polydatin attenuates cardiac hypertrophy through...
TRANSCRIPT

Polydatin attenuates cardiac hypertrophy through modulation of cardiacCa2� handling and calcineurin-NFAT signaling pathway
Wenwen Ding,2* Ming Dong,1* Jianxin Deng,2 Dewen Yan,3 Yun Liu,1 Teng Xu,1 and Jie Liu1,2
1Department of Pathophysiology, School of Medicine, Shenzhen University, Shenzhen, China; 2Department ofPathophysiology, Southern Medical University, Guangzhou, China; and 3Department of Endocrinology, The First AffiliatedHospital of Shenzhen University, Shenzhen, China
Submitted 14 January 2014; accepted in final form 20 June 2014
Ding W, Dong M, Deng J, Yan D, Liu Y, Xu T, Liu J. Polydatinattenuates cardiac hypertrophy through modulation of cardiac Ca2�
handling and calcineurin-NFAT signaling pathway. Am J PhysiolHeart Circ Physiol 307: H792–H802, 2014. First published July 11,2014; doi:10.1152/ajpheart.00017.2014.—Polydatin (PD), a resvera-trol glucoside extracted from the perennial herbage Polygonum cus-pidatum, has been suggested to have wide cardioprotective effects.This study aimed to explore the direct antihypertrophic role of PD incultured neonatal rat ventricular myocytes (NRVMs) and its thera-peutic effects against pressure overload (PO)-induced hypertrophicremodeling and heart failure. Furthermore, we investigated the mech-anisms underlying the actions of PD. Treatment of NRVMs withphenylephrine for 72 h induced myocyte hypertrophy, where the cellsurface area and protein levels of atrial natriuretic peptide and �-my-osin heavy chain (�-MHC) were significantly increased. The ampli-tude of systolic Ca2� transient was increased, and sarcoplasmicreticulum Ca2� recycling was prolonged. Concomitantly, calcineurinactivity was increased and NFAT protein was imported into thenucleus. PD treatment restored Ca2� handling and inhibited calcineu-rin-NFAT signaling, thus attenuating the hypertrophic remodeling inNRVMs. PO-induced cardiac hypertrophy was produced by trans-verse aortic constriction (TAC) in C57BL/6 mice, where the leftventricular posterior wall thickness and heart-to-body weight ratiowere significantly increased. The cardiac function was increased at 5wk of TAC, but significantly decreased at 13 wk of TAC. Theamplitude of Ca2� transient and calcineurin activity were increased at5 wk of TAC. PD treatment largely abolished TAC-induced hyper-trophic remodeling by inhibiting the Ca2�-calcineurin pathway. Sur-prisingly, PD did not inhibit myocyte contractility despite that theamplitude of Ca2� transient was decreased. The cardiac functionremained intact at 13 wk of TAC. In conclusion, PD is beneficialagainst PO-induced cardiac hypertrophy and heart failure largelythrough inhibiting the Ca2�-calcineurin pathway without compro-mising cardiac contractility.
polydatin; hypertrophy; heart failure; calcium transient; calcineurin;NFAT; transverse aortic constriction
CARDIAC HYPERTROPHY is an adaptive response most commonlyto hypertension and an independent risk factor for the devel-opment of heart failure and more generally an increased mor-bidity and mortality (1, 18). At the cellular level, enlargementof the cardiomyocyte involves multiple events, including genetranscription and protein translation/synthesis, which are reg-ulated by multiple signaling cascades. Among them, enhance-ment of calcium-regulated signaling pathways plays a majorrole in the development of pathological cardiac hypertrophy
(15, 26, 33). Accordingly, manipulation of cardiac Ca2� sig-naling represents a logical approach to treatment of cardiachypertrophy.
Cardiac Ca2� signaling is a delicate interplay of multipleCa2� handling proteins (2, 3, 12, 31). At systole, the activationof L-type Ca2� channel (LTCC) allows extracellular Ca2�
influx into cardiomyocytes upon membrane depolarization,triggering sarcoplasmic reticulum (SR) Ca2� release via Ca2�-induced Ca2� release (CICR) mechanism. At diastole, theincreased intracellular Ca2� is sequestered into the SR by theSR Ca2�-ATPase (SERCA) and extruded from cytoplasm bythe sodium/calcium (Na�/Ca2�) exchanger and sarcolemmalCa2�-ATPase. The activity of LTCC can be inhibited by Ca2�
channel blockers, which have been confirmed to be therapeu-tically effective for the treatment of cardiac hypertrophy inmany animal models (14, 29). However, clinical blockade ofLTCC has dubious benefit on cardiovascular mortality, despitethat calcium channel antagonists induce regression of hyper-trophy (19, 37). Given the essential role of intracellular Ca2�
in cardiac contraction, blockade of LTCC would decreasecardiac contractile function. Furthermore, blockade of noncar-diac Ca2� channels induces side effects, including systemichypotension, constipation, edema, etc. (7, 16). All these short-comings of LTCC antagonists counteract their beneficial ef-fects on the treatment of cardiac hypertrophy. Thus new agents(drugs) that can suppress cardiac Ca2� signaling without caus-ing the side effects are highly expected.
Polydatin (PD), a resveratrol glucoside with a 3,4=,5-trihy-droxystilben-3-�-D-mono-D-glucoside molecular structure, is anatural component extracted from the perennial herbage Poly-gonum cuspidatum Sieb. et Zucc (5, 10). So far, there is onlyone study reporting that PD attenuated ventricular remodelingin vivo by inhibiting the activation of neurohormone (13).Whether PD has a direct antihypertrophic effect and whether itmodifies the heart failure process in vivo remain unknown.Recently, we found that PD modulated Ca2� handling andcardiac contractility. When acutely applied to isolated ratventricular myocytes, PD significantly decreased LTCC cur-rent (ICa,L) and moderately decreased systolic Ca2� transient.Interestingly, PD slightly increased rather than decreasingcardiac contractility (8). The findings in normal cardiomyo-cytes lead us to the hypothesis that PD suppresses hypertrophicstimuli-induced upregulation of Ca2� signaling and thus exertsa direct antihypertrophic effect, without compromising cardiaccontractile function. If the hypothesis is true, combining withprevious findings that PD did not reduce blood pressure andinterfere with systemic hemodynamics in normal animals (30,38), it is rational to speculate that PD is a good candidate drugfor the treatment of cardiac hypertrophy.
* W. Ding and M. Dong contributed equally to this work.Address for reprint requests and other correspondence: J. Liu, School of
Medicine, Shenzhen Univ., Shenzhen, Guangdong 518000, China (e-mail:[email protected]).
Am J Physiol Heart Circ Physiol 307: H792–H802, 2014.First published July 11, 2014; doi:10.1152/ajpheart.00017.2014.
0363-6135/14 Copyright © 2014 the American Physiological Society http://www.ajpheart.orgH792
To test this hypothesis, we explored the therapeutic effect ofPD on attenuation of hypertrophy in cultured neonatal ratventricular myocytes stimulated with phenylephrine (PE) andin a pressure overload (PO)-induced hypertrophic mousemodel. Furthermore, we investigated the effects of PD oncardiac Ca2� handling and contractility, as well as the Ca2�/calmodulin-activated calcineurin-nuclear factor of activated Tcells (NFAT) signaling pathway, which has been shown to playa major role in the development of pathological cardiac hyper-trophy (6, 24, 25, 34). Here, we demonstrated that PD prohib-ited the development of cardiac hypertrophy in vitro as well asin vivo by inhibiting the Ca2� signal and calcineurin-NFATsignaling pathway.
METHODS
Animals. Animals were purchased from the Animal Center ofSouthern Medical University and handled according to a protocolapproved by the Institutional Care and Use Committee of ShenzhenUniversity that conforms to the Guide for the Care and Use ofLaboratory Animals published by the National Institutes of Health(NIH Publication No. 85–23, revised 1996).
Culture of neonatal rat ventricular myocytes (NRVMs) and treat-ment protocol. Neonatal rat ventricular myocytes (NRVMs) wereisolated from 2-day-old Sprague-Dawley rats and cultured as previ-ously described (23). In brief, hearts were obtained following decap-itation and immersed in PBS and minced with scissors. The smallpieces of heart tissue were digested with 0.25% trypsin-EDTA in PBS
at 37°C. The isolated cells were put in fetal bovine serum (FBS) andpelleted by centrifugation at 1,000 rpm for 5 min. The pelleted cellswere resuspended in DMEM containing 10% FBS, 1% penicillin-streptomycin, and then preplated for 30 min at 37°C to allow fibro-blasts to adhere to the plate. The unadhered cells were pelleted againand resuspended in DMEM containing 10% FBS, 1% penicillin-streptomycin, and bromodeoxyuridine (1:100, to inhibit fibroblastgrowth), which were finally plated at a concentration of about 1million cells per 35-mm plate. To produce hypertrophy, NRVMs weretreated with 20 �M phenylephrine (PE, Sigma) for 72 h after 24 h ofserum starvation in the absence or presence of PD (25–75 �M). PDwith a purity of 98.87% was kindly provided by Haiwang (Shenzhen,Guangdong, China).
Measurement of cell surface area. The surface area of a singlemyocyte was measured with Image-Pro Plus Data Analysis Pro-gram (Media Cybernetics, Silver Spring, MD). Quantification ofcell surface area was performed by measuring 50 random cellsfrom three experiments, and the average value was used foranalysis.
Calcineurin enzymatic assay. Calcineurin activity was determinedin myocyte extracts using a colorimetric Calcineurin Cellular ActivityAssay Kit following the manufacturer’s instructions (Enzo Life Sci-ences) (21). Calcineurin activity was determined as nanomoles phos-phate released at 620 nm using a SpectraMax 5 (Molecular Devices,Sunnyvale, CA) plate reader.
Western blotting. Cell lysates were resolved in SDS polyacryl-amide gel electrophoresis (PAGE) and transferred to polyvinylidenefluoride (PVDF) membranes (Millipore). Nuclear protein extraction
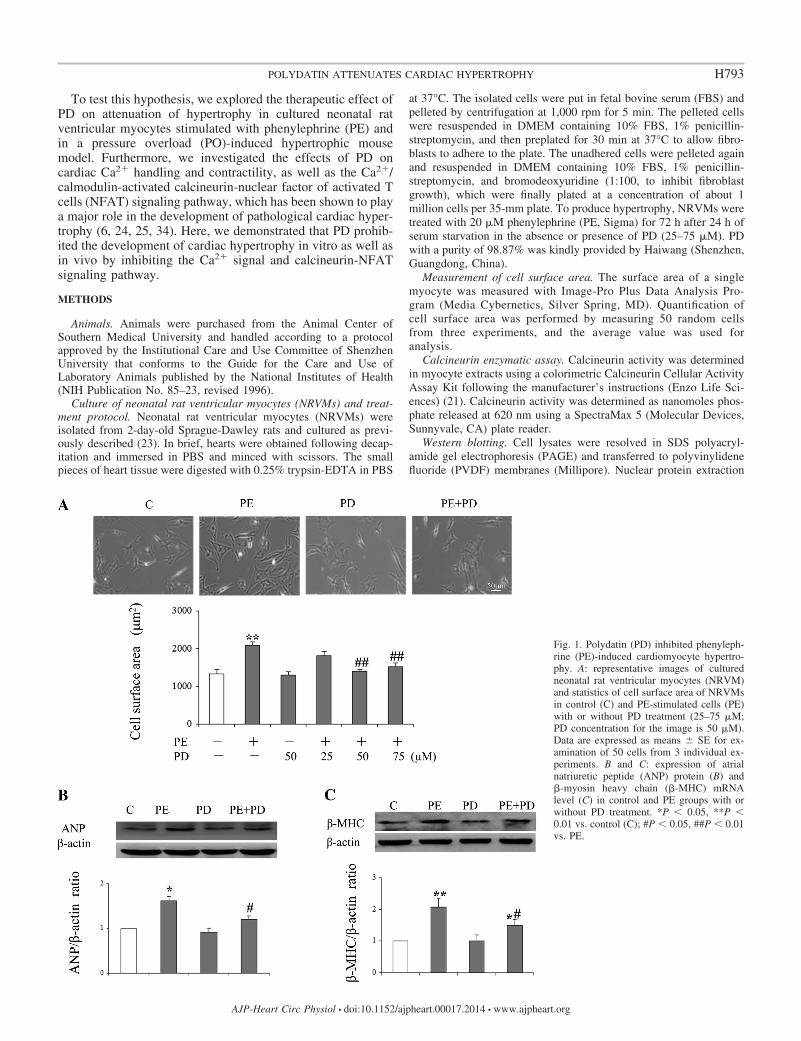
Fig. 1. Polydatin (PD) inhibited phenyleph-rine (PE)-induced cardiomyocyte hypertro-phy. A: representative images of culturedneonatal rat ventricular myocytes (NRVM)and statistics of cell surface area of NRVMsin control (C) and PE-stimulated cells (PE)with or without PD treatment (25–75 �M;PD concentration for the image is 50 �M).Data are expressed as means � SE for ex-amination of 50 cells from 3 individual ex-periments. B and C: expression of atrialnatriuretic peptide (ANP) protein (B) and�-myosin heavy chain (�-MHC) mRNAlevel (C) in control and PE groups with orwithout PD treatment. *P � 0.05, **P �0.01 vs. control (C); #P � 0.05, ##P � 0.01vs. PE.
H793POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
was performed using a Nuclear and Cytoplasmic Protein Extractionkit according to the manufacturer’s instruction (Pierce, Thermo Sci-entific). Ventricular tissue proteins were obtained by homogenizingwith lysis buffer. Target proteins were reacted with their respectiveantibodies, atrial natriuretic peptide (ANP; Santa Cruz Biotechnol-
ogy), �-MHC (Santa Cruz Biotechnology), calcineurin (Abcam) (28),LaminA/C (Cell Signaling Technology), NFAT3 (Cell SignalingTechnology), �-AMPK (Cell Signaling Technology), p-�-AMPK(Cell Signaling Technology),and then incubated with respective sec-ondary antibody. �-Actin (ZSGB-Bio, China) or GAPDH (Santa Cruz
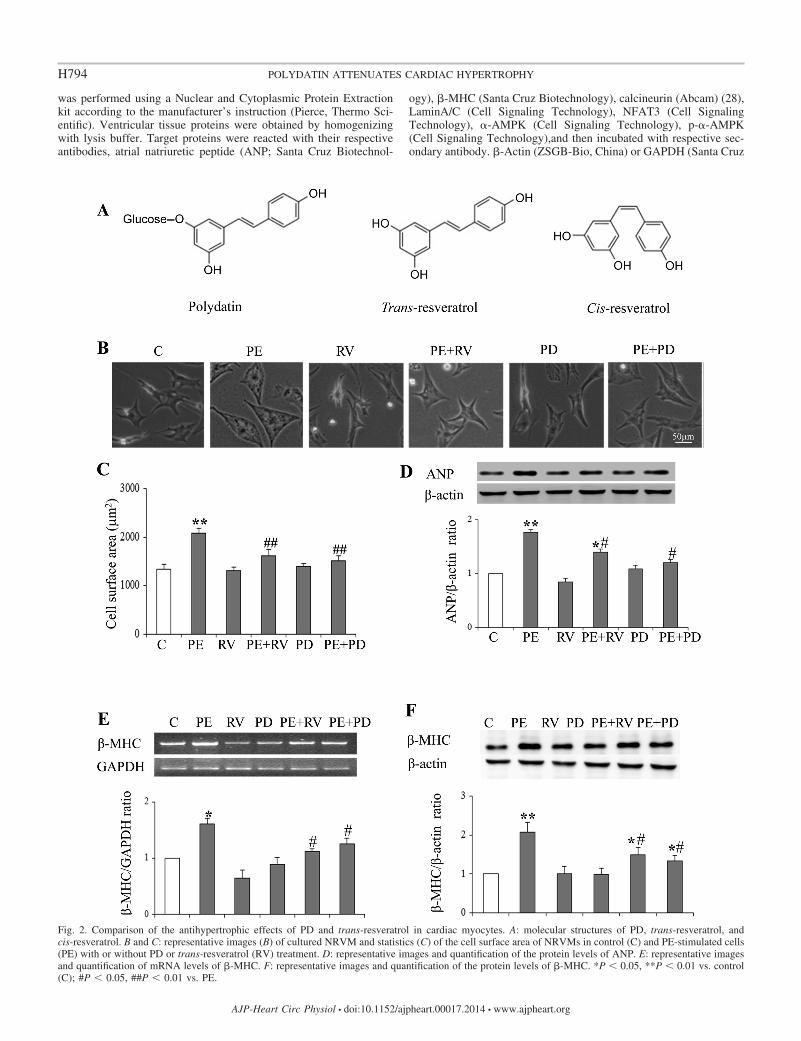
Fig. 2. Comparison of the antihypertrophic effects of PD and trans-resveratrol in cardiac myocytes. A: molecular structures of PD, trans-resveratrol, andcis-resveratrol. B and C: representative images (B) of cultured NRVM and statistics (C) of the cell surface area of NRVMs in control (C) and PE-stimulated cells(PE) with or without PD or trans-resveratrol (RV) treatment. D: representative images and quantification of the protein levels of ANP. E: representative imagesand quantification of mRNA levels of �-MHC. F: representative images and quantification of the protein levels of �-MHC. *P � 0.05, **P � 0.01 vs. control(C); #P � 0.05, ##P � 0.01 vs. PE.
H794 POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
Biotechnology) was used as a loading control for ANP and calcineurinproteins, and LaminA/C as a loading control for NFAT3 nuclearprotein.
Determination of �-MHC mRNA level. Total RNA was extractedusing TRIZOL (Invitrogen) according to the manufacturer’s instruc-tions. The mRNA level of �-MHC was determined with reversetranscription-polymerase chain reaction (RT-PCR) with GAPDH asan internal control. PCR conditions to amplify �-MHC and GAPDHgenes were as follows: denaturation at 94°C for 30 s, followed byannealing at 57°C for 30 s and elongation at 72°C for 1 min. Bothgenes were amplified for 35 cycles. The sequences of �-MHC primerswere forward 5=-TCGTGGAGCGCGCAACAAC-3= and reverse 5=-TCAAAGGCTCCAGGTCTCAGGGCT-3=. The sequences ofGAPDH primers were forward 5=-GCATGTCAGATCCACAA-CGG-3= and reverse 5=-GCATGTCAGATCCACAACGG-3=.
Transverse aortic constriction (TAC). C57BL/6 male mice (9–10wk, 18–22 g) were randomly divided into four groups: sham and TACwith or without PD treatment, 10 in each group. The TAC model wasproduced as described previously (22). In brief, the animal wasendotracheally intubated under anesthesia with a mixture of pento-barbital sodium (50 mg/kg ip). The chest cavity was entered in thesecond intercostal space at the left upper sternal border, and thetransverse aorta between the carotid arteries was isolated and con-stricted by a 7-0 silk suture ligature tied firmly against a 26-gaugeneedle. The latter was promptly removed to yield a constriction of0.45 mm in diameter. Sham-operated mice underwent a similarsurgical procedure without constriction of the aorta. PD was admin-istered by oral gavage (50 mg·kg�1·day�1) 3 days after the surgery fora period of 9 wk.
Two-dimensional guided M-mode echocardiography. Two-dimen-sional (2-D) guided M-mode echocardiography was performed inanesthetized mice (with 1.5% isoflurane) using a Vevo 2100 system(VisualSonics, Toronto, Ontario, Canada). The heart was imaged inthe 2-D mode in the parasternal short-axis view. From this view, thefollowing parameters were measured: percentage of left ventricular(LV) fractional shortening (LVFS), LV ejection fraction (EF), LV
internal dimensions at both diastole and systole (LVIDd and LVIDs,respectively), and LV posterior wall dimensions at both diastole andsystole (LVPWd and LVPWs, respectively). All measurements weredone from leading edge to leading edge according to the AmericanSociety of Echocardiography guidelines. The percentage of LVFS (%)was calculated as [(LVIDd � LVIDs)/LVIDd] 100, and EF (%)was calculated as [(LVIDd2 � LVIDs2)/LVIDd2] 100.
Isolation of adult mouse ventricular myocytes. Ventricular myo-cytes were isolated from anesthetized C57BL/6 mice at 5 wk ofoperation as described previously (39). Briefly, the heart was quicklyremoved from the chest and cleaned and flushed with a Ca2� freebuffer containing (in mM) 120 NaCl, 5.4 KCl, 1.2 MgSO4, 1.2NaH2PO4, 5.6 glucose, 20 NaHCO3, 10 2,3-butanedione monoxime(BDM; Sigma), and 5 taurine, 10 HEPES (pH 7.4), and perfusedusing a Langendorff apparatus. All solutions were bubbled with 100%O2. The enzymatic digestion was initiated by adding collagenase typeB (0.75 mg/ml; Worthington), and protease type XIV (0.02 mg/ml;Sigma) to the perfusion solution. When the heart became swollen andhard after 3 min of digestion, 50 �M Ca2� was added to the enzymesolution and perfused for about 30 min. Following the perfusionprocedure, the heart was minced into small chunks, and single cellswere shaken loose from the heart tissue and stored in HEPES-bufferedsolution containing (mM) 1 CaCl2, 137 NaCl, 5.4 KCl, 15 dextrose,1.3 MgSO4, 1.2 NaH2PO4, and 20 HEPES, adjusted to pH 7.4 withNaOH. Cells were used for the following experiments within 4 h afterisolation.
Ca2� transient detection and contraction measurement. Cellsloaded with Ca2� indicator fluo-4 AM (5 mmol/l, for 8 min) (Invit-rogen) were placed in a recording chamber. Confocal line-scan im-aging was carried out in cells at 488 nm excitation and 505 nmcollection with a Zeiss 710 inverted confocal microscope (Carl Zeiss,Oberkochen, Germany) with 40 oil immersion lens (NA 1.3). Afterthe cells were stimulated with field stimulation (1 Hz) to reach asteady state, confocal line-scan imaging was acquired at a samplingrate of 3.84 ms per line. Myocyte contraction was measured bydetecting the length of two edges of the cell along with the time of
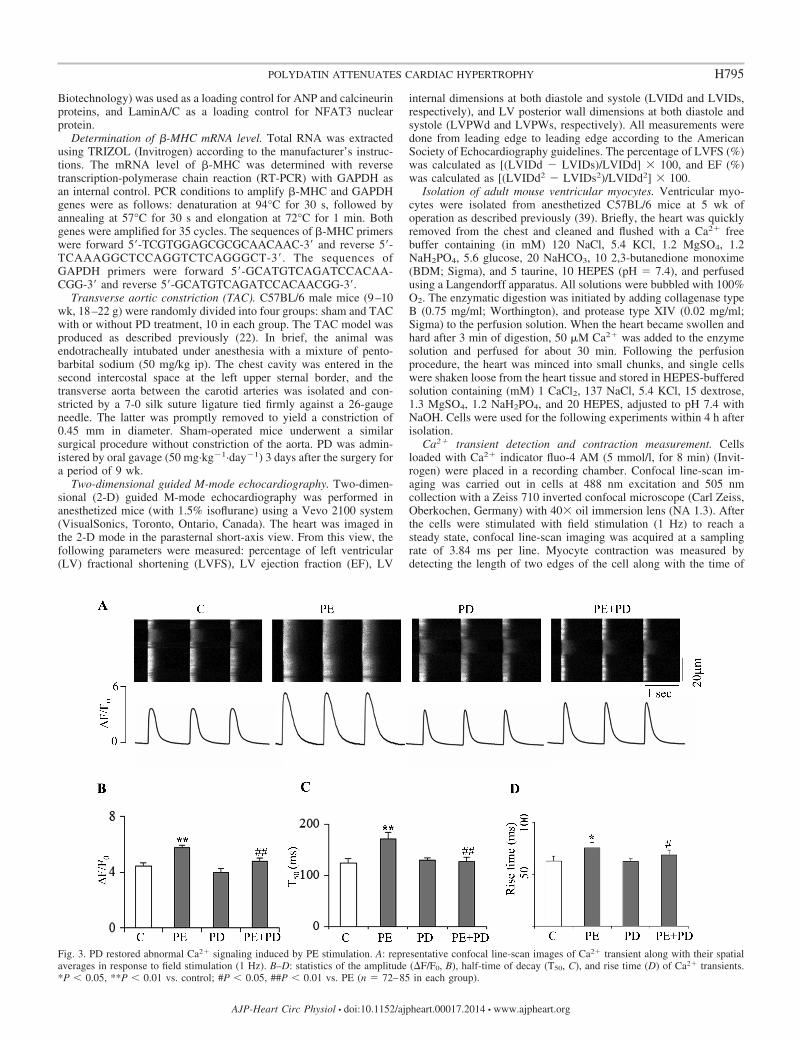
Fig. 3. PD restored abnormal Ca2� signaling induced by PE stimulation. A: representative confocal line-scan images of Ca2� transient along with their spatialaverages in response to field stimulation (1 Hz). B–D: statistics of the amplitude (�F/F0, B), half-time of decay (T50, C), and rise time (D) of Ca2� transients.*P � 0.05, **P � 0.01 vs. control; #P � 0.05, ##P � 0.01 vs. PE (n 72–85 in each group).
H795POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
stimulation. Myocytes were superfused with HEPES-buffered exter-nal solution during the experiment.
Statistical analysis. All values were expressed as means � SE.Statistical analysis was performed by one-way analysis of variance
(ANOVA) for multiple comparisons, followed by the Dunnett’s test toevaluate the difference between two groups through the software ofSPSS version 13.0. Values of P � 0.05 were considered statisticallysignificant.
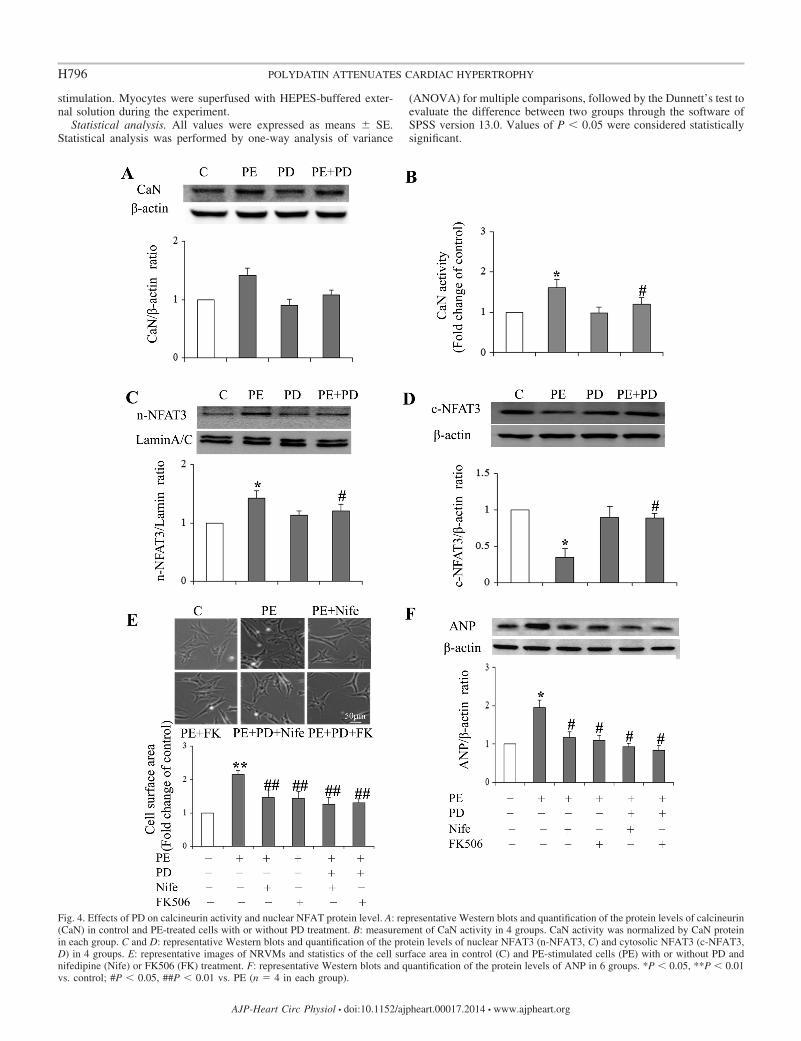
Fig. 4. Effects of PD on calcineurin activity and nuclear NFAT protein level. A: representative Western blots and quantification of the protein levels of calcineurin(CaN) in control and PE-treated cells with or without PD treatment. B: measurement of CaN activity in 4 groups. CaN activity was normalized by CaN proteinin each group. C and D: representative Western blots and quantification of the protein levels of nuclear NFAT3 (n-NFAT3, C) and cytosolic NFAT3 (c-NFAT3,D) in 4 groups. E: representative images of NRVMs and statistics of the cell surface area in control (C) and PE-stimulated cells (PE) with or without PD andnifedipine (Nife) or FK506 (FK) treatment. F: representative Western blots and quantification of the protein levels of ANP in 6 groups. *P � 0.05, **P � 0.01vs. control; #P � 0.05, ##P � 0.01 vs. PE (n 4 in each group).
H796 POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
RESULTS
PD inhibited phenylephrine (PE)-induced cardiomyocytehypertrophy. The in vitro cardiomyocyte hypertrophic modelwas successfully produced by treating the cultured neonatal ratventricular myocytes (NRVMs) with 20 �M PE for 3 days, inwhich the cell surface area was significantly increased by56.5 � 6.2% (2,089 �m2 in PE-treated cells vs. 1,334 �m2 incontrol, P � 0.01, Fig. 1A), and the protein levels of ANP and�-MHC were remarkably increased (Fig. 1, B and C). PDtreatment alone had no significant effects on the hypertrophicparameters observed. However, PD treatment (20–75 �M)dose-dependently reduced PE-induced hypertrophic remodel-ing. PD at the concentration of 50 �M caused significantinhibition of PE-induced enlargement of cell size (Fig. 1A). Inparallel, the increased protein levels of ANP and �-MHC werelargely reduced by 50 �M PD treatment (Fig. 1, B and C).
PD is an analog of trans-resveratrol. Figure 2A demonstratesthe molecular structures of PD, trans-resveratrol and cis-resveratrol. Previous studies have reported that resveratrol cansignificantly inhibit myocyte hypertrophy (9, 17). We thuscompared the effects of PD and trans-resveratrol (RV) on thehypertrophic parameters, including cell surface area and pro-tein levels of ANP and �-MHC, in PE-stimulated NRVMs andfound that the effect of PD (50 �M) on suppression of myocytehypertrophy was comparable to that of RV (50 �M, Fig. 2,B–F).
Effects of PD on PE modulation of cardiac Ca2� signaling.Although a previous study demonstrated PD acutely modulatedCa2� handling in isolated adult rat cardiomyocytes (8), itremains unknown how PD regulates the abnormal Ca2� han-dling induced by pathological PE stimulation. We thus inves-tigated action potential (AP)-elicited Ca2� transient in PE-stimulated cells with or without PD treatment. As shown inFig. 3, A and B, PE stimulation significantly increased theamplitude of AP-elicited Ca2� transient indexed by �F/F0. Incontrast to accelerating SR Ca2� recycling by acute applicationof PE, chronic application of PE (for 72 h) slowed SR Ca2�
recycling, where the half time decay (T50) of Ca2� transientwas prolonged (Fig. 3C). The result indicates that the SERCAfunction was impaired. Moreover, sustained PE stimulationincreased the rise time of Ca2� transient, suggesting the syn-chrony of intracellular Ca2� release was impaired (Fig. 3D).Consistent with the previous study, PD moderately decreasedthe amplitude of Ca2� transient in normal cells by 12.4%.However, PD treatment (50 �M) significantly decreased theamplitude of Ca2� transient which was increased by PE stim-ulation (by 20.7%, Fig. 3, A and B). Furthermore, PD treatmentnormalized the rise time and T50 of Ca2� transient prolongedby PE stimulation, indicating the restoration of the synchronyof intracellular Ca2� release and SR Ca2� recycling (Fig. 3, Cand D). The data collectively indicate that PD corrects abnor-mal Ca2� handling induced by long-term PE stimulation.
PD suppressed PE-activated calcineurin-NFAT signalingpathway. It is well established that an increase in intracellularCa2� activates calcineurin-NFAT signaling pathway, whichplays a major role in the pathogenesis of cardiac hypertrophy.The above finding suggests that PE activates this hypertrophicpathway and PD may inhibit it. As anticipated, PE significantlyincreased calcineurin activity by 60.8 � 7.2% (P � 0.05)without significantly changing the protein expression (P �
0.05, Fig. 4, A and B). Concomitantly, NFAT transferred intonuclear, whereby the nuclear NFAT protein level was signifi-cantly increased by 50.3 � 4.6% (P � 0.01) with PE stimu-lation (Fig. 4C), but cytosolic protein level of NFAT wassignificantly decreased (Fig. 4D). PD treatment (50 �M) hadno significant effect on calcineurin activity and NFAT nuclearprotein level in control cells. In contrast, PD significantlyreduced PE-induced upregulation of calcineurin activity andNFAT nuclear import (Fig. 4, B and C). The results implicatethat PD inhibits the activation of the calcineurin-NFAT signal-ing pathway, contributing to PE-induced myocyte hypertrophy.
To further confirm that PD attenuated myocyte hypertrophythrough inhibition of the Ca2�-calcineurin signaling pathway,we cotreated PE-stimulated cells with PD and nifedipine (20�M), the LTCC blocker, or FK506 (200 nM), the calcineurininhibitor. The results show that nifedipine and FK506 treat-ment alone significantly inhibited PE-induced increase of cellsize and ANP protein level (Fig. 4, E and F), suggesting theimportant role of the Ca2�-activated calcineurin pathway in thepathogenesis of hypertrophic remodeling. Cotreating PE-stim-ulated cells with PD and nifedipine or FK506 only slightlyincreased the effect of PD on cell size and ANP protein level(Fig. 4, E and F). The results further indicate the causalrelationship between inhibition of Ca2�-calcineurin pathwayand attenuation of myocyte hypertrophy by PD treatment.
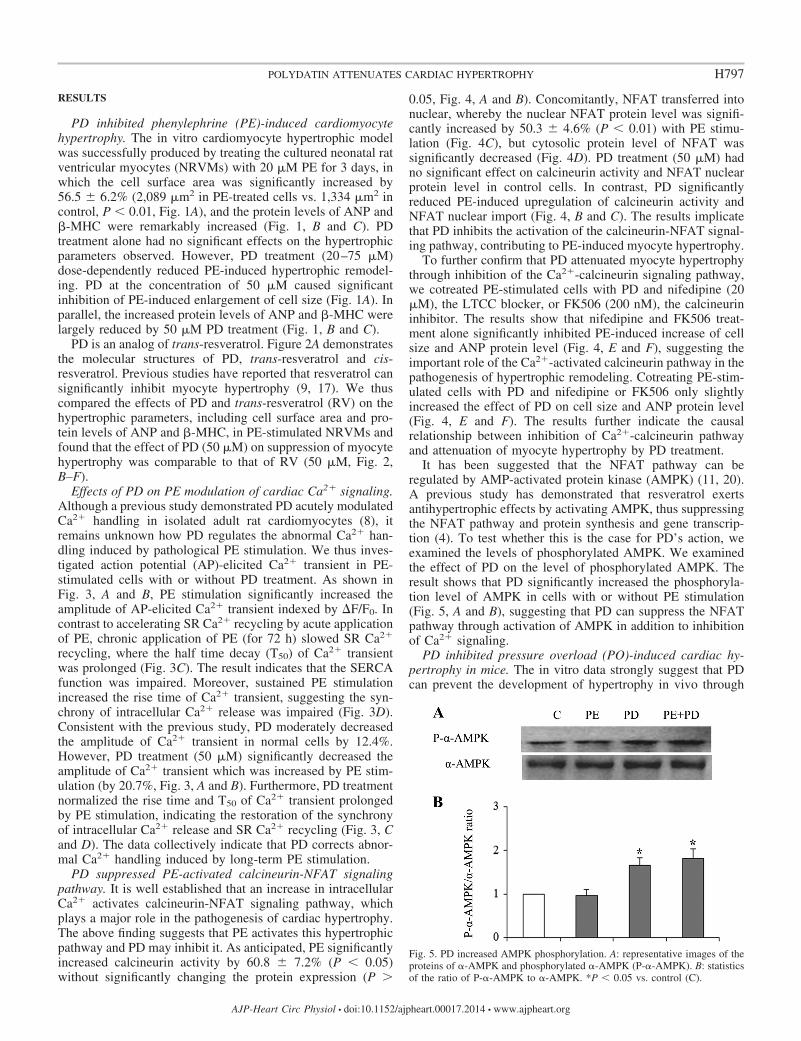
It has been suggested that the NFAT pathway can beregulated by AMP-activated protein kinase (AMPK) (11, 20).A previous study has demonstrated that resveratrol exertsantihypertrophic effects by activating AMPK, thus suppressingthe NFAT pathway and protein synthesis and gene transcrip-tion (4). To test whether this is the case for PD’s action, weexamined the levels of phosphorylated AMPK. We examinedthe effect of PD on the level of phosphorylated AMPK. Theresult shows that PD significantly increased the phosphoryla-tion level of AMPK in cells with or without PE stimulation(Fig. 5, A and B), suggesting that PD can suppress the NFATpathway through activation of AMPK in addition to inhibitionof Ca2� signaling.
PD inhibited pressure overload (PO)-induced cardiac hy-pertrophy in mice. The in vitro data strongly suggest that PDcan prevent the development of hypertrophy in vivo through
Fig. 5. PD increased AMPK phosphorylation. A: representative images of theproteins of �-AMPK and phosphorylated �-AMPK (P-�-AMPK). B: statisticsof the ratio of P-�-AMPK to �-AMPK. *P � 0.05 vs. control (C).
H797POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
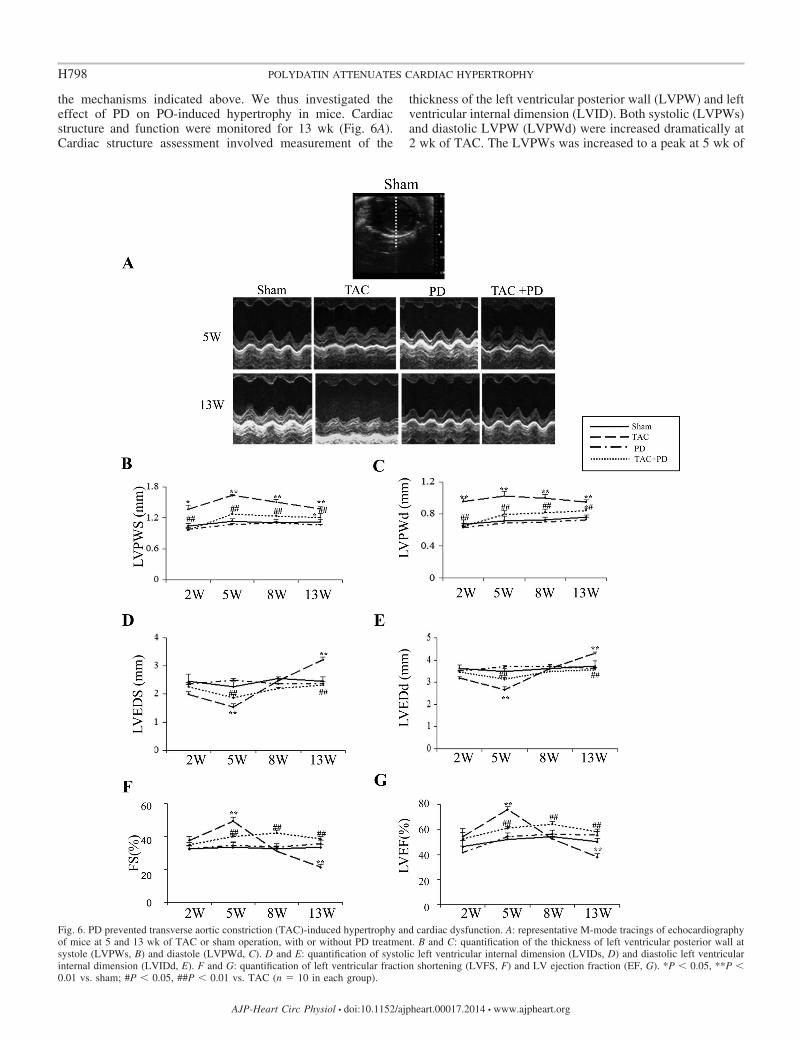
the mechanisms indicated above. We thus investigated theeffect of PD on PO-induced hypertrophy in mice. Cardiacstructure and function were monitored for 13 wk (Fig. 6A).Cardiac structure assessment involved measurement of the
thickness of the left ventricular posterior wall (LVPW) and leftventricular internal dimension (LVID). Both systolic (LVPWs)and diastolic LVPW (LVPWd) were increased dramatically at2 wk of TAC. The LVPWs was increased to a peak at 5 wk of
Fig. 6. PD prevented transverse aortic constriction (TAC)-induced hypertrophy and cardiac dysfunction. A: representative M-mode tracings of echocardiographyof mice at 5 and 13 wk of TAC or sham operation, with or without PD treatment. B and C: quantification of the thickness of left ventricular posterior wall atsystole (LVPWs, B) and diastole (LVPWd, C). D and E: quantification of systolic left ventricular internal dimension (LVIDs, D) and diastolic left ventricularinternal dimension (LVIDd, E). F and G: quantification of left ventricular fraction shortening (LVFS, F) and LV ejection fraction (EF, G). *P � 0.05, **P �0.01 vs. sham; #P � 0.05, ##P � 0.01 vs. TAC (n 10 in each group).
H798 POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
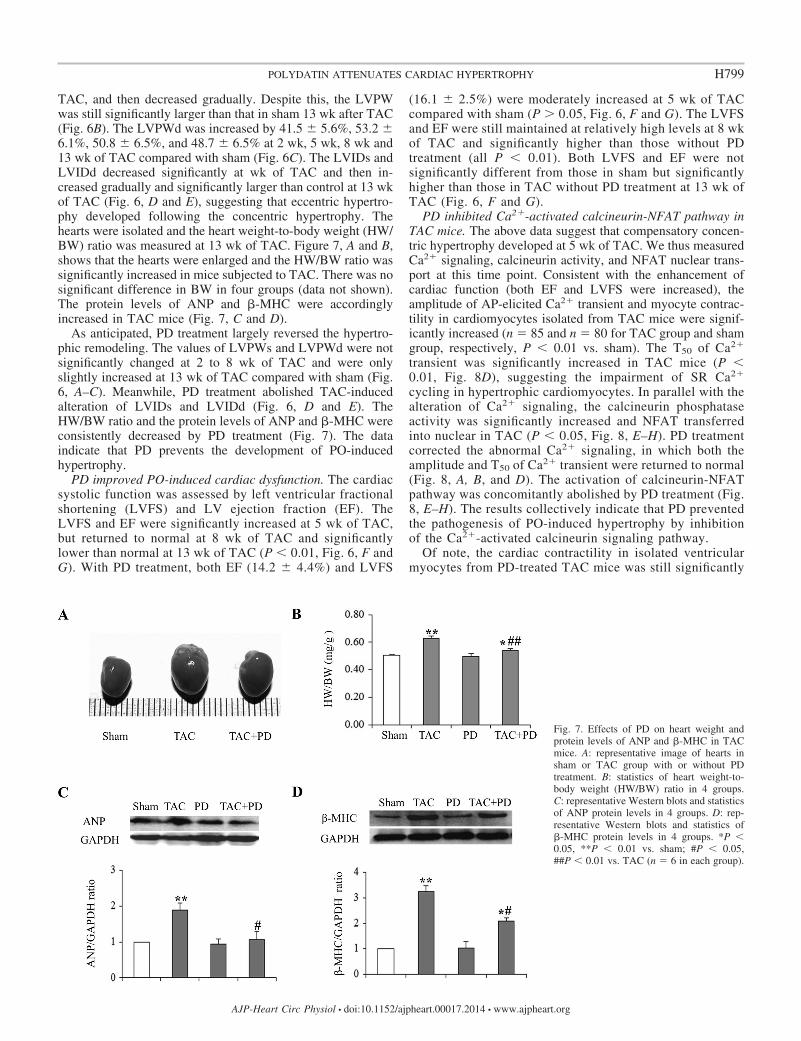
TAC, and then decreased gradually. Despite this, the LVPWwas still significantly larger than that in sham 13 wk after TAC(Fig. 6B). The LVPWd was increased by 41.5 � 5.6%, 53.2 �6.1%, 50.8 � 6.5%, and 48.7 � 6.5% at 2 wk, 5 wk, 8 wk and13 wk of TAC compared with sham (Fig. 6C). The LVIDs andLVIDd decreased significantly at wk of TAC and then in-creased gradually and significantly larger than control at 13 wkof TAC (Fig. 6, D and E), suggesting that eccentric hypertro-phy developed following the concentric hypertrophy. Thehearts were isolated and the heart weight-to-body weight (HW/BW) ratio was measured at 13 wk of TAC. Figure 7, A and B,shows that the hearts were enlarged and the HW/BW ratio wassignificantly increased in mice subjected to TAC. There was nosignificant difference in BW in four groups (data not shown).The protein levels of ANP and �-MHC were accordinglyincreased in TAC mice (Fig. 7, C and D).
As anticipated, PD treatment largely reversed the hypertro-phic remodeling. The values of LVPWs and LVPWd were notsignificantly changed at 2 to 8 wk of TAC and were onlyslightly increased at 13 wk of TAC compared with sham (Fig.6, A–C). Meanwhile, PD treatment abolished TAC-inducedalteration of LVIDs and LVIDd (Fig. 6, D and E). TheHW/BW ratio and the protein levels of ANP and �-MHC wereconsistently decreased by PD treatment (Fig. 7). The dataindicate that PD prevents the development of PO-inducedhypertrophy.
PD improved PO-induced cardiac dysfunction. The cardiacsystolic function was assessed by left ventricular fractionalshortening (LVFS) and LV ejection fraction (EF). TheLVFS and EF were significantly increased at 5 wk of TAC,but returned to normal at 8 wk of TAC and significantlylower than normal at 13 wk of TAC (P � 0.01, Fig. 6, F andG). With PD treatment, both EF (14.2 � 4.4%) and LVFS
(16.1 � 2.5%) were moderately increased at 5 wk of TACcompared with sham (P � 0.05, Fig. 6, F and G). The LVFSand EF were still maintained at relatively high levels at 8 wkof TAC and significantly higher than those without PDtreatment (all P � 0.01). Both LVFS and EF were notsignificantly different from those in sham but significantlyhigher than those in TAC without PD treatment at 13 wk ofTAC (Fig. 6, F and G).
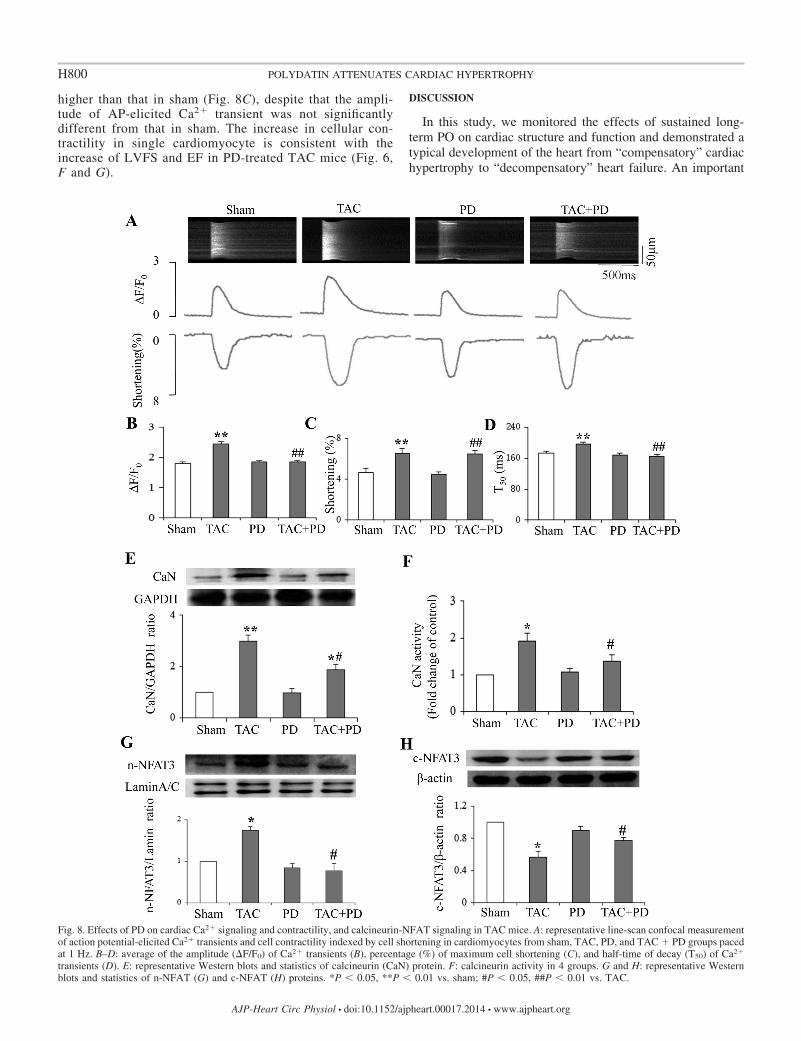
PD inhibited Ca2�-activated calcineurin-NFAT pathway inTAC mice. The above data suggest that compensatory concen-tric hypertrophy developed at 5 wk of TAC. We thus measuredCa2� signaling, calcineurin activity, and NFAT nuclear trans-port at this time point. Consistent with the enhancement ofcardiac function (both EF and LVFS were increased), theamplitude of AP-elicited Ca2� transient and myocyte contrac-tility in cardiomyocytes isolated from TAC mice were signif-icantly increased (n 85 and n 80 for TAC group and shamgroup, respectively, P � 0.01 vs. sham). The T50 of Ca2�
transient was significantly increased in TAC mice (P �0.01, Fig. 8D), suggesting the impairment of SR Ca2�
cycling in hypertrophic cardiomyocytes. In parallel with thealteration of Ca2� signaling, the calcineurin phosphataseactivity was significantly increased and NFAT transferredinto nuclear in TAC (P � 0.05, Fig. 8, E–H). PD treatmentcorrected the abnormal Ca2� signaling, in which both theamplitude and T50 of Ca2� transient were returned to normal(Fig. 8, A, B, and D). The activation of calcineurin-NFATpathway was concomitantly abolished by PD treatment (Fig.8, E–H). The results collectively indicate that PD preventedthe pathogenesis of PO-induced hypertrophy by inhibitionof the Ca2�-activated calcineurin signaling pathway.
Of note, the cardiac contractility in isolated ventricularmyocytes from PD-treated TAC mice was still significantly
Fig. 7. Effects of PD on heart weight andprotein levels of ANP and �-MHC in TACmice. A: representative image of hearts insham or TAC group with or without PDtreatment. B: statistics of heart weight-to-body weight (HW/BW) ratio in 4 groups.C: representative Western blots and statisticsof ANP protein levels in 4 groups. D: rep-resentative Western blots and statistics of�-MHC protein levels in 4 groups. *P �0.05, **P � 0.01 vs. sham; #P � 0.05,##P � 0.01 vs. TAC (n 6 in each group).
H799POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org
higher than that in sham (Fig. 8C), despite that the ampli-tude of AP-elicited Ca2� transient was not significantlydifferent from that in sham. The increase in cellular con-tractility in single cardiomyocyte is consistent with theincrease of LVFS and EF in PD-treated TAC mice (Fig. 6,F and G).
DISCUSSION
In this study, we monitored the effects of sustained long-term PO on cardiac structure and function and demonstrated atypical development of the heart from “compensatory” cardiachypertrophy to “decompensatory” heart failure. An important
Fig. 8. Effects of PD on cardiac Ca2� signaling and contractility, and calcineurin-NFAT signaling in TAC mice. A: representative line-scan confocal measurementof action potential-elicited Ca2� transients and cell contractility indexed by cell shortening in cardiomyocytes from sham, TAC, PD, and TAC � PD groups pacedat 1 Hz. B–D: average of the amplitude (�F/F0) of Ca2� transients (B), percentage (%) of maximum cell shortening (C), and half-time of decay (T50) of Ca2�
transients (D). E: representative Western blots and statistics of calcineurin (CaN) protein. F: calcineurin activity in 4 groups. G and H: representative Westernblots and statistics of n-NFAT (G) and c-NFAT (H) proteins. *P � 0.05, **P � 0.01 vs. sham; #P � 0.05, ##P � 0.01 vs. TAC.
H800 POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org

finding in this study was that PD has a potent therapeutic effecton preventing the development of cardiac hypertrophy anddeterioration to heart failure upon sustained PO stimulation.Furthermore, we found that PD dose-dependently preventedhypertrophic remodeling in cultured cardiomyocytes in vitro.The results indicate that PD can target directly on cardiomyo-cytes to prevent the pathogenesis of hypertrophy.
It is well established that Ca2�-regulated signaling pathwayscontribute to the pathogenesis of cardiac hypertrophy. For genetranscription, the Ca2�/calmodulin (CaM)-activated calcineu-rin-NFAT signaling pathway has been shown to play a majorrole in the development of pathological cardiac hypertrophy.Upon dephosphorylation by calcineurin, NFAT translocates tothe nucleus of the cardiomyocyte where it mediates the tran-scription of numerous targets involved in hypertrophic growth(27, 32). In this study, we found that calcineurin activity wasupregulated and the nuclear NFAT protein level was increasedin both PE-induced hypertrophic cardiomyocytes and in PO-induced hypertrophic heart. In accordance with the activationof the calcineurin-NFAT signaling pathway, the amplitude ofsystolic Ca2� transient in cardiomyocytes was significantlyincreased. Meanwhile, the SR Ca2� recycling indicated by T50
of Ca2� transient was slowed, indicating the disturbance ofintracellular Ca2� homeostasis. Of importance, blockage ofLTCC with nifedipine or inhibition of calcineurin activity withFK506 significantly suppressed PE-induced hypertrophic re-modeling. This in vitro and in vivo evidence is in support of thekey role of the Ca2�-activated calcineurin-NFAT signalingpathway in the development of cardiac hypertrophy.
Since upregulation of Ca2� signaling increases cardiac con-tractile function, which represents “compensatory” responseprojected to normalize wall stress and facilitates systolic per-formance, inhibition of Ca2� signaling with a LTCC blockersuppresses cardiac function and is not beneficial for the treat-ment of cardiac hypertrophy. In this study, we found that PDinhibited the calcineurin-NFAT signaling pathway. PD atten-uated the development of cardiac hypertrophy both in vitro andin vivo. Furthermore, we found that this effect was mediated byinhibition of cardiac Ca2� handling, for PD completely abol-ished the increase of the amplitude of systolic Ca2� transient incultured cardiomyocytes upon PE stimulation and in singlecardiomyocytes isolated from hearts challenged with PO. Ofparticular interest, PD, unlike Ca2�-channel blockers, did notimpair cardiac contractile function, despite that PD signifi-cantly decreased the amplitude of systolic Ca2� transients.Moreover, PD restored the impaired SR Ca2� recycling, whichhas been well documented, inducing cardiac dysfunction inheart failure by causing Ca2� overload during diastole. Theunique regulations of PD on Ca2� handling and contractility incardiomyocytes indicate that PD is superior to Ca2�-channelblockers on the treatment of cardiac hypertrophy.
Previous study has demonstrated that PD can also inhibitcalcineurin activity. However, the causal link between themodulations of Ca2� signaling and calcineurin activity remainsunknown. This present study reveals for the first time that PDmodulation of intracellular Ca2� signaling is an importantmechanism for PD inhibition of the activation of calcineurin-NFAT pathway, which may also be the case for the action ofresveratrol.
The ability of PD to inhibit Ca2� signaling and subsequentcalcineurin-NFAT signaling pathway may represent its pivotal
effect in terms of attenuating the hypertrophic response. How-ever, other mechanisms should also participate in the antihy-pertrophic effect of PD. In this study, we found that PDsignificantly increased AMPK activity in PE-stimulated cells,suggesting the involvement of this signal pathway in PDsuppression of cardiac hypertrophy.
In this study, we found PD only mildly regulated cardiacCa2� handling under physiological conditions, whereas it hadstrong suppression on abnormal Ca2� handling under patho-logical hypertrophic stimuli. Reports have confirmed safety ofPD administration orally up to 100 mg/kg in animals (13, 35,36), and PD has now obtained permission from the ChineseFood and Drug Administration for phase II clinical trials forthe treatment of severe hemorrhagic shock. All the evidencehighlights the beneficial role of PD in the treatment of cardiachypertrophy and heart failure.
In conclusion, our present study demonstrated that PD had apotent antihypertrophic effect in PE-induced cardiomyocytehypertrophy and PO-induced cardiac hypertrophy. Further-more, PD prevented the development of cardiac contractiledysfunction under sustained PO. The beneficial effects of PDwere largely attributed to its modulation of Ca2� handling andinhibition of subsequent activation of the calcineurin-NFATsignaling pathway without compromising cardiac contractilefunction.
ACKNOWLEDGMENTS
We thank Haiwang for kindly providing us polydatin.
GRANTS
This work was supported by the National Science Foundation of China (Nos.31171096, 31371159, and 81200122) and the Basic Research Foundation of SZ(JC201005250059A, JCYJ20120613115535998, GJHS20120621143653775, andGJHS20120621143627572).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: W.D. and M.D. performed experiments; M.D. ana-lyzed data; J.D. and T.X. interpreted results of experiments; D.Y. approvedfinal version of manuscript; Y.L. and J.L. prepared figures.
REFERENCES
1. Bauml MA, Underwood DA. Left ventricular hypertrophy: an over-looked cardiovascular risk factor. Cleve Clin J Med 77: 381–387, 2010.
2. Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205,2002.
3. Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. AnnNY Acad Sci 1047: 86–98, 2005.
4. Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE,Dyck JR. Resveratrol inhibits cardiac hypertrophy via AMP-activatedprotein kinase and Akt. J Biol Chem 283: 24194–24201, 2008.
5. Cheng Y, Zhang HT, Sun L, Guo S, Ouyang S, Zhang Y, Xu J.Involvement of cell adhesion molecules in polydatin protection of braintissues from ischemia-reperfusion injury. Brain Res 1110: 193–200, 2006.
6. Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T.Ca2� oscillation frequency decoding in cardiac cell hypertrophy: role ofcalcineurin/NFAT as Ca2� signal integrators. Proc Natl Acad Sci USA105: 2859–2864, 2008.
7. Delumeau JC, Bentue-Ferrer D, Saiag B, Allain H. Clinical neurophar-macology of calcium antagonists. Fundam Clin Pharmacol 3, Suppl:89s–102s, 1989.
8. Deng J, Liu W, Wang Y, Dong M, Zheng M, Liu J. Polydatin modulatesCa2� handling, excitation-contraction coupling and beta-adrenergic sig-naling in rat ventricular myocytes. J Mol Cell Cardiol 53: 646–656, 2012.
H801POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org

9. Dolinsky VW, Chakrabarti S, Pereira TJ, Oka T, Levasseur J, BekerD, Zordoky BN, Morton JS, Nagendran J, Lopaschuk GD, DavidgeST, Dyck JR. Resveratrol prevents hypertension and cardiac hypertrophyin hypertensive rats and mice. Biochim Biophys Acta 1832: 1723–1733,2013.
10. Dong J, Wang H, Wan L, Hashi Y, Chen S. Identification and deter-mination of major constituents in Polygonum cuspidatum Sieb. et Zucc. byhigh performance liquid chromatography/electrospray ionization-ion trap-time-of-flight mass spectrometry. Se Pu 27: 425–430, 2009.
11. Fassett JT, Hu X, Xu X, Lu Z, Zhang P, Chen Y, Bache RJ. AMPKattenuates microtubule proliferation in cardiac hypertrophy. Am J PhysiolHeart Circ Physiol 304: H749–H758, 2013.
12. Gao H, Wang F, Wang W, Makarewich CA, Zhang H, Kubo H,Berretta RM, Barr LA, Molkentin JD, Houser SR. Ca2� influx throughL-type Ca2� channels and transient receptor potential channels activatespathological hypertrophy signaling. J Mol Cell Cardiol 53: 657–667,2012.
13. Gao JP, Chen CX, Gu WL, Wu Q, Wang Y, Lu J. Effects of polydatinon attenuating ventricular remodeling in isoproterenol-induced mouse andpressure-overload rat models. Fitoterapia 81: 953–960, 2010.
14. Godfraind T. Antioxidant effects and the therapeutic mode of action ofcalcium channel blockers in hypertension and atherosclerosis. PhilosTrans R Soc Lond B Biol Sci 360: 2259–2272, 2005.
15. Houser SR, Molkentin JD. Does contractile Ca2� control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? SciSignal 1: pe31, 2008.
16. Johnson SH, Kraimer JM, Graeber GM. Effects of flunarizine onneurological recovery and spinal cord blood flow in experimental spinalcord ischemia in rabbits. Stroke 24: 1547–1553, 1993.
17. Juric D, Wojciechowski P, Das DK, Netticadan T. Prevention ofconcentric hypertrophy and diastolic impairment in aortic-banded ratstreated with resveratrol. Am J Physiol Heart Circ Physiol 292: H2138–H2143, 2007.
18. Kannel WB, Gordon T, Offutt D. Left ventricular hypertrophy byelectrocardiogram. Prevalence, incidence, and mortality in the Framing-ham study. Ann Intern Med 71: 89–105, 1969.
19. Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. Ameta-analysis of the effects of treatment on left ventricular mass inessential hypertension. Am J Med 115: 41–46, 2003.
20. Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, Dong YG. Long-termactivation of adenosine monophosphate-activated protein kinase attenu-ates pressure-overload-induced cardiac hypertrophy. J Cell Biochem 100:1086–1099, 2007.
21. Li J, Yatani A, Kim SJ, Takagi G, Irie K, Zhang Q, Karoor V, HongC, Yang G, Sadoshima J, Depre C, Vatner DE, West MJ, Vatner SF.Neurally-mediated increase in calcineurin activity regulates cardiac con-tractile function in absence of hypertrophy. Cardiovasc Res 59: 649–657,2003.
22. Liao Y, Ishikura F, Beppu S, Asakura M, Takashima S, Asanuma H,Sanada S, Kim J, Ogita H, Kuzuya T, Node K, Kitakaze M, Hori M.Echocardiographic assessment of LV hypertrophy and function in aortic-banded mice: necropsy validation. Am J Physiol Heart Circ Physiol 282:H1703–H1708, 2002.
23. Liu W, Deng J, Xu J, Wang H, Yuan M, Liu N, Jiang Y, Liu J.High-mobility group box 1 (HMGB1) downregulates cardiac transientoutward potassium current (Ito) through downregulation of Kv4.2 andKv43 channel transcripts and proteins. J Mol Cell Cardiol 49: 438–448,2010.
24. Molkentin JD. Calcineurin and beyond: cardiac hypertrophic signaling.Circ Res 87: 731–738, 2000.
25. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, RobbinsJ, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathwayfor cardiac hypertrophy. Cell 93: 215–228, 1998.
26. Nakayama H, Fujio Y, Yamaguchi O. Calcium dependent signaling incardiac hypertrophy and cell death. Clin Calcium 23: 505–515, 2013.
27. Rao A. Signaling to gene expression: calcium, calcineurin and NFAT. NatImmunol 10: 3–5, 2009.
28. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev 80:1483–1521, 2000.
29. Sanada S, Node K, Minamino T, Takashima S, Ogai A, Asanuma H,Ogita H, Liao Y, Asakura M, Kim J, Hori M, Kitakaze M. Long-actingCa2� blockers prevent myocardial remodeling induced by chronic NOinhibition in rats. Hypertension 41: 963–967, 2003.
30. Sheng C, Yu YH, Zhao KS, Qin W, Wang CH. Hypotensive resusci-tation combined with polydatin improve microcirculation and survival ina rabbit model of uncontrolled hemorrhagic shock in pregnancy. J SurgRes 168: 103–110, 2011.
31. Wang W, Zhang H, Gao H, Kubo H, Berretta RM, Chen X, HouserSR. �1 Adrenergic receptor activation induces mouse cardiac myocytedeath through both L-type calcium channel-dependent and -independentpathways. Am J Physiol Heart Circ Physiol 299: H322–H331, 2010.
32. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, JonesF, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participatesin pathological, but not physiological, cardiac hypertrophy. Circ Res 94:110–118, 2004.
33. Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regula-tion of cardiac hypertrophy. Biochem Biophys Res Commun 322: 1178–1191, 2004.
34. Wolska BM. Calcineurin and cardiac function: is more or less better forthe heart? Am J Physiol Heart Circ Physiol 297: H1576–H1577, 2009.
35. Zhang H, Yu CH, Jiang YP, Peng C, He K, Tang JY, Xin HL.Protective effects of polydatin from Polygonum cuspidatum against car-bon tetrachloride-induced liver injury in mice. PLoS One 7: e46574, 2012.
36. Zhang J, Tan Y, Yao F, Zhang Q. Polydatin alleviates non-alcoholicfatty liver disease in rats by inhibiting the expression of TNF-alpha andSREBP-1c. Mol Med Rep 6: 815–820, 2012.
37. Zhang W. Old and new tools to dissect calcineurin’s role in pressure-overload cardiac hypertrophy. Cardiovasc Res 53: 294–303, 2002.
38. Zhao KS, Jin C, Huang X, Liu J, Yan WS, Huang Q, Kan W. Themechanism of Polydatin in shock treatment. Clin Hemorheol Microcirc29: 211–217, 2003.
39. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B,Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviralinfection of adult mouse cardiac myocytes: methods for cellular geneticphysiology. Am J Physiol Heart Circ Physiol 279: H429–H436, 2000.
H802 POLYDATIN ATTENUATES CARDIAC HYPERTROPHY
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00017.2014 • www.ajpheart.org