nitrogÉn-monoxid szerepe t limfocita...
TRANSCRIPT

D o k t o r i é r t e k e z é s
NITROGÉN-MONOXID SZEREPE T LIMFOCITA
AKTIVÁCIÓBAN
Dr. Koncz Ágnes
Semmelweis Egyetem Molekuláris Orvostudományok Tudományági Doktori Iskola
Témavezető: Dr. Buzás Edit egyetemi docens, orvostudományok doktora
Hivatalos bírálók: Dr. Bajtay Zsuzsanna Ph.D., tudományos munkatárs
Dr. Igaz Péter Ph.D., egyetemi tanársegéd Szigorlati bizottság elnöke: Prof. Dr. Füst György egyetemi tanár, orvostudományok doktora Szigorlati bizottság tagjai: Prof. Dr. Sipka Sándor egyetemi tanár,
orvostudományok doktora Dr. Pállinger Éva Ph.D., tudományos munkatárs
Budapest
2008.

2
T A R T A L O M J E G Y Z É K I.RÖVIDÍTÉSEK JEGYZÉK ……………………………………………………….4
II.BEVEZETÉS …………………………………………………………………5
A. T limfociták jellemzői ……………………………………………………....5
B. T limfocita aktiváció és nitrogén-monoxid ………………………………….6
C. Nitrogén-monoxid …………………………………………………………8
D. Nitrogén-monoxid szerepe a mitokondrium bioszintézisben……………..11
E. Nitrogén-monoxid szerepe az immunregulációban ………………………13
F. Reaktív oxigén intermedierek és szerepük T limfociták aktivációjában…...15
G. Hisztamin ………………………………………………………………….19
H. Hisztamin hatása a T limfociták citokintermelésére és érésére…………….22
III. CÉLKITŰZÉSEK ……………………………………………………………….26
IV. MÓDSZEREK …………………………………………………………………27
A. T limfocita aktiváció mérése ……………………………………………..27
B. Hisztidin dekarboxiláz génkiütött (HDC-KO) állat………………………27
C. Áramlás citometriás mérések …………………………………………...28
D. Western-blot módszer ……………………………………………………..30
E. ELISPOT módszer ………………………………………………………31
F. Nitrit/nitrát mérés ……………………….......................................32
G. PCR alapú technikák ……………………………………………………...32
H. Statisztikai elemzések ……………………………………………………33
V. EREDMÉNYEK ……………………………………….........................................34
A. Nitrogén-monoxid szerepének vizsgálata T limfociták aktivációjában …….34
B. HDC-KO egér eltérő citokintermelése………………………………………42
C. Hisztamin hatása a limfociták nitrogén-monoxid termelésére ..…………….44
D. Eltérő T limfocita aktiváció a HDC-KO egerekben ………………………..46

3
E. Nitrogén-monoxid hatása az IFN-γ termelésre .…………………………….48
VI. MEGBESZÉLÉS ÉS KÖVETKEZTETÉSEK ……………………………50
VII. ÖSSZEFOGLALÁS …………………………………………………………56
VIII. SUMMARY …………………………………………………………………...58
IX. SAJÁT PUBLIKÁCIÓK LISTÁJA …………………………………………….59
X. KÖSZÖNTENYILVÁNÍTÁS …………………………………………………. 61
X. IRODALMI JEGYZÉK ……………………………………………………….62

4
I. RÖVIDÍTÉSEK JEGYZÉKE
TCR: T sejt receptor
Th: T helper sejtek
NO: nitrogén-monoxid
ZAP-70: ζ-associated protein-70
LAT: linker for activation of T cells
IP3: inozitol-1,4,5-trifoszfát
ROI: reaktív oxigén intermedierek
NOS: nitrogén-monoxid szintetáz; nNOS, neuronális NOS; eNOS, endotheliális NOS;
iNOS, indukálható NOS
2-APB: 2-aminoetoxidifenil borát
MnTBAP: mangán (III) tetrakis (4-benzoesav)porfirin klorid
C-PTIO: carboxi-2-fenil-4,4,5,5-tetrametil-imidazolin-1-oxil-3-oxid
HE: hidroetidin
TMRM: tetrametilrhodamin metil észter
DAF-FM: 4-amino-5-metilamino-2’,7’-difluorofloureszcein diacetát
Fluo-3: fluoro-3 acetoximetil-észter
NOC-18: (Z)-1-[2-(2-aminoetil)-N-(2-ammonioetil)amino]diazen-1-ium-1,2-diolát
dietilenetriamin
ConA: Concavalin A
CFA: komplett Freund’s adjuváns
HDC: hisztidin dekarboxiláz
IL: interleukin
IFN: interferon

5
II. BEVEZETÉS
A. T limfociták jellemzői
T limfociták központi szerepet játszanak az adaptív immunválaszban. A sejtfelszínükön
expresszálódó T sejt receptor (TCR) az antigén prezentáló sejt (APC) felszínén
exresszálódó fő hisztokompatibilitási komplex (MHC) által bemutatott antigénepitóp
specifikus felismerésére képes. T limfociták érése a thymusban történik, melynek során
az autoreaktív T limfociták kiszelektálódnak. A thymusból perifériára kerülő T sejtek
jellegzetes sejtfelszíni receporaik alapján több típusba sorolhatók.
A CD8 sejtfelszíni receptorral rendelkezőket citotoxikus T sejteknek (Tc) nevezzük.
Mint nevük is mutatja sejtkárosító hatással rendelkeznek, fontos szerepük van az
intracelluláris parazitával, vírussal fertőzött sejtek és a tumor sejtek elpusztításában,
illetve fő mediátorai a traszplantáció után fellépő kilökődési mechanizmusnak.
A CD4 sejtfelszíni receptort viselő sejteket T helper (Th) sejteknek nevezzük.
Jellemzőjük, hogy aktiváció során immunválaszt befolyásoló citokineket termelnek.
Ezeket a sejtek citokintermelése alapján több alcsoportba sorolhatjuk: Th1, Th2, Th17.
Th1 sejtek elsősorban IL-2, IFN-γ, és IL-12 citokineket termelnek, jellemző
transzkripciós faktoruk a T-bet. A Th2 sejtek által termelt citokinek az IL-4, IL-5, IL-6,
IL-10, IL-13; jellegzetes transzkripciós faktoruk a GATA-3. Az újabban leírt Th
alcsoport a Th17, melynek jellemző citokinje az IL-17, transzkripciós faktora pedig a
ROR-γ, irodalmi adatok szerint fontos szerepe van egyes autoimmun betegségek
patomechanizmusában. A CD4 sejtfelszíni receptorral rendelkező T limfociták egy
további csoportját alkotják az un. regulatorikus T sejtek (Treg). Ezeket a sejteket
szupresszor T sejteknek is nevezik mert legfontosabb feladatuk a T sejt által közvetített
immunválasz gátlása illetve a thymusból a perifériára kerülő autoreaktív T sejtek gátlása.
Az irodalom a Treg limfociták két csoportját különbözteti meg; a természetes Treg és az
adaptív Treg populációt. A természetes Treg sejtek jellemzője a sejtfelszínen
expresszálódó CD4 mellett magas CD25 (IL2Rα) receptor expresszió illetve a foxp3
transzkripciós faktor kifejeződése. Ezen sejtek érése a thymusban történik, míg az
adaptív Treg sejtek a periférián alakulnak ki az immunválasz során [1.]

6
A T limfociták felszínén expresszálódó molekulák valamint a sejtek
mikrokörnyezetében jelenlévő citokinek és szolubilis molekulák fontos részét képezik a
T sejt aktivációnak és érésnek. Munkánk során vizsgáltuk hogy két bioaktív molekula, a
nitrogén monoxid (NO) és a hisztamin milyen szerepet játszik a T sejtek aktivációjában,
jelátviteli folyamataiban és citokin termelésében.
B. T limfocita aktiváció és nitrogén monoxid
Az antigén kötődése a T sejt receptorhoz (TCR) Lck és Fyn tirozin kinázok
aktiválódásához vezet. Ezen tirozin kinázok felelősek a TCR ζ lánc és a CD3 alegység
foszforilációjáért, melyek ilyen formában a ζ- associated protein-70 (ZAP-70)
molekulát képesek lehorgonyozni és ezáltal aktiválni. A ZAP-70 protein foszforilálja
a ’linker for activation of T cells’ (LAT) proteint, mely direkt módon kötődik többek
között a foszfolipáz C-γ1-hez. A foszfolipáz C-γ1 katalizálja a foszfatidol-inozitol-4,5-
bifoszfát hidrolízisét inozitol-1,4,5-triszfoszfáttá (IP3) és diacilglicerollá (DAG). Az IP3
kötődése belső Ca2+ raktárakon lévő receptorához Ca2+-ot mobilizál, létrehozva ezzel a
citoplazmatikus Ca2+-szignált. A DAG protein kináz-C aktivációt okoz. Ezek a
útvonalak továbbiakban traszkripciós faktorok aktiválásáért, citokintermelés
beindításáért, T sejt proliferációért felelősek [2;3]. (1. ábra) A T sejt aktivációhoz
azonban a T sejt receptor (TCR) által közvetített folyamat mellett kostimulációs
szignálokra (CD28, CD40 ligand, LFA-1, CD2) is szükség van.
A T sejt aktiválással párhuzamosan jelentős strukturális változások is bekövetkeznek,
ami szintén nélkülözhetetlen részjelensége a hatékony T sejt aktivációnak. A T limfocita
antigénprezentáló sejt felé eső részén kialakuló szupramolekuláris aktivációs komplex
(SMAC) aktivált enzimeket, adaptor proteineket, szignáltranszdukcióban részvevő
proteineket tartalmaz és szabályozza ezek konformációváltozását. Ezen kívül a T sejt
membránja ezen a részen koleszterolban és szfinglipidben gazdaggá válik, un. lipid-
tutajok (raftok) alakulnak ki. A T sejt aktiváció során tehát létrejön az immunológiai
szinapszis, melyben a sejt polarizálttá válik [4;5;6].

7
1. Ábra: T sejt receptor aktivációt követő jelátviteli folyamatok sematikus rajza.
A T sejt immunszinapszisban történő polarizációjának részjelensége az az újabban leírt
szubcelluláris megfigyelés, miszerint a T sejt antigénprezentáló sejttel kapcsolódó
részében kétszer nagyobb mennyiségű NO termelődik mint a citoplazma többi részében.
Ennek hátterében az eNOS megváltozott lokalizációja áll. Az immunológiai
szinapszisban fokozott NO termelés új megvilágításba helyezi a T sejt aktiváció
folyamatát, a helyileg nagyobb mennyiségben termelődő NO ugyanis befolyásolja a
CD3 ζ lánc és a ZAP-70 foszforilációt [7]. Ezen eredmények is alátámasztják a NO T
sejt aktivációban betöltött szerepét [8].
Lck/Fyn
γ
ZAP-70
α β
ζζε
PP
P
P
kostimulációs szignálok
P P
P
LATP PP
PLCγ1 PIP2
IP3 DAG
CD3
ANTIGÉN
+
T sejt aktiváció, proliferáció
NO
Ca2+ szignál PKC aktiváció

8
C. Nitrogén-monoxid
A nitrogén-monoxid (NO) a sejtfunkciók fontos és sokrétű fiziológiás szabályozója.
NO-t emittáló gyógyszereket használunk egyes betegségek gyógyítására, továbbá az NO
a környezetünkben is jelentős mennyiségben megtalálható. Molekuláris oxigénből és
nitrogénből igen magas hőmérsékleten, például a légkörben villámlás során keletkezhet.
NO termelődik továbbá a belső égésű motorok, erőművek működése során is. NO és
ózon kemiluminescens reakciója során oxigén és nitrogén dioxid keletkezik, a
folyamatot kísérő fény fotodetektorral laboratóriumi körülmények között mérhető, ez
képezi a NO mérésére használható egyik módszer alapját is. Az ózonnal történő
reakciója miatt a NO az ózonréteg elvékonyodásában, továbbá a savas esők
kialakulásában is szerepet játszik.
A NO fontos intra-, trasz- és intercelluláris hírvivő molekula mely számos fiziológiás és
patológiás folyamatban szerepet játszik. A NO egyik legjobban ismert funkciója az
értónus szabályozása, vazodilatációt okoz, növeli az oxigén szükségletet [9;10].
A NO élettani hatásainak kutatásában elért eredményeiért Robert F Furchgott, Louis J
Ignarro és Ferid Murad 1998-ban Nobel díjat kapott, fő kutatási területük a NO szív és
érrendszeri szerepének vizsgálata volt. Megfigyeléseik szerint a NO számos jelátviteli
útvonalat szabályozó gáz, mely a legtöbb jelátvitelt szabályozó anyaggal szemben,
szabadon átjut a biológiai membránokon. A NO egyik fontos intracelluláris hatása tiol
és cisztein csoportok S-nitrozilációja. Ez a poszttranszlációs módosulás reverzibilis
módon befolyásolja a proteinek funkcióját. A másik ismert hatásmechanizmusa a guanil
cikláz aktivitásának befolyásolása révén a cGMP szint emelkedése [11].
Szervezetünkben NO L-argininből képződik, nitrogén monoxid szintetáz (NOS)
enzimek által katalizált folyamatban, melyhez NAPH és tetrabiopterin kofaktor jelenléte
szükséges [12]. Három NOS izoformát ismerünk: a neuronális NOS-t (nNOS, NOS1),
az indukálható NOS-t (iNOS, NOS2) és az endoteliális NOS-t (eNOS, NOS3). A három
izoforma génjei különböző kromoszómán helyezkednek el, nNOS a 12. kromoszóma

9
24.1-31 régiójában; az iNOS a 17. kromoszóm 11.2-12 régiójában; az eNOS pedig a 7.
kromoszóma 35-36 régiójában. Ghafourifar és Cadenas 2005-ben leírt a mitokondrum
belső membránjában elhelyezkedő, Ca2+ függő módon működő NOS izoformát, melyről
még kevés, ellentmondásos irodalmi adattal rendelkezünk [13]. Az eNOS és nNOS
folyamatosan expresszálódik, aktivációja Ca-calmodulin függő. Ezek az izoformák
felelősek a sejtek kis mennyiségű, alap szintű NO termelésért. Ezzel ellentétben az
iNOS működése transzkripciós szinten szabályozott és ez az izoforma nagy mennyiségű
NO termelésért felelős. eNOS érendothel sejtekben történő expressziója szerepet játszik
az értónus fenntartásában és a trombocita aggregáció gátlásban illetve endothel
sejtekhez történő adhéziójának megakadályozásában. Az idegsejtekben folyamatosan
expresszálódó nNOS által termelt NO a ’non-adrenerg non-cholinerg’ (NANC)
idegvégződések fontos neuromodulátora. Makrofágokban az iNOS elsősorban a
mikrobiális endotoxinok és proinflammatórikus citokinek hatására indukálódik [14]. A
NO élettani és patológiás folyamatokban betöltött szerepének tanulmányozásához a
különböző genetikailag módosított állatmodellek jelentős segítséget nyújtanak. Az
nNOS génkiütött állat jellemző betegsége a pylorus sphincter hipertrófia, az agresszív
magatartásforma és fokozott védelem az agyi ischemiával szemben. Az eNOS knock-
out állat jellemzői közé tartoznak a vasculáris léziók, hipertenzió és a fogékonyság az
agyi ischemiára. Az iNOS génkiütött állat legjellemzőbb fenotípusos elváltozása pedig a
fertőzésekkel szembeni fokozott érzékenység és a bakteriális lipopoliszacharidokra
adott csökkent hipotenzív válasz [15].
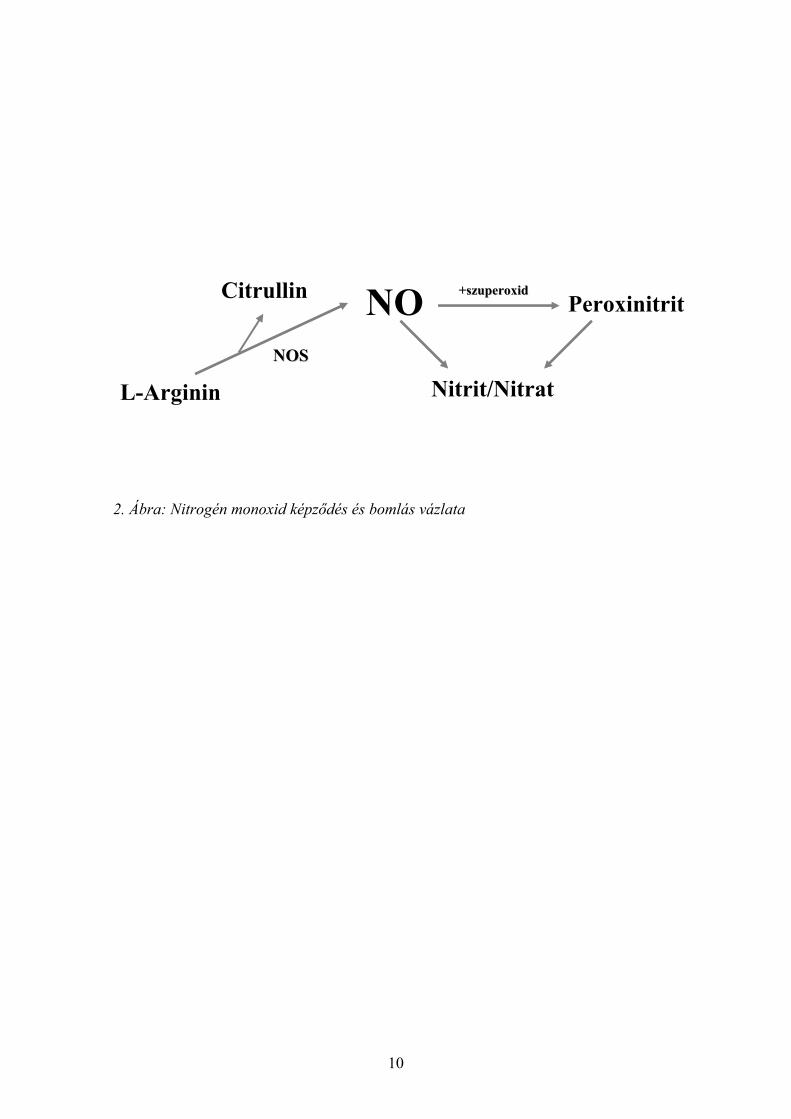
A NO rövid féléletidejű, (3-15 másodperc) szuperoxiddal (O2-) reagálva spontán
peroxinitritté (ONOO-) alakul. A peroxinitrit maga is oxidálószer, vagy átalakul nitráttá,
mely stabil, jól mérhető, a NO termeléssel arányos mennyiségben termelődő vegyület
[16] (2. ábra).
10
2. Ábra: Nitrogén monoxid képződés és bomlás vázlata
L-Arginin
NONNOOSS
Citrullin
Nitrit/Nitrat
Peroxinitrit++sszzuuppeerrooxxiidd

11
D. Nitrogén-monoxid szerepe a mitokondrium működésben és
bioszintézisében
A mitokondriumok képezik az oxidatív foszforiláció és a terminális oxidáció színhelyét
ahol a szervezetünk működéséhez energiát szolgáltató ATP több mint 90 százaléka
termelődik. Az oxidatív foszforiláció tulajdonképpen elektrontranszfer az elektron
donor (pl. NADH) és az elektron akceptor (oxigén) között. Az elektron transzfer redox
reakciók során valósul meg, amit a mitokondrium belső membránjában elhelyezkedő
protein-komplexek biztosítanak. Alapvetően öt protein komplex vesz részt a
folyamatban, melyek a következők: NADH-koenzim Q oxidoreduktáz (komplex I),
szukcinát-Q oxidoreduktáz (komplex II), elektron transzfer flavoprotein-Q
oxidoreduktáz, Q-citokróm-c oxidoreduktáz (komplex III), citokróm-c oxidáz (komplex
IV). Az elektron transzportláncon történő végighaladása során felszabaduló energia a
mitokondrium belső membránján keresztüli protontranszportra használódik. Ezt a
folyamatot kemiozmózisnak nevezzük, melynek során a membrán két oldala között
elektrokémiai gradiens alakul ki. Ez a grádiens két részből áll, pH grádiensből és
elektromos potenciál különbségből [17]. A mitokondrium belső membránján át mérhető
elektromos potenciálkülönbség mértéke 180-200 mV, negatív belül és potenciál-
szenzitív fluoreszcens festékekkel jól detektálható.
A kemiozmózis formájában tárolt energia végül ATP szintézist eredményez, a
mitokondrium belső membránjában elhelyezkedő ATP szintáz enzim segítségével. Ez
az enzim a fennálló proton gradiens rovására felszabaduló energiát ATP szintézisre
használja:

12
ADP + Pi + 4H+→ ATP 4H+mitok.matrix
Az utóbbi években derült fény arra hogy a NO a mitokondriális légzés fiziológiás
szabályozója. Szabályozza a mitokondriumok ATP szintézisét és oxigén felhasználását
azáltal hogy alacsony koncentrációban reverzibilisen gátolja a citokróm-c oxidázt. A
citokróm-c oxidáz az elektron transzportlánc utolsó enzimje, mely a sejt csaknem teljes
oxigén fogyasztásáért felelős. Alacsony koncentrációban a NO az oxigénnel
versenyezve reverzibilisen gátolja az enzimet, ATP depléciót okozva [18].
Ezenkívül a NO a mitokondrium bioszintézis fő regulátora is, hatását a cGMP- függő
peroxiszóma proliferator-activating receptor γ coactivator-1 molekulán (PPAR-γ
coactivator-1) keresztül fejti ki. Jelenlegi ismereteink szerint a barna zsírsejtek, U937
sejtek, HeLa sejtek és humán limfociták mitokondrium bioszintézise befolyásolható
NO-val[19].
A sejt pillanatnyi állapotától függően a NO egyaránt gátolhatja, vagy stimulálhatja a
programozott sejthalál folyamatát. A NO szuperoxiddal (O2-) reagálva spontán
peroxinitritté (ONOO-) alakul. A peroxinitrit erélyes oxidálószer, igen toxikus, nagyobb
mennyiségben apoptózist vagy nekrózist okoz, kisebb mennyiségben jelátviteli
útvonalak szabályozásában vesz részt. Ezenkívül a NO mitokondriális hiperpolarizációt
és ATP depléciót okoz astrocitákon, Jurkat sejteken. NO-t emittáló vegyületekkel (NO
donor) mitokondriális és citoplazmatikus Ca2+ szignál váltható ki. Jurkat sejteken a
FAS indukálta apoptózis során NO termelés mérhető [20;21].

13
E. Nitrogén-monoxid szerepe az immunregulációban
A NO sokrétű szabályozója az immunfolyamatoknak is. Az irodalomból jól ismert,
hogy az iNOS nagy mennyiségű NO termelése révén citotoxikus hatással rendelkezik.
Számos tanulmány foglalkozik a makrofágokban az iNOS által termelt NO
antimikrobiális és tumor sejt károsító hatásával. A sejtkárosító hatást különböző
enzimek gátlása révén fejti ki. A NO gátolja például a célsejtek mitokondriális légzési
láncának I, IV komplexét, a ribonukleotid reduktázt, gliceraldehid-3-foszfát
dehidrogenázt ezen kívül DNS-t módosító hatása is van. A makrofágok iNOS
termelését elsősorban a Th1 immunválasz citokinjei (INF-γ, TNF-α, IL-1β) valamint
bakteriális termékek (LPS, Staphylococcus enterooxin B) indítják be [22]. Mivel az
iNOS transzkripció órákon át tartó nagy mennyiségű NO termelést eredményez,
transzkripciója pontosan szabályozott. Az iNOS expresszió NF-κB útvonalon keresztül
szabályozott [23;24]. Az NF-κB transzkripciós faktor, homo- és heterodimer formában
fordul elő a citoplazmában, gátló proteinekhez, az IκB-hez kötődve. Megfelelő stimulus
hatására (citokinek, mikrobiális termékek, UV sugárzás) IκB kináz aktiválódik és az
IκB-k foszforilálódnak. Foszforilált IκB gyors proteolízisen megy keresztül aminek
eredményeként NF-κB felszabadul a komplexből és specifikus szekvenciájának
köszönhetően (5’-GGGRNNYYCC-3’) kötődni tud megfelelő DNS szekvenciához. Az
NF-κB kötő DNS szekvencia az indukálható gének promóter régiójában található mint
pl. citokinek, adhéziós molekulák, antioxidáns enzimek és iNOS. Érdekes módon az
NF-κB útvonalat maga a NO is befolyásolja. Citokin aktivált asztroglia és mikroglia
sejteken NO donorral gátolható az iNOS expresszió. Ennek hátterében több
mechanizmust írtak le, egyik szerint NO hatására nitrozilálódik az NF-κB DNS
kapcsolódásban szerepet játszó p50-es alegység 62 ciszteinje, így nem tud léterjönni a
DNS - NF-κB komplex [25]. Ezenkívül ismert az is hogy NO hatására fokozódik az
IκBα inhibitor protein stabilitása, szintén gátolva ezzel a NF-κB útvonalat. Ezek a
hatások azonban függnek a sejt pillanatnyi redox állapotától, nagy koncentrációjú
glutation és redox aktivitással rendelkező proteinek (thioredoxin) kivédik a NO ezen
hatásait [26]. Ugyanakkor bizonyos körülmények között NO fokozza a NF-κB által
szabályozott gének expresszióját, mint a COX-2, TNF-α, gluthation szintetáz [27;28].
Az NF-κB útvonalra kifejtett serkentő hatás molekuláris mechanizmusa kevésbé ismert.

14
Feltehetően az oxidatív stresszre aktiválódó p21ras nitrozilációja játszik szerepet ebben a
folyamatban [29;30]. Ezek az eredmények arra engednek következtetni hogy a NO
génexpresszió szabályozásban gátló és serkentő módon egyaránt részt vesz.
Egér makrofágokon végzett kísérlet szerint a NO koncentráció függő módon gátolni és
serkenteni is tudja az NF-κB útvonalat, ezáltal számos proinflammatorikus gén (pl.
citokinek, COX-2, iNOS) expresszióját ki- illetve bekapcsolja. Kis koncentrációjú NO
(30 nM – 3 μM) NF-κB-t aktiváló hatása érvényesül, míg nagy koncentrációban (30 –
300 μM) NF-κB útvonalat gátolja. A kis koncentrációjú NO termelésért eNOS és nNOS
izoformák felelősek, míg a nagy koncentrációjú NO termelés az iNOS izoformára
jellemző. A NO NF-κB útvonalra kifejtett kettős hatása magyarázatot ad az
irodalomban ellentmondásosnak ismert olyan eredményekre mint a Th1 sejtekre,
osteoclastokra, apoptózisra kifejtett serkentő és gátló hatás [31].

15
F. Reaktív oxigén intermedierek és szerepük a T limfociták
aktivációjában
Reaktív oxigén intermedierek (ROI) közé tartoznak: a szuperoxid anion, a
hidrogénperoxid, a hidroxil gyökök és a szabad gyökök (3. ábra). Jellemezőjük az
extrém mértékű instabilitás, mely a külső elektronpályán elhelyezkedő páratlan
elektronnak köszönhető. Instabil konfigurációjuk következtében gyorsan reagálnak más
molekulákkal vagy gyökökkel, miközben stabil molekulák képződnek. A szervezetben
többféle módon keletkezhetnek ROI-k. Képződhetnek külső ionizáló sugárzás hatására,
vagy a sejtlégzés melléktermékeként, mikor ez elektronok az elektron transzportláncon
történő áthaladásuk során direkt módon reakcióba lépnek az oxigénnel [32]. Ezeken
kívül, bizonyos sejtek ROI szintézisre is képesek NADPH oxidáz (makrofágok) vagy
mieloperoxidáz (neutrofil granulociták) enzimjeik révén.
3. Ábra: Legfontosabb reaktív oxigén intermedierek sematikus rajza, a külső
elekronhéjon lévő elektronok ábrázolásával.
o H o o - o H
H o oH
Szuperoxid anion Hidroxil gyök Hidroxil ion (OH-)
Hidrogén peroxid

16
A ROI-k erőteljes oxidáló hatásuknál fogva jól ismert sejtkárosítók, oxidálják a
proteineket, lipideket és a DNS-t. Sejtkárosító hatásaik közül legismertebb a lipid
peroxidációt okozó hatásuk. Lipidperoxidációs folyamatban a többszörösen telítetlen
zsírsavak kettős kötés mellet elhelyezkedő metilén csoporton (-CH2) lévő reaktív
hidrogén vesz részt [33] (4. ábra).
4. Ábra: Lipidperoxidáció folyamata.
A sejtek képesek védekezni a ROI károsító hatásaival szemben illetve képesek a ROI
által okozott károsodások helyreállítására. Egyik védekezési mechanizmus antioxidáns
enzimek segítségével történik. Ilyen enzimek a szuperoxid dizmutáz és a kataláz. A
szuperoxid dizmutáz (SOD) a következő reakciót katalizálja:
2 O2- + 2 H+ → O2 + H2 O 2
iniciáció
prolongáció
termináció
Molekuláris áterndeződés
Lipid gyök

17
A SOD a sejtekben kétféle lokalizációban található, a mitokondrium mátrixban, és
citoplazmában. A mitokondriális forma mangánt, a citoplazmatikus forma cinket és
rezet tartalmaz. A kataláz enzim a hidrogén peroxidot oxigénné és vízzé bontja:
2 H2O2 → 2 H2O + O2
A szervezetben ezenkívül számos antioxidáns hatással rendelkező molekula
megtalálható, mint például az E vitamin (alfa-tokoferol), C-vitamin, glutation, melanin.
Az antioxidánsok gyökfogó molekulák, melyek redukáló tulajdonságuknál fogva a ROI
oxidatív hatását kivédik.
Abban az esetben, ha a sejtekben az oxidatív és antioxidatív egyensúlya felborul
oxidatív stressz állapota alakul ki.
1950-es években Dehman Harman írta le először az ún. „free radical theories” (FRTA)
elméletet [34]. Lényege, hogy a normál sejtlégzés során termelődő szabad gyökök által
létrehozott károsodások a szervezetben évek során felhalmozónak és az öregedés
folyamatát eredményezik. Ez az elmélet később tovább bővült, ma már jól ismert a ROI
szomatikus mutációt, protein károsodást és lipidperoxidációt okozó hatása, melyek
mind részét képezik az öregedés folyamatának. Elsősorban a lassan bomló proteinek
(mint a kollagén, elasztin) reaktív aminosavainak oxidációja illetve glikozilációja tehető
felelőssé ezért a folyamatért. Ma már ismert az is, hogy a ROI által okozott oxidatív
károsodás nemcsak az öregedésben, hanem a neurodegeneratív betegségek
kialakulásában is szerepet játszik, mint például Alzheimer-kór, Parkinson kór, de
szerepe van más betegségek patomechanizmusában is (pl. atherosclerosis, diabetes
szövődményei, arthritis, tumoros betegségek) [35;36].
A ROI előbb ismertetett sejtkárosító hatásán túl nélkülözhetetlen szabályozója számos
fiziológiás folyamatnak. Hidrogén peroxid keletkezése szükséges a pajzsmirigy által
termelt tiroxin hormon szintéziséhez. Szintén ROI termelődik a makrofágokban és a
neutrofil granulocitákban a baktériumok fagocitózisa során, mely a sejtek antibakteriális
hatásához szükségesek. A makrofágok baktériumölő hatásában a NADPH oxidáz enzim
működése biztosítja ROI termelődését. NADPH oxidáz a következő reakciót katalizálja:
NADPH – 2e- + 2 O2 → NADP+ + H+ + 2 •O2-

18
Ha az enzim genetikai károsodás miatt nem működik az X kromoszómához kötött
chronicus granulomatosus betegség alakul ki. A betegség jellemzője a bakteriális
infekció, melyet elsősorban azok a baktériumok okoznak, melyek katalázt termelnek,
például E. coli, Salmonella, Staphylococcus törzsek.
A ROI ezenkívül a sejtek szignálfolyamataiban is részt vesz. Fontos szabályozója
például a programozott sejthalál, az apoptózis beindításának és folyamatának. A T
limfociták TCR-nek megfelelő specifikus antigénnel történő találkozásuk során
aktiválódnak, differenciálódnak és osztódnak. Ezen hatások eredménye az lesz, hogy
kialakul egy aktivált effektor T sejt populáció. Az aktivált T sejtek egy része memória T
sejtté alakul és a limfoid szövetet elhagyva a keringés segítségével nem limfoid
szövetekbe vándorol. Másik részük apoptózissal elpusztul, ezt a folyamatot aktiváció
indukálta sejthalálnak (AICD) nevezzük. Az AICD molekuláris mechanizmusának
hátterében a TCR aktivációt követő Fas és Fas ligand (FasL) expresszió áll. Számos
irodalmi adat támasztja alá a ROI központi szerepét a T limfocita aktiváció során
bekövetkező fokozott Fas és FasL expresszióban [37; 38; 39]. A TCR aktivációt követrő
15 percen belül fokozott ROI termelés detektálható [ 40 ]. Bár a TCR aktivációra
bekövetkező ROI szignálhoz vezető útvonal kevésbé ismert, a legújabb eredmények azt
mutatják hogy a ZAP-70, LAT, PLCγ1 deficiens sejtvonalon a TCR aktiváció indukálta
ROI szignál nem váltható ki. Kiváltható ugyanakkor a DAG mimetikus hatással
rendelkező phorbol 12-miristát 13-acetáttal (PMA), továbbá a PKC siRNS-el történő
blokkolása gátolta a TCR aktivációt követő ROI szignált és a Fas, FasL expresszió
fokozódását [41].
Ezek az eredmények arra utalnak hogy a T sejt aktiváció következtében kialakuló ROI
szignál fontos részét képezi a normál T limfocita funkciónak.

19
G. Hisztamin
A hisztamin (β-imidazolylethylamin) különböző fiziológás folyamatok és patológiás
állapotok fontos mediátora. Szerepet játszik az idegrendszer ingerület átviteli
folyamataiban, hipofizis hormonok szekréciójában, a gyomor-bélrendszer és a
kardiovaszkuláris rendszer működésében. Irodalmi adatok szerint a melanóma sejtek
proliferációját közvetlenül befolyásolja, ezáltal hatása van a melanóma progressziójára
[42;43]. Ezenkívül jól ismert immunregulátor, fő mediátora az allergiás reakciónak,
részt vesz a fertőzésekben, gyulladásos reakciókban.
Hisztamin L-hisztidinből keletkezik hisztidin dekarboxiláz enzim (HDC) által katalizált
folyamatban (5. ábra).
5. Ábra: Hisztamin képződés
Hisztidin dekarboxiláz (HDC)
L-Hisztidin Hisztamin

20
A hisztidin dekarboxiláz enzim (HDC) csaknem minden szöveti sejtben expresszálódik
[ 44 ], legnagyobb mértékben a hízósejtekben és bazofil granulocitákban, melyek a
hisztamint nagy mennyiségben tárolni is képesek granulumaikban. Lebontásáért a
diamin oxidáz és a hisztamin N-methyltraszferáz enzimek felelősek. A hisztamin
metabolizmusban illetve a hisztamin által kiváltott jelátviteli folyamatokban szerepet
játszó gének polimorfizmusa összefügg különböző patológiás folyamatok kialakulásával,
lezajlásával és a terápiára adott válasszal [45].
Számos irodalmi adat utal a hisztamin immunregulációban betöltött szerepére.
Különböző kísérletek eredményei a hisztamin mind gyulladást serkentő, mind
gyulladást gátló hatását leírják. A hisztamin farmakológiai hatását négy különböző
receptoron (HR) keresztül fejti ki: 1-es típusú hisztamin receptor (H1R), 2-es típusú
hisztamin receptor (H2R), 3-as típusú hisztamin receptor (H3R) és a legújabban
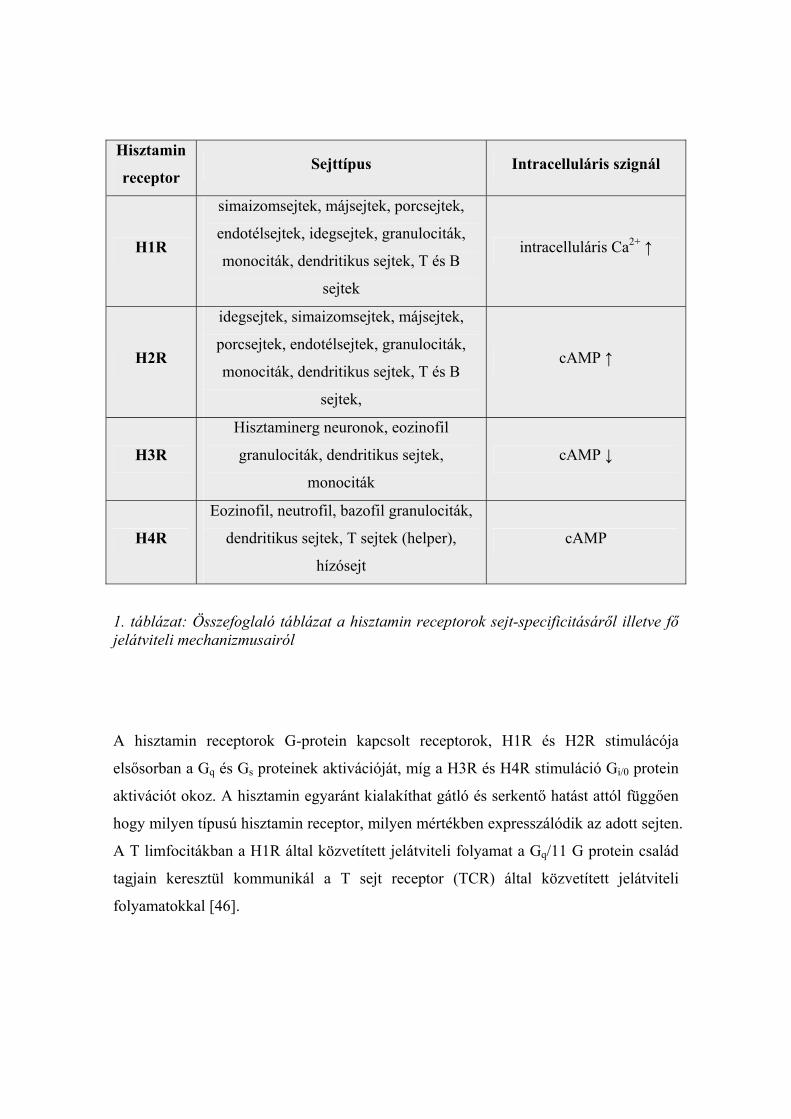
felfedezett 4-es típusú hisztamin receptor (H4R) (1. táblázat).
Hisztamin
receptor Sejttípus Intracelluláris szignál
H1R
simaizomsejtek, májsejtek, porcsejtek,
endotélsejtek, idegsejtek, granulociták,
monociták, dendritikus sejtek, T és B
sejtek
intracelluláris Ca2+ ↑
H2R
idegsejtek, simaizomsejtek, májsejtek,
porcsejtek, endotélsejtek, granulociták,
monociták, dendritikus sejtek, T és B
sejtek,
cAMP ↑
H3R
Hisztaminerg neuronok, eozinofil
granulociták, dendritikus sejtek,
monociták
cAMP ↓
H4R
Eozinofil, neutrofil, bazofil granulociták,
dendritikus sejtek, T sejtek (helper),
hízósejt
cAMP
1. táblázat: Összefoglaló táblázat a hisztamin receptorok sejt-specificitásáről illetve fő jelátviteli mechanizmusairól
A hisztamin receptorok G-protein kapcsolt receptorok, H1R és H2R stimulácója
elsősorban a Gq és Gs proteinek aktivációját, míg a H3R és H4R stimuláció Gi/0 protein
aktivációt okoz. A hisztamin egyaránt kialakíthat gátló és serkentő hatást attól függően
hogy milyen típusú hisztamin receptor, milyen mértékben expresszálódik az adott sejten.
A T limfocitákban a H1R által közvetített jelátviteli folyamat a Gq/11 G protein család
tagjain keresztül kommunikál a T sejt receptor (TCR) által közvetített jelátviteli
folyamatokkal [46].

22
H. Hisztamin hatása a T limfociták citokin termelésre és
érésére
A hisztamin immunfolyamatok szabályozásában betöltött szerepét bizonyítják azok az
eredmények is, melyek a citokin termelést befolyásoló hatásáról szólnak. Perifériás
humán mononukleáris sejteken és a CD19 depletált alpopulációban a hisztamin gátolja a
lipopoliszacharid (LPS) által okozott interferon-γ (IFN- γ) szintézist mRNS és protein
szinten egyaránt [ 47 ]. Ugyanakkor a hisztamin H2R-on keresztül fokozza az
interleukin-6 (IL-6) [48], interleukin-1 (IL-1) termelést [49],dózis függő módon pedig
gátolja a humán monociták interleukin-12 (IL-12) termelést [50]. A H2R-on keresztül
kifejtett hatása szelektív H2R antagonistával gátolható, míg szelektív H1R illetve H3R
antagonistával ez a hatás nem befolyásolható. A hisztamin T sejtek IL-2 termelésére
gyakorolt hatása kettős, gátló vagy serkentő hatás egyaránt lehetséges. Ez a kettős hatás
függ egyrészt a hisztaminnal történő kezelés módjától, a kezelt sejtek típusától, másrészt
a T sejt aktiváció időbeli lefutásától. Abban az esetben például, ha perifériás
mononukleáris sejtek vagy T sejtvonalak hisztaminnal történő kezelése az aktiváció
kezdeti szakaszában történik, erőteljes IL-2 és IFN-γ expresszió fokozódás jön létre, az
aktiváció későbbi szakaszában viszont ezen citokinek termelésére a hisztamin gátló
hatást fejt ki. [51].
A perifériás szövetekben elhelyezkedő éretlen dendritikus sejtek antigénnel történő
találkozásuk után érési folyamaton mennek keresztül, ezalatt történik az antigén
feldolgozása, miközben megváltozik a sejtfelszíni molekulák expressziós mintázata és a
sejtek citokineket kezdenek termelni. Ezután az érett dendritikus sejtek a
nyirokszövetekbe vándorolnak, ahol naiv T sejteknek bemutatják a feldolgozott antigént.
A dendritikus sejtek érésében szerepe van a sejtek mikrokörnyezetének is. Abban az
esetben, ha a dendritikus sejtek érése az elsősorban hízósejtek és bazofilek
degranulációjából származó hisztamin jelenlétében történik, csökken a dendritikus
sejtek IL-12 termelése, ugyanakkor fokozódik az IL-10 termelésük. Abban az esetben
viszont, ha az érés során hisztamin nincs jelen, a dendritikus sejtek IL-12 termelése
fokozódik. Antigén prezentáció során a dendritikus sejtek által termelt IL-12 fontos
szerepet tölt be a naív T sejtek T helper sejtekké érésében. Az elsősorban dendritikus
sejtekből származó IL-12 jelenlétében ugyanis a naív T sejtek T helper 1 típusú sejtekké

23
(Th1) érnek, melyek jellemző citokinjei az IFN-γ, TNF-β, IL-2; jellemző transzkripciós
faktoruk a T-bet. Ha az antigén prezentáció során gátolt a dendritikus sejtek IL-12
termelése, a T sejtek T helper 2 típusú sejtekké (Th2) érnek, melyek jellemző citokinjei
az IL-4, IL-5, IL-10, IL-13; jellemző transzkripciós faktoruk pedig a GATA-3 [52] (6.
ábra).

24
STAT1
Ca2+ signal
TCR
Antigen
MHC
PKC
T-bet
IFN-γ
cAMP
IL-2
GATA3 IL-4 IL-13
IL-5
p38 MAPK
PKA
H2R
IL-4R
IL-4
STAT6
IL-12
IL-12R
IL-12
STAT4
6. Ábra: T helper sejtek érését befolyásoló tényezők és jelátviteli útvonalak egyszerűsített rajza.(Szaggatott nyíl: gátlást jelöl)

25
A hízósejtek degranulációja során felszabaduló hisztamin tehát fontos szabályozója a
naiv T sejtek T helper 2 sejtekké történő differenciálódásának. Az allergén által
keresztkötött IgE molekula kapcsolódva a hízósejt felszínén expresszálódó FcεRI
receptorhoz a hízósejt granulumainak plazmamembránnal való összeolvadását idézi elő,
azaz létrejön a degranuláció folyamata [53].
Jól ismert hogy az allergiás, atópias betgségek Th2 túlsúllyal járnak. Allergiás
egyénekben a fokozott Th2 típusú immunválasz beindítójának az allergén által
keresztkötött IgE sejtfelszíni receptorhoz való kapcsolódását teszik felelőssé.
Összehasonlítva az egészséges egyének és allergiás egyének allergének által kiváltott T
sejt mintázatát, Akdis és munkatársai azt találták, hogy az egészséges egyénekben az
allergén- specifikus regulatorikus T (Treg) sejtek szubpopulációja dominál, míg
allergiásokban az allergén-specifikus IL-4 termelő Th2 sejtek jelenléte a meghatározó.
Ezek az eredmények azt mutatják hogy az allergén-specifikus Treg és Th2 sejtek
megváltozott egyensúlya fontos szerepet játszik az allergiás betegségek kialakulásában
[54]. Az újabban alkalmazott allergén-specifikus immunterápia (SIT) éppen a fokozott
Th2 immunválasz megfékezését célozza meg azáltal, hogy serkenti az allergén-
specifikus Treg sejtek által közvetített immunválaszt. Az allergén-specifikus
immunterápia hatására megjelenő Treg sejtek többféle szupresszor mechanizmus (IL-10,
TGF-β citokin termelés illetve a sejtfelszínükön megjelenő citotoxikus T-limfocita
antigen 4 és programmed death-1) révén csökkentik az IL-4, IL-5, IL-13-at termelő
CD4+ T sejtek (Th2) számát, létrehozva ezzel a perifériás T sejt toleranciát. Ezenkívül a
megnövekedett IL-10 és TGF-β termelés hatására megváltoztatják a ’pro-allergén’-IgE
és a ’protektív’-IgG4 arányt is [ 55 ; 56 ; 57 ]. Az allergén-specifikus immunterápia
legújabb eredményei szerint a hisztamin által közvetített jelátviteli folyamat interferál a
perifériás T sejt toleranciával. A terápia során adott H1R antagonista fokozza a
specifikus tolerancia mechanizmust és a vakcináció gyulladást gátló hatását [58].

26
II. CÉLKITŰZÉSEK
Célunk volt humán T limfociták CD3/CD28 aktivációra kialakuló intracelluláris
Ca2+ szignállal párhuzamosan végbemenő NO és ROI termelés mérése illetve a
mitokondriális membránpotenciál detektálása és vizsgálata.
Vizsgáltuk hogy az IP3 receptor antagonista 2-ABP és a NO kelátor C-PTIO és
a szuperoxid dizmutáz gátló MnTBAP önmagában alkalmazva hogyan
befolyásolja a CD3/CD28 stimuláció során létrejövő szignálfolyamatokat.
Választ kerestünk arra a kérdésre is, hogy a NO donor NOC-18 hogyan
befolyásolja az intracelluláris és mitokondriális Ca2+ szignált, ROI termelést és a
mitokondriális hiperpolarizációt.
Vizsgáltuk melyik NOS izoforma expresszálódik T limfocitákban nyugalmi
állapotban illetve CD3/CD28 stimulációra.
Vizsgáltuk továbbá a NO T sejt aktivációban és citokin termelésben betöltött
szerepét olyan genetikailag módosított állatmodellben, melyben hiányzik a T sejt
érésben és aktivációban szintén fontos szerepet játszó molekula, a hisztamin.
Összehasonlítottuk a HDC-KO egér és a vad típusú egerek limfocitáinak
citokin- és NO termelését, a Con-A stimulációra fellépő intracelluláris Ca2+
szignált, a ROI termelést és mitokondriális membránpotenciál változást.
Mivel szignifikáns eltérést találtunk a HDC-KO és vad típusú egér limfocitáinak
IFN-γ és NO termelésében, megvizsgáltuk hogy a NO és a hisztamin hogyan
befolyásolja a sejtek IFN-γ termelését.

27
III. MÓDSZEREK
A. Humán T limfocita aktiváció mérés Humán perifériás mononukleáris sejteket (PBMC) alvadásgátolt vénás vérből Ficoll-
Hypaque grádiensen centrifugálva nyertünk, majd a perifériás limfociták (PBL)
izolálása céljából autológ szérummal fedett Petri csészében tenyésztettük. Ezután a
perifériás limfocitákat 106/ml sejtkoncentrációban RPMI 1600-as médiumban vettük fel,
ami 10% fetal bovin szérumot (FBS), 2 mM L-glutamint, 100 IU/ml penicillint, 100
μg/ml gentamicint tartalmazott. A sejteket 37˚C-os 5% CO2-t tartalmazó termosztátban
tartottuk.
T sejt aktivációhoz a sejtek tenyésztése olyan plate-en történt, melyet először CD3
ellenes OKT3 monoklonális antitesttel (CRL 8001; American Type Culture Collection,
Manassas, VA) 1 órán keresztül inkubáltuk, majd PBS-el mostuk.. Az így előkezelt
lemezekre 106/ml koncentrációban limfocitákat helyeztünk. A CD28 kostimulációhoz
500 ng/ml monoklonális CD28 antitestet használtunk (BD PharMingen, San Diego, CA).
A sejteket a kísérlettől függően 4, 12, 24 vagy 48 órán keresztül stimuláltuk.
B. Hisztidin dekarboxkáz génkiütött (HDC-KO) állat
A genetikailag módosított HDC-KO egértörzset intézetünkkel együttműködésben Ohtsu
és társai homológ rekombinációval hozták létre. A HDC-KO CD1 egereket kilenc
generáción keresztül kereszteztük BALB/C egértörzsbe [59]. Az állatokat intézetünk
állatházában SPF körülmények között tarottuk. Kísérletekeinkhez 8-10 hetes hím
egereket használtunk, melyek legalabb két héttel a kísérletek megkezdése előtt
hisztamin mentes tápot (Altromin Gmbh., Németország) kaptak. Mivel a hisztamin
termelésért felelős egyetlen enzim, a hisztidin dekarboxiláz nem működik az állatokban,
illetve a hisztamin-mentes diéta az exogén hisztamin bevitel lehetőségét is
megakadályozza, az állatok immunfolyamatai hisztamin mentesen zajlanak.
Kísérleteinkhez HDC-KO és vad típusú egerek lépéből izolált sejteket használtuk.
Bizonyos kísérletekhez az állatokat CFA-val oltottuk, majd kilenc nap elteltével

28
izoláltuk a lépsejteket. A sejteket RPMI 1640 komplett médiumban vettük fel 106
sejt/ml koncentrációban. A sejtek stimulálásához 2 μg/ml ConA-t használtunk.
C. Áramlási citometriás mérések
Az intracelluláris és mitokondrális Ca2+ koncentráció, NO termelés, ROI termelés,
mitokondriális membránpotenciál, intracelluláris IFN-γ, sejtfelszíni CD markerek
mérését áramlási citometriával végeztük, BD FACS Calibour készülékeken. A műszer
20mV argon lézerrel (emisszió maximum: 488 nm) és 16 mV hélium-neon lézerrel
(emisszió max.: 634 nm) van felszerelve. Az elpusztult sejteket és sejttörmelékeket a
vizsgálatból forward- és side scatter mintázatuk alapján kizártuk. Mérésenként 10.000
sejtet számoltunk meg, kivétel a gyors Ca2+ szignál mérés, ebben az esetben a mérési
idő a stimulációt követő 10 percig tartott.
Intracelluláris Ca2+ koncentráció mérés
A citoplazmatikus Ca2+ koncentráció méréshez 1 μM Fluo-3-acetoxymthyl észter
(Molecular Probes) fluoreszcens festéket használtunk. A festék lipidoldékony, így a
sejtek könnyen felveszik. Miután a sejtekbe került, a citoplazmában lévő észterázok
hatására az észter csoport lehasad, miáltal vízoldékonnyá válik, így a sejtben marad. A
festékhez ilyen formában kötődik a Ca2+. A Ca2+-ot kötött festék fluoreszencia intezitása
sokszorosára fokozódik (excitációs hullámhossz maximuma: 506 nm, emissziós
hullámhossz maximuma: 526 nm) amit az áramlási citométer FL-1 csatornáján
detektálunk. T sejt aktiváció hatására kialakuló gyors Ca2+ szignál mérésénél a sejteket
25 percig inkubáltuk 37˚C-on 1 μM Fluo-3 festéket tartalmazó komplett RPMI 1600
médiumban, majd 3 percen keresztül mértük az alap fluoreszcencia intenzitást áramlási
citométeren. Ezután ConA-val, vagy CD3/28-al stimuláltuk a sejteket és további 10
percen át rögzítettük a fluoreszcencia intenzitást. Fenntartott Ca2+ szignált 4 órás, 24
órás és 48 órás stimuláció után mértünk.
A Ca2+ szignál gátlását a membrán permeábilis 2-ABP-t 10 μM és 1 mM
koncentrációkban teszteltük. Eredményes Ca2+ szignál gátlást 100 μM
végkoncentrációban kaptunk, így a kísérletek során ebben a koncentrációban használtuk.

29
Az egér limfocitákon végzett kísérletekben Ca2+ kelátor BAPTA-AM-el (Molecular
Probes) gátoltuk a Ca2+ szignált, 10 μM koncentrációban.
Mitokondriális membránpotenciál és tömeg mérés
A mitokondriális membránpotenciál mérésére a kationos lipidoldékony
tetramethylrhodamine-methylester-perchlorate (TMRM) fluoreszcens festéket
(excitációs hullámhossz 543 nm, emissziós hullámhossz 567 nm) használtunk 1 μM-os
koncentrációban (Molecular Probes). TMRM anélkül kötődik szelektíven az aktívan
működő mitokondrium membránhoz, hogy bármilyen citotoxikus hatást kifejtene. A
membránkötött forma fluoreszcencia intenzitása a mitokondriális membránpotenciállal
korrelál. Mérés megkezdése előtt a sejteket 15 percig inkubáltuk a festékkel 37˚C- os
CO2 termosztátban. Az áramlási citométer FL-2 csatornáján mért fluoreszcencia
intenzitás a mitokondriális membránpotenciállal korrelál.
A mitokondrium mennyiség meghatározásához membránpotenciáltól független
fluoreszcens festéket, nonyl acridine orange (NAO) használtunk 50 nM
végkoncentrációban. NAO a mitokondriumok kardiolipinjéhez kötődik függetlenül a
sejt energiaállapotától. Sejteket 15 percig inkubáltuk a festékkel 37˚C- os CO2
termosztátban, sötétben. A NAO excitációs hullámhossza 490 nm, emissziós
hullámhossza 540 nm, mely az áramlási citométer FL-1 csatornáján detektálható.
ROI termelés mérése
A reaktív oxigén intermedierek (ROI) termelésének mérésére hidroetidin (HE)
(Molecular Probes) oxidációra érzékeny fluoreszcens festéket használtunk. A sejtek 1
μM HE-el inkubáltuk 37˚C-os termosztátban 15 percig, majd áramlási citométerrel
megmértük a sejtek fluoreszcencia intenzitását (az oxidált HE emmissziós
hullámhossza: 605 nm; FL-2). A fluoreszcencia intenzitás a termelődött ROI
mennyiségével arányos. A ROI termelés gátlására illetve a T sejt aktivációra
bekövetkező ROI szignál gátlására szuperoxid-dizmutáz gátlót (superoxide dismutase
mimic manganese (III) tetrakis (4-benzoic acid) porphyrin chloride) (MnTBAP) 300
μM-os koncentrációban használtunk.

30
NO termelés mérése
Limfociták NO termelésének méréséhez 4-amino-5-methylamino-2',7'-difluoro-
fluorescein diacetát (DAF-FM) festéket használtunk (Molecular Probes). DAF-FM
passzív diffúzióval átjut a sejtmembránon, majd az intracelluláris észterázok
deacetilálják, így lipidoldékonyságát elveszti, a sejtből nem tud kijutni. A festék
fluoreszcens tulajdonságai megváltoznak mikor NO-hoz kötődik. NO kötődés hatására
benzotriazollá alakul, melynek excitációs hullámhossza 495 nm, emiszziós
hullámhossza 515 nm. A festék fluoreszcencia intenzitása arányos a sejt által termelt
NO koncentrációjával. A sejteket 2 órán keresztül inkubáltuk 1 μM DAF-FM festéket
tartalmazó komplett RPMI 1640 médiumban, 37˚C- on CO2 termosztátban.
Intracelluláris IFN-γ mérés és T sejt szubpopuláció meghatározás
A HDC-KO és a vad típusú egerek lépéből izolált sejteket 48 órán át 2 μg/ml ConA-val
stimuláltunk, majd GolgiPlug Protein Transpot inhibitorral (BD Biosciences) kezeltük 5
órán át 37˚C -os CO2 termosztátban. Ezután 0,5 μg/ml fluoreszcens festékkel konjugált
monoklonális antitesttel megfestettük a vizsgálni kívánt sejtfelszíni CD markerekkel, 30
percig 4˚C-on. Az antitestek a következők voltak: CD3-PE, CD8-PerCP, CD45-Per-CP
vagy CD25-PerCP-Cy5 (BD Biosciences). Mosási lépés után a sejteket fixáltuk és
permeabilizáltuk Fixation/permeabilization (BD Boisciences) oldattal 20 percig 4˚C-on.
Az újabb mosási lépés után (Perm/Wash Buffer BD Biosciences) 0,5 μg/ml FITC-el
konjugált monoklonális IFN-γ antitesttel (BD Biosciences) inkubáltuk 30 percig 4˚C-on,
sötétben. Végül a sejteket PBS-ben mostuk és analizáltuk három vagy négy csatornán az
előzőekben említett áramlási citométeren.
D. Western-blot módszer
NOS izoformák expresszióját humán perifériás limfocitákban Western-blot módszerrel
vizsgáltuk. A sejteket NOS szolubilizáló oldatban vettük fel, mely 10 mM HEPES-t (pH
7.4), 320 mM szacharózt, 100 μM EDTA-t, 1.5 mM DTT-t, 10 μg/ml leupeptint, 10

31
μg/ml aprotinint, 1 mg/ml PMSF-t, és 10 μM tetrahydrobiopterint tartalmazott. Ebben
az oldatban háromszori fagyasztás-olvasztás során lizáltuk a sejteket, majd 15000xg
sebességgel centrifugáltuk. A centrifugálás után a felülúszó proteinkoncentrációját
Bradford- módszerrel (Bio-Rad) határoztuk meg, majd 20 μg protein-lizátumot 7,5%
SDS- polyacrylamid gélen szétválasztottunk. A szétválasztás után a proteineket
nitrocellulóz membránra blottoltuk. A nitrocellulóz membránokat a három NOS
izoformának megfelelő monoklonális antitesttel detektáltuk, kontollként β-aktin ellenes
monoklonális antitestet használtunk. NOS izoformák detektálása ECL (Western
Lightning Chemiluminescence Reagent Plus; PerkinElmer) reagenssel, β-aktin
detektálása 4-chloronaphthol szubszráttal történt. Az egyes izoformák relatív
mennyiségét Kodak Image Station 440CF készüléken, Kodak 1D Image Analysis
software (Eastman Kodak) automatikus denzitometrás programjával határoztuk meg.
E. ELISPOT módszer
T sejteket egér lépszuszpenzióból negatív szelekcióval mágneses gyöngyökkel
(Miltenyi Biotech) szeparáltunk. 96 lyukú ELISPOT lemezt (MultiScreen; Millipore)
anti-IFN-γ, vagy anti-IL-10 vagy anti-IL-4 capture monoklonális antitestekkel (Duoset;
R&D Systems) inkubáltuk egy éjszakán át, majd 1% FCS-t tartalmazó PBS-el
blokkoltuk 1 órán át 37°C-on, CO2 termosztátban. Ezután helyeztük rá a szeparált
sejteket 105 sejt/lyuk koncentrációban, duplikátumban. A sejtek 2 μg/ml ConA- val
stimuláltuk, egyes kísérletekben a ConA- val történő stimulálással együtt NO donorral
60 μM [(Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate-
diethylenetriamine] (NOC-18) vagy NOS gátlószerekkel (600 μM 7-nitronidazolide
vagy 100 μM NG-mono-methyl – L-arginine) (Moleculáar Probes) is kezeltük a sejteket.
A stimuláció minden esetben 24 órán át tartott, 37°C-on, 5% CO2 termosztátban. Utána
a lemezeket PBS Tween-20-al 10-szer mostuk, majd streptavidin-HRP-vel konjgált
detektáló antitesttel inkubáltuk 1 órán át. Újabb 10 mosási lépés után hozzáadtuk a
kromogén szubsztrátot (aminoethyl carbazole és H2O2,Sigma-Aldrich), mellyel 20
percen át inkubáltuk, majd desztillált vízzel mostuk. A kiszáradt membránokon a spot-
számot ELISPOT reader (CTL) segítségével határoztuk meg.

32
F. Nitrit/nitrát mérés
A HDC-KO és vad típusú egerektől nyert szérumból nitrát/nitrit meghatározára High-
Sensity Nitrit Assay Kitet (Molecular probes) használtunk.
G. PCR alapú technikák
Reverz transzkritpáz PCR (RT-PCR)
Az RT-PCR vizsgálatokat CFA-val oltott HDC-KO és vad típusú egerek lépsejtjein
végeztük. A lépből teljes RNS-t preparáltuk TRI reagenssel (Sigma- Aldrich), majd
random hexamerekkel (Promega) cDNS-be írtuk át. Az RT-PCR reakció során a
következő primereket használtuk: egér IFN-γ szenz: CCT CAG ACT CTT TGA CT,
antiszenz primer: CAG CGA CTC CTT TTC CTC TT, (54°C, 35 ciklus); egér GADPH
szenz primer: CTG GTG CTG AGT ATG TCG TG, antiszenz primer: CAG TCT TCT
GAG TGG CAG TG (57°C, 30 ciklus). A PCR Perkin-Elmer thermal cycler készüléken
végeztük, a PCR terméket 3%-os agaróz gélen futtattuk.
Kvantitatív RT-PCR
T sejteket az egér lépszuszpenzióból negatív szelekcióval mágneses gyöngyök
segítségével (Miltenyi Biotech) szeparáltunk. Az így szeparált sejtekből RNS-t
prepaláltunk Rneasy Mini Kit (Qiagen) felhasználásával. A preparált RNS-t random
primerekkel (Promega) cDNS-be írtuk át. A NOS enzim mindhárom izoforma relatív
mennyiségének meghatározásához standard TaqMan real-time RT-PCR technikát
alkalmaztunk. A kvantitatív mérést ABI Prism 7000 Sequence Detector (Applied
Biosystems) készüléken mértük. A mennyiségi analízist a küszöbciklusok
összehasonlításával végeztük. A NOS enzimek mRNS-ének mennyiségét a mintákkal
párhuzamosan megmért hypxanthine phosphoribosyl transferase (HGPRT)
mennyiségének arányában adtuk meg.

33
H. Statisztikai elemzések
Az eredmények értékeléséhez Student t tesztet hasznaltunk, nem parametrikus eloszlást
mutató adatok esetén Mann-Whitney tesztet alkalmaztuk. Az eredményeket
szignifikánsnak tekintettük, ha p<0,05.

34
V. EREDMÉNYEK
A. Nitrogén-monoxid szerepének vizsgálata T limfociták
aktivációjában
Humán T limfociták CD3/CD28 antitesttel történő 4 órás és 24 órás stimuláció hatására:
• Ca2+ szignál (4 óránál 3,9 ± 0,75-szeres Ca2+cc. emelkedés p=0,001; 24 óránál
2.77+/-0.75-szeres Ca2+cc. emelkedés p= 0.0043 a nyugalmi szinthez képest)
• mitokondriális membránpotenciál szignál (4 óránál 1,73±0.44-szeres membrán-
potenciál emelkedés p=0,001; 24 óránál 1,53± 0.17-szeres membránpotenciál
emelkedés p=0,001 a nyugalmi szinthez képest)
• fokozott ROI termelés (4 óránál 1,53±0.36-szoros emelkedés p= 0.002; 24 óránál
4.57±1.75-szeres emelkedés p= 0.001 a nyugalmi szinthez képest)
• illetve ezekkel a folyamtokkal párhuzamosan NO szignál (4 óránál 6.09±2.98-szeres
p= 0.001; 24 óránál 4.9±1.8-szeres emelkedés) mérhető (7. ábra).

35
7. Ábra: A humán perifériás limfociták CD3/CD28 antitesttel történő stimuláció hatására bekövetkező citoplazmatikus Ca2+ koncentráció, mitokondriális membránpotenciál és nitrogén monoxid termelés változásának mérése áramlási citometriás módszerrel (n=6) .
IP3 receptor antagonista hatása T limfocita aktivációra
Jól ismert hogy TCR aktiváció során keletkező inozitol-1,4,5-tiszfoszfát (IP3)
intracelluláris receptorhoz (IP3R) történő kapcsolódása az endoplazmatikus retikulum
raktáraiból Ca2+ felszabadulást eredményez, ami fontos része a T sejt aktiváció hatására
bekövetkező Ca2+ szignálnak. Ezért megvizsgáltuk, hogy a membrán permeábilis IP3
receptor antagonista 2-aminoetoxidifenil borát (2-APB) hogyan befolyásolja a fent leírt
szignálfolyamatokat. Azt találtuk, hogy a 2-APB csökkenti a CD3/CD28 kostimulációra
fellépő Ca2+ szignált, NO szignált, továbbá csökkenti a mitokondriális hiperpolarizáció
mértékét, illetve csökkent ROI termelést is eredményez.
DAF-FM
266
DAF-FM
283
DAF-FM
50
497
788
952
965
115
358
Fluo3 Fluo3 Fluo3
CD3/CD28 24óra CD3/CD28 4 óra Kontroll
NITROGÉN MONOXID NITROGÉN MONOXID NITROGÉN MONOXID

36
Szuperoxid dizmutáz hatása a T limfocita aktivációra
A szuperoxid dizmutáz (MnTBAP) jelentősen csökkentette a CD3/CD28 stimulációra
fellépő ROI szignált, hatására kis mértékben csökken a Ca2+- és NO jel.
NO kelátor hatása a T limfocita aktivációra
A specifikus NO kelátor carboxi-2-fenil-4,4,5,5-tetrametil-imidazolin-1-oxil-3-oxid (C-
PTIO) viszont gátolja a CD3/CD28 kostimulációra fellépő intracelluláris Ca2+ szignált,
mitokondriális hiperpolarizációt, a ROI szignált és természetesen a NO szignált is. (8.
ábra)
8. Ábra: A humán perifériás limfocitákban CD3/CD28 + NO kelátor C-PTIO kezelés hatására bekövetkező citoplazmatikus Ca2+ koncentráció, mitkondriális membránpotenciál és a nitrogén monoxid termelés mérése áramlási citometriás módszerrel. (n=9)
NITROGÉN MONOXID NITROGÉN MONOXID
Kontroll CD3/CD28 4 óra
C-PTIO CD3/CD28 20 perc
C-PTIO

37
Thapsigargin, ionomycin hatása a T limfocita aktivációra
Mivel a CD3/CD28 kostimuláció hatására létrejövő mitokondriális hiperpolarizáció
gátolható 2-ABP-vel és C-PTIO-val, ezért megvizsgáltuk hogy az intracelluláris Ca2+
emelkedés önmagában hogyan hat a mitokondriális folyamatokra. Humán perifériás
limfocitákat Ca2+ ATPáz gátló thapsigarginnal illetve kalcium ionofor ionomycinnel
kezelve TCR aktivációtól független intracellulális Ca2+ koncentráció emelkedés jön
létre. Eredményeink szerint sem a thapsigargin sem az ionomycin hatására létrejövő
Ca2+ koncentráció emelkedés nem okoz mitokondriális memránpotenciál változást és
NO szignált, önmagában tehát nem elégséges a hatásos T sejt aktivációhoz (9.A, B.
ábra).
9A. Ábra: Ionomycin hatása a humán perifériás limfociták citoplazmatikus Ca2+ koncentráció, a mitokondriális membránpotenciál és a nitrogén monoxid termelés változására. A dot plot ábrák X tengelyén a citoplazmatikus Ca2+ koncentráció (Fluo-3), Y tengelyen a mitokondriális membránpotenciál (TMRM) látható. A hisztogrammok a NO (DAF-FM) termelést mutatják. (n=9)

38
9B. Ábra: Thapsigargin hatása a humán perifériás limfociták citoplazmatikus Ca2+ koncentráció, mitokondriális membránpotenciál és nitrogén monoxid termelésre. . A dot plot ábrák X tengelyén a citoplazmatikus Ca2+ koncentráció (Fluo-3), Y tengelyen a mitokondriális membránpotenciál (TMRM) látható. A hisztogrammok a NO (DAF-FM) termelést mutatják (n=9).
NO-donor hatása a T limfociták aktivációjára
A NO mitokondriális membránpotenciálra kifejtett hatásának vizsgálatához humán
perifériás limfocitákat NO donor - NOC-18 – molekulával kezeltünk. 60 μM NOC-18
hatására intracelluláris NO termelés figyelhető meg, mely a kezelést követő 4 óra múlva
3,13±0,8-szoros (p=0,04), 24 óra múlva pedig 3,7±0,6-szeres (p=0,003) emelkedést
mutat, a sejtek életképességét nem befolyásolva. Kísérleteink során azt találtuk, hogy 24
órán át 60 μM NOC-18-al történő kezelés hatására a limfociták mitokondriális
membránpotenciálja 3,5± 0,79-szörösére emelkedik (p=0,002). Ezzel párhuzamosan,
emelkedett intracelluláris, Ca2+ koncentrációt és ROI szintet mértünk (10. ábra).

39
10. Ábra: A NO donor (NOC-18) hatása a humán perifériás limfociták a citoplazmatikus Ca2+ koncentráció, a mitkondriális membránpotenciál és a nitrogén monoxid termelésre. A dot plot ábrák X tengelyén a citoplazmatikus Ca2+ koncentráció (Fluo-3), Y tengelyen a mitokondriális membránpotenciál (TMRM) látható. A hisztogrammok a NO (DAF-FM) termelést mutatják. (n=9)
NOS izoformák detektálása
Eredményeink szerint tehát NO szükséges a fiziológiás T sejt aktivációhoz,
megvizsgáltuk tehát milyen NOS izoforma felelős a megnövekedett NO termelésért.
Western-blot analízissel azt találtuk hogy humán perifériás limfocitákban eNOS és
nNOS izoforma expresszálódik. Ezen izoformák szintje 15-szörösére növekszik 24 órás
CD3/CD28 antitestekkel történő aktiváció során. Az iNOS izoforma nem detektálható
limfocitákban, még CD3/CD28 stimuláció hatására sem. (11.A és 11.B ábra) Az eNOS
és nNOS expresszió mértéke ugyanakkor fokozható 4 órán át 100 μM H2O2 –dal történő
kezeléssel.

40
11.A. Ábra: Humán perifériás T sejtek eNOS és nNOS proteinszintek változása 4 órás és
24 órás CD3/CD28 kostimulációra illetve 100 μM H2O2 hatására, Wsterd-blot
mószerrel. Konrollként aktint használtunk. A számok az aktinra normalizált
denzitometriás értékek.

41
11.B. Ábra: Humán perifériás T sejtek iNOS protein expressziója. CC: porcsejt mint
pozitív kontroll, C: stimulálatlan sejtek, S: 24 órás CD3/CD28 kostimuláció.

42
B. HDC-KO egér eltérő citokintermelése
Munkánk során összehasonlítottuk a HDC-KO és vad típusú egerek lépsejtjeinek ConA
stimulációra bekövetkező citokintermelését. Megmértük az IFN-γ, IL-4, IL-10 proteinek
szintjét. Eredményeink szerint a HDC-KO állat lépsejtjei több IFN-γ-t termelnek mind
mRNS mind protein szinten a vad típusú állatokéhoz képest. Az IL-4, IL-10 proteinek
szintjében nem találtunk szignifikáns különbséget (12.A. és B. ábra).
12.A. Ábra: HDC-KO és vad típusú állat lépsejtjeiből készült reprezentatív interferon-γ
RT-PCR. (n=3)
HDC-KO
interferon-γ
GADPH
VAD TÍPUS

43
12.B. Ábra: HDC-KO és vad típusú állat lépsejtjeiből készült interferon-γ ELISPOT.
(n=6)
Intracelluláris áramlási citometriás módszerrel megnéztük hogy az IFN-γ melyik T sejt
szubpopulációban mutat eltérést. Azt találtuk hogy a CD4 pozitív, CD8 pozitív, CD45
pozitív és CD4/CD25 kettős pozitív alpopulációkban nincs különbség a HDC-KO és
vad egerek limfocitáinak intracelluláris IFN-γ mennyiségében, valószínűleg a
megváltozott IFN-γ termelésért a teljes T sejt populáció eltolódása felelős (nem
ábrázoltuk).
0
1
2
3 p=0.001 Rel. spot szám
VAD TÍPUS HCD-KO

44
C. Hisztamin hatása a limfociták nitrogén-monoxid
termlésére
Előző eredményeinkből ismert hogy a NO fontos szerepet játszik a T sejt aktivációban
és érésben, megmértük tehát a limfociták NO termelését. A T limfociták NO termelése
szignifikánsan különbözött a HDC-KO és a vad típusú egerek limfocitáiban. A HDC-
KO egér sejtjei 2,5-ször több NO termelnek (p=0,0009) mint a vad típusú egerek
limfocitái. A ConA stimuláció hatására bekövetkező NO szignál is nagyobb mértékű a
HDC-KO egerek limfocitáiban.(13.A. és B. ábra)
13.A. Ábra: HDC-KO és vad típusú állat limfocitáinak NO termelése. (n= 9)
0
1
2
3
p=0.0009
VAD típus HDC-KO
Rel. DAF-FM fluoresc. int.
(NO)

45
13.B. Ábra: HDC-KO és vad típusú állat limfocitáinak ConA stimulációra bekövetkező
NO szignálja.(n=6)
A következőkben vizsgáltuk melyik NOS izoforma expresszálódik a CD3 pozitív
limfocitákban. Azt találtuk hogy mindhárom NOS izoforma (eNOS, nNOS, és iNOS)
mRNS-e megtalálható a limfocitákban, legnagyobb mértékű expressziót azonban a
nNOS mutat. Mindhárom izoforma nagyobb mennyiségben expresszálódik a HDC-KO
egerekből származó limfocitában, bár külön-külön vizsgálva az izoformákat nem
kaptunk szignifikáns különbséget.
Mivel a hisztamin hiányos állat limfocitái több NO-t termelnek és a ConA stimunációra
adott NO szignál is eltér a vad típusú állatokéhoz képest, megvizsgáltuk hogy a
hisztamin hogyan befolyásolja a limfociták NO termelését. Azt az eredményt kaptuk,
hogy mind a vad mind a HDC-KO limfociták NO termelése szignifikánsan csökkent, ha
a 24 órás ConA stimuláció 10-6 M hisztamin jelenlétében történt (p=0,004 HDC-KO
egérben, p=0,001 vad típus esetén).
A NO féléletideje nagyon rövid, stabil végtermékei a nitrit és nitrát. Az egerek
szérumának nitrit és nitrát szintjét megmértük, eredményünk szerint nincs szignifikáns
különbség a HDC-KO és vad típusú egerek szérum nitrit és nitrát szinjében.
0,5
1
1,5
2
2,5
3
3,5
4
0 óra 4 óra 24 óra idő
HDC KO
VAD típus
p=0.00086
p=0.00024
p=0.093
Rel. DAF-FM fluoresc. int.

46
D. Eltérő T limfocita aktiváció a HDC-KO egerekben
Előző munkánk eredményeiből ismert hogy T sejt aktiváció hatására Ca2+ szignál,
mitokondriális membránpotenciál szignál és ROI szignál mérhető, melyeket a NO
befolyásol. A HDC-KO egerek limfocitáiban a T sejt aktivációra bekövetkező gyors
Ca2+ szignál eltér a vad típusétól. Eredményeink szerint mind a nyugalmi, mind a ConA
stimuláció hatására bekövetkező gyors Ca2+ szignál szignifikánsan magasabb volt a
génkiütött állat limfocitáiban (p=0,02; p=0,04), bár a ConA hatására létrejövő gyors
Ca2+ szignál változásban (ΔCa2+) és a fenntartott Ca2+ szignálban nem találtunk
különbséget (14. Ábra).
14. Ábra: HDC-KO és vad típusú állat limfocitáinak ConA stimulációjára bekövetkező
gyors Ca2+ szignál.(n=3)
0,5
1
1,5
2
2,5
3
3,5
4
4,5
1 2 3 4 5 6 7 8 9 10
Vad típus
HDC KO
perc
ConA stimuláció
p=0.02
p=0.04 p=0.05
p=0.03
p=0.02 p=0.04
Rel. Fluo-3 fluoresc. int.

47
Mivel a HDC-KO egerek limfocitái szignifikánsan több NO-t termelnek, megvizsgáltuk
hogy a fokozott NO termelés hogyan befolyásolja a mitokondrium biogenezist. Azt
találtuk hogy a mitokondriális membránpotenciál nem különbözik a két állat
limfocitáiban. A ConA hatására létrejövő ROI szignál viszont 24 órás stimuláció után
szignifikánsan kisebb volt a HDC-KO egerek limfocitáiban.

48
E. Nitrogén-monoxid hatása az IFN-γ termelésre
Az eddig bemutatott eredmények szerint a HDC-KO egér limfocitái több IFN-γ –t
termelnek, ami együtt jár fokozott NO termeléssel is. Ismert, hogy a NO transzkripciós
szinten befolyásolja a citokinek termelését, ezért megvizsgáltuk hogyan hat a limfociták
IFN-γ termelésére. Vad típusú egerek limfocitáit ConA-val stimuláltuk NO donor (60
μM NOC-18) jelenlétében és jelenléte nélkül. ELISPOT módszerrel összehasonlítottuk
a NO donorral és az NO donor nélkül 24 órán át stimulált IFN-γ-t termelő sejetek
számát. Azt találtuk hogy 60 μM NOC-18 hatására az IFN-γ-t termelő sejtek száma
szignifikánsan megnő (p=0,0002) (15. ábra).
15. Ábra: Vad típusú állat limfocitáinak ConA stimulációra illetve ConA + 60 μM
NOC-18 (NO donor) hatására bekövetkező INF-γ termelő sejtek relatív mennyisége,
ELISPOT módszerrel mérve.(n=6)
0,4
0,8
1,2
1,6 p=0.0002
ConA 24 óra ConA + 60μM NOC-18 24h
Rel. spot szám

49
Megmértük a sejtek IFN-γ termlését NOS inhibitorokkal történő kezelés után is. Kétféle
NOS gátlót alkalmaztunk (600 μM 7-nitronidazolide és 100 μM NG-mono-methyl – L-
arginine), mindkettőt olyan koncentrációban hogy mindhárom NOS izoformát gátolja.
Eredményeink szerint a NOS gátlóval történt kezelés szignifikánsan csökkentette a
sejtek IFN-γ termelését (p=0,01 és p=0,002) (16. ábra).
16. Ábra: Reprezentatív ELISPOT ábra a vad típusú állat limfocitáinak ConA
stimulációra illetve ConA + nitronidazole vagy ConA + monomethyl- L-arginine (NOS
gátlók) hatására bekövetkező INF-γ termeléséről. (n=6)
10-6 M hisztaminnal történt kezelés a limfociták IFN-γ termelését nem befolyásolta,
szignifikánsan csökkentette viszont a NO termelésüket. A HDC-KO egér limfocitái
hisztamin hiányában több NO-t termelnek, a fokozott NO termelés pedig befolyásolja a
sejtek IFN-γ termelését. Így eredényeink szerint elmondható hogy a hisztamin a NO
termelés befolyásolásával is szerepet játszik a T sejt citokintermelés szabályozásában.
Nem stimulált ConA stimuláció ConA + 600 μM 7-nitronidazole stimuláció
ConA + 100 μM NG-Monomethyl-L-arginine
stimuláció
p=0,01
p=0,002

50
V I. MEGBESZÉLÉS ÉS KÖVETKEZTETÉSEK
A T sejt receptor (TCR) aktiváció ZAP-70 protein aktiválódásához vezet, mely felelős a
LAT foszforilációért aminek hatására a kialakuló aktivált PLCγ1 katalizálja az inozitol
1,4,5-trifoszfát (IP3) és diacilglicerol (DAG) keletkezését. Az IP3 továbbiakban a
citoplasmatikus Ca2+ szignálért, a DAG a protein kináz-C (PKC) aktivációért felelős
[ 60 ](1. ábra). A citoplazmatikus Ca2+ szignál korai jele a T sejt aktivációnak és
nélkülözhetetlen a transzkripciós faktorok (NF-AT) aktiválódásában. A TCR stimuláció
után perceken belül regisztrálható a gyors citoplazmatikus Ca2+ szignál, mely 5-10
percen át tart, illetve TCR aktiváció után 24-48 óra múlva a nyugalmi állapothoz képest
emelkedett Ca2+ koncentráció mérhető [61]. A tartósan emelkedett Ca2+ szignál a T sejt
proliferáció és differenciálódás jele.
Megfelelő antigénepitóppal történő találkozás során a T limfocita tehát nyugvó
állapotából egy nagyobb energiaigényű, aktivált állapotba kerül. A megnövekedett
energiaszükségletet a sejt fokozott ATP termeléssel biztosítja, aminek egyrészt fokozott
glukóz felhasználás, másrészt fokozott mitokondriális működés a következménye
[62;63]. Mitokondriális hiperpolarizáció ismert korai jele a T sejt aktivációnak [64].
Oxidatív foszforiláció során az elektronok egy része az oxigénnel direkt módon
reakcióba lép, ROI képződik. Több tanulmány is foglalkozik a T limfociták aktivációja
során keletkező ROI szignál jelentőségével [39; 40], ugyanakkor a fokozott ROI
termeléshez vezető útvonal kevésbé tisztázott. Egyik elmélet szerint a T sejt aktivációra
bekövetkező ROI szignálért a mitokondriumok fokozott oxidatív foszforilációs
tevékenysége tehető felelőssé. Ezt támasztja alá az az eredmény, miszerint a
mitokondrium elektrontranszportlánc I. komplexét gátolva – mely a ROI termelésben
részt vesz - TCR stimuláció indukálta ROI szignál nem jön létre [41]. Saját
eredményeink szerint a CD3/CD28 aktivációra fellépő ROI szignál kismértékben
csökkenthető ha a stimulációval párhuzamosan IP3 receptor antagonista 2-
aminoetoxidifenil boráttal (2-APB) is kezeltük a sejteket. Tehát a TCR felől érkező
szignálfolyamatok befolyással vannak a ROI szignál mértékére. Újabb irodalmi adatok
szerint ZAP-70, LAT, PLCγ1 deficiens sejtvonalon TCR aktiváció indukálta ROI
szignál nem váltható ki. Abban az esetben, ha limfociták intracelluláris Ca2+

51
koncentráció emelkedését tapsigarginnal vagy ionomycinnel idéztük elő, nem jött létre
ROI szignál. Ez az eredmény azt mutatja, hogy Ca2+ szignál önmagában nem elégséges
a ROI termelés kiváltásához. Mások eredményei szerint a TCR stimuláció során
aktiválódó PKC útvonal viszont jelentős szerepet tölt be a ROI szignál kialakulásában, a
PKC Ca2+ szignáltól független aktivációja során ugyanis kiváltható ROI szignál a T
limfocitákban [41].
Saját és irodalmi adatok alapján elmondhatjuk, hogy TCR stimuláció hatására kialakuló
ROI szignál és a fokozott mitokondrium működés szoros kapcsolatban áll. Az utóbbi
években a NO mitokondrium működés szabályozásában betöltött fiziológiás szerepének
egyre bővülő irodalma van [19]. Saját eredményeink szerint humán limfociták eNOS-t
és nNOS-t expresszálnak, iNOS izoformát nem. Az irodalomból ismert, hogy ezen
konstitutív NOS izoformák szabályozása Ca2+-calmodulin szignálon keresztül történik.
Érdekes módon eredményeink szerint az eNOS, nNOS proteinek szintje a humán T
sejtek 24 órás CD3/CD28 stimulálása során többszörösére fokozódott, ami ezeknek az
izoformáknak transzkripciós szinten történő szabályozását mutatja. Az iNOS izoformát
viszont a T limfociták aktivációja során sem tudtuk detektálni Western-bolt módszerrel.
Ugyancsak többszörösére fokozza az eNOS és nNOS expressziót, ha a sejteket 100 μM
H2O 2-vel kezeltük.
Megmérve a limfociták NO termelését, azt találtuk hogy TCR stimuláció hatására NO
szignál detektálható, mely a csúcsát 24 órás stimulációnál éri el és párhuzamos a
mitokokndriális hiperpolarizációval és a ROI szignállal. Részletesebben megvizsgálva a
NO mitokondriális és oxidatív folyamatokra kifejtett hatását T limfociták aktivációja
során, NO kelátor C-PTIO-t adtunk a sejtekhez. A NO gátlása során nem jön létre a T
sejt aktivációra jellemző Ca2+ szignál, ROI szignál és a mitokondriális változások.
Abban az esetben viszont, ha NO donorral kezeljük a sejteket, kialakul egy
sejtpopuláció melyben Ca2+ szignál, NO szignál és mitokondriális hiperoplarizáció
illetve ROI szignál mérhető.

52
Ezek az eredmények azt mutatják, hogy a TCR stimuláció hatására fellépő NO szignál
nélkülözhetetlen része a hatékony T sejt aktivációnak és szoros összefüggésben áll a
mitokondriális és oxidatív szignáltranszdukciós folyamatokkal (17 ábra).
17. Ábra. Sematikus ábra a NO T sejt aktivációban betöltött szerepéről.
T sejt aktiváció
Ca2+ szignál
NO szintézis
Emelkedett O2-
eNOS mitokondriumból T sejt szinapsziba
történő transzlokációja
SejthalálSejt túlélés
H2O2 MMaaggaass cccc..NNOO Szuperoxid dizmutáz
AAllaaccssoonnyy cccc..NNOO iNOS
Emelkedett O2-
OONO
AG
Monocita / Dendritikus sejt
TT sseejjtt NNOO NNOO
CD 80
CD 28
NNOO
NNOO
NNOO
Mitokondriális hiperpolarizáció és
bioszintézis
Mitokondriális hiperpolarizáció és
bioszintézis
transzkripció és protein szintézis
(IL-2)

53
Az irodalomban szintén jól ismert, hogy a hisztaminnak fontos szerepe van T limfociták
aktivációjában és érésében. A hisztamin immunfolyamatok szabályozásában betöltött
szerepének vizsgálatához jó modell a hisztidin dekarboxiláz génkiütött (HDC-KO)
egérmodell, mivel hisztamin termelésért csak a HDC enzim felelős. Abban az esetben
ha az állatokat hisztamin mentes diétán tartjuk, az immunfolyamatok in vivo hiszatamin
mentesen zajlanak. Laboratóriumunk előző eredményeiből ismert hogy a HDC-KO
egerek lépéből szeparált dendritikus sejtek fokozott antigénprezentáló képességgel
rendelkeznek illetve LPS stimulációt követően szignifikánsan több IFNγ-t és IL-12-t
termelnek [65]. Ezek az eredmények alátámasztják azt az irodalmi adatot miszerint a
hisztamin a naiv Th sejtek Th2 sejtekké történő differenciálódásában játszik szerepet.
Ugyanakkor az is ismert, hogy alacsony koncentrációjú NO a naiv Th sejtek Th1
irányban történő érését segíti elő. A NO cGMP útvonalat befolyásoló hatása révén
fokozza a naiv Th sejtek IL-12 receptor β2 (IL-12Rβ2) sejtfelszíni expresszióját, de
nincs hatással az IL-4 receptor expresszió mértékére [66]. Saját eredményeink szerint a
HDC-KO egerek limfocitái szignifikánsan több INF-γ-t termelnek mind mRNS, mind
protein szinten. Az IL-10 és IL-4 citokinek termelésében nem találtunk különbséget. A
fokozott IFN-γ termelésért a T sejt szubpopulációk eltolódása tehető felelőssé, mivel az
intracelluláris IFN-γ mennyiség nem különbözik a CD4, CD8, CD45 és CD4/CD25
pozitív szubpopulációkban. Ezek az eredmények is azt mutatják, hogy a HDC-KO egér
immunválasza hiszamin hiány következtében Th1 irányba tolódik el.
Kíváncsiak voltunk arra, hogy a HDC-KO állat Th1 irányban eltolt immunválaszában
szerepet játszik-e a NO, ezért megmértük a limfociták NO termelését. HDC-KO egér
lépéből szeparált T sejtek 2,5-ször több NO-t termelnek a vad típusú egér limfocitáihoz
képest. Ezenkívül a HDC-KO egerek limfocitái ConA stimuláció hatására fokozott NO
szignállal válaszolnak. Tovább vizsgálva a hisztamin NO termelésre kifejtett hatását, azt
találtuk, hogy mind a vad, mind a HDC-KO limfociták NO termelése csökkenthető
hisztaminnal történő kezeléssel.
Mivel a hisztaminkezelés nem befolyásolta a sejtek IFN-γ termelését, megvizsgáltuk a
NO termelés hogyan hat a limfociták citokintermelésére. Azt az eredményt kaptuk,
hogy NO donor hatására a sejtek IFN-γ termelése szignifikánsan nagyobb a kontroll,

54
csak ConA-val stimulált sejtekhez képest. Abban az estben viszont, ha a sejteket ConA
stimulációval párhuzamosan NOS inhibitorral kezeltük, az IFN-γ termelés
szignifikánsan csökkenthető volt. Ezen eredményeink szerint elmondhatjuk, hogy a
hisztamin irodalomból ismert citokintermelésre gyakorolt direkt hatásán túl, a NO
termelés befolyásolása révén is részt vesz a T sejt citokintermelés szabályozásában (18.
ábra).

55
18. Ábra. Sematikus ábra a hisztamin és a NO szerepéről a T helper sejtek érésében.
STAT1
Ca2+ szignál
TCR
Antigén
MHC
PKC
T-bet
IFN-γ
cAMP
IL-2
GATA3 IL-4 IL-13
IL-5
p38 MAPK PKA
H2R
IL-4R
IL-4 STAT6
IL-12
IL-12
NOS
IL-12R
ROI
-
-

56
VII. ÖSSZEFOGLALÁS
T limfociták aktivációjában, proliferációjában illetve a programozott
sejthalálban a mitokondriális membránpotenciál illetve reaktív oxigén intermedierek
(ROI) termelődése fontos szerepet játszik. A mitokondriális hiperpolarizáció egyik korai,
reverzibilis jele a T sejt aktivációnak. Munkánk során vizsgáltuk a humán T limfociták
CD3/CD28 kostimulációra bekövetkező citoplazmatikus és mitokondriális Ca2+ szignált,
a ROI és nitrogén-monoxid (NO) termelést illetve a mitokondriális membránpotenciál
változás mértékét. Abban az esetben, ha inozitol 1,4,5-trifoszfát receptor (IP3R)
antagonistával vagy a szuperoxid dizmutáz hatását utánzó szerrel kezeltük a sejteket a
citoplazmatikus Ca2+ szignál, a ROI szignál és a NO szignál mértéke csökkent.
Specifikus NO kelátorral történő kezelés hatására viszont a stimulált sejtekben nemcsak
az előbb említett szignálok mértéke csökkent, hanem a mitokondriális hiperpolarizáció
is gátolható volt. Ugyanakkor NO donor hatására ROI szignál, Ca2+ szignál, átmeneti
ATP depléció és markáns mitokondriális hiperpolarizáció detektálható. Az önmagában
adott ionomycinnel vagy thapsigarginnal létrehozott Ca2+ koncentráció emelkedés
azonban nem járt együtt sem a NO termelés fokozódásával, mitokondriális
membránpotenciál változással, sem a ROI szignállal. Western-blot analízissel humán
perifériás limfocitákban a NO szintetáz enzim (NOS) izoformái közül az endotheliális
és a neuronális izoformákat lehetett detektálni, ezekből a sejtekből hiányzik a Ca2+-
független indukálható NOS izoforma. CD3/CD28 kostimuláció hatására az eNOS és
nNOS izoformák expressziója többszörösére fokozódik. Eredményeink azt mutatják,
hogy a T limfociták aktivációja során bekövetkező mitokondriális hiperpolarizációban
és az oxidatív szignálfolyamatokban a Ca2+ -függő NO termelésnek fontos szerepe van.
Munkánk során szintén vizsgáltuk a hisztamin T sejt aktivációban és
citokintermelés befolyásolásában betöltött szerepét hisztidin dekarboxiláz génkiütött
(HDC-KO) és vad típusú állatmodellen. Eredményeink szerint hisztamin hiányában a
HDC-KO egerek lépsejtjeinek interferon-γ (IFN-γ) termelése mind protein, mind mRNS
szinten szignifikánsan nagyobb mértékű. A sejtek fokozott IFN-γ termelése ugyanakkor
fokozott NO termeléssel társul. Eredményeink szerint hisztaminnal történő kezelés a
limfociták NO termelését csökkenti mind a HDC-KO mind a vad típusú egérben. NO
donor hatására viszont a limfociták fokozott IFN-γ termeléssel válaszoknak, míg NOS

57
gátlószerek, (NG-monomethyl-L-arginine és nitronidazole) hatására a sejtek
szignifikánsan kevesebb IFN-γ-t termelnek, mely a NO IFN-γ termelés szabályozásában
betöltött szerepére utal. Eredményeink szerint tehát a hisztamin T limfocitákra
gyakorolt direkt hatásán túl, az NO termelés befolyásolása révén is szabályozza a T
sejtek citokintermelését és jelátviteli folyamatait.

58
VIII. SUMMARY
Activation, proliferation, or programmed cell death of T lymphocytes is
regulated by the mitochondrial transmembrane potential through controlling ATP
synthesis, production of reactive oxygen intermediates (ROI). Mitochondrial
hyperpolarization is an early and reversible event associated with T cell activation.
CD3/CD28 costimulation of human PBL elevated cytoplasmic and mitochondrial Ca2+
levels, ROI production, and NO production, and elicited mitochondrial
hyperpolarization. Although T cell activation-induced Ca2+ release, ROI levels, and NO
production were diminished by inositol 1,4,5-triphosphate receptor antagonist,
superoxide dismutase mimic and NO chelator, mitochondrial hyperpolarization was
selectively inhibited by NO chelator. Moreover, NO precursor NOC-18 elicited NO and
ROI production, Ca2+ release, transient ATP depletion, and robust mitochondrial
hyperpolarization. Ca2+ influx by ionomycin or Ca2+ release from intracellular stores by
thapsigargin alone failed to induce NO synthase expression, NO production, or
mitochondrial membrane potential elevation. Western blot analysis revealed expression
of Ca-dependent endothelial NO synthase and neuronal NO synthase isoforms and
absence of Ca-independent inducible NO synthase in PBL. CD3/CD28 costimulation
elicited severalfold elevations of endothelial NO synthase and neuronal NO synthase
expression.. The results suggest that T cell activation-induced mitochondrial
hyperpolarization is mediated by ROI- and Ca2+ dependent NO production.
In order to study the role of histamine in T cell activation, we investigated
cytokine production and T cell signal transduction in HDC-KO and wild type mice. In
the absence of histamine, an elevated INF-γ mRNA and protein levels of splenocytes
were associated with a markedly increased NO production, compared to wild type
animals. Furthermore, histamine treatment decreased the NO production of splenocytes
from both wild type and HDC-KO mice. NO precursor NOC-18 elicited IFN-γ
production, while NO synthase inhibitors, NG-monomethyl-L-arginine and nitronidazole
both inhibited IFN-γ production, suggesting the role of NO in regulating IFN-γ
synthesis. Our present data indicate that in addition to its direct effects on T lymphocyte
function, histamine regulates cytokine production and T cell signal transduction through
regulating NO production.

59
IX. SAJÁT PUBLIKÁCIÓK LISTÁJA
Ph.D. értekezés témájában megjelent publikációk
Koncz A, Pasztoi M, Mazan M, Fazakas F, Buzas E, Falus A, Nagy G. Nitric oxide
mediates T cell cytokine production and signal transduction in histidine dexarboxylase
knockout mice. J Immunol 2007 Nov. 15;179(10):6613-9. IF: 6,3
Nagy G, Koncz A, Fernandez D, Perl A.Nitric oxide, mitochondrial hyperpolarization,
and T cell activation. Free Radic Biol Med. 2007 Jun 1; 42(11): 1625-31. IF:5,4
Nagy G, Koncz A, Perl A Mitochondrial T cell signal transduction abnormalities in
systemic lupus erythematosus Curr Immunol Rev 2005, 1, 62-68.
Perl A, Gergely P Jr, Nagy G, Koncz A, Banki K. Mitochondrial hypepolarization: a
checkpoint of T-cell life, death and autoimmunoty. Trends Immunol 2004, 25, 360-7.
IF:13,1
Nagy G, Koncz A, Perl A. T cell activation induced mitochondrial hyperpolarization is
mediated by Ca2+ and redox dependent production of nitric oxide. J Immunol 2003,
171, 5188-97. IF: 6,7
A Ph.D. értekezés témájában megjelent közlemények összesített impakt faktora: 31,5
Ph.D. értekezés témájához nem kapcsolódó publikációk
Nagy G, Ward J, Mosser DD, Koncz A, Gergely P Jr, Stancato C, Qian Y, Fernandez D,
Niland B, Grossman CE, Telarico T, Banki K, Perl A.Regulation of CD4 expression via
recycling by HRES-1/RAB4 controls susceptibility to HIV infection. J Biol Chem.
2006 Nov 10; 281(45):34574-91 IF: 5,8

60
Nagy G, Koncz A, Perl A. T- and B-cell abnormalities in systemic lupus
erythematosus. Crit Rev Immunol. 2005, 25(2),123-40. IF: 3,2
Gergely P Jr, Pullmann R, Stancato C, Otvos L Jr, Koncz A, Blazsek A, Poor G, Brown
KE, Phillips PE, Perl A. Increased prevalence of transfusion-transmitted virus and
cross-reactivity with immunodominant epitopes of the HRES-1/p28 endogenous
retroviral autoantigen in patients with systemic lupus erythematosus.Clin Immunol.
2005 Aug;116(2):124-34. IF: 3,2
Nagy G, Clark J M, Buzas E, Gorman C, Pasztoi M, Koncz A, Falus A and Cope A P.
Nirtic oxide production of T lymphocytes is increased in rheumatoid arthritis.
Immunology Letters 2008 in press. IF: 2,3
Koncz A, Nagy G, Perl A. Nitrogén monoxid függő mitokondrium bioszintézis
systemas lupus erythematosusban Klinikai és kísérletes laboratóriumi medicina 2005.
szeptember
Nagy G, Géher P, Koncz A, Perl As. Jelátviteli defektusok systemás lupus
erythematosusban. Orvosi Hetilap. 2005 Jul 31.
Kumulatív impakt faktor: 46

61
X. KÖSZÖNETNYILVÁNÍTÁS
Köszönettel és hálával tartozom Falus András Professzor Úrnak és Dr. Buzás Editnek,
amiért lehetőséget adtak munkatervem és kísérleteim megvalósítására és mindenben
támogatták munkámat. Kutatómunkával a syracues-i egyetemen a Perl András
Professzor Úr által vezetett laboratóriumban ismerkedtem meg, köszönettel tartozom
azért, hogy számos laboratóriumi módszer elsajátításában segítséget és lehetőséget
kaptam tőle. Szintén ezúton szeretném megköszönni a Heim Pál Gyermekkórházban
dolgozó munkatársaimnak hogy türelmükkel és megértésükkel támogatták Ph.D.
munkám befejezését, Simonné Sebestyén Piroskának pedig külön köszönetet szeretnék
mondani bíztatásáért és lelkesítéséért.
Szerencsésnek érzem magam, hogy férjemmel, Nagy Györggyel együtt dolgozhattam. A
közösen végzett munka általa történő szakmai, szellemi irányítása nagymértékben
elősegítette fejlődésemet és fontos részét képezi Ph.D. munkámnak. Köszönet illeti
azért is, mert családfőként lehetőséget biztosított munkám végzéséhez és a legnehezebb
pillanatokban is mellettem állt, támogatására folyamatosan számíthattam.
Szeretnék köszönetet mondani Édesanyámnak is, aki gondoskodásával, szeretetével és
állandó segítőkészségével járult hozzá munkám eredményességéhez.

62
XI. IRODALOMI JEGYZÉK
1 . Falus A, Buzás E, Rajnavölgyi É. Az immunológia alapjai. Semmelweis
Kiadó 2007
2. Wange, R. L., and L. E. Samelson. Complex complexes: signaling at the TCR.
Immunity 1996. 5:197-205
3. Weiss, A., and D. R. Littman. Signal transduction by lymphocyte antigen
receptors. Cell 1994. 76:263-274.
4. Dustin, M. L., and J. A. Cooper. The immunological synapse and the actin
cytoskeleton: molecular hardware for T cell signaling. Nat. Immunol. 2000. 11:23
5. Bromley, S. K., W. R. Burack, K. G. Johnson, K. Somersalo, T. N. Sims, C.
Sumen, M. M. Davis, A. S. Shaw, P. M. Allen, and M. L. Dustin. The immunological
synapse. Annu. Rev. Immunol. 2001. 19:375
6. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin
ML. The immunological synapse: a molecular machine controlling T cell activation.
Science 1999;285:221–227.
7. Ibiza S, Victor VM, Bosca I, Ortega A, Urzainqui A, O'Connor JE, Sanchez-
Madrid F, Esplugues JV, Serrador JM. Endothelial nitric oxide synthase regulates T cell
receptor signaling at the immunological synapse. Immunity 2006;24:753–765
8. Vincenzo Di Bartolo1 Benjamin Montagne, Mogjiborahman Salek, Britta
Jungwirth1 Florent Carrette, Julien Fourtane, Nathalie Sol-Foulon, Frédérique Michel1
Olivier Schwartz2 Wolf D. Lehmann, and Oreste Acuto. A novel pathway down-
modulating T cell activation involves HPK-1–dependent recruitment of 14-3-3 proteins
on SLP- J Exp Med. 2007. March 19; 204(3): 681–691)

63
9. Brown-CG: Nitric oxide and mitochondrial respiration. Biochem Biophys
Acta.1999. 1411:351-369
10. Beltran-B, Mathur-A, Duchen-MR, Erusalimsky-JD and Moncada-S: The
effect of nitric oxide on cell respiration: a key to undertanding its role in cell survival, or
death. PNAS 2000. 26:14602-14607
11. Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular
system: a historical overview J Physiol Pharmacol. 2002. Dec. 53(4 Pt 1):503-14.
12 . Bredt-DS: Endogenous nitrice oxide synthesis: biological functions and
pathophysiology. Free Radic Res 1999. 31:577-596
13 . Ghafourifar P, Cadenas E Mitochondrial nitric oxide synthase. Trends
Pharmacol Sci. 2005. Apr;26(4):190-5.
14. T.J. Guzik, R. Korbut, T. Adamek-Guzik. Nitric oxide and superoxide in
inflammation and immune regulation. Journal of Phisiology AND Pharmacology 2003.
54, 4,469-487
15 Masato Tsutsui, Hiroaki Shimokawa, Tsuyoshi Morishita, Yasuhide
Nakashima, and Nobuyuki Yanagihara. Development of Genetically Engineered Mice
Lacking All Three Nitric Oxide Synthases. J Pharmacol Sci 102, 147 – 154
16 . Chung-HT, Pae-HO, Choi-BM, Billiar-TR, Kim-YM: Nitric oxide as a
bioregulator of apoptosis. Biochem Biophys Res Commun 2001. 282:1075-1079
17.Mitchell P, Moyle J "Chemiosmotic hypothesis of oxidative phosphorylation".
Nature 1967. 213 (5072): 137–9.)

64
18 . Félix Rodríguez-Juárez, Enara Aguirre, and Susana Cadenas. Relative
sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous
nitric oxide at physiological oxygen concentration. Biochem J. 2007 July 15; 405(Pt 2):
223–231.
19 . Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J. Cell
Sci. 2006. 119:2855–2862
20. Beltran-B, Quintero-M, Gracia-Zaragoza-E, O'Connor-E, Esplugues-JV and
Moncada-S: Inhibition of mitochondrial respiration by endogenous nitric oxide: a
critical step in Fas signalling. PNAS 2002. 13:8892-8897
21 . Kim-YM, B Ombeck-CA, Billiar-TR: Nitric oxide as a bifunctional
regulator of apoptosis. Circ Res 1999. 19: 253-256
22. MacMicking, J., Q. W. Xie, and C. Nathan. Nitric oxide and macrophage
function. Annu. Rev. Immunol. 1997. 15:323–350.
23 . Kleinert, H., C. Euchenhofer, I. Ihrig-Biedert, and U. Forstermann.
Glucocorticoids inhibit the induction of nitric oxide synthase II by down-regulating
cytokine-induced activity of transcription factor nuclear factor-kB. Mol. Pharmacol.
1996. 49:15
24. Liu, S. F., X. Ye, and A. B. Malik. In vivo inhibition of nuclear factor-kB
activation prevents inducible nitric oxide synthase expression and systemic
hypotensionin a rat model of septic shock. J. Immunol. 1997. 159:3976.
25. Song Kyu PARK, Hsin Lee LIN and Sean MURPHY. Nitric oxide regulates
nitric oxide synthase-2 gene expression by inhibiting NF-κB binding to DNA Biochem.
J. 1997. 322,

65
26. Peng, H. B., P. Libby, and J. K. Liao. Induction and stabilization of IκBα by
nitric oxide mediates inhibition of NF-kB. J. Biol. Chem. 1995. 270:14214
27. Habib, A., C. Bernard, M. Lebret, C. Creminon, B. Esposito, A. Tedgui, and
J. Maclouf. Regulation of the expression of cyclooxygenase-2 by nitric oxide in rat
peritoneal macrophages. J. Immunol. 1997. 158:3845;
28. Moellering, D., A. J. Mc, R. P. Patel, H. J. Forman, R. T. Mulcahy, H. Jo,
and V. M. Darley-Usmar. The induction of GSH synthesis by nanomolar concentrations
of NO in endothelial cells: a role for g-glutamylcysteine synthetase and g-glutamyl
transpeptidase. FEBS Lett. 1999. 448:292.
29. Lander, H. M., D. P. Hajjar, B. L. Hempstead, U. A. Mirza, B. T. Chait, S.
Campbell, and L. A. Quilliam. A molecular redox switch on p21ras: structural basis for
the nitric oxide-p21ras interaction. J. Biol. Chem.1997. 272:4323.
30. Sheffler, L. A., D. A. Wink, G. Melillo, and G. W. Cox. Exogenous nitric
oxide regulates IFN-g plus lipopolysaccharide- induced nitric oxide synthase expression
in mouse macrophages. J. Immunol. 1995. 155:886.
31. L. Connelly, M. Palacios-Callender, C. Ameixa, S. Moncada, and A. J.
Hobbs. Biphasic Regulation of NF-kB Activity Underlies the Pro- and Anti-
Inflammatory Actions of Nitric Oxide. The Journal of Immunology, 2001, 166: 3873–
3881
32. Han, D., Williams, E. and Cadenas, E. Mitochondrial respiratory chain-
dependent generation of superoxide anion and its release into the intermembrane space.
Biochem. J. 2001. 353, 411-416.

66
33 . Kappus, H. and Sies, H., Toxic drug effects associated with oxygen
metabolism: redox cycling and lipid peroxidation, Experientia, 37, 1233, 1981
34. Harman, D. "Aging: a theory based on free radical and radiation chemistry".
J Gerontology 1956. 11 (3): 298-300
35 . Tuppo EE, Forman LJ. Free radical oxidative damage and Alzheimer's
disease. J Am Osteopath Assoc. 2001 Dec;101(12 Suppl Pt 1):S11-5
36 . Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp
Diabetes Res. 2007: 43603
37 . Bauer, M. K., M. Vogt, M. Los, J. Siegel, S. Wesselborg, and K. Schulze-
Osthoff. Role of reactive oxygen intermediates in activation-induced CD95 (APO-
1/Fas) ligand expression. J. Biol. Chem. 1998. 273:8048–8055.
38 . Gulow, K., M. Kaminski, K. Darvas, D. Suss, M. Li-Weber, and P. H.
Krammer. HIV-1 trans-activator of transcription substitutes for oxidative signaling in
activation-induced T cell death. J. Immunol. 2005. 174:5249–5260.
39 . Peter, M.E, and Krammer, P.H. Mechanisms of CD95 (APO-1/Fas)-
mediated apoptosis. Curr. Opin. Immunol. 1998. 10:545–551.
40. Devadas S, Zaritskaya L, Rhee S. G, Oberley L and Williams M.S. Doscrete
genetation of superoxide and hydrogen peroxide by T cell receptor stimulation:
selective regulation of mitogen-activated protein kinase activation and fas ligand
expression. J. Exp. Med. 2002. 195:59-70
41 . Marcin Kaminski, Michael Kießling, Dorothee Su¨ss, Peter H. Krammer,
and Karsten Gulow. Novel Role for Mitochondria: Protein Kinase C -Dependent
Oxidative Signaling Organelles in Activation-Induced T-Cell Death. Molecular And
Cellular Biology, 2007 May, p. 3625–3639

67
42. Falus A, Hegyesi H, Lazar-Molnar E, Pos Z, Laszlo V, Darvas Z. Paracrine
and autocrine interactions in melanoma: histamine is a relevant player in local
regulation. Trends Immunol. 2001. 22(12):648-52.
43. Pos Z, Safrany G, Muller K, Toth S, Falus A, Hegyesi H. Phenotypic
profiling of engineered mouse melanomas with manipulated histamine production
identifies histamine H2 receptor and rho-C as histamine-regulated melanoma
progression markers. Cancer Res.2005. 65(10):4458-66.
44. Laszlo V, Falus A. Yet unrevealed aspects of histamine: questions from
outside--answers at inside? Semin Cancer Biol. 2000. 10(1):1-2.
45. Igaz P, Fitzimons CP, Szalai C, Falus A. Histamine genomics in silico:
polymorphisms of the human genes involved in the synthesis, action and degradation of
histamine.Am J Pharmacogenomics. 2002. 2(1):67-72
46. Kehrl JH: Heterotrimeric G protein signaling: roles in immune function and
fine-tuning by RGS proteins.Immunity. 1998. Jan;8(1):1-10
47. Horvath B, Szalai Cs, Mandi Y, Laszlo v, Radvanyi Zs, Darvas Zs, Falus A:
Histamine and histamine-receptor antagonists modify gene expression and biosynthesis
of interferon γ in priferial human blood mononuclear cells and in CD19-depleted cell
subsets. Immunol Let. 1999. 70: 95-9..
48. Mor S, Nagler A, Barak V, Handzel ZT, Geller-Bernstein C, Fabian I.
Histamine enhances granulocyte-macrophage colony-stimulating factor and interleukin-
6 production by human peripheral blood mononuclear cells. J Leukoc Biol. 1995
Oct;58(4):445-50

68
49. E Vannier and C A Dinarello Histamine enhances interleukin (IL)-1-induced
IL-1 gene expression and protein synthesis via H2 receptors in peripheral blood
mononuclear cells. Comparison with IL-1 receptor antagonist.J Clin Invest. 1993 July;
92(1): 281–287.
50. van der Pouw Kraan TC, Snijders A, Boeije LC, de Groot ER, Alewijnse AE,
Leurs R, Aarden LA. Histamine inhibits the production of interleukin-12 through
interaction with H2 receptors. J Clin Invest. 1998 Nov 15;102(10):1866-73
51. Igaz P, Novák I, Lázaár E, Horváth B, Héninger E, Falus A. Bidirectional
communication between histamine and cytokines. Inflamm Res. 2001 Mar;50(3):123-8.
52 . Mazzoni A, Young HA, Spitzer JH, Visintin A, Segal DM. Histamine
regulates cytokine production in maturing dendritic cells, resulting in altered T cell
polarization. J Clin Invest. 2001. 108(12):1865-73.
53. Mazzoni A, Siraganian RP, Leifer CA, Segal DM. Dendritic cell modulation
by mast cells controls the Th1/Th2 balance in responding T cells.J Immunol. 2006 Sep
15;177(6):3577-81.
54. Mübeccel Akdis, Johan Verhagen,Alison Taylor, Fariba Karamloo, Christian
Karagiannidis, Reto Crameri, Sarah Thunberg, Günnur Deniz, Rudolf Valenta,Helmut
Fiebig, Christian Kegel, Rainer Disch, Carsten B. Schmidt-Weber, Kurt Blaser, and
Cezmi A. Akdis. Immune Responses in Healthy and Allergic Individuals Are
Characterized by a Fine Balance between Allergen-specific T Regulatory 1 and T
Helper 2 Cells J. Exp. Med. June 7, 2004 Volume 199, Number 11,
55. Jayasekera NP, Toma TP, Williams A, Rajakulasingam K. Mechanisms of
immunotherapy in allergic rhinitis. Biomed Pharmacother. 2007 Jan;61(1):29-33

69
56. Jutel M, Akdis M, Blaser K, Akdis CA. Mechanisms of allergen specific
immunotherapy--T-cell tolerance and more Allergy. 2006 Jul;61(7):796-807
57. Alison Taylor, Johan Verhagen, Kurt Blaser, Mu¨beccel Akdis and Cezmi A.
Akdis Mechanisms of immune suppression by interleukin-10 andtransforming growth
factor-b: the role of T regulatory cells. Immunology, 2005 117, 433–442
58. Jutel M, Blaser K, Akdis CA Histamine receptors in immune regulation and
allergen-specific immunotherapy Immunol Allergy Clin North Am. 2006
May;26(2):245-59
59 . Ohtsu H, Tanaka S, Terui T, Hori Y, Makabe-Kobayashi Y, Pejler G,
Tchougounova E, Hellman L, Gertsenstein M, Hirasawa N, Sakurai E, Buzas E, Kovacs
P, Csaba G, Kittel A, Okada M, Hara M, Mar L, Numayama-Tsuruta K, Ishigaki-Suzuki
S, Ohuchi K, Ichikawa A, Falus A, Watanabe T, Nagy A. Mice lacking histidine
decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001;502:53-56
60. Williamson, J. R. 1986. Role of inositol lipid breakdown in the generation of
intracellular signals. State of the art lecture. Hypertension 8(Part II):140–156.
61. Droge, W. Free radicals in the physiological control of cell function. Physiol.
Rev.2002. 82:47–95.
62. Aulwurm, U.R., and Brand, K.A. 2000. Increased formation of reactive
oxygen species due to glucose depletion in primary cultures of rat thymocytes inhibits
proliferation. Eur. J. Biochem. 2000. 267:5693–5698.
63. Hildeman, D.A., et al. Reactive oxygen species regulate activation induced T
cell apoptosis. Immunity. 1999. 10:735–744.

70
64 . Banki K, Hutter E, Gonchoroff N, Perl A. Elevation of mitochondrial
transmembrane potential and reactive oxygen intermediate levels are early events and
occur independently from activation of caspases in Fas signaling. J.Immunol.
1999;162:1466–1479
65. Jelinek I, Laszlo V, Buzas E, Pallinger E, Hangya B, Horvath Z, Falus A.
Increased antigen presentation and T(h)1 polarization in genetically histamine-free mice.
Int Immunol. 2007;19:51-58.
66. Wanda Niedbala, Xiao-qing Wei, Carol Campbell, Duncan Thomson, Mousa
Komai-Koma, and Foo Y. Liew. Nitric oxide preferentially induces type 1 T cell
differentiation by selectively up-regulating IL-12 receptor β2 expression via cGMP.
Proc Natl Acad Sci U S A. 2002 Dec 10;99(25):16186-91.