off-label use of prescription drugs
TRANSCRIPT

Off-Label Use of Prescription Drugs
February 23, 2021
Congressional Research Service
https://crsreports.congress.gov
R45792

Congressional Research Service
SUMMARY
Off-Label Use of Prescription Drugs When the Food and Drug Administration (FDA) approves a drug for sale in the United
States, the approval includes a section entitled “Indications for Use.” This section lists
the one or more diseases, conditions, or symptoms for which the drug’s sponsor (usually
the manufacturer) has provided, to FDA’s satisfaction, evidence in support of the drug’s
safety and effectiveness. FDA approval is also based on its review of the drug’s dosage,
packaging, manufacturing plan, and labeling. Before changing any of those elements,
the sponsor must inform, and usually receive permission from, FDA.
In essence, FDA regulates all approval and post-approval aspects of a drug product. But FDA traditionally has not
regulated the practice of medicine. Physicians, therefore, may prescribe an FDA-approved drug for indications
that FDA has not reviewed for safety and effectiveness. Those uses, furthermore, are not addressed in the labeling
information regarding, among other things, dosing, warnings about interactions with other drugs, and possible
adverse events.
How Are Off-Label Prescription Drugs Used?
Prescribing for so-called off-label uses can be accepted medical practice, often reflecting cutting-edge clinical
expertise. For example, this is the case with oncology drug use, more than half of which is off-label. Off-label
prescribing can be a reasonable choice when labeling overlooks certain populations—for example, when a drug
tested in adults is prescribed to children. A drug may be used off-label when it was tested for the treatment of one
disease and prescribed in an attempt to prevent or treat another, when it was tested at one dose and used at higher
or lower doses, or when it was tested in an eight-week trial and prescribed for long-term use. Estimates for how
common off-label prescriptions are in the United States are hardly precise. Credible researchers have estimated
they make up as little as 12% and as much as 38% of doctor-office prescriptions.
What Are the Risks of Off-Label Prescriptions?
Prescriptions for off-label uses of FDA-approved drugs are made without the benefit of an FDA-reviewed
analysis of safety and effectiveness data. Physicians may resort to such prescribing to take advantage of new ideas
and treatment approaches when available information to support them is inadequate. However, despite the
potential risks associated with off-label uses, efforts to prohibit such uses might hurt the public. Some off-label
prescribing may result because manufacturers have chosen not to invest the resources needed to have FDA add
indications to the drug’s approval and labeling.
A worst-case scenario for the nation’s health would be the widespread acceptance of a drug for an off-label use
that sufficient research would have revealed to be ineffective, unsafe, or both. Aside from the drug’s direct harm,
the time spent waiting to see whether it worked would have been time not spent exploring other treatment options.
Unchecked off-label prescribing may also threaten the FDA gold standard of drug approval. If clinicians had
already accepted a new use into practice through off-label prescribing, a manufacturer may choose to not invest
resources to go through clinical trials and the FDA process to win approval.
Although manufacturers do share information on off-label uses, courts have sometimes found they had
overstepped allowable bounds. Congress has given permission for limited sharing. Are there other ways to share
clinical information that do not put the public’s health or FDA’s authority at risk?
What Role Can Congress Play in the Use of Off-Label Prescriptions?
How might Congress, in its legislative or oversight roles, consider the use of off-label drugs to protect the public’s
health? Legislators and health analysts have suggested both restrictive and permissive actions regarding off-label
use. Ideas—some of which conflict with others—include
R45792
February 23, 2021
Agata Bodie Analyst in Health Policy

Off-Label Use of Prescription Drugs
Congressional Research Service
disclosure to patients;
data collection, availability, and analysis;
dissemination of clinical data;
linking reimbursement and coverage to evidence of safety and effectiveness;
clinical research and research transparency;
clinical guidance;
congressional oversight through the Government Accountability Office, the Federal Trade
Commission, and the Department of Health and Human Services; and
consideration of other countries’ approaches to off-label use.
Some actions would require federal legislation. Other proposals would involve actions by other entities, such as
state authorities and professional organizations, which Congress could urge.

Off-Label Use of Prescription Drugs
Congressional Research Service
Contents
Introduction ..................................................................................................................................... 1
Drug Labeling and Off-Label Use ................................................................................................... 2
Labeling: History, Requirements, and Value ............................................................................. 3 Off-Label Use: Description and Examples ............................................................................... 4
Benefits and Risks of Off-Label Use ............................................................................................... 6
History of Congressional Interest and Action.................................................................................. 8
Possible Avenues of Future Congressional Interest ......................................................................... 9
Concluding Comments .................................................................................................................. 12
Tables
Table 1. Selected Off-Label Uses .................................................................................................... 5
Table A-1. Selected Congressional and FDA Actions to Expand Information in Drug
Labeling ...................................................................................................................................... 15
Appendixes
Appendix. Selected Actions to Expand Information in Drug Labeling ......................................... 15
Contacts
Author Information ........................................................................................................................ 16

Off-Label Use of Prescription Drugs
Congressional Research Service 1
Introduction Both legislators and regulators have expressed concern about the safety and effectiveness of
prescription drugs prescribed for “off-label uses”—purposes other than those for which the Food
and Drug Administration (FDA) has approved their sale.1 Two recent incidents illustrate the bases
for those concerns.
The first involves a drug already on the market. Safety experts raised concerns about ketamine, a
drug available as an injectable anesthetic.2 They noted that physicians have created outpatient
clinics to administer intravenous ketamine in an off-label use to treat depression and migraines.3
FDA has not reviewed clinical data that could support the clinics’ promotional claims of safety
and effectiveness.4
In August 2018, a second incident occurred in a Texas courtroom. Astra Zeneca settled a case
concerning its alleged promotion of Seroquel for uses other than those for which it had sought
and obtained FDA approval for sale in the United States.5 The core of the complaint by the state
of Texas was that the company promoted the drug’s use in children, although the FDA-approved
labeling of the drug was for adult use. It was one of a number of settlements since 2000 resulting
in payments by drug companies for the promotion of off-label uses.6
To help understand the issues involving off-label use, and how these issues might concern
Congress, this report addresses five questions:
What is drug labeling? What is off-label use?
How are off-label prescriptions used in medicine today?
What are the risks and benefits associated with off-label prescriptions?
1 FDA Memorandum, “Public Health Interests and First Amendment Considerations Related to Manufacturer
Communications Regarding Unapproved Uses of Approved or Cleared Medical Products” January 2017,
https://www.regulations.gov/document?D=FDA-2016-N-1149-0040. See Table A-1 for selected history of
congressional concerns.
2 Megan Thielking, “A STAT Investigation: Ketamine gives hope to patients with severe depression. But some clinics
stray from the science and hype its benefits,” STAT, September 24, 2018, https://www.statnews.com/2018/09/24/
ketamine-clinics-severe-depression-treatment; and Melvyn W. Zhang, Keith M. Harris, and Roger C. Ho, “Is Off-label
repeat prescription of ketamine as a rapid antidepressant safe? Controversies, ethical concerns, and legal implications,”
BMC Medical Ethics, vol. 17, no. 4, published online January 14, 2016, https://www.ncbi.nlm.nih.gov/pmc/articles/
PMC4714497/pdf/12910_2016_Article_87.pdf.
3 See, for example, Actify Neurotherapies, https://www.fda.gov/downloads/AdvisoryCommittees/
CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM631429.pdf.
4 In March 2019, FDA approved a new drug application for intranasal esketamine for treatment-resistant depression
(FDA, “FDA approves new nasal spray medication for treatment-resistant depression; available only at a certified
doctor’s office or clinic,” News Release, March 5, 2019, https://www.fda.gov/NewsEvents/Newsroom/
PressAnnouncements/ucm632761.htm?dom=prime&src=syn).
5 Ken Paxton, Attorney General of Texas, “AG Paxton Recovers $110 Million for Texas in Medicaid Fraud
Settlements,” News Release, August 7, 2018, https://www.texasattorneygeneral.gov/news/releases/ag-paxton-recovers-
110-million-texas-medicaid-fraud-settlements; https://www.texasattorneygeneral.gov/sites/default/files/files/epress/
Seroquel_SA_Fully_Executed.pdf.
6 For example, Department of Justice, “GlaxoSmithKline to Plead Guilty and Pay $3 Billion to Resolve Fraud
Allegations and Failure to Report Safety Data,” Office of Public Affairs, July 2, 2012, https://www.justice.gov/opa/pr/
glaxosmithkline-plead-guilty-and-pay-3-billion-resolve-fraud-allegations-and-failure-report; and Department of Justice,
“Fact Sheet: Significant False Claims Act Settlements & Judgments, Fiscal Years 2009-2016,” https://www.justice.gov/
opa/press-release/file/918366/download.

Off-Label Use of Prescription Drugs
Congressional Research Service 2
What concerns, if any, does Congress have about such prescriptions?
If Congress wanted to do something about off-label prescriptions, what would be
some of the options?
Drug Labeling and Off-Label Use To market a prescription drug in the United States, a manufacturer needs FDA approval.7 To
obtain that approval, the manufacturer must demonstrate the drug’s safety and effectiveness
according to criteria specified in law and agency regulations. It must also ensure that its
manufacturing plant passes FDA inspection.
Finally, it must obtain FDA approval for the drug’s labeling—a term that covers all written
material about the drug, including, for example, packaging, prescribing information for
physicians, and patient brochures. FDA, thus, approves the drug and its labeling for a specific
use. That use specifies the disease or condition, the population, and the way the drug is packaged
and administered.
When a physician prescribes a drug for reasons other than those specified in the FDA approval
and labeling, the medical profession considers this to be off-label use.
FDA regulates the drug and the manufacturer. Each state regulates clinicians and pharmacies.8 A
licensed physician may—except in highly restricted circumstances9—prescribe the approved drug
without limitation. A prescription to an individual whose demographic or medical characteristics
differ from those indicated in a drug’s FDA-approved labeling is accepted medical practice.
In a 2006 study of drug prescribing by office-based physicians, 21% of prescriptions were written
for off-label uses. Of those off-label prescriptions, the study’s authors found that 27% were
backed by strong scientific support.10 A 2016 Canadian study of primary care clinics found an
overall rate of 12% of prescriptions for off-label uses. The percentage varied, however, by
therapeutic class, ranging from 5% for ear, nose, and throat medications to 25% for central
7 FDA-approved drugs are designated by law into only two categories: prescription and nonprescription (also referred
to as over-the-counter). No drug was prescription-only until the 1951 Humphrey-Durham amendments [P.L. 82-215,
the Food, Drug, and Cosmetics Act Amendments Act, 1951], which stated, “A drug intended for use by man which ...
because of its toxicity or other potentiality for harmful effect, or the method of its use, or the collateral measures
necessary to its use, is not safe for use except under the supervision of a practitioner licensed by law to administer such
drug” (FFDCA §503(b)(1)). See CRS In Focus IF10463, Regulation of Over-the-Counter (OTC) Drugs.
8 The FFDCA does not give FDA authority to regulate the practice of medicine; that responsibility generally rests with
the states and medical professional associations.
9 FDA may place restrictions on prescribing or dispensing as “elements to assure safe use” (ETASU) as part of a risk
evaluation and mitigation strategy (REMS) that the agency may require (see FFDCA §505-1(f)).
10 David C. Radley, Stan N. Finkelstein, and Randall S. Stafford, “Off-label Prescribing Among Office-Based
Physicians,” Archives of Internal Medicine, vol. 166, May 8, 2006. The authors note, “[a]n indication was considered
to be scientifically supported if, according to DRUGDEX, its effectiveness has been shown in controlled trials or
observed in clinical settings.”

Off-Label Use of Prescription Drugs
Congressional Research Service 3
nervous system medications.11 An econometric model from the National Ambulatory Medical
Care Survey estimated a 38% rate of off-label use.12
Research has shown that more than half of oncology drug use is off-label. A 2018 study examined
43 FDA-approved cancer drugs and compared their 99 labeled uses with the acceptable uses
published by a national compendium Medicare relies on to make coverage decisions.13 Of the 451
compendium-accepted uses, 56% were off-label. Of the off-label uses, the authors deemed 91%
as “well-accepted off-label use.”14
Labeling: History, Requirements, and Value
History. Drug labeling has been central to FDA’s role as a protector of the public’s health since
1906. That year, Congress (1) required that sellers state on a drug’s label the “quantity or
proportion of any alcohol [or] opium” contained, and (2) considered as “misbranded” any drug
whose label was “false or misleading.” Requirements that drugs be safe were not established until
1938. Congress did not require they be effective until 1962.15
Requirements. Today, a drug’s labeling is more than the sticker the pharmacy places on the
amber vial it dispenses to a customer.16 The Federal Food, Drug, and Cosmetics Act (FFDCA)
and associated FDA regulations17 require and describe a product’s labeling as “a compilation of
information about the product, approved by FDA, based on the agency’s thorough analysis of the
new drug application (NDA) or biologics license application (BLA) submitted by the applicant.
This labeling contains information necessary for safe and effective use.”
11 Tewodros Eguale, David L. Buckeridge, Aman Verma, Nancy E. Winslade, Andrea Benedetti, James A. Hanley,
Robyn Tamblyn, “Association of Off-label Drug Use and Adverse Drug Events in an Adult Population,” JAMA
Internal Medicine, vol. 176, no. 1, January 2016, https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/
2467782.
12 W. David Bradford, John Turner, and Jonathan W. Williams, “Off-Label Use of Pharmaceuticals: A Detection
Controlled Estimation Approach,” presentation at the Innovation and Behavior in Health Markets Conference, 2016,
revised May 24, 2018, https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2230976.
13 Michael B. Shea, Mark Stewart, Hugo Van Dyke, Linda Ostermann, Jeff Allen, and Ellen Sigal, “Outdated
Prescription Drug Labeling: How FDA-Approved Prescribing Information Lags Behind Real-World Clinical Practice,”
Therapeutic Innovation & Regulatory Science, vol. 52, issue 6 (first published online March 5, 2018; issue published
November 1, 2018), https://www.focr.org/sites/default/files/pdf/FOCR-Labeling-Research-Article.pdf?eType=
EmailBlastContent&eId=067b86ca-de59-4767-86ab-8a4ad95a7978; abstract at https://journals.sagepub.com/doi/full/
10.1177/2168479018759662.
14 The authors (Shea et al.) defined “well-accepted off-label drug use” if the National Comprehensive Cancer Network
(NCCN) Drugs and Biologics Compendium graded the use in its top two categories of evidence.
15 Federal Food and Drug Act of 1906 (F&DA), P.L. 59-384, 1906; Federal Food, Drug, and Cosmetic Act (FFDCA),
P.L. 75-717, 1938; and Kefauver-Harris Drug Amendments to the FFDCA, P.L. 87-781, 1962.
16 For examples of labeling, search for a drug at Drugs@FDA (https://www.accessdata.fda.gov/scripts/cder/daf/).
17 FFDCA Section 201(m), 21 C.F.R. Part 201—Labeling, and FDA, “[Docket No. 2000N–1269] (formerly Docket No.
00N–1269) RIN 0910–AA94, Requirements on Content and Format of Labeling for Human Prescription Drug and
Biological Products,” Federal Register, vol. 71, no. 15, January 24, 2006, pp. 3922-3997, http://www.gpo.gov/fdsys/
pkg/FR-2006-01-24/pdf/06-545.pdf.

Off-Label Use of Prescription Drugs
Congressional Research Service 4
FDA requires that labeling begin with a highlights section that includes, if appropriate, black-box
warnings, so called because their black borders signify importance.18 The regulations list the
required elements of labeling:19
indications and usage
dosage and administration
dosage forms and strengths
contraindications
warnings and precautions
adverse reactions
drug interactions
use in specific populations
drug abuse and dependence
overdosage
description
clinical pharmacology
nonclinical toxicology
clinical studies
references
how supplied/storage and
handling
patient counseling information
Value. Labeling plays a major role in the presentation of safety and effectiveness information. For
clinicians, it is a primary source of prescribing information. The manufacturer submits the
approved labeling for publication in the widely used Physician’s Desk Reference. That labeling
also serves as the basis for several patient-focused information sheets that manufacturers,
pharmacy vendors, and many web-based drug information sites produce.20
Off-Label Use: Description and Examples
Off-label prescribing can reflect cutting-edge clinical expertise. It can also be a response to price:
a physician may choose to prescribe a lower-priced drug instead of a specifically labeled higher-
priced one. Or a physician may prescribe off-label in an attempt to try a different treatment
approach when other options have failed.
Sometimes an off-label use becomes so widespread that it becomes accepted practice. However,
without the backing of carefully designed clinical trials and expert analysis, it remains unknown
whether the drug is, in fact, safe and effective for the off-label use. Also unknown are dosing
details and systematically evaluated associated adverse events.
Examples of off-label use include
a drug tested for the treatment of one disease prescribed in an attempt to prevent
or treat another;
a drug tested at one dose used at higher or lower doses;
a drug tested in adults prescribed to children; and
a drug tested in an eight-week trial prescribed for long-term use.
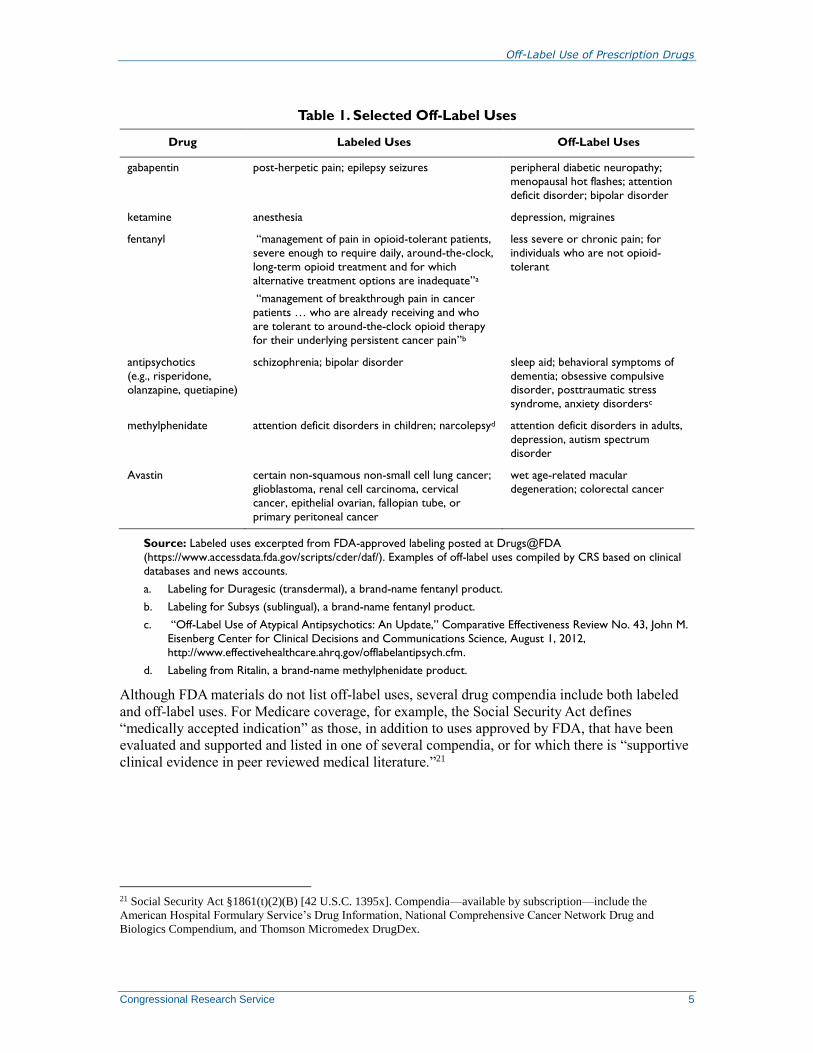
Table 1 lists several examples of FDA-approved drugs widely prescribed for off-label uses.
18 “Boxed warning. Certain contraindications or serious warnings, particularly those that may lead to death or serious
injury, may be required by the FDA to be presented in a box” (21 C.F.R. 201.57(c)(1)).
19 21 C.F.R. 201.56(d). For older drugs, labeling content requirements are listed in 21 C.F.R. 201.56(e).
20 PDR Network, http://www.pdr.net/about-pdr-network/. Examples of consumer medication information materials
include those by Wolters Kluwer Health, Inc. (used, for example, with Target dispensing,
http://www.consumerreports.org/health/best-buy-drugs/prescription-labels/patient-instructions/index.htm); First
Databank, Inc. (provides FDA Medication Guides, http://www.fdbhealth.com/solutions/retail-pharmacy-dispensing/);
and American Society of Health-System Pharmacists Consumer Medication Information
(http://www.ahfsdruginformation.com/products_services/ahfs_cmi.aspx).
Off-Label Use of Prescription Drugs
Congressional Research Service 5
Table 1. Selected Off-Label Uses
Drug Labeled Uses Off-Label Uses
gabapentin post-herpetic pain; epilepsy seizures peripheral diabetic neuropathy;
menopausal hot flashes; attention
deficit disorder; bipolar disorder
ketamine anesthesia depression, migraines
fentanyl “management of pain in opioid-tolerant patients,
severe enough to require daily, around-the-clock,
long-term opioid treatment and for which
alternative treatment options are inadequate”a
“management of breakthrough pain in cancer
patients … who are already receiving and who
are tolerant to around-the-clock opioid therapy
for their underlying persistent cancer pain”b
less severe or chronic pain; for
individuals who are not opioid-
tolerant
antipsychotics
(e.g., risperidone,
olanzapine, quetiapine)
schizophrenia; bipolar disorder sleep aid; behavioral symptoms of
dementia; obsessive compulsive disorder, posttraumatic stress
syndrome, anxiety disordersc
methylphenidate attention deficit disorders in children; narcolepsyd attention deficit disorders in adults,
depression, autism spectrum
disorder
Avastin certain non-squamous non-small cell lung cancer;
glioblastoma, renal cell carcinoma, cervical
cancer, epithelial ovarian, fallopian tube, or
primary peritoneal cancer
wet age-related macular
degeneration; colorectal cancer
Source: Labeled uses excerpted from FDA-approved labeling posted at Drugs@FDA
(https://www.accessdata.fda.gov/scripts/cder/daf/). Examples of off-label uses compiled by CRS based on clinical
databases and news accounts.
a. Labeling for Duragesic (transdermal), a brand-name fentanyl product.
b. Labeling for Subsys (sublingual), a brand-name fentanyl product.
c. “Off-Label Use of Atypical Antipsychotics: An Update,” Comparative Effectiveness Review No. 43, John M.
Eisenberg Center for Clinical Decisions and Communications Science, August 1, 2012,
http://www.effectivehealthcare.ahrq.gov/offlabelantipsych.cfm.
d. Labeling from Ritalin, a brand-name methylphenidate product.
Although FDA materials do not list off-label uses, several drug compendia include both labeled
and off-label uses. For Medicare coverage, for example, the Social Security Act defines
“medically accepted indication” as those, in addition to uses approved by FDA, that have been
evaluated and supported and listed in one of several compendia, or for which there is “supportive
clinical evidence in peer reviewed medical literature.”21
21 Social Security Act §1861(t)(2)(B) [42 U.S.C. 1395x]. Compendia—available by subscription—include the
American Hospital Formulary Service’s Drug Information, National Comprehensive Cancer Network Drug and
Biologics Compendium, and Thomson Micromedex DrugDex.

Off-Label Use of Prescription Drugs
Congressional Research Service 6
Benefits and Risks of Off-Label Use Why has Congress given FDA the authority to regulate whether a drug may be on the U.S.
market? Two key reasons: to protect patients and to encourage research in a competitive
pharmaceutical industry.22
By statute and regulation, FDA now approves a drug for a specific use once its sponsor (usually
the manufacturer) has provided sufficient evidence that the drug is safe and effective for that use.
FDA has developed procedures for the review of that evidence. The FDA-approved labeling,
which informs the clinician about dosing and likely and unlikely adverse events, helps protect the
individuals for whom the drug is prescribed.
Labeling also helps protect the interests of the manufacturers who invest in the clinical trials that
demonstrate safety and effectiveness. For new drugs and new uses of already approved drugs, the
sponsor receives a period of market protection, in the form of regulatory exclusivity for the sale
of the drug for those uses. Payors—such as private health insurers or Medicare—benefit from
FDA-approved labeling in their evaluation of whether to pay for a drug’s use.
But use of a drug evolves as clinicians (and the manufacturer) share their experiences regarding
off-label uses, which, by definition, were not part of the premarket clinical studies used to obtain
FDA approval. Off-label use can benefit patients. In some instances, such as in the treatment of
rare diseases, clinical practice may use drugs approved for other indications. A manufacturer may
choose not to invest in trials for such a small patient group.23 A patient whose physician is already
prescribing the drug off-label may not want to enroll in a clinical trial where there is a chance he
or she may be assigned to the placebo group. Once drugs are well-established in off-label uses,
manufacturers rarely design studies to determine or verify the safety and effectiveness of such
uses. Individuals and groups wanting to conduct such studies may find it hard to obtain funding.
Examples of adverse events (AEs) associated with the use of drugs for specific off-label uses
include heart valve damage from the use of fenfluramine and phentermine (fen-phen) for weight
loss, and seizures from the use of tiagabine hydrochloride for depression.24 Using a Canadian
primary care database that captured all prescriptions, the reason for each prescription, and adverse
events, researchers looked at the rate of AEs for on-label use, off-label use associated with
“strong scientific evidence,” and off-label use without such evidence. They found more AEs for
off-label prescriptions than for on-label prescriptions. However, off-label use associated with
strong scientific evidence had similar rates of AEs as did on-label uses. The increased risk of AEs
for off-label use was concentrated in those uses without strong scientific evidence.25
22 FDA statement at https://www.fda.gov/AboutFDA/Transparency/Basics/ucm194877.htm; and FFDCA §1003 [21
U.S.C. 393].
23 See, for example, testimony to FDA by the National Organization for Rare Disorders, November 9, 2016,
https://rarediseases.org/wp-content/uploads/2014/11/2016-11-09.-NORD-Statement-at-FDA-Off-Label-Public-
Hearing.pdf.
24 Tewodros Eguale, David L. Buckeridge, Aman Verma, Nancy E. Winslade, Andrea Benedetti, James A. Hanley,
Robyn Tamblyn, “Association of Off-label Drug Use and Adverse Drug Events in an Adult Population,” JAMA
Internal Medicine, vol. 176, no. 1, January 2016, https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/
2467782.
25 Tewodros Eguale, David L. Buckeridge, Aman Verma, Nancy E. Winslade, Andrea Benedetti, James A. Hanley,
Robyn Tamblyn, “Association of Off-label Drug Use and Adverse Drug Events in an Adult Population,” JAMA
Internal Medicine, vol. 176, no. 1, January 2016, https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/
2467782.

Off-Label Use of Prescription Drugs
Congressional Research Service 7
Manufacturers benefit from sales for off-label uses. However, they risk losing that market should
a competitor complete studies to obtain FDA approval and labeling for those uses. Researchers
who are not supported by the manufacturer who try to assess the safety and effectiveness of off-
label uses are hampered by the inexact nature of secondary data sources: the standard clinical trial
data collection in preparation for an FDA application is not available for off-label uses. What may
begin as hopeful and intermittent off-label use may gain momentum and offer opportunities for
planned studies.
Drug and device companies argue that current regulations prevent them from distributing
important information to physicians and payors about unapproved, off-label uses of their
products.26 In November 2016, FDA held a two-day public meeting to hear from various groups
regarding off-label uses of approved or cleared medical products.27 In June 2018, FDA issued
final guidances explaining the agency’s policy about medical product communications that
include data and information not contained in FDA-approved labeling.28
One FDA guidance document, in particular, described the types of information that a
manufacturer could provide payors and formulary committees about unapproved uses of
approved products.29 FDA made two points especially relevant to off-label uses.
First, FDA differentiates among its audiences in its presentation of information. The material it
allows in product labeling is directed to a clinical audience. FDA staff have reviewed the
information and require that it be presented in a way that is understandable by individual
clinicians, who often do not have the statistical sophistication or data analysis skills or resources
to fully evaluate the claims of manufacturers. This guidance notes, though, that payors and
formulary committees do have such expertise and resources. FDA also acknowledges that it is
useful for payors to have information in their decisions on coverage, but it wants to ensure that
the information manufacturers provide is not misleading.
Second, the FDA guidance describes what harm could come from allowing more sharing of
information about off-label use.
Some firm communications regarding unapproved products or unapproved uses of
approved/cleared/licensed medical products may potentially undermine substantial
government interests related to health and safety. These interests include motivating the
development of robust scientific data on safety and efficacy; maintaining the premarket
review process for safety and efficacy of each intended use in order to prevent harm, to
protect against fraud, misrepresentation, and bias, and to develop appropriate instructions
for use for medical products; protecting the integrity and reliability of promotional
26 STAT, “FDA to hold long-awaited meeting to review off-label marketing,” August 31, 2016,
https://www.statnews.com/pharmalot/2016/05/27/fda-hhs-free-speech-patient-safety/.
27 FDA, Docket No. FDA-2016-N-1149, https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-
21062.pdf. All prescription drugs must receive FDA approval for marketing. About 1% of medical devices go through
a comparable approval process; most devices receive FDA clearance for marketing. For a description of FDA medical
device regulation, see CRS Report R42130, FDA Regulation of Medical Devices.
28 FDA, “Guidance for Industry: Medical Product Communications That Are Consistent With the FDA-Required
Labeling—Questions and Answers,” Center for Drug Evaluation and Research, Center for Biologics Evaluation and
Research, Center for Devices and Radiological Health, Center for Veterinary Medicine, and Office of the
Commissioner, June 2018, https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/
guidances/ucm537130.pdf.
29 FDA, “Guidance for Industry and Review Staff: Drug and Device Manufacturer Communications With Payors,
Formulary Committees, and Similar Entities—Questions and Answers,” Center for Drug Evaluation and Research,
Center for Biologics Evaluation and Research, Center for Devices and Radiological Health, and Office of the
Commissioner, June 2018, https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/UCM537347.pdf.

Off-Label Use of Prescription Drugs
Congressional Research Service 8
information regarding medical product uses; and preventing the diversion of health care
resources toward ineffective treatments.
The concerns FDA raised in the June 2018 final guidance documents might explain why Congress
may be interested in exploring the issue of off-label use further.
History of Congressional Interest and Action Congress has developed a system to protect the public by ensuring that drugs sold in the United
States have met clinical and manufacturing standards of safety and effectiveness. Labeling is the
mechanism that bridges the regulated drug and the use of that drug in clinical practice. The
labeling establishes the uses for which the manufacturer has demonstrated safety and
effectiveness to FDA’s satisfaction. Congress and the FDA have tried several approaches, using
labeling as a tool, to reduce the risk to patients. Table A-1 includes examples of congressional
and FDA actions to expand the information provided in a drug’s labeling. The actions have
addressed topics such as
content labeling,
directions for use,
permissible and prohibited advertising,
research incentives to support labeling specific to population subgroups (e.g.,
children),
criteria for Medicare coverage,
required and permissible labeling changes, and
balance of benefit and risk information in labeling and advertising.
Manufacturers, meanwhile, want to be able to provide information to insurers and other entities
(such as the Centers for Medicare and Medicaid Services [CMS] and hospital pharmacy and
therapeutics committees) that decide whether to cover a drug in a policy and whether to limit
reimbursement for specific uses. Congress has provided some leeway relative to its earlier
prohibition on promotional activities. For example, in 2016, it broadened the types of health care
economic information drug and device manufacturers could provide to payors (e.g., insurance
companies).30 Although the underlying FFDCA section continues to exclude information related
only to an off-label use, it now requires “a conspicuous and prominent statement describing any
material differences between the health care economic information and the labeling approved for
the drug.” Several bills concerning off-label use have been introduced that would have expanded
the information that manufacturers could provide about off-label uses. For example, the
Subcommittee on Health of the House Committee on Energy and Commerce marked up H.R.
2026 (115th Congress), the Pharmaceutical Information Exchange Act, in January 2018, which
would have allowed the provision of scientific information to payors, in addition to health care
economic information.31 Then-Subcommittee Chair Michael Burgess, in commenting on the bill’s
effort to clarify how manufacturers can share “if it is based on competent and reliable evidence,”
expressed strong support for the bill. He noted “the importance of cutting edge information in
30 Sec. 3037 of P.L. 114-255, the 21st Century Cures Act.
31 Subcommittee on Health, House Committee on Energy and Commerce, mark-up of H.R. 2026, January 17, 2018,
https://energycommerce.house.gov/committee-activity/markups/markup-of-hr-over-the-counter-monograph-safety-
innovation-and-reform-act.

Off-Label Use of Prescription Drugs
Congressional Research Service 9
medicine and science to optimize patient care and outcomes … and [how the bill] could have the
potential to save patients’ lives.”32
Then-Ranking Member (now, Chair) Frank Pallone, Jr., though, argued that the ability to
communicate about off-label use “had great potential to undermine” FDA’s approval process and
to “hamstring” its enforcement efforts.33 H.R. 2026 passed the subcommittee, although it did not
reach the floor in the 115th Congress.
Possible Avenues of Future Congressional Interest Off-label use presents both opportunities and risks to clinicians, patients, manufacturers, and
researchers. At times, those interests clash. Academics, public health organizations, and journals
have suggested what actions Congress might take based on their particular concerns regarding
off-label prescriptions.34 These actions include both direct legislation and oversight activities to
encourage action by other entities. The 116th Congress might consider some of these varied
approaches, summarized in the issues described below.
Disclosure to patients. Most individuals, unaware of the nuances of FDA regulation, may not
know that physicians may prescribe drugs for uses that FDA has not reviewed for safety and
effectiveness. Several potential opportunities for providing this information exist. Congress could
require or work with the states to require that
the prescriber inform the patient about the off-label use and describe the meaning
of off-label use;
the prescriber note in the prescription why the drug is being prescribed; or
the pharmacist inform the patient that the use is off-label.
Data collection and availability. FDA, under federal law, determines whether a drug requires a
prescription. The states, under their individual laws, determine what information the prescription
order contains. Because clinicians do not need to note on the prescription order why they are
prescribing a drug (e.g., simvastatin for high cholesterol or citalopram for depression), the
information in pharmacy, administrative, and clinical databases often cannot directly identify off-
label uses. Therefore, as Congress and other public policy groups consider whether and how to
address off-label drug prescribing, they do not have adequate information on the scope and details
of the practice. Congress could require or work with the states to encourage
32 Subcommittee on Health, House Committee on Energy and Commerce, mark-up of H.R. 2026, January 17, 2018,
https://energycommerce.house.gov/committee-activity/markups/markup-of-hr-over-the-counter-monograph-safety-
innovation-and-reform-act.
33 Subcommittee on Health, House Committee on Energy and Commerce, mark-up of H.R. 2026, January 17, 2018,
https://energycommerce.house.gov/committee-activity/markups/markup-of-hr-over-the-counter-monograph-safety-
innovation-and-reform-act.
34 For example: Zachary Brennan, “New Report Investigates Drivers of Off-Label Prescribing in the EU,” Regulatory
Focus, March 3, 2017; Christine Fukada, Jillian Clare Kohler, Heather Boon, Zubin Austin, and Murray Krahn,
“Prescribing gabapentin off label: Perspectives from psychiatry, pain and neurology specialists,” CPJ/RPC, vol. 145,
no. 6, November/December 2012; Megan Thielking, “A STAT Investigation: Ketamine gives hope to patients with
severe depression. But some clinics stray from the science and hype its benefits,” STAT, September 24, 2018,
https://www.statnews.com/2018/09/24/ketamine-clinics-severe-depression-treatment; and Joshua D. Wallach and
Joseph S. Ross, “Gabapentin Approvals, Off-Label Use, and Lessons from Postmarketing Evaluation Efforts,” JAMA,
vol. 319, no. 8, February 27, 2018.

Off-Label Use of Prescription Drugs
Congressional Research Service 10
clinicians to note on prescriptions the reason for medication use (e.g., the specific
condition, disease, or symptom), thereby allowing that information to appear in
pharmacy databases, which would enable focused analysis of off-label uses;
the establishment of confidential registries of off-label prescribing and follow-up
information that FDA (or other designated scientifically appropriate agencies)
could use in its electronic surveillance systems to identify associated adverse
events and other drug use problems; or
FDA to increase its surveillance of available data sources, such as registries and
administrative and clinical databases, to identify patterns of off-label use and
evidence suggesting effectiveness and associated adverse events.
Dissemination. Because some information may be valuable to clinicians and entities that
influence prescribing decisions (such as insurers and pharmacy and therapeutics committees),
Congress could allow manufacturers to disseminate information about off-label uses that they
have developed or of which they are aware, perhaps subject to certain limitations or
accompanying reporting requirements. Possibilities include
broader sharing of clinical analyses of off-label use with coverage deciders (e.g.,
CMS, insurers, pharmacy and therapeutics committees) to support requests that
they cover a particular use of the drug; and
dissemination of clinical analyses of off-label use at clinical and pharmaceutical
conferences.
Oversight. With an estimated 12% to 38% of all prescriptions’ (and 56% of oncology
prescriptions’) being written for uses not listed on FDA-approved labeling,35 valid information on
the extent of off-label use and the effect of such use on manufacturer, insurer, and clinician
behavior could potentially better inform debate on how to best protect the public’s health.
Congress could direct
the Government Accountability Office (GAO) to study the extent of off-label use
in government-provided care (e.g., Department of Veterans Affairs, Bureau of
Prisons, and Indian Health Service) and in government-funded care (e.g.,
Medicare and Medicaid);
the Secretary of Health and Human Services (HHS) to contract with the National
Academy of Medicine or a similar organization to assemble management or
industrial policy experts to study the costs and rewards to industry of off-label
prescriptions; or
35 David C. Radley, Stan N. Finkelstein, and Randall S. Stafford, “Off-label Prescribing Among Office-Based
Physicians,” Archives of Internal Medicine, vol. 166, May 8, 2006; Tewodros Eguale, David L. Buckeridge, Aman
Verma, Nancy E. Winslade, Andrea Benedetti, James A. Hanley, Robyn Tamblyn, “Association of Off-label Drug Use
and Adverse Drug Events in an Adult Population,” JAMA Internal Medicine, vol. 176, no. 1, January 2016,
https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/2467782; W. David Bradford, John Turner, and
Jonathan W. Williams, “Off-Label Use of Pharmaceuticals: A Detection Controlled Estimation Approach,”
presentation at the Innovation and Behavior in Health Markets Conference, 2016, revised May 24, 2018,
https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2230976; Michael B. Shea, Mark Stewart, Hugo Van Dyke, Linda
Ostermann, Jeff Allen, and Ellen Sigal, “Outdated Prescription Drug Labeling: How FDA-Approved Prescribing
Information Lags Behind Real-World Clinical Practice,” Therapeutic Innovation & Regulatory Science, vol. 52, issue 6
(first published online March 5, 2018; issue published November 1, 2018), https://www.focr.org/sites/default/files/pdf/
FOCR-Labeling-Research-Article.pdf?eType=EmailBlastContent&eId=067b86ca-de59-4767-86ab-8a4ad95a7978;
abstract at https://journals.sagepub.com/doi/full/10.1177/2168479018759662.

Off-Label Use of Prescription Drugs
Congressional Research Service 11
FDA and the Federal Trade Commission to investigate drugs whose off-label
prescriptions account for a particularly high percentage of all prescriptions or that
generate a particularly high percentage or high dollar value of sales.
Pricing. The decision to use a drug off-label based solely or primarily on price introduces a new
element to study. For example, the pricing difference between Avastin and Lucentis, both made
by the same manufacturer with the same active ingredient, is so large that many ophthalmologists
use the lower priced Avastin in their treatment of macular degeneration, despite its being an off-
label use.36 The manufacturer included the treatment of macular degeneration in its application to
FDA for Lucentis. To explore the ramifications of a traditionally nonclinical element in
prescribing decisions, Congress could require
the HHS Secretary to study the relationship among pricing, access, and
prescribing and its effect on patient safety.
Reimbursement. Although FDA approval of a drug—not for each use of a drug—is a
requirement for sale in the United States, the decision to reimburse a physician or patient for a
drug is made by entities such as the CMS and private insurers. Such decisions, therefore,
influence what drugs are prescribed and, in part, for what uses drugs are prescribed. Congress
could direct
the HHS Secretary to study such coverage decisions or to contract with the
National Academy of Medicine or a similarly equipped entity to do so; or
HHS to form a task force to include CMS and FDA, along with private insurers
and others involved in coverage decisions, and patient and clinician groups
representing those affected by coverage decisions, to identify areas in need of
action and to recommend steps in those directions.
Congress also could encourage
payors to require safety and effectiveness evidence before covering off-label
uses.
Research. The traditional path toward adding an indication (reason for use) to the labeling of a
drug already approved for other uses has been for the sponsor of the drug to conduct clinical trials
and submit a supplemental new drug application (NDA) to FDA. The widespread extent of off-
label use suggests that relying on that model is not helping prescribers get better information.
Congress could consider
assigning responsibility (and funding) to the National Institutes of Health and the
Patient-Centered Outcomes Research Institute for safety and effectiveness
evaluations of off-label uses; or
requiring, for a drug that has substantial (to be defined) off-label sales, that the
manufacturer37 fund studies, such as clinical trials, to assess the safety and
effectiveness of the drug for the off-label use and submit evidence to the HHS
Secretary. Depending on the Secretary’s assessment of the evidence, the
Secretary could request that the manufacturer amend the drug’s labeling either to
36 Kierstan Boyd, “What Is Avastin?” American Academy of Ophthalmology, May 17, 2018, https://www.aao.org/eye-
health/diseases/amd-macular-degeneration.
37 For drugs on patent, this would be the brand-name sponsor of the new drug application (NDA). For drugs no longer
on patent that are only marketed as generic products, this would be the holder of the abbreviated NDA. For a
description of the regulation of generic drugs, see CRS Report R44703, Generic Drugs and GDUFA Reauthorization:
In Brief.

Off-Label Use of Prescription Drugs
Congressional Research Service 12
add the off-label use to the label as an approved use or to add a statement that
clinical evidence does not support the safety and effectiveness of the drug for the
off-label use.
Research transparency. FDA regulations describe standards for the design and analysis of
clinical trials that a sponsor uses in an NDA. Studies done by or for the manufacturer or by other
groups or individuals are not always made public, in which case their findings cannot be reviewed
and evaluated. Because the results of such studies may be used in support of off-label uses, by
providing positive and negative incentives, Congress could consider requiring or encouraging
prospective posting of the designs and statistical plans of studies of off-label
uses; or
public reporting of studies of off-label use.
Clinical guidance. Professional societies and other clinical groups often supplement the
information available to prescribers from FDA-approved labeling, medical journals, and
information from manufacturers. They can issue guidelines and recommend best practices.
Congress could engage such groups and encourage
professional societies to develop evidence-based clinical guidelines and training
regarding off-label use.
Precedents from other countries. The United States is not alone in facing the health care and
economic implications of off-label use. For example, the member countries of the European
Union have addressed measures involving reimbursement, guidance for prescribers, professional
standards, and informed consent.38 Congress could require
HHS to contract with the National Academy of Medicine or a similarly equipped
entity to review measures taken by the European Union and other regulatory
bodies and recommend legislative or administrative actions as appropriate.
Concluding Comments Concerns over off-label use overlap with questions raised by some legislators and regulators in
other contexts. In addition to clearly related issues such as what is allowable information in
direct-to-consumer advertising, promotion to clinicians, and material shared with payors and
insurers, off-label use also affects the entire basis of FDA regulation of drugs through its authority
to approve drugs. That means it directly or indirectly affects research, clinical innovation,
transparency, patents and exclusivities, pricing, and access.
Changes in law or regulation in any one area may have benefits in some areas and drawbacks in
others. For example, FDA’s initiative to encourage research in drugs used traditionally but never
reviewed and approved by FDA—so-called legacy drugs—stems from its desire for evidence of
safety and effectiveness.39 Such evidence helps protect patients from possible use of ineffective,
38 Zachary Brennan, “New Report Investigates Drivers of Off-Label Prescribing in the EU,” Regulatory Focus, March
3, 2017.
39 FDA, Unapproved Drugs Initiative, https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/
EnforcementActivitiesbyFDA/SelectedEnforcementActionsonUnapprovedDrugs/ucm118990.htm Guidance for FDA
Staff and Industry, and FDA “Marketed Unapproved Drugs—Compliance Policy Guide Sec. 440.100 Marketed New
Drugs without Approved NDAs or ANDAs,” September 19, 2011, https://www.fda.gov/downloads/drugs/
guidancecomplianceregulatoryinformation/guidances/ucm070290.pdf.

Off-Label Use of Prescription Drugs
Congressional Research Service 13
unsafe, or misdosed drugs. That initiative, in turn, helps enable sponsors to conduct the clinical
trials, submit new drug applications, obtain regulatory exclusivity with the new drug approval—
and then raise the price of the new branded product while expecting FDA to honor the exclusivity
and block the sale of the drug by others. For example, clinicians had been prescribing a
compounded version of progestin for use in preventing preterm delivery. Such use had never been
adequately tested in clinical trials. One company conducted those trials, and FDA approved its
new drug application. The initial price for the new drug, Makena, was $30,000 for a 20-week
course; patients had been paying pharmacists $200-$400 for the same course.40 Unanticipated
price increases can also arise from the apparent following of FDA procedures. For example, a
new owner of the FDA-approval of an old antiparasitic generic drug raised the price from $14 per
tablet to $750, Daraprim’s initial price.41 Such sudden and large price increases become a barrier
to patient access and, therefore, a potential threat to health.
In the case of off-label prescribing, actions in any direction—whether by Congress, FDA, or the
courts—could have both intended and unanticipated effects. Actions to limit off-label prescribing
could have the intended effects of reducing safety risks and the economic cost of using drugs
ineffective for their prescribed purposes. Such limits, however, could also stifle informed clinical
exploration. Similarly, incentives to mount clinical trials needed to add an indication to the label
could help identify and disseminate information on dosing and contradictions. Such incentives,
however, could also hurt. For example, a brand drug that added a new indication to its labeling
could prevent, through exclusivities, generics from similarly modifying their labeling. Patients
could need to pay more.
The recent debates over the right-to-try movement regarding use of investigational drugs by
terminally ill patients42 may preview discussion about any proposed restrictions on off-label use.
The Goldwater Institute, considered a key impetus to development and passage of the 2018
enactment of a right-to-try act,43 looks to diminish government’s role in an individual’s choice
and has supported dissemination of off-label information.44
40 Joanne Armstrong, “Unintended Consequences—The Cost of Preventing Preterm Births after FDA Approval of a
Branded Version of 17OHP,” New England Journal of Medicine, online March 16, 2011; and Gardiner Harris, “Drugs’
Cost and Safety Fuel a Fight,” New York Times, April 4, 2011, https://www.nytimes.com/2011/04/05/health/
05FDA.html?rref=collection%2Fbyline%2Fgardiner-harris&action=click&contentCollection=undefined®ion=
stream&module=stream_unit&version=search&contentPlacement=1&pgtype=collection. The compounded version of
the drug continues to be available for indications other than prevention of prematurity. FDA has not actively sought to
stop sales. See Yesha Patel and Martha M. Rumore, Controversies in Practice “Hydroxyprogesterone Caproate
Injection (Makena) One Year Later: To Compound or Not to Compound—That Is the Question,” P&T, vol. 37 no. 7,
July 2012, pp. 405-411, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3411212/pdf/ptj3707405.pdf, and FDA,
“Questions and Answers on Updated FDA Statement on Compounded Versions of hydroxyprogesterone caproate (the
active ingredient in Makena),” Press Announcement, Page Last Updated: 06/29/2012, http://wayback.archive-it.org/
7993/20170113105722/http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm310215.htm.
41 Andrew Pollack, “Drug Goes From $13.50 a Tablet to $750, Overnight,” New York Times, September 20, 2015,
https://www.nytimes.com/2015/09/21/business/a-huge-overnight-increase-in-a-drugs-price-raises-protests.html.
42 CRS Report R45414, Right to Try: Access to Investigational Drugs.
43 See, for example, ADI News Services, “Goldwater Institute’s Right To Try Bill Signed By California Governor,”
Arizona Daily Independent, September 29, 2016, https://arizonadailyindependent.com/2016/09/29/goldwater-institutes-
right-to-try-bill-signed-by-california-governor/; and Marie McCullough, “For the terminally ill, are ‘right-to-try’ laws
offering a lifeline, or false hope?” The [Philadelphia] Inquirer, April 4, 2017, https://www.philly.com/philly/health/
Do-Right-to-Try-laws-offer-a-lifeline-or-false-hope-to-the-terminally-ill.html.
44 Naomi Lopez Bauman, “Restoring Free Speech in Medicine,” Goldwater Institute, June 6, 2017,
https://goldwaterinstitute.org/article/restoring-free-speech-in-medicine; and Starlee Coleman, “Arizona becomes first
state to allow pharmaceutical companies to legally communicate off-label treatment uses to medical professionals,”
Goldwater Institute, March 23, 2017, https://goldwaterinstitute.org/article/arizona-becomes-first-state-to-allow-

Off-Label Use of Prescription Drugs
Congressional Research Service 14
Congress has built a process in which a robust FDA can regulate drugs to protect the public’s
health. Is there cause for concern that perhaps a third45 of prescriptions are for off-label uses and
that, in at least one study, three-quarters of those had minimal or no accepted scientific evidence
to support their use?46
Policy tension exists over the line between wanting to ensure individuals’ freedom to take drugs
for off-label uses and wanting to protect the public from the risk of unsafe or ineffective drugs.
Where to draw that line—and how to know when it may be time to move the line—is of
continuing interest to regulators and legislators.
pharmaceutical-companies-legally-communicate-off-label-treatment/.
45 W. David Bradford, John Turner, and Jonathan W. Williams, “Off-Label Use of Pharmaceuticals: A Detection
Controlled Estimation Approach,” presentation at the Innovation and Behavior in Health Markets Conference, 2016,
revised May 24, 2018, https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2230976.
46 David C. Radley, Stan N. Finkelstein, and Randall S. Stafford, “Off-label Prescribing Among Office-Based
Physicians,” Archives of Internal Medicine, vol. 166, May 8, 2006.

Off-Label Use of Prescription Drugs
Congressional Research Service 15
Appendix. Selected Actions to Expand Information
in Drug Labeling
Table A-1. Selected Congressional and FDA Actions to
Expand Information in Drug Labeling
Date Topic Law or Agency Action
1906 Required that sellers state on a drug’s label the
“quantity or proportion of any alcohol, opium, …”
contained; and considered as “misbranded” any drug
whose label was “false or misleading.”
Federal Food and Drug Act of 1906 (F&DA), P.L.
59-384
1938 Required that labeling include adequate directions
for use and evidence of safety of their labeled use.
Federal Food, Drug, and Cosmetic Act (FFDCA),
P.L. 75-717
1962 Would declare as misbranded a prescription drug
whose advertising did not adhere to required
labeling information.
Kefauver-Harris Drug Amendments to the
FFDCA, P.L. 87-781. Sec. 131 added FFDCA Sec.
502(n).
1975 Prohibited manufacturers from advertising and
otherwise promoting off-label uses of approved
drugs.
21 CFR 202.1, Prescription drug advertisements
[40 FR 14016, March 27, 1975]
1977–
2003
FDA issued a 1977 guidance and a 1998 rule, and
Congress took up related efforts in 2002 and 2003
to both require and encourage manufacturers to
conduct the research to support adequate labeling
concerning the use for children of approved drugs.
FDA, “Guidance for Industry: General
Considerations for the Clinical Evaluation of
Drugs in Infants and Children,” September 1977,
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/ucm071687.pdf; FDA, “Regulations
Requiring Manufacturers to Assess the Safety and
Effectiveness of New Drugs and Biological
Products in Pediatric Patients; Final rule,” Federal
Register, vol. 63, no. 231, December 2, 1998, pp.
66632-66672; Best Pharmaceuticals for Children
Act (BPCA, P.L. 107-109); and Pediatric Research
Equity Act (PREA, P.L. 108-155).
1993 Added Medicare coverage of off-label cancer drug
use, by defining “medically accepted indication” as,
for an FDA-approved drug, a use included in a cited
compendia or other Secretary-defined compendia
or which the carrier determines is acceptable based
peer-reviewed supportive clinical evidence
according to guidance from the Secretary.
P.L. 103-66 amended the Social Security Act Sec.
1861(t)(2)(B) [42 U.S.C. 1395x].
1993–
2007
FDA issued “Guideline for the Study and Evaluation
of Gender Differences in the Clinical Evaluation of
Drugs.”
Directed the Secretary of Health and Human
Services to review and develop guidance on
including women and minorities in clinical trials,
which would make appropriate labeling possible.
FDA, “Guideline for the Study and Evaluation of
Gender Differences in the Clinical Evaluation of
Drugs,” Federal Register, Vol. 58, No. 139, July
22, 1993.
FDA Modernization Act of 1997 IFDAMA),
P.L. 105-115
2006 Required that labeling include highlights of
prescribing information to help health care
providers use the information.
“Requirements on Content and Format of
Labeling for Human Prescription Drug and
Biological Products,” referred to as the “Physician
Labeling Rule,” 71 FR 3922, January 24, 2006,
21 CFR 201.56 and 201.57.

Off-Label Use of Prescription Drugs
Congressional Research Service 16
Date Topic Law or Agency Action
2007 Authorized the Secretary, upon learning of new
relevant safety information, to require a labeling
change.
FDA Amendments Act of 2007, P.L. 110-85.
See FFDCA 505(o).
2012 Directed the Secretary of Health and Human
Services to assess the extent to which demographic
subgroups (to include sex, age, race, and ethnicity)
participate in clinical trials and are included in safety
and effectiveness data submitted in marketing
applications. It also required the Secretary to
publish an action plan with recommendations to
improve the completeness, quality, and inclusion of
subgroup data in various analyses. Such information
could then be reflected in a drug’s labeling.
FDA Safety and Innovation Act (FDASIA),
P.L. 112-144
2014 Required that labeling include a summary of the
risks of using a drug during pregnancy and lactation
“Content and Format of Labeling for Human
Prescription Drug and Biological Products;
Requirements for Pregnancy and Lactation
Labeling,” referred to as the “Pregnancy and
Lactation Labeling Rule,”
79 FR 72063, December 4, 2014.
2016 Required FDA to establish a program to evaluate
the use of real world evidence (RWE) to support
approval of a new indication for an already approved
drug.
Section 3022 of the 21st Century Cures Act,
P.L. 114-255
Source: Compiled by CRS.
Author Information
Agata Bodie
Analyst in Health Policy
Acknowledgments
Susan Thaul, retired CRS Specialist in Drug Safety and Effectiveness, was the original author of this report.

Off-Label Use of Prescription Drugs
Congressional Research Service R45792 · VERSION 2 · NEW 17
Disclaimer
This document was prepared by the Congressional Research Service (CRS). CRS serves as nonpartisan
shared staff to congressional committees and Members of Congress. It operates solely at the behest of and
under the direction of Congress. Information in a CRS Report should not be relied upon for purposes other
than public understanding of information that has been provided by CRS to Members of Congress in
connection with CRS’s institutional role. CRS Reports, as a work of the United States Government, are not
subject to copyright protection in the United States. Any CRS Report may be reproduced and distributed in
its entirety without permission from CRS. However, as a CRS Report may include copyrighted images or
material from a third party, you may need to obtain the permission of the copyright holder if you wish to
copy or otherwise use copyrighted material.