ngs: current and future platforms elibrary by author © escmid
TRANSCRIPT

11/13/18
1
NGS: current and future platforms
Natacha Couto
Department of Medical Microbiology
University Medical Center Groningen, RUG
Groningen, 21-24th October 2018
Disclosure slide for speaker at further training events
(Potential) conflict of interest None
Potentially relevant company relationships in connection with
event
Nanopore, Qiagen– no personal benefits
Sponsorship or research funding Nanopore, Qiagen, Roche
© ESCMID eLibrary by a
uthor

11/13/18
2
Lab design
Sample collectionDNA/RNA extraction
Library Preparation
Sequencing Data analysisResults
interpretation
Different types of samples (tissues,
fluids, etc).
Sequencing equipment.
Read length.
Taxonomy.ARG detection.
Phylogeny.
Sensitivity.PPV.
Limits of detection.
Human DNA/RNA
removal.Microbial post-
enrichment.
Fragmentation.Random pre-
amplification.Target
enrichment.
Where: @ the Microbiology lab not in our core
facility
Why:
• Need for Speed
• Infectious samples
• Customized protocols
• Building experience and capacity
• Gaining knowledge
Ion Torrent MiSeq 1 MiSeq 2 NextSeq
4x MinION 1x MinIT
NGS in the Medical Microbiology laboratory
© ESCMID eLibrary by a
uthor

11/13/18
3
How do I choose the best
platform for my metagenomics experiments?Million dollar question…
How do I choose the best platform for my metagenomics experiments?
• It depends on what you want to do!
• Where are you performing your sequence? In a capacitated diagnostic lab or in remote areas?
• Do you want to sequence DNA or RNA metagenomes? Or both?
• Do you want to perform taxonomic classification only or do you wish to obtain
whole genomes with a high accuracy?
• Do you want to detect low frequency variants (e.g. viruses)?
• What is the time available for an answer?
• Do you want to look at the host-response?
© ESCMID eLibrary by a
uthor

11/13/18
4
Three major sequencing categories
•Sequencing by synthesis
•Sequencing by ligation
•Nanopore sequencing
Sequencing by synthesis
© ESCMID eLibrary by a
uthor

11/13/18
5
Sequencing by
synthesis
The process of sequencing DNA using a DNA
polymerase and a primer to create a new complementary DNA strand (natural process that occurs
during cell division).
Sequencing platforms
Manufacturer Amplification Detection Chemistry
Sanger Single molecule Optical Sequencing by synthesis
Roche 454 (GS FLX) Clonal Optical Sequencing by synthesis
Helicos BioSciences
(Heliscope)
Single molecule Optical Sequencing by synthesis
Ion Torrent
(ThermoFisher)
Clonal Solid state Sequencing by synthesis
Illumina Clonal Optical Sequencing by synthesis
PacBio Single molecule Optical Sequencing by synthesis
GeneReader (Qiagen) Clonal Optical Sequencing by synthesis
GenapSys (GENIUS) Clonal Solid state Sequencing by synthesis
Levy and Myers. Annu. Rev. Genomics Hum. Genet. 2016, 17: 95-115.
© ESCMID eLibrary by a
uthor

11/13/18
6
1st Step – Attachment to the flow cell
Adapter
DNA fragment
Dense lawnof primers
Adapter
DNA
Adapters
Prepare genomic DNA sample
Randomly fragment genomic DNAand ligate adapters to both ends ofthe fragments.
Attach DNA to surface
Bind single-stranded fragmentsrandomly to the inside surfaceof the low cell channels.
a
Mardis. Annu. Rev. Genomics Hum. Genet. 2008, 9: 387-402.
© ESCMID eLibrary by a
uthor

11/13/18
7
2nd Step – Cluster generation
Attached
Bridge ampliication
Add unlabeled nucleotides
and enzyme to initiate solid-
phase bridge ampliication.
Denature the doublestranded molecules
Nucleotides
Mardis. Annu. Rev. Genomics Hum. Genet. 2008, 9: 387-402.
Optimal cluster densities
Illumina Technical Note – Optimizing Cluster Density on Illumina Sequencing Systems.
© ESCMID eLibrary by a
uthor

11/13/18
8
Clustering
Blue boxes - raw
cluster density
range.
Green boxes - %PF
cluster density
range.
Red line – median
cluster density
values.
Illumina Technical Note – Optimizing Cluster Density on Illumina Sequencing Systems.
Clustering
• Cluster density is critical because it influences run quality, reads
passing filter, Q30 scores, and total data output.
• Underclustering leads to lower data output, although maintaining the high data quality.
• Overclustering leads to poor run performance, lower Q30 scores, possible introduction of sequencing artifacts and, in the end, lower
quality data output.
Illumina Technical Note – Optimizing Cluster Density on Illumina Sequencing Systems.
© ESCMID eLibrary by a
uthor

11/13/18
9
How does overclustering affect sequencing data?
• Overclustering acts on sequencing data in the following ways:
• Lower Q30 Scores—Due to overloaded signal intensities, the ratio of base
intensity to background for each base is decreased. This decrease often results in ambiguity during base calling, and leads to a decrease in data quality.
• Lower Clusters Passing Filter—The percentage of clusters passing filter (%PF) is an indication of signal purity from each cluster. Overclustered flow cells typically have higher numbers of overlapping clusters. This leads to poor template generation, which then causes a decrease in the %PF metric.
Illumina Technical Note – Optimizing Cluster Density on Illumina Sequencing Systems.
How does overclustering affect sequencing data?
• Lower Data Output—Reduced yield (gigabases [Gb] per flow cell) is a by product of lower %PF.
• Inaccurate Demultiplexing—Index reads usually have low diversity by design, which can lead to poor base calling. Overclustering exacerbates the potential
for poor base calling, which in turn, can lead to demultiplexing failures.
• Complete Run Failure—In cases of extreme overclustering, focusing can fail and the run may terminate at any cycle.
lllumina Technical Note – Optimizing Cluster Density on Illumina Sequencing Systems.
© ESCMID eLibrary by a
uthor

11/13/18
10
MetaNet
• Metagenomics for clinical microbiology
• Organize ring test and proficiency testing trials (EQA, QC)
• Develop or improve databases for pathogens, host genes and know
pathogen-host relations
MetaNet
1st Ring Trial
• For this first test run, 1 positive control, 1 negative control, and 6
mock samples were extracted in Salt Lake City (RNA and DNA).
• Extracted nucleic acids were shipped to Groningen and then sent to Copenhagen, Münster, and Tübingen.
MetaNet
© ESCMID eLibrary by a
uthor

11/13/18
11
Run QC
A. Cluster density and total yield.
B. Proportion of clusters passing filter (CI PF), bases ³Q30 (Q30), and the error rates based on the phiX control Error Rate).
Metanet study - kindly provided by Robert Schlaberg.
Metanet study - kindly provided by Robert Schlaberg.
© ESCMID eLibrary by a
uthor

11/13/18
12
Organism detection in mock samples
Metanet study - kindly provided by Robert Schlaberg.
b
Laser
First chemistry cycle:
determine irst base
To initiate the irst
sequencing cycle, add
all four labeled reversible
terminators, primers, and
DNA polymerase enzyme
to the low cell.
Image of irst chemistry cycle
After laser excitation, capture the image
of emitted luorescence from each
cluster on the low cell. Record the
identity of the irst base for each cluster.
Sequence read over multiple chemistry cycles
Repeat cycles of sequencing to determine the sequenceof bases in a given fragment a single base at a time.
Before initiating the
next chemistry cycle
The blocked 3' terminus
and the luorophore
from each incorporated
base are removed.
GCTGA...
• After clustering is completed, all of the
reverse strands are washed off the flow cell,
leaving only forward strands.
• Primers attach to the forward strands and add fluorescently tagged nucleotides to the DNA
strand. Only one base is added per round.
• A reversible terminator is on every nucleotide
to prevent multiple additions in one round.
• Using color chemistry, each of the four bases has a unique emission, and after each round,
the machine records which base was added.
3rd Step – Synthesis of DNA
Mardis. Annu. Rev. Genomics Hum. Genet. 2008, 9: 387-402.
© ESCMID eLibrary by a
uthor

11/13/18
13
Optical detection
• In four-channel SBS, bases are identified using four different
fluorescent dyes for each base and four images per sequencing cycle.
• With four-channel sequencing, every sequencing cycle requires four dyes and four images to determine the DNA sequence.
• The MiSeqTM, and HiSeqTM systems use four-channel chemistry.
Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
[9] Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
© ESCMID eLibrary by a
uthor

11/13/18
14
Optical detection
• Around 2014, Illumina changed from a 4-channel SBS method to a 2-
channel SBS method.
• Images are taken using red and green filter bands (2 images per cycle, instead of 4).
• This accelerated sequencing and data processing times, while delivering the same quality and accuracy.
• The MiniSeqTM, NextSeqTM, and NovaSeqTM systems use two-channel chemistry.
Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
[9] Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
Thymines are labeled with a green fluorophore, cytosines are
labeled with a red fluorophore, and adenines are labeled with
both red and green fluorophores. Guanines are permanently
dark.
© ESCMID eLibrary by a
uthor

11/13/18
15
Optical detection
• In 2017, Illumina combined SBS chemistry with CMOS technology to
deliver one-channel sequencing chemistry.
• Using a CMOS sensor embedded in the consumable is a simple and fast detection method.
• The iSeqTM 100 system uses one-channel chemistry.
Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
Illumina Technical Note – Illumina CMOS Chip and One-Channel SBS Chemistry.
© ESCMID eLibrary by a
uthor

11/13/18
16
Experiment 1
• It has been shown that different Illumina sequencers may have
different sequencing errors that might influence the final sequences.
• Therefore, here we evaluated the use of shotgun metagenomics and bioinformatics analyses to type DENV directly from sera and plasma samples.
• To optimize the workflow, we evaluated the effect of: i) the DNase I treatment to decrease the human DNA background; ii) two different library preparation methods and iii) two sequencing platforms, on the sequence data quality.
Schirmer et al. BMC Bioinf. 2016, 17: 125; Lizarazo et al. Under review 2018.
MiSeq vs NextSeq
37
Table 2. Sequence quality of the 4 runs performed using two different library preparation kits and two sequencing platforms. 722
Platform Library Prep
Raw density
(K/mm2)
%PF %≥ Q30 Total reads Total reads (PF) Yield Gbp
MiSeq NXT 1,082 ± 38 86.12 82.96 40,332,330 34,734,340 5.45
MiSeq TS 869 ±16* 91.51 92.95 32,919,292 30,123,438 4.59
NextSeq NXT 22 ± 4* 98.82 96.50 38,684,052 37,613,244 2.29
NextSeq TS 179 ± 4 89.75 84.08 114,917,472 103,134,945 41.66
Abbreviations: Gbp, giga base pair; PF, passing filter; Q30, quality score with base call accuracy of 99.9% (1 incorrect base in 1000 based calls); 723
NXT, Nextera XT library pep; TS, TruSeq v2 RNA library prep. 724
*Raw density was under the optimal range. 725
Lizarazo et al. Under review 2018.
© ESCMID eLibrary by a
uthor

11/13/18
17
MiSeq vs NextSeq
Table 3. Comparison of the effect of different library preparation methods and sequencing platforms in the proportion of DENV reads. 726
Platform
Library
Preparation
No.
Samples
Average
total
number
of reads
Average
human
mapped
reads
Average
unmapped
Reads
Average
DENV
mapped
reads
Average
proportion
of DENV
mapped*
Average
depth
coverage
(fold)
Average
assembled
consensus
length
(bp)
Average
mapped
consensus
length
(bp)
MiSeq
NXT 5 2,792,515 2,042,971 749,544 469,950 63% 5,603 7,667 10,683
TS 5 1,396,358 1,182,726 213,632 136,998 64% 1,867 10,202 10,680
NextSeq
NXT 12 677,692 155,878 521,814 175,820 34% 2,498 4,960 9,543
TS 12 6,179,416 4,054,813 2,124,603 1,547,456 73% 21,327 10,347 10,483
*Using the number of unmapped reads as denominator. 727
Abbreviations: bp, base pair; NXT, Nextera XT library pep; TS, TruSeq v2 RNA library prep. 728
Lizarazo et al. Under review 2018.
Conclusions
• No significant differences were found between equipment.
• The NextSeq allowed the multiplexing of more samples.
• The cost per sample was slightly higher (86€) with the MiSeq, compared to the NextSeq (70€).
Lizarazo et al. Under review 2018.
© ESCMID eLibrary by a
uthor

11/13/18
18
Click to edit Master title style
• High accuracy
• High throughput (depending on the system)
• Expensive platforms (depending
on the system)
• Long run time
• Short read length
• Not enable to detect large structural variations
Advantages Disadvantages
© ESCMID eLibrary by a
uthor

11/13/18
19
Ion Torrent/Ion PGM Technology
• Each nucleotide floods the chip subsequently.
• When a nucleotide is added to the DNA template and is
then incorporated into a strand of DNA, a hydrogen ion (H+)
is released.
• The charge from that ion changes the pH of the solution,
which can be detected by the ion sensor.
• The sequencer calls the base, going directly from chemical
information to digital information.
www.thermofisher.com
Ion Torrent/Ion PGM Technology
www.thermofisher.com
• If the next nucleotide that floods the chip is not a match,
there will be no voltage change recorded and no base will
be called.© ESCMID eLibrary by a
uthor

11/13/18
20
Ion Torrent/Ion PGM Technology
www.thermofisher.com
• If there are two identical bases on the DNA strand, the
voltage will be double, and the chip will record two identical
bases.
Sequencing at sea: challenges andexperiences in Ion Torrent PGMsequencing during the 2013 SouthernLine Islands Research Expedition
• The authors successfully sequenced 26 marine microbial genomes, and two
marine microbial metagenomes using the Ion Torrent platform on the Merchant
Yacht Hanse Explorer.
• Onboard sequence assembly, annotation, and analysis enabled us to investigate
the role of the microbes in the coral reef ecology of these islands and atolls.
Lim et al. PeerJ 2014, 2:e520.
© ESCMID eLibrary by a
uthor

11/13/18
21
Click to edit Master title style
• Very fast
• Highly scalable
• Different chips available
• Low cost
• High sequencing error rate
(namely in homopolymericregions)
Advantages Disadvantages
© ESCMID eLibrary by a
uthor

11/13/18
22
PacBio
1. Each nucleotide contains a specific fluorescent dye.
2. Once the correct nucleotide is added to the sequence by the DNA polymerase, it emits light at a
specific wavelength.
3. The detector inside the zero-mode waveguide (ZMW, nanophotonic visualization chamber), captures
the light emitted.
Study 1
• The objective was to investigate the utility of Pacific Biosciences
circular consensus sequencing (CCS) reads for metagenomic projects.
• They compared the application and performance of both PacBio CCS and Illumina HiSeq data with assembly and taxonomic binning algorithms using metagenomic samples representing a complex microbial community.
| : | DOI: . /srep
www.nature.com/scientificreports
Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence dataJ. A. Frank , Y. Pan , A. Tooming-Klunderud , V. G. H. Eijsink , A. C. McHardy ,
A. J. Nederbragt & P. B. Pope
function within microbial communities. (ere we investigate the utility of Paciic Biosciences long and
SMRT cells produced approximately Mb of CCS reads from a biogas reactor microbiome sample that averaged nt in length and . % accuracy. CCS data assembly generated a comparative number of large contigs greater than kb, to those assembled from a ~ x larger (iSeq dataset ~ Gb produced from the same sample i.e approximately % of total contigs . (ybrid assemblies
produced signiicant enhancements in taxonomic binning and genome reconstruction of two dominant
Department of emistr iotec no o an oo cience orwe ian ni ersit of ife ciences s omputationa io o of Infection esearc e m o t entre for Infection esearc In o enstra
raunsc wei erman . o utionar nt esis in ern orwa . orrespon ence an re uests for materia s s ou e a resse to
. . . emai : p i .pope nm u.no
ecei e : Octo er
ccepte : pri
u is e : a
Frank et al. Sci Rep 2016, 6: 25373.
© ESCMID eLibrary by a
uthor

11/13/18
23
Improved metagenome assemblies and
taxonomic binning using long-read circular
consensus sequence data
• The proportion of total DNA binned in
the major phyla (A) represented in
the Link_ADI microbiome was similar
for both PacBio CCS and HiSeqcontigs.
• Differences between the sequencing
methods were evident at a species
level where some abundant species
assembled and binned better with PacBio, whereas others produced
better results with HiSeq data (B).
• The incorporation of PacBio data
produced significant enhancements
in taxonomic binning and genome reconstruction of two dominant
phylotypes, which assembled and
binned poorly using HiSeq data alone.
Frank et al. Sci Rep 2016, 6: 25373.
Click to edit Master title style
• Long reads
• Enables detection of structural variants (e.g. gene duplications)
• Very fast (real time sequencing)
• High sequencing error rate
• Very expensive
• Lower throughput compared to other platforms, especially when increasing the number of passages (DNA polymerase saturation)
Advantages Disadvantages
© ESCMID eLibrary by a
uthor

11/13/18
24
Sequencing by ligation
Sequencing by
ligation
The process of sequencing DNA using a DNA ligase to
create a new complementary DNA strand.
© ESCMID eLibrary by a
uthor

11/13/18
25
Sequencing platforms
Manufacturer Amplification Detection Chemistry
SOLiD (ThermoFisher) Clonal Optical Sequencing by ligation
Dover (Polonator) Clonal Optical Sequencing by ligation
Complete Genomics Clonal Optical Sequencing by ligation
Levy and Myers. Annu. Rev. Genomics Hum. Genet. 2016, 17: 95-115.
Sequencing using a nanopore
© ESCMID eLibrary by a
uthor

11/13/18
26
Levy and Myers. Annu. Rev. Genomics Hum. Genet. 2016, 17: 95-115.
Sequencing platforms
Manufacturer Amplification Detection Chemistry
Oxford Nanopore Single molecule Nanopore Nanopore
Roche Genia Single molecule Nanopore Nanopore
Quantum Biosystems Single molecule Nanogate Nanogate
Sequencing with a nanopore
• A nanopore is a pore of nanometer size.
• It can be divided into three categories:
• Biological– also called transmembrane protein channels, usually inserted into a substrate (membrane). Well-defined and highly-reproducible nanopore size and structure.
• Solid-state – synthetic nanopores. They have many superior advantages over their biological counterparts, such as chemical, thermal, and mechanical stability, size adjustability, and integration.
• Hybrid – a mixture of both, taking advantage of the features of biological and solid-state nanopores.
Feng et al. Gen. Prot. Bioinf. 2015, 13: 4-16.
© ESCMID eLibrary by a
uthor

11/13/18
27
Biological nanopores
Feng et al. Gen. Prot. Bioinf. 2015, 13: 4-16.
2.6 nm
Top view
10 nm
7 nm
5.2 nm
Side viewCap
β-barrel
ASide viewB
4 nm
1.2 nm
Top view
9 nm
3.6 nm 6 nm
13.8 nm
7.5 nm
Top view
Side viewC
10 nm 9.6 nm
Alpha-hemolysin
S. aureus
MspA porin
M. smegmatis
Connector channel
Bacteriophage phi29
© ESCMID eLibrary by a
uthor

11/13/18
28
Oxford Nanopore
• The detection principle is
based on monitoring the
ionic current passing
through the nanopore as a voltage is applied across the
membrane.
Feng et al. Gen. Prot. Bioinf. 2015, 13: 4-16.
Challenges/Hurdles
• The Oxford Nanopore data (i.e.
provided by the MinION) presents a higher error rate than other sequencing platforms.
• It is improving, but it is still not at the level of short-read sequencing.
Ashton et al. Nat. Biotechnol. 2015, 33: 296-300; George et al. Microb. Genom. 2017, 3, 1-8.
© ESCMID eLibrary by a
uthor

11/13/18
29
Real-time sequencing
• We obtained results after 10 minutes.
• However, you need the right equipment, for example the MinIT(Nanopore).
• You also need bioinformatic skills.
NGS for Tuberculosis
• Routine full characterization of Mycobacterium tuberculosis (TB) is
culture-based, taking many weeks.
• Whole-genome sequencing (WGS) can generate antibiotic susceptibility profiles to inform treatment, augmented with strain information for global surveillance.
• Such data could be transformative if provided at or near the point of care.
Votintseva et al. J. Clin. Microbiol. 2017, 55(5):1285–98.
© ESCMID eLibrary by a
uthor

11/13/18
30
NGS for Tuberculosis
• Applied directly to clinical samples.
• Initial evaluation with Illumina, followed by Nanopore sequencing.
• With Illumina MiSeq/MiniSeq, the workflow from patient sample to results could be completed in 44h/16 h at a reagent cost of £96/£198 per sample.
• For Nanopore, the estimated turnaround time to detection of resistance was 7.5h (full profile 5h later).
• Antibiotic susceptibility predictions were fully concordant.
Votintseva et al. J. Clin. Microbiol. 2017, 55(5):1285–98.
• They designed an adapter of a highly conserved termini of the
influenza A virus genome to target the (-) sense RNA into a protein nanopore on the Oxford Nanopore MinION sequencing platform.
• The researchers used total RNA extracted from the allantoic fluid of influenza rA/Puerto Rico/8/1934 (H1N1) virus infected chicken eggs (EID50 6.8 × 109).
• They demonstrated successful sequencing of the coding complete influenza A virus genome with 100% nucleotide coverage, 99% consensus identity, and 99% of reads mapped to influenza A virus.
| DO): . /s - - -
www.nature.com/scientificreports
Direct RNA Sequencing of the Coding Complete )nluenza A Virus GenomeMatthew W. Keller , Benjamin L. Rambo-Martin , Malania M. Wilson , Callie A. Ridenour ,
Samuel S. Shepard , Thomas J. Stark , Elizabeth B. Neuhaus , Vivien G. Dugan ,
David E. Wentworth & John R. Barnes
For the irst time, a coding complete genome of an RNA virus has been sequenced in its original form. Previously, RNA was sequenced by the chemical degradation of radiolabeled RNA, a diicult method that produced only short sequences. )nstead, RNA has usually been sequenced indirectly by copying it into cDNA, which is often ampliied to dsDNA by PCR and subsequently analyzed using a variety of DNA sequencing methods. We designed an adapter to short highly conserved termini of the inluenza A virus genome to target the - sense RNA into a protein nanopore on the Oxford Nanopore Min)ON sequencing platform. Utilizing this method with total RNA extracted from the allantoic luid of inluenza rA/Puerto Rico/ / ( N virus infected chicken eggs E)D . , we demonstrate successful sequencing of the coding complete inluenza A virus genome with % nucleotide coverage, % consensus identity, and % of reads mapped to inluenza A virus. By utilizing the same methodology one can redesign the adapter in order to expand the targets to include viral mRNA and
sense cRNA, which are essential to the viral life cycle, or other pathogens. This approach also has the potential to identify and quantify splice variants and base modiications, which are not practically measurable with current methods.
)nluenza Division, National Center for )mmunization and Respiratory Diseases NC)RD , Centers for Disease Control and Prevention CDC , Atlanta, Georgia, USA. Matthew W. Keller and Benjamin L. Rambo-Martin contributed equally. Correspondence and requests for materials should be addressed to J.R.B. email: fzq @cdc.gov
Received: 23 April 2018
Accepted: 5 September 2018
Published online: 26 September 2018
OPENCorrection: Author Correction
Keller et al. Sci. Rep. 2018, 8:14408 .
| DOI: . /s - - -
ResultsRNA calibration strand: enolase )) mRNA.
Sequencing RNA from crude versus puriied inluenza rA/Puerto Rico/ /1 (1N1 virus.
© ESCMID eLibrary by a
uthor

11/13/18
31
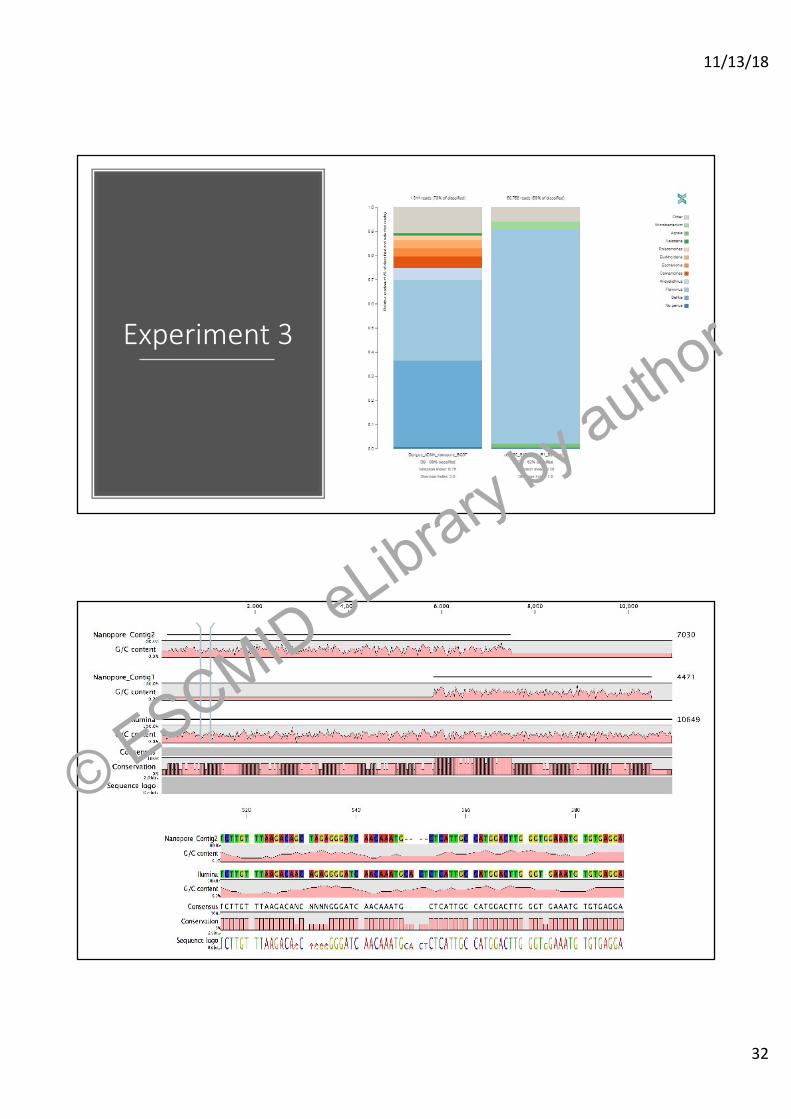
Experiment 3
• Comparison between Illumina sequencing and Nanopore sequencing.
• So far we have compared 3 metagenomes using both technologies.
Experiment 3© ESCMID eLibrary by a
uthor
11/13/18
32
Experiment 3
© ESCMID eLibrary by a
uthor

11/13/18
33
Click to edit Master title styleAdvantages
• Very portable
• Very fast (real time sequencing)
• Direct RNA sequencing is possible
• Very long reads
• Enables detection of structural variants (e.g. gene duplications)
• High sequencing error
• Chemistry is constantly changing
Disadvantages
Summary
• It is not an easy choice.
• It really depends on what you want to do and where you want to do
it!
• Keep in mind that none of the technologies is perfect and they all
have pros and cons.© ESCMID eLibrary by a
uthor

11/13/18
34
Any questions?
© ESCMID eLibrary by a
uthor