non-interventional studies: europe (part 1) - chcuk ltd€¦ · non-interventional studies: europe...
TRANSCRIPT

Non-Interventional Studies: Europe (Part 1)Considerations when Managing and Conducting Non-Interventional Studies in Europe
Belgium • Czech Republic • France• Germany • Greece • Netherlands • Poland • Portugal • Spain • Sweden • Switzerland
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
1

Table of Contents
Publisher! 24
PUBLISHER 24
COPYRIGHT 24
Disclaimer! 25
NIS Definitions! 26
EUROPEAN NIS DEFINITIONS 26
NIS DEFINITIONS 26
Non-interventional Study (NIS) 26
Post-authorisation Safety Study (PASS) 27
Post-authorisation Efficacy Studies (PAES) 27
Common NIS Terminology! 28
COMMONLY USED NIS TERMS 28
COMMON NIS TERMINOLOGY 28
Introduction! 31
NIS REGULATIONS & GUIDELINES 31
EUROPE - GENERAL CONSIDERATIONS 31
Figure 1 - NIS Regulatory Road Map 33
ETHICAL CONSIDERATIONS 34
DATA PRIVACY 36
EFPIA CODE OF PRACTICE 36
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
1

COUNTRY-SPECIFIC CONSIDERATIONS 38
Country-Specific Considerations for Conducting and Managing NIS 38
NON-INTERVENTIONAL STUDIES - USEFUL LINKS 39
Study Classification! 42
GENERAL CONSIDERATIONS WHEN PLANNING NIS 42
STEP 1 - DETERMINE WHAT TYPE OF STUDY YOU INTEND TO CONDUCT 43
Is your Study Interventional? 43
Is your Study a Post-Authorisation Safety Study (PASS)? 43
Is your study a Non-Safety Related Non-Interventional Study? 44
STEP 2 - DETERMINE THE COUNTRY-SPECIFIC REQUIREMENTS 44
STUDY CLASSIFICATION - USEFUL LINKS 46
NIS Summary! 47
SUMMARY OF THE COUNTRY-SPECIFIC CONSIDERATIONS WHEN CONDUCTING NON-INTERVENTIONAL STUDIES 47
SUMMARY TABLE 47
Belgium! 55
COUNTRY-SPECIFIC CONSIDERATIONS 55
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 56
DEFINITIONS 57
Non-Interventional Study 57
"non-interventional trial" 57
Clarification 57
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 58
REGULATORY BODIES 60
Competent Authority 60
Competent Authority Approval 60
Research Ethics Committees (RECs) 60
Ethical Approval 61
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
2

Pharmaceutical Self-regulation Body (Pharma.be) 61
Data Protection Agency (CPP) 61
Notification 62
REGULATORY FRAMEWORK 63
Applicable Legislation and Guidance 63
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 65
Purpose 65
Prohibition of Promotion 65
Quality Framework 65
Contract 65
Declaration of Conflicts of Interest/ Financial Support 67
Market Research 67
Remuneration 67
Study Protocol 68
Procedures for the Supply of Medicines 68
Justification for the Number of Patients and Number of Investigators 68
Future Use of Data 68
Study Design and Follow-up 68
Limited Involvement of Medical Reps 69
Competent Authority Approval 69
Favourable Ethics Opinion 69
Procedure for Submission to Ethics Committees 69
Approval of Substantial Amendments 71
Attendance at Scientific Events 71
Advance Visa Procedure 71
End of Study Notification 72
Analysis and Retention of Study Results 72
Dissemination of Study Results 72
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
3

Notification of Important Risk-Benefit Information 72
Retrospective & ‘Other’ Non-Interventional Studies 73
Visa Application 73
Dossier Requirements for a Visa Application 73
Visa Approval Timeframe 74
Ethical Review of Retrospective NIS & Applicability of the LEHP 75
Informed Consent in the Context of Retrospective Non-interventional Studies 75
ADDITIONAL RESOURCES 77
Country-specific eLearning Module 77
Course Format 77
Who Would Benefit from the Course? 78
Further Information and/or Free Demonstration 78
BELGIUM - USEFUL LINKS 79
Czech Republic! 82
COUNTRY-SPECIFIC CONSIDERATIONS 82
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 83
DEFINITIONS 84
Non-Interventional Study 84
Non-Interventional Studies 84
Non-interventional Post-Marketing Safety Study 84
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 85
REGULATORY BODIES 86
Competent Authority 86
Competent Authority Notifiction 86
Research Ethics Committees (RECs) 87
Ethical Approval 87
Pharmaceutical Self-regulation Body (AIFP) 87
Data Protection Agency (UOOU) 88
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
4

DPA Notification 88
REGULATORY FRAMEWORK 89
Applicable Legislation and Guidance 89
CRITERIA FOR THE CONDUCT OF NIS 90
Purpose 90
Prohibition of Promotion 90
Scientific Service 90
Scientific Service (Body Responsible for Approval & Supervision of NIS) 90
Limited Involvement of Sales Reps 90
Competent Authority 91
Competent Authority Approval 91
Competent Authority Notification 91
Favourable Ethics Opinion 91
Contract 92
Financial Compensation/ Remuneration 92
Meetings Organised or Sponsored by the Pharmaceutical Industry 92
Clinical Research 93
Consultancy Services and Cooperation 95
Analysis and Retention of Study Results 95
Dissemination of Summary Reports 95
NIS Registration 96
NIS Registration with the AIFP Executive Director before Study Start 96
Notification of Important Risk-Benefit Information 96
COMPLIANCE WITH THE AIFP CODE 96
ADDITIONAL RESOURCES 99
Country-specific eLearning Module 99
Course Format 99
Who Would Benefit from the Course? 100
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
5

Further Information and/or Free Demonstration 100
Czech Republic - USEFUL LINKS 101
France ! 103
COUNTRY-SPECIFIC CONSIDERATIONS 103
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 104
DEFINITIONS 108
“Non-interventional Studies” 108
Non-Interventional Study 108
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 109
REGULATORY BODIES 110
Competent Authority 110
Competent Authority Approval 111
Research Ethics Committees (CPPs) 111
Ethical Approval 111
Pharmaceutical Self-regulation Body (LEEM) 111
Data Protection Agency (CCTIRS & CNIL) 112
CCTIRS & CNIL Approvals 112
National Medical Council (CNOM) 112
CNOM Approval 113
REGULATORY FRAMEWORK 114
Applicable Legislation and Guidance 114
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 115
Purpose 115
Prohibition of Promotion 115
Limited Involvement of Medical Reps 115
Study Protocol 115
Company Oversight 116
Remuneration 116
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
6

Use of Consultants 117
Contracts 118
Market Research 118
Authorisation by the Drug Competent Authority (ANSM) 118
Compliance with Data Protection Legislation 119
Authorisation by the Data Protection Competent Authorities (CCTIRS & CNIL) 119
CCTIRS Approval 119
CNIL Approval 120
Simplified Procedure: CNIL 120
Other Approvals (CNOM) 122
Favourable Ethics Opinion 122
Adverse Event Reporting 123
Study Results 123
Applicability to Other Types of Studies 124
ADDITIONAL RESOURCES 125
Country-specific eLearning Module 125
Course Format 125
Who Would Benefit from the Course? 126
Further Information and/or Free Demonstration 126
FRANCE - USEFUL LINKS 127
Germany! 130
COUNTRY-SPECIFIC CONSIDERATIONS 130
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 131
DEFINITIONS 134
Non-Interventional Trial 134
Non-Interventional Study 134
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 135
REGULATORY BODIES 137
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
7

Competent Authority 137
Competent Authority Notification 138
Research Ethics Committees (RECs) 138
Ethical Approval 138
Pharmaceutical Self-regulation Body (vfa) 139
Data Protection Agency (BFDI) 139
Notification 140
Other Notification Requirements 140
REGULATORY FRAMEWORK 141
Applicable Legislation and Guidance 141
BFARM GUIDANCE ON NIS 142
How are clinical trials using investigational medicinal products distinguished from open post-marketing observations? 142
Which documents are necessary for the submission of a non-interventional trial pursuant to Section 67 sub-section 6 of the German Medicines Act? 142
Is there an official form for notification of non-interventional studies? 143
When does a non-interventional trial in accordance with Section 67 sub-section 6 of the German Medicines Act have to be notified? 143
Is the non-interventional trial limited to a particular number of patients per doctor or to a particular number of doctors? 143
Is the Announcement on the Authorisation and Registration of Medicinal Products - Recommendations for Design and Conduct of Observational Post-authorisation Safety Studies dated 12 November 1998) still up-to-date? 143
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 144
Purpose 144
Prohibition of Promotion 144
Head of Medical Department/ Scientific Service (Body Responsible for Approval & Supervision of NIS) 144
Limited Involvement of Medical Reps 145
Guidelines for Medical Reps 145
Internal Procedures 148
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
8

Quality Assurance 148
Study Plan/ Protocol 148
Authorisation by the Competent Authority (BfArM) 149
Notifying the Competent Authority (BfArM) 149
Other Notification Requirements 149
Favourable Ethics Opinion 150
Contract 150
Remuneration 151
Compliance with Data Protection Legislation/ Need for Informed Consent 151
Registration of Clinical Studies 152
Invitation to Job-Related, Science-Oriented Training Events 152
Analysis and Retention of Study Results 154
Dissemination of Summary Reports 155
Notification of Important Benefit-Risk Information 155
ADDITIONAL RESOURCES 156
Country-specific eLearning Module 156
Course Format 156
Who Would Benefit from the Course? 157
Further Information and/or Free Demonstration 157
GERMANY - USEFUL LINKS 158
Greece ! 160
COUNTRY-SPECIFIC CONSIDERATIONS 160
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 161
DEFINITIONS 164
Non-Interventional/Pharmacoepidemiological Clinical Trial of a Marketed Medicinal Product 164
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 165
REGULATORY BODIES 167
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
9

Competent Authority 167
Competent Authority Approval 168
Research Ethics Committees (RECs) 168
Ethical Approval 169
Pharmaceutical Self-regulation Body (Pharma.be) 169
Data Protection Agency (CPP) 169
Notification 170
REGULATORY FRAMEWORK 171
Applicable Legislation and Guidance 171
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 172
Purpose 172
Prohibition of Promotion 172
Scientific Service (Body Responsible for Approval and Supervision of NIS) 172
Involvement of Medical Reps 172
Authorisation by the Competent Authority (EOF) 172
Favourable Ethics Opinion 173
Contract 173
Provision of Counseling Services or similar collaborations by Healthcare Professionals to the Pharmaceutical Industry 173
Declaration of Conflicts of Interest 175
Financial Compensation/ Remuneration 175
Compliance with Data Protection Legislation 175
Registration of NIS 176
Scientific Events 176
Scientific Content Conferences (Type A events) 176
Provisions for the organization of ‘Type A’ scientific events 176
Scientific Information Events (Type B events) 178
Scientific Information Events on medicinal or other products (Type C events) 178
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
10

Legality of doctors’ participation 178
Provisions for the organization of Type B and Type C scientific events 178
Determination of ceilings for hospitality expenses of physicians for scientific events in Greece and abroad 180
Scientific Advisory Boards 180
Analysis and Retention of Study Results 181
Dissemination of Study Results 181
Notification of Important Risk-Benefit Information 181
‘Other’ Studies 181
Market Research 182
ADDITIONAL RESOURCES 183
Country-specific eLearning Module 183
Course Format 183
Who Would Benefit from the Course? 184
Further Information and/or Free Demonstration 184
GREECE - USEFUL LINKS 185
The Netherlands! 187
COUNTRY-SPECIFIC CONSIDERATIONS 187
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 188
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 189
REGULATORY BODIES 190
Competent Authority 190
Competent Authority Approval 191
Research Ethics Committees (METC) 191
Ethical Approval 191
Pharmaceutical Self-regulation Body (Nefarma) 192
CGR Approved Review Process for Non-WMO Research 192
Data Protection Agency (DPA) 192
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
11

Notification 192
REGULATORY FRAMEWORK 193
Applicable Legislation and Guidance 193
INVOLVEMENT OF HUMAN SUBJECTS IN SCIENTIFIC RESEARCH 194
WMO RESEARCH 194
Is the study Medical/scientific research? 194
Does the research involve subjecting people to treatment/ procedures or will it require them to follow specific rules of behaviour? 195
APPROVAL REQUIREMENTS FOR WMO RESEARCH 196
Protocol 196
Protocol Amendments 197
Patient Information 197
Consent Form 198
Independent Physician 199
Insurance for Research Subjects 199
Notification of Study Start 200
Progress Reports 200
End of Study Notification 200
Final Report 200
SCIENTIFIC RESEARCH USING PERSONAL DATA (NON-WMO) 201
SEPARATE REVIEW OF STUDIES NOT SUBJECT TO THE WMO (NON-WMO) 202
CGR Review Process for non-WMO Research Studies 202
IF IN DOUBT ABOUT THE STUDY CLASSIFICATION... 202
We are currently setting up a phase IV drugs trial. The drug is already registered. Does the trial still have to be reviewed? 203
Our research consists simply of a questionnaire that the subjects have to complete. Is it covered by the WMO? 203
What about clinical drugs trials that are not covered by the WMO, surely they have to undergo some kind of review? 203
SEPARATE REVIEW OF DRUG TRIALS NOT SUBJECT TO THE WMO 204
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
12

ARTICLE 16 OF THE CGR CODE OF CONDUCT: NON-WMO REALED RESEARCH 204
DETAILED SUPPLEMENT TO ARTICLE 16 OF THE CGR CODE OF CONDUCT 204
Introduction 204
CGR guidelines for research not subject to WMO 205
Demarcation and definition 205
Coverage 206
Requirements for all types of “research not subject to the WMO” 206
Assessment of Non WMO related research 207
EXPLANATION OF FURTHER DETAILS OF ARTICLE 16 208
General Explanation 208
Facts and backgrounds 210
Possible areas of tension 211
Explanation article by article 212
DIRECTIVES FOR INTERNAL PROCEDURE FOR ASSESSING RESEARCH NOT SUBJECT TO THE WMO 214
Ad 1. Which activities must be assessed? 214
Ad 2. Who is performing the assessment? 215
Ad 3. Criteria 216
Other 217
ADDITIONAL RESOURCES 218
Country-specific eLearning Module 218
Course Format 218
Who Would Benefit from the Course? 219
Further Information and/or Free Demonstration 219
THE NETHERLANDS - USEFUL LINKS 220
Poland! 222
COUNTRY-SPECIFIC CONSIDERATIONS 222
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 223
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
13

DEFINITIONS 224
Non-interventional Studies 224
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 225
REGULATORY BODIES 227
Competent Authority 227
Competent Authority Approval 227
Research Ethics Committees (RECs) 228
Ethical Approval 228
Pharmaceutical Self-regulation Body (INFARMA) 229
Data Protection Agency (GIODO) 229
Notification 229
REGULATORY FRAMEWORK 231
Applicable Legislation and Guidance 231
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 232
Scientific Objective 232
NIS Criteria 232
TRANSPARENCY OF STUDIES 233
Prohibition of Promotion 233
Prohibition of Using NIS to Compare Medicinal Products 233
NIS TO BE CONDUCTED IN LINE WITH THE PROTOCOL 233
PRIOR ETHICS APPROVAL 233
RESPONSIBILITIES OF THE MEDICAL DEPARTMENT 233
CONTRACTS 234
The obligation to conclude a contract for financing a trial/study 234
Remuneration 234
Services Rendered by Health Care Centres 234
Employing Consultants 235
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
14

PRINCIPLES OF ORGANISING SYMPOSIA, CONGRESSES AND OTHER MEETINGS 236
Objectivity criteria for selection of meetings participants 236
Venues of meetings 236
Hospitality 236
What rules govern the offering of hospitality to health professionals? Does it make a difference if the hospitality offered to those health professionals will take place in another country? 237
Is it possible to pay for a doctor in connection with attending a scientific meeting? If so, what may be paid for? Is it possible to pay for his expenses (travel, accommodation, enrolment fees)? Is it possible to pay him for his time? 237
To what extent will a pharmaceutical company be held responsible by the regulatory authorities for the contents of and the hospitality arrangements for scientific meetings, either meetings directly sponsored or organised by the company or independent meetings in respect of which a pharmaceutical company may provide sponsorship to individual doctors to attend? 238
Is it possible to pay doctors to provide expert services (e.g. participating in focus groups)? If so, what restrictions apply? 238
Is it possible to pay doctors to take part in post marketing surveillance studies? What rules govern such studies? 238
Is it possible to pay doctors to take part in market research involving promotional materials? 239
Is there a requirement in law and/or self-regulatory code for companies to make publicly available information about donations, grants, benefits in kind or any other support provided by them to health professionals, patient groups or other institutions? If so, what information should be disclosed, from what date and how? 239
STUDY REGISTRATION 239
Publishing information about a trial/study start-up 239
Considerations Studies that haven’t been Registered 240
MEDICAL SALES REPRESENTATIVES 241
The Role of Medical Reps in Non-interventional Studies 241
END OF STUDY OBLIGATIONS 241
Analysis and Presentation of Study Results 241
Communication of Summary Results to Participating Researchers 241
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
15

Communication to the Competent Authority of Results Significant for the Assessment f the Product 241
Archiving 242
CONDITIONS FOR CONDUCTING OTHER STUDIES 242
Epidemiological Trials, Registers and Health Economic Studies 242
Market Research 242
ADDITIONAL RESOURCES 243
Country-specific eLearning Module 243
Course Format 243
Who Would Benefit from the Course? 244
Further Information and/or Free Demonstration 244
POLAND - USEFUL LINKS 245
Portugal ! 247
COUNTRY-SPECIFIC CONSIDERATIONS 247
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 248
DEFINITIONS 249
Non-interventional Studies (NIS) 249
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 250
REGULATORY BODIES 251
Competent Authority 251
Competent Authority Approval 251
Research Ethics Committees (RECs) 252
Ethical Approval 252
Pharmaceutical Self-regulation Body (Pharma.be) 252
Data Protection Agency (CNPD) 252
CNPD Notification 253
CNPD Approval 253
REGULATORY FRAMEWORK 254
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
16

Applicable Legislation and Guidance 254
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 255
Purpose 255
Prohibition of Promotion 255
Data Protection 255
Compliance with the Apifarma Code of Ethics 255
Study Design & Follow-Up 255
Limited Involvement of Medical Reps 256
Favourable Ethics Opinion 256
Analysis and Retention of Study Results 256
Dissemination of Summary Reports 256
Notification of Important Benefit-Risk Information 257
Other NIS 257
ADDITIONAL RESOURCES 258
Country-specific eLearning Module 258
Course Format 258
Who Would Benefit from the Course? 259
Further Information and/or Free Demonstration 259
PORTUGAL - USEFUL LINKS 260
Spain! 262
COUNTRY-SPECIFIC CONSIDERATIONS 262
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 263
DEFINITIONS 265
Post-Authorization Studies (PAS) 265
Observational Study 265
Post-Authorization Observational Studies 265
REGULATORY CLASSIFICATION 266
Post-Authorization Observational Studies 266
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
17

REGULATORY SUBMISSION ROADMAPS 267
Post-Authorisation Studies (EPA) 267
Prospective Observational Studies (EPA-SP) 268
Retrospective Observational Studies (EPA-OD) and Disease Registries (Non-EPA) 269
Post-Approval Commitments (EPA-LA) 270
Publicly Funded Prospective Observational Studies (EPA-AS) 271
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 272
REGULATORY BODIES 274
Competent Authority 274
Competent Authority Approval 275
Competent Authority Classification 275
Research Ethics Committees (RECs) 275
Ethical Approval 276
Pharmaceutical Self-regulation Body (Pharma.be) 276
Data Protection Agency (CPP) 276
Notification 276
REGULATORY FRAMEWORK 278
Applicable Legislation and Guidance 278
NIS OBJECTIVES 279
ETHICAL CONSIDERATIONS 279
STUDY PROTOCOL 281
Identification of those Responsible for the study 281
Sponsor 281
Monitor 282
Researchers 282
Research Coordinator 282
Key Elements of the Study Protocol 283
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
18

Study Identification 285
Protocol Amendments 286
OPERATIONAL REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 287
Ethical Considerations 287
Notification of Start Date 287
Progress Reports 287
Monitoring 287
Study Protocol 287
Study Identification 288
CEIC Approval of Substantial Amendments 288
Notification of Related Serious Adverse Events 289
Inspections 289
Final Report 289
Archiving 289
BEST PRACTICE REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 290
Purpose 290
Prohibition of Promotion 290
Registration of the Study with the AEMPS 290
Contract 290
Services Provided by Healthcare Professional 291
Remuneration 293
Competent Authority Approval (AEMPS) 293
Classification of the Study by the AEMPS 293
Autonomous Community Approval 293
Favourable Ethics Opinion 294
Insurance 294
Protocol Amendments 294
Notification & Reporting 294
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
19

Archiving of Study Documents 295
Appointment of an Archivist 295
Dissemination of Summary Reports 295
Notification of Important Benefit-Risk Information 296
Safety Reporting 296
ADDITIONAL RESOURCES 297
Country-specific eLearning Module 297
Course Format 297
Who Would Benefit from the Course? 298
Further Information and/or Free Demonstration 298
Spain - USEFUL LINKS 299
Sweden! 302
COUNTRY-SPECIFIC CONSIDERATIONS 302
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 303
DEFINITIONS 304
Non-Interventional Study 304
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 305
REGULATORY BODIES 307
Competent Authority 307
Competent Authority Approval 307
Research Ethics Committees (RECs) 307
Ethical Approval 308
Pharmaceutical Self-regulation Body (LIF) 308
Data Protection Agency (Data Inspection Board) 308
Notification 309
Notification need not be made if there is a Personal Data Representative 309
Personal Data that are Handled within the Scope of Clinical Trials 309
REGULATORY FRAMEWORK 311
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
20

Applicable Legislation and Guidance 311
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 312
THE DIFFERENCE BETWEEN CLINICAL TRIALS & NON-INTERVENTIONAL STUDIES 313
WHEN ARE NON-INTERVENTIONAL STUDIES PERFORMED? 314
CRITERIA FOR NON-INTERVENTIONAL STUDIES 315
Standard Healthcare Provision 315
Purpose 316
Prohibition of Promotion 316
Limited Involvement of Medical Reps 316
Considerations when Engaging Healthcare Personnel in Research 317
Limited Market Research Studies 318
Contracts and Agreements 319
Ownership of data 319
Conditions for clinical trials/non-interventional studies that has been initiated by a pharmaceutical company 319
Financial Compensation/ Remuneration 322
Ethics Approval 322
The Swedish Medical Products Agency 322
Study Plan/ Protocol 322
The Company’s Internal Process 323
Company Approval and Monitoring of Non-Interventional Studies 323
Quality Assurance 324
The Swedish Data Protection Act (PUL) 324
Notification Duty 324
Notification need not be made if there is a Personal Data Representative 324
Reports 325
Industry Self-Regulation 325
Reporting of Results Likely to have an Impact on the Drugs Risk Profile 325
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
21

Publications 325
ADDITIONAL RESOURCES 326
Country-specific eLearning Module 326
Course Format 326
Who Would Benefit from the Course? 327
Further Information and/or Free Demonstration 327
sweden - USEFUL LINKS 328
Switzerland ! 330
COUNTRY-SPECIFIC CONSIDERATIONS 330
SUMMARY OF CHANGES SINCE PREVIOUS VERSION 331
DEFINITIONS 332
Non-interventional Studies using Authorized Medicinal Products 332
VKlin or Non-VKlin Studies 332
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS 334
REGULATORY BODIES 335
Competent Authority 335
Competent Authority Approval 335
Research Ethics Committees (RECs) 336
Ethical Approval 336
Pharmaceutical Self-regulation Body (SGCI) 337
Data Protection Agency (CPP) 337
Notification/Registration 337
REGULATORY FRAMEWORK 338
Applicable Legislation and Guidance 338
REGULATION OF RESEARCH INVOLVING HUMAN SUBJECTS 339
Regulations at the Swiss Federal Level 339
The Federal Law on Federal Law on Research on Humans 341
Chronology and Status of the Federal Law on Research on Humans 341
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
22

Cantonal Regulations 342
Human Research Requirements 342
Cantonal Ethics Commissions (CECs) 343
Cantonal Ethics Commission Submission Guidelines (Zurich CEC Example) 343
REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES 343
SGCI Code of Practice 343
PROSPECTIVE NON-INTERVENTIONAL STUDIES 343
Prohibition of Promotion 344
Scientific Service (Body Responsible for Approval & Supervision of NIS) 344
Limited Involvement of Medical Reps 345
Competent Authority Approval 345
Favourable Ethics Opinion 346
Ethics Approval Process - Multi-Centre Studies 346
Contract 346
Compliance with Data Protection Legislation 347
Compliance with Data Protection Legislation/ Need for Informed Consent 347
Insurance 347
Evidence of GCP Training 348
Safety Reporting 348
Dissemination of Study Reports 348
ADDITIONAL RESOURCES 349
Country-specific eLearning Module 349
Course Format 349
Who Would Benefit from the Course? 350
Further Information and/or Free Demonstration 350
switzerland - USEFUL LINKS 351
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
23

PublisherPUBLISHER
Published by Compliance Healthcheck Consulting UK Ltd
Address: Kylen, Craigdarroch Drive, Contin, Ross-Shire, IV14 9EL
Tel: +44 (0)1997 42 33 11
Email: [email protected]
Website: www.chcuk.co.uk
3rd Edition, September 2012
ISBN 978-0-9563319-4-6
COPYRIGHT
Copyright © 2012 Dr Stuart McCully. All rights reserved.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
24

DisclaimerAlthough this Compilation contains information of a legal nature, it has been developed for
informational purposes only and does not constitute legal advice or opinions as to the current
operative laws, regulations, or guidelines of any jurisdiction. In addition, because new
standards are issued on a continuing basis, this Compilation is not an exhaustive source of all
current applicable laws, regulations, and guidelines relating to market health research. While
reasonable efforts have been made to assure the accuracy and completeness of the information
provided, researchers and other individuals should check with local authorities and/or
research ethics committees before starting research activities.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
25

NIS DefinitionsConsiderations when providing access to unapproved drugs
EUROPEAN NIS DEFINITIONS
NIS DEFINITIONS
Non-interventional Study (NIS)
A non-interventional study is a study fulfilling
cumulatively the following requirements:
The medicinal product is prescribed in the
usual manner in accordance with the terms of
the marketing authorisation;
The assignment of the patient to a particular therapeutic strategy is not decided in
advance by a trial protocol but falls within current practice and the prescription of the
medicine is clearly separated from the decision to include the patient in the study; and
No additional diagnostic or monitoring procedures are applied to the patients and
epidemiological methods are used for the analysis of collected data.
Non-interventional studies are defined by the methodological approach used and not by the
scientific objectives. Non-interventional studies include database research or review of
records where all the events of interest have already happened (e.g. case-control, cross-
sectional and cohort studies). Non-interventional studies also include those involving primary
data collection (e.g. prospective observational studies and registries in which the data
collected derive from routine clinical care), provided that the conditions set out above are
met.
In this context, interviews, questionnaires and blood samples may be performed as normal
clinical practice.
(as per Annex I of the EMA Guideline on good pharmacovigilance practices (GVP), 2012)
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
26

Post-authorisation Safety Study (PASS)
Any study relating to an authorised medicinal product conducted with the aim of identifying,
characterising or quantifying a safety hazard, confirming the safety profile of the medicinal
product, or of measuring the effectiveness of risk management measures (Article 1(c)(15) as
2001/83/EC as amended by Directive 2010/84/EU)
Post-authorisation Efficacy Studies (PAES)
Any study conducted where concerns relating to some aspects of the efficacy of the medicinal
product are identified and can only be resolved after the medicinal product has been marketed
(Article 21(a) as 2001/83/EC as amended by Directive 2010/84/EU)
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
27
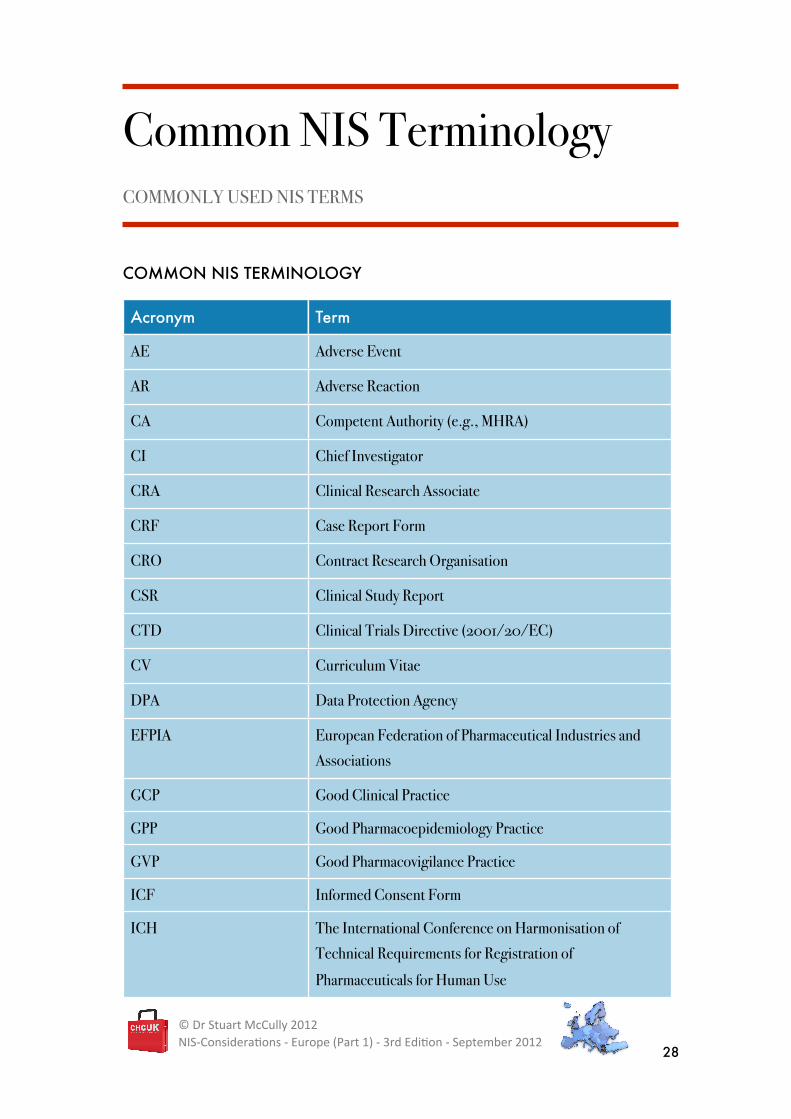
Common NIS TerminologyCOMMONLY USED NIS TERMS
COMMON NIS TERMINOLOGY
Acronym Term
AE Adverse Event
AR Adverse Reaction
CA Competent Authority (e.g., MHRA)
CI Chief Investigator
CRA Clinical Research Associate
CRF Case Report Form
CRO Contract Research Organisation
CSR Clinical Study Report
CTD Clinical Trials Directive (2001/20/EC)
CV Curriculum Vitae
DPA Data Protection Agency
EFPIA European Federation of Pharmaceutical Industries and
Associations
GCP Good Clinical Practice
GPP Good Pharmacoepidemiology Practice
GVP Good Pharmacovigilance Practice
ICF Informed Consent Form
ICH The International Conference on Harmonisation of
Technical Requirements for Registration of
Pharmaceuticals for Human Use
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
28
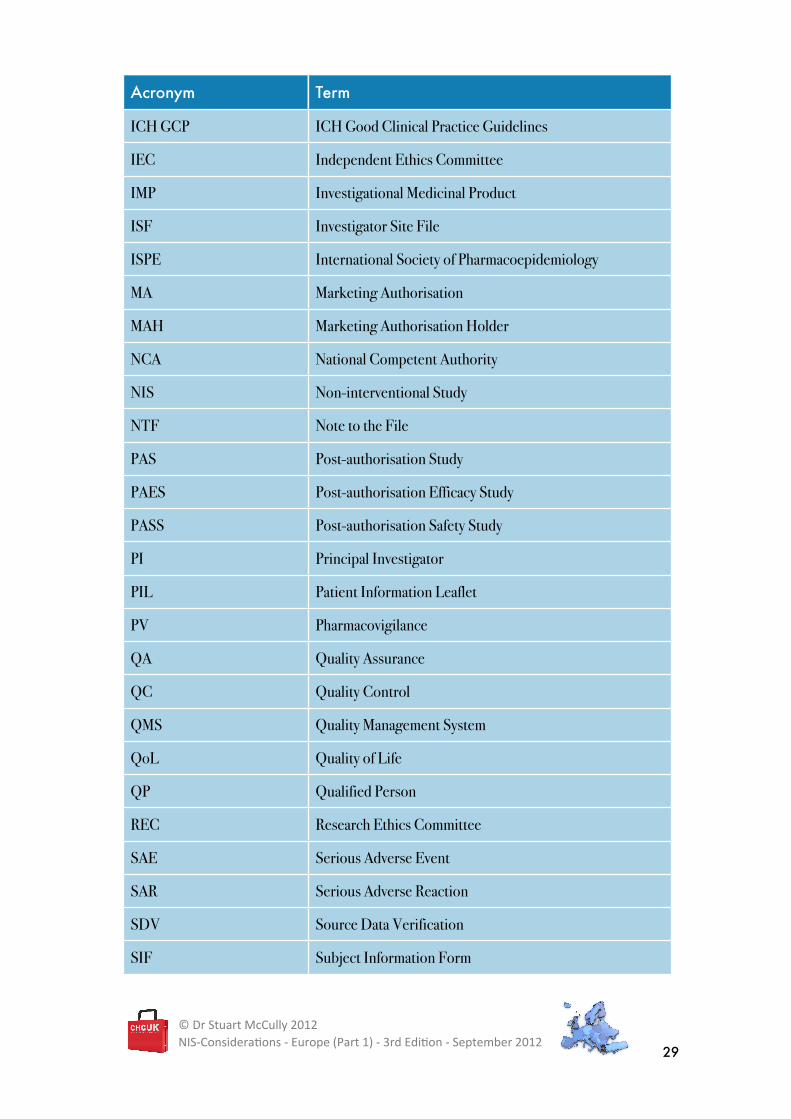
Acronym Term
ICH GCP ICH Good Clinical Practice Guidelines
IEC Independent Ethics Committee
IMP Investigational Medicinal Product
ISF Investigator Site File
ISPE International Society of Pharmacoepidemiology
MA Marketing Authorisation
MAH Marketing Authorisation Holder
NCA National Competent Authority
NIS Non-interventional Study
NTF Note to the File
PAS Post-authorisation Study
PAES Post-authorisation Efficacy Study
PASS Post-authorisation Safety Study
PI Principal Investigator
PIL Patient Information Leaflet
PV Pharmacovigilance
QA Quality Assurance
QC Quality Control
QMS Quality Management System
QoL Quality of Life
QP Qualified Person
REC Research Ethics Committee
SAE Serious Adverse Event
SAR Serious Adverse Reaction
SDV Source Data Verification
SIF Subject Information Form
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
29
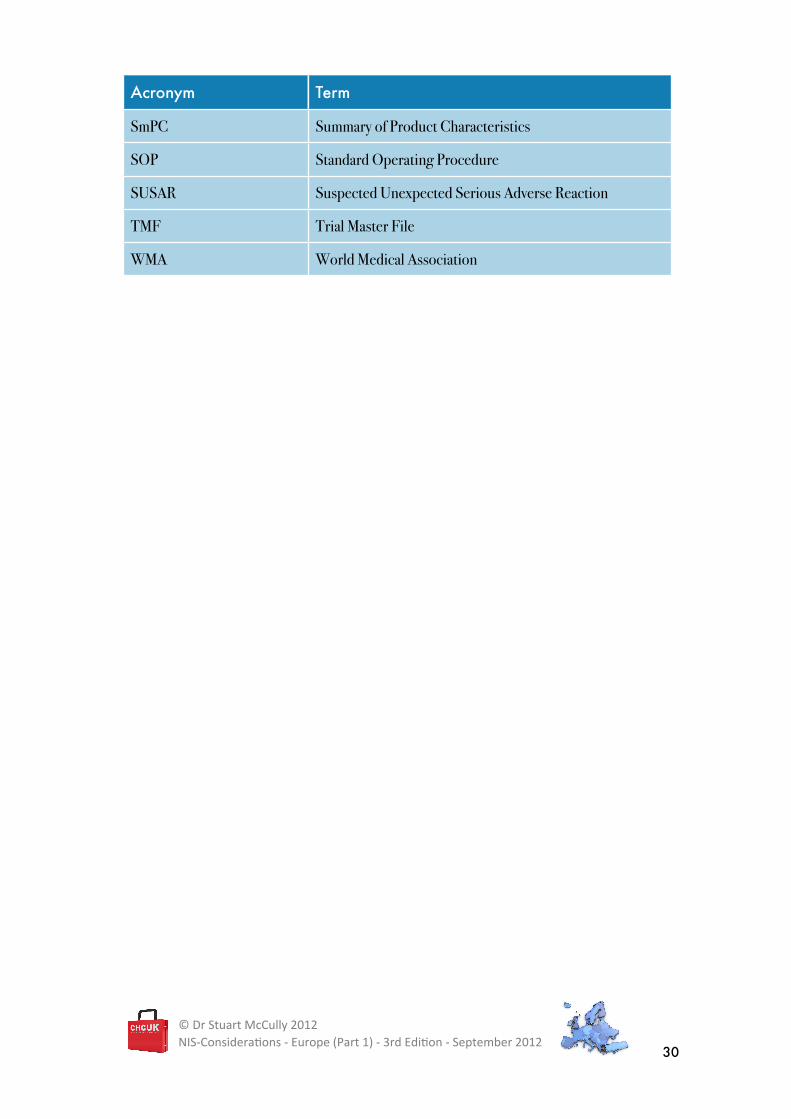
Acronym Term
SmPC Summary of Product Characteristics
SOP Standard Operating Procedure
SUSAR Suspected Unexpected Serious Adverse Reaction
TMF Trial Master File
WMA World Medical Association
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
30

Study ClassificationGENERAL CONSIDERATIONS WHEN PLANNING NIS
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
42
It Depends!
What are the regulatory
requirements for my NIS?

STEP 1 - DETERMINE WHAT TYPE OF STUDY YOU INTEND TO CONDUCT
Before starting your study you first need to determine what type of study it is:
Is your Study Interventional?
Is your study an interventional clinical trial?
➡ Refer to the Decision tree
Is your Study a Post-Authorisation Safety Study (PASS)?
Your study is a PASS if the objectives include at least one of the following conditions:
1. Characterization of the safety profile (e.g. identifying the most frequent adverse
reactions in a large population over time);
2. Providing reassurance about the absence of a safety concern related to a specific
adverse reaction;
Study !Classification!
Is#your#study#an#interven/onal#clinical#trial?#
Regulatory !Framework!
Interventional?!
PASS?!
Other NIS?!
Clinical Trials Directive !(2001/20/EC)!
Pharmacovigilance Directive !
(2010/84/EU)!
Country Specific!!
Is#your#study#a#post4authoriza/on#safety#study#(PASS)?#
Is#your#study#a#non4safety#related#non4interven/onal#study?#
��
��
��
��
��
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
43

3. Investigating potential or identified risks, e.g. to characterize the incidence rate,
estimate the rate ratio or rate difference in comparison to a non-exposed population
and investigate risk factors and effect modifiers;
4. Evaluating risks of a product used in authorised indications by patients groups not
studied in the pre-authorisation phase (e.g. pregnant woman, elderly patient);
5. Assessing patterns of drug utilisation and use of the product that may have an impact on
its safety (e.g. co-medication, medication errors);
6. Evaluating the effectiveness of a risk mitigation activity (e.g. drug utilisation study,
patient or physician survey)
Furthermore, if your study falls within the scope/definition of a PASS and it isn’t a study that
has been required by a Competent Authority as part of your Marketing Authorisation then it is
a ‘voluntary’ PASS. If you are the study sponsor you will need to determine, and document,
what your policy is with regards to the notification and reporting considerations for these
voluntary PASS. The EMA encourages Companies to notify and register these study in the
same way as Obligatory PASS. However, this is currently a business decision.
Is your study a Non-Safety Related Non-Interventional Study?
If so, the requirements are not standardised and you will need to determine the country-
specific considerations.
STEP 2 - DETERMINE THE COUNTRY-SPECIFIC REQUIREMENTS
The regulatory requirements for ‘Other’ NIS are very much dependent on a number of factors
(see below), which is why there isn’t generally a single answer to the question, “what are the
regulatory requirements for my non-interventional study?”
There are no short cuts. You will need to verify the country-specific regulations and
considerations for each of the countries where you intend to conduct your non-interventional
study.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
44
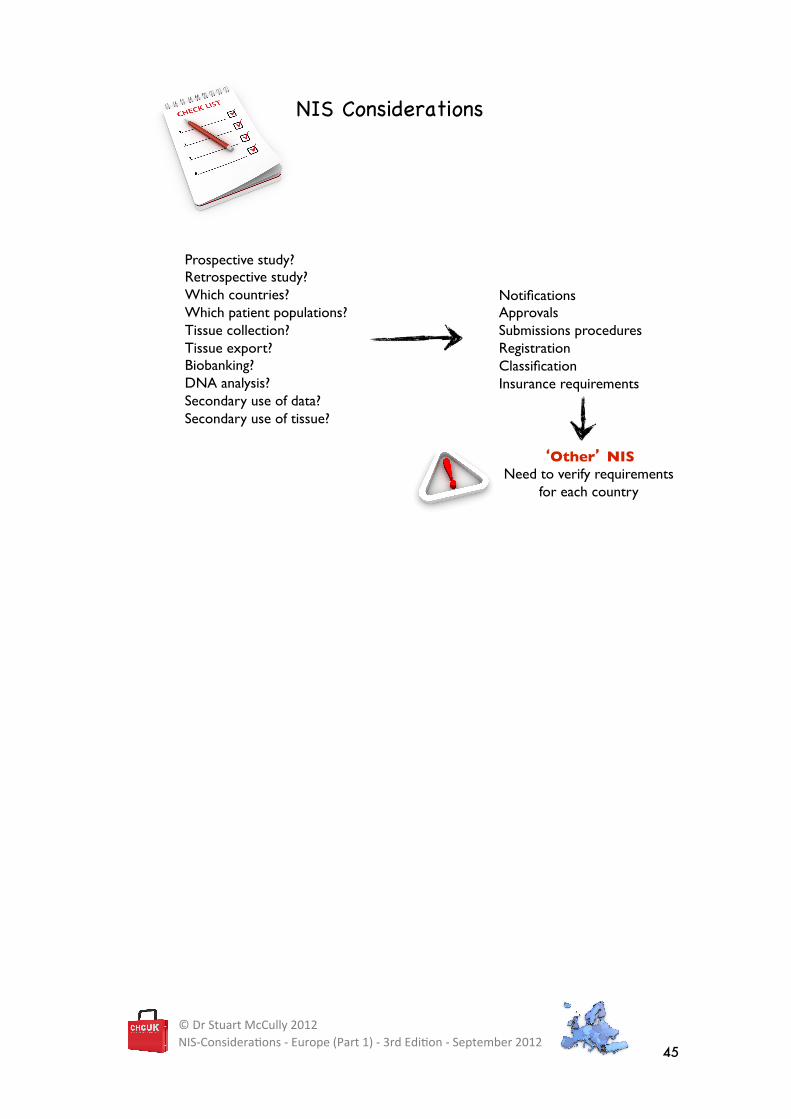
NIS Considerations�
Prospective study?!Retrospective study?!Which countries?!Which patient populations?!Tissue collection?!Tissue export?!Biobanking?!DNA analysis?!Secondary use of data?Secondary use of tissue?!
Notifications!Approvals!Submissions procedures!Registration!Classification!Insurance requirements!
�Other� NIS!Need to verify requirements
for each country!
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
45
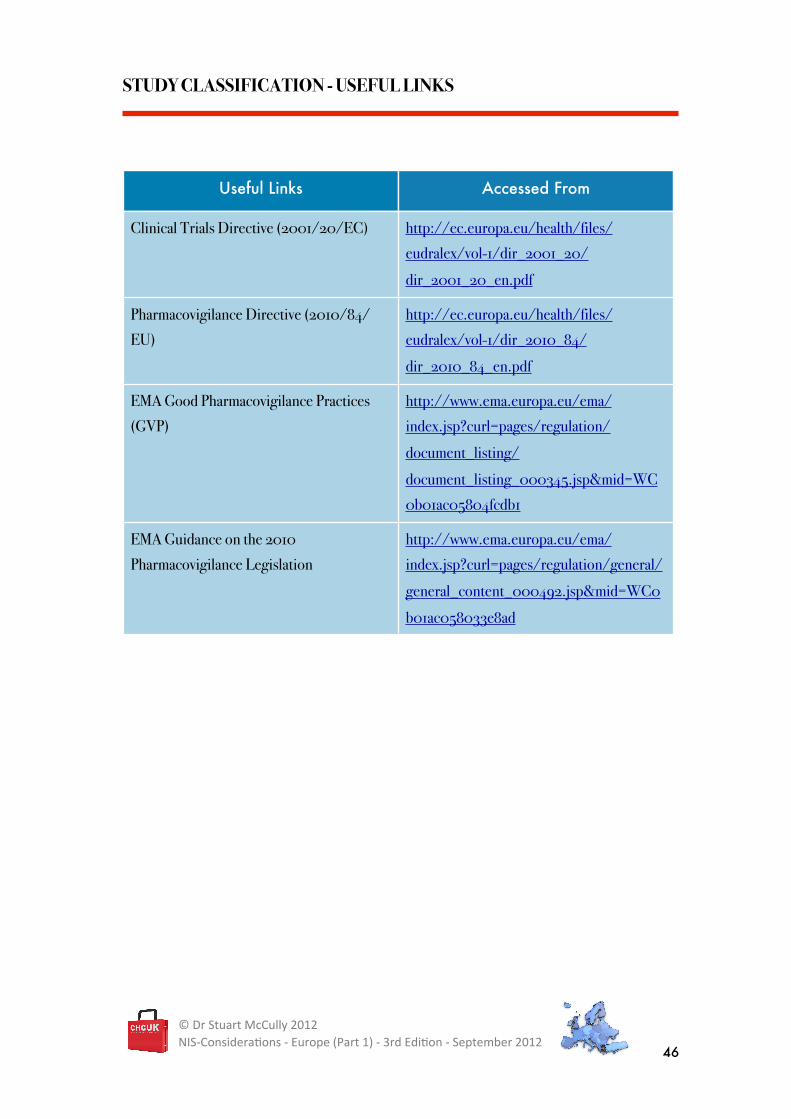
STUDY CLASSIFICATION - USEFUL LINKS
Useful Links Accessed From
Clinical Trials Directive (2001/20/EC) http://ec.europa.eu/health/files/
eudralex/vol-1/dir_2001_20/
dir_2001_20_en.pdf
Pharmacovigilance Directive (2010/84/
EU)
http://ec.europa.eu/health/files/
eudralex/vol-1/dir_2010_84/
dir_2010_84_en.pdf
EMA Good Pharmacovigilance Practices
(GVP)
http://www.ema.europa.eu/ema/
index.jsp?curl=pages/regulation/
document_listing/
document_listing_000345.jsp&mid=WC
0b01ac05804fcdb1
EMA Guidance on the 2010
Pharmacovigilance Legislation
http://www.ema.europa.eu/ema/
index.jsp?curl=pages/regulation/general/
general_content_000492.jsp&mid=WC0
b01ac058033e8ad
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
46
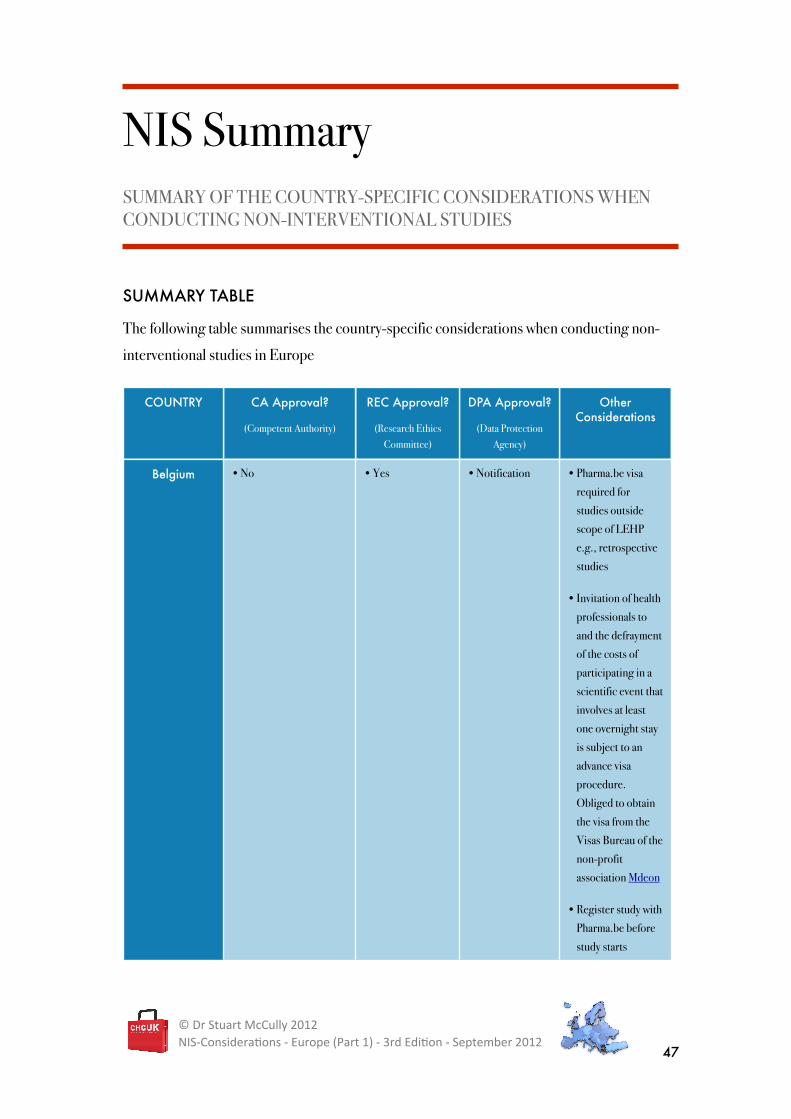
NIS Summary SUMMARY OF THE COUNTRY-SPECIFIC CONSIDERATIONS WHEN CONDUCTING NON-INTERVENTIONAL STUDIES
SUMMARY TABLE
The following table summarises the country-specific considerations when conducting non-
interventional studies in Europe
COUNTRY CA Approval?
(Competent Authority)
REC Approval?
(Research Ethics Committee)
DPA Approval?
(Data Protection Agency)
Other Considerations
Belgium • No • Yes • Notification • Pharma.be visa
required for
studies outside
scope of LEHP
e.g., retrospective
studies
• Invitation of health
professionals to
and the defrayment
of the costs of
participating in a
scientific event that
involves at least
one overnight stay
is subject to an
advance visa
procedure.
Obliged to obtain
the visa from the
Visas Bureau of the
non-profit
association Mdeon
• Register study with
Pharma.be before
study starts
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
47

BelgiumCOUNTRY-SPECIFIC CONSIDERATIONS
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
55
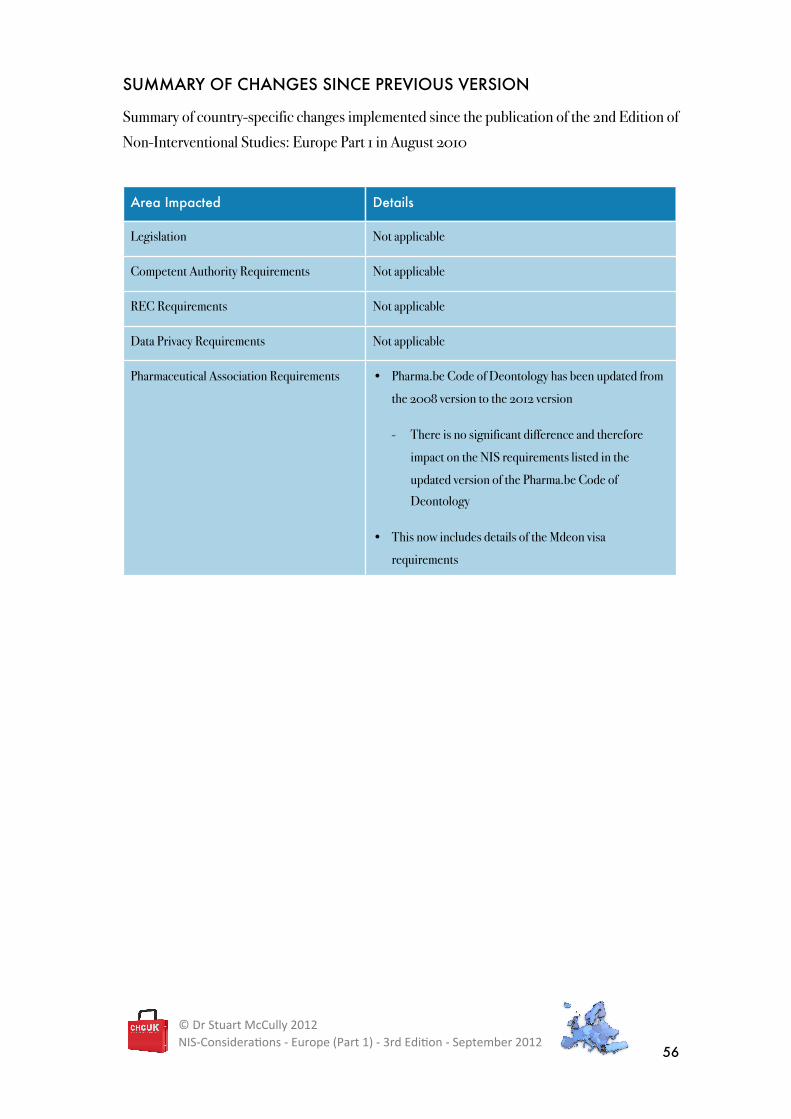
SUMMARY OF CHANGES SINCE PREVIOUS VERSION
Summary of country-specific changes implemented since the publication of the 2nd Edition of
Non-Interventional Studies: Europe Part 1 in August 2010
Area Impacted Details
Legislation Not applicable
Competent Authority Requirements Not applicable
REC Requirements Not applicable
Data Privacy Requirements Not applicable
Pharmaceutical Association Requirements • Pharma.be Code of Deontology has been updated from
the 2008 version to the 2012 version
- There is no significant difference and therefore
impact on the NIS requirements listed in the
updated version of the Pharma.be Code of
Deontology
• This now includes details of the Mdeon visa
requirements
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
56

DEFINITIONS
Non-Interventional Study
A non-interventional study is understood to mean a study in which the medicinal products are
prescribed in the usual manner, in accordance with the terms of the marketing authorisation.
The assignment of the patient to a particular therapeutic strategy is not decided in advance by
a trial protocol but falls within current medical practice and the decision to prescribe the
medicinal product is clearly separated from the decision to include a patient in the study. The
patient in question must not be subject to additional diagnostic or monitoring procedures and
epidemiological methods shall be used for the analysis of collected data (as per Article 43 of
the Pharma.be Code of Deontology, 2012).
"non-interventional trial"
A study where the medicinal products are prescribed in the usual manner in accordance with
the terms of the marketing authorization. The assignment of the patient to a particular
therapeutic strategy is not decided in advance by a trial protocol but falls within current
practice and the prescription of the medicine is clearly separated from the decision to include
the patient in the study. No additional diagnostic or monitoring procedures shall be applied to
the patients and epidemiological methods shall be used for the analysis of collected data (as
per Article 2.8 of the Law Concerning Experiments on the Human Person (LEHP) of 7th May
2004).
Clarification
For a non-interventional trial to come within the scope of application of the legislation, it also
must be a prospective study. The notification relating to the safety required for clinical trials is
not applicable and only the normal post-marketing drug monitoring system is applicable (as
per Annex 1 of Circular 455 of 28/11/2004).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
57
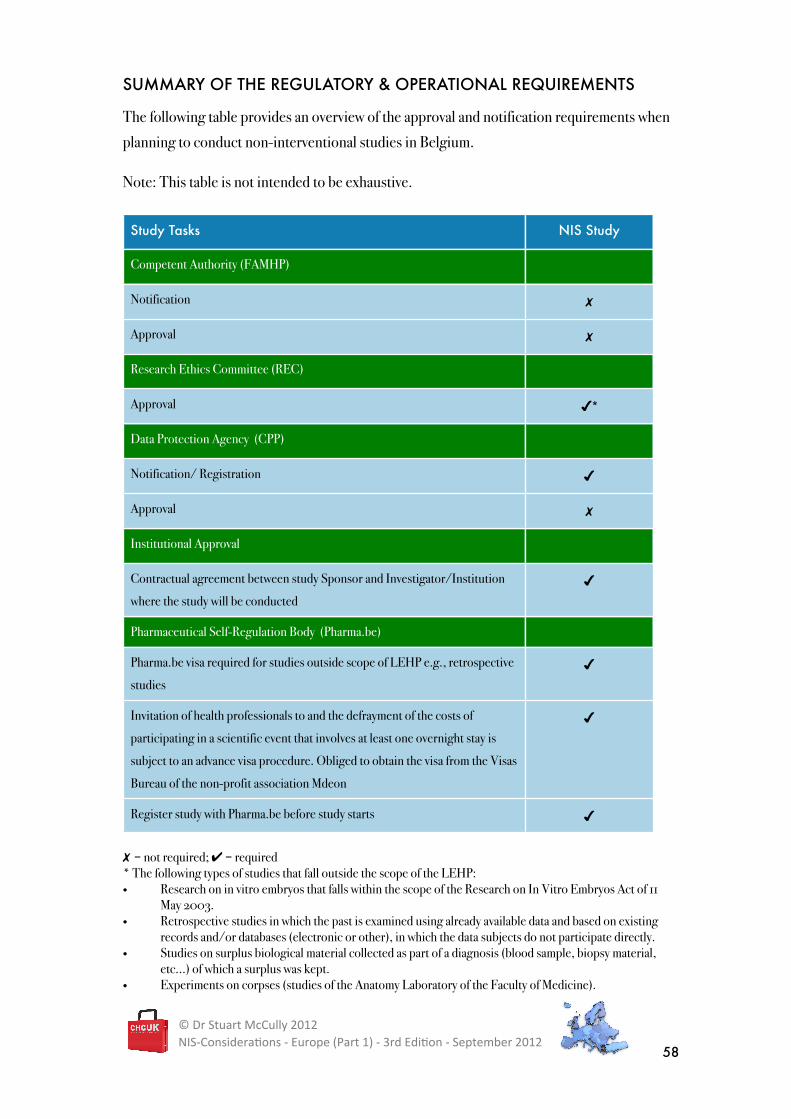
SUMMARY OF THE REGULATORY & OPERATIONAL REQUIREMENTS
The following table provides an overview of the approval and notification requirements when
planning to conduct non-interventional studies in Belgium.
Note: This table is not intended to be exhaustive.
Study Tasks NIS Study
Competent Authority (FAMHP)
Notification �
Approval �
Research Ethics Committee (REC)
Approval ��
Data Protection Agency (CPP)
Notification/ Registration �
Approval �
Institutional Approval
Contractual agreement between study Sponsor and Investigator/Institution
where the study will be conducted�
Pharmaceutical Self-Regulation Body (Pharma.be)
Pharma.be visa required for studies outside scope of LEHP e.g., retrospective
studies�
Invitation of health professionals to and the defrayment of the costs of
participating in a scientific event that involves at least one overnight stay is
subject to an advance visa procedure. Obliged to obtain the visa from the Visas
Bureau of the non-profit association Mdeon
�
Register study with Pharma.be before study starts �
��= not required; ✔ = required* The following types of studies that fall outside the scope of the LEHP:� Research on in vitro embryos that falls within the scope of the Research on In Vitro Embryos Act of 11
May 2003.� Retrospective studies in which the past is examined using already available data and based on existing
records and/or databases (electronic or other), in which the data subjects do not participate directly.� Studies on surplus biological material collected as part of a diagnosis (blood sample, biopsy material,
etc...) of which a surplus was kept.� Experiments on corpses (studies of the Anatomy Laboratory of the Faculty of Medicine).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
58

� Psychological studies: Psychologists are not mentioned in the Royal Decree n° 78 on the Practice of Healthcare Professions and the law of May 2004 only applies to "any trial, study or investigation carried out on a human being in order to gain knowledge to further the practice of healthcare professions as set out in Royal Decree n° 78 of 10 November 1967" as specified in Article 2, point 11. However, any psychological study that is based on medical examinations (EEG, brain scan, etc.) falls within the scope of the law through the intervention of the physician responsible for the medical examinations.
Source: http://www.erasme.ulb.ac.be/page.asp?id=11373&langue=EN
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
59

REGULATORY BODIES
Competent Authority
Research Ethics Committees
Data Protection Agency
Pharmaceutical Self-Regulation Body
Pharmaceutical Code of Practice
Federal Agency for Medicines and Health Products (FAMHP)
(Agence Fédérale pour les Médicaments et les Produits de Santé - AFMPS)
Ethical approval of “Experiments on Human Persons” is provided by the Leading Ethics Committees (LECs)
Commission for the Protection of Privacy
(CPP)
Association Générale de l’Industrie du Médicament (Pharma.be)
Pharma.be Code of Deontology (March 2012)
Competent Authority
The Belgian Federal Agency for Medicines and Health Products (FAMHP) was officially
created on the 1st January 2007. The FAMHP has completely taken over the role and fields of
competency of the Directorate-General for Medicinal Products (DG Medicinal Products) of
the Federal Public Service (FPS) Public Health (FAMP Annual Report 2007).
Competent Authority Approval
There is currently no legal requirement to seek FAMHP approval for non-interventional
studies or notify the FAMHP of an intention to perform a non-interventional study.
Research Ethics Committees (RECs)
The Royal Decree of 12th August 1994 (RD 12/08/1994) made RECs compulsory for every
hospital or group of hospitals. This law also defined the RECs aims, composition & function,
closely following the code of deontology of the Order of Doctors.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
60

There are currently about 215 research ethics committees. The majority are associated with
hospitals and some are associated with non-hospital institutions. Until the 31st of March 2012,
only 38 of these research ethics committees (see link) have full recognition to give a single
opinion (EFGCP Report, Belgium - 2011).
There is a central Consultative Committee for Bioethics, officially installed in 1996, which
formulates opinion & informs the public; however this is only a consultative body (EFGCP
Report, Belgium - 2011).
On 7 May 2004 the Law relating to Experimentation on Humans (LEHP) came into force,
which for the first time endowed Belgian RECs with a legal status. It covers all types of
medical experimentation, including prospective non-interventional studies.
Ethical Approval
Sponsors are legally required to obtain ethical approval prior to starting a prospective non-
interventional study.
Pharmaceutical Self-regulation Body (Pharma.be)
The ‘Association Générale de l’Industrie du Médicament’, otherwise known as “Pharma.be”
is the Belgian self-regulation Pharmaceutical body affiliated with EFPIA.
The website and most of the resources are currently only available in Dutch and French.
However, an English translation of the Pharma.be Code of Practice on the Promotion of
Medicines (Pharma.be Code of Deontology - 2012) is available through the Pharma.be
website.
Data Protection Agency (CPP)
The Commission for the Protection of Privacy (CPP), better known as the Privacy
Commission, is an independent authority ensuring the protection of privacy during the
processing of personal data.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
61

The Commission was established by the Belgian Federal House of Representatives with the
Act of 8 December 1992.
Notification
According to Article 17 of the Act of 8 December 1992 on the protection of privacy in relation
to the processing of personal data (Privacy Act or Data Protection Act - as amended), prior to
any wholly or partly automatic operation or set of operations intended to serve a single
purpose or several related purposes, the controller or his representative, if any, must notify
the Commission for the Protection of Privacy (CPP).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
62

REGULATORY FRAMEWORK
Applicable Legislation and Guidance
The laws and guidelines which are applicable to the management and conduct of non-
interventional studies in Belgium include (but are not limited to):
Regulations Applicable to NIS
Data Protection Regulations
Ethical Standards
NIS best Practice Considerations
• Law Concerning Experiments on the Human
Person (LEHP) of 7th May 2004
• Royal Decree of August 12th 1994 -Local
Hospital Ethics Committee (RD 12/08/1994)
• FAMHP Guide for the Evaluation of Non-
interventional Studies (May 2008)
• The Patient Rights Act (August 2002)
• Act of 8 December 1992 on the protection of
privacy in relation to the processing of
personal data (LVLP)
• Royal Decree of 13/2/2001
• Royal Decree of 17/12/2003
• CPP Guidance - How to apply the Privacy Act
in biomedical research (2011)
• World Medical Association's Declaration of
Helsinki (2008 version)
• Nuremberg Code
• ICH GCP
• Pharma.be Code of Deontology (March 2012)
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
63

©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
64

REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES
Purpose
The study should be conducted with a clear scientific purpose (as per Article 44 of the
Pharma.be Code of Deontology, 2012).
Prohibition of Promotion
The study must not constitute an inducement to recommend, prescribe, purchase or sell,
supply or administer medicinal products (as per Article 44 of the Pharma.be Code of
Deontology, 2012).
Quality Framework
Non-interventional studies shall be conducted within a quality framework (as per Article 43 of
the Pharma.be Code of Deontology, 2012).
Contract
A written contract shall provide a detailed description of the services expected from the
investigators as well as of the amount and the procedures for remunerating the investigators
(as per Article 44 of the Pharma.be Code of Deontology, 2012).
Notwithstanding article 40 of this Code and notwithstanding the legal provisions, contracts
between pharmaceutical companies and institutions, organisations or associations of
healthcare professionals by the terms of which such institutions, organisations or associations
provide services to companies, are only allowed if such services:
1) support healthcare or research,
2) do not constitute an inducement to recommend, prescribe, purchase, sell, supply or
administer medicinal products.
(as per Article 41 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
65

Notwithstanding article 40 of this Code and notwithstanding the legal provisions, a
pharmaceutical company may use one or more healthcare professionals as consultants or
advisors for services such as speaking at or chairing scientific meetings, involvement in
medical/scientific studies, clinical trials or training courses, participation in advisory board
meetings or participation in market research, in which the healthcare professionals concerned
receive a remuneration and/or are expected to travel (as per Article 42 of the Pharma.be Code
of Deontology, 2012).
The arrangements made in this respect, if relevant to this subject, must satisfy the following
conditions:
(a) a legitimate need for the services is clearly identified before retaining the healthcare
professionals and making arrangements in this respect;
(b) the criteria for selecting consultants are directly related to the identified legitimate
need as referred to in clause a and the persons responsible for selecting the consultants
have the expertise necessary to evaluate whether the contacted healthcare professionals
meet those criteria;
(c) the number of healthcare professionals retained is not greater than the number
reasonably necessary to achieve the identified need;
(d) a written contract must be drawn up before the commencement of the services which
specifies the nature of the services to be provided by the healthcare professionals as
well as the basis for payment for their services notwithstanding what is cited in clause g.
hereafter;
(e) the pharmaceutical company maintains records concerning the services provided and
makes appropriate use of them;
(f) the hiring of the healthcare professionals to provide the services is not an inducement
to recommend, prescribe, purchase, sell, supply or administer medicinal products and
(g) the compensation for the services is reasonable and reflects the usual market value of
the services provided.
(as per Article 42.1 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
66

Declaration of Conflicts of Interest/ Financial Support
For the purposes of transparency, pharmaceutical companies are strongly encouraged to
include in the written contract as mentioned above in paragraph 1.d. a provision regarding the
obligation of the healthcare professional in question to declare that he/she is fulfilling a
consultancy or advisory mission for the company whenever he/she speaks in public or
publishes on matters that are the subject of the agreement or any other issue relating to that
company (as per Article 42.2 of the Pharma.be Code of Deontology, 2012).
Similarly, pharmaceutical companies that employ healthcare professionals on a part-time basis
who are still practising their profession are strongly encouraged to impose on such persons
the obligation to declare their employment arrangement with the company whenever they
speak in public or publish on matters that are the subject of their employment arrangement or
any other issue relating to that company (as per Article 42.2 of the Pharma.be Code of
Deontology, 2012).
Market Research
The provisions mentioned under paragraph 1.d. do not apply to limited market research, such
as one-off phone interviews or surveys by post, e-mail or internet, provided that the healthcare
professionals concerned are not consulted repeatedly, whether in the context of the same or
another survey, and that the remuneration for their collaboration is of token value (as per
Article 42.3 of the Pharma.be Code of Deontology, 2012).
Remuneration
The remuneration is commensurate with the services requested and reflects the market value
thereof (as per Article 44 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
67

Study Protocol
A written scientific protocol shall provide a detailed description of the purpose sought and
methodology implemented; the aforementioned purpose and methodology shall always be
coherent with one another (as per Article 44 of the Pharma.be Code of Deontology, 2012).
The scientific protocol must be approved in advance by the company’s scientific service as
referred to under article 6 of this Code and this service has to supervise the conduct of the
study (as per Article 44 of the Pharma.be Code of Deontology, 2012).
Procedures for the Supply of Medicines
The procedures for supplying the medicines studied shall be described in detail in the
protocol; they shall be coherent in regard to the stated purpose and methodology (as per
Article 44 of the Pharma.be Code of Deontology, 2012).
Justification for the Number of Patients and Number of Investigators
The number of patients requested for inclusion as well as the number of investigators
included shall be justified in a scientific manner in the protocol, for example by means of a
biostatistical calculation (as per Article 44 of the Pharma.be Code of Deontology, 2012).
Future Use of Data
The future use of the data collected shall be stated clearly in the protocol (as per Article 44 of
the Pharma.be Code of Deontology, 2012).
Study Design and Follow-up
The holder of the marketing authorisation shall establish a permanent connection with a
scientific service charged with providing information on the medicinal products that it
markets and which is responsible for the approval and the supervision of non-interventional
studies which are carried out by or with the support of the holder of the authorisation (as per
Article 6 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
68

Regardless of the way the scientific service is organised, this service should include a medical
doctor or a pharmacist who will approve any promotional material before release (as per
Article 6 of the Pharma.be Code of Deontology, 2012).
In addition, the scientific service must include a medical doctor or a pharmacist who will be
responsible for the supervision of all non-interventional studies which are carried out or
sponsored by the company (as per Article 6 of the Pharma.be Code of Deontology, 2012).
Limited Involvement of Medical Reps
Medical informants (Medical or Sales Representatives) may only intervene in a study to
perform administrative tasks under the supervision of the Company’s Scientific Service; this
service will ensure that the medical informants are adequately trained for this purpose; their
involvement in scientific studies must not be linked to the promotion of medicinal products
(as per Article 44 of the Pharma.be Code of Deontology, 2012).
Competent Authority Approval
Not required.
Favourable Ethics Opinion
This type of study must be carried out in respect of the requirements set out by applicable
legislation – among others, the favourable opinion of an accredited Lead Ethics Committee
(LEC) must be granted before the study can start (Article 10 of the LEHP).
Procedure for Submission to Ethics Committees
The procedure for submission to the Ethics Committees of the sites concerned should be in
accordance with strict rules.
The sponsor designates a principal investigator (PI) for Belgium and an ethics
committee that has to issue a single opinion (CAISO = Committee authorised to issue a
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
69

single opinion or LEC = Leading Ethics Committee) as per Articles 11.2 and 11.3 of
LEHP.
The CAISO communicates with the local ethics committees of the different sites
involved in the research project and summarises the received opinions (as per Articles
11.7 & 11.8 of LEHP) to formulate:
• a first temporary opinion intended to obtain amendments of the protocol /
patient information leaflet and the informed consent form;
• a final opinion or approval, which applies to all the sites that participated in
the review process.
According to Article 11.5 of LEHP, the ethics committee has a maximum of 15 days to provide
an opinion for single centre Phase 1 studies and a maximum of 28 days for all other
experiments.
Further detailed information on the LEC submission requirements can be found via the
website of the Ethics Committee at the Erasmus Hospital in Brussels. This includes:
Remuneration for examining an application – Lists the fees which must be paid in
advance to the Ethics Committee.
Definitions and types of clinical research – Explains which types of studies fall within
the scope of the LEHP (e.g., prospective NIS) and further explains the process for
submitting applications to ethics committees.
Non-interventional clinical trial sponsored by the industry – Lists which documents
must be submitted to the ethics committee when applying for favourable opinion to
conduct an NIS.
Submitting an amendment – Explains the procedure to follow when submitting an
amendment to the ethics committee.
Information about the completion, premature discontinuation, temporary
discontinuation and restart of a trial.
Insurance and Clinical Investigation on Human Subjects – Explains the liability and
insurance requirements as laid down by Article 29 of LEHP.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
70

Data Privacy and the Processing of Medical Data – Provides a summary of the applicable
data privacy laws as applicable to processing medical data.
Approval of Substantial Amendments
Belgian law (Chapter X of LEHP) requires ethical approval (favourable opinion) of substantial
amendments to the NIS protocol.
Attendance at Scientific Events
Scientific events that are directly or indirectly supported or organised by pharmaceutical
companies and that are attended by health professionals shall take place within a framework of
quality, as required by articles 31 to 35. When a scientific event does not take place in
Belgium, it must also, in accordance with article 3, § 3 of this Code, comply with the criteria
laid down by the Code of Deontology that applies in the country where the event takes place
(as per Article 30 of the Pharma.be Code of Deontology, 2012).
This applies, for example, to events of an exclusively professional and scientific nature, events
to promote medicinal products, symposiums, international scientific congresses, advisory
board meetings, visits to research or manufacturing facilities, investigator meetings for
clinical or other scientific studies and any other form of scientific meeting held in Belgium or
abroad (as per Article 30 of the Pharma.be Code of Deontology, 2012).
When a healthcare professional attends a scientific event in the capacity of consultant or
advisor, the articles 30 to 35 of this Code apply (as per Article 42.3 of the Pharma.be Code of
Deontology, 2012).
Advance Visa Procedure
The invitation of health professionals to and the defrayment of the costs of participating in a
scientific event as referred to under article 30 that involves at least one overnight stay are
subject to an advance visa procedure (as per Article 72 of the Pharma.be Code of Deontology,
2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
71

In which case, pharmaceutical companies are obliged to obtain the visa from the Visas Bureau
of the non-profit association Mdeon (as per Article 72 of the Pharma.be Code of Deontology,
2012).
End of Study Notification
Within 90 days of the end of an experiment, the sponsor shall notify the ethics committee that
the experiment has ended. If the experiment must be terminated early, this term shall be
reduced to 15 days. The notification shall clearly explain the reasons for the early termination
(Article 21 of LEHP).
Analysis and Retention of Study Results
The study results must be analysed and reports thereof must be submitted within a reasonable
period of time to the company’s scientific service which shall maintain these reports for a
reasonable period of time (as per Article 44 of the Pharma.be Code of Deontology, 2012).
The study results should also be kept at the disposal of the bodies of pharma.be as referred to
under article 52, paragraph 1 of this Code and must be submitted following a request on their
part (as per Article 44 of the Pharma.be Code of Deontology, 2012).
Dissemination of Study Results
The company must send the study results to all healthcare professionals who participated in
the study (as per Article 44 of the Pharma.be Code of Deontology, 2012).
Notification of Important Risk-Benefit Information
If the study shows results that are important for the assessment of the benefit-risk ratio of the
studied medicinal product(s), these results should be immediately forwarded to the
competent authority (as per Article 44 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
72
Retrospective & ‘Other’ Non-Interventional Studies
The non-interventional studies referred to under article 43 of this Code , with the exception
of the experiments referred to in the law of 7 May 2004 concerning experiments on human
persons (LEHP) and that are subject to the advice of an ethics committee, are subject to an
advance visa procedure (as per Article 73 of the Pharma.be Code of Deontology, 2012).
Visa Application
To this end, pharmaceutical companies are bound to obtain the visa from the Visas Bureau of
pharma.be prior to the inclusion of the first investigator (as per Article 73 of the Pharma.be
Code of Deontology, 2012).
Only the studies cited in the first paragraph and that cumulatively meet the following
conditions are submitted to the visa procedure:
they are carried out or supported directly or indirectly by a pharmaceutical company;
they relate to one or more of the characteristics of the medicinal product(s) studied;
they involve one or more persons from outside the company.
Proceedings in regard to the Visas Bureau are conducted in writing only (as per Article 73 of
the Pharma.be Code of Deontology, 2012).
Dossier Requirements for a Visa Application
Companies introduce an application for visa by submitting the duly completed model dossier
as drawn up by the Secretariat, together with all information and documents attesting to
compliance with article 43 and 44 of the Code (as per Article 74 of the Pharma.be Code of
Deontology, 2012).
The model dossier drawn up by the Secretariat includes the following elements:
general identification data,
the starting and closing dates of the study,
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
73

the detailed scientific protocol including the following elements: the objective,
methodology, possible approval by the company’s scientific service referred to under
article 6 of this Code and added value,
the detailed financial protocol including the following elements: the amount and
methods of remuneration for the investigators, the detailed description of the services
requested of the investigators who receive the remuneration and the model contract
concluded with the investigators,
the methods for the supply and distribution of the medicinal products studied,
the description of the future use of the data collected,
the scientific justification, biostatistical if applicable, for the number of patients and
investigators included as well as their geographical distribution,
any other information deemed to be useful by the applicant company.
On pain of being declared inadmissible, five copies of the application file must be submitted
by post (as per Article 74 of the Pharma.be Code of Deontology, 2012).
The Secretariat acknowledges reception of each dossier received and at the same time issues a
dossier number to the applicant company. This dossier number may only be used by the
applicant company in accordance with the conditions laid down under article 75, para. 2 and
article 76, para. 2 (as per Article 74 of the Pharma.be Code of Deontology, 2012).
Visa Approval Timeframe
The Visas Bureau notifies the applicant company of its decision no later than the tenth
working day following that on which the Secretariat receives the application (as per Article 76
of the Pharma.be Code of Deontology, 2012).
If, at the end of the tenth working day following that on which the Secretariat receives the
application, the Visas Bureau has not notified the applicant company of its decision, the visa is
deemed to have been granted. In which case, the applicant company is bound to indicate the
dossier number referred to under article 74, last paragraph, of this Code, known as the “visa
number”, on all documents concerning the project in question drawn up after the expiry of
the aforementioned term (as per Article 76 of the Pharma.be Code of Deontology, 2012).
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
74

Ethical Review of Retrospective NIS & Applicability of the LEHP
If, during a retrospective study, the patient is contacted specifically in the context of the
study, this study is interventional and falls well within the law experiments (as per Section
1.4.2 of the FAMHP Guide for the Evaluation of Non-interventional Studies - May 2008).
Under general ethical principles, it is advisable to submit all non-interventional studies,
including those not falling within the scope of the law, for the opinion of a medical ethics
committee (as per Section 1.4.2 of the FAMHP Guide for the Evaluation of Non-
interventional Studies - May 2008).
Informed Consent in the Context of Retrospective Non-interventional Studies
Studies of records, studies based on questionnaires, etc.. do not fall within the scope of the
law concerning experiments on humans (LEHP). * The treatment team has a right of direct
access to uncoded data extracted from medical records of patients and this right is not open to
discussion. In this regard, it should be noted that when the therapeutic team consults and
processes data for its own use, the patient's informed consent is not required provided that
the rules of confidentiality are respected.
[* Therapeutic teams acting under the direct responsibility of the attending physician:
personnel (para-) medical external - prosthetic intervention, for example - must also be
included in the therapeutic team. It does not however apply to hospital pharmacists, for
example, who have partial knowledge of pharmaceutical personal data, but are bound by
professional secrecy. ]
Communication of data outside of the therapeutic team must be in compliance with the LPVP
and RD-LPVP (Chapter 2):
Where anonymised data are made available to third parties not part of the therapeutic
team, the patient's consent is not necessary.
When the encoded data are made available to third parties not part of the therapeutic
team, the obligation to inform the patient (or his legal representative in the case of a
minor or an incompetent patient) applies, except in exceptional cases for which
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
75

arrangements specific statement to the Commission for the Protection of life Private are
provided.
When uncoded data are made available to third parties not part of the therapeutic team,
the principle of informed consent (or its legal representative in the case of a minor or an
incompetent patient) applies, except in exceptional cases for which specific
arrangements for reporting to the Commission for the Protection of Privacy Act are
provided.
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
76

ADDITIONAL RESOURCES
Country-specific eLearning Module
CHCUK now offer an eLearning Module for
Belgium that explores the country-specific
requirements when conducting non-
interventional studies in Belgium and looks
specifically at:
Regulatory classification
Regulatory framework/applicable
legislation and guidelines
Approval requirements and timeframes
Submission documents
Who is responsible for what?
Practical considerations when conducting non-interventional studies
Requirements for non-interventional studies that intend to collect, store and/or
analyse human tissue samples
Industry best practice
Course Format
The course:
Includes reminders at the end of each module
Has an audio commentary
Includes PDFs of all the training materials
Includes a comprehensive tools and resources report that is an ideal reference guide for
anyone involved in the oversight or operations aspects of non-interventional studies
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
77
Includes a certificate of completion that is automatically issued to all participants who
complete the course
Who Would Benefit from the Course?
The course is intended for anyone involved in the oversight or operations aspects of non-
interventional studies, such as:
Medical Affairs groups/ Company Affiliates
Project Managers
CRAs
Quality Assurance personnel
Regulatory Affairs personnel
Further Information and/or Free Demonstration
If you would like further details, or would like to request a free demo then please contact us at:
Email: [email protected]
Phone: +44 1997 42 33 11
website: www.chcuk.co.uk
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
78
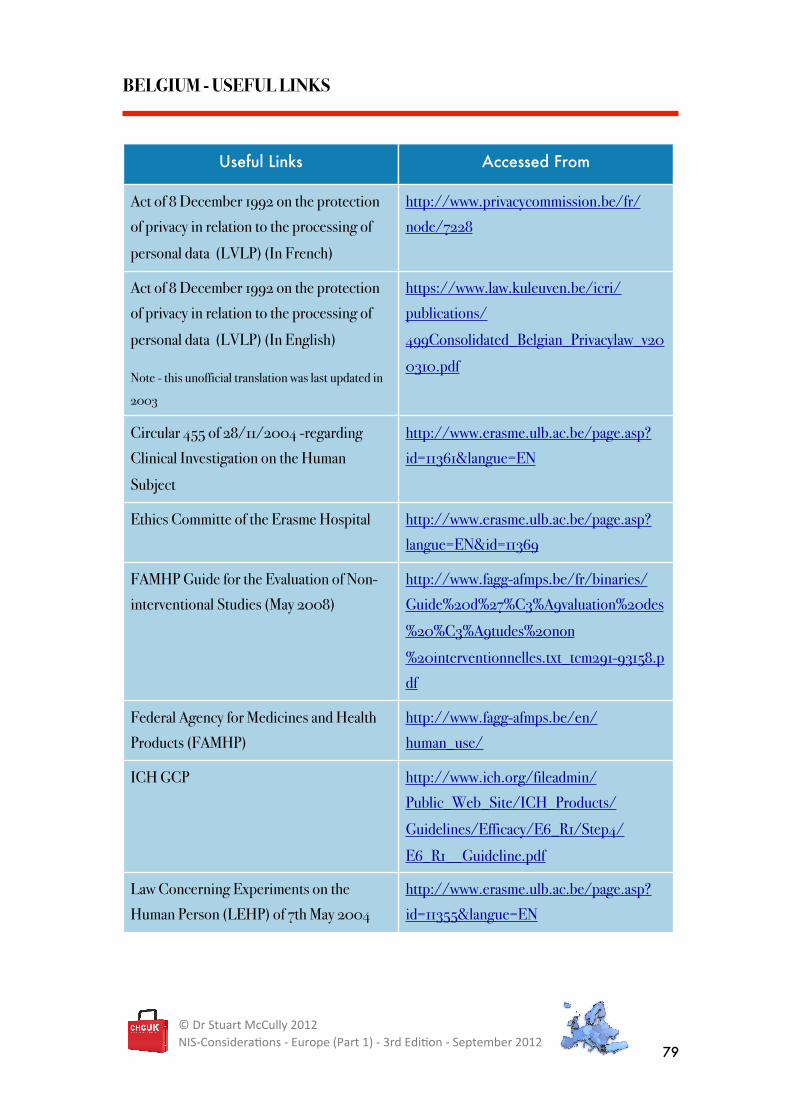
BELGIUM - USEFUL LINKS
Useful Links Accessed From
Act of 8 December 1992 on the protection
of privacy in relation to the processing of
personal data (LVLP) (In French)
http://www.privacycommission.be/fr/
node/7228
Act of 8 December 1992 on the protection
of privacy in relation to the processing of
personal data (LVLP) (In English)
Note - this unofficial translation was last updated in
2003
https://www.law.kuleuven.be/icri/
publications/
499Consolidated_Belgian_Privacylaw_v20
0310.pdf
Circular 455 of 28/11/2004 -regarding
Clinical Investigation on the Human
Subject
http://www.erasme.ulb.ac.be/page.asp?
id=11361&langue=EN
Ethics Committe of the Erasme Hospital http://www.erasme.ulb.ac.be/page.asp?
langue=EN&id=11369
FAMHP Guide for the Evaluation of Non-
interventional Studies (May 2008)
http://www.fagg-afmps.be/fr/binaries/
Guide%20d%27%C3%A9valuation%20des
%20%C3%A9tudes%20non
%20interventionnelles.txt_tcm291-93158.p
df
Federal Agency for Medicines and Health
Products (FAMHP)
http://www.fagg-afmps.be/en/
human_use/
ICH GCP http://www.ich.org/fileadmin/
Public_Web_Site/ICH_Products/
Guidelines/Efficacy/E6_R1/Step4/
E6_R1__Guideline.pdf
Law Concerning Experiments on the
Human Person (LEHP) of 7th May 2004
http://www.erasme.ulb.ac.be/page.asp?
id=11355&langue=EN
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
79
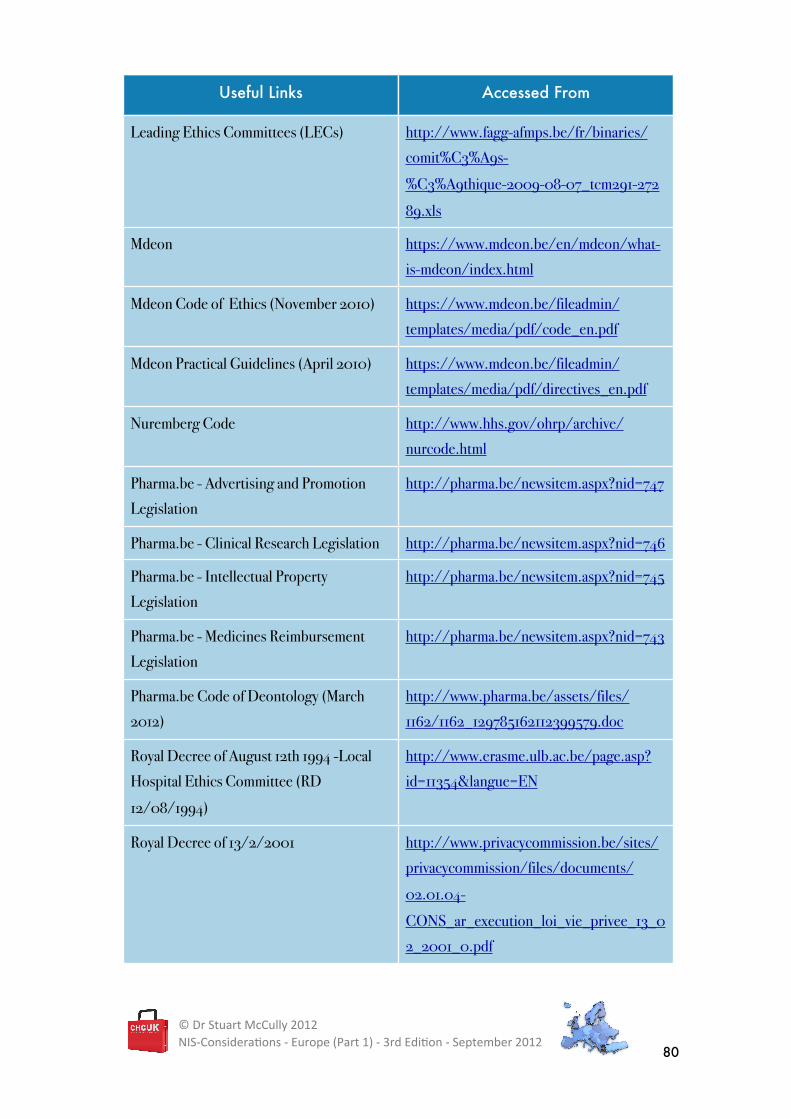
Useful Links Accessed From
Leading Ethics Committees (LECs) http://www.fagg-afmps.be/fr/binaries/
comit%C3%A9s-
%C3%A9thique-2009-08-07_tcm291-272
89.xls
Mdeon https://www.mdeon.be/en/mdeon/what-
is-mdeon/index.html
Mdeon Code of Ethics (November 2010) https://www.mdeon.be/fileadmin/
templates/media/pdf/code_en.pdf
Mdeon Practical Guidelines (April 2010) https://www.mdeon.be/fileadmin/
templates/media/pdf/directives_en.pdf
Nuremberg Code http://www.hhs.gov/ohrp/archive/
nurcode.html
Pharma.be - Advertising and Promotion
Legislation
http://pharma.be/newsitem.aspx?nid=747
Pharma.be - Clinical Research Legislation http://pharma.be/newsitem.aspx?nid=746
Pharma.be - Intellectual Property
Legislation
http://pharma.be/newsitem.aspx?nid=745
Pharma.be - Medicines Reimbursement
Legislation
http://pharma.be/newsitem.aspx?nid=743
Pharma.be Code of Deontology (March
2012)
http://www.pharma.be/assets/files/
1162/1162_129785162112399579.doc
Royal Decree of August 12th 1994 -Local
Hospital Ethics Committee (RD
12/08/1994)
http://www.erasme.ulb.ac.be/page.asp?
id=11354&langue=EN
Royal Decree of 13/2/2001 http://www.privacycommission.be/sites/
privacycommission/files/documents/
02.01.04-
CONS_ar_execution_loi_vie_privee_13_0
2_2001_0.pdf
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
80
Useful Links Accessed From
Royal Decree of 17/12/2003 http://www.privacycommission.be/sites/
privacycommission/files/documents/
02.01.01.01-
ar_comites_sectoriels_17_12_2003_0.pdf
World Medical Association's Declaration
of Helsinki
http://www.chcuk.co.uk/pdf/
Declaration_of_Helsinki_2008_Version.p
df
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
81

Published by Compliance Healthcheck Consulting UK Ltd
www.chcuk.co.uk
ISBN 978-0-9563319-4-6
©"Dr"Stuart"McCully"2012NIS3Considera:ons"3"Europe"(Part"1)"3"3rd"Edi:on"3"September"2012"
354