neoplastic phenotype of gap-junctional intercellular communication–deficient wb rat liver...
TRANSCRIPT

120 RAE ET AL. MOLECULAR CARCINOGENESIS 22:120–127 (1998)
© 1998 WILEY-LISS, INC.
Neoplastic Phenotype of Gap-Junctional IntercellularCommunication–Deficient WB Rat Liver Epithelial Cellsand Its Reversal by Forced Expression of Connexin 32Robert S. Rae,1 Parmender P. Mehta,2 Chia-Cheng Chang,3 James E. Trosko,3 and Randall J. Ruch1*1Department of Pathology, Medical College of Ohio, Toledo, Ohio2Department of Medicine, Sylvester Comprehensive Cancer Center, University of Miami School of Medicine and GeriatricResearch Education and Clinical Center, VA Medical Center, Miami, Florida3Department of Pediatrics and Human Development, Michigan State University, East Lansing, Michigan
Gap-junctional intercellular communication (GJIC) is involved in cellular growth control and is often reducedin neoplastic cells. In this study, four GJIC-deficient rat liver epithelial cell lines (WB-aB1, WB-bA2, WB-cD6, andWB-dA2) were examined for altered growth and tumorigenicity in comparison with their GJIC-competentparental cell line, WB-F344. WB-aB1 cells were also forced to express connexin 32 (Cx32) by transduction witha Cx32 cDNA retroviral expression vector to help determine whether the restoration of GJIC could reverse theirneoplastic phenotype. WB-aB1 and WB-bA2 cells had faster population doubling times (PDTs) and highersaturation densities (SDs) than did WB-F344 cells. In contrast, the growth of WB-cD6 and WB-dA2 cells was notsignificantly different from that of WB-F344 cells. WB-aB1 and WB-bA2 cells formed tumors in male F344 rats,but WB-cD6 and WB-dA2 cells did not. After transduction of WB-aB1 cells with Cx32, four stable clones (WB-a/32-3, -8, -9, and -10) were isolated that had GJIC levels of 5.2%, 44.5%, 69.8%, and 90.5%, respectively. Thegrowth of poorly coupled clones 3 and 8 was similar to that of parental WB-aB1 cells, but the growth of well-coupled clones 9 and 10 was similar to that of WB-F344 cells. The tumorigenicity of WB-a/32-9 and WB-a/32-10cells was also significantly lower than that of WB-aB1 cells. Our results suggest that reduced GJIC contributes toneoplastic transformation of WB cells, that additional changes are necessary, and that restoration of GJIC byforced Cx32 protein expression can suppress the neoplastic phenotype of these cells. Mol. Carcinog. 22:120–127, 1998. © 1998 Wiley-Liss, Inc.
Key words: tumorigenicity, cell growth, contact inhibition, transformation, retrovirus
INTRODUCTION
Cell growth is regulated by extracellular, intracel-lular, and intercellular communication pathways [1].One pathway of intercellular communication is me-diated by gap-junction channels that link the interi-ors of adjacent cells. These channels, formed ofproteins known as connexins that are encoded by afamily of related genes, enable small molecules (lessthan 1000 Da) to pass directly between cells, a pro-cess known as gap-junctional intercellular commu-nication (GJIC) [2,3]. Growth modulators, secondmessengers, sugars, nucleotides, amino acids, fattyacids, drugs, and carcinogens can pass through thesechannels [reviewed in 4]. Thus, GJIC has been impli-cated in tissue homeostasis, growth control, differ-entiation, embryogenesis, and bystander killing and,more recently, apoptosis [4–6]. In support of this ideais the fact that the expression and function of a vari-ety of gap-junction genes are altered in neoplasticcells [7,8] and during embryonic development [9].
This loss of GJIC may be an early event in neo-plastic transformation and may facilitate the clonalexpansion of neoplastically initiated cells [10–12].Direct evidence supporting the role of reduced GJIC
in the development of cancer has been obtained bystudies in which forced expression of connexins inconnexin-deficient neoplastic cells corrected theirneoplastic phenotypes [13–21]. Additional evidencestems from studies in which inhibition of GJIC innormal cells by antisense techniques dysregulatedtheir growth [22,23].
We have been studying the relationship betweenneoplasia and GJIC in WB-F344 rat liver epithelialcells and have previously described several GJIC-de-ficient mutants of these cells [24–26]. This defect inGJIC is not due to the lack of gap-junction proteinexpression or altered localization, because wild-typeand mutant cells express similar amounts ofconnexin 43 (Cx43) protein, which is located on theplasma membrane. Instead, the lack of GJIC in the
*Correspondence to: Department of Pathology, Medical Collegeof Ohio, 3000 Arlington Avenue, Toledo, OH 43699.
Received 6 October 1997; Revised 8 December 1997; Accepted12 December 1997
Abbreviations: GJIC, gap-junctional intercellular communication;Cx43, connexin 43; Cx32, connexin 32; Cx26, connexin 26; PDT,population doubling time; SD, saturation density.

PHENOTYPIC EFFECTS OF GAP JUNCTIONS 121
mutant cell lines may be due to defective Cx43 phos-phorylation [24–26].
In the study reported here, we investigated whetherthese GJIC-deficient mutants exhibited neoplasticproperties and whether this was due to the loss ofGJIC. We characterized the in vitro growth charac-teristics and tumor-forming abilities of the mutantsand investigated whether the restoration of GJIC byforced expression of connexin 32 (Cx32) proteinwould reverse their neoplastic phenotypes. We trans-duced GJIC mutant cells with Cx32 rather than Cx43for two reasons. First, restoration of GJIC to the wild-type level by transfection with Cx43 has beenachieved by using a methotrexate-amplifiabledihydrofolate reductase vector [25], but the responserequired high doses of methotrexate (5 mM) and wasnot stable upon methotrexate removal (unpublishedobservations). Because we wished to determine thetumorigenicity of GJIC-restored WB mutants, we didnot want to treat animals with high doses of meth-otrexate to maintain Cx43 protein expression, be-cause the drug might be toxic. Second, a previousstudy indicated that restoration of GJIC by forcedCx32 protein expression does not suppress the invitro growth of C6 glioma cells but does inhibit tu-mor growth [27]. This result also led us to examinewhether GJIC mediated by Cx32 had a growth-sup-pressive effect in hepatic cells.
MATERIALS AND METHODS
Cell Lines
The rat liver epithelial cell line WB-F344, which isdiploid and nontumorigenic and exhibits contact in-hibition of growth [28], was obtained from Dr. J. W.Grisham (University of North Carolina, Chapel Hill,NC). WB-F344 cells have a high capacity for GJICand express Cx43 as their predominant gap-junctionprotein [26,29]. Several GJIC-deficient cell lines—namely, WB-aB1, WB-bA2, WB-cD6, and WB-dA2—that were derived by mutation of WB-F344cells [25,26] were also used. All cell lines werecultured in Richter’s improved minimal essentialmedium (Irvine Scientific, Irvine, CA) supple-mented with 5% fetal bovine serum (SigmaChemical Co., St. Louis, MO) and 40 µg/mL gen-tamicin sulfate (Sigma Chemical Co.).
Several clones of WB-aB1 cells that expressed Cx32were generated by infecting WB-aB1 cells with therecombinant retrovirus LXSNRCx32. This vector wasconstructed by inserting a 1.4-kb rat Cx32 cDNA frag-ment into the EcoRI site of the retroviral vector LXSN[30]. The cDNA fragment contained the entire cod-ing sequence of Cx32 [31], and its expression wasdriven by the 5´-flanking long terminal repeat of thevector. The vector also contained a neomycin phos-photransferase gene whose expression was driven byan internal simian virus 40 promoter. Stably trans-duced clones were selected by culturing in medium
containing G418 (0.4 mg/mL; GIBCO/BRL, Gaithers-burg, MD) for 4 wk and then isolating and charac-terizing several independent clones. The followingclones were isolated and used in this study: WB-a/32-3, WB-a/32-8, WB-a/32-9, and WB-a/32-10.
Determination of GJIC by Fluorescent DyeMicroinjection (Dye-Coupling Assay)
GJIC was quantified by microinjecting a gap-junc-tion–permeable fluorescent tracer, Lucifer yellow CH,into test cells and determining the percentage of first-order neighbors to which dye had transferred (dyecoupling) [26]. In brief, test cells were first grown toconfluence in 35-mm dishes. Then, 10 cells per dishwere microinjected with dye, and their first-orderneighbors were evaluated for the presence or absenceof the dye 5 min later. At least four dishes weresampled per cell line.
Connexin Expression Studies
Northern blot analyses of connexin mRNA con-tent in WB cell lines were performed using Cx43,Cx32, and connexin 26 (Cx26) full-length cDNAprobes radiolabeled by random priming as previouslydescribed [26,32]. Western blot and immunohis-tochemical studies of Cx43 and Cx32 protein wereperformed with mouse monoclonal antibodies(Zymed, South San Francisco, CA) [26,32].
In Vitro Growth Studies
The population doubling time (PDT) and satu-ration density (SD) of each cell line were deter-mined by plating cells into 24-well plates (2.5 ×104 cells/well), allowing them to attach overnight,refeeding them with complete medium, and thencounting the cells with a hemocytometer aftertrypsinization of individual wells over the next12 d. The PDT was determined in the logarithmicphase of growth (days 2–4); the SD was defined asthe cell number at day 12 [33].
Tumorigenicity Studies
The tumorigenicity of these cells in rats was deter-mined after injection of cells into the rat liver. Inbrief, adult male F344 rats (150–175 g each) wereanesthetized with sodium pentobarbital, and a 1-cmincision was then made in each rat’s abdominal cav-ity to expose the median lobe of the liver. Five mil-lion cells per rat were suspended in 0.2 mL ofserum-free medium and then injected into the me-dian lobe with a tuberculin syringe fitted with a 27-gauge needle. After injection of the cells andensurance that any bleeding from the injection sitehad stopped, the abdominal wound was closed withsutures. The rats were observed for 6 mo and thenkilled; rats were killed sooner if they exhibited signsof distress. At necropsy, livers and tumors were re-moved and fixed in 10% buffered formalin. All speci-mens were processed for routine histological analysis
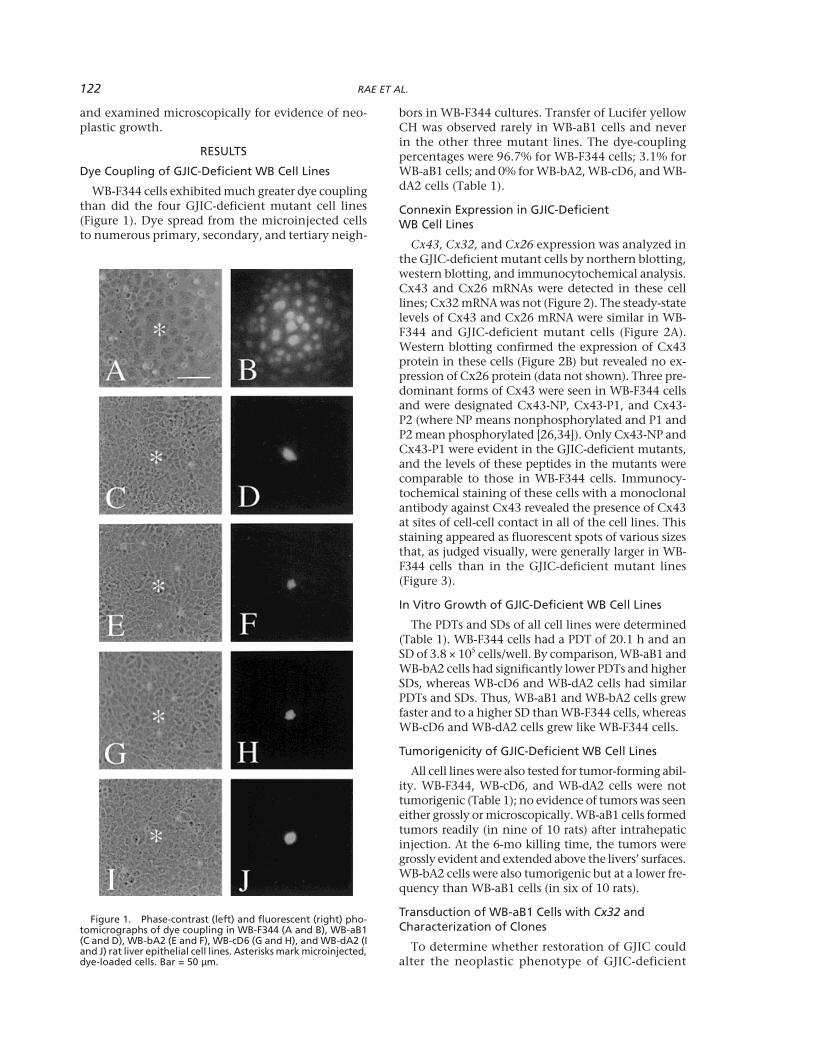
122 RAE ET AL.
and examined microscopically for evidence of neo-plastic growth.
RESULTS
Dye Coupling of GJIC-Deficient WB Cell Lines
WB-F344 cells exhibited much greater dye couplingthan did the four GJIC-deficient mutant cell lines(Figure 1). Dye spread from the microinjected cellsto numerous primary, secondary, and tertiary neigh-
bors in WB-F344 cultures. Transfer of Lucifer yellowCH was observed rarely in WB-aB1 cells and neverin the other three mutant lines. The dye-couplingpercentages were 96.7% for WB-F344 cells; 3.1% forWB-aB1 cells; and 0% for WB-bA2, WB-cD6, and WB-dA2 cells (Table 1).
Connexin Expression in GJIC-DeficientWB Cell Lines
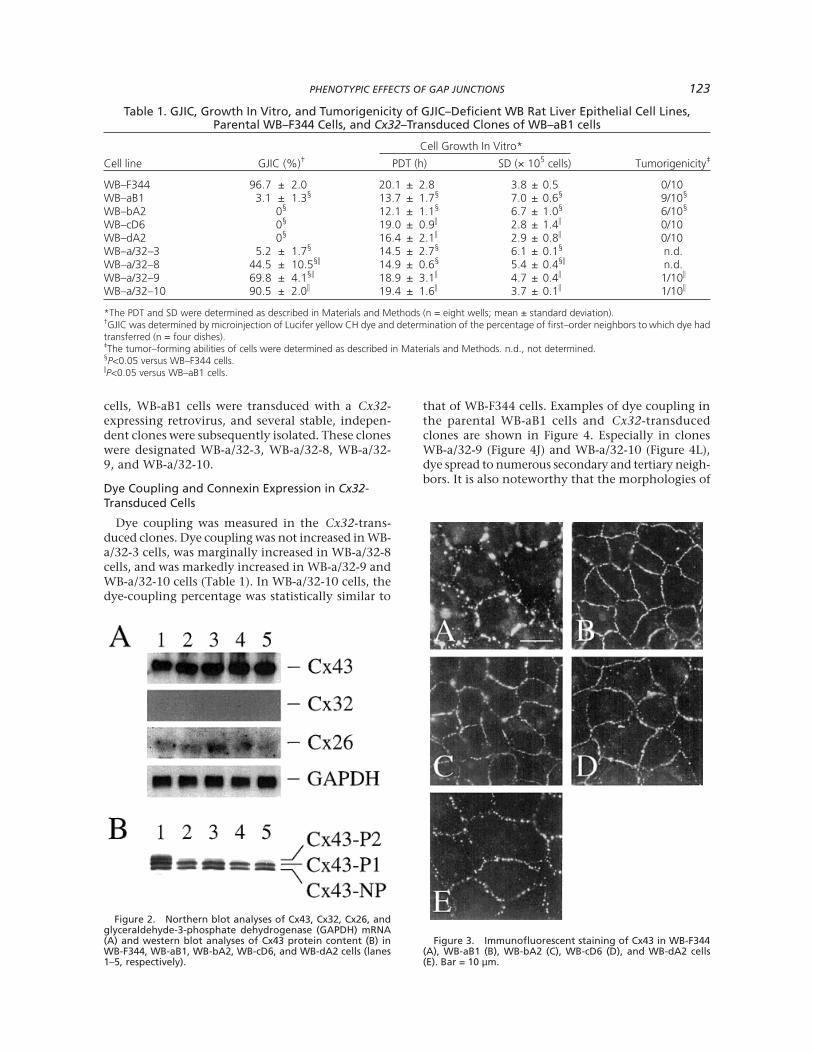
Cx43, Cx32, and Cx26 expression was analyzed inthe GJIC-deficient mutant cells by northern blotting,western blotting, and immunocytochemical analysis.Cx43 and Cx26 mRNAs were detected in these celllines; Cx32 mRNA was not (Figure 2). The steady-statelevels of Cx43 and Cx26 mRNA were similar in WB-F344 and GJIC-deficient mutant cells (Figure 2A).Western blotting confirmed the expression of Cx43protein in these cells (Figure 2B) but revealed no ex-pression of Cx26 protein (data not shown). Three pre-dominant forms of Cx43 were seen in WB-F344 cellsand were designated Cx43-NP, Cx43-P1, and Cx43-P2 (where NP means nonphosphorylated and P1 andP2 mean phosphorylated [26,34]). Only Cx43-NP andCx43-P1 were evident in the GJIC-deficient mutants,and the levels of these peptides in the mutants werecomparable to those in WB-F344 cells. Immunocy-tochemical staining of these cells with a monoclonalantibody against Cx43 revealed the presence of Cx43at sites of cell-cell contact in all of the cell lines. Thisstaining appeared as fluorescent spots of various sizesthat, as judged visually, were generally larger in WB-F344 cells than in the GJIC-deficient mutant lines(Figure 3).
In Vitro Growth of GJIC-Deficient WB Cell Lines
The PDTs and SDs of all cell lines were determined(Table 1). WB-F344 cells had a PDT of 20.1 h and anSD of 3.8 × 105 cells/well. By comparison, WB-aB1 andWB-bA2 cells had significantly lower PDTs and higherSDs, whereas WB-cD6 and WB-dA2 cells had similarPDTs and SDs. Thus, WB-aB1 and WB-bA2 cells grewfaster and to a higher SD than WB-F344 cells, whereasWB-cD6 and WB-dA2 cells grew like WB-F344 cells.
Tumorigenicity of GJIC-Deficient WB Cell Lines
All cell lines were also tested for tumor-forming abil-ity. WB-F344, WB-cD6, and WB-dA2 cells were nottumorigenic (Table 1); no evidence of tumors was seeneither grossly or microscopically. WB-aB1 cells formedtumors readily (in nine of 10 rats) after intrahepaticinjection. At the 6-mo killing time, the tumors weregrossly evident and extended above the livers’ surfaces.WB-bA2 cells were also tumorigenic but at a lower fre-quency than WB-aB1 cells (in six of 10 rats).
Transduction of WB-aB1 Cells with Cx32 andCharacterization of Clones
To determine whether restoration of GJIC couldalter the neoplastic phenotype of GJIC-deficient
Figure 1. Phase-contrast (left) and fluorescent (right) pho-tomicrographs of dye coupling in WB-F344 (A and B), WB-aB1(C and D), WB-bA2 (E and F), WB-cD6 (G and H), and WB-dA2 (Iand J) rat liver epithelial cell lines. Asterisks mark microinjected,dye-loaded cells. Bar = 50 µm.
PHENOTYPIC EFFECTS OF GAP JUNCTIONS 123
cells, WB-aB1 cells were transduced with a Cx32-expressing retrovirus, and several stable, indepen-dent clones were subsequently isolated. These cloneswere designated WB-a/32-3, WB-a/32-8, WB-a/32-9, and WB-a/32-10.
Dye Coupling and Connexin Expression in Cx32-Transduced Cells
Dye coupling was measured in the Cx32-trans-duced clones. Dye coupling was not increased in WB-a/32-3 cells, was marginally increased in WB-a/32-8cells, and was markedly increased in WB-a/32-9 andWB-a/32-10 cells (Table 1). In WB-a/32-10 cells, thedye-coupling percentage was statistically similar to
that of WB-F344 cells. Examples of dye coupling inthe parental WB-aB1 cells and Cx32-transducedclones are shown in Figure 4. Especially in clonesWB-a/32-9 (Figure 4J) and WB-a/32-10 (Figure 4L),dye spread to numerous secondary and tertiary neigh-bors. It is also noteworthy that the morphologies of
Table 1. GJIC, Growth In Vitro, and Tumorigenicity of GJIC–Deficient WB Rat Liver Epithelial Cell Lines,Parental WB–F344 Cells, and Cx32–Transduced Clones of WB–aB1 cells
Cell Growth In Vitro*
Cell line GJIC (%)† PDT (h) SD (× 105 cells) Tumorigenicity‡
WB–F344 96.7 ± 2.0 20.1 ± 2.8 3.8 ± 0.5 0/10WB–aB1 3.1 ± 1.3§ 13.7 ± 1.7§ 7.0 ± 0.6§ 9/10§
WB–bA2 0§ 12.1 ± 1.1§ 6.7 ± 1.0§ 6/10§
WB–cD6 0§ 19.0 ± 0.9j 2.8 ± 1.4j 0/10WB–dA2 0§ 16.4 ± 2.1j 2.9 ± 0.8j 0/10WB–a/32–3 5.2 ± 1.7§ 14.5 ± 2.7§ 6.1 ± 0.1§ n.d.WB–a/32–8 44.5 ± 10.5§j 14.9 ± 0.6§ 5.4 ± 0.4§j n.d.WB–a/32–9 69.8 ± 4.1§j 18.9 ± 3.1j 4.7 ± 0.4j 1/10j
WB–a/32–10 90.5 ± 2.0j 19.4 ± 1.6j 3.7 ± 0.1j 1/10j
*The PDT and SD were determined as described in Materials and Methods (n = eight wells; mean ± standard deviation).†GJIC was determined by microinjection of Lucifer yellow CH dye and determination of the percentage of first–order neighbors to which dye hadtransferred (n = four dishes).‡The tumor–forming abilities of cells were determined as described in Materials and Methods. n.d., not determined.§P<0.05 versus WB–F344 cells.jP<0.05 versus WB–aB1 cells.
Figure 2. Northern blot analyses of Cx43, Cx32, Cx26, andglyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA(A) and western blot analyses of Cx43 protein content (B) inWB-F344, WB-aB1, WB-bA2, WB-cD6, and WB-dA2 cells (lanes1–5, respectively).
Figure 3. Immunofluorescent staining of Cx43 in WB-F344(A), WB-aB1 (B), WB-bA2 (C), WB-cD6 (D), and WB-dA2 cells(E). Bar = 10 µm.

124 RAE ET AL.
the parental WB-aB1 cells and Cx32-transducedclones were similar.
All four clones expressed Cx43 mRNA at levels simi-lar to those in WB-F344 and WB-aB1 cells, but Cx32mRNA was detected only in the transduced clonesand was greatest in clones WB-a/32-9 and -10 (Fig-ure 5). This Cx32 expression correlated with the dye-coupling levels (Table 1). The Cx32 transcriptsexpressed by the clones varied in size and were largerthan the endogenous 1.6-kb Cx32 mRNA expressed
by rat hepatocytes (Figure 5). This may have beendue to differences in polyadenylation of viral andendogenous Cx32 mRNAs.
Cx43 and Cx32 protein were also detected in theclones by western blotting and immunocytochemicalanalysis. On western blots, only Cx43-NP and Cx43-P1 were evident in the clones and were present at lev-els similar to those in WB-aB1 cells. A band migratingslightly ahead of Cx43-NP, which may have been adegradation product of Cx43 [35], was also evident.Cx32 protein was also detected in the clones by west-ern blotting. The peptide migrated at 27 kDa, as oth-ers have reported [36]. Clone WB-a/32-9 expressed thehighest amount of Cx32 (Figure 6). Immunostainingrevealed that Cx32 was expressed in the clones andthat it was localized to regions of cell-cell contact (Fig-ure 7). Cx32 staining was not apparent on parentalWB-aB1 cells (Figure 7).
In Vitro Growth and Tumorigenicity of Cx32-Transduced Cells
The PDTs and SDs of the Cx32-transduced cloneswere measured to determine whether Cx32 expres-sion altered growth. The PDTs of clones WB-a/32-3and WB-a/32-8 were statistically similar to the PDTof parental WB-aB1 cells (Table 1). The SD of cloneWB-a/32-3 was similar to that of WB-aB1 cells, andthe SD of clone WB-a/32-8 was significantly less. Bothclones, however, grew faster and to a higher densitythan WB-F344 cells did. In contrast, WB-a/32-9 andWB-a/32-10 cells grew essentially like WB-F344cells (Table 1); there were no statistically signifi-cant differences in PDTs or SDs between thesethree cell lines. Thus, clones WB-a/32-3 and -8,which had low levels of GJIC, grew like poorlycommunicating WB-aB1 cells, whereas clones WB-
Figure 4. Phase-contrast (left) and fluorescent (right)photomicrographs of dye coupling in WB-F344 (A and B),WB-aB1 (C and D), WB-a/32-3 (E and F), WB-a/32-8 (G andH), WB-a/32-9 (I and J), and WB-a/32-10 (K and L) rat liverepithelial cell lines. Asterisks mark microinjected, dye-loaded cells. Bar = 50 µm.
Figure 5. Northern blot analyses of Cx43 and Cx32 mRNAcontent in WB-F344, WB-aB1, WB-a/32-3, WB-a/32-8, WB-a/32-9, and WB-a/32-10 cells (lanes 1–6, respectively) and infreshly isolated male F344 rat hepatocytes (lane 7). Lane –was empty. Cx32(v), Cx32 transcripts expressed by the trans-duced viral Cx32 gene; Cx32(e), Cx32 transcripts expressedby the endogenous Cx32 gene; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.

PHENOTYPIC EFFECTS OF GAP JUNCTIONS 125
a/32-9 and -10, which had much higher levels ofGJIC, grew like WB-F344 cells.
The tumorigenicity of WB-a/32-9 and WB-a/32-10cells was determined by injecting these cells into thelivers of adult male rats. This revealed that the twoclones caused significantly fewer tumors than didWB-aB1 cells (Table 1).
DISCUSSION
We investigated the relationship between reducedGJIC and the neoplastic phenotype in WB rat liver
Figure 6. Western blot analyses of Cx43 (A) and Cx32 (B)protein content in WB-F344, WB-aB1, WB-a/32-3, WB-a/32-8,WB-a/32-9, and WB-a/32-10 cells (lanes 1–6, respectively).
Figure 7. Immunofluorescent staining of Cx32 in WB-aB1(A), WB-a/32-3 (B), WB-a/32-8 (C), WB-a/32-9 (D), and WB-a/32-10 cells (E). Bar = 10 µm.
epithelial cells. We compared the in vitro growth andtumorigenic properties of four GJIC-deficient WB celllines with highly communicative parental WB-F344cells. We also investigated whether the restorationof GJIC by transduction of Cx32 into one of theseGJIC-deficient cell lines (WB-aB1) could alter its phe-notype. Two of the GJIC-deficient WB cell lines,namely WB-aB1 and WB-bA2, grew faster and to ahigher SD than did WB-F344 cells and were tumori-genic in adult F344 rats. WB-F344 cells were not tu-morigenic. The other two GJIC-deficient lines,WB-cD6 and WB-dA2, grew like WB-F344 cells andwere not tumorigenic. GJIC was restored in WB-aB1cells by Cx32 transduction; four Cx32-transducedclones of WB-aB1 cells that were identified had low(WB-a/32-3 cells), moderate (WB-a/32-8 cells), or high(WB-a/32-9 and WB-a/32-10 cells) levels of GJIC andCx32 expression. In vitro, the former two clones grewlike WB-aB1 cells, whereas the latter two clones grewlike WB-F344 cells and were not tumorigenic.
These results provide strong evidence that reducedGJIC is directly involved in the enhanced growth andtumorigenicity of WB-aB1 cells and possibly of WB-bA2 cells. Restoration of GJIC in WB-aB1 cells re-versed this phenotype in a way that correlated withthe level to which GJIC was restored. In WB-bA2 cells,the loss of GJIC was also associated with increasedgrowth and tumorigenicity, but connexin transfec-tion studies need to be performed to verify that thisphenotype involves reduced GJIC. Our data agreewith those from other studies demonstrating thatreduced GJIC is involved in the neoplastic pheno-type. The enhancement of GJIC by connexin-genetransfection and transduction reduces the growthand tumorigenicity of many types of neoplastic cells[13–21]. In addition, the inhibition of connexin ex-pression in nontransformed cells by antisense tech-niques enhances cell growth [22] and impairs theability of untransformed cells to suppress the growthof co-cultured neoplastic cells [23]. Thus, reducedGJIC appears to be involved in growth deregulationand expression of the neoplastic phenotype in manytypes of cells.
In contrast to the results we obtained with WB-aB1 and WB-bA2 cells, the data we obtained fromWB-cD6 and WB-dA2 cells suggest that reduced GJICdoes not result in a neoplastic phenotype. WB-cD6and WB-dA2 cell growth was unchanged comparedwith that of WB-F344 cells, and these two GJIC-defi-cient lines were not tumorigenic. This conflicts withthe data already mentioned above and may suggestthat GJIC is not involved in neoplasia. There are sev-eral possible explanations of this paradox, however.One is that because the GJIC-deficient lines werederived by treatment with 5-bromodeoxyuridine fol-lowed by black light exposures [24], several muta-tions probably exist in each line. Some of thesemutations may affect growth or other processesneeded for tumor formation.

126 RAE ET AL.
Another explanation is that several genetic andepigenetic changes must occur in cells before a neo-plastic phenotype can be expressed and that the lossof GJIC alone may not be sufficient. It is possiblethat WB-aB1 and WB-bA2 cells have undergonethese additional changes, whereas WB-cD6 and WB-dA2 cells have not. That reduced GJIC is only onecontributing factor in the development of neopla-sia is supported by the fact that restoration of neo-plastic-cell GJIC to normal-cell levels by connexintransfection and transduction did not completelyreverse the neoplastic phenotype (this study) [13–19] and that antisense inhibition of connexin ex-pression in normal cells does not make themneoplastic [22,23]. Thus, we view the reduction ofGJIC as one of several changes required for neoplas-tic transformation. Because many transformed cellsexpress functional gap junctions, one might also ar-gue that reduced GJIC has no role in neoplastictransformation. Previous connexin transfection/transduction and connexin antisense studies [13–23], however, provided direct evidence to the con-trary. In addition, studies from several groupsindicated that neoplastic cells can express functionalgap junctions among themselves (homologous GJIC)but be incapable of forming gap junctions with nor-mal cells (heterologous GJIC) [37–40]. As Loewen-stein [7] first noted, neoplastic cells may manifestdefective GJIC in several ways including the lack ofgap junctions or the formation of gap junctions withreduced channel permeability, the inability to formgap junctions with normal cells (lack of heterolo-gous GJIC), and the inability to respond to junc-tional signal molecules.
As noted above, there are several mammalianconnexins, and they are expressed in a cell-, tissue-, and developmentally specific manner [3]. Previ-ous studies suggested that connexins are also tissuespecific in their ability to reverse the neoplastic phe-notype. For instance, transfection of HeLa cells,which were derived from a human cervical carci-noma, with Cx26 but not with Cx40 or Cx43 sup-pressed growth and tumor formation [19]. Similarly,transfection of C6 glioma cells with Cx43 suppressedgrowth in vitro and tumor formation [13,16],whereas transfection with Cx32 suppressed onlytumor growth [27]. We have reported that transfec-tion of murine lung carcinoma cells with Cx43 sup-presses their in vitro growth and tumorigenicity butthat Cx32 transduction suppresses only in vitrogrowth [21]. In contrast, transfection of SK-Hep-1human hepatocarcinoma cells with Cx32 reducedtumor growth but not in vitro growth [14]. Thus,the ability of transfected connexins to suppress theneoplastic phenotype appears to be cell andconnexin specific. The data reported here, unlikethe data from other studies [14,21,27], indicated thatCx32 suppresses the growth of neoplastic cells invitro and in vivo.
Is Cx32 a tumor suppressor gene? Analyses ofchemically induced rat liver tumors indicate thatCx32 is rarely mutated or deleted in these tumors[41] but that its expression is often reduced or thatthe protein is abnormally located in liver cells[10,11,42]. This suggests that Cx32 might be a classII tumor suppressor gene that causes loss of GJICwhen a defect occurs in the regulation of the gene’sexpression or in trafficking of the peptide the geneencodes, rather than a Class I tumor suppressor genethat is inactivated by mutation or deletion [43].
ACKNOWLEDGMENTS
We thank Drs. Eric Beyer, David Paul, and BruceNicholson for providing the Cx43, Cx32, and Cx26cDNAs, respectively, and Dr. Joe Grisham for pro-viding the WB-F344 cells. This study was supportedby National Institutes of Health/National CancerInstitute grants CA 57612 (to RJR) and CA 21104(to JET) and by grants from the American CancerSociety, Florida Division, Inc. (to PPM); the Uni-versity of Miami Sylvester Comprehensive Can-cer Center (to PPM); and the Department ofVeteran Affairs (to PPM).
REFERENCES
1. Trosko JE, Madhukar BV, Chang CC. Endogenous and exog-enous modulation of gap junctional intercellular communica-tion: Toxicological and pharmacological implications. Life Sci53:1–19, 1993.
2. Loewenstein WR. The cell-cell channel of gap junctions. Cell48:725–726, 1987.
3. White TW, Bruzzone R, Paul DL. The connexin family of intercel-lular channel forming proteins. Kidney Int 48:1148–1157, 1995.
4. Ruch RJ. The role of gap junctional intercellular communicationin neoplasia. Ann Clin Lab Sci 24:216–231, 1994.
5. Yamasaki H, Naus CCG. Role of connexin genes in growth con-trol. Carcinogenesis 17:1199–1213, 1996.
6. Trosko JET, Goodman JI. Intercellular communication may facili-tate apoptosis: Implications for tumor promotion. Mol Carcinog11:8–12, 1994.
7. Loewenstein WR. Junctional intercellular communication and thecontrol of growth. Biochim Biophys Acta 560:1–65, 1979.
8. Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, TraubO. Expression of Cx26, Cx32, and Cx43 gap junction proteinsin normal and neoplastic human tissues. Int J Cancer 51:522–529, 1992.
9. Lo CW. The role of gap junction membrane channels in develop-ment. J Bioenerg Biomembr 28:379–385, 1996.
10. Klaunig JE, Ruch RJ, Hampton JA, Weghorst CM, Hartnett JA.Gap junctional intercellular communication and murine hepaticcarcinogenesis. Prog Clin Biol Res 331:277–291, 1990.
11. Neveu MJ, Hully JR, Babcock KL, et al. Multiple mechanisms areresponsible for altered expression of gap junction genes duringoncogenesis in rat liver. J Cell Sci 107:83–95, 1994.
12. Freidman EA, Steinberg M. Disrupted communication betweenlate-stage premalignant human colon epithelial cells by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res 42:5096–5105, 1982.
13. Zhu D, Caveney S, Kidder GM, Naus CCG. Transfection of C6glioma cells with connexin 43 cDNA: Analysis of expression, in-tercellular coupling, and cell proliferation. Proc Natl Acad Sci USA88:1883–1887, 1991.
14. Eghbali B, Kessler JA, Reid LM, Roy C, Spray DC. Involvement ofgap junctions in tumorigenesis: Transfection of tumor cells withconnexin 32 cDNA retards growth in vivo. Proc Natl Acad SciUSA 88:10701–10705, 1991.
15. Mehta PP, Hotz-Wagenblatt A, Rose B, Shalloway D, LoewensteinWR. Incorporation of the gene for a cell-cell channel protein intotransformed cells leads to normalization of growth. J MembrBiol 124:207–225, 1991.

PHENOTYPIC EFFECTS OF GAP JUNCTIONS 127
16. Naus CCG, Elisevich K, Zhu D, Belliveau DJ, Del Maestro RF. Invivo growth of C6 glioma cells transfected with connexin43cDNA. Cancer Res 52:4208–4213, 1992.
17. Rose B, Mehta PP, Loewenstein WR. Gap-junction proteingene suppresses tumorigenicity. Carcinogenesis 14:1073–1075, 1993.
18. Chen S-C, Pelletier D, Ao P, Boynton A. Connexin43 reverses thephenotype of transformed cells and alters their expression ofcyclin/cyclin-dependent kinases. Cell Growth Differ 6:681–690,1995.
19. Mesnil M, Krutovskikh V, Piccoli C, et al. Negative growth con-trol of HeLa cells by connexin genes: Connexin species specific-ity. Cancer Res 55:629–639, 1995.
20. Hirschi KK, Xu CE, Tsukamoto T, Sager R. Gap junction genesCx26 and Cx43 individually suppress the cancer phenotype ofhuman mammary carcinoma cells and restore differentiationpotential. Cell Growth Differ 7:861–870, 1996.
21. Ruch RJ, Cesen-Cummings KT, Malkinson AM. Role of gap junc-tions in lung neoplasia. Exp Lung Res, in press.
22. Ruch RJ, Guan XJ, Sigler K. Inhibition of gap junctional intercel-lular communication and enhancement of growth in BALB/c 3T3cells treated with connexin43 antisense oligonucleotides. MolCarcinog 14:269–274, 1995.
23. Goldberg GS, Martyn KD, Lau AF. A connexin 43 antisensevector reduces the ability of normal cells to inhibit the fociformation of transformed cells. Mol Carcinog 11:106–114,1994.
24. Oh SY, Dupont E, Madhukar BV, et al. Characterization of gapjunctional communication–deficient mutants of a rat liver epi-thelial cell line. Eur J Cell Biol 60:250–255, 1993.
25. Jou Y-S, Matesic D, Dupont E, et al. Restoration of gap-junc-tional intercellular communication in a communication-deficientrat liver cell mutant by transfection with connexin 43 cDNA. MolCarcinog 8:234–244, 1993.
26. Esinduy C, Chang C, Trosko J, Ruch R. In vitro growth inhibitionof neoplastically transformed cells by non-transformed cells: Re-quirement for gap junctional intercellular communication. Car-cinogenesis 16:915–921, 1995.
27. Bond SL, Bechberger JF, Khoo NKS, Naus CCG. Transfection ofC6 glioma cells with connexin32: The effects of expression of anonendogenous gap junction protein. Cell Growth Differ 5:179–186, 1994.
28. Tsao MS, Smith JD, Nelson KG, Grisham JW. A diploid epithelialcell lines from normal adult rat liver with phenotypic propertiesof “oval” cells. Exp Cell Res 154:38–52, 1984.
29. Matesic DF, Rupp HL, Bonney WJ, Ruch RJ, Trosko JE.Changes in gap-junction permeability, phosphorylation, andnumber mediated by phorbol ester and non–phorbol-ester
tumor promoters in rat liver epithelial cells. Mol Carcinog10:226–236, 1994.
30. Miller AD, Rossman GJ. Improved retroviral vectors for gene trans-fer and expression. Biotechniques 7:980–990, 1989.
31. Paul DL. Molecular cloning of cDNA for rat liver gap junctionprotein. J Cell Biol 103:123–134, 1986.
32. Ren P, Ruch RJ. Inhibition of gap junctional intercellular commu-nication by barbiturates in long-term primary cultured rat hepa-tocytes is correlated with liver tumour promoting activity.Carcinogenesis 17:2119–2124, 1996.
33. Mehta PP, Lokeshwar BL, Schiller PC, et al. Gap-junctional com-munication in normal and neoplastic prostate epithelial cells andits regulation by cAMP. Mol Carcinog 15:18–32, 1996.
34. Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Dif-ferential phosphorylation of the gap junction protein connexin43in junctional communication–competent and –deficient cell lines.J Cell Biol 111:2077–2088, 1990.
35. Laird DW. The life cycle of a connexin: Gap junction formation,removal, and degradation. J Bioenerg Biomembr 28:311–318,1996.
36. Hertzberg E. A detergent-independent procedure for the isola-tion of gap junctions from rat liver. J Cell Biol 259:9936–9943,1984.
37. Mesnil M, Asamoto M, Piccoli C, Yamasaki H. Possible molecularmechanism of loss of homologous and heterologous gap junc-tional intercellular communication in rat liver epithelial cell lines.Cell Adhes Commun 2:377–384, 1994.
38. Krutovskikh V, Mazzoleni G, Mironov N, et al. Altered homolo-gous and heterologous gap-junctional intercellular communica-tion in primary human liver tumors associated with aberrantprotein localization but not gene mutation of connexin 32. Int JCancer 56:87–94, 1994.
39. Garber SA, Fernstrom MJ, Stoner GD, Ruch RJ. Altered gap junc-tional intercellular communication in neoplastic rat esophagealepithelial cells. Carcinogenesis 18:1149–1153, 1997.
40. Neveu MJ, Hully JR, Babcock KL, et al. Multiple mechanisms areresponsible for altered expression of gap junction genes duringoncogenesis in rat liver. J Cell Sci 107:83–95, 1994.
41. Omori Y, Krutovskikh V, Mironov N, Tsuda H, Yamasaki H. Cx32gene mutation in a chemically induced rat liver tumour. Carcino-genesis 17:2077–2080, 1996.
42. Janssen-Timmen U, Traub O, Dermietzel R, Rabes HM, WilleckeK. Reduced number of gap junctions in rat hepatocarcinomasdetected by monoclonal antibody. Carcinogenesis 7:1475–1482, 1986.
43. Lee SW, Tomasetto C, Sager R. Positive selection of candidatetumor-suppressor genes by subtractive hybridization. Proc NatlAcad Sci USA 88:2825–2829, 1991.