organocatalytic synthesis of optically active organophosphorus compounds
TRANSCRIPT

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
MICROREVIEW
DOI: 10.1002/ejoc.201403184
Organocatalytic Synthesis of Optically Active Organophosphorus Compounds
Marek Dziegielewski,[a] Jakub Pieta,[a] Elzbieta Kaminska,[a] and Łukasz Albrecht*[a]
Keywords: Organocatalysis / Asymmetric synthesis / Organophosphorus reagents / Phosphonates / Phosphorylation
Owing to their wide reactivity profile, as well as a broadrange of biological activity, organophosphorus compoundsplay a pivotal role in modern organic chemistry. Recently,much effort has been devoted to their preparation in anenantioselective fashion. This microreview summarizes theuse of chiral organic molecules as catalysts in the synthesis
1. IntroductionModern organophosphorus chemistry is an exciting and
widely explored field of research.[1] The milestone discover-ies of excellent chemists such as Arbuzov, Wittig, Horner,Wadsworth, Emmons, Kabachnik, or Fields revealed theenormous potential of organophosphorus chemistry and itsunderlying principles, making it readily applicable in everyorganic chemistry laboratory all over the world.[2] Nowa-days, thanks to their reliability, accessibility, and generality,organophosphorus reagents play a pivotal role in contem-porary organic chemistry and are readily employed by syn-thetic chemists. In addition to fascinating reactivity profilesoffered by organophosphorus reagents, important aspectsof their chemistry also relate to the natural occurrence ofmany organophosphorus compounds and their intriguingbiological activity.[3] As such, organophosphorus com-pounds have become important synthetic targets in modernorganic synthesis.
Among the multitude of possible applications of organo-phosphorus compounds in contemporary organic chemis-try, one trend is particularly worth noting. They are moreand more commonly applied in asymmetric synthesis, ful-filling the increasing demand for optically active com-pounds. The research activity in this field can be dividedinto two main areas. Firstly, organophosphorus compoundscan serve as chiral catalysts or ligands, offering an efficientmeans for the introduction of chirality into target mol-ecules. Intensive studies relating to this aspect of organo-phosphorus chemistry have resulted in many exciting find-
ings summarized recently in various excellent review arti-
[a] Institute of Organic Chemistry, Faculty of Chemistry, ŁódzUniversity of Technology,Zeromskiego 116, 90-924 Łódz, PolandE-mail: [email protected]
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1
of optically active organophosphorus compounds. Particularattention is given to the catalytic activation modes employedin the devised synthetic strategies, with special emphasis onfactors governing stereochemical reaction outcomes.Furthermore, the importance of the products obtained andtheir selected applications are discussed.
cles.[4] Secondly, the unique electronic and structural char-acteristics of organophosphorus reagents, resulting in theirbroad and diverse reactivity, makes them particularly well-suited for application as reagents in asymmetric synthesis.In such a manner, access to enantiomerically enriched prod-ucts that can either serve as key synthetic intermediates orare in themselves biologically interesting targets is possible.In this respect, synthesis of naturally occurring organo-phosphorus compounds or their analogues is of particularimportance.[3d,3e]
Of the different procedures for obtaining enantio-merically enriched products, catalytic methods are the mostdesirable, due to their economic and ecological advan-tages.[5] In particular, the application of simple chiral or-ganic molecules (very often referred to as an organocata-lysis) as catalysts for various stereodifferentiating reactionshas been receiving increasing attention in recent years.[6] Asa result, organocatalysis has become a general and reliabletool in modern asymmetric synthesis, complementary toclassical metal and enzyme catalysis.
The aim of this microreview is to provide a general over-view and summary of the application of organophosphoruscompounds as reagents in asymmetric organocatalysis.Since 2010, when the last review article dedicated to thistopic was published,[6i] many new and fascinatingapproaches have been developed and disclosed in the litera-ture, and these are a topic of this article. This minireviewcovers the literature published from 2010 to date. Particularattention is given to the catalytic activation modesemployed in the devised synthetic strategies, with specialemphasis on factors governing stereochemical reaction out-comes. Furthermore, the importance and selected applica-tions of the products obtained are discussed. It should benoted that this review covers only examples in which anorganophosphorus reagent is directly involved in a keyenantiodifferentiating step.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
2. Organocatalytic Hydrophosphonylation ofAldehydes and Imines
Hydrophosphonylation of aldehydes or imines consti-tutes one of the most fundamental methods for the prepara-tion of α-hydroxy- or α-amino phosphonates. As such, thisprocess has received considerable attention, and variousenantioselective hydrophosphonylation protocols, includingorganocatalytic methods, have been developed. These stra-tegies are summarized in a few reviews.[6i,7] In this sectiononly recent papers dealing with this topic are outlined andsummarized.
One of the recently disclosed methods revealed thepossibility of employing the simple chincona alkaloidquinine (4a) as a catalyst for the addition of H-phosphonate2a to N-protected isatins 1a (Scheme 1).[8] The developedstrategy enabled various substituents to be introduced onthe nitrogen atom and in the C-5 position of the isatinframework 3. Reactions proceeded in high yields. However,enantioselectivities were at most moderate (up to 67% ee).It is postulated that quinine (4a) acted as a bifunctionalcatalyst, enabling activation of isatins 1a through H-bond-ing interactions with its free hydroxy group and activationof the H-phosphonate 2a through deprotonation.
Marek Dziegielewski was born in Kutno (Poland). He received his Ph.D. in organic chemistry from the University ofŁódz in 2013 under the supervision of Professor Jarosław Lewkowski. Currently he is a postdoctoral fellow at the ŁódzUniversity of Technology, where he is working on discovery of new organocatalysts and asymmetric methodologies inŁukasz Albrecht’s research group.
Jakub Pieta was born in Łódz (Poland). He received his M.Sc. in 2006 and his Ph.D. in 2012 from the Łódz Universityof Technology, both under the supervision of Professor Andrzej Małkiewicz. Primarily his area of interest was modifiednucleosides/tRNA synthesis. In 2012 he joined the group of Professor Marvin Caruthers at the University of Colorado inBoulder (USA), where he focused on modifications of the phosphodiester bond in the DNA backbone. Currently he isworking under supervision of Dr. Albrecht in the field of organocatalysis at the Łódz University of Technology.
Elzbieta Kaminska was born in Zyrardów (Poland). She obtained her M.Sc. degree in organometallic chemistry fromthe Warsaw University of Technology in 2006. Then she joined the research group of Prof. Marek Marczewski, andreceived her Ph.D. in the area of catalysis in 2012. In 2013 she was a postdoctoral researcher at the Łódz University ofTechnology in the Łukasz Albrecht’s research group. Her research focused on new biomimetic catalysts and technologiesin asymmetric synthesis.
Łukasz Albrecht was born in Łódz (Poland). He obtained his M.Sc. degree (2004) and then his Ph.D. (2009) from theŁódz University of Technology, Poland, under the guidance of Professor Henryk Krawczyk. Subsequently, he joinedProfessor Karl Anker Jørgensen’s research group as a postdoctoral fellow working on new applications of asymmetricorganocatalysis. In 2013 he started his independent research activity at the Łódz University of Technology, Poland.
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–02
Scheme 1.
Better results in hydrophosphonylation of simple alde-hydes 5 were achieved when a double H-bonding strategywas employed (Scheme 2).[9] The bifunctional catalyst 7a,enabling independent activation of aldehyde component 5(through double H-bonding interactions) and the H-phosphonate 2a (through deprotonation), turned out to bean effective promoter of this important transformation.Both aliphatic and aromatic aldehydes were successfullyutilized in the reaction. In the case of aromatic aldehydes,no significant influence either of electronic properties of thesubstituents on the aromatic ring or of their position wasobserved. However, when aliphatic aldehydes were em-
Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
ployed, increasing bulkiness of the substituents led to im-proved results.
Scheme 2.
In 2011, Barros and Phillips made a not fully successfulattempt to synthesize optically active tertiary β-chloro α-hydroxy phosphonates 9 by way of highly regioselectivemodified Pudovik reactions between dialkyl or diaryl phos-phites 2 and α-halo ketones 8 (Scheme 3).[10] In this studyan interesting synthetic protocol employing combinationsof quinine (4a) with a stoichiometric racemic base was used.Intensive optimization studies resulted in the developmentof general reaction conditions. However, despite rathergood yields and regioselectivities, the main drawbacks ofthe methodology are related to low enantioselectivities ofthe reaction (�36% ee) as well as to difficulties in purifica-tion of the desired products 9, considerably lowering overallyields.
Scheme 3.
In a series of experiments in 2013, Bhusare et al. exam-ined the possibility of synthesizing a variety of opticallyactive α-amino phosphonates 12 in three-component reac-tions between aromatic aldehydes 5a, anilines 10, and tri-ethyl phosphite (11, Scheme 4).[11] After thorough investi-gations it was found that 1-acetyl-N-tosylpyrrolidine-2-car-boxamide (13) was the catalyst of choice. To determine thereliability of the developed approach and to confirm itsgenerality a variety of aromatic aldehydes 5a and amines 10were treated with triethyl phosphite (11) in the presence of13 and glacial acetic acid in ethanol. In all cases the targetα-amino phosphonates 12 were efficiently formed in anenantioselective manner.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 3
Scheme 4.
In 2014, two research groups independently reported onhighly enantioselective phospha-Mannich reactions be-tween diphenyl phosphite (2a) and N-Boc ketimines 14, de-rived from N-protected, differently substituted isatins in thepresence of bifunctional H-bonding/Brønsted base catalysts16a or 16b (Scheme 5).[12,13]
Scheme 5.
In the first report, Reddy and co-workers demonstratedthat quinine-derived squaramide catalyst 16a was an ef-ficient promoter of this type of hydrophosphonylation reac-tion, providing biologically important α-amino phosphon-ates 15, each containing a quaternary stereogenic center atthe C3 position (Scheme 5, Conditions A).[12] Thus, underoptimal reaction conditions a variety of products 15 were

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEWprepared in very good yields and with high enantio-selectivities (up to 98 % ee). It is worth noting that amongthe N-substituted imines 14 tested, a N-benzyl-protectedketimine proved the best in terms of enantioselectivity.Surprisingly, a simple Pudovik reaction between diphenylphosphite (2a) and N-benzyl-isatin was not fully successfulunder the established reaction conditions. Although a 3-hydroxy-2-oxindolyl-3-phosphonate was smoothly obtainedin 95% yield the reaction turned out to be not at allenantioselective (0 % ee). This result showed that asymmet-ric addition of a diphenyl phosphite such as 2a was onlyapplicable to ketimines and unsuccessful with ketones.
Furthermore, the authors proposed a plausible transitionstate model responsible for the stereochemistry of this ad-dition. It was postulated that the quinuclidine moiety ofthe catalyst 16a activated diphenyl phosphite (2a) throughdeprotonation whereas the isatin imine 14 was activatedthrough a double H-bonding interaction with the squar-amide scaffold (Figure 1). Therefore, preferential attack ofa phosphite nucleophile from the Re-face of an azomethinebond of ketimine occurred, allowing the desired product 15with R configuration to be obtained.
Figure 1. Proposed transition state model explaining the stereo-selectivity in the addition of 2a to imines 14.
In the study by Chimni et al., a similar synthesis ofenantiomerically enriched α-amino phosphonates 15 fromvarious substituted N-Boc ketimines 14 was performed(Scheme 5, Conditions B).[13] A broad range of opticallyactive products 15 was isolated with good enantioselectivi-
Scheme 6.
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–04
ties (up to 97 % ee) and yields (71–88%) under optimizedreaction conditions involving bifunctional thiourea organo-catalyst 16b. Interestingly, the authors demonstrated that athree-component version of this reaction could be achieved(Scheme 6). Thus, a one-pot domino aza-Wittig/phospha-Mannich sequence allowed products 15 to be obtained withcomparable enantiomeric excesses and yields.
3. Acylphosphonates in AsymmetricOrganocatalysis
α-Keto phosphonates, commonly known as acyl-phosphonates, constitute a very useful class of reagentswidely employed in asymmetric synthesis.[14] The increasedelectrophilicity of the carbonyl groups in such systemsmakes them particularly well suited for application invarious addition reactions, leading to biologically relevantα-hydroxy phosphonates. Furthermore, the nucleophilicityof α-keto phosphonates, manifesting at the β-position, isalso enhanced, due to the presence of the electron-with-drawing phosphoryl moiety. Importantly, the introductionof alkenyl substituents at the β-position of α-keto phos-phonates, yielding β,γ-unsaturated systems, affords verygood Michael acceptors that are electrophilic in the γ-posi-tion. An important feature of α-keto phosphonates is theirability to act as carboxylic acid surrogates, owing to thelability of the phosphonate moiety. In this section differentreactivities of α-keto phosphonates promoted by varioustypes of organocatalysts are outlined and summarized.
3.1. Saturated Acylphosphonates in AsymmetricOrganocatalysis
In 2012, Zhao et al. reported the successful applicationof acetylphosphonate 18a in organocatalytic aldol reac-tions, in combination with the transformation of theoriginal aldol products into the corresponding acetates 19and 21 or imide 23 in one-pot fashion (Scheme 7).[15] Theauthors examined reactions between diisopropyl acetyl-phosphonate (18a) and N-trityl-isatins 1b, phenylglyoxalhydrates 20, or β,γ-alkynyl α-keto ester 22 in the presence

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
of cinchona alkaloid derivatives 16c or 4b. Importantly, theinitial aldol products were converted in situ with high yieldsand stereoselectivities into the corresponding acetates 19 or21 or into imide 23 in the presence of MeOH/DBU orMeNH2, respectively. For this reason, the developed syn-thetic strategy can be considered a highly enantioselectivesynthetic equivalent of an acetate or acetamide aldol reac-tion.
Scheme 7.
The possibility of employing the nucleophilic characterof α-keto phosphonates in asymmetric organocatalysis wasfurther demonstrated by Guang and Zhao (Scheme 8).[16]
The authors described asymmetric Michael reactions be-tween β-aryl α-keto phosphonates 18b and nitroalkenes 24,promoted by a chiral bifunctional Brønsted base catalyst.The initially formed adducts were transformed in situ intothe corresponding amides 25 in high yields and with goodstereoselectivities. The best results were achieved with cata-lyst 26a, possessing a 2,6-diisopropylphenyl group on thethiourea moiety, and dichloromethane as a solvent at lowtemperatures. The scope of the developed reaction was verybroad, with a change of the electronic properties of the sub-stituents in the starting materials from electron-donating toelectron-withdrawing not significantly influencing either theyield or the diastereoselectivity of the reaction.
The authors proposed a plausible mechanism for this re-action. They postulated initial deprotonation of the β-arylα-keto phosphonate 18b by the tertiary amine moiety of
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5
Scheme 8.
catalyst 26a. Subsequent ionic interactions (between thethus obtained enolate and the ammonium group in the cat-alyst) and recognition of the nitroalkene counterpart 24(through H-bonding interactions with the thiourea moietyof the catalyst) positioned the two reactants in close prox-imity, allowing the reaction to occur (Figure 2). As a resultof the employed activation strategy, attack from the Re-faceof the enolate was favored, and (2R,3S) diastereomers wereformed as the major products.
Figure 2. Proposed spatial arrangement of the two reactantsthrough the action of catalyst 26a.
Miao et al. investigated the enantioselective synthesis oftertiary α-hydroxy phosphonates 29 (Scheme 9).[17] Reac-tions between acylphosphonates 18 and trimethylsilyl cyan-ide (27) were performed in the presence of bifunctionalthiourea catalyst 16d, containing two chiral motifs basedon cinchona alkaloid and carbohydrate units. The initialproducts 28 were converted into α-hydroxy phosphonates29 under acidic conditions. The best results were obtainedin the presence of 4-nitrophenol as an additive. Modifica-tions of the R1 group in acylphosphonates 18, from aro-matic groups with electron-withdrawing to -donatinggroups, did not significantly influence either the yield or theenantioselectivity. Interestingly, the enantioselectivity de-creased with increasing bulkiness of the R2 groups.
Furthermore, the reaction between diethyl cinnamoyl-phosphonate and TMSCN was also studied. The desiredproduct 29a (R1 = PhCH=CH-) was obtained in good yieldand in a highly enantioselective manner. Importantly, thesite-selectivity of the reaction was very high, with noMichael addition product being observed.
The authors postulated a plausible mechanism involvingan aldol reaction between diethyl benzoylphosphonate and

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 9.
TMSCN in the presence of catalyst 16d and p-nitrophenol.In the first step, phosphonate 18 is recognized by the thio-urea moiety of the catalyst 16d through H-bonding interac-tions. p-Nitrophenol is involved in the generation of HCNfrom TMSCN prior to the attack of cyanide anion on thecarbonyl group of the activated α-keto phosphonate 18.
In 2011, Zhao et al. reported on highly enantioselectivecross-aldol reactions between enolizable aldehydes 5b andα-keto phosphonates 18 (Scheme 10).[18] After a thoroughscreening, 9-amino-9-deoxy-epi-quinine (31) in the presenceof 4-methoxybenzoic acid (MBA) was identified as a cata-lytic system of choice. Reactions were performed in tolueneat 0 °C. Scope studies of these cross-aldol transformationsrevealed that the size of the ester alkyl group in the phos-phonate moiety 18 had almost no influence either on theyield or on the enantioselectivity of the reaction. Moreover,in case of the benzoylphosphonates (R2 = aryl), both theelectronic nature of the substituents and their positions onthe aromatic ring seemed to be irrelevant. In view of thewell-known biological activity of the α-hydroxy phosphon-ates, initial biological tests on selected β-formyl α-hydroxyphosphonates were conducted, revealing that they cansuppress the proliferation of human and murine tumor cells.
An interesting strategy for the preparation of opticallyactive α-hydroxy-H-phosphinates 35 was reported in 2013by Yao and Yuan (Scheme 11),[19] who developed a highlyenantioselective synthesis of α-hydroxyphosphinates 35based on enamine-mediated addition of acetone (32a) in thepresence of l-proline (36a) as a catalyst. The dialk-oxymethyl groups in the initially obtained products 34
Scheme 11.
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–06
Scheme 10.
served as masking groups for the P–H bond. As demon-strated by the authors, the synthesis of highly reactive andunstable α-hydroxy-H-phosphinates 35 could be ac-complished under acidic conditions. The best results wereobtained with aryl phosphinates 33 with halogen substitu-ents in the meta or para positions of the aromatic ring assubstrates. The authors proposed a mechanism for these re-actions involving enamine-mediated addition of acetone(32a) to the carbonyl group in the α-acylphosphinate 33. Itis worth noting that phosphinates 33 employed in the reac-tion are chiral at the phosphorus stereogenic center and ra-cemic. Thus, products 34 were formed as mixtures of di-asteroisomers with different configurations at the phos-phorus center and predominantly the R absolute configura-tion at the carbon atom.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
The first enantioselective synthesis of diisopropyl α-amino α-cyano phosphonates 39 by means of organocata-lytic Strecker-type reactions was performed by Palacios andco-workers (Scheme 12).[20] The desired products 39 wereobtained in good yields and enantioselectivities under opti-mal reaction conditions with use of cinchonidine (4c) as abifunctional catalyst and pyruvonitrile (37) as a cyanidesource. The authors showed that only α-aryl α-ketiminophosphonates 38 can be employed in this reaction. Notably,the α-amino α-cyano phosphonates 39 obtained in this wayserved as direct precursors of the corresponding α-aryl-sub-stituted α-phosphonoglycine derivatives.
Scheme 12.
3.2. β,γ-Unsaturated Acylphosphonates in AsymmetricOrganocatalysis
The first application of β,γ-unsaturated α-keto phos-phonates in asymmetric organocatalysis was reported in2010 by Jørgensen et al.[21] Stereoselective addition of avariety of nucleophilic reagents including oxazolones,indoles, and 1,3-dicarbonyl compounds was performed withthe aid of H-bonding catalysis. Initially, the authors studiednucleophilic addition of oxazolones 40 to acylphosphonates18c in the presence of cinchona-alkaloid-based thiourea 16e(Scheme 13). It was found that catalyst 16e promoted selec-tive addition at the C-2 position of the oxazolone ring. Sub-sequent treatment of the obtained adducts with alcohols oramines in the presence of a base (DBU) gave the corre-sponding esters or amides 41 in high yields and in anenantioselective fashion. Additionally, the potential toconvert adducts 41 into γ-lactones or Stetter-type productsin the presence of NaBH4 or NaHCO3, respectively, wasdemonstrated.
The Friedel–Crafts alkylation of indoles 42 with dimethyl(E)-alkenoylphosphonates 18c in the presence of chiralthiourea catalyst 26b, based on an aminoindanol scaffold,was also described (Scheme 14).[21] The initial Friedel–Crafts products were subsequently treated with secondnucleophiles (alcohols or amines) in the presence of DBU.The authors observed that in the second step partial race-
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 7
Scheme 13.
mization of the originally obtained products took place, de-pending on the reaction temperature, time, and the type ofsecond nucleophile. The desired esters or amides 43 wereformed in good yields and enantioselectivities.
Scheme 14.
Finally, the authors investigated stereoselective additionof cyclic 1,3-dicarbonyl compounds 44 to dimethyl (E)-but-2-enoylphosphonate (18d, Scheme 15).[21] Chiral quinine-derived squaramide 16f was found to be the most efficientcatalyst for the reactions. The originally formed α-oxophosphonates were transformed into the correspondingesters or benzyl amides 45 under basic conditions.
Later on, Wang, Zhou, et al. studied the addition of 2-hydroxy-1,4-naphthoquinones 46 to β,γ-unsaturated α-ketophosphonates 18c (Scheme 16).[22] Reactions were per-formed at –25 °C in a 1:1 mixture of chloroform and tolu-ene as a solvent. The thiourea catalyst 16g was found to bethe most efficient, particularly in terms of enantioselectivity.Importantly, the resulting adducts were converted into β-(3-hydroxy-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-substitut-ed carboxylates 47 by simple treatment of the crude reac-tion mixtures with methanol in the presence of DBU.
Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 15.
Notably, despite the basic reaction conditions, no loss ofthe optical purity introduced in the first organocatalyticstep was observed.
Scheme 16.
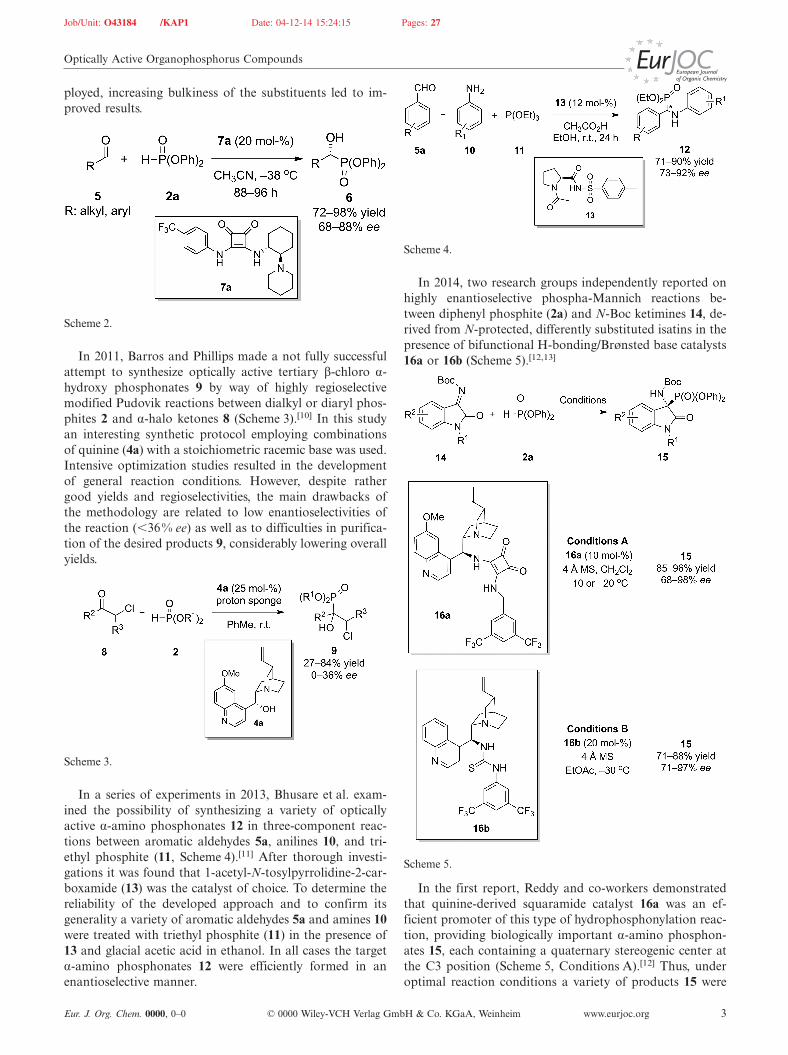
The authors proposed that the bifunctional nature of thecatalyst 16g enabled independent activation of both reac-tion partners in a transition state, with the 1,4-dioxo-1,4-dihydronaphthalen-2-olate 46 being activated through ionicpair formation and dialkyl (E)-alkenoylphosphonate 18cthrough H-bonding interactions (Figure 3).
This example again confirmed the usefulness of β,γ-un-saturated α-keto phosphonates 18c as very convenient andactive carboxylic acid surrogates.
In 2013, the same research group reported asymmetricMichael addition of 4-hydroxycoumarins 48 to β,γ-unsatu-rated α-keto phosphonates 18c (Scheme 17).[23] Among thecatalysts tested, bifunctional squaramide derivative 7b, con-taining a 9,10-dihydro-9,10-ethanoanthracene scaffold, gavethe best results. Analogously to previously described re-ports, the originally formed adducts, containing α-ketophosphonate moieties, were transformed in situ into the
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–08
Figure 3. Independent activation of both reaction partners in thetransition state by catalyst 16g.
corresponding carboxylates or amides 49 in the presence ofDBU as a base and methanol or morpholine as nucleophilicreagents.
Scheme 17.
To explain the stereochemical outcome of the reactionthe transition state model shown in Figure 4 was proposed.It was postulated that the tertiary amine moiety of 7b acti-vated the 4-hydroxycoumarine component 48 through de-protonation whereas dialkyl (E)-alkenoylphosphonate 18cwas involved in H-bonding interactions with the squar-amide moiety. Thus, the nucleophile attack on the unsatu-rated α-keto phosphonate occurred predominantly from itsRe-face.
Figure 4. Proposed transition state model explaining the stereo-chemical outcome of the asymmetric Michael addition.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Asymmetric [4+2] cycloaddition between allenic esters 50and β,γ-unsaturated α-keto phosphonates 18c was devel-oped in 2012 by Wei, Shi, et al.[24] In these reactions theformation of two products, 51 and 52, was observed in eachcase. The product distribution depended on the catalyst em-ployed. In the presence of the cinchona-alkaloid-derived or-ganocatalyst 4d, possessing an amide moiety, phosphonate-substituted pyran derivatives 51 were formed as majorproducts (Scheme 18). Reactions were performed in aceto-nitrile with 4 Å molecular sieves as an additive at low tem-perature. The reaction outcome was independent of the na-ture of the substituent in the γ-position of 18c. Both alkyland aryl substituents in 18c were tolerated well, givingaccess to compounds 51 in good yields and in a highlyenantioselective manner.
Scheme 18.
The formation of different products was observed whenthe reactions were performed in the presence of catalyst 4e(Scheme 19). Under these conditions phosphonate-substi-tuted dihydropyran derivatives 52 were obtained as soleproducts in high yields (up to 93%) and with excellentenantiomeric excesses (90–96 % ee). Differences in the be-havior of the two catalysts 4d and 4e were explained bytheoretical calculations, which revealed that the ability ofthe catalyst 4d to participate in H-bonding interactions with18c is crucial for obtaining products 51.
In 2013, inverse-electron-demand hetero-Diels–Alder re-actions between enolizable aldehydes 5c and β,γ-unsatu-rated α-keto phosphonates 18c were developed by Zhaoet al. (Scheme 20).[25a] The devised synthetic strategyutilized a concept of modularly designed organocatalysts(MDOs) introduced earlier by the same research group.[25b]
MDOs are formed by self-assembly of amino acids andcinchona alkaloids through ionic interactions. In pursuit ofan appropriate MDO system for the devised reactions itwas found that the presence of both a tertiary amine basicmoiety and a thiourea functionality was crucial for the reac-tivity as well as for the enantioselectivity. Therefore, the re-
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 9
Scheme 19.
actions were performed in the presence of quinidine-de-rived-thiourea 16h and octahydro-1H-indole-2-carboxylicacid (OIHC, 54) as the second MDO component. The ex-amined reactions were performed in toluene at ambienttemperature and proceeded with excellent yields andenantioselectivities. The authors postulated that a Re-faceattack of the (E)-enamine (formed in the reaction betweenan aldehyde 5c and amine 54) on α-keto phosphonate 18coccurred selectively due to H-bonding interactions betweenthe thiourea moiety of 16h and α-keto phosphonate 18c.
Scheme 20.
Recently, the possibility of employing β,γ-unsaturated α-keto phosphonates 18c as dienes in inverse-electron-demandhetero-Diels–Alder reactions was demonstrated byJørgensen et al. (Scheme 21).[26] Crucial for the success ofthis approach was the employment of bifunctional aminocatalyst 36b, enabling independent activation of α,β-unsatu-rated aldehydes 5d (through dienamine formation) and acyl-phosphonates 18c (by a H-bonding activation strategy).With such a dual activation strategy the [4+2] cycload-ditions proceeded efficiently to afford dihydropyran deriva-tives that were isolated as alcohols 55 after in situ re-duction. Notably, the obtained products could easily be em-

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEWployed for the synthesis of optically active tetrahydropyransand δ-lactones, with preservation of the enantioselectivityintroduced in the amino-catalytic step.
Scheme 21.
4. Phosphorus Ylides in AsymmetricOrganocatalysis
Phosphorus ylides are a highly useful group of organo-phosphorus reagents widely employed in Wittig olefinationof carbonyl compounds for the construction of doublebonds.[1d,1e] Catalytic asymmetric Wittig reactions were de-veloped by Bernardi et al. (Scheme 22).[27] In this approachthe absolute stereochemistry of the products 57 was con-trolled by the H-bonding catalyst 58. Olefination of thesymmetric 4-substituted cyclohexanones 32b afforded axi-ally chiral alkenes 57. The main focus of this research wason Wittig reactions with EWG-stabilized phosphorus ylides56a. The highest levels of conversion and enantioselectivitywere obtained with ylides bearing electron-rich aromaticgroups at the phosphorus atom. In a wide screening of anappropriate catalyst for this reaction the (R,R)-TADDOLderivative 58 was found to be the most efficient.
Scheme 22.
Another aspect of phosphorus ylide chemistry that hasbeen utilized in asymmetric organocatalysis is their abilityto act as nucleophiles in various reactions. Importantly, the
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–010
products thus obtained can still participate in Wittig reac-tions to allow introduction of olefinic moieties.
Such a reactivity pattern was employed in 2011 by Zhu,Cheng, et al. for the synthesis of optically active α-methyl-idene-δ-oxoalkanoates 59 (Scheme 23).[28] The authors re-ported enantioselective Michael-type reactions betweenphosphorus ylides 56b and α,β-unsaturated ketones 32c,followed by olefination with formaldehyde. The salt 60,derived from 9-amino-9-deoxy-epi-quinine and l-N-Boc-proline, was successfully applied as the catalyst in these re-actions. The developed reaction sequence proceeded withmoderate yields but excellent enantioselectivities. It wasfound that the electronic properties of aryl groups in theα,β-unsaturated ketones 32c did not influence either theyields or the enantioselectivities of the reactions. However,replacement of a methyl group on the ketone moiety witha longer chain (from two to eight carbon atoms) causedincreases in the enantioselectivity (from 82 to 95% ee).
Scheme 23.
In a similar manner, phosphorus ylides can participateas nucleophiles in substitution reactions, as demonstratedby Zhu and co-workers (Scheme 24).[29] The authorsstudied the synthesis of the alkanoates 63, each bearing twomethylidene moieties. The synthesis was initiated by doubleSN2� reactions of Morita–Baylis–Hillman (MBH) carb-onates 61 in the presence of tertiary amine catalyst 4f, to-gether with phosphorus ylides 56b as nucleophiles. In sucha manner the first methylidene moiety and a stereogeniccenter were introduced in a stereoselective fashion. Subse-quent Wittig reaction with formaldehyde afforded the ex-pected products 63 in good yields and with excellentenantioselectivities. The authors found that the nature ofaryl substituents in MBH-carbonates 61 slightly influencedthe enantioselectivities whereas increasing bulkiness of thealkyl groups in P-ylides 56b caused decreases.
To explain the stereochemical outcome of these reactionsthe transition state shown in Figure 5 was proposed. Ini-tially, the MBH-carbonate 61 underwent SN2� reaction withthe nitrogen atom of the quinuclidine ring of the catalyst4f serving as nucleophile. In the E-configured olefin thusobtained, the Si-face was efficiently blocked, allowing forthe P-ylide 56b to approach the system from its Re-face ina stereoselective fashion.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Scheme 24.
Figure 5. Proposed transition state explaining the stereochemicaloutcome of the reactions shown in Scheme 24.
5. Reactions Involving Phosphoryl-Group-Stabilized Carbanions
The ability of the phosphoryl group to stabilize an adja-cent carbanion is well established in organic chemistry andhas been widely employed in organic synthesis.[1] In the fieldof organocatalysis this phenomenon has also been success-fully utilized to achieve various new, enantioselective trans-formations. In this section these studies are outlined anddiscussed.
In 2011 Wang et al. studied enantioselective Mannich re-actions between Horner–Wadsworth–Emmons reagents 65aand imines generated in situ from compounds 64(Scheme 25).[30] The reactions proceeded with high yieldsand enantioselectivities in the presence of bifunctional thio-urea catalyst 26c. The initially formed adducts 66 were con-verted into aza-MBH-type products 67 by treatment withparaformaldehyde in the presence of sodium methoxide asa base. Importantly, despite the basic reaction conditions,the optical activity introduced in the Mannich reaction stepcould be preserved throughout the developed reaction se-quence. The authors discovered that imines possessing ali-phatic groups gave lower yields and enantioselectivities thanaromatic or heteroaromatic substrates. Furthermore, imineprecursors 64 with Boc-protected amino moieties neededlonger reaction times than derivatives with Cbz groups.Transformation of the Mannich adducts 66 into arylidene-substituted aza-MBH products 68 was also studied, withbenzaldehyde and cinnamaldehyde in the presence of so-
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 11
dium methoxide or Verkade’s superbase. It was found thatthe bulkiness of a base had a great influence on the geome-try of the obtained products 68. Whereas the use of sodiummethoxide led to the formation of (Z)-68 as the major prod-ucts, in the presence of Verkade’s superbase the E isomerspredominated.
Phosphoryl-group-stabilized carbanions were also re-cently utilized by Jørgensen and co-workers[31] for the prep-aration of biologically relevant α-methylidene-hetero-cycles[32] (Scheme 26). The syntheses were initiated byhighly enantioselective Michael reactions between trimethylphosphonoacetate (65b) and iminium-ion-activated α,β-un-saturated aldehydes 5d. The stereochemical outcome of theaddition was governed by the sterically demanding substitu-ent present in the C2-position of the catalyst 36c. The phos-phoryl-group-stabilized carbanion approached the corre-sponding iminium ion intermediate from the side oppositeto this directing group to avoid unfavorable steric interac-tions. The originally formed Michael adducts 69 were notisolated but were utilized in subsequent transformationsleading to biologically relevant β-substituted α-methyl-idene-δ-lactones 71 and -lactams 73 or 76. Transformationof 69 into β-substituted α-methylidene-δ-lactones 71 wasachieved through a reaction sequence involving reductionof the aldehyde moiety, cyclization, and Horner–Wad-sworth–Emmons reaction with formaldehyde. Lactams 73could be synthesized when reductive amination with meth-ylamine as the aminating reagent was performed in the firststep of the sequence. Interestingly, Pictet–Spengler reactionsbetween compounds 69 and tryptamines 74 were also per-formed, to achieve access to the indolo[2,3-a]quinolizinealkaloid framework 75. Initial Boc protection of the nitro-gen atom of the indole moiety and subsequent Horner–Wadsworth–Emmons olefination afforded the desired ole-finic derivatives 76. A noteworthy feature of the developedstrategy is the structural diversity of the olefinic hetero-cycles obtained.
Replacement of the ester moiety in a phosphonoacetatenucleophile of type 65 with the cyano group yields anotherphosphoryl-group-stabilized carbanion precursor of type77, which can be employed in asymmetric organocatalysisas demonstrated in 2010 by Jászay et al. (Scheme 27).[33]
The authors reported enantioselective addition of diethylcyanomethylphosphonate (77) to chalcones 32d in the pres-

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 25.
Scheme 26.
ence of cinchona-alkaloid-derived organocatalyst 16i. TheMichael adducts 78, which can be smoothly converted into
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–012
α-substituted β-amino phosphonates, were obtained inmoderate yields and with good enantiomeric excesses. Opti-

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
mization of the reaction conditions revealed that the solu-bility of the catalyst 16i had a crucial effect in terms bothof conversion and of enantioselectivity. Toluene was foundto be the most suitable and effective solvent.
Scheme 27.
In 2011, Cheng and co-workers reported the asymmetricα-sulfenylation of β-keto phosphonates 80, in which diaryl-prolinol catalyst 36d was employed (Scheme 28).[34] Duringscreening of the reaction conditions it was found that theenantioselectivity of the sulfenylation was dependent on thereaction temperature. Moreover, the bulkiness of the estergroups in the phosphonates 80 had a slight effect on theenantiomeric excess, and hexane was found to be the bestsolvent for these reactions. The scope of the developedstrategy proved broad: either electron-donating or electron-withdrawing substituents could be present on the aromaticmoiety in 80 without any influence either on the yield oron the enantioselectivity of the sulfenylation. The authorspostulated that the α,α-diarylprolinol catalyst 36d interactswith β-keto phosphonates 80 in the enol form through H-bonding, thereby facilitating their reaction.
Scheme 28.
The first asymmetric Michael additions of β-keto phos-phonates 82 to nitro olefins 24 were developed in 2011 byTang et al. (Scheme 29).[35] In the presence of cinchona-alkaloid-derived thiourea organocatalyst 16g the reactionswere found to proceed efficiently to afford α-substituted β-keto phosphonates 83 in good yields, with low diastereo-selectivity and moderate to good enantioselectivity. The
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 13
study revealed that both the size of the phosphonate moietyand the positions of the substituents on the aromatic ringsof nitro olefins 24 had a great impact on the stereochemicalreaction outcomes. On the other hand, the electronic char-acters of the aryl substituents in 24 did not have a signifi-cant role, because both electron-donating and -withdrawinggroups could be present. The authors postulated that stericeffects were crucial and that the dual activation mode wasoperating. Initially, the tertiary amine residue in 16g actedas a Brønsted base to deprotonate the β-keto phosphonate82. Secondly, the nitro olefin 24 was activated by double H-bonding interactions with the thiourea moiety of 16g.
Scheme 29.
Recently, Kwiatkowski and Lu presented their results re-lating to the enantioselective synthesis of highly function-alized α-acyl α-fluoro γ-nitro phosphonates 85 by means ofchiral-base-catalyzed Michael addition of α-fluoro β-ketophosphonates 84 to nitro olefins 24 (Scheme 30).[36a] Fluor-inated phosphonates are a very interesting class of biolo-gically active compounds that can exquisitely mimic bio-active phosphates.[36b] In the examined Michael addition,the tertiary amine-thiourea catalyst 16j, with an incorpo-rated l-tert-leucine-derived spacer, was employed. Theauthors claimed that the catalytic activity of this system canbe easily tuned by suitable choice either of the cinchona-alkaloid backbone or of the chiral amino-acid-derivedlinker. The research showed that the spectrum of substratesthat can be employed in this transformation is quite broad.The reactions proceeded efficiently for a variety of nitroolefins 24 and α-fluoro β-keto phosphonates 84 regardlessof the substituents or their electronic properties. However,when aliphatic nitro olefins 24 were applied, lower yieldswere obtained despite the fact that higher catalyst loadingswere used. Importantly, the excellent enantioselectivity ofthe reaction was maintained.
α-Nitro phosphonates are another group of compoundsthat can easily be deprotonated in the α-position under ba-sic conditions, yielding stabilized carbanions. Interestingly,these reactive intermediates are available under organocata-lytic conditions, as demonstrated recently by different re-search groups. An important feature of this class of nucleo-philes is their ability to serve as direct precursors of biolo-gically relevant α-amino phosphonates.[3f–3h]

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 30.
In 2010, Gajda and co-workers demonstrated the appli-cation of the α-nitro phosphonate 87a in organocatalyticMannich reactions (Scheme 31).[37] In the study, imineswere formed in situ from the corresponding N-Boc-substi-tuted α-aminoalkyl-p-tolylsulfones 86. Satisfactory enantio-selectivity (up to 67% ee) and low syn diastereoselectivitywere achieved with use of the bifunctional quinine-derivedthiourea catalyst 16e. However, long reaction times (8 d)were required in order to achieve high conversion. Import-antly, Mannich products 88 were easily transformed intoenantiomerically enriched 1,2-diamino phosphonates understandard reaction conditions.
Scheme 31.
In 2012, Bera and Namboothiri demonstrated thepossibility of employing α-nitro phosphonates 87b inorganocatalytic Michael reactions (Scheme 32, Condi-tions A).[38] Vinyl ketones 89a were applied as electrophiliccomponents and bifunctional thiourea 16k as a catalyst,affording α-nitro δ-oxo phosphonates 90a in high yields.The enantioselectivity was highly dependent on the struc-ture of the starting vinyl ketone 89a, with lower enantio-selectivities obained for aliphatic systems. Interestingly, be-cause the Michael adducts 90a bear nitro and carbonyl sub-stituents in a 1,4-relationship they can easily be transformedinto biologically relevant phosphorylated γ-lactam and di-hydropyrrole derivative.
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–014
Scheme 32.
Subsequent studies from various research groups iden-tified other Michael acceptors that can be employed in suchadditions. For instance, in 2013 Jaszay demonstrated thatacrylates 89b constitute a related group of Michaelacceptors that can be employed in the discussed Michaeladditions (Scheme 32, Conditions B).[39] In this case bifunc-tional catalyst 16l proved optimal. Disappointingly, thestudies were limited only to α-nitroethyl phosphonate 87b(R2 = CH3). The sizes of the ester groups both on theacrylate components and on the phosphonate moieties werethe only parameters evaluated in the scope study.
In 2012, Mukherjee et al. reported highly diastereo- andenantioselective conjugate addition of α-alkylnitro phos-phonates 87b to various aliphatic and aromatic nitro olefins24 (Scheme 33).[40] The reactions were catalyzed by quinine-derived bifunctional catalyst 16e. Optimization studies onthe catalyst employed and the reaction conditions revealedthat the reaction medium had a major influence on the di-asteroselectivity of the addition. Trifluorotoluene was foundto be the best solvent. Reaction temperature also played akey role. Because of reaction reversibility at 0 °C theauthors were unable to achieve complete conversion, anddeterioration in the diastereoselectivity and enantio-selectivity was observed. Therefore almost all reactionswere carried out at –10 °C. Importantly, the optimized reac-tion conditions were particularly well suited for aliphatic

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
nitro olefins. The best result was achieved with n-pentyl-substituted nitro olefin 24 (R1 = n-Pent), with the productbeing obtained in a very good yield and with excellentstereoselectivity (20:1 dr, � 99:1 er). Notably, the products91 served as direct precursors of α,β-disubstituted α,γ-di-aminophosphonic acids.
Scheme 33.
Recently, Namboothiri and Bera performed the firstenantioselective synthesis of α-nitro γ-sulfonyl phosphon-ates 93, by utilizing organocatalytic activation of α-alkyl-nitro phosphonates 87b through chiral ion pair formation(Scheme 34, Conditions A).[41] The authors studied theMichael addition of compounds 87b to phenyl vinyl sulfone(92). Screening of several cinchona-alkaloid-based organo-catalysts for this reaction allowed quinine-derived squar-amide catalyst 16m to be identified as the best reaction pro-moter. Under optimized reaction conditions the corre-sponding Michael adducts 93 were isolated in excellentyields and enantioselectivity. Moreover, the authors demon-strated the possibility of obtaining products 93 on a gramscale. Notably, nitrosulfonyl phosphonates 93 served as di-rect precursors of α-amino phosphonates. The sameorganocatalytic transformations were independently studiedby Chen and Lu in the same year (Scheme 34, Condi-tions B).[42] Their developed reaction protocol involvedthiourea derivative 16h as a catalyst and toluene as a sol-vent. Similar levels of stereoinduction were obtained, withthe reactions proceeding efficiently for a wide range of ali-phatic α-nitro phosphonates 87b.
A different reactivity profile of α-alkylnitro phosphon-ates 87b was observed when they were treated with 3-bromooxindoles 94 (Scheme 35).[43a] In this case the reac-tions were found to proceeded exclusively by the O-alkyl-ation pathway. Bifunctional organocatalyst 16n ensuredhigh levels of stereoinduction. However, the final products95 were formed as mixtures of Z/E isomers with the Zisomer predominating in all cases. From the mechanisticpoint of view a stereoablative[43b] reaction mechanism waspostulated by the authors. Disappointingly, the authors didnot demonstrate any transformation of the products 95 ob-tained, although their usefulness seems limited.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 15
Scheme 34.
Scheme 35.
6. Phospha-Michael Additions
The phospha-Michael reaction is an efficient means forthe construction of C–P bonds. Its enantioselective variantswith both PIII and PV species have been extensively studiedand summarized in reviews.[44] Recent examples of organo-catalytic phospha-Michael reactions involve mainly diaryland dialkyl phosphites or diarylphosphine oxides, and theseare discussed in this section.
An important example of enantioselective conjugate ad-dition of diphenyl phosphite (2a) to various nitroalkenes 24was disclosed by Rawal and co-workers in 2010(Scheme 36).[45] The authors demonstrated that squar-amide-moiety-containing catalyst 7a, based on a trans-1,2-

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEWdiaminecyclohexane scaffold, was a remarkably effectivepromoter for these reactions. Notably, thanks to thepresence of the tertiary amine moiety, 7a can act as aBrønsted base to activate 2a through deprotonation. Theauthors noticed that in the presence of 7a both aryl- andalkyl-substituted nitroalkanes 24 could easily be convertedinto the corresponding β-nitro phosphonates 96 with excel-lent yields and enantioselectivities. It is noteworthy that theselectivity of this phospha-Michael addition was not signifi-cantly sensitive to a change in the solvent used. Moreover,as the authors had expected, the enantioselectivity was im-proved by a decrease in the temperature. For this reason allreactions were carried out at 0 °C in dichloromethane,which was chosen as the best solvent. This paper indicatedgreat potential of H-bonding organocatalysis in such asimple, yet challenging organophosphorus transformation.
Scheme 36.
Further studies from different research groups on thisparticular Michael addition type enabled the identificationof other bifunctional H-bonding/Brønsted base catalyststhat can efficiently promote these reactions (Scheme 37).Herrera and co-workers[46] demonstrated that the commer-cially available bifunctional Takemoto’s catalyst (26d) wasan efficient rate enhancer of the phospha-Michael addition(Scheme 37, Conditions A). The established optimal reac-tion conditions allowed enantiomerically enriched β-nitrophosphonates 96 to be obtained in good overall yields, al-though the enantioselectivities were only moderate. To pro-vide an explanation for the stereochemical outcome of thereaction, the authors proposed that in the transition statethe β-nitrostyrene 24 was activated by double H-bondinginteractions with the thiourea moiety of 26d, enabling dis-crimination of the two enantiotopic faces.
Comparable results in the synthesis of optically active β-nitro phosphonates 96 were achieved by Zhao et al. withquinidine-derived thiourea organocatalyst 16h (Scheme 37,Conditions B).[47] Molecular sieves were found to act as apromoter, which dramatically improved the yields of thesereactions. It was postulated that the molecular sieves canact as a heterogeneous Lewis acid, shifting the H-phos-phonate/phosphite equilibrium and favoring the formationof the more reactive phosphite form.
Another interesting organocatalyst for the phospha-Michael addition of diphenyl phosphonate (2a) to nitro
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–016
olefins 24 was introduced by Nagasawa et al. (Scheme 37,Conditions C).[48] The unusually conformationally flexibleguanidinium organocatalyst 26e, bearing two thioureamoieties, turned out to be a very useful catalyst for thesetransformations. The authors noticed an increase in the re-action enantioselectivity under biphasic conditions. Thus, amixture of toluene and water (2:1 ratio) was found to be thebest solvent system for these conjugate additions. Potassiumcarbonate was used as a basic co-promoter. Under opti-mized reaction conditions the target products 96 wereformed with excellent enantiomeric excesses. Importantly,substrates 24 containing both aromatic and aliphatic sub-stituents in the β-position were successfully utilized.
A green protocol for the hydrophosphonylation of nitroolefins 24 was also recently developed (Scheme 37, Condi-tions D).[49] It was found that organocatalytic asymmetricMichael addition of 2a to 24 can be achieved underBrønsted base catalysis conditions with supercritical carbondioxide as a solvent. The reactions were found to proceedefficiently for a wide range of nitro olefins 24, includingaliphatic, aromatic, and heteroaromatic substrates. Interest-ingly, it was demonstrated that for the purpose of chiralcatalyst recovery and product isolation a supercritical ex-traction can be employed.
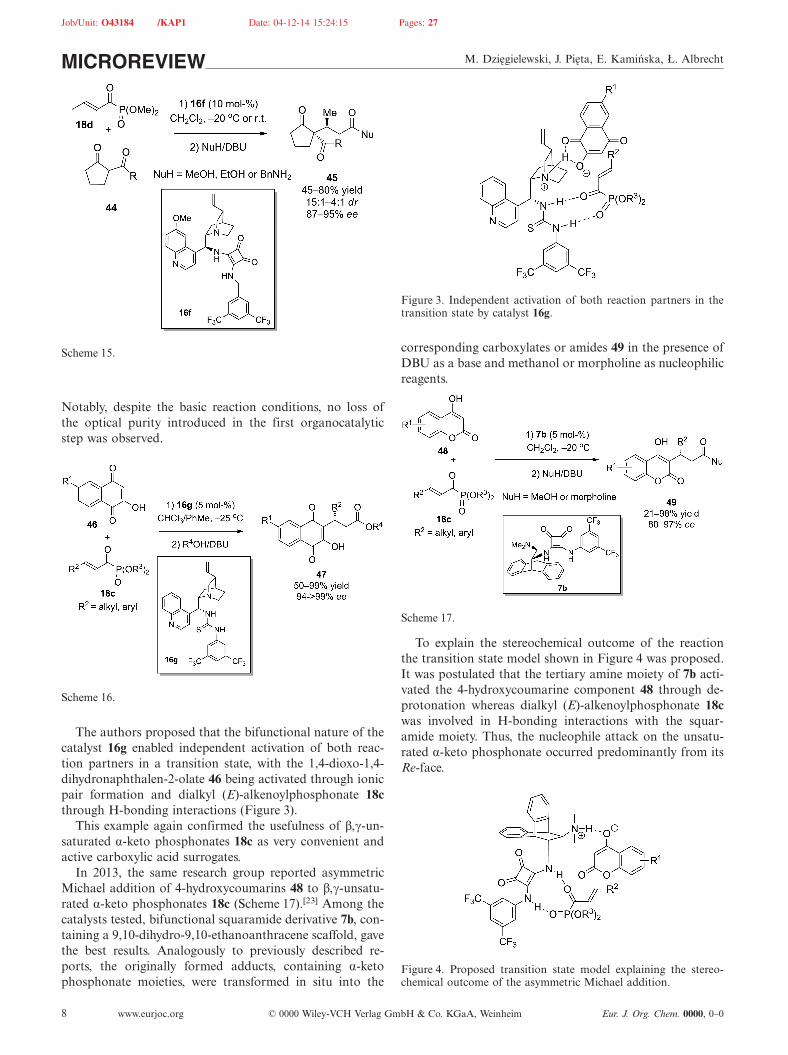
Asymmetric organocatalysis has also been employed inphospha-Michael additions of H-phosphonates to Michaelacceptors other than nitro olefins. For instance, an efficientand facile approach to enantiomerically enriched 3-phosphooxindoles 98, based on organocatalytic conjugateMichael addition of dialkyl phosphites 2 to isatylidene-sub-stituted malonitriles 97, was reported by Wang et al.(Scheme 38).[50] A variety of products 98 were obtainedwith high enantioselectivities and in good to excellent yieldsunder optimized reaction conditions in the presence of bi-functional thiourea catalyst 26f, derived from (R,R)-1,2-di-phenylethylenediamine. The authors noticed that the ad-dition of molecular sieves was beneficial for the reactivityand enantioselectivity. Interestingly, the position of the sub-stituent on the aromatic ring of oxindole framework 97 hadan influence on the reaction rate. Reaction with isatinsbearing substituents in the 4-position were much faster thanthose of substrates 97 with substituents in the C-6 or C-7positions.
Notably, two plausible transition states for the additionwere suggested (Figure 6). Attack of the phosphite 2 at theRe-face of the isatylidene-malonitrile 97 in the postulatedthree-component transition state is favored and responsiblefor the formation of the major enantiomer of 98. However,either the isatylidene-malonitrile moiety or the carbonylgroup of oxindole ring can be involved in the recognitionprocess.
Diarylphosphine oxides are a second group of organo-phosphorus reagents recently employed in organcatalyticphospha-Michael reactions. In 2010 the asymmetric conju-gate addition of diarylphosphine oxides 99 to chalcones 32dwas investigated for the first time by Russo and Lattanzi(Scheme 39).[51] Systematic studies on the mechanism andoptimization of these organocatalytic reactions were under-

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Scheme 37.
taken. Among the promoters evaluated, dihydroquinine(4g) showed the highest catalytic activity. Differently substi-tuted diarylphosphine oxides 99 were also examined, withthe commercially available diphenylphosphine oxide (99a)being found to be the most appropriate substrate. To ex-plain the stereochemical outcome of the reactions, theauthors postulated that the activation of trans-chalcones32d was possible through H-bonding interactions with thehydroxy group of the catalyst 4g. Additionally, due to the
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 17
presence of the quinuclidine nitrogen atom in 4g, the phos-phine oxide/phosphinous acid equilibrium is shifted towardthe phosphinous acid form possessing increased reactivity.
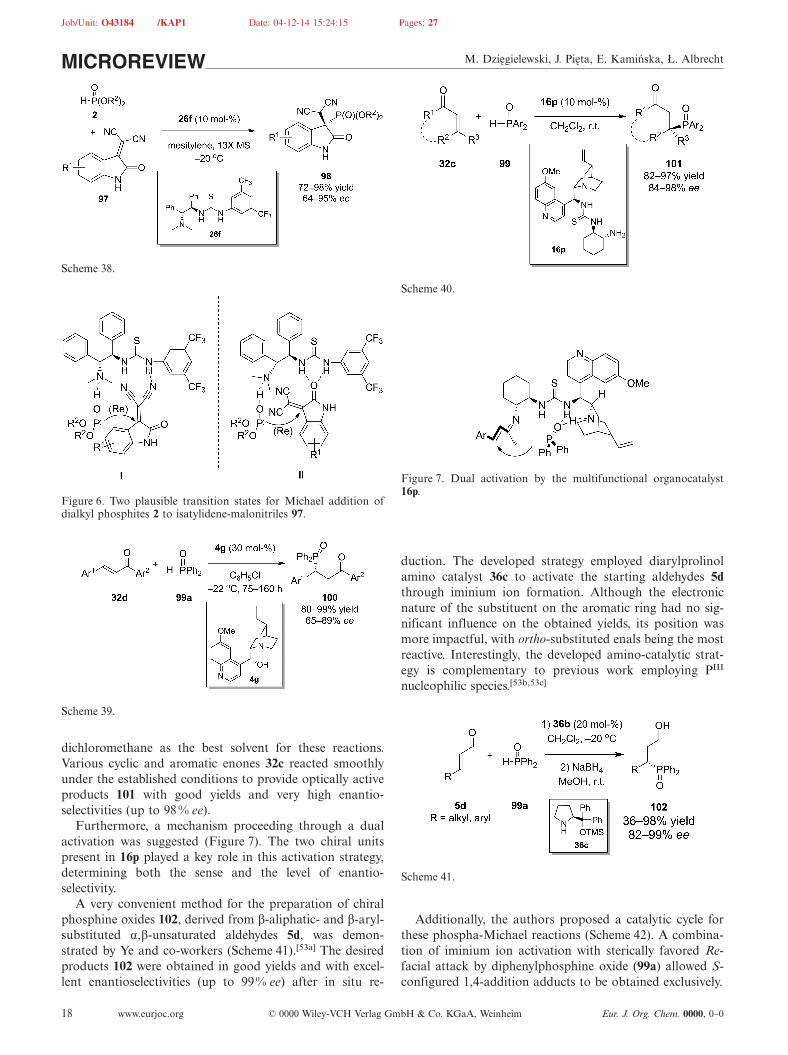
α,β-Unsaturated ketones 32c constitute another class ofMichael acceptors that can be utilized in organocatalyticphospha-Michael reactions with diarylphosphine oxides 99,as shown by Liang et al. (Scheme 40).[52] The authors per-formed optimization studies that identified thiourea-epi-quinine catalyst 16p as the most powerful catalyst and
Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 38.
Figure 6. Two plausible transition states for Michael addition ofdialkyl phosphites 2 to isatylidene-malonitriles 97.
Scheme 39.
dichloromethane as the best solvent for these reactions.Various cyclic and aromatic enones 32c reacted smoothlyunder the established conditions to provide optically activeproducts 101 with good yields and very high enantio-selectivities (up to 98 % ee).
Furthermore, a mechanism proceeding through a dualactivation was suggested (Figure 7). The two chiral unitspresent in 16p played a key role in this activation strategy,determining both the sense and the level of enantio-selectivity.
A very convenient method for the preparation of chiralphosphine oxides 102, derived from β-aliphatic- and β-aryl-substituted α,β-unsaturated aldehydes 5d, was demon-strated by Ye and co-workers (Scheme 41).[53a] The desiredproducts 102 were obtained in good yields and with excel-lent enantioselectivities (up to 99% ee) after in situ re-
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–018
Scheme 40.
Figure 7. Dual activation by the multifunctional organocatalyst16p.
duction. The developed strategy employed diarylprolinolamino catalyst 36c to activate the starting aldehydes 5dthrough iminium ion formation. Although the electronicnature of the substituent on the aromatic ring had no sig-nificant influence on the obtained yields, its position wasmore impactful, with ortho-substituted enals being the mostreactive. Interestingly, the developed amino-catalytic strat-egy is complementary to previous work employing PIII
nucleophilic species.[53b,53c]
Scheme 41.
Additionally, the authors proposed a catalytic cycle forthese phospha-Michael reactions (Scheme 42). A combina-tion of iminium ion activation with sterically favored Re-facial attack by diphenylphosphine oxide (99a) allowed S-configured 1,4-addition adducts to be obtained exclusively.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Scheme 42.
Heterocyclic α,β-unsaturated pyrazolone derivatives 106were also utilized for enantioselective phospha-Michael ad-dition of diphenylphosphine oxide (99a, Scheme 43).[54] Agreat number of sophisticated organocatalysts were testedin these reactions enabling thiourea 26g, featuring an iso-steviol motif, to be identified as the optimal organocatalystfor the transformation. The desired heteroaromatic prod-ucts 107 were efficiently obtained with moderate enantio-selectivities.
Scheme 43.
7. Michael Additions to Phosphorus-ContainingMichael Acceptors
Owing to the electron-withdrawing properties of thephosphoryl group, its introduction on a double bond resultsin polarization of the olefinic moiety and makes such asystem prone to undergo conjugate addition with selectednucleophiles. Of the different phosphoryl-group-containingMichael acceptors, vinyl bisphosphonate esters occupy a
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 19
prominent position because they can serve as direct precur-sors of biologically relevant geminal bisphosphonates.[3i,3j]
This group of compounds is used for treatment and preven-tion of several bone diseases such as osteoporosis, Paget’sdisease, and bone-related cancers.[3i,3j] In this section recentapplications of vinyl bisphosphonate esters as Michael ac-ceptors in asymmetric organocatalytic conjugate additionsare outlined.
In 2012, Shi et al. reported the enantioselective additionof 3-aryloxindoles 108 to electron-deficient tetraethyl vinyl-bisphosphonate (109a) for the first time (Scheme 44).[55]
The cinchonidine-derived thiourea catalyst 16b was recog-nized as a very effective promoter of these C–C bond-form-ing reactions. A wide range of bisphosphonate adducts 110were obtained in high yields and with good enantio-selectivities under optimized reaction conditions. Moreover,tetraethyl esters 110 were smoothly hydrolyzed to the corre-sponding bisphosphonic acids by treatment with iodotri-methylsilane (TMSI) without significant loss of enantio-selectivity.
Scheme 44.
In the same year, Barros and Phillips reported diastereo-and enantioselective Michael addition of 4-substitutedcyclohexanones 32b to tetraethyl vinylbisphosphonate(109a, Scheme 45).[56] The commercially available 1-(2-pyr-rolidinylmethyl)pyrrolidine (36e) was found to be the bestamino catalyst for this reaction. Importantly, the describedMichael addition constitutes an example of a desymmetri-zation reaction in which two new stereogenic centers areformed that originate from the achiral Michael donor 32b.Intensive studies on the effect of acidic co-catalyst and cata-lyst loading were undertaken by the authors. As a result,all reactions were carried out in dry dichloromethane with10 mol-% of amino catalyst 36e and in the presence ofbenzoic acid as the additive.
Additionally, under optimized reaction conditions 4-pip-eridones and tetrahydropyranone were also converted intocorresponding germinal bisphosphonates with good yields(58–88 %) and moderate to high enantioselectivities (40–90% ee).
Geminal bisphosphonate moieties have also recentlybeen incorporated into quaternary amino acid structures(Scheme 46).[57] The developed reaction sequence consisted

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 45.
of Michael addition of α-substituted oxazolones 40 to tetra-ethyl vinylbisphosphonate (109a) followed by oxazolonering opening under acidic conditions. The main focus of theresearch was the development of the racemic synthesis ofthe target quaternary amino acids. However, initial studieson the development of an enantioselective approach em-ploying chiral Brønsted base catalysis were also described.Of the catalysts tested, quinine (4a) proved the most effec-tive, affording product 112a in moderate enantoselectivity.
Scheme 46.
8. Application of Organophosphorus Reagents inOne-Pot and Domino Strategies
Domino and one-pot strategies in which more than onebond is formed in a reaction sequence without isolation orpurification of reaction intermediates occupy a prominentposition in modern organic chemistry.[58] In the case oforganophosphorus reagents containing phosphoryl groups,the ability of these groups to activate either a Michaelacceptor or a donor, in conjunction with the potential ofthe initially formed products to undergo Horner–Wadsworth–Emmons reactions, has been widely exploredfor a construction of various carbo- and heterocyclic scaf-folds. In this section the application of organophosphorusreagents in such strategies is discussed.
In 2012, Lu and co-workers reported an interestingdomino reaction sequence involving nitro-Michael additionof 2-diethoxyphosphoryl-4-nitrobutanoate (113) to α,β-un-saturated aldehydes 5d followed by an intramolecularHorner–Wadsworth–Emmons reaction (Scheme 47).[59]
Whereas the application of a well-established iminium ionactivation strategy with silyl-protected diarylprolinol cata-lyst 36f ensured high levels of stereocontrol, the presenceof co-promoters (DABCO and LiClO4) was crucial for thereaction efficiency. The reaction sequence worked well for
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–020
both aromatic and aliphatic enals 5d, affording access tocyclohexene derivatives 114 in moderate to good overallyields. Interestingly, the potential of the developed syntheticstrategy was further confirmed in the syntheses of the di-peptidyl peptidase 4 (DPP-4) inhibitor ABT-341, as well asof an influenza neuraminidase inhibitor.
Scheme 47.
A related reaction cascade involving enantioselectivesulfa-Michael addition followed by intramolecular Horner–Wadsworth–Emmons reactions was recently developed(Scheme 48).[60] The domino process was efficiently pro-moted by the urea catalyst 26h, enabling dual H-bonding/Brønsted base activation. In such a reaction setup, the di-ethoxyphosphoryl moiety served two purposes. Firstly, itactivated the starting Michael acceptors 115, making themmore prone to undergo Michael addition. Secondly, it madethe construction of the 2,3-disubstituted thiochromeneframework, as in compounds 116, possible through intra-molecular Horner–Wadsworth–Emmons olefination. Targetproducts 116 were formed in excellent yields and in a highlyenantioselective manner.
Scheme 48.
Recently, Jørgensen et al. described an interesting imple-mentation of vinylogous amino catalysis in an organocata-lytic one-pot synthesis of optically active compounds 117,six-membered carbocycles conjugated with various ringsystems (Scheme 49).[61] In this procedure, γ-alkylation ofenals 5f, proceeding through the intermediacy of dien-amines, was performed in the first step. α,β-Unsaturatedphosphonates 109 served as electrophilic components inthese reactions. With Michael addition accomplished,annulation through intramolecular Horner–Wadsworth–Emmons olefination was performed. The one-pot reaction

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
sequence was conducted at ambient temperature in thepresence of 5 mol-% of diphenylprolinol trimethylsilyl ether(36c), yields and enantiomeric excesses of the desired prod-ucts 117 were good to excellent regardless of the electronicnature or the position of substituents on the aromaticmoiety in 5f. However, there are also some limitations inthe presented γ-alkylation/HWE reaction sequence. β-Sub-stituted vinyl phosphonate analogues of 109, use of whichcould result in the introduction of an additional stereogeniccenter, turned out not to be good substrates. Furthermore,attempts to isolate Michael acceptors containing nitrogroups were unsuccessful due to their instability, in contrastto the corresponding ketone-, ester-, sulfone- orphosphonate-containing electrophiles.
Scheme 49.
The first organocatalytic electrophilic phosphination wasalso recently disclosed (Scheme 50).[62] These reactions con-stituted the first enantiodifferentiating step in the synthesisof β-aminophosphine oxides 121 and were catalyzed bycinchona-alkaloid-derived catalyst 4f. After the initialorganocatalytic step, subsequent oxidation of the thus in-troduced phosphine moiety and reduction of the cyanogroup, combined with its in situ protection, were carriedout, leading to the formation of the target products 121 inmoderate to good overall yields. All reactions were per-formed in a one-pot manner, thus increasing the impor-tance and practicality of the synthetic strategy. Interest-ingly, an unexpected activation strategy was reported on thebasis of 31P NMR studies. The authors postulated that thecatalyst 4f served as a nucleophilic catalyst that initiallyunderwent substitution reactions on the phosphorus elec-trophilic centers. Second substitution reactions with 118 asnucleophiles led to the formation of the desired products
Scheme 50.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 21
120. Furthermore, it is worth stressing that these reactionsconstitute the only examples of asymmetric organocatalyticreactions in which the electrophilic centers are located atthe phosphorus atoms.
Michael-induced ring closure (MIRC) constitutes an im-portant means for the preparation of cyclopropane deriva-tives.[63] Recently, MIRC reactions employing α-bromo-phosphonoacetates 122 with cinnamaldehyde (5g) havebeen reported (Scheme 51).[64] Initial iminium-ion-mediatedMichael addition of 122 to 5g afforded the correspondingenamines, which underwent intramolecular alkylation tofurnish cyclopropanes 123. Although diastereoselectivitieswere moderate, high enantiomeric excesses were obtainedfor both diastereoisomers of the products 123. Disappoint-ingly, the scope of the reaction was very narrow, because thereactions were studied only with the one α,β-unsaturatedaldehyde system 5g.
Scheme 51.
Trienamine-mediated reactions have recently emerged asa powerful tool for the construction of cyclohexene frame-works.[65] In 2013, Chen et al. demonstrated the possibilityof employing organophosphorus reagents in such chemistry(Scheme 52).[66] Phosphoryl-group-activated olefinic oxind-oles 124 were employed as dienophiles in reactions with tri-enamines derived from various linear and branched 2,4-di-enals 5h. TMS-protected diphenylprolinol 36c proved to bean efficient catalyst of the studied transformations. Bothphosphonates and phosphine oxides could be present onthe exocyclic double bond of the oxindole substrates 124.Of the other phosphorus-containing dienophiles evaluated,only diethyl (3-oxo-3-phenylprop-1-en-1-yl)phosphonateunderwent the reaction with hexa-2,4-dienal under trien-amine activation.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 52.
Among various biologically relevant organophosphoruscompounds, α-amino phosphonates occupy an importantposition.[3f–3h] Recently, two independent reports on thesynthesis of α-amino β-hydroxy phosphonates 128a weredisclosed, both relying on reactions between phosphoryl-ated α-substituted isothiocyanates 127 and aldehydes 5(Scheme 53).[67a,67b] The authors employed bifunctional cat-alysts 16e or 16m, enabling independent activation of alde-hydes 5 (through H-bonding activation) and α-substitutedisothiocyanates 127 (through chiral ion pair formation).They found that under these conditions the reactions pro-ceeded in a domino sequence involving nucleophilicaddition of a phosphoryl-group-stabilized carbanion to thealdehyde electrophile 5 followed by cyclization through asecond addition of hetero-nucleophile to the isothiocyanatemoiety. In such a manner α-amino β-hydroxy phosphonates128a were efficiently obtained in a stereoselective fashion.
Scheme 53.
Importantly, when aldehydes 5 were replaced with imines126a in the developed synthetic strategy, enantioselectivesynthesis of α,β-diamino phosphonates 128b provedpossible, as demonstrated by Wang et al.[67b]
In 2011, Xu et al. published a paper in which a sequentialone-pot enantioselective reaction sequence for the prepara-tion of highly functionalized cyclohexane derivatives con-taining five contiguous stereogenic centers was examined
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–022
(Scheme 54).[68a] A combination of asymmetric Michael ad-dition of aldehydes 5b to nitro olefin 24a, followed by annu-lation of the cyclohexene framework through dominoMichael/Horner–Wadsworth–Emmons reactions involving109b and terminating with sulfa-Michael reactions with 4-methylbenzenethiol (129), led to target compounds 130.The reaction sequence was catalyzed by secondary aminocatalyst 36g, enabling enamine activation of starting alde-hydes 5b. Notably, the sequence was based on previousstudies by the Hayashi group.[68b] Interestingly, modifica-tion of the structure of a diarylprolinol silyl ether aminocatalyst with a tertiary amine moiety allowed the efficient,water-soluble, and recyclable organocatalyst 36g to be ob-tained. Its application in the evaluated tandem reactionsimproved both the yields and the enantioselectivities of thesequence. The authors postulated that the enhancement ofenantioselectivity in the developed one-pot transformationsin the presence of catalyst 36g was caused by electronic ef-fects in its aryl substituents.
Scheme 54.
9. Miscellaneous Methods
A very elegant method for the synthesis of enantiomeri-cally enriched β-amino phosphonates 135, based onMannich-type reactions between dimethyl(diazomethyl)-phosphonate (134) and N-Boc imines derived from aro-matic aldehydes 126b, was demonstrated by Maruoka andco-workers (Scheme 55).[69] The authors performed ascreening of axially chiral BINOL-derived dicarboxylicacids and found catalyst 136, bearing 4-(tert-butyl)-2,6-di-methylphenyl groups, to be the most suitable for theseasymmetric transformations. It is worth noting that allproducts 135 were easily accessed with high enantio-selectivities (�91% ee) under optimal reaction conditions.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Importantly, addition of molecular sieves was crucial forobtaining high yields.
Scheme 55.
In 2012, Ooi, Johnson, et al. reported a new type of base-catalyzed direct glycolate aldol addition in which a pivotalrole was played by a [1,2]-phosphonate–phosphate re-arrangement (Scheme 56).[70] The nature of these transfor-mations can be simply explained by the presence of an adja-cent electron-withdrawing ester moiety, which facilitates mi-gration of a dialkyloxyphosphinyl group from the carbonatom to the oxygen owing to stabilization of the negativecharge. In the developed synthetic strategy, α-hydroxyphos-phonoacetates 137 were treated with different aldehydes 5in the presence of catalytic amounts of chiral iminophos-phorane catalyst 139 to give α-hydroxy-β-(dimethoxyphos-phoryloxy)esters 138 in good yields and with excellentenantioselectivities. Importantly, the phosphate moietiespresent in the products of this transformation, compounds138, could serve as leaving groups and were directly em-ployed in nucleophilic displacement chemistry, opening ac-cess to various interesting products in a stereoselective fash-ion.
Scheme 56.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 23
The asymmetric substitution of MBH-carbonates 61 withphosphine oxides 99 was developed by Wang et al.(Scheme 57, Conditions A).[71a] The MBH-carbonates 61were treated with diarylphosphine oxides 99 in the presenceof quinidine (4h) as a catalyst. The reactions proceeded bya double SN2�/SN2� mechanism, with the catalyst 4h actingas a nucleophile in the first displacement event, yieldingcompounds 143 as the only products. Reactions were per-formed with use of a mixture of xylenes as a solvent and4 Å molecular sieves as an additive. The products 143 wereobtained with high yields and enantiomeric excesses regard-less of the electronic properties of aryl groups in MBH-carbonates 61. However, in the case of a 2-furyl substituenta decrease in the reaction enantioselectivity (to 44% ee) wasobserved.
The same group continued research with dialkyl phos-phine oxides 99 as nucleophiles (Scheme 57, Condi-tions B).[71b] The authors discovered that the evaluated re-actions performed in the presence of quinidine 4h as thecatalyst did not work in the absence of inorganic base. Ad-dition of alkali metal carbonates caused the formation ofallylic phosphine oxides 143, with the best results beingachieved with Na2CO3. An interesting reaction outcomewas observed when sodium hydroxide was used as a base.In such a case, the formation of allylic phosphine oxides,resulting from direct SN2� reactions between 99 and 61,occurred.
To explain the stereochemistry of obtained products, theauthors proposed that the reactions proceeded via the tran-sition state depicted in Figure 8. It was postulated that thehydroxy group of the catalyst 4g and sodium dialkyloxo-phosphate can interact with each other and that this is veryimportant for the reaction. To confirm this postulate, theauthors performed a similar reaction but with use ofquinidine with its hydroxy group protected by a trimethyl-silyl group as a catalyst. The yield of obtained product wassignificantly lower (21 %).

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW
Scheme 57.
Figure 8. Proposed transition state for asymmetric substitution ofMBH-carbonates 61 with phosphine oxides 99.
10. Summary and Outlook
In conclusion, we have summarized recent progress in thefield of organocatalytic reactions employing organo-phosphorus reagents. The vast range of catalytic activationmethods applied to activate organophosphorus reagents isastonishing and mainly includes amino-catalytic ap-proaches, H-bonding strategies, and Brønsted base acti-vation. Importantly, one trend is particularly evident: appli-cation of bifunctional catalysis allowing for the independentactivation of two reaction partners. In terms of chiralscaffolds present in the catalysts employed, motifs derivedfrom cinchona alkaloids or amino acids are dominant. Fi-nally, to achieve high selectivities and yields, fine-tuning ofreaction conditions including solvent, temperature, and re-action time optimization proved inevitable in most of thecases. The research discussed in this microreview shows thetrue potential of organophosphorus compounds in theidentification of new reaction pathways and also as newbiologically active compounds. Recently developed organo-catalytic methods offer easy and efficient routes to differentclasses of biologically relevant organophosphorus com-pounds such as, for example, phosphonates or phosphineoxides. Owing to the high enantioselectivities offered bythese strategies they might become methods of choice whenthe synthesis of an organophosphorus compound is needed,replacing traditional approaches based on metal catalysis.We believe that in the years to come the application of or-ganophosphorus reagents in asymmetric organocatalysiswill still be receiving considerable attention and that newand inspiring findings in the field are only a matter of time.
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–024
Acknowledgments
This work was carried out within the Lider programme from theNational Center for Research and Development (NCBR) (grantnumber LIDER/01/87/L3/11/NCBR/2012).
[1] a) J. I. G. Cadogan, Organophosphorus Reagents in OrganicSynthesis Academic Press, London, 1979; b) R. Engel, Hand-book of organophosphorus chemistry, Marcel Dekker, NewYork, 1992; c) P. Savignac, B. Iorga, Modern phosphonateschemistry, CRC Press LLC, 2003; d) B. E. Maryanoff, A. B.Reitz, Chem. Rev. 1989, 89, 863–927; e) P. J. Murphy, Chem.Soc. Rev. 1988, 17, 1–30; f) M. Mikołajczyk, P. Bałczewski, Top.Curr. Chem. 2003, 223, 161–214.
[2] a) A. E. Arbuzov, J. Russ. Phys. Chem. Soc. 1906, 38, 687; b)G. Wittig, G. Geissler, Justus Liebigs Ann. Chem. 1953, 580,44; c) G. Wittig, U. Schollkopf, Chem. Ber. 1954, 87, 1318; d)L. Horner, H. Hoffmann, H. G. Wippel, Chem. Ber. 1958, 91,61; e) L. Horner, H. Hoffmann, H. G. Wippel, G. Klahre,Chem. Ber. 1959, 92, 2499; f) W. S. Wadsworth Jr., W. D. Em-mons, J. Am. Chem. Soc. 1961, 83, 1733; g) M. I. Kabachnik,T. Y. Medved, Dokl. Akad. Nauk SSSR 1952, 83, 689; h) M. I.Kabachnik, T. Y. Medved, Dokl. Akad. Nauk SSSR 1952, 84,717; i) E. K. Fields, J. Am. Chem. Soc. 1952, 74, 1528.
[3] a) M. Horiguchi, M. Kandatsu, Nature 1959, 184, 901–902; b)T. Hori, M. Horiguchi, A. Hayashi, Biochemistry of NaturalC–P Compounds, Maruzen, Tokyo, 1984; c) R. L. Hildebrand,The Role of Phosphonates in Living System, CRC Press, BocaRaton, Fl, 1983; d) D. F. Wiemer, Tetrahedron 1997, 53, 16609–16644; e) S. C. Fields, Tetrahedron 1999, 55, 12237–12273; f) P.Kafarski, B. Lejczak, Phosphorus Sulfur Silicon Relat. Elem.1991, 63, 193–215; g) V. P. Kukhar, H. R. Hudson, Aminophos-phonic and Aminophosphinic Acids, John Wiley & Sons, Chich-ester, UK, UK, 2000; h) J.-A. Ma, Chem. Soc. Rev. 2006, 35,630–636; i) H. Fleish, Bisphosphonates in Bone Disease: Fromthe Laboratory to the Patient, Parthenon Publishing Group,London, 1995; j) S. Zhang, G. Gangal, H. Uludag, Chem. Soc.Rev. 2007, 36, 507–531.
[4] a) A. Börner, Phosphorus ligands in asymmetric catalysis: syn-thesis and applications, Wiley-VCH, Weinheim, Germany, 2008;b) T. Akiyama, Chem. Rev. 2007, 107, 5744–5758; c) G. Adair,S. Mukherjee, B. List, Aldrichim. Acta 2008, 41, 31–39; d) M.Terada, Synthesis 2010, 1929–1982; e) M. Rueping, A. Kuen-kel, I. Atodiresei, Chem. Soc. Rev. 2011, 40, 4539–4549; f) M.Rueping, B. J. Nachtsheim, W. Ieawsuwan, I. Atodiresei, An-gew. Chem. Int. Ed. 2011, 50, 6706–6720; Angew. Chem. 2011,123, 6838.
[5] E. N. Jacobsen, A. Pfaltz, H. Yamamoto (Eds.), ComprehensiveAsymmetric, Catalysis I–III, Springer, New York, 1999.

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
[6] For recent, selected reviews on organocatalysis, see, for exam-ple: a) M. J. Gaunt, C. C. C. Johansson, A. McNally, N. C. Vo,Drug Discovery Today 2007, 2, 8–27; b) P. I. Dalko, Enantiose-lective Organocatalysis, Wiley-VCH, Weinheim, Germany,2007; c) D. Enders, O. Niemeier, A. Henseler, Chem. Rev. 2007,107, 5606–5655; d) S. Sulzer-Mossé, A. Alexakis, Chem. Com-mun. 2007, 3123–3135; e) A. Dondoni, A. Massi, Angew. Chem.Int. Ed. 2008, 47, 4638–4660; Angew. Chem. 2008, 120, 4716;f) P. Melchiorre, M. Marigo, A. Carlone, G. Bartoli, Angew.Chem. Int. Ed. 2008, 47, 6138–6171; Angew. Chem. 2008, 120,6232; g) M. Bella, T. Gasperi, Synthesis 2009, 1583–1614; h)Asymmetric Phase Transfer Catalysis (Ed.: K. Maruoka),Wiley-VCH, Weinheim, Germany, 2008; i) Ł. Albrecht, A. Al-brecht, H. Krawczyk, K. A. Jørgensen, Chem. Eur. J. 2010, 16,28–48; j) P. Melchiorre, Angew. Chem. Int. Ed. 2012, 51, 9748–9770; Angew. Chem. 2012, 124, 9886; k) K. L. Jensen, G.Dickmeiss, H. Jiang, Ł. Albrecht, K. A. Jørgensen, Acc. Chem.Res. 2012, 45, 248–264; l) A. M. Faisca Phillips, Mini-Rev. Org.Chem. 2014, 11, 164–185.
[7] a) P. Merino, E. Marqués-López, R. P. Herrera, Adv. Synth.Catal. 2008, 350, 1195–1208; b) S. Van der Jeught, C. V. Ste-vens, Chem. Rev. 2009, 109, 2672–2702.
[8] L. Peng, L.-L. Wang, J.-F. Bai, L.-N. Jia, Q.-C. Yang, Q.-C.Huang, X.-Y. Xu, L.-X. Wang, Tetrahedron Lett. 2011, 52,1157–1160.
[9] J. V. Alegre-Requena, E. Marqués-López, P. J. S. Miguel, R. P.Herrera, Org. Biomol. Chem. 2014, 12, 1258–1264.
[10] M. T. Barros, A. M. F. Phillips, Eur. J. Org. Chem. 2011, 20–21, 4028–4036.
[11] P. B. Thorat, S. V. Goswami, R. L. Magar, B. R. Patil, S. R.Bhusare, Eur. J. Org. Chem. 2013, 24, 5509–5516.
[12] J. George, B. Sridhar, B. V. S. Reddy, Org. Biomol. Chem. 2014,12, 1595–1602.
[13] A. Kumar, V. Sharma, J. Kaur, V. Kumar, S. Mahajan, N. Ku-mar, S. S. Chimni, Tetrahedron 2014, 70, 7044–7049.
[14] For selected examples, see: a) D. A. Evans, J. S. Johnson, J. Am.Chem. Soc. 1998, 120, 4895–4896; b) D. A. Evans, J. S. John-son, C. S. Burgey, K. R. Campos, Tetrahedron Lett. 1999, 40,2879–2882; c) D. A. Evans, K. A. Scheidt, K. R. Fandrick,H. W. Lam, J. Wu, J. Am. Chem. Soc. 2003, 125, 10780–10781;d) N. Takenaka, J. P. Abell, H. Yamamoto, J. Am. Chem. Soc.2007, 129, 742–743.
[15] J. Guang, Q. Guo, J. C.-G. Zhao, Org. Lett. 2012, 14, 3174–3177.
[16] J. Guang, J. C.-G. Zhao, Tetrahedron Lett. 2013, 54, 5703–5706.[17] S. Kong, W. Fan, G. Wu, Z. Miao, Angew. Chem. Int. Ed. 2012,
51, 8864–8867; Angew. Chem. 2012, 124, 8994.[18] S. Perera, V. K. Naganaboina, L. Wang, B. Zhang, Q. Guo, L.
Rout, C.-G. Zhao, Adv. Synth. Catal. 2011, 353, 1729–1734.[19] Q. Yao, C. Yuan, Chem. Eur. J. 2013, 19, 6080–6088.[20] J. Vicario, J. M. Ezpeleta, F. Palacios, Adv. Synth. Catal. 2012,
354, 2641–2647.[21] H. Jiang, M. W. Paixão, D. Monge, K. A. Jørgensen, J. Am.
Chem. Soc. 2010, 132, 2775–2783.[22] T. Liu, Y. Wang, G. Wu, H. Song, Z. Zhou, C. Tang, J. Org.
Chem. 2011, 76, 4119–4124.[23] X. Chang, Q. Wang, Y. Wang, H. Song, Z. Zhou, C. Tang, Eur.
J. Org. Chem. 2013, 11, 2164–2171.[24] C.-K. Pei, Y. Jiang, Y. Wei, M. Shi, Angew. Chem. Int. Ed.
2012, 51, 11328–11332; Angew. Chem. 2012, 124, 11490.[25] a) D. Sinha, S. Perera, J. C.-G. Zhao, Chem. Eur. J. 2013, 19,
6976–6979; b) T. Mandal, C.-G. Zhao, Angew. Chem. Int. Ed.2008, 47, 7714–7717; Angew. Chem. 2008, 120, 7828.
[26] C. F. Weise, V. H. Lauridsen, R. S. Rambo, E. H. Iversen, M.-L. Olsen, K. A. Jørgensen, J. Org. Chem. 2014, 79, 3537–3546.
[27] L. Gramigna, S. Duce, G. Filippini, M. Fochi, M. C. Franchini,L. Bernardi, Synlett 2011, 18, 2745–2749.
[28] A. Lin, J. Wang, H. Mao, H. Ge, R. Tan, C. Zhu, Y. Cheng,Org. Lett. 2011, 13, 4176–4179.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 25
[29] A. Lin, J. Wang, H. Mao, Y. Shi, Z. Mao, C. Zhu, Eur. J. Org.Chem. 2013, 28, 6241–6245.
[30] D. Zhao, D. Yang, Y. Wang, Y. Wang, L. Wang, L. Mao, R.Wang, Chem. Sci. 2011, 2, 1918–1921.
[31] A. Albrecht, F. Morana, A. Fraile, K. A. Jørgensen, Chem.Eur. J. 2012, 18, 10348–10354.
[32] For a recent review, see: A. Albrecht, Ł. Albrecht, T. Janecki,Eur. J. Org. Chem. 2011, 2747–2766.
[33] T. S. Pham, L. Balázs, I. Petneházy, Z. Jászay, Tetrahedron:Asymmetry 2010, 21, 346–351.
[34] A. Lin, L. Fang, X. Zhu, C. Zhu, X. Cheng, Adv. Synth. Catal.2011, 353, 545–549.
[35] K. Hu, T. Liu, A. D. Lu, Y. Liu, Y. Wang, G. Wu, Z. Zhou, C.Tang, Eur. J. Org. Chem. 2011, 19, 3507–3513.
[36] a) J. Kwiatkowski, Y. Lu, Asian J. Org. Chem. 2014, 3, 458–461; b) D. B. Berkowitz, M. Bose, J. Fluorine Chem. 2001, 112,13–33.
[37] R. Błaszczyk, A. Gajda, S. Zawadzki, E. Czubacka, T. Gajda,Tetrahedron 2010, 66, 9840–9848.
[38] K. Bera, I. N. N. Namboothiri, Org. Lett. 2012, 14, 980–983.[39] T. S. Pham, K. Gönczi, G. Kardos, K. Süle, L. Hegedus, M.
Kállay, M. Kubinyi, P. Szabó, I. Petneházy, L. Toke, Z. Jászay,Tetrahedron: Asymmetry 2013, 24, 1605–1614.
[40] C. B. Tripathi, S. Kayal, S. Mukherjee, Org. Lett. 2012, 14,3296–3299.
[41] K. Bera, I. N. N. Namboothiri, Adv. Synth. Catal. 2013, 355,1265–1270.
[42] G.-Y. Chen, Y. Lu, Synthesis 2013, 45, 1654–1658.[43] a) X. Xie, L. Jiang, D. Qin, W. He, S. Wu, L. Jin, G. Luo, RSC
Adv. 2014, 4, 11605–11609; for a review, see: b) J. T. Mohr,D. C. Ebner, B. M. Stoltz, Org. Biomol. Chem. 2007, 5, 3571–3576.
[44] For selected reviews, see: a) D. Zhao, R. Wang, Chem. Soc.Rev. 2012, 41, 2095–2108; b) D. Enders, A. Saint-Dizier, M.-I.Lannou, A. Lenzen, Eur. J. Org. Chem. 2006, 29–49; c) A. N.Pudovik, I. V. Konovalova, Synthesis 1979, 81–96.
[45] Y. Zhu, J. P. Malerich, V. H. Rawal, Angew. Chem. Int. Ed.2010, 49, 153–156; Angew. Chem. 2010, 122, 157.
[46] A. Alcaine, E. Marqués-López, P. Merino, T. Tejero, R. P. Her-rera, Org. Biomol. Chem. 2011, 9, 2777–2783.
[47] S. Abbaraju, M. Bhanushali, C.-G. Zhao, Tetrahedron 2011, 67,7479–7484.
[48] Y. Sohtome, N. Horitsugi, R. Takagi, K. Nagasawa, Adv.Synth. Catal. 2011, 353, 2631–2636.
[49] I. V. Kuchurov, A. G. Nigmatov, E. V. Kryuchkova, A. A. Kos-tenko, A. S. Kucherenko, S. G. Zlotin, Green Chem. 2014, 16,1521–1526.
[50] Z.-M. Liu, N.-K. Li, X.-F. Huang, B. Wu, N. Li, C.-Y. Kwok,Y. Wang, X.-W. Wang, Tetrahedron 2014, 70, 2406–2415.
[51] A. Russo, A. Lattanzi, Eur. J. Org. Chem. 2010, 35, 6736–6739.[52] S. Wen, P. Li, H. Wu, F. Yu, X. Liang, J. Ye, Chem. Commun.
2010, 46, 4806–4808.[53] a) X. Luo, Z. Zhou, X. Li, X. Liang, J. Ye, RSC Adv. 2011, 1,
698–705; b) A. Carlone, G. Bartoli, M. Bosco, L. Sambri, P.Melchiorre, Angew. Chem. Int. Ed. 2007, 46, 4504–4506; An-gew. Chem. 2007, 119, 4588; c) I. Ibrahem, R. Rios, J. Vesely,P. Hammar, L. Eriksson, F. Himo, A. Córdova, Angew. Chem.Int. Ed. 2007, 46, 4507–4510; Angew. Chem. 2007, 119, 4591.
[54] Z.-C. Geng, J.-X. Zhang, N. Li, J. Chen, X.-F. Huang, S.-Y.Zhang, H.-Y. Li, J.-C. Tao, X.-W. Wang, Tetrahedron 2014, 70,417–426.
[55] M.-X. Zhao, T.-L. Dai, R. Liu, D.-K. Wei, H. Zhou, F.-H. Ji,M. Shi, Org. Biomol. Chem. 2012, 10, 7970–7979.
[56] A. M. F. Phillips, M. T. Barros, Org. Biomol. Chem. 2012, 10,404–412.
[57] M. Dziegielewski, J. Hejmanowska, Ł. Albrecht, Synthesis2014, 46, DOI: 10.1055/s-0034-1378997.
[58] For selected reviews, see: a) K. C. Nicolaou, T. Montagnon,S. A. Snyder, Chem. Commun. 2003, 551–564; b) J.-C. Wasilke,S. J. Obrey, R. T. Baker, G. C. Bazan, Chem. Rev. 2005, 105,

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
M. Dziegielewski, J. Pieta, E. Kaminska, Ł. AlbrechtMICROREVIEW1001–1020; c) D. Enders, C. Grondal, M. R. M. Hüttl, Angew.Chem. Int. Ed. 2007, 46, 1570–1581; Angew. Chem. 2007, 119,1590; d) Ł. Albrecht, H. Jiang, K. A. Jørgensen, Angew. Chem.Int. Ed. 2011, 50, 8492–8509; Angew. Chem. 2011, 123, 8642;e) D. B. Ramachary, S. Jain, Org. Biomol. Chem. 2011, 9, 1277–1300; f) N. T. Patil, V. S. Shinde, B. Gajula, Org. Biomol. Chem.2012, 10, 211–224.
[59] J. Weng, J.-M. Li, F.-Q. Li, Z.-S. Xie, G. Lu, Adv. Synth. Catal.2012, 354, 1961–1970.
[60] A. R. Choudhury, S. Mukherjee, Adv. Synth. Catal. 2013, 355,1989–1995.
[61] B. S. Donslund, K. S. Halskov, L. A. Leth, B. M. Paz, K. A.Jørgensen, Chem. Commun. 2014, Advance Article DOI:10.1039/c4cc06556e.
[62] M. Nielsen, C. B. Jacobsen, K. A. Jørgensen, Angew. Chem.Int. Ed. 2011, 50, 3211–3214; Angew. Chem. 2011, 123, 3269.
[63] For selected reviews, see: a) H. Lebel, J. F. Marcoux, C. Molin-aro, A. B. Charette, Chem. Rev. 2003, 103, 977–1050; b) D.Caine, Tetrahedron 2001, 57, 2643–2684.
[64] A. M. F. Phillips, M. T. Barros, Eur. J. Org. Chem. 2014, 1,152–163.
[65] For a seminal report, see: a) Z.-J. Jia, H. Jiang, J.-L. Li, B.Gschwend, Q.-Z. Li, X. Ying, J. Grouleff, Y.-C. Chen, K. A.Jørgensen, J. Am. Chem. Soc. 2011, 133, 5053–5061; b) J.-L.Li, T.-Y. Liu, Y.-C. Chen, Acc. Chem. Res. 2012, 45, 1491–1500;c) I. Kumar, P. Ramaraju, N. A. Mir, Org. Biomol. Chem. 2013,
www.eurjoc.org © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 0000, 0–026
11, 709–716; d) H. Jiang, Ł. Albrecht, K. A. Jørgensen, Chem.Sci. 2013, 4, 2287–2300; e) I. D. Jurberg, I. Chatterjee, R. Tan-nert, P. Melchiorre, Chem. Commun. 2013, 49, 4869–4883; f) S.Reboredo, A. Parra, J. Alemán, Asymmetric Organocatalysis2013, 24–31.
[66] Q.-Q. Zhou, X. Yuan, Y.-C. Xiao, L. Dong, Y.-C. Chen, Tetra-hedron 2013, 69, 10369–10374.
[67] a) W.-Y. Han, J.-Q. Zhao, Z.-J. Wu, X.-M. Zhang, W.-C. Yuan,J. Org. Chem. 2013, 78, 10541–10547; b) Y.-M. Cao, F.-F. Shen,F.-T. Zhang, J.-L. Zhang, R. Wang, Angew. Chem. Int. Ed.2014, 53, 1862–1866.
[68] a) H. Shen, K.-F. Yang, Z.-H. Shi, J.-X. Jiang, G.-Q. Lai, L.-W. Xu, Eur. J. Org. Chem. 2011, 26, 5031–5038; for a seminalreport, see: b) H. Ishikawa, T. Suzuki, Y. Hayashi, Angew.Chem. Int. Ed. 2009, 48, 1304–1307; Angew. Chem. 2009, 121,1330.
[69] T. Hashimoto, H. Kimura, H. Nakatsu, K. Maruoka, J. Org.Chem. 2011, 76, 6030–6037.
[70] M. T. Corbett, D. Uraguchi, T. Ooi, J. S. Johnson, Angew.Chem. Int. Ed. 2012, 51, 4685–4689; Angew. Chem. 2012, 124,4763.
[71] a) L. Hong, W. Sun, C. Liu, D. Zhao, R. Wang, Chem. Com-mun. 2010, 46, 2856–2858; b) W. Sun, L. Hong, C. Liu, R.Wang, Org. Lett. 2010, 12, 3914–3917.
Received: September 8, 2014Published Online: �

Job/Unit: O43184 /KAP1 Date: 04-12-14 15:24:15 Pages: 27
Optically Active Organophosphorus Compounds
Enantioenriched Organophosphorus Compounds
Organophosphorus reagents play pivotal M. Dziegielewski, J. Pieta, E. Kaminska,roles in modern organic synthesis and have Ł. Albrecht* .................................... 1–27found many applications for the prepara-tion both of synthetically important com- Organocatalytic Synthesis of Optically Ac-pounds and of biologically relevant ones. tive Organophosphorus CompoundsThey are now widely employed in asym-metric organocatalysis to afford optically Keywords: Organocatalysis / Asymmetricactive organophosphorus compounds. This synthesis / Organophosphorus reagents /review summarizes recent progress in this Phosphonates / Phosphorylationfield of enantioselective synthesis.
Eur. J. Org. Chem. 0000, 0–0 © 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 27