for GMA) and 94 mol-% (for GA) after 120 h
whereas 2-glyceryl (meth)acrylate is obtained to a low
extent [1,3-dihydroxyisopropyl acrylate (DHIPA) with a
molar concentration of 3 mol-% and 1,3-dihydroxyisopro-
pyl methacrylate (DHIPMA) with 6 mol-%]. Only minor
amounts of bis(meth)acrylates (to a maximum 3 mol-%) are
formed. The different isomers of bis(meth)acrylates were
not determined.
Transacylation of MA and MMA with 1,2,6Htriol using
Novozyme 435 as catalyst was performed in 2Me2BuOH at
50 8C with a molar ratio MA/1,2,6Htriol of 1:3. Five products
were identified: three monosubstituted and two disubsti-
tuted hexanetriol derivates. Since the OH group in position
6 is the least hindered, 6-methacryloyl-1,2,6-hexanetriol
was found to be the major product (71 mol-%) followed by 1-
methacryloyl-1,2,6-hexantriol (19 mol-%) and 2-methacry-
loyl-1,2,6-hexanetriol (5 mol-%). The two bismethacrylates
identified were assigned to 1,6-dimethacryloyl-1,2,6-hex-
anetriol (4 mol-%) and 2,6-dimethacryloyl-1,2,6-hexanetriol
(1 mol-%) again based on the reactivity hypothesis. It was
assumed that 1,2-dimethacryloyl-1,2,6-hexanetriol is not
. 2011, 32, 559–572
H & Co. KGaA, Weinheim567

568
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
formed due to sterical hindrance. The same assumption was
made for MA as acyl donor. The following distribution
of products was found: 86 mol-% of the two major products
(6-acryloyl-1,2,6-hexanetriol and 1-acryloyl-1,2,6-hexane-
triol), 8 mol-% of 2-acryloyl-1,2,6-hexanetriol, 4 mol-% of
1,6-diacryloyl-1,2,6-hexanetriol, and 2 mol-% of 2,6-diacry-
loyl-1,2,6-hexanetriol.[100]
The concentrations of mono-(meth)acrylates obtained
after enzymatic transacylation are presented in the Table 2.
Polymerization of the Monomer Mixture Obtained byTransacylation
Free Radical Polymerization
For the synthesis of multifunctional polymers the concept
of cascade reactions was applied: starting with M(M)A and
a functional alcohol ROH, in the first step transacylation
was performed with the result of a mixture of two
monomers (M(M)A and R(M)A), which in the second step,
after removal of the enzyme by means of filtration and of
M(M)A by distillation and, in some cases, after addition of 2-
hydroxyethyl (meth)acrylate (HE(M)A—was subjected to
free radical polymerization. Thus, for example after
transacylation reaction between MMA and D-ol, the
mixture was cooled to room temperature, Novozyme 435
was filtered off and AIBN, as a free radical initiator was
added. After polymerization, the polymer remained soluble
in the reaction medium and was isolated by precipitation in
methanol. A polymer with Mn ¼ 47 000 and Mw=Mn ¼ 2.3
was obtained. Moreover, the composition of repeating units
in the copolymer corresponds to the composition of
monomers in the feed proving the formation of a random
copolymer.
Table 2. Enzymatic transacylation of M(M)A with diols and triols: co
Mono-methacrylateb) Concentrationc)
mol-%
HEMA 0
3HPMA 38.1
4HBMA 26.5
5HPMA 31.6
2HPMA 18.6
2M3HPMA 29
3HNPGMA 18
GMA 31.5
1,2DHHMA 71d)
a)All reactions were performed in 2Me2BuOH at T¼50 8C for t¼ 24 h u
major isomer resulting from the enzymatic transacylation; c)The conce
concentration was determined after 120 h.
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
Several polymerization reactions were performed in
bulk, in butyl acetate, and ethanol. In order to prepare more
complex copolymers two distinct transacylation reactions
with two different alcohols were performed. In one case,
dodecanol was used as the alcohol, in the other case,
DMAPA. The two reaction mixtures were combined in a
certain ratio and then polymerized in bulk using AIBN as the
radical initiator. SEC analysis performed in DMAc revealed
the formation of the polymer with Mn ¼ 11 500 and
Mw=Mn ¼ 2. In order to introduce a reactive group into
the copolymer beside the functional groups a transacyla-
tion reaction with two different alcohols, DMAPA and D-ol
was performed and in the second step, after removal of the
enzyme by filtration, HEMA was added to the mixture of
monomers and copolymerized in ethanol at 90 8C for 9 h,
using AIBN as the free radical initiator. Thus, a copolymer
bearing hydrophobic, tertiary amino, and reactive groups
was obtained. Furthermore, a higher degree of hydrophi-
licity was achieved by introducing into the copolymer
beside the hydrophilic acrylate DEGMA resulting from the
enzymatic step, HEA in a mixture of molar ratio 1:1. A
functional and reactive copolymer soluble in water was
obtained.
Free radical polymerization of 3HPA and 4HBA did not
lead to soluble poly(hydroxyl acrylate)s, but a gel was
formed after 15–20 min. The gel formation can be explained
by the high concentration of bisacrylates, namely 10 mol-%
in 3HPA and 12 mol-% in 4HBA, leading to crosslinking
reactions. However, the free radical polymerization of
2Me3HPA that contains 7 mol-% bisacrylates yielded
soluble poly(2Me3HPA), the size exclusion chromatography
of the isolated polymer, however, revealed a trimodal
distribution with a number average molecular weight Mn
of 21 500 and a polydispersity index of Mw=Mn ¼ 9
ncentration of mono-(meth)acrylates at equilibrium.a)
Mono-acrylateb) Concentrationc)
mol-%
HEA 7.1
3HPA 63.5
4HBA 63.3
5HPA 64.1
2HPA 42.9
2M3HPA 59.8
3HNPGA 39
GA 50.7
1,2DHHA 72c)
sing 10 wt.-% Novozyme 435 with respect to M(M)A; b)Refers to the
ntration was determined by means of 1H NMR spectroscopy; d)The
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Table 3. Free radical polymerization of hydroxy acrylates obtainedvia enzymatic transacylation.
Monomera) Free radical polymerization
Xpb) (%) Mn
c) PDIc)
3HPA n.d.d) n.d.d) n.d.d)
4HBA n.d.d) n.d.d) n.d.d)
2Me3HPA 97 21 500 9
NPGA 86 17 600 2.3
GA 96 5 100 2
2HPA 98 14 000 2.3
DHHA 98 6 100 1.3
a)Major isomer in the feed; b)Total monomer conversion calculated
from 1H NMR spectroscopy; c)Number average molecular weight
(Mn) and polydispersity index (PDI) of purified polymers deter-
mined by size exclusion chromatography (SEC) using N,N-
dimethylformamide as eluent. GA, was measured in water;d)Not determined due to gelation.
Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
suggesting the formation of coupled chains induced by the
bisacrylates.
In contrast to 3HPA, 4HBA, and 2Me3HPA, all other
hydroxy acrylates showed the expected free radical
polymerization behavior resulting in monomodal molecu-
lar weight distributions with polydispersity indices around
2 as can be seen from Table 3. These results indicate that up
to 6 mol-% bisacrylate may be present in the monomer for
the preparation of soluble poly(hydroxyl acrylate)s with a
monomodal molecular weight distribution. Nonetheless,
the resulting soluble poly(hydroxy acrylate)s will still
contain unreacted acrylate units in the side chains and,
thus, might be used as reactive intermediates for cross-
linking or further functionalization.
It was reported in the literature that P(DEGEA) exhibits a
cloud point at �9 8C for a concentration of 0.1 wt.-%.[113] To
increase the cloud point of the polymer, copolymerization
with a more hydrophilic monomer is required. Thus, DEGEA
was copolymerized with HEA in different molar ratios: 25,
50, and 75 mol-%, under the same conditions. According to
the same procedure P(2HPA) and P(DHHA) were prepared
starting with the stock solutions obtained after enzymatic
transacylation. Finally, in order to increase the hydro-
phobicity of P(DHHA) direct copolymerization of the MA–
DHHA monomer mixture resulting after the enzymatic
transacylation reaction was performed.
Nitroxide Mediated Polymerization
The nitroxide-mediated polymerization of the new
hydroxy functional acrylates was investigated using
www.MaterialsViews.com
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
the optimized conditions: 110 8C polymerization tempera-
ture and 10 mol-% excess SG-1 free nitroxide relative to the
Blocbuilder alkoxyamine initiator.[114,115] Since the acrylate
monomers have a high propagation rate constant, excess of
free nitroxide is required to reduce the polymerization rate
and retain control over the polymerization. Since
the monomer solutions are viscous due to the presence
of diol/triol that was used in excess during the monomer
synthesis, a certain amount of DMF, which is known to
increase the polymerization rate in NMP,[116] was added to
decrease the viscosity and to prevent the loss of control
over the polymerization due to limited diffusion.
The NMP of the hydroxy acrylates obtained by enzymatic
transacylation was studied taking into account the amount
of bisacrylate present in the monomers. Kinetic investiga-
tions of 3HPA (10 mol-% bisacrylate) and 4HBA (12 mol-%
bisacrylate) revealed controlled polymerizations up to a
monomer conversions of 40%, after which the control is lost
due to coupling reactions caused by the presence of
bisacrylates. In the case of 5,6DHHA (6 mol-% bisacrylate)
and 2Me3HPA (7 mol-% bisacrylate), a controlled NMP was
achieved up to 50% monomer conversion while in the
case of NPGA (containing 5 mol-% bisacrylate) and GA
(containing 3 mol-% bisacrylate) the first order kinetic plots
remained linear up to high conversions. Moreover, for these
two latter monomers, the polymerization rate increased by
a factor of three when the monomer concentration was
doubled which is ascribed to the accelerating hydrogen
bonding effect which was suppressed at lower concentra-
tions in DMF.
The copolymerization kinetics of DEGEA with 25, 50, and
75 mol-% HEA were investigated, too. It was demonstrated
that the monomer composition does not influence the
control over the polymerization. The apparent rate constant
increased by a factor 1.5 for each 25 mol-% of HEA added. An
explanation for this experimental result might be the
interaction of the hydroxyl groups with the nitroxide via
hydrogen bonding which was found to accelerate the
NMP.[117] This effect is more pronounced for the copoly-
merization compared to the HEA homopolymerization due
to the higher monomer concentration. The incorporated
HEA fraction (FHEA) was found to be close to the HEA fraction
in the feed ( fHEA) for all the copolymerizations indicating a
nearly ideal copolymerization of the two monomers.
The copolymerization kinetics of monomer stock
solutions resulting from three enzymatic transacylation
reaction of MA with 1,2,6Htriol performed at the same
reaction conditions but for different times (2, 4, and 8 h)
were investigated and a first conclusion after analyzing the
purified copolymers by 1H NMR spectroscopy is that the
DHHA has a higher reactivity than MA which agrees with
the acceleration of the polymerization rates in NMP due to
hydrogen bonding formed between the monomers and the
initiator.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim569

570
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
Polymer Modification
Via cascade reaction we obtained multifunctional polymers
with hydrophobic, hydrophilic, tertiary amine, and
hydroxy groups. The hydroxy groups are eligible for
esterification with phenyl chloroformate and the tertiary
amine groups for quaternization.
The conversion of HEMA repeating units into PCEMA
repeating units results in highly reactive repeating units;
with primary amines additional functionality can be
introduced. The functional tertiary amino groups
(DMAP(M)A) were quaternized with iodomethane in
methanol solution at room temperature to obtain a
multifunctional cationic polymer with a preferred adsorp-
tion ability to negatively charged surfaces. By these
procedures amphipathic polymers were prepared.[116]
Applications
The multifunctional polyacrylates can be used for
surface coating. The concentration of ionic groups will
determine the strength of adhesion to the surface. Due to
the cooperativity of these effects a good adhesion is
expected. Depending on the ratio of hydrophilic to
hydrophobic groups and the environment, switchable
surfaces may be generated.
In addition, polymers containing hydrophobic and
quaternary ammonium groups are known to have anti-
microbial properties.[118]
Polymers that undergo phase transitions in response to
external stimuli are of great interest. The LCST is the critical
temperature below which a mixture is miscible in all
proportion. This behavior is based on hydrogen bonding
between water molecules and groups of the polymer chain.
The polymers with LCST behavior show a sudden and
mostly reversible change from hydrophilic to hydrophobic
that makes them attractive for application in biotechnol-
ogy, drug delivery systems,[31] tissue engineering,[32] and
biomolecule separation[33,34] but are also of interest for
catalysis.[35] The poly(hydroxy acrylate)s synthesized both
by FRP and NMP following the cascade reaction were
analyzed also for the thermo response in aqueous media. All
observed phase transitions were fully reversible and almost
no hysteresis was observed for the copolymers of DEGEA
and HEA as well as for the homopolymer P(2HPA),
nonetheless, in the case of the copolymers of DHHA and
MA during the cooling process, the rehydration of the
polymer chains seems to be hindered by hydrophobic
interchain interactions, leading to a marked hysteresis.
The hydroxyl groups of HEMA, copolymerized with the
monomer mixture resulting from the enzymatic transacy-
lation, were later converted with phenyl chloroformate into
the reactive phenyl carbonate or para-nitro phenyl
carbonate repeating units. An interesting application of
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
these multifunctional and reactive polymethacrylates is
their ability to react with different peptides/proteins. The
covalent binding of different proteins like silk peptide,
lysozyme, and CALB with the reactive groups attached to
the polymethacrylate back-bone was recently studied.[119]
Thus different bioconjugates were prepared. If in the case of
silk peptide and lysozyme a protein content of 20 and 24%
was determined, in the case of CALB no more than 11% of
protein was determined in the bioconjugate. The bioconju-
gates between CALB and different polymers showed
enzyme activity. An interesting approach for the formation
of peptide/protein-polymer bioconjugates was the use
of a miniemulsion for the reaction of the polymer with
peptides.
Conclusion
The transacylation of MA and MMA with alcohols, diols,
and triols is carried out under mild conditions, is easy and
rapid in processing and suitable for the preparation of
sensitive monomers. The resulting monomers are ready for
polymerization without further purification, thus, a wide
range of multifunctional and reactive poly(meth)acrylates
can be synthesized for different applications like bacterio-
static polymers, thermoresponsive polymers as well as
polymers suitable for the preparation of bioconjugates.
Received: November 17, 2010; Revised: December 22, 2010;Published online: January 31, 2011; DOI: 10.1002/marc.201000725
Keywords: enzymatic transacylation; free radical polymeriza-tion; hydrophilic polyacrylates; monomers; multifunctionalpolyacrylates; nitroxide mediated polymerization; surfaces
[1] K. Takemoto, R. M. Ottenbrite, M. Kamashi, FunctionalMonomers and Polymers, 2nd Edition, Marcel Dekker Inc.,New York 1997, p. 537.
[2] D. C. Webester, Polym. News 1998, 23, 187.[3] K. S. Bagdasar’yan, Theory of Free-Radical Polymerization,
Davey, Hartford CT 1968, p. 317.[4] G. Moad, D. H. Solomon, The Chemistry of Free Radical
Polymerization, Pergamon Press, Tarrytown, NY 1995, p. 315.[5] G. Moad, D. H. Solomon, Aust. J. Chem. 1990, 43, 215.[6] V. Coessens, T. Pintauer, K. Matyjaszewski, Prog. Polym. Sci.
2001, 26, 337.[7] M. Szwarc, Nature 1956, 178, 1168.[8] M. van Beylen, S. Bywater, G. Smets, M. Szwarc, D. J. Wors-
fold, ‘‘Developments in Anionic Polymerization – A CriticalReview’’, in: Polysiloxane Copolymers/Anionic Polymeri-zation, Advances in Polymer Science, Vol. 86, SpringerVerlag,Berlin/Heidelberg 1988, p. 87.
[9] M. Miyamoto, M. Sawamoto, T. Higashimura, Macromol-ecules 1984, 17, 265.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com

Poly(meth)acrylates Obtained by Cascade Reaction
www.mrc-journal.de
[10] P. J. Kennedy, J. Polym. Sci., Part A: Polym. Chem. 1999, 37,2285.
[11] S. Penczek, J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 1919.[12] S. Penczek, M. Cypryk, A. Duda, P. Kubisa, S. Slomkowski,
Prog. Polym. Sci. 2007, 32, 247.[13] T. Endo, ‘‘General Mechanisms in Ring-Opening Polymeri-
zation’’, in: Handbook of Ring-Opening Polymerization (Eds.P. Dubois, O. Coulembier, J. M. Raquez), Wiley-VCH VerlagGmbH & Co. kGaA, Weinheim 2009, p. 53.
[14] H. Hocker, H. Keul, Adv. Mater. 1994, 6, 21.[15] K. Matyjaszewski, Chem.–Eur. J. 1999, 5, 3095.[16] T. E. Patten, K. Matyjaszewski, Acc. Chem. Res. 1999, 32, 895.[17] K. Matyjaszewski, Controlled Radical Polymerization, Amer-
ican Chemical Society, Washington DC 1998.[18] K. Matyjaszewski, J. H. Xia, Chem. Rev. 2001, 101, 2921.[19] J. S. Wang, K. Matyjaszewski, J. Am. Chem. Soc. 1995, 117,
5614.[20] M. Kamigaito, T. Ando, M. Sawamoto, Chem. Rev. 2001, 101,
3689.[21] C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 2001, 101,
3661.[22] S. Armido, S. Tobias, Chem. Rec. 2005, 5, 27.[23] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le,
R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E.Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
[24] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2005, 58, 379.[25] K. Matyjaszewski, J. Panswick, Controlled/Living Radical
Polymerization, Vol. 8, Oxford, United Kingdom 2005, pp.26–33.
[26] K. Matyjaszewski, D. A. Shipp, G. P. McMurtry, S. G. Gaynor,T. Pakula, J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2023.
[27] P. McCarthy, V. Tsarevsky Nicolay, L. Bombalski, K. Maty-jaszewski, C. Pohl, ‘‘Grafting Chromatographic StationaryPhase Substrates by Atom Transfer Radical Polymerization’’,in: Controlled/Living Radical Polymerization, AmericanChemical Society, Washington, DC 2006, p. 252.
[28] R. E. Richard, M. Schwarz, S. Ranade, A. K. Chan, K. Maty-jaszewski, B. Sumerlin, Biomacromolecules 2005, 6, 3410.
[29] H. Mori, H. Iwaya, A. Nagai, T. Endo, Chem. Commun. 2005,38, 4872.
[30] L. D. Taylor, L. D. Cerankowski, J. Polym. Sci., Part A: Polym.Chem. 1975, 13, 2551.
[31] D. Schmaljohann, Adv. Drug Delivery Rev. 2006, 58, 1655.[32] I. Lynch, I. A. Blute, B. Zhmud, P. MacArtain, M. Tosetto, L. T.
Allen, H. J. Byrne, G. F. Farrell, A. K. Keenan, W. M. Gallagher,K. A. Dawson, Chem. Mater. 2005, 17, 3889.
[33] M. D. Costioli, I. Fisch, F. Garret-Flaudy, F. Hilbrig, R. Freitag,Biotechnol. Bioeng. 2003, 81, 535.
[34] A. Kikuchi, T. Okano, Prog. Polym. Sci. 2002, 27, 1165.[35] D. E. Bergbreiter, B. L. Case, Y.-S. Liu, J. W. Caraway, Macro-
molecules 1998, 31, 6053.[36] M. Eberhardt, P. Theato, Macromol. Rapid Commun. 2005, 26,
1488.[37] M. Eberhardt, R. Mruk, R. Zentel, P. Theato, Eur. Polym. J.
2005, 41, 1569.[38] E. G. Gil, S. A. Hudson, Prog. Polym. Sci. 2004, 29, 1173.[39] B. R. Twaites, C. D. Alarcon, D. Cunliffe, M. Lavigne, S.
Pennadam, J. R. Smith, D. C. Gorecki, C. Alexander, J. Con-trolled Release 2004, 97, 551.
[40] J. T. Zhang, Y. N. Xue, F. Z. Gao, S. W. Huang, R. X. Zhuo, J. Appl.Polym. Sci. 2008, 108, 3031.
[41] J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau,S. P. Armes, M. Smallridge, P. McKenna, Macromolecules2004, 37, 2395.
www.MaterialsViews.com
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
[42] M. Save, J. V. M. Weaver, S. P. Armes, P. McKenna, Macro-molecules 2002, 35, 1152.
[43] M. T. Iglesias, J. Guzman, E. Riande, J. Polym. Sci., Part A:Polym. Chem. 1994, 32, 2565.
[44] M. T. Iglesias, J. Guzman, E. Riande, J. Polym. Sci., Part A:Polym. Chem. 1995, 33, 2057.
[45] F. Garcia, J. L. De la Pena, J. J. Delgado, N. Garcia, J. Guzman, E.Riande, P. Calle, J. Polym. Sci., Part A: Polym. Chem. 2001, 39,1843.
[46] B. A. Hill, J. Chem. Soc. -Trans. 1898, 634.[47] A. M. Klibanov, Chemtech 1986, 16, 354.[48] M. Iso, B. X. Chen, M. Eguchi, T. Kudo, S. Shrestha, J. Mol.
Catal. B: Enzym. 2001, 16, 53.[49] W. Du, Y. Y. Xu, D. H. Liu, Z. B. Li, J. Mol. Catal. B: Enzym. 2005,
37, 68.[50] S. Kobayashi, H. Uyama, S. Kimura, Chem. Rev. 2001, 101,
3793.[51] R. A. Gross, A. Kumar, B. Kalra, Chem. Rev. 2001, 101, 2097.[52] F. Binns, P. Harffey, S. M. Roberts, A. Taylor, J. Chem. Soc. -
Perkin Trans. 1 1999, 19, 2671.[53] S. Kobayashi, A. Makino, Chem. Rev. 2009, 109, 5288.[54] O. Kirk, M. W. Christensen, Org. Proc. Res. Dev. 2002, 6, 446.[55] J. Uppenberg, M. T. Hansen, S. Patkar, T. A. Jones, Structure
1994, 2, 293.[56] A. Heise, C. J. Duxbury, A. R. A. Palmans, ‘‘Enzyme-Mediated
Ring-Opening Polymerization’’, in: Handbook of Ring-Open-ing Polymerization (Eds. P. Dubois, O. Coulembier, J. M.Raquez), Wiley-VCH Verlag GmbH & Co. kGaA, Weinheim2009, p. 379.
[57] S. Bloomer, P. Adlercreutz, B. Mattiasson, Enzyme Microb.Technol. 1992, 14, 546.
[58] Z. Y. Li, O. P. Ward, J. Am. Oil Chem. Soc. 1993, 70, 745.[59] M. Y. Sen, J. E. Puskas, S. Ummadisetty, J. P. Kennedy,
Macromol. Rapid Commun. 2008, 29, 1598.[60] S. Ramaswamy, B. Morgan, A. C. Oehlschlager, Tetrahedron
Lett. 1990, 31, 3405.[61] X. F. Li, M. H. Zong, R. D. Yang, J. Mol. Catal. B: Enzym. 2006,
38, 48.[62] A. W.-Y. Chan, B. Ganem, Biocatal. Biotransform. 1993, 8, 163.[63] A. D. Ghogare, G. S. Kumar, J. Chem. Soc. 1989, 1533.[64] I. Ikeda, J. Tanaka, K. Suzuki, Tetrahedron Lett. 1991, 32, 6865.[65] A. B. Hajjar, P. F. Nicks, C. J. Knowles, Biotechnol. Lett. 1990,
12, 825.[66] R. Tor, Y. Dror, A. Freeman, EnzymeMicrob. Technol. 1990, 12,
299.[67] R. Derango, Y. F. Wang, R. Dowbenko, L. C. Chiang, Biotechnol.
Lett. 1994, 16, 241.[68] S. Warwel, G. Steinke, A. Rusch, M. R. Klaas, Biotechnol.
Technol. 1996, 10, 283.[69] V. Athawale, N. Manjrekar, M. Athawale, Tetrahedron Lett.
2002, 43, 4797.[70] V. Athawale, N. Manjrekar, J. Mol. Catal. B:-Enzym. 2000, 10,
551.[71] X. Ding, H. X. Yue, I. Ikeda, K. Suzuki, Sen’i Gakkaishi 1996, 52,
524.[72] US 0048927 (2010), BASF AG, invs.: H. Dietmar, M. Uwe, C.
Mathieu, L. Gunter.[73] US 5240835 (1993), Genencor International Inc., invs.: A. F.
Petrone, J. P. Grisdale, M. G. G. Whited, C. T. Paulson.[74] US 0084779 (2006), BASF AG, invs.: F. Dietsche, D. Haering, E.
Wagner, R. Schwalm, H.-P. Rink, E. Beck.[75] US 0009589 (2006), BASF AG, invs.: D. Haering, U. Meisen-
burg, B. Hauer.[76] US 0062500 (2010), BASF SE, invs.: D. Haering.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim571

572
www.mrc-journal.de
D. Popescu, H. Keul, M. Moeller
[77] US 0200631 (2008), invs.: D. Haering, U. Meisenburg, B.Hauer, F. Dietsche.
[78] US 0035341 (2006), BASF AG, invs.: D. Boeckh, B. Hauer, D.Haering.
[79] A. R. A. Palmans, A. Heise, Adv. Polym. Sci. 2011, 237, 79.[80] U. Meyer, A. R. A. Palmans, T. Loontjens, A. Heise, Macro-
molecules 2002, 35, 2873.[81] M. De Geus, L. Schormans, A. R. A. Palmans, C. E. Koning,
A. Heise, J. Polym. Sci., Part A: Polym. Chem. 2006, 44,4290.
[82] J. Peeters, A. R. A. Palmans, M. Veld, F. Scheijen, A. Heise, E. W.Meijer, Biomacromolecules 2004, 5, 1862.
[83] B. A. C. van As, P. Thomassen, B. Kalra, R. A. Gross, E. W.Meijer, A. R. A. Palmans, A. Heise, Macromolecules 2004, 37,8973.
[84] J. Peeters, A. R. A. Palmans, E. W. Meijer, C. E. Koning, A. Heise,Macromol. Rapid Commun. 2005, 26, 684.
[85] B. Yu, A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem. 2009, 47,1877.
[86] K. S. Karmee, C. Roosen, C. Kohlmann, S. Lutz, L. Greiner, W.Leitner, Green Chem. 2009, 11, 1052.
[87] R. D. Patil, S. J. Dordick, G. D. Rethwisch, Macromolecules1991, 24, 3462.
[88] D. B. Martin, A. S. Ampofo, J. R. Linhardt, S. Dordick, J.Macromol. 1992, 25, 7081.
[89] X. Chen, A. Johnson, S. J. Dordick, G. D. Rethwisch, Macromol.Chem. Phys. 1994, 195, 3567.
[90] G. H. Park, H. N. Chang, Biotechnol. Lett. 2000, 22, 39.[91] P. Potier, A. Bouchu, G. Descotes, Y. Queneau, Tetrahedron
Lett. 2000, 41, 3597.[92] J. Xie, Y.-L. Hsieh, J. Polym. Sci., Part A: Polym. Chem. 2001, 39,
1931.[93] L. Ubaghs, B. Sharma, H. Keul, H. Hocker, T. Loontjens, R. van
Benthem, e-Polymers 2003, 68, 1.[94] L. Ubaghs, N. Fricke, H. Keul, H. Hocker, Macromol. Rapid
Commun. 2004, 25, 517.[95] N. Pasquier, H. Keul, M. Moeller, Des. Monomers Polym. 2005,
8, 679.[96] D. Popescu, H. Keul, M. Moeller, Macromol. Chem. Phys. 2009,
210, 123.[97] M. M. Soumanou, U. T. Bornscheuer, Enzyme Microb. Tech-
nol. 2003, 33, 97.[98] W. Du, L. Wang, D. H. Liu, Green Chem. 2007, 9, 173.
Macromol. Rapid Commun
� 2011 WILEY-VCH Verlag Gmb
[99] D. Popescu, R. Hoogenboom, H. Keul, M. Moeller, J. Polym.Sci., Part A: Polym. Chem. 2010, 48, 2610.
[100] D. Popescu, R. Hoogenboom, H. Keul, M. Moeller, Polym.Chem. 2010, 1, 878.
[101] S. Chand, P. Adlercreutz, B. Mattiasson, Enzyme Microb.Technol. 1997, 20, 102.
[102] J. F. Kunzler, J. A. McGee, Chem. Ind. 1995, 16, 651.[103] M. Kurisawa, M. Yokoyama, T. Okano, J. Controlled Release
2000, 69, 127.[104] M. Hans, H. Keul, A. Heise, M. Moeller, Macromolecules 2007,
40, 8872.[105] M. Ferrer, J. Soliveri, F. J. Plou, N. Lopez-Cortes, D. Reyes-
Duarte, M. Christensen, J. L. Copa-Patino, A. Ballesteros,Enzyme Microb. Technol. 2005, 36, 391.
[106] J. E. Puskas, M. Y. Sen, J. R. Kasper, J. Polym. Sci., Part A: Polym.Chem. 2008, 46, 3024.
[107] Y. Watanabe, Y. Shimada, A. Sugihara, H. Noda, H. Fukuda, Y.Tominaga, J. Am. Oil Chem. Soc. 2000, 77, 355.
[108] K. L. Deng, H. Tian, P. F. Zhang, X. B. Ren, H. B. Zhong, ExpressPolym. Lett. 2009, 3, 97.
[109] I. Hilker, G. Rabani, G. K. M. Verzijl, A. R. A. Palmans, A. Heise,Angew. Chem., Int. Ed. 2006, 45, 2130.
[110] A. O. Magnusson, M. Takwa, A. Hamberg, K. Hult, Angew.Chem., Int. Ed. 2005, 44, 4582.
[111] V. Leonard, L. Fransson, S. Lamare, K. Hult, M. Graber,Chembiochem 2007, 8, 662.
[112] V. Leonard-Nevers, Z. Marton, S. Lamare, K. Hult, M. Graber,J. Mol. Catal. B: Enzym. 2009, 59, 90.
[113] K. Skrabania, J. Kristen, A. Laschewsky, A. z. r. Akdemir, A.Hoth, J.-F. o. Lutz, Langmuir 2006, 23, 84.
[114] R. Hoogenboom, D. Popescu, W. Steinhauer, H. Keul, M.Moller, Macromol. Rapid Commun. 2009, 30, 2042.
[115] T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R.Hoogenboom, U. S. Schubert, Macromolecules 2008, 41,5132.
[116] B. Kejian, F. C. Michael, J. Polym. Sci., Part A: Polym. Chem.2006, 44, 414.
[117] E. Harth, B. V. Horn, C. J. Hawker, Chem. Commun. 2001, 9,823.
[118] R. Adelmann, M. Mennicken, D. Popescu, E. Heine, H. Keul,M. Moeller, Eur. Polym. J. 2009, 45, 3093.
[119] D. Popescu, H. Keul, M. Moeller, React. Funct. Polym. 2010, 70,767.
. 2011, 32, 559–572
H & Co. KGaA, Weinheim www.MaterialsViews.com