nms-e973, a novel synthetic inhibitor of hsp90 with...
TRANSCRIPT

Cancer Therapy: Preclinical
NMS-E973, a Novel Synthetic Inhibitor of Hsp90 with Activityagainst Multiple Models of Drug Resistance to TargetedAgents, Including Intracranial Metastases
Gianpaolo Fogliatto1, Laura Gianellini1, Maria G. Brasca1, Elena Casale1, Dario Ballinari1, Marina Ciomei1,Anna Degrassi1, Anna De Ponti1, Massimiliano Germani2, Marco Guanci1, Mauro Paolucci1, Paolo Polucci1,Micaela Russo1, Francesco Sola1, Barbara Valsasina1, Carlo Visco1, Fabio Zuccotto1, Daniele Donati1,Eduard Felder1, Enrico Pesenti1, Arturo Galvani1, Sergio Mantegani1, and Antonella Isacchi1
AbstractPurpose: Recent developments of second generation Hsp90 inhibitors suggested a potential for
development of this class of molecules also in tumors that have become resistant to molecular targeted
agents. Disease progression is often due to brain metastases, sometimes related to insufficient drug
concentrations within the brain. Our objective was to identify and characterize a novel inhibitor of Hsp90
able to cross the blood–brain barrier (BBB).
Experimental Design:Here is described a detailed biochemical and crystallographic characterization of
NMS-E973. Mechanism-based anticancer activity was described in cell models, including models of
resistance to kinase inhibitors. Pharmacokinetics properties were followed in plasma, tumor, liver, and
brain. In vivo activity and pharmacodynamics, as well as the pharmacokinetic/pharmacodynamic relation-
ships, were evaluated in xenografts, including an intracranially implanted melanoma model.
Results:NMS-E973, representative of a novel isoxazole-derived class of Hsp90 inhibitors, binds Hsp90awith subnanomolar affinity andhigh selectivity towards kinases, aswell as other ATPases. It possesses potent
antiproliferative activity against tumor cell lines and a favorable pharmacokinetic profile, with selective
retention in tumor tissue and ability to cross the BBB. NMS-E973 induces tumor shrinkage in different
human tumor xenografts, and is highly active in models of resistance to kinase inhibitors. Moreover,
consistent with its brain penetration, NMS-E973 is active also in an intracranially implanted melanoma
model.
Conclusions:Overall, the efficacy profile of NMS-E973 suggests a potential for development in different
clinical settings, including tumors that have become resistant to molecular targeted agents, particularly in
cases of tumors which reside beyond the BBB. Clin Cancer Res; 19(13); 3520–32. �2013 AACR.
IntroductionThe molecular chaperone Hsp90 is essential for the
conformational stability and activity ofmanykeyoncogenicproteins, including kinases such as ErbB2, B-Raf, Alk, Flt3,EGFR, RET, KIT, PDGFR, MET, AKT but also transcriptionfactors, telomerase, and other proteins that participate inthe activation of the several biologic pathways whose dys-functional activation have been collectively described as
constituting the hallmark traits of cancer (1). On the basisof this mechanism of action it is possible, by using aspecific inhibitor of Hsp90, to induce the degradation ofdiverse client proteins and as a consequence to achieve aparallel block of multiple signaling pathways that results incell death and inhibition of the growth of a broad rangeof tumors, as already described in several preclinicalstudies (2).
For these reasons, Hsp90 emerged in the last decade as amajor therapeutic target and a great deal of efforts have beendedicated to the discovery of inhibitors, some of themcurrently under clinical evaluation (3). After the identifica-tion of the initial Hsp90 inhibitors, in particular 17-AAGand other semisynthetic ansamycin derivatives such asretaspimycin (4), a new group of fully synthetic second-generation inhibitors were developed with improved pre-clinical efficacy and "drug-likeness".
Among the most advanced compounds, retaspimycinand the second-generation representative ganetespib haveshown significant clinical benefit, measured as RECIST-
Authors' Affiliations: 1Nerviano Medical Sciences S.r.l. and 2AcceleraS.r.l., Nerviano (MI), Italy
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author: Gianpaolo Fogliatto, Department of Biotechnol-ogy, Nerviano Medical Sciences S.r.l., Viale Pasteur 10, 20014 Nerviano(MI), Italy. Phone: 39-0331-581294; Fax: 39-0331-581267; E-mail:[email protected]
doi: 10.1158/1078-0432.CCR-12-3512
�2013 American Association for Cancer Research.
ClinicalCancer
Research
Clin Cancer Res; 19(13) July 1, 20133520
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

based partial responses at tolerated dosing regimens in Alk-driven NSCLC and ErbB2-positive breast cancers, as singleagents, or in combination with trastuzumab (5–7). Themost frequent clinical toxicities described so far are hepaticfor 17-AAG and its derivatives (6), gastrointestinal andocular for many second-generation inhibitors (3), reportedto be dependent on the schedule of administration and onthe tissue distribution of the molecule (8).The clinical benefit observed with drugs targeting onco-
genic kinases, such as the recently approved crizotinib (forAlk) and vemurafenib (for B-Raf), is very often transient dueto the appearance of resistance linked to secondary muta-tions in the targetmolecules (9), to the activation of parallelpathways such as EGFR andC-Raf signaling (10–13), aswellas to the intrinsic heterogeneity of the tumors. Also, in arelevant number of Alk-related cases, disease progressiondue to brain metastases is suggested to be related to insuf-ficient concentrations of the drugwithin the brain (14). Thehigh dependency onHsp90 of Alk andB-Raf kinases, aswellas of other kinases that are drivers of escape pathways,provides an additional mechanism to prevent and/or over-come resistance.Here, we describe a novel potent and selective Hsp90
inhibitor, the isoxazole derivative NMS-E973, with antitu-mor efficacy against a broad range of preclinical tumormodels, including tumors resistant to vemurafenib. Its veryfavorable features in terms of biochemical and cellularpotency, broad efficacy spectrum, tumor retention, and BBBpenetration suggest a potential for clinical developmentalso as second line treatment in tumors that have becomeresistant to molecular-targeted agents, also in the presenceof brain metastases as proven by the activity obtained in anintracranial tumor model.
Materials and MethodsReagents and chemicals
NMS-E973,5-[2,4-Dihydroxy-6-(4-nitrophenoxy)phenyl]-N-(1-methylpiperidin-4-yl)-isoxazole-3-carboxamide wassynthesized at Nerviano Medical Sciences S.r.l. laboratories.The synthesis has been previously reported (WO 2010/121963). NMS-E973 and other reference Hsp90 inhibitorswere synthesized in house.
Hsp90 binding and selectivity assaysThe recombinant proteins have been expressed as His-
tagged versions of the human Hsp90 (full length for assaysand a.a. 9–236 for structure determination), human Grp94ATPase domain (a.a. 69–337), and human Trap1 (a.a. 26-end), following the published conditions (15–17). Briefly,they have been expressed in Escherichia coli BL21(DE3) at18�C for 16 hours, then the soluble proteins were purifiedby affinity chromatography on a Ni-Sepharose column (GEHealthcare). After the overnight cleavage of the hexahisti-dine tag by the site-specific protease PreScission (GEHealth-care), the final purification was achieved by size exclusionchromatography (Superdex 200, GE Healthcare), and theproteins were stored in PBS buffer containing 10% glyceroland 0.1 mmol/L EDTA.
A commercially available FITC-Geldanamycin (Invivo-Gen)was used as probe in a fluorescence polarization assay,after its reduction as described elsewhere (18).
For competition experiments, a protein concentration of5 nmol/L for Hsp90 and of 200 nmol/L for Trap1 weremixed with 0.5 nmol/L probe (final concentrations). Afterincubation, the dimethyl sulfoxide (DMSO) compoundsolution was added to the mixture. The plate was incubatedfor 18 hours at room temperature and then the fluorescencepolarization signal was measured. Data were fitted with theprogram Dynafit version 3.28.039 (BioKin Ltd) or Sigma-Plot (SSI) using the mathematical equation for competitivebinding of 2 ligands to the receptor (19).
We synthesized a Grp94 binding probe in house bycoupling NMS-E973 to the fluorophore Atto-610 (Atto-TecGmbH). For competition experiments, Grp94 wasmixed tothe probe at a final concentration of 50 and 20 nmol/Lrespectively.
Fluorescence polarization displacement assay for Hsc70-FL was set up using a commercially available probe N6-6-amino-Hexyl-EDA-ATTO-590-ATP (JenaBioScience).
Determination of NMS-E973 binding kinetics to Hsp90was conducted using Biacore T100 with the approach of thekinetic titration as previously described (20). The data werefitted using the 1:1 kinetic titration model of BIAevaluationsoftware.
NMS-E973 was tested against a panel of 52 diversekinases as previously described (21). The following inhouseproduced kinaseswere tested: ABL, ACK1, AKT1, Alk, AUR1,AUR2, BRK, CDC7, CDK2/CYCA, CHK1, CK2alpha/beta,eEF2K, EGFR1, ERK2, FAK, FGFR1, Flt3, GSK3beta, HaspinV473-K798, IGFR1, IKK2, IR, JAK 1, JAK2, JAK 3, KIT, LCK,MELK, MET, MK2, MPS1, MST4, NEK6, NIM, P38alpha,PAK4, PDGFRb, PDK1, PERK, PIM1, PIM2, PKAalpha,
Translational RelevanceThe clinical benefit observed with drugs targeting
oncogenic kinases is very often transient due to theappearance of resistance linked to secondary mutationsin the target molecules, or to the activation of parallelpathways. Also, brain metastases lead to the progressionof thediseasewhendrugs donot reach the tumor site dueto the presence of the BBB.This study presents original data on the biochemical
and biologic characterization of NMS-E973, a novelsecond generation inhibitor of Hsp90. This nonansa-mycin compound has antitumor efficacy against a broadrange of preclinical tumor models, including models ofresistance to some recently approved kinase inhibitorssuch as vemurafenib and crizotinib. Furthermore, NMS-E973 shows antitumor activity against intracraniallyimplanted tumor xenografts, consistent with its abilityto penetrate the BBB. This feature highlights the poten-tial of this new class of molecules with the ability to gainaccess to the brain, as the most advanced inhibitors ofHsp90 are not brain permeable.
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3521
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

PKCbetaII, PLK1, RET, SULU1, SYK, TRKA, TYK 2, VEGFR2,VEGFR3, ZAP70.
CrystallographyCrystallization studies were conducted using the N-ter-
minal domain of Hsp90a (a.a. 9–236). Crystals of the N-terminal domain of Hsp90 in complex with NMS-E973were grown using the hanging-drop vapor diffusion meth-od from a solution of 20% MPEG 2000K, 0.2 mol/L mag-nesium chloride, 0.1mol/L Cacodylate, pH 6.5, at 4�C. Theprotein was concentrated at 25 mg/mL and compound wasadded to a final concentration of 2 mmol/L. For datacollection, the crystals were transferred to drops containingthe equivalentmother liquorwith 25%glycerol. Diffractiondata were collected at the ESRF (Grenoble, France) onbeamline ID23-2. Data were processed using the HKLpackage (22). The space group is P21. Model building wasdone using Coot (23) and refinement was done withRefMac (24). The coordinates have been deposited in theProtein Data Bank with code 4b7p together with structurefactors and detailed experimental conditions.
Cell cultureHuman cancer cell lines were obtained either from the
American Type Culture Collection (ATCC) or from theEuropean Collection of Cell Culture. Cells weremaintainedin the media at serum concentrations recommended by thesuppliers in a humidified 37�C incubator with 5% CO2.
Cell line authenticationAll cell lines were profiled using DNA fingerprinting
technology (AmpFlSTR Identifiler Plus PCR Amplificationkit, Applied Biosystems) according to the manufacturer’sprotocol. The kit amplifies simultaneously 15 tetranucleo-tide repeated loci and the amelogenin gender marker in asingle assay. The short tandem repeat profiles of the ana-lyzed cell lines were compared with DNA fingerprintingdatabases (e.g., ATCC and DMSZ). All the analyzed profilesshowed a similarity more than 80%. In accord with manypublished studies (25, 26), this is the suggested threshold todeclare the correspondence between the examined cell lineand the external reference. All cell lines are routinely con-firmed at 6-month intervals.
Cell proliferation assayExponentially growing cells were seeded in complete
medium in a 384-well plate format. Twenty-four hoursafter seeding, cells were treated with compounds dissolvedin 0.1% DMSO, at different concentrations. The cells wereincubated at 37�C and 5% CO2 and at the end of treatmenttime, the plates were processed using Cell Titer-Glo assay(Promega) following the manufacturer’s instruction. Cell-Titer-Glo is a homogenous method based on the quantifi-cation of the ATP present, an indicator of metabolicallyactive cells. Briefly, 25 mL reagent solution is added to eachwell and after 5 minutes of shaking the microplates, theluminescent signal is read by Envision (PerkinElmer)luminometer. Inhibitory activity was evaluated comparing
treated versus control data using sigmoidal equation onAssay Explorer (Symix) program.
For combination assays, NMS-E973 and drug solutions,prepared immediately before use, were added to the cells inscalar doses, in 7 point dose–response curves at constantratios, using a separate pipetting step for each drug. Whensequential, rather than simultaneous combinations werestudied, the first drug was added to culture 24 hours aftercell seeding, and the second drug 48 hours after seeding (i.e.at time ¼ 24 hours). At time ¼ 72 hours, cells were lysedusing 50 mL/well of Cell Titer-Glo reagent solution(Promega).
The data were expressed as percentage of viability versusDMSO controls and analyzed using a proprietary software(Beremiz) according to the Chou Talalay equation (27).Combination indices (CI) were calculated for mutuallynonexclusive drugs where a CI <0.3 indicates strong syner-gism; 0.3–0.8 synergism, 0.8–1.2 additivity; 1.3–3 antago-nism; and >3 strong antagonism.
Cell-based assaysThe mechanism of action of the compound was investi-
gated using cell lines treated for 24 hours with compoundsat the indicated concentration of DMSO as control. Clientprotein degradation was determined by Western blot anal-ysis, using DMSO as a control. Immunoblotting was doneaccording to standard procedures (28) and using the fol-lowing antibodies: anti-Hsp70, anti-Hsp90 (Enzo Life Sci-ences), anti-Flt3, anti-B-Raf, anti-PARP1, anti-EGFR (SantaCruzBiotechnology), anti-ERK, anti-pThr201/204ERK, anti-Akt, anti-pSer473 Akt, anti-pTyr1068 EGFR, anti-STAT5,anti-pTyr694 STAT5, anti-C-Raf, Mek1/2, anti-pSer217/221Mek1/2, anti-S6, anti-pSer240/244 S6, anti-cleaved Cas-pase3, anti-pThr199 NPM (Cell Signaling); anti-HistoneH3 (Millipore). The SuperSignal Chemiluminescence Kit(Pierce) was used for detection.
Human xenograft models and antitumor efficacystudies
All procedures adopted for housing and handling ofanimals were in strict compliance with European andItalian Guidelines for Laboratory Animal Welfare. Atotal of 3 � 106 A375 or A2780 cells were transplantedsubcutaneously in athymic nu/nu mice (Harlan). Micebearing a minimal tumor mass (130–180 mm3) wererandomized into vehicle and treated groups made of 7animals each unless differently stated. Intravenous treat-ments started the day after randomization, with differentschedules as described. Tumor dimensions were mea-sured regularly using Vernier calipers and tumor volumewas calculated according to the following formula: length(mm) � width2 (mm)/2.
The percentage of tumor growth inhibition (%TGI) wascalculated as follows:
%TGI¼100�Mean tumor volume of treated group
Mean tumor volumeof control group�100
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3522
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

Toxicity was evaluated on the basis of body weightreduction. At the endof the experiment,micewere sacrificedand gross autopsy findings were reported. Tumor-free ani-mals at 90 days after tumor implant were considered cured.For the leukemia studies, female severe combined immu-
nodeficient (SCID) mice (Harlan) were used. Animals wereexposed to g-irradiation, 200 Rads of whole body gamma-irradiation, within 24 hours of injection of cells. To obtaingrowth as a solid tumor, 5 � 106 MOLM-13 cells wereinjected subcutaneously and treatment initiated whentumor size reached 130 to 200 mm3. Tumor dimensionswere monitored during the experiment and TGI wasassessed as described above. In the case of disseminatedmodel, 10 mice were injected intravenously with 5 � 106
MOLM-13 cells and treatment was initiated after 2 days.Mice were monitored daily for clinical signs of disease anddeaths were recorded for calculation of the median survivaltime.A pharmacokinetic and pharmacodynamic approach,
based on a previously published model (29, 30) wasapplied to the efficacy data of NMS-E973. This model linksthe dosing regimen to the tumor growth dynamics inde-pendently from the schedule and levels of dose. A quanti-tative estimate of the drug potency is assessed through theparameters k2, the proportionality factor linking the plasmaconcentration to the effect, and CT, which provides anestimate of the steady-state drug concentration in plasma(Css) to be maintained for observing tumor stabilization.
PharmacokineticsThe pharmacokinetics of NMS-E973 was investigated in
an ancillary group of 3 tumor-bearing mice. Blood samplesfor the pharmacokinetic assessment were collected, and thedrug was assayed in plasma using liquid chromatography/tandem mass spectrometry techniques (29). The pharma-cokinetics of NMS-E973 was evaluated using a 2 compart-ment model after single intravenous administration of 10mg/Kg.
Data analysisWinnonlin Program (Pharsight) was used both for com-
partmental pharmacokinetic and pharmacokinetic/phar-macodynamic analysis. Parameters were estimated by usingweighted nonlinear least squares (with weight 1/y2observed).
In vivo imaging of A375-LUC intracranially implantedIntracranial model. Balb/c male nude mice, aged 6 to 8
weeks, were anesthetized and stereotactically implanted inthe caudate nuclei region with 1 � 105 melanoma A375-LUCcells in 2mL.With amicrodrill, a small holewas done atthe chosen coordinates and cells were injected at the speedof 1 mL every minute. Hole was closed using bone wax andthe wound with sterile autoclips. After the end of surgery,miceweremonitored for recovery until completewakening.Bioluminescence imaging. Twelve animals were imaged
with an IVIS Lumina device (Xenogen, CaliperLS). Afterintraperitoneal injection of D-luciferin (150 mg/Kg), ani-mals were anesthetized in 2% to 3% isoflurane atmosphere
and 10 minutes after injection of luciferin, mice wereimaged. Images were analyzed and scaled after all acquisi-tions using appropriate computer software (Living Imageversion 3.00; Xenogen Corporation).
Magnetic resonance imaging. MRIwas conducted beforeand 6 days after last treatment on 3 control and 3 treatedanimals. The treatment started 5 days after the injection ofcells. A 7.0 T Bruker Pharmascan instrument was used;anesthetized animals (isofluorane gas anesthesia) werepositioned prone in the animal bed and inserted into theradiofrequency coil (diameter 38 mm) inside the magnet.T2-weighted coronal scans (RARE: TR ¼ 5,000 ms; TEeff ¼57 ms; rare factor ¼ 8; 4 averages; FOV ¼ 2.5�2.5 cm2; Sl.Thickn. ¼ 1 mm; matrix¼ 256�128; acq. time¼ 5 minutesand 20 seconds) were acquired to better visualize tumors. Amacro was used to calculate total tumor volume, startingfrom the bidimenisonal area of all contiguous slices andslice thickness.
ResultsNMS-E973 is a potent and selective inhibitor of HSP90
A fragment-based approach coupled to an intensivemedicinal chemistry effort led to the identification of theisoxazole derivative NMS-E973 (WO2010/121963), a nov-el potent Hsp90 inhibitor structurally unrelated to ansa-mycins (Fig. 1A and Supplementary Fig. S1).
NMS-E973 binds to the ATP binding site of Hsp90awith a DC50 of <10 nmol/L in a fluorescence polarizationdisplacement assay. As this assay has a sensitivity limitclose to the nanomolar concentration of the Hsp90protein used in the assay, we used a direct bindingmethod, Surface Plasmon Resonance analysis, to definea more accurate KD of 0.346 nmol/L. We then studied theselectivity of the compound towards Grp94 and Trap1,the structurally related endoplasmic reticulum and mito-chondrial isoforms of Hsp90, which are also reported tobe overexpressed in tumors and possibly involved incancer development (31–33). We calculated a KD of4.5 nmol/L (>10 fold selectivity) for Grp94, whereas aKD of 670 nmol/L (�200 fold selectivity) was obtainedfor Trap1. No activity was observed when the compoundwas tested against a panel of 52 diverse protein kinases, aswell as against the biologically related ATPase Hsc70(IC50 >10 mmol/L).
As expected, on the basis of its high biochemicalpotency, the crystal structure of NMS-E973 bound to theN-terminal domain of Hsp90 in the ATP binding site(Fig. 1A) shows a broad network of hydrogen bondinginteractions, in addition to those between Asp93 and thephenolic hydroxyl already described for other inhibitors.The resorcinol moiety forms a network of water-mediatedinteractions with the neighborly residues Gly97, Ser52,and Leu48. The 4-nitro-phenoxy group linked to theresorcinol residue is located near a flexible region formedby residues 103–111 and causes its partial rearrangementwithout, however, inducing the opening of the lipophilicpocket described for purine-based inhibitors. Additionalinteractions are provided by the isoxazole-3-carboxamide
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3523
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

group making hydrogen bonds with the carbonyl back-bone of Gly97 and with the nitrogen side chain atom ofLys58, and a supplementary binding contribution is givenby a salt interaction of the piperidine nitrogen with thecarboxyl of Asp102.
NMS-E973 inhibits cancer cell proliferation andinduces client protein degradation
When profiled against a panel of 140 human tumor celllines of various tissue origins and molecular background,NMS-E973 shows a widespread antiproliferative activity,
A
B
Cell line
Activity: Log (
IC50/ M
ean IC
50)
C A2780A375MOLM13
MOLM-13
A2780
A375
0.5
0
–0.5
–1
–1.5
–2
HSP70
H3
Caspase3Cleaved
AKT
Phospho-AKTSer473
Phospho-ERK
ERK
Hsp90
0 0.1 1
C-Raf
AKT
Phospho-AKTSer473
Hsp70
B-Raf
Phospho-MEK
MEK
PARP cleaved
GAPDH
Phospho-ERK
ERK
Phospho-S6
S6
μμmol/L
μmol/L
0 0.1 1
H3 Tot
Hsp70
AKT
Phospho-AKTSer473
Phospho-ERK
ERK
Phospho-STAT5
STAT5
FLT3
Phospho-S6
S6
μmol/L0 0.1 1
Figure 1. A, mode of binding ofNMS-E973 to the active site withinthe crystal structure of Hsp90a.NMS-E973 is shown with yellowcarbon atoms whereas the proteinis colored in green. Ordered watermolecules are shown by redspheres and hydrogen bonds asdashed lines. B, antiproliferativeactivity of NMS-E973 in a panel of140 diverse cell lines, whereactivity is represented as 1/IC50
(mmol/L). C, modulation of Hsp90dependent markers after 24 hourincubation of the specified cancercell line with the indicated dose ofNMS-E973.
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3524
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

with an average IC50 of 1.6 mmol/L and 15 cell lines with anIC50 < 100 nmol/L (Fig. 1B and Supplementary Table S1).As expected, because of the broadmechanismof action of
Hsp90 inhibitors, it was not possible to unambiguouslyassociate the pattern of sensitivity observed across the cellpanel with mutations in a single gene. However, weobserved that all of the most sensitive cell lines are drivenby the disregulated activation of known oncogenicpathways (Table 1), including Alk mutations and ErbB2overexpression, alterations well known to induce clinicalsensitivity to Hsp90 inhibitors, as well as Flt3, B-Raf, PI3K,and RAS mutations.We characterized the in vitro and in vivo effects and
mechanism of action of NMS-E973 in some of these sen-sitive molecular backgrounds.The acute myeloid leukemia (AML) cell line MOLM-13 is
known to be driven by the presence of an internal tandemduplication (ITD) in the Flt3 kinase, leading to constitutiveactivation of its downstream signaling pathway and toSTAT5 phosphorylation. Incubation of the MOLM-13 cellsfor 24 hours with NMS-E973 induced degradation of Flt3and inhibition of its signaling pathway, as shown by thedecrease of STAT5 phosphorylation, as well as of the AKTand MAPK pathways, as shown by the reduction of S6 andMAPK phosphorylation, with the typical induction ofHsp70 that follows Hsp90 inhibition (Fig. 1C).In A375melanoma cells NMS-E973 induced degradation
of B andC-Raf proteins, associatedwith a dual inhibition ofthe MAPK and AKT pathways and with induction of apo-ptosis as shown by PARP cleavage (Fig. 1C) and by time-lapsemicroscopy, which shows that the compound inducesan initial accumulation in mitosis (Supplementary Fig. S2,green arrowheads), followed by extensive pyknosis and
membrane blebbing damage (Supplementary Fig. S2, redarrowheads).
The AKT and MAPK pathways were also simultaneouslyinhibited in the A2780 ovarian cancer cell line upon treat-ment. Again, these modulations were associated with thefeedback induction of Hsp70 expression (Fig. 1C).
Pharmacokinetics and tissue distribution of NMS-E973To explore the possibility to use NMS-E973 for in vivo
experiments, its pharmacokinetic properties were investi-gated in mice. When administered intravenously to 3 miceat 10 mg/Kg, the compound resulted suitable for in vivoadministration with high exposure levels, compatible withbiologic activity, a half life of more than 5 hours and a highvolume of distribution, and associated to a moderate sys-temic clearance (Fig. 2A). The kinetics of tissue distributionof the compound in plasma, brain, liver, and tumor overtime were determined in mice xenografted with the A375tumors after treatment with the active dose of 60 mg/Kg(Fig. 2B). Significant levels of NMS-E973 were detected intumors for 3 days, with amore rapid elimination from liverand plasma. Interestingly, significant concentrations ofNMS-E973, exceeding those observed in plasma, wereobserved in brain, indicating the capacity of the compoundto penetrate the BBB.
NMS-E973 inhibits the growth of A375 tumorssubcutaneously or intracranially implanted in mice
NMS-E973 was administered at 60 mg/Kg twice daily i.v.in the A375 xenograft model according to 2 schedules: (i)every other day for 12 days and (ii) 3 days on/1 day off/3days on (3-1-3, one cycle). Both schedules resulted in tumorshrinkage and TGI of 74% and 89%, respectively (Fig. 2C),
Table 1. Tissue of origin and the keyoncogenic alterationspresent in themost sensitive cell lines (IC50<100nmol/L), with their antiproliferative activity
Cell line TissueNMS-E973IC50 (mmol/L)
Altered oncogenicpathway
DU-4475 Carcinoma breast 0.013 BRAFEVSA-T Carcinoma breast 0.016 HER2MV-4-11 Leukemia acute monocytic 0.029 FLT3HT-1080 Fibrosarcoma 0.034 NRASMOLM-13 Leukemia acute myeloid 0.035 FLT3L-363 Myeloma multiple 0.049 NRASSH-SY5Y Neuroblastoma 0.051 ALKCAL-51 Carcinoma breast 0.056 PIK3CAHCC1954 Carcinoma breast ductal 0.061 HER2A2780 Adenocarcinoma ovary 0.069 PTENBT-474 Carcinoma breast ductal 0.073 HER2HCC1419 Carcinoma breast ductal 0.076 HER2RKO Adenocarcinoma colon 0.084 BRAFHDQ-P1 Carcinoma breast 0.089 HER2A-375 Melanoma 0.133 BRAF
NOTE: The complete list of antiproliferative data is shown in Supplementary Table S1.
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3525
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

associated to amaximum body weight loss of 9% and 13%,which rapidly recovered at the end of the treatment. Thisactivity was superior to that of the inhibitor AUY922 (34),one of the second generation Hsp90 inhibitors currently inclinical phase II, administered at 50 mg/Kg every other dayfor 12 days (Fig. 2C). Interestingly, administration of asecond cycle of NMS-E973 with the intermittent schedule3-1-3 to large regrown tumors (0.5 g), was again efficacious,with tumor regression and 1 of 7 mice tumor free at day 92(Fig. 2C).
To study the ex vivomechanism of action, the compoundwas administered at 60 mg/Kg twice daily, and at differenttime points after the second administration, biomarkermodulationwas investigated in lysates fromA375 xenografttumors. Inhibition of Hsp90 resulted in decreased MAPKpathway activation at early time-points (up to 6–12 hours),followed at later time points (12–24 hours) by induction ofapoptosis, measured as caspase 3 activation and upregula-tion of Hsp70 levels. Most biomarkers returned to controllevels after 24 to 48 hours, although significant recovery ofAKT and BRAF was observed by 12 hours (Fig. 2D). Toachieve sustained modulation of these biomarkers, wetherefore conducted efficacy experiments in which we
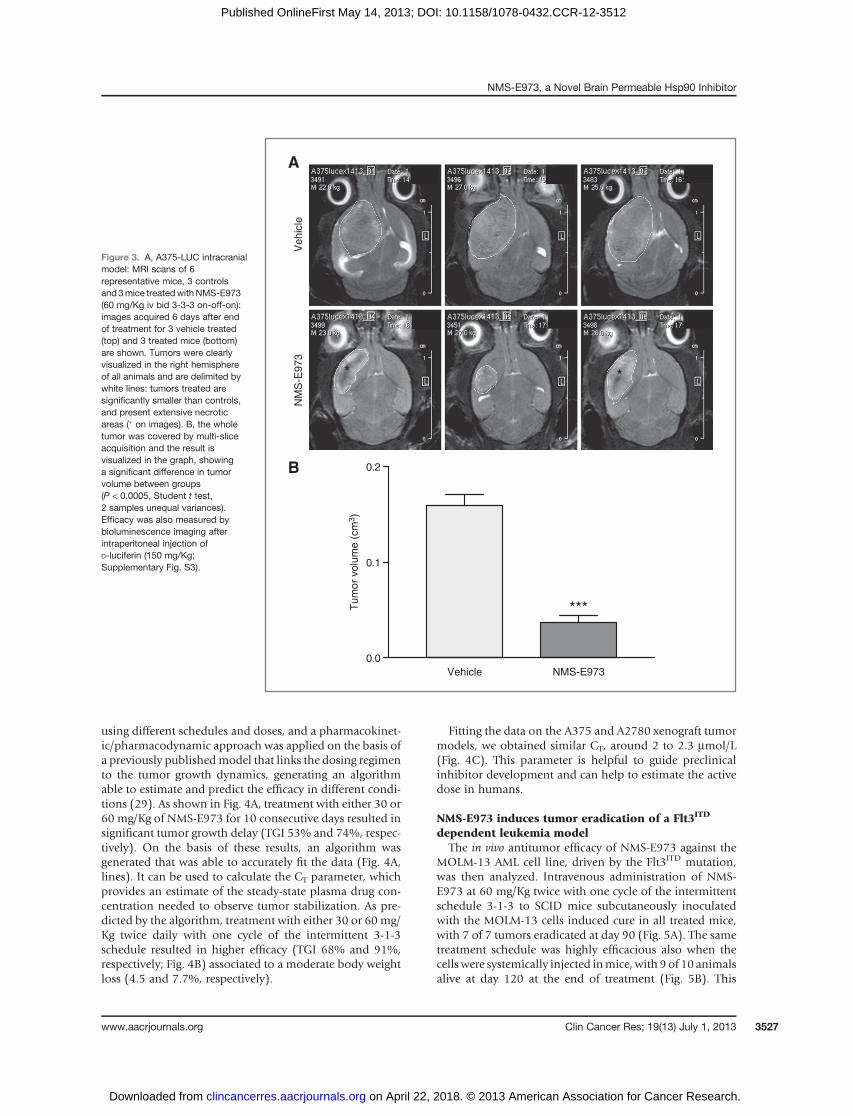
administered the compound every 12 hours (twice daily).On the basis of the observed presence of compound in thebrain, the efficacy ofNMS-E973was tested in an intracranialmodel in which luciferized A375 tumor cell lines wereinjected in the caudate nuclei region of the right brainhemisphere, and 5 days later, treated with 60 mg/Kg twicedaily with one cycle of the intermittent schedule 3-3-3 (3days on/3 days off/3 days on). Bioluminescence imaging ofthe luciferized tumor cells showed a clear signal decreaseafter treatment, with an inhibition of 92% of the lumines-cence signal in the treated group (Supplementary Fig. S3)compared with the control group. Magnetic resonance(MRI) was also used to monitor tumor localization andgrowth 6 days after the end of the treatment, showing 77%decrease in tumor volume in treated groups with respect tocontrols, associated with extensive necrosis visible as areasof lower signal intensity (Fig. 3).
Treatment with NMS-E973 is active in A2780 xenografttumors
As the ovarian carcinoma A2780 cell line is one of themost sensitive to treatment with NMS-E973 in vitro, theefficacy of the compoundwas tested in vivo in 2 experiments
BA
DC
5.83 ± 3.18Vdss L/kg
39.9 ± 1.70CL mL/min./kg
9.26 ± 0.36AUC μmol/L*h.
5.55 ± 1.07T ½ h.
13.23 ± 2.95Cmax μmol/L
NMS-E973
10 mg/kg IV Bolus
Parameter
Mean ± SD
NMS-E973
10 mg/kg i.v. Bolus
Parameter
Mean ± SD
0.01
0.10
1.00
10.00
100.00
7260483624120
Hours
NM
S-E
97
3 c
on
ce
ntr
atio
n (
μmo
l/L
)
PlasmaTumorBrainLiver
0.0
0.5
1.0
1.5
2.0
2.5
3.0
4035302520151050
Vehicle
60 mg/Kg 3-1-3 bid x 2
60 mg/Kg qod x 6 bid
AUY922 50 mg/Kg qod x 6
Mean tum
or
volu
me (
cm
3)
Time (d)
Vehicle 30′ 6 h 12 h 24 h 48 h 72 h
Hsp70
pMEK
Caspase 3 act
GAPDH
AKT tot
pS6
BRAF tot
pERK
Figure 2. A, average pharmacokinetic parameters after injection in 3 mice of 10 mg/mL of NMS-E973. B, kinetics of compound distribution in tumors,plasma, liver, and brain collected at the indicated times after injection of 60 mg/Kg of NMS-E973 in 3 different nude mice bearing A375 tumors.C, antitumor efficacy after subcutaneous injection of A375 cells in 7 nude mice/group. At day 10 NMS-E973 was administered either with the schedule60 mg/Kg iv twice daily, 3-1-3, on-off-on (white triangles, group A), or 60 mg/Kg i.v. twice daily every other day (qod, black triangles), for 6 treatments.At day 30, after tumor regrowth to an average volume of 0.5 cm3, one additional cycle of NMS-E973 60mg/Kg bid, 3-1-3was administered tomice of group A,achieving tumor regressions again. D, ex vivomodulation of markers measured in lysates from A375 tumors (3 tumors/group) isolated at the different post-treatment times as indicated, after single iv administration of 60 mg/Kg of NMS-E973.
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3526
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512
using different schedules and doses, and a pharmacokinet-ic/pharmacodynamic approach was applied on the basis ofa previously publishedmodel that links the dosing regimento the tumor growth dynamics, generating an algorithmable to estimate and predict the efficacy in different condi-tions (29). As shown in Fig. 4A, treatment with either 30 or60 mg/Kg of NMS-E973 for 10 consecutive days resulted insignificant tumor growth delay (TGI 53% and 74%, respec-tively). On the basis of these results, an algorithm wasgenerated that was able to accurately fit the data (Fig. 4A,lines). It can be used to calculate the CT parameter, whichprovides an estimate of the steady-state plasma drug con-centration needed to observe tumor stabilization. As pre-dicted by the algorithm, treatment with either 30 or 60mg/Kg twice daily with one cycle of the intermittent 3-1-3schedule resulted in higher efficacy (TGI 68% and 91%,respectively; Fig. 4B) associated to a moderate body weightloss (4.5 and 7.7%, respectively).
Fitting the data on the A375 and A2780 xenograft tumormodels, we obtained similar CT, around 2 to 2.3 mmol/L(Fig. 4C). This parameter is helpful to guide preclinicalinhibitor development and can help to estimate the activedose in humans.
NMS-E973 induces tumor eradication of a Flt3ITD
dependent leukemia modelThe in vivo antitumor efficacy of NMS-E973 against the
MOLM-13 AML cell line, driven by the Flt3ITD mutation,was then analyzed. Intravenous administration of NMS-E973 at 60 mg/Kg twice with one cycle of the intermittentschedule 3-1-3 to SCID mice subcutaneously inoculatedwith the MOLM-13 cells induced cure in all treated mice,with 7 of 7 tumors eradicated at day 90 (Fig. 5A). The sametreatment schedule was highly efficacious also when thecells were systemically injected inmice,with 9of 10 animalsalive at day 120 at the end of treatment (Fig. 5B). This
Vehic
leN
MS
-E973
* *
NMS-E973Vehicle
0.0
0.1
0.2
Tum
or
volu
me (
cm
3)
***
A
B
Figure 3. A, A375-LUC intracranialmodel: MRI scans of 6representative mice, 3 controlsand 3mice treatedwithNMS-E973(60 mg/Kg iv bid 3-3-3 on-off-on):images acquired 6 days after endof treatment for 3 vehicle treated(top) and 3 treated mice (bottom)are shown. Tumors were clearlyvisualized in the right hemisphereof all animals and are delimited bywhite lines: tumors treated aresignificantly smaller than controls,and present extensive necroticareas (� on images). B, the wholetumor was covered by multi-sliceacquisition and the result isvisualized in the graph, showinga significant difference in tumorvolume between groups(P < 0.0005, Student t test,2 samples unequal variances).Efficacy was also measured bybioluminescence imaging afterintraperitoneal injection ofD-luciferin (150 mg/Kg;Supplementary Fig. S3).
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3527
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

activity was superior to that observed for sunitinib, highlyactive against Flt3, when administered at 40mg/Kg daily for20 consecutive days (deathof all animals byday41; Fig. 5B).
Althoughwe did not conduct ex vivo biomarkermodulationstudies in this experiment, the ability of NMS-E973 toinduce degradation of Flt3ITD was confirmed in MOLM-13 cells in vitro (Fig. 1C)
NMS-E973 is active in resistance modelsEmergence of resistance is a common feature of targeted
therapy with kinase inhibitors, often resulting from muta-tions of critical residues in the ATP pocket, such as thoseobserved after treatment with imatinib in AML (Abl T315I)and in GIST (Kit D816V, T670I, and PDGFRa D842V), orcrizotinib in lung adenocarcinoma (Alk F1174L, L1196M,C1156Y, G1269A; ref. 35).
As expected for Hsp90 inhibitors, NMS-E973 was equallyactive in the inhibition of Ba/F3 cells dependent on Alk ormutants insensitive to crizotinib (F1174L, L1196M,C1156Y and F1174C, Supplementary Fig. S4).
In melanoma, resistance to treatment with vemurafeniband the short duration of responses have not been associ-ated to B-Raf kinase mutations so far, but they are linked tothe activation of alternative signaling pathways (10, 11).Also, in colon, an escape mechanism from B-Raf inhibitionthrough the activation of EGFR signaling has been sug-gested. Infact, B-Raf–mutant colorectal cancer cell lineshave been described to harbor higher basal levels ofEGFR/pEGFR compared with melanoma cells (12), withan increase in pEGFR after vemurafenib treatment (13). Asall the activated pathways identified so far are reported to besensitive to Hsp90 inhibition in vitro (36), treatment withHsp90 inhibitors as single agent or in combination with B-Raf inhibitors could potentially eradicate cancer cells thathave acquired resistance.
Combined in vitro treatment of A375 melanoma cellswith NMS-E973 and PLX-4720 shows a synergic antiproli-ferative activity (Fig. 6A), further supporting the rationale totest in vivo the combination of the 2 drugs. Simultaneousadministration of NMS-E973 at 60 mg/Kg i.v. twice dailywith one cycle of the 3-1-3 schedule, together with B-Rafinhibitor PLX-4720 at 100mg/Kg per os for 10 days, showedan efficacy superior to the treatments as single agents, asjudged by the delay observed in tumor regrowth after initialregression (Fig. 6B), supporting the rationale to combineHsp90 inhibitors with vemurafenib in melanoma.
A
B
C
Vehicle30 mg/kg x 1060 mg/kg x 10
Vehicle30 mg/kg 3-1-3 bid60 mg/kg 3-1-3 bid
Time (d)
28211470
Me
an
tu
mo
r vo
lum
e (
cm
3)
0.5
1.5
2.5
Time (d)
28211470
Me
an
tu
mo
r vo
lum
e (
cm
3)
0.5
1.5
2.5
Model PK/PD parameters
K2
CT
1/μmol/L/day 0.139 (5,8)
2.30 (3.47)
0.107 (16,2)
2.06 (13,7)
K2 1/μmol/L/day
μmol/L
CT μmol/L
Estimate (CV%)
A2780
A375
Figure 4. Antitumor efficacy of NMS-E973 in A2780 model (7 mice/group).NMS-E973 was administered 30 and 60 mg/Kg daily for 10 consecutivedays (A) or 30 and 60 mg/Kg bid, 3-1-3, on-off-on (B). Symbols with errorbars represent experimental measurements and the relative standarderrors; lines representpredictedcurves resulting fromthepharmacokinetic/pharmacodynamic model. The average pharmacokinetic/pharmacodynamic parameters for A2780 and A375models are shown (C).
BA
80200
0.0
0.5
1.0
1.5
2.0
2.5
3.0
VehicleNMS-E973
Mean tum
or
volu
me (
cm
3)
12040200
0
2
4
6
8
10
12
NMS-E973Vehicle
Surv
ival (n
°mic
e)
Time (d) Time (d)
Sunitinib
Figure 5. A, antitumor efficacy ofNMS-E973 administered 60 mg/Kg twice daily, 3-1-3, on-off-on, to7 SCID mice inoculatedsubcutaneously with the MOLM-13 cells. All the animals showcomplete tumor eradication at theend of the experiment (day 90). B,antitumor efficacy of NMS-E973after administration of 60 mg/Kgtwice daily, 3-1-3, on-off-on, to 10SCIDmice inoculated systemicallywith the MOLM-13 cells. Nineanimalswere still alive at day120atthe end of the experiment.
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3528
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

About 8% to 10% of colon carcinomas harbor the B-RafV600E oncogenic lesion and these patients show a verylimited response to vemurafenib (37, 38). Given the poorprognosis of these patients, there is a need of new effectivetreatments for this patient population and it is suggestedthat combining B-Raf inhibitors with drugs with differentmechanism of action could overcome treatment resistancein colon cancer.We found that RKO cells that are resistant to B-Raf
inhibitors (IC50 ¼ 8.5 mmol/L for vemurafenib, >10mmol/L for PLX-4720) are sensitive to Hsp90 inhibitors,with an IC50 of 84 nmol/L for NMS-E973. We analyzed themolecular basis for this sensitivity in RKO cells treated withPLX-4720 and NMS-E973, as single agents or in combina-tion.Weobserved that PLX-4720, even at the highest dose of10mmol/L only partially inhibits B-Raf activity in these cells,with a modest decrease of MEK and ERK phosphorylation
and lack of caspase activation, possibly due to the observedconcomitant increase in phospho-EGFR, which has beendescribed as a potential mechanism of resistance to B-Rafinhibition in colorectal carcinoma (12, 13). Significantly,treatment with NMS-E973 induces downregulation of bothtotal and phospho-EGFR, reduction of MEK, ERK, and S6phosphorylation, coupledwith caspase activationas a singleagent and is able to markedly enhance suppression of BRAFsignaling by PLX-4720 when combined with this agent.
Moreover, treatment with NMS-E973 induced a degra-dation of C-Raf and B-Raf proteins, which was partiallyabrogated by combined treatment with PLX-4720. Theseresults suggest that treatment with NMS-E973 represents apotential approach to bypass escape mechanisms from B-Raf inhibition in this tumor context.
Accordingly,we treatednudemicebearingRKOxenografttumors with NMS-E973 (60 mg/Kg twice daily) with the
50403020100
Vehicle
NMS-E973
Time (d)
CI @ 90%CI @ 70%Ratio of combination
1 : 0.5 0.360.561 : 1 0.430.651 : 2 0.540.751 : 4 0.660.83
Combination studies using NMS-E973 and
PLX4720 on A375 cell line.
Sequential treatment: PLX4720 administered
24 hr before NMS-E973
A
0.0
0.5
1.0
1.5
2.0
4035302520151050
Vehicle
NMS-E973
PLX-4720
NMS-E973 + PLX-4720
Mean tum
or
volu
me (
cm
3)
B
C
D
Mean tum
or
volu
me
(cm
3)
Time (d)
10.1010.1010.10 NMS-E973 (μmol/L)101010111000 PLX-4720 (μmol/L)
Phospo-ERK
Phospho-MEK
B-RAF
HSP70
MEK
C-RAF
Phospho-S6
ERK
S6
PARP
H3
EGFR
Phospho-EGFR
0
0.5
1
1.5
2
2.5
3
Figure 6. A, combination experiment of NMS-E973 with PLX-4720. Twenty-four hours after the seeding of exponentially growing A375 cells, the experimentwas started by addition of PLX-4720, followed 24 hours later by the treatment with scalar doses of NMS-E973. C.I. calculated for mutually nonexclusivedrugs are indicative of synergism. B, antitumor efficacy in A375 model (7 mice mice/group). NMS-E973 was administered with the schedule 60 mg/Kgtwice daily, 3-1-3, on-off-on, whereas PLX-4720 was given orally for 10 consecutive days. The combination resulted in an efficacy superior to the singletreatments, as judged by the delay observed in tumor regrowth after initial regression, and also by the increase in median survival time for the combination ofthe drugs (59 days), as compared to the MST of 32, 39, and 42 days registered for controls, PLX-4720 and NMS-E973, respectively. C, modulation ofHsp90-dependent markers after 24 hour incubation of RKO cells with the indicated doses of NMS-E973 and PLX-4720 as single agents or in combination.D, in vivo antitumor efficacy in the RKOmodel resistant to vemurafenib. RKOcells were inoculated subcutaneously in 7 nudemice/group, andNMS-E973wasadministered with the schedule 60 mg/Kg bid, 3-1-3, on-off-on.
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3529
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

intermittent schedule 3-1-3 and observed tumor shrinkagein all the animals with a TGI of 93%, with 2 of 7 animalsremaining tumor free at day90 after the endof the treatment(Fig. 6D).
DiscussionDespite the significant number of molecules that have
entered clinical studies (39–41), the development of Hsp90inhibitors hasbeendelayed so far by theweakpotencyof thefirst-generation drugs that resulted in modest clinical activ-ity, by the difficulty to optimize the treatment scheduletowards a suitable therapeuticwindow, andby the challengeto identify the most sensitive patient population. However,there is currently a new wave of interest in this field linkedto the promising clinical activity observed recently withretaspimycin and ganetespib in Alk-driven NSCLCs (5, 6)and in ErbB2-positive metastatic breast cancers (7, 42, 43),that allowed to identify clinical settings in which scheduleoptimization of agents with higher potency resulted inpermissive conditions for selected Hsp90 inhibitors.
Here, we showed the characterization of NMS-E973, alead compound belonging to a novel isoxazole series struc-turally unrelated to ansamycins.NMS-E973bindswithhighpotency and selectivity to the ATP binding pocket ofHsp90a, shows broad antiproliferative activity in vitro asso-ciated to the degradation of Hsp90 client proteins such asFlt3, B-Raf, AKT,with the resultant blockof keypathways fortumor cell progression, such as the Raf/MAPK, PI3K/AKT,and JAK/STAT signal transduction pathways.
In vivo, NMS-E973 showed high antitumor efficacy in allthe models tested, including A375 and A2780 xenografts,achieving tumor shrinkage also after rechallenge of largetumor masses. Schedule optimization supported by an effi-cient pharmacokinetic/pharmacodynamic model allowedidentification of the 3-1-3 treatment as an efficient way tomaximize the efficacy of the compound at tolerated doses.
NMS-E973 showed efficacy in models representative ofclinical settings with high medical need. The best approachto the treatment of Flt3ITD AML is currently undefined, andmultiple clinical trials are investigating inhibitors of the Flt3kinase. These agents showed, so far, clinical activity inpatients with Flt3-mutant AML that correlates with effectivein vivo suppression of the Flt3 target, although it is very oftentransient, possibly due to inadequate dosing or insufficientselectivity of these drugs that may reduce their tolerability(44). NMS-E973 treatment of the MOLM-13 Flt3ITD tumorcell line caused efficient target degradation and silencing ofthe pathway and resulted in complete eradication of thetumors after both subcutaneous and intravenous tumor cellimplantation. This activity was superior to that of themulti-kinase inhibitor sunitinib in the same experiment andcompared favorably with the activity of AC220 (45), themost potent and selective Flt3 inhibitor described so far,tested in the same model (data not shown). Resistancemutations to Flt3 inhibitors in clinics are beginning toemerge, such as the D835Y mutation in the tyrosine kinasedomain (45), raising the opportunity for future studies withNMS-E973 in this setting.
Metastases to vital organs of difficult surgical access arethe ultimate cause of death when not controlled, and brainmetastases are the most life-threatening among the second-ary localizations of cancers such asmelanoma, NSCLC, andbreast cancers for their unresponsiveness to chemothera-peutic treatments. Brain cancers represent another crucialunmet medical need, and although the presence and func-tion of the BBB in brain tumors is still poorly understood, itis likely that small tumors could be protected from thera-peutics by the barrier itself, creating a "sanctuary" site (46,47). AmongHsp90 inhibitors, SNX-5422does not reach thebrain (48), the efficacy of AUY-922 in brain cancers hasbeen restricted so far to subcutaneously implanted glioblas-tomas due to low BBB permeability (49), and only CUDC-305 (50) is reported to have the capacity to penetrate thebrain barrier. We showed that NMS-E973 enters the brainand slows the progression of an intracranially implantedmelanoma model and would suggest the opportunity ofinvestigating clinical efficacy on brain tumors while poten-tial CNS effects should clearly be closely monitored.
Vemurafenib and crizotinib are recently approved kinaseinhibitors that have shown unprecedented activity in mel-anoma and Alk-positive NSCLC, respectively. However, theefficacy of these drugs is limited by target mutations (forAlk), resistance mechanisms based on activation of addi-tional pathways and progression due to brain metastases.Also, vemurafenib has failed to show activity in B-Raf–mutated colon cancers. The preclinical features of NMS-E973 have the potential to overcome these limitations,based on its high activity towards the known Alk resistancemutations and theV600E vemurafenib resistant tumors, thebroad spectrum of Hsp90 target proteins, the ability topenetrate the blood brain barrier and its proven activity inintracranial tumor models. The observed synergy with theB-Raf inhibitor PLX-4720 in melanoma may also suggestadditional therapeutic combinations in some tumors.
The results obtained for NMS-E973 show the high poten-tial of this class in the Hsp90 arena, including the ability topass the BBB which is an uncommon feature among thecompounds described so far. We are currently completingthe lead optimization of the NMS-E973 class to allow thefinal selection of the candidate for clinical studies.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: G. Fogliatto, M.G. Brasca, E. Casale, P. Polucci, C.Visco, F. Zuccotto, D. Donati, E. Felder, S. Mantegani, A. IsacchiDevelopment of methodology: L.M. Gianellini, M. Paolucci, F. Sola, C.Visco, S. ManteganiAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): D. Ballinari, A. Degrassi, A.D. Ponti, M. Russo,E. Pesenti, S. ManteganiAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): L.M.Gianellini, E. Casale,D. Ballinari,M.Ciomei, A. Degrassi, M. Germani, M. Guanci, M. Paolucci, M. Russo, B.Valsasina, C. Visco, F. Zuccotto, D. Donati, E. Felder, A. IsacchiWriting, review, and/or revision of the manuscript: G. Fogliatto, M.G.Brasca, D. Ballinari, M. Ciomei, M. Germani, M. Paolucci, C. Visco, D.Donati, E. Felder, S. Mantegani, A. Galvani, A. Isacchi
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3530
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

Administrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): G. Fogliatto, F. Sola, C. Visco, A.GalvaniStudy supervision: G. Fogliatto, M. Ciomei, D. Donati, E. Pesenti, S.Mantegani, A. Isacchi
AcknowledgmentsThe authors thank Nadia Amboldi, Antonella Ciavolella, Paolo Cappella,
Claudio Dalvit, Ron Ferguson, Laura Mancini, Jan Malyszko, Gaia Sanga,
Alessio Somaschini, theNMSbiochemical and cellular screening groups, andFrancesco Caprera and the in vivo group for their excellent technicalassistance.
The costs of publicationof this articlewere defrayed inpart by thepaymentof page charges. This articlemust therefore be herebymarked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received November 13, 2012; revised April 6, 2013; accepted April 30,2013; published OnlineFirst May 14, 2013.
References1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation.
Cell 2011;144:646–74.2. Neckers L. Heat shock protein 90: the cancer chaperone. Heat Shock
Proteins in Cancer 2007;231–52.3. Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical
development of heat shock protein 90 (Hsp90) inhibitors in cancers.Biochim Biophys Acta 2012;1823:742–55.
4. Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, et al.Development of 17-allylamino-17-demethoxygeldanamycin hydro-quinone hydrochloride (IPI-504), an anti-cancer agent directed againstHsp90. Proc Natl Acad Sci U S A 2006;103:17408–13.
5. Wong K, KoczywasM, Goldman JW, Paschold EH, Horn L, Lufkin JM,et al. An open-label phase II study of the Hsp90 inhibitor ganetespib(STA-9090) as monotherapy in patients with advanced non-small celllung cancer (NSCLC). J Clin Oncol 2011;29(suppl):7500.
6. Neckers L,Workman P. Hsp90Molecular chaperone inhibitors: are wethere yet? Clin Cancer Res 2012;18:64–76.
7. Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, et al.HSP90 inhibition is effective in breast cancer: a phase II trial oftanespimycin (17-AAG) plus trastuzumab in patients with HER2-pos-itive metastatic breast cancer progressing on trastuzumab. Clin Can-cer Res 2011;17:5132–9.
8. Ying W, Du Z, Sun L, Foley KP, Proia DA, Blackman RK, et al.Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibitspotent antitumor activity and a superior safety profile for cancertherapy. Mol Cancer Ther 2012;11:475–84.
9. Sasaki T, KoivunenJ,OginoA,YanagitaM,NikiforowS,ZhengW, et al.A novel ALK secondarymutation andEGFR signaling cause resistanceto ALK kinase inhibitors. Cancer Res 2011;71:6051–60.
10. Poulikakos PI, ZhangC, Bollag G, Shokat KM, RosenN. RAF inhibitorstransactivate RAF dimers and ERK signaling in cells with wild-typeBRAF. Nature 2010;464:427–30.
11. Poulikakos PI, Rosen N. Mutant BRAF melanomas–dependence andresistance. Cancer Cell 2011;19:11–5.
12. Corcoran RB, Ebi H, Turke AB, Coffee EM, NishinoM, Cogdill AP, et al.EGFR-mediated reactivation of MAPK signaling contributes to insen-sitivity of BRAF-mutant colorectal cancers to RAF inhibition withvemurafenib. Cancer Discov 2012;2:227–35.
13. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D,et al. Unresponsiveness of colon cancer to BRAF (V600E) inhibitionthrough feedback activation of EGFR. Nature 2012;483:100–3.
14. CostaDB,Kobayashi S, PandyaSS, YeoWL,ShenZ, TanW, et al. CSFconcentration of the anaplastic lymphoma kinase inhibitor crizotinib.J Clin Oncol 2011;29:e443–5.
15. Panaretou B, ProdromouC, RoeSM,O'Brien R, Ladbury JE, Piper PW,et al. ATP binding and hydrolysis are essential to the function of theHsp90 molecular chaperone in vivo. EMBO J 1998;17:4829–36.
16. Leskovar A, Wegele H, Werbeck ND, Buchner J, Reinstein J. TheATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem2008;283:11677–88.
17. Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures ofGRP94-nucleotide complexes reveal mechanistic differencesbetween the hsp90 chaperones. Mol Cell 2007;28:41–56.
18. Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, et al.BIIB021, an orally available, fully synthetic small-molecule inhibitor ofthe heat shock protein Hsp90. Mol Cancer Ther 2009;8:921.
19. Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data:application to HIV proteinase. Anal Biochem 1996;237:260–73.
20. Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. Analyzing akinetic titration series using affinity biosensors. Anal Biochem2006;349:136–47.
21. Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L,et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitu-moral activity. Clin Cancer Res 2006;12:4080–9.
22. Otwinowski Z, Minor W. Processing of X-ray diffraction data collectedin oscillation mode. Methods Enzymol 1997;276:307–26.
23. Emsley P, Cowtan K. Coot: model-building tools for molecular gra-phics. Acta Crystallogr D Biol Crystallogr 2004;60(Pt 12 Pt 1):2126–32.
24. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecularstructures by the maximum-likelihoodmethod. Acta Crystallogr D BiolCrystallogr 1997;53(Pt 3):240–55.
25. Masters JR, Thomson JA, Daly-Burns B, Reid YA, DirksWG, Packer P,et al. Short tandem repeat profiling provides an international referencestandard for human cell lines. Proc Natl Acad Sci U S A 2001;98:8012–7.
26. Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y,Chanock SJ, et al. DNA fingerprinting of the NCI-60 cell line panel.Mol Cancer Ther 2009;8:713–24.
27. Chou TC, Talalay P. Analysis of combined drug effects: a new look at avery old problem. Trends Pharmacol Sci 1983;4:450–4.
28. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteinsfrom polyacrylamide gels to nitrocellulose sheets: procedure andsome applications. Proc Natl Acad Sci U S A 1979;76:4350–4.
29. SimeoniM,Magni P, CammiaC, DeNicolaoG,Croci V, Pesenti E, et al.Predictive pharmacokinetic-pharmacodynamic modeling of tumorgrowth kinetics in xenograft models after administration of anticanceragents. Cancer Res 2004;64:1094–101.
30. Magni P, Germani M, De Nicolao G, Bianchini G, Simeoni M, Poggesi I,et al. A minimal model of tumor growth inhibition. IEEE Trans BiomedEng 2008;55:2683–90.
31. McLaughlin M, Vandenbroeck K. The endoplasmic reticulum proteinfolding factory and its chaperones: new targets for drug discovery? BrJ Pharmacol. 2011;162:328–45.
32. Ni M, Lee AS. ER chaperones in mammalian development and humandiseases. FEBS Lett 2007;581:3641–51.
33. Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrialHsp90. Biochim Biophys Acta 2012;1823:767–73.
34. Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, et al.NVP-AUY922: a novel heat shock protein 90 inhibitor active againstxenograft tumor growth, angiogenesis, and metastasis. Cancer Res2008;68:2850–60.
35. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, WeickhardtAJ, et al. Mechanisms of resistance to crizotinib in patients with ALKgene rearranged non-small cell lung cancer. Clin Cancer Res 2012;18:1472–82.
36. Paraiso KH, Haarberg E, Wood E, Rebecca VW, Chen YA, Xiang Y,et al. The heat shock protein-90 inhibitor XL888 overcomes BRAFinhibitor resistance mediated through diverse mechanisms. Clin Can-cer Res 2012;18:2502–14.
37. Sullivan RJ, Flaherty KT. BRAF in Melanoma: Pathogenesis, Diagno-sis, Inhibition, and Resistance. J Skin Cancer 2011;2011:423239.
38. Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al.Antitumor activity of BRAF inhibitor vemurafenib in preclinical modelsof BRAF-mutant colorectal cancer. Cancer Res 2012;72:779–89.
39. Jhaveri K, Modi S. HSP90 inhibitors for cancer therapy and overcom-ing drug resistance. Adv Pharmacol 2012;65:471–517.
NMS-E973, a Novel Brain Permeable Hsp90 Inhibitor
www.aacrjournals.org Clin Cancer Res; 19(13) July 1, 2013 3531
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

40. Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery anddevelopment of heat-shock protein 90 inhibitors for cancer treatment.Expert Opin Drug Discov 2011;6:559–87.
41. Soga S, Akinaga S, Shiotsu Y. Hsp90 inhibitors as anti-cancer agents,from basic discoveries to clinical development. Curr Pharm Des2013;19:366–76.
42. Synta corporate presentation March 2013 [cited 2013 Apr 8]. Availablefrom: http://www.syntapharma.com/Documents/Synta_Corporate_presentation.pdf.
43. Arteaga CL. Why is this effective HSP90 inhibitor not being developedin HER2 þbreast cancer? Clin Cancer Res 2011;17:4919–21.
44. Fathi AT, Chabner BA. FLT3 inhibition as therapy in acute myeloidleukemia: a recordof trials and tribulations.Oncologist 2011;16:1162–74.
45. Moore AS, Faisal A, Gonzalez de Castro D, Bavetsias V, Sun C, AtrashB, et al. Selective FLT3 inhibition of FLT3-ITDplus; acute myeloidleukaemia resulting in secondary D835Ymutation: a model for emerg-ing clinical resistance patterns. Leukemia 2012;26:1462–70.
46. Deeken JF, Loscher W. The blood-brain barrier and cancer: trans-porters, treatment, and Trojan horses. Clin Cancer Res 2007;13:1663–74.
47. Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK.The biology of brain metastases—translation to new therapies. NatRev Clin Oncol 2011;8:344–56.
48. Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, et al.SNX2112, a synthetic heat shock protein 90 inhibitor, has potentantitumor activity againstHERkinase-dependent cancers.ClinCancerRes 2008;14:240–8.
49. Gaspar N, Sharp SY, Eccles SA, Gowan S, Popov S, Jones C, et al.Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922in adult and pediatric glioblastoma. Mol Cancer Ther 2010;9:1219–33.
50. Bao R, Lai CJ, Qu H,Wang D, Yin L, Zifcak B, et al. CUDC-305, a novelsynthetic HSP90 inhibitor with unique pharmacologic properties forcancer therapy. Clin Cancer Res 2009;15:4046–57.
Fogliatto et al.
Clin Cancer Res; 19(13) July 1, 2013 Clinical Cancer Research3532
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512

2013;19:3520-3532. Published OnlineFirst May 14, 2013.Clin Cancer Res Gianpaolo Fogliatto, Laura Gianellini, Maria G. Brasca, et al. Including Intracranial Metastasesagainst Multiple Models of Drug Resistance to Targeted Agents, NMS-E973, a Novel Synthetic Inhibitor of Hsp90 with Activity
Updated version
10.1158/1078-0432.CCR-12-3512doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2013/05/15/1078-0432.CCR-12-3512.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/19/13/3520.full#ref-list-1
This article cites 48 articles, 25 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/19/13/3520To request permission to re-use all or part of this article, use this link
on April 22, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 14, 2013; DOI: 10.1158/1078-0432.CCR-12-3512