ox26 modified hyperbranched polyglycerol-conjugated poly(lactic-co-glycolic acid) nanoparticles:...
TRANSCRIPT

OX26 modified hyperbranched polyglycerol-conjugatedpoly(lactic-co-glycolic acid) nanoparticles: synthesis,characterization and evaluation of its brain delivery ability
Hanmei Bao • Xu Jin • Ling Li • Feng Lv •
Tianjun Liu
Received: 18 February 2012 / Accepted: 21 April 2012 / Published online: 9 May 2012
� Springer Science+Business Media, LLC 2012
Abstract A novel nanoparticles-based brain drug delivery
system made of hyperbranched polyglycerol-conjugated
poly(lactic-co-glycolic acid) which was surface func-
tionalized with transferrin antibody (OX26) was prepared.
Hyperbranched polyglycerol-conjugated poly(lactic-co-
glycolic acid) was synthesized, characterized and applied to
prepare nanoparticles by means of double emulsion solvent
evaporation technique. Transmission electron micrograph
and dynamic light scattering showed that nanoparticles
had a round and regular shape with a mean diameter of
170 ± 20 nm. Surface chemical composition was detected
by X-ray photoelectron spectroscopy. Endomorphins, as a
model drug, was encapsulated in the nanoparticles. In vitro
drug release study showed that endomorphins was released
continuously for 72 h. Cellular uptake study showed that the
uptake of nanoparticles by the brain microvascular endo-
thelial cells was both time- and concentration-dependant.
Further uptake inhibition study indicated that the uptake of
nanoparticles was via a caveolae-mediated endocytic
pathway. In vivo endomorphins brain delivery ability was
evaluated based upon the rat model of chronic constriction
injury of sciatic nerve. OX26 modified nanoparticles had
achieved better analgesic effects, compared with other
groups. Thus, OX26 modified hyperbranched polyglycerol-
conjugated poly(lactic-co-glycolic acid) nanoparticles may
be a promising brain drug delivery carrier.
1 Introduction
The blood–brain barrier (BBB), formed by brain micro-
vascular endothelial cells (BMECs) sealed with tight
junctions, protects the brain from harmful substances and
fluctuations in blood [1–3]. The unique property of BBB
restricts various diagnostic and therapeutic agents to enter
the central nervous system (CNS) via the systemic route [4,
5]. The BBB allows only highly lipid-soluble molecules
under a threshold of 400–600 Da to penetrate [6, 7].
Approximately 100 % of large-molecular-weight drugs
including peptides, recombinant proteins, monoclonal
antibodies, RNA interference-based drugs and gene thera-
pies, and more than 98 % of small-molecular-weight drugs
do not cross the BBB [8]. Therefore, developing drug
delivery strategies across the BBB is important for the
diagnostic and therapeutic purposes.
Studies have demonstrated that the routes across the
BBB include paracellular aqueous pathway, transcellular
lipophilic pathway, transport proteins, receptor-mediated
transcytosis (RMT), or adsorptive-mediated transcytosis
[9]. With advantages of brain targeting, high incorporation
capacity, reduction of side effects and circumvention of the
multidrug efflux system, RMT seems to be one of the most
promising strategies [10]. Transferrin receptor (TfR),
which is highly concentrated in brain capillary endothe-
lium, may trigger RMT across the BBB via transcytosis
H. Bao � L. Li � F. Lv � T. Liu (&)
Institute of Biomedical Engineering, Chinese Academy of
Medical Sciences & Peking Union Medical College, Tianjin Key
Laboratory of Biomaterial Research, Tianjin 300192,
People’s Republic of China
e-mail: [email protected]
X. Jin
Department of Anesthesia, Beijing Tiantan Hospital, Capital
University of Medical Sciences, Beijing 100050,
People’s Republic of China
123
J Mater Sci: Mater Med (2012) 23:1891–1901
DOI 10.1007/s10856-012-4658-7

[11]. Transferrin or the antibodies against the TfR (for
example, OX26 monoclonal antibody) have been investi-
gated in a number of studies [12–14]. OX26 has been
used in the RMT system for the delivery of peptides
[15], liposome-containing digoxin [16] and nanoparticles
(NPs)-containing loperamide [17] across the BBB, which
manifest the extraordinary capacity of the receptor-medi-
ated routes. New drug carriers with better vesicle properties
are necessary for the construction of the RMT system with
more rational and effective brain delivery property. Among
the various drug containers, nanoparticulate carriers and
particularly polymeric NPs seem to be one of the most
interesting strategies [18]. Drugs can be loaded into the
NPs, adsorbed or chemically linked to their surface. What
is more, these carriers possess a higher stability in bio-
logical fluids and against the enzymatic metabolism than
other colloidal carriers, such as the liposomes or lipidic
vesicles [13, 19, 20].
NP vectors formulated with poly(lactic-co-glycolic
acid) (PLGA) for vaccines, peptides, proteins and mac-
romolecules have attracted much attention over the last
decades [21–23]. However, small amounts of moieties for
surface modification on PLGA makes it difficult for
antibodies to conjugate onto, which restricts the applica-
tion of PLGA in the RMT system. Hyperbranched poly-
glycerol (HPG), a polymer consisting of an inert polyether
backbone with functional hydroxyl groups at every
branch-end, is ready to be chemically modified. The end
groups of HPG are not altogether located at the same
distance from the core. Partial modification of HPG with
apolar groups, for example, esterification, etherification, or
transketalization, offers promising options for the prepa-
ration of amphiphilic core–shell structures, which can be
used as a vehicle for controlled drug release [24–26]. Up
to now, few reports of NPs combining advantages of both
PLGA and HPG as brain drug delivery vectors have been
published.
Here a novel brain targeting nanocarrier system was
prepared by modification the surface of NPs composed of
HPG-conjugated PLGA (HPG–PLGA) with OX26 (OX26–
HPG–PLGA). The copolymer of HPG–PLGA was syn-
thesized by esterification method and the NPs were pre-
pared by double emulsion solvent evaporation technique
[27]. The conjugation of OX26 onto the surface of NPs was
confirmed by X-ray photoelectron spectroscopy (XPS)
analysis. Endomorphins (EM) is an analgesic drug. Its main
target is located in the CNS and the presence of BBB
restricts its analgesic effect by peripheral administration
[28]. In this paper, EM, chosen as a model drug, was
encapsulated into the OX26–HPG–PLGA NPs to demon-
strate the brain delivery ability of OX26–HPG–PLGA NPs.
The analgesic effect was evaluated upon chronic constric-
tion injury (CCI) rats.
2 Experimental section
2.1 Materials
PLGA 75/25 was bought from Chinese Academy of Sci-
ence (China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodi-
imide (EDC), OX26, EM and N-hydroxysuccinimide
(NHS) were obtained from Sigma (USA). All other
chemicals and organic solvents were of analytical grade
and purchased from Tianjin Jiangtian Chemical Engineer-
ing Co. Ltd. (China). N,N-Dimethylformamide (DMF) was
dried over anhydrous magnesium sulphate for 2 days and
later with phosphoric anhydride overnight. After drying,
DMF was distilled under reduced pressure of nitrogen.
Other reagents were used without purification.
2.2 Polymerization and characterization of HPG
HPG was synthesized according to the literature [29] and
can be roughly described as follows. Polymerizations were
carried out in a reactor equipped with a mechanical stirrer
and dosing pump under nitrogen atmosphere. 1,1,1-
Tris(hydroxymethyl)propane (0.4 g) was partially depro-
tonated (10 %) with potassium methylate solution
(3.7 mol/l in methanol, 0.5 ml) by distilling off excess
methanol from the melt. A 50 ml aliquot of glycidol (17 g
in tetrahydrofuran) was slowly added at 95 �C over 12 h.
After completion of the reaction (absence of excess epox-
ide), the product was dissolved in methanol and neutralized
by filtration over cation-exchange resin. The polymer was
twice precipitated from methanol solution into acetone and
subsequently dried for 15 h at 80 �C in vacuo. Gel per-
meation chromatography (GPC, 1525–2414 Waters, USA)
was used for the determination of the polydispersity index.
The weight-average (Mw) and number-average (Mn)
molecular weight data were expressed with respect to
polystyrene standards. Carbon nuclear magnetic resonance
(13C NMR) spectrum was recorded on a Varina Mercury
spectrometer, operating at 75.4 MHz.
2.3 Synthesis and characterization of copolymer
HPG–PLGA
The copolymer of HPG–PLGA was synthesized by esteri-
fication. Briefly, HPG was completely dissolved in anhy-
drous DMF and a suitable amount of PLGA was added.
EDC and NHS were acted as catalysts. After reaction for
48 h at room temperature, the solution was evaporated and
was precipitated in distilled water three times to remove
the un-reacted HPG. The resultant products were lyophi-
lized for 2 days and then stored at a temperature of 4 �C.
The product was characterized by Fourier transform
infrared spectroscopy (FTIR, FTS 3000 Bio-Rad, USA)
1892 J Mater Sci: Mater Med (2012) 23:1891–1901
123
and hydrogen proton nuclear magnetic resonance spec-
troscopy (1H NMR, Varina Mercury, USA). The water
contact angle of the copolymer was analyzed at room
temperature on a DSA10 contact angle measuring system
from Kruss.
2.4 Formulation of EM-loaded HPG–PLGA
(EM–HPG–PLGA) NPs
NPs were prepared using a water-in-oil-in-water (w/o/w)
emulsion solvent evaporation technique [28]. In a typical
procedure, 90 mg HPG–PLGA copolymer was dissolved in
3 ml of dichloromethane (DCM). An aqueous solution of
EM (300 ll, 10 % w/v) was added into the HPG–PLGA
solution, followed by sonication (100 W, 1 min) to obtain
a water-in-oil emulsion. It was then added into 12 ml of
polyvinyl alcohol solution (2 %, w/v), and was sonicated
on an ice bath for 3 min to obtain the multiple w/o/w
emulsion. The w/o/w emulsion was stirred overnight at
room temperature to allow the evaporation of DCM and
formation of EM–HPG–PLGA NPs. The samples were
then washed 3 times by centrifugation (1,000 r/min),
lyophilized, and stored at -20 �C.
2.5 Preparation and characterization of OX26 modified
EM–HPG–PLGA (OX26–EM–HPG–PLGA) NPs
HPG–PLGA NPs were completely dissolved in 0.1 mol/l
phosphate buffered saline (PBS). Then, EDC was added,
followed by OX26 (1 ml, 100 lg/ml). After reaction in the
dark at room temperature for 2 h, the reaction mixture was
applied to a 1.5 9 20 cm Sepharose CL-4B column and
then eluted with PBS (0.01 mol/l), in order to remove the
un-conjugated OX26. NPs were analyzed to determine the
surface composition of C, O and N with XPS. The XPS
analysis was carried out on a PHI-5300 ESCA system
(Perkin Elmer, USA). The particle size and morphology of
the NPs were characterized by transmission electron
microscopy (TEM, JEM-100C VII, Japan) at an acceler-
ating voltage of 100 kV. Zeta-potential of the NPs were
measured with dynamic light scattering (DLS, ZEN3600,
UK).
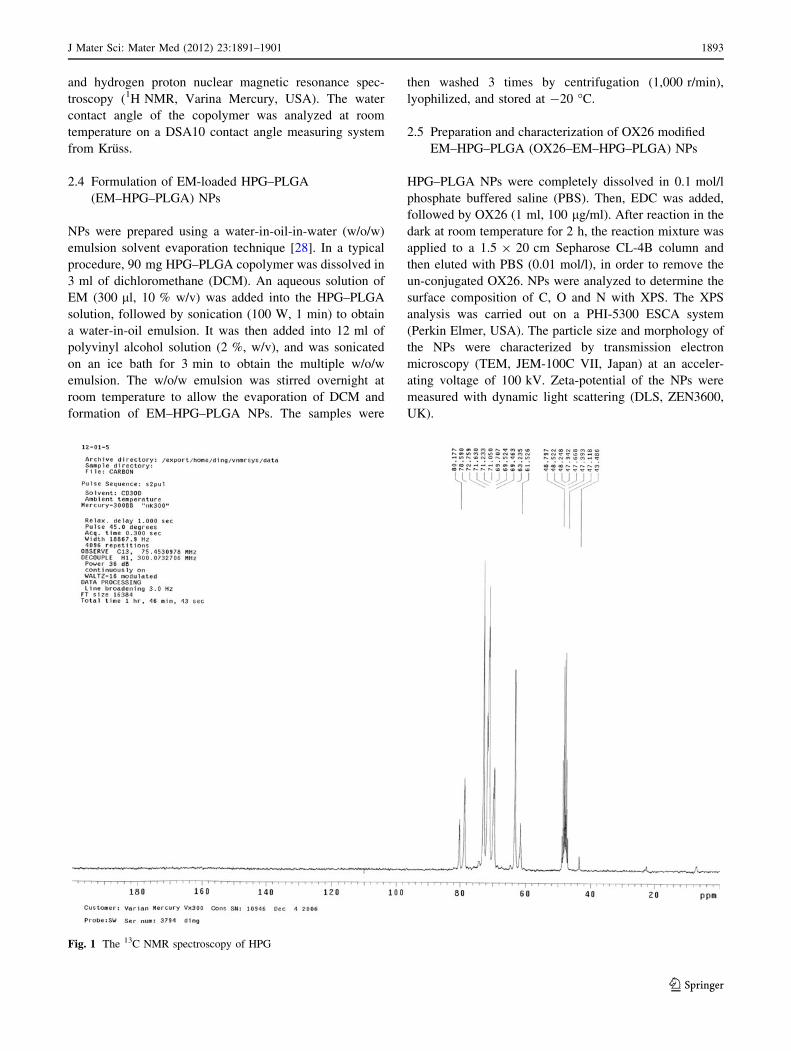
Fig. 1 The 13C NMR spectroscopy of HPG
J Mater Sci: Mater Med (2012) 23:1891–1901 1893
123

2.6 Drug release behavior in vitro
EM release behavior in vitro was studied by incubating
solution of NPs in a dialysis bag immersed in PBS, which
was placed in an air bath shaker at 150 rpm at
37 ± 0.5 �C. At certain time intervals, samples were col-
lected from the medium and the same volume of fresh
release medium was added. The amount of released EM
was determined by high performance liquid chromatogra-
phy (HPLC) method. To determine the drug loading
capacity (DLC) and encapsulation efficiency (EE), 5 mg
NPs were dissolved in 5 ml DCM and then 5 ml deionized
water was added. The mixture was stirred for 6 h and then
1 ml of water phase was taken to measure the drug con-
centration by HPLC method.
2.7 In vitro cellular uptake of NPs
BMECs culture was performed according to the literature
[30]. Briefly, BMECs were isolated and characterized as
described previously [31]. Cells were cultured in the
presence of Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10 % fetal bovine serum
(FBS), penicillin (100 U/ml) and streptomycin (100 mg/ml).
The time- and concentration-dependent internalization of
NPs by BMECs was investigated. BMECs were seeded
onto 24-well plates at a density of 104 cells/cm2. On the
second day, pretreated with Hank’s balanced salt solution
(HBSS) for 30 min, the medium was replaced with the
suspension of fluorescein isothiocyanate (FITC) labeled
OX26–HPG–PLGA (*O) NPs with concentrations of 100,
300, 600 and 900 lg/ml for 0.5, 1, 3 h at 37 �C, respec-
tively. For uptake inhibition experiment, The BMECs were
first pretreated with inhibitor filipin (1 mg/ml) for 15 min
at 37 �C in humified atmosphere with 5 % CO2. Then the
cells were washed with Ringer-Hepes buffer (RH) and
Fig. 2 The IR spectroscopy of HPG (a) and HPG–PLGA (b)
Scheme 1 Schematic synthesis of HPG and copolymer HPG–PLGA
1894 J Mater Sci: Mater Med (2012) 23:1891–1901
123

different concentrations of *O NPs were added. After dis-
carding the incubation medium, the cells were washed twice
with ice-cold PBS (pH 7.4). Incorporation of FITC-labeled
NPs was allowed for quantitative estimation of the cellular
uptake of NPs by flow cytometry (BD caliber, USA).
2.8 EM brain delivery efficiency in vivo
Male Sprague–Dawley rats (clean grade), weighing
180–200 g, purchased from Institute of Laboratory Animal
Sciences, Peking Union Medical College (Beijing, China),
were used in accordance with protocols approved by the
Animal Care Committee of Beijing Tiantan Hospital.
The surgery to produce CCI was performed according to
the literature [32]. Briefly, under sodium pentobarbital
anesthesia (1.5 %, 0.2 ml/100 g, i.p.), the right sciatic
nerve was exposed proximally to the trifurcation, approx-
imately 7 mm of the common sciatic nerve was freed from
underlying connective tissues. Four ligations with intervals
about 1 mm were placed around the nerve with 4–0 chro-
mic gut suture. A typical switch of the hind paw was seen
when the nerve was constricted. The surgical incision was
sutured with the same silk. After CCI model had been
successfully established, rats were divided into four groups
(n = 6): group C with intravenous (i.v.) injection of HPG–
PLGA NPs; group E with EM (100 lg/kg); group EP with
EM–HPG–PLGA NPs (EM loading, 100 lg/kg) and group
OEP with OX26–EM–HPG–PLGA NPs (EM loading,
100 lg/kg). Mechanical withdrawal threshold (MWT) was
determined by up-down method [33] before injection, and
15, 30, 60, 120 min after injection, respectively.
2.9 Statistical analysis
Statistical analysis was performed with Excel 2003 and
SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). All
values were expressed as the mean ± standard deviation.
Statistical significance was calculated using the t-test
(within groups) or one-way analysis of variance (between
groups). A value of P \ 0.05 was considered to be statis-
tically significant.
3 Results and discussion
3.1 Characterization of HPG
HPG was obtained as a transparent, highly viscous liquid.
The yield of the isolated polymer was 85.3 %. GPC result
showed that the polydispersity of HPG was 1.45 (Mw/Mn).
The 13C NMR (CD3OD-d4) analysis of HPG (Fig. 1) was
Fig. 3 The 1H NMR spectroscopy of HPG (a) and HPG–PLGA (b)
Fig. 4 The water contact angle of HPG (a), PLGA (b) and HPG–PLGA (c)
J Mater Sci: Mater Med (2012) 23:1891–1901 1895
123

as follows: (i) linear 1,3-unit (L13): CH2OH carbon
at 61.4 ppm, CH2 carbon at 69.3 ppm, and CH carbon at
80.4 ppm; (ii) linear 1,4-unit (L14): both CH2 carbons at
73.2 ppm, CHOH carbon at 69.3 ppm; (iii) terminal unit
(T): CH2OH carbon at 62.9 ppm, CHOH carbon at
71.7 ppm, and the CH2 carbon at about 71.8 ppm; (iv)
dendritic unit (D): CH carbon at 78.8 ppm, one CH2 carbon
at 72.0 ppm [29]. The absorption peak at 48.8 ppm was
attributed to the solvent (CD3OD-d4). The relative abun-
dance of each structural unit and the degree of branching
(DB) were calculated according to the literature [34].
Results showed that the DB of HPG was 64.07 %.
3.2 Characterization of copolymer HPG–PLGA
HPG, a flexible water-soluble molecule with rich hydroxyl
groups at every branch-end, can be copolymerization with
other polymers via end-functionalization. Synthetic
approach of copolymer HPG–PLGA is signified in
Scheme 1. PLGA moieties were grafted onto HPG
molecular by esterification of terminal carboxylic groups.
Figure 2 shows the IR spectra of HPG (a) and HPG–PLGA
(b), respectively. The peak assignment of HPG (Fig. 2a) is
as follows: strongest absorption peak at 3,396 cm-1
assigned as O–H bond stretch, overlapped with each other;
2,914 and 2,876 cm-1 as stretch movement of C–H bond,
1,475–1,300 cm-1 as bend of C–H bond; 1,045 cm-1 as
stretch of C–O bond. Compared with HPG, the absorption
intensity of HPG–PLGA (Fig. 2b) at 3,396 cm-1 (O–H
stretch) observably decreased. Moreover, the peak at
1,759 cm-1 assigned as C=O stretch appeared.1H NMR spectra of HPG (a) and HPG–PLGA (b) are
shown in Fig. 3. The proton assignment of HPG (Fig. 3a) is
as follows (ppm): peaks at 3.0–4.0 (CH2–O, CH–O),
4.4–4.9 (OH). In the 1H NMR spectrum of HPG–PLGA
(Fig. 3b), besides signals above, the new added character-
istic proton peak at around 1.44 ppm can be attributed to
the proton signal of methyl from PLGA, as evidence of
PLGA graft. Therefore, degree of substitute (DS) could be
calculated by comparing the ratio of methyl of PLGA
protons (1.44 ppm) to HPG protons (4.4–4.9 ppm, OH).
The DS could be calculated as follows:
DS ¼ I1:44=3ð Þ= I 4:4� 4:9ð Þð Þ � 100 %
The result of DS was 29.58 %.
The water contact angle of HPG (a), PLGA (b) and
HPG–PLGA (c) is 43.6�, 92.8� and 81.2�, respectively
(Fig. 4). The water contact angle of HPG is relatively small
for its high degree of OH groups. Whereas for PLGA, the
water contact angle is relatively large as a result of its
hydrophobic carbon. Combination of HPG with PLGA
causes parts of the OH groups of HPG to be substituted by
the hydrophobic moieties of PLGA. As a result, the water
contact angle of HPG–PLGA was smaller than HPG.
3.3 Surface analysis
XPS was carried out to investigate the elemental and
average chemical compositions of the materials on the
surface of NPs. Results in Table 1 showed that nitrogen
was only detected on the OX26–EM–HPG–PLGA NPs,
with the value of 0.4 %. Peak 6 corresponded to the N 1s
envelope at 399.3 eV with a low signal, confirming the
existence of OX26 conjugated onto the surface of EM–
HPG–PLGA NPs (Fig. 5e). There were three peaks pre-
sented to achieve the best fit in the C 1s envelope (Fig. 5a
and b). Peak 1 was attributed to the carbon in C–C or C–H
[35]. Peak 2 corresponded to the carbon of C–O. Peak 3
was generated by the carbon of C=O. Figure 5c, d revealed
the presence of two types of oxygen O=C (peak 4) at
532.0 eV and O–C (peak 5) at 533.5 eV, respectively [36].
Results indicated that OX26 is successfully conjugated
onto EM–HPG–PLGA NPs, thus available for the inter-
action with the BBB.
Table 1 The XPS analysis of PLGA, HPG–PLGA and OX26–HPG–PLGA
Sample XPS elemental ratios C 1s envelope ratios (%) O 1s envelop ratios (%)
C O N C–C/C–H C–O C=O O=C O–C
Binding energy (ev)
284.7 286.6 288.8 531.9 533.3
PLGA 61.6 38.4 – 39.4 30.3 30.3 52.3 47.7
HPG–PLGA 59.0 41.0 – 31.2 35.8 33.0 51.7 48.3
OX26–HPG–PLGA 64.9 34.7 0.4 38.2 34.9 26.9 53.4 46.6
‘‘–’’below the detection limit
Fig. 5 Carbon C 1s envelopes from XPS analysis of: (a) PLGA,
(b) HPG–PLGA. Oxygen O 1s envelope from XPS analysis of:
(c) PLGA, (d) HPG–PLGA. Nitrogen N 1s envelope from XPS
analysis of: (e) OX26–EM–HPG–PLGA
c
1896 J Mater Sci: Mater Med (2012) 23:1891–1901
123

J Mater Sci: Mater Med (2012) 23:1891–1901 1897
123

3.4 Characterization of NPs
Amphiphilic copolymer NPs have been gaining increasing
attention as drug carrier penetrating the BBB [37, 38]. In
this paper, the model medicine EM is a small water soluble
peptide. The w/o/w emulsion solvent evaporation tech-
nique chosen here provided the NPs forming the inner
aqueous phase, which could dissolve hydrophilic drugs,
including small molecules, proteins and gene drugs [39].
Scheme 2 illustrates the formation of NPs surface func-
tionalized with OX26. TEM photograph shows that NPs
are generally spherical and in relatively uniform size, with
average diameters of 170 ± 20 nm (Fig. 6). What’s more,
the hydrophilic core and the membrane stained by phos-
photungstic acid provide the contrast lateral and the
membrane thickness is homogeneous, with a measured
width d of 30 ± 1.2 nm (Fig. 6a). The zeta potential value
of NPs is -27 ± 1.6 mV. The particle size is an important
property that affects its endocytosis by the brain capillary
cells and the favorable size distribution is generally con-
trolled under 200 nm in diameter for NPs [28]. The size of
the NPs we prepared is regarded as favorable to brain
transport. What’s more, the membrane of the prepared NPs
was much thicker than liposomes (3–5 nm), suggesting that
the NPs may be electromechanically tough, have low
permeability and good stability [40].
3.5 Drug release study in vitro
In vitro release behavior of EM from NPs was carried out
under PBS (0.1 mol/l, pH 7.4) at 37 �C. As shown in
Fig. 7, EM appeared to be released in a biphasic way,
which was characterized by a rapid release period followed
by a step of slower release. EM was released
45.86 ± 2.95 % from HPG–PLGA NPs in the first 4 h.
After the fast release stage, EM was released in a contin-
uous way for 72 h, reaching a percentage of cumulative
release to 63.21 ± 1.84 % from NPs. The DLC of EM was
8.65 ± 1.27 % and EE 83.96 ± 2.60 %, respectively.
3.6 Uptake of NPs into BMECs
Cellular uptake experiment in vitro showed that the uptake
of NPs by BMECs was both time- and concentration-
dependant. As shown in Fig. 8, the uptake of NPs increased
as the concentration increased from 100 to 600 lg/ml. No
Scheme 2 Schematic
representation of the formation
of OX26-conjugated NPs with
encapsulation of EM
Fig. 6 TEM images of OX26–EM–HPG–PLGA NPs
1898 J Mater Sci: Mater Med (2012) 23:1891–1901
123

significant difference was seen between 600 and 900 lg/ml.
The uptake amount of *O NPs at a concentration of
300 lg/ml was 82.15 ± 13.17 at 3 h, about 1.5 times
higher than that at 0.5 h. The relative uptake efficiency,
which was calculated by dividing the uptake amount of
NPs with inhibitors by that without inhibitors, was used to
evaluate the inhibitory effect of filipin on NPs uptake. At
each time point, the uptake amount of *O NPs was much
higher than NPs pretreated with filipin (f*O NPs)
(P \ 0.01) for the same concentration, even about two
times higher than that of f*O NPs, which proved the sig-
nificant effect of filipin on NPs uptake. Flow cytometry
images of *O-3 (*O NPs at a concentration of 300 lg/ml)
and f*O-3 NPs (f*O NPs at a concentration of 300 lg/ml)
uptake into BMECs 0.5 and 3 h after incubation were
shown in Fig. 9. There was a high expression of caveolin in
the capillary endothelium. Cellular uptake could be inhib-
ited by filipin, suggesting a caveolae-mediated incorpora-
tion process [41]. However, the precise endocytosis
mechanism of NPs needs further investigation. Results
presented here demonstrated that OX26–HPG–PLGA NPs
we prepared can be an effective carrier for brain drug
delivery.
3.7 Analgesic effects of EM loaded NPs
In order to investigate the brain delivery ability of OX26
conjugated NPs, rat CCI model was constructed, followed
by i.v. injection of different kinds of NPs with EM loading
dose of 100 lg/kg. Prior to drug administration, there was
no significant difference of MWT among total four groups;
after drug administration, for group E and group C, there
was no significant difference of MWT in all of the time-
points (P [ 0.05). At the time point of 45 min, MWT in
group EP (10.3 ± 2.1 g) and group OEP (15.2 ± 4.1 g)
increased dramatically, with P \ 0.05 and P \ 0.01,
respectively. While at other time points, there was no
significant difference of MWT in group EP. As shown in
Fig. 10, MWT in group OEP was 10.1 ± 2.5 g 30 min post
injection, 15.2 ± 4.1 g 45 min post injection, 13.3 ± 3.3 g
60 min post injection and 11.9 ± 1.7 g 90 min post
injection, respectively. It is clearly that group OEP
enhanced MWT within 30–90 min following drug delivery
(P \ 0.05). MWT in group OEP was much higher than in
group EP 30, 45, 60 and 90 min post injection (P \ 0.05),
while no significant difference was seen between the two
groups before and 15 min post injection (P [ 0.05).
Dalargin, a synthetic analogue of leu-enkephalin with
analgesic activity, has been widely used as a model com-
pound for development of brain targeting delivery systems
[42, 43]. Dalargin exhibited no analgesic effect via i.v.
injection as a result of the BBB. The carrier system for
dalargin brain delivery was evidenced by the central
analgesic effect [44]. EM works in the same way as da-
largin, and no analgesic effect can be obtained via i.v.
injection as a result of the BBB. Therefore the successfully
occurrence of the analgesic effect of OX26 conjugated NPs
loaded with EM could be the evidence of passing across the
BBB. In the animal experiment, i.v. injection of EM yields
no effects, suggesting that the drug cannot penetrate into
CNS. Intravenous injection of EM–HPG–PLGA NPs
without OX26 conjugation (group EP) had some analgesic
effect, indicating that solely NPs could carry drugs through
the BBB as a result of their small sizes, which was
accompanied by passive diffusion or convection [45]. The
structure of HPG was similar to the surfactant and the
interactions between HPG and lipoids of BMECs may
Fig. 7 The in vitro release of EM from HPG-PLGA NPs under
0.1 mol/l PBS buffer at 37 �C of pH 7.4
Fig. 8 Time- and concentration-dependent uptake of NPs by
BMECs. Data are mean ± SD, n = 3. *O-1,*O-3, *O-6, *O-9: *O
NPs with concentrations of 100, 300, 600 and 900 lg/ml, respec-
tively. f*O-1, f*O-3, f*O-6, f*O-9: f*O NPs with concentrations of
100, 300, 600 and 900 lg/ml, respectively
J Mater Sci: Mater Med (2012) 23:1891–1901 1899
123

facilitate the NPs to pass across the BBB. However,
compared with OX26 conjugated NPs (group OEP), the
analgesic effect of group EP appeared late and weak,
suggesting that the ‘‘scale effect’’ and the surfactant effect
of NPs could not fully achieve the purpose. NPs surface
manipulation by OX26 was performed to increase cell
uptake and the potential delivery of the NPs in different
cell compartments. Furthermore, the drug delivery system
constituted by HPG–PLGA releasing EM for a long period
of time in a controlled manner increased its analgesic
efficacy.
4 Conclusions
In this paper, a novel brain drug delivery system was
constructed by conjugation of biodegradable HPG–PLGA
NPs with OX26, a promising brain targeting molecule. The
copolymer of HPG–PLGA was synthesized and applied for
the preparation of NPs by double emulsion solvent evap-
oration technique. The uptake of NPs by BMECs was both
time- and concentration-dependant. Further uptake inhibi-
tion experiment indicated a caveolae-mediated endocytic
process. With EM acted as a model medicine, the intra-
venous injection of OX26–EM–HPG–PLGA NPs showed
efficient analgesic effect in the rat CCI model, which
demonstrated OX26 modified HPG–PLGA NPs would be a
promising brain drug delivery carrier for therapeutic pep-
tides and proteins access to CNS.
Acknowledgments This study was financially supported by China
National Natural Scientific Found (30700780), Beijing Natural Sci-
entific Found (7102052) and Beijing Nova Program (2008A083).
References
1. Begley DJ, Brightman MW. Structural and functional aspects of
the blood–brain barrier. Prog Drug Res. 2003;61:39–78.
2. Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV,
Achim CL, et al. Blood–brain barrier tight junction disruption in
human immunodeficiency virus-1 encephalitis. Am J Pathol.
1999;155:1915–27.
3. Wolburg H, Lippoldt A. Tight junctions of the blood–brain bar-
rier: development, composition and regulation. Vasc Pharmacol.
2002;38:323–37.
4. Abbott NJ. Prediction of blood–brain barrier permeation in drug
discovery from in vivo, in vitro and in silico models. Drug Discov
Today Technol. 2004;1:407–16.
5. Teichberg VI. From the liver to the brain across the blood–brain
barrier. Proc Natl Acad Sci USA. 2007;104:7315–6.
6. Pardridge WM. The blood–brain barrier: bottleneck in brain drug
development. NeuroRx. 2005;2:3–14.
7. Levin VA. Relationship of octanol/water partition coefficient and
molecular weight to rat brain capillary permeability. J Med
Chem. 1980;23:682–4.
Fig. 9 Flow cytometry images of *O-3 (a, c) and f*O-3 (b, d) NPs uptaken by BMECs after 0.5 h (a, b) and 3 h (c, d) of incubation,
respectively
Fig. 10 Comparison of MWT (g) among four groups at different
time points. Compared to pre-injection group, *P \ 0.05 and
**P \ 0.01, respectively. Compared to EP group, #P \ 0.05
1900 J Mater Sci: Mater Med (2012) 23:1891–1901
123

8. Pardridge WM. Drug and gene delivery to the brain: the vascular
route. Neuron. 2002;36:555–8.
9. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial
interactions at the blood–brain barrier. Nat Rev Neurosci.
2006;7:41–53.
10. Calvo P, Gouritin B, Chacun H, Georgin D, Fattal E, Couvreur P,
et al. Long-circulating PEGylated polycyanoacrylate nanoparti-
cles as new drug carrier for brain delivery. Pharm Res. 2001;18:
1157–66.
11. Manoj R, Tracey B, Jennifer L, Ayman EK, Joanne B. Current
advances in delivery of biotherapeutics across the blood–brain
barrier. Curr Drug Discov Technol. 2011;8:87–101.
12. Kuo YC, Lin PI, Wang CC. Targeting nevirapine delivery across
human brain microvascular endothelial cells using transferrin-
grafted poly(lactide-co-glycolide) nanoparticles. Nanomedicine.
2011;6:1011–26.
13. Huwyler J, Wu DF, Pardridge WM. Brain drug delivery of small
molecules using immunoliposomes. Proc Natl Acad Sci USA.
1996;24:14164–9.
14. Zhang Y, Calon F, Zhu C, Boado RJ, Pardridge WM. Intravenous
nonviral gene therapy causes normalization of striatal tyrosine
hydroxylase and reversal of motor impairment in experimental
parkinsonism. Hum Gene Ther. 2003;14:1–12.
15. Karatas H, Aktas Y, Bodur E, Yemisci M, Caban S, Vural A,
et al. A nanomedicine transports a peptide caspase-3 inhibitor
across the blood–brain barrier and provides neuroprotection.
J Neurosci. 2009;29:13761–9.
16. Deeken JF, Loscher W. The blood–brain barrier and cancer:
transporters, treatment, and trojan horses. Clin Cancer Res.
2007;13:1663–74.
17. Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin- and
transferrin-receptor-antibody-modified nanoparticles enable drug
delivery across the blood–brain barrier (BBB). Eur J Pharm Bi-
opharm. 2009;71:251–6.
18. Chang J, Jallouli Y, Kroubi M, Yuan XB, Feng W, Betbeder D,
et al. Characterization of endocytosis of transferrin-coated PLGA
nanoparticles by the blood–brain barrier. Int J Pharmaceut.
2009;379:285–92.
19. Perez C, Sanchez A, Putnam D, Ting D, Langer R, Alonso MJ.
Poly(lactic acid)–poly(ethylene glycol) nanoparticles as new
carriers for the delivery of plasmid DNA. J Control Release.
2001;75:211–24.
20. Li Y, Ogris M, Wagner E, Pelisek J, Ruffer M. Nanoparticles
bearing polyethyleneglycol-coupled transferrin as gene carriers:
preparation and in vitro evaluation. Int J Pharmaceut. 2003;
259:93–101.
21. Betancourt T, Shah K, Brannon-Peppas L. Rhodamine-loaded
poly(lactic-co-glycolic acid) nanoparticles for investigation of in
vitro interactions with breast cancer cells. J Mater Sci Mater Med.
2009;20:387–95.
22. Yang H, Li K, Liu YY, Liu ZH, Miyoshi H. Poly(D,L-lactide-co-
glycolide) nanoparticles encapsulated fluorescent isothiocyanate
and paclitaxol: preparation, release kinetics and anticancer effect.
J Nanosci Nanotechnol. 2009;9:282–7.
23. Costantino L, Gandolfi F, Bossy-Nobs L, Tosi G, Gurny R, Rivasi
F, et al. Nanoparticulate drug carriers based on hybrid poly(D,L-
lactide-co-glycolide)-dendron structures. Biomaterials. 2006;27:
4635–45.
24. Sunder A, Kramer M, Hanselmann R, Mulhaupt R, Frey H.
Molecular nanocapsules based on amphiphilic hyperbranched
polyglycerols. Angew Chem Int Edit. 1999;38:3552–5.
25. Kainthan RK, Brooks DE. Comparison of hyperbranched and
linear polyglycidol unimolecular reverse micelles as nanoreactors
and nanocapsules. Macromol Rapid Commun. 2005;26:155–9.
26. Wilms D, Stiriba SE, Frey H. Hyperbranched polyglycerols: from
the controlled synthesis of biocompatible polyether polyols to
multipurpose applications. Accounts Chem Res. 2010;43:129–41.
27. Olivier JC, Huertas R, Lee HJ, Calon F, Pardridge WM. Synthe-
sis of pegylated immunonanoparticles. Pharm Res. 2002;19:
1137–43.
28. Tomboly C, Peter A, Toth G. In vitro quantitative study of the
degradation of endomorphins. Peptides. 2002;23:1573–80.
29. Sunder A, Hanselmann R, Frey H, Mulhaupt R. Controlled syn-
thesis of hyperbranched polyglycerols by ring-opening multi-
branching polymerization. Macromolecules. 1999;32:4240–6.
30. Dehouck MP, Jolliet-Riant P, Bree F, Fruchart JC, Cecchelli R,
Tillement JP. Drug transfer across the blood–brain barrier: cor-
relation between in vitro and in vivo models. J Neurochem.
1992;58:1790–7.
31. Meresse S, Dehouck MP, Delorme P, Bensaid M, Tauber JP,
Delbart C, et al. Bovine brain endothelial cells express tight
junctions and monoamine oxidase activity in long-term culture.
J Neurochem. 1989;53:1363–71.
32. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that
produces disorders of pain sensation like those seen in man. Pain.
1988;33:87–107.
33. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL.
Quantitative assessment of tactile allodynia in the rat paw.
J Neurosci Meth. 1994;53:55–63.
34. Holter D, Burgath A, Frey H. Degree of branching in hyper-
branched polymers. Acta Polym. 1997;48:30–5.
35. Scholes PD, Coombes AGA, Illum L, Davis SS, Watts JF, Davies
MC, et al. Detection and determination of surface levels of pol-
oxamer and PVA surfactant on biodegradable nanospheres using
SSIMS and XPS. J Control Release. 1999;59:261–78.
36. Quellec P, Gref R, Dellacherie E, Sommer F, Tran MD, Alonso
MJ. Protein encapsulation within poly(ethylene glycol)-coated
nanospheres. II. Controlled release properties. J Biomed Mater
Res. 1999;47:388–95.
37. Gan CW, Feng SS. Transferrin-conjugated nanoparticles of
poly(lactide)-D-a-tocopheryl polyethylene glycol succinate di-
block copolymer for targeted drug delivery across the blood–
brain barrier. Biomaterials. 2010;31:7748–57.
38. Baratchi S, Kanwar RK, Ashok C, Hittu M, Parratt A, Sahoo SK,
et al. Promises of nanotechnology for drug delivery to brain in
neurodegenerative diseases. Curr Nanosci. 2009;5:15–25.
39. Prabha S, Labhasetwar V. Critical determinants in PLGA/PLA
nanoparticle-mediated gene expression. Pharm Res. 2004;21:
354–64.
40. Discher BM, Won YY, Ege DS, Bates FS, Discher DE, Hammer
DA, et al. Polymersomes: tough vesicles made from diblock
copolymers. Science. 1999;284:1143–6.
41. Thole M, Nobmann S, Huwyler J, Bartmann A, Fricker G. Uptake
of cationized albumin coupled liposomes by cultured porcine
brain microvessel endothelial cells and intact brain capillaries.
J Drug Target. 2002;10:337–44.
42. Schroder U, Sabel BA. Nanoparticles, a drug carrier system to
pass the blood–brain barrier, permit central analgesic effects of
i.v. dalargin injections. Brain Res. 1996;710:121–4.
43. Das D, Lin S. Double-coated poly(butylcynanoacrylate) nanop-
articulate delivery systems for brain targeting of dalargin via oral
administration. J Pharm Sci. 2005;94:1343–53.
44. Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. Passage of
peptides through the blood–brain barrier with colloidal polymer
particles (nanoparticles). Brain Res. 1995;674:171–4.
45. Juillerat-Jeanneret L. The targeted delivery of cancer drugs across
the blood–brain barrier: chemical modifications of drugs or drug-
nanoparticles? Drug Discov Today. 2008;13:1099–106.
J Mater Sci: Mater Med (2012) 23:1891–1901 1901
123