neuromuscular development in the avian paralytic mutant crooked neck dwarf (cn/cn): further evidence...
TRANSCRIPT

Neuromuscular Development in the AvianParalytic Mutant Crooked Neck Dwarf(cn/cn): Further Evidence for the Role
of Neuromuscular Activityin Motoneuron Survival
RONALD W. OPPENHEIM,1* DAVID PREVETTE,1 LUCIEN J. HOUENOU,1
MARTINE PINCON-RAYMOND,2 VIOLETTA DIMITRIADOU,3 ANNE DONEVAN,4
MICHAEL O’DONOVAN,4 PETER WENNER,4 DAVID D. MCKEMY,5
AND PAUL D. ALLEN6
1Department of Neurobiology andAnatomy and The Neuroscience Program, Wake ForestUniversity, Bowman Gray School of Medicine, Winston-Salem, North Carolina 27157
2Group de Biologie et Pathologie Neuromusculaires, INSERM U153, 75005 Paris, France3Laboratoire de Physiologie, Faculte de Pharmacie, 75006 Paris, France
4Section on Developmental Neurobiology, Laboratory of Neural Control, National Institute ofNeurological Disease and Stroke, National Institutes of Health, Bethesda, Maryland 20892
5Department of Pharmacology, University of Nevada School of Medicine,Reno, Nevada 89557
6Department of Anesthesia, Brigham and Women’s Hospital, Boston, Massachusetts 02115
ABSTRACTNeuromuscular transmission and muscle activity during early stages of embryonic
development are known to influence the differentiation and survival of motoneurons and toaffect interactions with their muscle targets. We have examined neuromuscular developmentin an avian genetic mutant, crooked neck dwarf (cn/cn), in which a major phenotype is thechronic absence of the spontaneous, neurally mediated movements (motility) that arecharacteristic of avian and other vertebrate embryos and fetuses. The primary genetic defectin cn/cn embryos responsible for the absence of motility appears to be the lack ofexcitation-contraction coupling.Althoughmotility inmutant embryos is absent from the onsetof activity on embryonic days (E) 3–4, muscle differentiation appears histologically normal upto about E8. After E8, however, previously separate muscles fuse or coalesce secondarily, andmyotubes exhibit a progressive series of histological and ultrastructural degenerativechanges, including disarrayed myofibrils, dilated sarcoplasmic vesicles, nuclear membraneblebbing, mitochondrial swelling, nuclear inclusions, and absence of junctional end feet.Mutant muscle cells do not develop beyond the myotube stage, and by E18–E20 most muscleshave almost completely degenerated. Prior to their breakdown and degeneration, mutantmuscles are innervated and synaptic contacts are established. In fact, quantitative analysisindicates that, prior to the onset of muscle degeneration, mutant muscles are hyperinner-vated. There is increased branching of motoneuron axons and an increased number ofsynaptic contacts in the mutant muscle on E8. Naturally occurring cell death of limb-innervating motoneurons is also significantly reduced in cn/cn embryos. Mutant embryoshave 30–40% more motoneurons in the brachial and lumbar spinal cord by the end of thenormal period of cell death. Electrophysiological recordings (electromyographic and direct
Contract grant sponsor: NIH; Contract grant numbers: NS20402, AR43140; Contract grant sponsor: Muscular Dystrophy Association; Contract grantsponsor: B. Philippe Foundation.*Correspondence to: Ronald W. Oppenheim, Department of Neurobiology and Anatomy, Wake Forrest University, Bowman Gray School of Medicine,
Medical Center Boulevard, Winston-Salem, NC 27157.Received 6 July 1994; Revised 19 December 1996; Accepted 27 December 1996
THE JOURNAL OF COMPARATIVE NEUROLOGY 381:353–372 (1997)
r 1997 WILEY-LISS, INC.

records form muscle nerves) failed to detect any differences in the activity of control vs.mutant embryos despite the absence of muscular contractile activity in the mutant embryos.The a-ryanodine receptor that is genetically abnormal in homozygote cn/cn embryos is notnormally expressed in the spinal cord. Taken together, these data argue against the possibilitythat the mutant phenotype described here is caused by the perturbation of a central nervoussystem (CNS)-expressed a-ryanodine receptor. The hyperinnervation of skeletal muscle andthe reduction of motoneuron death that are observed in cn/cn embryos also occur ingenetically paralyzed mouse embryos and in pharmacologically paralyzed avian and ratembryos. Because a primary common feature in all three of these models is the absence ofmuscle activity, it seems likely that the peripheral excitation of muscle bymotoneurons duringnormal development is a major factor in regulating retrograde muscle-derived (or muscle-associated) signals that control motoneuron differentiation and survival. J. Comp. Neurol.381:353–372, 1997. r 1997 Wiley-Liss, Inc.
Indexing terms: chick embryo; spinal cord; cell death; muscle; neurons
During the normal development of most vertebrates, asubstantial portion (,50%) of all postmitotic spinal moto-neurons degenerate and die by a process of naturallyoccurring or programmed cell death (Oppenheim, 1991).Studies carried out primarily with avian embryos haveshown that the regulation of motoneuron survival ismediated by interactions between developing motoneu-rons and their synaptic targets, skeletal muscle cells(Oppenheim, 1981; Hamburger and Oppenheim, 1982;Landmesser, 1992).Although there is substantial evidencethat an important aspect of this interaction involves thesurvival promoting action of putative muscle-derived neu-rotrophic agents (see, e.g., Oppenheim et al., 1988, 1993;Bloch-Gallego et al., 1991), it is also thought that neuromus-cular activity plays a critical role in this process (Oppen-heim, 1987; Landmesser, 1992; Oppenheim et al., 1993).Chronic reductions in neuromuscular activity during thecell death period prevent motoneuron death (Pittman andOppenheim, 1978, 1979; Laing andPrestige, 1978), whereashyperactivity generated by direct electrical stimulation ofskeletalmuscle in ovo increasesmotoneuron death (Oppen-heim and Nunez, 1982). Although the cellular and molecu-lar mechanisms responsible for this activity-dependentregulation of motoneuron survival are not entirely clear, itappears that one previously suggested mechanism, theactivity-dependent regulation of muscle-derived trophicagents, might not be involved (Tanaka, 1987; Oppenheim,1989; Houenou et al., 1991). By contrast, there is increas-ing evidence that activity may regulate motoneuron sur-vival by controlling the extent of intramuscular motoraxon branching and synaptogenesis, which may, in turn,regulate the access of motoneurons to a muscle-derivedtrophic factor (Oppenheim and Chu-Wang, 1983; Oppen-heim, 1989; Oppenheim et al., 1989a,b; Houenou et al.,1991; Landmesser, 1992; Tang and Landmesser, 1993).Although the activity-dependent regulation of motoneu-
ron survival has been studied primarily in geneticallynormal avian embryos treated with paralytic neuromuscu-lar blocking agents such as curare and a-bungarotoxin(a-BTX), another potentially valuable source of informa-tion for studying this process is genetically mutant strainsof mice and chicken embryos that exhibit neuromuscularparalysis during development. For example, embryos ofthe paralytic mouse mutant muscular dysgenesis (mdg/mdg) exhibit excessive intramuscular motor axon branch-ing, increased neuromuscular synaptogenesis, and a greatly
reduced programmed death of spinal motoneurons (Riegerand Pincon-Raymond, 1981; Rieger et al., 1984; Oppen-heim et al., 1986), all of which are characteristics similarto what occurs in genetically normal, but pharmacologi-cally paralyzed, chick embryos. Considerable evidencesupports the idea that the primary defect in themdg/mdgmutant is myogenic and involves mechanisms mediatingexcitation-contraction coupling (Pincon-Raymond et al.,1985) and, therefore, that it is the absence of musclecontraction, per se, that induces the phenotypic changes inneuromuscular development.Another genetic mutant that is phenotypically similar to
the mdg/mdg mouse is the autosomal recessive avianmutant crooked neck dwarf (cn/cn), first described byAsmundson (1945). This mutant also exhibits embryonicparalysis and skeletal muscle dysgenesis, and all embryosdie prior to hatching owing to respiratory failure.Althoughthere have been a few studies of myogenesis in cn/cnembryos (Rosenberg, 1945; Wick and Allenspach, 1978;Pautou et al., 1982;Mauger et al., 1983; Kieny et al., 1988),very little information is available on neuromuscular ormotoneuron development in this mutant. Additionally,whereas the results of previous studies suggested that theonset of paralysis in cn/cn was a relatively late event(Kieny et al., 1983), in our preliminary studies we observedthat mutant embryos fail to initiate neuromuscular activ-ity on embryonic days (E) 3–4 (i.e., the age when suchactivity begins in control embryos) and that neuromuscu-lar activity is absent for the remainder of the incubationperiod (Oppenheim and Prevette, 1986). Because the com-plete absence of neuromuscular activity in avian cn/cnmutants is similar to the situation in themdg/mdgmousemutant and in pharmacologically paralyzed chick em-bryos, we have examined the extent to which neuromuscu-lar andmotoneuron developmentmay be similarly affectedin all three models.
MATERIALS AND METHODS
Embryos
Eggs from a cross of heterozygous carriers of the cn genewere obtained from the Department of Animal Genetics,University of Connecticut, and were incubated in thelaboratory at 37.5°C and 60% relative humidity. On E3, awindow was made in the shell through which the embryocould be observed, after which the window was sealed with
354 R.W. OPPENHEIM ET AL.

Parafilm (Sigma) and the eggs were returned to theincubator. Beginning on E4, all embryos were observedevery other day for 5 minutes with a dissecting microscope(320), and those embryos failing to exhibit spontaneous orinduced neuromuscular activity (embryonic motility; seeHamburger, 1963) for three alternate days (E4, E6, E8)were provisionally designated as homozygous cn/cn mu-tants. Observations on subsequent days (E10–E18) con-firmed that these embryos never exhibited the spontane-ous neuromuscular movements characteristic of normal orcontrol chick embryos. By E8–E10, all of the immobileembryos also exhibited generalized edema, and, whenexamined histologically on E10, all immobile embryosexhibiting edema also had perturbations of muscle differ-entiation (see below). Because heterozygous embryos(cn/1) are indistinguishable from homozygous wild-typeembryos (1/1; see Asmundson, 1945), in the presentreport control embryos are designated 1/cn?.As describedin more detail below, embryos of different ages wereremoved from the shell, killed, staged according to thechick embryo stage series (Hamburger and Hamilton1951), and prepared for analysis of various aspects ofneuromuscular development.
Cell counts and cell size
For the analysis of cell numbers and cell size, thebrachial and lumbosacral spinal cords with vertebra anddorsal root ganglia (DRG) attached were removed andplaced in Carnoy’s or Bouin’s fixative for 10–24 hours,processed for paraffin histology, sectioned on a rotarymicrotome (6–10 µm), and stained with thionin or hema-toxylin and eosin. Limb-innervating motoneurons in thelateral motor column (lmc) were counted in every tenthsection. For analysis of sensory neurons, cells in the thirdlumbar dorsal root ganglion (L3 DRG) were counted inevery fifth section. The criteria for including cells in thecounts were as previously described (Oppenheim et al.,1989a,b; Clarke and Oppenheim, 1995). With these crite-ria, it is not necessary to employ correction factors orsterological methods to compensate against double count-ing. Motoneuron size was determined by measuring thediameter of the nucleus with a drawing tube at 31,200(Oppenheim et al., 1988).
Muscle histology, muscle weight,and creatine kinase activity
For light microscopic examination of general features ofmuscle histogenesis, the hindlimb of embryos at differentages was fixed in Bouin’s, processed for paraffin histology,sectioned transversely (6–8 µm), and stained with hema-toxylin and eosin. Muscle wet weights (hindlimb) weredetermined following removal of the skin and dissection ofmuscles from the bones (Hermann et al., 1963). Creatinekinase activity was determined by a modification of themethod of Rosalski (1967) and expressed as specific activ-ity per milligram of protein (Oppenheim et al., 1986).Protein concentrationwas determined by using themethodof Lowry et al. (1951).
Electron microscopy
Muscles (gastrocnemius or muscles in the same generallocation in the ventral shank of older mutant embryos)were fixed in situ by immersion of the skinned shank in2.5% glutaraldehyde, 0.5% tannic acid at room tempera-ture. After 15–20 minutes, muscles were dissected and
placed overnight into a solution of 0.6% glutaraldehyde,0.5% tannic acid in 0.1 M phosphate buffer (pH 7.4) at 4°C.Postfixation was performed in 2% osmic acid in 0.1 Mphosphate buffer for 1 hour. Samples were dehydrated in agraded alcohol series and embedded in Epon, and ultra-thin sections (70 nm) were obtained by using an LKBultratome. Sections were stained with a saturated solutionof uranyl acetate in 50% acetone, followed by 0.2% leadcitrate. Observations were made with a Philips EM 410.
Muscle innervation
Two different methods were used to examine innerva-tion patterns and end-plate formation (synaptogenesis) incontrol and mutant muscles. In the first method, muscleswere prepared and sectioned and intramuscular nerveswere stained with silver, and the end plates were visual-ized with a histochemical stain for acetylcholinesterase(AChE). This double-staining technique was first de-scribed by Toop (1976) and has been commonly used in ourlaboratory (see, e.g., Pittman and Oppenheim, 1979). Theother technique described below was modified from Dahmand Landmesser (1991).Control and cn/cn embryos (E8–E11) were killed by
decapitation, and the hindlimbs, still attached to thepelvis, were removed and placed in phosphate-bufferedsaline (PBS) at room temperature. The posterior iliotibi-alis, iliofibularis, and sartorius muscles were revealed byremoving overlying skin and connective tissue. Tissueseither were stained with C2, a mouse IgG monoclonalantibody to a chick-specific neuronal cytoskeletal element(Dahm and Landmesser, 1988, 1991), to label intramuscu-lar nerves, or were double stained with a-bungarotoxin(a-BTX) to visualize postsynaptic acetylcholine receptors(AChR) andwith SV2, amouse IgGmonoclonal antibody toa presynaptic vesicle protein (Buckley and Kelly, 1985;Dahm and Landmesser, 1991).Muscles were stained with C-2 (a gift from Dr. L.
Landmesser, Case Western Reserve University) as de-scribed previously (Dahm and Landmesser, 1991). Briefly,muscles were prefixed in cold (220°C) acetone for 5–7minutes, washed (3 3 5 minutes) in PBS, and incubated inC2 antibody diluted 1:4 in 1% Triton X-100 for 1 hour atroom temperature (or overnight at 4°C). Tissues werewashed in PBS, fixed in 3.7% formaldehyde for 30minutes,washed in PBS, incubated in rhodamine isothiocyanate(RITC)-labeled secondary (i.e., goat anti-mouse) antibodyfor 1 hour at room temperature or overnight at 4°C, andwashed in PBS. The posterior iliotibialis, iliofibularis, andsartorius muscles were then individually dissected outfrom the limb and mounted in 50% glycerol containing (1mg/20 ml) phenylenediamine.To examine synaptic junctions, muscles were double
stained with tetramethylrhodamine-labeled aBTX (Rh-aBTX; Molecular Probes, Inc., Eugene, OR) and SV2antibody (a gift from Dr. K. Buckley, University of Califor-nia, San Francisco) as described previously (Dahm andLandmesser, 1991). Briefly, tissues were fixed (,45 min-utes) in 3.7% formaldehyde, washed in PBS, and incubatedin 25 nM Rh-aBTX for 1–2 hours at room temperature (orovernight at 4°C). Samples were washed in PBS andincubated in SV2 diluted 1:100 in 0.3% Triton X-100 for 2hours at room temperature (or overnight at 4°C) beforethey were washed and incubated in fluorescein isothiocya-nate (FITC)-labeled rabbit anti-mouse antibody (Cappel,Durham, NC) diluted 1:100 in PBS for 2 hours at room
DEATH OF AVIAN EMBRYONIC MOTONEURONS 355

temperature. Tissues were then washed in PBS, andindividual muscles were dissected from the limb andmounted as described above.The pattern of intramuscular nerve sprouting was deter-
mined by directly counting in the microscope the numberof side branches permillimeter of length of the two or threemajor intramuscular nerve trunks from slides of whole-mount posterior iliotibialis, iliofibularis, and sartoriusmuscles stained with the C2 antibody (see Dahm andLandmesser, 1988). Whole-mounted posterior iliofibularisand iliotibialis muscles double stained with SV2 anda-BTX were used to examine synapse formation. AChRclusters were counted in control and mutant iliofibularismuscle on E8 and E10. Synaptic contracts were defined asregions in which AChR clusters (a-BTX labeling) werecolocalized with presynaptic (SV2-labeled) profiles. Quan-tification of AChR clusters and synaptic contacts wasassessed from several visual fields defined by the 360objective of the microscope from comparable regions ofcontrol and mutant muscles.Muscle innervation was also assessed by examining the
retrograde transport of horseradish peroxidase (HRP) tolumbar motoneurons following injections into the hind-limb. The methods used were as described by Oppenheimand Heaton (1975). Briefly, HRP injections were madethrough a window in the shell by using a 5 µl Hamiltonsyringe. Approximately 1 µl of a 50% HRP solution wasinjected and embryos were allowed to survive for 10–12hours. Spinal cords were removed and processed for 3,83-diaminobenzidine tetrahydrochloride (DAB) histochem-istry as described previously (Oppenheim and Heaton,1975).
Expression of the a-ryanodine receptor
Using a monoclonal antibody (110F) that recognizes theavian a-ryanodine receptor (Airey et al., 1993a), we exam-ined by immunofluorescence the expression of this recep-tor in E16–E18 skeletal muscle and lumbar spinal cord ofcn/cn and control embryos. The methods employed werethose described byAirey et al. (1993a). Briefly, mutant andcontrol muscle (gastrocnemius) and spinal cord (lumbar)were removed from E16–E18 embryos and placed in 4%paraformaldehyde at 4°C for 30 minutes, followed by two
Fig. 2. Mean (6SD) number of lumbar (A) and brachial (B)motoneurons in control (solid circles; 1/cn?) and mutant (open circles;cn/cn) embryos between E6 and E21. *P , 0.01 E10; *P , 0.001,E8–E9 (t-tests) in lumbar region. *P, 0.01 E8; *P, 0.001 E10–E15 inbrachial region. Sample sizes for each data point were 6–8 for both Aand B, except for E21, for which only two surviving embryos wereassessed.
Fig. 1. Mean (6SD) number of trunk (E4) and hindlimb (E6–E18)movements (motility) in control (CON; solid circles) and mutant(CN/CN; open circles) embryos from embryonic days (E) 4 to 18.Mutant embryos were completely immobile throughout the incubationperiod. Sample sizes at each age were 6–10.
Fig. 3. Mean (6SD) diameter of the nucleus of lumbar motoneu-rons in control (CON) and mutant (cn/cn) embryos on E10 and E16.The values in the bars refer to the numbers of neurons examined.
356 R.W. OPPENHEIM ET AL.

Fig. 4. Histology of lumbar spinal cord and motoneurons on E8(A,B), E10 (C,D), and E15 (E,F) in mutant (B,D,F) and control (A,C,E)embryos. Insets are higher magnifications of motoneurons in the lmc.lmc, Lateral motor column (ventral horn); d, dorsal root ganglia. The
arrows indicate the medial border of the lmc. Scale bars 5 40 µm inA,B, 50 µm in C, D, 30 µm in E, F, 13 µm in A, B insets, 17 µm in C, D,insets, 16 µm in E, F, insets.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 357

10 minute washes in PBS and then cryprotection insucrose/OCT solutions. Frozen longitudinal (muscle) andtransverse (spinal) sections (10 µm) were cut on a cryostatat 220°C and processed for immunofluorescence. Theprimary antibody was diluted 1:5 and added to the sec-tions, followed by a secondary antibody diluted 1:500conjugated to biotin. Sections were then incubated withstreptavidin conjugated to HRP followed by (FITC) conju-gated tyramides according to the Tyramide Signal Amplifi-cation kit from NEN-Dupont.
Electrophysiological recordings in ovo
Initially, embryos (E8–E9) were observed through asmall window in the shell to establish whether theyexhibited neuromuscular motility. Embryos that exhibitedno movement over a 5 minute period of observation wereprovisionally classified as cn/cn. The window in the shellwas then expanded, and the amnion was opened to exposethe hindlimb. When necessary, the amniotic fluid wassupplemented with warmed Tyrode’s and the temperaturein ovo was maintained at 34–36°C using a heating coilaround the egg. A small incision was made in the skin overthe lateral aspect of the thigh to expose the surface of theposterior iliotibialis muscle. A flexible suction electrodewas applied to the muscle surface. In actively movingcontrol embryos, it was sometimes necessary to repositionthe electrode to maintain a good recording. Muscle electri-cal activity was amplified (1,000–10,0003; A and M Sys-tems model 1700), filtered (10 Hz to 2 khz), and recordedon tape (Neuro-corder model DR-886). Recordings of spon-taneous electrical activity were analyzed and quantified bymeasuring the frequency of bursts and of synchronizedspikes. The smaller-amplitude, synchronous bursts werecounted as synchronized spikes. The smaller-amplitude,asynchronous bursts were counted as single episodes whenseparated by more than 30 seconds of electrical silence.The large, synchronized spikes were easily distinguishedon the basis of their amplitude.
Electrophysiological recordings in vitro
In vitro experiments were performed using some of theembryos from the in ovo studies and additional cn/cn or1/cn? embryos. Embryos (E8–E9) were dissected in aperfusion bath of cooled Tyrode’s solution to isolate thespinal cord with intact muscle nerves (Landmesser andO’Donovan, 1984).Muscle nerves supplying the femorotibi-alis (a knee extensor) and sartorius (a hip flexor) muscleswere drawn into suction electrodes and the signals ampli-fied (1,0003; WPI DAM50) and filtered (100 Hz to 10 khz).The perfusion bath was warmed to 27°C, and spontaneousactivity was recorded on tape and displayed on a chartrecorder (Gould model TA11). The activity patterns wereanalyzed by measuring the frequency of episodes and thenumber of cycles per episode.
Ryanodine receptor deleted mice
Fetal mice with a targeted deletion of the skeletalmuscle isoform of the mouse a-ryanodine receptor (RyR-1)gene are immobile owing to the absence of excitation-contraction coupling (Nakai et al., 1996). The spinal cordsof E18.5 homozygote (2/2) heterozygote (1/2) and wild-type (1/1) fetuses were prepared as described above forchick embryo tissue. Motoneurons in the thoracic andlumbar regions were counted in every 10th section as
described previously (Oppenheim et al., 1986; Clark andOppenheim, 1995).
RESULTS
Neuromuscular activity (motility)
CNS generated embryonic movements begin on E3.5 inthe chick embryo, reach a peak frequency at E10–E15, andthen decline towards the time of hatching (Oppenheim,1975). The first movements involve neck and trunkmuscu-lature, whereas hindlimb movements begin by about E6(Hamburger and Balaban, 1963; Landmesser and Morris,1975). Control embryos (1/cn?) exhibited a similar trendin the development of neuromuscular activity as describedin these earlier reports, whereas mutant embryos (cn/cn)were completely inactive during the entire observation period(Fig. 1), and not a single embryo exhibited detectable activemovements of the trunk or limbs at any stage of developmentfrom E3.5 to E18. Amniotic contractions appeared normal inmutant embryos. Virtually all mutant embryos died priorto the normal time of hatching on E21–E22.
Neuronal survival and growth
OnE6, prior to the onset of naturally occurringmotoneu-ron death in either the brachial or the lumbar region(Hamburger, 1975; Oppenheim andMajors-Willard, 1978),control and mutant embryos had similar numbers ofneurons in the lmc (Fig. 2). However, at later ages (E8–E10lumbar, E8–E15 brachial), there was considerably lessmotoneuron death in the mutant embryos. By E12 for thelumbar region and by E18 for the brachial region,motoneu-ron numbers in mutant embryos were again comparable tocontrol values. Despite the differences in early motoneu-ron survival, motoneuron size (nuclear diameter, lumbar)was comparable at both E10 and E15 in control andmutant embryos (Fig. 3). There also were no differences inthe number of sensory neurons in the L3 DRG betweencontrol and mutant embryos on either E10 (9,875 6 1,017control vs. 10,192 6 977 mutant, n 5 5) or E16(8,687 6 881 control vs. 9,270 6 904 mutant, n 5 5). Al-though this was not examined systematically, there wereno obvious differences in spinal cord histology, organiza-tion or size between control and mutant embryos (Fig. 4).
Fig. 5. Wet weight in milligrams (mean 6 SD) of hindlimbmusclesof control (solid circles) and mutant (open circles) embryos from E8 toE16. Sample sizes for each data point are 10. *P , 0.01; **P , 0.0001(t-tests).
358 R.W. OPPENHEIM ET AL.

Mouse embryos with a targeted deletion of the RyR-1gene (2/2) exhibit a significant increase in the number ofboth thoracic (5715 6 650 n 5 5 vs. 3073 6 421 n 5 5, P ,
0.0025 t-test) and lumbar (34986 606 n5 5 vs. 20106 212n 5 5, P , 0.0025 t-test) motoneurons on E18.5 comparedto wild type (1/1) controls. This represents a reduction in
Fig. 6. Transverse sections of the leg of control (A,C,E) and mutant (B,D,F) embryos on E8 (A,B), E10(C,D), and E15–E16 (E,F). t, Tibia; f, fibula. Scale bars 5 1 mm inA, B, 1.5 mm in C, D, 2.2 mm in E, F.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 359

the amount of normal MN death that occurs between E13and E18 (Oppenheim et al., 1986) of approximately 60% forthe lumbar and 64% for the thoracic spinal cord. Thenumbers of motoneurons in heterozygotes (1/2) was simi-lar to wild-type control (1/1) values (data not shown).
Muscle development
Based on gross morphology, mutant embryos couldusually be reliably distinguished from controls by E9–E10owing to their overall small size and by apparent differ-ences in muscle size. Prior to this, mutant embryos weregrossly indistinguishable from controls (except for theabsence of motility), andmuscle weights were normal (Fig.5). However, by E10, leg muscle weights were significantlyreduced (25%), and, by E16–E17, there was a .80%reduction in muscle weight. It is only after E10 thatmutant embryos exhibit the fusion of joints (ankylosis),including fused cervical vertebra that give rise to theabnormally short, crooked necks that are the basis fortheir name. Because the differentiation of muscles in thecrooked neck dwarf have been described in detail at thelightmicroscopic level in several previous studies (Asmund-son, 1945; Pun, 1954; Harris, 1962; Elson, 1972; Wick andAllenspach, 1978; Kieny et al., 1983; Mauger et al., 1983);we have not attempted systematically to repeat thoseobservations. However, we have confirmed these previousreports in observing that, prior to E8, muscle developmentappears similar in control and mutant embryos (Fig. 6).After this time, previously distinct muscle groups disap-pear and become fused, connective tissue (epimysia, peri-mysia, etc.) and tendons are absent or abnormal, there areincreased numbers of mononucleated muscle cells, myo-tubes are reduced and wavy in appearance, and at laterages (after E15) there are increasing numbers of dyingcells in remaining muscle. Despite these morphologicalchanges, creatine kinase-specific activity, a biochemicalindex of muscle differentiation, was not reduced in E15mutant muscle (Fig. 7).
Electron microscopy
In general, we have confirmed previous descriptions ofthe ultrastructural development of mutant skeletal muscle(Elson, 1972; Kieny et al., 1988; Airey et al., 1993a,b).These reports (and our own observations) indicate that, asdevelopment proceeds, increasing numbers of mutantmyo-tubes exhibit the following characteristics: disarray ofmyofilaments and myofibrils, dilated sacroplasmic reticu-lum, blebbing of the nuclear membrane, nuclear inclu-sions, mitochondrial swelling, retraction clots, and muscledegeneration. In addition, we have also observed that,already by E8, mutant muscle exhibits disorganization ofsarcomeres owing to the absence or abnormal accumula-tion of the Z-line (Fig. 8) and that, by E14–E15 (Figs. 9, 10),contractile elements are aberrantly scattered in manydirections in virtually all surviving myotubes. However,despite the obvious disorganization of myotubes, the basallamina appears normal at all ages examined (Figs. 8, 10).Although triadic junctions also form and appear normal inmutant muscle (Figs. 8–10), they generally develop 1–2days later than in control muscle, and at high magnifica-tion the junctional feet in mutant muscle are absent orundifferentiated. Despite the apparent absence of junc-tional feet, some dense material can still be observedbetween the membranes of the T-tubule and the sacroplas-mic reticulum at all ages (Figs. 8F, 9F, 10F).
Muscle innervation
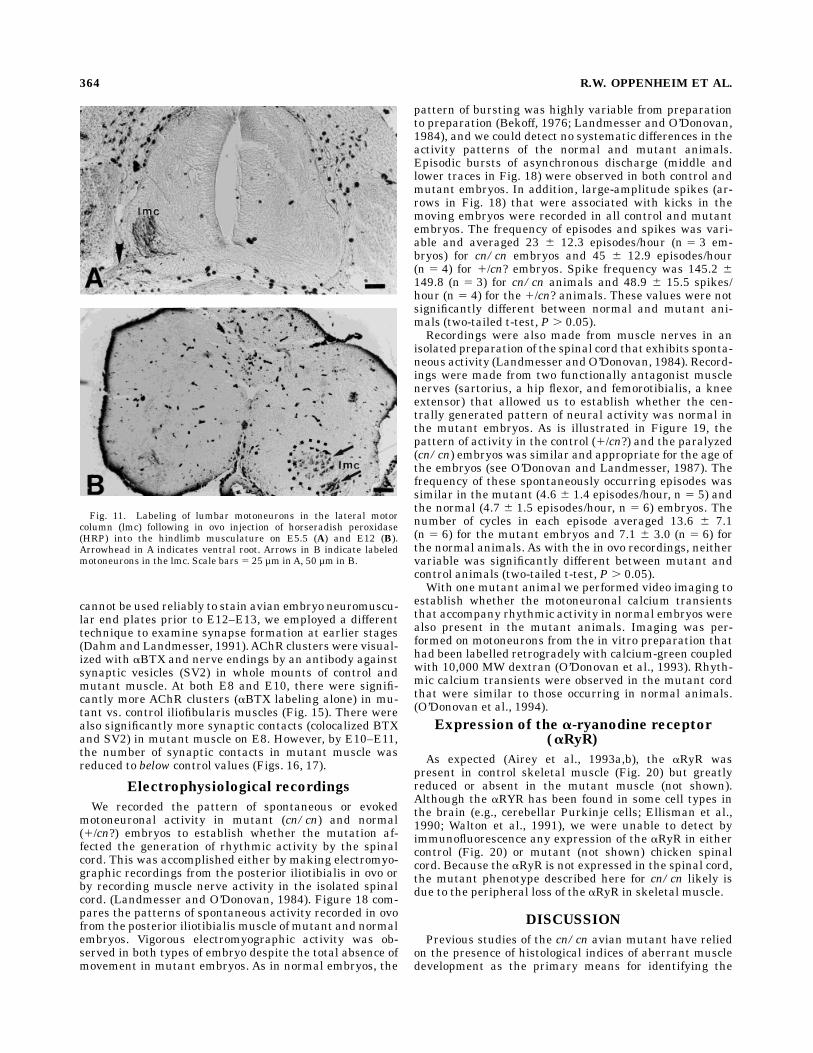
Following the in ovo injection of HRP into control andmutant hindlimb muscles on E6 and E12, embryos in bothgroups exhibit extensive retrograde labelling of specificgroups of motoneurons in the ipsilateral lumbar lmc (Fig.11). Following silver staining of histological sections ofhindlimb muscles on E10, E12, and E15, mutant muscle atall ages exhibitedmore tortuous and extensive innervationpatterns than controls (Fig. 12). Whereas nerve fibers incontrol muscles were generally well fasciculated and exhib-ited stereotyped branching patterns, nerve fibers in mu-tant muscle were less well fasciculated and showed morerandom branching patterns. Because it was not possible toquantify reliably the extent of axonal branching in silver-stained preparations, we have utilized a different methodfor assessing axonal branching, one in which whole-mounted muscles are stained with a cytoskeletal antibody(C2) for immunocytochemical visualization of neurofila-ments within nerve fibers and axonal branches (Dahm andLandmesser, 1991). Using this technique, not only were weable to confirm our observations obtained with the silverstain (Fig. 13), but it was also possible to quantify theextent of intramuscular axonal branching by counting thenumber of side branches per unit length of axon. Thesedata indicate that there are substantially more sidebranches in mutant vs control iliofibularis muscles on E9(Fig. 14). Similar differences were observed between con-trol andmutant sartorius and posterior iliotibialis muscles(data not shown).Examination of E14–E15 muscle doubly stained for
silver and AChE (Toop, 1976) demonstrated that, even atthis relatively advanced stage of the progressive musculardysgenesis in mutant embryos, innervated neuromuscularend plates were still present (Fig. 12). However, eventhough end plates were present in E15 mutant muscle,they appeared greatly reduced in number and in theintensity of AChE staining. Because the Toop technique
Fig. 7. Creatine kinase (C.K.) specific activity (mean 6 SD) ofentire leg musculature on E15 in control (CONT) andmutant (CN/CN)embryos. Numbers in histogram 5 sample size.
360 R.W. OPPENHEIM ET AL.

Fig. 8. Electron micrographs of gastrocnemius muscles from E8control (A–C) and mutant (D–F) embryos. A: The myofibrils begin toorganize, with well-defined Z-lines (arrows) in the periphery of themyotube. The nucleus is still in a central position. 317,750. B: At themyotube periphery, the basal lamina is accumulating but is notcontinuous. The arrow indicates the basal lamina. 362,500. C:A triadcomposed of the opposing membranes of one T-tubule (star) betweentwo sarcoplasmic reticulum cisternae. Between the two membranes,
the junctional feet are differentiated (arrows). 3142,500. D: In thecn/cn mutant myofibrils, the sarcomeres are not very well defined,and they are not joined together by a definite I-band; the Z-line (arrow)is rarely found. 317,750. E: Despite the sarcomeric disorganization,the basal lamina is present as in a normal myotube (arrow). 362,500.F: The sarcoplasmic reticulum is readily recognizable but is notassociated with a T-tubule. 3142,500. Scale bars 5 1 µm in A, D, 0.1µm in B, C, E, F.

Fig. 9. Electron micrographs of mutant gastrocnemius musclefrom E10 (A), E13 (B,C), and E14 (D,E,F) embryos. A,B: At E10 andE13, the muscle is totally disorganized, with longitudinal filaments indisarray. 317,750. C:At E13, some sarcomeres are found in very raremyotubes with aberrant Z-lines (arrows) between two sarcomeres;I-band material seems to be absent or poorly defined. 317,750. D: Arare myotube in E14 mutant muscle with sarcomeres in register and
normal Z-lines (arrow). 362,500. E: The more common appearance ofE14 mutant muscle, with disorganized internal morphology but intactbasal lamina (arrow). 362,500. F: At E14, a triad recognizable by theelectron-lucid central T-tubule (star) opposed to two electron-denseterminal cisternae but with no visible junctional feet. 3142,500. Scalebars 5 1 µm inA–C, 0.1 µm in D–F.
362 R.W. OPPENHEIM ET AL.

Fig. 10. Electronmicrographs of control (A,E) andmutant (B,C,D,F)gastrocnemius muscle on E15. A: General aspect of a myofiber in anormal embryo. The muscle is quite mature with sarcomeres inregister and long myofibrils; the nucleus is in a peripheral position.The arrows indicate Z-lines. 317,750. B: In the mutant muscle, someindividual normal sarcomeres are still present, with normal Z-lines(see curved arrows in B and C), but most of the myotube is disorga-nized, with contractile filaments in complete disarray, scatteredtransversely (open arrows in B) or longitudinally. 317,750. C: In the
cn/cnmuscle, the basal lamina (straight arrow) is present despite themuscle disorganization. 362,500. D: A representative myotube show-ing contraction clots with thick and thin filaments. 362,500. E: Atriadic junction in a normal myotube. The junctional feet (arrows) areclearly differentiated between the T-tubule (star) and the sarcoplasmicreticulum membranes. 3142,500. F: In the mutant muscle, the triadappears normal, but junctional feet are absent. 3142,500. Scalebars 5 1 µm inA, B, 0.1 µm in C–F.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 363
cannot be used reliably to stain avian embryo neuromuscu-lar end plates prior to E12–E13, we employed a differenttechnique to examine synapse formation at earlier stages(Dahm and Landmesser, 1991).AChR clusters were visual-ized with aBTX and nerve endings by an antibody againstsynaptic vesicles (SV2) in whole mounts of control andmutant muscle. At both E8 and E10, there were signifi-cantly more AChR clusters (aBTX labeling alone) in mu-tant vs. control iliofibularis muscles (Fig. 15). There werealso significantly more synaptic contacts (colocalized BTXand SV2) in mutant muscle on E8. However, by E10–E11,the number of synaptic contacts in mutant muscle wasreduced to below control values (Figs. 16, 17).
Electrophysiological recordings
We recorded the pattern of spontaneous or evokedmotoneuronal activity in mutant (cn/cn) and normal(1/cn?) embryos to establish whether the mutation af-fected the generation of rhythmic activity by the spinalcord. This was accomplished either by making electromyo-graphic recordings from the posterior iliotibialis in ovo orby recording muscle nerve activity in the isolated spinalcord. (Landmesser and O’Donovan, 1984). Figure 18 com-pares the patterns of spontaneous activity recorded in ovofrom the posterior iliotibialis muscle of mutant and normalembryos. Vigorous electromyographic activity was ob-served in both types of embryo despite the total absence ofmovement in mutant embryos. As in normal embryos, the
pattern of bursting was highly variable from preparationto preparation (Bekoff, 1976; Landmesser and O’Donovan,1984), and we could detect no systematic differences in theactivity patterns of the normal and mutant animals.Episodic bursts of asynchronous discharge (middle andlower traces in Fig. 18) were observed in both control andmutant embryos. In addition, large-amplitude spikes (ar-rows in Fig. 18) that were associated with kicks in themoving embryos were recorded in all control and mutantembryos. The frequency of episodes and spikes was vari-able and averaged 23 6 12.3 episodes/hour (n 5 3 em-bryos) for cn/cn embryos and 45 6 12.9 episodes/hour(n 5 4) for 1/cn? embryos. Spike frequency was 145.2 6149.8 (n 5 3) for cn/cn animals and 48.9 6 15.5 spikes/hour (n 5 4) for the 1/cn? animals. These values were notsignificantly different between normal and mutant ani-mals (two-tailed t-test, P . 0.05).Recordings were also made from muscle nerves in an
isolated preparation of the spinal cord that exhibits sponta-neous activity (Landmesser andO’Donovan, 1984). Record-ings were made from two functionally antagonist musclenerves (sartorius, a hip flexor, and femorotibialis, a kneeextensor) that allowed us to establish whether the cen-trally generated pattern of neural activity was normal inthe mutant embryos. As is illustrated in Figure 19, thepattern of activity in the control (1/cn?) and the paralyzed(cn/cn) embryos was similar and appropriate for the age ofthe embryos (see O’Donovan and Landmesser, 1987). Thefrequency of these spontaneously occurring episodes wassimilar in the mutant (4.6 6 1.4 episodes/hour, n 5 5) andthe normal (4.7 6 1.5 episodes/hour, n 5 6) embryos. Thenumber of cycles in each episode averaged 13.6 6 7.1(n 5 6) for the mutant embryos and 7.1 6 3.0 (n 5 6) forthe normal animals. As with the in ovo recordings, neithervariable was significantly different between mutant andcontrol animals (two-tailed t-test, P . 0.05).With one mutant animal we performed video imaging to
establish whether the motoneuronal calcium transientsthat accompany rhythmic activity in normal embryos werealso present in the mutant animals. Imaging was per-formed on motoneurons from the in vitro preparation thathad been labelled retrogradely with calcium-green coupledwith 10,000 MW dextran (O’Donovan et al., 1993). Rhyth-mic calcium transients were observed in the mutant cordthat were similar to those occurring in normal animals.(O’Donovan et al., 1994).
Expression of the a-ryanodine receptor(aRyR)
As expected (Airey et al., 1993a,b), the aRyR waspresent in control skeletal muscle (Fig. 20) but greatlyreduced or absent in the mutant muscle (not shown).Although the aRYR has been found in some cell types inthe brain (e.g., cerebellar Purkinje cells; Ellisman et al.,1990; Walton et al., 1991), we were unable to detect byimmunofluorescence any expression of the aRyR in eithercontrol (Fig. 20) or mutant (not shown) chicken spinalcord. Because the aRyR is not expressed in the spinal cord,the mutant phenotype described here for cn/cn likely isdue to the peripheral loss of the aRyR in skeletal muscle.
DISCUSSION
Previous studies of the cn/cn avian mutant have reliedon the presence of histological indices of aberrant muscledevelopment as the primary means for identifying the
Fig. 11. Labeling of lumbar motoneurons in the lateral motorcolumn (lmc) following in ovo injection of horseradish peroxidase(HRP) into the hindlimb musculature on E5.5 (A) and E12 (B).Arrowhead in A indicates ventral root. Arrows in B indicate labeledmotoneurons in the lmc. Scale bars 5 25 µm inA, 50 µm in B.
364 R.W. OPPENHEIM ET AL.

mutant phenotype. With these criteria, mutant embryoshave first been identified on E8 (e.g., see Kieny et al., 1983,1988). Although some prior studies have also noted theabsence of neuromuscular activity, our observations ap-pear to be the first to demonstrate that cn/cn embryosnever exhibit neuromuscular activity. Beginning at E3.5,when normal embryos first exhibit such activity, andthroughout the remainder of the incubation period, mu-tant embryos are completely paralyzed. In this regard,they are similar to the muscular dysgenic mouse (mdg/mdg), which also is paralyzed throughout fetal develop-ment (Pai, 1965). The electrophysiological data presentedhere argue strongly for a selective loss of excitation-contraction coupling as the basis for the lack of neuromus-cular movements in mutant embryos. Direct recordings ofspinal cord activity as well as EMG recordings failed toreveal any significant differences between control andmutant embryos. Unpublished observations by Lynn Land-messer (personal communication) also indicate that, onE10, 1) spinal cord stimulation evokes action potentials incn/cn limb muscle nerves in a manner indistinguishablefrom controls; 2) spontaneous miniature end-plate poten-tials (mepps) can be recorded from mutant muscle, and,following spinal cord stimulation, compound postsynapticpotentials (PSPs) are observed, and these can be blockedby curare; 3) direct stimulation of mutant muscle fibers atone end of the sartorius muscle evokes action potentialsthat can be recorded from the opposite end of the muscle;
and 4) spontaneous bursts of motoneuron activity insartorius and adductor motor pools (on E7) are of normalduration and appear to be indistinguishable from thepatterned activation of these same motor pools in controlembryos. Collectively, these observations indicate that,similar to the situation with the muscular dysgenic mouse(Pincon-Raymond et al., 1985; Franzini-Armstrong et al.,1991; Flucher et al., 1992), the primary defect in cn/cnembryos involves a failure of normal muscle contraction(excitation-contraction coupling) and thus that the ab-sence of motility is not due to a failure of spinal cordactivity or synaptic transmission in the CNS. That is, theprimary defect appears to be myogenic rather than neuro-genic in origin (see below). The observation that theprimary genetic defect in cn/cn involves the aRyR and thefact that the aRyR is not normally expressed in the spinalcord are also consistent with the cn/cn phenotype beingprimarily myogenic in origin.The ultrastructural observations reported here are also
consistent with a primary myogenic defect in excitation-contraction coupling. In normal muscle, the intracellulartriadic junction plays a critical role in the coupling be-tween the electrical signal from themuscle surface and therelease of Ca21 from the sacroplasmic reticulum, whichleads to contraction. In the mdg/mdg mouse mutant,triadic junctions aremarkedly decreased in skeletalmuscle,junctional tetrads are absent, and these structural abnor-malities are associated with the absence (or decreased
Fig. 12. Longitudinal sections through the gastrocnemius muscle of control (A,C) and mutant (B,D)embryos on E15–E16. Large arrowheads indicate nerve trunks and branches; small arrowheads indicateacetylcholinesterase (AChE) staining of end plates. Scale bars 5 30 µm inA, B, 15 µm in C, D.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 365

numbers) of dihydropyridine receptors that act as voltage-sensitive Ca21 channels (Pincon-Raymond et al., 1985;Flucher et al., 1992). By contrast, whereas triads arepresent in apparently normal numbers in skeletal musclefrom cn/cn embryos, junctional end feet are absent oraberrant. Additionally, recent studies have shown that,whereas there are essentially normal levels of dihydropyri-dine receptor protein and dihydropyridine binding as wellas normal sacroplasmic reticulum Ca21-ATPase and calse-questrin protein in cn/cn mutant skeletal muscle, theaRyR associated with the sacroplasmic reticulum calciumrelease channel is absent (Airey et al., 1993a,b). Accord-ingly, the absence of normal end feet in the mutant isassociated with the loss of aRyR and with the inability ofmutant muscle to contract despite apparently normal CNSactivity and neuromuscular transmission. Although addi-tional studies are needed, it seems likely that virtually allof the phenotypic changes seen in cn/cn embryos can beaccounted for by the chronic absence of muscle contraction(see also Airey et al., 1993a,b). Many of these samephenotypic changes in neuromuscular development alsooccur in themdg/mdgmouse and in chronically paralyzedbut genetically normal avian and rat embryos (see below),in which the only common factor among all three models isthe loss of muscle contraction.
Fig. 13. Intramuscular nerve branching demonstrated with the neurofilament antibody C2 in wholemounts of control (A,C) and mutant (B,D) sartorius (A,B) and iliotibialis (C,D) muscles on E9–E10.Arrows indicate side branches. Scale bar 5 100 µm.
Fig. 14. Mean (6SD) number of side branches on intramuscularnerves in iliofibularis muscle of control (CONT) and mutant (CN/CN)embryos on E8 and E11. Numbers in histograms are sample sizes.*P , 0.03, **P , 0.0001, t-tests.
366 R.W. OPPENHEIM ET AL.

Although there are substantial morphological alter-ations in mutant muscle that become apparent afterE8–E9, many biochemical and molecular properties ap-pear normal at least up to E15. In addition to the lack of achange in creatine kinase activity as observed here, mu-tant muscle expresses normal levels of b-ryanodine recep-tor, the a1-subunit of the dihydropyridine receptor, thesarcoplasmic reticulum Ca21-ATPase, and calsequestrin(Airey et al., 1993a,b). In addition, calcium transientselicited by electrical stimulation, acetylcholine, and caf-feine appear normal, and comparable L- and T-type cal-cium currents are present in control and mutant muscle.(Airey et al., 1993a,b). Accordingly, mutant muscle doesnot exhibit general, nonspecific alterations in muscledifferentiation. Rather, only specific morphological andmolecular changes are observed that are related to theabsence of the aRyR and the chronic absence of musclecontraction during development.The absence of muscle contraction in the mdg/mdg
mousemutant and in genetically normal but pharmacologi-cally and chronically paralyzed avian embryos is associ-ated with abnormal muscle differentiation (including late-onset muscle degeneration), increased survival of spinalmotoneurons, increased intramuscular branching of motoraxons, and increased numbers of neuromuscular synapses
Fig. 15. The numbers (mean 6 SD) of acetylcholine receptor (AChR)clusters (a-bungarotoxin; aBTX immunofluorescent profiles) in con-trol (CONT; 1/cn?) and mutant (CN/CN) iliofibularis muscle on E8and E10. Numbers in histogram are sample sizes (number of musclesexamined). **P , 0.001, *P , 0.0005, t-test.
Fig. 16. Immunofluorescence of SV2 (A–C) and aBTX (D–F) label-ing of control (A,D) and mutant (B,C,E,F) iliofibularis muscle on E10.In mutant tissue preparation, SV2 immunoreactivity was found alongnerve trunks (C) in addition to nerve terminals in control tissue
(arrows in A). Scale bar 5 2 µm. The arrows in B and C indicate SV2labeling in nerve trunks (lower arrow in B) and nerve terminals (upperarrow in B, and both arrows in C). The arrows in D–F indicatepostsynaptic (aBTX-labeled) structures.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 367

(Pittman and Oppenheim, 1979; Rieger and Pincon-Raymond, 1981; Oppenheim and Chu-Wang, 1983; Powellet al., 1984; Rieger et al., 1984; Oppenheim et al., 1986,1989; Dahm and Landmesser, 1988, 1991; Tang and Land-messer, 1993). Similar changes have also been observed ingenetically normal but pharmacologically paralyzedmouseand rat embryos (see, e.g. Harris and McCaig, 1984;Houenou et al., 1990). As reported here, we have observeda similar array of alterations in the neuromuscular devel-opment of cn/cn embryos which lack excitation-contrac-tion coupling. The failure to observe increased numbers ofsynapses on E10 despite increased intramuscular axonalbranching and increased clusters of postsynaptic AchRs,however, is difficult to explain in this context. One possibil-ity is that the critical postsynaptic molecules that main-tain the presence of the presynaptic terminal becomealtered in the mutant muscle on E10, resulting in with-drawal of the terminal or the loss of differentiated proper-ties such as SV2 expression.A major rationale for assessing neuromuscular develop-
ment in cn/cn embryos was to examine the role of neuro-muscular activity in regulating motoneuron survival. Be-cause the loss of motoneurons by naturally occurring celldeath is markedly reduced in all of the situations in whichneuromuscular activity is absent or reduced (i.e.,mdg andcnmutants and in paralyzed but genetically normal avianand mammalian embryos), it seems reasonable to arguethat the normal CNS-generated muscle activity that oc-curs in all vertebrate embryos plays a critical role inregulating motoneuron numbers. In this regard, it is ofinterest that a targeted deletion of the skeletal muscle-specific RyR-1 gene in themouse which causes paralysis byan absence of excitation-contraction coupling (Nakai et al.,1996) also results in a dramatic reduction in the normaldeath of spinal motoneurons that occurs between E13 andE18.
Fig. 17. Mean percentage (6SEM) of AChR clusters (a-BTX-labeled profiles) that are colocalized with nerve terminals (SV2-labeled profiles) in control (CONT) and mutant (CN/CN) iliofibularismuscle on E8 and E10. Numbers in bars represent the total number ofAChR clusters examined from three different muscles in each group.*P , 0.05, **P , 0.02, t-test.
Fig. 18. In ovo suction electrode recordings from posterior iliotibi-alis muscle in cn/cn and 1/cn? embryos. In both the cn/cn and the1/cn? embryos, the activity consists of a variable combination oflow-amplitude, episodic bursts (asterisks) and large, synchronizedspikes (arrows). The records were filtered from 10 Hz to 2 kHz.
Fig. 19. Activity patterns recorded from muscle nerves of isolatedspinal cords from cn/cn and 1/cn? embryos. Upper traces are neuro-grams from the sartorius (SART) and femorotibialis external (FEM)muscle nerves in an embryo exhibiting spontaneous movements in ovo(1/cn?, E8). Lower traces are recordings from the same muscle nervesin a paralyzed mutant embryo (cn/cn, E9). These activity patterns arecharacteristic of normal embryos of the same age. Neurograms werefiltered from 100 Hz to 10 kHz.
368 R.W. OPPENHEIM ET AL.

Because previous studies (e.g., Pittman and Oppenheim,1978, 1979) have reported that the rescue of motoneuronsby activity blockade persists as long as the embryosremain paralyzed, it was somewhat surprising that in boththe brachial and lumbar regions motoneurons in themutant embryos eventually decreased to control values orbelow control values despite the absence of muscle activity.Although it could be argued that this indicates that celldeath was only delayed or developmentally retarded, wefavor an alternative explanation. At the time when moto-
neurons begin to decrease in number in the mutantembryos, muscle atrophy is much greater than is observedin most curare-treated embryos. However, very high dosesof curare also result in massive muscle atrophy that isaccompanied by a loss of motoneurons (Pittman andOppenheim, 1979). Accordingly, we favor the idea that,once muscle mass decreases below some critical threshold,motoneuron survival is impaired perhaps owing to a loss ofsufficient muscle-derived trophic factor.One way in which neuromuscular activity may be in-
volved in neuronal survival is by regulating the productionor release of muscle-derived neurotrophic agents that arenecessary for survival. According to this view, normallevels of activity would result in the production of levels oftrophic factor sufficient to maintain only one-half of allpostmitotic motoneurons (the number that normally sur-vive the cell death period), whereas reduced activity wouldresult in an up-regulation of trophic factor and increasedmotoneuron survival. Although it is not possible to excludethis possibility entirely (see, e.g., Houenou et al., 1991),the available evidence is not consistent with this idea.Partially purified skeletal muscle extracts derived fromeither genetically paralyzed avian (cn/cn) or mouse (mdg/mdg) embryos or from pharmacologically paralyzed nor-mal avian embryos are just as effective as extracts derivedfrom normally active avian or mouse embryonic muscle inpromoting motoneuron survival in vitro and in vivo(Tanaka, 1987; Houenou et al., 1991). That is, based onthese admittedly rather crude assays, active and inactivemuscle appears to contain similar amounts of a putativetrophic agent that rescues motoneurons from normal celldeath.Therefore, if one assumes that inactive muscle is not
producing more trophic factor, what might be an alterna-tive explanation for the increased survival of motoneuronsfollowing the loss of neuromuscular activity? One possibil-ity, for which there is now considerable support, is thatneuromuscular activity regulates a muscle-derived signalthat modulates axonal branching and synapse formationby developing motoneurons (Oppenheim, 1989; Land-messer, 1992). According to this view, axonal terminalsand nerve-muscle contacts are thought to be uptake sitesfor muscle-derived trophic (survival) factors. Therefore,perturbations of axonal branching and synaptogenesiswould be expected to alter motoneuron survival by modify-ing access to muscle-derived trophic factors. Several linesof evidence are consistent with the access hypothesis: 1)Developing avian motoneurons have been shown to de-pend on muscle-derived trophic factors for their survivalboth in vitro and in vivo (Dohrmann et al., 1986; Oppen-heim et al., 1988, 1993; Bloch-Gallego et al., 1991). 2) Thisdependence on target-derived trophic support begins atthe same time that muscle innervation and synapticcontacts are being established (Dahm and Landmesser,1988, 1991; Oppenheim, et al., 1989a,b). 3) As was notedabove, neuromuscular paralysis achieved by a variety ofmeans during the period of naturally occurring motoneu-ron death rescues motoneurons and results in increasedaxonal branching and synaptogenesis. 4) There are alsocertain perturbations of motoneuron activity that fail torescue motoneurons from cell death, and importantlythese also fail to alter axonal branching and synaptogen-esis (Oppenheim et al., 1989a,b; Houenou et al., 1992). 5)Reductions in axonal branching and synaptogenesis by
Fig. 20. Immunofluorescence of control E18 gastrocnemius muscle(A) and lumbar spinal cord (B) by using a monoclonal antibody specificfor the a-ryanodine receptor (aRyR). Immunofluorescence was alsocompletely absent frommutant spinal cord (not shown). Scale bar530µm.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 369

means that do not alter neuromuscular activity nonethe-less result in increased motoneuron death (Tang andLandmesser, 1993). These last two findings are especiallycompelling in that they strongly support the notion thatactivity regulates motoneuron survival only to the extentthat it also affects axonal branching and synaptogenesis.The present results with the cn mutant are entirelyconsistent with this conclusion. The observation that thereis increased axonal branching and motoneuron survival inmutant embryos on E8 and E10, whereas synaptic con-tacts are increased only on E8, suggests that the increasein axonal branches alone may be sufficient to promotemotoneuron survival at least transiently in this situation(see Dahm and Landmesser, 1988, 1991). Taken collec-tively, then, these various lines of evidence, when consid-ered together with the negative results on activity-dependent regulation of a muscle-derived trophic agent(Tanaka, 1987; Houenou et al., 1991), provide considerablesupport for the access hypothesis (Oppenheim, 1989).The present results with the cn/cn chick embryo, as well
as the related results from mdg/mdg mice and 2/2aRyR-deleted mice, also indicate that a failure of musclecontraction alone is sufficient to rescue motoneurons fromcell death. As was noted above, CNS activity and synaptictransmission in both the mdg/mdg mouse and the cn/cnchicken mutant appear normal, whereas muscle contrac-tion is absent. Therefore, it seems likely that, in thosecases where pharmacological agents and neurotoxins thatblock neuromuscular transmission were also found to bindto neuronal nicotinic AchRs (nAchRs; Renshaw et al.,1993; Renshaw and Goldie, 1996) and to perturb spinalcord (CNS) activity (Landmesser and Szente, 1987), it isactually the absence of peripheral muscle contraction andnot the reduction in synaptic or CNS activity in spinal cordthat is critical in rescuing motoneurons. The failure ofchronic electrical stimulation of the spinal cord in ovo toalter motoneuron survival is also consistent with this idea(Fournier LeRay et al., 1993).Against this evidence are theresults of a recent study using chronic activity blockade inthe chick embryo, which suggest that classic neuromuscu-lar-blocking agents such as curare and aBTX may, in fact,rescue motoneurons by acting centrally to block neuronalnAchRs rather than by the peripheral blockade of muscleAchRs (Hory-Lee and Frank, 1995). Hory-Lee and Frankreport that nonparalytic doses of curare and aBTX that arewithout any apparent affect on embryonic movementsnonetheless rescue motoneurons, and they suggest thatthe mechanism may involve the blockade of neuronalnAchRs that normally act to modulate calcium entry intomotoneurons rather than by perturbing synaptic transmis-sion (see, e.g., Vijayaraghavan et al., 1992; Rathouz andBerg, 1994; Rathouz et al., 1995; Role and Berg, 1996).Although we have so far been unable to confirm thatnonparalytic doses of curare rescue motoneurons (Oppen-heim et al., 1996), we cannot exclude the possibility thatneuronal nAchRs play some role in the effects of curare oraBTX on motoneuron survival. However, in the presentstudy, we have shown that increased motoneuron survivalin the cn/cn mutant is mediated by perturbation ofperipheral muscle activity without affecting either CNSsynaptic activity or calcium transients in motoneurons.The lack of expression of the aRyR in either normal ormutant spinal cord also argues strongly against a CNS-mediated mechanism in the mutant cn/cn phenotype.However, we cannot exclude the possibility that the rescue
of motoneurons in the cn/cn mutant vs. that followingactivity blockade by curare is mediated by a distinct mech-anism (e.g., peripheral vs. central). Studies are in progressto examine the possible role of neuronal nAchRs in theevents mediating motoneuron survival in normal andcurare-treated chick embryos.Important studies by Lynn Landmesser and her associ-
ates indicate that a significant event in the cascade ofsignals linking muscle activity with motoneuron survivalis the activity-dependent modulation of cell surface adhe-sion molecules that act to regulate axonal branching(Landmesser et al., 1990). Based on the informationpresently available, one can formulate a working model ofactivity-dependent neuromuscular development in thechick embryo, in which innervation, synaptic transmis-sion, and muscle activity, beginning between E5 and E6,regulate the production or release of a muscle-derivedsprouting or branching signal that then modulates theexpression of adhesion molecules on the surface of growingmotoneuron axons and ultimately affects axonal growthvia the regulation of growth-associated cytoskeletal pro-teins. Branching and neuromuscular contacts would beincreased following reductions in muscle activity andwould provide motoneurons with increased access tomuscle-derived trophic agents that promote their survival.Although neither the putative muscle-derived sproutingsignal nor the muscle-derived motoneuron survival agenthave been identified, promising candidates exist for both(Ishii, 1989; Caroni and Gandes, 1990; Oppenheim et al.,1992, 1993; Gurney et al., 1992; Caroni and Becker, 1992;Neff et al., 1993; Henderson et al., 1993; Caroni et al.,1994; Funakoshi et al., 1995; Oppenheim, 1996). Addition-ally, activity-dependent adhesion molecules that modulateaxonal branching in vivo have been identified (Land-messer et al., 1990; Landmesser, 1992; Tang and Land-messer, 1993). Therefore, it seems likely that in the nearfuture the cascade of cellular and molecular events thatpromotemotoneuron survival following activity blockade—and which, by inference, regulate these events duringnormal development—will, after more than 20 years ofinvestigation, finally be identified.
ACKNOWLEDGMENTS
This work was supported by NIH grants NS20402(R.W.O.) and AR43140 (P.D.A.), and grants from the Mus-cular DystrophyAssociation (L.J.H. and P.D.A.) and the B.Phillipe Foundation (L.J.H.).The authors are grateful to Lynn Landmesser for shar-
ing her unpublished data from electrophysiological studiesof cn/cn embryos and to Kathy Paull for technical assis-tance.
LITERATURE CITED
Airey, J.A., M.D. Baring, C.F. Beck, Y. Chelliah, T.J. Deerinck, M.H.Ellisman, L.J. Houneou, D.D. McKenny, J.L. Sutko, and J. Talvenheimo(1993a) Failure to make normal a-ryanodine receptor is an early eventassociated with the crooked neck dwarf (cn) mutation in chicken. Dev.Dynam. 197:169–188.
Airey, J.A., T.J. Deerinck, M.H. Ellisman, L.J. Houenou, A. Ivanenko, J.L.Kenyon, D.D. McKenny, and J.L. Sutko (1993b) Crooked neck dwarf(cn) mutant chicken skeletal muscle cells in low density primarycultures fail to express normal a-ryanodine receptor and exhibit apartial mutant phenotype. Dev. Dynam. 197:189–202.
Asmundson, V.S. (1945) Crooked neck dwarf in domestic fowl. J. Hered.36:173–176.
370 R.W. OPPENHEIM ET AL.

Bekoff,A. (1976) Ontogeny of legmotor output in the chick embryo:Aneuralanalysis. Brain Res. 106:271–291.
Bloch-Gallego, E., M. Huchet, H. ElM’Hamdi, F.R. Xie, H. Tanaka, and C.E.Henderson (1991) Survival in vitro of motoneurons identified or puri-fied by novel antibody based methods is selectively enhanced bymuscle-derived factors. Development 111:221–232.
Buckley, K., and R. Kelly (1985) Identification of a transmembraneglycoprotein specific for secretory vesicles of neuronal and endocrinecells. J. Cell Biol. 100:1284–1294.
Caroni, P., and M. Becker (1992) The down regulation of growth associatedproteins in motoneurons at the onset of synapse elimination is con-trolled by muscle activity and IGF-1. J. Neurosci. 12:3849–3861.
Caroni, P., and P. Grandes (1990) Nerve sprouting in innervated adultskeletal muscle induced by exposure to elevated levels of insulin-likegrowth factors. J. Cell Biol. 110:1307–1317.
Caroni, P., C. Schneider, M.C. Kiefer, and J. Zapf (1994) Role of muscleinsulin-like growth factors in nerve sprouting: suppression of terminalsprouting in paralyzed muscle by IGF-binding protein 4. J. Cell Biol.125:893–902.
Clarke, P.G.H., and R.W. Oppenheim (1995) Neuron death in vertebratedevelopment: In vivo methods. In B. Osborne and L. Schwartz (eds):Methods in Cell Biology, Cell Death, Vol 46. New York: Academic Press,pp. 277–321.
Dahm, L., and L. Landmesser (1988) The regulation of intramuscular nervebranching during normal development and following activity blockade.Dev. Biol. 130:621–644.
Dahm, L., and L. Landmesser (1991) The regulation of synaptogenesisduring normal development and following activity blockade. J. Neuro-sci. 11:238–255.
Dohrmann, U., D. Edgar, M. Sendtner, and H. Thoenen (1986) Muscle-derived factors that support survival and promote fiber outgrowth fromembryonic chick spinalmotor neurons in culture. Dev. Biol. 118:209–221.
Ellisman, M.H., T.J. Deerinck, Y. Ouyang, C.F. Beck, S.J. Tanksley, P.D.Walton, J.A. Airey, and J.L. Sutko (1990) Identification and localizationof ryanodine binding proteins in the avian central nervous system.Neuron 5:135–146.
Elson, S.K. (1972)An ultrastructural study of skin andmuscle developmentin the crooked neck dwarf chick embryo. MS thesis, Miami University,Oxford, Ohio.
Flucher, B.E., J.L. Phillips, J.A. Powell, S.B. Andrews, and M.P. Daniels(1992) Coordinated development of myofibrils, sarcoplasmic reticulumand transverse tubules in normal and dysgenic mouse skeletal muscle,in vivo and in vitro. Dev. Biol. 150:266–280.
Fournier LeRay, C., D. Prevette, R.W. Oppenheim, and J. Fontaine-Perus(1993) Interactions between spinal cord stimulation and activity block-ade in the regulation of synaptogenesis and motoneuron survival in thechick embryo. J. Neurobiol. 24:1142–1156.
Franzini-Armstrong, C., M. Pincon-Raymond, and F. Rieger (1991) Musclefibers from dysgenic mouse in vivo lack a surface component ofperipheral couplings. Dev. Biol. 146:364–376.
Funakoshi, H., N. Belluardo, E. Arenas, Y. Yamamoto, A. Casabona, H.Persson, and C.F. Ibanez (1995) Muscle-derived neurotrophin-4 as anactivity-dependent trophic signal for adult motor neurons. Science268:1495–1499.
Gurney, M.E., H. Yamamoto, and Y. Kwon (1992) Induction of motor neuronsprouting in vivo by ciliary neurotrophic factor and basic fibroblastgrowth factor. J. Neurosci. 12:3241–3247.
Hamburger, V. (1963) Some aspects of the embryology of behavior. Q. Rev.Biol. 38:342–365.
Hamburger, V. (1975) Cell death in the development of the lateral motorcolumn of the chick embryo. J. Comp. Neurol. 160:535–546.
Hamburger, V., and M. Balaban (1963) Observations and experiments onspontaneous rhythmical behavior in the chick embryo. Dev. Biol.7:533–545.
Hamburger, V., and H.L. Hamilton (1951) A series of normal stages in thedevelopment of the chick embryo. J. Morphol. 88:49–92.
Hamburger, V., and R.W. Oppenheim (1982) Naturally occurring neuronaldeath in vertebrates. Neurosci. Comment. 1:38–55.
Harris,A.J., and C.D. McCaig (1984) Motoneuron death andmotor unit sizeduring embryonic development of the rat. J. Neurosci. 4:13–24.
Harris,M.J. (1962)Apreliminary study ofmorphological and neurohistologi-cal effects of the cn lethal mutation in the crooked neck dwarf chickembryo. MS thesis, University of Kansas, Lawrence, Kansas.
Henderson, C.E., W. Camu, C. Mettling, A. Gouin, K. Poulsen, M. Kariha-loo, J. Ruilamas, T. Evans, S.B. McMahon, M.P. Armanini, L. Berke-
meier, H. Philips, and A. Rosenthal (1993) Neurotrophins promotemotor neuron survival and are present in embryonic limb bud. Nature363:266–270.
Hermann, H., E.M. Clark, and W. Landauer (1963) Muscle development inthe crooked neck dwarf mutant and in the acetylpyridine-treated chickembryo. Acta Embryol. Morphol. Exp. 6:169–174.
Hory-Lee, F., and F. Frank (1995) The nicotinic blocking agents d-tubocurare and a-bungarotoxin savemotoneurons from naturally occur-ring death in the absence of neuromuscular blockade. J. Neurosci.15:6453–6460.
Houenou, L.J., M. Pincon-Raymond, L. Garcia, A.J. Harris, and F. Rieger(1990) Neuromuscular development following tetrodotoxin-induced in-activity in mouse embryos. J. Neurobiol. 21:1249–1261.
Houenou, L.J., J.L. McManaman, D. Prevette, and R.W. Oppenheim (1991)Regulation of putative muscle-derived neurotrophic factors by muscleactivity and innervation: In vivo and in vitro studies. J. Neurosci.11:2829–2837.
Houenou, L.J., D. Prevette, and R.W. Oppenheim (1992) Effects of differentneuromuscular blocking agents on naturally occurring motoneurondeath during development. Soc. Neurosci. Abstr. 18:575.
Ishii, D. (1989) Relationship of insulin-like growth factor II gene expressionin muscle to synaptogenesis. Proc. Natl. Acad. Sci. USA 86:2898–2902.
Kieny, M., A. Mauger, I. Hedayat, and P.F. Goetinck (1983) Ontogeny of theleg muscle tissue in the crooked neck dwarf mutant (cn/cn) chickembryo. Arch. Anat. Microsc. Morphol. Exp. 72:5–17.
Kieny, M., A.M. Boutineau, M.P. Pautou and P.F. Goetinck (1988) Musculardysgenesis in fowl: Ultrastructural study of skeletal muscles in thecrooked neck dwarf (cn/cn) mutant. Biol. Struct. Morphol. 1:15–27.
Laing, N., and M. Prestige (1978) Prevention of spontaneous motoneurondeath in chick embryos. J. Physiol. 282:34P.
Landmesser, L. (1992) The relationship of intramuscular nerve branchingand synaptogenesis to motoneuron survival. J. Neurobiol. 23:1131–1139.
Landmesser, L., and D. Morris (1975) The development of functionalinnervation in the hindlimb of the chick embryo. J. Physiol. 249:301–326.
Landmesser, L.T., and M.J. O’Donovan (1984) Activation patterns ofembryonic chick hindlimb muscles recorded in ovo and in an isolatedspinal cord preparation. J. Physiol. 347:189–204.
Landmesser, L., and M. Szente (1987) Activation patterns of embryonicchick hindlimb muscles following blockade of activity and motoneuroncell death. J. Physiol. 380:157–174.
Landmesser, L., L. Dahm, J. Tang, and U. Rutishauser (1990) Polysialicacid as a regulator of intramuscular nerve branching during embryonicdevelopment. Neuron 4:655–667.
Lowry, O.H., N.J. Rosebrough, A.L. Farr, and R.J. Randall (1951) Proteinmeasurementwith the Folin phenolmethod. J. Biol. Chem. 193:265–275.
Mauger, A., M. Kieny, I. Hedayat, and P.F. Goetinck (1983) Tissue interac-tions in the organization and maintenance of the muscle pattern in thechick limb. J. Embryol. Exp. Morphol. 76:199–215.
Nakai, J., R.T. Dirksen, H.T. Nguyen, I.N. Pessah, K.G. Beam, and P.D.Allen (1996) Enhanced dihydropyridine receptor channel activity in thepresence of ryandodine receptor. Nature 380:72–75.
Neff, N.T., D. Prevette, L.J. Houenou, M.E. Lewis, M.A. Glickman, Q.W.Yin, and R.W. Oppenheim (1993) Insulin-like growth factors: Putativemuscle-derived trophic agents that promote motoneuron survival. J.Neurobiol. 24:1578–1588.
O’Donovan,M.J., and L.T. Landmesser (1987) The development of hindlimbmotor activity studies in an isolated preparation of the chick spinalcord. J. Neurosci. 7:3256–3264.
O’Donovan, M.J., S. Ho, G. Sholomenko and W. Yee (1993) Real-timeimaging of neurons retrogradely and anterogradely labelled withcalcium sensitive dyes. J. Neurosci. Methods 46:91–106.
O’Donovan, M.J., S. Ho, and W. Yee (1994) Calcium imaging of rhythmicnetwork activity in the developing spinal cord of the chick embryo. J.Neurosci. 14:6354–6369.
Oppenheim, R.W. (1975) The role of supraspinal input in embryonicmotility: A reexamination in the chick. J. Comp. Neurol. 160:37–50.
Oppenheim, R.W. (1981) Neuronal cell death and some related regressivephenomena during neurogenesis: A selective historical review andprogress report. In W.M. Cowan (ed): Studies in Developmental Neuro-biology: Essays in Honor of Viktor Hamburger. New York: Oxford, pp.74–133.
DEATH OF AVIAN EMBRYONIC MOTONEURONS 371

Oppenheim, R.W. (1987) Muscle activity and motoneuron death in thespinal cord of the chick embryo. In G. Bock and M. O’Connor (eds):Selective Neuronal Death. NewYork: Wiley, pp. 96–112.
Oppenheim, R.W. (1989) The neurotrophic theory and naturally occurringmotoneuron death. Trends Neurosci. 12:252–255.
Oppenheim, R.W. (1991) Cell death during development of the nervoussystem.Annu. Rev. Neurosci. 14:453–501.
Oppenheim, R.W. (1996) Neurotrophic survival molecules for motoneurons:An embarrassment of riches. Neuron 17:195–197.
Oppenheim, R.W., and I.W. Chu-Wang (1983)Aspects of naturally occurringmotoneuron death in the chick spinal cord during embryonic develop-ment. In G. Burnstock and G. Vrbova (eds): Somatic and AutonomicNerve-Muscle Interactions. Amsterdam: Elsevier, pp. 57–107.
Oppenheim, R.W. and M.B. Heaton (1975) The retrograde transport ofhorseradish peroxidase from the developing limb of the chick embryo.Brain Res. 98:291–302.
Oppenheim, R.W., and C. Majors-Willard (1978) Neuronal cell death in thebrachial spinal cord of chick is unrelated to the loss of polyneuronalinnervation in wing muscle. Brain Res. 154:148–152.
Oppenheim, R.W., and R. Nunez (1982) Electrical stimulation of hindlimbincreases neuronal cell death in the chick embryo. Nature 295:57–59.
Oppenheim, R.W., and D. Prevette (1986) Neuromuscular development inthe crooked neck dwarf mutant (cn/cn) chick embryo. Soc. Neurosci.Abstr. 12:983.
Oppenheim, R.W., L.J. Houenou, M. Pincon-Raymond, J.A. Powell, F.Rieger, and L.J. Standish (1986) The development of motoneurons inthe embryonic spinal cord of the mouse mutant, muscular dysgenesis(mdg/mdg): Survival, morphology and biochemical differentiation. Dev.Biol. 114:426–436.
Oppenheim, R.W., L.J. Haverkamp, D. Prevette, J.L. McManaman, and S.Appel (1988) Reduction of naturally occurringmotoneuron death in vivoby a target-derived neurotrophic factor. Science 240:919–922.
Oppenheim, R.W., S. Bursztajn, and D. Prevette (1989a) Cell death ofmotoneurons in the chick embryo spinal cord. XI: Acetylcholine recep-tors and synaptogenesis in skeletal muscle following the reduction ofmotoneuron death by neuromuscular blockade. Development 107:331–341.
Oppenheim, R.W., T. Cole, and D. Prevette (1989b) Early regional varia-tions in motoneuron numbers arise by differential proliferation in thechick embryo spinal cord. Dev. Biol. 133:468–474.
Oppenheim, R.W., Q.W. Yin, D. Prevette, and Q. Yan (1992) Brain-derivedneurotrophic factor (BDNF) rescues developing avian motoneuronsfrom cell death. Nature 360:755–757.
Oppenheim, R.W., D. Prevette, L.J. Haverkamp, L.J. Houenou, Q.W. Yin,and J. McManaman (1993) Biological studies of a putative avianmuscle-derived neurotrophic factor that prevents naturally occurringmotoneuron death in vivo. J. Neurobiol. 24:1065–1079.
Oppenheim, R.W., D. Prevette, and S.W. Wang (1996) The rescue of avianmotoneurons by activity blockade at the neuromuscular junction. Soc.Neurosci. Abstr. 22:44.
Pai, A.C. (1965) Developmental genetics of a lethal mutation, musculardysgenesis (mdg) in the mouse. Dev. Biol. 11:82–109.
Pautou, M.P., I. Hedayat, and M. Kieny (1982) The pattern of muscledevelopment in the chick leg. Arch. Anat. Microsc. Morphol. Exp.71:193–206.
Pincon-Raymond, M., F. Rieger, M. Fosset, and M. Lazdunski (1985)Abnormal transverse tubule system and abnormal amount of receptorsfor Ca21 channel inhibitors of the dihydropyridine family in skeletalmuscle from mice with embryonic muscular dysgenesis. Dev. Biol.112:458–466.
Pittman, R., and R.W. Oppenheim (1978) Neuromuscular blockade in-creases motoneuron survival during normal cell death in the chickembryo. Nature 271:364–366.
Pittman, R., and R.W. Oppenheim (1979) Cell death of motoneurons in thechick embryo spinal cord. IV. Evidence that a functional neuromuscularinteraction is involved in the regulation of naturally occurring celldeath and stabilization of synapses. J. Comp. Neurol. 187:452–446.
Powell, J.A., F. Rieger, B. Blandet, P. Dreyfus, and M. Pincon-Raymond(1984) Distribution and quantification of ACh receptors and innerva-tion in diaphragmmuscle of normal andmdgmouse embryos. Dev. Biol.101:168–180.
Pun, C.F. (1954) The crooked neck dwarf lethal syndrome in the domesticfowl. J. Exp. Zool. 126:101–132.
Rathouz, M.M., and D.K. Berg (1994) Synaptic type acetylcholine receptorsraise intracellular calcium levels in neurons by two mechanisms. J.Neurosci. 14:6935–6945.
Rathouz, M.M., S. Vijayaraghavan, and D.K. Berg (1995) Acetylcholinedifferentially affects intracellular calcium via nicotinic and muscarinicreceptors on the same population of neurons. J. Biol. Chem. 270:14366–14375.
Renshaw, G.M.C., and R. Goldie (1996) Neuronal bungarotoxin displaces[125I]a-bungarotoxin binding at the neuromuscular junctions as well asto the spinal cord during embryogenesis. Brain Res. 709:316–318.
Renshaw, G., P. Rigby, G. Self, A. Lamb, and R. Goldie (1993) Exogenouslyadministered alpha-bungarotoxin binds to embryonic chick spinal cord:Implications for the toxin-induced arrest of naturally occurringmotoneu-ron death. Neuroscience 53:1163–1172.
Rieger, F., and M. Pincon-Raymond (1981) Muscle and nerve in musculardysgenesis in the mouse at birth: sprouting and multiple innervation.Dev. Biol. 87:35–101.
Rieger, F., J.A. Powell, and M. Pincon-Raymond (1984) Extensive nerveovergrowth and paucity of the tailed asymmetric form of acetylcholines-terase in the developing skeletal neuromuscular system of the dysgenic(mdg) mouse. Dev. Biol. 101:181–192.
Role, L.W., andD.K. Berg (1996) Nicotinic receptors in the development andmodulation of CNS synapses. Neuron 16:1077–1085.
Rosalski, S.B. (1967)An improved procedure creatine phosphokinase deter-mination. J. Lab. Clin. Med. 69:696.
Rosenberg, L.E. (1945) Histological studies of muscle from crooked neckdwarf fowl. Anat. Rec. 97:277–282.
Tang, J., and L. Landmesser (1993) Reduction of intramuscular nervebranching and synaptogenesis is correlated with decreasedmotoneuronsurvival. J. Neurosci. 13:3095–3103.
Tanaka, H. (1987) Chronic application of curare does not increase the levelof motoneuron survival-promoting activity in limb muscle extractsduring the naturally occurring motoneuron cell death period. Dev. Biol.124:347–357.
Toop, J. (1976) A rapid method for demonstrating skeletal muscle innerva-tion in frozen sections. Stain Technol. 51:1–6.
Vijayaraghavan, S., P.C. Pugh, Z.W. Zhaug, M.M. Rathouz, and D.K. Berg(1992) Nicotinic receptors that bind a-bungarotoxin on neurons raiseintracellular free Ca21. Neuron 8:353–362.
Walton, P.D., J.A. Airey, J.L. Sutko, C.F. Beck, G.A. Mignery, T.C. Sudhof,T.J. Deerinck, and M.H. Ellisman (1991) Ryanodine and inositoltriphosphate receptors coexist in avian cerebellar neurons. J. Cell Biol.113:1145–1159.
Wick, R.A., and A.L. Allenspach (1978) Histological study of muscularhypoplasia in the crooked neck dwarf (cn/cn) chick embryo. J. Morphol.158:21–30.
372 R.W. OPPENHEIM ET AL.