partie theorique -...
TRANSCRIPT

- 1 -
PARTIE THEORIQUE INTRODUCTION .......................................................................................................... 5 HISTORIQUE ............................................................................................................... 7 ANATOMIE PATHOLOGIQUE....................................................................................... 11
A. LES LESIONS DU SYSTEME NERVEUX PERIPHERIQUE : ............................................ 11 B. LA TOPOGRAPHIE DES LESIONS ............................................................................... 13 C. LES LESIONS ASSOCIEES ........................................................................................... 13 D. LES CONSEQUENCES DE LA DEMYELINISATION AIGUE.......................................... 14 E. LA CORRELATION ANATOMO-CLINIQUE ENTRE DEMYELINISATION ET SGB ...... 14
PHYSIOPATHOLOGIE-ETIOPATHOGENIE .................................................................... 15 A. INTRODUCTION ....................................................................................................... 15 B. PHYSIOPATHOLOGIE ................................................................................................. 15 C. FACTEURS DECLENCHANTS ........................................................................................ 16
EPIDEMIOLOGIE ..................................................................................................... 25 ASPECTS CLINIQUES ET PARACLINIQUE ..................................................................... 27
A. Généralités ................................................................................................................ 27 B. Le diagnostic de l'insuffisance respiratoire aigue ................................................ 28 C. La symptomatologie: ................................................................................................ 29 D. La chronologie ......................................................................................................... 36 E. L'analyse du LCR ....................................................................................................... 38 F. Le bilan électrophysiologique ................................................................................ 39 G. La radiographie pulmonaire ................................................................................... 42
LE BILAN ETIOLOGIQUE .............................................................................................. 43 LES FORMES CLINIQUES ............................................................................................ 45
A. Les formes selon le terrain ...................................................................................... 45 B. Les formes symptomatiques ................................................................................... 48 C. Les PRNA secondaires .............................................................................................. 50 D. Les pathologies associées au SGB ......................................................................... 55
LE DIAGNOSTIC DIFFERENTIEL .................................................................................. 57 PRISE EN CHARGE GLOBALE ....................................................................................... 60
A. INTRODUCTION ....................................................................................................... 60 B. APPROCHE PRATIQUE .............................................................................................. 60
TRAITEMENT SYMPTOMATIQUE ................................................................................ 62 A. Défaillance du carrefour aéro-digestif ................................................................. 62

- 2 -
B. La défaillance respiratoire ...................................................................................... 62 C. Défaillance cardiovasculaire ................................................................................... 65 D. Assistance gastro-intestinale ................................................................................ 66 E. Assistance vésico-sphinctérienne .......................................................................... 66 F. Le traitement de la douleur .................................................................................... 66 G. L’équilibre nutritionnel et hydro électrolytique ................................................... 67 H. Le soutient psychologique ..................................................................................... 68 I. Les complications ..................................................................................................... 69
LES TRAITEMENTS SPECIFIQUES ................................................................................ 70 A. Pourquoi ? ................................................................................................................. 70 B. Buts ........................................................................................................................... 71 C. Concept général ...................................................................................................... 71 D. Les moyens .............................................................................................................. 72
1. Effet de la corticothérapie .................................................................................. 72 2. Effet des échanges plasmatiques ...................................................................... 72 3. Effet de fortes doses d'immunoglobulines (IgG) utilisées seules
ou en association avec les échanges plasmatiques ....................................... 80 E. Utilisation des mesures spécifiques ....................................................................... 81
REHABILITATION ET COMMUNICATION .......................................................................... 83 A. La réhabilitation ....................................................................................................... 83 B. La communication ................................................................................................... 85
EVOLUTION ET PRONOSTIC ....................................................................................... 87 A. L’évolution ................................................................................................................ 87 B. Le pronostic ............................................................................................................. 88
CONCLUSION ............................................................................................................ 90
PARTIE PRATIQUE MATERIELS ET METHODES ............................................................................................ 93
A. Choix des patients ................................................................................................. 93 B. Critères d’inclusion ................................................................................................. 93 C. Fiche d’exploitation ................................................................................................ 94 D. Nos résultats ............................................................................................................. 96 E. Méthodologie statique ............................................................................................. 96
RESULTATS ................................................................................................................ 97 v CHAPITRE I : « Les données épidémiologiques de l’étude » ............................... 97 v CHAPITRE II : « L’histoire de la maladie » ............................................................. 105 v CHAPITRE III : « L’examen physique » ................................................................... 108

- 3 -
v CHAPITRE IV : « Les examens complémentaires » ............................................... 115 v CHAPITRE V : « La prise en charge thérapeutique » ............................................ 120 v CHAPITRE VI : « Le profil évolutif »........................................................................ 128
DISCUSSION ................................................................................................ 132 FIGURES ...................................................................................... 144 TABLEAUX ................................................................................... 153 ANNEXES..................................................................................... 157 RESUMES ..................................................................................... 163 BIBLIOGRAPHIE ............................................................................ 166

- 4 -
ADH : Hormone anti-diurétique.
AIDP : Acute Inflammatoiry Demyelinisating Polyneuropathy.
AMAN : Acute Motrice Axonal Neuropathy.
AMSAN : Acute Motrice and Sensitif Axonal Neuropathy.
BHM : Barrière hémato-méningée.
CJ : Campylobacter Jejuni.
CMV : Cytomégalovirus.
CV : Capacité vitale.
DAC : Dissociation albumino-cytologique.
EH : Echelle d’handicap.
EMG : Electromyogramme.
Ig : Immunoglobulines.
Ig IV : Immunoglobulines intraveineuses.
L.C.R : Liquide céphalo-rachidien.
LED : Lupus érythémateux disséminé.
LPS : Lipopolysaccharides.
MI : Membres inférieurs.
MP : Mycoplasma pneuminae.
MS : Membres supérieurs.
PRNA : Polyradiculonévrites aigues.
ROT : Reflexes ostéo-tendineux.
SGB : Syndrome de Guillain Barré.
SNP : Système nerveux périphérique.
VAS : Voies aériennes supérieures.
VHB : Virus de l’hépatite B.

- 5 -
INTRODUCTION:
Les polyradiculonévrites aigues (PRNA) constituent un groupe hétérogène de
maladie neurologique périphérique dont le syndrome de Guillain Barré (SGB) représente
la forme la plus fréquente.
Elles correspondent à une atteinte inflammatoire démyélinisante diffuse du
système nerveux périphérique (SNP) d'origine probablement immunologique,
déterminant un syndrome neurogène périphérique. [1]
Cependant, le SGB se distingue des PRNA par sa relative bénignité, évoluant
habituellement vers la guérison.
Certes, le diagnostic du SGB est clinique, mais, il ne pourra souvent être établi de
façon formelle qu'au terme d'une évolution caractéristique en 3 phases:
• Phase d'extension.
• Phase de plateau.
• Phase de récupération.
Précédés souvent par une phase prodromique, non nécessaire pour porter le
diagnostic, mais très évocatrice de SGB. [2]
Les examens complémentaires, comme la ponction lombaire, sont surtout
indispensables pour écarter une autre étiologie, plus particulièrement une méningo-
radiculite. [2]
Bien que décrite historiquement comme bénigne vu son évolution spontanément
favorable, il reste une urgence médicale dont la gravité peut justifier une
hospitalisation en réanimation. [1,3]

- 6 -
Un élément essentiel de la prise en charge est l'identification des patients à
risque de développer une insuffisance respiratoire aigue, et nécessitant alors une
ventilation mécanique. L'autre volet thérapeutique est le choix entre les échanges
plasmatiques et les immunoglobulines intra vasculaires; étant donné l'inefficacité
démontrée des corticoïdes.[2] Ces traitements ont permis de réduire considérablement
la mortalité du syndrome, dont les séquelles sensitivomotrices sont en fait assez
fréquentes.
Décrit au début de siècle, par Guillain, Barre, et Ströhl, ce syndrome qui porte
leur nom, a fait l'objet de nombreux rapports, mises au point et controverses.
Le but de notre étude est de :
Identifier le profil épidémiologique des polyradiculonévrites aiguës au sein de
service de NEUROLOGIE au CHU Hassan II de FES.
Faire une étude clinique et paraclinique détaillée des polyradiculonévrites
aiguës.
Détailler les éléments de prise en charge des polyradiculonévrites aiguës.

- 7 -
HISTORIQUE :
A. La « paralysie ascendante aigue » de Landry : Une des intéressantes descriptions de ce que l'on nomme aujourd'hui SGB a été
retrouvé dans les rapports de Jean Baptiste Octave Landry (Fig.3) de Thézillat ; (1829-
1865) qui publie, en 1859, le tableau d’ « une paralysie ascendante aigue et
symétrique » :
"Après une phase prodromique de six semaines environ,..., les fourmillements
des extrémités gagnent de proche en proche les parties les plus élevées des membres,
remplacés par l'engourdissement, puis par la paralysie des parties qu'ils abandonnent
successivement. La paralysie, qui frappe surtout la motilité, se propage avec rapidité
des pieds au reste des membres inférieurs, puis aux membres supérieurs, au tronc,
aux muscles respiratoires, à la langue..." Ce malade décède 8 jours après le début de la
paralysie "en manifestant des signes d'asphyxie". [4,5]
Cette forme nommée à l'époque: Syndrome de Landry, n'est cependant rattaché
au SGB que tardivement.
B. Une origine infectieuse est évoquée : Après recherche bibliographique, Landry réunit dix cas de paralysie ascendante
ou centripète aigue, deux cas s'étaient manifestés "pendant la convalescence de
maladies aigues (pneumopathie probable et longue fièvre typhoïde, syphilis)".

- 8 -
Mais il conclut ainsi: "Dans tous les cas, ces influences ne peuvent être
considérées que comme causes éloignées, et il reste à déterminer la cause prochaine
des désordres fonctionnels". [4,5]
C. La description par Guillain, Barré, et Ströhl : Le 13 Octobre 1916, George Guillain (1876-1961), Jean-Alexandre Barré (1880-
1967) et André Ströhl (1887-1977) publient un article où ils décrivent une observation
originale réalisée sur les militaires de la VIéme armée:
" Des troubles moteurs, l'abolition des réflexes ostéo-tendineux (ROT) avec
conservation des réflexes cutanés, des paresthésies avec troubles légers de la
sensibilité objective, des douleurs de pression des masses musculaires, des
modifications peu accentués des réactions électriques des nerfs et des muscles, de
l'hyper albuminose très notable du LCR avec absence de réaction cytologique
(Dissociation Albumino-Cytologique (DAC))". [4,5]
D. Guillain, Barré, et Ströhl : [6] En cette période ; Guillain et Barré collaborèrent au centre neurologique de la
VIéme armée à Amiens, Ströhl était radiologue dans ce même hôpital militaire.
Guillain (Fig.1) fut l’élève des professeurs Raymond puis Pierre Marie, à qui il
succéda comme professeur à la Salpêtrière, en 1923. Il prit sa retraite en 1947, et son
successeur fut Alajouanine.
Barré (Fig.2) était un breton, interne à Paris de Babinski, et travailla avec Guillain
pendant la Première Guerre mondiale. Il fut professeur de neurologie à Strasbourg de
1919 à 1950.

- 9 -
André Ströhl devint professeur de médecine physique en 1925, à Paris, et
contribua à plus de 200 publications, dont plusieurs sur les réflexes et la conduction
nerveuse. Il prit sa retraite en 1957.
E. Le terme « SYNDROME DE GUILLAIN BARRE » apparaît en 1927 : Après 1917 il restait deux maladies : la paralysie ascendante de Landry, et la
maladie décrite par Guillain, Barré et Ströhl. Draganescu et Claudian ont introduit pour
la première fois le terme de “syndrome de Guillain-Barré” en 1927 à Paris, en
présentant à la Société Française de Neurologie, une observation sur radiculonévrite
survenue après une ostéomyélite staphylococcique. [6]
Après la communication princeps de 1916, George Guillain publie en 1936 dans
les "archives of neurology and psychiatry" un mémoire consacré à l'étude de syndrome
de "radiculoneuritis with acellular hyperalbuminosis of the cerebrospinal fluid"; où il
reprend la symptomatologie globale du SGB. [4,5]
Dans son chapitre: "les problèmes étiologiques et pathogéniques" Guillain
précise:
" L’affection m'a toujours paru d'origine infectieuse, précédée souvent d'une
légère angine, de troubles intestinaux, il semble s'agir d'une infection spéciale dont,
comme pour nombre d'autres en nosographie, nous ne connaissons pas encore le
germe".
F. De l’histoire récente :
§ En 1943, Bauwarth émet l'hypothèse d'une origine allergique, idée reprise en
1949 par Colares et Coll.

- 10 -
§ En 1949, Haymaker W. avait mené une étude anatomopathologique à partir de
50 cas de PRN mortelles, et avait retrouvé malgré les tableaux cliniques très
divers, une base commune anatomopathologique: l'existence d'un œdème puis
d'une fragmentation et d'une irrégularité des gaines de myéline et des
cylindres, au niveau des nerfs médullaires, de la partie proximale des nerfs
périphériques et des racines. [7]
§ Ce Haymaker W, avec Kernohon J.W proposent de grouper dans le générique
de Landry Guillain Barre, un certain nombre de tableaux neurologiques [7]
dont :
-Les PRNA infectieuses ou idiopathique.
-Les PRN.
-Toutes les formes cliniques de paralysie ascendante.
-Les encéphalomyélites.
§ En 1960, Osler et Sidell proposent de définir le SGB par 12 critères
diagnostiques ; et en 1966, Mac Farland et Col ne retiennent que 6 critères.
§ En 1978, Asbury publie d’autres critères de SGB [8]. Ces critères comprennent
des données cliniques et para cliniques, ils sont réactualisés en 1990 et sont
les plus utilisés actuellement.
§ La première étude tentant de prouver l’efficacité des échanges plasmatiques
s’est déroulée en 1984.
à Ces différentes étapes retracent les difficultés à cerner ce syndrome.

- 11 -
ANATOMIE PATHOLOGIQUE :
A. LES LESIONS DU SYSTEME NERVEUX PERIPHERIQUE : La lésion inflammatoire et la démyélinisation aigue sont les caractéristiques
anatomopathologiques fondamentales du SGB.
1. La lésion inflammatoire : (Fig.4)
La lésion principale dans le SGB est la présence d’infiltrats inflammatoires
lymphocytaires et macrophagiques au niveau du SNP. Ces infiltrats sont disposés en
manchons autour des petits vaisseaux de l’endonerve, et accessoirement de l’épinerve
[9,10]. Notons que ces infiltrats inflammatoires ne présentent aucun caractère de
spécificité, contrairement aux PRN des affections [11], telles que la sarcoïdose, ou les
collagénoses, où les lésions sont spécifiques de la maladie générale en cause.
2. La démyélinisation segmentaire : (Fig. 5, 6, 7, 8)
La réaction inflammatoire détruit la gaine de myéline entrainant une
démyélinisation segmentaire [12] qui met à nu, sur une partie de sa longueur, un axone
qui reste souvent intact ; sauf en cas de réaction inflammatoire importante ou
prolongée, où il peut y avoir une dégénérescence axonale [13], dite « dégénérescence
wallérienne », qui serait le témoin de lésion axonale sévère.
3. La dégénérescence Wallérienne : [13] (Fig. 9)
Les neurones endommagés passent en mode réparation, en grossissant le
volume des segments proximaux, et en changeant leur patron d’expression protéique,
pour favoriser la production de protéines associées à la croissance.

- 12 -
Par contre, les axones atteints qui sont déconnectés des pericaryons neuronaux
se fragmentent, la vague de cette fragmentation se propage en direction rétrograde,
jusqu’à la fragmentation totale, ce qui complète le processus de dégénérescence
Wallérienne.
4. Prolifération des cellules de Schwann : [13-15] (Fig. 9, 10)
La perte de contact entre les cellules de Schwann et les axones en
dégénérescence, force les cellules de Schwann myélinisantes à modifier leur
composition protéique en se dé-différenciant pour former les neurolemmocytes
capables de produire : ü des facteurs de transcription qui favorisent la prolifération des cellules de
Schwann qui s’alignent en colonnes cellulaires formant les bandes de Büngner ou tubes
de régénération
ü des molécules d’adhésion cellulaires, qui permettent d’installer un pont
tissulaire (le neuroma), tentant ainsi de rétablir la jonction entre les segments
proximaux et distaux
ü des neurotrophines, et des facteurs de croissance réputés d’accroître la
survie et la régénération axonale.
5. Régénération axonale et remyélinisation : [13-15] (Fig. 10)
Il se forme aux bouts des axones atteints du SNP, des cônes de croissance
(structures effilées à forte concentration mitochondriale et micro tubulaire)
constituants ainsi de véritables unités d’avancée axonale assurant la régénération et la
guidance axonales. Ces cônes de croissance sont attirés vers les bandes de Büngner
qui offrent des conditions propices pour favoriser la régénération axonale.

- 13 -
Les neurotrophines produites par les macrophages induisent la remyélinisation
des fibres nouvellement régénérées par les cellules de Schwann. Ce processus est
primordial pour rétablir la conduction saltatoire.
6. Au total :
Notons que des lésions d’âges différents peuvent coexister, en raison de la durée
de la phase d’extension de la maladie inflammatoire.
La prédominance des lésions myéliniques par rapports des lésions axonales, et la
présence de processus de régénération des axones et leur subséquente remyélinisation
dans le SNP, expliquent la régression habituelle de ce syndrome, et la restauration
anatomo-fonctionnelle relativement rapide. [11-13]
B. LA TOPOGRAPHIE DES LESIONS : [11]
Le processus lésionnel se caractérise par sa dispersion et son extrême diffusion
au niveau du SNP : racines antérieures et postérieures, plexus, troncs nerveux sur toute
leur longueur jusqu’à leurs extrémités, ganglions des racines dorsales, terminaisons
nerveuses intramusculaires, ganglions et chaînes sympathiques.
C. LES LESIONS ASSOCIEES :
Les lésions du SNC peuvent exister, elles consistent en infiltrats lymphocytaires
péri-vasculaires. Un infiltrat méningé inconstant et modéré peut expliquer la discrète
pléiocytose du LCR. [11]

- 14 -
D. LES CONSEQUENCES DE LA DEMYELINISATION AIGUE : Quand la démyélinisation se produit, le potentiel d’action est incapable de faire
un pont sur les lacunes entre « ce qui était » nœuds de Ranvier (Fig. 11). Par
conséquent, les impulses ne peuvent pas atteindre leurs cellules cibles pour déclencher
une réponse, ainsi, il en résulte un déficit sensitif et/ou moteur (Fig. 12). Ceci entraîne
une faiblesse musculaire non seulement dans le SGB, mais aussi dans autres maladies
démyélinisantes, comme par exemple : sclérose en plaque, myélite transverse. [16]
Dés lors qu’il s’agit d’une déperdition de la myéline qui est responsable de SGB,
les nerfs périphériques, qui sont fortement myélinisés (nerfs moteurs), sont atteints de
façon plus sévère que ceux non myélinisés (nerfs de médiation algique, ou de thermo-
contrôle). [17]
E. LA CORRELATION ANATOMO-CLINIQUE ENTRE DEMYELINISATION ET
SGB :
Les lésions histologiques et les conséquences électro-physiologiques expliquent
l’évolution de la maladie :
Le caractère aigue de la démyélinisation explique le début brutal.
La démyélinisation est le plus souvent ascendante, car elle touche
préférentiellement l’extrémité distale des fibres les plus longues en premier.
La récupération motrice spontanée est la conséquence de l’arrêt du
processus de démyélinisation, et la reprise de celui de remyélinisation.
Les séquelles motrices sont observées lorsque la phase de plateau est
prolongée, ou lorsque la récupération n’a pas été optimale.

- 15 -
PHYSIOPATHOLOGIE-ETIOPATHOGENIE
A. INTRODUCTION: Le SGB a probablement une origine « auto-immune » ; c'est-à-dire il se présente
comme le résultat d’une réponse immunitaire inappropriée, cette hypothèse est
supportée par :
v La démonstration expérimentale que le sérum des malades souffrant de
SGB détériore la myéline périphérique et les cellules de Schwann. [18]
v Dépôt des immunoglobulines le long des gaines de myéline [18], et
l’invasion lymphocytaire dans le ganglion dorsal des racines [19] des nerfs périphériques
des porteurs de SGB.
v La déposition des anti-corps, et du complément, et l’infiltration par des
cellules mononucléaires dans les nerfs périphériques et leurs racines [20] retrouvées
lors des recherches anatomo-pathologiques et expérimentales.
v La multitude d’anomalies des paramètres immunitaires trouvés durant la
phase aigue de la maladie, tel que le niveau élevé de la concentration sérique en Ig et
en complément chez les porteurs de SGB. [21]
v Sa réponse favorable aux traitements immunitaires. [21]
B. PHYSIOPATHOLOGIE : [22] (Fig. 13)
Les observations sur les modèles animaux de polyradiculonévrite suggèrent que
la première étape de la maladie serait une activation d'effecteurs lymphocytaires qui du
fait de leur état d'activation peuvent passer à travers la barrière hémato-nerveuse

- 16 -
(BHN), et pénétrer dans le SNP, pour aller interagir avec ses cellules. Ils induisent
ensuite une inflammation, une altération de la BHN, un recrutement inflammatoire
constitué de lymphocytes T, NK, B et de macrophages et la diffusion de facteurs
sériques : anticorps, cytokines, différents médiateurs inflammatoires, dont le rôle
physiopathologique est soutenu par le dépôt de fractions du complément et la
présence du complexe d'attaque membranaire (C5b9). Ainsi, une démyélinisation
segmentaire inflammatoire se créée, pouvant aller jusqu'à l'interruption de l'axone.
C. FACTEURS DECLENCHANTS :
Les causes de l'agression immunologique sont moins connues. On suspecte très
fortement une origine infectieuse à cette pathologie, et beaucoup d’autres événements
sont impliqués comme facteurs déclenchants la survenue de SGB. Cependant, une
diverses études ont tenté d’établir une relation de cause à effet entre un ou plusieurs
de ces événements, et l’apparition secondaire du syndrome neurologique, mais elles
restent, ces études, contradictoires.
1) Les infections :
a. Généralités :
L’origine infectieuse est fortement suspecte vue les faits suivants :
v L’association de SGB avec un antécédent infectieux dans les 2semaines
précédents l’installation des symptômes, dans plus de 50% des cas [18,23], voire 60% des
cas, si on élargie la période à 6 semaines avant l’apparition des signes neurologiques.
[14,15,24,25]
v Des petits groupes de malades atteints de SGB ont été associés à des
entérites bactériennes dues à la contamination des eaux [26],

- 17 -
v L’infection à Campylobacter jejuni définie par étude sérologique et culture
[27-29] était trouvée chez 66% des cas de SGB au cours de l’épidémie estivale rapportée
en Chine de nord [29].
b. Les agents causals :
Le plus communément retrouvé est l’infection des voies aériennes supérieures
(VAS), mais aussi des troubles digestifs [11,15], et on a pu identifier quelque germe
responsable (Tableau I), néanmoins, l’agent causale, est souvent non identifiable.
b. 1. Campylbacter jejuni : [30-33]
i. Généralités :
Le CJ est un petit bacille Gram négatif, spiralé et flagellé, génétiquement très
variable, déterminant ainsi plusieurs souches (plus de 70 sérotypes à antigène
thermo_stable du système Penner, et plus de 100 sérotypes à antigène thermo_labil du
système Lior) [34] . C’est un hôte fréquent du tractus digestif des volailles, tout
particulièrement du poulet, et qui est considéré comme la source majeure de l’infection
humaine à CJ. On estime généralement que 50 à 70% des cas d’infection à CJ sont liés à
la consommation de produits avicoles.
L’infection à CJ chez l’Homme peu être asymptomatique, mais les symptômes
habituels sont ceux d’une gastro-entérite avec une diarrhée liquidienne ou sanglantes.
Elle est considérée actuellement comme la première cause de diarrhée bactérienne
aigue dans le monde. [33]
ii. Relation CJ et SGB :
Cette relation est, actuellement bien établie. Ce qui en est en faveur est :
v A peu près 20% des patients rapportent, comme antécédent, un épisode
diarrhéique. [35]

- 18 -
v Une saisonnalité des infections à CJ a été démontrée pendant la période
estivale ou au début de la période automnale, alors que les SGB liés au CJ seraient
observés, de façon décalée, en période automnale.
v Le délai moyen entre le début des signes neurologiques et la diarrhée est
d’environ 9 jours.
v Sur la base des données sérologiques et bactériologiques, des études
réalisées en Europe et aux USA estiment que 26% à 36% des SGB seraient consécutifs à
une infection à CJ [35,36-38], habituellement dans le mois précédant le début des signes
cliniques. Une pareille étude faite au Japon [39] en a trouvé 45% à peu près. Ce qui
représente la première cause de SGB.
iii. Diagnostic de SGB associé au CJ :
Le diagnostic du SGB associé à une infection à CJ va reposer sur la mise en
évidence soit d’une séroconversion, soit d’une culture positive dans les selles.
iv. Particularités du SGB lié au CJ :
Le SGB secondaire à CJ se distingue par rapport au SGB survenant après les
autres facteurs déclenchants, par :
• Sa survenue majoritairement chez les hommes d’âge mûr (âge moyen : 51,3
ans ; sex-ratio H/F : 1,76) [39]
• Sa gravité particulière [35] : les patients développent plus souvent une forme
axonale (AMAN ou AMSAN), et moins encore la forme démyélinisante (AIDP)
[24% vs 2% et 40% vs 76% respectivement] et présentent le plus souvent une
dégénérescence axonale avec des anomalies électrophysiologiques
particulièrement de mauvais pronostic.
• Une réponse immunitaire type Ig G Anti GM1 (44%) [39]

- 19 -
• Une récupération motrice plus tardive, et des séquelles résiduelles sévères à
un an d’évolution. [35]
v. Pathogénie du CJ en association avec SGB :
Cette pathogénie est expliquée par un phénomène dit de : « mimétisme
moléculaire ». Les gangliosides constituent des molécules de surface importantes du
système nerveux (la membrane neurale, synapses, les noeuds de Ranvier et le myéline),
et selon le concept de mimétisme moléculaire, les anti-corps dirigés contre un épitope
« ganglioside-like » ; c'est-à-dire de structure similaire à celle des gangliosides du SN ;
dans le lipopolysaccharide (LPS) de CJ, ces anti-corps interagissent avec les nerfs
périphériques entraînant ainsi leur destruction. [38]
Ce mimétisme moléculaire était décrit pour la première fois entre le
lipopolysaccharide de sérotype O : 49, et le ganglioside GM1 de tissu nerveux [40].
vi. Autres intervenants pathogéniques :
Le sérotype du micro-organisme (CJ) semble jouer un rôle primordial dans la
pathogénie. Il y a divers sérotypes du CJ :
ü Au Japon, Penner sérotype 19 (O:19) est le plus communément retrouvé en
association avec SGB (aux alentours de 80%) mais, il est rarement responsable de
gastro-entérite sporadique à CJ [41,42].
ü Dans l’Afrique du sud, le sérotype du CJ le plus fréquent est O:41 [43], et le
sérotype O:2 est le plus commun en association au syndrome de Miller Fisher [44,45].
La susceptibilité individuelle semble, elle aussi, être importante dans la
détermination de devenir suivant l’infection à CJ : il a été démontré que le CJ isolé de 2
groupes de malades (un groupe avec SGB, l’autre avec une entérite) contient un épitope

- 20 -
« ganglioside-like » similaire, sauf que les patients ayant développé un SGB, ont des
titres très élevés d’Ac anti-gangliosides GM1 et GD1a [46].
b. 2. CMV :
i. Généralités : [30]
L’infection par le CMV est une maladie fréquente touchant entre 40% et 100% de
la population adulte. La maladie se manifeste le plus souvent de façon
asymptomatique, mais un syndrome grippal d’évolution prolongée est possible.
ii. CMV et SGB :
La présence d’infection à CMV dans les antécédents était mise en évidence chez
11-13% des patients par des études européennes [47,37], et ce taux dépasse même 22%
dans une étude belge. [48] Ainsi, les infections à CMV sont le second facteur étiologique
du SGB, après le CJ.
iii. Diagnostic de l’infection à CMV : [30]
L’infection à CMV est habituellement diagnostiquée devant une élévation des
IgM avec la mesure de l’index d’avidité pour les IgG pour affirmer la séroconversion et
exclure une infection datant de plus de 3mois.
iv. Les particularités clinico-biologiques de SGB lié au CMV :
Le SGB suivant une infection à CMV semble être une entité distincte, car il se
distingue par : [39]
• Sa survenue plus communément chez les femmes d’âges plus jeunes,
• Son début plus sévère avec des difficultés respiratoires, donc un recours plus
fréquent à une ventilation.

- 21 -
• Son tableau clinique marqué très fréquemment par une atteinte des nerfs
crâniens (usuellement paralysie faciale bilatérale), et par des troubles sensitifs
sévères, mais avec une neuropathie motrice moins fréquente
• La réduction du potentiel d’action des nerfs sensitifs est trouvée plus chez ce
groupe, mais la vitesse de conduction nerveuse et l’activité de dénervation ne
différent pas significativement des autres groupes.
• Pour ce qui est évolution, ce groupe de malade a une récupération retardée
par rapport aux autres SGB, mais le pronostic à long terme reste bon.
v. La pathogénie :
La pathogénie exacte n’est pas encore claire, le concept de mimétisme
moléculaire est proposé comme mécanisme possible, car des Ac anti-GM2 ont été
retrouvés plus fréquemment (de façon significative) chez les sujets à SGB associé au
CMV, que chez le groupe témoin. [49]
b. 3. Autres associations infectieuses de SGB :
v Les associations de SGB avec les infections à Epstein-Barr virus ou à
Mycoplasma pneumoniae (MP) sont les plus retrouvées (respectivement 10% et
5%) en comparaison avec les patients témoins. [37,47]
v L’infection à HIV est bien connue en association avec SGB qui peut survenir
durant la séroconversion. [50]
v Le SGB a été également rapporté après la maladie de Lyme. [51]
v Mais la mise en évidence sérologique des infections à Haemophilus influenzae,
parainfluenzae type1 virus, influenza A et B virus, adénovirus, Varicella Zoster
Virus, et parvovirus B19, chez les porteurs de SGB, n’est pas plus que chez le
groupe témoin. [37,47]

- 22 -
v De nombreuses autres associations ont été citées, comme les hépatites A, B, C,
D, la fièvre typhoïde, paludisme, mais restent limitées à des anecdotiques cas
rapportés.
2) LES VACCINS :
a. Généralités :
Plusieurs sont les rapports qui ont tirés l’attention sur la possible association
entre SGB et plusieurs vaccins, mais l’association temporelle entre 2 événements
(vaccin et SGB) ne signifie pas obligatoirement la présence de relation de cause à effet.
b. Le vaccin de la grippe :
Durant le programme de vaccination contre « grippe porcine » à New Jersey en
1976-77 aux USA, il a été rapporté l’augmentation du nombre de cas de SGB, ce qui a
conduit une étude épidémiologique qui a montré que le risque relatif de développer un
SGB après vaccination passe de 4 à 7.8 après une période de 6 semaines [52] , Après, un
groupe d’expert a montré que 8,6 cas de SGB/1million de vaccination à Michigan et
9,7cas/1million à Minnesota sont attribués à la vaccination. [53]
Pour conclure, l’association est statistiquement significative, néanmoins, l’effet
bénéfique de ce vaccin dépasse largement le risque de survenue de SGB.
c. Polio orale :
Une étude rétrospective effectuée en Finlande montre une augmentation
significative de l’incidence de SGB durant la compagne de vaccination nationale par la
polio orale [54]

- 23 -
De plus, l’étude analytique d’Amérique latine, trouve une association entre le SGB
et la compagne de vaccination de masse par polio orale entre 1989 et 1991, et qui est
NON temporale. [55]
d. Vaccin TTC :
Le risque de développer SGB après vaccin TTC a été évalué en utilisant des
données retirées de 2 études épidémiologiques à grande échelle; et on a trouvé
quelques cas de SGB après vaccination qu’on peut expliquer par simple hasard, et les
auteurs ont conclu qu’il n’y avait pas d’association signifiante. [56]
e. Vaccins Rougole-Oreillon-Rubeole, et vaccin des oreillons :
La relation entre ces vaccins et le SGB ne sont qu’à titre anecdotique, car des
larges études épidémiologiques, intéressants plus de 2000 enfants sud-américains
porteurs de SGB après vaccination de masse contre les oreillons en 1992-1993, ont
échoué d’établir une relation causale statistiquement irréfutable. [57]
f. Vaccin contre VHB:
Le SGB était bien rapporté après vaccination contre l’hépatite B [58]. Une étude de
pharmaco-vigilance réalisée après sa mise sur le marché, a rapportée 9 cas parmi
850000 à peu près de destinataires de ce vaccin. Cette étude n’a pas montré de
relation épidémiologiquement convaincante entre les effets secondaires neurologiques
et le vaccin. [59]

- 24 -
3) Les médicaments :
i. Gangliosides :
Les rapports sur les neuropathies aigues suivant le traitement parentéral par
gangliosides cérébraux ont été enregistrés en Espagne, l’Allemand, et en Italie [60-62],
cependant, une étude épidémiologique rétrospective de cas témoin menée sur un très
large échantillon de la population de la région Latium de l’Italie [63] n’a pas supporté la
présence d’association entre le traitement par les gangliosides et SGB.
ii. Autres :
Plusieurs sont les médicaments qui ont été impliqués comme facteurs
favorisants : une pénicilline, un antispasmodique, et les contraceptifs oraux [64] ; mais
une revue des rapports publiés sur les médicaments associés au SGB conclue
qu’aucune relation de cause à effet ne peut être établie définitivement. [65]
4) Autres associations « anecdotiques » :
Divers associations et plusieurs facteurs déclenchants ont été rapporté comme
favorisants la survenue de SGB, dans la majorité des cas, ils correspondent à des cas
isolés et individuels, par exemple :
-Chirurgie.
-Anesthésie péri durale.
-transplantation rénale.
-Lupus Erythémateux Aigue Disséminé.
-Lymphome.
-Sarcoïdose.
-Morsure de serpent.
-Contact avec les animaux.
La signification statistique de ces relations n’est pas claire, comme elles ne
peuvent être que des associations hasardeuses.

- 25 -
EPIDEMIOLOGIE:
A. Incidence : àCette affection est devenue la cause la plus fréquente des paralysies aigues
flasques, après l'éradication de la poliomyélite antérieure aigue, dans les pays
développés. Cependant, elle reste encore une rare maladie. [66]
àDans la population générale, des études rétrospectives ont permis d’évaluer le
taux d’incidence de ce syndrome entre 0,9 et 1,9 pour 100.000 habitant par an ; ce qui
fait que le taux moyen de 1,5cas/an/100.000 habitants est habituellement retenu. [66]
àLa plupart des cas sont sporadiques, mais on note trois épisodes
internationaux pouvant être qualifiés d’épidémiques [67-68] :
v En Colombie : 1968.
v En jordanie : 1968.
v Aux USA : après une compagne de vaccination contre la grippe.
B. Age : Tous les âges peuvent être touchés par ce syndrome, mais, on distingue une
distribution bimodale avec de 2 pics de fréquence [69,70] : 15-34 ans et 50-74ans.
L’incidence semble augmenter progressivement avec l’avancée dans l’âge : En
Europe et le nord d’Amérique, l’incidence passe de moins de 1/100.000 chez les
patients plus jeune que 30ans, à environ 4/100.000 des patients plus âgés que 75ans.
[71]

- 26 -
C. Sexe : Tous les rapports sont d’accords que l’homme est 1,5 fois plus touché que la
femme.
D. Répartition saisonnière : Les résultats sont discordants, dans la population générale, certains auteurs ne
retiennent aucune tendance saisonnière, mais certaines séries notent une variation
saisonnière, et dégagent comme saisons prédominants : l’hiver et le printemps. [72,73]
E. Répartition annuelle : Dans les îles Caraïbes, l’incidence a passé spectaculairement de 1,62/100.000
entre 1987 et 1991, à 3,10/100.000 entre 1992 et 1999 [74] ; alors que des études
similaires, dans d’autres régions rapportent une stable incidence de SGB avec la
succession des années.
F. Répartition géographique : Il n’y a pas de variation géographique consistante.
G. La variation raciale : Aucune prédominance de race n’est mise en évidence.

- 27 -
LES ASPECTS CLINIQUES ET PARACLINIQUES DU SGB
A. Généralités : La présentation clinique la plus classique du SGB est celle d'une paralysie
extensive d'évolution aigue et ascendante, avec un déficit globalement symétrique,
troubles sensitifs modérés (le plus souvent subjectifs), une atteinte des paires
crâniennes et une abolition des réflexes ostéo-tendineux. Ce tableau clinique est quasi
pathognomonique d'un SGB quand il est complet, mais les polymorphismes cliniques de
ce syndrome (en rapport avec la distribution multifocale des lésions) sont courants, ce
qui rend compte de quelques difficultés diagnostiques. Mais pour le diagnostic positif
du SGB, il existe actuellement des critères diagnostiques établis. [75]
L'éléctroneuromyogramme (EMG) permet, de classer le SGB selon les différentes
formes incluses sous un seul générique, un unique prototype, qui est le SGB. Ces
formes sont:
• Polyradiculoneuropathie inflammatoire aigue: AIDP=Acute Inflammatoiry
Demyelinisating Polyneuropathy.
• Neuropathie aigue axonale motrice: AMAN= Acute Motrice Axonal Neuropathy.
• Neuropathie aigue axonale motrice et sensitive: AMSAN=Acute Motrice and
Sensitif Axonal Neuropathy.
• forme inexcitable.
Cette distinction entre les différentes entités est adoptée pour les auteurs anglo-
saxons, mais la majorité des auteurs français s'accordent pour définir ce syndrome en
restant le plus fidèle possible à sa description originelle en 1916 par Guillain, Barre et
Ströhl.

- 28 -
Bien que le traitement spécifique de ces entités électrophysiologiques soit
identique, leur distinction revêt une pertinence physiopathologique, mais également
pronostique. En effet, la survenue d'une insuffisance respiratoire aigue est plus
fréquente dans les formes démyélinisantes [76] ; et les séquelles sont plus sévères dans
les formes inexcitables et axonales [77,78]
B. Le diagnostic de l'insuffisance respiratoire aigue: L’insuffisance respiratoire (due à la paralysie des muscles expiratoires et
inspiratoires, notamment du diaphragme) demeure la complication la plus grave du
SGB, sa prévalence est en moyenne de 20 % des cas (de 6 à 44 % selon les séries). [79,80]
Il est capital d’évaluer de la fonction respiratoire :
Sur le plan clinique, l’insuffisance respiratoire se manifeste par une limitation de
l'ampliation thoracique, avec une respiration superficielle ou abdominale et des signes
de lutte "tirages intercostal, sus claviculaire et xiphoïdien, battement des ailes du nez"
la dyspnée est souvent discrète, la tachypnée est habituelle, la bradypnée signe la
phase de pré arrêt respiratoire, témoin de la fatigue des muscles respiratoires.
La gazométrie artérielle est le plus souvent normale, l'hypercapnie et l'hypoxémie
n'étant observées que dans les formes sévères. [81] L'atteinte respiratoire peut aussi
résulter de l'atteinte de la Xème paire crânienne, ou se voir au décours d'un trouble de
déglutition.
La mesure de la capacité vitale [82] est un outil de surveillance capital, elle doit
être effectuée à l'admission, et réitérée au cours de l'hospitalisation. A côté de la
spiromètrie, la surveillance clinique et para clinique est importante.

- 29 -
C. La symptomatologie:
1) Les signes de début :
Les signes de début sont variés, la symptomatologie débute habituellement par
des paresthésies des extrémités, à types d'impressions d'engourdissement, de
fourmillements, de picotements, ou par des douleurs, telles que : myalgies, dorsalgies,
lombalgies, radiculalgies.
Le déficit moteur peut apparaître d’emblée, et être le premier signe, donnant des
parésies qui intéressent initialement, dans la plupart des cas, les MI, mais là aussi, elles
peuvent d'emblée concerner les quatre membres, ou plus rarement les MS seuls.
L’atteinte des paires crâniennes est rarement inaugurale.
2) Le déficit moteur : [83]
La présentation typique consiste en une difficulté à marcher, à se relever, le
déficit moteur, telle une vague ascendante, s’étends alors à l'ensemble de la
musculature réalisant une quadriplégie flasque touchant à la fois les racines et les
extrémités des membres, de même pour les muscles du tronc, de la nuque, du
pharynx et les muscles respiratoires.
Il est primordial de codifier l'intensité des déficits, car beaucoup d'auteurs
corrèlent la sévérité de l'atteinte motrice, au risque de ventilation assistée et à la
fréquence des séquelles, et ce, d'autant que l'installation des troubles est rapide.
Evaluation de la sévérité de la fonction motrice :
La mesure du déficit moteur et son retentissement fonctionnel peuvent
s'effectuer à l'aide d'un score fonctionnel "MRC-Sun score", des échelles d’handicap

- 30 -
(EH), et plus récemment le Guillain Barre syndrome disability score.[5,83,84] [Annexes:1,3]
Ces échelles permettent d'une part, de suivre de manière simple l'évolution du
déficit moteur, et d'autre part, de distinguer précocement des stades cliniques de
gravité et de pronostic différents.
3) Les signes sensitifs : [15,83] Les signes sensitifs précèdent souvent les anomalies motrices mais ils peuvent se
manifester concomitamment ou à posteriori. Classiquement, on différencie entre :
Les signes subjectifs: sont très fréquents, voir quasi constants, il s'agit:
*de paresthésie ou sensations d'engourdissement, qui touchent initialement les
extrémités et gagnent progressivement l'ensemble du corps et la face.
*de douleurs, plus ou moins diffuses, intéressant les muscles et les articulations.
*des névralgies sont parfois retrouvées, pouvant être de type radiculaire et alors
mises en évidence par la manœuvre de Lasègue.
*D’autres part, l'existence de céphalées, de douleurs de la nuque, et/ou de
dorsalgie d'autant qu'elles s'accompagnement de manifestations digestifs
"vomissements" et de fièvre peuvent engendrer des erreurs diagnostiques.
Les signes objectifs: l'examen clinique est, par contre, pauvre.
Une étude globale relate l'existence de paresthésie au niveau des MS dans 66%
des cas, et des MI dans 75% des cas, contrastant avec l'absence de signes objectifs.
Dans le cas contraire, les déficits portent alors essentiellement sur la sensibilité tactile
et profonde.

- 31 -
4) Les ROT: [83] Les données classiques montrent une aréflexie généralisée dans 60 à 80% des
cas, les ROT peuvent rester conservés, cette persistance ne doit pas faire récuser le
diagnostic, elle est le fait de la distribution multifocale des lésions démyélinisantes, et
donc de la préservation de certains territoires. Les réflexes cutanéo-plantaires sont
habituellement normaux, ainsi que les réflexes cutanés abdominaux.
Les ROT des MI disparaissent souvent avant ceux des MS.
5) L'atteinte des nerfs crâniens : [15,85,86,87]
Les VIIème (facial), IXème (glosso-pharyngien), et la Xème (vague) paires sont le
plus souvent touchées, plus rarement la XIIème paire (grand hypoglosse), en outre, une
ophtalmoplégie, témoignant d'une atteinte des nerfs oculomoteurs, peut survenir dans
15% des cas. [15]
Le nerf facial: dans 53% des cas.
Il est responsable d'une paralysie faciale périphérique souvent bilatérale,
lorsqu’elle est asymétrique, elle se manifeste par une dysarthrie, une incapacité à
siffler, à gonfler les joues et à occlure complètement les paupières.
Sa présence constitue un critère diagnostique majeur, mais ne fait pas partie des
critères obligatoires d'ASBURY. Par contre, elle représente un critère du syndrome
FISHER. Donc, elle doit conduire à la recherche d'une DAC dans le LCR, si des signes
moteurs lui sont associés.
Les nerfs oculomoteurs III et VI : dans 9 à 30% des cas
Les nerfs oculomoteurs commun (III) et le moteur oculaire externe (VI) sont à
l'origine d'une ophtalmoplégie partielle ou plus rarement complète.
L'atteinte du III se traduit cliniquement par un ptosis, un strabisme externe, une

- 32 -
perte d'élévation et d'adduction du globe oculaire, une diplopie croisée, une mydriase
paralytique.
La paralysie du VI, entraîne une perte de l'abduction du globe, un strabisme
interne, une diplopie homonyme.
Le nerf glosso-pharyngien et le nerf pneumogastrique :
Leur atteinte réalise une paralysie du carrefour aéro-digestif qui constitue
l'éventualité redoutable présente dans 20 à 30% des cas.
La recherche des troubles de la déglutition et de la phonation, doit être
pluriquotidienne dès que le diagnostic d'une PRNA est posé, car ces troubles
(exprimant l'atteinte des nerfs mixtes du bulbe) précèdent ou accompagnent souvent
les troubles respiratoires.
Les manifestations cliniques peuvent se limiter à un enrouement de la voix ou
discret nasonnement, une dysphagie aux liquides, et aux solides devant imposer l'arrêt
de toute alimentation orale, dans le but d’éviter les fausses routes, et ainsi, la
pneumopathie d’inhalation.
La paralysie du nerf Grand Hypoglosse IX et du nerf Vague X:
La paralysie du IX est affirmée par l'abolition du réflexe nauséeux, l'anesthésie du
tiers postérieur de la langue avec perte des propriétés gustatives, et le signe du rideau
de Vernet. La voix n'est modifiée que lorsque la paralysie est bilatérale.
En ce qui concerne le nerf X, l'examen peut objectiver une parésie vélo-palatine,
source d'une asymétrie du voile au repos et inductrice du risque de rejet de liquide par
le nez lors de la déglutition. La voix est bitonale du fait de la parésie laryngée, par
inégalité des cordes vocales. Il existe également une anesthésie du voile, du pharynx,
du larynx, multipliant le risque de fausses routes. Il faut considérer, par ailleurs, les
manifestations générales secondaires aux lésions du X : accélération du rythme

- 33 -
cardiaque, et gène respiratoire.
Les autres paires crâniennes:
Au total, chacune des paires crâniennes peut être affectée, hormis le nerf olfactif:
• L'enregistrement électrophysiologique (potentiels évoqués auditifs) a pu
mettre en évidence l'atteinte démyélinisante du nerf acoustique ou VIII.
• Les paresthésies faciales témoignent de l'extension du processus à la Vème
paire crânienne ou le nerf Trijumeau.
• Les anomalies d'articulation ou de mastication dans le cas d'un déficit
bilatéral, et de déviation de la langue, dessinent l'atteinte du XII.
6) Les signes dysautonomiques : [66] a) Généralités:
Les manifestations dysautonomiques, qui reflètent un excès ou une activité
inadéquate des systèmes sympathique et parasympathique, sont fréquentes
(fréquence=65% dans diverses séries), et sont d'autant plus présentes que l'atteinte
neurologique est sévère.
Leur durée est en moyenne de 2 à 3 jours, donc, elles sont souvent transitoires,
et par conséquent, elles peuvent passer inaperçus, mais leur récidive est fréquente et
souvent plus longue.
Elles sont reconnues comme complications du SGB, car ils sont mal contrôlées et
sont responsables de 3 à 14% des décès, notamment dans les unités de soins intensifs.

- 34 -
b) Expression clinique :
Ø Cardiovasculaire : (25% des cas)
à Les anomalies de la fréquence cardiaque :
Les tachycardies, habituellement sinusales, sont signalées dans 10 à 50% des cas,
et sont conséquentes à des dommages vagaux et non à une atteinte cardiaque toxique.
Plus préoccupantes, car pouvant précéder un arrêt cardiaque, sont les
bradycardies. Spontanées, pouvant paradoxalement succéder ou parvenir au décours
d'un accès de tachycardie, ou bien provoquées lors des changements de position ou
lors d'aspirations trachéales.
à Les anomalies de la pression artérielle:
L'HTA, qui est secondaire à une hyperactivité sympathique, est souvent labile,
transitoire, dure quelques heures à quelques semaines, elle persiste rarement après
résolution des symptômes neurologiques. Elle peut s'associer, surtout chez l’enfant, à
des crises convulsives, dans le cadre d’encéphalopathie hypertensive secondaire.
à Les arythmies :
Elles sont variables dans leurs formes (tachycardie atriale paroxystique,
fibrillation auriculaire, tachycardie nodale, et pauses cardiaques...), mais très
dangereuses, car mettent en jeu le pronostic vital.
Un monitoring cardiaque continu est indispensable, voir si besoin la pose d'un
pace-marker.
à Les anomalies de l'électrocardiogramme ECG :
Sont décrits essentiellement des troubles de la repolarisation : sous décalage ST,
onde T plate ou inversée. Plus rarement sont retrouvés des ondes T amples ou
l'allongement du segment ST.

- 35 -
Ø Manifestations systémiques :
à Les troubles vasomoteurs:
Ils sont fréquents, et se traduisent par des sueurs profuses, d'une hypersécrétion
salivaire et bronchique (concourant ainsi à aggraver l'état respiratoire par
encombrement bronchique).
à Les troubles intestinaux et urinaires :
Les signes urinaires sous forme de rétention urinaire, et digestifs, tel l'iléus
intestinal, ne sont pas rares et habituellement transitoires. Leur fréquence est entre 27
à 32 % des cas. Néanmoins, ces signes sphinctériens conservent une grande valeur dans
le diagnostic différentiel.
7) Les autres manifestations : v Atteinte de la musculature intrinsèque de l'œil : [86]
Elle est responsable de pupilles aréactives à la lumière et à l'accomodation.
Un œdème papillaire a été également décrit dans plusieurs observations. On
l'impute alors à l'hyper-protéinorachie et à une diminution de la résorption du LCR. Son
pronostic est tout à fait favorable, il disparaît habituellement lors de la phase de
récupération motrice.
v Signes encéphaliques : [88]
L'association de signes centraux est encore plus rare. De tels cas seront désignés
sous le terme de neuropathie radiculo-myélo-encéphalique.
Des crises convulsives peuvent survenir, avec des troubles de consciences,
hydrocéphalie, dont l'origine est controversée. Il faut éliminer un déséquilibre ionique
tel une hyponatrémie, une poussée hypertensive, il peut s'agir d'hypoxie sur
insuffisance respiratoire aigue.

- 36 -
v Méningisme:
Un syndrome méningé est parfois rapporté au tout début de l'évolution.
v Ataxie : [89]
Il s’agit plutôt d'une pseudo-ataxie par atteinte déficitaire motrice purement
proximale et modérée, ou bien d'une ataxie de type sensitif. Cette dernière s'explique
par la perte de l'orientation de la position des membres, et de la direction du
mouvement en rapport avec une atteinte de la sensibilité profonde.
D. La chronologie: Le profil évolutif classiquement décrit dans le SGB se subdivise selon 3 phases que nous
étudierons successivement :
• la phase D'EXTENSION
• la phase de PLATEAU
• la phase de RECUPERATION
1. La phase d'extension des paralysies : [66, 86,90-92]
Le début de cette phase est brutal ou rapidement progressif. Il correspond
théoriquement à l'apparition du premier signe neurologique. Sa durée est en moyenne
de 12 jours, mais elle peut être brève 2 à 3 jours ou prolongée à 30 jours.
L'évolution de cette phase, caractérisée par la progression des déficits tant en
intensité qu'en territoires touchés, se fait vers l'aggravation à tout moment, par
l'apparition de signes d'insuffisance respiratoire ou de signes dysautonomiques.
Ainsi, une surveillance étroite de ces patients est primordiale, pour détecter la
survenue imprévisible de paralysie des muscles respiratoires et pharyngés, ainsi que
les troubles cardio-circulatoires, nécessitant le transfert immédiat en réanimation, d'où

- 37 -
les PRNA doivent être considérées comme une urgence.
2. La phase de plateau : [90-92]
La phase de plateau ou de maximum des paralysies est ainsi qualifiée car les
signes déficitaires sont effectivement à leur acmé. Elle dure de quelques jours à 2
semaines dans les formes habituelles, elle peut également durer plusieurs semaines, se
terminant au premier signe de récupération motrice. A ce stade, le degré de sévérité
des déficits est très variable.
Le "calme" évolutif de cette phase contraste avec la possibilité de mise en jeu du
pronostic vital, risque présent à tout instant. Ce pronostic est engagé de par:
• l'atteinte des muscles respiratoires dont la déficience est responsable
d'insuffisance respiratoire plus ou moins grave, pouvant conduire à la
nécessité de la VA.
• la survenue brutale de manifestations dysautonomiques, notamment
cardiovasculaire, pouvant être cause d'arrêt cardiaque imprévisible.
• des complications surajoutées tels les problèmes infectieux "pneumopathies,
infections urinaires, septicémie..." et thrombo-emboliques dus à
l'immobilisation prolongée.
3) La phase de récupération : [93,94]
Elle correspond à la troisième phase évolutive du SGB, elle se caractérise par la
régression des signes déficitaires dans l'ordre inverse de leur installation, de la partie
proximale à la partie distale.
Sa durée minimale est en moyenne de 30 jours et le plus souvent elle dure
plusieurs mois, mais il ne faut pas oublier que la détermination de cette durée est

- 38 -
entravée par les difficultés d'appréciation de ses limites. Elle débute au premier signe
de récupération, essentiellement moteur, qui est difficilement identifiée. La
confirmation est apportée par un fait plus marquant, comme la restauration d'une
certaine autonomie de marche.
Le pronostic fonctionnel est, quant à lui, péjorativement dépendant des séquelles
plus ou moins invalidantes, dont la persistance est possible.
E. L'analyse du LCR: Le LCR des patients porteurs de SGB a toutes les qualités d'un transsudat (par
altération de la barrière hémato-méningée) : eau de roche, parfois xanthochromique,
sa pression est normale.
L'analyse du LCR constitue un critère diagnostic, selon la classification proposée
par Asbury. [Annexe 2] ; et la DAC, bien que non pathognomonique des PRNA, est un
signe biologique fondamental. Elle peut manquer lors de la première PL, justifiant ainsi
son absence chez 60% des patients. Par ailleurs, il est recommandé de renouveler ce
geste dès le 10ème jour.
La DAC du LCR se caractérise dans sa forme typique par des constantes : [14,24]
ü une protéinorachie élevée supérieure à 0.45g/1, et dont les valeurs
moyennes sont généralement comprises entre 0.80 et 2g/1.
Elle témoigne de l'inflammation radiculaire multiple, et est généralement décalée
de quelques jours par rapport au début des manifestations neurologiques. Elle est
évidente vers le 15ème jour, et elle est maximale 4 à 6 semaines après le début des
signes neurologiques.
La valeur de la protéinorachie n’est pas corrélée à la sévérité de la maladie et

- 39 -
n'est pas considérée comme un facteur pronostique.
Son absence n'écarte pas le diagnostic si les autres signes sont typiques.
Le profil électrophorétique est de type sérique, dans 80% des cas, on note une
augmentation du taux des gammaglobulines. Et à la phase initiale du SGB, on rapporte
une ascension significative des alpha 2 globulines.
ü une réaction cellulaire inférieure à 3 éléments/mm 3 [95]
La limite supérieure admise pour la cytorachie fait l'objet d'une controverse, fixée
initialement à moins de 3 éléments/mm3, puis par Franz à 30 éléments, un taux plus
élevé oriente vers une méningo-radiculite d'origine soit infectieuse, notamment une
maladie de Lyme, soit inflammatoire ou néoplasique, en particulier un lymphome, et
doit faire récuser le diagnostic de SGB. L'analyse des éléments cellulaires peut montrer
des lymphocytes ou plasmocytes, cellules immunocompétentes.
ü la glycorachie reste toujours normale.
Bien entendu, la PL a un autre intérêt, son analyse est un atout supplémentaire
pour le diagnostic différentiel ou étiologique.
F. Le bilan électrophysiologique :
1. Généralités :
L’EMG constitue un appoint non négligeable pour les diagnostic positif et
différentiel du SGB grâce aux critères électrophysiologiques définis par Asbury en 1990
(annexe 2), il permet aussi d’approcher le mécanisme physiopathologique axonal ou
myélinique, et d’apprécier l’étendue des atteintes et donc d’évaluer le pronostic. (Fig.
12)

- 40 -
2. Les méthodes :
a) Le premier EMG :
Il montre l’aspect classique d‘une neuropathie démyélinisante radiculaire ou
tronculaire. [96]
b) L’EMG de stimulodétection :
Les anomalies traduisant un processus démyélinisant sont les blocs de
conduction, la dispersion temporelle de la réponse motrice, l’augmentation de la
latence distale motrice (traduisant une atteinte des fibres nerveuses à conduction
rapide), la réduction de la vitesse de conduction motrice (traduisant une
démyélinisation étagée) et l’allongement des latences de la réponse F et H (traduisant
l’atteinte radiculaire).
Les anomalies les plus précoces sont le retard de réponse des ondes F et
concernent également l’amplitude des réponses motrice distales.
Les vitesses de conduction motrice et les latences distales sont altérées
secondairement.
Les potentiels sensitifs peuvent être normaux en raison e l’atteinte motrice
prédominante. [97]
Une étude récente BRADCHAW [98] rappelle que les signes électrophysiologiques
les plus précoces permettant de retenir le diagnostic de SGB sont l’abolition du réflexe
H (97% des cas), l’allongement des latences F (84% des cas) et à un degré moindre la
diminution d’amplitude des latences distales motrice (71% des cas).
Le phénomène de bloc de conduction est la caractéristique électrophysiologique
majeure de la forme démyélinisante, il est à l’origine du déficit moteur et sensitif.

- 41 -
Dans les deux premières semaines, ce phénomène de bloc de conduction est
noté dans 30 à 40 % des cas avec les méthodes classique. Les stimulations proximales
en particulier radiculaires à l’aiguille monopolaire améliorent la sensibilité puisque les
blocs notés dans 75% des cas. [93]
Dans une étude électrophysiologique, des stimulations électriques sont utilisées
à la phase initiale du SGB, pour améliorer les possibilités du diagnostic
électrophysiologique. 22 patients ayant un SGB sont examinés avec cette technique
dans un intervalle de 3 à 17 jours à partir de la date du premier signe. Auparavant, un
examen conventionnel a permis le diagnostic d’une forme démyélinisante chez 12 cas.
Pour 10 autres patients, cet examen standard est resté négatif ou insuffisant, et les
stimulations électriques radiculaires ont alors permis d’établir le diagnostic, mettant en
évidence des blocs de conduction. Cette étude confirme la possibilité de lésions
démyélinisantes uniquement proximales du système nerveux périphérique au début du
SGB, et l’intérêt de recherche initialement l’atteinte radiculaire par tous les moyens
possibles : onde F, réflexes H et stimulations proximale à l’aiguille monopolaire. [99]
c) L’EMG de détection :
L’EMG de détection se caractérise par des tracés pauvres avec des unités
motrices déchargeant à des fréquences élevées. L’existence d’activité de dénervation
(fibrillations, potentiels lents de dénervation) traduit la survenue d’une atteinte axonale
primitive ou secondaire à une démyélinisation prolongée.
L’étude de la série italienne montre que le premier EMG effectué trouve une
atteinte axonale primitive chez 30 patients (48.4%) [24], par contre dans la série de

- 42 -
Birouk [100], cette proportion est réduite, l’atteinte axonale ayant concerné uniquement
15% des patients (27 malades).
d) L’EMG de contrôle : [101]
L’intérêt d’un EMG de contrôle est diversement apprécié. La plupart des auteurs
pensent que la répétition des examens électriques n’a pas un grand intérêt, car
l’évolution des anomalies électrophysiologiques n’est pas corrélée à la clinique. [14]
Cependant certains auteurs pensent, qu’effectué à distance de la phase
d’installation ou durant la phase de plateau, l’EMG de contrôle permet de faire un bilan
lésionnel plus complet et donc plus objectif que l’examen initial (étendue des atteintes,
atteinte axonale secondaire). Ceci souligne l’intérêt de mener des études dans ce sens,
afin d’étayer l’apport de L’EMG de contrôle effectué à des intervalles précis pour
confirmer ou infirmer la corrélation électro-clinique.
G. La radiographie pulmonaire :
C’est un élément essentiel et un examen réalisé quotidiennement pour les
patients hospitalisés en milieu de réanimation, dans un but de suivi et de surveillance
de l’évolution de l’état pleuro pulmonaire des patients, il est impératif surtout en
présence en signes cliniques d’encombrement trachéo-bronchiques à l’admission.
Il permet à ce moment de montrer :
- Des foyers d’atélectasie.
- Des foyers de pneumopathie.

- 43 -
LE BILAN ETIOLOGIQUE Les testes classiquement demandés lors de l’hospitalisation sont normaux.
Cependant quelques précisions doivent être apportées.
1. L’hémogramme : Il retrouve une hyperleucocytose souvent à prédominance neutrophile.
2. Le syndrome inflammatoire biologique : La vitesse de sédimentation est accélérée et le taux de fibrinogène élevé. Dans la
plupart des cas, ceci traduit une infection récente (phase prodromique) ou patente
(surinfection broncho-pulmonaire ou infection urinaire).
3. L’ionogramme sanguin : Il peut révéler un syndrome de SHWARTZ-BARTTER, défini comme une sécrétion
inappropriée de l’hormone anti diurétique (ADH), responsable d’une hyponatrémie
(inférieure à 130mmol/1) à natriurèse conservée. Le traitement se limite à la restriction
hydrique.
On observe également une légère hyperglycémie ; ce qui explique certains cas de
diabète déstabilisé lors d’un SGB évolutif sur ce terrain.
4. Le bilan hépatique : Les enzymes hépatiques peuvent être perturbés transitoirement, ainsi que les
enzymes musculaire surtout s’il existe cliniquement des myalgies importantes.

- 44 -
5. Le bilan immunologique : ü L’électrophorèse des protéines sériques :
Sur le plan quantitatif, on ne décèle pas d’anomalies. Mais sur le plan qualitatif,
les fractions alpha 1 et alpha 2 sont souvent significativement élevées, et plus
rarement on observe un pic monoclonale des gammas globulines.
ü Les anticorps anti-système nerveux :
Les anticorps anti-myéline et les complexes immuns circulants sont observés
dans 45 à 50% des cas de SGB. Ces anticorps composés d’IgM et IgG ont pour cible la
fraction protéique P1L du SNP.
ü Les agglutinines froides :
Des auto- anticorps anti-globules rouges responsables d‘anémie hémolytique
sont parfois détectés.
ü Les fractions C3c et C4 du complément :
Plusieurs études admettent comme valeurs pronostiques négatives, des taux
élevées des fractions C3c et C4 du complément.
Ces diverses remarques pourraient se résumer ainsi : les dosages des alpha 1 et
surtout des alpha 2 sériques serait intéressants à titre diagnostique, et ceux des C3c et
C4 à visée pronostique.
6. L’enquête infectieuse : a. Les sérologies :
Elles regroupent les sérologies du CJ, du CMV, du MP, du virus de Epstein Barr
responsable de la mononucléose infectieuse et du virus Herpès Varicelle Zona.

- 45 -
Plus accessoirement, sont retrouvés les virus de la rubéole, de la rougeole, des
oreillons, de l’hépatite A, B, C, les virus inflenzae.
Par contre la PRN peut être révélatrice de la primo-infection HIV et de la maladie
de Lyme (Borrelia Burgdorferi).
b. les vaccins antérieurs :
Sont incriminés les vaccins de la Diphtérie Tétanos Polio, de la Variole et de la
Grippe.
7. Les autres facteurs à recherche selon le contexte clinique et
l’interrogatoire : Il s’agit notamment du LED, des maladies thyroïdiennes, da la myasthénie, de
l’existence d’un cancer, de certains médicaments comme les antidépresseurs, la
chimiothérapie…

- 46 -
LES FORMES CLINIQUES :
A. Les formes selon le terrain : SGB chez l'enfant :
Diverses études cliniques ont prouvé qu'il n'existe pas de différences
fondamentales entre le SGB de l'adulte et celui de l'enfant, à l'exception d'un temps de
récupération plus court et un taux de séquelles moins important chez les enfants par
rapport aux adultes. [102-106]
SGB et grossesse :
Les PRNA représentent 1.5% de la pathologie neurologique chez la femme
enceinte, et dans un grand nombre de cas, les symptômes apparaissent au dernier
trimestre. [107]
Sur les 45 cas relevés dans la littérature, il est impossible de conclure si les
modifications immunologiques engendrées par la grossesse favorisent ou protègent
d'une PRNA.
L'accouchement par voie basse est d'indication large, et sans difficultés
particulières chez les parturientes paraplégiques.
Aucune malformation foetale n'a été décrite à la naissance, même lorsque la PRN
survient au premier trimestre, et aucun nouveau né n'a présenté un syndrome
neurologique évocateur de PRNA.
La grossesse ne semble pas influencer l'évolution des séquelles neurologiques et
n'aggrave pas le pronostic vital. Sur le plan thérapeutique, les auteurs affirment l'effet
bénéfique des plasmaphérèses chez les femmes enceintes présentant une PRNA, et

- 47 -
aucune complication particulière n'a été décrite. [108]
SGB néonatal:
Un seul cas est retrouvé dans la littérature [109], celui d'un nouveau né de sexe
masculin dont la maman a présenté à la 29ème semaine d'aménorrhée un SGB confirmé
par une concordance clinique, biologique (analyse du LCR) et électrophysiologique une
avec une sérologie du CMV est positive, et une détresse respiratoire croissante,
imposant une hospitalisation en unité de soins intensifs.
L'administration d'une forte dose d'IgIV à raison de 0.4g/kg/j pendant 5 jours n’a
entraîné aucune amélioration clinique notable, d'où l'indication d'un traitement par EP,
suite auquel l'amélioration était lentement progressive au delà de 4 mois.
L’accouchement a eu lieu par voie basse, à la 38ème semaine d'aménorrhée, chez
une parturiente toujours tétraplégique et mise sous VM, et il a donné naissance à un
nouveau né bien portant, avec un score d'APGAR à 10 à la 1ère et la 5ème minute.
Cependant au 12ème jour de vie, ce nouveau-né présente une hypotonie et une
détresse respiratoire nécessitant une hospitalisation en réanimation néonatale.
A l'examen, le nouveau-né ouvre spontanément les yeux, grimace au stimulus, il
présente une paralysie flasque profonde des membres et des ROT abolis.
L'analyse du LCR montre une DAC avec hyper-proteinorrachie à 2.43g/1 sans
pleiocytose.
L'EMG réalisé le 12ème jour de vie objective un ralentissement de la vitesse de
conduction nerveuse, avec bloc de conduction.
La sérologie au CMV montre une infection évolutive avec des IgG positifs.
La récupération est complète au bout de 15 jours après un traitement par IgIV.

- 48 -
B. Les formes symptomatiques : En dehors du tableau classique, d’autres formes du SGB seront appelées
également variantes cliniques. Leur incidence est comprise entre 0.14 et 0.35
cas/an/100000 habitants. Il n’existe pas de différence statistiquement significative
concernant le sexe, l’âge moyen des patients, la distribution saisonnière, la durée de la
phase d’extension et la protéinorachie entre les variantes cliniques du SGB et les SGB
classiques. [93]
1) Les formes axonales motrices ou motrice pures (AMAN) [110]
Cette forme est caractérisée par l’absence de trouble sensitifs subjectifs et
objectifs, elle est étayée essentiellement à partir des études faites en Chine du Nord où
il existe une forme de PRNA motrice pure survenant sur un mode épidémique,
particulièrement en été, et touchant surtout les enfants et les adultes jeunes. Ces
formes seraient préférentiellement associées à une infection par le CJ et aux anticorps
anti-GM1 [93, 111]. Le pronostic est plutôt bon, avec récupération de la marche dans la
grande majorité des cas.
2) Le syndrome de Miller Fisher :
En 1956, Ficher observe chez trois malades un syndrome partageant les
caractéristiques évolutives du SGB classique, mais sans déficit moteur, et caractérisé
par l’association d’une ataxie, d’une aréflexie et d’une atteinte prédominante des
paires crâniennes, avec notamment une ophtalmoplégie totale ou partielle. Les troubles
sensitifs sont moins observés, mais la DAC est quasi constante,
C’une forme rare du SGB représente moins de 5% en Europe et aux Etats-Unis,
alors qu’elle est fréquente au Japon [93]. Le pronostic de cette forme est bon et la
récupération est totale.

- 49 -
Contrairement au SGB typique, il est caractérisé par des marqueurs biologiques :
les anticorps anti-gangliosides antiGQ1b sont présents à la phase aigue de la maladie
dans 81% à 94% des cas. [112-115]
Ropper et Widjick ont proposé en 1991 des critères cliniques pour le syndrome
de Miller Ficher [112] . (Tableau II)
3) Les formes sensitives pures :
Une forme sensitive pure se caractérise par des paresthésies distales touchant
initialement les MI, plus rarement les mains ou les quatre extrémités, l’atteinte de la
sensibilité superficielle est généralement discrète, alors que la faiblesse musculaire est
rare pendant la maladie [14,116]. La protéinorachie est augmentée dans tous les cas. Il y a
très peu de complications, et le pronostic fonctionnel est bon.
4) La pandysautonomie aigue idiopathique :
En 1969, Young et al. rapportent une forme rare qu’ils nomment
« pandysautonomie aigue pure » et proposent de le rattacher au SGB du fait de son
évolution aigue, de la présence de la DAC, de sa précession par un épisode infectieux
et l’abolition fréquente des ROT.
Cliniquement, cette forme se manifeste par une fatigue, des vomissements, des
douleurs abdominales, des troubles du transit, une hypotension orthostatique avec
parfois une impuissance, des troubles de la sudation et de la salivation, et des
anomalies pupillaires [14]
5) Les formes purement crâniennes :
Elles sont rares, elles représentent 7.8% de la série de Govoni (1999)

- 50 -
Deux signes sont constants : l’aréflexie ostéo-tendineuse et la DAC. L’imagerie
cérébrale par résonance magnétique nucléaire devrait être normale. La récupération est
généralement complète. [93]
C. Les PRNA secondaires : Le SGB est par définition un PRNA idiopathique, d’où la recherche systématique
d’une étiologie pour les autres PRNA dites secondaires.
Ces PRNA doivent faire l’objet d’une enquête étiologique approfondie, car
souvent, une affection spécifique est découverte et traitée. En outre, le pronostic,
dépendant de l’étiologie, n’est pas nécessairement aussi favorable que celui du SGB.
[12, 117]
1) les PRN infectieuses :
• Maladie de Lyme ou Borréliose :
Le diagnostic devrait être évoqué devant la présence d’un tableau de PRNA
asymétrique, la notion de piqûre de tiques, d’érythème migrant et de poly arthralgie
dans les antécédents ; une hypercytose lymphocytaire à la PL, et souvent une atteinte
axonale à l’EMG.
On rapproche le diagnostic par la recherche d’anticorps anti-Borrélia dans la
sang et le LCR, étant donné l’absence de moyen fiable pour la mise en évidence de
l’agent pathogène
Un traitement antibiotique (tétracycline ou pénicilline G) sera à entreprendre
devant tout tableau évocateur. L’évolution se fait habituellement vers la guérison sans
séquelles. [96,117,118]

- 51 -
• Rickettsiose :
Au Maroc, elle est représentée essentiellement par la fièvre boutonneuse
méditerranéenne, et la PRNA constitue une rare complication qui survient surtout en
absence de traitement.
L’examen clinique permet le diagnostic dans la majorité des cas, la confirmation
est biologique. On utilise le sérodiagnostic de Weil-Felix, la microagglutination de
Giroud, ou plus récemment des réactions d’immunofluorescence, de fixation du
complément et d’ELISA.
Le traitement repose sur le chloramphénicol et les tétracyclines. [96,117]
• PRN et infection par le VIH : |19,120]
L’atteinte du SNP au cours de l’infection par le VIH est fréquente (30 à 35%) et
peut survenir à tous les stades de la maladie, les aspects cliniques sont très
polymorphes.
Les PRNA démyélinisantes surviennent tôt dans l’infection, avant même la
séroconversion et avant l’apparition du déficit immunitaire acquis.
L’étude du LCR peut mettre en évidence une hyperprotéinorachie et une
pléiocytose lymphocytaire (10 à 50 cellules/mm3), cette dernière associée à un tableau
de PRNA doit faire suspecter une infection par le VIH.

- 52 -
• Mononucléose infectieuse :
Le tableau clinique regroupe des atteintes variables du SNP, il pourra s’agir à côté
des PRNA d’une atteinte isolée d‘un ou de plusieurs nerfs crâniens, d’une paralysie uni
ou bilatérale du plexus brachial, ou d’un tableau de multinévrite. [93]
Mais, la survenue de façon subaiguë, au décours d’un syndrome infectieux
associé à une poly-adénopathie, d’une atteinte du SNP doit faire recherche une
mononucléose infectieuse par les tests d’inflammation et le « mononucléose
infectieuse » test.
2) les PRN carentielles et toxiques :
a) les neuropathies carentielles :
Les neuropathies carentielles, comme le béri béri et la pellagre, sont rares, à
l’exception des formes secondaires à l’intoxication par l’isoniazide par carence en
vitamine B6, et touchent surtout l’enfant.
b) les neuropathies toxiques :
Ces intoxications sont le plus souvent accidentelles, et on citera les toxiques les
plus importants. [93]
- l’arsenic
- les composés organiques, industriels à l’origine parfois d’intoxication
collective (huiles frelatées…)
- Les composés chimiques de certains herbicides, insecticides, raticides tels le
triorthocrésylphosphate, les sels de lithium.
- Les hexacarbones sont les principaux toxiques capables d’engendrer une
neuropathie périphérique. Notamment le N-héxane et le méthyl-N-
butylkétone sont des solvants utilisés dans l’industrie des laques et colles, ils

- 53 -
peuvent entraîner une PRNA. Ces PRNA toxique professionnelles surviennent
surtout chez des travailleurs utilisant ces produits dans des endroits peu
ventilés.
L’intoxication peut aussi se voir chez des sujets reniflant la colle (glue sniffers)
contenant ces solvants.
- Le plomb :
L’intoxication est souvent accidentelle chez l’enfant par ingestion de produits
« contaminés » : peinture au plomb, vernis, contrairement à l’adulte où elle est
chronique par exposition prolongée professionnelle.
- Certains médicaments, notamment la chimiothérapie. [93]
3) Les PRN associées à un cancer :
La survenue des neuropathies périphériques paranéoplasiques est rare (1 à 5%).
La neuropathie est parfois révélatrice.
L’atteinte du SNP au cours d’un cancer relève de mécanismes multiples. Les nerfs
ou les racines nerveuses peuvent être comprimés ou infiltrés par le processus
néoplasique ; il s’agit alors d’une méningoradiculite carcinomateuse particulièrement
douloureuse : le diagnostic repose sur la mise en évidence de cellules atypiques dans le
LCR. [93,121,122]
4) Les PRN et pathologie inflammatoire :
Cette association se rencontre volontiers chez l’adulte. La biopsie du nerf
périphérique peut objectiver des lésions spécifiques.
• LEAD : environ 10% des cas de LEAD se compliqueraient de neuropathie
périphérique.

- 54 -
Le déficit immunitaire engendré par le lupus peut être responsable d’une réaction
immunologique contre la myéline du SNP. [[123]
• Autres : Il s’agit de la périatérite noueuse, la sarcoïdose, la sclérodermie et la
polyarthrite rhumatoïde. [93]
5) Les PRN associées à une hémopathie :
• Les lymphomes : selon les études, l’incidence de l’atteinte clinique du SNP au
cours des lymphomes varie de 0.1 à 8% [93]
• Les leucémies : souvent l’atteinte du SNP est liée à une infiltration, plus
rarement à un infarctus, plus rarement à un infarctus ou des hémorragies
intraveineuses. La PRNA est rare au cours d’une leucémie myéloïde chronique.
[93]
6) Les PRN et dysglobulinémie :
• Maladie de Waldenström et gammapathie monoclonale bénigne à IgM :
La clinique, stéréotypée, permet de suspecter le diagnostic : PRN surtout
sensitive et démyélinisante d’évolution lente associée à un tremblement d’attitude et
d’action des MS.
L’étude en immunofluorescence de la biopsie nerveuse objective une fixation
anormale de l’IgM, le plus souvent à chaînes légères kappa au niveau des gaines
myéliniques. [93]

- 55 -
• Myélome :
C’est une PRNA distale sensitivo-motrice secondaire à une atteinte axonale
aspécifique. Une PRNA peut révéler un myélome solitaire. Sa découverte permettra son
traitement et donc une régression des troubles neurologiques. [93]
7) Les polyneuropathies métaboliques :
• L’amylose primitive et héréditaire: les paralysies amyloïdes sont rares.
• L’insuffisance rénale chronique : la neuropathie urémique, souvent latente, est
améliorée par la correction des troubles métaboliques par des hémodialyses.
• Le diabète :
La neuropathie diabétique, de fréquence probablement sous-estimée en raison
de formes mineures ou latentes de cette neuropathie, se rencontre habituellement chez
l’adulte de plus de 50 ans.
• La paralysie périodiques hypokalièmique
D. Les pathologies associées au SGB : Le SGB peut être associé à plusieurs pathologies, essentiellement à des maladies
auto-immunes, des maladies métaboliques ou endocriniennes ou autres. Ces
associations sont décrites sans que pour autant une relation de causalité soit établie.
o La première observation est celle d’un homme de 60 ans ayant comme
antécédent pathologique un SGB dans sa forme axonale, qui développe 9 ans
plus tard une sclérose latérale amyotrophique. Sa survenue peut être fortuite,

- 56 -
mais la similitude clinique entre ces deux tableaux suggère un mécanisme
physiopathologique commun. [124]
o La deuxième observation décrit un SGB associé à une hypothyroïdie chez une
patiente de 81 ans. Signalons que les rares cas de pathologies thyroïdiennes
associées au SGB concernaient les thyroïdites de Hashimoto, ou des maladies
de Basedow. [125]
o La troisième observation est celle d’un patient de 25 ans, qui a présenté une
méningite purulente associée à une myélite transverse, le syndrome infectieux
évolue rapidement sous antibiotiques, mais le patient garde une paraplégie
spastique séquellaire, et quatre mois après, il développe un SGB en même
temps que des signes clinique et biologique d’un lupus. [126]
o La quatrième observation est celle d’une femme agée de 40 ans, non
immunodéprimée, hospitalisée pour des troubles récents de la marche.
L’examen neurologique et électromyographique est celui d’un SGB. Durant
l’hospitalisation, une pan cytopénie apparaît, à l’exploration il s’agit de
leshmaniose viscérale.
L’apparition d’un SGB au cours d’une leshmaniose viscérale pourrait être la
conséquence de dérèglement d’un terrain immunologique sous jacent propre
au patient. Il paraît intéressant de poser la question de l’induction d’un SGB
par un parasite connu : la leshmanie. [127]

- 57 -
LE DIAGNOSTIC DIFFERENTIEL :
v La porphyrie aigue intermittente : C’est une affection génétique autosomique dominante qui est rare, elle est
caractérisée par un trouble du métabolisme des porphyrines.
L’accès peut être déclenché par certains médicaments (barbituriques). Les
manifestations neurologiques s’installent de façon aigue décrivant des paralysies à
prédominance motrice, proximale plus ou moins étendue.
Le tableau clinique se caractérise aussi par des douleurs abdominales pseudo
chirurgicales prodromiques et des troubles psychiques, à type de confusion mentale ou
convulsions. Les urines, anormalement rouges, deviennent noires à la lumière.
Le diagnostic repose sur la mise en évidence des dérivés porphyriques dans les
urines. [96,118,128]
v La poliomyélite antérieure aigue : Appelée encore paralysie infantile, la poliomyélite est éradiquée au Maroc, aucun
cas n’a été rapporté depuis 1993. [117,118] Mais, il faut y penser devant un tableau
exclusivement moteur, asymétrique, et survenant dans un contexte fébrile.
v Les atteintes médullaires :
Cliniquement, peut prêter à confusion une quadriplégie flasque rapidement
progressive, avec une aréflexie telle que peut la réaliser une compression de la moelle
cervicale, un ramollissement médullaire, une myélite aigue ou le saignement d’un
angiome [14]. Il existe en général des troubles sphinctériens avec à l’examen

- 58 -
neurologique, un niveau sensitif et un syndrome pyramidal dont les différentes
manifestations apparaissent en cours d’évolution. [85]
v Le syndrome de la queue de cheval :
Il doit faire rechercher en urgence une cause compressive par les examens
morphologiques appropriés (IRM lombo-sacrée). Comme pour les syndromes
médullaires, l’attention est attirée par les troubles sphinctériens précoces et les
troubles sensitifs objectifs.
L’EMG apporte les arguments clés, et pose le diagnostic. [85]
v La myasthénie : Il s’agit en général d’une fatigabilité musculaire qui augmente à l’effort, avec
diplopie ou ptôsis à bascule. Il n’y a pas d’amyotrophie au moins au début, ni de
troubles sensitifs, ni d’atteinte sphinctérienne, et les ROT sont en général préservés.
Cette maladie peut engager le pronostic vital vu les troubles de déglutition et
respiratoire qu’elle peut engendrer. Devant un déficit musculaire pur d’installation
rapide, avec ou sans atteinte oculaire, il faut évoquer une myasthénie et pratique un
test à la Prostigmine®.
v Le botulisme : Le botulisme est une toxi-infection alimentaire décrivant une neuropathie
particulière, résultant d’un blocage acétycholinergique au niveau des terminaisons
nerveuses et du système nerveux autonome.

- 59 -
Succédant à des troubles digestifs, elle est singulière par l’atteinte prédominante
des paires crâniennes.
Les oculomoteurs sont préférentiellement touchés, ainsi que la motilité
intrinsèque de l’œil responsable d’une mydriase fixe et aréactive.
S’y associent des troubles de la phonation, de la déglutition, une sécheresse
buccale. Le déficit moteur est de gravité variable. Le traitement comporte l’injection
d’antitoxine, la guanidine qui favorise la sortie de l’acétylcholine hors des terminaisons
nerveuse. [93]
v les PRN chroniques : Selon la littérature, un SGB aigu, peut évoluer secondairement sous forme d’une
PRN chronique à rechutes. Ces derniers cas posent un problème de diagnostic
différentiel avec les SGB récidivants. Cependant les cas de SGB récidivants présentent
des intervalles asymptomatiques longs (plusieurs années), et à chaque récidive une
phase d’installation des symptômes inférieure à 4 semaines. Par conséquent, tous les
autres cas devraient être considérés comme des PRN chroniques à rechutes. Il est
parfois difficile au début de l’affection de les différencier d’un SGB, et ce n’est que le
suivi des patients qui permet de les rattacher définitivement aux formes chroniques.
Cette précision diagnostique est importante car l’attitude thérapeutique est bien
différente, puisque les corticoïdes sont efficaces dans les PRN chroniques et les PRN
subaiguës. [129]

- 60 -
PRISE EN CHARGE GLOBALE DE SGB :
A. INTRODUCTION : Du faite que c’est une urgence neurologique thérapeutique qui met en jeu aussi
bien le pronostic fonctionnel que vital, le SGB impose une re-évaluation continue du
patient, et des interventions précoces pour une gestion bien réussite.
B. APPROCHE PRATIQUE : [130]
1. Diagnostic :
C’est vrai que le diagnostic de SGB est relativement facile, il se fait en se basant
sur des critères cliniques, et l’exclusion des autres causes de PRNA.
2. Evaluation primaire :
Le patient avec un SGB suspect doit être évalué de façon systématique et
prioritaire. L’évaluation primaire inclue un examen rapide pour identifier les situations
immédiatement menaçantes :
• AIRWAY : détresse respiratoire, arrêt respiratoire.
• BREATHING : fréquence respiratoire, efficacité respiratoire, effets de
respiration inadéquate (fréquence respiratoire, état mental, épuisement).
• CIRCULATION : fréquence respiratoire (arythmie), pression artérielle, temps
de recoloration.
• DISABILITY : état de conscience.

- 61 -
3. Réanimation :
Tout problème menaçant la vie du patient doit être traité immédiatement, une
fois il est identifié lors de l’évaluation primaire.
4. Evaluation secondaire :
Une fois les mesures de réanimation sont mises en place, un examen physique
méthodique et complet doit être fait, pour évaluer la sévérité du SGB suspect, et
adapter des investigations et des thérapeutiques appropriés.
5. Investigations :
Les investigations seront demandées une fois le malade est stabilisé, dans le but
de confirmé le diagnostic, éliminer les diagnostics différentiels, et rechercher
éventuellement une cause sous jacente.
6. Hospitalisation :
Une fois le diagnostic du SGB posé, la plupart des patients sont hospitalisés, s'ils
ne le sont pas déjà.
7. Traitement :
Le SGB est une urgence neurologique et thérapeutique, car il s’agit d’éviter le
décès du patient à la phase aigue de la maladie, mais aussi de limiter l’extension des
paralysies, de favoriser la récupération motrice, et par conséquent, de réduire la
fréquence de survenue des complications, et la gravité des séquelles. Les volets
thérapeutiques comportent :
àTraitement symptomatique et assistance des organes.
àTraitement spécifique.
àRéhabilitation et communication.

- 62 -
TRAITEMENT SYMPTOMATIQUE :
A. Défaillance du carrefour aéro-digestif : [131] Les troubles du carrefour aéro-digestif sont d’autant plus probables que le
déficit moteur est sévère, avec notamment atteinte des paires crâniennes. Cette
catégorie des malades bénéficie de soins intensifs.
Il ne faut pas sous estimer les petits problèmes de déglutition, souvent
précurseurs de paralysies des muscles respiratoires. Ils doivent d’emblée poser
l’indication d’une sonde gastrique (même s’ils ne sont qu’intermittents), et proscrire
l’alimentation per os, afin d’éviter les fausses routes et les surinfections pleuro
pulmonaires, sans oublier qu’il faut établir un équilibre nutritionnel (voir chapitre :
équilibre nutritionnel).
B. La défaillance respiratoire : Le clinicien doit savoir anticiper la survenue d’une dégradation respiratoire et
connaître les critères stricts d’indication de l’intubation endotrachéale afin que la
surveillance soit adaptée et qu’un recours trop tardif à la ventilation mécanique soit
évité (Tableau III).
1. Prédiction et indication de la ventilation mécanique invasive :
Nous avons récemment identifié trois facteurs prédictifs indépendants du recours
à la ventilation mécanique [132] :

- 63 -
• Un délai entre le début du déficit moteur et l’hospitalisation inférieur à 7jours
• L’impossibilité à relever la tête du plan du lit ;
• Une CV à l’admission inférieure à 60 % de la valeur théorique.
La probabilité d’être ultérieurement ventilé est de 85 % chez les patients ayant
ces trois signes. En l’absence de mesure de la CV, quatre facteurs doivent être pris en
compte :
• Un délai entre le début du déficit moteur et l’hospitalisation inférieur à 7jours
• L’impossibilité de se tenir debout ;
• L’impossibilité de soulever les coudes ;
• La présence d’une cytolyse hépatique.
En dehors de la situation de détresse respiratoire aiguë, la ventilation mécanique
invasive est indiquée par la présence d’un critère majeur ou de deux mineurs [133,134].
Les premiers regroupent une hypercapnie (PaCO2 > 6,4 kPa), une hypoxémie (PaO2 <
7,5 kPa en air ambiant), une CV inférieure à 15 ml/kg ou la combinaison des pressions
inspiratoire et expiratoire maximales (PImax et PEmax) inférieures à 25cm H2O et
40cm H2O ; les seconds comportent une toux inefficace, des troubles majeurs de la
déglutition, l’existence d’une atélectasie.
La place de la ventilation mécanique non-invasive (VNI) nous semble dans ce
contexte limitée en raison de la fréquence mais également de la difficulté à
diagnostiquer un déficit oropharyngé qui, avec la gastroparésie, en est une contre-
indication formelle [135]. L’autre inconvénient majeur de la VNI est qu’elle peut procurer

- 64 -
à tort au clinicien un sentiment de sécurité alors que l’état respiratoire continue de se
dégrader ou qu’apparaissent des troubles de la déglutition « de manière masquée ».
L’examen électrophysiologique pourrait aider à l’individualisation des patients à
risque de décompensation respiratoire. Nous avons ainsi mis en évidence que les
formes démyélinisantes nécessitent plus fréquemment d’être subséquemment ventilés
[136]. L’étude électrophysiologique des nerfs phréniques pourrait également être utile
[137].
2. La durée de la ventilation, et moment du sevrage :
Le délai médian entre l’admission et le début de la ventilation mécanique est de
deux jours et la durée médiane de celle-ci de 21 jours [138] ; mais, la ventilation peut
être prolongée ; dans ce sens, Lawn et al. ont proposé de calculer le rapport des
sommes, mesurées le jour de l’intubation et 12 jours plus tard, de la CV (mL/kg),
PImax et PEmax (cmH2O). Un ratio inférieur à un aurait une valeur prédictive positive
de ventilation prolongée de 100 % [139].
Le moment optimal du début du sevrage du ventilateur doit être choisi en
fonction de la récupération à la fois des capacités respiratoires, des troubles de la
déglutition, difficiles à évaluer chez les patients de réanimation, et du déficit moteur.
Au contraire des autres patients de réanimation, les patients atteints de SGB doivent
être sevrés progressivement, préférentiellement sur pièce en T, l’aide inspiratoire
n’ayant pas été rigoureusement explorée dans cette indication. La persistance de
troubles de déglutition peut être une cause d’échec tardif de l’extubation. Chevrolet et
Deléamont ont décrit une méthode de sevrage qui était débutée lorsque la CV
atteignait la valeur minimale de 7 ml/kg et consistait en des épreuves de respiration

- 65 -
spontanée sur pièces en T de plus en plus prolongée puis une extubation lorsque la CV
dépassait 15 ml/kg et la durée de respiration libre 24 heures d’affilée [140].
On procède à une extubation lorsque la CV est supérieure à 40%de la valeur
théorique et que le patient tolère un débranchement du respirateur pendant plus de
huit heures consécutives.
C. Défaillance cardiovasculaire : [66] Comme les troubles cardio-vasculaires sont surprenants par leur brutalité de
survenue, il faut donc s’attacher à les dépister. Pour cela, un monitoring cardiaque et
tensionnel est nécessaire.
Pour la prise en charge de tels troubles, il est capital de conserver un état
physiologique basal, c'est-à-dire une hydratation, une oxygénation normale afin de
minimiser la fréquence et l’intensité de ces incidents.
L’atropine reste très efficace pour traiter une bradycardie, mais la nécessité de
recourir à des méthodes d’entraînement électro-systolique peut se révéler, dans
certains cas, indispensable (pauses cardiaques).
Une tachycardie isolée ne nécessite aucun traitement anti-arythmique.
On se heurte parfois aux fluctuations fréquentes et rapides de situations telles
des accès de tachycardie puis de bradycardie, des variations tensionnelles importantes.
Traiter au coup à coup n’est pas donc facile.
L’extrême prudence est de rigueur concernant les manipulations des malades au
cours du nursing, des soins kinésithérapeutiques ; les aspirations trachéales, dont
beaucoup d’auteurs soulignent l’influence dans la survenue de tels symptômes.
Néanmoins, les bradycardies extrêmes lors des aspirations peuvent être évitées par une
hyperoxygénation transitoire à FiO2 100%.

- 66 -
En cas d’accès hypertensif, on ne doit pas user des béta-bloqueurs aux
propriétés cardio-freinatrices, un traitement par Labétolol (Trandate®) est préconisé.
D. Assistance gastro-intestinale : [141] Une série de méthodes peuvent être utilisées pour traiter la constipation : entre
autres le lait de magnésium, les laxatifs, comme le sulfosuccinate de sodium, ou les
agents tels que le psyllium et même certains produits n'ayant pas été initialement
conçus pour traiter la constipation, comme le lactulose. La dose de départ proposée est
de 3 cuillerées à soupe ou 45 cc, 4 fois par jour, jusqu'à ce que les intestins du patient
se remettent à bouger, ensuite de 1 à 3 cuillerées à soupe par jour.
E. Assistance vésico-sphinctérienne : [142,143] Les troubles vésico-sphinctériens nécessitent une prise en charge spécifique à la
phase initiale et jusqu'à récupération complète. Ils doivent être traités de façon
conservatrice, car, ils vont régresser totalement dans la majorité des cas lors de la
résolution des symptômes neurologiques.
F. Le traitement de la douleur : Des douleurs son rapportées par 89 % des patients, dont la moitié décrivent des
douleurs sévères [144]. Les antalgiques doivent être rationnellement prescrits en
fonction de la sémiologie de la douleur et afin d’éviter leur cumul.
En cas de douleurs neuropathiques, nous préconisons la gabapentine
(Neurontin®), dont il ne faut pas hésiter à administrer rapidement les doses maximales.
Le clonazépam (Rivotril®) peut lui être associé, mais il entraîne souvent une
somnolence.

- 67 -
En cas d’échec, un traitement par opiacés est alors requis [145,146]. Les troubles du
sommeil sont fréquents et peuvent être accompagnés d’hallucinations, le plus souvent
visuelles. Celles-ci imposent l’interruption des traitements hallucinogènes et
éventuellement la prescription d’un neuroleptique.
G. L’équilibre nutritionnel et hydro électrolytique : Les patients souffrant de SGB peuvent avoir des difficultés pour maintenir une
nutrition et hydratation adéquate, ce problème s’ajoute à celui de l’hyper-catabolisme
induit l’état de stress, et aussi, au risque d’amyotrophie qui accompagne les formes
sévères du SGB.
La prise en charge nutritionnelle des patients porteurs de SGB dépend de
l’extension des déficits, ainsi :
àL’alimentation par voie orale doit être établie tant qu’il n’y a pas de signe en
faveur de paralysie bulbaire ou pseudo-bulbaire, ni de détresse respiratoire. Car, les
patients présentant une paralysie bulbaire ou pseudo-bulbaire seront incapables de
déglutir, par atteinte déficitaire des muscles qui assurent la coordination du complexe
acte, qui est la déglutition. Par ailleurs, ceux qui sont mis sous ventilation mécanique,
seront incapables d’avaler normalement, à cause de la présence du tube endo-trachéal.
àMais si l’alimentation par voie orale n’est pas possible, un gavage par sonde
naso-gastrique s’avère nécessaire. On le préfère de l’alimentation parentérale pour les
raisons suivantes [18]:
§ Maintient de l’intégrité intestinale et de la fonction immunitaire.
§ Prévention de l’ulcère de stress.

- 68 -
§ Réduction du risque de sepsis et de pneumothorax liés à l’alimentation
parentérale.
§ Son faible coût.
Cette sonde naso-gastrique peut être laissée en place pendant quelques
semaines, mais si on anticipe que ça serait pour une très longue durée, il faut
envisager une alimentation à travers une gastrostomie per-cutanée.
Quelque soit la méthode d’alimentation ; on doit assurer un apport calorique,
protidique et vitaminique adéquat ; une hydratation correcte, afin de maintenir le sujet
dans un état d’homéostasie optimale. Cet apport sera adapté selon les circonstances.
La surveillance de la natrémie doit être stricte pour guetter le syndrome de
sécrétion inappropriée d’ADH, et ceci par des ionogrammes sanguins quotidiens à la
recherche d’hyponatrémie de dilution. Les traitements pour ce genre de troubles
peuvent être l'utilisation de diurétiques, comme par exemple la furosémide (Lasilix®),
ou tout simplement par une restriction hydrique.
H. Le soutient psychologique :
L’évolution des paralysies et la perte progressive d’autonomie crée un état
d’anxiété et d’inquiétude. L’état du malade peut se dégrader très vite, nécessitant des
mesures de réanimation impressionnantes (intubation, monitoring du rythme
cardiaque…). Le découragement et le renoncement peuvent s’installer, surtout lorsque
les progrès tardent à arriver, et qu’il existe des séquelles potentielles. Un soutien
psychologique est donc nécessaire pour le malade et ses proches.

- 69 -
I. Les complications : La prise en charge comporte aussi la prévention et le traitement des
complications de décubitus.
1. Les complications thrombo-emboliques :
L’incidence des thromboses veineuses profonde et de l’embolie pulmonaire est
d’environ 5 % [147].
La prévention thromboembolique par héparines de bas poids moléculaire ou non
fractionnée est systématique chez les patients alités [148].
Mais il ne faut pas oublier que parmi les moyens, il y a la mobilisation passive
des MI, et les changements positionnels pluri quotidiennement.
2. Les infections pulmonaires :
Les aspirations des sécrétions bronchiques, bien que délicates dans ce contexte,
seront effectuées régulièrement, tout en conservant la liberté des voies aériennes dans
le but de réduire le risque infectieux.
De même, toutes les précautions concernant l’asepsie devront être prises, car
malgré des consignes strictes, des pneumopathies nosocomiales sont encore observés,
concomitamment au début des aspirations trachéales [149].
3. Les autres infections :
L’alitement est favorable au développement d’infections urinaires, qu’il faut
systématiquement traquer devant un état fébrile.
Les escarres, heureusement rares grâce au nursing préventif, peuvent également
se surinfecter.

- 70 -
LES TRAITEMENTS SPECIFIQUES :
A. Pourquoi ? : Pour des raisons historiques [150], le SGB a longtemps été considéré comme
une pathologie bénigne. Il a fallu plus d’un demi-siècle pour que, dans le monde, on
comprenne que cette maladie « bénigne » pouvait entraîner le décès ou laisser des
séquelles motrices graves.
L’appréciation de la sévérité de cette maladie, trouve une morbi-mortalité
extrêmement effrayante avant l’instauration de traitement spécifique :
§ 50% des malades seulement récupèrent leur totale force musculaire à 1an
[151,152]. Ce taux est couramment entre 60% et 70%.
§ Le taux de mortalité est entre 20 et 25% entre 1960-65, et 10% entre 1970-
1980 [150,151].
§ La durée moyenne des paralysies, mesurée par le délai de reprise de la marche
avec appui, est de 65 jours.
Choses qui expliquent la nécessité d’un traitement spécifique, qui limiterait
l’extension et minimiserait la durée des paralysies.
Cependant, il est difficile de juger objectivement les résultats et l’efficacité
réelle d’un traitement, en raison de l’évolution naturelle de la maladie en faveur d’une
récupération spontanée dans la majorité des cas.

- 71 -
B. Buts : Le traitement spécifique se doit de limiter au maximum l’engagement des
pronostics vital et fonctionnel, qui éliminent tout caractère de bénignité lié à cette
maladie.
Grâce à de tel traitement spécifique, on a pu réduire :
§ La proportion des malades qui garderont des séquelles résiduelles sévères 1an
après, et suffisamment invalidantes pour entraver la marche, à 10% [152-154].
§ Le taux des patients qui décèdent de cette maladie, qui est actuellement
proche de 5% seulement. [153-156]
Pourtant, SGB est encore une grave maladie.
C. Concept général :
Le concept étiopathogénique général du SGB, actuellement admis, est celui d’un
conflit immunologique au niveau des nerfs périphériques.
De ce fait, les thérapeutiques proposées se sont toutes orientées dans le
domaine d’immunothérapie, dans le but d’arrêter le processus démyélinisant par
isolement des facteurs immunologiques démyélinisants sériques des patients atteints
de SGB, permettant ainsi au phénomène de réparation ou remyélinisation de reprendre.

- 72 -
D. Les moyens :
1) Effet de la corticothérapie :
Les corticoïdes ou l'ACTH ont été les premiers traitements évalués. Cela se
justifiait d'une part par la constatation de lésions inflammatoires au niveau des
racines nerveuses et, d'autre part, par le fait que des fortes doses de corticoïdes étaient
efficaces sur un modèle animal [157,158].
Récemment [159], une étude multicentrique a comparée l’utilisation des
corticoïdes à un placebo, mais aucune différence significative n'apparaît pour
l'amé1ioration motrice jugée sur l'échelle de Hughes, à la 4° semaine, ni pour les
séquelles après 1 an d’évolution. Une étude similaire montrait que la durée
d’hospitalisation paraît plus longue chez le groupe mis sous corticoïdes.
Ø La conclusion est que la corticothérapie est inutile dans le SGB.
2) Effet des échanges plasmatiques :
a) Définition : [160]
L’échange plasmatique ou plasmaphérèse thérapeutique est une technique de
circulation extra corporelle (CEC) permettant de séparer le plasma du sang total, dans
le but de retirer de l’organisme des molécules responsables de pathologies,
notamment des anticorps et de restituer les éléments figurés du sang.
Le plasma extrait est remplacé volume pour volume par des liquides de
substitution de composition variable selon les indications ou les centres.
b) Technique : [161]
Les échanges plasmatiques nécessitent la mise un place d’un circuit extra-
corporel (CEC) qui ramène le sang au système séparateur des cellules, et le rendre,

- 73 -
après extraction plasmatique et adjonction de solution colloïdale substitutive, au
patient. Ce CEC impose une anti-coagulation qui se fait par l’héparine ou les solutions
citratées.
Le plasma peut être séparé des globules par 2 principes :
àTechnique de filtration, habituellement utilisée dans les services de
réanimation.
àTechnique de centrifugation généralement réalisée dans les centres
d’hémobiologie.
² Technique de filtration : (Fig. 13)
La technique de filtration, qui est possible avec toutes les machines réalisant des
reins artificiels, utilise un module séparateur dont le paramètre le plus important est la
membrane microporeuse dont il existe plusieurs constituants (polycarbonate,
polypropylène, acétate de cellulose,...). Par ailleurs, ces membranes différent par la
taille des pores variant de 0.3 à 0.5µm, l’épaisseur de la membrane et la surface
choisie en fonction du volume de plasma à épurer.
Ä Ce système a pour avantage l’obtention d’un plasma acellulaire, un faible
volume extra-corporel et un faible volume résiduel érythrocytaire.
Ä Par contre, il a l’inconvénient d’avoir une extraction plasmatique moins
performante, chose qu’on peut palier par l’augmentation du débit sanguin.
Cette technique est plus onéreuse que la centrifugation en raison du coût élevé
du matériel à usage unique.
² Technique de centrifugation :
Il existe 2 procédés de séparation plasma / cellules par centrifugation :
*à flux continu : utilisant un bol de centrifugation avec cycles aller/retour
successifs. (Fig. 14)

- 74 -
Ä Avantage : ne nécessite qu’un seul accès veineux,
Ä Inconvénients :
§ un volume extra-corporel élevé de l’ordre de 400 à 800ml, parfois mal toléré
au point de vue hémodynamique,
§ demande un temps de 20% supérieur aux autres techniques.
*à flux discontinu : avec anneau de centrifugation alimenté et vidé en
permanence (Fig. 15).
Ä Avantages :
§ un volume sanguin extracorporel qui est faible, de l’ordre de 170 à 350ml,
assurant une très bonne tolérance hémodynamique.
§ un volume plasmatique séparé par unité de temps qui est élevé, permettant
des séances beaucoup plus courtes que par la technique à flux discontinu.
§ les moniteurs fonctionnent en mode automatique contrôlant les débits
d’extraction et de réinjection, et l’administration de l’anti-coagulant.
Ä Inconvénients :
§ absence d’un réchauffeur à sang, qui permettrait d’éviter l’hypo-thermie
dans le CEC,
§ Nécessité de deux accès veineux.
c) Voies d’abords : [160]
Il est impératif de favoriser un abord veineux périphérique pour éviter les
complications septiques, thrombotiques ou hémorragiques, sauf en cas d’échec
d’accès, ou d’EP par filtration qui nécessite un débit sanguin élevé, où le recours à une
voie veineuse centrale s’impose.
Deux voies sont indispensables (ligne aller dite « artère » dans le sens
patientàmachine ; et ligne retour dite « veine » dans le sens machineàpatient) sauf en

- 75 -
centrifugation discontinue où une voie unique peut desservir les cycles d’extraction et
de restitution de façon alternée.
Les Shunts et fistules artério-veineuses sont peu utilisés dans les échanges
plasmatiques en réanimation. Leur indication peut être portée dans le cas d’EP pour
pathologie chronique nécessitant des échanges répétitifs au long cours.
d) Anticoagulation : [160]
Pour la plupart des centres, elle est indispensable. Elle repose sur l’héparine
seule ou associée à l’ACD-A (citrate agissant par chélation calcique) dans la technique
de filtration, et sur l’ACD-A seul en cas de centrifugation.
L’héparine de bas poids moléculaire utilisée en bolus IV ne semble pas donner
plus de complications de thrombose des circuits, mais aucune étude comparative ne l’a
démontré. Certains centres effectuent des séances courtes à haut débit de sang,
autorisant l’absence d’anticoagulation avec de bons résultats.
e) Calcul du volume à échanger :
Aucune base scientifique ne peut déterminer le volume idéal de plasma à
échanger. Selon les équipes, le volume préconisé est compris entre 1 à 1,5 masse
plasmatique, avec une élimination de 60% des immunoglobulines (Ig) pour une masse
et de 70% pour une masse et demi [154].
Cette dernière est évaluée par la formule suivante :
(100–Hématocrite)×0,7×poids
Les appareils de centrifugation continue paramétrables effectuent
automatiquement les calculs.
f) Fluides de substitution : [160]
Le produit de substitution idéal devrait avoir les propriétés suivantes :
§ Un pouvoir d’expansion volémique soutenue.

- 76 -
§ Une sécurité infectieuse.
§ Une action non délétère (pas d’effet secondaire, pas d’effet déplétif ou
d’interférence avec les protéines plasmatiques.
§ Une facilité d’emploie.
§ Un prix raisonnable.
En pratique ; le volume de plasma épuré est remplacé par un colloïde naturel
(albumine humaine à 4%) ou par un colloïde synthétique (gélatine, HEA).
g) Epuration des composants plasmatiques :
L’effet épurateur est le seul effet recherché des EP dans le cadre des PRNA. Le
coefficient de tamissage des molécules de poids moléculaire atteignant un million de
daltons est égale à 1, impliquant un passage libre des molécules à travers le filtre d’EP.
Ainsi, les Ig et les fragments du complément sont épurés par l’EP. Cette
épuration expose à des risques infectieux. Ainsi, tout état infectieux contre indique
formellement la pratique d’EP.
En revanche, il n’y a pas de recommandation quant à l’usage systématique d’Ig
au décours d’EP.
h) Effets secondaires :
Ø Effets secondaires communs :
ü La surcharge volémique (œdème pulmonaire) au cours des séances d’EP est
observée devant une mauvaise appréciation du degré d’hypo volémie et /ou
la présence de cardiopathie.
ü De plus, les facteurs de coagulation sont perdus. Au décours immédiat de
l’EP, le taux de prothrombine est abaissé (30%). Il existe secondairement un
bref pic d’hypercoagulabilité. En pratique, chez un malade sous anti
coagulant, le traitement doit être repris 8heures après la fin des EP.

- 77 -
Ø Effets secondaires spécifiques :
ü L’installation d’une hémodilution peut être due à une perfusion de grand
volume de colloïde.
ü Certains effets peuvent être observés sur l’hémostase :
-L’albumine peut être responsable d’un état d’hyperviscosité induit par une
hémodilution à 20%, avec l’albumine à 4%.
-Les gélatines diminueraient l’agrégation plaquettaire induite par la ristocétine,
la fixation du vWF par la gélatine modifiant les rapports vWF-GR 1b, ainsi que
l’agrégation impliquant les récepteurs plaquettaires GP IIb-IIIa, réduisant la formation
de caillot et la synthèse de thrombine. En revanche, les conséquences cliniques de ces
effets semblent modérées.
-Les HEA sont à l’origine des accidents hémorragiques rares en rapport avec un
allongement du temps de saignement, une diminution du temps de thrombine et une
diminution de la concentration plasmatique du fibrinogène.
ü Le risque allergique n’est pas rare : une incidence des réactions
anaphylactoïdes de l’albumine est de 0.01% par flacon et de 0.099% par
patient, ces chiffres étant inférieurs à ceux des gélatines, mais comparables à
ceux des amidons. Des réactions d’hypothermie et de frisson ont été décrites.
ü Des cas de nécrose tubulaire rénale ont été observés après perfusion d’HEA.
ü Sur le plan infectieux, le risque vital n’existe plus en théorie en raison des
techniques d’élaboration. Les parvovirus qui résistent à la pasteurisation de
l’albumine n’ont pas, jusqu’à présent, provoqué d’effet pathogène majeur. Le
risque de transmission d’agent pathogène, de type prion, ne semble que
théorique dans les conditions normales d’utilisation d’albumine. L’origine

- 78 -
animale des gélatines ne permet pas de considérer le risque de transmission
de l’agent de l’encéphalopathie spongiforme bovine comme nul.
ü Des erreurs d’appréciation du facteur rhésus peuvent survenir en présence de
gélatines, ainsi qu’un gène dans l’interprétation du groupe sanguin en
présence d’HEA, imposant le prélèvement de sang pour groupage avant la
perfusion de ces solutés.
i) La place des échanges plasmatiques dans SGB :
Plusieurs travaux ont évalué l’efficacité des EP dans le SGB (Tableau IV), et si
certains d’entre eux ont aboutit à un effet bénéfique nul (Greenwood et al.) [162] ou
médiocre (Osterman et al.) [163], c’est parce qu’ils avaient des critères d’inclusion
divergents (collectif réduit, délai d’inclusion trop long, forme particulièrement sévère à
l’inclusion, patients peu graves).
Mais, deux larges études (Guillain Barre Study Group[164] , French Cooperative
Group on GBS[165]) confirment que les malades ayant reçus le traitement précocement
(moins d’une semaine après l’installation de la maladie) ont un meilleur devenir et des
plus bons résultats : les délais nécessaires sont réduits aussi bien pour observer la
récupération motrice, que pour la reprise d’une marche sans assistance, leur sevrage
de la ventilation mécanique est plus précoce, ainsi que leur durée d’hospitalisation.
La diminution de la sévérité et la réduction de la durée de progression aigue de la
maladie dues aux EP expliquent probablement [165]:
/La réduction de l’incidence de certaines complications telles que :
pneumopathies, troubles végétatifs.

- 79 -
/L’augmentation du taux de bonne évolution, c'est-à-dire un nombre de sujets
plus grand ait récupéré une force musculaire normale après un recul d’un an,
grâce aux EP.
Par ailleurs, la French Cooperative Group on GBS[165] a comparé l’effet des deux
solutés de remplacement : albumine diluée à 4% et le plasma frais congelé, du fait que
ce dernier contient des Ig et des compléments, chose qui pourrai modifier l’effet
clinique du traitement de SGB pour lequel un processus immunologique a été suggéré.
Il s’avérait que le plasma est tout aussi efficace que l’albumine, mais compte tenu des
risques infectieux, il doit être abandonné.
Ce même groupe a mené une autre étude [166] pour standardiser les procédures
dans le but de minimiser les risques des EP. Pour cela :
o le volume de plasma épuré a été réduit de 2 à une masse plasmatique et
demie,
o le PFC était évité, et le soluté de remplacement était un mélange d'albumine
diluée à 4% et de gélatine, à parts égales.
Elle a conclut que la morbidité des EP était nettement plus basse que celle
observé dans les protocoles antérieurs ; ainsi, le pourcentage d’au moins un accident,
par chaque EP, est passé de 65%, à seulement 24%, et pour le pourcentage de patients
ayant nécessité l’interruption des EP est réduit de 14%, à 7%.
Quoiqu’on a montré précédemment que 4EP est plus bénéfique que les soins
intensifs standards ; plusieurs questions se posaient : ce bénéfice serait-il amélioré par
l’augmentation ou diminution du nombre de séances? Quel est le nombre optimal de
séance d’EP pour chaque groupe de sévèrité ? (Tableau). Pour répondre à ces questions,
« French cooperative group » recommande l’utilisation, le plus précocement possible,

- 80 -
de 2EP pour les formes légères, et 2 EP additionnels doivent être ajoutés en cas de
détérioration aux formes avancées de ce syndrome (perte de la capacité de marcher, ou
ventilation mécanique). [166]
3) Effet de fortes doses d'immunoglobulines (IgG) utilisées seules ou en
association avec les échanges plasmatiques (tableau V) :
L'utilisation de fortes doses d'IgG a été proposée dans certaines maladies
auto-immunes [167,168]. C'est sans doute par analogie que ce traitement fut propose
dans les formes chroniques de polyradiculonévrite [169,170] puis, dans une étude pilote,
dans le syndrome de Guillain Barré [171].
Comme les EP étaient devenus le traitement de référence du SGB, il était
logique d'organiser un essai comparant l'effet d’EP à la perfusion d'lgG, dont la dose
était arbitrairement fixée à 0,4 g/kg/j pendant 5 jours[155]. Cette étude aboutit à la
conclusion que les IgG sont au moins aussi efficaces que les EP.
Dans cette étude plus de complications ont été observées avec les EP qu'avec les
IgG. Cela justifie la conclusion de ce travail, qui est de préconiser les IgG chez les
patients atteints d'un SGB.
Dans un dernier travail [154], 3 stratégies ont été définies par tirage au sort:
-EP (cinq EP pendant 8 à 13 jours),
-IgG (0,4 g/kg/j pendant 5 jours)
-ou association des deux, la perfusion des IgG suivant le dernier EP.
383 adultes ont été inclus dans cet essai multicentrique international dont le
résultat principal est l'absence de différence entre les trois stratégies. Par ailleurs, cet
essai notait que la morbidité des EP et des IgG était identique, et elle était doublée

- 81 -
dans le groupe qui a reçu les deux traitements. Cette étude a des conséquences
pratiques importantes. Elle confirme l'équivalence entre l'effet des EP et celui des IgG,
sans toutefois confirmer que le premier traitement a une morbidité supérieure au
second. Dans l'état actuel des connaissances, il n'est pas utile d'associer les deux
traitements.
E. Utilisation des mesures spécifiques : Le tableau VI résume le choix des mesures spécifiques qui résultent des
différents essais publiés.
Dans les formes bénignes (groupe A), l'effet des IgG n'a jamais été étudié,
il est donc logique de préconiser deux EP; deux EP supplémentaires devant être
pratiqués en cas d'aggravation. Dans les formes initialement vues aux stades B ou
C, il n'existe pas d'argument pour privilégier l'un ou l'autre de ces traitements.
Les EP sont une technique contraignante, mais on a vu que leur morbidité
diminue au fur et à mesure des années, probablement parce que la technique est
mieux maîtrisée et que la posologie optimale mieux connue.
Les IgG, a priori plus simples, ne sont pas dénuées de morbidité à des
posologies élevées [172]. L'erreur fondamentale serait que la perfusion d'IgG devienne
un geste de routine, effectué sans respecter les contre-indications des deux
traitements doivent être respectées.

- 82 -
Les complications infectieuses, notamment pulmonaires, sont la contre-
indication principale des EP, l'insuffisance rénale étant la contre-indication
principale des IgG. Les contraintes économiques sont également à envisager. Le
coût d'un EP a été estimé en France 4 000 francs et le coût d'une perfusion
d'IgG à 0,4g/kg/j à 5 000 francs par jour [173]. Le coût direct d'une perfusion d'IgG
paraîtrait donc supérieur. D'autres paramètres doivent néanmoins être pris en
compte pour calculer le coût réel des EP (nombre de séances réalisées par an par
un service, amortissement du matériel, coût du personnel qui assure ce traitement,
formation des équipes, etc).
Le SGB est en principe d'évolution monophasique. Les rechutes sont toutefois
possibles dans environ 5 % des cas. Dans les études précédentes ce pourcentage et sa
gravité ne paraissent pas être influencés par le traitement utilisé, il est même
comparable à ce qui est observe dans le groupe témoin. La survenue d'une rechute
peut donc ne pas être traitée. Néanmoins, si sa gravité le justifie, il est logique de
reprendre le premier traitement utilisé.
Une autre question, souvent posée par les cliniciens, concerne la conduite à
tenir vis-à-vis des patients qui, malgré le traitement, continuent à s'aggraver ou
ne s'améliorent pas. Dans I'état actuel des connaissances, il n'est pas utile ni
d'augmenter la dose du traitement, ni d'associer les traitements, ce qui en accroît la
morbidité et le coût.

- 83 -
Réhabilitation et communication :
A. La réhabilitation : [174-177] La rééducation est un élément majeur du traitement. Elle sera adaptée en
fonction des phases de la maladie. Elle a pour objectif de prévenir des complications,
de favoriser la récupération et de limiter les séquelles, puis éventuellement d'en limiter
les conséquences fonctionnelles.
1. Pendant la phase d'installation et pendant la phase d'état :
L'installation au lit doit être régulièrement surveillée. Les mobilisations passives
ont pour but de maintenir la trophicité et la mobilité des articulations. Elles doivent
être manuelles, douces, lentes, respectant les douleurs et mobilisant chaque
articulation plusieurs fois dans toutes les amplitudes anatomiques. Elles doivent être
quotidiennes, voire pluriquotidiennes.
En cas d'atteinte respiratoire, la ventilation assistée ou contrôlée est mise en
route et une trachéotomie est souvent nécessaire. Le drainage des sécrétions
bronchiques est facilité par les postures latérales et déclives, prudentes mais
prolongées, ainsi que par les vibrations thoraciques. Les aspirations bronchiques sont
prudentes en raison du risque d'arrêt cardiaque.
Si l'état général et ventilatoire le permettent, l'adaptation à l'orthostatisme est
maintenue par la mise au fauteuil. Le port d'une gaine de contention compense le

- 84 -
déficit de la sangle abdominale et améliore les conditions ventilatoires et
hémodynamiques.
2. Rééducation à la phase de récupération :
Elle vise à renforcer prudemment le potentiel moteur par un travail comportant
des exercices analytiques, globaux et fonctionnels. Le programme de réadaptation sera
adapté à l'état général du patient, respectant la fatigue, l'importance et l'évolution de
l'atteinte.
Le travail moteur analytique peut être effectué sur tous les groupes musculaires
dont on guette et exploite le moindre réveil. Il est toujours guidé par le
kinésithérapeute en travail actif aidé, puis contre résistance manuelle.
La réadaptation à l'orthostatisme, si l'alitement est prolongé, doit être prudente,
progressive au plan incliné avec port de bas de contention et de gaine abdominale.
Au lit, le renforcement du contrôle du tronc dans les exercices de retournement
et de mise en position assise, est réalisé. Le travail des muscles stabilisateurs du bassin
(moyen fessier) et des membres inférieurs (grand fessier, quadriceps, soléaire), au lit
puis au tapis, prépare la station debout et la marche.
Progressivement la marche est entraînée dans les barres parallèles au
déambulateur, avec cannes anglaises puis sans canne. Le port d'orthèse releveur est
souvent indispensable au début. Une orthèse verrouillant les genoux peut être
transitoirement nécessaire. L'autonomie fonctionnelle est ensuite renforcée.
Parallèlement au travail moteur, le réveil sensitif, proprioceptif en particulier est
essentiel. La pression des mains du kinésithérapeute lors du travail passif et actif est
un élément important, ainsi que la mobilisation active et passive de tous les segments
sous contrôle de la vue.

- 85 -
Le travail analytique et global des membres supérieurs suit les mêmes règles,
particulièrement aidé par le contrôle de la vue.
Certains proposent à long terme un programme de ré-entraînement à l'effort qui
améliorerait les performances quotidiennes même en cas de séquelles motrices [178].
B. La communication : [179] Au cours des premiers stades de la maladie, plus particulièrement pour le patient
en soins intensifs, les manifestations peuvent être très effrayantes. La plupart des
patients atteints du SGB étaient auparavant en très bonne santé et le fait de se
retrouver tout à coup paralysés, désemparés, sous perfusion, avec un cathéter vésical
et un moniteur cardiaque qui fait bip de manière continue et monotone, peut
provoquer un choc émotionnel.
Donc, la communication est primordiale, et voici quelques suggestions destinées
au personnel hospitalier et à la famille:
1. On peut expliquer la maladie au patient et l'informer de ses chances de
guérison. Si la famille, les amis, le personnel médical comprennent également
la maladie, ils peuvent adopter une attitude plus positive face au patient.
2. Il est utile d'avoir un personnage central sur qui le patient et les membres de
sa famille peuvent compter pour obtenir des explications sur l'état du patient
ou de la thérapie à suivre.
3. Des visites fréquentes de la famille et des amis s'avéreront réconfortantes et
apporteront un soutien moral.
4. Particulièrement dans une salle de soins intensifs dépourvue de fenêtres, une
horloge, un calendrier électrique, un radio, une lumière de nuit peuvent aider

- 86 -
le patient à suivre les heures du jour et de la nuit, à garder un contact avec le
monde extérieur, à améliorer son orientation et à minimiser son désarroi.
5. Si des sensations anormales venaient à se présenter, le patient peut se sentir
soulagé s'il sait que celles-ci sont normales et qu'elles peuvent être souvent
contrôlées.
6. Si l'on permet au patient de parler de ses réactions émotionnelles, comme la
colère, la frustration et la crainte, cela l'aidera à faire face à ces sentiments.
7. Si la famille et des amis passent du temps au chevet du patient et pratiquent
avec lui certaines activités (soins, lecture, jeux de cartes, discussions au sujet
des dernières nouvelles de la famille, etc), ceci réduira le sentiment
d'isolement et d'inutilité qu'un séjour prolongé à l'hôpital peut susciter chez
un patient.

- 87 -
EVOLUTION ET PRONOSTIC :
A. L’évolution : [179,180] L'évolution du SGB est aigüe et monophasique, avec une phase d'extension des
paralysies qui ne dépasse pas 4 semaines.
• Le degré d'extension des paralysies est très variable: tous les intermédiaires
existent entre des formes ambulatoires avec déficit minime et des formes grabataires.
Les formes nécessitant une assistance respiratoire représentent 10 à 20% des
cas. Elles s'inscrivent dans un contexte de paralysie, en général, extensive mais,
parfois, modérée des membres.
L'insuffisance respiratoire est due au déficit des muscles respiratoires
(intercostaux et diaphragme) avec hypoventilation et inefficacité de la toux ainsi qu'au
déficit des muscles de la déglutition avec fausses routes et encombrement. Elle doit
être redoutée à la moindre défaillance de la toux, de la phonation ou de la déglutition.
Le malade doit, alors, être confié à une unité de soins intensifs pour bénéficier de la
surveillance et des soins appropriés.
• Le cap critique reste fatal dans une proportion non négligeable de cas (10%
dans les séries à recrutement hospitalier; 2% dans les séries tout - venant), même avec
la meilleure prise en charge.
Le décès est le fait d'une défaillance respiratoire aigüe, de troubles paroxystiques
du rythme cardiaque ou de la régulation tensionnelle et des complications thrombo -
emboliques du décubitus.

- 88 -
• Le cap critique passé, la récupération est la règle même pour les formes qui
ont nécessité des mesures de réanimation lourde.
La guérison complète est, ainsi, obtenue en 3 à 6 mois dans la majorité des cas.
Cependant, des séquelles existent dans 10 à 20% des cas, le plus souvent limitées à
une aréflexie ostéotendineuse. Elles sont rarement invalidantes et, exceptionnellement,
sévères.
Enfin, des rechutes s'observent dans environ 1% des cas.
B. Le pronostic : [180,184-186] La perspective générale pour le patient victime du SGB est relativement optimiste.
Et les valeurs suivantes donnent une estimation du pronostic à long terme :
àLa guérison peut être totale chez 50 à 90 % des patients. Certains de ces
patients peuvent avoir des anomalies persistantes légères qui n'interféreront pas avec
la fonction à long terme. Celles-ci peuvent être des sensations anormales telles que
des fourmillements, des muscles endoloris ou la fatigue de certains muscles qui
rendent la marche ou tout autre activité maladroite ou difficile.
àSeulement 5 à 15 % des patients atteints du SGB resteront gravement invalides
à long terme, ce qui les empêchera de reprendre un style de vie normale.
àUn patient ne sera que rarement contraint de se déplacer dans une chaise
roulante pour une période de temps prolongée.
La guérison peut s'étendre de six mois à deux ans ou plus. Certaines généralités
concernant la rapidité de guérison peuvent être établies en se fondant sur les données
du Hopkins-based GBS Study Group, publiées en 1988. Ces données indiquent que les
patients présentant les caractéristiques sous décrites ont d'excellentes chances (95 %)

- 89 -
de guérir totalement (être capable de marcher sans la moindre assistance) dans les
trois mois suivant le début de leur maladie. Les caractéristiques de ces patients sont :
1. une étude VCN-EMG relativement normale (activité électrique musculaire
normale [amplitude de la motricité])
2. traitement par échange plasmatique ou plasmaphérèse dans les 4 semaines
suivant l'apparition des premiers symptômes.
3. aucune ventilation artificielle
4. aucune consultation d'un médecin avant le 7ème jour des symptômes (ce qui
signifie que leur maladie évoluait plus lentement, c'est-à-dire sur une à trois
semaines, plutôt que rapidement, c'est-à-dire en quelques jours).
5. le patient approche de la trentaine plutôt que de la soixantaine.

- 90 -
CONCLUSION :
Epidémiologie : § Le syndrome de Guillain Barré a été décrit par messieurs Guillain Barré et
Strohl en 1916. L'incidence actuelle en France est de 1.5 cas pour 100 000 habitants,
sans qu'il y ait d'augmentation saisonnière statistiquement prouvée. Elle atteint plus
fréquemment les hommes (ratio 1.5), avec un age moyen d'environ 40 ans.
§ Dans 2/3des cas, on retrouve à l'interrogatoire du patient un épisode
infectieux respiratoire ou digestif (d'origine virale ou bactérienne) dans les semaines
précédant l'apparition des troubles neurologiques.
PHYSIOPATHOLOGIE ET ATTEINTE CLINIQUE : La polyradiculonévrite aiguë correspond à une atteinte quasi-exclusive du
système nerveux périphérique. On peut rencontrer 3 formes, qui se distinguent par la
structure atteinte:
àDes formes démyélinisantes, responsables de polyradiculoneuropathie
démyélinisante inflammatoire aiguë.
àDes formes axonales, responsables de neuropathie axonale aiguë motrice et
sensitive, ou motrice seule.
àLe syndrome de Miller-Fisher correspondant à 5% des SGB. Il s'agit d'une
variante du SGB avec une atteinte probable du système nerveux central, puisque le
tableau clinique comprend une ophtalmoplégie et un syndrome cérébelleux.

- 91 -
Cliniquement, le SGB se manifeste par l'installation rapide et ascendante d'une
paralysie symétrique des membres inférieurs, puis supérieurs avec aréflexie ostéo-
tendineuse et de façon inconstante de troubles sensitifs. Dans 65% des cas, on retrouve
une atteinte du système nerveux autonome, qui se manifeste notamment par une
instabilité hémodynamique, qui peut être sévère, une rétention urinaire, une
constipation, pouvant aller jusqu'à l'iléus et une hypersudation. L'atteinte des nerfs
crâniens est fréquente, pouvant entraîner notamment des troubles de la déglutition.
Le SGB, qui est une PRNA idiopathique, évolue classiquement en 3 phases :
- la phase d'installation de la paralysie, qui ne dépasse pas 3 semaines
- la phase de plateau, de durée courte
- et la phase de régression des troubles, qui peut durer plusieurs mois (à 1 an,
70% des patients ont complètement récupéré).
Le diagnostic de la maladie repose sur l'examen clinique, sur les résultats de la
ponction lombaire (dissociation albumino-cytologique typique) et de
l'électromyogramme.
Traitement :
o Le traitement du conflit immunologique :
Il est efficace s'il est mis en jeu au stade précoce d'installation des déficits. Deux
types de traitement ont montré une efficacité au cours d'essais contrôlés.
· Les échanges plasmatiques
Ils sont réalisés sur 10 à 14 jours pour un volume total de 200 à 250 ml/kg. Leur
efficacité a été prouvée sur la réduction de la durée de ventilation assistée, de la durée

- 92 -
d'hospitalisation et l'amélioration du pronostic fonctionnel.
· Les immunoglobulines IV polyvalentes
400 mg/kg/j pendant 5 j Leur efficacité est comparable à celle des échanges
plasmatiques.
·Les traitements par corticoïdes, ACTH ou des fortes doses de méthyl-prednisolone
n'ont pas montré d'efficacité.
o La prise en charge de décubitus au stade aigu :
Elle ne doit pas être négligée. Elle repose sur les soins de nursing ; la prévention
des thromboses veineuses, des troubles trophiques cutanés, des infections
nosocomiales ; le traitement de la douleur ; la surveillance de la déglutition, de la toux,
de l'encombrement bronchique ; la réanimation hydro-électrolytique et respiratoire si
nécessaire.
o Rééducation.
Pronostic :
30% des patients vont avoir besoin d'une ventilation mécanique ; la mortalité est
d'environ 5%, alors qu'elle était de 25% dans les années 60.

- 93 -
A. Choix des patients : Il s’agit d’une étude rétrospective, elle concerne une série de patient ayant
présenté une PRNA, hospitalisés au service de neurologie du C.H.U Hassan II, et ceci
depuis 2003 jusqu’au 31 Mars 2009.
La recherche des dossiers a été effectuée à partir des registres d’hospitalisation
du service.
B. Critères d’inclusion :
• LES PRNA :
L’installation d’un déficit moteur extensif bilatéral.
Une aréflexie ostéo-tendineuse.
Une évolution selon une phase d’extension puis une phase de plateau ; et
enfin une phase de régression.
• Chez l’adulte : à partir de l’âge de 15ans.
• Le diagnostic de SGB a été conforté par :
La présence de DISSOCIATION ALBUMINO-CYTOLOGIQUE.
Etude EMG en faveur de SGB.

- 94 -
C. Fiche d’exploitation :
Nous avons adopté pour l’ensemble de nos patients une fiche d’exploitation
standardisée |Voir annexe 4], qui a permis l’étude des paramètres suivants :
1. Données épidémiologiques :
Les paramètres notés sont l’âge, le sexe, la scolarité, la couverture sociale, la
profession, le niveau socio-économique, le lieu d’habitat, la saison, la répartition
mensuelle, la répartition annuelle, et les antécédents.
2. Données cliniques :
Les paramètres étudiés comprennent : les modalités de début, les signes de
début (signes fonctionnels sensitivo-moteurs, troubles respiratoires, troubles de la
déglutition, troubles sphinctériens, ou neuro-végétatifs…) et leurs modes d’évolution,
et la présence d’autres signes associés.
On a noté aussi les données de l’examen physique : le déficit moteur, sa
distribution, sa vitesse de progression, son sens d’extension, sa symétrie, son
intensité, sa prédominance éventuelle, les anomalies de la sensibilité superficielle et
profonde, le tonus, les ROT, l’atteinte des paires crâniennes, l’état de conscience, et la
présence des signes de détresse respiratoire à l’admission.
3. Données para cliniques :
a. Biologiques :
Précisant pour chaque patient la protéinorachie, la cytorachie, ainsi que
l’existence d’une DISSOCIATION ALBUMINO-CYTOLOGIQUE.

- 95 -
On note aussi la présence de syndrome inflammatoire, anomalie de
l’ionogramme sanguin, ou du bilan d’hémostase, ou du bilan lipidique, ou celui
infectieux éventuellement.
b. Électro physiologiques :
Elles sont représentées par l’EMG qui précise l’atteinte myélinique et/ou axonale.
4. Complications et leurs bilans :
Sont notés : l’atteinte pleuro-pulmonaire au cours de l’hospitalisation, les
infections urinaires, les troubles neuro-végétatifs, les troubles de la déglutition, les
phlébites, l’embolie pulmonaire, et le retentissement psychique.
Le bilan comprend le cliché radiologique pulmonaire, le scanner cérébral si le
patient présente des troubles de la conscience ou des signes atypiques, voire même
une IRM, l’échographie abdominale pour les patients ayant une anomalie de l’examen
abdominal, et l’étude cytobactériologique des urines (ECBU).
5. Données thérapeutiques :
Nous avons mentionné les mesures de réanimation, les thérapeutiques
spécifiques, avec éventuellement la survenue d’effets secondaires, et la conduite
entreprise devant un tel incident, et les traitements symptomatiques.

- 96 -
6. Données évolutives :
Nous avons relevé la durée de la phase d’extension et de plateau, l’intensité du
handicap à l’entrée et à la sortie du malade, celui-ci a été évalué grâce à l’échelle du
Hughes (EH) [Annexe1], et le score MRC.
Sont également notés tous les cas décédés, ou les transferts vers d’autres
services, ainsi que le suivi des patients.
D. Nos résultats : Toutes les données recueillies sont mentionnées sur les tableaux [Voir annexe 5]
E. Méthodologie statistique :
Dans notre étude, nous avons utilisé le logiciel SPSS et sphinx.

- 97 -
CHAPITRE I : « LES DONNÉES EPIDEMIOLOGIQUES DE L’ETUDE »
A. L’effectif de notre étude :
Notre étude a concerné 40 patients admis en service de neurologie du CHU
Hassan II, entre 2003 jusqu’au 31 Mars 2009.
B. L’incidence hospitalière annuelle :
Le SGB chez l’adulte représente 1,55% des causes d’hospitalisation dans le
service de neurologie du CHU Hassan II, depuis son inauguration en 2003.
L’incidence annuelle la plus élevée était calculée 2006, avec un chiffre qui atteint
32/1.000 hospitalisations, mais ce taux va être dépassé cette année, car, déjà après les

- 98 -
3premiers mois, l’incidence touche 32,5/1.000hospitalisations. En dehors de ces deux
années, cette proportion n’excède pas 16/1.000 hospitalisation.
C. La répartition mensuelle : Le SGB se présente majoritairement dans le mois de Janvier, puis dans les mois
de Mars et Juillet, avec des taux qui correspondent respectivement à 27,5%, 15% et
15%.
D. La répartition saisonnière : Il existe des variations saisonnière de la fréquence de survenue du SGB, ainsi on
remarque que le pic da la maladie se fait ressentir pendant les deux saisons de l’hiver
et l’été, au cours desquels, le SGB s’est présenté respectivement chez 37,5% et 25% de
nos patients.

- 99 -
E. Le sexe : On note une importante prédominance masculine, dont le taux représente plus
du double du taux des malades de sexe féminin. Le taux des Hommes et celui des
Femmes représentent respectivement 70% et 30%, donnant un sex-ratio de 2,33.
F. L’âge : L’âge des patients inclus dans cette étude variait entre 15ans et 81ans, avec un
âge moyen de 37,21ans ; et nous avons pu remarquer que la grande majorité des
patients atteints de cette maladie (55% ; n=22) ont un âge jeune, entre 15ans et 35ans.
Par contre, seulement 10% (n=4) de nos malades était des vieillards, avec un âge
supérieur à 65ans.

- 100 -
G. La répartition géographique : Durant la période, objet de notre étude, seulement 40% des patients hospitalisés
pour SGB était originaire de Fès, pour le reste, les patients étaient originaires
majoritairement de Taza, et de Taounat, avec une proportion qui atteint respectivement
40% et 17,5%.

- 101 -
H. Le niveau socio-économique : Presque la totalité de nos patients sont d’un niveau socio-économique moyen
(90%), les 10% restants sont démunis.
I. La couverture sociale : Nous notons que plus des trois quarts de la population étudiée (78%) n’avaient
pas de couverture sociale.

- 102 -
J. La scolarité : Il faut signaler que seulement 65% de nos patients étaient scolarisés.
K. Les antécédents : Globalement, parmi les 40 malades de la série, 47.5% (n =19) était sans
antécédent particulier, le reste avait un antécédent infectieux et/ou un événement
prodromique.
Dans les 4 semaines précédant l’installation des signes neurologiques, un
événement infectieux était rapporté chez la moitié (n=20) de nos patients.

- 103 -
Il s’agissait par ordre de fréquence de :
ü Syndrome grippal : 50%.
ü Gastro-entérite : 30%.
ü Pneumopathie : 15%.
ü Infection urinaire : 5%.
Alors qu’un événement prodromique n’a été rapporté que chez 10% (n=4) de nos
malades.

- 104 -
Il s’agissait d’une grossesse dans 75% des cas (n=3), et de cardiothyréose dans
le reste (n=1).

- 105 -
CHAPITRE II : « L’HISTOIRE DE LA MALADIE »
A. Le délai de consultation :
Si un nombre important de malade a consulté dans la première semaine suivant
l’installation des signes neurologiques, un nombre non négligeable a tardé pour
consulter, dépassant même dans certain cas 2semaines.
Seulement 52,5% de la population étudiée a consulté dans la première semaine
suivant l’installation des signes neurologiques, et même, 27,5% des patients, ont
dépassé le délai de 15 jours pour consulter un médecin.

- 106 -
B. Le mode d’installation : La moitié des patients observés sont sujets à un début brutal (n=20), alors que
l’installation de la maladie était progressive chez 27,5% (n=11) ; le reste (22,5%)
décrivait un début rapidement progressif.

- 107 -
C. Les signes fonctionnels de début : Le début de la symptomatologie était tout le temps marqué par la présence d’une
lourdeur des membres (100%), et il s’agissait majoritairement d’atteinte des 4 membres
(87,5%-n=35).
Par contre, seulement 62,5% de nos malades (n=25) ont présentés des signes
fonctionnels sensitifs à la phase initiale, tels que des paresthésies, des myalgies, des
radiculalgies, ou des dorso-lombalgies.
Les signes de gravité, tels que des difficultés de la déglutition, les troubles
neuro-végétatifs, des gênes respiratoires, se sont présentés moins fréquemment, avec
des proportions égales respectivement à : 32,5% ; 22,5% et 9%.
Mais il faut noter qu’on n’a pas observé de trouble de conscience en aucun cas.
Par ailleurs, des signes centraux comme des céphalées, étaient rapportés par un
seul malade (2,5%).

- 108 -
CHAPITRE III : « L’EXAMEN PHYSIQUE »
A. Le déficit moteur :
1) La distribution du déficit : Tous nos patients avait un déficit moteur (100%), il concernait très fréquemment
les 4 membres (90%), dans ces cas, le déficit était essentiellement complet réalisant
une tétraplégie (52,5% de toute la population étudiée) ; par ailleurs, les 10% des cas
restants, réalisaient tous un déficit partiel de 2 membres seulement (MS ou MI),
réalisant une paraparésie.

- 109 -
2) La symétrie :
Le déficit moteur était majoritairement symétrique (dans 77,5% de nos malades),
par contre, l’asymétrie était remarquée seulement chez 22,5% de nos malades.
3) la prédominance :
Le caractère proportionnel se voit moins fréquemment, avec un taux de 17,5%. La
prédominance soit proximale, soit distale était notée respectivement chez 42,5% et 40%
des malades de notre série.

- 110 -
4) Le sens de progression du déficit :
Dans notre étude, Il s’agissait souvent d’un déficit moteur extensif ASCENDANT
(82,5%), depuis les membres inférieurs aux muscles du tronc et/ou des membres
supérieurs.
Néanmoins, des modes d’extension moins typiques de SGB, tels que
DESCENDANT ou EN « U » ; étaient très peu rapportés, dans 12,5% et 5% des cas,
respectivement.
5) La vitesse d’extension :
La progression de déficit était souvent très rapide (en 24h) ou rapide (en moins
de 5jours), avec des taux respectifs qui correspondaient à : 42,5% et 37,5% ; et ce n’est
que dans 20% des cas que l’extension du déficit s’est faite en plus de 6 jours.

- 111 -
B. Le déficit sensitif : Dans notre étude, l’examen de la sensibilité était souvent normal, et on n’a pas
noté d’atteinte de la sensibilité superficielle ou profonde que chez 20% et 22,5%
respectivement.

- 112 -
De même, le signe de Lasègue n’a été objectivé que chez 30% de nos patients.
C. Les ROT :
Les ROT, dont la diminution est un signe majeur du syndrome neurogène
périphérique, étaient faibles, ou carrément abolis, dans presque la totalité des cas
(97,5%).

- 113 -
D. Le tonus : L’hypotonie qui constitue un élément très important dans la définition du
syndrome neurogène périphérique, était objectivée dans 90% de nos patients, par
contre, le tonus était considéré normal chez le reste (10%).
E. Atteinte des nerfs crâniens : L’atteinte des nerfs crâniens était remarqué dans, à peu près, plus d’un tiers des
cas (37,5%).

- 114 -
Il s’agissait essentiellement de l’atteinte du VII° paire crânienne (dans 35,71% des
cas), suivie des nerfs bulbaires : X ; IX ; XI dont l’atteinte représente respectivement
41,42%, 14,28% et 14,28%.
Par ailleurs, si l’atteinte des nerfs III, IV, V, VI était rare, on n’a pas noté d’atteinte
des nerfs VIII et XII.

- 115 -
CHAPITRE IV : « LES EXAMENS COMPLEMENTAIRES »
A. La ponction lombaire :
a. La Dissociation albumino-cytologique :
La PL était réalisée chez 72,5% de nos malades.
La DISSOCIATION ALBUMINO-CYTOLOGIQUE, qui est pathognomonique du SGB,
était retrouvée dans un peu plus de deux tiers de ces malades (68,96%).

- 116 -
b. La corrélation délai de réalisation de la PL et DISSOCIATION
ALBUMINO-CYTOLOGIQUE :
Pour les PL réalisées dans la 1ière semaine suivant l’installation des symptômes, la
DISSOCIATION ALBUMINO-CYTOLOGIQUE était trouvée dans la moitié des cas
seulement, ce taux augmente jusqu’à 75% lorsque la PL était réalisée au-delà du 8ème
jour d’évolution.
Il faut signaler que la PL n’a été refaite en aucun cas, même devant l’absence de
DISSOCIATION ALBUMINO-CYTOLOGIQUE.
c. Les résultats de l’étude cyto-chimique du L.C.R :
La protéinorachie mesurée lorsque la DISSOCIATION ALBUMINO-CYTOLOGIQUE
est présente varie entre 0.49g/l et 2.63g/l, avec une moyenne de 0,80g/l ; mais la
cellularité dans ces cas ne dépassait pas 10 éléments/mm3. Par contre, la cellularité
atteignait même 200 éléments (PL traumatique) sans que la protéinorachie ne dépasse
pas 0,33 g/l.

- 117 -
B. L’examen électro-physiologique :
L’EMG n’a été réalisé que chez 12,5% de nos malades, et aucune donnée n’a été
retrouvée dans les observations, concernant les signes électro physiologiques décelés
lors de l’EMG, il a été noté seulement la conclusion.
Dans les cas où on a réalisé d’EMG, on n’a pas noté de forme axonale pure, mais
plutôt, une forme démyélinisante chez 40% (n=2), une forme mixte (démyélinisante et
axonale) chez 40% (n=2), et une forme séquellaire chez 20% (n=1).

- 118 -
C. Bilan biologique :
1. Syndrome inflammatoire : VS+/-CRP :
Le syndrome inflammatoire biologique était souvent remarqué (62%), mais il
s’agissait tout le temps d’un léger syndrome inflammatoire.
2. Sérologies :
Toutes les sérologies réalisées (TPHA-VDRL, VHB, VHC, HIV, Borréliose) étaient
négatives. Mais, il faut noter comme même qu’elles n’étaient pas tout le temps
réalisées.

- 119 -
D. Bilan radiologique : La radiographie pulmonaire réalisée chez la moitié de nos malades était souvent
normale (90% ;n=18), néanmoins, elle a trouvé une pneumopathie d’inhalation chez les
10% restant (n=2).

- 120 -
CHAPITRE V : « La prise en charge thérapeutique »
A. L’hospitalisation et les services de référence :
L’hospitalisation des patients présentant un SGB était systématique (100%).
La plupart de ces malades étaient hospitalisés par le biais de service des
URGENCES, et ceci était dans 77,5% des cas. En 2ème position, c’est le service de
réanimation qui nous a adressé 7,30% de nos malades.
B. La mise en condition : /Une sonde gastrique pour gavage était posée chez 12 malades (30%).
/La sonde vésicale était mise chez 22,5% de nos malades.
/L’oxygénothérapie était nécessaire pour 8 malades (20%).

- 121 -
/Mais la voie veineuse périphérique pour réhydratation n’était mise en place que
dans 5% des cas (2malades).
C. Le traitement de la douleur : Les antalgiques ont été utilisés par nos patients pour lutter contre la douleur,
dans 70% des cas.

- 122 -
Les médicaments qui ont été prescrits dans la plupart des temps correspondaient
aux antidépresseurs dans 38,90% des cas, puis aux corticoïdes (en bolus ou per os)
dans 27,7% des cas, contre 16,7% et 11,15% des cas pour l’utilisation des
indométacines, paracétamol associé à la dextropropoxiphene et/ou la codéine. Par
ailleurs, un paracétamol était prescrit efficacement, tout seul, seulement chez 5,55% de
nos patients.
D. Le traitement antibiotique : L’utilisation des antibiotiques se fait dans le cadre de traitement curatif des
infections communautaires ou nosocomiales.
On a eu recours à une antibiothérapie chez 20% de la population étudiée (n=10).

- 123 -
Ceci essentiellement pour traiter une pneumopathie (12,5%-n=5), puis une
infection urinaire (7,5%-n=3), et enfin pour traiter des leucorrhées survenant chez
2malades, ce qui correspond à un taux de 5%.
E. Laxatifs : Les laxatifs osmotiques ou lubrifiants ont été utilisés comme traitement de la
constipation pour 10 malades (25%).

- 124 -
F. La protection gastrique :
L’association d’une protection gastrique a été effectuée chez 8 malades (20%)
pour prévenir l’ulcère de stress utilisant des inhibiteurs de la pompe.

- 125 -
G. Les autres traitements symptomatiques : Tous nos patients ont bénéficié systématiquement d’un nursing, d’une
kinésithérapie quotidienne, et d’une héparinothérapie préventive à base d’héparine de
bas poids moléculaire.
H. Le traitement spécifique : 80% de nos malades (32malades) n’ont reçu qu’un traitement symptomatique
bien mené, et seulement 20% (8malades) ont bénéficié d’un traitement spécifique.
Le traitement spécifique utilisé était tout le temps à base d’immunoglobuline
intraveineuse pendant 5jours ; par contre, les autres moyens thérapeutiques, aussi bien
la plasmaphérèse et la corticothérapie ; n’ont pas été instauré en aucun cas.

- 126 -
L’utilisation des immunoglobulines intraveineuses pendant 5jours a était
confronté par la survenue d’effets secondaires dans 27,5% des cas traités, et ceci vers
le 2ème, 3ème jour d’instauration d’Ig IV, et il s’agissait exclusivement de réaction
hyperthermie-frisson, qui régresse immédiatement après une fenêtre thérapeutique de
8h et perfusion de paracétamol.
I. La prise en charge des formes graves : 12,5% de nos patients (5malades) avaient des signes de gravité imposant un
transfert en milieu de réanimation.

- 127 -
Dans le milieu de soins intensifs, le recours à une trachéotomie pour détresse
respiratoire était nécessaire chez 3 malades (7,5%) ; et le recours à une intubation
ventilation pour troubles de la déglutition était inéluctable pour 2 malades (5%).

- 128 -
CHAPITRE VI : « Le profil évolutif »
A. La durée de la phase d’extension :
La durée de la phase d’extension était inférieure à 7jours, donc rapide, chez
77,5% des patients, mais il faut signaler que le quart de nos malades avait une
extension très rapide, en moins de 3jours.
La durée moyenne de cette phase dans notre série, était de 5,5 jours.
B. La durée de la phase de plateau : La durée de cette phase dans notre étude variait en fonction des cas, et elle était
aussi bien courte (<=14jours) que prolongée (>14jours), avec des proportions égales
de 50%.

- 129 -
La durée moyenne de cette phase était de 16,85 jours.
C. La durée des hospitalisations :
Bien que 72,5% de nos malades était hospitalisé durant moins d’un mois, une
importante proportion de malade était hospitalisée durant plus de 2mois (7,5%). Par
ailleurs, 20% de nos malades ont séjournés dans notre service entre 1 et 2mois.
Mais, globalement, la durée moyenne d’hospitalisation était de 25jours.

- 130 -
D. L’évolution de l’handicap fonctionnel : Dans notre série, mise à part le nombre restreint des patients qui avaient des
signes mineurs (2,5%seulement) ; la plupart de nos malades était, à l’admission,
confiné au lit (82,5%-n=33), et 15% (n=6) des malades était dépendant d’un autrui ou
d’un déambulateur pour marcher.
Par ailleurs, aucun malade n’a été capable de marcher sans aide.
Après certaines périodes, le score de Hughes s’améliore selon le tableau suivant :

- 131 -
C’est vrai qu’on n’a pas noté de décès dans les observations exploitées, mais il
ne faut pas oublier de considérer le nombre important des patients perdus de vue
avant toute déduction.
E. Score Sun MRC Parmi la population étudiée, l’inaptitude fonctionnelle évaluée par le score Sun MRC était
essentiellement légère, car dans 47,5% des cas, le score Sun MRC était inclus entre 41-60. Il
vient ensuite, par ordre de fréquence, une inaptitude fonctionnelle moyenne (Sun MRC entre
21-40) avec un taux de 37,5%. Néanmoins, l’inaptitude était sévère chez 15% des patients.
A l’acmé d’évolution, les formes légères d’inaptitude se sont réduit presque de moitié
(22,5%) ; pour devenir des formes modérée à sévères, dont les taux se sont devenus
respectivement 50%, et 27,5%.

- 132 -
Il est probable que le nombre de 40 malades étudiés dans cette série est sous
estimé, vue les critères d’inclusion stricts appliqués dans cette étude.
L’âge de nos patients variait entre 15 et 81ans, avec un âge moyen de 37,21ans,
une moyenne qui concordent avec celle rapportée dans d’autres séries.
La PRNA touche les deux sexes avec, dans les séries comparatives, une nette
prédominance masculine. Les résultats de notre série ne sortent pas de cette règle,
avec une importante prépondérance masculine, comme il en témoigne le sex-ratio qui
atteint 2,33.

- 133 -
Le tableau ci-dessous montre la distribution des patients selon le sexe et l’âge :
On en déduit que le plus grand nombre des cas de SGB, survient entre 15-25ans
et 25-35ans. La majorité des patients était jeune (67,5% au dessous de l’âge de 45ans,
55% au dessous de l’âge de 35ans, et seulement 32,5% au-delà de l’âge de 45ans) ;
reflétant la réalité que le Maroc, comme la plupart des pays en voies de
développement, a une population qui est jeune.
Notre série montre un modèle de distribution selon l’âge qui ressemble à celui
des séries de Rong Juo Cheng et de Bahemuka, avec une grande fréquence de survenue
chez le jeune adulte avant l’âge de 40ans.

- 134 -
C’est vrai que les grandes séries ne trouvent pas de tendance saisonnière, mais
quand on a regroupé les patients selon le moment d’installation de leur maladie
neurologique (saison), une importante variation saisonnière était manifeste. Un
regroupement saisonnier modéré était noté en hiver et en été ; et occasionnellement
en automne. La prépondérance saisonnière s’est remarquée en printemps dans les
séries de Bahemuka et de Ramzia.
De même, une variation mensuelle manifeste était notée dans notre série qui
parle d’une importante fréquence de survenue le mois de janvier (27,5%), et d’absence
de cas rapporté le mois de mai. Par contre, les variations mensuelles rapportées dans
les séries comparatives restent modestes, et statistiquement non significatives. La
fréquence varie entre 11,28% (Mai) et 3,76% (Juillet) dans la série israélienne de Dov
Soffer, et elle est entre 7,92% (Janvier et février) et 0% (Juillet) dans la série canadienne
de Winner.

- 135 -
La fréquence de la présence de facteur déclenchant (événement infectieux ou
prodromique) était de 52,5%, une proportion qui reste comparable aux chiffres
rapportés par divers centres :

- 136 -
L’antécédent infectieux le plus fréquemment retrouvé est le syndrome grippal
(50%), ce taux concorde avec les études dr Rong Kuo Chang et de Ramzia, or, la série
chinoise de Qi Cheng, trouve plutôt l’antécédent de pneumopathie plus fréquemment.
Dans notre série, on a remarqué qu’il y a souvent un retard de prise en charge
imputable au retard de consultation des malades, car seulement 52,5% des patients ont
consulté un médecin en moins d’une semaine après installation des signes
neurologiques, et 27,5% des patients ont dépassé un délai de 2semaines pour
consulter. Ceci pourrait être expliqué par :
/le taux élevé des personnes non scolarisées, qui est de 35%.
/un problème financier du fait que presque tout nos patients était de moyen
niveau socio-économique (90%), voir démunis (10%).
/une couverture sociale restreinte, car seulement 23% des malades était
mutualiste.

- 137 -
/Accès au soin difficile, vu qu’une grande partie de nos patients provenait d’une
région situé au-delà du périmètre urbain, et seulement 40% des cas, était habitant Fès.
L’analyse des signes fonctionnels rapportés par nos patients montre que le signe
qui a été constamment présent à l’admission était la lourdeur d’un ou plusieurs
membres (100%), par contre, les signes sensitifs étaient moins fréquents (62,5%), ce
qui concordent avec les chiffres présentés par des séries africaines de Bahemuka et
Ramzia, contrairement aux séries européennes, asiatiques, et nord américaine, dont les
patients rapportaient souvent plus de signes sensitifs que de faiblesse musculaire, avec
une lourdeur d’un ou plusieurs membres moins constante, n’excédant même pas le
taux de 43%.
L’atteinte oculomotrice, avec l’asymétrie faciale et les signes bulbaires étaient
rapportés dans 27,5% des cas, et il s’agissait majoritairement de troubles bulbaires

- 138 -
(20%), cette remarque concorde à celle rencontrée dans la série de Bahemuka, alors
qu’on rapporte la prépondérance d’atteinte oculomotrice dans la série de Ramzia, et
d’atteinte faciale fréquente dans les autres séries comparatives.
Les autres signes (ataxie, céphalées) étaient rapportés moins fréquemment, aussi
bien dans notre série que celles de Bahemuka et Bouche P.
Lors de l’examen à l’admission, la PRNA se présente, aussi bien dans notre étude
que celle de Bahemuka, sous forme de paralysie hypotonique hypo-aréflectique, dans
presque 100% des cas ; mais si nos patients avaient un déficit plutôt distal (42,5%), ou
proximal (40%), les patients de la série comparative, ont eu un déficit plutôt mixte
(38,9%). Par ailleurs, l’examen physique de nos malades trouvait un déficit sensitif
distal, ou une détresse respiratoire imposant une ventilation assistée, ou une
fluctuation tensionnelle, moins fréquemment.
Pour conclure, le tableau clinque typique de nos malades correspondait à
l’installation brutale (50%) d’un déficit moteur (100%) total (52,5%) intéressant les 4
membres (90%) ; symétrique (77,5%), ascendant (82,5%), à prédominance soit

- 139 -
proximale (42,5%) ou distale (40%), avec aréflexie (97,5%) et hypotonie (90%) ;
progressant en moins de 24 heures (42,5%), associées à des signes sensitifs surtout
subjectifs (paresthésies, myalgies, radiculalgies,..) (62,5%) contrastant avec une
sensibilité souvent normale à l’examen physique (77,5%). L’atteinte des nerfs crâniens
s’est vue dans presque un tiers des cas (37,5%), touchant préférentiellement le nerf
facial (35,71%) et les nerfs bulbaires (X : 21,42% - IX : 14,28%- XI : 14,28%).
L’étude cyto-chimique du L.C.R était réalisée chez 29 patients (72,5%) ; 20
(68,96%) avait la dissociation albumino-cytologique définie comme une protéinorachie
supérieure à 0,5g/l et une cytorachie inférieure à 10/mm3. Cette fréquence de
DISSOCIATION ALBUMINO-CYTOLOGIQUE est similaire à celle rapportée dans les autres
séries.
La moyenne des protéinorachies trouvées est 0.80g/l, mais le niveau de
protéinorachie le plus élevé est 2,63g/l.

- 140 -
Notre étude montre que la fréquence de la présence de DISSOCIATION
ALBUMINO-CYTOLOGIQUE augmente de 50% à plus de 70%, lorsque la PL était réalisée
au-delà de la première semaine d’évolution de la maladie (à 75% après 8jours
d’évolution, à 71,42% après le 15ème jour d’évolution, et à 75% après 3semaines). Ce
qui rejoint le délai de réalisation de PL rapporté dans la littérature, est qui est entre 3
et 10 jours.
Entre les 5 malades (12,5%) qui ont bénéficié d’une étude électro-physiologique,
on n’a pas trouvé de forme axonale pure, il s’agissait soit de forme démyélinisante (2
malades), soit de forme mixte (2malades). Pour le cas restant, l’EMG parlait de forme
séquellaire de PRN ancienne. Ce nombre est surement sous estimé, et ne reflète pas le
nombre réel des malades ayant bénéficié de l’EMG, mais il s’agit seulement du nombre
des EMG dont on n’a pas oublié de mentionner le résultat sur les dossiers, ou dont le
document n’a pas été perdu lors de l’archivage.

- 141 -
L’absence des résultats détaillés de l’EMG ne permet pas une étude analytique
comparative avec les autres séries.
Entre nos 40 patients, aucun malade n’a bénéficié de plasmaphérèse, par contre,
les Ig IV étaient utilisés pour 8 malades (20%). Par ailleurs, la majorité de nos patients
(80%) n’ont reçu aucune immunothérapie, et c’est les thérapeutiques symptomatiques
qui gardent leur grand intérêt.
Ce taux réduit d’utilisation des traitements spécifiques est dû à leur coût trop
élevé avec le bas niveau socio-économique de la population, à la non disponibilité du
matériel spécialisé pour réaliser les EP.
Il faut noter comme même, la fréquence des effets secondaires (62,5%) chez les
patients ayant reçu IG IV, mais, ils restent comme même des simples incidents faits de
réactions d’hyperthermie frissons, efficacement traitées par le paracétamol en IVL, et
une fenêtre thérapeutique de 8h.

- 142 -
La plupart de nos malades avait une phase d’extension inférieure à 7jours, avec
un taux qui avoisine 78%, un résultat qui concorde avec celui trouvé dans les séries de
Ramzia et Tourabi, par contre, les séries de Masschusetts et de Bouche P rapportent un
plus petit pourcentage (11% et 12% respectivement).
La durée moyenne de la phase de plateau dans notre série reste proche de celle
rapportée par les études comparatives.
Les pourcentages du recours à la ventilation assistée et de la mortalité sont les
plus bas dans notre série ; contrairement à ceux trouvés dans les séries de Ramzia et
Tourabi, et ceci s’explique par leurs population étudiée qui est particulièrement
sévères, du fait qu’elle regroupe les malades du milieu de réanimation. D’autre part, la
durée moyenne d’hospitalisation était relativement courte (moins d’un mois) aussi bien
pour notre série que celle de Ramzia et Tourabi, alors quelle arrivait jusqu’à 2mois
dans les séries de Massachusets et Bouche P, car les patients de ces deux dernières
séries restent hospitalisés le plus longtemps possible pour complément de

- 143 -
rééducation, alors que nos malades, par défaut de place, étaient obligés de quitter le
service une fois ils dépassent le cap sévère, en prenant un rendez-vous pour
kinésithérapie.
Seulement 10% de nos malades étaient capables de marcher sans aide après
8semaines d’évolution, de plus, une proportion non négligeable (2,5%) restait confinée
au lit après 3mois d’évolution de la maladie. Par contre, dans les autres séries
comparatives, la grande partie des populations étudiées était devenu indépendant lors
de la marche après 8semaines d’évolution (37% pour la série de Bahemuka ; 41% pour
celle de Qi Cheng). Ceci pourrait être expliqué par l’utilisation des traitements
spécifiques, permettant ainsi une récupération fonctionnelle précoce.

- 144 -
Figure 1: Georges Guillain
Figure 2: Jean Alexendre Barré
Figure 3: Jean Baptiste Octave Landry

- 145 -
Figure 4: Fibre nerveuse d'un patient présentant une AIDP Microscopie électronique montrant un macrophage (M) infiltrant la membrane de cellule de Schwann.
Reproduit de la référence : Hughes RAC. Guillain-Barré Syndrome. Heidelberg: Springer- Verlag; 1990.
avec permission de Spinger Science et Business Media.
Figure 5:Démyélinisation segmentaire au cours du SGB
Segments consécutifs d’une fibre myélinique provenant d’un patient ayant une PRNA. De haut en bas, la fibre, dont la myéline est colorée
en noir par postfixation par la tétraoxyde d’osmium, présente des segments démyélinisés(espaces clairs) avec des débris myéliniques
témoignant de la progression des lésions.

- 146 -
Figure 6: Coupe semi-fine d'une biopsie de nerf musculo-cutané d'un patient ayant un SGB en phase de plateau prolongée
Notez les nombreuses fibres démyélinisées(Flèches) Echelle :20µm.
Figure 7: Microscopie électronique. SGB atypique.
Biopsie de nerf montrant un axone démyélinisé entouré de nombreuses cellules macrphagiques (M).
Ax : axone ;S : Cellule de Schwann

- 147 -
Figure 8: Fibre nerveuse d'un patient ayant une AMAN
La coupe d’en bas est un élargissement de la zone délimitée dans la coupe d’en haut.
Microscopie électronique montrant des macrophages qui s’infiltrent dans l’espace péri-axonale et la membrane cytoplasmique
axonale(Flèches), entourant ainsi l’axone (A).mcp :macrophage.
Reproduit de la référence : Kieseier BC, Kiefer R, Gold R, Hemmer B, Willison HJ, Hartung HP. Advances in understanding and treatment of
immune-mediated disorders of the peripheral nervous system. Muscle Nerve 2004; 30: 131–56., avec la permission de Spinger Science et
Business Media.

- 148 -
Figure 9:Lésion et réparation d'un neurone du SNP
Figure 10: mécanismes cellulaires de la régénération axonale et de réparation.

- 149 -
Figure 12 : Mécanismes des déficits après démyélinisation.
Cytoplasme
Noyau
Corps cellulaire
Dendrites
Axone
Gaine de myéline
(blanche)
Fibres musculaires
Terminaison axonique
Plaque motrice
Synapse neuromuscul
aire
Figure 11: Organisation d’un neurone

- 150 -
Figure 12 : Mécanismes immunologiques dans AIDP (A), et AMAN, AMSAN et syndrome de Fisher
(A) L’épitope protéique bacterien est présenté par un macrophage à une lymphocyte T qui est ainsi activée, cette cellule traverse
l’endothélium et reconnanit un antigene par réaction croisée,dans la section d’en bas, relâche des cytokines qui activent des
macrophages endoneuriaux. Ces derniers relâchent des enzymes, et des radicaux libres de monoxyde d’azote qui détruisent la
myéline. Dans la section d’en haut, les cellules T actives relâchent des cytokines qui aident les lymphocytes B dans la production
des anticorps qui traverse la barrière hémato-nerveuse endommagée, ces anticorps reconnaissent un épitope (non encore identifié)
sur la surface abaxonale des cellules de Schwann, par réaction croisée, et après fixation du complément, les cellules de Schwann
seront détruites. avec dissolution de myéline.
(B) Un épitope “anglioside-like” bactérien stimule les cellules B pour des anticorps pour interagir avec des antigènes axonales, et
après fixation du complement, les macrophages s’infiltrent dans l’espace périaxonal pour bloquer la conduction ou entrainer une
dégénérescence axonale. Dans le syndrome de Fisher, même les cellules de Schwann péri-synaptiques des nerfs moteurs, sont pris
pour cibles.

- 151 -
Figure 12: Electromyogramme
Figure 13: Technique de filtration

- 152 -
Figure 14: Anneau de centrifugation continue
Figure 15: Bol de centrifugation à flux discontinu

- 153 -
Tableau I: Germes incriminés comme facteurs favorisants de la survenue de SGB
Hollande 1987-96 (n=476)
Amérique du nord et Europe
1993-95 (n=383)
Campylobacter Jejuni
32 23
Cytomégalovirus 18 8 Ebstein Barr Virus 7 2
M. pneumonie 9 Non testé
Tableau II: Les critères cliniques du syndrome de Miller Fisher
a) Déficit des muscles oculomoteurs sévère, bilatérale, relativement
symétrique, associé à un ptôsis.
b) Ataxie avec tremblement.
c) Aréflexie généralisée pendant au moins une semaine.
d) Installation de ces trois premiers symptômes entre quelques jours et trois
semaine.
e) Déficit des membres, de la face ou oropharyngé absent ou discret.
Paresthésies absentes ou discrètes.
f) Etat e conscience normal, absence de dysarthrie, sauf secondaire aux
déficits précédents
g) ; absence de signe de Babinski.

- 154 -
Tableau III: Critères prédictifs de ventilation et indications de la ventilation mécaniques au cours de SGB
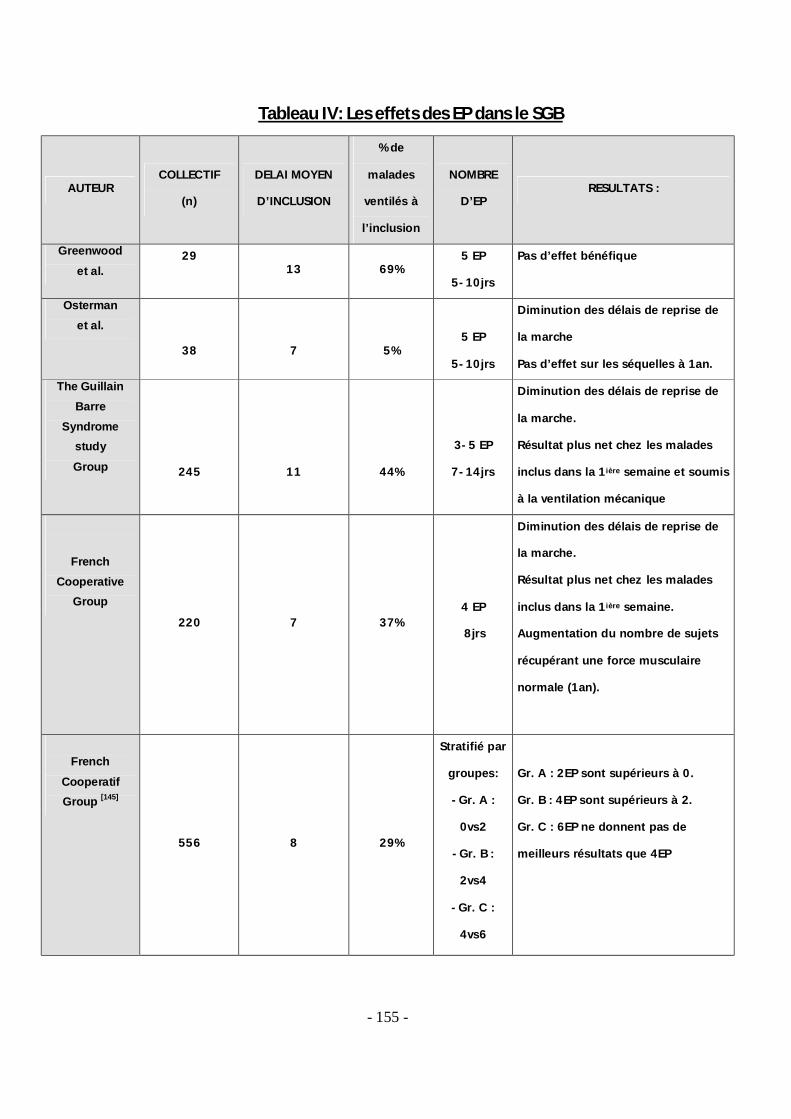
- 155 -
Tableau IV: Les effets des EP dans le SGB
AUTEUR COLLECTIF
(n)
DELAI MOYEN
D’INCLUSION
% de
malades
ventilés à
l’inclusion
NOMBRE
D’EP RESULTATS :
Greenwood et al.
29
13 69%
5 EP
5-10jrs
Pas d’effet bénéfique
Osterman et al.
38
7
5%
5 EP
5-10jrs
Diminution des délais de reprise de
la marche
Pas d’effet sur les séquelles à 1an. The Guillain
Barre Syndrome
study Group
245
11
44%
3-5 EP
7-14jrs
Diminution des délais de reprise de
la marche.
Résultat plus net chez les malades
inclus dans la 1ière semaine et soumis
à la ventilation mécanique
French Cooperative
Group
220 7 37%
4 EP
8jrs
Diminution des délais de reprise de
la marche.
Résultat plus net chez les malades
inclus dans la 1ière semaine.
Augmentation du nombre de sujets
récupérant une force musculaire
normale (1an).
French
Cooperatif Group [145]
556 8 29%
Stratifié par
groupes:
-Gr. A :
0vs2
-Gr. B :
2vs4
-Gr. C :
4vs6
Gr. A : 2EP sont supérieurs à 0.
Gr. B : 4EP sont supérieurs à 2.
Gr. C : 6EP ne donnent pas de
meilleurs résultats que 4EP

- 156 -
Tableau V:Effet comparé ou cumulé de EP et des Ig IV dans le SGB
AUTEURS :
Nombre
de cas
Délai
moyen
d’inclusion
(J)
Malades
ventilés à
l’inclusion
(%)
Traitements :
Résultats :
Van der Meché
Et al 150
≤7 chez 73%
des patients 19
5EP entre 7 et 14j
ou IgG (0,4g/Kg/j)
pendant 5j
Les IgG sont au
moins aussi efficaces
que les EP et
entrainent moins de
complications.
PE/Sandoglobin
Group 383 7 12
5EP pendant 5à13j
ou IgG (0,4g/Kg/j)
pendant 5j ou
association des
deux.
Pas de différence des
résultats
Tableau VI: Schéma d'utilisation des traitements spécifiques dans le SGB de
l'adulte
Formes bénignes (Groupe A) Formes intermédiaire ou sévère (Groupes B et C)
Traitement
initial
2EP
4EP ou IgG(0,4g/kg/j pdt 5j)Respect des contre-
indications dans les deux cas.
Aggravation 2EP supplémentaires Pas de traitement
Rechutes Pas de traitement ou reprise du premier traitement utilisé.

- 157 -
ANNEXE 1: ECHELLE DE HUGHES
0 Examen normal 1 Signes mineurs, Peut courir. 2 Capable de marcher 5min dans un espace ouvert sans aide. 3 Capable de marcher 5min dans un espace ouvert, avec l’aide d’une personne, d’un
déambulateur, ou 2cannes. 4 Confiné au lit, ou dans un fauteuil, ne peut pas marcher même avec une aide. 5 Patient mis sous ventilation assistée. 6 Décès
ANNEXE 2: Critères diagnostiques du SGB selon Asbury (1990)
ANNEXE 3: Score fonctionnel (Medical Research Council (MRC-Sun score)
Abduction du bras
Cotation de la force musculaire de 0 à 5, pour chaque élément, de
façon bilatérale, pour une cotation entre 0 et 60.
La flexion de l’avant bras
L’extension du poignet
La flexion de la cuisse.
L’extension du genou
La flexion dorsale du pied.

- 158 -
ANNEXE 4: Fiche d’exploitation
THESE : Syndrome de Guillain Barre
Identité :
*NE: *Age:
*NO : *Sexe : M □ / F □
*Date d’entrée : *Origine :
*Date de sortie : *Habitat :
*Profession :
Antécédents particuliers :
Evénement infectieux récent : oui □ non □ non définie□
àIl y a : .......jours.
àLequel? : VAS□ respiratoire□ digestif□ urinaire□ autres□ ...………………
à Identifié ? Oui □ non □ àlequel ? Virose□ Mycoplasma□ Campylobacter jejuni□
Evénement prodromique : oui□ non□
àIl y a : ......jours.
àLequel? : Traumatisme□ acte chirurgical□ autres□
Sérothérapie□ lequel ?.......................................
Vaccination□ laquelle ?...................................
Cas similaire dans l’entourage : oui□ non□ famille□ autre□......................
Clinique :
Délai de consultation après l’apparition du 1er symptôme :.....jrs imprécis□
Quelle phase ? d’extension□ plateau□ de récupération□

- 159 -
Phase d’extension ou d’installation :
Déficit moteur Déficit sensitif Nerfs
crâniens ROT Tonus
Amyotrophie ? S.N. végétatif
ui
MS □ Dt□ Gch□
MI □ Dt□ Gch□
Symétrie ?
Oui □ non □
Paresthésie □
- Fourmillement□
- Picotements □
-
Engourdissement□
Myalgie □
Dorsalgie □
Lombalgie □
Radiculalgie □
- LASEGUE □
Sensibilité profonde ?
Nle□ altérée□ imprécis□
........
........
........
........
........
........
Aux MS
..........
..........
Aux MI
...........
...........
HTA□
Hypotension
orthostatique□
Tachycardie□
Tr. De rythme□
Tr vasomoteurs□
Constipation□
Rétention
urinaire□
Alie de la
sudation□
Imprécis□
on
à Durée :........jrs.
àComplications ? Tr. Respiratoire □ compte expiratoire jusqu’à .......... ou Imprécis □
Tr. De la déglutition □
Tr. Végétatif □
Autres □.............................
à Recours à la ventilation assistée ou le transfert en réanimation :
Oui□ non□ préciser........................

- 160 -
Phase de plateau :
Déficit moteur Déficit sensitif Nerfs crâniens
ROT Tonus Amyotrophie
S.N. végétatif
oui MS □ Dt□ Gch□ MI □ Dt□ Gch□ Symétrie ? Oui □ non □
Paresthésie □ Fourmillement□ Picotements □ Myalgie □ Dorsalgie □ Lombalgie □ Radiculalgie □ Sensibilité profonde ? Nle□ altérée□ imprécis□
........
........
........
........
........
........
Aux MS .......... .......... Aux MI ........... ...........
HTA□ Hypotension orthostatique□ Tachycardie□ Tr. De rythme□ Tr vasomoteurs□ Constipation□ Rétention urinaire□ Alie de la sudation□ Imprécis□
non à Durée :........jrs.
àComplications ? Tr. Respiratoire □ compte expiratoire jusqu’à .......... ou Imprécis □
Tr. De la déglutition □
Tr. Végétatif □
Complications de décubitus□ : phlébite□ EP□ Escarres□ Infection urinaire□ infection
pulmonaire□ Autre□.....
à Recours à la ventilation assistée ou le transfert en réanimation :
Oui□ non□ préciser........................
Phase de récupération :
à Durée :.......jrs.
à Dans le sens inverse de l’installation des déficits ? oui □ non □
à Complications ?
Complications de décubitus□ : phlébite□ EP□ Escarres□ Infection urinaire□ infection
pulmonaire□ Autre□.....
Rétraction tendineuse □
Au total :
Forme typique□ motrice pure□ purement sensitive□ descendante□

- 161 -
Syndrome de Miller Fisher ataxie cérébelleuse aréflexie diffuse et
ophtalmoplégie□
Paraclinique :
LCR : faite au .......° jours après le début des symptômes.
Aspect ? clair□ non□
Cellularité :....
Proteinorachie :.......
Refaite ? Oui □ non □ ; si oui, pourquoi ?......................ou imprécis □
Étude électrophysiologique :
o Signes de démyélinisation un ralentissement des vitesses de conduction, prédominant sur la
partie proximale (retard des ondes F) et distale (temps de latence distale augmenté) des nerfs, de
distribution bilatérale, symétrique et diffuse. De véritables blocs de conduction sont, parfois, observés.
Oui□ non□ les souligner !!!!!!!
o Signes d’atteinte axonale, donnant un tracé neurogène, avec diminution du nombre des
unités motrices fonctionnelles oui□ non□
Sd inflammatoire ? oui□ non□
Sérologie HIV ? faite□ non□ si oui, positive□ négative□
Radio pulmonaire ? faite□ non□ si oui, normal□ ou anormal□
préciser□..............................................
Autres ?résultats ?...............................................................................................................
..................................................................................................................................................................
...........................
PEC
² Hospitalisation ? oui□ non□ si non prq ?........................

- 162 -
² TTT symptomatique :
*Ventilation assistée □ * Alimentation entérale □
*Sonde nasogastrique □ *Nursing des tr. Sphinctériens □
*Kinésithérapie □ délai après hospitalisation?....jrs. Rythme journalier □ Durée ?.................
*Ttt de la douleur □ quel médicaments...............................................ou imprécis□
*ttt des troubles de la régulation cardiaque et tensionnelle. □
*Prévention des phlébites par héparinothérapie □ des escarres par mobilisation□ ou matelas à
eau□ ou non□
² TTT étiologique :
*Corticothérapie ? oui□ non□
*Plasmaphérèse ? oui□ non□ si oui, à j.......... de la phase........... ; avec □ ou non □ de
l’albumine.
*immunoglobulines intraveineuses ? oui□ non□ si oui, à j..... de la phase.................
*Association Plasmaphérèse – immunoglobulines intraveineuses ? oui□ non□
Evolution :
* guérison ? oui□ non□ en combien de temps ?............
*séquelles ? oui□ non□ quel type ? aréflexie tendineuse□ autre□...............
*récidives ? oui□ non□ après combien de temps ?.............
*Décès ? oui□ non□ Si oui, la cause ?
Défaillance respiratoire aiguë □
Troubles paroxystiques du rythme cardiaque ou de la régulation tensionnelle □
complications thrombo - emboliques du décubitus □
En quelle phase ?.............................

- 163 -
Durant la période d’étude qui s’étend de 2003 au 31Mars 2009, la polyradiculonévrite aigue était diagnostiquée chez 40 marocains adultes.
Le tableau prototype de nos malades était celui d’une forme paralysante (100%)
des 4 membres (90%), ascendante (82,5%), hypotonique (90%) hypo-aréflectique (97,5%) et symétrique (77,5%), avec une installation plutôt brutale (50%), chez des malades d’âge jeune (67,5% <45ans ; âge moyen : 37ans), et de sexe surtout masculin (Sex-ratio :2,33). Par ailleurs, si les signes sensitifs subjectifs étaient rapportés assez souvent (62,5%), l’examen physique ne trouvait l’atteinte sensitive que dans 22,5% des cas. Ce tableau était fréquemment précédé par syndrome grippal dans 50%.
La dissociation albumino-cytologique est retrouvée chez presque 70% des
malades ayant bénéficié de ponction lombaire, alors que l’électromyogramme était rarement réalisé (12,5%).
Seulement 37,5% des malades présentait une atteinte des nerfs crâniens,
représentée surtout par l’atteinte bulbaire (20%) ; en plus, seulement 7,5% de nos malades ont présenté une détresse respiratoire, choses qui expliqueraient :
• Un taux de transfert en milieu de réanimation réduit (12,5%). • Un recours à une trachéotomie ou une ventilation assistée moins fréquent (7,5% et
5% respectivement). • Une mortalité à la phase aigue qui est nulle. • La durée d’hospitalisation est prolongée (au-delà d’un mois) chez 27,5%. • La récupération fonctionnelle est retardée, • 65% de nos malades était confiné au lit à la sortie du service.
Ceci est dû probablement à l’absence du traitement de fond, qui est les
immunoglobulines

- 164 -
During the period of our study which extends from 2003 to the 31Mars 2009, the
acute polyradiculoneurotis was diagnosed at 40 Moroccan adults. The clinical features of our patients are characterized by a paralysis (100 %) of 4
members (90 %), ascending (82,5 %), flaccid (90 %), aréflexic (97,5 %), and symmetric (77,5 %), with a rather rough installation (50 %). Patients was younger (67,5 % < 45ans; average age: 37ans), and males appear to be affected more commonly (Sex-ratio 2,33). Besides, if the sensory disturbance were rather often reported (62,5 %), clinical examination failed to show abnormal sensory signs in 77,5 % of the cases. Symptoms are preceded frequently by the flu-like syndrome in 50 %.
The cyto-albuminological dissociation is found at almost 70 % of the patients
having a cerebrospinal fluid analysis, while the neurophysiological testing was rarely realized (12,5 %).
Only 37,5 % of the patients had a cranial nerves involvement, paralysis of the
lower cranial nerves( chiefly the 9th and 10TH) is the commonest (20 %); moreover, only 7,5 % of our patients had a respiratory failure, which would explain:
• A reduced rate of transfer to Intensive Care Unit (12,5 %). • An appeal to a tracheotomy or mechanical ventilation less frequent (7,5 % and
5 % respectively). • No mortality in the acute stage. • A long duration of hospitalisation (beyond one month at 27,5 % of cases) • A slower functional recovery, • A great rate of patients confined on the bed at the exit of the service (65%). It would be explained by the fact that specific treatment was not used, especially
the intravenous immunoglobulin.

- 165 -
حالة 40 تشخيص تم ،2009 مارس 31 غاية إلى 2003 سنة من امتدت والتي دراستنا، فترة خالل .فقط البالغين األشخاص عند وذلك المتعددة، ولجذورها لألعصاب الحادة االلتهابات مرض من
( شلل ودوج في وتتمثل ،%) 50( وحاد سريع بشكل تظهر المرضى، هؤالء عند نمطية األكثر األعراض
بصفة%) 90( األطراف جميع يصيب ،% )82.5( األعلى إلى األسفل من صاعد%) 90( رخو%) 100 بين النسبة( الذكور من غالبا هم المرضى هؤالء %) .97.5( الوثرية للمنعكسات غياب مع ،%) 77.5( متماثلة كانت وإذا) . سنة 37= رالعم متوسط ، سنة 45 من أقل% 67.5( الشباب سن في) 2.33= الجنسين
اإلحساس في خالل يجد لم السريري الفحص فإن ،%) 62.5( الحضور غالبة الحسية الوظيفية االضطرابات . الحاالت من% 22.5 في إال العميق أو السطحي
المرض ظهور تسبق وهي الحادة باإلنفلوانزا شبيهة فيروسية التهابات وجدنا ما غالبا البحث وخالل)50(% .
السائل تحليل من استفادوا الذين المرضى من% 70 عند وجد فقد ، الخلوي_األبوميني التفارق أما %) .12.5( ناذرا إال للعضالت الكهربي التحليل يستعمل لم حين في النخاعي،
، التاسعة األعصاب بالخصوص همت وقد%) 37.5( تأثرت ما فقليال القحفية، األعصاب بخصوص أما
وقد ، التنفس في حاد لقصور تعرضوا المرضى من% 7.5 فقط ذلك، على زيادة%) 20( عشر والحادية رةالعاش : تفسر التي األمور هي هاته تكون
% 12.5 اإلنعاش مصلحة في ناذرا استشفاء - %) 5( األصطناعي التنفس مع الرغامي والتنبيب ،%) 7.5( الرغامي للخزع نسبيا ضعيفا لجوءا - . المرض من الحادة المرحلة في وفاة حاالت دوجو عدم - % . 27.5 عند) واحد شهر من أكثر( طويلة استشفاء دةم - .متأخر وظيفي استرجاع - .الفراش يطريح وهم المصلحة غادروا%) 65( المرضى أغلب - .الوريد عبر" الغاماغلوبولين: " الخاص العالج استعمال لعدم ذلك يرجع وقد

- 166 -
[1] : Bouche P : Polyradiculonévrite aigue inflammatoire (Syndrome de Guillain Barré).
Impact internat, Septembre 1997. [2] : Sharshar T , Siami S, Orlikowski D. Does this patient present a Guillain-Barré
syndrome?. Réanimation (2007) 16, 504-510. [3] : Guidet B, Gallouedec G : Polyradiculonévrite aigue. Anesthésie Réanimation, 2000 ;
36-913. [4] : Pritchard J, Hughes RAC, Guillain Barre syndrome. Lancet 2004 ; 363 ; 2186-88. [5] : Raphaël JC, Sharshar T, Bourdain F, Léger JM, Le syndrome de Guillain Barré : de la
description princeps aux concepts modernes. Ann Med Interne 1999 ; 150(1) : 33-41.
[6] : Créange A. Le syndrome de Guillain Barré, Bréve histoire… . Neurologies. Juin-Juillet2006. Vol.9 ; n°87. 428-430.
[7] : Rossignol V. Polyradiculonévrites aigues d’origine infectieuse probable. A propos de 18 observations. Thèse : Med : Faculté de Nancy (France) 1992 ; 11271.
[8] : Asbury AK, Arnasson BG, Karp Herbert R. Criteria for diagnosis of Guillain Barre syndrome. Ann Neurol 1978; 3(6): 565-66.
[9] : Prineas JW. Acute idiopathic polyneurotys. An electronmicroscop study. Lab Invest 1972 ; 26 : 133-47.
[10]: Prineas JW. Pathology of the Guillain-Barré syndrome. Ann Neurol 1981; 9suppl:6-19.
[11]: Cambier J.Polyradiculonévrites inflammatoires. Encyclopédie Médico-chirurgicale Paris système nerveux 1975 ;17097D-10.
[12] :Dubas F, Berger F. Polyradiculonévrites. Encyclopédie Médico-chirurgicale Paris 1987 ; 1215-12.
[13] : Créange A, Sharshar T, Raphaël JC, Gherardi R. Aspects cellulaires de la neuroinflammation au cours du syndrome de Guillain Barré : une clé pour une nouvelle voie thérapeutique ? Rev Neurol (Paris) 2002 ; 158 (1) : 15-27.
[14] : Said G, Goulon GC. Syndrome de Guillain Barre. EMC2002.17-095-A-10. [15] Raphaël JC. Polyradiculonévrites aiguës inflammatoires (Syndrome de Guillain
Barré). La revue du praticien 2001 ; 51 :2119-24.

- 167 -
[16]: Chipps E, Clanin N, CampbeIl V 1992 Neurologlc disorders Mosby Clinical Nursing Series, London Editorial 1988 Lancet Sept 17(2). 659-661.
[17]: Kelly B 1992 Landry-Guillain-Barré syndrome. In Parsons P, Wiener-Kronish J (eds) Critical Care Secrets. Hanley & Belfus/Mosby-Year Book, Philadelphia, ch. 72.
[18]: Hund E, O'Borel C, Cornblath D, Hanley D, McKhann G 1993 ICU management of Guillain-Barré. Critical Care Medicine 21 (3): 433-446.
[19]: Murray D 1993 Impaired mobility: Guillain-Barré syndrome Journal of Neuroscience Nursing 25 (2):100-104.
[20]:Hughes RAC, Cornblath DR. Guillain Barré syndrome. Lancet2005 ; 366 :1653-66. [21]: Pritchard J. What’s new in Guillain Barre syndrome ? Postgrad. Med.J. 2008 ; 532-
538. [22]: http://www.assim.refer.org/8122.htm [23]: Mlshu B, Blaser M 1993 Role of infection due to Campylobacter jejuni in the
initiation of Guillain-Barré syndrome. Clinical Infectious Diseases 17(July) 104-108
[24] :Chio A, Cocito D, Leone M, Giordana MT, Mora G, Mutani R. Guillain Barré syndrome : A prospectivf, population based incidence and outcome survey. Neurology 2003;60:7.
[25]: Guidet B, Gallouedec G. Polyradiculonévrite aiguë. Traité d’anesthésie-réanimation2000 :36-913-A-10
[26]: Roman GC. Tropical neuropathies. In: Hartung H-P, editor. Peripheral Neuropathies: Part 1. London: Baillière Tindall; 1995:469–87.
[27] : Jacobs BC, Rothbarth PH, van der Meché FG, et al. The spectrum of antecedent infections in Guillain Barré syndrome : a case contrôle study. Neurology1998:1110-15.
[28] :Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in Guillain Barré syndrome. Neurology 2001; 56:758-65.
[29] :Ho TW, Mishu B, Li CY, et al. Guillain-Barré syndrome in northern China, Relationship to Campylobacter jejuni infection end anti-glycolipid antibodies. Brain1995; 118:597-605.
[30]: Orlikowski D, Quijano RS, Sivadon, TV, Raphaël JC, Guaillard JL. Infections à campylobacter jéjuni et à cytomégalovirus associé au syndrome de Guillain Barré. Pédiatrie au quotidien 2006 :2-4

- 168 -
[31] : Kei F, Michiaki K, Masaki T, Koichi H, Nobuhiro Y. Campylobacter coli enteritis and Guillain Barré syndrome : No evidence of molecular mimicry and serological relationship. Journal of the neurological sciences. Short communication 2006; 246:163168.
[32]: Koga M, Takahashi M, Masuda M, Hirata K, Yuki N, Neurology 2005;65:1376-1381.
[33]: Diagana M, Khalil M, Preux PM, Dumas M, Jauberteau MO, Polyradiculonévrites et Campylobacter Jéjuni: Aspects cliniques et physiologiques. Méd trop 2003 ;63 :68-74.
[34] : Patton, C.M., Wachsmuth, I.K., Evins, G.M., Kiehlbauch, J.A., Plikaytis, B.D., Troup, N., Tompkins, L., Lior, H., 1991. Evaluation of 10 methods to distinguish epidemic-associated Campylobacter strains. J.Clin. Microbiol. 29, 680–688.
[35] :Rees JH, Soudain SE, Gregson NA, et al. Campylobacter jejuni infection and Guillain Barré syndrome. N Engl J Med 1995a ;333 :1374-9.
[36] : Mishu B, Ilyas AA, Koski CL, et al. Serologic evidence of previous Campylobacter Jejuni infection in patients with the Guillain Barre syndrome. Ann Intern Med 1993; 118; 947-53
[37]: Jacobs BC, Rothbarth PH, Van der Meche FG, et al. The spectrum of antecedent infections in Guillain Barre syndrome: a case control study. Neurology 1998;51:1110-15.
[38]: Jacobs BC, Van Doorn PA, Schmitz PI, et al. Campylobacter Jejuni infections and anti-GM1 antibodies in Guillain Barre Syndrome.Ann Neurol1996;40:181-7.
[39]: Sivadon V, Orlikowski D, Rozenberg F, et al. Prevalence and caracteristics of Guillain Barre syndromes associeted with Campylobacter Jejuni and Cytomegalovirus in greater Paris.Path biologie 53(2005): 536-8.
[40]: Yuki N, Taki T, Inagaki F, et al. A bacterium lipopolysaccharide that elicits Guillain Barre syndrome has a GM1 ganglioside-like structure. Exp Med 1993, 178:1771-5.
[41]:Hao Q, Saïda T, Kuroki S, et al. Antibodies to gangliosides and galactocerebrosides in patients with Guillain Barre syndrome with preceding Campylobacter Jejuni and other identified infections. Neuroimmunol1998; 81:116-26.

- 169 -
[42]:Kuroki S, Saïda T, Nukina M, et al. Campylobacter jejuni strains from patients with Guillain Barre syndrome belong mostly to Penner serogroup 19 and contain β-N-acetylglucosamine residues. Ann Neurol1993; 33:243-7.
[43]:Goddard EA, Lastovica AJ, Argent AC. Campylobacter O:4 isolation in Guillain Barre syndrome.Arch Dis Child 1997; 76:526-8.
[44]:Yuki N, Takahashi M, Tagawa Y, et al. Association of Campylobacter Jejuni serotype with antiganglioside antibody in Guillain Barre syndrome and Fisher’s syndrome. Ann Neurol 1997; 42: 28-33.
[45]:Ohtsuka K, Nakumora Y, Hashimoto M et al. Fisher syndrome associated with Ig G anti-GQ1b antibody following infection by a specific serotype of Campylobacter Jejuni. Ophtalmology 1998; 105:1281-5.
[46]:Sheikh KA, Nachamkin I, Ho TW, et al. Campylobacter Jejuni lipopolysaccharides in Guillain Barre syndrome: molecular mimicry and host susceptibility. Neurology 1998; 51:371-8.
[47]: Winer J, Hughes RAC, Anderson MJ, et al. A prospective study of acute idiopathic neuropathy. II. Antecedent events. Journal of Neurology, Neurosurgery and Psychiatry1988; 51:613-18.
[48]: Boucquey D, Sindic CJ, Lamy M, et al. Clinical and serological studies in a serie of 45patients with Guillain Barre syndrome. J Neurol Sci 1991;104:56-63.
[49]: Jacobs BC, Van Doorn PA,Groeneveld JH, et al. Cytomegalivirus infections and anti-GM2 antibodies in Guillain Barre syndrome.J Neurol Neurosurg Psychiatry 1997;62:641-3
[50]: Hagberg L, Malmvall BE, Svennerrholm L,et al. Guillain Barre syndrome as an early manifestation of HIV central nerveus system infection. Scand J Infect Dis 1986;18:591-2.
[51]: Bouma PAD,Carpay HA, Rijpkema SGT. Antibodies to Borrelia burgdorferi in Guillain Barre syndrome [letter]. Lancet 1989; ii:739.
[52] :Langmuir AD, Bregman DJ, Kurland LT, et al. An epidemiologic and clinical evaluation of Guillain Barre syndrome reported in association with the administration of swine influenza vaccines. Am J Epidemiol 1984; 119:841-79.

- 170 -
[53] : Safranek TJ, Lawrence DN, Kurland LT, et al. Reassessement of the association between Guillain Barre syndrome and receipt of swine influenza vaccine in 1976-1977: results of a two-state study. Ewpert Neurology Group. Am J Epidemiol 1991;133:940-51.
[54] : Kinnunen E, Farkkila M, Hovi T, et al. Incidence of Guilain Barre syndrome during a nationwide oral poliovirus vaccine compaign. Neurology 1989; 39:1034-6.
[55]: Olive JM, Castillo C, Castr RG, et al. Epidemiologic study of Guillain Barre syndrome in children <15years of age in Latin America. J Infect Dis 1997; 175 (suppl1):S160-4.
[56]: Tuttle J, Chen RT, Rantal H, et al. The risk of Guillain Barre syndrome after tetanus-toxoïde-containing vaccines in adults and children in the United States. Am J Public Health 1997; 87:2045-8.
[57]: da Silveira CM, Salisburg DM, de Quadros CA. Measles vaccination and Guillain Barre syndrome. Lancet 1997; 349: 14-16.
[58]: Tuohy PG, Guillain Barre syndrome following immunisation with synthetic hepatitis B vaccine [letter]. NZ Med J 1989;102:114-115.
[59]: Shaw FE, Graham DJ, Guess HA et al. Postmarketing surveillance for neurologic adverse events reported after hepatitis B vaccinationExperience of the first three years. Am J Epidemiol 1988; 127:337-52.
[60]: Granieri E, Casetta I, Gononi V, Tola, et al. Ganglioside therapy and Guillain Barre syndrome. Neuroepidemiology 1991 ;10 :161-169
[61]: Schönhöfer, P.S., 1991. Guillain–Barre syndrome and parenteral gangliosides. Lancet 338, 757–757.
[62]: Figueras, A., Morales-Olivas, F.J., Capella, D., Palop, V., Laporte, J.-R., 1992. Bovine gangliosides and acute motor polyneuropathy. Br. Med. J. 305, 1330–1331.
[63]: Raschetti, R., Maggini, M., Popoli, P., Caffari, B., Da Cas, R., Menniti, I., Spila-Alegiani, S., Traversa, G., 1995. Gangliosides and Guillain– Barre syndrome. J. Clin. Epidemiol. 48, 1399–1405.
[64] : Stricker BH, van der Klaw MM, Ottervanger JP, et al. A case-controle study of drugs and other determinants as potential causes of Guillain Barre syndrome. J Clin Epidemiology1994; 47:1203-10

- 171 -
[65] : Awong IE, Dandurand KR, Keeys CA, et al. Drur associated Guillain Barre syndrome : a literature review. Ann Pharmacother1996 ;30 : 173-80.
[66]: Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barré syndrome. J Infect Dis 1997;176(Suppl 2):92-8
[67]: Said G, Goulon GC. Syndrome de Guillain Barre. Editions techniques. EMC (Paris-France), Neurologie1993, 5p.
[68] : Guidet B, Gallouedec G, Korach JM. Polyradiculonévrites et myasthenia grave en reanimation . SFAR éd. Confèrences d’actualisation . 40ème Congrès national d’anesthésie et deréanimayion. Paris : Elsevier 1998 ; 399-415.
[69] : Halls J, Bradkjaer C, Friis ML. Guillain Barre syndrome: diagnostic criteria, epidemiology, clinical course and prognosis. Act Neurol Scand 1988;78:118-22.
[70]: Kaplan JE, Schonberger LB, Hurwitz ES, Katona P.Guillain Barre syndrome in the United States 1978-1981: Additional observations from the national surveillance system. Neurol 1983;33:633-37.
[71]: Bogliun G, Beghi E. Incidence and clinical features of acute inflammatory polyradiculoneuropathy in Lombardy, Italy, 1996. Acta Neurol Scand 2004; 110: 100–06.
[72] : Jiang GX, Cheng Q, Link H, et al. Epidemiological features of Guillain Barre syndrome in Sweden; 1978-93. J Neurol Neurosurg Psychiatry 1997; 62:447-53.
[73]: de Pedro Cuesta J, Abraira V, Jiang GX, et al. Guillan Barre syndrome in south-west Sockholm, 1973-1991. 3. Clinicoepidemilogical subgroups. Acta Neurol Scand 1996; 93:175-83.
[74]: Van Koningsveld R, Rico R, Gerstenbluth I, et al. Gastroenteritis associated Guillain-Barré syndrome on the Caribbean island Curacao. Neurology 2001; 56: 1467–72.
[75]:Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol 1990; 27 (suppl): S21–24.
[76]: Durand MC, Porcher R, Orlikowski D, Aboab J, Devaux C, Clair B, et al. Clinical and electrophysiological predictors of respiratory failure in Guillain-Barré syndrome: a prospective study. Lancet Neurol 2006;5:1021—8.
[77] Hadden RD, Karch H, Hartung HP, Zielasek J, Weissbrich B, Schubert J, et al. Preceding infections, immune factors, and outcome in Guillain-Barré syndrome. Neurology 2001;56:758—65.

- 172 -
[78] Ho TW, Li CY, Cornblath DR, Gao CY, Asbury AK, Griffin JW, et al. Patterns of recovery in the Guillain-Barré syndrome. Neurology 1997;48:695—700.
[79]: Ropper AH. The Guillain-Barre syndrome. N Engl J Med 1992;326: 1130–6. [80]: Orlikowski D, Ratrimoson T, Prigent H, Annane D, Raphael JC, Clair B. Pneumonia
in ventilated Guillain Barre syndrome. Crit Care Med 2003;31(supplement):A87. [81]: Ropper AH, Kehne SM. Guillain-Barre syndrome: management of respiratory
failure. Neurology 1985;35:1662–5. [82]: Chevrolet JC, Deleamont P. Repeated vital capacity measurements as predictive
parameters for mechanical ventilation need and weaning success in the Guillain-Barre syndrome. Am Rev Respir Dis 1991; 144:814–8.
[83]: Sharshar T, Raphaël JC. Le syndrome de Guillain Barre. La lettre neurologique n°4 2001 ; V : 185-91.
[84] : Koningsveld R, Schmitz PIM, Van der Meché FGA, et al. For the Dutch GBS study group. Effect of methylprednisolone when added to standard treatement with IV Ig for Guillain Barre syndrome : randomised trial. Lancet 2004; 363:192-96.
[85]:Confavreux C, Moreaut. Sémiologie neurologique. Traité de médecine. Flammarion 2004 ;2 : 2399-408
[86]: Andrew S, Gurwood OD, Drake J. Guillain Barre syndrome. Optometry2006;77:540-46.
[87]: David HR, Oleszek JL, Cha-kim A, Guillain Barre syndrome [consulté le 16/01/2009]. Disponible à partir de : URL : http://www.emedecine.com/pmr/topic48.htm.
[88] : Liu CY, Kao CD, Chen JT, et al. Hydrocephalus associated with Guillain Barre syndrome. J Clin Neurosc 2006 ; 13 :866-69
[89] : Guy Sanders. A case of Guillain Barre syndrome presenting as ataxia. Am J Emerg Med 2004; 22:137-8.
[90]: Orlikowski D, Prigent H, Raphaël JC, Sharshar T.L’insuffisance respiratoire aigue du syndrome de Guillain Barre et de la myasthénie auto-imune. De la détection au sevrage de la ventilation mécanique. Science direct. Réanimation 2005 ; 14 : 118-25.
[91] :McGillicuddy DC, Walker O, Shapiro NI, Edlow JA. Guillain Barre syndrome in the emergency department. Ann Emerg Med 2006; 47: 390-93

- 173 -
[92] :Sharshar T, Durant MC, Orlokowski D, et al. Atteintes mécaniques respiratoires et électriphysiologiques phréniques à la phase précoce du syndrome de Guillain Barre. Réanimation 2001 ; 10 :133.
[93] : Azulay JP, Verschueren A, Attarian S, Pouget J. Le syndrome de Guillain Barre et ses frontières. Rev neurol (Paris) 2002 ; 158suppl 12 : 6S21-6S26.
[94] : Rees JH, Thompson RD, Smeeton NC, Hughes RAC. Epidemiological study of Guillain Barre syndrome in south east Englend. J Neurol Neurosurg Psychiatry1998; 64:74-7.
[95]: Franz GA, Van Der Meché, Pieter AV.Guillain Barre syndrome. Current treatmentd options in neurology 2000 ; 2 : 507-16.
[96] : Vallat JM, Hugon J, Tabaraud F et al. Quatre cas de syndrome de Guillain Barre avec lésion axonales.Rev Neurol (Paris) 1990 ;146(6-7) :420-4.
[97] : Fournier E.Actualités : Que peuvent attendre l’un de l’autre , syndrome de Guillain Barre et l’électrophysiologie ? Rev Neurol (Paris)2000 ;156(10) :925-32.
[98] : Flodrops H, Houdon HP, Plesiat Trommsdroff V et al. Apport de l’électromyogramme dans le syndrome de Guillain Barre atypique. Arch pédiatr. 2004 ; 11 :460-9.
[99] : Boulesteix JM, Tabaraud F, Salle JY et al. Diagnostic précoce du syndrome de Guillain Barre par stimulations électriques radiculaires. Rev. Neurol (Paris) 1995 ; 151(10) : 569-75
[100] : Birouk N, Belaidi H, Berramdane M, et al. Etude clinique et physiologique de 180 cas de syndrome de Guillain Barre . Rev Neurol (Paris) 2004 ; 3suppl : 102.
[101] : Flodrops H, Houdon HP, Plesiat Trommsdroff V et al. Apport de l’électromyogramme dans le syndrome de Guillain Barre atypique. Arch pédiatr. 2004 ; 11 :463-5.
[102] :Gire C, Perez N, Lamoureux S, Mancini et al. Syndrome de Guillain Barre chez l’enfant : une étude rétrospective de 31 cas. Ann Pédiatr 1997 ;44 : 593-600.
[103]Hung P, Wen-Neng C, uang L et al. A clinical and électrophysiologic survey of childhood Guillain Barre syndrome. Pédiatr Neurol 2004;30: 2.
[104]: Quarmouchi A, Approche diagnostique du syndrome de Guillain Barre chez l’enfant (20cas). Thèse : Méd : Faculté de médecine de Casablanca.1984 ;87.

- 174 -
[105] : Rantala H, James D, Donald Shield W, Uhari M. Epidemiology of Guillain Barre syndrome in children : relationship of oral polio vaccine adinistration to occurrence. J Pédiatr 1994; 124:220-3.
[106] : Tabarki B, Othmani K, Oubicha F et al. Sydrome de Guillain Barre chez l’enfant, étude de 39cas. La tunisie médicale 2001 ;79 :183-7.
[107] : Bouaggad A, Bouderka MA, Laraki M et al. Syndrome de Guillain Barre et grossesse. Rev Fr Gynecol Obstét 1994 ; 89(2) : 86-7.
[108] : Berteau P, Morvan J, Bernard AM. Association polyradiculonévrite aiguë, diabète insipide et grossesse. A propos d’un cas et revue de la littérature. J Gynécol Obstét1990 ; 19 : 739-802.
[109] : Luijckx GJ, Vles J, de Baets M, Buchwald B, Troost J. Guillain Barre syndrome in mother and newborn cild. Lancet 1997;349:27.
[110]: Tekgul H, Serdaroglu G, Tutuncuoglu S. Outcome of axonal and demyeliniting forms of Guillain Barre syndrome in children. Pediatr Neurol 2003;28: 295-9.
[111]: Lefève C, Martinez L, AbdulnayefA et al. Cas clinique : la neuropathie axonale motriceaiguë sous type du syndrome de Guillain Barre. Rev Neurol (Paris)2002 ;158 :616-8.
[112] : Durant MC, Goulon GC, Schewitzer A et al. Etude électrophysiologique de 10 cas de syndrome de Miller Fisher.Rev Neurol Paris 2001 ; 157(1) :72-9.
[113] : Masahiko K, Fujioka T, Miura H et al. The relation of the clinical symptoms and anti-ganglioside antbodies to MEPPs frequency increased in 8 cases of variant type Guillain Barre syndrome. J Peripher Nerv syst2003(8): 82-90.
[114]: Susuki K, Atsumi M, Koga M et al. Acute facial diplegia and hyperreflexia. A Guillain Barre syndrome variant. Neurology 2004;62:5.
[115]: Trojaborg W. Acute and chronic inflammatory demyelinating polyneuropathy, an overview and un update. Electroencephalography and clinical neurophysiology1998;107(5):303-16.
[116]: Créange A. mise au point: diagnostic des neuropathies sensitives acquises. Rev Neurol (Paris)2004 ; 160(3) : 363-70.
[117] : Labauge P, Neuropathies périphériques : Approche diagnostique. Traité de médecine Akos 1998 ;5-0920.
[118] :Kerry HL. Variant and mimics of Guillain Barre syndrome. The neurologist2004; 10:2,61-74.

- 175 -
[119] : Moulonguet A. Actualités: Septièmes journées des maladies du système nerveux périphèriques : neuropathies périphériques au cours de l’infection par le VIH. Rev Neurol (Paris) 2003 ; 159(12) : 1223-6.
[120] : Thomas H, Brannagan III, Yili Z. HIV associated Guillain Barre syndrome. J neurol Sci 2003 ; 208 : 39-42.
[121] :Antoine JC. Actualités Cinquièmes Journées du système nerveux périphérique : les neuropatie périphériques paranéoplasiques. Rev Neurol (Paris)2001 ;157(12) : 1557-60.
[122] :Antoine JC, Camdesanche JP. Les neuropathies périphériques paranéoplasiques. Rev Neurol (Paris) 2004 ; 160(2) :188-98.
[123] : Aït Benhaddou E, Birouk N, El Alaoui Faris et al. Polyradiculonévite aigue de type Guillain Barre révélant un lupus érythémateux aigu disséminé : Etude de deux cas et revue de la littérature. Rev Neurol (Paris)2003 ;159(3) :300-3.
[124] : Corcia P, Giraud P, Guennoc AM et al. Syndrome de Guillain Barre axonal, entérovirus et sclérose latérale amyotrophique : peut-il y avoir une relation ? Rev Neurol (Paris)2003 ; 159(1) :80-82.
[125] : Aissaoui M, Helier JP, Mathelier FP, P Leynadier P. Syndrome de Guillain Barre : une autre association ? Maghreb Médical 1996 ;35 :24-25.
[126] : Mouti O, Harmouch H, El Alaoui Faris M, et al. Méningomyélite et polyradiculonévrites aigues révélant un lupus érythémateux disséminé. Rev Neurol (Paris) 2002 ;158(1) : 81-3.
[127] : Attarian Sn Serratrice J, Mazodier C et al. Syndrome de Guillain Barre révélateur d’une leishmaniose viscérale chez une patiente non immunodéprimée. Rev Neurol (Paris) 2003 ; 159(11) : 1046-8.
[128] : Thibault L, Adams D. Syndrome de Guillain Barre. Traité de médecine Akos1998 ;5-0930.
[129] : Maisonobe T, Léger JM. Polyradiculonévrites chroniques. EMC Paris Neurologie 1999 ; 17-095-7.
[130] : Deirdre Peake A, William P, Whitehouse B, Sunny Philip C. The management of Guillain Barre syndrome. Current Pediatrics (2004) ; 14: 252-257.
[131]: Cole GF, Matthew DJ. Prognosis in severe Guillain Barre Syndrome. Arch Dis Chlid 1987; 62(3): 288-91.

- 176 -
[132]: Sharshar T, Chevret S, Bourdain F, Raphael JC. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med 2003;31:278–83.
[133]: Wijdicks EF, Borel CO. Respiratory management in acute neurologic illness. Neurology 1998;50:11–20.
[134]: Ropper AH. Intensive care of acute Guillain-Barré syndrome. Can J Neurol Sci 1994;21:S23–7.
[135] : Chevrolet JC. Ventilation non invasive à pression positive dans les défaillances neuromusculaires. In: Actualités en réanimation et urgences. Paris: Elsevier Ed; 2000. p. 128–39.
[136] : Durand MC, Lofaso F, Lefaucheur JP, Chevret S, Gajdos P, Raphael JC, et al. Electrophysiology to predict mechanical ventilation in Guillain-Barré syndrome. Eur J Neurol 2003;10:39–44.
[137] : Zifko U, Chen R, Remtulla H, Hahn AF, Koopman W, Bolton CF. Respiratory electrophysiological studies in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry 1996;60:191–4.
[138]: Sharshar T, Chevret S, Bourdain F, Raphael JC. Early predictors of mechanical ventilation in Guillain-Barré syndrome. Crit Care Med 2003;31:278–83.
[139]: Lawn ND, Wijdicks EF. Post-intubation pulmonary function test in Guillain-Barre syndrome. Muscle Nerve 2000;23:613–6.
[140]: Chevrolet JC, Deleamont P. Repeated vital capacity measurements as predictive parameters for mechanical ventilation need and weaning success in the Guillain-Barre syndrome. Am Rev Respir Dis 1991; 144:814–8.
[141]: Joel Steinberg. Polyneuropathie aigue idiopathique. Une publication de la Fondation Internationale du Syndrome de Guillain-Barré. 1ère ed. Française 1993.15-16
[142]: Kogan BA, Solomon MH, Diokno AC. Urinary retention secondary to Landry-Guillain-Barre syndrome. J Urol 1981;126:643-4
[143]: Chung K, Tokibonu H, Tanabe H, Tomoishi J. Unnary disturbance in Guillain- Barré syndrome. Autonomic Nervous System 1988;25:626-32.
[144] : Moulin DE, Hagen N, Feasby TE, Amireh R, Hahn A. Pain in Guillain-Barré syndrome. Neurology 1997;48:328—31.

- 177 -
[145]: Pandey CK, Bose N, Garg G, Singh N, Baronia A, Agarwal A, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg 2002;95:1719—23.
[146] Tripathi M, Kaushik S. Carbamezapine for pain management in Guillain-Barré syndrome patients in the intensive care unit. Crit Care Med 2000;28:655—8.
[147]: Henderson RD, Lawn ND, Fletcher DD, McClelland RL, Wijdicks EF. The morbidity of Guillain-Barré syndrome admitted to the intensive care unit. Neurology 2003;60:17—21.
[148]: Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet 2005;366:1653—66. [149] : Ropper AH, Kehn SM. Guillain Barre syndrome. Management of respiratory
failure.Neurology1985 ; 35 :1662-5. [150] : Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barre syndrome. Contemporary
Neurology Series, 1 volume. Philadelphia: FA Davis Company, 1991 [151] : Raphael JC, Masson C, Morice Vet al. Le syndrome de Landry-Guillain-
Barré. L’étude des facteurs pronostiques dans 223 cas. Rev Neurol 1986; 142:613-24
[152] : French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Plasma exchange in Guillain- Barré syndrome: one year follow-up. Ann Neurol 1992; 32:94-7
[153]: The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Appropriate number of plasma exchanges in Guillain-Barr6 Syndrome. Ann Neurol 1997; 41:298-306
[154]: Plasma Exchange/Immunoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barr6 syndrome. Lancet 1997; 349:225-30
[155]: Van Der Meché FGA, Schmitz PIM and the Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med 1992;326:1123-9
[156]: FeasbyTE, Gilbert J J, Brown WF et al. An acute axonal form of Guillain-Barré polyneuropathy. Brain 1986; 109: 1115-26.
[157]: Hartung HP, Schafer B, Heininger K, Stoll G, Toyka KV. The role of macrophages and eicosanoids in the pathogenesis of experimental allergic neuritis. Brain 1988;11 I: 1039-59

- 178 -
[158]: Watts PM, Taylor WA, Hughes RAC. High-dose methyl-prednisolone suppresses experimental allergic neuritis in the Lewis rat. Exp Neurol 1989; 103:101-4
[159]: Guillain-Barr6 Syndrome Steroid Trial Group. Double- blind trial of intravenous methylprednisolone in Guillain- Barr6 syndrome. Lancet 1993;341:586-90.
[160] : Abel J J,Rowntree LG, Turner BB.Plasma removal with return of Corpuscles (plasmaphaeresis) .The Journal of Pharmacology and Experimental Therapeutics 1914 Vol.V.No.6.July Transfus Sci 1990; 11:166–77.
[161] : Gurland HJ. Status of therapeutic apheresis in Europe. J Clin Apheresis1991;6:93–4.
[162]: Greenwood RJ, Newsom-Davis J, Hughes RAC et al. Controlled trial of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet 1984;i:877-9
[163]: Osterman PO, Fagius J, Lundemo Get aL Beneficial effects of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet 1984;ii: 1296-9
[164]: The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barr6 syndrome. Neurology 1985;35: 1096-104.
[165]: The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Plasma exchange in Guillain- Barré syndrome: one year follow-up. Ann Neurol 1992; 32:94-7
[166]: The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Appropriate number of plasma exchanges in Guillain-Barré Syndrome. Ann Neurol 1997; 41:298-306.
[167] : Imbach P, Barandum S, d'Apuzzo V et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981 ;i: 1128-31
[168] : Sultan Y, Kazatchkine MD, Maisonnneuve P, Nydegger UE. Anti-idiotypic suppression of autoantibodies to factor VIII (anti-haemophilic factor) by high-dose intravenous gamma-globulin. Lancet 1984;ii:765-8
[169]: Vermeulen M, Van der Mech6 FGA, Speelman JD, Weber A, Busch HFM. Plasma and gamma-globulin infusion in chronic inflammatory polyneuropathy. J Neurol Sci 1985; 70:317-26

- 179 -
[170]: Van der Meché FGA, Vermeulen M, Busch HFM. Chronic inflammatory demyelinating polyneuropathy: conduction failure before and during immunoglobulin or plasma the- rapy. Brain 1989;112:1563-71
[171]: Kleyweg RP, Van der Mechr"FGA, Meulstee J. Treatment of Guillain-Barr6 syndrome with high-dose gammaglobulin. Neurology 1988;38:1639-41
[172]: Thornton CA, Griggs RC. Plasma exchange and intravenous immunoglobulin treatment of neuromuscular disease. Ann Neurol 1994;35:260-8
[173] : Minvielle E, Viens-Bitker C, Rochant H et al. La maîtrise de la diffusion d'une innovation en santé: le cas des immunoglobulines intraveineuses polyvalentes ~ l'Assistance publique Hôpitaux de Paris. Ann Méd Interne 1995; 146:19-24
[174] : Washburn, K.B. Physical Medicine and Rehabilitation, 2nd ed. Medical Examination Publishing Co., Garden City, N.Y., 1981, p. 104.
[175] : Nichols, P.J.R. Rééducation Medicine, 2nd ed. Butterworth & Co., London, 1980, pp. 144, 148-153.
[176] : Kaplan, P.E. & Materson, R.S. The Practice of Rééducation Medicine. Charles C. Thomas, Springfield, Ill. 1982, pp. 464-468.
[177] : Kottke, F.J. et al. sen's Handbook of Physical Medicine and Rééducation, 3rd ed. W.B. Saunders, Philadelphia, 1982, pp. 686, 693-694, 734.
[178]: Conomy, J.B. Physical Ther. 51:517-523 (1971) [179]: Eisendrath, S.J. et al. Psychosomatics, 24:465-475 (Mai 1983) [180]: de la Cour CD, Jakobsen J. Residual neuropathy in long-term population-based
follow-up of Guillain-Barré syndrome. Neurology 2005; 64: 246–53. [181]: Hiraga A, Mori M, Ogawara K, et al. Recovery patterns and long term prognosis
for axonal Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2005; 76: 719–22.
[182]: Bernsen RAJAM, Jacobs HM, De Jager AEJ, van der Meché FGA. Residual health status after Guillain-Barré Syndrome. J Neurol Neurosurg Psychiatry 1997; 62: 637–40.
[183]: Bernsen RAJAM, De Jager AEJ, Schmitz PIM, van der Meché FGA. Residual physical outcome and daily living 3 to 6 years after Guillain-Barré syndrome. Neurology 1999; 53: 409–10.

- 180 -
[184]: Visser LH, Schmitz PIM, Meulstee J, van Doorn PA, van der Meché FGA. Prognostic factors of Guillain-Barré syndrome after intravenous immunoglobulin or plasma exchange. Neurology 1999; 53: 598–604.
[185]: Hughes RAC, Hadden RDM, Rees JH, Swan AV. The italien Guillain study group. The prognosis and main prognosis indicators of Guillain Barré syndrome: a multicentre prospectif study of 297patients 1997 [consulté le 16/01/09]. Disponible à partir du URL: www.mdconsult.com.
[186]: Pélissier J, Maire O, Benaim C, Tintrelin I, et al. Le syndrome de Guillain Barré d’évolution prolongée. Facteurs prédictifs et pronostic fonctionnel. Ann Readapt Med Phys 1994 ; 37 : 25-31.