palladium-catalyzed asymmetric allylic alkylation
TRANSCRIPT

1
B. M. Trost, J. E. Schultz ReviewSyn thesis
SYNTHESIS0 0 3 9 - 7 8 8 1 1 4 3 7 - 2 1 0 XGeorg Thieme Verlag Stuttgart · New York2019, 51, 1–30reviewen
Palladium-Catalyzed Asymmetric Allylic Alkylation Strategies for the Synthesis of Acyclic Tetrasubstituted StereocentersBarry M. Trost* 0000-0001-7369-9121 Johnathan E. Schultz 0000-0002-8261-7711
Department of Chemistry, Stanford University, 335 Campus Drive, Stanford, CA 94305, [email protected]@stanford.edu
Published as part of the 50 Years SYNTHESIS – Golden Anniversary Issue Pd
L L
R'
R
NO2
OH
EtO2C
O
O
i-PrNO O
OH
Ph O
O
CO2Me
OH
OO
OH
OPMB
OH
N O
Br
NC
OHOH
PdL L
O
NHAcMeO2CPh
CHO
F
Ph Ot-Bu
O
i-Bu NO2
Ph
O
F
CHO
OTBSO
N
O
Bu
MeO2C
Ph
NH3+OH
O
Cl–
HO
HO
Stereocontrol on prochiral electrophiles
OH
O
Ar
Stereocontrol on prochiral nucleophiles
Received: 23.10.2018Accepted: 24.10.2018Published online: 05.12.2018DOI: 10.1055/s-0037-1610386; Art ID: ss-2018-z0713-r
License terms:
Abstract Over the past 20 years, the asymmetric synthesis of acyclictetrasubstituted stereocenters by Pd-catalyzed asymmetric allylic al-kylation (Pd-AAA) strategies has seen considerable growth. Despite theinherent difficulty in accessing acyclic tetrasubstituted stereocenters,creative approaches toward this problem have resulted in high stereoin-duction on both electrophilic and nucleophilic reaction partners. Muchof this chemistry has paved the way for unique solutions in Mo-, Ir-, andRh-AAA, with many complimentary methods arising due to the uniqueregiochemical outcomes of AAA outside of Pd catalysis.1 Introduction2 Stereocontrol on Prochiral Electrophiles3 Stereocontrol on Prochiral Nucleophiles4 Temporary Cyclic Pronucleophiles5 Allylic Alkylation with Other Metals6 Conclusions and Outlook
Key words allylic alkylation, acyclic stereocontrol, tertiary alcohols, -tertiary amines, quaternary carbon stereocenters
1 Introduction
Palladium-catalyzed asymmetric allylic alkylation (Pd-AAA) stands as a unique reaction due to the high number ofstereocenters that may be created by this process.1 Argu-ably, few transition-metal-catalyzed reactions offer thesynthetic chemist the ability to form C–C, C–N, C–O, C–F,and C–S bonds by such a variety of mechanisms for asym-metric induction (Scheme 1). The development of Pd-AAAhas been reviewed elsewhere; however, a specific aspect ofPd-AAA has significantly matured over the past 20 years,namely the synthesis of acyclic tetrasubstituted stereocen-ters.2
Pd-AAA has been shown to be successful for inducingasymmetry on both electrophilic and nucleophilic partners.Consequently, this review is divided into these two classesof reactions, with an emphasis on vinyl epoxides as electro-philes and enolates as prochiral nucleophiles. We concludewith a discussion on cyclic pronucleophiles that are readilyconverted into acyclic tetrasubstituted stereocenters subse-quent to the alkylation reaction, as well as some importantadvances made with other metals.
Scheme 1 Examples of stereocenters formed in Pd-AAA reactions
2 Stereocontrol on Prochiral Electrophiles
In the case of Pd-allylic alkylation, the most commonmechanism operative involves an initial coordination of achiral Pd(0) catalyst with an olefin bearing a leaving groupat the allylic position (Scheme 2). Ionization occurs with in-version, where the η2--allyl–Pd(II) complex is situated onthe opposite face to the leaving group. Alkylation of the η3--allyl–Pd(II) complex occurs with inversion of the stereo-chemistry about Pd(II), with simultaneous regeneration ofthe chiral Pd(0) catalyst. Because both the ionization andnucleophilic addition occur with inversion, the nucleophilic
Ph
O
SO2t-Bu
PhNNBn
O
BnO
F
OH
Ph Ph
PPh2
PdLn*
R1 R2
C–FC–C
C–S C–O
C–PC–N
Ph
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

2
B. M. Trost, J. E. Schultz ReviewSyn thesis
substitution occurs with net retention of the stereochemis-try of the leaving group. An outer-sphere attack of a nucleo-phile is the most common pathway for soft nucleophiles(pKa <25). For hard nucleophiles, the nucleophile attacksthe metal directly and undergoes an inner-sphere reductiveelimination with retention of stereochemistry about Pd(II),and overall net inversion of stereochemistry.
Nucleophiles such as ketone enolates alkylate at the -carbon, producing a stereocenter on the nucleophile. In thiscase, asymmetric induction can be difficult to achieve sincethe C–C bond forming event most commonly occurs via anouter-sphere mechanism and the nucleophile remains dis-tal to the chiral information about the metal–ligand envi-ronment. This mode of asymmetric induction will be dis-cussed in Section 3.
Alkylation of allyl electrophiles bearing substituents, al-though typically much easier substrates for obtainingasymmetric induction, operate via more complicatedmechanisms involving –– equilibration of the -allyl in-termediate (Scheme 3d). Additionally, the enantiodeter-
mining step of the reaction may occur during any step ofthe catalytic cycle, save for dissociation of the alkylationproduct. In Pd-AAA, only two mechanisms are likely to be
Scheme 2 Mechanism of regio- and stereospecific Pd-catalyzed AA
X
R
Complexation
IonizationNucleophilicAttack
ProductDissociation
Nu
R'
Pd0Ln
Pd(II)Ln
R R'
R
Pd0Ln
Nu
R'R
Pd0Ln
X
R'
Nu
R
X
R'I
III
IV
V
VI II
Biographical Sketches
Barry M. Trost was born inPhiladelphia, Pennsylvania. Heobtained a BA degree from theUniversity of Pennsylvania in1962 and Ph.D. degree justthree years later at the Massa-chusetts Institute of Technology(1965). He directly moved tothe University of Wisconsinwhere he was promoted to Pro-fessor of Chemistry in 1969 andsubsequently became the VilasResearch professor in 1982. Hejoined the faculty at Stanford asProfessor of Chemistry in 1987and became Tamaki Professor ofHumanities and Sciences in1990. In addition, he has beenVisiting Professor in Germany(Universities of Marburg, Ham-burg, Munich, and Heidelberg),Denmark (University of Copen-
hagen), France (Universities ofParis VI and Paris-Sud), Italy(University of Pisa), and Spain(University of Barcelona). He re-ceived honorary degrees fromthe Université Claude-Bernard(Lyon I), France (1994), andTechnion – Israel Institute ofTechnology, Haifa, Israel (1997).In recognition of his many con-tributions, Professor Trost hasreceived a large number ofawards, a few among which arethe ACS Award in Pure Chemis-try (1977), the Dr. Paul JanssenPrize (1990), the ASSU GraduateTeaching Award 91991), BingTeaching Award (1993), the ACSRoger Adams Award (1995), thePresidential Green ChemistryChallenge Award (1998), theBelgian Organic Synthesis Sym-
posium Elsevier Award (200),The ACS Nobel Laureate Signa-ture Award for Graduate Educa-tion in Chemistry (2002), theACS Cope Award (2004), theNagoya Medal (2008), the RyojiNoyori Prize (2013), the Interna-tional Precious Metals Insti-tute’s Tanaka DistinguishedAchievement Award (2014), theGerman Chemical Society’sAugust-Wilhelm-von-HofmannDenkmünze (2014), the Tetra-hedron Prize (2014), and theACS Linus Pauling Award(2015). Professor Trost has beenelected a fellow of the AmericanAcademy of Sciences (1982)and a member of the U.S. Na-tional Academy of Sciences(1980).
Johnathan E. Schultz grewup in the southwestern NorthDakota and west-central Minne-sota. He attended North DakotaState University in Fargo, NDwhere he graduated SummaCum Laude with a BS degree inChemistry and a minor in Math-
ematics. He conducted under-graduate research in thelaboratories of Gregory R. Cookwith research studies on thesynthesis of isoform selectivehistone deacetylase inhibitors aswell as nucleophilic allylationmethodology. He is currently
pursuing his Ph.D. studies atStanford University as a re-search assistant in the laborato-ries of Barry M. Trost. Hisresearch has focused on Pd-cat-alyzed allylic alkylation strate-gies for the synthesis of acyclictetrasubstituted stereocenters.
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

3
B. M. Trost, J. E. Schultz ReviewSyn thesis
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30
operative in the synthesis of acyclic tetrasubstituted stereo-centers on the electrophile, namely differentiation of enan-tiotopic olefin faces (Scheme 3b) and –– equilibration(Scheme 3d). In most cases, the chiral ligands employed inPd-AAA are phosphines, and a variety of scaffolds have aris-en from the many creative approaches toward acyclic ste-reocontrol in allylic alkylation (Figure 1).
Scheme 3 Modes of asymmetric induction on electrophile
Differentiation of enantiotopic termini
PdLn
R R Nu
– PdLn*
R R
Nu
Differentiation of enantiotopic leaving groups
R
X
X PdLn*
– X PdLn
R X
X
X PdLn*
– X
X
PdLn
Differentiation of enantiotopic olefin faces
Pd
R'
Nu
– PdLn*
R'
Nu
– X
π−σ−π Equilibration
PdL L
R'
R R
R'
PdLL
Ha
Hb
R
R'Pd
L
LHaHb
PdL L
R'
R
R'X
Pd R
R
R*Ln *Ln
a) b)
c) d)
Figure 1 Structures of chiral ligands used in Pd-AAA strategies toward acyclic tetrasubstituted stereocenters
HNNH
PPh2 Ph2P
O O
(R,R)-TrostL1
HNNH
PPh2 Ph2P
O O
(R,R)-TrostL2
Ph Ph
NH HNO O
Ph2PPPh2
(R,R)-TrostL3
NH HNO
PPh2
O
Ph2P
(R,R)-TrostL4
HNNH
N N
O O
(R,R)-TrostL5
PPh2
PPh2
(R)-BINAPL13
PPh2 N
O
PHOXL21
Ph
CH2OH
O
OP
O
OH
i-Pr
i-Pri-Pr
i-Pr
i-Pri-Pr(S)-TRIP L15
O
OMe
2-naphthyl
2-naphthyl
Ar2P
Me3N
NH HNO
PAr2
O
Ar2P CF3F3C
Ar = 4-CF3C6H4
(R,R)-Trost
L16
N
PPh2
(R)-QUINAPL22
PPh2 N
O
(S)-t-Bu-PHOXL19
t-Bu
P N
O
t-Bu
Di-CF3-(S)(t-Bu)-PHOX L20
CF3
F3C
CF3
PAr2 Ar2P
L17 (R,R,R)-Xyl-SKP
SNH
OH
O
MeO
F
F
F
F
O O
L18
Ar = 3,5-Me2C6H3
MeO
MeO
P
P
O
O
2
2
(R)-L7
Fe PPh2
PPh2
NMe
(R)-(S)-L9
O
N
OO
N
2
Me
FePh2P
Ph2P
N
3
O
N
O
O
Me
(S)-(R)-L10
Fe FeR
Ar2P
(S,S)-(R,R)-TRAP
Ar = 4-MeOC6H4, L11Ar = Ph, L12
HR =
Fe
N OP
Et2N
O
OH
(S,RPHOS,R)-L8
Fe
N OP
Et2N
O
OH
i-Pr(S,SPHOS,R)-L14
Ph
Ar = 4-ClC6H4
L6

4
B. M. Trost, J. E. Schultz ReviewSyn thesis
2.1 Reactions of Isoprene Monoepoxide
The potential of achieving Pd-AAA with isoprene mon-oepoxide (1) was an attractive pursuit, due to the prospectof achieving high enantioinduction for alkylation at thebranched position (Scheme 4). The Trost group hasachieved branch-selective alkylation for carbon,3 oxygen,4,5
and nitrogen6–8 nucleophiles, affording valuable chiral acy-clic tetrasubstituted building blocks containing orthogonalhydroxymethyl and vinyl functional group handles.
Scheme 4 Pd-AAA strategy for the synthesis of acyclic tetrasubstitut-ed building blocks using electrophilic isoprene monoepoxide
These reactions constitute a dynamic kinetic asymmet-ric transformation (DYKAT), whereby racemic isoprenemonoepoxide is ionized by a palladium catalyst, with theinitially formed -allyl species bearing an opposite sense ofchirality or opposite syn/anti configuration9 (Scheme 5).Since the unbranched terminus of the -allyl contains twohydrogen substituents, the η1--Pd(II) complex allows forrotation from one stereoisomer isomer to the other withoutsyn/anti isomerization (Scheme 5a). Although –– equil-
ibration may occur at the branched carbon (Scheme 5b),this pathway is unproductive since both the -allyl stereo-chemistry and the syn/anti configuration have isomerized.
An important structural consideration for this reactionalso involves the equilibrium between syn and anti isomers(Scheme 6). Upon ionization of butadiene monoepoxide (2),diastereomeric -allyl complexes are formed. The hy-droxymethylene substituent is sterically more hinderedthan the hydrogen substituent; consequently, complex 2a isthe most favored complex due to the least amount of stericclash with the ligand environment. The regioselectivity ofnucleophilic addition runs counter to that typically ob-served for Pd-AAA, where the branched isomer is formed inhigh selectivity. The ‘wall and flap model’ shows the ap-proximate chiral environment about each terminus of theelectrophile based on chelation of a C2-symmetric ligandbearing diarylphosphines (i.e. Trost ligand, BINAP).
In the case of isoprene monoepoxide (1), the methyl andhydroxymethylene substituents are sterically very similar;however, the presence of an alkoxide from the epoxideopening renders the two electronically dissimilar (Scheme7a). Two factors dictate alkylation at the branched termi-nus, namely: 1. The alkoxide leaving group often acts as abase for the pronucleophile and engages in hydrogen bonddirected alkylation of the nucleophile. 2. Ligand and addi-tive effects can alter the most favorable trajectory of the in-coming nucleophile toward the branched product.10 The
O
(+/–)
Pd(0) cat.
L* cat
OHNu
+ NuHNu
OH+
1 linear achiralproduct
branched chiralproduct
Scheme 5 Mechanism of dynamic kinetic asymmetric transformation (DYKAT) of isoprene monoepoxide
PdL L
OHOH
PdLL
Ha
Hb
OHPd
L
LHaHb
PdL L
OH
PdL L
OHOH
Pd LL OH
Pd LL
PdL L
OH
H
syn syn
synanti
a.)
b.)
H
HH
Scheme 6 Wall and flap model as a rationale for regio- and enantioselectivity in allylic alkylation with butadiene monoepoxide
Pd Pd
Pd R
Ionize:
PdR
π−σ−π
π−σ−π
π−σ−π π−σ−π
H
O
O O
(+/–)
2
2a 2b
2c 2d
R = CH2O–
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

5
B. M. Trost, J. E. Schultz ReviewSyn thesis
outer-sphere nucleophilic attack occurs with inversion ofthe stereochemistry of the -allyl, and each enantiomer ofproduct can be obtained from two of the stereoisomericcomplexes (Scheme 7b).
Scheme 7 Mechanistic pathways for Pd-AAA of isoprene monoepox-ide
Methanol was shown to alkylate at the branched carbonwith high enantioselectivity in the Pd-AAA of isoprenemonoepoxide (Scheme 8).4 The use of catalytic borane wascrucial for reactivity, as the reaction proceeded with littleconversion in its absence. Initially, stoichiometric trimethylborate [B(OMe)3] was used in the reaction, affording theproduct in 80% yield and 2% ee. It was envisioned that thestoichiometric use of borane results in too rapid a reaction,such that equilibration of isoprene monoepoxide cannotoccur prior to nucleophilic attack. Use of catalytic triethyl-borane in the presence of methanol produced diethylme-thoxyborane as catalyst, and these conditions proved opti-mal for enantioselectivity.
Because of the lower acidity of aliphatic alcohols, theiruse in Pd-AAA has remained limited; however, the kineticeffect imparted by the boron additive renders this reactionfavorable in alkylation of vinyl epoxides (Scheme 9a).4 Ad-ditionally, the unimolecular mechanism accounts for the
high selectivity for the branched product. Primary alcohols3 undergo the alkylation chemoselectively over secondaryalcohols. Additionally, the high yields indicate the productalcohol 6 is not competitive with primary alcohol substrate3, presumably due to either a slow alkylation reaction or asmall population as the alkoxydiethylborane species 4(Scheme 9b).
The reaction scope shows several primary alcohols 3were competent nucleophilic partners in this reaction(Scheme 10).4 For substrates with low enantioselectivityusing triethylborane as catalyst, it was reasoned that ether-ification was occurring faster than the required -allylequilibration. Use of the more bulky tri-sec-butylboraneproved useful for solving this problem (6b, 6d, 6g). With ahampered rate of etherification, the products were formedwith higher enantioselectivity. Spontaneous cyclization to ahemiketal (6d) or lactone (6b) occurred when the nucleop-hilic alcohol contained a pendant ketone or methyl ester re-spectively. The etherification product of 4-methoxybenzylalcohol addition 6c is a particularly useful synthetic precur-sor due to the ubiquity of PMB-protected alcohols in syn-thetic sequences.
A net addition of nucleophilic hydroxide was discoveredfor this reaction using a mixed system of triethylborane andsodium hydrogen carbonate (Scheme 11).5 Upon ionization,a borinic ester intermediate is formed that directs the al-kylation of hydrogencarbonate to the branched positionwith high enantioselectivity. The unstable carbonate prod-uct 7 undergoes decarboxylation to directly afford the ter-tiary alcohol product 8.
NuOH
Nu
OH
PdL L
OH
PdL L
PdL L
PdL L
OH
OHOH
OHNu
OHNu
A B
C D
A
B
C
D
NuOH
Nu
OH
OHNu
OHNu
+
+
+
+
a.) Diastereomeric Pd(II) allyl complexes b.) Alkylation products by Pd(II) diastereomer
Scheme 8 Optimization of allylic etherification with isoprene monoep-oxide
O(+/–)
Pd2dba3·CHCl3 (1%)(S,S)-L1 (3%)
OMe
OH
Conditions
B(OMe)3 (1 equiv)B(OMe)3 (1%), MeOH (1 equiv)
Et2B(OMe) (1 equiv)Et2B(OMe) (1%), MeOH (1 equiv)
BEt3 (1%), MeOH (1 equiv)
% ee
2495890941 6a
conditionsCH2Cl2, r.t
Scheme 9 Borane co-catalyst in Pd-catalyzed regio- and enantioselective allylic etherification of isoprene monoepoxide
PdL L
PdL L
O OBB
RO RO
Et Et Et Et
OBEt2
OR
OBEt2
OR
a.) Alkoxydiethylborane-directed allylic etherification of isoprene monoepoxide
OBEt2
OR
ROH R
OBEt2
BEt3
– C2H6
Pd(0)L* cat
1
3OH
OR
RO
BEt2+
b.) Mechanism of alkoxydiethylborane catalytic turnover
3 4 5 6 4
5 ent-5
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

6
B. M. Trost, J. E. Schultz ReviewSyn thesis
Scheme 11 Pd-AAA of isoprene monoepoxide with sodium hydrogen carbonate in the presence of borane co-catalyst
In the absence of borane additive; however, the carbon-ate product 9 was isolated (Scheme 12).5 The difference inreaction outcomes was attributed to the attack of carbondioxide by the alkoxide leaving group. This system provedmore challenging for optimization due to the lability of 9 toracemization. In the presence of tetrabutylammoniumchloride, the product was isolated with high enantioselec-tivity at low conversion, but was nearly racemic upon com-pletion of the reaction. However, sodium hydrogen carbon-ate in phosphate buffer resulted in high enantioselectivityat high conversion. Under the optimized conditions, lowcatalyst loading in the absence of buffer proved effective forachieving high enantioselectivity.
Scheme 12 Pd-AAA of isoprene monoepoxide with sodium hydrogen carbonate in the absence of borane co-catalyst.
Carbon nucleophiles were successfully applied in thischemistry using nitromethane (10) or -keto esters(Scheme 13).3 These reactions were pioneering for the met-al-catalyzed synthesis of acyclic all-carbon quaternary ste-reocenters, which has been an active area of pursuit sincethe development of this chemistry.11 The product from ni-
tromethane addition 11 is particularly valuable, as the re-sulting nitromethylene substituent serves as an acyl anionequivalent, orthogonal to the two other functional grouphandles (Scheme 13a). In the case of -keto ester 12, the hy-droxymethyl group spontaneously cyclizes to form a he-miketal 13 upon allylic alkylation (Scheme 13b). A retro-aldol reaction of 13 proceeds cleanly in the presence oftetrabutylammonium fluoride (TBAF) to furnish acyclic 14.
Scheme 13 Pd-AAA of isoprene monoepoxide with carbon nucleo-philes
Isoprene monoepoxide has been successfully alkylatedby phthalimide (15) to afford the branched, -tertiary im-ide 16 (Scheme 14).7 The regioselectivity is invoked to bethe result of both hydrogen bonding and ligand-directed ef-fects. Achiral dppe afforded exclusively the linear product.The reaction with chiral ligand L2 proved to be solvent,base, and temperature sensitive. High enantioselectivitycould be achieved without exogenous base; however, thisresulted in exceedingly long reaction times. Prolonged reac-tion time after completion of the reaction resulted in ahigher proportion of branched product, likely due to re-ion-ization of 16.
Scheme 14 Pd-AAA of isoprene monoepoxide with phthalimide
Enantiopure amino esters 17 were reacted with iso-prene monoepoxide in a catalyst-controlled regio- and di-astereoselective allylic alkylation (Scheme 15).8 A subse-quent cyclization by KCN afforded 2-oxomorpholines 18with high diastereoselectivity. Although the products of thesequence are cyclic, the product of the first step of the reac-tion sequence is an acyclic -tertiary amine, demonstratingthat amines react in analogy to phthalimide with isoprenemonoepoxide. It was shown that catalyst control could dic-tate diastereodivergent pathways when using the naturalamino acids. Multiple ligands were employed, due to the
Scheme 10 Select examples of tertiary ether synthesis with isoprene monoepoxide
O
(+/–)+
Pd2dba3·CHCl3 (1%)(S,S)-L1 (3%)
1.0 equiv 1.0 equivO
OHROH
RHNNH
PPh2Ph2P
O O
(S,S)-L1
MeOOH
OOH
OPMBOH
OOH
NC
OOH
OH
O
O
OO
O
OHMe
88% yield, 94% ee(BEt3)
83% yield95% ee(BEt3)
91% yield, 94% ee(BEt3)
77% yield,98% ee(B(s-Bu)3), DMAP 5%
81% yield90% ee
(B(s-Bu)3)40 °C
63% yield85% ee1.1 d.r.(BEt3)40 °C
75% yield, 94% ee(B(s-Bu)3)
1 3 6
6a
6e
6c 6d6b
6f 6g
BR3 (1%)CH2Cl2, r.t. or 40 °C
O(+/–)
+
Pd2dba3·CHCl3 (1%)(S,S)-L1 (3%)
BEt3 (1%)CH2Cl2/H2O, 40 °C1.0 equiv 1.0 equiv
NaHCO3
OCO2–
OHdecarboxylation
– CO2OH
OH
91% yield97% ee1 7 8
HNNH
PPh2 Ph2P
O O
(S,S)-L1
O
(+/–)+
Pd2dba3·CHCl3 (0.5%)(S,S)-L1 (1.5%)
CH2Cl2/H2O, r.t.1.0 equiv 1.2 equiv
NaHCO3 O
O
O
88% yield93% ee1 9
O
(+/–)Pd2dba3·CHCl3 (1%)
(S,S)-L2 (3%)CH2Cl2
+ MeNO2NO2
OHHNNH
PPh2 Ph2P
O O
(S,S)-L251% yield97% ee
i-Pr
O O
OEt
Pd2dba3·CHCl3 (1%)(S,S)-L2 (3%)
1 (1.3 equiv)CH2Cl2
1.0 equiv
74% yield, 97% ee
O
EtO2CHO
i-Pr
TBAF
THF, r.t. EtO2C
O
O
i-Pr
97% yield
1 10 11
12 13 14
a.)
b.)
[Pd(allyl)Cl]2 (2.5%)(R,R)-L2 (7.5%)
1 (1 equiv)Cs2CO3 (5%)THF, 55 °C
1.1 equiv
HNNH
PPh2 Ph2P
O O
(R,R)-L2
NO O
OHHNO O
72% yield, 87% ee
15 16
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

7
B. M. Trost, J. E. Schultz ReviewSyn thesis
presence of match-mismatch effects unique to each aminoester. Since the enantiomeric amino acids are commerciallyavailable, all four stereoisomeric products are accessible bythis route. This is a valuable feature, especially in diversity-oriented approaches to compound libraries.12
Scheme 15 Pd-AAA of isoprene monoepoxide with enantiopure ami-no esters
2.2 Applications in Complex Molecule Synthesis
Isoprene monoepoxide was successfully used in the keysteps in the synthesis of hyperolactone C (26) and (+)-biy-ouyanagin A (29) (Scheme 16).13 The nucleophilic precursor24 was accessed by dianion functionalization of methyl ace-toacetate (21) with benzaldehyde. The necessary diazo pre-
cursor 23 was synthesized in two steps from 22, and thePd-AAA pronucleophile 24 was accessed by rhodium car-bene insertion into an enol O–H bond. The two vicinal tet-rasubstituted stereocenters of 25 were formed by a regio-,enantio-, and diastereoselective Pd-AAA between 24 andisoprene monoepoxide (1). The short reaction time provedcrucial to the success of this reaction, as a host of side reac-tions occurred upon completion of the reaction. The 59%isolated yield of 25 is reflective of a modest 2.1:1branched/linear ratio; however, the product was isolated asa single enantiomer in 26:1 d.r.
Lactonization of 25 catalyzed by 4-toluenesulfonic acidproduced hyperolactone C (26) in a single step. For the syn-thesis of (+)-biyouyanagin A (29), ent-hyperolactone C wasreadily accessed by simply changing the enantiomer ofTrost L2. The efficiency of the sequence is outstanding, withthe synthesis of hyperolactone C requiring a longest linearsequence of six steps, and (+)-biyouyanagin A synthesizedby an additional photocycloaddition with zingiberene (27)in the presence of 2′-acetonaphthone (28).
In the total synthesis of (–)-terpestacin14 (Scheme 17),the key Pd-AAA reaction between a diosphenol 30 and ra-cemic isoprene monoepoxide (1) was observed to proceedrapidly and in high yield, but with low enantioselectivity(~50% ee). It was reasoned that the low enantioselectivitypotentially resulted from a short lifetime of the -allyl in-termediate due to rapid nucleophilic attack. Addition of ha-lide additives has been shown to increase enantioselectivityby increasing the rate of –– equilibration.15 Increasingthe concentration of tetrabutylammonium chloride dis-played this effect; however, slow addition of 30 provedmost effectual for increasing the lifetime of the -allyl in-termediate. Under optimized conditions, silylation of theproduct alcohol was performed in a single operation, af-fording product 31 in 93–95% yield and 88–96% ee. The chi-ral information of the newly formed stereocenter could betranslated via a [3,3]-sigmatropic rearrangement, and di-
O
(+/–)+
1) [Pd(allyl)Cl]2 (2.5%)Ligand (3.75%)
NEt3 (1 equiv), CH2Cl2, r.t.
2) KCN, THF or MeCN80 °C1.0 equiv 1.0 equiv
CO2Me
NH4Cl
RH
HN
ORH
O
HN
O
HO
HN
O
HO
HN HN
HN
O
HO
HN
O
HO
HO HO
HNNH
PPh2 Ph2P
O O
(R,R)-L2
HNNH
PPh2 Ph2P
O O
(R,R)-L1
Ph Ph
NH HNO O
Ph2PPPh2
(R,R)-L3
HN
ORH
O
+
1 17 18a 18b
Ligand
(R,R)-L1(S,S)-L1(R,R)-L2(S,S)-L2(R,R)-L3(S,S)-L3
Yield
395175755045
19a/19b
1:2.8 6.7:1
1:6.112.4:1
1:11.5 14:1 19a 19b
Ligand
(R,R)-L1(R,R)-L2(S,S)-L2(R,R)-L3(S,S)-L3
Yield
6985602859
20a/20b
1:21:54:1
1:2.83:1 20a 20b
Scheme 16 Pd-AAA of isoprene monoepoxide in the total synthesis of hyperolactone C and (+)-biyouyanagin A
Ph O
O
CO2Me
Pd2dba3·CHCl3 (1%)(R,R)-L2
(+/–)-1CH2Cl2, r.t. 10 mins
Ph O
O
CO2Me
OH
2.1:1 b/l, 26:1 d.r.99% ee, 59% yield
PTSA (20%)
CH2Cl2, r.t. Ph O
O
O
O85% yield
hyperolactone C
O
CO2Me
NaH, THF, 0 °Cthen BuLi
then PhCHO
O
CO2Me
OH
Ph
1) TsN3, NEt3, MeCN88% yield
2) DMP, CH2Cl291% yield
O
CO2Me
O
Ph
N2
[Rh2(OAc)4]
CH2Cl2, r.t.
92% yield
54% yield
Ph O
O
O
O
ent-hyperolactone C(1.0 equiv)
H
H
+O
O
O
OPh
H
H
H H
H
O
zingiberene(6.0 equiv)
2'-acetonaphthone(1.0 equiv)
43% yield(+)-biyouyanagin A
hv, CH2Cl2
5 °C, 8 h+
21 22 23
2425
26
27 ent-26 28 29
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

8
B. M. Trost, J. E. Schultz ReviewSyn thesis
rect oxidation of the Claisen rearrangement product afford-ed the desired diketone 33 in 78% yield over the sequence,with 4:1 selectivity in the formation of the resultant olefin.A Sakurai allylation of 33 installed an allyl functional grouphandle to form 34. This precursor was used in the construc-tion of macrocycle 35. An additional catalyst-controlled Pd-AAA/sigmatropic rearrangement in the late stages of thesynthesis afforded 38, which was efficiently elaborated tothe target.
Scheme 17 Pd-AAA of isoprene monoepoxide in the total synthesis of (–)-terpestacin
The synthesis of tipranavir was accomplished by settingthe stereochemistry of a tetrasubstituted and tertiary ste-reocenters by Pd-AAA (Scheme 18) and Mo-AAA, respec-tively (Scheme 19).16 A propyl-substituted vinyl epoxide 41was accessed in two steps from 1-chloropentan-2-one (40).After addition of vinylmagnesium bromide, the tertiary al-cohol product was efficiently cyclized to form 41 in 86%yield over two steps. A borane co-catalyzed Pd-AAA of thevinyl epoxide with 4-methoxybenzyl alcohol proved highlyenantioselective in the construction of acyclic tertiary ether42. Synthesis of the phenethyl side chain was readily ac-
complished by Heck arylation and hydrogenation of the vi-nyl functional group handle. The hydroxymethylene func-tional group handle was homologated via an oxidation/Wit-tig olefination/hydroboration/oxidation sequence.
Scheme 18 Synthesis of acyclic tertiary ether fragment in the total synthesis of tipranavir
The tertiary stereocenter was synthesized by a branch-selective cinnamylation of dimethyl malonate sodium salt48 and 3-nitrocinnamyl electrophile 47 under chiral molyb-denum catalysis (Scheme 19). Decarboxylation of a methylester afforded nucleophilic precursor 48, which was cou-pled with the previously prepared aldehyde 46 via an esterenolate aldol reaction. Oxidation of the resulting alcohol,followed by PMB deprotection provided substrate 50 for asodium hydroxide mediated lactonization. Hydrogenationconditions afforded both nitro and olefin reduction, and theproduct 52 was completed by sulfonamide formation. Asimilar strategy was employed in the total synthesis of (–)-malyngolide.17 Additionally, synthetic studies on amphidin-olide B1,18 as well as (+)-pleuromutilin19 have employedisoprene monoepoxide in Pd-AAA for the synthesis of earlychiral building blocks.
2.3 Other Acyclic Electrophiles
In the work of the Trost group on the prenylation of ox-indoles, conditions were developed to selectively affordboth the branched and linear prenylation products (Scheme20).20 This strategy culminated in a unified approach to-ward flustramine natural products. For the reverse pre-nylated product 56, vicinal quaternary carbons are formedwhile setting the cyclic stereocenter in high enantioselec-tivity. Formation of the linear product 55 using the Trost L1was typical of most Pd-AAA; however, the unique structur-al features of Trost L2 provide the branched product. Addi-tionally, tetrabutylammonium difluorotriphenylsilicate(TBAT) as a halide additive improved the reaction outcome,due most likely to a required rate increase in –– equili-bration.
OOH
O
(+/–)
1) Pd2dba3·CHCl3 (2%)(R,R)-L1 (6%)
Bu4NCl (0.5 equiv)CH2Cl2, r.t., 6 h
2) TIPSOTf2,6-lutidine
+O
O
OTIPS
CHCl3
100 °C0.25 h
HO
O
OTIPS
Pd(OAc)2 (1 equiv)Cs2CO3 (1.5 equiv)
MeCN, r.t.
O
O
OTIPS
allylTMSMgBr2·Et2O
CH2Cl2 –78 °C to r.t.
78% yield over two stepsE/Z 4:1
1.0 equiv 2.0 equiv
93-95% yield, 88-96% ee
HO
O
OTIPS
86% yield, 5.7:1 d.r.
O
HO
OHOH
(–)-terpestacin
O
HO
OH
Pd2dba3·CHCl3 (2.5%)(S,S)-L1 (7.5%)
CH2Cl2, r.t.
OBoc
O
O
OH
1) DME, 150 °C2) PMBCl, Cs2CO3
TBAI (cat.) DMF
3) Ac2O, Py
O
PMBO
OAc
89% yield, d.r. > 15:1
steps
5 steps
69% over 3 steps
(+/–)
32 33
34
35
36
37
38
39
30 1 31
O
Cl
O
OPMB
OH1) vinylMgBrTHF, 0 °C
2) NaOHEt2O, r.t.
Pd2dba3·CHCl3 (1%)(S,S)-L1 (3%)
PMBOH (1.1 equiv)BEt3 (1%), CH2Cl2, r.t.
Pd(OAc)2 (10%)P(o-Tol)3 (40%)
NEt3, PhItoluene, reflux
OPMB
OH
Pd/C, H2
MeOH, Py25 °C
Ph
OPMB
OH
Ph
1) Dess–MartinCH2Cl2, 25 °C
2) Ph3PCH2THF reflux
OPMBPh
catechol borane(Ph3P)RhCl (1%)
THF, then
NaOH, H2O2, r.t.
OPMB
CHO
Ph
86% yield 69% yield, 98% ee
92% yield99% yield
94% yield 88% yield
40 41 42
43 44
45 46
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30
9
B. M. Trost, J. E. Schultz ReviewSyn thesis
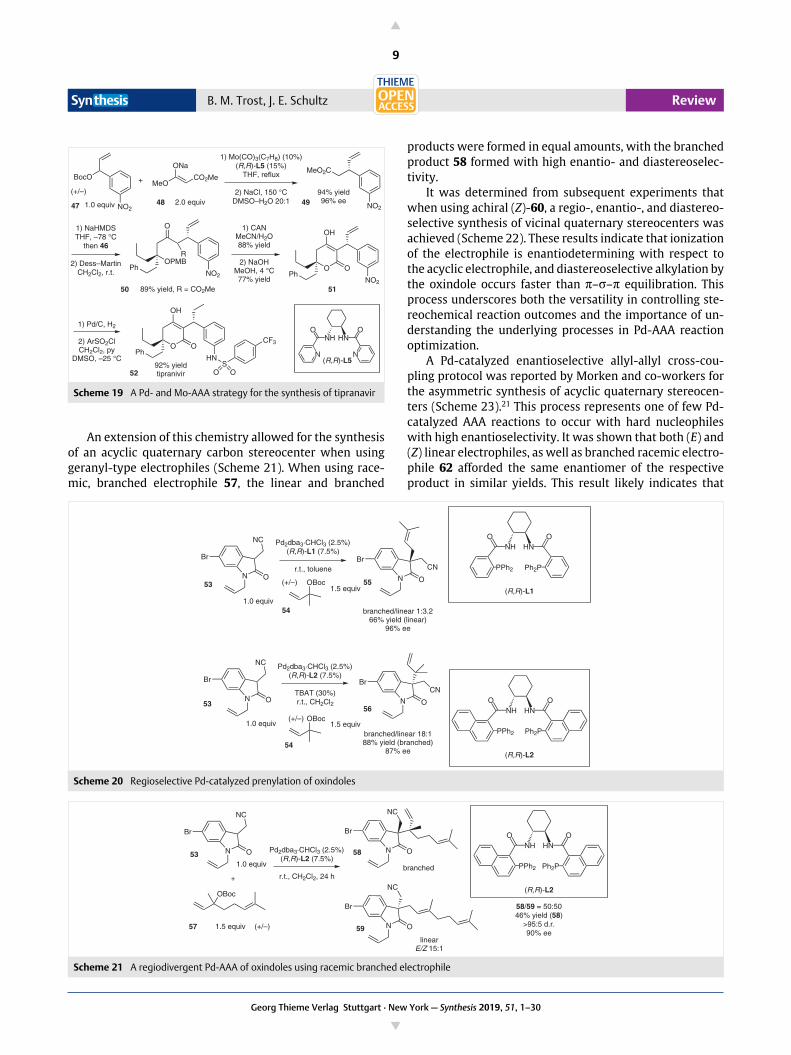
An extension of this chemistry allowed for the synthesisof an acyclic quaternary carbon stereocenter when usinggeranyl-type electrophiles (Scheme 21). When using race-mic, branched electrophile 57, the linear and branched
products were formed in equal amounts, with the branchedproduct 58 formed with high enantio- and diastereoselec-tivity.
It was determined from subsequent experiments thatwhen using achiral (Z)-60, a regio-, enantio-, and diastereo-selective synthesis of vicinal quaternary stereocenters wasachieved (Scheme 22). These results indicate that ionizationof the electrophile is enantiodetermining with respect tothe acyclic electrophile, and diastereoselective alkylation bythe oxindole occurs faster than –– equilibration. Thisprocess underscores both the versatility in controlling ste-reochemical reaction outcomes and the importance of un-derstanding the underlying processes in Pd-AAA reactionoptimization.
A Pd-catalyzed enantioselective allyl-allyl cross-cou-pling protocol was reported by Morken and co-workers forthe asymmetric synthesis of acyclic quaternary stereocen-ters (Scheme 23).21 This process represents one of few Pd-catalyzed AAA reactions to occur with hard nucleophileswith high enantioselectivity. It was shown that both (E) and(Z) linear electrophiles, as well as branched racemic electro-phile 62 afforded the same enantiomer of the respectiveproduct in similar yields. This result likely indicates that
Scheme 19 A Pd- and Mo-AAA strategy for the synthesis of tipranavir
BocOMeO
ONa
CO2Me
2.0 equiv1.0 equiv
1) Mo(CO)3(C7H8) (10%)(R,R)-L5 (15%)
THF, reflux
2) NaCl, 150 °CDMSO–H2O 20:1
+
94% yield96% ee
MeO2C
NO2
(+/–)
NO2
1) NaHMDSTHF, –78 °C
then 46
2) Dess–MartinCH2Cl2, r.t.
O
R
NO2
OPMBPh
89% yield, R = CO2Me
1) CANMeCN/H2O88% yield
2) NaOHMeOH, 4 °C77% yield
OH
NO2Ph
O O
1) Pd/C, H2
2) ArSO2ClCH2Cl2, py
DMSO, –25 °C
OH
HNPh
O O
SO O
92% yieldtipranivir
HNNH
N N
O O
(R,R)-L5
47 48 49
50 51
CF3
52
Scheme 20 Regioselective Pd-catalyzed prenylation of oxindoles
N O
NC
Br
Pd2dba3·CHCl3 (2.5%)(R,R)-L2 (7.5%)
TBAT (30%)r.t., CH2Cl2 N O
Br
1.5 equiv1.0 equiv
HNNH
PPh2 Ph2P
O O
(R,R)-L2
branched/linear 18:188% yield (branched)
87% ee
OBoc(+/–)
CN
N O
NC
Br
Pd2dba3·CHCl3 (2.5%)(R,R)-L1 (7.5%)
r.t., tolueneN O
Br
1.5 equiv
1.0 equiv
HNNH
PPh2 Ph2P
O O
(R,R)-L1
branched/linear 1:3.266% yield (linear)
96% ee
OBoc(+/–)
CN
53
54
55
53
54
56
Scheme 21 A regiodivergent Pd-AAA of oxindoles using racemic branched electrophile
N O
NC
+
Br
Pd2dba3·CHCl3 (2.5%)(R,R)-L2 (7.5%)
r.t., CH2Cl2, 24 h
N O
Br
NC
1.5 equiv
1.0 equiv
N O
Br
NC
branched
linearE/Z 15:1
HNNH
PPh2 Ph2P
O O
(R,R)-L2
58/59 = 50:5046% yield (58)
>95:5 d.r.90% ee
OBoc
(+/–)
53
57
58
59
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

10
B. M. Trost, J. E. Schultz ReviewSyn thesis
rapid equilibration of the -allyl intermediate occurs beforereductive elimination, and it is ligand control of the -allylstereochemistry that dictates the enantioselectivity. In thereaction optimization it was observed that -hydride elimi-nation was a significant side product. Addition of fluoridein a THF/water mixture was shown to minimize this sidereaction by acceleration of allyl transmetalation.
Scheme 23 Catalytic enantioselective branch selective allyl-allyl cross coupling
The substrate scope revealed high enantioselectivitiesfor the synthesis of acyclic benzylic quaternary stereocen-ters. High enantioselectivity was observed when placingbranched aliphatic substituents in addition to the methylgroup (64f, 64g, 64i). A modest, but appreciable enantioin-duction was observed for a geranyl-type substrate (64j).
The C–C bond-forming event is invoked to occur via a 7-membered 3,3′ inner-sphere reductive elimination ofbis(η1-allyl)Pd(II) complex 67 (Scheme 24). In this scenario,palladium is bound to the unsubstituted terminus of thetrisubstituted allyl partner. The alternative mechanismwould involve a more classical 3-membered reductive elim-ination; however, calculations place this pathway at ~15kcal/mol higher in energy.22 The source of enantioinductionin this reaction is not immediately clear. The stereocenter iscreated in the reductive elimination step to form 68; how-ever, the pre-equilibrium of the (E)- and (Z)-isomers of 66may be enantiodetermining if the Re or Si pathway is sig-nificantly favored for both isomers.
The branch-selective Pd-AAA reaction has beenachieved using ligands developed by the Hou group.23 Race-mic tertiary allylic acetates were alkylated with modest en-antioselectivity and branch/linear selectivity using dimeth-yl malonate as the pronucleophile (Scheme 25). Extensiveoptimization of ligand, solvent, additive, and base was re-quired in the process. This ligand class offers modulation ofthe BINOL, phosphorus, and oxazoline chirality for achiev-ing branch-selective Pd-AAA. The presence of high (E)-se-lectivity in the linear product 71 indicates that high syn/an-
Scheme 22 Pd-catalyzed regio-, diastereo-, and enantioselective synthesis of vicinal quaternary stereocenters
N O
NC
TrocO
+
Br
CpPd(allyl) (5%)(R,R)-L2 (7.5%)
60 °C, CH2Cl2, 36 h
N O
Br
NC
1.5 equiv
1.0 equiv
N O
Br
NC
branched
linear
HNNH
PPh2 Ph2P
O O
(R,R)-L2
58/61 = 13:192% yield>95:5 d.r.91% ee
53
60
58
61
MeO
MeO
P
P
O
O
2
2
R1
OBoc
R2
Bpin+
1.0 equiv 1.2 equiv
Pd2dba3 (2%)(R)-L7 (4%)
CsF (3 equiv)THF–H2O, 60 °C
R1R2(+/–)
(R)-MeO-furyl-biphep L7
Br Me NCl
90% yield92% ee
90% yield92% ee
76% yield92% ee
81% yield90% ee
97% yield84% ee
Et
97% yield92% ee
78% yield92% ee
45% yielda
86% ee
n-Bu
58% yield80% ee
MOMO
96% yieldb
52% ee
a) Tertiary allylic chloride electrophile was used. b) Linear geranyl electrophile was used.
62 63 64
64a 64b 64c 64d 64e
64f 64g 64h 64i 64j
Scheme 24 Mechanism of enantioselective allyl-allyl cross coupling
PdLn*
RL
RS
PdLn*
RS
RL
RL
RS PdLn*
RS
RL PdLn*
PdRS
RL
Ln*
PdRL
RS
Ln*
RLRS
RSRL
Re addition
Si addition
65 66 67 68
anti-65 (Z)-66 (Z)-67 ent-68
63
63
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

11
B. M. Trost, J. E. Schultz ReviewSyn thesis
ti control is achieved in the -allyl formation. Enantioinduc-tion is achieved by facial discrimination of the stericallydifferentiated methyl and aryl substituents.
Scheme 25 Pd-catalyzed branch-selective cinnamylation of dimethyl malonate for the synthesis of acyclic quaternary stereocenters
3 Stereocontrol on Prochiral Nucleophiles
In Pd-AAA, achieving high enantiocontrol on nucleo-philes remains inherently more difficult than achieving en-antiocontrol on the electrophilic partner, due to the outer-sphere mechanism for nucleophilic addition. In the alkyla-tion event, the chiral information about the metal–ligandsphere must be successfully relayed distal to the metal inorder to observe catalyst differentiation of enantiotopic fac-es of the nucleophile. This problem is compounded by thepresence of enantioconvergent and enantiodivergent allyla-tion pathways (Scheme 26). In the case of enantioconver-gent allylation (Scheme 26a), the chiral catalyst recognizesthe substituents at the site of reaction. Although the restric-tion of controlling the enolate geometry has been removed,R1 and R2 are limited to sterically differentiable substituents(i.e. alkyl vs. aryl).
When an enantiodivergent pathway is operative(Scheme 26b), the catalyst recognizes the substituents vici-nal to the site of reaction. This pathway is desirable if R1 and
R2 are of limited steric differentiation; however, the enolategeometry must be strictly controlled in order to observehigh enantioselectivity. The enantiodivergent pathway hasan important advantage over the enantioconvergent path-way, since the identity of X can be exploited as an achiralauxiliary and a tunable parameter for reaction optimiza-tion.
As will be seen, both the substituents and the enolategeometry are commonly recognized by the catalyst in theallylation event. In this case, the presence of match-mis-match effects will be apparent when both enolate isomerscan be synthesized in geometrically pure form. The stereo-fidelity of the enolate is assumed to remain intact duringenantiodivergent allylations; however, oxygen-to-carbonmigration may provide a mechanism for erosion of the eno-late geometry if palladium enolates are formed as interme-diates (Scheme 27a). Isomerization of palladium enolateshas been exploited for the Pd-AAA DYKAT of butenolideelectrophiles with phenolic nucleophiles (Scheme 27b).24 Inthis scenario, the palladium enolate is formed on thebutenolide electrophile (Scheme 27c), and the mechanismfor isomerization of the -allyl species involves carbon-to-oxygen migration.
[Pd(allyl)Cl]2 (2%)(S,SPHOS,R)-L8 (4%)
KOAc (6%)
dimethyl malonate (3 equiv)BSA (3 equiv), Et2O, 25 °C
Fe
N OP
Et2N
OOH
(S,RPHOS,R)-L8
Ar
OAc
1.0 equiv
∗∗
Ar
CO2Me
CO2Me Ar
CO2Me
CO2Me+
(+/–)
Aromatic
Ph4-CNC6H41-Naphthyl4-MeOC6H44-MeC6H44-ClC6H4
Yield
668646319090
70/71
96:489:1176:2443:5795:595:5
%ee
737878797869
Entry
70a70b70c70d70e70f
69 70 71
Scheme 26 Structural considerations for catalyst recognition of prochiral enolates
R2
R1
O
X
Enantiodivergent allylation
X
O
R1 R2
R1
R2
O
X X
O
R2 R1
R2
R1
O
X
Enantioconvergent allylation
X
O
R1 R2
R1
R2
O
X
Catalyst differentiation of X and O–Catalyst differentiation of R1 and R2
b)a)
Scheme 27 Potential mechanism for the erosion of enolate stereochemistry
R2
R1
O
X
Pd(II)
X
O
R1
Pd(II)R2
X
O
R2
R1
Pd(II) R1
R2
O
X
Pd(II)
O to C
migration
C to O
migration
rotation
a)
b)
O OBocO+
MeO
OH
Pd2dba3 (1%)(R,R)-L1 (3%)
TBAC (30%)CH2Cl2, r.t.
1.0 equiv 1.0 equiv
O OO
MeO
74% yield, 84% ee
(+/–)
O O
LnPd
O O
PdLn
O OPdLn
O O
PdLn
O O
LnPd
c)
72 73 73 74
75 76 77
78 79 80 ent-79 ent-78
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

12
B. M. Trost, J. E. Schultz ReviewSyn thesis
3.1 Intermolecular Alkylation of Prochiral Nucleo-philes
Many of the first syntheses of acyclic tetrasubstitutedstereocenters by reactions of prochiral nucleophiles werediscovered by the Ito group. In 1992, allylation of an acyclic1,3-diketone 81 (Scheme 28a) was reported in modest en-antioselectivity by employing a point and planar chiral fer-rocenyl ligand scaffold bearing an aza-crown ether.25 In1996, allylation of -nitro ester 83 was achieved by a simi-lar strategy (Scheme 28b).26 In order to enhance enantiose-lectivity upon nucleophilic attack of a -allyl palladium(II)complex, it was envisioned that linking a crown ether tochiral phosphine ligands would allow for recognition of thenucleophile counterion. In this case, the chiral informationof the catalyst could be relayed distal the site of the reac-tion, thus solving the problem of poor selectivity when us-ing prochiral nucleophiles in Pd-AAA. In both cases, signifi-cant optimization of the crown ether and counterion wasrequired to obtain the enantioselectivity.
Scheme 28 Asymmetric allylation of a 1,3-diketone and an -nitro es-ter by counterion recognition of a chiral phosphine ligand
A dual catalytic approach to Pd-AAA was achieved byexploiting chiral rhodium enolate chemistry (Scheme 29).27
It had been shown that rhodium salts form a trans bis-
phosphino complex that is bound to the nitrogen of cyano-derived enolates.28 This chemistry had been previously ap-plied to Michael additions and aldol reactions.29 Using fer-rocenyl ligands developed in the Ito group, they were ableto achieve high enantioselectivity in the allylation of cyanoester 85, cyanoamide 88, and -cyanophosphonate 90 un-der -allyl–Pd catalysis. Potentially, the ligand forms com-plexes with both rhodium and palladium, and the chiralityof each species is synergistic in the alkylation event.
Scheme 29 Dual catalytic Pd-AAA of chiral cyano-rhodium enolates
An early report on asymmetric induction utilizing acy-clic nucleophiles was provided by the Ito group30 in theirPd(0)/BINAP-catalyzed Pd-AAA of -acetamido--keto es-ters 92 (Scheme 30). High levels of enantioselectivity werereported when using potassium tert-butoxide as base. Ad-ditionally, high geometric control of the resulting olefinicproducts 94 was reported, even for commonly difficult ali-phatic allylic acetates (94g, 94h, 94i). The highest enantio-selectivities were observed for cinnamylation reactions(94c, 94d, 94e, 94f), although simple allylations proceededin good enantioselectivity.
Ph
O OPd2(dba)3·CHCl3 (0.5%)
(S)-(R)-L9 (1.1%)
1.0 equiv
(+/-)Fe PPh2
PPh2
NMe
(R)-(S)-L9
O
N
OO
N
2
Me
Ph
O
∗∗
93% yield72% ee
Ac
O2N
O
Ot-Bu
Pd2(dba)3·CHCl3 (0.5%)(S)-(R)-L10 (1%)
1.0 equiv
O
Ot-Bu
92% yield80% ee
O2N FePh2P
Ph2P
N
3
O
N
O
O
Me
(S)-(R)-L10
(+/-)
a)
b)
81 82
83 84
allyl acetate (1.5 equiv)KF (2 equiv) mesitylene
–37 °C
allyl acetate (1.5 equiv)RbF (2 equiv)
RbClO4 (1 equiv)CH2Cl2, –40 °C, 70 h
Fe FeR
Ar2P
(S,S)-(R,R)-TRAP
Ar = p-MeO-Ph
L11
HR =CO2i-PrNCRh(acac)(CO)2 (1%)(Cp)Pd(allyl) (1%)
(S,S)-(R,R)-L11 (2%)THF, –40 °C, 6 h
1.0 equiv
CN
O
Oi-Pr
93% yield99% eeO
O
O
CF3
CF3
2.0 equiv
+
Fe FeR
Ph2P
(S,S)-(R,R)-TRAP
L12
HR =
NC
Rh(acac)(CO)2 (1%)[(allyl)Pd(cod)]BF4 (1%)
(S,S)-(R,R)-L12 (2%)86 (2 equiv)
THF, –25 °C, 40 h1.0 equiv
CN
O
N
94% yield87% ee
(+/–)
O
N
OMe
Me
OMe
Me
PNC
Rh(acac)(CO)2 (1%)[(allyl)Pd(cod)]BF4 (1%)
(S,S)-(R,R)-L12 (2%)86 (2 equiv)
THF, –25 °C, 72 h1.0 equiv
∗∗ P
CN
O
91% yield, 92% ee(+/–)
O
OEtOEt
OEtOEt
a)
b)
c)
85
86 87
88 89
90 91
Scheme 30 Pd/BINAP-catalyzed allylation of -acetamido--keto esters
R1
O
CO2Me
NHAc
+R2 OAc
[Pd(allyl)Cl]2 (1%)(R)-BINAP-L13 (1.05%)
1.0 equiv 1.5 equiv
R1
O
NHAcMeO2CR2 PPh2
PPh2
(R)-BINAP-L13
O
NHAcMeO2CPh
O
NHAcMeO2C
O
NHAcMeO2Cn-Pr Ph
O
NHAcMeO2Cn-Pr Ph
O
NHAcMeO2Cn-Pr
O
NHAcMeO2C
Ph
O
NHAcMeO2CPh i-Bu
O
NHAcMeO2CPh i-Pr
O
NHAcMeO2CPh
Ph
84% yield76% ee
92% yield80% ee
96% yield87% ee
40% yield89% ee
87% yield91% ee
87% yield94% ee
71% yield95% ee
86% yield92% ee
85% yield91% ee
92 93 94
94a 94b 94c 94d 94e
94f 94g 94h 94i
KOt-Bu (1.2 equiv)toluene –30 °C
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

13
B. M. Trost, J. E. Schultz ReviewSyn thesis
Curiously, the reaction outcome was largely indepen-dent of whether the branched 96 or linear (Z)-95 electro-philic substrate was used (Scheme 31). It was proposed thatany stereo- or regiochemistry present in the starting elec-trophiles is lost due to a rapid –– equilibration relativeto intermolecular nucleophilic alkylation.
Scheme 31 Independence of electrophile regio- and stereochemistry on reaction outcome
A similar catalytic system was shown to provide allyla-tion products for -acetamido--ketophosphonate nucleo-philes 97 (Scheme 32).31 A possible explanation for the highenantioselectivity in this acyclic system is control of theenolate geometry by chelation between the phosphonateand enolate oxygen atoms. The highest enantioselectivitywas observed for 3-substituted allylic electrophiles 93; al-lylation proceeded with only modest enantioselectivity.
Other work by the Ito group demonstrated acyclic con-trol in the cinnamylation of 1,3-diketones (Scheme 33).32
Two examples demonstrated cinnamylation of acyclic 1,3-diketones in good enantioselectivity. Similar levels of asym-metric induction on cyclic substrates were also reported.Cryogenic temperatures were required for high enantiocon-trol in this reaction, however, in conjunction with theirwork on -acetamido--keto esters and -acetamido--ke-tophosphonates, these reactions serve as some of the earli-est examples of Pd-AAA with stereocontrol on acyclic nu-cleophiles under simple catalyst conditions.
Scheme 33 Examples of acyclic 1,3-diketone cinnamylation
Hou and co-workers have demonstrated Pd-AAA of acy-clic diphenylamides33 using the ferrocenyl-based ligandsthat they developed (Scheme 34). In most cases, -tertiarycarbon centers were synthesized, however, the report con-tains two examples of the asymmetric synthesis of acyclictetrasubstituted stereocenters. The N,N-diphenylamide wasfound to be optimal in the initial optimization. Subsequentvariation of the chirality on the oxazoline, phosphorus, andthe BINOL moieties of the ligand proved crucial for achiev-ing high enantioselectivity.
Scheme 34 Synthesis of acyclic tetrasubstituted stereocenters by -al-kylation of N,N-diphenylamides
List and Jiang were able to demonstrate the asymmetric-allylation of -arylpropanals 107 under triple chiral ac-id/enamine/palladium catalysis (Scheme 35).34 An achiralpalladium source was used in the reaction, and asymmetricinduction was achieved by achiral amine catalyst (e.g., 106)in the presence of (S)-TRIP phosphoric acid L15. Optimiza-tion of the amine catalyst showed benzhydrylamine (106)to be differential among other achiral benzylic amines. Inaddition to allyl alcohol, more substituted alcohols 108were effective in the reaction. Although the reactionshowed impressive display of control for asymmetric qua-ternary carbon synthesis, the method was limited to -aryl-propanals, as an elongation of the aliphatic aldehyde sidechain resulted in diminished enantioselectivity (109f).
O
CO2Me
NHAc+
n-Pr
[Pd(allyl)Cl]2 (1%)(R)-BINAP-L13 (1.05%)
KOt-Bu (1.2 equiv)toluene, –30 °C1.0 equiv
1.5 equiv
O
NHAcMeO2Cn-Pr
OAc
n-Pr
OAc(+/–)
86% ee from (Z)-95
85% ee from 9694g
92g
95
96
Scheme 32 Pd-AAA of -acetamido--ketophosphonates
R1
O
P(OMe)2
NHAc+
R2 OAc
[(allyl)Pd(cod)]BF4 (0.5%)(R)-BINAP-L13 (1.1%)
KOt-Bu (1.2 equiv)toluene, –30 °C1.1 equiv 1.0 equiv
PPh2
PPh2R1
O
P(OMe)2
NHAc
O
R2
O
P(OMe)2
NHAcPh
Et
O
P(OMe)2
NHAcPh
Ph
O
P(OMe)2
NHAcPh
O
P(OMe)2
NHAcn-Pr
O
P(OMe)2
NHAc
87% yield87% ee
72% yield78% ee
78% yield88% ee
27% yield79% ee
80% yield65% ee
9397 98(R)-BINAP-L13
98a 98b 98c 98d 98e
O
O O O O O
O
+Ph OAc
[Pd(allyl)Cl]2 (0.5%)(R)-BINAP-L13 (1.05%)
1.2 equiv1.0 equiv
O
R
O O
R Ph
R = H 90% yield, 83% eeR = OMe 96% yield, 80% ee
99100
101102
KOt-Bu (1.2 equiv)toluene, –60 °C
Fe
N OP
Et2N
O
OH
i-Pr
Ph2N
O
Ph
R
OAc
[Pd(allyl)Cl]2 (1%)(S,SPHOS,R)-L14 (2%)
Ph2N
O
∗∗
Ph R
1.0 equiv
2.0 equiv
R = OPh 99% yield 93% eeR = Me 75% yield 93% ee
(S,SPHOS,R)-L14
103 104
105
LiHMDS (1 equiv)LiCl (1 equiv), THF, r.t.
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

14
B. M. Trost, J. E. Schultz ReviewSyn thesis
Scheme 35 The direct -allylation of -arylpropanals with allylic alcohols
Steric elements that result in high geometric control ofthe transiently formed chiral enamine were invoked to ac-count for the stereoselectivity observed (Scheme 36a), andchiral information is transferred by hydrogen bonding bythe enamine to the chiral phosphate (Scheme 36b). Sincethis species is counterion-paired with the cationic -allyl-palladium(II) species, asymmetric induction can be realizedin the alkylation event. Additionally, it cannot be ruled outthat the phosphoric acid acts as a ligand to the -allylPd(II)species. Interestingly, under the reaction conditions, allylalcohols can be used directly, due to the activation of thehydroxyl leaving group by the phosphoric acid (Scheme36c).
Scheme 36 Proposed mechanistic feature of triple catalysis
Another dual-catalytic approach was showcased by Ooiand co-workers for the -cinnamylation of -nitro esters(Scheme 37).35 Like the system developed by List and Jiang,the phosphine ligands employed in the method are achiral.However, a linked cationic ammonium functional group al-lows for self-assembly with a chiral BINOL counterion. APd/ligand ratio of 1:2 was used, as it was believed that a C2-symmetric palladium complex would form upon cis-com-plexation of the phosphine ligands. Excellent yields and en-antioselectivities were observed for the reaction when us-
ing cinnamyl carbonate electrophiles 111, although no en-antioselectivity was observed for the simple allyl system(112d).
Scheme 37 Chiral counterion strategy for the cinnamylation of -nitro esters
The use of dual chiral palladium/chiral boron catalysiswas applied to the -allylation of carboxylic acids (Scheme38).36 A chiral boron species is generated by chelation of aboron Lewis acid with the chiral amino acid catalyst L18.Upon ionization of the allyl ester by the chiral palladiumspecies (L17), a carboxylate ene-diolate species is readilyformed, presumably due to the Lewis acidity of the coordi-nated boron catalyst and the presence of DBU as base. Al-though the enolates of carboxylic acids possess no geome-try, the large steric differences of the -aryl substituentsrender the two enantiotopic faces of the nucleophile suffi-ciently distinguishable. The ability to use two chiral cata-lysts enables excellent enantioselectivity for both allyl and
Ar CHO
R1
HO
R2
R3
+
(S)-TRIP-L15 (3%)Pd(PPh3)4 (1.5%)
5 Å MS
Ar
R2
R3R1CHO
O
OP
O
OH
i-Pr
i-Pri-Pr
i-Pr
i-Pri-Pr
Ph
NH2
Ph
40 mol%
1 equiv
2 equiv
(S)-TRIP-L15
CHO CHO CHO CHO CHO
MeO Cl97% yield97:3 e.r.
95% yield97:3 e.r.
98% yield95:5 e.r.
94% yield96:4 e.r.
F
94% yield96:4 e.r.
Me
EtCHO
77% yield81:19 e.r.
CHO
96% yield94:6 e.r.
CHO
96% yield99:1 e.r.
CHO
66% yield96:4 e.r.
CHO
95% yield94:6 e.r.
Ph
Ph
106
107
108
109
109a 109b 109c 109d 109e
109f 109g 109h 109i 109j
toluene, 40 °Cthen 2N HCl
OH O
PO
OR*
OR*
Pd
Pd(0)
Ar
NH
Ph
Ph
OP
O
*ROOR*
H
Acid-catalyzedalcohol activation
Phosphate-mediatedpreorganization
Ar
N
H
Ph
Ph
H
Ar
N
H
Ph
Ph
H
Steric control of enamine geometry
a) b) c)
CO2t-BuO2N
R1
R2 OCO2Me
+
Pd2dba3 (2.5%)L16 (5%)
R2 Ot-Bu
O
R1 NO2
OOMe
2-naphthyl
2-naphthyl
Ar2P
Me3N
L16
PhAr - 4-ClPh
Ph Ot-Bu
O
Et NO2
97% yield93% ee
Ph Ot-Bu
O
i-Bu NO2
96% yield97% ee
Ph Ot-Bu
O
Bn NO2
94% yield97% ee
Ot-Bu
O
NO2
91% yield1%< ee
Ot-Bu
O
NO2
90% yield90% ee
Cl
Ot-Bu
O
NO2
99% yield93% ee
MeO
Ot-Bu
O
NO2
94% yield91% ee
S
110
111112
112a 112b 112c 112d
112e 112f 112g
toluene/H2O(20:1)
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

15
B. M. Trost, J. E. Schultz ReviewSyn thesis
3-substituted allyl systems. The scope of the reaction waslargely limited to -aryl substrates, although and -benzylsubstrate gave 114b with 80% ee.
The DAAA of cyclic -fluoro--keto esters proceedswith high enantioselectivity.37 Acyclic systems affordedproducts with high efficiency, although the enantioselectiv-ities were low. More promising enantioselectivities wereobtained when generating geometrically pure (Z)-lithiumenolates (Scheme 39).38 These species were obtained bydeprotonation of -fluoropropiophenones by LiHMDS at 0°C, and the transient species were subjected to Pd-AAA con-ditions to provide -allyl--fluoropropiophenones with60–90% ee.
3.2 Decarboxylative Allylic Alkylation Strategies
Unlike intermolecular allylic alkylation reactions, Pd-catalyzed decarboxylative allylic alkylations occur via alargely unimolecular reaction pathway (Scheme 40).39 That
is, upon ionization of an allyl moiety, the Pd–-allyl speciesis counterion-paired to its pronucleophile. Upon a decar-boxylation event, alkylation occurs via an outer-sphere at-tack of the nucleophile on the reactive Pd–-allyl species,effectively furnishing the desired product and the regener-ated chiral Pd(0) species.
The question of whether this reaction occurs via an in-ner- or outer-sphere mechanism is complicated by differ-ences in the experimental outcomes of these reactions andthe computed lowest energy pathway.40 It is worth notingthat computational work is for phosphino-oxazoline(PHOX) ligands, and experimental work is with structurallydifferent bisphosphino Trost ligands. Experimental workshowed that enol carbonate 118 underwent Pd-DAAA withkinetic resolution of the enantiomeric starting material(Scheme 41). The alkylation product 119 was formed withnet retention of the stereochemistry, indicating an outer-sphere mechanism.
Scheme 38 Select examples in chiral Pd-AAA/chiral borane-catalyzed -allylation of carboxylic acids
O
O
R1
R2
R3
[Pd(allyl)Cl]2 (2.5%)(AcO)4B2O (10%)(R,R,R)-L17 (5%)
(+/–)
OH
O
R3
R1 R1
PAr2 Ar2P
(R,R,R)-L17Ar = 3,5-Me2C6H3
SNH
OH
O
MeO
FF
FF
O O
L18
OH
O
Ph
91% yield90% ee
OH
O
Et Ph
69% yield99% ee
OH
O
Ph
93% yield87% ee
OH
O
Ar1
60% yield93% ee
OH
O
93% yield80% ee
Ph
OH
O
MeO Ph
65% yield84% ee
OH
O
AcHN Ph
54% yield84% ee
OH
O
Ar2
96% yield92% ee
OH
O
Ar3
45% yield99% ee
Ar2 = 3-ClC6H4 Ar3 = 2-ClC6H4Ar1 = 3-thienyl
113 114
114a 114b 114c 114d
114e 114f 114g 114h 114i
L18 20%, DBU (1.5 equiv)toluene, r.t., 12 h
Scheme 39 Pd-AAA of -fluoropropiophenones
PPh2 N
O
(S)-t-Bu-PHOXL19
t-Bu
O
F
LiHMDS (1.2 equiv)THF 0 °C, then
R2
OCO2Me
1.0 equiv
1.1 equiv
(+/–)R1
O
∗∗
FR1
R2
O
F
O
F
O
F
O
FMeO
86% yield78% ee
84% yield88% ee
88% yield73% ee
74% yield77% ee
O
F
O
F
O
F
O
F
91% yield84% ee
44% yield90% ee
93% yield85% ee
30% yield60% ee
F3C
Ph
Br
115
116 117
117a 117b 117c 117d
117e 117f 117g 117h
[Pd(allyl)Cl]2 (1.25%)(S)-L19 (3.1%)NaOAc (10%)
THF, 0 °C to r.t.
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

16
B. M. Trost, J. E. Schultz ReviewSyn thesis
It was observed by the Trost group in work on the -al-lylation of acyclic ketones that control of the enolate geom-etry was crucial for high asymmetric induction (Scheme
42).41 It was shown that opposing enolate geometries di-verged to opposite enantiomers of product, with matchedand mismatched cases. This problem was easily overcomein the case of -tertiary ketone formation, since either eno-late isomer could be synthesized by judicious choice ofbase. However, the synthesis of acyclic tetrasubstituted ste-reocenters would prove more difficult, due in large part tothe small steric differences in the -substituents.
The Trost group developed Pd-DAAA reactions for thesynthesis of acyclic tetrasubstituted stereocenters by thesynthesis of protected -tertiary hydroxyaldehydes.42 Theallyl carbonate precursors were synthesized with high geo-metric purity (Scheme 43). A highly flexible route to sub-strates allowed for -hydroxy 122 or -bromo ketones 124to be used as substrates. Either regioisomer could be syn-thesized using hard or soft enolization conditions. Undersoft enolization using TBSOTf and triethylamine, acyl trans-fer was suppressed, presumably due to the absence of anyenolate formed, and 127a was isolated with high regiose-lectivity. Under hard enolization with sodium hexameth-yldisilazanide, acyl transfer occurs to form the more stablealdehyde enolate. Trapping with TBSCl allows for synthesisof the opposite regioisomer 128a.
A DAAA reaction proceeds smoothly with high regio-and enantioselectivity when the alcohol protecting groupcan undergo alkoxide transfer (Scheme 44). The processwas found to be regioconvergent; that is, either regioiso-meric starting material afforded the tertiary aldehyde prod-uct with nearly identical enantioselectivities. When analkoxide transfer is not required (substrate 127a), the prod-uct was formed in 15 minutes in 93% yield compared to a 1hour reaction for the regioisomeric enol carbonate 128a.
Scheme 44 A regioconvergent Pd-DAAA strategy toward -tertiary hydroxyaldehydes
Scheme 40 Mechanism of the Pd-catalyzed decarboxylative allylic al-kylation
CO2
Nu
Complexation& Ionization
Decarboxylation
ReductiveElimination
ProductDissociation
Pd0Ln
NuPd0Ln
Nu
ONu
O
Pd(II)LnNu O
O
Pd(II)Ln
I
II
III
IV
V
VI
Scheme 41 Experimental evidence for an outer-sphere mechanism
Pd2dba3·CHCl3 (2.5%)(R,R)-L4 (5.5%)
39% yield99% ee
O O
O
Ph
(+/–)
O
H H
H H
PhO O
O
PhH H
37% yield99.5% ee
+118
119 (R,R)-118
dioxane, r.t., 12 h
Scheme 42 Enantiodivergent allylations of (E)- and (Z)-enol carbon-ates
O
O
O
Pd2dba3·CHCl3 (2.5%)(R,R)-L4 (5.5%)
dioxane, r.t., 16 h
O
72% yield, 60% ee
O
O
OPd2dba3·CHCl3 (2.5%)
(R,R)-L4 (5.5%)
dioxane, r.t., 2 h
O
94% yield, 97% ee
(Z)-120
(E)-120
NH HNO
PPh2
O
Ph2P
(R,R)-L4
(S)-121
(R)-121
Scheme 43 An efficient regiodivergent synthesis of Pd-DAAA substrates
Ph
O
OH Cl
O
O
pyridine, CH2Cl2, 0 °C85% yield
Ph
O
BrNaO
O
O
DMF, r.t.60% yield
Ph
O
OCO2allyl
TBSOTf, NEt3CH2Cl2, –78 °C to r.t.
83% yield
NaHMDS, THF–78 °C, then
TBSCl83% yield
Ph
OTBS
OCO2allyl
Ph
OCO2allyl
OTBS
122
123
124
125126
127a
128a
Ph
OTBS
OCO2allyl
Pd2dba3·CHCl3 (2.5%)(R,R)-L4 (5.5%)dioxane, 23 °C
Ph CHO
OTBS
Ph
OCO2allyl
OTBS 1 h, 86% yield, 91% ee
0.25 h, 93% yield, 92% ee
NH HNO
PPh2
O
Ph2P
(R,R)-L4
127a
128a
129a
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

17
B. M. Trost, J. E. Schultz ReviewSyn thesis
The substrate scope revealed aromatic, vinyl, andalkynyl substituents provided the product with high enan-tioselectivity (Scheme 45). High yields and a rapid reactionrate were achieved by employing substrates 127 with a tert-butyldimethylsiloxy group at the benzylic position, as thesesubstrates undergo Pd-AAA without silyl transfer. Addition-ally, decarboxylation likely occurs more rapidly with theseregioisomeric substrates, since the resulting aldehyde eno-late is more stabilized than the initially formed ketone eno-late when using the regioisomeric substrate.
Scheme 45 A Pd-DAAA strategy toward -tertiary hydroxyaldehydes
This strategy was applied to the synthesis of a protected-hydroxy ketone 132 (Scheme 46). Displacement of the al-kyl bromide 130 occurred smoothly in 87% yield. Underhard enolization conditions, acyl transfer was not observeddue to the trans relationship of the oxygen atoms. This iso-mer 131 was observed to be the major one, and it was sepa-rated from the minor isomeric products in 67% yield. ThePd-DAAA proceeded smoothly with regiospecificity in 94%yield and 80% ee.
Scheme 46 Synthesis and reaction of ketone-derived -alkoxy enol carbonate
Additionally, electrophiles bearing stereochemistrycould be subjected to the reaction conditions to produce al-kylation products with high diastereoselectivity. This strat-egy was applied to the formal synthesis of (S)-oxybutynin(Scheme 47). The sodium carbonate of 133 was formed bytreatment with sodium hydride, followed by carbon dioxide
capture. The transiently formed species was then treatedwith 2-bromoacetophenone and was subsequently con-verted into DAAA substrate 135 by regioselective silylation.Although the starting enol carbonate 135 existed as a mix-ture of enantiomers, the stereochemistry about the electro-phile was lost upon ionization, as the -allyl species be-comes symmetrical. The DAAA reaction proceeded smooth-ly in 18 hours to afford the hydroxyaldehyde 136 in 99%yield and 11:1 d.r. For substrate 135, the tert-butyldimeth-ylsilyl protecting group transfer occurred efficiently, as in-dicated by the high yield in the DAAA reaction. Hydrogena-tion of the olefin afforded product 137 with 84% ee, reflect-ing the stereoselectivity about the tetrasubstituted carbon.Upon desilylation of 137 and Pinnick oxidation, hydroxyacid 139 was obtained as a single isomer after a single re-crystallization, completing the formal synthesis of (S)-oxy-butynin.
Scheme 47 Formal synthesis of (S)-oxybutynin by Pd-DAAA
A method for the synthesis of acyclic quaternary stereo-centers by DAAA was investigated by Tunge and Ariyarath-na.43 Racemic 141 was used with the hope that stereoin-duction would be observed, even in the absence of enolatecontrol (Scheme 48). In this system, the two -substituentsare of considerable steric difference, and it could be imag-ined that this could result in stereocontrol of the formedenolate. Under conditions of catalyst control, only modestenantioselectivity could be observed, with Trost Ligand L4affording the product with the highest enantioselectivity.
Scheme 48 Synthesis of quaternary stereocenters by Pd-DAAA of acy-clic allyl -oxo esters
R
OTBS
O O
O
Pd2dba3·CHCl3 (2.5%)(R,R)-L4 (5.5%)
dioxane, 23 °C
CHO
OTBS
R CHO
OTBS NH HNO
PPh2
O
Ph2P
(R,R)-L4
CHO
OTBS
CHO
OTBS
MeO
CHO
OTBS
NO2
CHO
OTBS
CHO
OTBS
Me
CHO
OTBS
CHO
OTBS
Ph
O
93% yield92% ee
94% yield92% ee
92% yield85% ee
89% yield98% ee
69% yield79% ee
93% yield98% ee
81% yield93% ee
76% yield89% ee
127
129a
129
Me129b 129c 129d
129e 129f 129g 129h
Ph
O
Br
1. sodium allylcarbonateDMF, r.t.
2. NaHMDS, THF–78 °C, then
TBSCl
Ph
OTBS
OCO2allyl58% yield
over two steps
Pd2dba3·CHCl3 (2.5%)(R,R)-L4 (5.5%)
dioxane, 23 °C
O
TBSO Ph
94% yield80% ee
(+/–)
130 131 132
OHNaH, CO2THF, then
PhCOCH2Br
Ph
O
O
O
ONaHMDS
TBSCl, THF
–78 °C to 23 °C Ph
O
OTBS
O
O
42% yield 83% yield
Pd2dba3·CHCl3 (2.5%)
(R,R)-L4 (5.5%)Dioxane, 23 °C
Ph
OHC OTBS
99% yield, 11:1 d.r.
H2, Pd/C
EtOH, 23 °CPh
OHC OTBS
96% yield, 84% ee
NaClO2NaH2PO4
2-methylbut-2-ene
t-BuOH, H2O
Ph
HO2C OH
95% yield(S)-oxybutynin
133 134 135
136 137
139
(+/–)
(+/–)
(+/–)
Ph
OHO
O
Et2N
Ph
OHC OH
44% yield
138
TBAF
THF, r.t.
140
N
O
PhO
O Pd2dba3 (5%)Ligand* (13%)
solvent, temp.
N∗∗
O
Ph
Ligand
Quinap L22t-Bu-PHOX-L19
Trost L4Trost L4Trost L4
Solvent
benzenebenzene
THFDME
toluene
Temp °C
25 25–40 –40–40
%Yield
9397– –
90
141 142(+/–)
% ee
115
242049
Entry
12345
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

18
B. M. Trost, J. E. Schultz ReviewSyn thesis
Additionally, a series of chiral auxiliary containing sub-strates 143 were employed as mixtures of diastereomers atthe -carbonyl position (Scheme 49). Products were formedwith some level of stereoinduction observed, and one auxil-iary proved useful for recrystallization of the product in di-astereomerically pure form.
Scheme 49 Auxiliary approach for the Pd-DAA of acyclic allyl -oxo es-ters
A more elaborate method of synthesizing geometricallydefined enolates was developed by Marek and co-workers.44
An auxiliary-containing ynamide was carbometallated andoxidized to give geometrically pure enolates. This work wasoriginally employed in aldol reactions; however, Starkov,Marek, Stoltz, and co-workers demonstrated that the tran-siently formed enolate can be successfully trapped as theenol carbonate (Scheme 50).45 Two protocols were devel-oped for this reaction, due to limitations in substrate scopeusing a one-pot method when employing acyclic carbamatesubstrates. In the second method (Scheme 50b), the iodoenamide 149 was lithiated with t-BuLi, and the enol car-bonate 150 was isolated with geometric purity after oxida-tion and acylation.
Optimization with a variety of PHOX and Trost-style li-gands revealed electron-deficient Trost ligand L6 bearingthe anthracene-9,10-diamine backbone provided the prod-uct with the highest enantioselectivity (Scheme 51). Sub-strate 151a was shown to provide 152a with higher enantio-selectivity compared to substrates bearing an oxazolidi-none.
The subsequent Pd-DAAA reaction provided allylationproducts with high enantioselectivity, even in cases wherethe two -substituents are of similar steric bulk (Scheme52). This likely indicates that the chiral catalyst recognizesthe steric and electronic differences of the enolate oxygenand the achiral auxiliary as opposed to the differentiationof the -substituents. A two-step allylation/cross-metathe-sis protocol was employed to better facilitate assay of theenantioselectivity. The acrylate moiety provided products153 with higher differentiability of substituents and betterabsorptivity for UV detection.
For substrates bearing two -substituents with consid-erable steric difference (i.e., aryl vs. alkyl), high enantiose-lectivity can be achieved for aryl ketones (Scheme 53).46 Us-ing a protocol developed for the synthesis of analogous enoltosylates,47 enol carbonate 155a was synthesized with (E)selectivity. Solvent optimization with an electron-deficientPHOX ligand L20 provided high enantioselectivity with amixed hexane/toluene solvent system.
Although high geometric control could be achieved inthe enol carbonate formation, it was observed that this wasunnecessary when employing the PHOX L20, as similar lev-els of enantioselectivity were observed for a mixture ofenolate isomers and -keto ester 157 (Scheme 54). Howev-er, high levels of enantioselectivity (86% ee) were observedwhen using Trost ligand L4 for the geometrically definedenol carbonate under the optimized conditions for the
*Xc
O
PhO
OPd(PPh3)4 (10%)
benzene, 25 °C *Xc∗∗
O
Ph
N
O
PhO
O
i-Pr
N
O
PhO
O
t-Bu
N
O
PhO
O
CHPh2
N
O
PhO
O
Bn88% yield, 85:15 d.r. 83% yield, 84:16 d.r.*88% yield, 38:62 d.r. 87% yield, 26:74 d.r.
*53% yield, <5:>95 d.r. after recrystallization
143 144
144a 144b 144c 144d
Scheme 50 One-pot carbometalation/oxidation/acylation synthesis of tetrasubstituted amide enol carbonates and two-pot carbometalation/iodina-tion followed by lithiation/oxidation/acylation
N
R3
N
Cu
R4
R3
R4
(R4)2CuLi (1.2 equiv)
N
O
R4
R3
O
Oallyl1.) t-BuOOH (1.2 equiv)
–78 °C, 0.5 h
N
CO2R2
R1
R3N
R4
R3
R2O2C
R1
R4MgBr (2 equiv)CuI (1 equiv)
N
O
R4
R3
R2O2C
R1
O
Oallyl1) t-BuLi (2 equiv)
Et2O, –78 °CI
148
146
149
147
150
O
O
O
O
145
O
O
a)
b)
–30 °C to –10 °CEt2O, 0.5 h
2.) allyl chloroformate
2.) t-BuOOLi, THF3.) allyl chloroformate
–30 °C to –10 °CEt2O, then I2
Scheme 51 Optimized substrate for Pd-DAAA of tetrasubstituted amide enol carbonates
NH HNO
PAr2
O
Ar2P
(R,R)-L6
CF3F3C
Ar = 4-CF3Ph
N
O
Bu
MeO2C
O
OPd(dba)2 (4%)(R,R)-L6 (7%)
EtOAc
N
O
Bu
MeO2C
85% yield94% ee
PhPh
E/Z < 2:98151a 152a
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

19
B. M. Trost, J. E. Schultz ReviewSyn thesis
PHOX system, but selectivity (50% ee) was diminishedwhen using a mixture of isomers. It was speculated thatrapid C–O equilibration of the palladium(II) enolate may ac-count for this effect when using the PHOX system. Poten-tially, optimization with the PHOX ligand resulted in condi-tions where the facial differentiability of the -substituentsdictates the stereochemical outcome of the reaction.
A reaction involving the DAAA of -acetamido--ketoesters 158 has been described for an acyclic system(Scheme 55).48 Interestingly, with little optimization, highlevels of enantioselectivity were realized in this reaction,albeit for a limited substrate scope. Potentially, there is con-trol of the enolate geometry due to hydrogen bonding be-tween the amide hydrogen and the transient enolate ion.Phenol and naphthol were found to improve the enantiose-lectivity, although the origin of this effect is not well under-stood.
A Pd-AAA of nitroalkanes has been described by Shiba-saki and co-workers (Scheme 56).49 Although most of thereactions described in the method involve stereocontrol onthe electrophile, a single example of Pd-AAA with a racemicsecondary nitroalkane 160 and allyl carbonate 161 was re-ported, albeit with low enantioselectivity.
Scheme 56 Asymmetric allylation for the synthesis of -tertiary ni-troalkanes
4 Temporary Cyclic Pronucleophiles
4.1 Reactions of Azlactones
Alkylation of azlactones directly furnishes productswith cyclic tertiary amino functionality, and the subse-quent hydrolysis of the products has proven useful for pro-ducing acyclic -tertiary amino acids (Scheme 57). Thisprovides an efficient method for the synthesis of unnaturalamino acids, as the synthesis and elaboration of azlactonescan be readily accomplished using commercially available
Scheme 52 Substrate scope of Pd-DAAA of tetrasubstituted amide enol carbonates
N
O
R4
R3
MeO2C
Bn
O
O
1. Pd(dba)2 (4%)(R,R)-L6 (7%)EtOAc or THF
2. Grubbs' II (6%)methyl acrylate
40 °C, CH2Cl2, 3 h
N
O
R4R3
MeO2C
Bn
CO2Me
X
O
n-hexCO2Me X
O
CO2Me X
O
PhCO2Me
TBSO
69% & 84% yield94% ee
76% & 79% yield94% ee
77% & 78% yield76% ee
X
O
Etn-BuCO2Me X
O
Etn-hexCO2Me X
O
n-hexCO2Me
76% & 85% yield82% ee
64% & 85% yield94% ee
56% & 84% yield76% ee
Ph
151 153
153a 153b 153c
153d 153e 153f
Scheme 53 A Pd-DAAA of stereodefined tetrasubstituted 1,2-diaryl enol carbonates
Ar1
O
R
Ar2
O
O
Pd2dba3 (0.5%)(S)-L20 (1.2%) Ar1
O
Ar2
R
R = alkyl
95–99% yield67–92% ee
21 examples
P N
O
t-Bu
Electron-Deficient(t-Bu)-PHOX
CF3
F3C
CF3
Ph
O
Et
PhPh
O
Et
Ph
O
O
> 98:2 E/Z
LiHMDS, Me2NEt
> 98:2 E/Z
(+/–)154 155a
155
156
L20toluene, 23 °C, thenallyl chloroformate
hexane–toluene(3:1), 25 °C, 12 h
Scheme 54 Independence of enolate geometry on enantioselectivity for an electron-deficient (t-Bu)-PHOX ligand
Ph
O
Et
Ph
O
OPd2dba3 (0.5%)(S)-L20 (1.2%)
hexane–toluene(3:1), 25 °C
Ph
O
Ph
Et
> 98:2 E/Z
97% yield91% ee
Ph
O
Et
Ph
O
OPd2dba3 (0.5%)(S)-L20 (1.2%)
hexane–toluene(3:1), 25 °C
Ph
O
Ph
Et
25:75 E/Z
95% yield90% ee
Ph
O
O
O
Ph Et (+/–)
Pd2dba3 (0.5%)(S)-L20 (1.2%)
hexane–toluene(3:1), 25 °C
Ph
O
Ph
Et70% yield90% ee
155a
155a
157
156a
156a
156a
Scheme 55 Pd-DAAA of -acetamido--keto esters
R1
O
AcHN R2O
O Pd2dba3·CHCl3 (2.5%)(R,R)-L2 (5%)
R1
O
AcHN R2
HNNH
PPh2 Ph2P
O O
(R,R)-L2
O
AcHN Me 79% yield87% ee
Et
O
AcHN Me 81% yield90% ee
O
AcHN Et 55% yield80% ee
O
AcHN Bn 81% yield71% ee
158 159
159a 159b 159c 159d
1-naphthol (0.5 equiv)DCE, r.t.
Pd2dba3·CHCl3 (0.5%)L21 (1.3%)NO2
Bn
NO2
Bn
94% yield, 49% ee
OBoc+ PPh2N
O
PHOX Ligand-L21
Ph
CH2OH160 161
162toluene, r.t.TBD (10%)
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

20
B. M. Trost, J. E. Schultz ReviewSyn thesis
amino acids. Additionally, the identity of the carboxylicacid template may serve as a point for reaction optimiza-tion.
Scheme 57 Azlactones as precursors to acyclic -tertiary amino acids
The Trost group has shown a breadth of Pd-AAA reac-tions with azlactones for a variety of allyl electrophiles.50
Impressively, some of the first asymmetric Pd-catalyzedbenzylation reactions were developed using azlactones aspronucleophiles.51 For electron-deficient electrophiles 163,a diphenyl phosphate leaving group in conjunction with el-evated temperatures allowed for the difficult ionization tooccur (Scheme 58).
In the case of electron-rich benzyl electrophiles 166, thediethyl phosphate leaving group proved sufficient at ambi-ent temperatures. This methodology was used in a short
asymmetric synthesis of -methyl-D-DOPA (Scheme 59a).Under conditions for the asymmetric benzylation of elec-tron-rich benzylic phosphates, the key alkylation step af-forded -tertiary azlactone 167 in 83% yield and 90% ee. Thedesired acyclic target 168 was synthesized in 96% yield byhydrolysis of the azlactone. Conveniently, the methylenedi-oxy group was hydrolyzed under these conditions. Addi-tionally, triflic acid hydrolysis in the presence of methanolallows for access to the acyclic protected amino acids 171(Scheme 59b).
Similarly, allyl electrophiles allow for the installation ofa functional group handle for synthetic modification(Scheme 60).50a Interestingly, symmetrical allyl and 2-me-thallyl electrophiles yielded products with low enantiose-lectivity. However, unsymmetrical 3-substituted electro-philes and prenyl electrophiles afforded the -tertiary al-kylated products with high enantioselectivity. It isimportant to note that prenyl and cinnamyl electrophilesproduce alkylation products with an opposite sense of chi-rality.
R
NH3
O–
O1. ArCOCl
2. DCC N
O
Ar
R
Oasymmetric
alkylation N
O
Ar
R
OR'
hydrolysis
NH3
O–R
OR'
α-aminoacids
azlactonepronucleophiles
α-tertiaryazlactones
α-tertiaryamino acids
Scheme 58 Pd-catalyzed benzylation of azlactones
OP
O
OPhOPh
1.0 equiv
+
NO
O
Ph
R2
R1
1.5 equiv
(Cp)Pd(allyl) (5%)(R,R)-L3 (6%)
NEt3 (1.2 equiv)t-BuOH (5 equiv)Dioxane, 50 °C
NO
O
Ph
R2
R1
Ph Ph
NH HNO O
Ph2PPPh2
(R,R)-L3
NO
O
PhN
O
O
Ph
MeS
NO
O
Ph
F
88% yield93% ee
80% yield85% ee
74% yield94% ee
NO
O
Ph
91% yield95% ee N
O
O
Ph
MeO
79% yield78% ee
NO
O
Ph
MeO
82% yield84% ee
MeO2CCl
163 164 165
165a 165b 165c
165f165e165d
Scheme 59 Pd-catalyzed benzylation strategy for the synthesis of -methyl-D-DOPA and benzylation strategy toward acyclic -tertiary amino acids
OP
O
OEtOEt
1.0 equiv
+ NO
O
Ph
1.5 equiv
(Cp)Pd(allyl) (5%)(R,R)-L3 (6%)
Cs2CO3 (0.6 equiv)t-BuOH (5 equiv)
CH2Cl2, 25 °C
NO
O
Ph
PhOH, AcOH
6 M HCl, reflux
O
O
OO 83% yield
90% ee
NH3
OH
O
94% yield
Cl
α-methyl-D-DOPA
HO
HO
OP
O
OEtOEt
1.0 equiv
+ NO
O
Ph
1.5 equiv
(Cp)Pd(allyl) (5%)(R,R)-L3 (6%)
Cs2CO3 (0.6 equiv)t-BuOH (5 equiv)
CH2Cl2, 25 °C
NO
O
Ph
TfOH (3 equiv)
MeOH, 80 °C
90% yield96% ee
NHBzOMe
O
86% yieldMeO
MeO
MeO
166164a
164a
167
168
169
170
171
a)
b)
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

21
B. M. Trost, J. E. Schultz ReviewSyn thesis
Scheme 60 Enantiodivergent alkylation with reverse prenyl and cinnamyl electrophiles
For unsymmetrical allyl systems, diastereomeric -allylcomplexes are possible (Scheme 61). This feature likely dic-tates a distinct mode of attack by the nucleophile, and inthe case of azlactones, this trajectory of approach results inbetter recognition of the prochiral faces of the incoming nu-cleophile. When the linear product is favored, it can be seenthat by placing the 3-allyl substituent under the less hin-dered flap (Scheme 61a), nucleophilic attack occurs at theterminus where chiral information is better relayed fromthe metal center.
Scheme 61 Rationale for higher enantioselectivity for alkylation of 3-substituted electrophiles
In addition to reactions that form stereochemistry onthe nucleophile, the Trost group has utilized this transfor-mation for simultaneous stereoinduction on the electro-philic partner (Scheme 62).50b A Pd-AAA with cyclic elec-trophile 177 afforded the product 178 with high diastereo-and enantioselectivity. The remarkably high level of enantio-selectivity observed in this reaction is likely a result of sta-tistical enrichment, since the Pd(0)/chiral ligand system hasbeen shown to induce high levels of asymmetric inductionon each partner independently.
A geminal diacetoxyallylic electrophile 179 was appliedto the synthesis of sphingofungin F (Scheme 63).52 In thisreaction, the stereochemistry on the electrophile resultsfrom selective ionization of enantiotopic leaving groups in-dependent of the presence of the nucleophile. The stereo-chemistry on the azlactone in 180 was additionally ob-tained with high diastereoselectivity. The relative stereo-chemistry of the amino and acetoxy functional groups wasstrategically inverted in subsequent steps in the synthesisof sphingofungin F.
HNNH
PPh2 Ph2P
O O
(R,R)-L1
OAc
+ NO
O
Ph
Bn
1.0 equiv 1.2 equiv
(R,R)-L1 (1.5%)[Pd(allyl)Cl]2 (0.5%)
NEt3toluene, r.t.
173 75% yield, 99% ee
NO
O
Ph
Bn
+ NO
O
Ph
Bn
1.0 equiv 2.25 equiv
(R,R)-L1 (7.5%)[Pd(allyl)Cl]2 (2.5%)
NEt3toluene, r.t.
176 91% yield, 90% ee
NO
O
Ph
Ph
BnPh OAc
174 15% yield, 20% ee
NO
O
Ph
Bn+
172 164b
164b175
a)
b)
Pd Pd
π−σ−π
R R
∗∗EWGR
R1 R2
∗∗ EWGR
R1R2
low enantioselectivityhigh enantioselectivity
a) b)
Scheme 62 Diastereo- and enantioselective Pd-AAA of azlactones with cyclic prochiral electrophiles
HNNH
PPh2 Ph2P
O O
(R,R)-L1
+ NO
O
Ph
(R,R)-L1 (7.5%)[Pd(allyl)Cl]2 (2.5%)
NEt3, MeCN
178 77% yield, 13:1 d.r.99% ee
NO
O
Ph
OAcH
(+/–)177 164c (+/–)
Scheme 63 A diastereo- and enantioselective synthesis of sphingofungin F
TBDPSO
OAc
OAc NO
O
Ph
+
(R,R)-L1 (1.5%)[Pd(allyl)Cl]2 (0.5%)
NaH, THF NO
O
Ph
OAc
TBDPSO
180 70% yield11:1 d.r., 89% ee
n-C6H13 CO2
OH
OH
OH
NH3Osphingofungin F
HNNH
PPh2 Ph2P
O O
(R,R)-L1
179 164a
181
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

22
B. M. Trost, J. E. Schultz ReviewSyn thesis
4.2 Cyclic -Alkoxy-Bearing Substrates
A similar strategy as that used for -tertiary amino acidscould be envisioned for asymmetric -tertiary alcohol syn-thesis, whereby a cyclic template could serve to controlenolate geometry (Scheme 64). The Stoltz group haveshown that dioxanones 182 derived from 1,3-dihydroxyac-etone can be used for this purpose.53
Scheme 64 Acyclic -tertiary hydroxy ketones, acids, and esters from dioxanone
Alkylation of unsubstituted dioxanones, followed by si-lyl enol ether formation provided substrates 184 for allylicalkylation. The trimethylsilyl enol ethers proved too unsta-ble; however, the regioisomeric triethylsilyl enol ethers 183and 184 could be separated by silica gel chromatography. Inthe presence of TBAT as a desilylation reagent, allylationproceeded in high yield and enantioselectivity using sym-metrical unsubstituted and 2-substituted allyl carbonates185 as electrophiles. A 4-toluenesulfonic acid catalyzed hy-drolysis of 186 in the presence of methanol gave the acyclichydroxy ketone products 187. Periodate cleavage, followedby esterification provided -tertiary hydroxyl esters 188.This method was used in an efficient synthesis of (+)-eu-comic acid.54
A Pd-DAAA of 5- and 6-membered lactam precursorswas developed by the Stoltz group (Scheme 65).55 An -hy-droxy ester 191 was accessed by hydrolysis of the DAAAproduct 190 in the presence of catalytic methanol and sul-furic acid.56 The method additionally was also performed
using a sulfur-containing lactam, an allyl 2-methyl-3-oxo-thiomorpholine-2-carboxylate, although derivatization ofthe product to the acyclic precursor was not demonstrated.
Likewise, thiopyranones were used for the synthesis ofacyclic quaternary stereocenters (Scheme 66).57 Acylationof thiopyranone and subsequent alkylation of the resulting-keto ester provides substrates 192 for Pd-DAAA. The reac-tion proceeds with high yield and enantioselectivity inmost cases. Direct extrusion of the sulfur atom was at-tempted, but hydrogenation of the allyl olefin of 193a couldnot be avoided. A hydroboration/oxidation proved usefulfor avoiding this problem, as Raney nickel in ethanol afford-ed the desired acyclic -quaternary ketone 195 in 94% yieldover the sequence. Although this method represents an ap-plication of cyclic structure for creating acyclic stereocen-ters, it suffers from the drawback of being limited to thesynthesis of -methyl ethyl ketones only.
Scheme 66 Thiopyranones as precursors of acyclic quaternary stereo-centers
5 Allylic Alkylation with Other Metals
The first reactions within the area of allylic alkylation todemonstrate stereocontrol in the synthesis of acyclic ste-reocenters were in Pd-AAA, followed by reactions in Mo-catalyzed AAA; however, within the last few years, majoradvances have been made employing other metals, mostnotably rhodium and iridium. Copper-catalyzed allylic sub-stitution reactions58 nicely complement methods with oth-er metals due to the unique SN2′ pathway and the ability toemploy hard nucleophilic precursors. The structures of li-gands used in Mo-, Ir-, and Rh-catalyzed AAA are shown inFigure 2.
O O
O
TESCl, NaI
NEt3, MeCN O O
OTES
O O
OTES
+
R1 R1
(S)-(t-Bu)-PHOX-L19 (5.5%)Pd(dmdba)2 (5%)
TBAT (1 equiv)toluene, 25 °C, 5–10 h
O O
OR1
R2
6–15%yield
46–78%yield
R2 = H, Me, Ph, Cl
186 59–93% yield85–94% ee
TsOH·H2O
MeOH OH OH
OR1
R2
1) H5IO6THF–H2O
2) K2CO3MeI, DMF
MeO
OH
OR1
R2
80–90% yield 54–85% yield
R1 = alkyl
R1
182 183 184
187 188
O
R2
O
O
R2185
Scheme 65 Lactam precursor to -hydroxy tertiary esters
NO
O O
OBz
Pd2(pmdba)3 (5%)(S)-PHOX-L20 (12.5%)
toluene, 60 °C
NO
O
Bz H2SO4
MeOH, 65 °CMeO
O
OH
191 71% yield190 82% yield
96% ee(+/–)189
S
O
O
OR Pd2(dmdba)3 (1%)
(R,R)-Trost L4 (2.4%)
TBME, 25 °C, 12 hS
OR
R = alkyl 70–92% yield50–94% ee
NH HNO
PPh2
O
Ph2P
(R,R)-L4
S
OBn BH3.THF
cyclohexene
then NaBO3·H2OS
OBn
OHRaney Ni
EtOH, 70 °CEt
OBn
OH
94% yield over 2 steps94% ee
(+/–)192193
193a 194 195
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

23
B. M. Trost, J. E. Schultz ReviewSyn thesis
Figure 2 Structures of ligands used in Mo-, Ir-, and Rh-catalyzed AAA
5.1 Molybdenum-Catalyzed AAA
The ability to selectively access branched allylic alkyla-tion products is an attractive feature for creating stereo-chemistry on electrophilic partners. Among metals thattypically afford branched products selectively (Mo and Ir),selectivity on the nucleophilic partner was first achieved inMo-AAA for azlactones59 and oxindoles.60
In the case of branch-selective azlactone alkylation,58
the reaction proceeded in excellent yield as well as diaste-reo- and enantioselectivity (Scheme 67). Similar levels ofyield and selectivity were observed when using a carbonateor phosphate leaving group; however, the use of lithiumbase was found to afford higher branch selectivity com-pared to sodium or potassium hexamethyldisilazanide. Ad-ditionally, adopting a one-pot protocol for hydrolysis of theazlactone afforded 197 in higher yield compared to thestepwise process.
The scope of the reaction showed the best results forcinnamyl methyl carbonate electrophile (197a–e), withslightly diminished enantioselectivities for heteroaromatic
as well as 2-substituted phenyl systems (197f–j). High se-lectivity was achieved for aliphatic -substituents with var-ied substitution patterns. It is important to note that linear,achiral electrophiles 196 performed better than branched,racemic allylic carbonates, presumably due to slow ––equilibration relative to nucleophilic attack. The mode ofasymmetric induction on the electrophile therefore likelyresults from differentiation of the enantiotopic faces of theelectrophile in olefin coordination and/or ionization.
With success using azlactones as precursors for acyclic-tertiary amino acids, the Trost group studied the isomer-ic oxalactims 19861 as precursors to acyclic -tertiary alco-hols (Scheme 68).
Indeed, these pronucleophiles undergo Mo-AAA withregio-, diastereo-, and enantioselectivity. In addition to thestandard cinnamyl-type electrophiles, vinyl-substitutedsystems perform as well (199c,d). An efficient hydrolysis ofthe products directly affords acyclic -tertiary hydroxy am-ides (Scheme 69).
HNNH
N N
O O
(S,S)-TrostL5
HNNH
N N
O O
(R,R)-TrostL23
MeO OMe
O
OP(OMe)
(R)-BINOL-MeOPL25
N
N
NH2
C1
N
N
NH2
C2
O
OP
(R)-L24
N
Scheme 67 One-pot Mo-catalyzed regio-, diastereo-, and enantioselective allylic alkylation of azlactones followed by hydrolysis
NO
Ph
OR2
R1 OCO2Me +
NHBz
R2i) (S,S)-L5 (15%)
Mo(CO)3C7H8 (10%)
LiHMDSTHF, 65 °C
ii) K2CO3–MeOH
R1
(+/–)
HNNH
N N
O O
(S,S)-TrostL5
CO2Me
NHBz
Ph
CO2Me
NHBz
Ph
CO2Me
SMe
NHBz
Ph
CO2Me
NHBz
i-PrPh
CO2Me
NHBz
Ph
CO2Me
92% yield97:3 d.r.99% ee
86% yield>98:2 d.r.92% ee
85% yield>98:2
96% ee
76% yield>98:2 d.r.96% ee
82% yield>98:2 d.r.97% ee
NHBz
CO2Me
84% yield96:4 d.r.91% ee
S
NHBz
CO2Me
84% yield>98:2 d.r.92% ee
O
NHBz
CO2Me
86% yield>98:2 d.r.94% ee
S
Ph
NHBz
CO2Me
82% yield>98:2 d.r.85% ee
Br
NHBz
CO2Me
90% yield>98:2 d.r.94% ee
MeO
OMe
Ph
196 164 197
197a 197b 197c 197d 197e
197f 197g 197h 197i 197j
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

24
B. M. Trost, J. E. Schultz ReviewSyn thesis
Scheme 69 Conversion of oxalactim into acyclic -tertiary hydroxy amides
The synthesis of acyclic quaternary stereocenters wasachieved using -substituted -cyano esters 200 as pronu-cleophiles (Scheme 70).62 Interestingly, the process affordsacyclic quaternary carbon products in excellent enantiose-lectivity in cases where the allyl electrophile bears no ste-
reochemistry (201a,b). Both alkyl and aryl -substituentsresulted in high regio-, diastereo-, and enantioselectivity.Additionally, -tertiary ether product 201h was accessedwith good diastereo- and enantioselectivity.
Scheme 70 Mo-catalyzed regio-, diastereo-, and enantioselective allylic alkylation of acyclic -cyano esters
Scheme 68 Mo-catalyzed regio-, diastereo-, and enantioselective allylic alkylation with oxalactims
ON
Ph
OR2
R1 OCO2Me +
ON
Ph
OR2Trost L5 (15%)Mo(CO)3C7H8
LiHMDSTHF, 65 °C
R1
(+/–)
HNNH
N N
O O
(S,S)-TrostL5
ON
Ph
OMeO
OMe
ON
Ph
OBr
ON
Ph
O
ON
Ph
O
ON
Ph
Oi-Bu
ON
Ph
Oi-PrPh
S
ON
Ph
OBuPh
ON
Ph
OPh
ON
Ph
OCyPh
ON
Ph
OBnPh
82% yield12:1 b/l18:1 dr99% ee
78% yield27:1 b/l24:1 dr99% ee
54% yield12:1 b/l12:1 dr98% ee
77% yield14:1 b/l12:1 dr89% ee
89% yield14:1 b/l10:1 dr99% ee
70% yield5.5:1 b/l20:1 dr99% ee
86% yield49:1 b/l9:1 dr
99% ee
97% yield8:1 b/l10:1 dr99% ee
74% yield9:1 b/l12:1 dr99% ee
84% yield20:1> b/l7.4:1 dr99% ee
196 198 199
199a 199b 199c 199d 199e
199f 199g 199h 199i 199j
ON
Ph
Oi-Bu
S
NaOH
EtOH/H2O60 °C
OHNH2
Oi-Bu
S
86% yield
ON
Ph
Oi-Bu Trost L5 (15%)
Mo(CO)3C7H8
LiHMDSTHF, 65 °C
(+/–)OCO2Me +
S 89% yield14:1 b/l10:1 dr99% ee
O
EtNC
BocNS
t-BuO
O
OMeNC
t-BuO
O
PhNC
Ph
t-BuO
O
EtNCt-BuO
O
NC
Br NO2
99% yield99% ee
96% yield98% ee
>20:1 b/l, 16.8:1 dr93% ee, 96% yield
>20:1 b/l, 29:1 dr98% ee, 93% yield
>20:1 b/l, 20:1 dr99% ee, 95% yield
>20:1 b/l, 13.7:1 dr97% ee, 92% yield
>20:1 b/l, 11.1:1 dr92% ee, 83% yield
>20:1 b/l, 11.1:1 dr88% ee, 79% yield
>20:1 b/l, 11:1 dr97% ee, 99% yield
>20:1 b/l, 20:1 dr98% ee, 93% yield
2.2 equiv
196
200
201
201a 201b 201c 201d 201e
201f 201g 201h 201i 201j
t-BuO
O
R2
CN
R1 OCO2Me
+
Trost L23 (15%)Mo(CO)6 (10%)
NaH (10%), BSATHF, 60 °C
HNNH
N N
O O
(R,R)-TrostL23
MeO OMe
t-BuO
O R1
R2NC
t-BuO
O
NCt-BuO
O
EtNCt-BuO
O
NC
Ph
t-BuO
O
BnNC
Ph
t-BuO
O
EtNC
t-BuO
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

25
B. M. Trost, J. E. Schultz ReviewSyn thesis
5.2 Iridium-Catalyzed AAA
In iridium-catalyzed allylic alkylation, the ability to con-trol stereochemistry on the nucleophilic partner simulta-neous to stereocontrol was first demonstrated by Takemotoand co-workers63 in 2003. Since that first report, enantio-and diastereoselective Ir-AAA reactions have been reportedfor several systems. This topic has been reviewed else-where,64 so we will focus on two systems that demonstratethe synthesis of acyclic quaternary carbons on the nucleo-philic and electrophilic partners, respectively.
In an impressive display of enantio- and diastereodiver-gence, Carreira and co-workers (Scheme 71)65 were able todemonstrate the ability to access all four stereoisomers inthe -cinnamylation of aldehydes to afford vicinal acyclicquaternary and tertiary stereocenters. Borrowing from thesuccess of dual-catalysis in Pd-AAA, they employed cincho-na alkaloids with chiral phosphoramidite ligands. The pseu-do-enantiomeric catalyst was employed to access the otherdiastereomer. Despite a small match-mismatch effect, allfour diastereomers can be accessed in near perfect selectiv-ity by this method.
The branch-selective Ir-AAA of trisubstituted cinnamylelectrophiles was demonstrated by the Stoltz group(Scheme 72).66 In this reaction, acyl anion equivalent 205 isused to allow for easy access to -aryl--vinyl carbonylcompounds 207. This method demonstrates a new land-mark in Ir-AAA, as more highly substituted electrophiles206 have proven difficult in Ir-AAA. In a single operation,the acyl anion was cleaved to directly afford carboxylic acid
207. Additionally, conditions were developed to obtain es-ters and primary and secondary amides directly from theallylic alkylation products.
The reaction was limited in electrophile scope, as re-placement of the methyl group with ethyl (207j), isopropyl,or butyl groups or the para- or mono-meta-substitutedphenyl group with di-meta- (207i) or ortho-substitutedphenyl groups (207l) resulted in lower, or no, reactivity, butnot lower enantioselectivity. Additionally, excess electro-phile over the acyl anion equivalent was required for highyields. Triethylborane proved to be crucial for reactivity, asno reaction proceeded in its absence, and lower yields wereobserved for lithium bromide as an additive. Presumably,triethylborane assists by Lewis acid activation in the other-wise difficult ionization reaction. The mode of asymmetricinduction of this reaction is selective ionization from enan-tiotopic faces. This was demonstrated by observing no en-antioselectivity when using the branched, racemic electro-phile.
5.3 Rhodium-Catalyzed AAA
Exploiting the stereospecificity of the Rh-allylic alkyla-tion, the Evans group was able to alkylate enantiopure ter-tiary allylic carbonates with regio- and stereospecificity(Scheme 73).67 An electron-deficient phosphite ligand wasfound to afford the product with highest enantiospecificity(denoted as es) compared to more electron-neutral phos-phite ligands.68 All ligands screened showed complete re-giospecificity in the reaction. It was reasoned that the elec-
Scheme 71 Regio-, diastereo-, and enantioselective -cinnamylation of aldehydes by dual chiral amine/chiral iridium catalysis
O
OP
(S)-L24
N
N
N
NH2
C1
N
N
NH2
C2
H
O
Ph
OH
S
+
[(Ir(cod)Cl)2] (2%)(R)-L24 (8%)
C1 (10%)Cl3CCO2H (0.5 equiv)
DCE, 25 °C
H
O
Ph
S
1.1 equiv 64% yield, dr >20:1ee >99%
H
O
Ph
OH
S
+
[(Ir(cod)Cl)2] (2%)(R)-L24 (8%)
C2 (10%)Cl3CCO2H (0.5 equiv)
DCE, 25 °C
H
O
Ph
S
70% yield, dr 11:1ee >99%
H
O
Ph
OH
S
+
[(Ir(cod)Cl)2] (2%)(S)-L24 (8%)
C1 (10%)Cl3CCO2H (0.5 equiv)
DCE, 25 °C
H
O
Ph
S
64% yield, dr 10:1ee >99%
H
O
Ph
OH
S
+
[(Ir(cod)Cl)2] (2%)(S)-L18 (8%)
C2 (10%)Cl3CCO2H (0.5 equiv)
DCE, 25 °C
H
O
Ph
S
70% yield, dr >20:1ee >99%
O
OP
(R)-L24
N
1.0 equiv202
1.1 equiv202
1.1 equiv202
1.1 equiv202
203
1.0 equiv203
1.0 equiv203
1.0 equiv203
204a
204b
204c
204d
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

26
B. M. Trost, J. E. Schultz ReviewSyn thesis
tron deficiency of the ligand facilitated a more rapid inter-molecular alkylation event relative to –– equilibration.Additionally, more bulky ligands resulted in significantlydiminished enantiospecificity, presumably due to high co-ordinative unsaturation of the Rh complex, thus favoringthe η1-allyl species necessary for isomerization.
The reaction scope revealed high regioselectivity in allcases (Scheme 74). The stereospecificity of the reaction washigh for most substrates, although disubstituted aromaticsfor Ar1 resulted in diminished enantiospecificity. The elec-tronics of Ar2 showed high stereospecificity for electron-withdrawing substituents at the 4-position, and diminishedspecificity for electron-donating groups. This likely resultsfrom a more electrophilic -allyl species with electron-
Scheme 72 Branch selective Ir-AAA of trisubstituted electrophiles by an acyl anion equivalent nucleophile
NC
OMOM
CN Ar
R
OCO2Me+
i) BEt3 (2 equiv)[Ir(cod)Cl]2 (2%)
(S)-L24 (4.2%), TBD (10%)THF, 60 °C
ii) 6 M HCl, 80 °C1.0 equiv 2.0 equiv
HO
O
Ar R
O
OP
(S)-L24
N
HO
O
HO
O
HO
O
HO
O
HO
O
HO
O
Cl F3C77% yield95% ee
NO2
80% yield93% ee
83% yield92% ee
65% yield94% ee
93% yield87% ee
*66% yield92% ee
HO
O
HO
O
HO
O
HO
O
EtHO
O
HO
O
F
*68% yield93% ee
Br
90% yield90% ee
*32% yield85% ee
*61% yield92% ee
69% yield92% ee
0% yield
R = Me or Et
*Double the Ir and ligand loading was used.
205 206 207
207a 207b 207c 207d 207e 207f
207g 207h 207i 207j 207k 207l
Scheme 73 Regio- and enantiospecific Rh-allylic alkylation of tertiary allylic carbonates with a cyanohydrin acyl anion equivalent
OCO2Me
Ph
i) [RhCl(cod)]2 (2.5%)P(OCH2CF3)3 (10%)
ii) LiHMDS (1.8 equiv)209 (1.3 equiv)THF, –10 °C
Ph CN
OTBS+
1.0 equiv 1.3 equiv
Ph
O
PhPh
TBSO
Ph
CN TBAT
THF, r.t.
87% yield>19:1 b/l, 91% es
208 209 210211a
Scheme 74 Scope of Rh-allylic alkylation of tertiary allylic carbonates with a cyanohydrin acyl anion equivalent
OCO2Me
Ar1
i) [RhCl(cod)]2 (2.5%)P(OCH2CF3)3 (10%)
ii) LiHMDS (1.8 equiv)213 (1.3 equiv)THF, –10 °C
iii) TBAT, THF, r.t.
Ar2 CN
OTBS+
1.0 equiv 1.3 equiv
Ar2
O
Ar1
O
Ph
87% yield, b/l >19:191% es
O
Ph
80% yield, b/l >19:193% es
F
O
Ph
83% yield, b/l >19:187% es
Br
O
Ph
78% yield, b/l >19:181% es
MeO
OMe
Ph
O
Cl
Ph
O
OMe
Ph
O
F
Ph
O
Ph
O
F
76% yield, b/l >19:199% es
69% yield, b/l >19:190% es
73% yield, b/l >19:190% es
86% yield, b/l >19:193% es
80% yield, b/l >19:194% es
211212 213
211a 211b 211c 211d
211e 211f 211g 211h 211i
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

27
B. M. Trost, J. E. Schultz ReviewSyn thesis
withdrawing substituents, favoring a rapid intermolecularreaction with cyanohydrin nucleophile relative to isomeri-zation.
This work was extended to vinyl acyl anion equivalent215 (Scheme 75).69 The product 217 can be chemoselective-ly reduced to afford the formal product from an aliphaticacyl anion equivalent addition, affording acyclic -quater-nary -vinyl ketone 218. This reaction was highlighted for atertiary allylic carbonate bearing two aliphatic substitu-ents, with the reaction proceeding with high regio- and ste-reospecificity.
The Evans group demonstrated the first direct asym-metric alkylation reaction of -tertiary nitriles 219 by Rh-catalyzed AAA (Scheme 76).70 The process improves step-economy in the -functionalization of nitriles by generatingthe reactive anion in situ, thus side-stepping the synthesisand isolation of racemic N-silylketenimines. The reactionwas shown to proceed smoothly in the absence of addi-tives; however, improved enantioselectivity was observedin the presence of a crown ether. High enantioselectivitywas demonstrated for substrates bearing methyl, isopropyl,and benzyl substituents.
Additionally, the Evans group have shown a direct inter-molecular Rh-catalyzed allylic alkylation of -aryl alde-hydes (Scheme 77).71 Deprotonation with lithium hexa-methyldisilazanide affords lithium enolates that undergoallylic alkylation with allyl benzoate (222) with high enan-tioselectivity. Although the reaction requires a ligand load-ing of 40 mol%, ligand L25 is conveniently accessed in onestep from BINOL.
Strangely, control experiments showed that both eno-late isomers converge to the same enantiomer of product(Scheme 78). The respective silyl enol ethers are convertedinto the lithium enolates by treatment with methyllithium.The enantioselectivity in the catalytic reaction is signifi-cantly higher. It is worth noting that the presence ofhexamethyldisilazanide is absent in the control experi-ments. The fact that both enolate isomers converge to thesame product is all the more surprising considering thesmall steric differences between the -substituents forproducts 223b, 223c, 223d, 223h, 223i, 223j. It could be en-visioned that there is electronic differentiation due to thestrong dependence of enantioselectivity on the 4-substitu-tion of the aromatic substituent.
Scheme 75 Rh-allylic alkylation of tertiary allylic carbonates with a vinyl cyanohydrin acyl anion equivalent
OCO2Me[Rh(cod)Cl]2 (2.5%)P(OCH2CF3)3 (10%)
LiHMDS (1.8 equiv)215 (1.3 equiv)THF, –10 °C
NC
OTBS
OBn
1.0 equiv 1.3 equiv
+ OBn
NC OTBS
TBAF
–40 °COBn
O
98% ee
82% yield, 92% ee
[PPh3CuH]6
benzene–H2Or.t.
OBn
O
82% yield, 92% ee
214 215216
217 218
Scheme 76 Rh-catalyzed asymmetric alkylation of secondary benzylic nitriles
R2
CN
R1
i) 15-crown-5 (1.2 equiv)LiHMDS, THF, –30 °C
ii) Rh(cod)2OTf (5%)(R)-L25 (20%)
iii) allyl benzoate (1 equiv)THF, –30 °C
R2
CN
R1
O
OP(OMe)
L25(R)-BINOL-
MeOP
CN CN CN CN CN
CN CN CN CN CN
86% yield, 92% ee 77% yield, 92% ee 81% yield, 84% ee
Ph
MeO
88% yield, 81% ee
Me
83% yield, 90% ee
Br
87% yield, 92% ee 92% yield, 95% ee
Br Br
Ph
85% yield, 92% ee
Ph
79% yield, 95% ee 81% yield, 92% ee
Ph
F3C F3C
219 220
220a 220b 220c 220d 220e
220j220i220h220g220f
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

28
B. M. Trost, J. E. Schultz ReviewSyn thesis
Scheme 78 Effect of enolate geometry on stereochemical outcome
6 Conclusions and Outlook
An understanding of the mechanistic features of Pd-AAA has allowed for a variety of methods for the asymmet-ric synthesis of acyclic tetrasubstituted stereocenters in-cluding tertiary alcohols and ethers, -tertiary amines, andall-carbon quaternary carbons. Isoprene monoepoxide andrelated electrophiles have served as excellent precursors forforming difficult stereocenters, many of which appear innatural products. Additionally, the presence of hy-droxymethylene and vinyl functional group handles in theresulting products allow for the synthesis of highly valuablesources of chirality. Stereocontrol on other electrophiles hasbeen demonstrated in the branch-selective reverse-pre-nylation of oxindoles. This remains an exciting precedent in
control of acyclic tetrasubstituted stereocenters on electro-philes; however, reactions with other nucleophiles is stillunderdeveloped in this area of research.
Despite the difficulty of achieving asymmetric induc-tion on nucleophiles in Pd-AAA, important advances havebeen made over the past 20 years. The ability to controlacyclic geometric elements of enol carbonates has beenachieved by many well-designed substrate syntheses. Thisstrategy works well in conjunction with strategies employ-ing cyclic precursor such as azlactones, as evidenced bymethods for the synthesis of tertiary alcohols, -tertiaryamines, and all-carbon quaternary stereocenters. The addi-tion of dual and triple catalysis has enabled the chemist tointroduce new modes of stereocontrol as well as the abilityto exploit multiple chiral catalysts within the same reactionfor enhanced stereoselectivity.
Following the success of Pd- and Mo-AAA strategies forstereocontrol in the synthesis of acyclic stereocenters, re-cent advances in Ir- and Rh-AAA nicely demonstrate acyclicstereocontrol on both nucleophilic and electrophilic reac-tion partners. Undoubtedly, more efficient strategies in thisbroad area of research will continue to emerge, further en-riching the options of synthetic chemists for stereocontrolin asymmetric synthesis.
Funding Information
Funding was provided in part by the Tamaki Foundation. ()
Acknowledgment
Dr. Yu Bai is thanked for reviewing the manuscript.
Scheme 77 Rh-catalyzed asymmetric allylic alkylation of -aryl aldehydes
R2
CHO
R1
RhCl(PPh3)3 (10%)(R)-L25 (40%)
DMPU, THF, r.t., thenLiHMDS
OBz+R2
CHO
R1
O
OP(OMe)
L25(R)-BINOL-
MeOP
Et
CHO CHO CHO CHO
Et
CHO
Et
CHO
Et
CHO CHO CHO CHO
74% yield, 92% ee
Br
79% yield, 88% ee
MeO
66% yield, 94% ee
F
73% yield, 87% ee 75% yield, 92% ee
Br
83% yield, 77% ee 79% yield, 91% ee
MeO MeO
61% yield, 90% ee 64% yield, 92% ee 62% yield, 91% ee
221 222 223
223a 223b 223c 223d 223e
223f 223g 223h 223i 223j
Et
CHO
RhCl(PPh3)3 (10%)(R)-L25 (40%)
DMPU, THF, r.t., thenLiHMDS
OBz
Et
CHOMeO
MeO
79% yield, 91% ee
a.)
MeO
Et
OTMSi) MeLi, THF, 0 °C
ii) RhCl(PPh3)3 (10%)(R)-L25 (40%)
DMPU, THF, r.t.
OBz
Et
CHO
MeO
81% yield, 71% eeE/Z 94:6
MeO
Et i) MeLi, THF, 0 °Cii) RhCl(PPh3)3 (10%)
(R)-L25 (40%)
DMPU, THF, r.t.
OBz
Et
CHO
MeO
84% yield, 68% eeE/Z 7:93
b.)
c.)
OTMS
H
H
221g
222
223g
223g
223g
222
222
224
225
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

29
B. M. Trost, J. E. Schultz ReviewSyn thesis
References
(1) (a) Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395.(b) Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921.(c) Trost, B. M. J. Org. Chem. 2004, 69, 5813. (d) Trost, B. M.;Machacek, M. R.; Aponick, A. Acc. Chem. Res. 2006, 39, 747.(e) Trost, B. M.; Fandrick, D. R. Aldrichimica Acta 2007, 40, 59.
(2) Although the term quaternary stereocenter is sometimes usedinterchangeably with tetrasubstituted stereocenter, for the sakeof this review, quaternary stereocenters will only be applied toall-carbon quaternary stereocenters.
(3) Trost, B. M.; Jiang, C. J. Am. Chem. Soc. 2001, 123, 12907.(4) Trost, B. M.; McEachern, E. J.; Toste, F. D. J. Am. Chem. Soc. 1998,
120, 12702.(5) Trost, B. M.; McEachern, E. J. J. Am. Chem. Soc. 1999, 121, 8649.(6) Trost, B. M.; Jiang, C.; Hammer, K. Synthesis 2005, 3335.(7) Trost, B. M.; Bunt, R. C.; Lemoine, R.; Calkins, T. L. J. Am. Chem.
Soc. 2000, 122, 5968.(8) Trost, B. M.; Calkins, T. L.; Oertelt, C.; Zambrano, J. Tetrahedron
Lett. 1998, 39, 1713.(9) The -allyl complex that places the higher priority group cis to
the central hydrogen is denoted as the syn isomer(10) Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1999, 121, 4545.(11) Feng, J.; Holmes, M.; Krische, M. J. Chem. Rev. 2017, For a review
on metal-catalyzed synthesis of acyclic quaternary stereocen-ters: 117, 12564.
(12) For a review on diversity-oriented synthesis: Galloway, W. R. J.D.; Isidro-Llobet, A.; Spring, D. R. Nat. Commun. 2010, 1, 80;DOI: 10.1038/ncomms1081.
(13) Du, D.; Li, L.; Xie, Z. Angew. Chem. Int. Ed. 2009, 48, 7853.(14) Trost, B. M.; Dong, G.; Vance, J. A. Chem.–Eur. J. 2010, 16, 6265.(15) Fagnou, K.; Lautens, M. Angew. Chem. Int. Ed. 2002, 41, 26.(16) Trost, B. M.; Andersen, N. G. J. Am. Chem. Soc. 2002, 124, 14320.(17) Trost, B. M.; Tang, W.; Schulte, J. L. Org. Lett. 2000, 2, 4013.(18) Mandal, A. K.; Schneekloth, J. S.; Crews, C. M. Org. Lett. 2005, 7,
3645.(19) Zeng, M.; Murphy, S. K.; Herzon, S. B. J. Am. Chem. Soc. 2017,
139, 16377.(20) Trost, B. M.; Malhotra, S.; Chan, W. H. J. Am. Chem. Soc. 2011,
133, 7328.(21) Zhang, P.; Le, H.; Kyne, R. E.; Morken, J. P. J. Am. Chem. Soc. 2011,
133, 9716.(22) Ardolino, M. J.; Morken, J. P. Tetrahedron 2015, 71, 6409.(23) Hou, X.-L.; Sun, N. Org. Lett. 2004, 6, 4399.(24) (a) Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1999, 121, 3543.
(b) Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 2003, 125, 3090.(25) Sawamura, M.; Nagata, H.; Sakamoto, H.; Ito, Y. J. Am. Chem. Soc.
1992, 114, 2586.(26) Sawamura, M.; Nakayama, Y.; Tang, W.-M.; Ito, Y. J. Org. Chem.
1996, 61, 9090.(27) Sawamura, M.; Sudoh, M.; Ito, Y. J. Am. Chem. Soc. 1996, 118,
3309.(28) Mizuho, Y.; Kasuga, N.; Komiya, S. Chem. Lett. 1991, 2127.(29) Sawamura, M.; Hamashima, H.; Ito, Y. J. Am. Chem. Soc. 1992,
114, 8295.(30) Kuwano, R.; Ito, Y. J. Am. Chem. Soc. 1999, 121, 3236.(31) Kuwano, R.; Nishio, R.; Ito, Y. Org. Lett. 1999, 1, 837.(32) Kuwano, R.; Uchida, K.; Ito, Y. Org. Lett. 2003, 5, 2177.(33) Zhang, K.; Peng, Q.; Hou, X.-L.; Wu, Y.-D. Angew. Chem. Int. Ed.
2008, 47, 1741.(34) Jiang, G.; List, B. Angew. Chem. Int. Ed. 2011, 50, 9471.(35) Ohmatsu, K.; Ito, M.; Kurieda, T.; Ooi, T. Nat. Chem. 2012, 4, 473.
(36) Fujita, T.; Yamamoto, T.; Morita, Y.; Chen, H.; Shimizu, Y.; Kanai,M. J. Am. Chem. Soc. 2018, 140, 5899.
(37) Belanger, E.; Houze, C.; Guimond, N.; Cantin, K.; Paquin, J.-F.Chem. Commun. 2008, 3251.
(38) Wang, W.; Shen, H.; Wan, X.-L.; Chen, Q.-Y.; Guo, Y. J. Org. Chem.2014, 79, 6347.
(39) Weaver, J. D.; Recio, A. III.; Grenning, A. J.; Tunge, J. A. Chem. Rev.2011, 111, 1846.
(40) Keith, J. A.; Behenna, D. C.; Sherden, N.; Mohr, J. T.; Ma, S.;Marinescu, S. C.; Nielsen, R. J.; Oxgaard, J.; Stoltz, B. M.;Goddard, W. A. J. Am. Chem. Soc. 2012, 134, 19050.
(41) Trost, B. M.; Xu, J.; Schmidt, T. J. Am. Chem. Soc. 2009, 131,18343.
(42) Trost, B. M.; Xu, J.; Reichle, M. J. Am. Chem. Soc. 2007, 129, 282.(43) Ariyarathna, J.; Tunge, J. A. Org. Biomol. Chem. 2014, 12, 8386.(44) Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I.
Nature (London) 2012, 490, 522.(45) Starkov, P.; Moore, J. T.; Duquette, D. C.; Stoltz, B. M.; Marek, I.
J. Am. Chem. Soc. 2017, 139, 9615.(46) Alexy, E. J.; Zhang, H.; Stoltz, B. M. J. Am. Chem. Soc. 2018, 140,
10109.(47) (a) Li, B. X.; Le, D. N.; Mack, K. A.; McClory, A.; Lim, N.-K.;
Cravillion, T.; Savage, S.; Han, C.; Collum, D. B.; Zhang, H.;Gosselin, F. J. Am. Chem. Soc. 2017, 139, 10777. (b) Mack, K. A.;McClory, A.; Zhang, H.; Gosselin, F.; Collum, D. B. J. Am. Chem.Soc. 2017, 139, 12182.
(48) Kuwano, R.; Naoki, I.; Murakami, M. Chem. Commun. 2005,3951.
(49) Maki, K.; Kanai, M.; Shibasaki, M. Tetrahedron 2007, 63, 4250.(50) (a) Trost, B. M.; Ariza, X. J. Am. Chem. Soc. 1999, 121, 10727.
(b) Trost, B. M.; Ariza, X. Angew. Chem., Int. Ed. Engl. 1997, 36,2635.
(51) (a) Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2012, 134,5778. (b) Trost, B. M.; Czabaniuk, L. C. Chem.–Eur. J. 2013, 19,15210.
(52) Trost, B. M.; Lee, C. B. J. Am. Chem. Soc. 1998, 120, 6818.(53) Seto, M.; Roizen, J. L.; Stoltz, B. M. Angew. Chem. Int. Ed. 2008, 47,
6873.(54) Estipona, B. I.; Pritchett, B. P.; Craig, R. A.; Stoltz, B. M. Tetrahe-
dron 2016, 72, 3707.(55) Behenna, D. C.; Liu, Y.; Yurino, T.; Kim, J.; White, D. E.; Virgil, S.
C.; Stoltz, B. M. Nat. Chem. 2012, 4, 130.(56) Numajiri, Y.; Jiménez-Osés, G.; Wang, B.; Houk, K. N.; Stoltz, B.
M. Org. Lett. 2015, 17, 1082.(57) Alexy, E. J.; Virgil, S. C.; Bartberger, M. D.; Stoltz, B. M. Org. Lett.
2017, 19, 5007.(58) Hartwig, J. F.; Stanley, L. Copper-Catalyzed Allylic Substitution, In
Organotransition Metal Chemistry: From Bonding to Catalysis;Hartwig, J. F., Ed.; University Science Books: Mill Valley, 2010,999–1008.
(59) Trost, B. M.; Dogra, K. J. Am. Chem. Soc. 2002, 124, 7256.(60) (a) Trost, B. M.; Zhang, Y. J. Am. Chem. Soc. 2006, 128, 4590.
(b) Trost, B. M.; Zhang, Y. J. Am. Chem. Soc. 2007, 129, 14548.(61) Trost, B. M.; Dogra, K.; Franzini, M. J. Am. Chem. Soc. 2004, 126,
1944.(62) Trost, B. M.; Miller, J. R.; Hoffman, C. M. Jr. J. Am. Chem. Soc.
2011, 133, 8165.(63) Kanayama, T.; Yoshida, D.; Miyabe, H.; Takemoto, K. Angew.
Chem. Int. Ed. 2003, 42, 2054.(64) Hethcox, J. C.; Shockley, S. E.; Stoltz, B. M. ACS Catal. 2016, 6,
6207.(65) Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science
(Washington, D. C.) 2013, 340, 1065.
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30

30
B. M. Trost, J. E. Schultz ReviewSyn thesis
(66) Shockley, S. E.; Hethcox, J. C.; Stoltz, B. M. Angew. Chem. Int. Ed.2017, 56, 11545.
(67) Evans, P. A.; Oliver, S.; Chae, J. J. Am. Chem. Soc. 2012, 134,19314.
(68) Evans, P. A.; Oliver, S. Org. Lett. 2013, 15, 5626.
(69) Turnbull, B. W. H.; Oliver, S.; Evans, P. A. J. Am. Chem. Soc. 2015,137, 15374.
(70) Turnbull, B. W. H.; Evans, P. A. J. Am. Chem. Soc. 2015, 137, 6156.(71) Wright, T. B.; Evans, P. A. J. Am. Chem. Soc. 2016, 138, 15303.
Georg Thieme Verlag Stuttgart · New York — Synthesis 2019, 51, 1–30