pneumorepaso - sogapar.info · • enfermedad intersticial pulmonar (sobre todo en icv) enfermedad...
TRANSCRIPT

PNEUMOREPASO

INMUNODEFICIENCIAS Y ENFERMEDADES RESPIRATORIAS
HÉRFANAS

ENFERMEDAD PULMONAR CON SÍNDROME DE INMUNODEFICIENCIA
PRIMARIA

Concepto de inmunodeficiencia primaria
• Alteración del sistema inmune por defecto genético generalmente hereditario
• Engloba más de 130 enfermedades
• Afecta tanto al sistema inmune innato como al adaptativo
• Predisposición a las infecciones

IP más frecuentes y tipo de defecto inmunitario
• Déficit selectivo de IgAa
• Inmunodeficiencia común variable (ICV)a
• Déficit de subclases de IgGa
• Agammaglobulinemia ligada al cromosoma X (ALX)a
• Inmunodeficiencia combinada graveb
• Síndrome de Wiskott-Aldrichab
• Síndrome de DiGeorgeb
• Síndrome hiper IgMa
a humoralb celular

Epidemiología y tipos
• Incidencias más frecuentesDéficit selectivo de IgA 1: 5.000 nacidosICV 1: 75.000 nacidos
• Tipos:Déficit de anticuerposAlteración inmunidad celularCombinadasDefectos granulocitos o complemento

Manifestaciones pulmonares de IP
• Infecciones agudas y crónicas
• Anormalidades estructurales (BQ)
• Malignidad (linfomas, carcinoma gástrico metastásico)
• Inflamación desregulada (inflamación granulomatosa, fibrosis pulmonar)

Microorganismos en IP de acuerdo al tipo de defectoGermen Anticuerpos Combinado Fagocitos Complemento
Virus Enterovirus CMV, VRS, EBV No No
Bacterias S. PneumoniaeH. InfluenzaeP. AeruginosaS. AureusCampilobacter
Además de las de anticuerposSalmonella typ.Listeria monoc.
S. AureusP. AeruginosaNocardia aster.Salmonella typ
Además de las de anticuerposNeiseriameningitidis
Micobacterias No Ambientales Ambientales No
Hongos No Cándida sppAspergilus sppPneumocitysC neoformans
Cándida sppAspergilus spp
No
Protozoos Giardia lamblia Toxoplasma gondii No No
Notarangelo. J Allerg Clin Immunol 2010

Manifestaciones no infecciosas de IP
• Bronquiectasias, especialmente en ICV, ALXPredominan en lóbulos inferiores y medios
• Enfermedad intersticial pulmonar (sobre todo en ICV)Enfermedad granulomatosa (ICV)Neumonía intersticial linfoideNeumonía intersticial linfocítica y granulomatosaNeumonía organizativaFibrosis

Déficit selectivo de IgA
• Definición:Disminución o ausencia de IgA (< 7 mg/dl)Niveles normales de IgG e IgM
• Déficit parcial de IgA IgA > 7 mg/dl con 2 DE menores de lo normal
• PatogeniaDefecto de maduración de células B
• IncidenciaVariablePredominio en raza caucásicaEspaña 1: 163 habitantes

Déficit selectivo de IgA
• 2/3 asintomáticos
• Clínica: Infecciones respiratorias BQ, especialmente cuando se combina con subclases de IgG Infecciones gastrointestinales Enf. alérgicas (asma, alimentos) Enf. autoinmunes (púrpura trombocitopénica, AR, LED)
• Evolución: Posibilidad de desarrollar inmunodeficiencia común variable Asociación con déficit de subclases de IgG (IgG2-IgG4) refuerza la
posibilidad de infecciones

ICV
• Incidencia 1: 75.000 nacidos• Prevalencia 1: 50.000 – 200.000 hab.• 30% de todas las IP• No predominio de sexos• Mayoría esporádicos• 10 – 25% herencia autosómica dominante• Edad media Inicio de clínica a los 25 añosDiagnóstico 31 años

ICV. Características clínicas (I)• Infecciones (90%)
SinusitisOtitis mediaBronquitisNeumonía IntestinalesHepatitis
• Enfermedades autoinmunesAnemia hemolíticaPúrpura trombocitopénicaTiroiditis
• Enf. pulmonar intersticial (10 – 25%)TACAR y biopsia pulmonar

ICV. Características clínicas (II)• Granulomatosis (8%)
Pulmonar y mediastínica (10-22%)HepáticaEsplénicaGastrointestinalCutánea
• Enfermedades gastrointestinales (14%)Diarrea crónica con malabsorción (40-50%)Enfermedad de CrohnCelíacaLinfangiectasia intestinal
• Neoplasias (16%)Linfomas células B (10%)Carcinoma gástrico

ICV. Criterios diagnósticos
• CardinalesHipogammaglobulinemiaIgG + IgA y/o IgM
Infecciones sinopulmonares de repeticiónAnomalías en la respuesta funcionalNula o disminuida respuesta a vacunas
o Difteria, tétanos, neumococo
• Considerar procesos asociadosGranulomatosisEnfermedades autoinmunesBronquiectasias

Enfermedad granulomatosa pulmonar en ICV (I)
• Incrementa la morbilidad y mortalidad• En ocasiones precede al diagnóstico de ICV• Histología granulomas similares a los de la sarcoidosis
• Asociada con una disminución de células B de memoria switch y déficit funcional células T
• Posibilidad de confundir con sarcoidosis determinar inmunoglobulinas en todos los casos
• Prevalencia mayor de enfermedades autoinmunes con respecto a no granulomatosis
• Asociada con infiltración intersticial linfoide Enf. intersticial pulmonar granulomatosa linfocítica (GLILD)Mal pronóstico

Enfermedad granulomatosa pulmonar en ICV (II)
• Carácterísticas radiológicas (TACAR)Patrón reticular difuso de predominio en lóbulos
inferioresNódulos difusos asociados al anteriorAdenopatías mediastínicas junto a las anteriores Fibrosis pulmonar en estadios avanzadosBronquiectasias por tracción
• Características funcionales Síndrome ventilatorio restrictivo Insuficiencia respiratoria en fase avanzadaCor pulmonale

Enf. pulmonar intersticial linfocítica granulomatosa (GLILD)
• Granulomas no necrotizantes
• Neumonía intersticial linfoide (NIL)
• Bronquiolitis folicular en la histopatología
• Granulomas en otros órganos (MO, bazo, hígado, piel, tracto gastrointestinal)

Enf. pulmonar intersticial linfocítica granulomatosa (GLILD)
• Entre 20 – 50 años
• Opacidades nodulares y en vidrio deslustrado y/o adenopatía mediastínicas (se confunde con sarcoidosis)
• Etiología desconocida. Infección pulmonar por HHV8
• Causa más común de enf. pulmonar parenquimatosa difusa en ICV
• Pacientes con ICV y GLILD presentan riesgo incrementado de enfermedades autoinmunes y malignidad, particularmente LNH
• Mal pronóstico. Responde mal a GC. Antagonistas TNF, ciclosporina, combinación rituximab y azatioprina

Agammaglobulinemia ligada a X (ALX)
• Mutación de la tirosin kinasa de Bruton que conduce a defecto de desarrollo de células B, resultando en carencia de células B maduras y profundo descenso de producción de Ig.
• Infecciones recurrentes tracto respiratorio por gérmenes encapsulados
• BQ de lóbulos medios e inferiores
• No asociada con incremento de incidencia de EPID y linfoma, a diferencia de la ICV.

ID combinada severa
• Infancia
• Linfopenia de células T
• Extrema susceptibilidad a infecciones con bacterias, virus y hongos (P. jirovecii, CMV, VSR, adenovirus)
• Trasplante hematopoiético de células madre antes de 4 meses

Tratamiento (I)• Déficit selectivo de IgA
Tratamiento de las enfermedades asociadas
• Reemplazar IgG en ICV y ALX400 – 600 mg/kg mensual
• Infecciones agudas o crónicasAntimicrobianosConsiderar vía nebulizada en infección crónica por P. aeruginosa
• BronquiectasiasBroncodilatadoresEsteroides inhaladosFisioterapia
• Insuficiencia respiratoria y/o ventilatoriaOxigenoterapiaVMNI

Tratamiento (II)• Granulomatosis
Gammaglobulina a dosis habitual Prednisona 10 mg/díaValorar esteroides inhalados en la pulmonar
• GLILDAumentar la dosis de IgGAdministrar al menos 10 mg/día de prednisona Ciclosporina a dosis bajas si mal control Considerar inhibidores de TNF-α
• Trasplante de médula ósea en inmunodeficiencias combinadas
Profilaxis y tratamiento frecuente infecciones víricas y fúngicas• Cotrimoxazol en inmunodeficiencias combinadas
Prevenir infección por P. jirovecii• Valoración individual
Vacuna antigripalVacuna antineumocócica

Conclusiones. Claves para sospechar una IP
• Infecciones recurrentes no habituales difíciles de tratar• Alteración en el peso y estatura de los niños• Infecciones repetidas
Vías altasNeumoníasAbscesos
• Presencia de AdenopatíasEsplenomegalia
• Enfermedades autoinmunes

Bibliografía
• Park MA, J Li J, Hagan JB, et al. Common variable immunodeficiency: a new look at an olddisease. Lancet. 2008; 372: 489 – 502.
• Notarangelo L. Primary immunodeficiencies. J All Clin Immunol. 2010; 125: 5182 – 94.• Hampson FA, Chandra A, Screaton NJ, et al. Respiratory disease in common variable
immunodeficiency and other primary immunodeficiency disorders. Clinical Radiology. 2012; 67: 587 – 95.
• Leman Y. Selective IgA deficiency. J Clin Immunol. 2010; 30: 10 – 6.• Ardeniz A, Cunningham-Rundles CH. Granulomatous disease in common variable
immunodeficiency. Clinical Immunology. 2009; 133: 198 – 207.• Cunningham-Rundles CH. How I treat common variable immune deficiency. Blood. 2010; 116:
7 – 15.• Al-Herz, Bousfiha A, Casanova JL, et al. Perimary immunodeficiency diseases: an updat on the
classification from the International Union of Immunological Societies Expert Committee forPrimary Immunodeficiency. Frontiers in Immunology. 2011; 2: 1 – 26.
• Park JH, Levinson AI. Granulomatous-lynphocytic interstitial lung disease (GLILD) in commonvariable immunodeficiency (CVID). Clin Immunol. 2010; 134: 97 – 103
• Mannina A, Chung JH, Swigris JJ, et al. Clinical Predictors of a Diagnosis of Common Variable Immunodeficiency-realted Granulomatous-Lynphocytic Interstitial Lung Disease. Ann Am Thorac Soc. 2016; 13: 1042 – 9.

HISTIOCITOSIS DE CÉLULAS DE LANGERHANS (HCL)

HCL. Definición
• Enfermedad respiratoria minoritaria
• Granuloma eosinófilo del pulmón, Histiocitosis Xpulmonar
• Caracterizada por la proliferación de células de Langerhans a nivel bronquiolocéntrico
• En niños, forma más agresiva y sistémica
• Etiología desconocida

HCL. Clasificación en adultos
• HCL de un único sistema (HCL-US) Un órgano/sistema afectado (uni o multifocal):
o Óseo: unifocal (único hueso) o multifocal (> de 1 hueso)o Pielo Ganglio linfático (diferente de la zona de drenaje de otra lesión de
HCL)o Pulmóno Hipófisis-hipotálamo / Sistema nervioso centralo Otros: tiroides, timo
• HCL multisistémica (HCL-MS) Dos o más órganos/sistemas afectados:
o Incluyendo órganos de riesgo (sistema hematopoyético, hígado, bazo)o Sin incluir órganos de riesgo

HCL. Epidemiología
• Prevalencia: < 1 caso por 200.000 habitantes
• Edad de presentación: 20 a 40 años
• Raza blanca
• Género: sin predominancia
• Factores de riesgo: tabaco (prácticamente 100% casos), QT

HCL. Patología
• Células de Langerhans: célula dendrítica que se diferencia de otros histiocitos por la presencia de gránulos de Birbeck en su citoplasma y la expresión de CD1 o CD207 en su membrana.
• Lesión inicial: granulomas que engloban progresivamente el bronquiolo hasta producir su dilatación. Están compuestos por células inflamatorias, entre las que destacan las células de Langerhans y, en menor medida, los eosinófilos. Otras lesiones presentes en otras enfermedades asociadas al tabaco (NID o bronquiolitis respiratoria asociada a enfermedad intersticial).
• Lesión final: conforme progresa se sustituye la lesión inflamatoria por una fibrosis, que adquiere una forma estrellada característica, y a su alrededor enfisema intersticial. Su confluencia constituye las lesiones quísticas habituales de la enfermedad avanzada.



HCL. Hallazgos clínicos (I)
• Inespecíficos:Síntomas más frecuentes: tos y disnea de esfuerzoNeumotórax espontáneo: 10 – 20%En ocasiones hallazgo incidental
• Localización:Pulmonar exclusivamente (80% casos)Multisistémica:
o Ósea: dolor, fracturas espontáneas (lesiones osteoblásticas, bien delimitadas, que pueden afectar costillas)
o Eje hipofisario-hipotalámico: diabetes insípida (poliuria/polidipsia)
o Piel: tumoraciones

HCL. Hallazgos clínicos (II)
• Asociación con tumores benignos y malignoscarcinoma broncogénicoLH y LNHtumor carcinoide pulmonarganglioneuroma mediastínico
• Hipertensión pulmonar es común

HCL. Radiología• RX tórax:
afectación reticular y micronodulilar que respeta bases fases avanzadas: desaparición de nódulos y aparición de signos
compatibles con enfisema
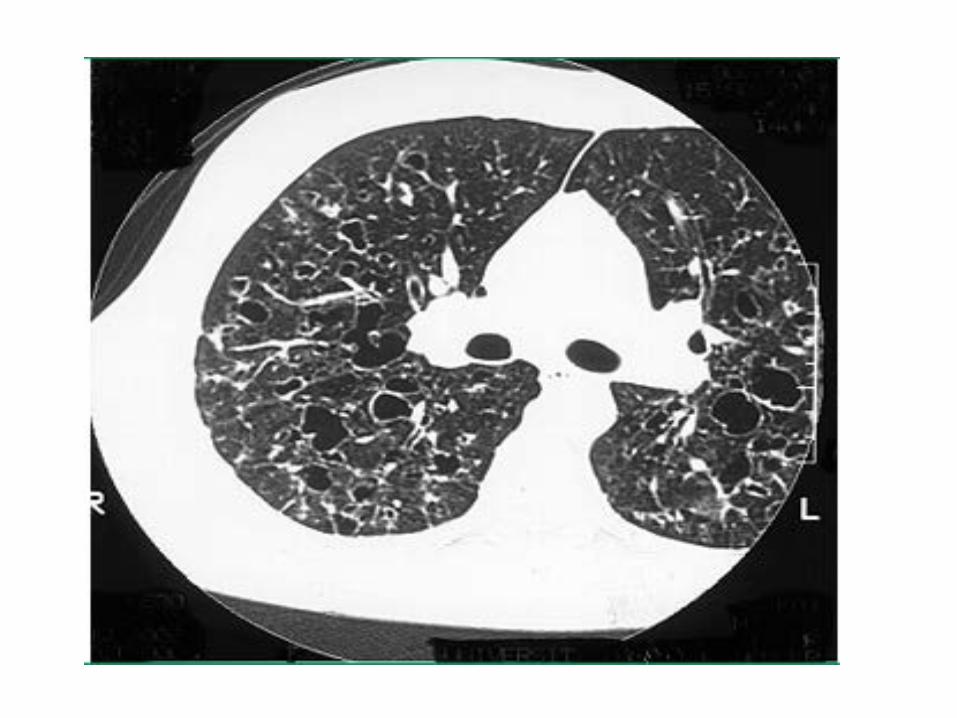
• TACAR: Tríada clásica:
o Nódulos infracentimétricos que en ocasiones se cavitan y pueden desaparecer durante el seguimiento
o Quistes finos con paredes de distinto grosor y morfología, que pueden acabar confluyendo simulando hallazgos de enfisema
o Senos costofrénicos preservados
Hallazgos ocasionales: arteria pulmonar alargada si HTP asociada
Hallazgos excepcionales: derrame pleural y adenopatías mediastínicas
Diagnóstico diferencial: enfisema, linfangioleiomiomatosis (LAM), síndrome de Birtt-Hogg-Dubé (con mutaciones del FCLN), síndrome de Sjögren y enfermedad por depósito de inmunoglobulinas






HCL. Función pulmonar
• Fase inicial: Parámetros espirométricos normales o levemente
afectadosDisminución de la DLCO, incluso en casos leves
(parámetro más sensible)
• Fase avanzada: En 1/3 de casos se desarrolla, en ocasiones de forma
intensa: limitación al flujo aéreo, atrapamiento aéreo y disminución de la DLCOSi la progresión no se frena, puede provocar
insuficiencia respiratoria
HCL. Diagnóstico (I)• Biopsia pulmonar:
Prueba con mayor rentabilidad diagnóstica En ocasiones se realiza durante tratamiento de un neumotórax (pleurodesis)
• BTB: rendimiento limitado por la distribución focal de las lesiones BAL: validez limitada Aumento del recuento celular, mayoritariamente macrófagos. Puede haber aumento de
eosinófilos. Disminución de la relación CD4/CD8 Presencia de > 5% de células positivas para CD1a es carácterístico de la enfermedad,
pero la sensibilidad es baja. Valores intermedios (1-5%) también pueden estar presentes en fumadores
• Otras exploraciones complementarias no contribuyen al diagnóstico de HPCL, pero si hay sospecha de enf. multisistémica, se ha de completar el estudio mediante serie ósea, eje hipofisario-hipotalámico y biopsia lesiones cutáneas sugestivas
En casos de enfermedad avanzada, con predominio de quistes y pérdida severade la función pulmonar, el rendimiento de las pruebas puede disminuir por laausencia de lesiones granulomatosas


HCL. Diagnóstico (II)
• Definitivo: Tejido pulmonar que muestra los hallazgos característicos de la enfermedad y
la presencia predominante de células de Langerhans, confirmada mediante inmunohistoquímica (proteína S-100, CD1a o CD207) o microscopía electrónica (gránulos de Birbeck)
• Presunción: Joven fumador con hallazgos característicos en la TACAR y afectación clínica y
funcional leve BTB (CD1a o S-100 positivos) y BAL (>5% de células de Langerhans-CD1a
positivo) el diagnóstico es altamente probable
El diagnóstico definitivo es fundamental en casos con afectación clínica y funcional significativa o si precisan tratamiento

HCL. Tratamiento (I)
NO EVIDENCIA DE EFICACIA DE NINGÚN TRATAMIENTO
• Recordar que muchos pacientes con HPCL no precisan tratamiento porque regresan espontáneamente.
• No se disponen de ensayos clínicos aleatorizados. Por lo tanto, la información respecto a la eficacia de los tratamientos es limitada.

HCL. Tratamiento (II)• Abandono tabaco: dada su relación con la enfermedad y para evitar
complicaciones futuras (EPOC, cáncer)
• Corticoides: en caso de progresión clínica o funcional se ha utilizado Prednisona a dosis de 0,5-1 mg/kg que se va reduciendo progresivamente en 6 – 12 meses. No obstante, no está demostrada su eficacia
• Citotóxicos: en casos severos, se había utilizado clásicamente Vinblastina. Recientemente se ha propuesto el uso de Cladribina, que parece tener mejores resultados. Uso limitado a centros de referencia
• Vasodilatadores pulmonares: en caso de HTP asociada
• Oxigenoterapia crónica: en caso de insuficiencia respiratoria
• Trasplante pulmonar: en enfermedad avanzada. Supervivencia a 10 años de un 50%. Puede recidivar en 1 de cada 5 casos.

HCL. Pronóstico
• Supervivencia a 5 años > 75% (Mayo Clinic: 12,5 años de media)
• Historia natural variable remisión espontánea de síntomasprogresión a enf. pulmonar fibrótica en estadio final
• Puede regresar con la cesación del tabaco
• La HTP está independientemente asociada con disminución de supervivencia


Bibliografía
• Tazi A. Adul pulmonary Langerhan´s cell histiocytosis. Eur Respir J. 2006; 27(6): 1272 – 85.• Xaubet A, Ancochea J, Blanquer R, et al. Diagnóstico y tratamiento de las enfermedades
pulmonares intersticiales difusas. Arch Bronconeumol. 2003; 39(12): 580 – 600.• Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans
Cell Histiocytosis: recommendations from an expert panel on behalf of Ruro-Histio-Net. Orphanet J Rare Dis. 2013; 8: 72.
• Arico M, Girschikofsky M, Genereau T, et al. Langerhans cell histiocytosis in adults. Reportfrom the International Registry of the Histicyte Society. Eur J Cancer. 2003; 39(16): 2341 – 8.
• Abbott GF, Rosado-de-Christenson ML, Franks TJ, et al. From the archives of AFIP: pulmonaryLangerhans cell histiocytosis. Radiographics. 2004; 24(3): 821 – 41.
• Lorillon G, Bergeron A, Detourmignies L, et al. Cladribine is effective against CysticPulmonary Langerhans Cell Histiocytosis. Am J Respir Crit Care Med. 2002; 186(9): 930 – 2.
• Castillo D, Martin I, Moreno A, et al. Histiocitosis pulmonar de células de Langerhans en adultos: aproximación a la realidad española. Med Clin. 2012; 143: 433 – 9.
• Harari S, Torre O, Cassandro R, et al. Bronchoscopic diagnosis of Langerhans cell histiocytosisand lymphangioleiomyomatosis. Respir Med. 2012; 106: 1286 – 92.

LINFANGIOLEIOMIOMATOSIS

LAM. Definición• Pertenece a familia de neoplasias con diferenciación epitelioide perivascular
• Rara, afecta a mujeres jóvenes en edad fértil
• Prevalencia: 1/400.000 mujeres adultas
• Se caracteriza por la proliferación de células musculares lisas atípicas (“células LAM”) en el intersticio pulmonar y alrededor de las estructuras broncovasculares y por la formación de quistes a nivel del parénquima
• La formación de “quistes pulmonares” conlleva la pérdida progresiva de función pulmonar, y en fases avanzadas, insuficiencia respiratoria
• Formas clínicas: LAM esporádica LAM asociada al Complejo Esclerosis Tuberosa (TSC)
• Etiología desconocida, aunque hay evidencia de implicación de determinados factores genéticos en la patogenia

LAM. Patogenia (I)• Estrecha relación entre LAM y ET
• La ET es una enf. dominante caracterizada por mutaciones en los genes supresores de tumores TSC1 y TSC2
• LAM se asocia a mutaciones somáticas en los genes TSC1 y TSC2
• Las proteínas TSC1 y TSC2 regulan su señal a través de la vía de señalización mTOR (mammalian target of rapamycin) controlando el crecimiento celular, la apoptosis y la autofagia
• Las mutaciones en TSC1 y TSC2 conllevan una activación anómala de mTOR, dando lugar a un crecimiento celular descontrolado (“células LAM”)
• Que afecte a mujeres jóvenes, pueda empeorar durante el embarazo o con la toma de anticonceptivos, y la identificación de receptores de estrógenos en el tejido pulmonar, sugiere una regulación hormonal en su patogenia
• Niveles elevados en suero de VEGF-D (> 800 pg/ml)

Receptor de tirosin-kinasa
P13 Kinasa
AktMutación TSC1 Mutación TSC2
Hamartina /Tuberina
Rheb
mTOR
Síntesis de proteínas y crecimiento celular
4EBP S6 kinasa

LAM. Patogenia (II)
• El tabaco puede empeorar la progresión de la enfermedad pero no parece ser un factor de riesgo para la inducción
• En LAM esporádica casi todos los casos son debidos a mutaciones TSC2
• Más altos niveles de prolactina sérica están asociados con tasas más grandes de declinación de función pulmonar y neumotórax
• El VEGF-D está elevado en suero en 2/3 de pacientes ( niveles> 800 picg/ml distingue LAM de otras enfermedades pulmonares quísticas) y ha sido postulado que puede promover linfangiogénesisy siembra de clusters de células LAM
• Si hay cambios característicos de LAM en TAC y niveles de VEGF-D > 800 no necesitan sufrir biopsia pulmonar

LAM. Clínica• Manifestaciones respiratorias:Síntoma más frecuente: disnea de esfuerzoComplicación más frecuente: neumotórax (afecta a
2/3 de pacientes). Permite sospechar la enfermedadQuilotórax: 7 – 31% pacientes. Por obstrucción o
rotura de conducto torácicoOtros síntomas: tos, hemoptisis
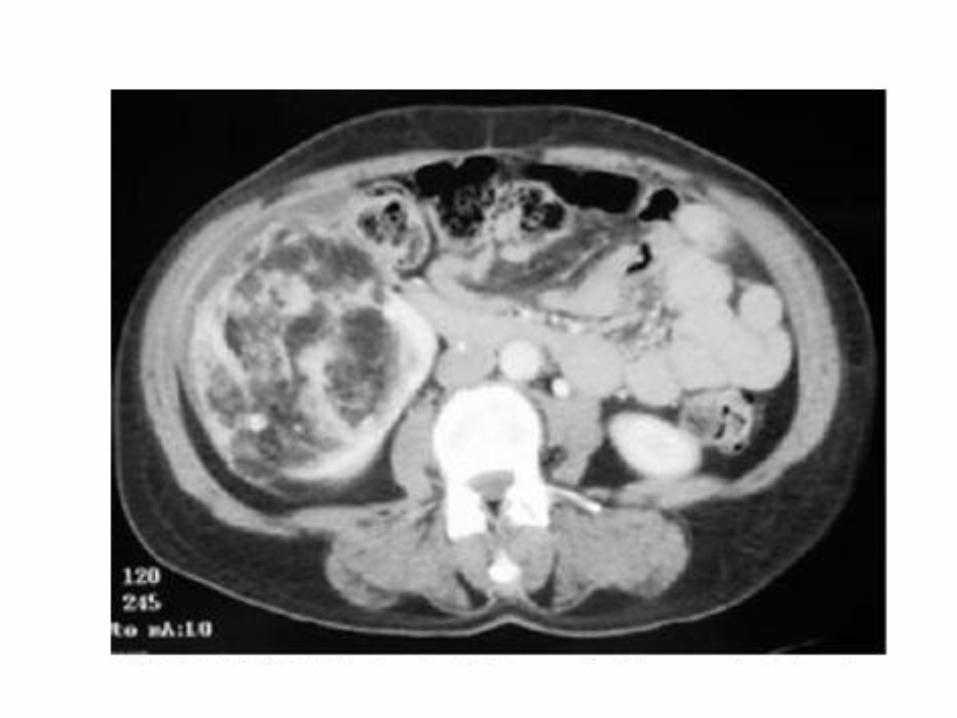
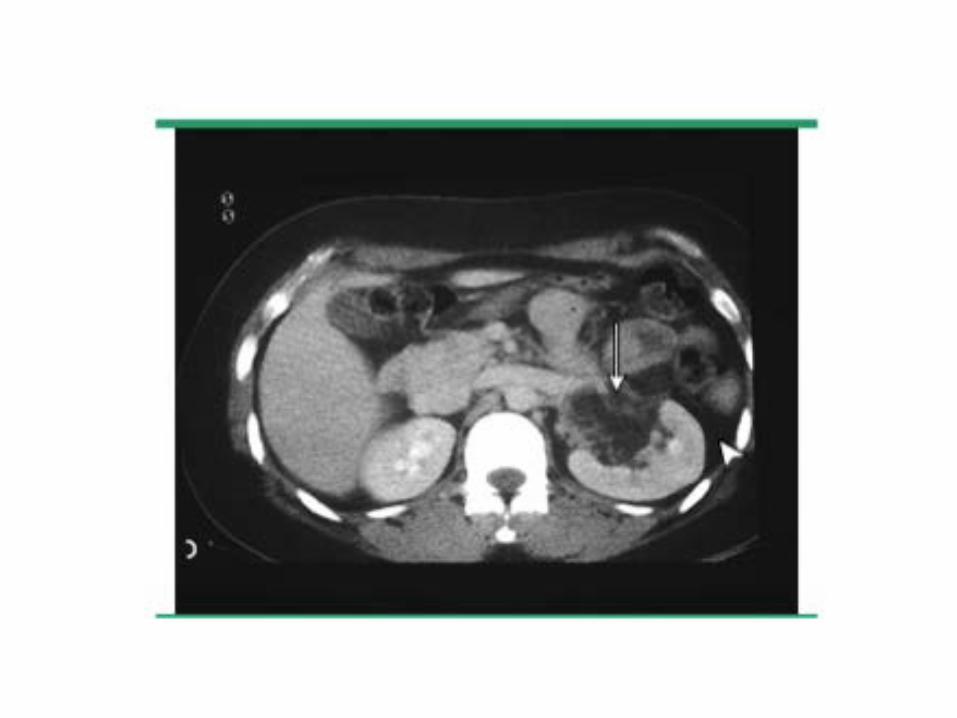
• Manifestaciones extrapulmonares: Angiomiolipomas renales: en 40% de pacientes con
LAM esporádica y 80% de LAM-ETLinfadenopatías retroperitoneales y pélvicasLinfangioleiomiomas






LAM. Diagnóstico (I)
• Sospecha: mujer joven con disnea, quistes pulmonares e historia de neumotórax
• RX tórax: puede ser normal
• TACAR: quistes pulmonares de pared fina distribuidos de forma difusa y homogénea por todos los campos pulmonares
• Eco abdominal/TAC abdomen: linfangiomiomas, angiomiolipomas renales
• PFR: predomina patrón obstructivo, con disminución de FEV1 y DLCO hipoxemia suele aparecer en fases avanzadas
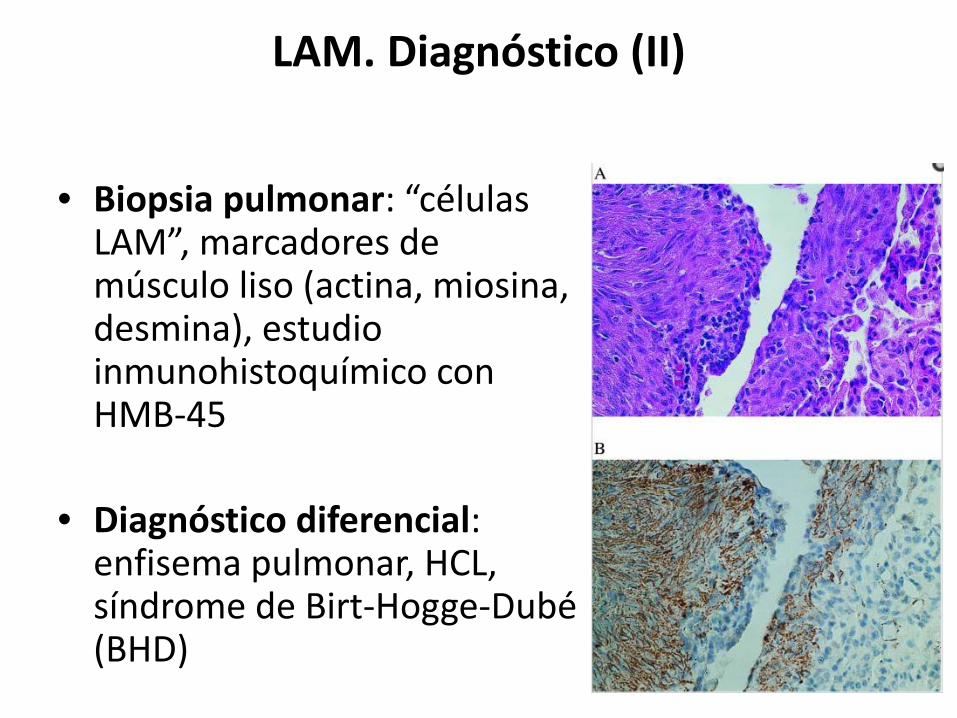
LAM. Diagnóstico (II)
• Biopsia pulmonar: “células LAM”, marcadores de músculo liso (actina, miosina, desmina), estudio inmunohistoquímico con HMB-45
• Diagnóstico diferencial: enfisema pulmonar, HCL, síndrome de Birt-Hogge-Dubé(BHD)

A B

Tinción HMB-45 (B)Tinción actina positiva (A)

LAM. Criterios diagnósticos de la ERS

Algoritmo diagnóstico de LAM

Diagnóstico diferencial de enfermedad quística pulmonar

LAM. Pronóstico
• La progresión es variable. Algunos estables durante largos períodos y otros experimentan deterioro rápido de la función pulmonar
• Supervivencia del 70 – 90% a los 10 años del diagnóstico
• Los pacientes con LAM-ET con síntomas leves tienen menos riesgo de desarrollar “LAM grave” que los pacientes con LAM esporádica
• DLCO y FEV1 son los mejores indicadores de progresión de la enfermedad y supervivencia
• La monitorización de la enfermedad debe evaluarse con PFRs cada 3-6 meses el primer año. Luego a intervalos de 3-12 meses según la progresión y gravedad de la enfermedad

LAM. Tratamiento (I)
• Medidas generales: Abandono del tabaco Tratamiento BD (en pacientes con prueba BD positiva) Evitar tratamientos que contengan estrógenos (anticonceptivos) Tratamiento de las complicaciones:
o Neumotóraxo Quilotóraxo Angiomiolipomas
Tratamiento de la osteoporosis:o Suplementos de calcio (Natecal D, Ideos)
Bifosfonatos (Fosamax, Actonel) Rehabilitación respiratoria: ejercicio físico, caminar, natación,
yoga.. Oxigenoterapia: pO2 < 60 mmHg, Sat. O2 < 90% Vacunación: gripe y neumococo

LAM. Tratamiento (II)
• Trasplante pulmonar: fases avanzadas. Aumenta supervivencia
• Terapias antiestrógenos:Ooforectomía, progesterona, tamoxifeno, análogos de
la hormona liberadora de gonadotropinasNo han demostrado ventajas significativasEstudios retrospectivos concluyen que el tratamiento
con progesterona no ralentiza el deterioro de la función pulmonar

LAM. Tratamiento (III)
• Inhibidores mTOR: Sirolimus, Everolimus Disminuyen el tamaño de los angiomiolipomas Disminuyen el tamaño de los astrocitomas en pacientes con ET Sirolimus estabiliza la función pulmonar (FEV1 y FVC) tras 1 año
de tratamiento comparado con placebo (ensayo fase III) Los inhibidores mTOR podrían estar indicados en pacientes con
LAM con una alteración moderada-grave de la función pulmonar tras una evaluación cuidadosa de la relación riesgo/beneficio
• Otros tratamientos en investigación: Inhibidores de las metaloproteinasas: Doxiciclina Inhibidor de la aromatasa: Letrozol

Bibliografía• Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006; 27: 1056 – 65.• Prino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med.
2006; 355: 1345 – 56.• Goncharova EA, Goncharov DA, Eszterhas A, et al. Tuberin regulates p70 56
activation and ribosomal protein 56 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem. 2002; 277: 30958 – 67.
• Antón E, Casanova A, Xaubet A, et al. Lymphangioleiomyomatosis: a study 72 patients from the spanish registry. Sarcoidosis Vasc Diffuse Lung Dis. 2009; 26: 85 –91.
• Johnson SR, Cordier JK, Lazor R, et al. European Respiratory Society guidelines forthe diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010; 35: 14 – 26.
• Taveira-DaSilva AM, Stylianou MP, Hedin CJ, et al. Decline in lung function in patients with lymphangioleiomyomatosis trated with or without progesterone. Chest. 2004; 126: 1867 – 74.
• McCormack FX, Inoue I, Young LR. Efficacy and safety of Sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011; 364(17): 1595 – 606.
• Casanova A, Girón RM, Acosta O, et al. Tratamiento de la linfangioleiomiomatosiscon sirolimus. Arch Bronconeumol. 2011; 47(9): 470 – 2.

PROTEINOSIS ALVEOLAR

Proteinosis alveolar (PAP)
• Es una enfermedad minoritaria caracterizada por la acumulación de surfactante en el espacio alveolar, produciendo alteración en el intercambio gaseoso que desencadena insuficiencia respiratoria
• Prevalencia: 1 – 3,7 casos/1.000.000 habitantes

PAP. Epidemiología • 3 Formas clínicas: Autoinmune o adquirida: es la más frecuente (90%).
o Se produce por alteración en la función de los macrófagos causados por la presencia de anticuerpos anti GM-CSF (factor estimulador de colonias granulocíticas)
o Produce una disminución del “clearence” del surfactante alveolar Secundaria: 10%. Está asociada a otras enfermedades:
o Hematológicas: el síndrome mielodisplásico es el más frecuente, MM, LMC, Macroglobulinemia de Waldenström
o Inmunodeficiencia (ICS, agammaglobilinemia..)o Infecciones (Nocardia, P. Jirovecii, CM)o Intolerancia a la proteína lisinúrica
Congénita: diagnosticada en la infancia. Suele asociarse a mutaciones en los genes que codifican:o Proteínas B y C del surfactante (SPB, SPC)o Cadenas α y β del receptor GM-CSFo ABCA3

PAP. Clínica
• Debut alrededor de la 5ª década de la vida (excepto formas congénitas)
• Forma autoinmune es más frecuente en hombres (2:1)
• Clínica progresiva, caracterizada por tos y disnea, según se va produciendo la ocupación alveolar por surfactante
• Formas paucisintomáticas, sobre todo en forma autoinmune (30% asintomáticos)

PAP. Diagnóstico
• 3 criterios diagnósticos o bien biopsia pulmonar
• Criterios:Hallazgos en TACAR compatibles, incluyendo patrón
geográfico difuso con infiltrados en vidrio deslustrado y/o engrosamiento de los septos interlobulillares en múltiples lóbulos
BAL con material eosinófilo con tinción PAS + Existencia de anticuerpos anti GM-CSF plasmáticos en la
forma autoinmune

PAP. Radiología (I)
• RX tórax: Infiltrados difusos bilaterales con distribución similar
al edema pulmonar (“alas de mariposa”)
• TAC torácica: Infiltrados pulmonares en vidrio deslustrado por la
ocupación alveolar, con márgenes angulados que dan apariencia de infiltrados “geográficos” (patrón en crazy paving)Diferencias entre PAP autoinmune y secundaria:
o Autoinmune: más frecuente la distribución geográfica, en empedrado, preservando zonas subpleurales
o Secundaria: es más frecuente la forma difusa

PAP. Función pulmonar
• Disminución precoz de la DLCO
• La hipoxemia moderada (PaO2 < 70 mmHg) es más frecuente y precoz en las formas autoinmunes





PAP. Radiología (II)
Apariencia de infiltrado
Autoinmunen= 21
Secundaria n= 21
Total n= 42
p
Geográfica 71% 24% 48% 0.02
Difusa 19% 62% 40% 0.005
Mixta 10% 14% 12% NS
Preservación subpleural
71% 33% 52% 0.01
Apariencia en empedrado
71% 14% 43% 0.001
Comparación entre hallazgos en TACAR de forma autoinmune y secundaria

PAP. Diagnóstico (I)
• Laboratorio:Anticuerpos anti GM-CSF: de gran utilidad
diagnóstica en las formas autoinmunes, con una sensibilidad y especificidad próximas al 100%
KL-6, CEA, SP-A, SP-B, LDH: pueden estar elevados sus niveles plasmáticos pero sin correlación diagnóstica o pronóstica con la enfermedad
• BAL:Aspecto característicamente lechoso por el
alto contenido fosfolipídico

PAP. Diagnóstico (II)• Anatomía patológica:Técnica gold standardSe caracteriza por presentar una
estructura pulmonar preservada, conservándose los espacios alveolares que están ocupados por un material lipoproteináceo, eosinofílico con tinción PAS +Aparecen macrófagos de
apariencia “espumosa” con inclusiones citoplasmáticas, producto del fallo del catabolismo de las proteínas del surfactante


Algoritmo diagnóstico de PAP

PAP. Tratamiento (I)
• Lavado broncoalveolar masivo Técnica bajo anestesia en quirófano Instilación entre 15-20 litros de suero salino utilizando un
tubo de doble luz (ventilación selectiva) de forma secuencial
Único tratamiento que ha demostrado disminuir la mortalidad
• Lavado broncoalveolar selectivo En caso de importante deterioro gasométrico que impida
realizar el lavado broncoalveolar totalMediante fibrobroncoscopia en un único lóbulo pulmonar

PAP. Tratamiento (II)
• Estimulador de colonias granulocíticas (GM-CSF)En caso de PAP autoinmunesEs el tratamiento más novedoso, aún en
investigación y desarrolloAdministración inhalada y subcutánea
• Otras en estudio: rituximabplasmaféresis

PAP. Pronóstico
• 8% remisión espontánea
• Supervivencia a 5 años: 75%
• Muertes por:72% fallo respiratorio20% infecciones (nocardia, criptococo,
histoplasma…)

Bibliografía• Ohashi K, Sato A, Takada T, et al. Reduced GM-CSF autoantibody in
improved lung of autoimmune pulmonary alveolar proteinosis. EurRespir J. 2012; 39(3): 777 – 80.
• Ohashi K, Sato A, Takada T, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir Med. 2012; 106(2): 284 – 93.
• Khan A, Agarwall R, Aggarwal AN. Effectiveness of granulocyte-macrophage colony-stimulating factor therapy in autoimmunepulmonary alveolar proteinosis: a meta-analysis of observationalstudies. Chest. 2012; 141(5): 1273 – 83.
• Luisetti M. Call for an international survey on therapeutic lavage forpulmonary alveolar proteinosis. Eur Respir J. 2012; 39(4): 1049 -
• Punatar AD, Kusne S, Blair JE, et al. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect. 2012; 65(2): 173 – 9.

ENFERMEDADES PULMONARES RELACIONADAS CON EL VIH

Introducción (I)
• Enf. pulmonar es una causa significativa de morbilidad y mortalidad ( > 50% de pacientes VIH padecen un episodio respiratorio en su evolución)
• La Terapia Combinada Retroviral (TCR) y terapia profiláctica ha disminuido de forma importante las infecciones oportunistas
• La neumonía bacteriana (NAC) es más común en los infectados por VIH que en población no infectada

Introducción (II)
• La infección fúngica es menos común desde la mejora de la TCR
• La TRC también está asociada a disminución de complicaciones pulmonares no infecciosas (sarcoma de Kaposi, LNH)
• El Síndrome inflamatorio de reconstitución inmune es una respuesta inmune a la TCR incontrolada y exhuberante frente a antígenos exógenos

Introducción (III)
• TCR puede desarrollar recrudescencia de enfermedades que han estado latentes (enf. micobacteriana latente, lesiones sarcoide-like)
• Los ADVP infectados por VIH tienen más riesgo de desarrollar TBC y NAC
• Profilaxis con TMP-SMX disminuye riesgo de NAC , mientras que vacuna antineumocócica no parece reducir el riesgo
• La secuencia de infecciones pulmonares es paralela a la disminución de linfocitos CD4
• Riesgo incrementado de desarrollar infección sistémica y pulmonar causada por Rhodococcus equi, organismo Nocardia-like, asociado con neumonía cavitaria y derrame pleuropericárdico

Estadios de VIH
• Temprano: CD4 > 500 cél/microL
• Intermedio: CD4 200 – 500 cél/microL
• Avanzado: CD4 100 -200 cél/microL
• Estadio final: CD4 < 100 cél/microL
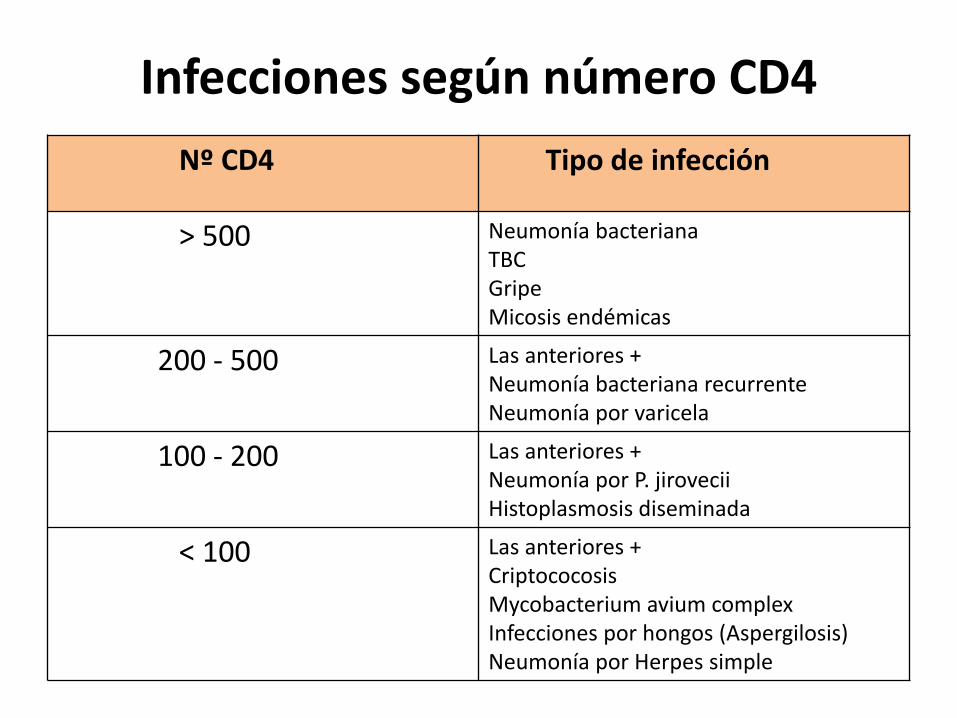
Infecciones según número CD4Nº CD4 Tipo de infección
> 500 Neumonía bacterianaTBCGripeMicosis endémicas
200 - 500 Las anteriores +Neumonía bacteriana recurrenteNeumonía por varicela
100 - 200 Las anteriores +Neumonía por P. jiroveciiHistoplasmosis diseminada
< 100 Las anteriores +CriptococosisMycobacterium avium complexInfecciones por hongos (Aspergilosis)Neumonía por Herpes simple

Enfermedades respiratorias asociadas a VIH
Enfermedades respiratorias infecciosas Enfermedades respiratorias no infecciosas
Infecciones tracto respiratorio alto• Bronquitis agudas• Sinusitis agudas• Sinusitis crónicas
BronquiectasiasNeumonías bacterianas
• Neumococo• Haemophilus
TuberculosisInfecciones por hongos
• Pneumocystis• Histoplasma• Criptococo
Cánceres• Sarcoma de Kaposi• Linfomas• Carcinoma de pulmón
No malignas• EPOC• Pneumonitis asociada a VIH: NIP –
LIP• Hipertensión arterial pulmonar• Neumotórax
Síntomas asociados a TCR• Síndrome inflamatorio de
reconstitución inmune

Radiología más comúnRX o TAC Comienzo agudo o subagudo Comienzo crónico
Consolidación focal Cualquier organismo, especialmente bacterias piógenasLegionella
Micobacterias, Nocardiosis, hongos
Infiltrados difusos bilaterales P. JiroveciiBacterias: H. InfluenzaeInfluenzaCMV
MicobacteriasNeumonías por hongos: criptococoToxoplasmosisCMV
Nódulo TBCHongos: aspergilus, criptococoBacterias
NocardiosisHongos
Adenopatías TBC MicobacteriosisHongos endémicos
Infiltrados cavitarios TBCS. AureusHongosAnaerobiosPseudomonaLegionella
MicobacteriasNocardiosisHongosRhodococcus equi
Derrame pleural Bacterias piógenasHongosTBC
HongosNocardiosis
Neumotórax TBCP. jirovecii


Infiltrado pulmonar lobar unilateral Infiltrado pulmonar múltiple, bilateral o difuso
• Hemocultivos x 2• Esputo: tinción y cultivo para
bacterias, hongos y micobacterias• Esputo inducido: tinción BK y cultivo• Ag urinario neumococo y legionella• Derrame pleural: cultivos aerobios,
anaerobios, hongos y micobacterias
Diagnóstico y/o mejoría clínica o RX con el tratamiento inicial
No diagnóstico y no mejoría con el tratamiento inicial
Completar tratamiento
• Test específicos en infiltrados lobares unilaterales• Esputo inducido: detectar P. Jirovecii• BF: tinción para P. Jirovecii, cultivo para bacterias
aerobias y anaerobias, hongos, micobacterias y virus. Test detección Ag virales
• Diagnóstico molecular para virus y micobacterias• Diagnóstico citológico
No diagnóstico y no mejoría con el tratamiento inicial
• PAAF guiada por TAC en infiltrados periféricos
• Biopsia pulmonar por VATS o toracotomía: estudios AP estudios microbiológicos
Tratamiento específico Diagnóstico etiológico
Algoritmo diagnóstico VIH

Infecciones bacterianas
• Las infecciones del tracto superior son más frecuentes que en población general
• Más frecuentes: bronquitis, sinusitis y neumonías
• BQ, cada vez más frecuentes debido a neumonías de repetición por PCP o bacterias

Neumonías bacterianas• En pacientes con TCR son 6 veces más frecuentes que en población
general
• Patógenos más frecuentes: neumococo y H. Influenzae
• En estadio avanzado: S. aureus y gram negativos (P. Aeruginosa)
• Presentación clínica usual pero la RX puede ser atípica (simula PCP en 50% casos)
• Bacteriemia 100 veces más frecuente, independientemente del nº de CD4
• Responden bien al tratamiento
• Se recomienda vacunación antineumocócica aunque eficacia menor con cifras de CD4 < 200. Indicada la nueva vacuna conjugada

Infección por P. jirovecii (PCP)• Cuadro de varios días o semanas, con disnea progresiva y tos no
productiva, con/sin fiebre. Auscultación: estertores teleinspiratorios
• Rx tórax normal en 10%
• Frecuente infiltrados intersticiales perihiliares que pueden evolucionar a patrón alveolar difuso
• RX atípicas: infiltrado LL.SS (simula TBC), linfadenopatía hiliar o mediastínica, nódulos pulmonares, consolidación lobar (20%), neumotórax
• Profilaxis con pentamidina aerosolizada aumenta riesgo de RX atípica
• La DLCO puede estar disminuida antes del desarrollo de infiltrados intersticiales en la RX


Infección por P. jirovecii (PCP)(signos de mal pronóstico)
• Edad avanzada
• Más de 1 episodio previo
• Hipoxemia
• Anemia
• Sarcoma de Kaposi pulmonar concomitante
• Comorbilidad médica
• Neumotórax
• Necesidad de ingreso en UCI
• Necesidad de ventilación mecánica

PCP. Estratificación clínica
Sat. O2 PaO2 mmHg PaO2 KPa
PCP leve > 95% > 82 > 11,0
PCP moderada 91 – 96% 60 - 82 8.0 – 11.0
PCP severa < 91% < 60 < 8.0

Tratamiento de la PCPTratamiento de elección Tratamiento alternativo Comentarios
Cotrimoxazol, tanto en formas graves (PaO2 < 70 mmHg o gradiente alveolo-arterial de O2 > 35 mmHg) como formas leves-moderadas
Formas graves:Cotrimoxazol: 15-20 mg/kg/día TMP + 75-100 mg/kg/día SMX, IV o VO, cada 6-8 h., 21 días
Formas leves-moderadas:Cotromoxazol misma dosis, VO, 21 días
oCotrimoxazol comp. (160 mg TMP/800 mg SMX): 2 comp/ 8 h, VO, 21 días
Formas graves:Pentamidina 3-4 mg/kg/día, IV, 21 días
oClindamicina 600-900 mg/6-8 h, IV o VO
+Primaquina 30 mg/día, VO, 21 días
Formas leves-moderdas:Dapsona 100 mg/día, VO
+TMP 15-20 mg/kg/día, VO (en 3 tomas), 21 días
oClindamicina 300-450 mg/6-8 h,VO + Primaquina 15-30 mg/día, VO, 21 días
En formas graves añadir corticoides:Prednisona 40 mg/8 horas, 5 días; 40 mg/día, 5 días; 20 mg/día, 11 días

Profilaxis primaria y secundaria de PCPIndicaciones para iniciar profilaxis
Profilaxis primaria o secundaria Indicaciones para retirar profilaxis
Profilaxis primaria:• CD4 < 200• Candidiasis oral• FOD durante > 20 días• CD4 < 14%• Enf. definitoria de SIDA
Profilaxis secundaria:• Neumonía por P.
jirovecii
Elección:TMP/SMX 160/800 mg, 1 comp., 3 días a la semanaAlternativas:• TMP/SMX 40/80 mg, 1 comp. día• Dapsona 100 mg/día, • Dapsona 100 mg/día +
Piremetamina 50 mg + ác folínico15 mg, 2 días a la semana
• Pentamidina 300 mg en aerosol o 300 mg IV cada 28 días
• Atovacuona 1500 mg+/-Piretamina 25 mg/día y ác. folínico15 mg/día
• Sulfadiacina 1 g/12 h + Piretamina25 mg/día + ac. folínico 15 mg/día
• Sulfadoxina-piretamina (Fansidar) 1 comp. + ac. folínico 15 mg, 1 o 2 días a la semana
Pacientes con TCRdurante más de 6 meses que tengan una carga viral plasmática del VIH indetectable y unos CD4 > 200 mm3 al menos durante 3 meses

Tuberculosis
• Todos los pacientes con TBC deben ser testados para descartar una coinfección con VIH
• La TBC activa es 20 - 40 veces más frecuente en infectados por VIH
• Alrededor de un 15% de los nuevos casos de TBC se producen en infectados por VIH
• La mayoría de casos de TBC representan infección recientemente adquirida más que reactivación

Tuberculosis
• La TBC es la complicación pulmonar más común del VIH en África, puede aparecer en cualquier momento de la infección VIH y constituye un problema de salud tanto personal como público. En USA es más común en ADVP
• La RX tórax tiende a correlacionarse con el nº de CD4. Con CD4 > 200 cél/microL comúnmente presentan patrón pot-primario (“típico”); con CD4 < 200 cél/microLlas RX suelen ser normales o compatibles con infección TBC primaria


Bibliografía• Benito N, Moreno A, Miro JM, et al. Pulmonary infections in HIV-infected
patients: an update in 21st century. Eur Respir J. 2012; 39: 730 – 45.
• Morris A, Crothers K, Beck JM, Huang L on behalf of the American ThoracicSociety Committee on HIV Pulmonary Disease. An official ATS WorkshopReport: Emerging Issues and Current Controversies in HIV-AssociatedPulmonary Diseases. Proc Am Thorac Soc. 2011; 8: 17 – 26.
• Corbett EL, Watt CJ, Walker N, et al. The growing burden of tuberculosis: global trends and interactions withthe HIV epidemic. Arch Intern Med. 2003; 163: 1009 – 21.
• Girardi E, Palmieri F, Cingolani A, et al. Changing clinical presentation and survival in HIV-associated tuberculosis after higly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2001; 26: 326 – 31.