o princípio da justiça, os ensaios clínicos e o registro
TRANSCRIPT

FUNDAÇÃO OSWALDO CRUZ
ESCOLA NACIONAL DE SAÚDE PÚBLICA SÉRGIO
AROUCA
MESTRADO ACADÊMICO EM BIOÉTICA
BIOÉTICA, ÉTICA APLICADA E SAÚDE COLETIVA
CECILIA FERREIRA DA SILVA
O Princípio da Justiça, os ensaios clínicos e o registro
de anticorpos monoclonais e biomedicamentos
oncológicos no Brasil.
Rio de Janeiro
2014

O Princípio da Justiça, os ensaios clínicos e o registro
de anticorpos monoclonais e biomedicamentos
oncológicos no Brasil.
CECILIA FERREIRA DA SILVA
.
Rio de Janeiro
2014
Dissertação apresentada ao curso de Bioética, Ética aplicada e Saúde Coletiva. da Escola Nacional de Saúde Pública Sergio Arouca como requisito parcial para obtenção de título de Mestre em Bioética. Orientadoras: Claudia Garcia Serpa Osoriode Castro e Miriam Ventura da Silva

ii
Agradecimentos
A concretização deste trabalho não seria possível sem a intervenção e sem a
presença de algumas pessoas especiais na minha vida. A começar pelos meus pais. Estes
representam peça fundamental na construção não só deste trabalho, mas do que eu sou
hoje como pessoa, como profissional, como amiga e, sobretudo como agente moral. Sou
e serei eternamente grata pelo carinho, amor, dedicação e estímulos que estes dois me
deram ao longo da minha vida.
Ao meu noivo, Frederico Campos, pelo amor e respeito de nosso
relacionamento, que me faz hoje uma mulher feliz e realizada. Pela sua cumplicidade e
companheirismo em todo o percurso do mestrado.
As professoras Claudia Osorio e Miriam Ventura pelas inúmeras contribuições
na construção deste trabalho.
Aos meus amigos, pela compreensão das minhas inúmeras ausências sociais e
por compartilharem a minha ansiedade e estresse durante estes dois anos.
Ao Hamilton Kai e Thalita da Matta pelas contribuições nas análises estatísticas.
A minha chefia do Instituto Nacional de Câncer por me apoiar na realização
deste Programa de Mestrado.

iii
“Os direitos, a segurança e o bem-estar dos sujeitos de pesquisa
são as mais importantes considerações e devem prevalecer sobre
os interesses da ciência e sociedade.”
(ICH, 1996)

iv
RESUMO
O Brasil vem se destacando no cenário das pesquisas clínicas. Porém, a maior parte dos ensaios clínicos realizados refere-se à participação do país como centro condutor de pesquisas com novos fármacos, promovidas por instituições estrangeiras. Na área de oncologia, que se destaca em função das altas taxas de incidência e morbidade, o surgimento de novos antineoplásicos, como os anticorpos monoclonais e biomedicamentos, é sempre acompanhado de grandes interesses pelas diferentes partes envolvidas. Algumas problemáticas éticas se configuram neste cenário, face às necessidades da população e ao desenvolvimento científico e tecnológico do país. Dentre os requerimentos éticos expressos na Resolução CNS 466/12 está o princípio da justiça e equidade. O presente trabalho se propôs a analisar os aspectos bioéticos relacionados à aplicação do princípio da justiça e equidade no percurso da realização dos ensaios clínicos com anticorpos monoclonais e biomedicamentos oncológicos no Brasil, até seu registro no país, nos últimos 10 (dez) anos. Após aplicação de critérios de eligibilidade nove ensaios foram selecionados nas bases ReBEC e Clínical Trials, e caracterizados. A trajetória do registro desses medicamentos no país (ANVISA) e nos principais órgãos regulatórios internacionais (FDA e EMA) foi mapeada, assim como os gastos com compras federais no período do estudo. Diferentes concepções do princípio da justiça, obtidas por busca sistemática, foram agrupadas em três categorias analíticas (relevância social, Equidade de acesso à saúde; Distribuição equitativa de encargos e benefícios) que subsidiaram a discussão dos resultados empíricos sobre os ensaios clínicos resgatados. No Brasil os ensaios estão concentrados na Região Sudeste. Todos os ensaios têm patrocínio estrangeiro e a maioria é de fase III. Grande parte restringe a inclusão de pacientes, baseado em características genéticas. Os gastos com os medicamentos examinados revelaram grande variação de preço ao longo do tempo, o que implica em questionamentos sobre justiça e equidade no acesso pelo SUS. Os resultados obtidos permitiram visualizar fragilidades no sistema regulatório brasileiro e falhas na transparência de informações sobre os estudos/medicamentos, impactando sobre a justiça na distribuição dos benefícios e relevância social. O Brasil precisa estimular pesquisas autóctones; não obstante, as pesquisas analisadas estavam adequadas quando examina-se indicação clínica e incidência de câncer no país. Recomenda-se que os órgãos reguladores proponham formas de fiscalização dos requerimentos e princípios éticos expressos nas normativas nacionais sobre ética em pesquisa com seres humanos. Palavras-chave: 1.Pesquisa Clínica; 2.Medicamentos; 3.Oncologia; 4.Justiça; 5.Ética em pesquisa.

v
ABSTRACT
In Brazil, the scenario of clínical trials has expanded. However, most of them are sponsored by foreign institutions, leaving to Brazilian institutions the role of mere trial conductor. Because of high cancer incidence and morbidity, new antineoplastic agents, such as monoclonal antibodies and biologics are very much in demand and accompanied by a variety of conflicting interests. Certain ethical issues arise, given the needs of the population and technological and scientific development in the country. Among the ethical requirements expressed in Resolution CNS 466/12, is the principle of justice and equity. This study aims to analyze bioethical aspects related to the application of the principle of justice and equity in the pathway from trials to market approval,followed by monoclonal antibodies and biologics, during the last ten years. After applying eligibility criteria, nine trials were selected from ReBEC and Clínical Trials, and characterized. Market approval history in the country regulatory agency (Anvisa) and in foreign regulatory bodies (FDA and EMA) was described, as were federal expenditures with these medicines during the study period. Different conceptions of the principle of justice and equity were obtained from a systematic search of the literature and were grouped in three categories (social relevance, equity of access to health care; Equitable distribution of burdens and benefits) which, in turn, sustained discussion of empiric data on trials. In Brazil, trials are concentrated in the Southeast. All trials have foreign sponsors and the majority are Phase III. Most restrict subject eligibility, based on their genetic characteristics. Medicines expenditures throughout the period revealed great price variation, bringing to light problems regarding justice and equity of access in the Brazilian Health System. Our findings showed frailties in the Brazilian regulatory system and lack of transparency in disclosure of information on trials and medicines, causing an unfavorable impact on the justice of distribution of benefits and on social relevance. Brazil needs to stimulate local research. In spite of this, results showed that the nine clínical trials were adequate in regard to clínical indication and cancer incidence. We propose that the regulatory agencies may implement inspection as to the adoption ethical principles expressed in national norms on ethics in research with human subjects. Key-words: 1.Clínical Research; 2. Medicines; 3.Oncology; 4.Justice; 5.Ethics research.

vi
SUMÁRIO
Conteúdo
LISTA DE FIGURAS ................................................................................................... viii
LISTA DE TABELAS .................................................................................................... ix
LISTA DE GRÁFICOS.................................................................................................. xii
LISTA DE SIGLAS.......................................................................................................xiii
INTRODUÇÃO.............................................................................................................. 15
1. OBJETIVO GERAL....................................................................................................19
JUSTIFICATIVA........................................................................................................... 20
REFERENCIAL TEÓRICO E NORMATIVO............................................................... 21
1. INOVAÇÃO E INCORPORAÇÃO DE TECNOLOGIAS NO SISTEMA DE SAÚDE
.........................................................................................................................................21
2. PESQUISA CLÍNICA E O DESENVOLVIMENTO DE NOVOS MEDICAMENTOS
.........................................................................................................................................27
3. ENSAIOS CLÍNICOS NO BRASIL...........................................................................30
3.1. Registro de novos medicamentos no Brasil..............................................................32
3.2. Ensaios clínicos em Oncologia.................................................................................35
3.3. Registro público de ensaios clínicos.........................................................................39
4. MARCO REGULATÓRIO DA ÉTICA EM PESQUISA COM SERES
HUMANOS.....................................................................................................................45
5. O PRINCÍPIO DA JUSTIÇA E EQUIDADE.............................................................55
6. JUSTIÇA E ÉTICA EM PESQUISA..........................................................................59
MÉTODO....................................................................................................................... 63
1. OBJETO ......................................................................................................................63
2. DESENHO ..................................................................................................................63
3. ETAPAS METODOLÓGICAS...................................................................................63
3.1. Etapa 1: Busca bibliográfica: revisão do estado da arte .................................. 63

vii
3.2. Etapa 2: Busca e coleta de dados empíricos .................................................... 64
3. VARIÁVEIS EMPÍRICAS DE INTERESSE.............................................................69
4. ANÁLISE ....................................................................................................................70
5. ASPECTOS ÉTICOS ..................................................................................................72
RESULTADOS .............................................................................................................. 73
1. BUSCA SISTEMÁTICA: JUSTIÇA E PESQUISA...................................................73
2. TRAJETÓRIA DOS ENSAIOS CLÍNICOS AO REGISTRO DE ANTICORPOS
MONOCLONAIS E BIOMEDICAMENTO E AS IMPLICAÇÕES ÉTICAS..............78
2.1. Resultados da seleção de estudos .................................................................... 78
2.2. Características gerais do conjunto de estudos ................................................. 83
2.2.1. Ano de aprovação dos medicamentos e duração dos estudos ...................... 83
2.2.2. Desenho do estudo........................................................................................ 84
2.2.3. Distribuição geográfica................................................................................. 85
2.2.4. Indicação Clínica .......................................................................................... 89
3. ANÁLISES DAS COMPRAS FEDERAIS DOS ANTICORPOS MONOCLONAIS E
BIOMEDICAMENTOS ONCOLÓGICOS ....................................................................95
DISCUSSÃO.................................................................................................................. 98
1. RELEVÂNCIA SOCIAL ........................................................................................98
2. EQUIDADE DE ACESSO À SAÚDE......................................................................105
3. DISTRIBUIÇÃO EQUITATIVA DE ENCARGOS E BENEFICIOS REFERENTE À
PARTICIPAÇÃO NA PESQUISA. ..............................................................................111
CONSIDERAÇÕES FINAIS E RECOMENDAÇÕES ............................................... 118
APÊNDICE A .............................................................................................................. 127
APÊNDICE B............................................................................................................... 128
APÊNDICE C............................................................................................................... 133
APÊNDICE D .............................................................................................................. 138
REFERÊNCIAS ........................................................................................................... 141

viii
LISTA DE FIGURAS
Figura 01. Ciclo de vida das tecnologias........................................................................ 22
Figura 02. Mapa dos ensaios clínicos na América do Sul registrados no
Clínicaltrials.gov até 2013.............................................................................................. 31
Figura 03. Distribuição proporcional dos dez tipos de câncer mais incidentes estimados
para 2012 por sexo, exceto pele não melanoma. ............................................................ 36
Figura 04. Representação espacial das taxas brutas de mortalidade por todas as
neoplasias, por 100.000 mulheres/homens, nas Unidades da Federação, entre 2000 e
2010. ............................................................................................................................... 36
Figura 05. Taxas de mortalidade das 5 localizações primárias mais frequentes em 2011,
ajustadas por idade, população mundial, por 100.000 homens, Brasil, entre 2003 e 2011.
........................................................................................................................................ 93
Figura 06. Taxas de mortalidade das 5 localizações primárias mais frequentes em 2011,
ajustadas por idade, população mundial, por 100.000 mulheres, Brasil, entre 2003 e
2011 ................................................................................................................................ 93
Figura 07. Número de estudos registrados no ClínicalTrials que possuem resultados
disponíveis de 2009 a 2014. ........................................................................................... 99
Figura 08. Representação espacial das taxas brutas de incidência por 100 mil homens e
100 mil mulheres, estimadas para o ano de 2014, segundo Unidade da Federação (todas
as neoplasias malignas). ............................................................................................... 107
Figura 09. Organograma com as categorias analíticas sobre justiça e ética em pesquisa e
suas implicações. .............................................................................................................. 1

ix
LISTA DE TABELAS
Tabela 01. Número e tipos de estudos registrados no ClinicalTrials.gov até 2013........ 24
Tabela 02. Descrição das variáveis do estudo e seus objetivos de coleta. ..................... 69
Tabela 03. Distribuição da frequência das características gerais dos estudos................ 85
Tabela 04. Identificação de alguns critérios de seleção dos estudos clínicos................. 94

x
LISTA DE QUADROS
Quadro 01. Lista de Jornais, Organizações e representantes do ICMJE em 2004. ........ 41
Quadro 02. Registros primários e parceiros da International Clínical Trials Registration
Plataform/World Health Organizartion (ICTRP/WHO)................................................. 42
Quadro 03. Bases de registros de Ensaios Clínicos aceitáveis para ICMJE................... 43
Quadro 04. Principais normas (em vigor) no contexto da pesquisa clínica no Brasil. ... 52
Quadro 05. Antineoplásicos e agentes imunomoduladores pelo ATC/DDD Index........ 67
Quadro 06. Categorias analíticas e variáveis relacionadas.............................................71
Quadro 07. Critérios de seleção da busca e os resultados preliminares. ........................ 73
Quadro 08. Descrição do fluxo de elegibilidade dos artigos da busca sistemática sobre
Justiça e Pesquisa clínica................................................................................................ 73
Quadro 09. Artigos da busca sistemática sobre justiça e ética em pesquisa, seus
objetivos e as respectivas categorias analíticas relacionadas. ........................................75
Quadro 10. Identificação dos estudos envolvendo anticorpos monoclonais e
biomedicamento, segundo ClínicalTrials.gov ................................................................ 79
Quadro 11. Monografias resumidas de anticorpos monoclonais e biomedicamento,
segundo bases de consulta (FDA, EMA e Anvisa). ....................................................... 80
Quadro 12. Identificação e localização de alguns centros de pesquisas envolvidos na
condução dos nove estudos clínicos da amostra............................................................. 87
Quadro 13. Resultados clínicos dos estudos com indicação clínica nova. ..................... 91

xi
Quadro 14. Distribuição dos anticorpos monoclonais e biomedicamentos da amostra
comprados pelo Governo Federal entre 2004 a 2012. .................................................... 97

xii
LISTA DE GRÁFICOS
Gráfico 01. Gráfico de linhas exibindo a tendência em relação ao ano de aprovação dos
anticorpos monoclonais/biomedicamentos e as agências reguladoras FDA, EMA e
ANVISA. ........................................................................................................................ 83
Gráfico 02. Linha do tempo da trajetória do estudo do recrutamento aos resultados. ... 84
Gráfico 03. Distribuição da frequência absoluta e percentual dos países envolvidos na
condução dos estudos. .................................................................................................... 86
Gráfico 04. Distribuição da frequência absoluta e percentual das Cidades e Estados
envolvidos na condução dos estudos. ............................................................................. 89
Gráfico 05. Distribuição da frequência dos tipos de câncer avaliados nos 09 estudos. . 92
Gráfico 06. Distribuição de itens comprados pelo Governo Federal, por região do país,
entre 2004 a 2012, de acordo com o SIASG. ................................................................. 96
Gráfico 07. Distribuição dos gastos federais, nos anos 2004 a 2012, com os
biomedicamentos e anticorpos monoclonais em oncologia identificados em nossa
amostra, de acordo com o SIASG. ................................................................................. 96

xiii
LISTA DE SIGLAS
Anvisa- Agência Nacional de Vigilância Sanitária
ATC/DDD - Anatomical Therapeutic Chemical and Defined Daily Dose
ATS - Avaliação de Tecnologias em Saúde
BVS - Biblioteca Virtual de Saúde
CE – Comunicado Especial
CEP – Comitê Ética em Pesquisa
CIOMS - Council for International Organizations of Medical Sciences
CMED - Câmara de Regulação do Mercado de Medicamentos
CNS - Conselho Nacional de Saúde
CONEP – Comissão Nacional de Ética em Pesquisa
CORP - Council of Public Representattives
CRO - Contract Research Organization
DCB - Denominações Comuns Brasileiras
DECIT - Departamento de Ciência e Tecnologia
DeCS - Descritores em Ciência em Saúde
EMA - European Medicines Agency
ENSP - Escola Nacional de Saúde Pública Sérgio Arouca
EUA – Estados Unidos da América
FDA – Food and Drug Administration
FDAAA - Food and Drug Administration Amendments Act
Finep - Financiadora de Estudos e Projetos
FMI - Fundo Monetário Internacional
GCP - Good Clínical Practice
GEMEG - Gerência de Medicamentos Genéricos
GEMES - Gerência de Medicamentos Similares
GEPEC - Gerência de Medicamentos Novos, Pesquisa e Ensaios Clínicos
GGMED - Gerência-Geral de Medicamentos
GMEFH - Gerência de Medicamentos Isentos, Fitoterápicos e Homeopáticos
GPBEN - Gerência de Pesquisas, Ensaios Clínicos Medicamentos Biológicos e Novos
HIV - Human Immunodeficiency Virus
Icesp - Instituto do Câncer do Estado de São Paulo Octavio Frias de Oliveira
ICH – International Conference on Harmonisation
ICMJE - International Committee of Medical Journal Editors

xiv
ICTRP -International Clínical Trials Registry Platform
IMD - Incrementally modified Drugs
INCA - Instituto Nacional de Câncer
INPI - Instituto Nacional da Propriedade Industrial
MCT – Ministério da Ciência e Tecnologia
MeSH - Medical Subject Headings
MS – Ministério da saúde
NCI - National Cancer Institute
NDA - New Drug Approval
NIH – National Institute of Health
NME - New molecular entities priority
OMS – Organização Mundial de Saúde
OPAS- Organização Panamericana de Saúde
ORPC - Organizações de Pesquisa Contratada
P&D - Pesquisa e desenvolvimento
PDP - Parcerias para o Desenvolvimento Produtivo
PMDA - Pharmaceutical and Medical Devices Agency
RDC – Resolução da diretoria colegiada
ReBEC - Registro Brasileiro de Ensaios Clínicos
RNPC - Rede Nacional de Pesquisa Clínica
RNPCC - Rede Nacional de Pesquisa Clínica em Câncer
SAS - Secretária de Atenção à Saúde
SCTIE - Secretaria de Ciência e Tecnologia e Insumos Estratégicos
SIASG - Sistema Integrado de Administração de Serviços Gerais
SUS – Sistema Único de Saúde
TCLE - Termo de Consentimento Livre e Informado
TGA - Therapeutic Goods Administration
UFARM - Unidade de Farmacovigilância
UPBIH - Unidade de Produtos Biológicos e Hemoderivados
UPROC - Unidade de Produtos Controlados
WHO - World Health Organizartion

15
INTRODUÇÃO
Indiscutivelmente as pesquisas com novos fármacos são importantes para a
saúde pública e potencialmente benéficas à saúde da população. O desenvolvimento
destas pesquisas envolvem a realização de ensaios não-clínicos (testes in vitro, in silico
e animais) e clínicos, estes em humanos.
Para a realização das pesquisas clínicas no Brasil, têm-se duas avaliações
importantes, uma de caráter ético, realizada pelas instâncias Comitê de Ética em
Pesquisa (CEP) e, quando aplicável, a Comissão Nacional de Ética em Pesquisa
(CONEP) e outra de caráter técnico, realizada pela Agência Nacional de Vigilância
Sanitária (Anvisa). A análise realizada pela Anvisa aplica-se a todos os ensaios clínicos
de fases I, II e III, com medicamentos e/ ou produtos para saúde, que poderão,
posteriormente, subsidiar o registro de medicamentos ou qualquer alteração pós-registro
do mesmo (Anvisa, 2008).
A trajetória célere do fármaco/medicamento da execução de pesquisas clínicas
até o seu registro e incorporação pelo setor público e/ou privado, tem sido considerada
um fator importante para o desenvolvimento do país. Esta trajetória conhecida como o
ciclo de tecnologias é composto por quatros fases. Inicialmente há a “inovação”
caracterizada pelo surgimento de uma nova tecnologia. A partir da sua utilização inicia-
se a fase da “difusão inicial”, fase que determinará o grau de aceitação desta tecnologia
dentro do setor saúde. Entre estas duas fases, usualmente são realizadas avaliações
econômicas, e estudos sobre benefícios e os riscos da nova tecnologia. A fase de
“incorporação” ocorre quando esta tecnologia passa a ser reconhecida, normalmente
pelo governo ou seguradoras, como tecnologia estabelecida para determinada
terapêutica. Eventualmente, uma tecnologia será abandonada, fase do “abandono”,
completando o seu ciclo de vida (Brasil, 2009).
No Brasil, a maior parte dos ensaios clínicos realizados refere-se à participação
dos centros do país como centros condutores de Pesquisas clínicas para o
desenvolvimento de novos fármacos sob a supervisão de instituições de pesquisa
estrangeiras (Lousana, 2002). Há duas décadas, os estudos clínicos eram desenvolvidos
principalmente em países desenvolvidos, porém, com as pressões para acelerar o
processo de desenvolvimento de fármacos, os estudos passaram a ser conduzidos em
outros países/regiões, e a América Latina se destacou, compondo 7,5% dos estudos

16
desenvolvidos do mundo (Rodrigues, 2007). Atualmente (2014), de acordo com o
ClínicalTrials.gov, a América Latina compõe 5,88% dos estudos registrados na base.
No processo de desenvolvimento de novos fármacos no Brasil, um aspecto
importante é a capacitação de profissionais para realizarem ensaios clínicos de
medicamentos. A capacitação é tida como o único caminho para o desenvolvimento de
medicamentos destinados a enfermidades nacionais e negligenciadas pela indústria
farmacêutica (Quental e Salles, 2006). Entretanto, a realização dos ensaios ocupam o
espaço rarefeito entre a pesquisa cientifica e a produção de medicamentos no país e a
maioria dos treinamentos em pesquisa clínica são promovidos pelas patrocinadoras ou
Organizações de Pesquisa Contratada (ORPC) (Rodrigues, 2007).
Uma estratégia importante para a inovação no âmbito da Saúde Pública é a
alteração das características dos centros de pesquisas de condutores para proponentes de
pesquisas clínicas, desenvolvendo capacitação local para o desenvolvimento de estudos
e sobretudo, pesquisas voltadas às necessidades da população.
A política nacional reconhece a importância do diálogo entre a ciência,
tecnologia e inovação no âmbito da Saúde Pública, sendo este indispensável ao
desenvolvimento econômico e progresso social de uma nação. Para o Brasil alcançar
este cenário de desenvolvimento desejável, voltado às necessidades da população, é
fundamental estreitar as relações entre pesquisa acadêmica e institucional e a gestão
pública, aproximando então, as atividades científicas às ações de prevenção e controle
dos problemas de saúde da população (Decit, 2006 a).
No Brasil, o perfil epidemiológico do câncer tem conquistado espaço nas
agendas políticas e técnicas das esferas de governo, pois, o “conhecimento sobre a
situação dessa doença permite estabelecer prioridades e alocar recursos de forma
direcionada para a modificação positiva desse cenário na população brasileira” (INCA,
2011 a). O câncer é uma das principais causas de morte no mundo (WHO, 2008) e nas
últimas décadas, vem ganhando dimensões maiores, convertendo-se em um evidente
problema de saúde pública mundial (INCA, 2011 a), o que tem despertado a atenção das
indústrias farmacêuticas pelo seu potencial perfil mercadológico.
Na oncologia, em função das altas taxas de morbidade e mortalidade, o
surgimento de novos antineoplásicos é sempre acompanhado de grande interesse,
expectativa e de pressões por parte dos pacientes, seus familiares, médicos, e ainda da
própria indústria farmacêutica (ANVISA, 2013 b). O investimento em terapias com
alvo molecular para o tratamento do câncer, por meio de fármacos que agem
seletivamente sobre as células tumorais, tem sido a principal estratégia das pesquisas

17
que buscam definir esquemas terapêuticos mais eficazes e menos tóxicos (INCA, 2011
b). Assim os biomedicamentos e anticorpos monoclonais tornam-se o alvo das
indústrias farmacêuticas.
Sabidamente, o Brasil tem se tornado um campo próspero para pesquisas clínicas
envolvendo os medicamentos, porém, não focados nas necessidades/prioridades no
âmbito do Sistema Único de Saúde - SUS, visto que boa parte destes estudos/pesquisas
têm origens internacionais. Destaca-se então, uma problemática ética envolvendo as
tecnologias, pois muitas vezes estas são vistas como moralmente neutras porque os
valores não estão intrinsecamente conectados à tecnologia, mas sim relacionados à sua
aplicação. Neste sentido, problemas de ordem moral podem ser gerados quando se
avaliam seus efeitos como benefício ou prejuízo, quando se estabelecem prioridades
e/ou alocam-se recursos, uma vez que estas avaliações são dotadas de juízo de valor
(Brasil, 2009).
Os estudos multicêntricos internacionais colocam grandes desafios éticos para os
países em desenvolvimento relacionados à condução dos ensaios clínicos, capacitação
dos centros para a pesquisa nacional e disponibilidade final do medicamento no sistema
de saúde local, entre outros. No Brasil, o princípio da justiça e equidade está expresso
na Resolução CNS 466/12 que determina as diretrizes e normas regulamentadoras de
pesquisas envolvendo seres humanos. Este princípio é tido como fundamental para
garantir a eticidade das pesquisas e implica na análise ética da “relevância social da
pesquisa, [que deve garantir] a igual consideração dos interesses envolvidos, não
perdendo o sentido de sua destinação sócio-humanitária” (Brasil, 2012, IIId). A partir
desta diretriz ética, a injustiça pode ser interpretada como a omissão ou negação de
benefícios, direitos e/ou encargos que são devidos às pessoas (Beauchamp & Childress,
2002 b) no contexto atual, social e científico em que vivemos.
Diante do exposto suscitam-se os seguintes questionamentos: Qual a participação
do Brasil na trajetória de desenvolvimento de novos fármacos em oncologia (anticorpos
monoclonais e biomedicamentos)? Quando ocorre o registro destes novos fármacos no
Brasil? Quais são as implicações bioéticas do princípio da justiça e equidade imbricadas
neste contexto? Será que este princípio está verdadeiramente sendo aplicado nas ações
quem envolvem as pesquisas ao registro e acesso ao medicamento? Qual é a efetividade
do referencial ético da justiça e equidade na trajetória dos ensaios clínicos de anticorpos
monoclonais e biomedicamentos até o registro e a disponibilidade do produto no
sistema público de saúde?
A bioética, como campo de conhecimento, integra uma reflexão acerca da justiça

18
nas políticas públicas de saúde que se justifica no sentido que:
“A vigência simultânea do paradigma biotecnocientífico (que incentiva a incorporação tecnológica) e da cultura dos limites (que seleciona as tecnologias) constitui um desafio para os sistemas sanitários, solicitados seja pelas demandas crescentes de seus usuários seja pela racionalização dos recursos imposta a seus gestores. Isso suscita debates éticos e políticos sobre quais seriam as escolhas mais razoáveis, moralmente legítimas e politicamente aceitáveis a serem feitas” (Schramm e Escosteguy, 2000, p.952).
Assim, a dissertação propõe analisar a trajetória entre os ensaios clínicos de
anticorpos monoclonais e biomedicamentos nos quais existe participação brasileira e os
registros desses fármacos, utilizados no tratamento oncológico, à luz do debate sobre
justiça e equidade, no que se refere à disponibilidade destes no sistema público de
saúde.

19
OBJETIVOS
1. OBJETIVO GERAL
Analisar os aspectos bioéticos relacionados à aplicação do princípio da justiça e
equidade no percurso da realização dos ensaios clínicos com anticorpos monoclonais e
biomedicamentos utilizados em oncologia no Brasil, até seu registro no país.
2. OBJETIVOS ESPECÍFICOS
1. Identificar e caracterizar os ensaios clínicos internacionais e nacionais envolvendo
anticorpos monoclonais e biomedicamentos oncológicos com participação brasileira,
nos últimos 10 (dez) anos nas bases de registro do Clínical Trials e Registro
Brasileiro de Ensaios Clínicos (ReBEC).
2. Investigar retrospectivamente a trajetória do registro desses medicamentos no país
(ANVISA) e nos principais órgãos estrangeiros internacionais de registro de
medicamentos (FDA e EMA) e verificar os gastos federais com a compra destes
medicamentos.
3. Analisar as questões bioéticas que envolvem a aplicação do princípio de justiça e
equidade, incorporado na regulamentação brasileira em pesquisa com seres humanos,
identificadas neste percurso.

20
JUSTIFICATIVA
Na área de oncologia, a trajetória do fármaco/medicamento, desde a execução de
pesquisas clínicas até o seu registro e incorporação pelo setor público e/ou privado
envolve inúmeras questões, entre elas, a crescente incidência de vários tipos de câncer
no país, as expectativas e necessidades dos pacientes dos pacientes oncológicos de
acesso a serviços e tratamentos, e os altos custos dos novos medicamentos para o
sistema público.
Dentro deste cenário, pode-se ainda verificar o crescimento no Brasil das
pesquisas clínicas tendo como objeto medicamentos oncológicos, e muitas destas
realizadas com a classe dos anticorpos monoclonais e biomedicamentos, ambos de
altíssimo custo para o sistema de saúde.
Considerando esta crescente participação brasileira nos estudos clínicos com
novos fármacos em oncologia e a ampla discussão sobre os estudos de duplo padrão
(double standard1) nos países em desenvolvimento, faz-se necessário o conhecimento
atual do desenvolvimento desses ensaios. Em especial, das implicações éticas
envolvidas concernentes à efetividade do referencial ético da justiça e equidade,
obrigatório na regulamentação brasileira, no percurso dos ensaios clínicos até o registro,
com vistas à disponibilidade dos produtos no sistema público de saúde.
O presente estudo pode contribuir na descrição deste panorama das pesquisas
dos novos fármacos (anticorpos monoclonais e biomedicamentos) com participação
brasileira na área de oncologia, na identificação e análise das principais questões
bioéticas de justiça e equidade envolvidas neste processo. Adicionalmente poderá
contribuir com as discussões sobre as melhorias nas políticas públicas de
desenvolvimento nacional de novos fármacos.
1 É a aplicação de padrões éticos diferentes, definidos em função das características socioeconômicas do país hospedeiro (Garrafa e Lorenzo, 2009).

21
REFERENCIAL TEÓRICO E NORMATIVO
1. INOVAÇÃO E INCORPORAÇÃO DE TECNOLOGIAS NO
SISTEMA DE SAÚDE
A inovação biofarmacêutica representa uma parte importante na busca de
soluções para os problemas e desafios na área da saúde. O desenvolvimento e as
descobertas contínuas de novas opções terapêuticas representam diretamente benefícios
aos pacientes e influi positivamente sobre a qualidade de vida dos mesmos. Entretanto,
o incentivo à inovação no setor de pesquisa biofarmacêutica deve ser acompanhada de
políticas e estruturas regulatórias consistentes e voltadas às prioridades dos pacientes
(Phrma, 2013a).
Assim, conjugar saúde pública, ciência, tecnologia e inovação é avançar no
sentido do desenvolvimento econômico e do progresso social. Para esta conjugação
virtuosa, o Brasil deve buscar responder às necessidades da população, estreitando as
relações entre pesquisa acadêmica e institucional e a gestão pública, aproximando então,
as atividades científicas às ações de prevenção e controle dos problemas de saúde da
população (Decit, 2006 a).
A tecnologia médica empregada tem sido em parte muito útil, porém, a
incorporação tecnológica na área da saúde, por vezes, ocorre de maneira acrítica, sem
avaliação cautelosa dos dados de eficácia, efetividade e eficiência dessa incorporação
(Schramm e Escosteguy, 2000), o que pode vir a refletir negativamente sobre os gastos
públicos no campo da saúde e, consequentemente na distribuição de recursos de saúde
para a população.
“A incorporação desordenada de tecnologias tem certamente implicações morais, resultantes de seu acesso reduzido e da perda de efetividade quando investimentos tecnológicos de alta complexidade são realizados em detrimento de soluções menos dispendiosas e sem resultados esperados” (Schramm e Escosteguy, 2000, p.956).
A avaliação tecnológica, por vezes, torna-se fonte de conflitos, visto que, os
atores envolvidos, como exemplos os pesquisadores, usuários e indústria, geram
diferentes expectativas acerca da difusão desta determinada tecnologia e desta forma,
podem expressar diferentes interesses e preocupações quanto à eficácia, efetividade,
custo e a relação custo-efetividade, impactos sociais e econômicos (Schramm e

22
Escosteguy, 2000, p. 953).
A Avaliação de Tecnologias em Saúde (ATS) configura-se como um
instrumento de auxílio técnico para regulação do ciclo de vida das tecnologias (Krauss
Silva, 2003), amparando o processo de tomada de decisões em saúde. O ciclo (Figura
01) é composto por quatros fases, inicialmente tem-se a “inovação” caracterizada pelo
surgimento de uma nova tecnologia. À medida que esta se difunde a partir da sua
utilização inicia-se a fase da “difusão inicial”, fase que determinará o grau de aceitação
desta tecnologia. Entre estas duas fases, usualmente são realizadas avaliações
econômicas, estudos de benefícios e riscos da nova tecnologia. A fase de
“incorporação” ocorre quando esta tecnologia passa a ser reconhecida, normalmente
pelo governo ou seguradoras, como tecnologia estabelecida. Eventualmente, uma
tecnologia será abandonada, fase do “abandono”, completando o seu ciclo de vida
(Brasil, 2009).
Figura 01. Ciclo de vida das tecnologias
Fonte: Anvisa, 2009 apud Banta, Behney, Willwams, 1981
Além do papel benéfico das tecnologias, existe um potencial obscuro de
consequências nocivas à utilização destas, por exemplo, os efeitos adversos aos
produtos, baixa resolutividade dos serviços e/ou má utilização dos recursos. Desta
forma, a avaliação tecnológica foi estabelecida como uma ferramenta para se
“identificar tanto os efeitos de ‘primeira ordem’ das tecnologias quanto os de ‘ordem
superior’, não intencionais ou imprevistos” (Schramm & Escosteguy, 2000 p.953).
É importante refletir sobre os potenciais riscos do encurtamento do ciclo das
tecnologias, que podem evoluir com sérias consequências econômicas e sociais. Em
estudo de Vieira e Zucchi (2007) sobre a avaliação dos processos movidos para o

23
fornecimento de medicamentos em 2005 por cidadãos contra a Secretária de Saúde de
São Paulo, verificou-se que os antineoplásicos corresponderam a 7,2% do total de itens
solicitados, porém, isto representou gastos de R$ 661 mil, o equivalente a 75% do gasto
total com a aquisição de medicamentos em decorrência de ações judiciais. Entre estes
medicamentos, a maioria carece de mais ensaios clínicos que fundamentem sua eficácia
e dois não estavam registrados na Anvisa.
Para Daniel Callahan (1998) uma das estratégias para se alcançar um acesso
equitativo de atenção à saúde é desencorajar o desenvolvimento e disseminação de
tecnologias alto custo que apresentam estatísticas marginais de seus benefícios para a
saúde populacional. O autor afirma que o progresso ilimitado da medicina é o maior
obstáculo para a estabilidade desta ciência, e trabalha contra a noção de que todo o
progresso implica em benefícios e acessibilidade da população, de forma que a intensa
busca pelo progresso levanta dúvidas quanto à moralidade.
Motivados pelo cenário de custos crescentes em saúde, o reconhecimento da
existência de desperdício de recursos, a necessidade de garantir direitos do cidadão e a
crescente intervenção do Poder Judiciário no setor de saúde, o Ministério da Saúde por
meio do Departamento de Ciência e Tecnologia (Decit) e Secretaria de Ciência e
Tecnologia e Insumos Estratégicos (SCTIE), em 2003 organizou uma oficina para
elaborar uma proposta para ATS no âmbito do SUS, visando aprimorar o processo de
decisão à incorporação e ao uso das tecnologias em saúde. Paralelamente, a Secretária
de Atenção à Saúde (SAS/MS) e a Anvisa passaram a elaborar ações complementares
aos trabalhos de fomento à pesquisa, capacitar gestores e melhorar o processo de
incorporação de tecnologias no setor da Saúde (Brasil, 2009).
No Brasil houve uma expansão orçamentária para os investimentos em projetos
de pesquisa e a construção e implementação da Agenda Nacional de Prioridades de
Pesquisa em Saúde, um processo político que busca a ampla participação de atores com
experiências e linguagens distintas tanto da pesquisa como da saúde, tendo como
pressuposto o respeito às necessidades nacionais e regionais de saúde e a indução
seletiva para a produção de conhecimentos e bens materiais e processuais nas áreas
prioritárias para o desenvolvimento das políticas sociais, visando à consonância das
prioridades de pesquisa em saúde com os princípios do SUS (Decit, 2006 b).
A maior proporção de recursos destinados à ATS é direcionada a avaliações de
medicamentos (Brasil, 2009, p.40). A base de registro de estudos clínicos Clínical
Trials, uma das mais relevantes bases do mundo, apresenta do seu total de estudos
registrados, até fevereiro de 2013, 140.702 (81%) estudos com intervenção e destes o

24
maior número, 76.886 envolvem o uso de medicamentos, conforme a Tabela 01.
Tabela 01. Número e tipos de estudos registrados no ClinicalTrials.gov até 2013.
Estudo e tipo de intervenção Número de estudos registrados e o
percentual do total
Total 140.702
Intervenção 114.061 (81%)
Fármacos ou biológicos 76,886
Comportamental e outros 27,747
Procedimentos cirúrgicos 12,841
Tipos de
intervenção*
Dispositivos 9,810
Observacional 25.997 (18%)
Acesso expandido 196
Fonte: Adaptado de Clínicaltrials.gov [Acesso em 15 fev 2013] *Um estudo pode incluir mais do que um tipo de intervenção (isto é, um único estudo pode ser contado mais de uma vez). Devido a isso, a soma das contagens por tipo de intervenção não será igual ao número total de estudos de intervenção.
No Brasil, a responsabilidade de prover o fornecimento dos medicamentos é
principalmente do Estado, entretanto, outros atores, como a indústria farmacêutica,
também podem dividir essa responsabilidade (Dainesi & Goldbaum, 2012). As
principais empresas farmacêuticas são mundiais e atuam nos países desenvolvidos e em
desenvolvimento, para a produção e/ou comercialização de medicamentos. Alguns
países em desenvolvimento figuram entre os principais mercados farmacêuticos
mundiais, porém, há uma expressiva concentração do mercado no âmbito dos países
desenvolvidos (Gadelha, Quental, & Fialho, 2003).
Nos grupos farmacêuticos, que investem no desenvolvimento de novos
fármacos, existe o desejo intrínseco de se descobrir ‘um sucesso terapêutico’, que seja
patenteável e, para isto, deve ser seguro, eficaz e/ou melhore a qualidade de vida dos
pacientes. Consequentemente houve um aumento do número de estudos pré-clínicos e
clínicos com a finalidade de se obter o registro do medicamento e concomitantemente
houve o aumento dos gastos orçamentários. Em 1950 o custo médio da pesquisa e
desenvolvimento (P&D) de um novo fármaco era de USD 1.500.000 (um milhão e meio
de dólares) e em 2003, a média de custo era de USD 800.000.000 (oitocentos milhões
de dólares) (Lima, 2003). Os custos são crescentes. Desde 2000, as empresas associadas
PhRMA investiram cerca de USD 550.000.000.000 (quinhentos e cinquenta bilhões de

25
dólares) na busca de novos tratamentos e curas, incluindo a estimativa de USD
48.500.000 (quarenta e oito milhões e quinhentos mil dólares) só em 2012 (Phrma, 2013
b).
Algumas indústrias farmacêuticas justificam que as patentes são necessárias para
reembolsá-los dos altos custos empregados para P&D e aprovação regulamentar do
medicamento. Entretanto, alguns especialistas em saúde pública rejeitam estas
reivindicações do setor. O monopólio das patentes permite as indústrias farmacêuticas
aumentarem os preços de seus medicamentos e por vezes, se popularizarem no mercado
(Carter, 2012).
James Love (2012) defende a necessidade da desvinculação dos custos de P&D
dos preços dos produtos, visando à expansão simultânea da inovação e do acesso,
principalmente dos países em desenvolvimento, assim como defende a expansão dos
investimentos em P&D nas áreas de maior necessidade. Tal desvinculação
possivelmente desmistificaria que os altos custos dos produtos/medicamentos são
reflexos dos altos custos da P&D, o que poderia estimular o processo de inovação dos
países em desenvolvimento, e a busca por transferência de tecnologias, ampliando então
o acesso ao conhecimento cientifico.
A solicitação de patente é uma etapa paralela na trajetória do desenvolvimento
de um medicamento. De acordo com o artigo 229-C da Lei 9.279/96 (modificada pela
Lei de Propriedade Industrial, Lei nº 10.196 em 2001), a concessão de patentes para
produtos e processos farmacêuticos dependerá da prévia anuência da Anvisa. Este fluxo
foi criado visando compatibilizar o direito à propriedade intelectual com os interesses
públicos de ampliação das políticas públicas de saúde e redução da desigualdade social
vigente no Brasil. A partir da Portaria Interministerial 1.065/12, a anuência prévia da
Anvisa deverá ser feita antes da avaliação do Instituto Nacional da Propriedade
Industrial (INPI). Assim, os pedidos de patentes farmacêuticas feitos ao INPI deverão
ser encaminhados à Anvisa e em casos de concessão pela Agência, o pedido retornará
ao INPI para que seja dado seguimento ao processo de avaliação. Nos casos de não
concessão pela Agência o pedido de patente será arquivado e a decisão publicada na
Revista da Propriedade Industrial.
Diante das necessidades de melhor articular as prioridades do SUS e ações do
INPI, assim como acelerar este processo, recentemente foi aprovada a Resolução
número 80/2013, que regulamenta o exame prioritário de pedidos de patentes
envolvendo produtos estratégicos para o SUS, entre eles os produtos para profilaxia,
diagnóstico e tratamento de câncer (INPI, 2013). A expectativa é que o tempo de análise

26
seja reduzido de nove anos para nove meses (INCA, 2013). Tal estratégia foi lançada,
pois o longo período para análise de patentes no Brasil pode atuar como um fator de
dificuldade no processo de desenvolvimento e na produção de medicamentos no país.
Além disso, tal produção poderia potencialmente estimular a indústria nacional.
O estudo de Pucca e colaboradores (2011) sobre a relação entre os estudos com
anticorpos monoclonais (scFv - single-chain variable fragment) e os números de
patentes publicadas no período de janeiro de 1996 a julho de 2009, mostraram a
presença de patentes de scFv em vários países, com destaque para os Estados Unidos,
China e Reino Unido. No Brasil, apesar de identificados artigos envolvendo estudos
com scFv, não foram encontrados as patentes relacionadas. Vale ressaltar que a maioria
dos medicamentos era destinada ao tratamento de câncer.
Muito se fala sobre o potencial inovador das indústrias farmacêuticas
multinacionais, porém, o número de fármacos introduzidos no mercado vem declinando
desde 1960. Na última década, cerca de 70% dos novos medicamentos lançados são na
verdade, novos arranjos moleculares de fármacos já conhecidos, os chamados
medicamentos “me too” (Freitas, 2009).
A Anvisa entende “me too” como:
“...um medicamento que embora seja apresentado como inovador não acrescenta nenhum benefício claro, no que diz respeito aos seus perfis de eficácia e segurança, em relação a outros medicamentos já registrados. Por esta definição não são, portanto, considerados como me-toos os medicamentos genéricos ou similares.” (Anvisa, 2004).
Apesar de algumas correntes defenderem o não registro destes medicamentos, a
Anvisa não faz restrição ao analisar pedidos de registro de medicamentos que pareçam
ser me-too, pois existem dificuldades de classificação destes produtos como tal no
momento do registro e visto que alguns atributos que permitiriam a qualificação só
podem ser verificados depois da comercialização e utilização em larga escala, assim
impossibilitando a Agência de estabelecer regras de registro destes medicamentos
(Anvisa, 2004).
A avaliação desses “medicamentos novos” ocorre no momento do registro
sanitário pela agência reguladora do país, no qual serão avaliadas a eficácia e a
segurança do produto. Tal momento é oportuno para avaliação do possível ganho
terapêutico deste medicamento (Gava e colaboradores, 2010).
Diante do exposto ao longo do capítulo, percebe-se a complexidade do processo
desenvolvimento científico, tecnológico e de inovação em saúde.

27
2. PESQUISA CLÍNICA E O DESENVOLVIMENTO DE NOVOS
MEDICAMENTOS
A palavra Pesquisa é definida como o “processo formal e sistemático que visa à
produção, ao avanço do conhecimento e/ou à obtenção de respostas para problemas
mediante emprego de método Científico” (Brasil, 2012, art.II.12). Dentro deste campo
de investigação, apresenta-se a Pesquisa envolvendo seres humanos, como a pesquisa
que, “individual ou coletivamente, tenha como participante o ser humano, em sua
totalidade ou partes dele, e o envolva de forma direta ou indireta, incluindo o manejo de
seus dados, informações ou materiais biológicos” (Brasil, 2012, art.II.14).
No Documento das Américas (OPAS, 2005), o manual de Boas Práticas Clínicas
acerca das Pesquisas envolvendo seres humanos, no qual o Brasil é signatário, não há a
definição do termo Pesquisa Clínica, porém há a utilização frequente do termo no corpo
do texto, sugerindo semelhanças entre os termos Pesquisa Clínica e Pesquisa
envolvendo seres humanos. Em 2008 a publicação da Resolução da Anvisa número 39
que dispõe sobre o regulamento para a realização de pesquisa clínica em território
nacional, clarificou esta impressão ao definir o termo Pesquisa Clínica como:
“Qualquer investigação em seres humanos, envolvendo intervenção terapêutica e diagnóstica com produtos registrados ou passíveis de registro, objetivando descobrir ou verificar os efeitos farmacodinâmicos, farmacocinéticos, farmacológicos, clínicos e/ou outros efeitos do(s) produto(s) investigado(s), e/ou identificar eventos adversos ao(s) produto(s) em investigação, averiguando sua segurança e/ou eficácia, que poderão subsidiar o seu registro ou a alteração deste junto a Anvisa” (ANVISA, 2008, art.8° XVIII).
Para que os benefícios das Pesquisas clínicas sejam extensivos é fundamental
que estas sejam desenvolvidas de forma a garantir a acurácia e qualidade dos dados,
bem como a sua integridade, confiabilidade e reprodutibilidade, assegurando
principalmente a segurança dos sujeitos envolvidos (Lima, 2003, p.8).
Comumente os termos Pesquisa Clínica, Ensaios Clínicos e Estudos clínicos são
utilizados no campo prático-teórico e por vezes, geram confusões em relação às
definições. O manual de Boas Práticas Clínicas Internacionais (ICH, 1996), do qual, os
Estados Unidos, Japão e União Europeia são signatários, assume os termos Ensaios
Clínicos e Estudos Clínicos como sinônimos. Para alguns autores, a singularidade destes
termos refere-se apenas ao emprego destes às pesquisas científicas que envolvem seres

28
humanos, mas apresentam metodologias diferenciadas dependendo da área de pesquisa.
Os ensaios clínicos e os métodos estatísticos neles empregados enquadram-se no âmbito
da pesquisa clínica, porém, nem toda pesquisa clínica enquadra-se na definição de
ensaio clínico e como exemplo, Lima (2003, p.01) cita “Um estudo transversal ou um
estudo de coorte, em que não há intervenção do pesquisador, são exemplos de pesquisa
clínica que não podem ser definidos como ensaios clínicos, pois não envolvem uma
experiência”.
O termo Ensaio Clínico refere-se a:
“Qualquer pesquisa conduzida em sujeitos humanos com o objetivo de descobrir ou confirmar os efeitos clínicos e/ou farmacológicos e/ou qualquer outro efeito farmacodinâmico do(s) produto(s) sob investigação e/ou identificar qualquer reação adversa ao(s) produto(s) sob investigação e/ou estudar a absorção, distribuição, metabolismo e excreção do(s) produto(s) sob investigação para verificar sua segurança e/ou eficácia” (OPAS, 2005, p.50).
Diante do exposto, explicitamos que os ensaios clínicos ou estudos clínicos
estão contidos na Pesquisa clínica e esta por sua vez, está contida necessariamente em
pesquisa envolvendo seres humanos. No entanto, nem toda pesquisa clínica é um ensaio
clínico.
Um ensaio clínico é um estudo sistemático que segue estritamente as diretrizes
do método científico para determinar a eficácia e a segurança do produto investigado ou
de seus ingredientes ativos, configurando-se como importante ferramenta na descoberta
de novas respostas terapêuticas às doenças (OPAS, 2005). Entretanto, estes ensaios
apresentam algumas limitações relacionadas ao alto controle na sua execução, como
exemplo, doses fixas e curto período de tratamento, condições estas que obstaculizam a
reprodutibilidade quando expandido ao uso na população em geral (Osorio-de Castro,
2012).
A execução dos ensaios clínicos com medicamentos experimentais, tratamentos
ou dispositivo de intervenção comportamental podem proceder por meio de quatro
fases: Fase I para testar uma nova intervenção biomédica ou comportamental em um
pequeno grupo de pessoas, por exemplo, 20-80 pela primeira vez para avaliar a
segurança; Fase II para estudar a intervenção biomédica ou comportamental em um
grupo maior de pessoas (várias centenas) para determinar a eficácia e adicionar
informações sobre a segurança; Fase III para estudar a eficácia da intervenção
biomédica ou comportamental em grandes grupos de pacientes humanos (a partir de
várias centenas a vários milhares), comparando a intervenção com outras intervenções
padrão ou experimentais, bem como para monitorizar os efeitos adversos e recolher

29
informações que irão permitir que a intervenção estudada seja utilizada com segurança;
Fase IV são estudos realizados após a comercialização do medicamento e são
desenhados para monitorar a eficácia da intervenção aprovada na população em geral e
para coletar informações sobre os efeitos adversos associados com o uso generalizado
(NIH, 2001).
Todos os resultados dos estudos não clínicos (realizados em animais e sistemas
biológicos in vitro e in silico) e clínicos (da fase I à fase III) farão parte de um dossiê
que é enviado às agências reguladoras aplicáveis a cada país, para a solicitação de
aprovação para a comercialização do medicamento.
Desta forma, o valor terapêutico dos fármacos se consolida ao longo do
desenvolvimento das fases do ensaio clínico e se fortalecem na relação positiva
estabelecida entre benefícios versus riscos. Posteriormente, o fármaco é incorporado a
uma determinada formulação e dose, surgindo então, o medicamento (Osorio-de-Castro,
Esher e Chaves, 2012).

30
3. ENSAIOS CLÍNICOS NO BRASIL
Os medicamentos no âmbito dos ensaios clínicos são avaliados em relação à
segurança e eficácia, normalmente são multicêntricos, envolvendo diferentes países. A
indústria farmacêutica multinacional concentrava seus esforços de inclusão de pacientes
em protocolos nos Estados Unidos e Europa, porém, passou a expandir seus horizontes
em busca de centros de pesquisa capacitados no Leste Europeu, na América Latina e na
Ásia, ampliando sua capacidade de recrutamento de pacientes (Glickman, 2009), o que
permitiu a crescente participação do Brasil no cenário da pesquisa clínica internacional.
Em função da redução de produtividade em P&D das indústrias farmacêuticas
nos últimos anos, consequência de algumas dificuldades técnicas (complexidade das
doenças), regulatórias (número crescente de exigências para o desenvolvimento do
medicamento) e financeiras (demanda por retorno dos investimentos), as indústrias
laçaram mão de algumas estratégias como o processo denominado internacionalização
(offshoring), que é o deslocamento de etapas de P&D para países emergentes e/ou a
terceirização (outsourcing) da condução dos testes clínicos ou partes destes (Gomes,
Pimentel, Landim e Pieroni, s.d).
Os ensaios clínicos realizados em ambientes multinacionais e multiculturais são
cada vez mais representativos com a inclusão de centros de pesquisa considerados "Não
tradicionais", como os localizados na Ásia, América Latina e Europa Central e do Leste.
Parece haver algumas vantagens econômicas para realização de ensaios nestes mercados
emergentes, estes centros atraem os patrocinadores com promessas de redução de custos
em relação ao recrutamento e compensação do investigador, porém, outros aspectos
podem custar mais, como por exemplo, investimentos na construção de novas infra-
estruturas, a formação pessoal, e viajar com mais frequência para monitorar o estudo,
assim como, para construir relacionamentos com os órgãos reguladores locais (Stober,
2003, p.24).
Segundo dados da Anvisa, dos estudos clínicos submetidos à apreciação da
agência no período de 2011, 30% das solicitações partiram de empresas terceirizadas, as
chamadas CRO2 e 70% dos próprios patrocinadores (Anvisa, 2011). Um levantamento
da relação de faturamento de 2011 das principais CRO demonstrou que as cinco
maiores CRO (Quintiles, Covance, PPD, Parexel e Charles River) respondem por 47%
2 De acordo o Manual de Boas Práticas Clínicas (GCP- Guideline for Good Clínical Practice, 1996), CRO corresponde “a pessoa ou a organização (Comercial, acadêmica ou outras) contratada pelo patrocinador para realizar uma ou mais tarefas e funções relacionadas ao estudo clínico”.

31
do mercado global e entre as oito maiores empresas, apenas uma não era de origem
norte-americana (Gomes, Pimentel, Landim e Pieroni, s.d).
Os medicamentos utilizados pela população brasileira, num passado recente,
eram avaliados por ensaios clínicos realizados em países do hemisfério norte, e suas
aprovações baseadas em dossiês locais com os dados de segurança e eficácia. A partir
da década de 90 no Brasil, os estudos internacionais, como participação de centros de
pesquisa brasileiros, passaram a ser submetidos à aprovação no país pela CONEP
(instância ética), seguida pela Anvisa para avaliação sanitária do estudo e fornecimento
de autorizações para importação de materiais e medicamentos que serão necessários à
condução do estudo (Dainesi & Goldbaum, 2012).
De acordo com a plataforma de registros Clínical trials.gov dos estudos
registrados3 até 2013 na América do Sul (4936), o Brasil é o mais representativo com
3339 estudos, seguido por Argentina (1542) e Chile (832). Os dados estão detalhados na
Figura 02.
Figura 02. Mapa dos ensaios clínicos na América do Sul registrados no Clínicaltrials.gov até 2013. Fonte: Clínicaltrials.gov [Acesso em 31 mai 2013]
3 Os números fornecidos pela Base de dados sobre os estudos registrados incluem estudos com diferentes status de recrutamento: ativos, completados, ativos mas não recrutando e finalizados.

32
Algumas vantagens são apontadas por Lima (2003, p.230) em relação a inclusão
do Brasil no cenário internacional de Pesquisa Clínica, entre elas, o intercâmbio de
informações entre os centros de excelência, criando um ambiente crítico que pode
subsidiar o aprimoramento dos métodos de ensino e pesquisa e a possibilidade dos
centros brasileiros e latino-americanos se tornarem competitivos, incorporando
melhorias na assistência aos pacientes.
3.1. Registro de novos medicamentos no Brasil
O conceito de medicamento adotado pelo Brasil, de acordo com a Lei Federal nº
5.991 de 1973, é todo produto farmacêutico, tecnicamente obtido ou elaborado com
finalidade profilática, curativa, paliativa ou para fins diagnósticos (Brasil, 1973, Art.4).
O termo “medicamento novo”, é utilizado para se referir a medicamentos novos com
princípios ativos sintéticos e semi-sintéticos, associados ou não, que são avaliados pela
GEPEC - Gerência de Medicamentos Novos, Pesquisa e Ensaios Clínicos da Anvisa
(Anvisa, 2012 a).
O FDA (Food and Drug Administration), órgão governamental dos Estados
Unidos da América, responsável pelo controle de medicamentos e equipamentos para
saúde, entre outras atribuições, classifica novos fármacos diferentemente do Brasil. O
FDA classifica-os em duas dimensões: por natureza química (refere-se à formação do
composto ativo do produto, podendo ser novo ou ser ingrediente ativo ou derivado de
outro medicamento que já tenha sido aprovado) e por potencial terapêutico (refere-se à
capacidade do fármaco com base em evidências disponíveis no momento da revisão
regulamentar, para melhorar o desempenho clínico de produtos disponíveis para
diagnosticar, tratar ou prevenir doenças) (NIHCM, 2002).
O registro de medicamentos no Brasil está sob a responsabilidade da Anvisa.
Entre as atribuições deste órgão estão a autorização de funcionamento dos laboratórios
farmacêuticos e demais empresas da cadeia farmacêutica, regulação de ensaios clínicos
e de preços, por meio da Câmara de Regulação do Mercado de Medicamentos (CMED),
vigilância pós-comercialização, aplicação das ações de farmacovigilância, regulação da
promoção de medicamentos e análise dos pedidos de patentes relacionados a produtos e
processos farmacêuticos, em atribuição conjunta com o Inpi (Anvisa, 2013a).
Os registros de medicamentos na Anvisa ocorrem através da sua Gerência-Geral
de Medicamentos (GGMED). Esta inclui a Gerência de Medicamentos Novos, Pesquisa
e Ensaios Clínicos (GEPEC), a Gerência de Medicamentos Similares (GEMES), a

33
Gerência de Medicamentos Genéricos (GEMEG), a Gerência de Medicamentos Isentos,
Fitoterápicos e Homeopáticos (GMEFH), a Unidade de Produtos Biológicos e
Hemoderivados (UPBIH), a Unidade de Produtos Controlados (UPROC) e a Unidade
de Farmacovigilância (UFARM). Os novos medicamentos podem ser registrados em
diferentes partes da GGMED, pois, à exceção das duas últimas unidades, as demais são
responsáveis pelo registro, avaliação de alterações, inclusões pós-registro e a renovação
do registro de medicamentos (Anvisa, 2012 a).
A GMED publicou recentemente que a partir de 24/02/2014, os setores
reguladores deverão notificar à Anvisa, por meio de petições eletrônicas secundárias, as
datas de início e término dos ensaios clínicos em até 30 dias corridos das respectivas
datas. Tal medida visa maior controle dos ensaios clínicos conduzidos no Brasil através
do monitoramento do status de estudos de forma mais ágil (Anvisa, 2014).
No Brasil, para a execução de um protocolo de pesquisa ou, quando há a
solicitação de Licenciamento de Importação do(s) produto(s) necessário(s) para a
condução desta pesquisa, é necessária a emissão pela Anvisa por meio da Coordenação
de Pesquisas e Ensaios Clínicos da Gerência de Pesquisas, Ensaios Clínicos
Medicamentos Biológicos e Novos (GPBEN), do documento de caráter autorizador
intitulado Comunicado Especial (CE). Emite-se um único CE por pesquisa clínica
submetida à apreciação pela área competente da Anvisa, e neste deverá conter todos os
centros participantes da referida pesquisa. Esta regulamentação se aplica a todas as
pesquisas clínicas com medicamentos e produtos para saúde, incluindo pesquisas
envolvendo intervenções terapêuticas ou diagnósticas não registradas no Brasil, fases I,
II e III que pretendam futuramente subsidiar junto a Anvisa o registro de medicamentos
ou realizar qualquer alteração pós-registro do mesmo (Anvisa, 2008, art.5°).
A RDC nº 36 de 2012, que altera a RDC nº 39, em seu artigo 2º inclui os artigos
8°- A e 8º - B. Estes dispõem sobre a adoção pela Anvisa do processo simplificado de
análise para os pedidos de aprovação de pesquisas clínicas nos seguintes casos4:
“I - quando a pesquisa clínica indicada no pedido de anuência já houver sido avaliada e aprovada por pelo menos uma das autoridades reguladoras a seguir: Estados Unidos da América (Food and Drug Administration - FDA), da Europa (European Medicines Agency - EMA), do Japão (Pharmaceutical and Medical Devices Agency -
4 O processo simplificado previsto no caput não se aplica às análises de pesquisas clínicas com vacinas e antiretrovirais.

34
PMDA), da Austrália (Therapeutic Goods Administration - TGA) ou do Canadá (Health Canada); ou II - quando a pesquisa clínica a ser avaliada pela Anvisa haja iniciado recrutamento de sujeitos de pesquisa em outro país participante do estudo” (ANVISA, 2012 b)
Normalmente a avaliação de um dossiê de registro é dividida em três partes:
análise farmacotécnica, análise de eficácia, e análise de segurança. Quanto às avaliações
de eficácia e segurança, feitas por meio da análise de estudos pré-clínicos e clínicos
(fases I, II, III e, eventualmente, IV) e nos casos de medicamentos já registrados em
outros países para os quais dados de farmacovigilância pós-mercado já são disponíveis,
tais avaliações dependem de consultores externos, em geral organizados em câmaras
técnicas, assim, o papel de técnicos da Anvisa torna-se limitado, embora desde meados
de 2003 a forma de encaminhamento dos pedidos de pareceres e de tomada de decisão
tenha sofrido modificações (ANVISA, 2012 a).
Após o registro do medicamento no Brasil, o comércio, a dispensação, a
representação, distribuição e a importação ou exportação deste deve ser exercido
somente por empresas e estabelecimentos licenciados pelo órgão sanitário competente
dos Estados, do Distrito Federal e dos territórios (Brasil, 1973, Art.21).
Acerca da temática de “novo medicamento” há uma discussão sobre o termo
novo, que não representa necessariamente inovação. Inovação de acordo com a Lei
Federal n.º 10.973 de 2004, conhecida como a “lei da inovação”, é a “introdução de
novidade ou aperfeiçoamento no ambiente produtivo ou social que resulte em novos
produtos, processos ou serviços” (Brasil, 2004 a, Art. 2º IV). Esta lei reflete claramente
a necessidade do país em ter dispositivos legais que contribuam para um cenário
favorável ao desenvolvimento científico, tecnológico e ao incentivo à inovação (Brasil,
s.d).
Fármacos altamente inovadores (New Molecular Entities - priority) que são, de
acordo com classificação do FDA, medicamentos que contêm novos ingredientes ativos
nunca antes comercializados dos Estados Unidos e que fornecem melhora clínica
significativa são raros. Durante o período de 12 anos analisados (1989 a 2000), apenas
153 (15%) de um total de 1.035 novas aprovações de fármacos pelo FDA enquadraram-
se nesta classificação (NIHCM, 2002).
Tais dados apontam que dos 1.035 novos fármacos - NDA (New Drug Approval)
aprovados no período supracitado, apenas um terço (35%) foram produtos com novos
ingredientes ativos ou novas moléculas – NME (New molecular entities priority – 15%
e standard 20%) que nunca foram aprovadas anteriormente no mercado norte-

35
americano. Os outros 65% eram de ingredientes ativos que já estavam disponíveis em
produtos comercializados. Destes 65%, 54% eram fármacos modificados – IMD
(Incrementally modified Drugs) e 11% eram fármacos que continham a mesma
substância ativa dos idênticos produtos comercializados (NIHCM, 2002).
Estudo de Gava e colaboradores (2010) que teve por objetivo analisar os
medicamentos novos registrados pela Anvisa entre os anos de 2000 a 2002 e
comercializados em 2003 em paralelo com outras autoridades sanitárias, como o FDA e
EMA, verificou que embora a maioria dos medicamentos de sua amostra tivessem em
sua composição novas moléculas, o ganho terapêutico adicional não foi significativo
quando comparado ao potencial terapêutico dos medicamentos previamente
comercializados.
Esta estratégia, chamada Obsolescência artificial, é utilizada por indústrias
farmacêuticas, na qual um número considerável de tecnologias é forçado para fora do
mercado para dar lugar a pequenas inovações ao invés de invenções radicais (Brasil,
2009).
3.2. Ensaios clínicos em Oncologia
O câncer por muitos séculos foi amplamente considerado como uma doença dos
países desenvolvidos. Porém, há aproximadamente quatro décadas, a situação vem
mudando, e a maior parte do ônus global do câncer hoje pode ser observada em países
em desenvolvimento. Desta forma, o câncer ganhou uma dimensão maior, convertendo-
se em um evidente problema de saúde pública mundial (INCA, 2011a).
No Brasil, as estimativas para o ano de 2012 apontam a ocorrência de
aproximadamente 518.510 casos novos de câncer, incluindo os casos câncer de pele não
melanoma, reforçando a magnitude do problema do câncer no país. Os tipos mais
incidentes de câncer estimados para 2012 por sexo estão descritos na Figura 03. Diante
do cenário, é importante que a prevenção e o controle do câncer adquiram o mesmo
foco e a mesma atenção que a área de serviços assistenciais, para que o câncer não se
torne um obstáculo para o desenvolvimento socioeconômico de países (INCA, 2011a).

36
Figura 03. Distribuição proporcional dos dez tipos de câncer mais incidentes estimados para 2012 por sexo, exceto pele não melanoma. Fonte: Estimativa, 2012. Incidência de Câncer no Brasil.
O percentual de projetos em desenvolvimento na área da oncologia que são
potencialmente first-in-class medicines (medicamentos que utilizam um mecanismo de
ação diferente de qualquer outro medicamento já aprovado) é elevado, representando
80% do total, acompanhados por 84% para neurologia e 81% para cardiologia (Phrma,
2013a). Entretanto, nem todos estes medicamentos concluem as fases clínicas de teste e
entram para o mercado. Usualmente, para cada 5.000 a 10.000 potenciais medicamentos
que são pesquisados, apenas cinco chegam às fases dos ensaios clínicos e apenas um aos
pacientes (Phrma, 2013 b).
Na oncologia, em função das altas taxas de morbidade e mortalidade (ver Figura
04), o surgimento de novos antineoplásicos é sempre acompanhado de grande interesse,
expectativa e de pressões por parte dos pacientes, seus familiares, médicos, e ainda da
própria indústria farmacêutica, para que tais medicamentos se tornem disponíveis
mesmo antes de serem registrados (Anvisa, 2013 b).
Figura 04. Representação espacial das taxas brutas de mortalidade por todas as neoplasias, por 100.000 mulheres/homens, nas Unidades da Federação, entre 2000 e 2010. Fonte: Atlas de Mortalidade por Câncer. Link: http://mortalidade.inca.gov.br/Mortalidade/

37
Estudo de Chieffi e Barata (2009), em análise das demandas judiciais em São
Paulo em 2006, verificou que 33% eram solicitações de antineoplásicos e agentes
imunomoduladores. Considerando os altos custos destes medicamentos e o
deslocamento dos recursos públicos para o atendimento das demandas individualizadas,
percebe-se que estas ações possuem possivelmente consequências orçamentárias
importantes.
Considerando entre outros aspectos, a meta do Complexo Econômico Industrial
da Saúde de redução do déficit da balança comercial da saúde através do incentivo à
produção nacional de fármacos e medicamentos de acordo com as prioridades
estabelecidas pelo Ministério da Saúde e com intuito de diminuir a dependência do
mercado externo e elevar a competitividade da indústria, na área da oncologia, a
estratégia lançada pela SCTIE, em 2011, foi instituir a Rede Nacional de Pesquisa
Clínica em Câncer (RNPCC), no âmbito da Rede Nacional de Pesquisa Clínica (RNPC)
(SCTIE, 2011).
A RNPCC tem “...o objetivo de articular instituições de pesquisa clínica em
câncer, no âmbito do Sistema Único de Saúde, visando a realização de ensaios clínicos e
a qualificação profissional”, sendo a gestão e a operacionalização financeira da RNPCC
de responsabilidade do Instituto Nacional de Câncer (INCA) (SCTIE, 2011).
No Brasil, o INCA é Centro de Referência de Alta Complexidade do Ministério
da Saúde que auxilia na formulação e na execução da Política Nacional de Atenção
Oncológica, instituída pela Portaria GM/MS nº 2.439, de 08 de dezembro de 2005, a
qual tem por objetivo a promoção, prevenção, diagnóstico, tratamento, reabilitação e
cuidados paliativos na área da oncologia (Brasil, 2005, art. 4°).
Além das atribuições acima, compete ao INCA coordenar, programar e realizar
pesquisas clínicas, epidemiológicas e experimentais em cancerologia (Brasil, 2004 (b),
IV, Art. 58) e à Coordenação de Pesquisa compete planejar, coordenar e dirigir o
desenvolvimento de pesquisas básicas, clínicas e aplicadas na área de cancerologia e
afecções correlatas (Brasil, 2004 b, Art. 172).
O investimento em terapias com alvo molecular para o tratamento do câncer, por
meio de fármacos que agem seletivamente sobre as células tumorais, tem sido a
principal estratégia das pesquisas que buscam definir esquemas terapêuticos mais
eficazes e menos tóxicos (INCA, 2011 b). Assim, os biomedicamentos e anticorpos
monoclonais tornam-se o alvo das indústrias farmacêuticas.
Os avanços científicos da década de 70 e 80 levaram os pesquisadores a explorar
anticorpos monoclonais como uma opção terapêutica. O foco inicial foi sobre as células

38
cancerosas e esta abordagem ajudou a tornar possível o combate ao câncer com menos
efeitos adversos. Com os resultados positivos, em 2008, um total de 21 anticorpos
monoclonais foram aprovados nos Estados Unidos e mais de 200 estavam no pipeline
das indústrias farmacêuticas (Phrma, 2013a).
A RDC n°55/10 da Anvisa define anticorpos monoclonais como
“imunoglobulinas derivadas de um mesmo clone de linfócito B, cuja clonagem e
propagação se efetuam em linhas de células contínuas”. Os anticorpos monoclonais são
moléculas capazes de reconhecer/localizar e ligar-se a um antigéno podendo inativá-lo
ou facilitar o processo de eliminação do mesmo. A estutura do anticorpo é única, sendo
específica para o seu respectivo antígeno (Pucca e colaboradores, 2011). Os
biomedicamentos são “medicamentos obtidos a partir de fluidos biológicos ou de
tecidos de origem animal ou medicamentos obtidos por procedimentos biotecnológicos”
(ANVISA, Art 2°, 2010).
Embora alguns medicamentos de terapia alvo-molecular registrados no Brasil
estejam disponíveis na rede pública, estes estão em apenas em algumas instituições,
como o INCA e o Instituto do Câncer do Estado de São Paulo Octavio Frias de Oliveira
(Icesp). A fim de ampliar o acesso da população a estes fármacos, o Governo Federal e
o Ministério da Saúde vêm desenvolvendo uma política de Estado para o fortalecimento
do Complexo Econômico-Industrial da Saúde, incrementando a produção nacional de
insumos para a saúde com vistas ao desenvolvimento e à produção de medicamentos,
sendo os esforços voltados aos os anticorpos monoclonais (mAb, na sigla em inglês) e
as pequenas moléculas inibitórias (INCA, 2011 b).
Um dos desafios para garantir acesso a esses medicamentos é o custo elevado,
imposto pela complexa infraestrutura necessária à sua produção, além de outros
determinantes. Assim, enquanto não for possível efetivar a produção nacional, outras
estratégias foram traçadas, como o “Acordo Brasil-Cuba” assinado em 2004, o qual
prevê acordos de transferência de tecnologia para a produção de anticorpos monoclonais
(INCA, 2011 b). Cuba é o único país da América Latina que produz anticorpos
monoclonais e a ideia central da cooperação é fortalecer o desenvolvimento de mAbs,
orientado pelas necessidades sociais desses produtos. De acordo com o INCA, 80% da
produção mundial de anticorpos monoclonais são consumidos exclusivamente nos
Estados Unidos, 15% na Europa e apenas 5% pelo resto do mundo (INCA, 2010 a).
A visita do INCA a Cuba em 2010 consolidou um projeto de cooperação
científica entre os dois países. A parceria compreende um grupo de projetos, nas áreas
de câncer de pulmão, colo do útero, mama e cólon e reto. O acordo de cooperação em

39
diferentes áreas assinado pelo presidente Luis Inácio Lula da Silva, em 2009, prevê o
financiamento para a concretização destes projetos (INCA, 2010 b).
Outras estratégias do Governo estão sendo lançadas, como exemplo, o Projeto
Inova Saúde, que tem por objetivo promover inovação em medicamentos de combate ao
câncer, este envolve R$ 2 bilhões de recursos. O ministério vai selecionar os melhores
projetos que envolvam produtos prioritários para o SUS, com foco nos
biomedicamentos, além de medicamentos tradicionais e equipamentos de saúde (INCA,
2013).
Tendo em vista que a capacidade de desenvolvimento de medicamentos
inovadores está diretamente relacionada às competências para realização de ensaios
clínicos, é fundamental que o Brasil trace estratégias e ações públicas de incentivo e
ampliação dos investimentos em P&D no país, principalmente na área dos
medicamentos biológicos de empresas nacionais. Estas estratégias constituem uma
oportunidade para consolidar as competências necessárias ao cenário e condensar a
cadeia de P&D de saúde (Gomes, Pimentel, Landim e Pieroni, s.d).
O financiamento federal em P&D na área biomédica é requerido para a
promoção da ciência biomédica assim como das necessidades de atenção à saúde
(Renisk, 2001). Entretanto, apesar de todos os esforços do governo e algumas
instituições de saúde, as mudanças no cenário da inovação tecnológica e da indústria
nacional ainda são incipientes.
3.3. Registro público de ensaios clínicos
Em 2004 a Organização Mundial de Saúde (OMS) convocou, em conjunto com
o Governo do México, um encontro Ministerial sobre Pesquisa em Saúde, onde foi
abordado o papel vital da pesquisa na melhoria e desenvolvimento sustentável da saúde
da população, com ênfase sobre a forma de traduzir o conhecimento em ação, com a
finalidade de melhorar a cooperação global sobre pesquisa em saúde e estreitar as
disparidades no desempenho dos sistemas de saúde entre países em desenvolvimento e
desenvolvidos. Assim, via consulta pública com representantes regionais da OMS,
sociedade civil, agências internacionais e governamentais, meios de comunicação
internacionais, revistas científicas, fundações, e outros, definiram quatro propostas para
melhoria dos países e da capacidade global de gerar, difundir e utilizar o conhecimento,
entre estas, a criação de um sistema global de registro de ensaios clínicos (WHO, 2004).

40
Em resposta ao encontro Ministerial supracitado, a OMS em 2006 lançou a
Plataforma de Registro Internacional de Ensaios Clínicos (ICTRP -International
Clínical Trials Registry Platform), a qual tem como missão garantir a acessibilidade às
informações da pesquisa a todos os envolvidos na tomada de decisão relacionada ao
cuidado e a saúde, garantindo uma melhor transparência da investigação e, em última
instância fortalecendo a validade e o valor da base de evidência científica. A OMS
ressalta que o registro de todos os ensaios clínicos é uma responsabilidade científica,
ética e moral (WHO, 2006).
Historicamente houve outros movimentos na luta para o registro público dos
ensaios clínicos. Em 2004, os Institutos Canadenses de Pesquisa em Saúde organizaram
uma reunião aberta em Ottawa, Canadá, composta por pesquisadores da área,
consumidores, editores de revistas, políticos e representantes da indústria, que
discutiram um conjunto de princípios orientadores para o desenvolvimento de sistema
global de registro de ensaios clínicos, a partir de então, foram publicados como parte 1:
The Ottawa Statement, Part One. Esta declaração é um documento de consenso que visa
orientar a implementação do registro dos estudos globais, sendo endossado por
indivíduos e organizações em toda a comunidade de pesquisa internacional. A
Declaração reconhece que a disponibilidade pública de informações sobre todos os
ensaios em saúde é essencial para garantir a integridade ética e científica em pesquisas
médicas (OTTAWA, 2005).
Em setembro de 2004, foi publicado simultaneamente um editorial em todas as
revistas-membros do Comitê Internacional de Editores de Jornais de Medicina -
International Committee of Medical Journal Editors (ICMJE) promovendo os registros
de ensaios clínicos de forma abrangente e anunciando que todas as 11 revistas-membros
do ICMJE (Quadro 01) adotariam uma política de ensaios-registro, exigindo como
condição de consideração para a publicação, o registro de ensaios em base pública de
registro. Os ensaios devem ser registrados antes ou no início da inscrição do primeiro
paciente no ensaio. Esta política se aplica a qualquer ensaio clínico com inscrição de
paciente após 01 de Julho de 2005. Para os ensaios que começaram anteriormente a esta
data, os membros do ICMJE exigiram o registro até 13 de setembro de 2005, antes de
considerar para o julgamento para publicação (De Angelis, 2004).

41
Quadro 01. Lista de Jornais, Organizações e representantes do ICMJE em 2004.
Participantes
Annals of Internal Medicine New Zealand Medical Journal
Canadian Medical Association Journal The Lancet
Croatian Medical Journal The Medical Journal of Australia
Journal of the American Medical
Association
Ugeskrift para Laeger (Journal of the
Danish Medical Association)
Nederlands Tijdschrift voor geneeskunde
(The Dutch Medical Journal)
U.S. National Library of Medicine
New England Journal of Medicine
Fonte: De Angelis, 2004.
O ICMJE determinou que os registros dos ensaios sejam feitos em bases que
sigam determinados critérios, entre eles, ser acessível ao público sem nenhum custo, ser
gerido por uma organização sem fins lucrativos, deve haver um mecanismo para
garantir a validade dos dados de registro e o registro deverá ser eletronicamente
pesquisável. Adicionalmente, para o registro seja aceitável este deve incluir, no mínimo,
as seguintes informações: um número único de identificação, uma declaração da
intervenção (ou intervenções) e comparação (ou comparações) estudadas, uma
declaração da hipótese de estudo, definição das medidas de resultado primário e
secundário, critérios de elegibilidade, datas-chave (data de registro, data de início
prevista ou real, data prevista ou real de último acompanhamento, a data prevista ou real
de fechamento de entrada de dados, e os dados de data para o julgamento consideradas
completas), o número alvo de assuntos, a fonte de financiamento e informações de
contato com o investigador principal. No período desta determinação, apenas a
plataforma Clínical Trials, promovido pela Biblioteca Nacional de Medicina do
National Institutes of Health (NIH) nos Estados Unidos, atendia aos requisitos (De
Angelis, 2004).
A primeira versão do ClínicalTrials.gov foi disponibilizada ao público em 29 de
fevereiro de 2000. O número total de estudos registrados nesse ano foi de 5.646, em
2005 foi de 25.033 e cresceu para 138.520 em 2012. Hoje o site apresenta uma lista de
140.146 estudos distribuídos em 50 estados e 182 países, e recebe mais de 95 milhões
de visualizações de páginas por mês e representa uma das bases mais relevantes bases
do mundo (Clínical Trials, 2013).

42
Tal base tem por objetivo garantir a acessibilidade às informações das pesquisas
envolvendo seres humanos, configurando-se como uma importante ferramenta de
controle social ao garantir uma melhor transparência das investigações que ocorrem no
mundo todo (Clínical Trials, 2013).
Hoje o número de bases de registro aceitáveis para o ICMJE expandiu e
atualmente para atender com requerimentos de transparência e publicação é necessário
registrar o ensaio clínico em base pública e esta pode ser realizada de duas maneiras,
nas bases de registro primário da International Clínical Trials Registration
Plataform/World Health Organizartion (ICTRP/WHO) (Quadro 02) ou nas bases de
registros aprovadas pelo ICMJE (Quadro 03).
Quadro 02. Registros primários e parceiros da International Clínical Trials Registration
Plataform/World Health Organizartion (ICTRP/WHO).
Nome do registro País/Região Situação
Australian New Zealand Clínical Trials Registry (ANZCTR)
Austrália
Brazilian Clínical Trials Registry (ReBec) Brasil
Chinese Clínical Trial Registry (ChiCTR) China
Clínical Research Information Service
(CRiS), Republic of Korea
Corréia do Sul
Clínical Trials Registry - India (CTRI) Índia
Cuban Public Registry of Clínical
Trials(RPCEC)
Cuba
EU Clínical Trials Register (EU-CTR) União Européia
German Clínical Trials Register (DRKS) Alemanha
Iranian Registry of Clínical Trials (IRCT) Irã
ISRCTN.org Inglaterra
Japan Primary Registries Network (JPRN) Japão
Registros
primários
UMIN Clínical Trials Registry (UMIN-CTR) Japão
JapicCTI - Japan Pharmaceutical
Information Center
Japão
JMACCT CTR -Center for Clínical trials,
Japan Medical Association
Japão
The Netherlands National Trial Register
(NTR)
Holanda
Membros da
rede de
Registros
primários

43
Pan African Clínical Trial Registry
(PACTR)
Africa
Sri Lanka Clínical Trials Registry (SLCTR) Sri Lanka
Clínical Trial Registry of the University
Medical Center Freiburg
Alemanha
DeReG - German Registry for Somatic
Gene-Transfer Trials
Alemanha
Centre for Clínical Trials, Clínical Trials
Registry Chinese
China
Registros
parceiros5
Fonte: WHO/ICTPR. Disponível em: http://www.who.int/ictrp/network/primary/en/index.html
Quadro 03. Bases de registros de Ensaios Clínicos aceitáveis para ICMJE.
Links de acesso para as bases de registros de Ensaios Clínicos
www.anzctr.org.au www.clínicaltrials.gov www.ISRCTN.org www.umin.ac.jp/ctr/index/htm www.trialregister.nl https://eudract.ema.europa.eu/ http://www.who.int/ictrp/network/primary/en/index.html 6
Fonte: ICMJE. Disponível em: http://www.icmje.org/faq_clínical.html
A declaração de Helsinque traz em seu artigo 30, que:
“Autores, editores e as editoras têm obrigações éticas no que diz respeito à publicação dos resultados da pesquisa. Os autores têm o dever de tornar públicos os resultados de suas pesquisas em seres humanos e são responsáveis pela integridade e precisão de seus relatos....Os resultados negativos e inconclusivos, bem como os positivos, devem ser publicados ou de alguma outra forma disponibilizados ao público. Fontes de financiamento, afiliações institucionais e conflitos de interesse devem ser declarados na publicação. Os relatórios de pesquisa em desacordo com os princípios desta Declaração não devem ser aceitos para publicação” (WMA, 2008).
No Brasil, uma das responsabilidades do pesquisador previstas pela Resolução
Brasileira 466/12 é “encaminhar os resultados da pesquisa para publicação, com os
devidos créditos aos pesquisadores associados e ao pessoal técnico integrante do
projeto”, e justificar perante o CEP a não publicação dos resultados (Brasil, 2012, Art.
5 Os registros parceiros não cumprem com os requesitos da ICMJE. 6 A partir de Junho de 2007 o ICMJE passou a aceitar os registros dos Ensaios Clínicos em qualquer base Primária de registro que participa do WHO International Clínical Trials Portal.

44
XI.2).
Fortalecendo esta tendência mundial sobre a política de registro de ensaios
clínicos, em 2008 no Brasil, entrou em vigor o regulamento (RDC nº39) para a
elaboração de dossiês e obtenção de Comunicado Especial para a realização de pesquisa
clínica com medicamentos em território nacional que dispõe sobre a necessidade de
submissão de Dossiê para Anvisa para Anuência7 em pesquisa clínica. Este dossiê deve
ser composto por dezenove documentos listados na citada resolução, entre estes,
encontra-se o comprovante, para os estudos de confirmação terapêutica (fase III), de que
a pesquisa clínica está registrada na base de dados de registro de pesquisas clínicas
International Clínical Trials Registration Plataform / World Health Organizartion
(ICTRP/WHO) ou outras reconhecidas pelo International Commite of Medical Journals
Editors (ICMJE) (ANVISA, 2008, Inciso XIX, Art. 1, anexo I).
Em 2012 a Resolução RDC nº 36, de 27 de junho de 2012 alterou a RDC nº 39,
de 05 de junho de 2008, incluindo como documento número 18 do dossiê a seguinte
descrição:
“Para estudos clínicos fase I, II, III e IV, deve-se apresentar comprovante de registro da pesquisa clínica na base de dados do Registro Brasileiro de Ensaios Clínicos [ReBEC] ou comprovante de submissão; ou, no caso de pesquisas já registradas em outros registros primários da International Clínical Trials Registration Platform (ICTRP/OMS) antes da vigência desta Resolução, este comprovante de registro será aceito para atender ao disposto neste inciso." (ANVISA, 2012 b, Art.3).
O Registro Brasileiro de Ensaios Clínicos (ReBEC) é um projeto conjunto do
Ministério da Saúde (DECIT/MS), da Organização Panamericana de Saúde (OPAS) e
da Fundação Oswaldo Cruz (FIOCRUZ) e foi lançado em Brasília em dezembro de
2010. É uma plataforma virtual de propriedade pública com acesso livre para registro de
estudos experimentais e não-experimentais realizados em seres humanos e atende ao
conjunto de dados mínimos requeridos pelo ICMJE e pela OMS. O ReBEC esclarece
que a responsabilidade pelo registro e informação dos estudos é do patrocinador. Em
2013, a plataforma apresentava 198 ensaios clínicos registrados (ReBEC, 2013),
enquanto o Clínicaltrials apresentava, no mesmo período, um registro de 3.339 estudos
no país, conforme Figura 02 (página 33).
Percebeu-se uma mudança na conduta dos investigadores ao verificarmos que
7 De acordo com a RDC 39/2008 o Dossiê é a "coletânea de documentos protocolizados na ANVISA, dentre eles: os formulários de petição, a descrição das etapas da pesquisa e seus aspectos fundamentais, informações relativas ao sujeito da pesquisa, à qualificação dos pesquisadores e da equipe responsável pelo estudo"

45
atualmente a ReBEC conta com 1948 estudos cadastrados (ReBEC, 2014), um aumento
significativo em relação ao ano anterior e o Clínicaltrials.gov aumentou para 3.749
estudos no Brasil.
4. MARCO REGULATÓRIO DA ÉTICA EM PESQUISA COM
SERES HUMANOS
No cenário internacional, a primeira discussão mundial sobre os aspectos éticos
de pesquisas envolvendo seres humanos ocorreu pós Segunda Guerra Mundial, com a
elaboração do Código de Nuremberg em 1947 (Nuremberg, 1947). A primeira
intervenção para novas regras que regeriam a pesquisa com novos fármacos partiu do
FDA norte-americana em 1962, seguidas pelo NIH em 1966, que publicou as normas
sobre as pesquisas clínicas que envolvem seres humanos ao introduzir a obrigação dos
protocolos de pesquisa de serem revisados por um comitê de ética da instituição na qual
se desenvolvia a pesquisa, criando o primeiro dispositivo institucional de controle
destas pesquisas (Schramm; Palácios e Rego, 2008).
Em 1964, surge a Declaração de Helsinque, documento proposto pela
Associação Médica Mundial em resposta às atrocidades (experimentos sem critérios
éticos e em populações vulneráveis) ocorridas durante o regime nazista. Ao longo da
história, este tornou-se um dos documentos mais importantes de regulação ética em
pesquisa, orientando as pesquisas médicas e os aspectos éticos que devem ser garantidos
em qualquer pesquisa com seres humanos (Diniz & Sugai, 2008).
O Conselho de Organizações Internacionais de Ciências Médicas (CIOMS -
Council for International Organizations of Medical Sciences), em parceria com a OMS,
iniciou seus trabalhos sobre a ética na pesquisa biomédica no final de 1970. A intenção
de trabalho era delinear diretrizes práticas de como aplicar os princípios éticos na
conduta das pesquisas biomédica em seres humanos, conforme já estabelecido pela
Declaração de Helsinque, como especial atenção aos países em desenvolvimento. Em
1982 propôs o Manual Ético Internacional para Pesquisas Biomédicas Envolvendo
Seres Humanos: International Ethical Guidelines for Biomedical Research Involving
Human Subjects (CIOMS, 2002)8.
O Manual de Boas Práticas Clínicas (GCP - Good Clínical Practice),
desenvolvido em 1996 durante a Conferência Internacional de Harmonização dos
8 Este documento sofreu algumas revisões em função das discussões a cerca da temática e das transformações no campo da ética em pesquisa. A última revisão ocorreu em 2002.

46
requerimentos técnicos para o registro de produtos farmacêuticos para Humanos
(International Conference on Harmonisation of Technical Requirements for
Registration of Pharmaceuticals for Humans) foi criado com o objetivo de estabelecer
um padrão de qualidade ética e científica internacional para a concepção, condução e
registro de estudos com participação de seres humanos, está em consonância com os
princípios estabelecidos pela Declaração de Helsinki e com as considerações das Boas
Práticas Clínicas da União Europeia, Japão e Estados Unidos, Austrália, Canadá, os
países nórdicos e da OMS (ICH, 1996). Desde a sua publicação tornou-se um
documento de referência para muitos países, incluindo o Brasil.
No Brasil, na década de 80 surge à primeira resolução em pesquisa clínica,
Resolução n°01/88 do Conselho Nacional de Saúde, a qual tinha por objetivo
normatizar a pesquisa na área de saúde em todo o território nacional e regulamentar o
credenciamento de centros de pesquisa no país, assim como recomendar a criação de um
CEP em cada centro de pesquisa (Brasil, 1988).
O Governo, entendendo que a Resolução n°01/88 do CNS não teve muito
impacto, em 1995 definiu a formação de um Grupo Executivo de Trabalho para revisão
desta, sendo o grupo composto por pesquisadores, representantes dos Ministérios da
Saúde e da Ciência e Tecnologia, Conselho Federal de Medicina, Ordem dos
Advogados do Brasil, representante de usuários do SUS, Organizações não
governamentais e outros (Brasil, 1995). Após um ano de trabalho, surge a resolução
196/96, que regulamenta as pesquisas envolvendo seres humanos.
A Resolução CNS n.º 196/96 instituiu o sistema CEP-CONEP, que constitui o
sistema brasileiro de revisão ética das pesquisas envolvendo seres humanos. A partir
desta, toda pesquisa envolvendo seres humanos deverá ser submetida à apreciação de
um CEP. Esta Resolução representou a principal regulamentação em pesquisa clínica
com seres humanos desde a sua publicação até a sua revogação em junho de 2013.
Em 2005 durante a IV Conferência Pan-americana para Harmonização da
Regulamentação Farmacêutica, na República Dominicana, foi elaborado o manual
“Boas Práticas Clínicas: Documento das Américas” com o objetivo de estabelecer e
harmonizar as diretrizes para as Boas Práticas Clínicas para os países não signatários do
GCP-ICH de 1996, fornecendo subsídios às agências regulatórias, investigadores,
comitês de ética, universidades e empresas (OPAS, 2005)9.
9 O Grupo de trabalho em Boas Práticas Clínicas foi composto por membros representantes do Brasil, Argentina, Chile, Costa Rica, Cuba, México, Venezuela e EUA.

47
Em 2013, passa a vigorar a nova resolução sobre ética em pesquisa, a 466/12,
que reforça o trabalho cooperativo do sistema CEP-CONEP através de mecanismos e
ferramentas próprios de inter-relação, com foco na proteção dos participantes de
pesquisa do Brasil (Brasil, 2012).
Os Comitês de Ética em Pesquisa são definidos como “colegiados
interdisciplinares e independentes, de relevância pública, de caráter consultivo,
deliberativo e educativo, criados para defender os interesses dos participantes da
pesquisa em sua integridade e dignidade e para contribuir no desenvolvimento da
pesquisa dentro de padrões éticos (Brasil, 2012, V2.II).
Recomenda-se que as instituições que realizem pesquisas envolvendo seres
humanos constituam um CEP vinculado à instituição, porém, na impossibilidade de
constituí-lo, caberá à CONEP a indicação de um CEP para proceder à análise (Brasil,
2012).
Atualmente, a apreciação de projetos de pesquisa com novos fármacos,
medicamentos e testes diagnósticos é de responsabilidade do CEP, entretanto, devem
ser encaminhados pelo CEP à CONEP e à SVS/MS para ciência (Brasil, 1997, Art.
V.2). A CONEP “é uma instância colegiada, de natureza consultiva, deliberativa,
normativa, educativa e independente, vinculada ao Conselho Nacional de
Saúde/Ministério da Saúde” (Brasil, 2012, VII.3). A responsabilidade de emissão de
parecer justificado pela CONEP é previsto apenas para algumas áreas temáticas
especiais, listadas abaixo:
1) Genética humana;
2) Reprodução humana;
3) Equipamentos e dispositivos terapêuticos, novos ou não registrados no país;
4) Novos procedimentos terapêuticos invasivos;
5) Populações indígenas;
6) Projetos de pesquisa que envolvam organismos geneticamente modificados;
7) Protocolos de constituição e funcionamento de biobancos para fins de pesquisa;
8) Pesquisas com coordenação e/ou patrocínio originados fora do Brasil, executadas
aquelas como copatrocínio do Governo Brasileiro;
9) Projetos que, a critério do CEP, devidamente justificado, sejam julgados merecedores
de análise pela CONEP;
A Resolução brasileira adota basicamente quatro princípios morais na

48
formulação das normas e diretrizes éticas a serem seguidas para o desenvolvimento de
pesquisas envolvendo seres humanos. São eles: Autonomia, Beneficência, Não-
maleficência e Justiça e equidade (Brasil, 2012). Na ética biomédica, tais princípios são
adotados pela teoria "O Principialismo" de Beauchamp & Childress.
Para a Resolução n°466/12 do CNS, Autonomia requer o respeito quanto à
dignidade e autonomia dos participantes da pesquisa. Este príncipio pode ser
contemplado no processo de aplicação do consentimento livre e esclarecido.
Beneficência diz respeito à promoção máxima de benefícios, individuais e/ou coletivos,
a partir da ponderação com os riscos existentes. Tal requerimento é expresso no item d,
o qual estabelece o dever de “buscar sempre que prevaleçam os benefícios esperados
sobre os riscos e/ou desconfortos previsíveis”. Não-maleficência refere-se a obrigação
de se evitar os danos previsíveis, expresso por exemplo, na requisição de justificar
plenamente o uso de placebo quando na existência de método terapêutico eficaz. Justiça
e equidade correspondem à relevância social da pesquisa, assim como a igual
consideração dos interesses dos envolvidos. Tal princípio é contemplado em vários itens
da resolução, como por exemplo, "garantir que as pesquisas em comunidade, sempre
que possível, traduzir-se-ão em benefícios cujos efeitos continuem a se fazer sentir após
sua conclusão" (Brasil, 2012, item l).
Esta teoria admite tais princípios como prima facie, ou seja, um não possui
prioridade sobre outro, devendo todos serem respeitados igualmente. A reflexão ética
desta teoria está centrada no controle social da pesquisa em seres humanos e
historicamente configurou um novo paradigma de pensamento sobre as questões éticas
na comunidade médico-científica (Beauchamp & Childress, 2002).
Considerando as atuais práticas em pesquisa envolvendo seres humanos nas
diferentes realidades sociais e o pluralismo moral, a objetividade da aplicação dos
quatro princípios pode ser insuficiente para a proteção dos participantes das pesquisas, e
pode requerer outros princípios éticos não contemplados pelo modelo principialista.
Apesar destas críticas à teoria principialista se constitui “...uma ferramenta [importante]
para abordar a eticidade da pesquisa envolvendo seres humanos na medida em que
permite reduzir incertezas no campo das ações biomédicas. É nisso que reside sua
pertinência” (Schramm; Palácios e Rego, 2008, p. 368).
A recente Resolução CNS n.º 466/2012, sensível a essas críticas, expressamente
incluiu como disposições preliminares, item I, a possibilidade de serem utilizados outros
princípios éticos (Brasil, 2012).

49
“Toda pesquisa com seres humanos envolve risco em tipos e gradações
variados” (Brasil, 2012, V), sejam eles, riscos físicos, morais, sociais ou de qualquer
outra natureza. Sabidamente, na condução de um estudo, mesmo havendo boas
intenções e esforços dos investigadores, podem ocorrer desvios não previstos no
decorrer do processo acarretando em riscos, os quais potencialmente poderão violar um
ou mais alicerces éticos da pesquisa. Resultados imprevistos são comuns em pesquisas
envolvendo humanos, visto que os eventos humanos raramente ocorrem exatamente
conforme o planejado (Owonikoko, 2013). Assim, a exigência ética é que quanto
maiores e potenciais riscos previsíveis, maiores devem ser as medidas de proteção aos
participantes da pesquisa.
Algumas questões éticas são identificadas com frequência no universo das
pesquisas farmacológicas multicêntricas, referentes aos sujeitos de pesquisa (Ex.:
guarda de material biológico para estudos adicionais sem consentimento específico10),
ao pesquisador (Ex.: limitação da liberdade e subordinação aos patrocinadores), à
instituição (Ex.: contato direto investigador-patrocinador com não inclusão da
instituição) e ao país (Ex.: Não transferência de tecnologia visto que muitos exames são
realizados no exterior) (Freitas, 2009).
Outra questão ética,
“...é a tentativa de introduzir um duplo padrão ético na pesquisa: um para países que garantem um grau satisfatório de empoderamento da população, necessário para que o sujeito da pesquisa possa consentir livremente e após todos os esclarecimentos necessários sobre a pesquisa; outro para países onde isso não é garantido e a participação pode ser enviesada pelas carências coletivas, como, por exemplo, a falta de acesso ao sistema de saúde e à assistência ” (Schramm; Palácios e Rego, 2008).
A expressão duplo padrão ou “double standard” nas pesquisas clínicas surgiu
nos anos 90 no contexto internacional a partir da publicação de dois estudos que
apontaram sérios desvios éticos em ensaios clínicos multicêntricos. Um deles testava a
eficácia de um novo esquema terapêutico antiviral na prevenção da transmissão
materno-fetal do vírus da imunodeficiência humana adquirida (HIV/AIDS) versus
placebo, em países dos continentes África e Ásia, mesmo já existindo no país de origem
do estudo, um regime terapêutico eficaz na redução desta transmissão (Garrafa e
10 A Resolução brasileira n°441/11 do CNS dispõe sobre a necessidade de apreciação do CEP, e CONEP quando aplicável, das pesquisas com intenção de armazenamento de material biológico humano, no País e/ou no exterior, incluindo apreciação das pesquisas com possibilidade de uso futuro das amostras para estudos adicionais. O uso e/ou armazenamento do material deve estar claramente expresso no TCLE do estudo, o qual será submetido a anuência dos participantes da pesquisa.

50
Lorenzo, 2009).
As discussões acerca do duplo padrão repercutiram por todo o mundo e algumas
revisões nos principais documentos internacionais de ética em pesquisa foram
realizadas, havendo modificações dos artigos da Declaração de Helsinque.
Em 2000 o Brasil, através da publicação da Resolução nº301 do Conselho
Nacional de Saúde, explicita sua postura contrária ao duplo padrão, reafirmando-se
como signatário do Item II.3 da Declaração de Helsinque que discorre sobre a garantia
do melhor tratamento diagnóstico ou terapêutico comprovado e adicionalmente
manifesta-se contrário às alterações propostas na referida declaração sobre o uso de
placebo quando na existência de métodos terapêuticos comprovados (Brasil, 2000).
Com o aumento da terceirização e a internacionalização dos ensaios clínicos
em países em desenvolvimento é fundamental refletir que “independentemente da
instituição financiadora, há sempre motivações comerciais, de carreiras ou científicas
envolvidas, que podem eventualmente entrar em conflito com questões ligadas à
proteção do paciente de pesquisa” (Gomes, Pimentel, Landim e Pieroni, s.d).
Os hospitais públicos representam o principal sítio dos estudos clínicos
internacionais patrocinados por empresas farmacêuticas na América Latina e comumente
estes hospitais prestam atenção á saúde para uma população com características
econômicas, sociais e educacionais de uma população vulnerável. Assim, a
vulnerabilidade dos pacientes da América Latina incita uma atenção especial das pessoas
envolvidas na análise das questões éticas, porém, por vezes, as considerações éticas são
entendidas pelos patrocinadores como a lentificação do processo pelos comitês de ética
locais (Rodrigues, 2007). Porém, vale ressaltar que o papel de revisão e
acompanhamentos dos projetos de pesquisa, assim como o papel educativo do sistema
CEP-CONEP é fundamental para controle social das pesquisas e defesa dos sujeitos
envolvidos (Freitas, 2009).
“A função prioritária da ética em pesquisa é proteger o participante, um indivíduo que se submete voluntariamente a um risco, vivenciando com freqüência condições de vulnerabilidade ou por razões sociais – pobreza, subnutrição, falta de poder – ou por ser portador de doenças que podem ou não ser o motivo de seu recrutamento para o estudo” (Kottow, 2008, sup.8).
Alguns grupos, em função da sua capacidade de autodeterminação reduzida ou
de seu status de dependente, como as “...minorias raciais, os economicamente
desfavorecidos, o muito doente, e os institucionalizados podem continuamente ser

51
procurados como sujeitos de pesquisa, devido à sua pronta disponibilidade em
ambientes onde a pesquisa é realizada” (HHS, 1979, item C.3.). O International Ethical
Guidelines for Biomedical Research Involving Human Subjects discute sobre a
exploração de comunidades e indivíduos vulneráveis, condenando o paternalismo por
parte dos países mais ricos e defende que os fatores econômicos envolvidos nas
pesquisas clínicas não devam influenciar as considerações éticas (CIOMS, 2002).
Embora a exploração seja um tópico bastante presente nas discussões em ética
em pesquisa, não se percebe uma definição muito clara do que de fato é exploração e,
portanto, o termo pode ser utilizado de forma errônea (Renisk, 2003). Renisk traz a
argumentação das análises de Alan Wertheimer11, o qual afirma que a exploração
envolve sempre promoção de vantagens injustas para o benefício próprio. Tal vantagem
é composta por três elementos básicos: o dano, o desrespeito ou injustiça, de forma que,
todo o caso de exploração haverá pelo menos um destes elementos. Por exemplo, uma
exploração pode ocorrer por desrespeito a dignidade e autonomia, sem necessariamente
ocorrer um dano, assim como as ações paternalistas podem ser interpretadas como
exploração mesmo que haja beneficio gerados aos explorados (Renisk, 2003).
A vulnerabilidade do indivíduo, muito discutida dentro da temática exploração,
refere-se à capacidade de autodeterminação reduzida, por quaisquer razões ou
motivação (Brasil, 1996, Item II.15). A vulnerabilidade nos remete ao princípio da
autonomia. A palavra deriva do grego autos (“próprio”) e nomos (“regra”, “governo”)
que se estende aos indivíduos e ao longo da história, adquiriu sentidos diversos, tais
como autogoverno, direitos de liberdade, escolha individual e privacidade (Beauchamp
& Childress, 2002 a).
Existem algumas teorias sobre a autonomia, e praticamente todas consideram
essenciais duas condições, a liberdade – estar livre de influências e a qualidade do
agente - capacidade de agir intencionalmente (Beauchamp & Childress, 2002, a).
Talvez, motivados por tais características, os documentos nacionais e internacionais em
pesquisa clínica tenham definido o processo de aplicação do Termo de Consentimento
Livre e Esclarecido como documento de caráter autorizador da participação voluntária
do sujeito de pesquisa.
O código de Nuremberg, o primeiro código de ética em pesquisa, traz em seu
primeiro artigo: “Consentimento voluntário do ser humano é absolutamente essencial”
(Nuremberg, 1947). Porém, a introdução do Termo de Consentimento Livre e
Informado (TCLE) não é suficiente para garantir o princípio da autonomia, na ética em
11 Wertheimer, A. Exploitation. Princeton: Princeton University Press, p.10, 1996.

52
pesquisa. O paradigma da autonomia é consentimento informado, documento no qual
legitima formas de autoridade, contudo, este ocorre sob várias condições, pode ser
maquinal ou relutante e, muitas vezes, ocorre sob pressões (Beauchamp & Childress,
2002 a).
Dentre as pesquisas clínicas, os ensaios clínicos são os que demandam maiores
cuidados do ponto de vista ético, especialmente os conduzidos em países em
desenvolvimento, em função de alguns pontos, como o consentimento esclarecido, os
benefícios devidos e o padrão de assistência apropriado. A população a ser recrutada
destes países geralmente possui acesso limitado aos serviços de saúde e imbuída da
expectativa da assistência médica, são facilmente pressionadas a participar das
pesquisas (Cabral, Schindler e Abath, 2006).
Problemas com o consentimento, como coerção, lacunas de informações,
influência indevida e fraude, entre outros, estão envolvidas em alguns tipos de
exploração (Resnik, 2003). “A bioética dos países em desenvolvimento necessita de um
caminho...que reconheça, defina e indique claramente as práticas impróprias e as
transgressões à ética em pesquisa, como a exploração, a coerção...a manipulação do
consentimento livre e esclarecido” (Kottow, 2008).
A elaboração, discussão e implementação de manuais e/ou regulamentações
éticas no contexto das pesquisas clínicas é essencial para a proteção e para salvaguardar
os direitos dos sujeitos de pesquisas.
O Brasil apresenta uma série de exigências regulatórias que visam previnir os
desvios das condutas éticas nas pesquisas com seres humanos. As principais
regulamentações em pesquisa clínica no Brasil estão descritas no Quadro 04.
Acredita-se que:
“ O [grande] desafio para a pesquisa ética internacional é aplicar os princípios éticos universais para a pesquisa biomédica em um mundo multicultural com uma multiplicidade de sistemas de cuidados de saúde e variação considerável nos padrões de cuidados de saúde” (CIOMS, 2002, p.11).
Quadro 04. Principais normas (em vigor) no contexto da pesquisa clínica no Brasil.
Ano Documento Órgão emissor Tema abordado 1976 Lei n°6.360 de
23/09/1976 Congresso Nacional
Dispõe sobre a vigilância sanitária de medicamentos, drogas, insumos farmacêuticos, correlatos, cosméticos, saneantes e outros produtos.
1995 Resolução n°173
Conselho Nacional de Saúde
Define o Plano de trabalho de revisão da Resolução CNS 01/88, incluindo a normatização de áreas temáticas especiais.
1995 Resolução n°170 Conselho Nacional de Saúde
Define a formação de um Grupo Executivo de Trabalho para revisão da Resolução CNS

53
01/88 (compuseram o grupo: pesquisadores, representantes dos Ministérios da Saúde e da Ciência e Tecnologia, CFM, OAB, CNBB, representante de usuários do SUS, ONGs e etc.)
1996 Lei n° 9.279 Congresso Nacional
Regulamenta direitos e obrigações relativos à propriedade intelectual.
1996 Resolução n°129 MERCOSUL/GMC Regulamento técnico sobre a verificação de boas práticas de pesquisa clínica.
1997 Resolução n°251 Conselho Nacional
de Saúde Contempla a norma complementar para a área temática especial de novos fármacos, vacinas e testes diagnósticos e delega aos CEPs a análise final dos projetos nessa área, que deixa de ser especial.
1997 Resolução 240 Conselho Nacional de Saúde
Define representação de usuários nos CEPs e orienta a escolha.
1999 Lei n°9782 Congresso Nacional
Define o Sistema Nacional de Vigilância e cria Agência Nacional de Vigilância Sanitária, e dá outras providências.
1999 Resolução n° 292
Conselho Nacional de Saúde
Estabelece normas específicas para a aprovação de protocolos de pesquisa com cooperação estrangeira, mantendo o requisito de aprovação final pela CONEP, após aprovação do CEP.
1999 Lei 9787 Congresso Nacional
Dispõe sobre a vigilância sanitária, estabelece o medicamento genérico, dispõe sobre a utilização de nomes genéricos em produtos farmacêuticos, e dá outras providências.
2000 Resolução n° 301
Conselho Nacional de Saúde
Contempla o posicionamento do CNS e CONEP contrário as modificações da Declaração de Helsinque.
2000 Resolução n° 304
Conselho Nacional de Saúde
Contempla norma complementar para a área de Pesquisas em Povos Indígenas.
2003 Resolução n°133 Anvisa Aprova o regulamento técnico para registro de medicamento similar.
2003 Resolução n°134 Anvisa Dispõe sobre a adequação dos medicamentos já registrados.
2003 RDC n°136 Anvisa Regulamenta sobre o registro de medicamento Novo.
2004 RDC n°210 Anvisa Altera artigos nas RDC 136, 134 e 133, de 29 de maio de 2003.
2004 Resolução n° 340
Conselho Nacional de Saúde
Aprova as Diretrizes para Análise Ética e Tramitação dos Projetos de Pesquisa da Área Temática Especial de Genética Humana.
2005 Resolução n°346
Conselho Nacional de Saúde
Regulamenta os projetos multicêntricos.
2007 Resolução n° 370
Conselho Nacional de Saúde
O registro e credenciamento ou renovação de registro e credenciamento do CEP.
2008 Resolução n°1885
Conselho Federal de Medicina
Veda ao médico participar de pesquisa envolvendo seres humanos utilizando placebo, quando houver tratamento disponível eficaz já reconhecido.
2008 Resolução n°34 Anvisa Institui o Sistema de Informações de estudos

54
de equivalência (Sineb) e o cadastro nacional de voluntários em estudos de Bioequivalência.
2008 RDC n° 38 Anvisa Aprova o regulamento para os programas de acesso expandido, uso compassivo e fornecimento de medicamento pós-estudo.
2008 RDC n° 39 Anvisa Regulamenta a realização de pesquisa clínica e dá outras providências.
2008 RDC n° 81 Anvisa Regulamenta a importação de bens e produtos, incluindo os medicamentos e insumos farmacêuticos, seja para a pesquisa clínica, uso hospitalar ou por estrita recomendação médica, como pelos serviços públicos para atender a demandas judiciais.
2009 Resolução n°421
Conselho Nacional de Saúde
Instituir a reestruturação na composição da Comissão Nacional de Ética em Pesquisa – CONEP.
2009 Resolução n° 4 Anvisa Determina a realização de farmacovigilância pelos detentores de registro de medicamentos.
2009 Instrução Normativa 04
Anvisa
Dispõe sobre o Guia de Inspeção em Boas Práticas Clínicas.
2009 Instrução Normativa 14
Anvisa Aprova os Guias de Farmacovigilância.
2010 RDC n°17 Anvisa Dispõe sobre as Boas Práticas de Fabricação de Medicamentos. Farmacovigilância.
2011 RDC n°24 Anvisa Dispõe sobre o registro de medicamentos específicos.
2011 Resolução n° 441
Conselho Nacional de Saúde
Aprovar as seguintes diretrizes para análise ética de projetos de pesquisas que envolvam armazenamento de material biológico humano ou uso de material armazenado em pesquisas anteriores.
2011 Resolução n° 446
Conselho Nacional de Saúde
Composição da Comissão Nacional de Ética em Pesquisa.
2012 RDC n°11 Anvisa Dispõe sobre o funcionamento de laboratórios analíticos que realizam análises em produtos sujeitos à Vigilância Sanitária e dá outras providências.
2012 Instrução Normativa n° 2
Anvisa
Dispõe sobre as solicitações e procedimentos de avaliação de licenciamentos de importação para pesquisas clínicas regulamentadas pela RDC 39/2008.
2012 Resolução n° 466
Conselho Nacional de Saúde
Aprovar as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos.
2013 Resolução nº 38 Anvisa Regulamenta os programas de acesso expandido, uso compassivo e fornecimento de medicamento pós-estudo.
2013 Norma operacional
nº001
Conselho Nacional de Saúde
Norma Operacional dispõe sobre a organização e funcionamento do Sistema CEP/CONEP.
Fonte: Elaboração própria. Atualizada em 25/02/2014.

55
Após a aprovação da pesquisa pelas instâncias éticas do país, CEP e/ou CONEP,
estas assumem coresponsabilidade pela execução do projeto. O acompanhamento do
desenvolvimento dos projetos deve ser realizado através da análise dos relatórios anuais
e/ou semestrais submetidos ao CEP pelos pesquisadores. “O Sistema CEP/CONEP
deverá ser informado de todos os fatos relevantes que alterem o curso normal dos
estudos por ele aprovados...” (Brasil, 2013, item V.5).
O CEP como órgão consultivo, deliberativo e educativo, deve estar aberto para
receber os participantes da pesquisa, denúncias de abusos ou qualquer outra natureza, de
qualquer parte envolvida, que possam alterar o curso normal do estudo, decidindo então,
pela continuidade, modificação ou suspensão da pesquisa (Brasil, 2013).
5. O PRINCÍPIO DA JUSTIÇA E EQUIDADE
Existem várias teorias acerca da Justiça, entretanto, na filosofia moral, todas as
concepções “interpretam a justiça como um tratamento justo, equitativo e apropriado,
levando em consideração aquilo que é devido às pessoas.” (Beauchamp & Childress,
2002 b).
Na filosofia do direito, o conceito de justiça envolve três elementos: a alteridade
-vinculada sempre a um outro sujeito, a igualdade - busca da simetria na distribuição ou
retribuição e a exigibilidade do débito – exigência daquilo que é devido (Barreto, 2006).
Na ética da pesquisa envolvendo seres humanos, o princípio da Justiça refere-se
principalmente à Justiça distributiva, a qual exige a distribuição equitativa dos encargos
e benefícios da participação na pesquisa aos sujeitos envolvidos (CIOMS, 2002).
A expressão “Justiça distributiva” vem originalmente de Aristóteles, e
posteriormente muitas teorias sobre tal foram propostas para sistematizar como os
encargos, bens e serviços sociais, incluindo a área da saúde, devem ser distribuídos ou
redistribuídos igualitariamente entre os indivíduos. Tais teorias podem ser classificadas
como (Beauchamp & Childress, 2002 b):
1. Utilitaristas: O enfoque está na maximação da utilidade pública, e por vezes, podem
sacrificar liberdades básicas em função de interesses públicos. Na área da saúde a
justiça envolveria a ponderação das vantagens e desvantagens dos benefícios ou
direitos.
2. Comunitaristas: O enfoque está na análise das concepções de justiça que evoluem
dentro da comunidade, ou seja, dependem dos padrões oriundos destas comunidades.

56
Neste sentido, adotam os princípios da justiça como plurais com diferentes origens de
concepção de bem. Compartilham as responsabilidades entre os indivíduos e a
comunidade. Na área da saúde, a justiça é garantida pela implementação de serviços
compatíveis com as concepções e necessidades particulares de cada comunidade.
3. Liberais: O enfoque está nos direitos humanos universais, em especial, a liberdade,
igualdade, e os procedimentos democráticos. Uma sociedade justa para estas teorias são
aquelas aonde há a proteção dos direitos de liberdade, propriedade e livre mercado, de
forma que as pessoas livremente busquem por seus ideais e bem-estar. Na perspectiva
destas teorias, a saúde não deve ser um bem público dado como direito dos indivíduos, e
sim um bem privado para que os indivíduos possam livremente e democraticamente
utilizarem ou não os serviços.
4. Igualitárias: O enfoque está na distribuição igual dos benefícios e encargos sociais. O
igualitarismo para estas teorias não é total, exige apenas algumas igualdades básicas
entre os indivíduos, permitindo algumas desigualdades desde que resultem em mais
benefícios aos menos favorecidos.
A regulamentação brasileira sobre ética em pesquisa com seres humanos traz o
princípio da “Justiça e equidade” entre os requisitos para se garantir a eticidade das
pesquisas. Buscando uma aproximação desses princípios, lançamos mão da concepção
de John Rawls de “justiça como equidade”, dentro da perspectiva igualitária, onde esta é
“entendida como as normas comuns de cooperação reconhecida por pessoas livres e
iguais que participam nas atividades sócias em respeito mútuo” (Beauchamp &
Childress, 2002 b).
O termo equidade, utilizado na dissertação, se refere a um tipo de julgamento
que associa igualdade e justiça, é definido como “um dos princípios imutáveis de justiça
que levem o juiz a um critério de moderação e igualdade, mesmo com prejuízo do
direito objetivo” (Ferreira, 2004). Para fins de esclarecimento é importante
introduzirmos os conceitos de justiça formal e a material dentro da perspectiva da
Justiça. O dito formal representa que os iguais devem ser tratados de maneiras iguais
independentemente das circunstâncias e o dito material, especifica quais aspectos são
relevantes para um tratamento igual que permite identificar características importantes
para o desenho de políticas públicas de distribuição (Beauchamp & Childress, 2002 b).
A justiça formal é aquela normalmente garantida por lei, mas o que se deseja é alcançar
a igualdade material através da redução das diferenças de qualquer ordem entre

57
indivíduos da sociedade, o que implica muitas vezes no tratamento diferente entre eles,
neste sentido a justiça material se aproxima mais do termo equidade.
“Uma das expressões do pluralismo moral das sociedades complexas contemporâneas é o conflito entre uma concepção de justiça entendida como igualdade de oportunidades para todos, que defende a saúde como um direito humano universal, e o ideal de justiça como eqüidade, que sustenta que, em casos de conflitos, se devem privilegiar as exigências dos menos favorecidos” (Ribeiro & Schramm, 2004, p. 1142).
Rawls traz a ideia de sociedade como um sistema equitativo de cooperação
social que se perpetua por gerações. Os termos equitativos de cooperação incluem a
reciprocidade ou mutualidade e também contém a ideia de vantagem ou bem racional
(Rawls,1996 ). Ou seja, se um membro da sociedade cumpre sua parte de acordo com as
regras previamente acordadas e reconhecidas, este deve-se beneficiar de tal cooperação.
A partir desta cooperação, da reciprocidade no trato entre os homens e instituições
sociais é que se preserva a sociedade como um empreendimento cooperativo (Rabelo
Junior, 2011).
Assim, apesar da teoria de Rawls não ser voltada para área da saúde e não ter o
foco no indivíduo, e sim na sociedade, é possível transpor tais ideias para o campo da
pesquisa clínica, supondo que a participação do sujeito de pesquisa é uma maneira de
cooperação social, este deve se beneficiar diretamente desta.
Rawls reconhece a obrigação social de eliminar ou reduzir os obstáculos à
equitativa igualdade de oportunidade - as formas injustas de distribuição - através de
programas, políticas que compensem as tais desvantagens, sejam elas de origem
biológica (doenças) e/ou da loteria da vida social (deficiências). A regra é que se não
deve conceder benefícios sociais a propriedades favoráveis imerecidas ou desfavoráveis
imerecidas (Beauchamp & Childress, 2002 b).
Nesta linha de pensamento pode-se inferir, por exemplo, que os sujeitos de
pesquisa recrutados em países sub ou em desenvolvimento, normalmente indivíduos
desfavoráveis e classificados como vulneráveis, devem ser tratados com atenção
especial, de maneira diferenciada dos demais sujeitos, devido ao risco aumentado de se
violar algum princípio ético.
Na Resolução n°466/12 do CNS/MS que regulamenta a pesquisa com seres
humanos no Brasil é possível verificar inclusão do princípio da Justiça e equidade como
referencial ético nos itens abaixo destacados que formulam exigências de pesquisa,
independente da área de conhecimento, visando a justa distribuição tratada pelos autores

58
supramencionados:
• “se houver necessidade de distribuição aleatória dos participantes da
pesquisa em grupos experimentais e de controle, assegurar que, a priori, não seja
possível estabelecer as vantagens de um procedimento sobre outro, mediante revisão
de literatura, métodos observacionais ou métodos que não envolvam seres humanos”;
• “Garantir que as pesquisas em comunidades, sempre que possível,
traduzir-se-ão em benefícios cujos efeitos continuem a se fazer sentir após sua
conclusão”;
• “Comunicar às autoridades competentes, bem como aos órgãos
legitimados pelo Controle Social, os resultados e/ou achados da pesquisa, sempre que
estes puderem contribuir para a melhoria das condições de vida da coletividade”;
• “Assegurar aos participantes da pesquisa os benefícios resultantes do
projeto, seja em termos de retorno social, acesso aos procedimentos, produtos ou
agentes da pesquisa”;
• “Comprovar, nas pesquisas conduzidas no exterior ou com cooperação
estrangeira, os compromissos e as vantagens, para os participantes das pesquisas e
para o Brasil, decorrentes de sua realização”;
• “Os estudos patrocinados no exterior também deverão responder às
necessidades de transferência de conhecimento e tecnologia para a equipe brasileira,
quando aplicável e, ainda, no caso do desenvolvimento de novas drogas, se
comprovadas sua segurança e eficácia, é obrigatório seu registro no Brasil”;
• “Assegurar a todos os participantes ao final do estudo, por parte do
patrocinador, acesso gratuito e por tempo indeterminado, aos melhores métodos
profiláticos, diagnósticos e terapêuticos que se demonstraram eficazes” (Brasil, 2013,
III.2).
Da maneira como é contextualizado o princípio, pode-se deduzir a injustiça
como a omissão ou negação de benefícios, direitos e/ou encargos que são devidos às
pessoas (Beauchamp & Childress, 2002 b). Vale ressaltar que o processo de revisão e
monitoramento ético das pesquisas envolvendo seres humanos por parte dos Comitês de
Ética deve ser tratado como um exercício de Justiça social, onde os interesses de todas
as partes envolvidas devem ser reconhecidos e levados em consideração (Pullman,
2002).
Diante dos pressupostos básicos estabelecidos por Rawls para uma sociedade
mais justa - igualdade de oportunidades em condições de plena equidade e que os

59
benefícios devem ser repassados preferencialmente aos menos favorecidos – e das
discussões na literatura científica sobre a aplicação do princípio de justiça na ética em
pesquisa, pretende-se analisar as implicações bioéticas que envolvem o princípio da
justiça e equidade na trajetória da pesquisa ao registro de anticorpos monoclonais e
biomedicamentos submetidos a ensaios clínicos em oncologia com participação do
Brasil.
Alguns pontos podem ser previamente elencados: igualdade de acesso aos bens e
serviços de saúde, garantia de benefícios oriundos das pesquisas nos quais os sujeitos
participaram e acesso facilitado aos resultados das pesquisas.
6. JUSTIÇA E ÉTICA EM PESQUISA
As questões de justiça foram por muito tempo comumente associados com as
práticas sociais ou a representação política, e as associações com a investigação
científica foram amadurecendo a posteriori. Durante o século 19 e início do século 20 o
recrutamento de sujeitos de pesquisa era focado nos sujeitos vulneráveis, como
pacientes de alas pobres, negros ou desfavorecidos, enquanto o retorno/ benefícios
destas pesquisas eram voltados à outra parcela da população, não envolvida no
processo. A partir destes contextos históricos e entre muitos exemplos, percebe-se quão
relevante são as concepções de justiça nas pesquisas envolvendo seres humanos (HHS,
1979).
Informações sobre o diagnóstico, prevenção e tratamento das doenças em
diferentes populações são consideradas benefícios e devem ser distribuídas
homogeneamente à sociedade, porém, quando se excluí determinados grupos da
participação nas pesquisas, as consequências de tais exclusões são as limitações de
informações naquele grupo, o que representa uma grave injustiça de classe (CIOMS,
2002).
É sabido que os gastos federais com P&D na área biomédica são escassos e em
vários países ficam aquém dos investimentos privados neste setor. David Resnik (2001)
expõe que em ambiente de escassez, questões de Justiça distributiva serão suscitadas
nos momentos das divisões dos recursos e das definições de quais condições médicas
deverão ser prioridades. Para o autor a participação dos cidadãos nestes processos
decisórios que afetam a saúde pública e o bem-estar dos indivíduos é fundamental.

60
Uma das estratégias utilizadas pelo NIH dos Estados Unidos, para melhorar a
participação pública, foi a expansão desta nas decisões de financiamento em P&D,
através do Council of Public Representattives (CORP), que é composto por pacientes e
pessoas leigas. Entretanto esta decisão é criticada por alguns autores e para David
Resnik (2001) para se alcançar uma efetiva e justa distribuição dos recursos federais
para P&D é necessária uma mistura equilibrada entre conselhos/decisões públicas
(cidadãos/pacientes) e de especialistas na área.
O princípio da Justiça requer que a pesquisa seja sensível na análise das
condições ou necessidades de saúde da população, em especial a da população
vulnerável a ser recrutada, pois os riscos aos sujeitos vulneráveis são mais justificáveis
quando as intervenções ou procedimentos a serem realizados promovem uma
perspectiva de benefícios diretos à saúde destes indivíduos (CIOMS, 2002).
Durante a seleção dos sujeitos da pesquisa dois níveis de justiça podem ser
expressos, o social e o individual, o que representa respectivamente, não fazer distinção
de qualquer ordem – raça, gênero, etc – para seleção de sujeitos, estabelecendo
preferências e oferecendo igualmente os benefícios da investigação a todos pacientes.
Entretanto, mesmo atentando-se para estes aspectos na seleção, algumas injustiças
podem surgir neste contexto, visto que existem alguns padrões sociais injustos
(preconceitos sociais) já institucionalizados na sociedade e que podem afetar a
distribuição geral dos encargos e benefícios da pesquisa aos indivíduos (HHS, 1979).
Questões de justiça distributiva também estão implicadas ao término do estudo.
O Manual Internacional de Pesquisa com Seres Humanos (Guideline for international
research on human subjects) da CIOMS requer que ao término do estudo com
resultados de sucesso, o patrocinador garanta disponibilidade razoável a tal produto ou
intervenção aos habitantes das comunidades que participaram do estudo e que as
exceções devem ser justificadas e acordadas entre ambas as partes antes do início do
estudo. Adicionalmente o protocolo de pesquisa deve especificar, no processo de
consentimento, por quanto tempo e quais serviços de cuidados à saúde serão
disponibilizados durante e após a pesquisa para os sujeitos de pesquisa e comunidade
envolvida ou país anfitrião (CIOMS, 2002).
No Brasil, questões relacionadas ao fornecimento de medicamentos pós-estudo
estão dispostas na Resolução n°38/11 da Anvisa. No capítulo V de tal resolução, fica
evidente o acesso gratuito, após o término do estudo, ao medicamento do estudo no qual
o participante da pesquisa está envolvido, sempre que houver benefícios (Anvisa, 2013
c).

61
A revisão da Declaração de Helsinque ocorrida em 2008 na reunião da
Associação Médica Mundial reforça este requerimento, ao suscitar o princípio da
Justiça, em seu artigo 33, onde discorre sobre os benefícios dos sujeitos de pesquisa
após o término de um estudo. A alteração exige que os participantes dos protocolos de
pesquisa tenham acesso aos resultados do estudo assim como aos benefícios oriundos
destas pesquisas (WMA, 2008).
Cooley (2001) faz várias críticas a este requerimento do CIOMS, baseado na
versão de 1993, relacionadas à sua ambiguidade e imprecisão. No texto do CIOMS é
utilizado o termo “reasonably available”12, que significa razoavelmente disponível, que
para o autor é vago e não define quem deve determinar o que é razoável ou não.
Adicionalmente, há outras questões relacionadas aos custos destes produtos,
distribuição local e serviços essenciais para o uso correto do produto. O autor cita um
exemplo onde uma companhia industrial forneceu a fórmula do produto a uma
comunidade, mas não os serviços necessários para o consumo, como refrigeradores e
água própria para consumo, o que prejudicou vários sujeitos envolvidos. Partindo deste
contexto, que alguns produtos não podem ser apropriadamente utilizados sem bens e
serviços auxiliares, Cooley (2001) julga importante determinar se a justiça distributiva
deveria demandar também dos patrocinadores a suplementação destes bens.
O autor também critica a imprecisão dos termos “community” (comunidade),
pois esta pode ser definida apenas como a comunidade abrigada na área geográfica onde
o estudo foi conduzido como ser expandida ao país e critica quanto à indefinição do
número de habitantes que seriam contemplados, poderiam ser todos ou uma parcela
(Cooley, 2001).
A CIOMS ressalta que embora o patrocinador, em geral, não seja obrigado a
providenciar serviços de cuidados à saúde para além das necessárias para a condução da
pesquisa, a execução de tal ação é moralmente louvável (CIOMS, 2002). Solomon
Benatar (2001) em resposta ao artigo de Cooley, afirma que embora a requisição do
CIOMS seja vaga, esta possui um papel importante na luta para atingir os objetivos da
justiça distributiva.
A Justiça possui um valor central na avaliação de ações com respeito ao outro.
Qualificar um ato como “ (...) `justo´ significa determinar os critérios do que vale,
merecer ser aprovado, na área da ação social (...)” (Perelman, 2005, p.68). E embora os
conceitos de Autonomia, Beneficência e Justiça estejam consagrados no âmbito das 12 “…any product developed will be made reasonably available to inhabitants of the underdeveloped community in wich the research was carried out” (Bankowski Z, Levine RJ, eds. Ethics and research on human subjects: international guidelines. Geneva: CIOMS, 1993, p.26)

62
Pesquisas clínicas, através das resoluções 196/96 e 466/12 e manuais internacionais de
ética em pesquisa, integrar estes pilares éticos à prática do mundo real é uma tarefa
contínua (Owonikoko, 2013). Assim a análise constante das implicações práticas destes
princípios faz-se necessária.

63
MÉTODO
1. OBJETO
O objeto do estudo é o conjunto de questões éticas de justiça envolvidas no
percurso da realização dos ensaios clínicos com anticorpos monoclonais e
biomedicamentos oncológicos no Brasil até o registro em nosso país.
2. DESENHO
Trata-se de um estudo retrospectivo e descritivo. O período do estudo
selecionado foi um período de dez anos, de 2003 a 2012, perpassando 2005, marco
histórico, quando o International Committee of Medical Journal Editors (ICMJE)
adotou a política de ensaios-registro, exigindo como condição de consideração para a
publicação em suas revistas, o registro de ensaios em base pública de registro.
3. ETAPAS METODOLÓGICAS
3.1. Etapa 1: Busca bibliográfica: revisão do estado da arte
Visando a necessidade de integrar as informações acerca da temática Justiça e
Ética em Pesquisa realizamos uma busca sistemática sobre o tema. Adicionalmente, o
objetivo desta revisão foi identificar as relações estabelecidas entre os dois conceitos.
Para tal busca foram selecionados os seguintes descritores em Ciência em Saúde
(DeCS): Justiça, Justiça social, ética em pesquisa e ensaio clínico. O termo equidade
expressa um critério de aplicação do princípio da justiça e, portanto, para a busca
bibliográfica supramencionada utilizamos apenas o termo Justiça.
Os DeCS são largamente utilizados como uma linguagem única na indexação de
artigos científicos, anais de congressos, livros e outros materiais. Foi utilizada também a
palavra-chave “medicamentos”. A fim de ampliar o conjunto de estudos para as línguas
espanhol e inglês, alguns descritores foram selecionados a partir do MeSH (Medical
Subject Headings) desenvolvido pela Biblioteca Nacional de Medicina dos Estados

64
Unidos (National Library of Medicine) com o objetivo de permitir o uso de
terminologia comum para a recuperação de informações independentemente do idioma.
Os MeSH selecionados foram: Social justice, Ethics research e clínical trials. Os
termos Justiça e Medicamentos não foram identificados no MeSH, porém, foram
mantidos na busca como palavras-chave.
As Bases de dados eletrônicas (BVS - Biblioteca Virtual de Saúde, PubMed e
Scopus) foram consultadas retrospectivamente até o ano de 1996, usando o filtro
"humano" e as seguintes chaves de busca: Justiça (Justice) ou Justiça social (Social
justice) combinado com os descritores ética em pesquisa (Ethics research) e ensaio
clínico (clínical trials) e medicamentos (medications). A busca se limitou aos artigos
escritos em inglês, português e espanhol.
A partir da leitura dos artigos selecionados na busca sistemática, buscou-se
identificar a aplicação e compreensão do princípio da justiça e equidade13 dos autores
no contexto da pesquisa clínica. O resultado foi agregado, de acordo com a percepção
da pesquisadora, em três categorias analíticas que serão descritas no Quadro 09 (página
77).
3.2. Etapa 2: Busca e coleta de dados empíricos
Busca e identificação dos estudos
Foram selecionadas duas importantes bases de registros listadas pelo ICTRP e
OMS, uma internacional, Clínical Trials e outra nacional, ReBEC. Os critérios de
elegibilidade foram utilizados para o refinamento da busca nestas bases de dados. O
produto extraído foi organizado e categorizados em tabelas e quadros.
Adotaram-se como critérios de elegibilidade as seguintes características:
Critérios de inclusão:
1. Ensaios com registro nas bases de ensaios públicos: Clínical Trials e Rebec, no
período de 2003 a 2012.
2. Estudos Clínicos fase II e fase III com intervenção terapêutica específica para o
tratamento de câncer e com participação do Brasil.
13 A maioria dos autores identificados na busca utilizava apenas o termo “principio da justiça” ou “Justiça”. O termo equidade era utilizado nas definições e situações descritas no contexto da justiça e pesquisa.

65
3. Estudos clínicos que envolvam a experimentação de anticorpo monoclonal ou
biomedicamentos destinados ao tratamento de câncer.
Critérios de exclusão:
1. Não ter publicação de ‘resultado do estudo’ disponível na base de dados pública.
2. Recrutamento de sujeitos de pesquisa ativo, não iniciado ou em andamento.
3. Não possuir dados suficientes para suportar a análise.
Os levantamentos dos ensaios clínicos junto às bases de dados do ClínicalTrials
e REBEC foram realizados em 05/10/2013 utilizando as seguintes palavras-chave:
“Cancer”, “ oncology”, “ neoplasm” e “neoplasia”. Para a construção da chave de busca
foi utilizado entre as palavras supracitadas o operador boleado “OR” (“ou”), a fim de
ampliar a margem de estudos selecionados, entretanto, a base de dados da ReBEC não
utiliza tais operadores, e desta forma, as palavras-chaves nesta plataforma foram
utilizadas separadamente em cada busca.
Os demais critérios de busca utilizados na base de dados do ClínicalTrials foram
para as seguintes características:
1. Tipo de Estudo (“Study Type”): estudos de intervenção
2. Localização do estudo (“Location”): Brasil
3. Fase do estudo (“Phase”): Estudos Clínicos fase II e fase III
4. Período de entrada na base (“First received”): período de 01/01/2003 a 31/12/2012.
Na busca avançada na base de dados da REBEC não existem os campos “Fase
de estudo” e “Período de entrada na base”. Assim, tais parâmetros foram avaliados após
os resultados da busca. Os demais campos selecionados foram: “Tipo de estudo”
(intervenção), “País de recrutamento” (Brasil) e “Gênero” (ambos).
Foram selecionados ao todo 492 estudos na base de dados do ClínicalTrials e 12
estudos na ReBEC. Dos 12 estudos da ReBEC, 09 foram identificados utilizando na
busca a palavra-chave “cancer”, 02 com “neoplasia” e 01 com “neoplasm”. Nenhum
estudo foi identificado utilizando a palavra “oncologia” ou “oncology”.
Dos 12 estudos, havia a sobreposição de um estudo em três buscas (cancer,
neoplasia e neoplasm), assim as repetições foram removidas, totalizando 10 estudos.
Destes, 05 estudos também estavam cadastrados no ClínicalTrials e sobrepostos aos
resultados nesta base. O demais 05 estudos foram excluídos da amostra por não
envolverem a experimentação de anticorpo monoclonal ou biomedicamentos. A
descrição dos estudos excluídos encontra-se no Apêndice A.

66
Dos 492 estudos extraídos do Clínicaltrials, 112 estudos foram excluídos da
amostra por não apresentarem câncer/neoplasia maligna como a condição médica prévia
ou por não apresentarem a intervenção terapêutica com anticorpo monoclonal ou
biomedicamento. As descrições destes medicamentos encontram-se no Apêndice B.
A condição de recrutamento dos sujeitos de pesquisa também foi utilizada como
critério de exclusão, uma vez que, o interesse do estudo é avaliar a trajetória dos
anticorpos monoclonais e biomedicamentos até o seu registro e possível incorporação
no país, para tanto os estudos que não iniciaram o recrutamento “not yet recruiting”,
com recrutamento ativo “recruiting”, ativo - mas não recrutando “active, not
recruiting”, suspensos precocemente “suspended” e interrompidos antes do primeiro
paciente ser recrutado “Withdrawn” foram excluídos da amostra. Assim, apenas os
estudos com a condição de finalizado “completed”14 e encerrado “terminated”15 serão
analisados.
Dos 380 estudos restantes, 238 foram excluídos após a aplicação do critério de
exclusão mencionado acima. Dos 142 estudos remanescentes, 92 estudos foram
excluídos por não apresentarem os seus resultados indexados no clínicaltrials.gov, uma
vez que, os resultados de um estudo, sejam eles positivos ou negativos, impactam
diretamente no processo de registro do medicamento ou intervenção avaliado, assim, a
publicação dos resultados nos ajudará a entender melhor sobre a trajetória destes
medicamentos.
Identificação dos medicamentos
Após análise primária dos estudos extraídos das bases de dados, conforme
metodologia descrita acima, os medicamentos foram identificados e categorizados. Para
a captação das informações sobre os biomedicamentos e anticorpos monoclonais
extraídos desta análise primária foi utilizada como ferramenta o dicionário de
medicamentos do National Cancer Institute- NCI do National Institutes of Health –
NIH: The NCI Drug Dictionary. Este dicionário contém definições técnicas e os
sinônimos dos fármacos e seus agentes utilizados para o tratamento de câncer ou de
condições relacionadas ao câncer.
Complementando as informações/definições dos medicamentos utilizamos o
14 De acordo com o ClínicalTrials o termo "completed” significa que o o recrutamento de sujeitos de pesquisa do estudo clínico terminou normalmente, e os participantes não estão sendo mais examinados ou tratados. 15 De acordo com o ClínicalTrials o termo “terminated” significa que o recrutamento de sujeitos de pesquisa do estudo foi interrompido ou foi encerrado precocemente.

67
sistema de classificação de fármacos – ATC/DDD Index (Anatomical Therapeutic
Chemical and Defined Daily Dose) da Organização Mundial de Saúde e o Manual de
Denominações Comuns Brasileiras (DCB). O ATC/DDD Index atua como uma unidade
de medição utilizada desde 1970 para a troca e comparação de dados sobre os fármacos
a nível internacional, nacional ou local e tornou-se padrão-ouro para pesquisas
internacionais sobre a utilização de fármacos (WHO, 2012).
Esta ferramenta divide as substâncias ativas em diferentes grupos de acordo com
o organismo ou sistema no qual ela tem ação e de acordo com as suas propriedades
terapêuticas, farmacológicas e químicas. Os fármacos são classificados em cinco
grandes níveis, o 1°nível classifica o grupo anatômico principal, o 2°nível classifica o
subgrupo terapêutico, o 3° nível classifica o subgrupo farmacológico, o 4° nível
classifica o subgrupo químico e o 5°nível a própria substância química. Segue abaixo o
Quadro 05 demonstrando a organização dos antineoplásicos e agentes
imunomoduladores de acordo como o ATC/DDD Index:
Quadro 05. Antineoplásicos e agentes imunomoduladores pelo ATC/DDD Index.
Nível Código Descrição do fármaco
1 L Antineoplásicos e agentes imunomoduladores
2 L01 Agentes antineoplásicos
3 L01X Outros agentes antineoplásicos
4 L01XC Anticorpo monoclonal
L01XC01 Edrecolomab
L01XC02 Rituximab L01XC03 Trastuzumab L01XC04 Alemtuzumab L01XC05 Gemtuzumab
L01XC06 Cetuximab
L01XC07 Bevacizumab
L01XC08 Panitumumab
L01XC09 Catumaxomab
L01XC10 Ofatumumab
L01XC11 Ipilimumab
L01XC12 Brentuximab vedotin
5
L01XC13 Pertuzumab

68
Fonte: http://www.whocc.no/atc_ddd_index/?code=L01XC&showdescription=no
O Manual das Denominações Comuns Brasileiras (DCB) é um guia de consultas
de substâncias farmacêuticas que representa um importante papel dentro da política
nacional de medicamentos genéricos ao estabelecer a padronização de regras de
nomenclatura e tradução de termos relacionados a substâncias de interesse
farmacêutico, proporcionando informações unificadas para os profissionais da área da
saúde e a sociedade em geral (Brasil, 2013b).
Busca e identificação dos registros dos medicamentos
Com base nos dados dos fármacos identificados e estudados foi realizada uma
investigação da trajetória do registro desses medicamentos no país, através da base de
dados da Anvisa e dos principais órgãos internacionais de registro de medicamentos,
FDA e EMA.
Para a busca na Anvisa foi necessário acessar dois ambientes virtuais: a data de
aprovação do medicamento foi baseada na “Consulta ao produto” (disponível em
http://www7.anvisa.gov.br/datavisa/Consulta_Produto/consulta_medicamento.asp) e
data da última revisão foi baseada no “Bulário Eletrônico” (disponível em
http://www.anvisa.gov.br/datavisa/fila_bula/index.asp), ambas disponíveis no website
da própria Agência.
A busca na agência FDA ocorreu atráves da ferramenta FDA Approved drug
Products disponível em seu Website (disponível em
http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) e busca na agência
EMA ocorreu através da ferramenta Find Medicine disponível em seu Website
(disponível em http://www.ema.europa.eu/ema/). Esta etapa permitiu a análise da
trajetória destes medicamentos do momento da investigação ao registro no Brasil e nas
principais agências reguladoras.
Identificação das compras Federais
A partir das informações acerca dos registros dos biomedicamentos e anticorpos
monoclonais oncológicos no país foi realizada uma investigação sobre o processo de
aquisições e gastos do Governo Federal com estes medicamentos pelo Sistema Único de
Saúde. Foi utilizada como fonte de dados a base SIASG - Sistema Integrado de
Administração de Serviços Gerais, que é um sistema informatizado de apoio às
atividades operacionais do Sistema de Serviços Gerais na Administração Federal que

69
atua como importante ferramenta na promoção da transparência dos atos do governo
sobre os processos licitatórios.
3. VARIÁVEIS EMPÍRICAS DE INTERESSE
A partir das bases de registro foram consideradas variáveis de interesse: Número
de registro do estudo na base, disponibilidade de resultados do estudo, nome do fármaco
avaliado, número estimado de pacientes, data de início, data do registro na base, data
última atualização no site, data estimada de término do estudo, patrocinador, topografia
do câncer, condição de recrutamento, países com recrutamento, modelo de intervenção e
uso de placebo.
No dicionário de medicamentos do National Cancer Institute- NCI do National
Institutes of Health foram considerados variáveis de interesse: a descrição do fármaco,
nome sinônimo, nome comercial e abreviações.
No índice ATC/DDD da Organização Mundial de Saúde a classificação da
substância ativa será considerada a variável de interesse.
Na base de dados da Anvisa, FDA e EMA serão consideradas como variáveis de
interesse: nome do fármaco, ingrediente ativo, companhia de origem, data de aprovação
pelos respectivos órgão e as indicações clínicas dos fármacos/medicamentos.
Na base de dados do SIASG será considerado como variáveis de interesse: nome
do medicamento, quantidade de itens comprados, valor unitário, data de compra e
localidade do órgão responsável pela compra.
Tabela 02. Descrição das variáveis do estudo e seus objetivos de coleta.
Variável Objetivo
Bases de registros de Ensaios Clínicos
Número de registro do estudo na base Identificação do estudo
Fase do estudo
Identificação do medicamento (forma
farmacêutica terminada) e aplicabilidade do
estudo
Randomização Conhecer o desenho do estudo
Cegamento Conhecer o desenho do estudo
Data de início Recorte temporal
Modelo de intervenção e uso de placebo Conhecer o desenho do estudo
Topografia do câncer Verificar emprego clínico/relevância
epidemiológica

70
Data do registro na base Analisar a rastreabilidade e publicidade dos
estudos
Data estimada de término do estudo Recorte temporal
Países com recrutamento Verificar quais os países envolvidos nos
estudos (abrangência)
Condição de recrutamento Ter possibilidade de avaliar os resultados dos
estudos
Número estimado de pacientes Verificar a amplitude da execução do estudo
Patrocinador Verificar a origem do patrocínio
Resultados do estudo Verificar a disponibilidade de resultados
Agências regulatórias
Nome do fármaco avaliado Identificação do medicamento
Sinônimo Identificação do medicamento
Nome comercial Identificação do medicamento
A descrição do fármaco Identificação e emprego clínico
Classificação da substância ativa (ATC) Recorte do estudo
Data do registro nas agências reguladoras Verificar a disponibilidade no mercado
Data da última atualização Verificar a disponibilidade no mercado
Indicações clínicas registradas Verificar emprego clínico/relevância
epidemiológica
SIASG
Quantidade de itens comprados Verificação da demanda
Valor do item Verificação dos gastos federais
Ano da compra Recorte temporal
Estado responsável Identificação da localidade da compra
4. ANÁLISE
A partir de uma abordagem qualitativa, a análise ética foi dividida em dois
momentos. Primeiramente foi feita análise de natureza teórica que empreendeu a
compreensão e exploração da discussão sobre o princípio da justiça e equidade nas
principais regulamentações da ética em pesquisa em seres humanos e na literatura
cientifica, a partir da revisão documental e bibliográfica. A análise relativa ao princípio
normativo de justiça e equidade na trajetória dos ensaios clínicos até o registro dos
anticorpos monoclonais e biomedicamentos no Brasil foi construída a partir dos
referenciais teóricos identificados na literatura científica. Neste momento foram

71
delimitadas três categorias analíticas que estruturaram a integração da análise de dados
empíricos e análise ética.
As análises dos dados provenientes das bases de registros de ensaios, de
registros de medicamentos e do banco SIASG foram de natureza quantitativa,
utilizando-se de análises da informação agregada, estatística descritiva e medidas de
tendência central.
No segundo momento, uma análise ética de natureza prática visou verificar a
aplicabilidade do princípio da justiça e da equidade, tal qual proposta na literatura, no
contexto real das pesquisas, utilizando como base de argumentação as categorias
analíticas.
Ao relacionarem-se as variáveis de interesse com as categorias analíticas,
emergiram as diferents possibilidades e dimensões da análise dos dados empíricos,
conformando um plano de análise preliminar. O Quadro 06 mostra esta relação.
Quadro 06. Categorias analíticas e variáveis relacionadas
Categorias analíticas Variáveis empíricas relacionadas
Relevância social
Fase do estudo; Randomização; Cegamento;
Data de início; Modelo de intervenção e uso
de placebo; Topografia do câncer; Data
estimada de término do estudo; Países com
recrutamento; Condição de recrutamento;
Número estimado de pacientes; Patrocinador;
Resultados do estudo; Nome do fármaco
avaliado; A descrição do fármaco;
Classificação da substância ativa (ATC); Data
do registro nas agências reguladoras; Data da
última atualização; Indicações clínicas
registradas.
Equidade no acesso à saúde
Países com recrutamento; Condição de
recrutamento; Número estimado de paciente;
Topografia do câncer.
Distribuição equitativa de encargos e
benefícios
Quantidade de itens comprados; Valor do
item; Ano da compra; Modelo de intervenção
e uso de placebo; Topografia do câncer.

72
5. ASPECTOS ÉTICOS
O projeto foi formulado à luz da Resolução n°466/12 do Conselho Nacional de
Saúde e demais critérios éticos adotados pelo país. Ressalvo que o presente estudo
não envolve pesquisa com seres humanos. Para a execução deste projeto foram
utilizadas bases de dados secundárias e de domínio público. Desta forma, não foram
identificados riscos de nenhuma ordem para os pesquisadores e instituições
envolvidas na análise do projeto.
Este projeto foi submetido à apreciação do Comitê de Ética em Pesquisa da
Escola Nacional de Saúde Pública Sérgio Arouca – ENSP e aprovação do mesmo
ocorreu em 03/10/2013 (Anexo A). Os resultados serão apresentados à sociedade no
formato de artigo científico e eventuais apresentações em eventos científicos.

73
RESULTADOS
1. BUSCA SISTEMÁTICA: JUSTIÇA E PESQUISA
Os resultados da busca com os números de publicações seguem descritos no
Quadro 07. A descrição completa dos artigos identificados encontra-se no Apêndice C.
Quadro 07. Critérios de seleção da busca e os resultados preliminares.
Bases de dados eletrônicas Descritores/palavras-chave BVS Pubmed Scopus
Chave de busca 1: justiça OR Justiça social AND ética em pesquisa AND ensaio clínico AND medicamentos
1 0 0
Chave de busca 2: justice OR Social justice AND Ethics research AND clínical trials AND medications
10 29 8
Nota: A busca nas bases de dados foi realizada no dia 04/10/2013.
Foram identificados 09 artigos repetidos nas bases utilizadas, sendo 05 artigos
encontrados nas bases Pubmed e BVS, 02 artigos nas bases Pubmed, BVS e Scopus, 01
artigo na Scopus e BVS e 01 artigo no Pubmed e BVS.
Dos 08 artigos identificados na base Scopus, 03 foram excluídos por não estarem
acessíveis ao público. Dos 11 artigos identificados na base BVS, 02 foram excluídos por
serem considerados inadequados ao objeto da pesquisa; 03 excluídos por estarem em
duplicidade nas demais bases e 01 não estava disponível na base. Dos 29 artigos da
Pubmed, 08 foram excluídos por estarem em duplicidade nas demais bases; 01 não
estava disponível e 01 foi excluído por ser considerado inadequado ao objeto da
pesquisa. Assim, a seleção final foi de 29 artigos. No entanto, após leitura completa, 07
foram excluídos por conteúdo não conforme com os objetivos da busca. Ao final foram
utilizados 22 artigos. A descrição do fluxo de elegibilidade encontra-se no Quadro 08.
Quadro 08. Descrição do fluxo de elegibilidade dos artigos da busca sistemática sobre Justiça e Pesquisa clínica.
Resumos capturados na pesquisa em bases de dados 48 Resumos restantes após a remoção das duplicidades 37 Resumos considerados inadequados ao objeto da pesquisa 03 Artigos completos não acessíveis 05

74
Resumos classificados considerados adequados ao objeto da pesquisa para leitura completa dos artigos
29
Artigos completos excluídos após a leitura 07
Na leitura dos artigos encontramos diversas problemáticas no contexto das
pesquisas envolvendo questões de Justiça, porém, poucos se aprofundaram sobre o
debate ético com o princípio da Justiça. Após a leitura, foi possível extrair dos textos a
compreensão do conceito do princípio da justiça e equidade. As diferentes
interpretações foram, de acordo com a percepção da pesquisadora, agrupadas em três
categorias analíticas, que são apresentadas no Quadro 09.
Os resumos dos objetivos dos artigos da busca sistemática sobre justiça e ética
em pesquisa também foram incorporados no Quadro 09. Os conteúdos dos artigos serão
utilizados na discussão dos resultados.

75
Quadro 09. Artigos da busca sistemática sobre justiça e ética em pesquisa, seus objetivos e as respectivas categorias analíticas relacionadas.
Categorias
Analíticas
Autores e datas de
publicação Objetivos dos artigos
Relevância social Kurihara C (2011)
Esclarecer as implicações éticas, legais e sociais de um estudo exploratório de mínimo risco com
humanos, com foco nos estudos de microdose.
Ballantyne A (2005)
Demonstrar que para redução dos níveis de exploração presentes em alguns ensaios clínicos
internacionais, os patrocinadores de tais pesquisas devem promover benefícios esperados e distribuir
eqüitativamente os benefícios pós estudos aos participantes da pesquisa.
Beran RG (2006) Discutir a conduta dos ensaios clínicos com medicamentos epilépticos em mulheres frente a gravidez
ou potencial de engravidar e os riscos teratogenicidade.
Clark PA (2002) Examinar o processo e as controvérsias relacionadas aos ensaios clínicos controlados por cirurgia-
placebo com terapia celular para doença de Parkinson, assim como os aspectos éticos envolvidos.
Haire BG (2011)
Analisar as relações entre a bioética e as pesquisas com tratamento padrão para prevenção do HIV
em países em desenvolvimento, com foco no encerramento das profilaxias pré-exposição no inicio
dos anos 2000.
Hawkins JS (2006) Discutir os problemas morais envolvidos nos estudos com placebo á luz do principio da justiça.
Mayss A (2000) Analisar os dilemas éticos e legais associados com os testes de transmissão vertical de HIV no
mundo em desenvolvimento.
Resnik DB (2002) Discutir o conceito e as implicações da exploração no contexto dos ensaios clínicos.
Distribuição equitativa de
encargos e beneficios referente
à participação na pesquisa.
Varmus H. Satcher D
(1997) Discutir as complexidades éticas na condução de pesquisas em países em desenvolvimento.

76
Kölch M. Ludolph AG.
Plener PL. Fangerau
H.Vitiello B. Fegert JM
(2010)
Revisar e discutir eticamente as principais alterações legislativas em pesquisa pediátrica,
especialmente em psicofarmacologia envolvendo menores com transtornos mentais (grupo
vulnerável), ao longo da última década nos EUA e Europa.
Oquendo MA. Stanley
B. Ellis SP. Mann JJ
(2004)
Discutir as abordagens das pesquisas clínicas intervencionistas com os pacientes de comportamento
suicida, em relação aos princípios de autonomia, beneficência e Justiça.
McMillan JR e Conlon
C (2004) Discutir os padrões éticos adotados para as pesquisas em países em desenvolvimento.
Bayer A. Fish M (2003) Discutir o acesso dos idosos aos ensaios clínicos, adequação do consentimento informado e as
responsabilidades médicas envolvidas.
Thomas J (1998) Explorar as questões éticas envolvidas na condução de ensaios clínicos em países em
desenvolvimento.
Smart A. Martin P.
Parker M (2004)
Discutir as relações entre os princípios da diferença e justiça no uso da farmacogenética nos ensaios
clínicos.
Van Delden J. Bolt I.
Kalis A. Derijk J e
Leufkens H (2004)
Discutir as implicações éticas da farmacogenômica na prática da medicina.
Booth KM (2010) Discutir sobre as pesquisas biomédicas com intervenção medicamentosa financiadas pelos EUA e
conduzidas em países sub-desenvolvidos ou em desenvolvimento, especialmente a África.
Equidade de acesso à saúde
Edwards SJ (2006) Demonstrar que certas restrições ao acesso público à possíveis novos tratamentos, via ensaios
clínicos, é moralmente problemática em algumas circunstâncias (ex. pacientes graves).
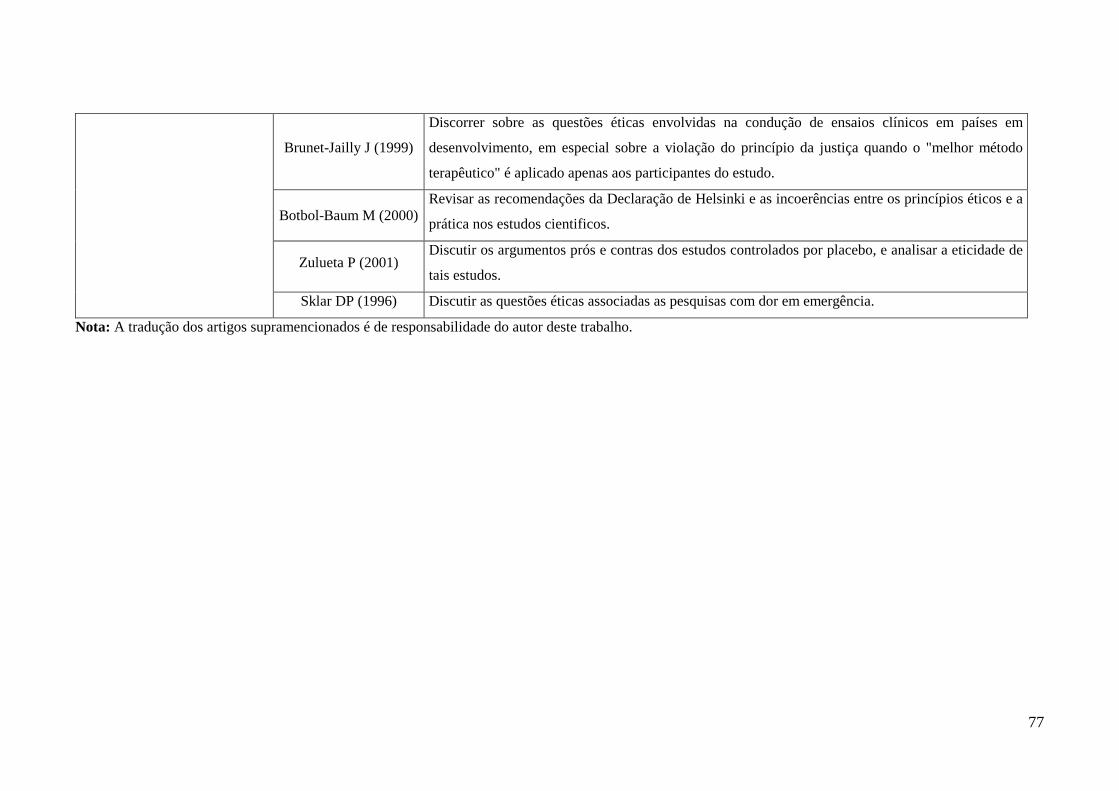
77
Brunet-Jailly J (1999)
Discorrer sobre as questões éticas envolvidas na condução de ensaios clínicos em países em
desenvolvimento, em especial sobre a violação do princípio da justiça quando o "melhor método
terapêutico" é aplicado apenas aos participantes do estudo.
Botbol-Baum M (2000) Revisar as recomendações da Declaração de Helsinki e as incoerências entre os princípios éticos e a
prática nos estudos cientificos.
Zulueta P (2001) Discutir os argumentos prós e contras dos estudos controlados por placebo, e analisar a eticidade de
tais estudos.
Sklar DP (1996) Discutir as questões éticas associadas as pesquisas com dor em emergência.
Nota: A tradução dos artigos supramencionados é de responsabilidade do autor deste trabalho.

78
2. TRAJETÓRIA DOS ENSAIOS CLÍNICOS AO REGISTRO DE
ANTICORPOS MONOCLONAIS E BIOMEDICAMENTO E AS
IMPLICAÇÕES ÉTICAS
2.1. Resultados da seleção de estudos
Após a aplicação dos critérios de seleção descritos no capítulo Método, a
amostra remanescente foi de 50 estudos; entretanto, apenas 08 (16%) estudos
apresentavam intervenção com anticorpos monoclonais e 01 (2%) estudo envolvia
intervenção com biomedicamento. Assim, 09 estudos foram incluídos para exame.
Destaca-se que 01 estudo não poderá ser plenamente avaliado por apresentar entre os
fármacos de intervenção, um codificado com letras e números, não passível de
identificação. As identificações de tais estudos e de seus medicamentos estão descritos
no Quadro 10. Os demais medicamentos estão descritos no Apêndice D.
Em relação aos medicamentos identificados nos 09 estudos, observamos a
participação de 05 anticorpos monoclonais (bevacizumabe, cetuximabe, figitumumabe,
ipilimumabe e rituximabe) e 01 biomedicamento (interferona-alfa 2a). As descrições
desses medicamentos (monografias resumidas) seguem no Quadro 10.

79
Quadro 10. Identificação dos estudos envolvendo anticorpos monoclonais e biomedicamento, segundo ClínicalTrials.gov
Código de
identificação do
estudo na base
Clínicaltrials.gov
Condição médica avaliada
Anticorpo
monoclonal e
biomedicamento
Código de
identificação
no ATC index
Código de
identificação
na DCB
Braço
experimental Braço controle
NCT00154102 Câncer Colorretal metastático
cetuximabe L01XC06 01975 cetuximabe e FOLFIRI FOLFIRI
NCT00439517 Câncer Colorretal mestástico
cetuximabe L01XC06 01975 UFOX e cetuximabe FOLFOX4 e
cetuximabe
NCT00678535 Câncer gástrico cetuximabe L01XC06 01975 cetuximabe,
capecitabina e cisplatina
capecitabina e
cisplatina
NCT00148798 Câncer de pulmão não pequenas células avançado
cetuximabe L01XC06 01975 cetuximabe, cisplatina e
vinorelbina
cisplatina e
vinorelbina
NCT00673049 Câncer de pulmão refratário figitumumabe Não há Não há figitumumabe e
erlotinibe erlotinibe
NCT00094653 Melanoma mestástico ou irressecável
ipilimumabe L01XC11 09642 ipilimumabe e MDX-
1379 (gp100)*
ipilimumabe e placebo
ou MDX-1379
(gp100) e placebo
NCT00312845 Linfoma folicular refratário rituximabe L01XC02 07758 bortezomibe e
rituximabe rituximabe
NCT00486759 Linfoma difuso de grandes células B
bevacizumabe e
rituximabe
L01XC07 e
L01XC02
01236 e
07758
bevacizumabe,
rituximabe e CHOP
Placebo, rituximabe e
CHOP
NCT00083889 Câncer de células renais interferona alfa-
2a** L03AB04 00515 sunitinibe interferona alfa-2a
*O estudo envolvia um dos fármacos de intervenção codificado com letras e números não passível de identificação. ** Este é o único biomedicamento. Nota: FOLFIRI (5-fluorouracil, leucovorin e irinotecan); CHOP (ciclofosfamida, doxorubicina, vincristine e prednisona); UFOX (oxaliplatina, UFT - tegafur com uracil - e ácido folínico); FOLFOX4 (oxaliplatin, 5-fluorouracil e ácido folínico).

80
Quadro 11. Monografias resumidas de anticorpos monoclonais e biomedicamento, segundo bases de consulta (FDA, EMA e Anvisa).
Anticorpo monoclonal (DCB): bevacizumabe
Nome de marca do medicamento Avastin Avastin Avastin
Produtor Genentech Roche Registration Ltd. Produtos Roche químicos e farmacêuticos S.A..
Descrição Anticorpo monoclonal anti-VEGF consistindo de anticorpo murino humanizado com ligação de antígeno.
Agência reguladora FDA EMA ANVISA *
Data de aprovação original 26 de fevereiro de 2004 12 de Janeiro de 2005 06 de maio de 2013
Data da última revisão 26 de março de 2013 08 de novembro de 2013 16 de setembro de 2013
Indicação de uso inicial 1 - Carcinoma metastático de colón ou reto Não disponível Não disponível
Indicação de uso atual 1 - Câncer colorretal metastático. 2 - Câncer de pulmão não-pequenas células não-escamosas, irressecável, localmente avançado, recorrente ou doença metastática. 3 - Glioblastoma em pacientes adultos com doença progressiva 4 - carcinoma de células renais metastático;
1 - Câncer colorretal metastático. 2 - Câncer da mama metastático. 3 - Câncer epitelial do ovário, da trompa de Falópio ou câncer peritoneal primário, avançados; 4 - carcinoma de células renais metastático;
Idem EMA
Anticorpo monoclonal (DCB): cetuximabe
Nome de marca do medicamento Erbitux Erbitux Erbitux
Produtor Imclone Merck KGaA Merck S/A
Descrição Um anticorpo que bloqueia os receptores de fator de crescimento epidérmico
Agência reguladora FDA EMA ANVISA
Data de aprovação original 12 de fevereiro de 2004 29 de junho de 2004 13 de fevereiro de 2012
Data da última revisão 04 de março de 2013 29 de abril de 2013 03 de abril de 2013
Indicação de uso inicial 1- Carcinoma colorretal metastático com expressão de EGFR
Não disponível Não disponível

81
Indicação de uso atual 1 - Carcinoma colorretal metastático com expressão de EGFR e K-Ras mutação negativa (tipo selvagem). 2 - Carcinoma de células escamosas de Câncer de cabeça e pescoço locorregional avançado, recorrente ou metastático.
Idem FDA Idem indicação atual do FDA e
EMA
Anticorpo monoclonal (DCB): ipilimumabe
Nome de marca do medicamento Yervoy Yervoy Yervoy
Produtor Bristol Myers Squibb Bristol Myers Squibb Bristol Myers Squibb
Descrição Um anticorpo anti-linfócito T citotóxico 4-antigénio associado
Agência reguladora FDA EMA ANVISA
Data de aprovação original 25 de março de 2011 13 de Julho de 2011 11 de dezembro de 2012
Data da última revisão 22 de maio de 2013 11 de Julho de 2013 17 de maio de 2013
Indicação de uso inicial 1- Melanoma metastático ou irressecável Não disponível Não disponível
Indicação de uso atual 1- Melanoma metastático ou irressecável Idem FDA Idem FDA
Anticorpo monoclonal (DCB): rituximabe
Nome de marca do medicamento Rituxan MabThera MabThera
Produtor Genentech Roche Registration Ltd Roche Registration Ltd
Descrição Um anticorpo anti-CD20
Agência reguladora FDA EMA ANVISA
Data de aprovação original 26 de novembro de 1997 02 de junho de 1998 07 de fevereiro de 2001
Data da última revisão 24 de setembro de 2013 24 de setembro de 2013 21 de junho de 2013
Indicação de uso inicial 1 – Linfoma de baixo grau ou folicular recidivado ou refratário, CD20 positivo, de Linfoma não-Hodgkin de células B.
Não disponível 1 - Linfoma não-Hodgkin
2- leucemia linfocítica crônica
3 - Artrite Reumatóide
Indicação de uso atual 1 - Linfoma não-Hodgkin 2- leucemia linfocítica crônica 3 - Artrite Reumatóide 4 - Granulomatose com Poliangeíte e Poliangeíte microscópica
Idem FDA 1 - Linfoma não-Hodgkin
2- leucemia linfocítica crônica
3 - Artrite Reumatóide

82
Biomedicamento (DCB): interferona-alfa 2a
Nome de marca do medicamento Roferon A Alpheon Interferona Alfa 2A
Produtor Hoffman-LA Roche BioPartners GmbH Merck S/A
Descrição Um recombinante não glicosilado de interferon alfa humano, subtipo 2a, produzido na bactéria E. coli.
Agência reguladora FDA EMA ANVISA
Data de aprovação original 04 de junho de 1986 05 de setembro de 2006** 30 de junho de 2000
Data da última revisão 16 de junho de 2011 Não aplicável Não disponível***
Indicação de uso inicial Não disponível Hepatite C crônica Não disponível
Indicação de uso atual Não disponível Não aplicável Não disponível
* A data de aprovação original não é válida para os dados proveninentes do sítio de internet da Anvisa. A data disponibilizada pela agência é a data da última aprovação do medicamento. **Data que o medicamento foi recusado pelo EMA. ***Apesar do medicamento estar listada na base de dados da Anvisa na Consulta aos produtos, este não está registrado no Bulário eletrônico da Agência. Fonte: Os dados foram verificados nos sites do FDA, EMA e Anvisa em 16/11/2013. Nota: O medicamento Figitumumabe não está registrado em nenhuma das três agências reguladoras estudadas.

83
2.2. Características gerais do conjunto de estudos
2.2.1. Ano de aprovação dos medicamentos e duração dos estudos
Em relação à linha do tempo para a aprovação de registro dos anticorpos
monoclonais e biomedicamentos nas diferentes agências regulatórias (ver Quadro 11)
observa-se uma linha com tendência levemente crescente. O FDA aparece como
primeira agência a realizar o registro dos medicamentos estudados, retratando o seu
pioneirismo na condução de estudos clínicos pelo mundo, seguido pelo registro no
EMA (órgão europeu), conforme Gráfico 01. À exceção do interferon-alfa 2a que foi
recusado pelo EMA16. A diferença de tempo entre as aprovações FDA e da EMA pouco
variaram, sendo a variação máxima de 01 ano e a média de 0,5 anos, demonstrando uma
sincronia entre as agências.
Gráfico 01. Gráfico de linhas exibindo a tendência em relação ao ano de aprovação dos anticorpos monoclonais/biomedicamentos e as agências reguladoras FDA, EMA e ANVISA.
Os dados da Anvisa não foram avaliados para a variável Ano de aprovação dos
medicamentos, pois a data disponibilizada pela Agência em seu site corresponde à data
da última aprovação/renovação do medicamento. Adicionalmente, não há
disponibilização de um histórico de registro.
Em relação ao tempo para condução dos estudos, a realização destes durou em
16 Para maiores informações sobre a recusa do interferon alfa 2a pelo EMA, consultar o relatório “Refusal Assessment Report for Alpheon”. Disponível em: http://www.ema.europa.eu

84
média 5,48 anos (desvio padrão: 0,9441), variando entre 4,08 a 6,83 anos entre o início
do recrutamento de sujeitos de pesquisa e a divulgação dos primeiros resultados de tais
estudos. Ver Gráfico 02.
Gráfico 02. Linha do tempo da trajetória do estudo do recrutamento aos resultados.
2.2.2. Desenho do estudo
Com relação ao desenho de estudo, dos 09 estudos avaliados todos eram
randomizados controlados com recrutamento para ambos os sexos, com idade mínima de
18 anos e apenas 01 estudo apresentava idade máxima (79 anos). A média de
recrutamento foi de 985,22 pacientes, variando de 302 a 1861. Dois estudos eram
duplos-cego e os demais abertos. Apenas 01 estudo era de fase II, os demais de fase III.
Apenas um estudo envolvia três braços de tratamento; os demais, dois braços. Todos
eram patrocinados por indústrias farmacêuticas. As variáveis estão descritas na Tabela
03.

85
Tabela 03. Distribuição da frequência das características gerais dos estudos.
Variável Frequência absoluta Percentual
Fase do estudo
Fase III 8 88.8 Fase II 1 11.1
Total 9 100.0
Desenho do estudo Randomizado controlado, aberto 7 77.7 Randomizado controlado, duplo-cego
2 22.2
Total 9 100.0 Uso de Placebo 2 22.2 Não uso de placebo 7 77.7
Total 9 100.0 Número de braços de tratamento
Dois 8 88.8 Três 1 11.1
Total 9 100.0 Estágio do estudo
Finalizado 7 77.7 Encerrado 2 22.2 Razões: Relação desfavorável entre benefício/risco Probabilidade alta de não cumprimento do objetivo primário17
Total 9 100.0
Desfecho clínico primário
Sobrevida livre de progressão 5 55.5 Sobrevida global 3 33.3 Taxa de melhor resposta 1 11.1
Total 9 100.0 Patrocinador
Merck KGaA 4 44.4 Pfizer 2 22.2 Bristol-Myers Squibb 1 11.1 Hoffmann-La Roche 1 11.1 Millennium Pharmaceuticals, Inc. 1 11.1
Total 9 100.0
2.2.3. Distribuição geográfica
Os 09 estudos analisados contaram com a participação de 58 diferentes países. O
17 Este estudo foi encerrado em 08 de marco de 2010. Por motivos éticos alguns pacientes, nos
quais observou-se benefício, continuaram a receber o tratamento proposto.

86
Brasil, a França e a Inglaterra tiveram uma participação em 100% dos estudos (n=09),
Alemanha, Argentina, Austrália, Itália e Polônia tiveram uma participação de 88,9%
(n=08). Os Estados Unidos apresentou uma participação inferior, representados por
55,6% (n=05). A descrição dos países encontra-se no Gráfico 03.
Gráfico 03. Distribuição da frequência absoluta e percentual dos países envolvidos na condução dos estudos.
Focando a avaliação para o Brasil, buscamos a identificação dos centros de
pesquisas no país com participação nos estudos. Dos nove estudos, cinco apresentavam
variáveis que permitem a identificação dos centros de pesquisa: dois estudos
(NCT00312845 e NCT00094653) reportaram o nome do centro e Código de

87
Endereçamento Postal (CEP) e três estudos (NCT00673049, NCT00486759 e
NCT00083889) reportaram a cidade sede e o CEP. Entretanto, quatro estudos
(NCT00154102, NCT00678535, NCT00148798 e NCT00439517) disponibilizavam
apenas as cidades sedes de tais Centros, o que impossibilita a identidicação dos centros
de pesquisas envolvidos em tais estudos.
Considerando o número do CEP registrado para os centros envolvidos nos cinco
estudos supramencionados foram realizadas buscas via internet para verificação das
respectivas localizações e identificações dos mesmos, contudo, não foi possível
identificar alguns centros de pesquisas. Dos 40 centros de pesquisas18 que participaram
da condução dos estudos, foi possível a identificação de 22 centros, conforme disposto
no Quadro 12.
Quadro 12. Identificação e localização de alguns centros de pesquisas envolvidos na condução dos nove estudos clínicos da amostra.
Localização Nome do centro de pesquisas
Estado de São Paulo
Centro Pesquisa Clínica Medicina Avançada São Paulo – SP. CEP 01224-010
Faculdade de Medicina do ABC Santo André – SP. CEP 09060-870
Faculdade de Medicina do ABC Santo André - SP. CEP 09060-650
Fundação Hospital Amaral Carvalho Jaú – SP.
CEP 17210-080
Fundação Pio XII - Hospital de Câncer de Barretos Barretos – SP.
CEP 14784-400
Hospital Brigadeiro São Paulo – SP. CEP 01404-901
Hospital das Clínicas da Faculdade de Medicina da USP São Paulo – SP. CEP 05403-000
Hospital Sírio Libanês São Paulo – SP. CEP 01308-050
Instituto do Câncer Dr. Arnaldo São Paulo – SP. CEP 01221-020
Santa Casa de São Paulo São Paulo – SP. CEP 01221-020
Santo Andre Diagnósticos a Tratamentos Santo André – SP. CEP 09090-780
Sociedade Beneficente de Senhoras do Hospital Sírio Libanês São Paulo – SP. CEP 01308-000
Universidade Estadual de Campinas Campinas – SP. CEP 13083-970
18 Houve a citação de 40 centros de pesquisas para a condução dos 09 estudos, porém, vale resaltar que alguns centros participaram de mais de um estudo, isto é, um único centro pode ser contado mais de uma vez.

88
Estado do Rio Grande do Sul
Fundação Central Sul-Americana para o Desenvolvimento de Drogas Anticancer (SOAD)
Porto Alegre – RS. CEP 90035-003
Hospital da Criança Nossa Senhora da Conceição Porto Alegre – RS.
CEP 91350-200
Hospital de Caridade de Ijuí Ijuí - RS.
CEP 98700-000
Hospital de Clínicas Porto Alegre – RS.
CEP 90035-903
Hospital São Lucas – PUC do Rio Grande do Sul Porto Alegre – RS.
CEP 90610-000
Estado de Goiás
Hospital Araujo Jorge Goiânia – GO.
CEP 74605-070
Estado de Minas Gerais
Biocor - Hospital de Doenças Cardiovasculares Belo Horizonte – MG.
CEP 34000-000
Estado do Paraná
Pró Onco Centro Tratamento Oncológico Londrina – PR. CEP 86050-190
Estado do Rio de Janeiro
Instituto Nacional de Câncer Rio de Janeiro – RJ.
CEP 20231-050
Quando estratificado os centros de pesquisas por cidades, verificamos a
participação de 13 cidades diferentes na condução dos 09 estudos no Brasil. A cidade de
São Paulo foi identificada com participação em 100% (n=09) dos estudos, ou seja, pelo
menos um centro de pesquisas sediado na cidade de São Paulo participou em cada
estudo. Seguida por Porto Alegre (88,9%, n=8) e Santo André (55,6%, n=5). O Rio de
Janeiro aparece com 33,3% (n=3).
Quando agrupados as cidades por estados, verificou-se que 47,5% (n=19) dos
centros envolvidos na condução dos estudos pertenciam ao Estado de São Paulo e 25%
(n=10) pertenciam ao Estado do Rio Grande do Sul. A descrição detalhada das Cidades
e Estados encontra-se no Gráfico 04.

89
Gráfico 04. Distribuição da frequência absoluta e percentual das Cidades e Estados
envolvidos na condução dos estudos.
2.2.4. Indicação Clínica
Com relação à indicação de uso dos medicamentos avaliados, o cetuximabe,
conforme descrição do Quadro 13 está sendo testado para duas novas indicações: câncer
gástrico e pulmão não-pequenas células. Os outros dois estudos com cetuximabe se
mantêm na indicação inicial (Câncer colorretal), porém, com novas combinações de
tratamentos, adicionando o cetuximabe ao esquema considerado padrão (FOLFORI ou
FOLFOX).
Os medicamentos ipilimumabe e rituximabe foram estudados para as mesmas
condições clínicas nas quais foram aprovados, porém, foram adicionados novos
medicamentos ao braço experimental versus o tratamento padrão. O bevacizumabe foi

90
estudado para uma nova condição clínica, o Linfoma difuso de grandes células B. Tal
medicamento foi adicionado no braço experimental juntamente com rituximabe e
CHOP, considerados como tratamento padrão, versus o tratamento padrão isolado.
Não é possível estudar a indicação clínica do medicamento figitumumabe, pois,
este não está registrado em nenhuma das três agências reguladoras estudadas e da
interferona-alfa 2a, por não apresentar sua indicação inicial disponível no FDA e
Anvisa.
Os resultados clínicos dos estudos com indicação clínica nova estão disponíveis
no Quadro 13.

91
Quadro 13. Resultados clínicos dos estudos com indicação clínica nova.
Código de identificação
do estudo na base
Clínicaltrials.gov e
condição médica avaliada
Anticorpo monoclonal e
biomedicamento Resultados dos estudos
Braço experimental:
Cetuximabe, Capecitabina e Cisplatina
Taxas de sobrevida livre de progressão (4.4 meses) e a sobrevida global (9.4
meses);
Taxa de melhor resposta (29.9%). NCT00678535:
Câncer gástrico Braço controle:
Capecitabina e Cisplatina
Taxas de sobrevida livre de progressão (5.6 meses) e a sobrevida global (10.7
meses);
Taxa de melhor resposta (29.2%).
Braço experimental:
Cetuximabe, Cisplatina e Vinorelbine
Taxas de sobrevida global (11.3 meses) e taxa de melhor resposta (36.4%);
Taxa livre de progressão (4.8 meses). NCT00148798:
Câncer pulmão não-
pequenas células Braço controle:
Cisplatina e Vinorelbine
Taxas de sobrevida global (10.1 meses) e taxa de melhor resposta (29.2%);
Taxa livre de progressão (4.8 meses).
Braço experimental:
Bevacizumabe, Rituximabe e CHOP
Taxa de sobrevida livre de progressão (40.2 meses);
Taxa de resposta Global (63.1 %) NCT00486759:
Linfoma difuso de grandes
células B Braço controle:
Rituximabe e CHOP
Taxa de sobrevida livre de progressão (42.9 meses);
Taxa de resposta Global (70.5%)

92
Ao avaliarmos os tipos de câncer envolvidos nos 09 estudos de nossa amostra,
identificamos: Colorretal (N=02), Gástrico19(N=01), Linfoma não-Hodking20 (N=02),
Melanoma (N=01), Pulmão não-pequenas células (N=02) e Renal (N=01).
Gráfico 05. Distribuição da frequência dos tipos de câncer avaliados nos 09 estudos.
Conforme Figuras 05 e 06, os cânceres de pulmão, gástrico e colorretal estão
entre os 05 tipos de câncer com maior mortalidade entre os homens e mulheres nos
últimos anos no Brasil. Quando verificamos a incidência, os tipos de câncer mais
incidentes estimados para 2012 no Brasil para o sexo masculino (ver Figura 03) foram:
pulmão em segundo lugar, cólon e reto em terceiro, estômago em quarto, esôfago em
sexto e linfoma não-Hodgkin em nono lugar. Para as mulheres, cólon e reto em terceiro
lugar, pulmão em quinto, estômago em sexto e linfoma não-Hodgkin em nono lugar. O
câncer renal não aparece entre os dez mais incidentes no Brasil. O melanoma representa
apenas 4% das neoplasias malignas do órgão, embora o câncer de pele seja o mais
frequente no Brasil, os estudos com melanoma se justificam por representar o tipo mais
grave entre neoplasias de pele em função da sua alta possibilidade de metástase.
19 Adenocarcinoma gástrico, incluindo adenocarcinoma da junção gastro-esofágica. 20 Linfoma difuso de grandes células B e linfoma folicular.

93
Figura 05. Taxas de mortalidade das 5 localizações primárias mais frequentes em 2011, ajustadas por idade, população mundial, por 100.000 homens, Brasil, entre 2003 e 2011. Fonte: Atlas de mortalidade por Câncer do INCA. Disponível em: http://mortalidade.inca.gov.br/Mortalidade/
Figura 06. Taxas de mortalidade das 5 localizações primárias mais frequentes em 2011, ajustadas por idade, população mundial, por 100.000 mulheres, Brasil, entre 2003 e 2011 Fonte: Atlas de mortalidade por Câncer do INCA. Disponível em: http://mortalidade.inca.gov.br/Mortalidade/
Ao analisarmos os critérios de seleção para participação nos estudos, percebe-se
que dos 09 estudos da amostra final, 05 (55,5%) determinavam como um dos critérios
de inclusão alguma característica genética do participante de pesquisa a ser recrutado.
Nos demais estudos, 02 (22,2%) apresentavam restrições em seus critérios de exclusão
quanto ao uso prévio de terapias envolvendo a característica genética a ser estudada, 01
(11,1%) apresentava em seu critério de inclusão a assinatura de TCLE para a realização

94
dos testes de farmacogenômica e apenas 01 (11,1%) não apresentava nenhuma restrição
genética em seus critérios, conforme tabela abaixo. Assim, 77,7% dos estudos
apresentavam algum tipo de restrição genética em seus critérios de seleção.
Tabela 04. Identificação de alguns critérios de seleção dos estudos clínicos.
Código de identificação do
estudo na base
Clínicaltrials.gov
Critério de inclusão
NCT00154102
Imunohistoquímica evidenciando expressão tumoral do receptor
de fator de crescimento epidermal (EGFR- Epidermal Growth
Factor Receptor).
NCT00678535 Amostra tumoral com pelo menos uma expressão de EGFR
avaliado.
NCT00148798 Imunohistoquímica evidenciando expressão tumoral do EGFR.
NCT00094653 HLA-A*0201 positivo
NCT00486759 CD20-positivo.
NCT00312845
Nos países em que as autoridades de saúde tiverem aprovado os
testes de farmacogênomica, os sujeitos de pesquisa ou seus
representantes legais devem assinar separadamente o TCLE para
a participação da etapa do estudo referente aos testes de
proteínas e genético. A participação nos testes de proteína e
genético é mandatória para os testes de farnacogênomica, mas
são opcionais para os testes de soro de proteína e futuros testes.
Código de identificação do
estudo na base
Clínicaltrials.gov
Critério de exclusão
NCT00673049 Terapia investigacional prévia anti IGF IR (Inhibition of insulin-
like growth factor-I receptor).
NCT00439517 Terapia sistêmica imunológica crônica, terapia-alvo, terapia
anti-EGFR ou com alvo em EFGR.
NCT00083889 Não há restrições genéticas nos critérios de inclusão ou exclusão

95
3. ANÁLISES DAS COMPRAS FEDERAIS DOS ANTICORPOS
MONOCLONAIS E BIOMEDICAMENTOS ONCOLÓGICOS
A partir das informações acerca dos registros dos biomedicamentos e anticorpos
monoclonais identificados na amostra, foi realizada uma investigação sobre o processo
de aquisições e gastos do Governo Federal brasileiro com estes medicamentos, com
base nos dados do Sistema Integrado de Administração de Serviços Gerais (SIASG). A
descrição dos achados encontra-se no Quadro 14.
O acesso aos dados do ano de 2003 estava indisponível. Nos anos de 2004 e
2005, o Governo Federal através de diferentes órgãos, como Universidades e
Ministérios, adquiriu os medicamentos inferona alfa 2a e rituximabe. Em 2006,
adicionou a esta lista de compras o bevacizumabe e em 2007, o cetuximabe. As
informações sobre a indicação de uso não estão disponíveis neste banco de dados,
exceto em algumas justificativas de compra onde a patologia em questão foi inserida;
porém, a maioria das justificativas são vagas/não informativas como, “Para atender o
ensino de graduação” e “atender paciente internado em caráter de extrema necessidade”
ou não informada. Das 1.013 justificativas registradas, 385 (38%) eram por demanda
judicial e 191 (18,5%) foram registradas como “Não informado”.
A Anvisa não disponibiliza o histórico de registro dos medicamentos, o que está
disponível é a data de publicação da última atualização de registro daquele produto. Tal
limitação impede a análise de concomitância entre os momentos das compras dos
medicamentos em questão e o período de registro dos mesmos.
Em relação às regiões responsáveis pelas compras, conforme Gráfico 06, a
Região Nordeste foi a maior aquisitora dos medicamentos bevacizumabe, cetuximabe e
rituximabe no período estudado. As maiores compras de interferona foram realizadas
pela Região Centro-Oeste. A Região Norte, caracterizada como uma região pouco
assistida pelas pesquisas clínicas, se destacou pelo baixo consumo destes medicamentos.
Entretanto afirmativas e discussões a cerca de tais achados não são consistentes, uma
vez que, a adesão ao sistema não é abrangente e homogênea.
Os usuários do SIASG são compulsoriamente os usuários do Sistema de
Serviços Gerais (SISG), criados ambos em 1994 pelo Governo Federal. O Decreto nº
1.094/94 dispõe que o SISG é integrado pelos órgãos e unidades da Administração
Federal direta, autárquica e fundacional, os quais possuem a responsabilidade de utilizar

96
o sistema SIASG em seus procedimentos de compras e contratações. Porém, a utilização
do sistema é facultativa para os comandos militares, e livres para os demais órgãos.
Fontes dos MS (Secreataria Executiva, Departamento de Economia da Saúde),
informam que, a partir de 2012 a utilização do SIASG seria obrigatória não apenas por
órgãos federias, mas de qualquer órgão público vinculado á administração direta. No
entanto, o sistema ainda estaria em construção e, portanto, dados específicos de
governos estaduais e municipais não seriam passíveis de consolidação no momento da
análise.
Gráfico 06. Distribuição de itens comprados pelo Governo Federal, por região do país, entre 2004 a 2012, de acordo com o SIASG.
Um dado que nos chama a atenção no Quadro 14 são as colossais variações de
preços dos produtos adquiridos. Os gastos totais por ano estão dispostos no Gráfico 07.
Gráfico 07. Distribuição dos gastos federais, nos anos 2004 a 2012, com os biomedicamentos e anticorpos monoclonais em oncologia identificados em nossa amostra, de acordo com o SIASG.

97
Quadro 14. Distribuição dos anticorpos monoclonais e biomedicamentos da amostra comprados pelo Governo Federal entre 2004 a 2012.
rituximabe
Variáveis/Medicamentos 2004 2005 2006 2007 2008 2009 2010 2011 2012
Média de valores por item (minimo-máximo)
R$ 3.669,54 (926,15-7.729,00)
R$ 3.129,95 (983,48-6.461,51)
R$ 3.054, 28 (876,51 - 6.313,15)
R$ 2.889,05 (801,26-5.311,67)
R$ 2.820,31 (798,81-5.800,00)
R$ 3.141,92 (818,93 - 5.825,00)
R$ 3.303,41 (772,54 – 6.023,43)
R$ 2.943,04 (610,82 - 5.176,17)
R$ 2.540,92 (546,06 - 5.176,18)
Total de itens
comprados
Quantidade da compra (item)
515 839 6.333 3.099 7.150 6.346 14.432 33.140 31.860 103.714
interferona-alfa 2a
Variáveis/Medicamentos 2004 2005 2006 2007 2008 2009 2010 2011 2012
Média de valores por item (minimo-máximo)
R$ 81,28 (6,02 -1.229,00)
R$ 362,19 (6,50-3.700)
R$ 519,02 (4,98 -
1.313,64)
R$ 201,65 (4,47 -
1.096,52)
R$243,46 (5,44-
1.135,40) R$ 402,73
(6,50 - 2.535)
R$ 139,35 (6,30 - 968,91)
R$ 255,89 (10,00 - 983,32)
R$ 354,00 (22,00 -
1.013,42)
Total de itens
comprados
Quantidade da compra (item)
66.279 18.002 82.585 212.528 344.447 249.208 565.579 646.247 23.995 2.208.870
bevacizumabe Variáveis/ Medicamentos 2004 2005 2006 2007 2008 2009 2010 2011 2012
Média de valores por item (minimo-máximo)
0 0 R$ 2.673,18 (1.312,23 -
5.800)
R$ 1.499,18 (317,33-4.208,11)
R$ 2.287,72 (815,99-4.314,15)
R$2379,76 (836,55– 4.568,69)
R$ 2.285,28 (432,21 –
4.772)
R$ 2.838,53 (964,98 - 4.940,93)
R$ 2.653,39 (830,12 - 4.940,93)
Total de itens
comprados Quantidade da compra
(item) 0 0 165 232 557 3.334 2.675 13.199 9.118 29.280
cetuximabe Variáveis/ Medicamentos 2004 2005 2006 2007 2008 2009 2010 2011 2012
Média de valores por item (minimo-máximo) 0 0 0
R$ 576,44 (477,21 - 633,66)
R$ 610,95 (475,75 - 649,62)
R$666,04 (649,62 - 687,94)
R$ 674,59 (562,12 - 701,30)
R$ 1.031,69 (562,11 - 3.500)
R$ 1.408,91 (453,59 - 3.592,65)
Total de
itens
comprados
Quantidade da compra (item)
0 0 0 251 935 2.623 3.102 6.217 11.880 25.008

98
DISCUSSÃO
Após a apresentação dos resultados da busca sistemática sobre Justiça e
Pesquisa clínica e dos resultados empíricos, os discutiremos sob a luz da Justiça
na perspectiva das três categorias analíticas elencadas anteriormente. Os
conteúdos dos artigos da busca sistemática complementaram a discussão.
1. RELEVÂNCIA SOCIAL
A categoria relevância social é uma categoria percebida através da leitua
do texto de Kurihara (2011), e envolve acesso aos resultados da pesquisa, acesso
às informações sobre o medicamento, a participação brasileira nos ensaios
clínicos e a cientificidade de tais ensaios.
Um dos critérios de exclusão para seleção dos estudos, descrito na
metodologia, foi a ausência de publicação dos resultados na base de dados
pública, pois o uso da publicação destes ao término dos estudos é importante no
escopo desta pesquisa, por se traduzir em relevância social, uma das implicações
da Justiça. A publicidade dos resultados pode contribuir para a melhoria das
condições de vida da população e servir como alerta sobre as melhores condições
de tratamento e/ou profilaxia. A não publicação dos resultados implica em
violação da Resolução CNS n°466/12 sobre pesquisas envolvendo seres humanos.
Caso isto ocorra, uma justificativa deve ser submetida à apreciação do CEP ou
CONEP.
O movimento mundial em prol da publicidade dos resultados dos estudos é
relativamente recente. O ClínicalTrials.gov, como já citado, é uma das principais
plataformas de registro de ensaios clínicos no mundo, e esta lançou sua base de
dados de resultados apenas em setembro de 2008, possibilitando a partir de então,
que os investigadores e/ou patrocinadores enviassem seus resultados. O banco de
dados de resultados foi desenvolvido para atender as requisições do Food and
Drug Administration Amendments Act de 2007- FDAAA21 (Clínical Trials, 2014).
21 Em 27/09/2007 foi assinada a Lei pública nº110-85 nos Estados Unidos da América que inclue uma seção sobre as bases de dados de ensaios clínicos, ampliando os tipos de ensaios clínicos que

99
A partir do lançamento da base de dados de resultados, o crescimento da
publicidade dos resultados nesta plataforma cresceu gradativamente ao longo dos
anos, conforme Figura 07. Apesar da figura abaixo não contemplar o período
completo de nosso recorte temporal, 2003 a 2012, é possível inferir que houve um
ganho importante para sociedade em termos de publicidade e acesso de
informações em relação aos estudos e seus resultados ao longo dos últimos anos.
Figura 07. Número de estudos registrados no ClínicalTrials que possuem resultados disponíveis de 2009 a 2014.
Um outro aspecto importante sobre o acesso às informações foi percebido
durante a coleta de dados sobre os registros dos medicamentos nas respectivas
agências reguladoras estudadas. Observou-se dificuldades nesta etapa com relação
à Anvisa: limitação de informações, clareza e as vezes, ausência de informações
sobre os medicamentos em nosso país, o que deve ser encarado negativamente do
ponto de vista macro, ou seja, da sociedade. A transparência nas informações
sobre os estudos, seus resultados e medicamentos visa homogenizar o
conhecimento dos indivíduos, sejam eles usuários e/ou pesquisadores.
Um dado crítico foi em relação à tentativa de identificação do
medicamento figitumabe, envolvido em um dos estudos da amostra. Não foi
possível verificar nenhuma informação sobre a indicação clínica e/ou dados de
registro de tal medicamento em nenhuma das três agências regulatórias
pesquisadas. O estudo era de fase III, o que pressupõe a existência de dados das
fases anteriores de estudo. Foi encerrado prematuramente, pela alta probabilidade
de não cumprimento do objetivo primário. Talvez por esta razão o medicamento
devem ser registrados no ClínicalTrials.gov, aumentando o número de informações requeridas e exigindo a apresentação de certos dados referente aos resultados dos estudos (NIH, [s.d]).

100
não tenha evoluído para a fase de registro em alguma agência reguladora.
Entretanto, essa insuficiência de informações sobre o medicamento é nociva à
sociedade científica e civil.
No Quadro 11 (página 82) é possível observar as datas de aprovações dos
medicamentos pelas Agências reguladoras FDA e EMA. A busca destas
informações e da indicação clínica dos medicamentos nestas agências, ocorreu
diferentemente da busca na Anvisa. As duas primeiras, centralizam as
informações sobre o registro e indicações em um único ambiente virtual e traz um
histórico dos registros e suas respectivas bulas. Para a busca na Anvisa foi
necessário acessar dois ambientes virtuais: a data de aprovação do medicamento
foi baseada na “Consulta ao produto” e data da última revisão foi baseada no
“Bulário Eletrônico”, ambas disponíveis no website da própria Agência.
Adicionalmente, a data oficial de registro do medicamento não é clara. O que é
disponibilizado é a data da publicação da rotulagem do medicamento. No Bulário
Eletrônico não são disponibilizadas as versões anteriores da bula do medicamento,
o que nos impossibilita avaliar a indicação inicial do medicamento.
O momento do registro sanitário do medicamento junto as agências
reguladoras representa uma etapa fundamental na avaliação do medicamento e
sobretudo, momento de atuação de tais autoridades “...como mediadoras entre os
interesses dos fabricantes de medicamentos e as necessidades da saúde pública,
visando, sobretudo, ao dever de proteção da saúde” (Gava e colaboradores, 2010,
p.3409). Portanto, a falta de acesso às informações de registros dos medicamentos
é uma questão crítica, a qual deve ser discutida junto às autoridades regulatórias.
A transparência pública das informações sobre o registro dos medicamentos é
fundamental para o fortalecimento da legitimidade do processo de registro no
país.
A participação brasileira nos ensaios clínicos mundiais traz vários
benefícios ao país, como o desenvolvimento econômico, possibilidade de
transferência de tecnologias, agregar conhecimento aos pesquisadores, e
sobretudo, disponibilizar tratamentos e tecnologias novas à população.
Sabidamente a descoberta e o desenvolvimento de novos medicamentos é
um processo longo e complexo. O processo de ensaios clínicos, incluindo as fases
I, II e III, leva em média, de seis a sete anos (Pharma, s.d), porém, a média de
tempo de execução dos estudos de nossa amostra mostrou-se superior (5,48 anos)

101
ao da literatura, considerando que estes contemplavam apenas uma das fases do
processo.
A demora no processo regulatório do nosso país é uma das hipóteses para
tal resultado. As implicações deste cenário são negativas, pois esta demora
representa tardança no acesso dos participantes de pesquisa aos novos tratamentos
ofertados pelas pesquisas e em casos de resultados positivos do estudo, demora no
processo de registro e acessibilidade da população ao medicamento.
Por vezes, os períodos de aprovação dos processos regulatórios na
América Latina são vistos como um problema, um ponto negativo para realização
de estudos clínicos nesta região, pois há extensos prazos e às vezes, o desrespeito
aos prazos pré-estabelecidos neste processo, o que afeta diretamente a fase de
recrutamento de sujeitos de pesquisa (Rodrigues, 2007). Para Sharif (1998) um
dos maiores problemas da execução de ensaios clínicos nos países em
desenvolvimento é a fragilidade dos Comitês científicos e de Ética que revisam os
estudos científicos.
Acompanhando a tendência de crescimento da pesquisa clínica no país, a
Anvisa e a CONEP vêm trabalhando no sentido de ampliar o debate ético da
investigação científica no campo da saúde com o objetivo de aprimorar e agilizar
o processo de aprovação de estudos clínicos no país, porém, mantendo os rigores
ético e processual necessários (Dainesi & Goldbaum, 2012).
Ainda em relação à participação brasileira nos ensaios clínicos mundiais, o
que se observou foi que a maioria dos estudos era de fase III (88,8%), o que é
visto por muitos autores sob um ponto de vista negativo, pois ratifica o fato dos
testes realizados no Brasil concentrarem-se em fases de menor complexidade
tecnológica (Gomes, Pimentel, Landim e Pieroni, s.d). Dados da Anvisa
corroboram com esta visão: no ano de 2011 foram divulgados pela Agência que
apenas 4 % de estudos no Brasil eram de fase I, que são estudos de alta
complexidade, enquanto a maior parte das aprovações eram estudos de fase III (63%),
seguidos por 22 % em fase II e 11 % de fase IV (ANVISA, 2011).
A variável “Patrocinador” do estudo deve ser avaliada conjuntamente com
as fases do estudo, pois conjeturam o mesmo cenário. Verificamos que 100% dos
estudos eram patrocinados por indústrias farmacêuticas estrangeiras, o que por
vezes, compromete a credibilidade da pesquisa em função dos conflitos de
interesse. Tal dado reflete a fragilidade da participação brasileira, sobretudo

102
acadêmica, na construção e realização de ensaios clínicos em oncologia. A Anvisa
divulgou em 2011 que 80% dos estudos de pesquisa clínica para desenvolvimento
de novos medicamentos desenvolvidos no Brasil eram conduzidos por empresas
multinacionais (ANVISA, 2011).
Diante deste repetido cenário de monopólio dos laboratórios farmacêuticos
atuando como principal financiador de pesquisas clínicas no país, o Decit/MS e
Finep/MCT implementaram ações de consolidação da rede de laboratórios de
pesquisa clínica direcionados as pesquisas de interesse do SUS, podendo atuar na
redução de tal monopólio sobre os cientistas na área da saúde (Brasil, 2009).
Entretanto, poucas mudanças são observadas neste contexto.
Adicionalmente, o Brasil adotou como exigência em sua Resolução CNS
n°466/12 que os estudos envolvendo seres humanos e “...patrocinados no exterior
também deverão responder às necessidades de transferência de conhecimento e
tecnologia para a equipe brasileira...” (Brasil, 2012, III.2.p).
Por outro lado, é inegável que o progresso na luta contra o câncer não seria
possível sem o substancial investimento em P&D por parte das indústrias
farmacêuticas. Tal setor é o principal financiador das pesquisas, e não o governo
ou academia, como se almejaria. Em termos de gastos, dados recentes da IMS
Health22 demostram que os medicamentos contra o câncer representam menos de
um por cento do total das despesas nacionais (governamentais) da saúde em 2012
(Phrma, 2013b).
Uma variável importante na avaliação da Relevância Social do estudo é a
qualidade de seu desenho, da sua metodologia. Estudos científicos bem propostos
possuem probabilidade aumentada de responder às questões/hipóteses levantadas
previamente sem exposição aumentada dos sujeitos de pesquisa e com planos de
mitigação traçados. Adicionalmente, traduzir-se-á em benefícios diretos aos
sujeitos de pesquisa e à comunidade científica. O desenho dos ensaios clínicos,
sejam eles, diagnósticos, terapêuticos ou preventivos suscitam questões científicas
e éticas inter-relacionados aos patrocinadores, investigadores e comitês de ética
(CIOMS, 2002).
Para Kurihara (2011), algumas novas estratégias para o desenvolvimento
de novos fármacos, como os desenhos de estudos com microdoses (microdose
22 IMS Health é a principal empresa de informação, serviços e de tecnologia dedicada à saúde no mundo, vinculada as empresas farmacêuticas.

103
clínical trials)23, apesar de despertar discussões éticas por não possuírem efeito
terapêutico e envolverem alguns riscos e ônus sem agregar benefícios diretos aos
sujeitos de pesquisa, entre outras discussões éticas, trazem benefícios voltados a
sociedade – retorno social- e contribuições para a medicina.
Os estudos randomizados controlados (RC) são considerados amplamente
pela comunidade cientifica como o padrão ouro “gold standard” para as pesquisas
de avaliação de segurança e eficácia de tratamento (Zulueta, 2001). Em nossa
amostra, observamos que dos 09 estudos, 100% eram randomizados controlados.
Este dado reflete positivamente sobre a ótica da Justiça, pois, a atribuição
aleatória de sujeitos de pesquisa (randomização) para os braços de tratamento
promove uma igualdade das chances na distribuição das intervenções. Oferecendo
de forma equivalente a todos os sujeitos, os benefícios previsíveis e os riscos de
participação naquele estudo (CIOMS, 2002). Adicionalmente, do ponto de vista
científico remove o potencial de viés na atribuição de pacientes para uma
intervenção ou outra através da introdução de imprevisibilidade (Kunz e Oxman,
1998), eliminando, por exemplo, viéses de seleção relacionados à vulnerabilidade
de alguns sujeitos.
A adoção da randomização é também importante para a segurança dos
pacientes, uma vez que, comprovadamente os estudos não-randomizados ou com
alocação inadequada, em média, superestimam os efeitos aos tratamentos, porém,
também podem mascarar os efeitos (Kunz e Oxman, 1998), afetando
negativamente os sujeitos de pesquisa e o dado científico.
Entretanto, em relação ao cegamento do estudo, a maioria era aberto
(77.7%) e apenas dois eram duplo-cego (22.2%). O cegamento é frequentemente
utilizado nos ensaios clínicos, sobretudo o duplo-cego24, por minimizar os
potenciais viéses resultantes das variações na administração do tratamento ou do
gerenciamento dos pacientes ao longo do estudo. A estratégia tem por objetivo
garantir que as avaliações e decisões médicas/clínicas não sejam influenciadas
pelo conhecimento sobre o tratamento o qual foi designado ao paciente (HHS,
2001).
23 “É uma espécie de fase exploratória inicial de ensaio clínico que administra o composto em doses estimadas que não tenham efeitos farmacológicos ou toxicológicos, visando selecionar candidatos para desenvolvimento posterior dos ensaios clínicos.” (Kurihara, 2011). 24 Estudo duplo-cego (ou duplo-mascarado) significa que os envolvidos, paciente, investigador e patrocinador não têm conhecimento sobre o tratamento administrado ao sujeito de pesquisa.

104
Os dois estudos duplo-cego da amostra utilizavam placebo em um dos
braços de tratamento. Comumente a presença destas duas características, placebo
e duplo-cego, estão associadas nos desenhos de estudo para reduzir os viéses
relacionados aos sujeitos de pesquisa e aos investigadores (HHS, 2001).
Dentro do contexto do desenho de estudo, a variável “Estágio do estudo”
também nos permite a avaliação de um aspecto importante no contexto dos
ensaios clínicos: quando é eticamente inviável seguir com a condução do ensaio?
Dois estudos tiveram o encerramento precoce de suas atividades
justificadas eticamente: um apresentou uma relação desfavorável entre
benefício/risco aos sujeitos de pesquisa e o outro pela alta probabilidade de não
cumprimento de seu objetivo primário.
Ambas as interrupções ocorreram corretamente. A resolução brasileira
sobre ética em pesquisa com seres humanos (n°466/12) determina como uma das
exigências para a realização de estudos nesta área a busca continua da prevalência
dos benefícios esperados sobre os riscos e/ou desconfortos previsíveis. Desta
maneira, quando há uma inversão nesta balança, ou seja, uma relação
desfavorável para o sujeito, o estudo deve findar a favor da segurança dos sujeitos
envolvidos e da população que possivelmente, no futuro, usufruiria da intervenção
estuda.
Outra recomendação da Resolução n°466/12 é em relação à adequação dos
estudos aos princípios científicos que os justifiquem e aos métodos utilizados, de
maneira que estes apresentem possibilidades concretas de se responder as
incertezas que motivaram à realização do estudo e a responder às questões
estudadas. Logo, quando é percebido algum problema no desenho de estudo que
impeça a obtenção de respostas aos objetivos primários ou obtenção de dados
cientificamente confiáveis, não é justificável a manutenção da condução do
estudo.
Dos desfechos clínicos dos estudos avaliados, 55,5% visava sobrevida
livre de progressão e, 33,3%, sobrevida global25. Apenas 01 estudo (11,1%)
visava a taxa de melhor resposta ao tratamento (11,1%). Em oncologia, em função
das altas taxas de morbidade e mortalidade, os tratamentos na área normalmente
25 O desfecho sobrevida global tem como interesse o evento Morte, por quaisquer causas. Já a sobrevida livre de progressão engloba os eventos Progressão de doença ou morte por quaisquer causas (Machado, Katz, Buyse e Saad, 2010).

105
são focados para a melhoria da sobrevida e da qualidade de vida. Assim, os
desfechos clínicos mais usados em oncologia são a sobrevida global e sobrevida
livre de progressão, principalmente nas fases avançadas de desenvolvimento
clínico de um fármaco (Machado, Katz, Buyse e Saad, 2010).
Um levantamento dos desfechos utilizados em estudos randomizados
controlados para o tratamento do câncer de mama avançado no período de 1998 a
2007, realizado por Machado, Katz, Buyse e Saad (2010), observou que 51%
(n=39) dos estudos avaliaram como desfecho primário a sobrevida livre de
progressão, tempo para progressão ou tempo para falha do tratamento, 40%
(n=31) avaliaram taxa de resposta objetiva e 5% (n=04) a sobrevida global. Este
último desfecho apresentou-se em 84,2% da amostra como desfecho clínico
secundário entre os cinco primeiros desfechos avaliados. Tais dados corroboram
com os achados em nossa amostra.
A expectativa de vida para pacientes com câncer aumentou em três anos
desde 1980 e aos novos tratamentos, o que inclui os novos medicamentos, são
atribuíveis 83% destes ganhos (Phrma, 2013b). Infelizmente, a cura do câncer
ainda parece um caminho longo a ser percorrido, mas hoje, com a inclusão de
novos medicamentos na saúde temos observado as transformações na vida dos
pacientes com câncer, com as melhorias nas taxas de expectativa e de qualidade
de vida.
2. EQUIDADE DE ACESSO À SAÚDE
Equidade de acesso à saúde é uma categoria percebida através da leitua
dos textos de Bayer e Fish (2003), Booth (2010), Botbol-baum (2000), Brunet
(1999), Edwards (2006), Kolch e colaboradores (2010), Mc Millan e Conlon
(2004), Oquendo, Stanley, Ellis e Mann (2004), Sklar (1996), Smart, Martin e
Parker (2004), Thomas (1998) , Van Delden J e colaboradores (2004) e Zulueta
(2001), e engloba a distribuição geográfica dos estudos e os critérios de seleção
dos ensaios clínicos.
Umas das implicações da Equidade de acesso à saúde é a distribuição
equitativa das oportunidades de acesso aos estudos clínicos, que pode ser
mensurada pela distribuição geográfica dos ensaios clínicos. O que observamos
em nossa amostra, conforme Gráfico 03 (página 88) é uma participação

106
homogênea dos países, variando entre países emergentes e com economias em
desenvolvimento (de acordo com a classificação do Fundo Monetário
Internacional-FMI), como o Brasil e Argentina, a países com economia avançada,
como Austrália, França, Inglaterra e Itália. Os países, Alemanha e Polônia,
destacados também com grande participação nos estudos da amostra, são
considerados pelo FMI países com economia de transição (IMF,s.d.).
Apesar do equilíbrio na distribuição dos estudos por países, a participação
reduzida dos EUA em apenas 05 estudos (55,5 %) surpreendeu. Estes representam
o país com maior participação em estudos clínicos no mundo, de acordo com o
cadastro do Clínicaltrials.gov, participando de 46,32% ( n=7.5937) dos estudos
mundiais cadastrados na base de dados (n=163.936).
Seria justo que os países desenvolvidos, em sua maioria, propulsores das
pesquisas, participassem equilibradamente dos estudos tanto quanto os países
hospedeiros, comumente os em desenvolvimento. Evitando-se assim o duplo
padrão e as injustiças com as populações locais.
A ampla dispersão de estudos pelo mundo é vantajosa, uma vez que,
diferentes etnias e raças estarão expostas as intervenções estudadas, aumentando
as chances de uma melhor reprodutibilidade de tal intervenção quando
extrapolada para a população em geral.
Contudo, quando avaliada a frequência de participação nos estudos por
estados no Brasil, percebe-se uma má distribuição geográfica das pesquisas pelo
país. Quando agrupamos os Estados por Regiões, verificamos que 60% dos
estudos estavam concentrados na Região Sudeste (São Paulo, Rio de Janeiro e
Minas Gerais), enquanto as Regiões Sul abrigavam 27,5% (Rio grande do Sul e
Paraná), Centro-Oeste com 7,5% e Nordeste com apenas 5%. Nenhuma
participação nos estudo foi identificada na Região Norte do país.
As taxas de incidência das neoplasias malignas por regiões representam
importante parâmetro para avaliação da distribuição dos estudos no país, pois
estas deveriam caminhar em consonância. Entretanto, verificamos que as maiores
taxas incidência de câncer concentram-se nas Regiões Sul e parte das Regiões
Centro-Oeste e parte da Sudeste, conforme Figura 08.
A Região Norte apresenta as menores taxas de incidência
comparativamente às demais regiões, o que não justifica a não participação desta
região na condução de estudos clínicos em oncologia. Sabidamente nesta região

107
há uma precariedade de estrutura social e econômica, o que talvez justifique a
baixa a taxa de incidência em função da provável subnotificação de casos de
câncer. Este cenário qualifica injustiça à população local.
Figura 08. Representação espacial das taxas brutas de incidência por 100 mil homens e 100 mil mulheres, estimadas para o ano de 2014, segundo Unidade da Federação (todas as neoplasias malignas). Fonte: Estimativa 2014. Incidência de Câncer no Brasil.
A dispersão homogênea dos estudos pelo mundo é uma luta de muitos
autores e pesquisadores. Muito tem se discutido sobre a execução estudos clínicos
em países em desenvolvimento, em especial no campo do HIV/AIDS. Thomas
(1998) discorre sobre os desafios éticos destes estudos, e afirma que a maior parte
dos manuais de ética para pesquisas com HIV em seres humanos são baseados em
requerimentos éticos mínimos (Respeito à pessoa - autonomia, Beneficência e
Justiça distributiva), o chamado “Ethics of minimal standards”, enquanto
deveriam ser estendidas as perspectivas maximalistas da bioética e levar em
consideração as questões relacionadas ao acesso continuo dos participantes das
pesquisas aos tratamentos e tecnologias avançadas, as questões de equidade e
direitos humanos como componentes essenciais ao desenvolvimentos dos estudos
clínicos.
Mc Millan e Conlon (2004) corroboram com a idéia acima, ao afirmarem
que o que se percebe da literatura e prática em ética em pesquisa não é uma
escassez de manuais gerais sobre ética em pesquisa, mas sim de manuais que
discutam as complexidades da realização de pesquisas em países em
desenvolvimento.

108
Botbol-baum (2000) traz alguns exemplos de estudos anti-éticos em HIV
conduzidos em países em desenvolvimento, com características de estudos com
duplo-padrão ético, e critica a revisão de 1999 da Declaração de Helsinque que
defende as pesquisas nesses países. Em seu olhar, a revisão estimula a tendência
das pesquisas não orientadas aos cuidados de saúde, mas sim orientadas pelos
lucros e interesses privados das empresas, ônus de alguns em prol de benefícios de
outros, encorajando então, a discriminação contra os pacientes economicamente
vulneráveis dos países em desenvolvimento, desrespeitando a equidade de acesso.
Para o autor o dilema não está na participação ou não de tais cidadãos nas
pesquisas terapêuticas, mas está em reconhecer as responsabilidades éticas da
sociedade para não permitir a exclusão destes sujeitos como seres humanos por
razões puramente econômicas e não permitir injustiças e abusos.
Um dos requerimentos da Declaração de Helsinque é que “Os benefícios,
riscos, encargos e eficácia de uma nova intervenção devem ser testados
comparativamente com a intervenção melhor comprovada...”, porém, há uma
dificuldade de se avaliar qual é o melhor método profilático, diagnóstico ou
terapêutico no contexto de países em desenvolvimento, visto que estes métodos
não são os mesmos por todo o mundo. Entretanto, em termos de Justiça, tal
princípio não nos exige o tratamento exatamente igual para todas as pessoas.
Assim, se há uma razão moralmente relevante, o tratamento diferente por meios
diferentes para alguns grupos é defensável (Millan e Conlon, 2004).
Brunet (1999), Mayss (2000), Zulueta (2001) e Booth (2010) discutem
problemas bioéticos em estudos internacionais aplicados em países em
desenvolvimento, especialmente sobre o estudo de prevenção da transmissão
perinatal do HIV conduzido na África, patrocinado pelos Estados Unidos da
América (USA), onde o grupo controle recebia placebo, enquanto o AZT já estava
consagrado nos USA como medicamento eficaz contra a transmissão vertical.
Para Booth (2010) estes estudos refletem e legitimam uma política neo-
imperialista e anti-reprodutiva da ideologia da Justiça, pois violam o direito da
pessoa, em qualquer lugar do mundo, de receber a melhor opção terapêutica
disponível para a condição estudada.
Não oferecer a melhor opção terapêutica aos sujeitos de pesquisa, para
Brunet (1999), é uma violação dos requerimentos do princípio de justiça. Ele
defende a o acesso eqüitativo á saúde a toda a comunidade, a qual é compreendida

109
por sujeitos de pesquisa no braço controle, experimental e os demais membros da
sociedade. Zulueta (2001) argumenta que os investigadores deste estudo falharam
com seus deveres de proteger os interesses dos sujeitos de pesquisa e na promoção
da Justiça distributiva. A Justiça endossa a equidade internacional, e portanto, não
poderia admitir que a definição da conduta e dos deveres dos pesquisadores
fossem determinados pelas condições locais, sejam elas econômicas ou outras.
Outro fator que nos remete a falhas na Equidade de acesso á saúde é a
verificação de que algumas indicações clínicas, como para o uso de alguns de
cetuximabe e rituximabe, são restritas a populações com específicas mutações ou
expressões genéticas. A Tabela 04 (página 96) apresenta estas especificações de
nossa amostra.
Este afunilamento é reflexo do progresso no campo da
farmacogenética/famarcogenômica. Comumente se percebe a inclusão, nas fases
avançadas do processo de teste de medicamentos, apenas de sujeitos com
expectativas de responderem bem baseado em informações prévias do seu perfil
genético, ou seja, os sujeitos são triados geneticamente antes da inclusão nos
estudos, o que representa uma estratégia utilizada pelas indústrias farmacêuticas
para reduzir as falhas dos testes de medicamento, e consequentemente ter ensaios
clínicos mais baratos e mais eficientes (Van Delden J, Bolt I, Kalis A, Derijks J e
Leufkens H., 2004).
O artigo de Smart, Martin e Parker (2004) traz a preocupação relacionada
ao uso da farmacogenética, que objetiva a estratificação genética individual e
consequentemente proporciona a agilidade na tomada de decisão médica acerca
do tratamento a ser oferecido (maior potencial para redução efeitos tóxicos e
melhora de eficácia), porém, o mau uso desta tecnologia pode conduzir a
injustiças.
Esta “inclusão seletiva” tem potencial para gerar maior benefício aos
participantes da pesquisa, uma vez que, aumentam as chances de maior
efetividade de tratamento, porém, há que se ponderar que há a exclusão de parte
da população e que consequentemente os resultados de tais estudos não
representam a população em geral.
É inegável que o maior conhecimento sobre o genoma humano aumenta as
expectivativas para melhor conhecimento e classificação das doenças, e
consequentemente a possibilidade de seleção de fármacos mais específicos para

110
determinadas doenças. Entretanto, Van Delden e colaboradores, (2004)
compartilham a preocupação acerca do uso da
farmacogenômica/Farmacogenética.26
A farmacogénetica tem potencial para estabelecer novos desafios éticos
sobre as implicações da estratificação genética para a equidade em saúde, ao
supormos que tal tecnologia poderá limitar a pesquisa de novos fármacos a certos
grupos genéticos. Alguns sub-grupos genéticos poderão sofrer discriminação
social/estigmas; as implicações desta seleção poderá direcionar a criação de novas
estratificações, como a social, caso o acesso a este cuidado seja de alto custo
monetário (Smart, Martin e Parker, 2004).
Kolch e colaboradores (2010) também apontam preocupações quanto ao
potencial das pesquisas com farmacogenética de gerar injustiças/iniqüidades,
apesar de assumirem que o conhecimento sobre tal é crucial para as pesquisas em
pediatria, foco de seu artigo.
Van Delden J e colaboradores (2004) reforçam a preocupação do aumento
da farmácia sob-medida (“tailor-made pharmacy”), que amplia os problemas de
equidade e distribuição justa de medicamentos/ tratamentos, ao disponibilizar no
mercado medicamentos específicos para um sub-grupo genético. Questiona como
será o acesso aqueles sub-grupos diferentes ou raros na população e qual será a
estratégia de priorização de pesquisas.
Oquendo e e colaboradores (2004) apontam problemas de injustiça na
aplicação dos critérios de seleção/método de alguns estudos sobre o
comportamento suicida que incluem seletivamente seus pacientes, excluindo os
pacientes com alto risco, como os com histórico suicida, comorbidades e doenças
severas. Estas ações diminuem a equidade na seleção, além de prejudicar os
resultados, uma vez, que não poderam ser generalizados a população em geral.
Sklar (1996) introduz o debate ético sobre Justiça nos estudos clínicos para
o alívio de dor ao indagar sobre tratamentos diferenciados variando de acordo
com as características étnicas, idade ou sexo. Há uma linha argumentativa, a qual
defende que a cultura do indivíduo interfere na graduação e na expressão da dor,
porém, para o autor os objetivos e desenhos de estudos deveriam criar limites para
o alivio da dor independentemente das características individuais.
26 Apesar dos termos Farmacogenética e Farmacogênomica serem utilizados amplamente indistintamente, os autores optam por utilizar o termo Farmacogênomica.

111
Injustiças no processo de seleção de sujeitos de pesquisa também são
apontadas por Bayer e Fish (2003), especificamente na discriminação etária para a
participação em estudos clínicos. Os autores demonstram a inadequada
representação da população idosa nos estudos clínicos e defendem que apesar da
vulnerabilidade desta população e da complexidade de alguns estudos, a inclusão
destes sujeitos deveria ser encorajada quando o estudo for relevante para a
condição clínica existente.
Alguns pacientes em estágios graves da doença, independentemente da
idade, já receberam os tratamentos padrões disponíveis ou não os podem receber,
pela inefetividade dos mesmos ou pela alta toxicidade. Uma alternativa seria testar
novos tratamentos através de pesquisas. Edwards (2006), porém, aponta algumas
problemáticas morais nas restrições de acesso, ora cientificas, ora geográficas, ora
econômicas aos novos tratamentos, e a configuração de exploração de
vulneráveis. O acesso a tratamentos restritivos pode alterar a capacidade de
consentimento dos pacientes, induzindo sua participação. Tal cenário configura
injustiça, pela redução ou não proteção da liberdade individual de escolha e
exploração, que não necessariamente precisa ser coerciva; mas, já a redução das
opções e da qualidade destas, possibilita injustiça.
Reflexões acerca da equidade e justiça social devem ser iniciadas ao
verificarmos exclusões ou sub-representações de grupos populacionais nos
ensaios clínicos, pois, tais indivíduos deixam de se beneficiar diretamente das
pesquisas (Smart, Martin e Parker, 2004).
3. DISTRIBUIÇÃO EQUITATIVA DE ENCARGOS E
BENEFICIOS REFERENTE À PARTICIPAÇÃO NA
PESQUISA.
Quanto á distribuição equitativa de encargos e benfícios, esta emergiu após
leitura de Ballantyne (2005), Beran (2006), Clark (2002), Haire (2011), Hawkins
(2006), Mayss (2000), Resnik (2002) e Varmus e Satcher (1997), e envolve o
acesso aos métodos profiláticos e diagnósticos disponíveis, as análises das
necessidades das pesquisas de acordo com a população e uso de placebo.

112
Quando os resultados de um estudo são favoráveis, o acesso a este
medicamento posteriormente aos sujeitos de pesquisa é visto como justo, pois
caracteriza um dos benefícios fornecidos a estes em retorno as participações. A
Resolução nº466/12 define Benefício da pesquisa como “Proveito direto ou
indireto, imediato ou posterior, auferido pelo participante e/ou sua comunidade
em decorrência de sua participação na pesquisa” (Brasil, 2012, II.4).
Varmus e Satcher (1997) discutem que a realização de protocolos
investigacionais terapêuticos que indevidamente envolvam populações em seus
estudos com altas probabilidades de não receberem os benefícios diretos da
aplicação da pesquisa, configuram violação do princípio da Justiça.
Um cenário de injustiça no fornecimento de medicamentos aos sujeitos de
pesquisa é apontado por Beran (2006) em estudos com medicamentos anti-
epiléticos (AEM – anti epileptic medications), os quais são potencialmente
teratogênicos. Por cumprimento aos requerimentos do Manual de Boas Práticas
Clínicas, as pacientes são advertidas a respeito no início do estudo. Porém, um
conflito entre os principios éticos da autonomia e justiça se estabelece, quando
uma paciente engravida inadvertidamente durante tais estudos, é imediatamente
retirada da pesquisa, mesmo sendo beneficiada pelo tratamento e desejando
permanecer no estudo.
Os medicamentos identificados em nossa amostra com registro no Brasil
foram comprados pelo Governo Federal, à exceção do ipilimumabe, entre o
período de 2004 a 2012, conforme descrição do Quadro 14 (página 99).
Entretanto, outras variáveis estão imbricadas ao processo de compra de
medicamento, como o processo de registro no país e as análises de custos.
O processo e prazo para o registro de medicamentos no Brasil não pode ser
comparado aos das agências FDA e EMA, conforme descrição na sessão
Resultados. Entretanto, na literatura muito se fala sobre a burocracia no processo
regulatório brasileiro e seus potenciais entraves para a participação do Brasil no
contexto das pesquisas clínicas mundiais.
A burocracia no Brasil é vista como fator dificultador do processo de
registro de medicamentos. Diante do gargalo, a Anvisa no primeiro semestre de
2013 adotou uma série medidas para agilizar e modernizar a análise do registro de
novos produtos através da criação do Sistema de Registro Eletrônico de
Medicamentos. A meta principal da implementação deste sistema é reduzir o

113
prazo final de registro para até seis meses para os produtos considerados
estratégicos para o SUS ou que represente inovação tecnológica (ENSP, 2013).
Além, da estratégia de tentar reduzir a morosidade do processo de registro
de medicamentos no país, a resolução brasileira lança mão de um requerimento
que configura uma tentativa de se garantir que os riscos aos quais os sujeitos serão
expostos, sejam contrabalanceados pela garantia de benefícios, assim prevê que
para as pesquisas experimentais na área biomédica com seres humanos, o
patrocinador deva “assegurar a todos os participantes ao final do estudo, [...] o
acesso gratuito e por tempo indeterminado, aos melhores métodos profiláticos,
diagnósticos e terapêuticos que se demonstraram eficazes”.
Em relação à análise de custo das compras destes medicamentos,
observamos uma variação colossal entre os preços dos itens dos medicamentos no
mesmo ano. A variação máxima foi de até 569,23 vezes o menor valor de compra,
para o interferona alfa 2a em 2005. As variações máximas para cada medicamento
nos seus respectivos períodos de compra foram: bevazicumabe variou em até
13,26 vezes em 2007; em 2012 o rituximabe variou em 9,47 e cetuximabe em 7.09
vezes o menor valor de compra.
A primeira hipótese para as grandes variações de preço seria a
característica da compra, emergencial ou por processo judicial, o que sabidamente
pode elevar o valor da compra, porém, ao se avaliar as justificativas de compra
para os itens de maiores valores para cada medicamento em questão, observou-se
que a inexistência desta. Os campos foram preenchidos como “Não informado”. A
justiça é inerente ao procedimento de compras públicas, as quais deveriam ser
eficientes e transparentes para permitir o acesso equitativo da população aos
medicamentos. As variações elevadas dos preços podem implicar em compras em
quantidades menores do que a planejada ou realocação dos recursos, o que afeta
consequentemente a distribuição destes medicamentos para as instituições/regiões.
Os anticorpos monoclonais rituximabe, bevacizumabe e cetuximabe,
comprados pelo Governo Federal no período previamente descrito, estão entre os
seis medicamentos biológicos que estão previstos para serem produzidos
nacionalmente a partir de 2014 através de um acordo recentemente liderado pelo
Ministério da Saúde com as multinacionais Merck Serono e a Bionovis com
intuito de impulsionar as Parcerias para o Desenvolvimento Produtivo (PDP)
nacional de biofármacos. Tais medicamentos possuem alto custo para o

114
tratamento de câncer no país e a execução destas estratégias governamentais é
essencial para o desenvolvimento nacional. Um exemplo real é o mesilato de
imatinibe, utilizado para o tratamento de Leucemia Mieloide Crônica, que foi o
primeiro oncológico nacional produzido por uma PDP, apresentando uma
economia em 2013 de R$ 31,5 milhões comparada com o ano de 2012. As PDP
também envolvem a transferência de tecnologia entre instituições públicas e
privadas, e são voltadas às demandas de produtos estratégicos para SUS (INCA,
2013 b), o que representa uma ação de respeito para com as necessidades da
população e de retorno social.
Entretanto, há controvérsias sobre a eficácia das PDP. As formas de
escolha de quais medicamentos devem ser objeto de PDP ainda não está clara.
Diversos medicamentos sob PDP podem ser considerados obsoletos ao final do
tempo dos contratos; outrossim, os preços ao final das PDP podem não
representar a economia tão decantada. Ainda, durante o tempo do contrato, a
empresa que detém a patente tem monopólio nas compras públicas. Como o Brasil
não se habilita a receber preços realmente diferenciados (uma vez que tem status
de ‘upper middle-income country’) e não possui acesso a versões genéricas
produzidas no exterior (pelo mesmo motivo), fica refém de PDP como saída
aparentemente atraente. Contudo é preciso analisar essas PDP sob o ponto de vista
ético.
Para que os estudos clínicos sejam verdadeiramente direcionados às
necessidades da população, há que se avaliar, entre outros aspectos, o impacto
daquela determinada doença sobre a população a ser estudada. Consequentemente,
as definições das prioridades de financiamento em P&D na área biomédica devem
ser proporcionais às necessidades de redução da carga da doença (burden of
disease), definida pelo NIH (1998), a partir da avaliação de alguns parâmetros
como: incidência, mortalidade, grau de incapacidade causada pela doença,
impacto na expectativa de vida, aspectos sociais e econômicos da doença e as
considerações de saúde pública. Caso a distribuição de recursos para as P&D das
doenças não seja proporcional à carga desta doença, podemos dizer que esta
distribuição foi injusta (Resnik, 2001).
As condições de uso de placebo nos ensaios clínicos é uma variável
importante para avaliação da balança entre os riscos e benefícios da pesquisa.
Habitualmente, os sujeitos de pesquisa do grupo de controle recebem uma

115
intervenção de eficácia comprovada versus o grupo que receberá a intervenção
experimental. Entretanto, em algumas circunstâncias pode ser eticamente
aceitável a utilização de um comparador alternativo, como placebo.
Hawkins (2006) discute os problemas morais em alguns estudos clínicos
com placebo, apontando injustiças cometidas contra os sujeitos de pesquisa
quando estes estão em uso de placebo, em momentos nos quais a terapia para
aquela condição estudada já está provada. A autora discute amplamente a
viabilidade de execução de tais estudos e fortemente a responsabilidade moral dos
pesquisadores para com estes sujeitos. Haire (2011) corrobora com a discussão
sobre a obrigação moral dos pesquisadores de fornecer o tratamento adequado a
população assistida no estudo, na tentativa de se evitar a exploração e atender ao
princípio de justiça.
Em nossa amostra apenas 02 estudos (NCT00094653 e NCT00486759)
utilizaram placebo em seus desenhos. A divisão de braços do estudo
NCT00094653 é eticamente questionável: Foram considerados dois braços como
controle, “ipilimumab e placebo” ou “fármaco experimental (MDX-1379) e
placebo”, e como braço experimental “ipilimumabe ou fármaco experimental”. A
proposta do estudo era determinar a segurança e eficácia do ipilimimabe em
combinação com o fármaco experimental em pacientes com melanoma
metastáticos ou irressecáveis versus as monoterapias com os respectivos
fármacos. Entretanto, o ipilimumabe é comprovadamente eficaz para a patologia
estudada, o que permitiria a avaliação deste versus “ipilimumabe em adição a um
fármaco experimental”, mas não “experimental e placebo” como braço controle.
Os sujeitos randomizados para este braço deixaram de receber o tratamento
padrão para a doença, ou seja, os potenciais benefícios da pesquisa, o que é
eticamente condenável. O julgamento mais denso deste caso, infelizmente não
será possível, pois as informações disponíveis na plataforma de dados do
ClínicalTrials.gov são limitadas e não fornecem informações sobre as
justificativas ponderadas pelo patrocinador junto aos CEP/CONEP para a
realização deste estudo.
No contexto das pesquisas internacionais, Ballantyne (2005) conclui que
mesmo havendo a promoção de benefícios mútuos (pesquisadores e sujeitos), o
estudo pode ser considerado exploratório quando a distribuição dos encargos e
benefícios é injusta, como ocorre nos estudos com emprego errôneo de placebo. O

116
autor refere que a base da definição normativa de justiça também envolve a
distribuição dos riscos, e adicionalmente, chama a atenção para a dificuldade de se
definir/quantificar/qualificar o que é distribuição justa de benefícios, visto que os
riscos e as incertezas de resultados são inerentes aos estudos clínicos, e diante da
impossibilidade de predizer completamente os resultados é que se percebe a
impossibilidade de garantir os benefícios ex-post.
As definições de justiça e exploração se entrelaçam. Tais definições são
abordadas por Resnik (2002) ao discuti-las em alguns estudos clínicos. Para o
autor o tratamento justo a um sujeito de pesquisa implica em distribuição
equitativa de encargos e benefícios, independentemente de razões sociais,
econômicas ou outras, e o que caracteriza a exploração é a tomada de vantagem
injusta de uma pessoa. Assim, a realização de estudos com populações vulneráveis
por exemplo, não seria um problema em si, mas o seria se houvesse a violação dos
princípios de justiça, beneficência e respeito à pessoa.
Ballantyne (2005), baseado em alguns conceitos da Teoria de Rawls,
divide o conceito de Justiça em três princípios: (1) a pessoa não pode ser forçada
pelas circunstâncias (econômica, social ou outras) a permutar para alcançar ou
proteger um bem-básico27 (exemplo, a saúde); (2) nos casos em que uma das
partes permutar com um bem-básico (exemplo, a vida) e a outra não, a
distribuição de encargos e benefícios devem ser organizados de modo que eles
beneficiem mais o menos favorecido28; (3) nos casos em que uma das partes
permutar em busca de um bem-básico e a outra não, deve ser assegurado ao
primeiro os benefícios garantidos (previstos antes do estudo) além de alguns
benefícios potenciais (pós-estudo). O cumprimento destes três princípios confere
o caráter de justo a pesquisa a ser realizada.
Uma outra discussão dentro do contexto do uso de placebos é elencada por
Clark (2002) ao fazer uma análise ética sob a luz do princípio da Justiça das
questões envolvidas na condução de estudos controlados com cirurgia-placebo,
em especial para a Doença de Parkinson. O princípio da Justiça, na concepção
adotada pelo autor, implica em tratamento equitativo das pessoas, fornecendo a
elas o que lhe é devido, ou seja, os benefícios da participação da pesquisa.
27 São os bens fundamentais para o florescimento completo da vida dos membros de uma sociedade. 28 Baseado no principio da Diferença de Rawls.

117
Visando a redução das explorações nas pesquisas com seres humanos em
países subdesenvolvidos e uniformização das recomendações, entre outros
objetivos, o CIOMS em 2002 através da publicação do International Ethical
Guidelines for Biomedical Research Involving Human Subjects determinou em
quais ocasiões o placebo poderia ser utilizado, e aparentemente o estudo em
questão não se aplica aos critérios abaixo:
“Quando não há intervenção de eficácia comprovada; Quando ocultado uma intervenção eficaz estabelecida, isso exporia o sujeito à, no máximo, um desconforto temporário ou atraso no alívio dos sintomas; Quando o uso de uma intervenção eficaz estabelecida como comparador, não produziria resultados cientificamente confiáveis e uso de placebo não acrescentaria qualquer risco grave ou danos irreversíveis aos sujeitos” (CIOMS, 2002, p.54).
O estudo NCT00486759 não apresentou problemas em relação ao uso de
placebo, pois, ofereceu aos dois braços de tratamento os medicamentos
considerados eficazes para a condição médica estudada em adição ao placebo ou
fármaco experimental.
Mayss (2000) observa que embora haja muitos manuais e declarações com
princípios éticos no contexto da pesquisa clínica, existe uma lacuna nas leis
nacionais que faça cumprir tais princípios, salvaguardando os direitos humanos
dos sujeitos de pesquisa.

118
CONSIDERAÇÕES FINAIS E RECOMENDAÇÕES
O presente trabalho identificou e caracterizou os ensaios clínicos
internacionais e nacionais envolvendo anticorpos monoclonais e biomedicamentos
oncológicos com participação brasileira, nos últimos 10 (dez) anos. Foram
consultadas as bases de registro Clínical Trials e Registro Brasileiro de Ensaios
Clínicos (ReBEC), e foi mapeada a trajetória de registro desses medicamentos no
país (ANVISA) e nos principais órgãos estrangeiros internacionais de registro de
medicamentos (FDA e EMA). Foram avaliados os gastos federais com a compra
destes medicamentos no período do estudo.
Após a leitura dos artigos selecionados na busca sistemática sobre justiça e
ética em pesquisa clínica foi possível, de acordo com a percepção da
pesquisadora, agrupar as concepções do principio de justiça em três categorias
analíticas, como descritas anteriormente. Diante destes resultados e após avaliação
global deste cenário, foi possível analisar os aspectos bioéticos relacionados à
aplicação do princípio da justiça e equidade, previsto na resolução brasileira sobre
ética em pesquisa com seres humanos, no percurso iniciado com a realização de
ensaios para biomedicamentos e anticorpos monoclonais no Brasil, até seu
registro no país, no período de 2003 a 2012. Portanto, entende-se que foi possível
cumprir os objetivos do estudo.
Algumas limitações e dificuldades deste processo foram identificadas:
1. Poucos estudos selecionados em nossa amostra envolviam a experimentação de
anticorpos monoclonais e biomedicamentos oncológicos (apenas 09), formando
uma base limitada para discussão;
2. A base de dados ReBEC não possui filtros em sua busca avançada para as
características (i) fase do estudo e (ii) período de cadastro do estudo na base de
dados, o que delonga e dificulta o processo de seleção dos estudos;
3. A base de dados Clínical Trials não identifica todos os centros de pesquisa
envolvidos na condução do estudo, dificultando a avaliação e caracterização
dos centros de pesquisa no país envolvidos;
4. A Anvisa não disponibiliza na sua interface de WEB o histórico de registro dos
medicamentos. O que está disponível é a data de publicação da última
atualização de registro daquele produto, impossibilitando a rastreabilidade e o

119
acesso transparente às informações acerca dos medicamentos. Além disso,
impede a análise de concomitância entre os momentos das compras federais e
os períodos reais de registro;
5. Os dados de 2003 da base de dados do Sistema Integrado de Administração de
Serviços Gerais (SIASG) não estavam disponíveis, inviabilizando a análise
completa de 2003 a 2012, como previsto;
6. A maioria das justificativas das compras federais do banco SIASG eram
vagas/não informativas e 18,5% eram “não informado”, impossibilitando o
conhecimento sobre as razões que motivaram as compras.
Apesar destas limitações, foi posspivel realizar o estudo em função da
cuidadosa verificação das informações, aliada à restrição intencional de discussão
e conclusões que não puderam ser consubsanciadas pelos dados empíricos.
Na primeira etapa do estudo, a busca bibliográfica sobre a temática justiça
e ética em pesquisa, foi identificada 48 potenciais artigos; porém, a amostra válida
final foi composta por 22 artigos. Na leitura dos artigos encontramos diversas
problemáticas no contexto das pesquisas envolvendo questões de Justiça, como
pesquisas em países em desenvolvimento, farmacogênomica e uso de placebos.
Entretanto, poucos se aprofundaram sobre o debate ético baseado no princípio da
Justiça.
A partir das diferentes percepções destes autores sobre o principio, foi
possível agrupá-las em três categorias, utilizadas na análise dos aspectos bioéticos
intrínsecos ao percurso da realização dos ensaios clínicos de nossa amostra. O
organograma com as categorias analíticas e suas implicações encontra-se na
Figura 09.
Na segunda etapa do estudo, a busca dos ensaios clínicos nas bases de
dados Clínical Trials e ReBEC observou-se que poucos estudos estavam
cadastrados na base brasileira (n=12) em comparação à base internacional Clínical
Trials (n=492). O achado é compreensível pelo pouco tempo de vigência da
ReBEC (2010) e além disto, a Anvisa instituiu a obrigatoriedade do registro dos
ensaios clínicos nesta base apenas em 2012, limite temporal de nossa amostra.
Apesar de justificável, é recomendável a divulgação da base e de suas
contribuições para a sociedade.

120
Busca sistemática
sobre Justiça e
Pesquisa Clínica
Relevância Social Equidade de acesso à
saúde
Distribuição
equitativa de
encargos e benefícios
Resultados empíricos
Acesso aos
resultados da
pesquisa
Acesso às
informações sobre o
medicamento
Participação
Brasileira nos
Ensaios Clínicos
Cientificidade dos
Ensaios Clínicos
Distruibuição
geográfica dos
Ensaios Clínicos
Critérios de seleção
dos Ensaios Clínicos
Acesso aos métodos
profiláticos, diagnósticos e
terapêuticos pós estudo
Estudos direcionados
as necessidades da
população local
Uso adequado de
placebo
Figura 09. Organograma com as categorias analíticas sobre justiça e ética empesquisa e suas implicações.

121
A amostra final de estudos foi composta por 09 estudos, os quais
envolviam a participação de 05 anticorpos monoclonais (bevacizumabe,
cetuximabe, figitumumabe, ipilimumabe e rituximabe) e 01 biomedicamento
(interferona-alfa 2a). A média de tempo de execução dos estudos de nossa amostra
foi de 5,48 anos, superior ao descrito na literatura. Em relação à linha do tempo
para a aprovação de registro dos anticorpos monoclonais e biomedicamentos nas
diferentes agências regulatórias, observou-se que a diferença de tempo entre as
aprovações do FDA e da EMA pouco variaram, sendo a variação máxima de 01
ano, demonstrando uma sincronia entre as agências. Não foi possível avaliar esta
variável para a Anvisa.
Os 09 estudos analisados contaram com a participação de 58 diferentes
países, em uma distribuição homogênea. O Brasil, a França e a Inglaterra tiveram
uma participação em 100% dos estudos (n=09). Os Estados Unidos apresentou
uma participação inferior, representados por 55,6% (n=05). Focando na
participação do Brasil, quando agrupados as cidades brasileiras por estados e os
estados por região verificamos uma má distribuição geográfica dos estudos pelo
país. A Região Sudeste concentrava mais de 50% dos estudos, enquanto nenhuma
participação foi identificada na Região Norte. Adicionalmente, as taxas de
incidência dos cânceres não estavam em consonância com a distribuição dos
estudos no país.
Verificamos que 100% dos estudos eram patrocinados por indústrias
farmacêuticas estrangeiras, o que reflete a fragilidade da participação brasileira,
sobretudo acadêmica, na construção e realização de ensaios clínicos em
oncologia.
Ao avaliarmos os tipos de câncer envolvidos em nossa amostra,
identificamos: Colorretal, Gástrico, Linfoma não-hodking, Melanoma, Pulmão
não-pequenas células e Renal. Tais patologias possuem altas taxas de mortalidade
e incidência no Brasil, justificando a realização destas pesquisas no país.
Uma variável importante na avaliação da Relevância Social do estudo é a
qualidade de seu desenho, da sua metodologia. Dos 09 estudos avaliados todos
eram randomizados controlados, considerado padrão ouro pela comunidade
cientifica, com recrutamento para ambos os sexos, com idade mínima de 18 anos e
apenas 01 estudo apresentava idade máxima (79 anos), em sua maioria de fase III.
Apenas 01 estudo era de fase II. A média de recrutamento foi de 985,22 pacientes,

122
variando de 302 a 1.861. Dois estudos eram duplos-cego e os demais abertos. Dois
estudos utilizavam placebo. A divisão de braços de um destes estudos é eticamente
questionável, porém, em função da insuficiência de dados sobre a apreciação ética
pelo CEP, a análise mais profunda foi impedida.
Em relação aos critérios de seleção para participação nos estudos, percebe-
se que dos 09 estudos da amostra final, 77,7% dos estudos apresentavam algum
tipo de restrição genética em seus critérios de seleção. Este afunilamento da
seleção dos participantes de pesquisa é reflexo do progresso no campo da
farmacogenética/famarcogenômica. Devemos estar atentos para os potenciais
desafios éticos que poderão surgir pela estratificação da população e exclusão de
alguns sub-grupos.
Na busca pela identificação e caracterização dos medicamentos da amostra
nas agências regulatórias - Anvisa, FDA e EMA – percebeu-se, durante a coleta
de dados, dificuldades no acesso às informações na base da Anvisa em relação aos
registros dos medicamentos. Observou-se limitação de informações, clareza e por
vezes, ausência de informações sobre os medicamentos em nosso país. Este é uma
aspecto crítico dos resultados, pois a transparência nas informações sobre os
estudos, seus resultados e medicamentos visa homogenizar o conhecimento dos
indivíduos, sejam eles usuários e/ou pesquisadores, trazendo benefícios ás
sociedades científica e civil.
Nas análises das compras federais dos anticorpos monoclonais e
biomedicamentos oncológicos a partir da base de dados SIASG, verificou-se os
medicamentos identificados com registro no Brasil foram comprados pelo
governo federal, à exceção do ipilimumabe, entre o período de 2004 a 2012. Um
dado que nos chama a atenção são as colossais variações de preços dos produtos
adquiridos. A maior variação identificada foi de 569,23 vezes em relação ao
menor valor de compra para o interferona alfa 2a, em 2005. As variações elevadas
os preços podem implicar em redução de itens comprados ou realocação dos
recursos, o que afeta consequentemente a distribuição destes medicamentos para
as instituições/regiões.
Os resultados obtidos a partir deste estudo nos permitiram visualizar
algumas fragilidades em nosso sistema regulatório. A principal resolução
brasileira sobre ética em pesquisa com seres humanos, por muitos anos a 196/96 e
hoje a 466/12, ambas do Conselho Nacional de Saúde, incorporou como exigência

123
ética o princípio da Justiça e equidade. Este princípio possui várias implicações no
percurso dos ensaios, partindo das pesquisas ao registro; porém, são implicações
pouco palpáveis, de difícil mensuração.
Reitero o pensamento de Mayss (2000) ao afirmar que embora haja muitos
manuais e declarações com requerimentos éticos na área da pesquisa clínica,
existe uma lacuna nas leis nacionais. Esta lacuna diz respeito aos mecanismos de
fiscalização da aplicação das normas, situação a qual, desfavorece o cumprimento
de tais requisitos e princípios éticos por parte dos pesquisadores e envolvidos.
As bases de dados para registro de ensaios clínicos são a principal fonte de
informações sobre o andamento dos estudos, porém, possuem limitações de
informações, como por exemplo, o acesso ao medicamento fornecido pelo
patrocinador no pós-estudo. A transparência na disponibilidade de informações
sobre as pesquisas realizadas no país são indispensáveis, assim algumas
informações poderiam ser adicionadas as bases sem violar o sigilo do estudo e
confidencialidade dos participantes do estudo.
É altamente recomendável que a Anvisa, nossa agência reguladora, reveja
suas ferramentas e banco de dados, a fim de melhorar a transparência nas
informações disponíveis ao público a cerca dos medicamentos registrados no
Brasil, como o histórico dos registros e as indicações clínicas iniciais e atuais dos
medicamentos.
Os altos custos verificados na base de dados SIAGS são alarmantes. A
introdução de novos medicamentos é benéfica à população, porém, quanto feita de
maneira cautelosa. Para tanto, o acompanhamentos dos ensaios clínicos com a
participação brasileira e seus resultados é fundamental para subsidiar as decisões
dos orgãos reguladores/governo federal a cerca das compras de medicamentos.
Por fim, acredita-se que as informações apresentadas neste trabalho
possam contribuir para o debate sobre as questões éticas envolvidas nos ensaios
clínicos no Brasil e também contribuir para o aprimoramento e o fortalecimento
da agência reguladora local e instituições envolvidas no processo.

124
ANEXO A

125

126

127
APÊNDICE A
Apêndice A: Descrição dos estudos excluídos dos resultados da base de dados da REBEC.
Código de
identificação do
estudo na base
Título Científico
RBR-3s39jp Efeito da Eletroanalgesia no controle da Disestesia em pacientes submetidas à Linfadenectomia axilar.
RBR-2jtbyw Avaliação da influência de um programa de tratamento fisioterapêutico no pré-operatório de correção de Megaesôfago
sobre a função pulmonar no pós-operatório.
RBR-237v4b Avaliação da eficácia da calêndula na prevenção e tratamento de Radiodermite de cabeça e pescoço.
RBR-3nm5bv Impacto da reabilitação pulmonar na incidência de complicações respiratórias no pós-operatório de ressecção pulmonar
por neoplasia de pulmão.
RBR-4y59tq Ensaio clínico randomizado, duplo-cego, sobre o efeito da Folha Informativa em pacientes e familiares atendidos no
Ambulatório de Oncogenética de um hospital universitário: avaliação do grau de informação e da percepção de coerção.

128
APÊNDICE B
Apêndice B: Descrição dos estudos clínicos excluídos da amostra por não apresentarem câncer/neoplasia maligna como a condição médica
prévia ou por não apresentarem a intervenção terapêutica com anticorpo monoclonal ou biomedicamento.
Código de identificação do estudo na base Clínicaltrials.gov Condição médica
NCT01137682 e NCT00600886 Acromegalia
NCT01001156 Alterações fisiológicas do envelhecimento
NCT01215747 Amiloidose
NCT01168505 Anemia em Câncer de mama
NCT00338286 Anemia em Câncer de mama metastático
NCT00858364 Anemia em Câncer de Não-pequenas células do pulmão
NCT01540019 Anorexia em pacientes oncológicos
NCT00960440, NCT01350804, NCT00122382 e NCT01202773 Artrite rematóide
NCT00412893 Aspergilose
NCT01459393 Ceratose actínica
NCT01405144 Ceratose actínica e foto envelhecimento
NCT01337934 Choque séptico
NCT00582426 Diarréia
NCT01463943 Diarréia associada ao uso de antibiótico

129
NCT01369329; NCT00406653 e NCT00552058 Doença de Crohn
NCT01582061, NCT01374906 e NCT00434148 Doença de Cushing
NCT00943111 Doença de Gaucher
NCT00054600 Doença do enxerto versus hospedeiro
NCT00634049 Doença fúngica invasiva
NCT00097110 Eclâmpsia e pré eclâmpsia
NCT00062803 Embolia pulmonar
NCT00862459 Encefalopatias e doenças da medula espinhal
NCT00340834, NCT00548405, NCT00930553 e NCT00530348 Esclerose Múltipla
NCT01209689 e NCT01209702 Espondilite Anquilosante
NCT00615316 Fadiga em Câncer de mama
NCT01043913 Fadiga induzida por quimioterapia para câncer de mama
NCT00412984 Fibrilação atrial
NCT00989599 Flebite
NCT01072045 Hemangioma
NCT01454739 Hemofilia A
NCT00065507 Hepatite B
NCT01254630 Herpes zoster em tumores sólidos ou hematológicos
NCT00697814 Hipogonadismo hipogonadotrófico; Prolactinoma
NCT01451411 Hiponatremia
NCT00478231, NCT00272779, NCT01461096 e NCT00604175 HIV/Aids

130
NCT01410175 Infecção de ferida operatória
NCT00413218 Infecção por candida
NCT00937950, NCT01418937, NCT00689741, NCT00120848 e
NCT00849381 Infecções por Papillomavirus
NCT00098319 Lábio leporino
NCT01757899 Lesão pulmonar aguda; Síndrome da Angústia Respiratória Aguda
NCT00389181 Malformação arteriovenosa cerebral não rompida
NCT00925639 Menopausa
NCT00874302 Mioma uterino
NCT01523574 Neuropatia periférica
NCT01079676 Neutropenia em Câncer de mama
NCT00141323 e NCT00205777 Osteoporose
NCT00171340, NCT00056407 Prevenção do Câncer
NCT00522873 Pós menopausa
NCT01163253 e NCT01186744 Psoríase
NCT00876642 Radiodermite
NCT01150097 Receptor de transplante hepático
NCT00774930 Síndrome carcinóide
NCT00630747 Síndrome de Hunter
NCT00427700 Síndrome do ovário policístico
NCT00171210 Talassemia

131
NCT00457002 e NCT00071279 Tromboembolismo e embolismo pulmonar
NCT00694382, NCT00643201 e NCT00633893 Tromboembolismo venoso
Código de identificação do estudo na base Clínicaltrials.gov Intervenção
NCT00534664 Campos eletromagnéticos
NCT01581918 Carbamazepina (Anticonvulsivante)
NCT01724450 Caverdilol (Beta-bloqueador)
NCT00156026 Cirurgia com alça
NCT00518336 Coleta de amostra de sangue e cervical
NCT01929993 Conização eletrocirurgica
NCT01468779 e NCT01168206 Dieta suplementar
NCT01267552 Drenagem por aspiração cirúrgica
NCT00254683 Exame de imagem: PET/CT
NCT00959400 Fentanil transdérmico (analgésico)
NCT00995020 Hetero Excisão de fio da Zona de Transformação; Excisão da zona de transformação (biópsia em cone)
NCT00614211 Histerectomia abdominal radical; Laparoscopia total ou histerectomia radical robótica
NCT01258413 Histerectomia laparoscópica Radical; linfadenectomia pélvica; histerectomia radical Abdominal
NCT01280045 Histerectomia vaginal
NCT01439724 Laser terapia
NCT00525967 Metadona/paracetamol; Metadona/placebo (opióde e analagésico)

132
NCT01570218 Musicoterapia
NCT01169129 Radiocirurgia ou cirurgia
NCT01169116, NCT00858741 NCT01450449, NCT00287196,
NCT01464177 e NCT00610272 Radioterapia
NCT00242567 Terapia hormonal
NCT01648946 Transfusão de hemácias
NCT01502215 Transfusão de sangue
NCT01391988 Uso de bisturi elétrico convencional e bisturi harmônico em Mastectomia
NCT01031719 Vacina com adjuvante gripe A (H1N1); Sem adjuvante A vacina contra a gripe (H1N1)

133
APÊNDICE C
Apêndice C: Descrição dos artigos encontrados nas respectivas bases eletrônicas.
Bases de dados
eletrônicas Título dos artigos Autores Dados da publicação
Klinische studies in ontwikkelingslanden [Clínical studies in developing countries]*
Munkhof, H.E.V.D. Nederlands Tijdschrift voor
Geneeskunde,157 (38), art.A6413, 2013.
Safeguarding children's rights in psychopharmacological research: Ethical and legal issues***
Kölch, M., Ludolph,A.G., Plener, P.L., Fangerau, H., Vitiello, B., Fegert, J.M.
Current Pharmaceutical Design, 16 (22):2398-2406, 2010.
Ethics of clínical trials*, ***
Iyalomhe,G.B., Imomoh, P.A Nigerian journal of medicine: journal of the National Association of Resident Doctors
of Nigeria, 16 (4):301-306, 2007. The ethics of excluding women who become pregnant while participating in clínical trials of anti-epileptic medications***
Beran, R.G. Seizure, 15 (8): 563-570, 2006.
Protection of human subjects in intervention research for suicidal behavior
Oquendo, M.A., Stanley, B., Ellis, S.P., Mann, J.J.
American Journal of Psychiatry, 161 (9):1558-1563, 2004.
Scopus
The ethics of research related to health care in developing countries***
McMillan,JR e Conlon, C. Journal of Medical Ethics, 30 (2): 204-206,
2004.

134
The Doctor's Duty to the Elderly Patient in Clínical Trials
Bayer, A., Fish, M. Drugs and Aging, 20 (15): 1087-1097,
2003.
Informed consent in pediatric research*
Harjaček, M. Paediatria Croatica, 47 (1): 15-18, 2003.
Ethical challenges of HIV clínical trials in developing countries***
Thomas, J. Bioethics; oct, 12(4): 320-7, 1998.
Pharmaceutical information systems and possible implementations of informed consent - developing and heuristic**,***
Ploug, T. e Holm, S.
BMC Medical Ethics, Nov 16: 13-30, 2012.
Tailored Medice: Whom will it fit? The ethics of patient and disease stratification***
Smart, A., Martin, P. e Parker, M.
Bioethics; 18 (4): 322 -343, 2004.
Tailor-made Pharmacotherapy: Future developments and Ethical Chanllenges in the Field of Pharmacogenomics***
Delden, JV., Bolt, I., Kalis, A., Derijk, J. e
Leufkens, H. Bioethics; Aug, 18 (4): 303 -321, 2004.
The pharmaceutical industry and the German National Socialist Regime: I.G. Farben and pharmacological research**
Lópes-Muñoz, F, García-García, P e Alamo, C.
Journal of Clínical Pharmacy and Theprapeutics, 34: 67-77, 2009.
The ethics of research related to health care in developing countries***
McMillan, JR e Conlon, C Journal of Medical Ethics, 30 (2): 204-206,
2004. The ethics of excluding women who become pregnant while participating in clínical trials of anti-epileptic medications***
Beran, R.G. Seizure, 15: 563-570, 2006.
Safeguarding Children´s right in psychopharmacological research: Ethical and legal issues***
Kolch, M; Ludolph, A.G; Plener, P.L;
Fangerau, H.; Vitiello, B; Fegert, J.M;
Current Pharmaceutical Design, 16 (22): 2398-2406, 2010.
BVS
Ethical, social and legal implications of pharmacogenomics: a critical review*,***
Moldrup, C. Community Genet; (4): 204-14, 2001.

135
Unpredictable Drug Shortages: An Ethical Framework for Short-Term Rationing in Hospitals**
Rosoff, PM. The American Journal of bioethics, v. 12, n.
1, 2012.
Avaliação da implantação da Clínica Ampliada e Equipes de referência em um serviço especializado em DST/AIDS, utilizando-se a triangulação de métodos**
Pinto, CAG. Tese de doutorado, FCM/UNICAMP, 2010.
Pharmaceutical information systems and possible implementations of informed consent - developing and heuristic**,***
Ploug, T. e Holm, S.
BMC Medical Ethics, Nov 16: 13-30, 2012.
Because we can: clashes of perspective over researcher obligation in the failed PrEP trials
Haire BG.
Dev World Bioeth. 2011 Aug;11(2):63-74,2011.
Ethical, legal, and social implications (ELSI) of microdose clínical trials.
Kurihara C.
Adv Drug Deliv Rev. Jun,19;63(7):503-10, 2011.
A magic bullet for the “African” mother? Neo-Imperial reproductive futurism and the pharmaceutical “solution” to the HIV/AIDS Crisis.
Booth KM.
Soc Polit., 17(3):349-78, 2010.
For-profit clínical trials in developing countries--those troublesome patient benefits*
Schuklenk U.
Am J Bioeth. Jun;10(6):52-4, 2010.
Safeguarding Children´s right in psychopharmacological research: Ethical and legal issues***
Kolch, M; Ludolph, A.G; Plener, P.L;
Fangerau, H.; Vitiello, B; Fegert, J.M;
Current Pharmaceutical Design, 16 (22): 2398-2406, 2010.
Pubmed
Ethics of clínical trials*,***
Iyalomhe,G.B., Imomoh, P.A Nigerian journal of medicine: journal of the National Association of Resident Doctors
of Nigeria, 16 (4):301-306, 2007.

136
Medical and ethical considerations of sham operation**
Antal J.
Magy Seb. 2007 Oct;60(5):233-8.
Justice and placebo controls
Hawkins JS.
Soc Theory Pract. Jul;32(3):467-96, 2006.
Restricted treatments, inducements and research participation.
Edwards SJ.
Bioethics. Apr; 20 (2):77-91, 2006.
HIV international clínical research: exploitation and risk.
Ballantyne A. Bioethics. 2005 Oct;19(5-6):476-91
Transnational medicine in public arenas: AIDS treatments in the South.**
Dodier N
Cult Med Psychiatry. Sep;29(3):285-307, 2005.
Physician, do no harm.**
Srinivasan S. Issues Med Ethics. Jan,6(1):22, 1998.
Defining standard of care in the developing world: the intersection of international research ethics and health systems analysis.**
Hyder AA, Dawson L.
Dev World Bioeth. May;5(2):142-52,2005.
Tailored Medice: Whom will it fit? The ethics of patient and disease stratification***
Smart, A., Martin, P. e Parker, M.
Bioethics; Aug, 18 (4): 322 -343, 2004.
Tailor-made Pharmacotherapy: Future developments and Ethical Challenges in the Field of Pharmacogenomics***
Delden, JV., Bolt, I., Kalis, A., Derijk, J. e
Leufkens, H. Bioethics; Aug, 18 (4): 303 -321, 2004.
The ethics of research related to health care in developing countries***
McMillan,JR e Conlon, C. Journal of Medical Ethics, 30 (2): 204-206,
2004.
Drug-testing on seropositive pregnant women in the developing world: moral and legal implications
Mayss A.
Med Law Int.;4(3-4):183-210, 2000.
Ethical, social and legal implications of pharmacogenomics: a critical review*,***
Moldrup, C. Community Genet; (4): 204-14, 2001.
Pubmed
The ethics of clínical research in developing countries
Brunet-Jailly J. IRB. Sep-oct; 21(5):8-11, 1999.

137
Exploitation and the ethics of clínical trials
Resnik DB. Am J Bioeth. Spring; 2(2):28-30, 2002.
After Helsinki: unresolved issues in international research**
Macklin R.
Kennedy Inst Ethics J. Mar;11(1):17-36, 2001.
I need a placebo like I need a hole in the head** Weijer C J Law Med Ethics. Spring;30(1):69-72,
2002.
Placebo surgery for Parkinson's disease: do the benefits outweigh the risks?
Clark PA.
J Law Med Ethics. Spring;30(1):58-68, 2002
The shrinking of human rights: the controversial revision of the Helsinki Declaration.
Botbol-Baum M.
HIV Med. Oct; 1(4):238-45, 2000.
Randomized placebo-controlled trials and HIV-infected pregnant women in developing countries. Ethical imperialism or unethical exploitation?
Zulueta P. Bioethics. Aug; 15(4):289-311, 2001.
Ethical challenges of HIV clínical trials in developing countries***
Thomas, J. Bioethics; oct, 12(4): 320-7, 1998.
Ethical complexities of conducting research in developing countries.
Varmus H, Satcher D.
N Engl J Med. 1997 Oct 2;337(14):1003-5.
Pubmed
Ethical issues associated with pain research in emergency medicine.
Sklar DP.
Ann Emerg Med. 1996 Apr;27(4):418-20.
Nota 1: *Artigo excluído por não estar disponível na base de dados.
Nota 2: ** Artigos excluídos da amostra por serem considerados inadequados ao objeto da pesquisa
Nota 3: *** Artigos em duplicidade nas diferentes bases.

138
APÊNDICE D
Apêndice D: Identificação dos demais medicamentos identificados na coleta de dados.
Medicamentos
Número de
identificação
DCB
Classificação dos antineoplásicos
pelo ATC index Código de identificação do estudo no Clínicaltrials.gov
Ácido folínico 00196 Não identificado NCT00439517
Aflibercepte 10019 Não identificado NCT00561470, NCT00519285 e NCT00532155
Bortezomibe 01335 Outros agentes antineoplásicos NCT00312845
Cabazitaxel 09820 Taxanos NCT00417079
Capecitabina 01686 Análogos da Pirimidina NCT00337103, NCT00080301, NCT00082433, NCT00373113,
NCT00678535 e NCT00069121
Carboplatina 01754 Compostos de Platina NCT00356525, NCT00363415, NCT00520676,
NCT00300885 e NCT00191854
Ciclofosfamida 02018 Análogos da mostarda Nitrogenada NCT00688740, NCT00486759 e NCT00117598
Cisplatina 02156 Compostos de Platina NCT00449033, NCT00678535, NCT00415194, NCT00148798,
NCT00191854 e NCT00565448
Cladribina 02193 Análogos da Purina NCT00117598
Clorambucila 02335 Análogos da mostarda Nitrogenada NCT00117598
Dasatinibe 09550 Inibidores da proteína quinase NCT00101647 e NCT00103844
Diaspartato de pasireotida
10620 Não identificado NCT00690430
Docetaxel 03167 Taxanos NCT00617669, NCT00520676, NCT00498797, NCT00565448,

139
NCT00283062,
NCT00532155 e NCT00688740
Doxorrubicina 03228 Antraciclinas e substâncias
relacionadas
NCT00113607, NCT00688740 e NCT00486759
Erlotinibe 03502 Inibidores da proteína quinase NCT00606502, NCT00550173, NCT00673049 e NCT00457392
Etoposídeo 03741 Derivados do Podofilotoxin NCT00363415
Fludarabina 04112 Análogos da Purina NCT00117598
Fluoruracila 04174 Análogos da Pirimidina
NCT00561470, NCT00154102, NCT00154102 NCT00457691,
NCT00565448 e NCT00688740, NCT00457691, NCT00069121,
NCT00439517, NCT00565448 e NCT00688740
Gencitabina 04419 Análogos da Pirimidina NCT00356525, NCT00449033, NCT00191854 e NCT00117598
Irinotecano 05051 Outros agentes antineoplásicos NCT00561470, NCT00154102 e NCT00457691
Ixabepilona 09614 Outros antibióticos citotóxicos NCT00080301 e NCT00082433
Leucovorin Não identificado Não identificado NCT00561470, NCT00154102, NCT00457691 e NCT00069121
Leuprolide acetate Não identificado Não identificado NCT00283062
Malato de sunitinibe 09465 Inibidores da proteína quinase NCT00373113, NCT00676650, NCT00457392, NCT00338884 e
NCT00083889
MDX-1379 Droga não identificada NCT00094653
Mesilato de eribulina 10068 Outros agentes antineoplásicos NCT00388726, NCT00337103
Mesilato de
imatinibe 04821 Inibidores da proteína quinase NCT00519090, NCT00124748 e NCT00103844

140
Mitoxantrona 06022 Antraciclinas e substâncias relacionadas
NCT00417079
Nilotinibe 09604 Inibidores da proteína quinase NCT00519090
Octreotida 06570 Não identificado NCT00690430
Oxaliplatina 06669 Compostos de Platina NCT00069121 e NCT00439517
Paclitaxel 06786 Taxanos NCT00300885 e NCT00191854
Pemetrexede 06897 Analógo de ácido fólico
NCT00356525, NCT00550173, NCT00363415, NCT00520676 e
NCT00415194
Pralatrexate Não identificado Analógo de ácido fólico NCT00606502
Prednisolona 07333 Não identificado NCT00498797
Prednisona 07341 Não identificado NCT00676650, NCT00417079 e NCT00486759
Tegafur 08347 Análogos da Pirimidina NCT00439517
Tensirolimo 09616 Inibidores da proteína quinase NCT00117598
Tosilato de
sorafenibe 09536 Inibidores da proteína quinase
NCT00449033, NCT00457691, NCT00863746, NCT00073307,
NCT00300885 e NCT00105443
Trabectedina 09648 Outras plantas alcalóides e produtos
naturais NCT00113607
Uracila 09016 Não identificado NCT00439517
Vandetanibe 09966 Inibidores da proteína quinase NCT00498797
Vincristina 09149 Analógos e alcaloídes da Vinca NCT00486759
Vinorelbina 09163 Analógos e alcaloídes da Vinca NCT00148798
Zibotentan Não identificado Não identificado NCT00617669, NCT00554229 e NCT00626548

141
REFERÊNCIAS
Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Medicamentos [acesso em 05 jan 2013] 2013a Disponível em: http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Esclarecimento sobre a posição da Anvisa quanto ao registro de medicamentos antineoplásicos novos. 2013 b. [acesso em 05 jan 2013] Disponível em: http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Interesse/Medicamentos+novos/Esclarecimento+sobre+a+posicao+da+Anvisa+quanto+ao+registro+de+medicamentos+antineoplasicos+novos Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Anvisa divulga perfil de pesquisa clínica de medicamentos no Brasil. 2011. [acesso em 31 de mai de 2013] Disponível em: http://portal.anvisa.gov.br/wps/portal/anvisa/anvisa/busca Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Como a Anvisa avalia o registro de medicamentos novos no Brasil, 2012a [acesso em 23 dec 2012] Disponível em: http://portal.anvisa.gov.br/wps/content/Anvisa+Portal/Anvisa/Inicio/Medicamentos/Assunto+de+Interesse/Medicamentos+novos/Como+a+Anvisa+avalia+o+registro+de+medicamentos+novos+no+Brasil. Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Posicionamento da Anvisa quanto ao registro de medicamentos novos considerados como me-toos, 2004 [11/06/04]. [acesso em 20 abr 2013] Disponível em: http://www.anvisa.gov.br/medicamentos/registro/metoos.htm
Agência Nacional de Vigilância Sanitária (Brasil) [Internet]. Notificação de Início e Término de Ensaios Clínicos conduzidos no Brasil, 2014 [24/02/2014] [acesso em 25 fev 2014] Disponível em: http://portal.anvisa.gov.br/wps/content/anvisa+portal/anvisa/inicio/medicamentos/assunto+de+interesse/pesquisa+clínica/notificacao+de+inicio+e+termino+de+ensaios+clínicos+conduzidos+no+brasil Agência Nacional de Vigilância Sanitária (Brasil). Resolução da Diretoria Colegiada nº 39 de 5 de junho de 2008. Aprova o regulamento para a realização de pesquisa clínica e dá outras providências, Diário Oficial da União de 06 jun 2008; Secção I. Agência Nacional de Vigilância Sanitária (Brasil). Resolução da Diretoria Colegiada nº 36 de 27 de junho de 2012. Altera a RDC nº 39, de 05 de junho de 2008, e dá outras providências, Diário Oficial da União de 28 jun 2012; Secção I. 2012 (b). Agência Nacional de Vigilância Sanitária (Brasil). Resolução nº55 de 16 de dezembro de 2010. Dispõe sobre o registro de produtos biológicos novos e produtos biológicos e

142
dá outras providências. Diário Oficial da União de 17 de dez de 2010; Secção I. Agência Nacional de Vigilância Sanitária (Brasil). Resolução da Diretoria Colegiada nº38, Anvisa de 12 de agosto de 2013. Dispõe sobre o regulamento para os programas de acesso expandido, uso compassivo e fornecimento de medicamento pós-estudo, 2013 (c). Diário Oficial da União de 13 de ago de 2013; Secção I. Ballantyne A. HIV international clínical research: exploitation and risk. Bioethics. 2005 Oct;19(5-6):476-91. Barreto VP. (coord) Dicionário da Filosofia do Direito. Justiça. Ed. Unisinos e Renovar. RS e RJ. 2006. Bayer A, Fish M. The Doctor's Duty to the Elderly Patient in Clínical Trials. Drugs and Aging 2003;20(15):1087-1097. Beauchamp T, Childress J. O respeito à autonomia. In: Princípios de Ética Biomédica. São Paulo: Edições Loyola; 2002 (a). p. 137-207. Beauchamp T, Childress J. Justiça. In: Princípios de Ética Biomédica. São Paulo: Edições Loyola; 2002 (b). p. 351- 422. Beauchamp T, Childress J. Tipos de teoria ética. In: Princípios de Ética Biomédica. Edições Loyola; 2002 (c). p. 59 - 135. Benatar S. Distributive justice and clínical trials in the third world. Review article. Theoretical Medicine, 2001;(22):169-171. Beran R.G. The ethics of excluding women who become pregnant while participating in clínical trials of anti-epileptic medications. Seizure, 2006;15(8):563-570. Booth KM. A magic bullet for the “African” mother? Neo-Imperial reproductive futurism and the pharmaceutical “solution” to the HIV/AIDS Crisis. Soc Polit, 2010;17(3):349-78. Brasil. Lei nº 5.991, de 17 de dezembro de 1973. Dispõe sobre o controle sanitário do comércio de drogas, medicamentos, insumos farmacêuticos e correlatos, e dá outras providências. Diário Oficial da União de 19 dez 1973 (Retificado em 21 dez 1973). Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº 01/88 de 13 de junho de 1988. Regulamenta o credenciamento de centros de pesquisa no país e recomenda a criação de um Comitê de Ética em Pesquisa em cada centro, 1988. Diário Oficial da União de 14 de jun 1988. Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº 170/95 de 09 de novembro de 1995. Define a formação de um Grupo Executivo de Trabalho para revisão da Resolução CNS 01/88, 1995. Diário Oficial da União de 24 nov de 1995;nº225. Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº

143
196/96 de 10 de outubro de 1996. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos, 1996. Diário Oficial da União de 16 de out de 1996;nº201. Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº 251 de 07 de agosto de 1997. Aprovar as seguintes normas de pesquisa envolvendo seres humanos para a área temática de pesquisa com novos fármacos, medicamentos, vacinas e testes diagnósticos, 1997. Diário Oficial da União de 23 de set 1997;nº183. Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº 301/00 de 16 de março de 2000. Contempla o posicionamento do CNS e CONEP contrário as modificações da Declaração de Helsinque, 2000. Brasil. Ministério da Saúde. Conselho Nacional de Saúde. Resolução MS/CNS nº 466/12 de 12 de dezembro de 2012. Aprova as diretrizes e normas regulamentadoras de pesquisas envolvendo seres humanos, 2013. Diário Oficial da União de 13 de jun de 2013. Brasil. Ministério da Saúde. Secretaria-Executiva. Área de Economia da Saúde e Desenvolvimento. Avaliação de tecnologias em saúde: ferramentas para a gestão do SUS, Brasília: Editora do Ministério da Saúde, 2009. Brasil. Ministério da Saúde. Portaria GM/MS n°2.123 de 07 de outubro de 2004. Aprova os regimentos internos dos órgãos do Ministério da Saúde. 2004 (b). Brasil. Ministério da Saúde. Portaria GM/MS n°2.439 de 08 de dezembro de 2005. Institui a Política Nacional de Atenção Oncológica, 2005. Diário Oficial da União de 09 de dez de 2005; Secção I, p.80-81. Brasil. Ministério da Casa Civil. Lei N° 10.973 de 02 dezembro de 2004. Dispõe sobre incentivos à inovação e à pesquisa cientifica e tecnológica no ambiente produtivo e dá outras providências, 2004 (a). Diário Oficial da União de 03 de dez de 2004;p.2. Brasil. Ministério da Ciência, Tecnologia e Inovação (MCTI). Marco legal da Inovação. Lei da Inovação. [s.d] [acesso em 23 abr 2013] Disponível em: http://www.mct.gov.br/index.php/content/view/8477.html Brasil. Manual das Denominações Comuns Brasileiras - DCB, Agência Nacional de Vigilância Sanitária. Brasília: Anvisa, 2013 (b). Botbol-Baum M. The shrinking of human rights: the controversial revision of the Helsinki Declaration. HIV Med. 2000 Oct;1(4):238-45. Cabral MML, Schindler HC, Abath FGC. Regulamentações, conflitos e ética da pesquisa médica em países em desenvolvimento. Rev. Saúde Pública, 2006;40(3):521-7. Callahan D. Equity and a steady-state medicine. In: Callahan D. False Hopes: Why america´s quest for perfect health is a recepe for failure. New York: Simon & Schuster; 1998. p.240-274.

144
Carter Z. US trade position protecting high drug prices blasted by UN agencies. Huffington Post 1 June 2012. [ acesso em 18 mai 2013]. Disponível em: http://www.huffingtonpost.com/2012/06/01/us-trade-drug-prices-un_n_1560481.html Chieffi AL e Barata RB. Judicialização da política de assistência farmacêutica e equidade. Cad. Saúde Pública, Rio de Janeiro 2009 Ago;25(8):1839-1849. Chieffi AL e Barata RB. Ações judiciais: estratégia da indústria farmacêutica para introdução de novos medicamentos. Rev. Saúde Pública 2010, 44(3):421-429.
Clark PA. Placebo surgery for Parkinson's disease: do the benefits outweigh the risks? J Law Med Ethics. Spring 2002;30(1):58-68. Clínical Trials, Trends, Charts and Maps [Internet]. [acesso em 07 fev 2013 e 27 fev 2014] Disponível em: www.clínicaltrials.gov/ct2/resources/trends#MapOfStudies Colley DR. Distributive Justice and clínical Trials in the third word. Theoretical medicine 2001; 22:151-167. Council for International Organizations of Medical Sciences (CIOMS). World Health Organization (WHO). International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva: 2002. Dainesi SM; Goldbaum, M. Pesquisa clínica como estratégia de desenvolvimento em saúde. Rev Assoc Med Bras 2012; 58(1):2-6. De Angelis C, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Clínical trial registration: a statement from the International Committee of Medical Journal Editors [Internet]. Ann Intern Med. 2004;141:477-8. Epub 2004 Sep 8. [acesso em 19 jan 2013] Disponível em: http://annals.org/article.aspx?articleid=717828 Departamento de Ciência e Tecnologia – Decit (Brasil). Ministério da Saúde. Da política à ação institucional de pesquisa no Ministério da Saúde. Rev. Saúde Pública 2006 (a);40(3):548-52. Departamento de Ciência e Tecnologia – Decit (Brasil). Ministério Da Saúde. Secretaria de Ciência, Tecnologia e Insumos estratégicos. Processo de definição de prioridades de pesquisa em saúde: a experiência Brasileira. Brasília – DF: 2006 (b). Edwards SJ. Restricted treatments, inducements and research participation. Bioethics 2006 Apr; 20(2):77-91. Escola Nacional de Saúde Pública Sérgio Arouca. [Internet]. Fiocruz. Anvisa adota série de medidas para agilizar registro de medicamentos. [25/03/2013]. [acesso em 28 fev 2014]. Disponível em: http://www6.ensp.fiocruz.br/visa/?q=node/5612 Ferreira ABH. Novo Dicionário Eletrônico Aurélio da Língua Portuguesa. São Paulo: Editora Positivo; 2004.

145
Freitas CBD. Bioética e Pesquisa com novos medicamentos. Com. Ciências e Saúde 2009;20 (1):75-78. Gadelha CAG, Quental C, Fialho BC. Saúde e inovação: uma abordagem sistêmica das indústrias da saúde. Cad. Saúde Pública, Rio de Janeiro 2003 jan-fev;19(1):47-59. Gava CM. Bermudez JAZ. Pepe VLE. Reis ALA. Novos medicamentos registrados no Brasil: podem ser considerados como avanço terapêutico? Ciência & Saúde Coletiva, 2010; 15(3):3403-3412. Gomes RP, Pimentel VP, Landim AB, Pieroni J. P. Ensaios clínicos no Brasil: competitividade internacional e desafios. Complexo Industrial da Saúde. BNDES. [s.d] Setorial 36, p.45-84. Garrafa V e Lorenzo C. Helsinque 2008: redução de proteção e maximização de interesses privados. Rev Assoc Med Bras 2009; 55(5): 497-520. Glickman SW, et al. Ethical and scientific implications of the globalization of clínical research. N Engl J Med 2009;(360):816-23. Haire BG. Because we can: clashes of perspective over researcher obligation in the failed PrEP trials. Dev World Bioeth 2011 Aug;11(2):63-74,2011. Hawkins JS. Justice and placebo controls. Soc Theory Pract 2006 Jul;32(3):467-96. International Conference on Harmonisation (ICH) of technical Requirements for registration of harmaceuticals for human Use. Tripartite guideline. Guideline for good clínical practice, version dated 10 june 1996. ICMJE. Uniform Requirements for Manuscripts Submitted to Biomedical Journals: Publishing and Editorial Issues Related to Publication in Biomedical Journals: Obligation to Register Clínical Trials.[s.d] [acesso em 19 jan 2013]. Disponível em: http://www.icmje.org/publishing_10register.html Instituto Nacional de Câncer José de Alencar Gomes da Silva. [Internet]. Ministério da Saúde. Estimativa, 2012. Incidência de Câncer no Brasil, 2011(a). [acesso em 13 abr 2013]. Disponível em: http://www.inca.gov.br/estimativa/2012 Instituto Nacional de Câncer José de Alencar Gomes da Silva. [Internet]. Ministério da Saúde. Estimativa, 2014. Incidência de Câncer no Brasil, 2014. [acesso em 02 mar 2014]. Disponível em: http://www.inca.gov.br/estimativa/2014/index.asp?ID=1 Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Alta tecnologia para o controle do câncer. Ciência. Revista Rede Câncer 2011(b) Dez;(16): 36-37. Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Notícias. Acordo Brasil-Cuba. 2010 (a). [acesso em 21 abr 2013]. Disponível em:

146
http://www2.inca.gov.br/wps/wcm/connect/acoes_programas/site/home/internacional/acordo_brasil_cuba/ Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Notícias [24/03/2010]. Brasil e Cuba vão desenvolver projetos na área de Oncologia. Seminário no Rio será o próximo passo. 2010 (b). [acesso em 21 abr 2013]. Disponível em: http://www2.inca.gov.br/wps/wcm/connect/agencianoticias/site/home/noticias/2010 Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Notícias [12/04/2013]. Governo lança Inova Saúde para promover inovação em medicamentos. [acesso em 21 abr 2013]. Disponível em: http://www2.inca.gov.br/wps/wcm/connect/agencianoticias/site/home/noticias/2013 Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Notícias [07/11/2013]. Ministério da Saúde fecha parceria para fabricação nacional de medicamentos contra o câncer. 2013 (b). [acesso em 14 nov 2013]. Disponível em: http://www2.inca.gov.br/wps/wcm/connect/agencianoticias/site/home/noticias/2013/ministerio_fecha_parceira_para_fabricacao_nacional_de_medicamentos_contra_cancer Instituto Nacional de Câncer José de Alencar Gomes da Silva. Ministério da Saúde. Atlas de mortalidade por Câncer. [acesso em 03 fev 2014]. Disponível em: http://mortalidade.inca.gov.br/Mortalidade/ Instituto Nacional da Propriedade Industrial. Ministério do desenvolvimento, indústria e comércio exterior. Resolução nº80/2013 de 19 de março de 2013. Resolução Disciplina a priorização do exame de pedidos de patente de produtos e processos farmacêuticos, bem como equipamentos e materiais relacionados à saúde pública. International Monetary Fund. [internet]. [s.d] [acesso em 02 mar 2014]. Disponível em: http://www.imf.org/external/ Brunet-Jailly J. The ethics of clínical research in developing countries. IRB 1999 Sep-oct; 21(5):8-11. Krauss Silva L. Avaliação tecnológica e análise custo-efetividade em saúde: a incorporação de tecnologias e a produção de diretrizes clínicas para o SUS. Ciência & Saúde 2003;8(2):501-520. Kölch M, Ludolph AG, Plener PL, Fangerau H, Vitiello B, Fegert J.M. Safeguarding children's rights in psychopharmacological research: Ethical and legal issues. Current Pharmaceutical Design 2010;16(22):2398-2406. Kottow, M. História da ética em pesquisa com seres humanos. RECIIS – R. Eletr. de Com. Inf. Inov. Saúde. Rio de Janeiro, Dez 2008;(2),Sup.1, Sup.7-Sup.18. Kunz R, Oxman AD. The unpredictability paradox: review of empirical comparisons of randomised and non-randomised clínical trials. BMJ Oct 31, 1998;317(7167):1185–1190. Kurihara C. Ethical, legal, and social implications (ELSI) of microdose clínical trials.

147
Adv Drug Deliv Rev 2011 Jun;63(7):503-10. Lima J.S. Reza D de La. Teixeira S. Costa C. Pesquisa clínica: fundamentos, aspectos éticos e perpectivas. Revista da SOCERJ 2003 Out/nov/Dez: 225-233. Lousana, G (org). Pesquisa Clínica na Brasil. Rio de Janeiro: Ed. Revinter; 2002. Love, J. Balancing options for health research and development. Bull World Health Organ 2012 November;90(11):796–796A. Machado KK, Katz A, Buyse M, Saad ED. Sobrevida global e outros desfechos clínicos em câncer de mama: situação atual e controvérsias. Rev Assoc Med Bras 2010; 56(5): 493-516. Maio, G. Research ethics and the principle of justice as fairness, a restatement. Theoretical Medicine 2003;(24):395-406. Mayss A. Drug-testing on seropositive pregnant women in the developing world: moral and legal implications. Med Law Int 2000;4(3-4):183-210. McMillan, JR e Conlon, C. The ethics of research related to health care in developing countries. Journal of Medical Ethics 2004;30(2):204-206. National Institutes of Health. NIH/OD/OER/OEP. Glossary of Terms For Human Subjects Protection And Inclusion Issues. [25 abr 2001]. [acesso em 19 jan 2013]. Disponível em: http://grants.nih.gov/grants/peer/tree_glossary.pdf National Institutes of Health. Setting Research Prioritues at the Nacional Institutes of Health. Bethesda, MD; 1998. National Institutes of Health. [Internet]. FDAAA - Further Resources for NIH Grantees. [s.d] [acesso em 28 fev 2014] Disponível em: http://grants.nih.gov/ClínicalTrials_fdaaa/faq.htm#1211 National Institute for Health Care Management (NIHCM). Changing patterns of pharmaceutical innovation; 2002. Nuremberg, Código de Nuremberg; 1947. OPAS, Organização Pan-Americana da saúde. IV Conferência Pan-Americana para Harmonização da regulamentação farmacêutica. Boas práticas clínicas: documento das americas; 2005. Oquendo, M.A., Stanley, B., Ellis, S.P., Mann, J.J. Protection of human subjects in intervention research for suicidal behavior. American Journal of Psychiatry 2004;161(9):1558-1563. Osorio-de-castro, C.G.S. Esher, A. Chaves, G.C. Pesquisa Clínica. In: Comitês de Ética em Pesquisa: Teorias e práticas. Ed. Fiocuz; 2012:179-208.

148
Ottawa Statement on Trial Registration. The Ottawa Statement, Part One: Principles for international registration of protocol information and results from human trials of health-related interventions; 2005. [acesso em 05 jan 2013]. Disponível em: http://ottawagroup.ohri.ca/index.html Organização Mundial de Saúde. World Health Organization. International Clínical Trials Registry Platform (ICTRP). How to Register a Trial; [s.d]. [acesso em 05 jan 2013]. Disponivel em: http://www.who.int/ictrp/trial_reg/en/index1.html Owonikoko, T.K. Unholding the principles of Autonomy, Beneficence and Justice in Phase I Clínical Trials. The Oncologist 2013;(18):242-244. Perelman, C. A Justiça. Primeira parte: A ética. In: Ética e direito. São Paulo: Martins Fontes; 2005. Pharmaceutical Research and Manufacturers of America (Phrma). [Internet]. The biopharmaceutical Pipeline: Evolving Science, Hope for Patients. Washington -DC: 2013(a) Jan. [acesso em 08 jul 2013]. Disponivel em: http://www.phrma.org/sites/default/files/pdf/phrmapipelinereportfinal11713.pdf Pharmaceutical Research and Manufacturers of America (Phrma). Cost of Cancer Medicines: What ABC News Missed. By Brewer J. [19 dec 2013(b)] [acesso em 02 mar 2014]. Disponível em: http://www.phrma.org/catalyst/what-abc-news-missed Pharmaceutical Research and Manufacturers of America (Phrma). Clínical Trials: The Phases of Drug Testing and Approval. [s.d] [acesso em 26 fev 2014]. Disponivel em: http://www.phrma.org/innovation/clínical-trials Pucca MB. Bertolini TB. Barbosa JE. Galina, SVR. Porto, GS. Therapeutic monoclonal antibodies: scFv patents as a marker of a new class of potential biopharmaceuticals. Braz. J. Pharm. Sci. 2011 Jan./Mar; 47(1): 31-39. Pullman D. Conflicting interests, Social Justice and proxy consent to research. Journal os Medicine and Philosophy 2002; 27(5):523-545. Quental C, Salles Filho S. Ensaios Clínicos: capacitação nacional para avaliação de medicamentos e vacinas. Revista Brasileira de Epidemiologia, 2006;9(4):408-24.
Rabelo Junior, LA. A justiça como equidade em Jonh Rawls. In: Âmbito Jurídico, Rio Grande, XIV, nov 2011; (94) [acesso em 30 mai 2013]. Disponível em: http://www.ambitojuridico.com.br/site/?n_link=revista_artigos_leitura&artigo_id=10755&revista_caderno=15
Rawls J. Parte I: Idéias fundamentais. In: Justiça como equidade: uma reformulação. São Paulo: Martins Fontes; 1996.p. 1-53. Registro Brasileiro de Ensaios Clínicos – ReBEC. [acesso em 13 fev 2013] e [acesso em

149
24 fev 2014]. Disponível em: http://www.ensaiosclínicos.gov.br/ Resnik, D.B. Setting Biomedical Research priorities: Justice, Science and Public Participation. Kennedy Institute of Ethics Journal 2001 June; l.11(2):181-2004. Resnik, D.B. Exploitation in biomedical research. Theoretical Medicine 2003;(24): 233-259. Resnik DB. Exploitation and the ethics of clínical trials. Am J Bioeth. Spring 2002; 2(2):28-30. Ribeiro CDM; Schramm FR. A necessária frugalidade dos idosos. Cad. Saúde Pública, Rio de Janeiro. set-out 2004;20(5):1141-1159. Rodrigues DG. Clínical Research and Drug Development in Latin America: Weighing the Pros and Cons, Talking about the Future. Journal of investigative Medicine 2007;(55):223-229. Stober M. Multinational Clínical Trials Breaking Language. Applied Clínical Trials Set 2003. [acesso em 27 jan 2013] Disponivel em: http://www.appliedclínicaltrialsonline.com/appliedclínicaltrials/data/articlestandard/appliedclínicaltrials/352003/68127/article.pdf nd Schramm FR; Escosteguy CC. Bioética e avaliação tecnológica em saúde. Cad. Saúde Pública, Rio de Janeiro, out-dez 2000;16(4):951-961. Schramm FR; Palácios M; Rego S; O modelo bioético principialista para a análise da moralidade da pesquisa científica envolvendo seres humanos ainda é satisfatório? Ciênc. saúde coletiva, Rio de Janeiro. Mar/Apr 2008;13(2):361-370. Sharif SK. ‘Letter to the Editor’. New England Journal of Medice. 1998 March 19;1.338(12). Sklar DP. Ethical issues associated with pain research in emergency medicine. Ann Emerg Med. 1996 Apr;27(4):418-20. Thomas, J. Ethical challenges of HIV clínical trials in developing countries. Bioethics 1998;12(4): 320-7. U.S. Department of Health and Human Services (HHS). [Internet]. The Belmont Report: Ethical principles and Guidelines for the protection of Human Subjects of Research, 1979. [acesso em 30 mai 2013]. Disponível em: http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html U.S. Department of Health and Human Services (HHS). Food and Drug Administration. [Internet]. Guidance for Industry E 10 Choice of Control Group and Related Issues in Clinical Trials. May, 2001. World Health Organization. Ministerial Summit on Health Research, 2004. [Internet]. [acesso em 05 jan 2013]. Disponível em: http://www.who.int/rpc/summit/en/

150
World Health Organization. Internacional Clínical trials Registry Platform, 2006. [Internet]. [acesso em 05 jan 2013]. Disponível em: http://www.who.int/ictrp/en/ World Health Organization. Cancer mortality and morbidity, 2008. Global Health Observatory. [scesso em 19 abr 2013]. Disponível em: http://www.who.int/gho/ncd/mortality_morbidity/cancer/en/ World Health Organization. Collaborating Centre for Drug Statistics Methodology. International language for drug utilization research: ATC/DDD [s.d.] [acesso em 20 dec 2012]. Disponível em: http://www.whocc.no/ World Medical Association. Declaration of Helsinki. Seoul: WMA, 2008. [acesso em 30 mai 2013]. Disponível em: http://www.wma.net/en/30publications/10policies/b3/index.html Van Delden J, Bolt I, Kalis A, Derijks J e Leufkens H. Tailor-made pharmacotherapy: future developments and ethical challenges in the field of pharmacogenomics. Bioethics 2004;18(4):304-321. Varmus H, Satcher D. Ethical complexities of conducting research in developing countries. N Engl J Med. Oct 2, 1997;337(14):1003-5.
Vieira, F.S.; Zucchi, P.; Distorções causadas pelas ações judiciais à política de medicamentos no Brasil. Revista de Saúde Pública 2007;41(2):214-222. Zulueta P. Randomised placebo-controlled trials and HIV-infected pregnant women in developing countries: Ethical imperialism or unethical exploitation? Bioethics Aug 2001;15(4):289-311.