n r t l: an empirical equation or an inspiring model for fluids mixtures properties?
TRANSCRIPT

Fluid Phase Equilibria, 24 (1985) 87-114 Elsevier Science Publishers B.V., Amsterdam - Printed in The Netherlands
87
N R T 1: AH EMPIRICAL EQUATION DR AN IWSPIRIWG MDDEL FOR FLUIDS MIXTURES
PROPERTIES?
Henri RENON
Ecole Nationale Superieure des Mines de Paris
CNRS UA 878
60 Boulevard Saint-Michel
75006 PARIS (FRANCE)
and
MASSACHUSSETTS INSTITUTE OF TECHNOLOGY
Department of Chemical Engineering
CAMBRIDGE, MA 02139 (USA)
(Received and accepted July 26, 1985)
ABSTRACT
The NRTL and other local composition models give good empirical repre-
sentation of phase equilibria. They are currently extended to electrolyte
solutions, and high pressure phase equilibria and transformed into group
contribution models. They inspire theoretical work to find out about real
randomness i.e. distribution of nearest neighbors, in fluids, potentially
leading to improved understanding of fluid state and better empirical models.
INTRODUCTION
The NRTL model was originally developped and presented (RENON, 1966;
RENON and PRAUSNITZ, 1968 a, 1968 b, 1969) as an empirical model which could
improve over the WILSON equation for the simultaneous representation of
Vapor-Liquid Equilibria (VLE), Liquid-Liquid Equilibria (LLE), heat of
0378~3812/85/$03.30 0 1985 Elsevier Science Publishers B.V.

mixing, limiting activity coefficients in binary and multicomponent
mixtures. Many experimental data were considered and a systematic compari-
son with other models for gE was presented for the deviations between
experimental and calculated values of properties of binary mixtures and
multicomponent systems.
In this work, the empirical advantages and shortcomings of the NRTL and
other local composition models will be reviewed. The industry in accepting
these models widely recognized their practical advantages.
Since 1966, the academic research has taken two main directions:
l- extending the usefulness of the local composition models by reducing the
number of their parameters, transforming them into group contribution
models, introducing them into an equation of state approach or applying them
to electrolyte solutions
2 - questioning the reasons of the success and the limitations of these
model. Presently the criticism of the hypotheses which were presented ori-
ginally to explain the model, are turning very precise and based on the most
advanced methods of theoretical study of liquid and liquid mixtures. These
efforts will be critically reviewed and some suggestions for future develop-
ment will be made.
EMPIRICAL ADVANTAGES AND LIUITATIONS OF THE LOCAL COMPOSITIONS MODELS
The NRTL model can be considered as an equation for the excess GIBBS
energy gE of multicomponent mixtures with adjustable parameters :
C
CJE/RT = if, xi3$ 'j 'ji Gji
k?I 'k Gki
G ij = exp (- ~1.. T..)
Jl Jl
(1)
where Xi is the mole fraction of component i, R the gas constant, T the
absolute temperature and 'ijs 'jfs T ij = 'ji
temperature
dependent parameters.
The important practical advantages of the model are in order of decrea-
sing importance :
- It represents fairly well - within one or two per cent the mole frac-

89
tions in absolute value and the pressure in relative value - the VLE and LLE
of binary mixtures by adjusting three parameters at ane temperature.
- It predicts VLE of ternary and multicomponent mixtures from binary
parameters obtained by fitting binary data.
- It represents multicomponent LLE if one adjusts the binary parameters
to ternary data. There is usually not enough information about binary data
to obtain three parameters per binary. But one may adjust nine parameters to
data for a ternary system at one temperature.
- It gives a consistent representation of VLE, LLE and heat of mixing
if one accepts to make all parameters temperature dependent .
The main problems (disadvantages) encountered .in the application of the
model are:
- the two parameters TIP and 721 are strongly correlated, when
the maximun value of gE is relatively low.
- There are problems of multiple solutions : it is possible to find
several sets of parameters which will represent equally well given sets of
data. There are also sometimes many solutions for mutual solubilities of
two components for a given set of parameters. In that case JACQ and
ASSELINEAU (19831 did provide a method to select the single physical
solution.
- The model cannot be used to assess the quality of experimental data
using the maximum likelihood principle to obtain the error in measurements
from very precise measurements. In this case, polynomial expression of the
excess GIBBS energy, like REDLICH-KISTER (19481 equation, should be used as
recommended by PENELOUX and coworkers (PEEIELOIJX et al., 19761.
- The third parameter o12 which appears in the exponential cannot be
fitted for low values of the maximum of the excess GIBBS energy of a binary
mixture (gEFIAX < 400 J/mole) because it has no influence in that case.
However, a12 is very useful for strongly nonideal mixture especially in
case of phase splitting and should be fitted (figure Il.
- A very precise simultaneous representation of hE and gE is not
always possible. Even if the general trend of variation of gE and hE
versus temperature can be taken care of by the temperature dependence of the
parameters, the shape of both gE and hE versus composition cannot neces-
sarily be represented by an empirical equation with only three temperature

dependent parameters.
- 2 0.6
> “- 0.4
x
2 0.2
-0.2 -I 0 I 2 4 5 6
m ACTIVITY COEFFICIENT FOR SYMMETRIC EPUIMOLAR MIXTURE AS
A FUNCTION OF ACTIVITY COEFFICIENT AT INFINITE DILUTION
- There are limitations of the applicability of the model in the
critical region. Especially, the very flat maximum of the binary miscibility
curve (T vs x1, x2) cannot be represented (NICOLAIDES and ECKERT, 1978).
Although a minority of authors addressed directly the empirical pro-
blems of application of the local composition models and the majority looked
more towards theoretical developments to be reviewed later, there are a num-
ber of models developed using the local composition concept which have limi- tations similar to the NRTL equation. But it is necessary to explain now the common feature to all these models which originate in WILSON (1974) idea to
take into account empirically the local order in solutions according to the
equation.
X.. x. exp (-g.i/RT) J1= J (3) 'ii xi exp (-gii/RT)
with a normalization relation on the local mole fraction which is for a
binary mixture
‘3 + ‘ii = 1 (4)

91
All the family of local compositions equations are obtained by introdu-
cing these local compositiorainto excess functions expressions. It is known
that molecules first neighbours are not randomly distributed but that there
are influences of sizes, shapes and energies of interaction on the distribu-
tion of species around a given molecule. Equation (3) can be considered as a
quantitative assumption on this effect. It has two conspicuous features: non
randomness depend on the difference of energies Sjf-gii only and
not on size, shape, or energy gjj.
The way authors introduce these "local mole fractions" into an
expression of gE is often arbitrary and can be considered
hypothetical. WILSON (1964) introduced them in FLORY's equation.
gE/RT = CXi In (@ii/Xi)
where the local volume fraction oii is given by :
Q.. = vi xii
11 vi xji +v i 'ii
very much
(5)
(61
Without turning yet to theoretical arguments, let us consider what are
the relative empirical advantages of some local composition equations.
The original WILSON equation had two parameters only fgji - gii) and
(Sij-Sjj)' it was unable to represent phase splitting. The three
parameter WILSON equation (SCATCHARD and WILSON, 19641 for a binary
mixture :
gE/RT = C (XI ln(@II/xl) + x2 ln (@&x2) ) (7)
has a third parameter C and is able to represent partially miscible
mixtures. It was shown IRENON and PRAUSNITZ, 19691 that equation (7) can
be obtained by integration with respect to temperature of the NRTL equa-
tion used to represent the excess enthalpy with temperature independent
energy parameters (gji - gii) = ~~~ x RT and C = 1 / o... 1J
It was not originally proposed to extend it to multicomponent mixtures.
The UNIOUAC equation (ABRAklS and PRAUSNITZ, 19751 is another extension
of the original WILSON equation where phase splitting can be obtained by
adding a "combinatorial", size and shape dependent term to a modified three
parameter "residual" WILSON term.

g E- E
- g combinatorial E
+ g residual
with
(8)
gE @i 8.
= c qi xi In - + f C qi xi In $ combinatorial i 'i i
gE residual = i - r. qi xi In (C ej ~~~~ (8.b)
(8.4
The advantage / disadvantage of the UNIQUAC equation is that it has
only two binary parameters T . . and T.. but it requires informa-
tion (not fitted) on size and shapgof molecu es. '1' It has the same other
advantages / disadvantages as NRTL equation.
Many other equations were proposed, some on theoretical arguments (they
will be reviewed later), other mainly an empirical grounds. The idea to add
a NRTL term and a WILSON term was many times visited. HEIL and PRAUSNITZ
(1966) applied such a model to polymer solution even before the NRTL equa-
tion was invented. WANG and CHAO (1983) proposed recently a very similar sum
of terms. ROWLEY and BATTLER (1984) discuss it in relation with simultaneous
representation of hE and gE. My suggestion (a little different from
theirs) is to use NRTL with temperature dependent parameters to represent
hE as a function of temperature and add an empirical "athermal" term to
the proper integral of hE with respect to T.
hE(T,X) dT
T* (3”
This problem was encountered in the development of the UNIOUAC equation
where the combinatorial term is independent of temperature and was
considered by SAYEGH and VERA (1980).
HIRANUMA (1974 - 1981) described a modified form of the three parameter
WILSON equation for multicomponent mixtures.
N gE / RT. = - CCi Xi In (J~I Afj Xj)
He modified it again in 1983 (HIRANUMA, 1983).
To conclude this rapid and incomplete review, it should be emphasized
that the empirical value of all suggestions should be proven by extensive
application to all properties (hE, gE, LLE, VLE) of binary and multicom-

93
ponent mixtures that the of multiple and correlations
parameters should checked for new modification.
is doubtful the' various will succeed improve
significantly the most used local equations WILSON,
and UNIQUAC if they the correlated parameters
T21 ’ ~12 and do not add other parameters.
EXTENSIONS OF THE LOCAL COMPOSITION APPROACH
Before reaching the "theory" underlying the local composition equation,
it is good to consider the extensions of this type of equation to equation
of state, electrolyte solution and group contributions.
Equations of state
The realization that equations of state can be used to represent devia-
tions from ideality in both vapor and liquid phase is not new. (It can be
attributed to VAN OER WAALS at the end of the 19th century). These
methods became popular among engineers with the article of SOAVE (19721
using the REDLICH-KWONG (1949) equation of state for both vapor and liquid
phase. We understood that the equation of state can be used mainly to repre-
sent vapor pressure of pure components by letting the energy parameter a
vary with reduced temperature and adjusting it on saturation pressure of
pure components, actually exactly as wanted. Using the one liquid VAN DER
WAALS model, one represents fairly well the vapor-liquid equilibria of
multicomponent mixture of hydrocarbons and not too polar molecules by
adjusting one or two parameters of interaction between unlike molecules
aij or bij ( SOAVE (19721, PENG and ROBINSON (1976), MATHIAS
(1983), EVELEIN and MOOR (1979) 1. VIDAL (19781 and HURON and VIDAL (1979)
went a step further by introducing expressions of excess GIBBS energy into
the mixing rules. They opened the so-called "symmetric" method of calcula-
tion of VLE to local composition models by demonstrating that, under
limiting conditions, the excess GIBBS energy of the fluid at infinite
pressure is related to the'mixing rule for a , assuming that b is given by
a linear mixing rule for a class of cubic equations of state :
p=% - (-1 (10)
including the REDLICH-KWONG (19491, PENG-ROBINSON (19761, where $, X2 are
non adjustable constants. Recently, SOAVE (1984) introduced a NRTL excess
Gibbs energy at infinite pressure into the mixing rule of a VAN DER WAALS
equation with temperature dependent of parameters. He applied it success-

94
fully to equilibria with polar components.
The more general relation, without taking the limit of infinite
pressure:
gE = RT [ In Q (T, 1
T, PI 1 (111
is not so easy to use because the equation of state is pressure explicit
and the fugacity coefficient are analytic functions of volume, not pressure.
Their contribution has some of the limitations of earlier work by BEHAR
(1970) and CHAUDRON (1973) using an infinite pressure excess GIBBS energy in
the liquid phase. The cubic equations of state do not represent well the
liquid volume, except if a volume translation is applied as described by
RAUZY (1982). Yet it is not known if they can represent well enough the
excess volume, the role of which is important to obtain the contribution of
non ideality at operating pressure from the same at infinite pressure.
MOLLERUP (1981), WHITING and PRAUSNITZ (1982 a.1 introduced density
dependent mixing rules into the attractive part of cubic equation of state
in order to eliminate the shortcoming they found in the rules introduced by
VIDAL and HURON that the second virial coefficient of a mixture is not a
quadratic function of mole fraction when using local composition or any non-
quadratic mixing rule. The P,V,T properties of the gas phase are not too
well represented by cubic equation of state and even the fugacity coeffi-
cients at saturation may be in error when the pressure is high enough,
making the representation of VLE an empirical achievement. It seems that
the use of density dependent NRTL or UNIOUAC mixing rule was not too
successful (MOLLERUP, 1983) with VAN DER WAALS type equations and a nex
formulation is offered by MOLLERUP in the same article: the random-non random
mixture equation. The current orientation seems to be towards either
simpler density dependent local composition (MATHIAS and COPEMAN, 1983),or
equation based on results of computer simulation to be reviewed hereafter.
Group contributions
Another major empirical development of local composition use was the
group contribution models. The principle of these models is very simple, the
excess GIBBS energy is divided into two parts, one (Sl depends on size and
shape of components (configurational), the other (G) (residual) is a group
contribution. One can write for the activity coefficient i:
1nYi = lny: + lnyf (12)

the group contribution
95
term is expressed as :
- Itirk')) (13)
where VKi is the number of structural group K in component i, and 1nI'K
respectively the group contribution calculated in the solution
and in pure liquid ', (,)
calculated with the same equation :
1nCK = f (Xi' T)
where Xi is the fraction of groups, depending of the composition when cal-
culated for the solution.
The ASOG (Analytical Solution of Groups) equation, introduced by
WILSON and DEAL (1962) is based on WILSON equation for lnry . It was
developed by DERR and DEAL (1969), TOCHIGI and KOJIMA (1976). A monograph
was published by the same authors (KOJIMA and TOCHIGI (1979)).
The UNIFAC (UNIQUAC Functional Group Activity Coefficients) equation
published by FREDENSLUND, JONES and PRAIJSNITZ (1975) uses the UNIQUAC equa-
tion to calculate both the shape and group contributions to ln,! . It is
described in a monograph by FREDENSLUND, GMEHLING and RASMUSSEN (1977). The
group of the Institute of Technical Chemistry of the Technical University of
Denmark and of the Technical University of Dortmund publishes updates of the
method with list of parameters for application to VLE (SKJOLD - JORGENSEN
et al (19791, GMEHLING et al (1982), MACEDO et al (1983)), and LLF:
(MAGNUSSEN (1981)).
The sets of parameters are different for VLE and LLE. Empirical
improvements and extensions are constantly taking place (SKJOLD, JORGENSEN
et al, 1980 - 1982, KIKIC et al, 1980, JENSEN et al, 1982), (SANDER et al,
1983, BEN YAIR, 1983).
In both the UNIFAC and ASOG method the configurational term is indepen-
dent of temperature.
VERA applied a"group contribution method derived from ASOG for heat of
mixing, calling it AGSM (Analytical Group Solution Method) (LA1 et al,
1978). Parameters are different from ASOG, he combined both methods in a
Simplified Group Method Analysis (SIGMA) able to represent both types of
data (ASHRAF and VERA, 1980) without combinatorial term lnv; . But the
method was not fully developed and extensively applied (VERA and VIDAL,
19841.

One of the latest development is the use of a simple group contribution
rule based on the NRTL equation in an equation of state (SKJOLO - JORGENSEN,
1984). The second virial coefficient has the correct form because the
BYLTZMAN exponential in the definition of local fraction contain the factor
V . This equation is used to calculate gas solubilities of gases including H2S and CO2 in various solvents. The accuracy of the K values is 15 per cent except when large amounts of water are present.
Electrolytes
The NRTL model was extended to electrolytes by CRUZ and RENON (1978)
and CHEN et al (1982). BALL et al (1985) made a comparison of these models,
using two parameters for representing the osmotic coefficient of solutions
of simple strong electrolytes in water in the range of molality 0 - 6 M.
The results are similar to those obtained with the simpler PITZER equation. The prediction for mixed salts is not quite as good. But the advantage of
the NRTL models is the possibility of treatment of mixtures of electrolytes and non electrolytes (non aqueous solvents, weak electrolytes). Other
extensions are u&er way (SANDER et al 1985).
THEDRETICAL DEVELDPWEMTS
A theoretical framework 'using radial distribution functions comes
naturally to mind in order to explore the role of order in the theory of
fluids. This is even more true when simulation methods (Molecular Dynamics
and Monte-Carlo) or theoretical method offer the possibility to obtain radial distribution functions and thermodynamic properties of mixtures of
molecules interacting through a given intermolecular potential. There are
however difficulties to define local composition and to derive the residual thermodynamic properties from them. Then, the older cell-lattice theories
look a little obsolete in this context although they may still provide use-
ful methods of deriving empirical models if the assumptions usually taken
for granted in building models are modified.
Radial Distribution Function
The number of molecule i in a spherical shell of radius L centered at
the center of a molecule j is :
n.. =- '3
r2 gij(r) dr (15)

97
If Lij is suitably chosen after the first peak of the distribution
function, ni j can be interpreted as the number of molecules i around
central molecule j, the coordination number of j is :
Zj = ; nij 1
The number of interactions ij counted from central molecule i or j
should be equal
Nj nij = Ni nji
this condition is fullfilled if Lij = Lji because gij(r) = gji(r).
The local mole fraction of i in the volume r< Lij around molecule j
is:
n..
'ij = 1J
+n "ij jj
(16)
The configurational energy of a mixture at given volume and temperature
is:
UCoNF (N1,...NC, V, T)
where N = i N is1 i
4,r2uij(r) gij(N1,...NC, V, T, c) dr
(17)
Uij is the potential of interaction between molecules i and j.
The radial distribution functions gij are related to the potentials
ufj(r), they are functions of density, composition and temperature and
cannot be obtained in any simple way -in the general case.
Simulation methods for the interactions of molecules exist. They
provide distribution functions for given Uij(r), composition, density
and temperature, and also the thermodynamic properties. They are very useful
to physicists to find which approximation can be used to solve the equations
of the statistical mechanics relating the radial distribution functions to
the potentials uij. The problem is essentially solved for fluids of hard
sphere, and some rigid hard bodies. Therefore, we can consider that a fair
approximation of the thermodynamic properties of molecular systems at very
high temperature is available and the configurational Helmholtz energy
A~O"F (N l,...NC,V,T) from which all other can be derived is
available by integration of U CONF:

98
TT
AcoNF(NI, . ..NC. V,T)= ACoNF(NI,...NC, V,T) -T /
UCoNF(N,,...NC.V,T)
T=m T* dt (18)
where
ACoNF(N1,...NC, V, T) = A (N1,...NC, V, T) - AIG(N1,...NC, V, T) (19)
The equation of state is readily obtained from ACoNF by :
p (N, v, Tl =t$?,q..NC + y
The excess properties are obtained through :
GE (N1,...NC, p, T)= AE(Nl,.+ v, T) = A coNF(Nl,...Nc, V, T)
- ‘$ ACoNF (ipir, Vxi, Ni) - RT Xxi In xi 1
(20)
(21)
There is no basic difference between the equation of state approach and
the excess properties approach.
Using this formalism, it is clear that properties can be separated into
two terms: altemperature independent term (sometimes called "free volume")
which is 'v_b in VAN DER WAALS or REDLICH-KWONG equations of state and gE(comb) in UNIOUAC , and a temperature dependent term which depends on
attractive forces, the --$$ term in the REDLICH-KWONG-SOAVE equation of state.
Perhaps because the temperature independent term is known only at infi-
nite temperature, errors in this term are not too important empirically (ASHRAF and VERA, 1980), except in the extreme case of solutions of polymers
where molecules differ strongly in size, shape and conformation (the last
property can in this case be temperature dependent and the approach may fail).
RESULTS OF SIWIATIONS
NAKANISHI and his coworkers performed Molecular Dynamics (MD) and Monte
Carlo (MC) calculations for liquid mixtures of molecules interacting
according to the LENNARD-JONES potential. They investigated strong size and
energy differences between molecules.

The first MD results (TOUKUBO and NAKANISHI, 19761 clearly show that
the number of solvent molecules around a solute molecule vary with the
solute solvent attractive energy parameter E 12 (simulation of 108 sol-
99
vent molecule around 1 solute molecule). The first peak of the distribution
function is higher and positioned further away if e 12 is larger. The results are for solute and solvent molecules of same size, a number density
of 0.75 and kT/cll=l(typical liquid State). They extended this work to molecules differing in size (TOUKUBO and and NAKANISHI, 1977 a.) and in
size and energy (NAKANISHI et al, 1978). The results are given in the following table 1.
"12 ?2
Position of maximum Height of Coordination
of first peak first peak number at
(r/u) r = 1.41 o
?l 0.5 El1 1.07 2.27 9.2
o11 %l 1.09 2.52 9.6
0.5 ;; 2 ?l
1.13 3.36 10.4
?l 0.83 2.84 2
Q El1 1.57 2.26 0.5 “11 0.5 El1 0.81 2.31 2
Tl 2 ?l 1.65 2.72
Table 1: Results of MD calculations of NAKANISHI et al.
The radial distribution is shown in figure 2. The number of solvent mole- cules within the distance r of the solute is also plotted, calculated accor-
ding to equation (15). It depends strongly on distance r. This coordination
number calculated for r >1.136 increases with increasing energy ~~~~ It
does not change any more with ~12 if r>1.706.
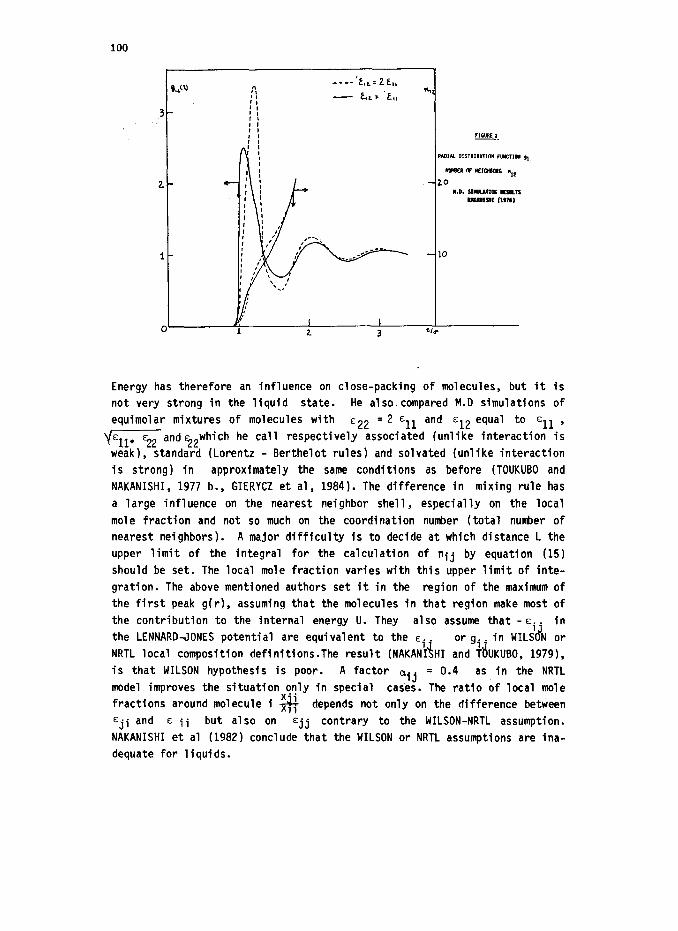
100
Energy has therefore an influence on close-packing of molecules, but it is
not very strong in the liquid state. He also compared M.D simulations of
equimolar mixtures of molecules with c22 = 2 EII and EI~ equal to EII ,
-and?2 which he call respectively associated (unlike interaction is
weak), standard (Lorentz - Berthelot rules) and solvated (unlike interaction
is strong) in approximately the same conditions as before (TOUKUBO and
NAKANISHI, 1977 b., GIERYCZ et al, 1984). The difference in mixing rule has
a large influence on the nearest neighbor shell, especially on the local
mole fraction and not so much on the coordination number (total number of nearest neighbors). A major difficulty is to decide at which distance L the
upper limit of the integral for the calculation of nij by equation (15)
should be set. The local mole fraction varies with this upper limit of inte-
gration. The above mentioned authors set it in the region of the maximum of
the first peak g(r), assuming that the molecules in that region make most of
the contribution to the internal energy U. They also assume that -E.. in
the LENNARD-JONES potential are equivalent to the e.. or gi. in WILS N 'd or NRTL local composition definitions.The result (NAKAN\iHI and TdUKUBO, 19791,
is that WILSON hypothesis is poor. A factor oij = 0.4 as in the NRTL
model improves the situation only in special cases. The ratio of local mole fractions around molecule i g depends not only on the difference between
eji and E ii but also on ejj contrary to the WILSON-NRTL assumption.
NAKANISHI et al (19821 conclude that the WILSON or NRTL assumptions are ina-
dequate for liquids.

101
The excess Gibbs energies obtained have the features of the experimen-
tal values for strongly non ideal mixture : it goes from large negative
(solvationl to large positive (association) with phase splitting in that
last case. For spherical molecules of equal size the excess GIBBS energy is
always nearly symmetrical (NAKANISHI et al, 19821. This is in contradiction
with NRTL, WILSON and other local composition models which attribute
dissymmetry of excess functions to differences in energy parameters.
Increase in size ratio between molecules makes the excess energy more endo-
thermic and dissymmetrfc, assuming the geometric rule for E~~(GIERYCZ et al,
1984).
Unfortunately the authors did not give the complete radial distribution
data and it is difficult i!iassEss the accuracy of their conclusion (espe-
cially that the product V . 4 does not vary with bulk composition)
because there is an arbitrary and perhaps inconsistent (as we shall see
later) choice of L in the calculation of number of neighbors.
HOHEISEL and KOHLER (1984) extended the work of NAKANISHI and coworkers
by aiming at better accuracy and studying composition dependence (T = 120 K,
liquid density). Local composition are calculated with the same value of L
IL/d = 1.35 after the decay of the first peak of g). They find that the
ratios g , 2 and g / '3 for molecules of same size are (see figure 3) ,2-,i'
bulk composition dependent 'and very different from the WILSON equation but
close to the quasi-chemical modeirto be described later. From the results
for molecules of different sizes, the local composition is harder to define
because the same value of L is chosen beyond all the first peaks. The local
composition x11 from MD for an equimolar mixture is, in accordance with
perturbation theory, getting alternatively lower and higher than 0.5 as r/o
varies. HOHEISEL and KOHLER conclude that local composition are caused
mainly by packing effects and that WILSON type formulas cannot be used at
all.
.*

102
APPLICATION OF RADIAL DISTRIBUTIDN FUNCTION
LEE, CHUNG and STARLING (1983) remarked that the energies of interac-
tion to be used in WILSON definition of local mole fraction should not be
directly the ~~~ of the LENNARD-JONES potential or any average of uij(r)
but the average of the potential of mean force Wij defined from the
radial distribution function by :
/ Lij
/
L
4 TI r2gij(r) dr = ij
0 0 4 TI r2 exp (- DWij) dr = Vij exp (- BWij)(2B)
with
V ij
= 5 L3 3 ij
But g and therefore W are composition dependent (and temperature and
density dependent as well). This confirms the work of HOHEISEL and KOHLER
(1984) in its conclusion that the ratios :
xij / 2 = > exp [ - fl(iiij - Pi,) ] 'ii 'i ii
(23)
are composition dependent. There remains the problem of the choice of Lij
which should be equal to Lji in order to keep consistency between the
nji as shown above. Another problem is the calculation of thermodynamic
properties : the exact value of the internal energy is generally not
obtained by integration of the product uij gij over the range of r ’ limited the region of the first peak of g, because the term arising from the
second or third peak may be important.
Nevertheless, LEE et al (1983) found that by adjusting parameters in
WILSON equation on results of AE from M.D. simulation by NAKANISHI, the
values of local composition in one case are qualitatively in agreement with
the values obtained from the radial distribution function resulting from the
simulation.
SANDLER (1985) suggests the overcome the problem of integration of the
energy equation by using a square-well potential and integrating over the
range of the potential well. Equation (17) becomes :
UCoNF (N1, . ..NC. V, T) = -+ z ij Nj clj "ij

103
(25)
L ij is selected as the upper limit of the range of the square well
potential. SANDLER explores the low density limit in this case.
gij (Nl,...NC,V ~ m, T, r) = exp(- uij(r)/kT) = exp(eij/kT) (26)
and
N. n.. = - 1J
Vi +o:j (R3 _ 1) eeij'kT (27)
The local mole fractions are in this case given nearly exactly by the
expression proposed by WILSON :
N ij =
xi v.. - N Jj
Yj * exp .-% )
(28)
The internal energy is :
“CONF = _ E. .N.N 1Jlj V
ij 2v ij exp (cij/kT) (29)
which would lead for the attractive term in VAN DER WAALS or REDLICH equa-
tion to :
amixture = C C Xi Xj aij(T) 1 J
(30)
aij(T) = RTNAV Vij (e oij/kT
- 1) /2
This shows that non randomness
(31)
is not negligible at low density and
this is not in contradiction with the quadratic form of the second virial
coefficient. The exact result is valid only for the square well potential
which is perhaps not the best suited potential to calculate low density
properties. In the case of other potentials approximations are needed like
truncation of the integration of Uij in equation (17) and taking average
of the potential over the range of integration.
Another important observation is that the coordination
selected range is composition dependent. For a binary mixture
Z2 q n12 + n22 = r N2
v12 exp(s12/kT) +-Q- V22 eXp(s22/kT) N1
number in the
(32)

104
It is the assumption of causes the violation of cient with non density
of state models.
constant z in the usual local composition model that the quadratic character of the second virial coeffi- dependent local composition mixing rule in equation
'1'
SANDLER (1985) transposes the local composition concept into a more
precise framework, but many problems remain to be solved : validity of
calculation of energy from first neighbor interaction through the truncated
energy equation, choice of the upper limit of integration of g(r) to obtain
number of neighbors. It is clear that more should be learned on the beha-
vior of radial distribution functions by simulation, or application of
theories.
The exclusive dependence of local composition on energy parameters is probably limited to low density. When the structure of the fluid depends
mainly of the shape and volume of interacting particles as it is the case in
the dense fluids and liquids, the local compositions defined by WILSON are
probably the least applicable. Even for molecule of similar size, the effect of energy differences is small in the liquid state, much smaller
than predicted by WILSON if the energy parameter is the potential energy, and in better agreement with the first approximation of GUGGENHEIM (quasi-
chemical theory) which involves exchange energies
Many other authors refer to NAKANISHI simulation results and improve
empirical formulation of local composition concept and its utili>zation in
equations of state and excess Gibbs energy equations. Among the,!latest
published articles, HU, LUDECKE and PRAUSNITZ (1984) present a new empirical
model taking into account the observations reported before : the local composition term in the case of high densities takes into account primarily
the molecular geometry (packing effect) and weakly the energy differences.
The coordination number of molecule i varies with composition,, and local mole fraction are optimized through GUGGENHEIM's method which wai‘ found to
give weaker effect of local order more in agreement with simulation results
at high density. The local mole fraction tends to the value given'by WILSON equation at low density.
A completely new formulation of local composition models will probably
result from the observations made through simulation and theory. The role of the structure of the fluid resulting from size and shape of molecules
will have to be taken into account, without neglecting the effects of density dependence. The more rigourous approach of ,model development using equations (17) and (18) can be used, once the hypothesis on u.. and
1J gij are chosen.

105
CELL OR LATTICE THEORIES
A very large amount of work was devoted to development of cell or
lattice models of solutions using the concept of local composition. First
there was criticism of the equations proposed by WILSON and attempts to
rationalize the derivation of empirical models. The lattice theory was used
to obtain the combinatorial part of gE. Finally, authors used the approach
of GUGGENHEIM to apply or develop "quasi-chemical" models.
All this work will be very shortly and incompletely reviewed here, just
citing a few typical publications.
The most often cited criticism of local composition models (WILSON,
NRTL, UNIDUAC) was written by FLEMR (1976) and MC DERMOTT and ASHTON (1977).
It is based on the balance of interactions 12 counted from cells centered
around molecules 1 and 2 respectively (assuming equal coordination number).
X1 91 = x2 52 (33)
This equation makes inconsistent the WILSON or NRTL definition of local
mole fraction :
x12 _ x1 --- x22 x2
w12 (341
taking into account the definitions :
x12 + x22 = Xl1 + x21 = 1
if W12 is constant it should
models cannot be justified as
(35)
be 1 (random solution) and therefore the
one fluid, lattice or cell models. Or W12
should be composition dependent, (what is confirmed by simulation results),
models can be developed in this way for binary systems as proposed by
PANAYIOTOU and VERA (1980). The way to get out of this difficulty is to assume different numbers of nearest neighbors around molecules 1 and 2, and
these numbers depending on the bulk composition as suggested by radial
distribution function theory.
Derivations of the UNIQUAC model using a lattice model was given by
MAURER and PRAUSNITZ (19781 and discussed by KEMENY and RASMUSSEN (19811,
VERA (1982) and FISCHER (19831,based on the two fluid model where the above
condition (equation 331 is not fulfilled. In connection with the UNIGUAC
model, there were efforts to develop lattice models to obtain the combina-

106
torial entropy of liquid mixtures as reviewed by SAYEGH and VERA (1980).
But in view of the theory developed above and the results of comparison
between simulation results with various model, the quasi-chemical method of
GUGGENHEIM (1952) seems theoretically the most promising. It is based on
the writing of the partition function for a model based on specified
assumptions and constraints. The local compositions are obtained by minimi-
zation. The disadvantage of that method is that it requires solutions of
many implicit equations in the case of multicomponent systems. This advan-
tage isthatit uses only one binary parameter on exchange energy for a binary
mixture.
Modifications of the quasi-chemical models were proposed by RENON
(1972) NAGATA and NAGASHIMA (19761, PANAYIOTOU and VERA (1980, 1981), KNOX,
VAN NESS and HOLLINGER (1984). The approach of GUGGENHEIM can be applied in
a variety of manners depending on the assumption on number of neighbors,
constraints taken into account. It would be interesting to develop such a
model for dense fluids with choices of assumptions making the results of
simulation likely to be obtained : coordination numbers varying with compo-
sition, good description of the effect of structure inspired by considera-
tions on geometrical number of nearest neighbors as discussed by DEITERS
(1982, 1983), SANDLER (1983), EDULJEE (1983). The advantage would be to
take into account the fact that the local composition are very much depen-
ding on the relative size of molecules in the case of dense fluid. The
correction term depending on energies of interaction is provided by the
quasi-chemical theory is small. DEITERS (1982) develops such a term in his
semi-empirical equation of state for fluids.
The method of BARKER (1952) is a group contribution form of the quasi
chemical theory of GUGGENHEIM. It was presented by KEHIAIAN, GROLIER AND
BENSON (1978) and applied to the representation of excess Gibbs energy,
enthalpy and heat capacities of many organic mixtures. KEHIAIAN and co-
workers published many articles of application of that method (T.O.M.
project). The method relies on considerations of the structure of molecules.
The adjustable parameters are group interchange energy parameter. Often it
is not necessary to take into account the non randomness effect which is
perhaps small enough to be developed analytically and explicitly as sugges-
ted by MICKELEIT and LACMAN (1983). The method has not been sufficiently
validated for the engineering applications, especially for the prediction
of the properties of multicomponent systems or liquid-liquid equilibria.
Another group contribution molecular model for liquids mixtures was
developed by GREENKORN, CHAO and coworkers (NITTA et al (19771, CHIEN et al
(19811, KOUKIOS et al (19841).

co
and
107
NCLUSION
The usefulness of the local composition models (WILSON, NRTL, UNIQUAC)
derived group contribution models (ASOG, IJNIFAC) in engineering applica-
tions has been proved. They can be considered as empirical. The unified
approach of using one equation of state for gas and liquid is gaining ground.
We should look for simple models in spite of the apparent complexity of
interactions. There is a need for critical evaluation of any new equation
in comparison with existing ones, based on treatment of data on many systems
including polar, aqueous, non polar components and also reacting systems and
electrolytes, if possible, with all types of equilibria, vapor-liquid,
liquid-liquid, solid-liquid, heat capacities, volumes in a wide range of
conditions. Test on prediction of multicomponent systems properties should
be performed.
There is a need of data.
- Data obtained by simulation in order to establish the theoretical basis of
the models. It is useful to report the whole radial distribution function
and all properties in a wide range of temperature, pressure and composition,
using simple but meaningful potential, with various combinations of parame-
ters and shapes of hard cores.
- Data in the gas phase are needed (experimental data, especially densities
or thermal properties of mixtures in a wide range of pressure) to evaluate
the fugacities at saturation. Phase equilibria which yield properties needed
to model the liquid phase, heat of mixing, excess heat capacity and compres-
sibility of the liquid, are also needed.
Theoretical development for the representation of the thermodynamic
properties of mixtures will probably use more and more the unified approach
deriving all properties from ACoNF (N,V,T) and decomposition into a
temperature independent "repulsive" part and a temperature dependent-part
resulting from attracting forces. In this approach, it will be necessary to
refine the representation of the properties of pure substances including
improvement for the critical region, the repulsive part, the intermediate
density region. But most of the effort is to be devoted to the understanding
of the mixtures, using the results of simulation to further understand the
role of local order and to check the validity of the approximations needed
in the rigorous statistical mechanics equations relating thermodynamic
properties to intermolecular potentials. The role of energy difference in
the local order is especially important at low density and the role of
shape, size and structure in group from interacting molecules especially
important at high density.

108
LIST OF SYHBOLS
C
Cscf
!tiE
gij
gij
)j
K
L ij
"ij
Ni N av
P
qi r
R
T
TR u.. 1J
u
V
vi V Vij
W ij
W ij
xi
'ji
xi z
a.. 'J
yi
Energy parameter in cubic equations of state
Helmholtz energy
Covolume parameter in cubic equations of state
Number of component in mixture
Parameters in WILSON and HIRANUMA equations
Moiar excess Gibbs energy
Radial distribution of molecule i around j
Parameter in NRTL equation
Parameter in NRTL equatian
Molar excess enthalpy
Distrjhution factor in vapor liquid equilibria, ratio of mole
fraction of Component i in vapor and liquid
Truncation distance in integral of equation (15)
Number of molecules i among first neighbors molecules of j
Total number of nearest neighbors molecules around j
Avogadro number
Pressure
Area parameter in UNIQUAC model
Inte,rmolecular distance
Gas constant
Absolute temperature
Reference temperature
Potential of interaction between molecules i and j
Internal energy
Molar volume of mixture
Molar volume of pure component i
Volume,characteristic volume for interaction ij
Potential of mean force
Average of potential of mean force
Mole fraction of component i
LOCal mole fraction of component j around i
Fraction of group i
Coordination number
Coordination number of species j
Parameter in NRTL equation
Activity coefficient of species i
Contribution of group K to activity coefficient
Contribution of group K to activity coefficient from pure substance
group-composition

109
?j
'i
hl' A2 A ij
"ki
'ij
'ij
Qj
Intermolecular energy parameter
Area fraction
Constants in cubic equation of state (10)
Parameter in WILSON and HIRANUMA equations
Number of structural group K in component i
Intermolecular distance parameter
Parameter in NRTL equation
Local volume fraction
SUPERSCRIPT
CdNF
IG
G
S
Configurational property
Ideal gas property
Residual, group contribution term in group contribution model
Size and shape term in group contribution model
SUBSCRIPT
i Component i
REFERENCES
ABRAMS, D.S. and PRAUSNITZ, J.M., 1975. Statistical Thermodynamics of Liquid Mixtures: A new Expression for the Excess Gibbs Energy of Partly or Completely Miscible Systems. A.1.Ch.E. J. 21: 116-128
ASHRAF, F.A. and VERA, J.H., 1980. A Simplified Group Method Analysis. Fluid Phase Equilibria. 4: 211
BALL, F.X., FURST, W. and RENON, H. 1985. Evaluation of Another NRTL Model for Representation and Prediction of Deviation from Ideality in Pure and Mixed Aaueous Electrolvtes: Comparison with CHEN's and PITZER's model. A.1.Ch.E'. J. 31: 392-399 ’
BARKER, J. 1952. Cooperative Orientation Effects in Solutions. J. Chem. Phys. 20: 1526-1532
BEHAR, E., ASSELINEAU, L., RENON, H. 1970. Equilibres liquide-vapeur sous haute pression - Systemes binaires contenant un corps non condensable. Chem. Engng. Sci. 25: 77-85
BEN YAIR, 0. and FREDENSLUND, A. 1983. Extension of the UNIFAC Group Contribution Method for the Prediction of Pure Component Vapor Pressures. Ind. Eng. Chem. Proc. Des. Devt. 22: 433-435
CHAUDRON, J., ASSELINEAU, L., RENON, H. 1973. A New Modification of the REDLICH-KWONG Equation of State Based on the Analysis of a Large Set of Pure Component Data. Chem. Engng. Sci. 28: 839-846

110
CHEN, C.C., BRITT, H.I., BOSTON, J.K. and EVANS, L. B. 1982. Local Composition Model of Excess Gibbs Energy of Electrolyte Systems. A.1.Ch.E. J. 28: 588-596
CHEN, C.H., GREENKORN, R.A., CHAO, K.C. 1981. A Group Contribution Molecu- lar Model of Liquid and Solutions: II Groups and their Interactions in Water and Aqueous Solutions of Paraffins, Ketones and Alcohols. A.1.Ch.E. J. 27: 303
CRUZ, J.C. and RENON, H. 1978. A New Thermodynamic Representation of Binary Electrolyte Solution Nonideality in the Whole Range of Concentration. A.1.Ch.E. J. 24: 817-830
DERR, E.L. and DEAL, C.J. 1969. Analytical Solutions of Groups: Correla- tion of Activity Coefficients through Structural Group Parameters. I. Chem. Eng. Symp. Series 32: 3-40
DEITERS, U.K. 1982. Coordination Numbers for Rigid Spheres of Different Size-Estimating the Number of Next Neighbour Interactions in a Mixture. Fluid Phase Equilibria. 8: 123-129
DEITERS, U.K. 1983. Coordination Numbers for Rigid Spheres of Different Size-Estimating the Number of Next Neighbour Interactions in a Mixture. Fluid Phase Equilibria. 12: 193-197
DEITERS, U.K. 1982. A New Semi-empirical Equation of State for Fluid III. Application to Phase Equilibria in Binary Mixtures. Chem. Eng. Sci. 6: 855-861
EDULJEE, G.H. 1983. Coordination Numbers for Rigid Spheres of Different Sizes Estimating the Number of Next Neighbour Interaction in the Mixture. Fluid Phase Equilibria. 12: 190-192
EVENLEIN, K.A. and MOORE, R.C. Some Natural Gas Systems Using I.E.C. P.D.D. 18: 618-624
FISCHER, J. 1983. Remarks on Fluid Phase Equilibria 10 : 1
1979. Prediction of Phase Equilibria in the SOAVE-REDLICH-KWONG Equation of State.
Molecular Approaches in Liquid Mixtures.
FLEMR, V. 1976. A Note on the Excess Gibbs Energy Equations Based on Local Composition Concepts. Coll. Czech. Conanun. 41: 3347
FREDENSLUND, A., GMHELING, J., MICHELSEN, M.L., RASMUSSEN, P. and PRAUSNITZ, J.M. 1977. Computerized Design of Multicomponent Distillation Columns Using the UNIFAC Group Contribution Method for Calculation of Activity Coefficients. Ind. Eng. Chem. Proc. Res. Devt. 16: 450-462
FREDENSLUND, A., GMEHLING, J. and RASMUSSEN, P. 1977. Vapor-Liquid Equili- bria Using UNIFAC, a Group Contribution Method. Elsevier, AMSTERDAM
FREDENSLUND, A., JONES, R.L., PRAUSNITZ, J.M. 1970. Group Contribution Estimation of Activity Coefficients in Nonideal Liquid Mixtures. A.1.Ch.E. J. 21: 1086-1099
GIERYCZ, P., TANAKA, H. and NAKANISHI, K. 1948 a. Molecular Dynamics Studies of Binary Mixtures of LENNARD-JONES Fluids with Differing Component Sizes. Fluid Phase Equilibria. 16: 241-253
GIERYCZ, P. and NAKANISHI, K. 1984. Local Compositon in Binary Mixtures of

111
LENNARD-JONES Fluids with Different Sizes of Components. Fluid Phase Equilibria. 16: 255-273
GMEHLING, J. RASMUSSEN, P., FREDENSLUND, A. 1982. Vapor-Liquid Equilibria by UNIFAC Group Contribution. Revision and Extension 2. Ind. Eng. Chem. Proc. Res. Devt. 21 : 118
GUGGENHEIM, E.A. 1952. Mixtures. Clarendon Press, Oxford, G.B.
HEIL, J.F. and PRAUSNITZ, J.M. 1966. Phase Equilibria in Polymer Solutions A.1.Ch.E. J. 12: 678
HIRANUMA, M. 1974. A New Expression Similar to the Three Parameter WILSON Equation. Ind. Engng. Chem. Fundamentals 13: 214
HIRANUMA, M. 1981. Significance and Value of the Third Parameter in the WILSON Equation. Ind. Engng. Chem. Fundamentals 20: 25
HIRANUMA, M. 1983. Equation suitable for Estimation of Ternary Liquid- Liquid Equilibria with Binary WILSON Parameters. Ind. Engng. Chem. Fundamentals. 22: 364-366
HOCHEISEL, C. and KOHLER, F., 1984. Local Composition in Liquid Mixture. Fluid Phase Equilibria. 16: 13-34
HU, Y., LUDECKE, D. and PRAUSNITZ, J.M., 1984. Molecular Thermodynamics of Fluid Mixtures Containing Molecules Differing in Size and Potential Energy. Fluid Phase Equilibria. 17: 217-241
HURON, M.J. and VIDAL, J., 1979. New Mixing Rules in Simple Equations of State for Representing Vapor-Liquid Equilibria of Strongly Nonideal Mixtures. Fluid Phase Equilibria. 3: 255-271
JACQ, J. and ASSELINEAU, L., 1983. Binary Liquid-Liquid Equilibria. Multi- ple Solutions for the NRTL Equation. Fluid Phase Equilibria. 14: 185-192
JENSEN, T., FREDENSLUND, A., RASMUSSEN, P., 1982. SUPERFAC: an Extended UNIFAC Model. SEP 8202, Instituttet for Kemiteknik, Lyngby DANEMARK
KEHIAIAN, H.V., GROLIER, J.P.E., BENSON, G.L., 1978. Thermodynamics of Organic Mixtures - A generalized Quasi-Chemical Theory in Terms of Group Surface Interactions. J. Chem. Phys. 75: 1031-1048
KEMENY, S. and RAMUSSEN, P., 1981. A Derivation of Local Composition Expressions from Partition Functions. Fluid Phase Equilibria. 7: 197-203
KIKIC, I., ALESSI, P., RASMUSSEN, P., FREDENSLUND, A., 1980. On the Combi- natorial Part of the UNIFAC and UNIQUAL Models. Can. J. Chem. Engng. 58: 253
KNOX, P.E., VAN NE S, tation of gE and
H.C. and HOLLINGER, H.B., 1984. A Model of Represen- h 8 . Fluid Phase Equilibria 15: 267-285
KOJIMA, K. and TOCHIGI, K., 1979. Prediction of Vapor-Liquid Equilibria by the ASOG Method. KODANSHA-ELSEVIER, Amsterdam.
KOUKIOS, E.G., CHIEN, C.H., GREENKORN, R.A., CHAO, K.C., 1984. A Group Contribution Molecular Model of Liauids and Solutions. Part III. Grouos and Interactions in Aromatics, Cycloparaffins, Ethers, Amines and Their' Solutions. A.1.Ch.E. J. 30: 662

112
L I, T.T., H a
NGUYEN, H.T.D., VERA, J.H., RATCLIFF, C.A., 1978. Prediction of of Liquid Mixtures Containing Alkane, Chloroalkane and Alcohols by the
AGSM. Can. J. Chem. Engng. 56: 358
MC DERMOTT, C., ASHTON? N., 1978. Note on the Definition of Local Composi- tion. Fluid Phase Equilibria. 1: 33
MACEDO, E.A., WEIDLICH, U., GMEHLING, J.C., RASMUSSEN, P., 1983. Vapor Liquid Equilibria by UNIFAC Group Contribution. Revision and Extension 3. Ind. Eng. Chem. Proc. Res. Devt. 22: 676-678
MAGNUSSEN, T., RASMUSSEN, P., FREDENSLUND, A., 1981. UNIFAC Parameter Table for Prediction of Liquid Liquid Equilibria. Ind. Eng. Chem. Proc. Des. Devt. 20: 331-339
MATHIAS, P.M., 1983. A Versatile Phase Equilibrium Equation of State. Ind. Eng. Chem. Proc. Res. Devt. 22: 385-391
MATHIAS, P.. and COPEMAN, T.W., 1983. Extension of the PENG-ROBINSON Equa- tion of State to Complex Mixtures. Evaluation of the Various Forms of the Local Composition Concept. Fluid Phase Equilibria 13: 91-108
MAURER, G. and PRAUSNITZ, J.M., 1978. On the Derivation and Extension of the UNIQUAC Equation. Fluid Phase Equilibria 2: 91-99
MICKELEIT, M., LACMAN, R., 1983. Statistical Mechanics and Thermodynamics Properties of Liquid Multicomponent Mixtures: Part 1 - The TAYLOR seriees for Quasichemical Equilibrium of Ternary Mixtures. Fluid Phase Equilibria 12: 201-206
MOLLERUP, J., 1981. A Note on Excess Gibbs Energy Models, Equations of State and the Local Composition Concept. Fluid Phase Equilibria 7: 121-138
MOLLERUP, J., 1983. Correlation of the Thermodynamic Properties of Mixtures Using a Random Mixture Reference State. Fluid Phase Equilibria 15: 189-207
NAGATA, I., NAGASHIMA, M., OGURA, M., 1975. A Comment on an Extended Form of the WILSON Equation in Correlation of Partically Miscible Systems. J. Chem. Engng. of Japan 8 (5): 406-408
NAGATA. I, NAGASHIMA, M., OGURA, L., 1959. An Extension of the WILSON Equa- tion to Partially Miscible Systems. Proc. Des. Devt. 14: 500
NAGATA. I., NAGASHIMA, M., 1976. Correlation and Prediction of Vapor-Liquid and Liquid-Liquid Equilibria. J. Chem. Eng. Japan 9: 6-11
NAKANISHI, K., TOUKUBO, D., WATANABE, N., 1978. Molecular Dynamics of LENNARD-JONES Liquid Mixtures. IV - Further Calculations on the Behavior of one Different Particle as a Model of Real Fluid Systems. J. Chem. Phys. 88: 2041
NAKANISHI, K. and TOUKUBO, k., 1979. Molecular Dynamics Studies of LENNARD- JONES Liquid Mixtures. V. Local Composition in Several Kinds of Equimolar Mixtures with Different Combinary Rules. J. Chem. Phys. 70 (121: 5848
NAKANISHI, K., OKAZAKI, S., IKARI, K., HIGUSHI, T. and TANAKA, H., 1982. Free Energy of Mixing, Phase Stability and Local Composition in LENNARD- JONES Liquid-Mixtures. J. Chem. Phys. 76: 629
NICOLAIDES, G.C. and ECKERT, C.A., 1978. Optimal Representation of Binary

113
Liquid Mixture Nonidealities. Ind. Eng. Chem. Fundamentals 17: 331
NITTA, T., TUREK, E.A., GREENKORN, R.A., CHAO, K.C., 1977. A Group Contri- bution Molecular Model of Liquids and Solutions A.1.Ch.E. J. 23(2): 144
PANAYIOTOU, C., VERA, J.H., domness in Liquid Mixtures.
1980. The Quasi Chemical Approach for Nonran- Expressions for local Surfaces and Local Compo-
sition with an Application to Polymer Solution. Fluid' Phase Equilibria 5: . 55-80
PANAYIOTOU, C., VERA, J.H., 1981. Local Composition and Local Surface Area Fraction. A Theoretical Discussion. Can. J. Chem. Eng. 59: 501
PENELOUX, A., DEYRIEUX, R., R., CANALS, E., NEAU, E., 1976. The Maximum Likelihood Test and the Estimation of Experimental Inaccuracies. Applica- tion to Data Reduction for Liquid-Vapor Equilibrium. Journal de Chimie Physique 73: 706
PENG, D.Y. and ROBINSON, P.B., 1976. A New Two Constant Equatio of State. Ind. Engng. Chem. Fundam. 15: 59-64
RAUZY, E., 1982. Les methodes simples de calcul des dquilibres liquide- vapeur sous pression. These, Universite Aix Marseille II
REDLICH, O., KWONG, J.N.S., 1949. On the thermodynamics of Solutions. V. An Equation of State. Fugacities of Gaseous solutions. Chem. Reviews 44: 233-244
REDLICH, O., KISTER, A.T., 1948. Algebraic Representation of Thermodynamics Properties and the Classification of Solutions. Ind. Eng. Chem. 40: 345-348
RENON, H. 1974. Extension de la theorie quasi-chimique I des melanges de mol&zules de tailles et de formes diffelrentes. C.R. Acad. Sci. Paris 278: 13147
RENON, H. and PRAUSNITZ, J.M., 1968. Local Composition Thermodynamic Excess Functions for Liquid Mixtures. A.1.Ch.E. J. 14: 135-144
RENON, H. and PRAUSNITZ, J.M., 1968. Liquid-Liquid and Vapor-Liquid Equilibria from Binary and Ternary Systems with Dibutylketone, Dimethyl Sulfoxide, n-Hexane and l-Hexane. IEC Proc. Res. Devt. 7: 221
RENON, H. and PRAUSNITZ, J.M., 1969. Estimation of Parameters for the NRTL Equation for Excess Gibbs Energes of Strongly Nonideal Liquid Mixtures. Ind. Engng. Chem. Proc. Res. Devt. 8: 413
RENON, H. and PRAUSNITZ, J.M., 1969. Derivation of the Three-Parameter WILSON Equation for the Excess Gibbs Energy of Liquid Mixtures. A.1.Ch.E. J. 15: 785
ROWLEY, R.C. and BATTLER, J.R., 1984. Local Composition Models and Prediction of Binary Liquid-Liquid Binodal Curves from Heat of Mixing. Fluid Phase Equilibria 18(21: 111
SANDER. B., JgGENSEN, S.S., RASMUSSEN, P., 1983. Gas Solubility Calculation I - UNIFAC. Fluid Phase Equilibria 11: 105-126
SANDER, B., RASMUSSEN, P., FREDENSLUND, A. 1985. Personal Communication.

114
SANDLER, S.I., 1985. The Generalized Van I - Basic Theory. Fluid Phase Equilibria
SANDLER, S.I., 1985. The Generalized Van I - Basic Theory. Fluid Phase Equilibria
SAYEGH, S.G. and VERA, J.H. 1980. Review
der Waals Partition Function 12: 189-190
der Waals Partition Function 19(31
Paper: Lattice Model Expressions for the Combinatorial Entropy of Liquid Mixtures. A Critical Discussion. Chem. Engng. J. 19: l-10
SCATCHARD, G. and WILSON, C.M., 1964. Vapor-Liquid Equilibrium 13. The System Water-Butyl-Glycol from 5 to 85" C. J. Am. Chem. Sot. 86: 133
SKJOLO-JBGENSEN, S., RASMUSSEN, P., FREDENSLUND, A., 1982. On the Concentration Dependence of the UNIQUAL / UNIFAC Models. Chem. Engng Sci. 37: 99-111
SOAVE, G., 1972. Equilibrium Constants from a Modified REDLICH-KWONG Equation of State. Chem. Engng. Sci. 27: 1197
SOAVE, G. 1984. Improvement of the Van der Waals Equation of State. Chem. Engng. Sci. 39: 357-369
TOCHIGI, K.K., and KOJIMA, K.K., 1976. The Determination of Group WILSON Parameters from Activity Coefficient Obtained by Ebulliometry. J. Chem. Engng. Japan. 9: 267-273
TOUKUBO, K. and NAKANISHI, K., 1937. Molecular Dynamics Studies of LENNARD-JONES Liquid Mixtures. I - Role of Attractive Term in Behavior of One Different Particle. J. Chem. Phys. 65: 1937
TOUKUBO, K.K., NAKANISHI, K.K. and WATANABE, N., 1977 a. Molecular Dynamics Studies of LENNARD-JONES Liquid Mixtures. II - Mass and Size Dependance in the Behavior of one Different Particle. J. Chem. Phys. 67: 4162
TOUKUBO, K.K., and NAKANISHI, K., 1977 b. Molecular Dynamics Studies of LENNARD-JONES Liquid Mixtures. III of Several Equimolar Mixtures.
- Structural and Thermodynamic Property Berichte der Bunsen-Gesellschaft ftir
Physikalische Chemie 81: 1046
VERA, J.H., 1982. On the Two-Fluid Local Composition Expressions. Fluid Phase Equilibria 8: 315-318
VERA, J.H. and VIDAL, J., 1984. An Improved Group Method for Hydrocarbon-Hydrocarbon Systems Arising from a Comparative Study of ASOG and UNIFAC. Chem. Engng. Sci. 39(4): 325-661
VIDAL, J., State.
1978. Mixing Rules and Excess Properties in Cubic Equations of Chem. Engng Sci. 33: 787-791
WANG, W. and CHAO, K.C., 1983. The Complete Local Concentration Model Activity Coefficients. Chem. Engng. Sci. 38: 1483
WHITING, W.B. and PRAUSNITZ, J.M., 1983. Equations of State for Strongly Nonideal Fluid Mixtures: Application of Local Composition towards Density-Dependent Mixing Rules. Fluid Phase Equilibria 9: 119-147
WILSON, G.M., 1964. Vapor Liquid Equilibrium. XI - A New Expression for the Excess Free Energy of Mixing. J. Amer. Chem. Sot. 86: 127
WILSON, G.M. and DEAL, C.H., 1962. Activity Coefficient and Molecular Structure Activity Coefficients in Changing Environment - Solutions of Group. Ind. Engng. Chem. Fundamentals. 1: 20-23