mutational analysis of human thioredoxin reductase 1: effects on
TRANSCRIPT

1
Manuscript M2:02286 (revised) April 10, 2002
Mutational analysis of human thioredoxin reductase 1: Effects on p53 mediated gene expression and Interferon and
Retinoic acid induced cell death
Xinrong Ma, Junbo Hu1, Daniel J. Lindner2, and Dhananjaya V. Kalvakolanu*
Greenebaum Cancer Center, Department of Microbiology & Immunology, Molecular and Cellular Biology Program, University of Maryland School of Medicine, Baltimore, MD 21201. Running title: thioredoxin reductase in tumor suppression
* Corresponding author
Phone: 410-328-1396 Fax: 410-328-1397 E-mail: [email protected] 1 Contributed Equally to this work
Current address: 1 Department of Surgery, Tongji Medical Center, Wuhan, P.R. China 2Taussig Cancer Center, Cleveland Clinic Foundation, Cleveland, OH 44195
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on April 12, 2002 as Manuscript M202286200 by guest on N
ovember 24, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
Abstract:
The IFN-β and all-trans retinoic acid (RA) combination suppresses tumor growth by inducing
apoptosis in several tumor cell lines. A genetic technique permitted the isolation of human
thioredoxin reductase (TR) as a critical regulator of IFN/RA induced cell death. Our recent
studies have shown that TR1:Trx1 regulated cell death is effected in part through the activation
of p53 dependent responses. To understand its death regulatory function, we have performed a
mutational analysis of TR. Human TR1 has 3 major structural domains, the FAD binding (FAD),
NADPH binding (NBD) and an Interface domain (ID) Here, we show that the deletion of the C-
terminal interface domain results in a constitutive activation of TR dependent death responses
and promotes p53 dependent gene expression. TR mutant without the ID still retains its
dependence on Trx for promoting these responses. Thus, our data suggest that TR-ID acts as a
regulatory domain.
Abbreviations used:
GRIM: Gene associated with Retinoid-IFN induced mortality; IFN: Interferon; RA: all trans
retinoic acid; SRB: Sulforhodamine B; STAT: Signal transducing activator of transcription; TR:
thioredoxin reductase; Trx: thioredoxin.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

3
Introduction:
Interferons (IFN) exert antitumor effects by inducing the expression of a number of
cellular genes using the Janus tyrosine kinase (JAK)-Signal Transducing Activator of
Transcription (STAT) pathways (1,2). A higher susceptibility of IFN-γ receptor-/- and STAT1 -/-
mice to chemical carcinogenesis than their wildtype counterparts, and a failure of syngeneic mice
to reject the IFN-γ receptor-/- and STAT1 -/- tumors underscore the importance of IFNs in tumor
growth control (3). Similarly, two IFN regulated transcription factors, IRF-1 and IRF-8 (ICSBP),
act as tumor growth suppressors (4,5) since mutations in these genes cause leukemias (6,7). In
rodent cells IFN-stimulated transcription factors of the p200 family control cell cycle progression
(8,9). IFNs also downregulate c -myc expression, activate tumor suppressor pRb and inhibit E2F
to inhibit cell cycle progression in human cell lines (10-12). Although a great deal is known
about IFN signaling pathways and the transcription factors involved, very little is known about
the gene products that mediate the tumor suppressive pathways employed by IFNs. Additionally,
despite their beneficial therapeutic effects in certain leukemias, IFNs are marginally active in the
therapy of solid tumors (13,14). Clinical and experimental models have shown that combination
of IFNs with retinoids, a class of vitamin A derivatives, yields a highly effective growth
suppressive effect in several solid tumors (15-17). All trans Retinoic Acid (RA), a vitamin
metabolite, inhibits the growth of promyelocytic leukemias, and teratocarcinomas in vitro (18).
Two structurally similar but genetically distinct classes of transcription factors, the retinoic acid
receptor (RAR) and the retinoid X receptor (RXR) mediate retinoid induced growth suppression
(19). One such receptor RARβ appears to be a tumor suppressor (20,21). However, the identity
of retinoid-regulated growth inhibitory gene products is unknown.
Our earlier studies showed that the IFN−β/RA combination, but not single agents, causes
cell death in vitro and suppresses tumor growth in vivo (17). Using a genetic technique we have
identified several Genes associated with Retinoid-IFN induced Mortality (GRIM) recently (22).
Human thioredoxin reductase 1 (TR1), a redox regulatory enzyme (23,24), was identified as one
of the GRIMs (22). Subsequent studies have shown that TR and its substrate Trx activate cell
death by modulating the activity of caspase-8 and tumor suppressor p53 (25-27). To further
understand the structure-function relationship of TR to these processes, we have performed a
mutational analysis. A comparative analysis of primary structure of this enzyme with other redox
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

4
enzymes led to the assignment of 3 major modules, the FAD, NBD and ID (23,24). While the
FAD and NBD are critical for redox function and well conserved among the TRs from all
sources, the ID is unique to mammalian TR. ID has been suggested to act as a dimerization
surface to generate functional TR (23,24). However, the functional significance of ID has not
been fully appreciated. Here we show that removal of ID enhances death stimulatory activity of
TR and p53 dependent gene expression.
Materials and Methods:
Reagents: Restriction and DNA modifying enzymes (NE Biolabs); G418 Sulfate, IPTG and
Lipofectamine plus (Life Technologies); nylon membranes, ECL reagents and horseradish
peroxidase coupled to anti-rabbit or anti mouse antibodies (Amersham Pharmacia Inc); human
IFN−βser (Berlex Inc.); and mouse monoclonal antibodies against actin, flag epitope (Sigma Inc),
p53 (Oncogene Science Inc) and myc-epitope (Zymed Inc) were employed in these studies.
Rabbit polyclonal antibody against the C-terminal peptide ofTR1 was described earlier (22).
Fresh stocks of all-trans retinoic acid (Sigma) were prepared in ethanol and added to cultures
under subdued light.
Cell culture: MCF-7 cells were cultured in phenol red free EMEM supplemented with 5%
charcoal stripped fetal bovine serum (CSFBS) and 10-11M estradiol during treatment with IFN-β
and all trans retinoic acid (RA). MCF-7 cells stably transfected with wildtype and mutant forms
of Trx were described earlier (25). The mutant Trx bears serine residues in the place of cysteines
at positions 32 and 35 (28). MCF-7 cells stably transfected with mammalian expression vector
pCMV-neo (MCF-7 neo) or the same vector with the E6 gene of human papilloma virus type-16
(MCF-7 E6) were provided by A.J. Fornace Jr., National Cancer Institute, Bethesda, MD (29).
The loss of p53 function in MCF-7 E6 cells has been demonstrated in earlier (29,30). These cells
were grown in phenol red free media 24h before treatments were initiated. DLD human
carcinoma cells, which lack the endogenous p53, were a gift from Bert Vogelstein, Johns
Hopkins University Oncology Center, Baltimore, MD.
Plasmids: Mammalian expression vector, pCMV-flag bears a flag epitope sequence in its
multiple cloning region. An in frame insertion of any cDNA lacking the N-terminal methionine
into this vector generates the protein with a flag epitope tag at the N-terminus. Mammalian
expression vector pCXN2-myc vector carries a C-terminal myc epitope tag. Cloning of an insert
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

5
without a “stop” codon between the 5’ EcoRI and 3’ KpnI sites of this vector permits the
addition of a myc-tag to the expressed protein. p53-luc carries p53 binding sites (8X) cloned
upstream of SV40 early promoter in the pGL3 basic vector (Promega Inc) was described earlier.
Wildtype and mutant p53 (R175 H) cloned in the pCMV expression vector were described
elsewhere (31,32). A luciferase reporter driven by human Bax promoter, Bax-Luc, was provided
by Carol Prives, Columbia University, New York, NY (33).
Generation of TR mutants: Gene specific primers bearing specific restriction enzyme cutting
sites (for facilitating the sub-cloning) and AmpliTaq gold enzyme (Roche-Perkin Elmer) were
employed in PCR for generating TR mutants. All primers used in this study are listed in table 1.
Two separate sets of primers were used for generating myc- and flag- epitope tagged constructs.
Construction of a myc-tagged full length TR was described in our earlier publication (26). At
first the myc-tagged mutants were generated, which served as templates for generating flag-
tagged mutants. Twelve cycles of PCR was performed to avoid the emergence of unwanted
mutants, owing to polymerase errors. Mutants were sequenced to verify their identity.
Mutant ∆-FAD was amplified using myc-primers -2 and -3 with wildtype TR cDNA as
template. ∆-ID was generated using myc primers -1 and -4. For generating the ∆-NBD mutant, 4
primers were used. The myc-primer 1 and ∆-NBD primer 1were used for amplifying the FAD
domain. The myc-primer2 and ∆-NBD primer2 were used for amplifying the ID region.
Oligonucleotides used for the amplification of FAD and ID regions have a BamHI site at their 3’
and 5’ ends, respectively. The myc primers -1 and -2 bore EcoRI and Kpn I sites. The FAD
product was digested with EcoRI and BamHI; and the ID product was digested with BamHI and
KpnI and then purified. The products were combined with pCXN2-myc vector pre-digested with
EcoRI and KpnI in a three-way ligation reaction. The final ∆-NBD construct has two non-
template derived aminoacids, a glycine and a serine, at the junction of FAD and ID, due to a
BamHI site present in amplifying primers. Constructs f -TR and f -∆-NBD were generated using
flag-primers -1 and -2, with the corresponding myc-tagged constructs as templates. The f-∆-ID
and f -∆-FAD mutants were generated using flag primer pairs –1 and –4 and flag primer pairs –3
and –2, respectively. f-tagged truncated mutants of ∆-ID were generated using the indicated
reverse primers and flag primer-1, with wildtype TR as template. Point mutants were generated
using ∆-ID80 as a template. For example, for generating the T193A mutant a reverse and a
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

6
forward primer bearing the same mutation were used in a 3 step PCR. Flag primer 1 and reverse
primer (mutagenic) were used in first PCR reaction. In the other forward primer (mutagenic) and
∆-ID-80 R primer were used for the second PCR. Purified PCR products from 1 st and 2 nd PCR
were mixed, denatured and annealed. This mixture now served as template for flag primer 1 and
∆-ID-80 R primer to generate the final product. The final PCR product was digested with EcoRI
and KpnI and ligated to pCMV-flag. The other mutants were generated in a very similar manner,
using appropriate mutant primers.
Cell growth assay: Cells (2000/well) were seeded into 96-well plates. Drugs were added and
growth was monitored using a colorimetric assay (34). Each group of treatments had 8 replicates.
Cells were fixed with 10% trichloroacetic acid at the end of the experiment and stained with
0.4% Sulforhodamine B (Sigma). The bound dye was eluted with 100 µl of Tris-HCl (pH 10.5)
and absorbance was monitored at 570 nm. One plate was fixed with TCA, 10h after plating.
Absorbance obtained with this plate was considered as 0% growth. Absorbance obtained with
untreated cells was considered as 100% growth. An i ncrease and decrease of A570 values in the
experimental wells relative to the 0% value indicates cell growth and death, respectively.
Death assays: Cell death was determined using Annexin-V binding assays. Following treatment
with IFN/RA, cells were stained using a commercially available kit (Trevigen Inc) per
manufacturer’s recommendation. FITC positive cells were considered as apoptotic and were
quantified using flow cytometry.
Gene expression analyses: Transfection, β-galactosidase, luciferase assays, SDS-PAGE,
Electrophoretic mobility shift analyses (EMSA) were performed as described in our earlier
publications (25-27). Total amount of transfected DNA (1.0 µg) was kept constant by adding
corresponding empty expression vector DNA, where required. In general, 0.2 µg of luciferase
and 0.2µg of TR mutant were cotransfected. CMV β-galactosidase reporter (0.1 µg) was used as
an internal control for normalizing variations in transfection efficiency. Electrophoretic mobility
shift assay with p53 oligonucleotides was performed as described earlier (25-27).
Western blot analysis: Equal quantities of cell extracts were separated on 12% SDS-PAGE and
western blotted onto nylon membranes. Specific first antibodies were incubated with the blots as
described in our previous publications (22). These blots were washed and incubated with an
appropriate second antibody tagged with horseradish peroxidase. Protein bands were visualized
using a commercially available enhanced chemiluminescence (ECL) kit (Amersham Inc).
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

7
Results:
Generation of TR mutants: We have shown earlier that over expression of a catalytically
inactive TR1or a redox inactive Trx1 in human tumor cell lines imparts resistance to IFN/RA
induced cell death (25,26). In contrast, a wildtype TR1 and Trx1 promoted cell death under the
same conditions. However, the role of other TR domains in cell growth control is unknown. To
understand the relationship between structural domains of TR and cell death regulation, we have
generated new TR mutants. Using PCR we generated 3 mutants, ∆-FAD, ∆-NBD and ∆-ID, each
lacking the FAD binding, NADPH binding and the interface domains, respectively. Since no
domain specific antibodies are available for TR1, we have cloned the PCR products into
mammalian expression vectors, pCMV-flag or pCXN2-myc. Proteins expressed from pCMV-
flag and pCXN2-myc will bear a N-terminal flag- and a C-terminal myc- epitope tag,
respectively. Wildtype TR produces a polypeptide with a theoretical Mr of 54.7 kDa. However, it
migrates as a ~58 kDa protein on SDS-PAGE due to post-translational modifications. The ∆-
FAD, ∆-NBD and ∆-ID constructs are expected to yield 40.1, 33.2 and 27.6 and kDa peptides,
respectively. The mutants were transiently transfected into human breast carcinoma cell line
MCF-7 to check for the production of proteins of proper size. Cell lysates were prepared and
western blotted using either Flag- or myc- epitope specific monoclonal antibodies. Indeed, all
mutants can be expressed to a comparable level upon transfection (Figure 1B and C). Both tags
were used only to demonstrate that either N-terminal or C-terminal tags have no effect on protein
function. Furthermore, the pCXN2-myc has a G418 resistance marker for selecting the stably
transfected cells, which is absent from pCMV-flag. The flag tag is shorter than the myc-tag by
about 8 aminoacids.
Removal of ID enhances the cell death effects of TR: To test the effect of TR
mutants on cell growth, we first generated cell lines that stably express them. Since the pCXN2-
myc vector also carries a G418r marker it permits the selection of stable transfectants, we have
used the myc-tagged constructs for this purpose. As observed earlier, transfection of a wildtype
TR resulted in the formation of fewer colonies than the vector. The ∆-FAD construct gave rise to
marginally more colonies than the control vector transfected cultures indicating its inhibitory
effect on cell death. The ∆-NBD mutant produced comparable number of colonies to that of
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

8
vector. Interestingly, the ∆-ID mutant yielded 75% and 50% fewer G418r colonies than the
vector and TR transfected cultures, respectively (Fig. 2A). The G418r colonies in each group
were pooled and used in the experiments described below to avoid a clonal bias. In the next
experiment the effect of TR mutants on cell growth was determined using a colorimetric assay
(34), where cell growth is quantified on the basis of the amount of sulforhodamine B dye bound
to cells (Fig. 2B). This method correlates well with Coulter counting and determination of cell
number. While the ∆-FAD expressing cells grew slightly faster than the vector transfected
cultures, the TR and ∆-ID transfectants grew relatively slower. Expression of ∆-ID caused
significantly slower growth compared to TR. Growth of ∆-NBD transfectants was comparable to
that of vector-transfectants. These observations suggest that removal of ID converts TR into a
significantly more potent inhibitor of cell growth. Similar results were obtained with f -tagged
TR mutants (data not shown).
To demonstrate that these differential effects on cell growth were due to the expression of
mutants in the stable transfectants, cell lysates were examined for the expression of transgenes
by western blot analysis with myc-tag specific antibodies (Fig. 2C). Although all mutants
expressed in the transfectants, Wildtype TR and ∆-ID expressed to a lesser extent. In fact, the
expression of ∆-ID was lost with further passages of the transfectants (data not shown),
indicating its strong anti-cellular effects. To demonstrate a functional relationship between cell
death and the mutant expression, the stable transfectants were exposed to IFN/RA combination
(IFN/RA) and then stained with FITC-labeled annexin-V, a maker for apoptotic death (35). A
higher FITC positive staining indicates higher apoptosis. FITC positive cells were quantified
using flow cytometry (Fig. 2D). The TR and ID transfectants exhibited a significantly higher
sensitivity to IFN/RA induced cell death compared to the other mutants. ∆-ID expressing cells
became FITC 2 -2.5 fold higher than TR expressing ones. The FAD mutant acted as an inhibitor
of apoptosis, because cells expressing it were less FITC-positive, compared to the vector
expressing ones. Together these data indicate that ID of TR attenuates its pro-apoptotic effects.
Expression of ∆∆ -ID has no effect on endogenous TR and p53: To rule out a
possibility that the expression of TR or ∆-ID somehow altered the endogenous TR levels to
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

9
mediate these differential effects, we next determined the levels of endogenous TR protein levels
by western blotting with antibodies specific for TR. Since only ∆-ID exhibited hyper death
stimulatory effects, we have selected it for further studies and compared its effects to full length
TR. As shown in Fig.3A neither the ∆-ID nor wildtype TR had an effect on the endogenous TR,
since its level was comparable between the vector and mutant transfected cells. In wildtype TR
transfected cells, a slow migrating band above the TR band was detected. It corresponds to the
TR protein derived from the transgene and migrates slower owing to the presence of an epitope
tag. This band is absent in the vector or ∆-ID expressing cells. Since the TR antibody was
directed against a peptide in the C-terminus, the ∆∆ -ID could not be detected.
We have shown earlier that the cell death effects of TR were in part due to its ability to
modulate tumor suppressor p53 dependent responses (25-27). Therefore, we examined the
possibility that a rise in endogenous p53 levels of mutant transfectants relative to vector
expressing cells occurred. Expression of ∆-ID or wildtype TR did not significantly affect p53
levels as revealed by a western blot analysis with an antibody that specifically detects a wildtype
p53 protein (Fig. 3B).
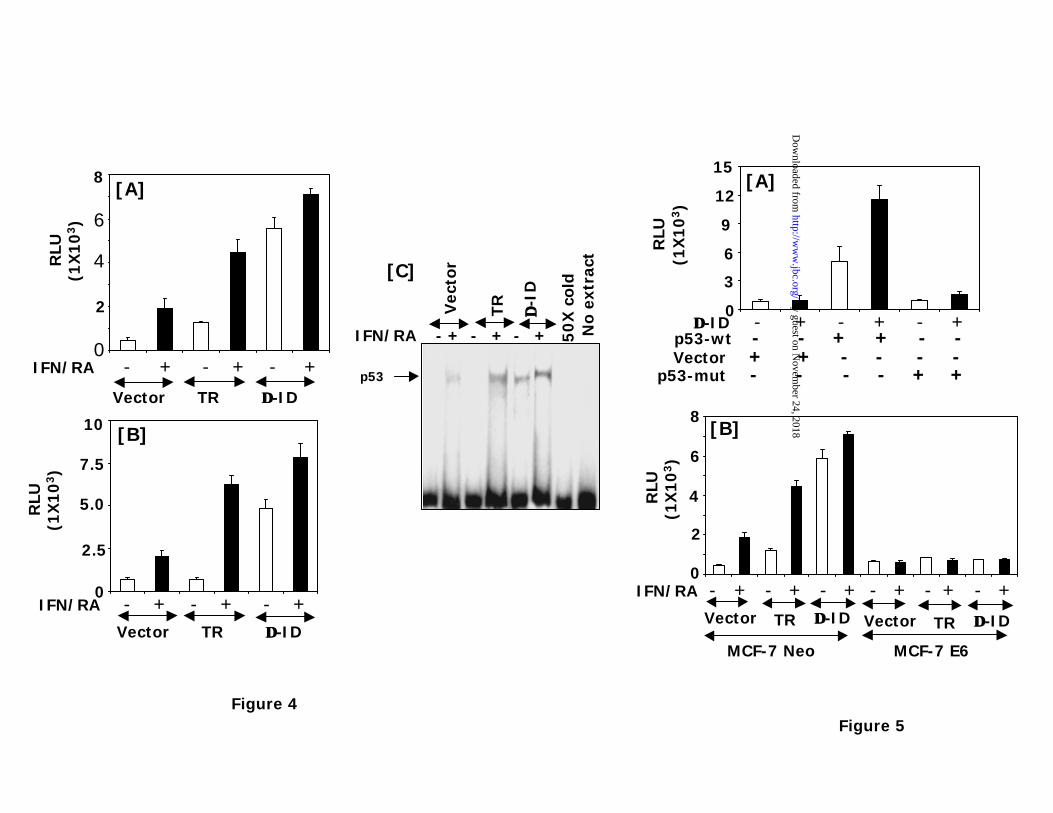
Effect of IFN/RA combination on p53 regulated gene expression: Earlier, we
have reported that TR modulates p53 dependent cell death via an upregulation of gene
expression (27). To test the influence of TR mutants on p53 stimulated gene expression we have
performed the following experiments. First we wanted to know whether ∆-ID induces the
expression a reporter gene driven by p53 response element. MCF-7 cells were transfected with a
p53RE-Luc reporter. Along with the reporter pCMV-flag, wildtype f -TR or f -∆-ID mutant were
cotransfected (Fig. 4A). Since flag tag is shorter and yielded a slightly better expression in
transient assays, we have used the flag tagged mutants for the following studies. Nevertheless,
the myc-tagged mutants exhibited similar properties like the flag-tagged ones (data not shown).
Following transfection cells were treated with IFN/RA and luciferase activity was measured.
IFN/RA induced luciferase expression in the vector transfectants. In TR transfectants basal
luciferase activity was elevated and it was further strongly induced by IFN/RA. ∆-ID co-
expression strongly enhanced and it was only slightly but significantly stimulated further by
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

10
IFN/RA. ∆-ID induced the luciferase expression slightly higher than the IFN/RA treated, TR co-
expressed control. Previously we have shown that Bax, a p53 responsive mRNA and its protein
are induced in the presence of wildtype TR and inhibited in the presence of a catalytically
inactive mutant. Since p53 -Luc used in this experiment contained a synthetic promoter, we next
explored whether ∆-ID exerted a similar effect on a native promoter. A luciferase reporter driven
by human Bax has been shown to respond p53 (33,36). Therefore, we have employed it in the
next experiment. Fig.4B shows the data obtained. ∆-ID constitutively activated this promoter in a
manner similar to p53-Luc. TR on the other hand required treatment with IFN/RA to exert a
similar effect. Thus, a synthetic and the native promoter respond to ∆-ID similarly.
Previously we have shown that in the presence of a wildtype TR IFN/RA treatment
enhances the DNA binding of p53 and a catalytically inactive TR blocks it. Therefore, we
examined the DNA binding of p53 in cells stably expressing vector, wildtype TR and ∆-ID. Our
previous studies have shown that exposure of cells to IFN/RA for 24-28h, a time when
significant apoptosis can be detected is optimal for detecting p53 binding by EMSA, without
causing a rise in p53 levels (25-27). While an IFN/RA dependent induction of p53 binding
occurred in the TR expressing cells, a constitutive binding of p53 to the response element was
observed in cells expressing ∆-ID (Fig. 4C). It was slightly enhanced by IFN/RA. This
observation is consistent with the luciferase expression data. This band was competed out by a
cold p53binding element. We have shown earlier that this band is not competed out by a mutant
p53 oligonucleotide and it is supershifted by a polyclonal antibody against p53 (25-27).
p53 is necessary for a hyper stimulating effect of ∆∆ -ID on gene expression: A
critical role for p53 in TR regulated cell death effects was examined using DLD human colon
carcinoma cells, which lacked the endogenous p53 (37). Re-introduction of p53 causes death of
these cells (37). Introduction of a p53-Luc reporter along with an empty vector did not cause an
upregulation of luciferase activity. ∆-ID alone had no effect on luciferase expression. In the
wildtype p53 transfected cells luciferase expression was stimulated 3.5-4.0 fold over the vector-
transfected cells. It was induced further significantly enhanced in the presence of ∆-ID.
Furthermore, the same reporter was not induced in cells transfected with a mutant p53 and
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

11
cotransfection of D-ID had no effect on (Fig. 5A). These data show that p53 is obligatory for an
inductive effect of ∆-ID on the luciferase reporter.
Human breast carcinoma cell line MCF-7 carries a wildtype p53 allele (38). The
endogenous p53 can be inactivated by targeting it to ubiquitin dependent proteolysis, upon stable
expression of the human papilloma viral (HPV)-E6 gene (29,30). Such epigenetic
downregulation confers a p53 null property to MCF-7-E6 cell line. The control cell line MCF-7
Neo carries an empty expression vector. Both cell lines were transfected with a p53RE-Luc
reporter along with empty vector, TR or ∆-ID construct. As shown in Fig.5B, transfection of
wildtype TR caused an elevation of luciferase gene expression in MCF7-Neo cells. ∆-ID also
induced luciferase expression, which was significantly higher than the wildtype TR. p53
dependent gene expression was induced by IFN/RA treatment in the vector-transfected cells. In
the presence of wildtype TR, IFN/RA further induced luciferase activity strongly. IFN/RA
caused only a marginal increase in luciferase expression in the ∆-ID transfected cells. Thus,
deletion of ID permits a constitutive activation of p53 dependent gene induction. The same
mutant when introduced into MCF7-E6 cells failed to promote luciferase expression. These
results show that p53 is critical for TR mediated gene induction.
Trx is required for hyperactivating p53 dependent gene expression: Since
mammalian TR exhibits a wide substrate range (23,24), we next examined whether the
hyperactivation of p53 dependent responses by ∆-ID was a result of its shift from the use of its
native substrate Trx1. Therefore, we next determined whether the ∆-ID mutant activates p53
dependent gene expression in MCF-7 cells stably expressing a mutant Trx, which lacks the
critical cysteines (at positions 32 and 35) for its redox function (25). For this purpose we have
employed three MCF-7 cell lines each stably expressing the vector (V), wildtype Trx1 (W) and
mutant Trx1 (M). Co-transfection of pCMV2 -flag with p53-Luc had no effect on luciferase
activity in the V, W and M cell lines (Fig. 6). However, co-transfection of wildtype TR elevated
the basal expression of p53-luc in V cells, which was further stimulated in W cells. However, TR
failed to augment p53 dependent gene expression in M cells. A similar pattern of gene regulation
was obtained with ∆-ID mutant in V, W and M cells. The only major difference between the ∆-
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

12
ID and TR is that ∆-ID enhanced the gene expression to a higher level than TR. Thus, Trx is
required for ∆-ID’s stimulatory effect on p53 inducible expression.
Minimal region of TR protein required for stimulating p53 dependent gene
expression: Based on the above results we next determined the minimal region of ∆-ID
required for the stimulating p53 dependent gene expression. Serial deletion mutants each lacking
specific number of aminoacids from the C-terminus of ∆-ID were generated using PCR. These
mutants: ∆-ID80, ∆-ID70, ∆-ID60, ∆-ID50 and ∆-ID30, which lacked 34, 44, 54,64 and 95
aminoacids, respectively, were expressed as N-terminal flag tagged proteins using pCMV-flag.
These mutants yielded 23.6, 22.6, 21.6, 20.6, and 17.1 peptides, respectively. A western blot
analysis of transfected cell extracts with flag-tag specific antibodies showed the expression of
these mutants (Fig.7A). All mutants expressed equivalently. We next determined the effect of
these mutants on p53 dependent gene expression (Fig.7B). The ∆-ID80 mutant was better than ∆-
ID70 at augmenting the reporter gene expression constitutively, although ∆-ID70 still retained a
significant amount of stimulatory effect. The other mutants lost their stimulatory effects on p53
dependent gene expression. Although a slight stimulatory effect of IFN/RA was found on the ∆-
ID80 mutant, it was lost with the ∆-ID70. These data suggest that a domain between ∆-ID80 and
∆-ID60 regulates the constitutive stimulatory effect on p53 regulated gene expression.
Residues critical for the stimulatory effect on p53 dependent transcription: In
the next set of experiments we determined the residues critical for the hyperstimulatory effect of
∆-ID on p53 dependent gene expression. Based on the fact that ∆-ID80 exhibits a strong
constitutive effect on p53 dependent gene expression (Fig. 7B), we engineered new point
mutants that lack specific residues. Since the ∆-ID-70 mutant has significant stimulatory effect,
we reasoned that residues might lie within it. Primary sequence analysis of this region revealed a
NADPH binding domain. There are potential residues that can be phosphorylated in this region.
These include a serine at 199, a threonine at 193, two tyrosine residues at 187 and 200. Two of
these are present within the NADPH binding motif. While mutating tyrosine 187, we have also
converted the adjacent cysteine residue at 189 into an alanine, simultaneously. We have, thus,
generated 3 new mutants: 1) Y187F & C189A; 2) T193A; and 3) S199A & Y200F. These
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

13
mutants were cloned into pCMV-flag and their expression was verified by western blot analysis
of the transfected cell extracts (Fig. 8A). The mutants were co-transfected along with a p53-
luciferase reporter and analyzed for their stimulatory effect on the promoter. Mutants Y187F &
C189A; and T193A completely lost their stimulatory effect on p53 dependent gene expression.
However, mutant S199A & Y200F retained its stimulatory effect, comparable to that of ∆-ID80
(Fig. 8B). Lastly, single mutants of Y187F and C189A were activated the p53 dependent gene
expression like WID indicating that both amino acids play a crucial in the regulation of p53
dependent gene expression.
The influence of point mutants on p53 activation was analyzed by EMSA. Stable cells
lines expressing flag-tagged mutants were generated (see below in Fig. 9). Four cell lines each
expressing ∆-ID; Y187F & C189A; T193A; and S199A & Y200F were utilized for EMSA (Fig.
8C). While ∆-ID and the S199A & Y200F mutant caused an elevation of p53 binding to the
response element, the Y187F & C189A and T193A mutants did not. IFN/RA did not cause an
activation of p53 in cells expressing the Y187F & C189A and T193A mutants. IFN/RA had a
slight stimulatory e ffect on ∆-ID and the S199A & Y200F stimulated p53 binding to its response
element. These data are consistent with the luciferase expression data.
To examine the role of these mutants in IFN/RA induced cytotoxicity, stable transfectants
expressing the flag tagged mutants were plated and selected with IFN/RA and G418 (0.5 mg/ml)
fro 3 weeks. The Y187F & C189A; and T193A mutants resisted IFN/RA unlike TR, ∆-ID, and
S199A & Y200F which were killed by the combination (Fig. 9A). This property is consistent
with their respective p53 augmenting functions. Expression of these mutants in the cells was
confirmed by a western blot analysis with flag-tag specific antibodies (Fig. 9B and 9C).
However, the expression of S199A & Y200F was lost over several passages, indicating its
growth inhibitory effect.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

14
Discussion:
Thioredoxin (Trx), a ubiquitous redox protein, regulates a wide-array of cellular activities
including growth, transcription and immune responses (23). Its redox status is controlled by a
cytosolic enzyme thioredoxin reductase (TR). In mammals 3 different TR genes, TR1, TR2 and
TR3, which express in an organelle and tissue specific manner, have been identified to date
(39,40). The founding member of this family, TR1, is expressed ubiquitously (23). Physiologic
roles of the new members of TR family are unclear at present. Mammalian TR has broader
substrate specificity than its prokaryotic homologues (23). For example, it can reduce unrelated
compounds such as selenite, alloxan, 5,5’ dithiobis(2-nitorbenzoic acid), vitamin-K,
selenocysteine, selinodiglutathione and S-nitrosoglutathione, in addition to Trx. Since TR
modifies its substrates extremely rapidly and no stable prosthetic groups are involved in this
process (23), it has been technically difficult to define its intracellular targets. Trx is also
implicated in maintaining the redox status of transcription factors, AP1, PEBP2, NF-κB and
HIF1α (28,41-44).
Based on chemical inhibition and other correlative data Trx1 is implicated in cell growth
promotion in some cell types (24). However, it is not a universal phenomenon. Several studies
showed that Trx and TR participate in growth inhibitory actions. The Trx homologue of
Drosophila, deadhead, is essential for female meiosis and early embryonic development (45) but
not for DNA synthesis in vivo (46). Deletion of TR1 in Drosophila affects survival (47). It codes
for a cytoplasmic and a mitochondrial isoform both of which are necessary for the viability of the
fly. Similarly Trx1 null mouse embryos do not implant at all (48). Thioredoxin inhibits DNA
synthesis in the fertilized Xenopus eggs (49). In yeast, deletion of Trx gene causes an increase in
the frequency of mitotic cell cycles (50). TR is necessary for p53-dependent growth suppression
in fission and budding yeasts (51,52). Inhibition of Trx1 causes resistance to IFN-γ induced
apoptosis (53) and Trx producing hepatomas grow slow (54). Using a genetic tool we have
shown earlier that TR1 is critical for the cell death controlled by IFN/RA combination (22,55).
We and others have shown that tumor suppressor p53, caspases-8 and –3 are also regulated by
TR:Trx (25-27,56,57).
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

15
Tumor suppressor p53 inhibits cell growth either by aborting the division cycle or by
inducing death (58); and is frequently inactivated gene in several human cancers. Its activity is
controlled by several post-translational mechanisms such as stability, phosphorylation by protein
kinases, ubiquitination, SUMOylation, acetylation and redox factors (59). We have reported
earlier that the IFN/RA combination promotes p53 dependent gene expression and cell death
using TR1 and Trx1 (27) without elevating the levels of p53 protein. p53 has two cysteines
(residues 242 and 176), which along with two histidines coordinate Zn2+ for DNA binding
(60,61). Oxidation of these cysteines ablates its transcriptional activity (60,62). Wild type human
p53, but not a cysteine mutant, inhibits growth in yeast (63,64). Redox control of p53 activity is
further substantiated by studies that showed tms1, a dehydrogenase, suppresses p53 induced
growth arrest (65). Lastly, studies using chimeric p53 proteins in yeast have revealed that in
addition to t he DNA binding site, the transactivation domain of p53 is also subject TR dependent
redox regulation (66). Retinoids are known to activate oxidative stress through an increased
synthesis of reactive oxygen species (67-70). Presence of ROS is a signal for p53 activation (58).
IFN/RA treatment induces TR and Trx in cell undergoing apoptosis (22,26). p53 is kept in a
reduced form by redox factor-1 (Ref-1), a down stream effector of TR:Trx system. Indeed, recent
studies have shown that Ref-1 promotes p53 dependent gene expression and cell death (33). Ref-
1 has been suggested to prevent the oxidation of cysteine residues of p53. Trx1 augments Ref-1
dependent gene expression through p53 (57). More importantly, the failure of ∆-ID to promote
gene expression in the absence of p53 (Fig. 5) clearly indicates an obligatory role for p53 in this
process. Since ∆-ID fails to i nduce the p53 responsive reporter in cells expressing a mutant Trx,
its effects are Trx-dependent (Fig. 5).
The present observations raise a question: what is the role of ID? Mammalian TR is a
dimeric selenoprotein. Mutational analyses have revealed that the mammalian TR has two redox
centers: one at the N-terminus in the FAD binding domain and the other at its C-terminus.
Phylogenetically well conserved of these, N-terminal redox center, is constituted by cysteines
residues at 59 and 64. A selenocysteine at the C-terminus forms the C-terminal redox center. In
an unusual manner, one of the two in frame stop codons at the 3’ end of the ORF in conjunction
with a stem-loop structure formed by a selenocystine incorporation sequence of the 3’
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

16
untranslated region of TR mRNA is proposed to act as an acceptor for selenocysteinyl tRNA
leading to a co-translational incorporation of selenocysteine into the mature protein (24,71).
Recent studies showed that bovine and rat TRs lacking this selenocysteine have an extremely
reduced kcat value in vitro (72,73). However, this enzyme did not completely lose its enzymatic
activi ty. Furthermore, depending on the substrate used for the assay, its activity is normal as long
as the N-terminal primary redox center is retained (73). In fact, incubation of the mutant derived
protein with selenocysteine restored the activity dramatically in vitro (72). These data suggest
that selenocysteine in trans can restore the enzyme activity. However, a low occurrence of free
selenocysteine (74) suggests that such modulation is a less frequent event in vivo. It is possible
that stress conditions such as apoptosis alter the physiologic availability of selenocysteine. Such
selenocysteine may be derived from an apoptotic degradation of other selenoproteins or its
biosynthesis. Atleast, 10-12 proteins that contain selenocysteine (75) have been identified to
date, degradation of which may provide free selenocysteine.
That yeast TR, which lacks a selenocysteine (76,77), can promote p53 dependent cell
growth arrest (51,52) and a truncated human TR lacking its ID also promotes p53 dependent
transcriptional response (this study) are consistent with the proposition that TR-stimulated p53-
mediated responses are selenocysteine-independent. Similarly, Drosophila and plant TRs do not
require selenium for their function (47,78). In fact, plant TR behaves like a prokaryotic TR (78).
Furthermore, the prokaryotic TRs do not reduce other substrates, like the mammalian TR. Recent
crystallographic data on rat TR (79) have shown that the enzyme forms a head-to-tail dimer, with
the N-terminal redox center buried inside. Reducing power is first transferred from NADPH to
the N-terminal redox center. The C-terminus of one subunit is inserted to the charged cleft (N-
terminal redox center) of the other subunit to tap electrons from the active site to selenocysteine.
The reduced selenocysteine then donates electrons to Trx/other substrates at the surface of TR
dimer, after emerging from the catalytic cleft. In this model, the ID extends like a robotic arm to
transfer electrons between the enzyme and its various substrates. In the case of prokaryotic TR,
which also acts as a dimer, after the receipt of electrons at the redox center from NADPH, the
NADPH binding domain undergoes a 66o rotation for providing an access to oxidized Trx to the
redox center (23,24). ∆-ID may behave like prokaryotic, yeast, and plant TRs with strict Trx
reductive properties. This would be sufficient for activating p53. Thus, it would appear that a
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

17
selenocysteine at the C-terminus has evolved to enhance catalysis and broaden substrate r ange
and is an optional accessory. Although an augmentation of TR activity by selenium has been
reported in mammalian cells, these studies used a supra-nutritional concentration of selenium
and are not physiologically relevant. In fact, an inverse correlation between selenium and TR
activity has also been reported (80). Lastly, selenium metabolites can activate cell cycle arrest in
the absence of p53 and DNA damage (80), indicating the existence of a separate mechanism of
action. In the light of these data, we suggest that selenium/selenocysteine plays a limited role in
TR mediated growth suppressive pathways in vivo, but are required for other redox reactions
during normal growth.
Alternatively, TR bearing alkylated C-terminal Selenocysteine and Cysteine residues
exhibits 30-fold higher NADPH oxidase activity compared to the wildtype enzyme, and is
capable of producing superoxides (81). These superoxides can oxidize intracellular environment,
thus tilting the balance towards p53 activation and cell death. Consistent with this suggestion,
deletion of NADPH binding domain (mutants ∆-ID60 and ∆-ID50) prevents the stimulatory
effects of TR on p53 dependent gene expression (Fig. 7). It is interesting to note that the
mutation of cysteine residue at 189 depletes the p53-augmenting function of ∆-ID (Fig. 8). This
suggests that Trx transiently interacts with this site during enzymatic modification, since Trx1 is
still necessary for promoting p53-dependent gene expression. The threonine and tyrosine
residues may undergo post-translational modifications, which in turn contribute to full activity of
∆-ID. Thus, ID, which is unique to mammalian TR, appears act as a regulatory switch in cell
growth control. Its presence attenuates the growth inhibitory effect of TR and its removal
promotes cell death. Since prokaryotic/unicellular organisms do not undergo apoptosis and
primary role of TR is only to maintain the redox functions they have a much simpler modular
structure. Because TR and Trx can promote pro- and anti- growth processes depending on the
physiologic status of cells and mammalian TR has expanded physiologic roles, evolution of a
regulatory switch (ID) is critical for preventing an inadvertent operation of these divergent
processes. Thus, ID may act as a decision switch to mediate such “Yin-Yang” reactions in vivo.
In the meanwhile, one potential use for the ∆-ID mutant will be in gene therapy along with
wildtype p53 in p53 null tumors.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

18
Acknowledgments: These studies are supported by the National Cancer Institute grants CA
78282 and CA 71401 to DVK. The authors thank Peter Gutierrez for helpful discussions.
References:
1. Kalvakolanu, D. V., Choi, K., and Borden, E. C. (2001) in The Molecular Basis of Cancer (Mendelsohn, J., Howley, P. M., Israel, M. A., and Liotta, L. A., eds), pp. 503-534, W.B Saunders Company, Philadelphia
2. Stark, G. R., Kerr, I. M., Williams, B. R. G., Silverman, R. H., and Schreiber, R. D. (1998) Annu Rev Biochem 67, 227-264
3. Kaplan, D. H., Shankaran, V., Dighe, A. S., Stockert, E., Aguet, M., Old, L. J., and Schreiber, R. D. (1998) Proc Natl Acad Sci U S A 95, 7556-7561
4. Kroger, A., Koster, M., Schroeder, K., Hauser, H., and Mueller, P. P. (2002) J Interferon Cytokine Res 22, 5-14
5. Tamura, T., and Ozato, K. (2002) J Interferon Cytokine Res 22, 145-152 6. Willman, C. L., Sever, C. E., Pallavicini, M. G., Harada, H., Tanaka, N., Slovak, M. L.,
Yamamoto, H., Harada, K., Meeker, T. C., List, A. F., and Taniguchi, T. (1993) Science 259, 968-971
7. Holtschke, T., Lohler, J., Kanno, Y., Fehr, T., Giese, N., Rosenbauer, F., Lou, J., Knobeloch, K. P., Gabriele, L., Waring, J., Bachmann, M. F., Zinkernagel, R. M., Morse, H. C., Ozato, K., and Horak, I. (1996) Cell 87, 307-317
8. Choubey, D., Li, S. J., Datta, B., Gutterman, J. U., and Lengyel, P. (1996) EMBO Journal 15, 5668-5678
9. Wen, Y., Yan, D. H., Wang, B., Spohn, B., Ding, Y., Shao, R., Zou, Y., Xie, K., and Hung, M. C. (2001) Cancer Res 61, 7142-7147
10. Kimchi, A. (1992) Journal of Cellular Biochemistry 50, 1-9 11. Resnitzky, D., Tiefenbrun, N., Berissi, H., and Kimchi, A. (1992) Proc Natl Acad Sci U S
A 89, 402-406 12. Tiefenbrun, N., Melamed, D., Levy, N., Resnitzky, D., Hoffman, I., Reed, S. I., and
Kimchi, A. (1996) Mol Cell Biol 16, 3934-3944 13. Gutterman, J. U. (1994) Proc. Natl. Acad. Sci. USA 91, 1198-1205 14. Lindner, D. J., Kalvakolanu, D. V., and Borden, E. C. (1997) Semin Oncol 24, S9-99-
S99-104 15. Moore, D. M., Kalvakolanu, D. V., Lippman, S. M., Kavanagh, J. J., Hong, W. K.,
Borden, E. C., Paredes-Espinoza, M., and Krakoff, I. H. (1994) Semin Hematol 31, 31-37 16. DiPaola, R. S., Rafi, M. M., Vyas, V., Toppmeyer, D., Rubin, E., Patel, J., Goodin, S.,
Medina, M., Medina, P., Zamek, R., Zhang, C., White, E., Gupta, E., and Hait, W. N. (1999) J Clin Oncol 17, 2213-2218
17. Lindner, D. J., Borden, E. C., and Kalvakolanu, D. V. (1997) Clin Cancer Res 3, 931-937 18. Love, J. M., and Gudas, L. J. (1994) Curr Opin Cell Biol 6, 825-831 19. Mangelsdorf, D. J., and Evans, R. M. (1995) Cell 83, 841-850 20. Faria, T. N., Mendelsohn, C., Chambon, P., and Gudas, L. J. (1999) J Biol Chem 274,
26783-26788 21. Lin, B., Chen, G. Q., Xiao, D., Kolluri, S. K., Cao, X., Su, H., and Zhang, X. K. (2000)
Mol Cell Biol 20, 957-970
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

19
22. Hofmann, E. R., Boyanapalli, M., Lindner, D. J., Weihua, X., Hassel, B. A., Jagus, R., Gutierrez, P. L., and Kalvakolanu, D. V. (1998) Mol Cell Biol 18, 6493-6504
23. Arner, E. S., and Holmgren, A. (2000) Eur J Biochem 267, 6102-6109 24. Mustacich, D., and Powis, G. (2000) Biochem J 346 Pt 1, 1-8 25. Ma, X., Karra, S., Lindner, D. J., Hu, J., Reddy, S. P., Kimchi, A., Yodoi, J., and
Kalvakolanu, D. V. (2001) Oncogene 20, 3703-3715 26. Ma, X., Karra, S., Guo, W., Lindner, D. J., Hu, J., Angell, J. E., Hofmann, E. R., Reddy,
S. P., and Kalvakolanu, D. V. (2001) J Biol Chem 276, 24843-24854 27. Hu, J., Ma, X., Lindner, D. J., Karra, S., Hofmann, E. R., Reddy, S. P., and Kalvakolanu,
D. V. (2001) Oncogene 20, 4235-4248 28. Hirota, K., Matsui, M., Iwata, S., Nishiyama, A., Mori, K., and Yodoi, J. (1997) Proc
Natl Acad Sci U S A 94, 3633-3638 29. Fan, S., Smith, M. L., Rivet, D. J., Duba, D., Zhan, Q., Kohn, K. W., Fornace, A. J., and
O'Connor, P. M. (1995) Cancer Res 55, 1649-1654. 30. Potapova, O., Gorospe, M., Dougherty, R. H., Dean, N. M., Gaarde, W. A., and
Holbrook, N. J. (2000) Mol Cell Biol 20, 1713-1722. 31. Baker, S. J., Markowitz, S., Fearon, E. R., Willson, J. K., and Vo gelstein, B. (1990)
Science 249, 912-915 32. Kern, S. E., Pietenpol, J. A., Thiagalingam, S., Seymour, A., Kinzler, K. W., and
Vogelstein, B. (1992) Science 256, 827-830 33. Gaiddon, C., Moorthy, N. C., and Prives, C. (1999) EMBO Journal 18, 5609-5621 34. Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., Warren, J.
T., Bokesch, H., Kenney, S., and Boyd, M. R. (1990) Journal of the National Cancer Institute 82, 1107-1112
35. van Engeland, M., Ramaekers, F. C., Schutte, B., and Reutelingsperger, C. P. (1996) Cytometry 24, 131-139
36. Miyashita, T., and Reed, J. C. (1995) Cell 80, 293-299 37. Yu, J., Zhang, L., Hwang, P. M., Rago, C., Kinzler, K. W., and Vogelstein, B. (1999)
Proc Natl Acad Sci U S A 96, 14517-14522. 38. Balcer-Kubiczek, E. K., Yin, J., Lin, K., Harrison, G. H., Abraham, J. M., and Meltzer, S.
J. (1995) Radiat Res 142, 256-262 39. Lee, S. R., Kim, J. R., Kwon, K. S., Yoon, H. W., Levine, R. L., Ginsburg, A., and Rhee,
S. G. (1999) J Biol Chem 274, 4722-4734 40. Sun, Q. A., Wu, Y., Zappacosta, F., Jeang, K. T., Lee, B. J., Hatfield, D. L., and
Gladyshev, V. N. (1999) J Biol Chem 274, 24522-24530 41. Akamatsu, Y., Ohno, T., Hirota, K., Kagoshima, H., Yodoi, J., and Shigesada, K. (1997)
J Biol Chem 272, 14497-14500 42. Hayashi, T., Ueno, Y., and Okamoto, T. (1993) J Biol Chem 268, 11380-11388 43. Hirota, K., Matsui, M., Murata, M., Takashima, Y., Cheng, F. S., Itoh, T., Fukuda, K.,
and Junji, Y. (2000) Biochem Biophys Res Commun 274, 177-182 44. Ema, M., Hirota, K., Mimura, J., Abe, H., Yodoi, J., Sogawa, K., Poellinger, L., and
Fujii-Kuriyama, Y. (1999) EMBO Journal 18, 1905-1914 45. Salz, H. K., Flickinger, T. W., Mittendorf, E., Pellicena-Palle, A., Petschek, J. P., and
Albrecht, E. B. (1994) Genetics 136, 1075-1086 46. Pellicena-Palle, A., Stitzinger, S. M., and Salz, H. K. (1997) Mech Dev 62, 61-65
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

20
47. Missirlis, F., Ulschmid, J. K., Hirosawa-Takamori, M., Gronke, S., Schafer, U., Becker, K., Phillips, J. P., and Jackle, H. (2002) J Biol Chem
48. Matsui, M., Oshima, M., Oshima, H., Takaku, K., Maruyama, T., Yodoi, J., and Taketo, M. M. (1996) Dev Biol 178, 179-185
49. Hartman, H., Wu, M., Buchanan, B. B., and Gerhart, J. C. (1993) Proc Natl Acad Sci U S A 90, 2271-2275
50. Muller, E. G. (1991) J Biol Chem 266, 9194-9202 51. Casso, D., and Beach, D. (1996) Mol Gen Genet 252, 518-529 52. Pearson, G. D., and Merrill, G. F. (1998) J Biol Chem 273, 5431-5434 53. Deiss, L. P., and Kimchi, A. (1991) Science 252, 117-120 54. Rubartelli, A., Bonifaci, N., and Sitia, R. (1995) Cancer Res 55, 675-680 55. Lindner, D. J., Hofmann, E. R., Karra, S., and Kalvakolanu, D. V. (2000) Biochim
Biophys Acta 1496, 196-206 56. Ueda, S., Nakamura, H., Masutani, H., Sasada, T., Yonehara, S., Takabayashi, A.,
Yamaoka, Y., and Yodoi, J. (1998) J Immunol 161, 6689-6695 57. Ueno, M., Masutani, H., Arai, R. J., Yamauchi, A., Hirota, K., Sakai, T., Inamoto, T.,
Yamaoka, Y., Yodoi, J., and Nikaido, T. (1999) J Biol Chem 274, 35809-35815 58. Vogelstein, B., Lane, D., and Levine, A. J. (2000) Nature 408, 307-310 59. Jayaraman, L., and Prives, C. (1999) Cell Mol Life Sci 55, 76-87 60. Hainaut, P., and Milner, J. (1993) Cancer Res 53, 4469-4473 61. Cho, Y., Gorina, S., Jeffrey, P. D., and Pavletich, N. P. (1994) Science 265, 346-355 62. Delphin, C., Cahen, P., Lawrence, J. J., and Baudier, J. (1994) Eur J Biochem 223, 683-
692 63. Epstein, C. B., Attiyeh, E. F., Hobson, D. A., Silver, A. L., Broach, J. R., and Levine, A.
J. (1998) Oncogene 16, 2115-2122 64. Bischoff, J. R., Casso, D., and Beach, D. (1992) Mol Cell Biol 12, 1405-1411 65. Wagner, P., Grimaldi, M., and Jenkins, J. R. (1993) Eur J Biochem 217, 731-736 66. Merrill, G. F., Dowell, P., and Pearson, G. D. (1999) Cancer Res 59, 3175-3179 67. You, K. R., Wen, J., Lee, S. T., and Kim, D. G. (2001) J Biol Chem 68. Chen, Y., Buck, J., and Derguini, F. (1999) Cancer Res 59, 3985-3990 69. Zang, Y., Beard, R. L., Chandraratna, R. A., and Kang, J. X. (2001) Cell Death Differ 8,
477-485 70. Dal-Pizzol, F., Klamt, F., Benfato, M. S., Bernard, E. A., and Moreira, J. C. (2001) Free
Radic Res 34, 395-404 71. Gasdaska, J. R., Harney, J. W., Gasdaska, P. Y., Powis, G., and Berry, M. J. (1999) J Biol
Chem 274, 25379-25385 72. Zhong, L., and Holmgren, A. (2000) J Biol Chem 275, 18121-18128 73. Fujiwara, N., Fujii, T., Fujii, J., and Taniguchi, N. (2001) J Biochem (Tokyo) 129, 803-
812 74. Stadtman, T. C. (1996) Annu Rev Biochem 65, 83-100 75. Allan, C. B., Lacourciere, G. M., and Stadtman, T. C. (1999) Annu Rev Nutr 19, 1-16 76. Gladyshev, V. N., Krause, M., Xu, X. M., Korotkov, K. V., Kryukov, G. V., Sun, Q. A.,
Lee, B. J., Wootton, J. C., and Hatfield, D. L. (1999) Biochem Biophys Res Commun 259, 244-249
77. Miranda-Vizuete, A., Damdimopoulos, A. E., and Spyrou, G. (2000) Antioxid Redox Signal 2, 801-810
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

21
78. Dai, S., Saarinen, M., Ramaswamy, S., Meyer, Y., Jacquot, J. P., and Eklund, H. (1996) J Mol Biol 264, 1044-1057
79. Sandalova, T., Zhong, L., Lindqvist, Y., Holmgren, A., and Schneider, G. (2001) Proc Natl Acad Sci U S A 98, 9533-9538
80. Ganther, H. E. (1999) Carcinogenesis 20, 1657-1666 81. Zhong, L., Arner, E. S., and Holmgren, A. (2000) Proc Natl Acad Sci U S A 97, 5854-
5859
Legends to Figures:
Figure 1: Expression of TR mutants. Mutants were generated using PCR as described in
materials and methods section. Panel A: diagrammatic representation of the structures of TR
mutants. Panels and B show the western blot (WB) analyses. MCF-7cells were transiently
transfected with the indicated plasmids (1µg) and equal amounts of lysates were separated on a
10% SDS-PAGE and western blotted. The blots were probed with indicated antibodies.
Figure 2: Effects of TR mutants on cell growth. (A) Effect of TR mutants on G418r colony
formation. MCF-7 cells were transfected with equimolar amounts of pCXN2-myc vector
expressing various mutants After 3 weeks of selection with G418 (1 mg/ml) in growth medium,
surviving colonies were counted. Each bar represents mean ± SE of triplicates. (B) Effects of TR
mutants on cell growth. Equal number of cells (2000/well), stably transfected with various TR
mutants, were plated and cell growth was monitored after 5 days using the sulforhodamine B
binding assay (34). Each bar represents mean ± SE of 8 replicates. Cell growth was monitored
quantified by measuring the absorbance of bound dye at 575 nm. (C) Expression of TR mutants
after stable transfection in MCF-7 cells. Total protein (65 µg) from the indicated cell lines was
western blotted and probed with anti-myc antibody. This blot was stripped and probed (shown
below) with actin specific antibodies. (D) Cells treated with IFN/RA were stained with annexin-
V as described under materials and methods. Percentage of annexin-V positive cells was
quantified using FACS analysis. Each bar represents mean ± SE of triplicates.
Figure 3: Equal amount of total cell lysate (85 µg) from the indicated cell lines were
immunoblotted and analyzed with s pecific antibodies. (A) Expression of endogenous TR protein.
Arrowhead indicates the position of transgene derived TR. This antibody does not recognize
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

22
protein derived from ∆-ID. This blot was stripped and probed with anti-actin antibodies (shown
below). (B) Expression of endogenous p53 protein. WB: western blot.
Figure 4: Effect of IFN/RA on p53 dependent gene expression. (A) MCF-7 cells were treated
with IFN/RA after transfecting with a p53-Luc, CMV-β-galactosidase, and TR mutants. Cell
extracts were measured for luciferase and β-galactosidase activity. Luciferase activity was
normalized to the β-galactosidase activity. Each bar represents mean ± SE of triplicates. A plus
(+) sign indicates treatment with IFN-β (500 U/ml) and RA (1 µM) for 16h. (B) Effect of
IFN/RA on the Bax promoter. MCF-7 cells were transfected with Bax-Luc and CMV-β-
galactosidase plasmids. Cells were stimulated with IFN/RA where indicated with a plus sign. (C)
Effect of TR mutants on p53 binding to DNA. Cell extracts from IFN/RA stimulated cells (24h)
were incubated with a 32P-labeled oligonucleotide bearing the consensus p53 binding site. Plus
and minus signs indicate no treatment and IFN/RA treatment, respectively. Where indicated 50X
cold: the D-ID cell extract was incubated with an excess (50X) unlabeled oligonucleotide, prior
to use in EMSA. Thirty micrograms of nuclear extract was used in this experiment.
Figure 5: p53 is required for the gene stimulatory effect of ∆-ID. In panel A, human DLD colon
carcinoma cell line (p53 null) was transfected with the indicated plasmids along with p53-Luc
and CMV-β-galactosidase reporters. A plus sign indicates the presence of the indicated plasmid
in the transfection mixture. MCF-7 neo and MCF-7E6 cells were transfected with p53RE-
Luciferase in the presence of various indicated plasmids. Total amount of DNA transfected into
the cells was kept constant (1.0 µg) by adding the pCMV-flag vector, where required.
Figure 6: Trx1 is required for the hyperstimulation of p53 dependent gene expression.
MCF-7 cells stably expressing control vector (V), Wildtype (W) and redox inactive Trx1 (M)
were transfected with TR mutants along with p53-Luc and CMV-β-galactosidase reporters.
Luciferase activity was quantified as described in figure 4. Plus sign indicates the presence of
that specific plasmid in transfection mixture.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

23
Figure 7: Minimal region of ∆-ID required for its hyperstimulatory effects on p53 responsive
gene expression. (A) Expression of mutants in MCF-7 cells. Cells in 6 well dishes were
transfected with 0.5 µg of indicated plasmids and whole cell lysates were prepared. A
comparable quantity of protein (50µg) from each sample was employed for western blot analysis
using flag specific antibodies. (B) Effect of the mutants shown in panel A on p53-dependnent
luciferase reporter. Indicated plasmids were co-transfected with p53-Luc and CMV-β-
galactosidase reporters and luciferase expression was analyzed. A plus (+) sign indicates
treatment with IFN-β (500 U/ml) and RA (1 µM) for 16h.
Figure 8: Residues required for the hyperstimulatory effect of ∆-ID on p53 mediated gene
expression. Panel A shows the expression of the point mutants indicated above it. This
experiment is similar to Fig. 7A. Panel B shows the effect of point mutants on p53 dependent
gene expression. Transfection and reporter gene analysis was similar to fig. 7B. (C) EMSA for
p53 activation. Stable cell lines expressing flag-tagged TR mutants (indicated above the lanes)
were treated with IFN/RA for 30h and EMSA was performed. A plus (+) sign indicates treatment
with IFN-β (500 U/ml) and RA (1 µM). Location of p53 band is indicated.
Figure 9: Effect of IFN/RA on cells expressing TR mutants. (A): Cells stably expressing flag-
tagged TR mutants were selected for 3 weeks with IFN-β (500 U/ml) and RA (1 µM) for 3
weeks. Cells were then stained with sulforhodamine B to visualize surviving cells. (B) & (C):
Expression of TR mutants in the stable transfectants. Extracts from the cells transfected with the
indicated plasmids (70µg) were employed for western blot analysis using anti-flag antibodies.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

pC
MV
-fla
g
f-T
R
f-∆∆
-FA
D
f-∆∆
-NB
D
f −−∆∆
-ID
Figure 1
WB:anti-flagWB:anti-myc
TR
∆∆-FAD
∆∆-NBD
∆∆-ID
[A]
[B]
pC
XN
2-m
yc
TR
-myc
∆∆-F
AD
-myc
∆∆-N
BD
-myc
∆∆-I
D-m
yc
[C]
FAD IDNBD
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

0
0.2
0.6
0.4
Ve
cto
r
Wt
∆∆-F
AD
∆∆-N
BD
∆∆-I
D
Ab
sorb
ance
Ve
cto
r
Wt
∆∆-F
AD
∆∆-N
BD
∆∆-I
D
800
0
200
400
600
G1
48
rco
lon
ies
Ve
cto
r
TR ∆∆-F
AD
∆∆-N
BD
∆∆-I
D
actin
anti-myc
10
50
40
30
20
0
Ve
cto
r
Wt
∆∆-F
AD
∆∆-N
BD
∆∆-I
D
Pe
rce
nt
An
ne
xin
-VP
osi
tive
[A] [B]
[C][D]
Figure 2
WB: Anti-p53
Ve
cto
r
TR ∆∆-I
D
WB: Anti-actin
[B]
WB: Anti-TR
Ve
cto
r
TR
∆∆-I
D
E
[A]
Figure 3
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from
RLU
(1X
10
3)
0
8
4
6
2
IFN/RA - + - + - +
Vector TR ∆∆-ID
Figure 4
0
10
5.0
7.5
2.5
IFN/RA - + - + - +Vector TR ∆∆-ID
[A]
[B]
RLU
(1X
10
3)
IFN/RA - + - + - + - + - + - +
RLU
(1X
10
3)
8
4
6
2
0
Vector TR ∆∆-ID Vector TR ∆∆-ID
MCF-7 Neo MCF-7 E6
[B]
Figure 5
15
9
12
6
Vector + + - - - -p53-wt - - + + - -
3
0∆∆-ID - + - + - +
p53-mut - - - - + +
[A]
RLU
(1X
10
3)
Ve
cto
r
TR
∆∆-I
D
IFN/RA - + - + - + No
ex
tra
ct5
0X
co
ld
p53
[C] by guest on N
ovember 24, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure 6
∆∆ID - - - - - - + + +
RLU
(1X
10
3)
12
3
9
0
6
pCMV-flag + + + - - - - - -TR - - - + + + - - -
Cell line with: V W M V W M V W M
∆∆-I
D-8
0
∆∆-I
D-7
0
∆∆-I
D-6
0
∆∆-I
D-5
0
∆∆-I
D-3
0
∆∆-I
D
Ve
cto
r
10
20
30
4060
[A]
RLU
(1X
10
3)
10
4
8
6
2
0IFN/RA - + - + - + - + - + - + - +
Vector ∆∆-ID ∆∆-ID80
∆∆-ID70
∆∆-ID60
∆∆-ID50
∆∆-ID30
[B]
Figure 7
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

∆∆-ID80
Vector
8
4
6
2
0
10
Y187F C189A
T193A S199A Y200F
RLU
(1X
10
3)
Y1
87
F C
18
9A
T1
93
A
S1
99
A
Y2
00
F
Ve
cto
r
Figure 8
WB: anti-flag
[A]
[B]
IFN/RA - + - + - + - + - +
IFN/RA - - + - + - + - +
[C]
Y1
87
F C
18
9A
T1
93
A
S1
99
A
Y2
00
F
∆∆-I
D
p53
No
ne
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

Y1
87
F C
18
9A
T1
93
A
S1
99
A
Y2
00
F
Ve
cto
rVe
cto
r
TR ∆∆-I
D[B] [C]
Y187F C189A
Vector TR ∆∆-ID
[A]
T193A S199A Y200F
Figure 9
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

Table 1: Primers used in this study
Primers for FLAG tagged constructs:
Flag primer1: 5’-CGGAATTCCATGAACGGCCCTGAAGAT –3’Flag primer2: 5’-GGCCATGGTCAGCAGCCAGCCTGGAGGAT-3’Flag primer 3: 5’-CCGGAATTCGCAATTTATTGGTCCTCAC-3’Flag primer4: 5’-GGCCATGGTCAATTGGTCTGTTCTTCATC-3’
∆∆-ID-80 R-primer : 5’-CGG GGT ACC TCA GAC GTT TAA ACC AAT ACC-3’∆∆-ID-70 R-primer: 5’-CGG GGT ACC TCA AGC GCA CTC CAA AGC GAC-3’∆∆-ID-60 R-primer : 5’-CGG GGT ACC TCA AAC AAC CAG GGT CTT ACC-3’∆∆-ID-50 R-primer : 5’-CGG GGT ACC TCA CAA GGA GAA AAG ATC ATC-3’∆∆-ID-30 R-primer : 5’-CGG GGT ACC TCA TCT CTC TGC TGA ATA AAT-3’
Y187F & C189A Forward primer: 5’-TTC TCC TTG CCT TTT GCT CCG GGT AAG ACC-3’Reverse primer: 5’-GGT CTT ACC CGG AGC AAA AGG CAA GGA GAA-3’
T193A - Forward primer: 5’-TGC CCG GGT AAG GCT CTG GTT GTT GGA GCA-3’Reverse primer: 5’-TGC TCC AAC AAC CAG AGC CTT ACC CGG GCA-3’
S199A & Y200F Forward primer: 5’-GTT GTT GGA GCA GCT TTT GTC GCT TTG GAG-3’Reverse primer: 5’-CTC CAA AGC GAC AAA AGC TGC TCC AAC AAC-3’
Y187F: Forward primer:5’-TTC TCC TTG CCT TTT TGC CCG GGT AAG ACC-3’Reverse primer: 5’-GGT CTT ACC CGG GCA AAA AGG CAA GGA GAA-3’
C189A: Forward primer: 5’-TTC TCC TTG CCT TAC GCA CCG GGT AAG ACC-3’Reverse primer: 5’-GGT CTT ACC CGG TGC GTA AGG CAA GGA GAA-3’
Primers for expressing myc-tagged mutants:
Myc-Primer 1: 5’-CGGAATTCGCCACCATGAACGGCCCTGAAGAT-3’Myc-Primer 2: 5’-GGCCATGGGCAGCCAGCCTGGAGGAT-3’Myc-Primer 3: 5’-CCGGAATTCCAATTTATTGGTCCTCAC-3’Myc-Primer 4: 5’-GGCCATGGATTGGTCTGTTCTTCATC-3’∆∆-NBD primer1: 5’-CGCGGATCCATAAGCATTCTCATAGAC-3’∆∆-NBD primer 2: 5’-CGCGGATCCGTGCCTTACATCTATGCCATT-3’
Mutated sites have been shown with bold face. All myc primers contained EcoRI and Kpn I at the 5’ and 3’ ends respectively. The 5’ and 3’ ends of flag primers bear EcoRI and BamHI sites respectively.
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from

Xinrong Ma, Junbo Hu, Daniel J. Lindner and Dhananjaya V. Kalvakolanuexpression and interferon and retinoic acid induced cell death
Mutational analysis of human thioredoxin reductase 1: Effects on p53 mediated gene
published online April 12, 2002J. Biol. Chem.
10.1074/jbc.M202286200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on Novem
ber 24, 2018http://w
ww
.jbc.org/D
ownloaded from