need for an integrated approach to formulation research and knowledge management
TRANSCRIPT

Ajaz S. Hussain, Ph.D., President
The National Institute of Pharmaceutical Technology & Education, Inc.
FDA’s FY 2016 Regulatory Science Initiatives Part 15 Public Meeting

Outline
• A note on NIPTE
• US FDA’s strategic response to maximizing how generics meet public health needs
• Some considerations for navigating multifaceted challenges
• Summary
• References
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 2

About NIPTEA 501(c)(3) Non-profit organization
Founded in 2005
Incorporated in 2007
Headquarters: Minneapolis, MN
12 Schools of Pharmacy, 3 Schools of Engineering, 1 Medical School
Improving Quality and Lowering Costs with Confidence
May 20, 2016 3FDA CDER Bldg. 31, Room 1503 (Great Room)

Outline
• A note on NIPTE
• US FDA’s strategic response to maximizing how generics meet public health needs
• Some considerations for navigating multifaceted challenges
• Summary
• References
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 4

US FDA’s strategic response to maximizing how generics meet public health needsDr. Woodcock’s testimony to the US Congress, 4 February 2016
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 5
“First Generics”
Public Health Priority
GADUFA goal date (15
months – to shorter).
GDUFA research
funding prioritization
(this meeting)
Need for research;
Research to policy to
practice - time and
effectiveness.
GDUFA II negotiations
a pre-ANDA process?
Submission quality -
Multiple review cycles
(on average 4)
Several programs in
OPQ, One Quality Voice
Need for additional
quality regulation to
“better assure quality in
an increasingly globalized
industry“.

Outline
• A note on NIPTE
• US FDA’s strategic response to maximizing how generics meet public health needs
• Some considerations for navigating multifaceted challenges
• Summary
• References
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 6

Recognizing multifaceted challengesTopics and Points of View
Topics for FY 2016 Regulatory Science Initiatives
1. Opportunities for scientific or technical advancements that would help to overcome specific barriers for industry that currently limit the availability of generic drug products.
2. Innovative approaches to pre-approval development of generic drugs, including new methodologies for product design and manufacturing, and design and conduct of in vitro, ex vivo, and clinical studies and identification of scientifically robust strategies for demonstration of bioequivalence for various product classes.
3. Innovation in scientific approaches to evaluating the therapeutic equivalence of generic drug products throughout their lifecycle.
4. Identification of high-impact public health issues involving generic drugs that can be addressed by the prioritized allocation of FY 2017 funding for regulatory science research.
5. Identification of specific issues related to generic drug products where scientific recommendations and/or clarifications are needed in developing and/or revising FDA's guidance for industry.
6. Strategies for enhancing quality and equivalence risk management during generic drug product development, during regulatory review, and/or throughout the drug product's lifecycle.
NIPTE point of view
1. Integrated approach for evolving standard for analytical characterization - case example excipient variability (Eric Munson)
2. Integrated approach for evolving standards for formulation design - case example NTI's (Ken Morris)
3. Two talks (above) also relevant to this topic
4. Confidence in Generics: Need for an Integrated approach to Formulation Research and Knowledge Management (Ajaz Hussain)
5. The talk above also illustrates some challenge
6. Mechanism for an integrated approach to Formulation Research, Knowledge Management, & Knowledge sharing with FDA & Industry (Steve Byrn)
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 7

Describing multifaceted challengesNeed for integration with clarity & consistency
Integrated Analysis & Synthesis
• Public perceptions are shaped by the few errors, recalls, etc.
• Stark reminders & reasons to pay attention to perceptions (e.g., color, shape, etc.)
• ‘Totality of Evidence’ is increasingly the dominant path for complex generics; complexity is increasing, generally; there is a need for integration with clarity & consistency
Needed to optimally address multifaceted challenges
• Therapeutic equivalence increasingly demands notable attention to integration of product/process design & development, orthogonal analytical characterization, in vitro and, when necessary, evidence of in vivo equivalence
• Knowledge bases and decision-making processes pertaining to integration of evidence (specifically – product/process design & development and orthogonal analytical characterization), need to grow, mature and progress.
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 8

Need for integration with clarity & consistency Illustrative examples
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 9
Nov. 2014 Draft Guidance on
Methylphenidate Hydrochloride Subject-by-Formulation Interactions?
Sept 2012 Draft Guidance on
Mesalamine Applicant should provide evidence of
high variability in the bioequivalence
parameters
Sept 2015 Draft Guidance on
Mometasone Furoate
Monohydrate In vitro BE, Pharmacokinetic (PK) BE
and Clinical Endpoint BE
Formulation Science?
Does subject-by-formulation interaction
variance, derived from a general
population, provide adequate assurance ?.
Evidence based on
underlying mechanisms
(e.g., failure modes) Delayed release tablet dissolution related
to coating thickness by terahertz pulsed
image mapping. Journal of
pharmaceutical sciences, 97(4),
pp.1543-1550. (2008).
Right Question at the Right TimeTimely integration of formulation and process
design, analytics & in vivo evidence can and
must be facilitated
To facilitate more optimal policy considerations,
to more rapidly recognize innovative proposals by
companies, to reduce scientific disputes,…

Considering potential impactsComplexity is increasing generally
• Increasingly a more complex environment; a disproportionally higher risk posed to maintaining/improving confidence in generic drugs
• Protracted and costly development, multiple review cycles, and/or delayed launch dates (for reasons beyond IP issues)
• Limited competition, even among generics
• Increased risk of continued challenges to approved generics (e.g., based on comparisons utilizing novel analytics)
• More reasons for significant delays; particularly in approval of “first generic”
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 10

Specific consideration
• In their allocation of FY 2017 funding for regulatory science research, the FDA is urged to consider prioritizing efforts towards development of knowledge bases and standards to guide optimal development & integration of multifaceted scientific evidence of Therapeutic Equivalence.
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 11

Outline
• A note on NIPTE
• US FDA’s strategic response to maximizing how generics meet public health needs
• Some considerations for navigating multifaceted challenges
• Summary
• References
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 12

Summary Confidence in generic drugs is built upon an optimal integration of evidence derived from formulation and process design, analytical characterization and, when necessary, in vivo assessment
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 13
Several programs in OPQ,
One Quality Voice
Need for additional quality regulation to
“better assure quality in an increasingly
globalized industry“.
“First Generics” Public Health
Priority
GADUFA goal date (15 months – to
shorter).
GDUFA research funding prioritization
(this meeting)
Need for research; Research to policy to
practice - time and effectiveness.
GDUFA II negotiations a
pre-ANDA process
Submission quality - Multiple review
cycles (on average 4)
On
e Q
uality
Vo
ice
Rig
ht Q
ue
stio
n @
Rig
ht T
ime
Firs
t, on
-time
?
I. In their allocation of FY 2017
funding for regulatory science
research, the FDA is urged to
consider prioritizing efforts
towards development of
knowledge bases and
standards to guide optimal
integration of multifaceted
scientific evidence of
Therapeutic Equivalence.

Outline
• A note on NIPTE
• US FDA’s strategic response to maximizing how generics meet public health needs
• Some considerations for navigating multifaceted challenges
• Summary
• References
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 14

References
• FY 2016 Regulatory Science Initiatives Part 15 Public Meeting. • http://www.fda.gov/ForIndustry/UserFees/GenericDrugUserFees/ucm489572.htm (accessed 5/12/2016)
• The National Institute for Pharmaceutical Technology & Education.• http://nipte.org/ (accessed 5/12/2016)
• Implementation of the Generic Drug User Fee Amendments of 2012 (GDUFA) - House Testimony.• http://www.fda.gov/NewsEvents/Testimony/ucm485057.htm (accessed 5/12/2016)
• Draft Guidance on Methylphenidate Hydrochloride.• http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320007.pdf (accessed 5/12/2016)
• Draft Guidance on Mesalamine.• http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320002.pdf (accessed 5/12/2016)
• Spencer, J.A., Gao, Z., Moore, T., Buhse, L.F., Taday, P.F., Newnham, D.A., Shen, Y., Portieri, A. and Hussain, A., 2008. Delayed release tablet dissolution related to coating thickness by terahertz pulsed image mapping. Journal of pharmaceutical sciences, 97(4), pp.1543-1550.
• Draft Guidance on Mometasone Furoate Monohydrate.• http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM461141.pdf (accessed 5/12/2016)
May 20, 2016 FDA CDER Bldg. 31, Room 1503 (Great Room) 15

Mechanism for an integrated approach to Formulation Research, Knowledge
Management, & Knowledge sharing with FDA & Industry
1
Steve Byrn
Purdue University

The Complexity of Formulation Science
• Performance
– Scientific Reliability
– Formulation Stability & State of Control
– Bioavailability/Bioequivalence
– Safety, Efficacy & Therapeutic Equivalence
• Processes
– Design/Design Control
– Characterization & Assessment
– Utility of prior knowledge
– Approval and compliance decisions
2

Issues
• Fundamental understanding and knowledge of formulations
• Development report describes operationally what happens but may not provide adequate understanding
– Structure
– Solid state chemistry/Solution chemistry
– Reactions & interaction of components in the composition
– Design and mechanism of operation/structure/performance/behavior/function of formulation
– Examples
• Acid-base reactions/salt switches
• Nanoparticles – Abraxane, coated nanoparticles
• Emulsion formulations
• Controlled release
3

Issues
• Specific populations; e.g., Pediatric formulations
• Nuances and critical aspects of manufacturing and how processes influence formulations
• Stability and potential stability issues
• Failure modes often not fully explored
• Frame the right questions in QbR
4

Example Complex/Problem Formulations
• Controlled/Sustained Release
– Bupropion/Wellbutrin
– Methylphenidate/ADHD drugs
• Emulsion- Based
– Neoral
• Nanoparticles/Abraxane
• BCS Class 2
– Ritonavir
– Efavirenz
• Failure Mode Analysis
– Acid-base reactions in formulations
– Abuse deterrent formulations
5
Understanding these formulations requires fundamental scientific understanding

Neoral – microemulsion formulations
• Neoral – a microemulsion formulation
• SangCya – a non-microemulsion formulation
• QbR – Importance of QTPP and structure differences of two formulations
6

Buproprion/Wellbutrin 300 mg Product
• Bupropion XL300 –matrix release product prone to dose dumping
• Wellbutrin XL300 –membrane technology releasing bupropion over 5 hours. Not prone to dose dumping.
• QbR – Importance of BE study for highest dose
7

Full Ad
8

9

Ritonavir
• Soft Gel – Form 2 precipitated• Reformulated SEC• Reformulated as melt
extrudedtablet with special surfactant –sorbitan monolaureate
• QbR – importance of structure and manufacturing method (CQA)
• Importance of CQA - excipient
10

Abraxane
• Structure
• Manufacturing method
• QbR - CQAs
11

Salt Disproportionation in Pioglitazone HCl Tablets
12

Steps to outlining a mechanism
• Case studies– Analysis: What didn’t occur as it should
have (e.g., identification of failure modes)
– What if/should: Experimental research (e.g., evaluating options for optimal integration of product/process design with analytics)
• Transdisciplinary synthesis– What: Right questions @ right time– How: Right questions @ right time– Classification systems (e.g., NTI Quality)
and decision trees, orthogonal methods (e.g., Utility of R&D Analytics, QC Tests and Effective Investigations)
• Process & system for knowledge acquisition & curation– Identify knowledge and/or resource
gaps and process to fill these– Building the knowledge base
• Deliverables (Examples)
– Targeted white papers & scientific publication
– What if/should research to evaluate option for optimal integration
– System for transdisciplinary elaboration to inform Question Based Review and recommendations on product specific recommendations for regulatory guidance
– Training programs
– Curated knowledge base
13

Summary• In their allocation of FY 2017 funding for
regulatory science research, the FDA is urged to consider prioritizing efforts towards development of knowledge bases and standards to guide optimal integration of multifaceted scientific evidence of Therapeutic Equivalence.
• Mechanism for such an integrated approach can/should include: Analysis (Looking back), Synthesis (Looking forward) and building of knowledge bases
• Deliverables, for example, may include:
– Targeted white papers & scientific publication
– What if/should research to evaluate option for optimal integration
– System for transdisciplinary elaboration to inform Question Based Review and recommendations on product specific recommendations for regulatory guidance
– Training programs, and
– Curated knowledge base
NIPTE Point of View:
Confidence in Generics: Need for an Integrated approach to Formulation Research and Knowledge Management (Ajaz Hussain)
Integrated approach for evolving standard for analytical characterization - case example excipient variability (Eric Munson)
Integrated approach for evolving standards for formulation design - case example NTI's (Ken Morris)
Mechanism for an integrated approach to Formulation Research, Knowledge Management, & Knowledge sharing with FDA & Industry (Steve Byrn)
14

QBR as an Organizing Principle for the Pre-approval Development of
Generic Drugs
1
Ken Morris Ph.D. University Professor
Director Lachman Institute for Pharmaceutical Analysis Long Island University – Brooklyn Campus
Arnold & Marie Schwartz College of Pharmacy and Health Sciences
FY 2016 Regulatory Science Initiatives Part 15 Public Meeting

Implementation of the Generic Drug User Fee Amendments of 2012 (GDUFA)
Testimony of Janet Woodcock, M.D. Before the Committee on Oversight and Government Reform
United States House of Representatives, February 4, 2016
Ongoing Challenges …”Second, there is a need for more research in the generics space. Some drugs lack generic competition because there is no convincing bioequivalence test method available. … Similarly, methods for showing chemical sameness for certain complex drugs are not available…”
2

What does QBR as an Organizing Principle Mean? QbR and QbD are complimentary not independent QbD is the framework/control strategy and QbR is the
logistics/execution • A Development History captured in the Report is the key to both
guiding development and being ready for QbR. • Essential aspects of the DH include:
– The rationale for all phases of development • Fundamental principles, prior knowledge, heuristics • Q8 and Q6 principles are implicit in DH
– The knowledge base created • Makes the DH an electronic living document with all changes
captured • Usage of new and prior knowledge to make decisions • Capturing of failure modes and sharing of knowledge
between FDA review and inspection
3

FDA Advisory Committee for Pharmaceutical Science—March 13, 2003
Carlos R. Hamilton, Jr. MD, FACE American Association of Clinical Endocrinologists—VP
UT Houston, Executive Vice-President for Clinical Affairs
Dosage Changes of as Little as 12.5 to 25 Micrograms of Oral L-Thyroxine Daily Have
Significant Effects on Serum TSH
“Historical” Example:
4

Eric Duffy – CDER-FDA
5

Impact of the Solid State on Chemical Stability: Dehydration (Solid State Chemistry of Drugs, Byrn 1999)
16. 11 OXI DATI ON REACTI ONS PRECEDED BY LOSS OF SOLVENT
Although not extensively studied, there are several solid-state oxidation reactions that are preceded by and may indeed require prior loss of solvent of crystallization.
A. Di hydr ophenyl al ani ne
O2CO2
H NH3
–
+
CO2
H NH3
–
+
dihydrophenylalanine phenylalanine
6
Impact of the Solid State: Crystalline Hydrate Classification System (Morris and Rodriguez 1993, Morris and Brittain1998)
Dehydration can lead to amorphous material, new
crystal structures, dehydrated hydrates which maintain the packing motif of the original structure, or
mixed structures

7
Dehydration of LTH can produce a dehydrated hydrate which maintain the packing motif of the original structure as shown
Katrusiak and Katrusiak, 93(12), JPS 2004

Does Dehydration Precede
Degradation?
8
Pharm Dev and Tech Vol. 20(3), pgs.314-319 May 2015
0 2 4 6 8 10 1260
65
70
75
80
85
90
95
100
105
Time (Days)
% L
evo
thyro
xin
e S
od
ium
Re
ma
inin
g
Control
fit to Control
RT / 0% O2
fit to RT / 0% O2
60oC / 0% O2
fit to 60oC / 0% O2
RT / 21% O2
fit to RT / 21% O2
60oC / 21% O2
fit to RT / 21% O2Does O2 Cause Degradation?

So a lot was known or discoverable about levo’s properties and issues
– NTI (narrow therapeutic index drug) – Very low dose, 25-300ug – Chemically labile: formulation failure modes
• Stable if hydrated but oxidized if dehydrated/disordered • Processing effected stability (ala structure) – Patel Cincinati thesis • Excipient interaction (often via pH), Mansoor’s papers
– Long PK t½ of approximately 7 days
Yet many products were developed and approved apparently oblivious of the prior knowledge and/or logical
concerns
9
Note - this pre-dates most of the “Q’s”
QbD, QbR, Q8 etc…so no Dev. Report in US

What does the example teach? • The dosage form quality specs need to be developed at the
preclinical or pre-bio-study stage (QTPP – Q8 -ala Q6A) to know what variability is due to the patient variability or PD – An opportunity to leverage pre-ANDA meetings – Specs established as a basis for product design not decided after
the fact • The development process has to be INTEGRATED to understand how
fundamental property changes propagate variability downstream – Orthogonal analytics and BE must be “connected” to formulation
design – Scaling-up amplifies but seldom solves problems (paraphrased
from Steve Byrn) • Designing a process/product and creating a sound Development
History is its own reward in product quality and for efficient review 10

An Integrated NTI Quality Classification • The problems facing NTI development and use are based on the
general hypothesis that the lack of IVIVC (and the phenomenon of a NTI itself) is a broad category, – i.e., the lack of IVIVC is a symptom with many different possible
root causes. – It is proposed that these root causes fall into a finite number of
predictable categories with variable but assignable contributions to the observed effect.
• Data mining and creation of an NTI quality-clinical response, adverse event, metabolism, and drug physico-chemical properties knowledge base will be a major element of classification. The data will be compiled into a fully relational knowledge base (FDA as a partner). This will not only serve to inform (right question at the right time) but to direct experimentation and modification strategies
11

NTI quality classification: knowledge base development
Drug Oral Dose (mg) Stability Cp t1/2
(hr) Solubility (mg/mL)
Product Recall Basis (2012 - 2016)
Mol. Wt (g/mol)
Prazosin 1 Light Sensitive 2.5 0.5 (HCl salt) - 383.4
Warfarin 1 Temperature sensitive 40 0.017 Super potetnt, CU,
Stability 308.33
Clonidine 0.1 + 14 50 - 230.09
Valproic Acid 125 Light and
Temperature Sensitive
12.5 50 Failed Dissolution 144.21
Digoxin 0.05 Light Sensitive 42 0.0648 - 580.94
Levothyroxine 0.025 Light Sensitive, Oxidation 168 0.000105
0.15 (salt) Subpotent, stability,
(45) 776.87
Phenytoin 30 Temperature sensitive 17 0.032 Failed dissolution 252.27
Isoetharine Mesylate 0.35 Temperature
sensitive 3.18 - 335.42
Disopyramide 100 + 6.7 0.0449 Failed dissolution 339.47
12
Many more descriptors are relatively straight forward to obtain or generate with concerted effort of teams of graduate students!

Summary: Opportunities for FDA Support • Research on integrated product development by category,
across disciplines/investigators, for pharmaceutical and therapeutic equivalence and guidance contribution – NTI quality matrix example – Complex Dosage Forms – Combination products – Complex APIs …
• Support for Knowledge Base R&D for formulation design – Elucidate fundamental phenomena associated with Dosage Form
categories – Facilitate modeling, e.g. ontologies; FDA-NIPTE excipient database – Prepare the ground for pre-ANDA meetings
• Development of programs for training and expert support for Generic Companies and Reviewers – Identify knowledge and/or resource gaps – Development report rationale and design
13

Integrated Approach for Evolving Standards for Analytical Characterization:
Case Example – Excipient Variability
Eric J. Munson
Department of Pharmaceutical Sciences University of Kentucky

FDA Quality Risk Management
2
“The only difference between an innovator product and a generic product is the formulation” – paraphrase from a
comment made at last year’s GDUFA meeting
Intrinsic – Ingredients and Process • Formulation – what’s in it • Manufacturing – how’s it made Extrinsic – What is the Product? • Ingredient variability • Drug-excipient interactions • Impact of processing
Analyze the Product Analyze the Performance
Functional Properties • in vitro composition,
disintegration, dissolution, Bioequivalence • in vivo clinical performance

FDA Quality Risk Management
3
“The only difference between an innovator product and a generic product is the formulation” – paraphrase from a
comment made at last year’s GDUFA meeting
Advanced Analytical Methodology • Drug Substance • Excipients • Drug Product - Interactions • Impact on physical properties • Physical/chemical stability • Transformations
Analyze the Product and its Performance
Functional Properties • in vitro composition,
disintegration, dissolution, Bioequivalence • in vivo clinical performance
è

FDA Quality Risk Management
4
Risk Reduction Opportunities: two “very” common causes - Deficient Facilities and Processes
“Oral powder for suspension product failed dissolution due to glyceryl behenate acid value”
“Dissolution failure in soft gel capsules as a result of cross-linking of short chain aldehydes and other liquid components”
“Tablet lots had significant dissolution failures due to variation in the coating agent, Zein NF…”
“Extended release tablet failed dissolution due to variability of ethyl cellulose excipient”
Recalls due to Excipient Variability:
Richard Friedman, CDER, PDA/FDA Executive Management Workshop, Baltimore, MD (September 12-13, 2012)

Magnesium Stearate: SSNMR, DSC,and TGA correlations
5
DSC
TGA
13C SSNMR

SSNMR Spectroscopy of Formulations Containing MgSt
6
Universal Problem – How do you see a complex excipient at 1% in a formulation?
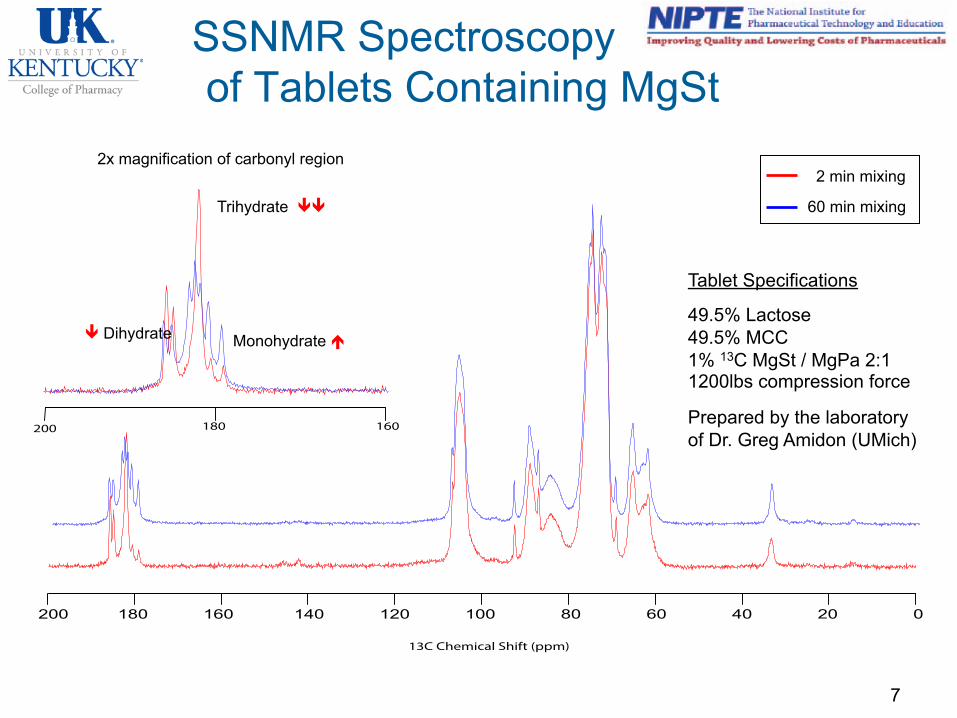
SSNMR Spectroscopy of Tablets Containing MgSt
7
200 180 160 140 120 100 80 60 40 20 0
13C Chemical Shift (ppm)
160200 180
2 min mixing
60 min mixing
2x magnification of carbonyl region
Tablet Specifications
49.5% Lactose 49.5% MCC 1% 13C MgSt / MgPa 2:1 1200lbs compression force
Prepared by the laboratory of Dr. Greg Amidon (UMich)
ê Dihydrate Monohydrate é
Trihydrate êê

Impact of MgSt on Dissolution – Mixing Variability
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60
Cumula&
ve%drugdissolved
Time(min)
Dissolu&onProfileusingMgStbyFisher
Batch1
Batch2
Batch3
Batch4
Batch5
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60
Cumula&
ve%drugdissolved
TIme(min)
Dissolu&onProfileusingbyChem-ImpexInt’l
Batch1
Batch2
Batch3
Batch4
Batch5

Impact of MgSt Form on Dissolution – Consistent Mild Mixing
0
10
20
30
40
50
60
70
80
90
100
00:00:00 00:07:12 00:14:24 00:21:36 00:28:48
% D
isso
lutio
n
Time (hh/mm/ss)
MgSt % Dissolution >300µ PS, 30min Turbula mix
Avg. CM 37-3 Monohydrate
Avg. CM 37-5 Trihydrate corrected

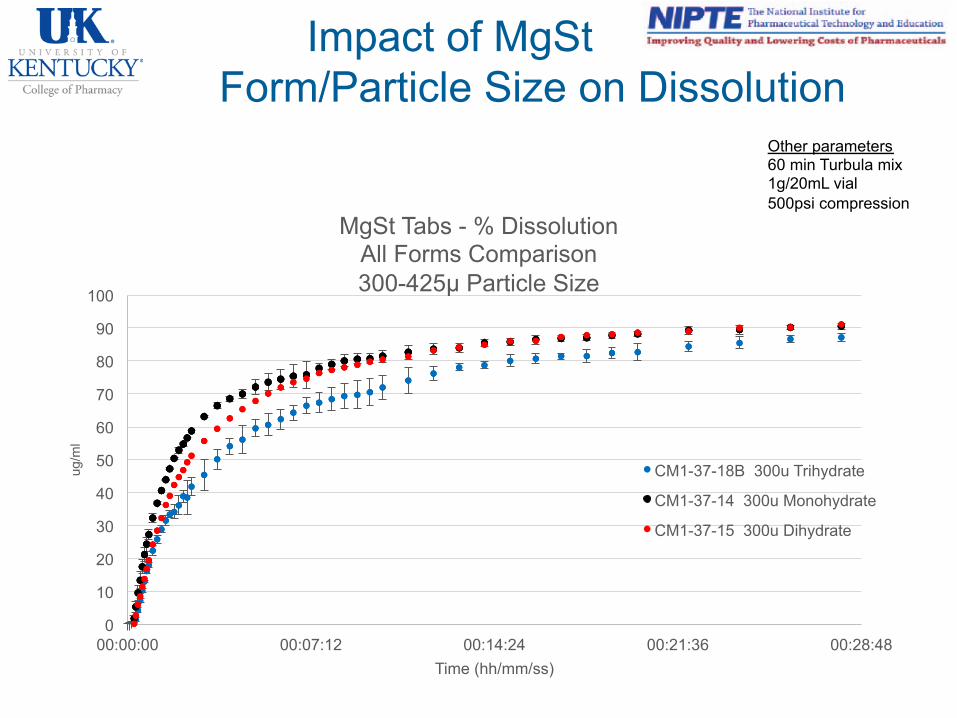
Impact of MgSt Form/Particle Size on Dissolution
0
10
20
30
40
50
60
70
80
90
100
00:00:00 00:07:12 00:14:24 00:21:36 00:28:48
% D
isso
lutio
n
Time (hh/mm/ss)
MgSt Tabs - % Dissolution All Forms Comparison
<45u Particle Size
CM1-37-17 45u Trihydrate
CM1-37-13 45u Monohydrate
CM1-37-23 45u Disordered
CM1-37-15 45u Dihydrate
Other parameters 60 min Turbula mix 1g/20mL vial 500psi compression
Impact of MgSt Form/Particle Size on Dissolution
0
10
20
30
40
50
60
70
80
90
100
00:00:00 00:07:12 00:14:24 00:21:36 00:28:48
ug/m
l
Time (hh/mm/ss)
MgSt Tabs - % Dissolution All Forms Comparison 300-425µ Particle Size
CM1-37-18B 300u Trihydrate
CM1-37-14 300u Monohydrate
CM1-37-15 300u Dihydrate
Other parameters 60 min Turbula mix 1g/20mL vial 500psi compression

Summary FDA Quality Risk Management
12
Recommendations for FDA Support – Establish Research Priorities for Generic Drug Product Characterization
• What is the optimum portfolio of orthogonal analytics that are needed for product characterization?
• How should these be integrated in design/development space? • What should be the validation criteria for R&D analytics? • What is the utility across dosage forms for these analytics? • What is the relationship between R&D analytics, QC testing, and
effective methods for root cause investigations?
Integrated approach to understand complex dosage forms, convert it to a knowledge base that is accessible, and translate
that to reviewers through education
Analyze the Product and its Performance – Advanced Analytical Characterization of Dosage Forms

Page 1 of 4
Generic Drug User Fee Amendments of 2012 Regulatory Science Initiatives: Request for Public Input for FY 2016 Generic Drug Research
Part 15 Public Hearing
May 20, 2016 FDA White Oak Campus,
10903 New Hampshire Ave. Bldg. 31, Rm. 1503
Silver Spring, MD 20993
Agenda 9:00 – 10:00 am Opening Remarks Robert Lionberger, Ph.D. Presiding Officer Director, Office of Research and Standards
Office of Generic Drugs (OGD), CDER, FDA “GDUFA Regulatory Science Update”
10:00 – 10:10 am
Michael Fischer, MD., MS Associate Professor of Medicine Brigham & Women’s Hospital Harvard Medical School “Regulatory science for generic drugs”
10:10 – 10:15 am Questions from Panel 10:15 – 10:25 am
Gordon L. Amidon Charles R. Walgreen Jr. Professor of Pharmacy & Pharmaceutical Sciences University of Michigan
10:25 – 10:30 am Questions from Panel 10:30 – 10:45 am Break 10:45 – 10:55 am
Duxin Sun, Ph.D. Professor of Pharmaceutical Science The University of Michigan “Potential New Method to Improve BE of Modified Release (MR) Drug Products by in vivo Dissolution Studies In Human GI tract”
10:55 – 11:00 am Questions from Panel

Page 2 of 4
11:00 – 11:10 am Chetan Pujara, Ph.D. Vice President, Small Molecule Product Development Allergan “Non-Biological Complex Drugs: Challenges in the assessment of Similarity or Equivalence of Ophthalmic Emulsions”
11:10 – 11:15 am Questions from Panel 11:15 – 11:25 am
Catherine M.T. Sherwin, PhD., MSCI., FCP Assistant Professor, Pediatrics and Pharmacy Interim Chief Division of Clinical Pharmacology University of Utah School of Medicine “Issues associated with generic drugs used in children”
11:25 – 11:30 am Questions from Panel 11:30 – 11:40 am
Ajaz S. Hussain, Ph.D. President, National Institute for Pharmaceutical Technology and Education (NIPTE) “Confidence in Generics: Need for an Integrated approach to Formulation Research and Knowledge Management”
11:40 – 11:45 am Questions from Panel 11:45 – 11:55 am
Stephen R. Byrn, Ph.D. Professor, National Institute for Pharmaceutical Technology and Education (NIPTE) and Purdue University “Mechanism for an integrated approach to Formulation Research, Knowledge Management, & Knowledge sharing with FDA & Industry”
11:55 am – 12:00 pm Questions from Panel 12:00 – 1:00 pm Lunch 1:00 – 1:10 pm
Kenneth R. Morris, Ph.D. Professor, National Institute for Pharmaceutical Technology and Education (NIPTE) and Long Island University “Integrated approach for evolving standards for formulation design – case example NTI’s”
1:10 – 1:15 pm Questions from Panel

Page 3 of 4
1:15 – 1:25 pm Eric J. Munson, Ph.D. National Institute for Pharmaceutical Technology and Education (NIPTE) and Patrick DeLuca Endowed Professor in Pharmaceutical Technology, University of Kentucky “Integrated approach for evolving standard for analytical characterization – case example excipient variability”
1:25 – 1:30 pm Questions from Panel 1:30 – 1:40 pm
Amy Barton Pai, PharmD, BCPS, FASN, FCCP, FNKF Professor and Chair, Department of Pharmacy Practice Director, ANephRx Core Laboratory Chair, NYS CKD Coalition Albany College of Pharmacy and Health Sciences "Relevant challenges in determination of bioequivalence of generic IV iron formulations"
1:40 – 1:45 pm Questions from Panel 1:45 – 1:55 pm
Diane J. Burgess, Ph.D. Board of Trustees Distinguished Professor of Pharmaceutics University of Connecticut "In Vitro-In Vivo Correlation for Complex Drug Products and In Vitro/In Vivo Stability Issues".
1:55 – 2:00 pm Questions from Panel 2:00 – 2:20 pm
David Gaugh, R.Ph. Senior Vice President for Sciences and Regulatory Affairs Generic Pharmaceutical Association (GPhA) “FY 2017 Regulatory Science Priorities GPhA’s Perspective”
2:20 – 2:25 pm Questions from Panel 2:25 – 2:35 pm
Nikunjkumar Patel Senior Research Scientist Simcypt(a Certara Company) “PBPK modelling in Generic Product Assessment”.
2:35 – 2:40 pm Questions from Panel 2:40 – 2:50 pm
Russ Rackley Global Head, PKDM Mylan Inc. “Challenges with the Demonstration of Statistical Non-Inferiority of Adhesion and Irritation for Transdermal Drug Delivery Systems Using the OGD Bioguidance Method”

Page 4 of 4
2:50 – 2:55 pm Questions from Panel 2:55 – 3:05 pm
David R. Schoneker Vice Chair for Scientific and Regulatory Affairs - IPEC Americas Director of Global Regulatory Affairs – Colorcon “The Need for Science and Risk-based Excipient Safety Assessment during generic drug review – Impact on formulation quality and performance”
3:05 – 3:10 pm Questions from Panel 3:10 – 3:25 pm Break 3:25 – 3:35 pm
Bahman Asgharian Research Scientist Applied Research Associates, Inc. “Reconstruction of the airway tree, airflow and drug delivery calculations in the lungs of children with disease”
3:35 – 3:40 pm Questions from Panel 3:40 – 3:50 pm
Tracy Rupp, PharmD / Paul Brown Director of Public Health Policy Initiatives National Center for Health Research “Protecting the public health through improved generic drug regulation”
3:50 – 3:55 pm Questions from Panel 3:55 – 4:05 pm
James E. Polli, Ph.D. Professor and Ralph F. Shangraw Endowed Chair in Industrial Pharmacy & Pharmaceutics University of Maryland, School of Pharmacy “Considerations in Excipients”
4:05 – 4:10 pm Questions from Panel 4:10 – 4:15 pm Closing Remarks
Kathleen “Cook” Uhl, M.D. Director Office of Generic Drugs (OGD), CDER, FDA