monensin is a potent inducer of oxidative stress and inhibitor of
TRANSCRIPT

Therapeutic Discovery
Monensin Is a Potent Inducer of Oxidative Stressand Inhibitor of Androgen Signaling Leadingto Apoptosis in Prostate Cancer Cells
Kirsi Ketola1, Paula Vainio1, Vidal Fey2, Olli Kallioniemi1,2,3, and Kristiina Iljin1,2
AbstractCurrent treatment options for advanced and hormone refractory prostate cancer are limited and responses
to commonly used androgen pathway inhibitors are often unsatisfactory. Our recent results indicated that
sodium ionophoremonensin is one of themost potent and cancer-specific inhibitors in a systematic sensitivity
testing of most known drugs and drug-like molecules in a panel of prostate cancer cell models. Because
monensin has been extensively used in veterinary applications to build muscle mass in cattle, the link to
prostate cancer and androgen signaling was particularly interesting. Here, we showed that monensin effects
at nanomolar concentrations are linked to induction of apoptosis and potent reduction of androgen receptor
mRNA and protein in prostate cancer cells. Monensin also elevated intracellular oxidative stress in prostate
cancer cells as evidenced by increased generation of intracellular reactive oxygen species and by induction of a
transcriptional profile characteristic of an oxidative stress response. Importantly, the antiproliferative effects
of monensin were potentiated by combinatorial treatment with the antiandrogens and antagonized by
antioxidant vitamin C. Taken together, our results suggest monensin as a potential well-tolerated, in vivo
compatible drug with strong proapoptotic effects in prostate cancer cells, and synergistic effects with
antiandrogens. Moreover, our data suggest a general strategy by which the effects of antiandrogens could
be enhanced by combinatorial administration with agents that increase oxidative stress in prostate cancer
cells. Mol Cancer Ther; 9(12); 3175–85. �2010 AACR.
Introduction
Prostate cancer is the most frequent cancer and thesecond leading cause of cancer deaths in the male popu-lation of the Western world. There is no efficient cure foradvanced hormone refractory prostate cancer and novelways to inhibit prostate cancer cell growth are needed.Because the majority of prostate tumors from patientswith an androgen-independent disease overexpressandrogen receptor (AR), AR and its coregulators arepotential drug targets for the disease. Recently, a frequentgene fusion between the androgen-regulated prostate-specific protease TMPRSS2 and the ERG transcriptionfactor has been discovered (1). In addition, other drivergenes and oncogenic ETS factors (e.g., ETV1, ETV4,
and ETV5) have been identified as gene fusions in pros-tate tumors (1–3).
To identify novel therapeutic opportunities forpatients with prostate cancer, we have recently appliedhigh-throughput screening to systematically exploremost currently marketed drugs and drug-like moleculesfor their efficacy against a panel of prostate cells. Theresults indicated that most of the effective compounds,including antineoplastic agents, were nonselective andinhibited both cancer and nonmalignant prostateepithelial cells in equal amounts. Only histone deace-tylase inhibitor trichostatin A, thiram, disulfiram, andmonensin were identified as potent and selective anti-neoplastic agents (4). The molecular mechanisms oftrichostatin A–induced and disulfiram-induced growthinhibition in prostate cancer cells have been previouslystudied (4, 5). However, the antiproliferative effect ofmonensin in prostate cancer cells remains to beexplored.
Monensin is a monocarboxylic acid ionophore veter-inary drug produced by Streptomyces cinnamonensis withantibiotic and antimalarial activity (6–8). In addition tomedicinal use, monensin has been shown to promotemuscle growth in cattle (9), suggesting potential andro-genic effects, which is interesting in the light of theanti–prostate cancer effects. Because monensin is oneof the most potent prostate cancer–specific drugs and
Authors’ Affiliation: 1Turku Centre for Biotechnology, University of Turku,and 2Medical Biotechnology, VTT Technical Research Centre of Finland,Turku, Finland; and 3Institute for Molecular Medicine, Finland (FIMM),University of Helsinki, Helsinki, Finland
Note: Supplementary material for this article are available at MolecularCancer Therapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Kristiina Iljin, Medical Biotechnology, VTT Tech-nical Research Centre of Finland, PL 106, 20521 Turku, Finland. Phone:35-840-032-0642; Fax: 35-820-722-2840. E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-10-0368
�2010 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org 3175
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

drug-like molecules, and it has an excellent safety recordin veterinary medicine, this provides an exciting oppor-tunity for novel cancer treatment. In this study, weexplored the mechanisms of monensin-induced growthinhibition in cultured prostate cancer cells.
Materials and Methods
CellsThe TMPRSS2-ERG gene fusion and AR-positive
prostate carcinoma cell line VCaP was received fromDrs. Adrie van Bokhoven (University of ColoradoHealth Sciences Center, Denver, CO) and KennethPienta (University of Michigan, Ann Arbor, MI) andwere grown in Dulbecco’s Modified Eagle’s Medium(10). LNCaP cells have a mutated AR (T877A) enablingprogestagens, estradiol, and antiandrogens to activateandrogen signaling (11) and were received from Dr.Marco Cecchini (University of Bern, Bern, Switzerland)and grown in T-Medium (Invitrogen Molecular Probes).The nonmalignant RWPE-1 prostate epithelial cells (12)were purchased from American Type Culture Collec-tion and grown according to provider’s instructions.The nonmalignant EP156T prostate epithelial cells werereceived from Dr. Varda Rotter (Weizmann Institute ofScience, Rehovot, Israel) and grown in the media recom-mended by the distributor (13). The identity of thecells was confirmed by array comparative genomichybridization (Agilent Technologies), and the cells werepassaged no longer than 6 months after receipt orresuscitation of frozen aliquots.
CompoundsMonensin (monensin sodium salt) and flutamide were
purchased from Sigma-Aldrich and diluted in ethanol.Bicalutamide was purchased from Sequoia Research Pro-ducts and diluted in ethanol. Disulfiram was purchasedfrom Fluka and diluted in dimethyl sulfoxide.
Cell viability and apoptosis assaysCell viability and apoptosis assays were done on 384-
well plates (Falcon). A total of 2,000 cells per well wereplated in 35 mL of their respective growth media andleft to attach overnight. Compound dilutions wereadded to the cells and incubated for 48 hours. Cellviability was determined with CellTiter-Blue (CTB) orCellTiter-Glo (CTG) cell viability assay (Promega, Inc.).Induction of caspase-3 and caspase-7 activity wasdetected with homogenous Apo-ONE assay (Promega,Inc.). Cell viability and apoptosis assays were then doneaccording to the manufacturer’s instructions. Thefluorometric signal from CTB (excitation FITC485 nm, emission FITC 535 nm) and luminescencesignals (700 nm) from CTG and apoptosis assays werequantified using Envision Multilabel Plate Reader (Per-kin-Elmer).
RNA extraction and quantitative reversetranscriptase PCR
Total RNA was extracted from cultured cells usingRNeasy (Qiagen) according to the manufacturer’sprotocol. Reverse transcription using 500 ng of totalRNA was done with Applied Biosystems’ cDNA synth-esis kit. TaqMan gene expression probes and primersfrom the Universal Probe Library (Roche Diagnostics)were used to study AR, ERG, MYC, Kruppel-like factor6 (KLF6), activating transcription factor 3 (ATF3), metal-lothioneins MT1G and MT1F, thioredoxin binding pro-tein (TXNIP), DNA damage-inducible transcripts 3 and4 (DDIT3 and DDIT4), and b-actin mRNA expression.Primer sequences are listed in Supplementary Table S1.Real-time quantitative PCR was done using ABI Prism7900 (Applied Biosystems). Quantitation was carriedout using the DDCT method with RQ manager 1.2 soft-ware (Applied Biosystems). b-Actin was used as anendogenous control. Average expression of theuntreated control samples was considered for the cal-culation of the fold changes. Two to four replicatesamples were studied for quantitation of mRNA expres-sion.
Gene expression analysis using bead arraysVCaP cells were grown into approximately 70% con-
fluence and treated with 1 mmol/Lmonensin for 3, 6, and24 hours before harvesting. Total RNAwas extracted andRNA integrity was monitored prior to amplification andhybridization using an Experion electrophoresis station(Bio-Rad Laboratories) according to manufacturer’sinstructions. The RNA quality indicator value for allsamples was 10, indicating high-quality, intact RNA.Purified RNA (300 ng) was used for amplification withthe Illumina RNA TotalPrep Amplification kit (Ambion,Austin, TX) and the biotin-labeled cRNA washybridized to Sentrix HumanRef-8 vs.3 Expression Bead-Chips (Illumina). The arrays were scanned with theBeadArray Reader (Illumina). Microarray data are avail-able in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-TABM-1049.
Statistical analysis of gene expression dataThe raw gene expression data were quantile-normal-
ized and analyzedwith theR/Bioconductor software (14).Statistical analysis of differential gene expression after thecompound treatmentswasdoneusing the empirical Bayesstatistics implemented in the eBayes function of the limmapackage (15). Gene expression profiles of the compoundtreated samples were compared with the negative controlsamples. The threshold for differential expression wasq < 0.05 after the Benjamini–Hochberg multiple testingcorrection. The functional gene ontology and pathwayannotations were analyzed for the sets of differentiallyexpressed genes using DAVID (16, 17). To identify drugswith similar or opposite effects on gene expression, Con-nectivity Map 02 was used (18).
Ketola et al.
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3176
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

Western blot analysis and subcellular proteomeextractionFor protein extraction andWestern blot analysis, VCaP
andLNCaP cellswere plated at 70% confluency and left toattach overnight before treatments with indicated com-pounds. Subcellular protein extraction was done with theCompartment Protein Extraction Kit (Chemicon). Thecytoplasmic and nuclear protein fractions were analyzedfrom VCaP cells. Protein concentrations were determinedusing a DC Bradford assay kit (Bio-Rad Laboratories).Tenmgof total proteinwasdenaturedat 95�Cfor 5minutesin Laemmli buffer, separated on a 7% SDS-polyacryla-mide gel, and transferred to a Protran nitrocellulosetransfer membrane (Schleicher & Schuell). Western blotanalysis was done using specific antibodies against AR(1:1,000dilution,mousemonoclonal; Labvision), prostate-specific antigen [(PSA) 1:1,000, rabbit polyclonal; Dako-Cytomation], histone H3 (1:1,000, rabbit polyclonal;Abcam), and b-actin (1:4,000dilution,mouse-monoclonal;Becton Dickinson). Signal was detected with 1:4,000 dilu-tions of appropriate HRP-conjugated secondary antibo-dies (all from Invitrogen Molecular Probes) followed byvisualization with the enhanced chemiluminescencereagent (Amersham Biosciences).
Subcellular localization of androgen receptorFor immunofluorescence stainings, VCaP cells were
androgen deprived by culturing cells in starvation med-ium with 10% charcoal-stripped serum for 18 hours inabsence or presence of 10 mmol/L flutamide prior tosubsequent addition of 35 nmol/L monensin for 24 hoursand stimulation with R1881 for the last 18 hours. Forcontrols, samples without monensin and/or R1881 wereprepared. Cells were fixed with 3.7% paraformaldehydein PBS, permeabilized with 0.2% Triton X-100 in PBS for15 minutes, and blocked with 2% bovine serum albuminor PBS for 30 minutes. Cells were stained with AR anti-body (1:33 dilution, mouse monoclonal; Labvision) andAlexa-conjugated secondary anti-mouse antibody wasused for secondary staining (1:300 dilution; InvitrogenMolecular Probes). Cell nucleus was stained with Vecta-shieldmountingmedium (Vector Laboratories, Ltd.) con-taining 40,6-diamidino-2-phenylindole and images weretaken with Zeiss Axiovert 200M Microscope with thespinning disc confocal unit Yokogawa CSU22 and a ZeissPlan-Neofluar�63 oil 1.4 NA objective (Carl Zeiss Micro-Imaging). Z-stacks with 1 airy unit optical slices wereacquiredwith a step size of 0.5 mmbetween slices, and themaximum intensity projections were created with Slide-Book 4.2.0.7 software (Intelligent Imaging InnovationsInc.).
Endocytosis of transferrin receptorTo study whether monensin affects transferrin inter-
nalization to the cell, cells were grown on cover slips in6-well plates. Monensin (10 mmol/L) or ethanol controlwas added to the cells prior to incubation with 546-labeled transferrin (5 pg/mL, Molecular Probes Europe
BV) for 0, 15, or 30 minutes. Total incubation time formonensin in every sample was 1.5 hours. The amount oftransferrin in the cells was detected with the confocalmicroscope described above.
Determination of combinatorial drug effectsThe nature of interaction and the degree of synergy
between monensin and antiandrogen flutamide or bica-lutamidewere analyzed using the combination index (CI)method (19). The concentration dependence of antiproli-ferative effects was determined for both compounds,either aloneor in combination. Synergywas thenanalyzedaccording to the fixed concentration method, that is, fixedconcentration of flutamide or bicalutamide together withincreasing concentrations of monensin (18, 35, 70, and 140nmol/L), thus providing ameasure of the enhancement ofmonensin activity at various levels of diminished ARactivity. The data were analyzed with Calcusyn software(Biosoft), and the combination index (CI) was calculatedfrom the median effect plots according to the followingequation: CI = D1/DX1 þ D2/DX2, where DX1 and DX2are the concentrations of compounds 1 and 2 needed toproduce a given level of antiproliferative effectwhenusedalone, whereas D1 and D2 are their concentrations thatproduce the same effect when used in combination. A CIof 0.9 to 1.1 indicates additive interaction, values below0.9indicate synergism, and values more than 1.1 indicateantagonism.
Reactive oxygen species detectionReactive oxygen species (ROS) assay was done on 384-
well plates (Falcon). Two thousand cells per well wereplated in 35 mL of medium and left to attach overnight.Monensin or hydrogen peroxide (H2O2) as positive con-trol was added to the cells and incubated for 48 hours.Medium was removed and cells were washed with PBS.Carboxy-H2DCFDA (100 mmol/L) was added to the cellsin 10 mL of medium and incubated in the dark at 37�C for30 minutes. Reagents were removed, cells washed withPBS, and oxidation of the probe was measured in PBS bymonitoring the increase in fluorescence with EnvisionMultilabel Plate Reader (Perkin-Elmer).
Determination of DNA double-strand breaksThe amount of DNAdouble-strand breaks in VCaP and
LNCaP cells was measured by monitoring the inductionof serine 139 phosphorylation of histone variant H2AX(gH2AX). Cells were grown on cover slip slides andincubated for 3, 6, or 24 hours with 1 mmol/L monensinor vehicle control. Cells were stained with the rabbitpolyclonal phosphorylated histone variant H2AX (Ser139) antibody (Abcam). Secondary staining was donewith anti-rabbit Alexa555 antibody (Abcam) and theresults were obtained with confocal microscope.
Determination of aldehyde dehydrogenase activityThe activity of aldehyde dehydrogenase (ALDH)
was determined with Aldefluor reagent (Stemcell Tech-
Monensin Induces Apoptosis in Prostate Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3177
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

nologies) according to the manufacturer’s instructions,but downscaled to 384-well format. Briefly, 2,000 cellsper well were plated in 35 mL of media in 384-well platesand incubated overnight. Monensin, disulfiram, andappropriate controls were added in 15 mL to final con-centration of 1 mmol/L in 50 mL and incubated for 48hours. After incubation, mediumwas removed and cellswere washed with 20 mL of PBS; 10 mL of Aldefluor orAldefluor with DEAB (ALDH inhibitor diethylamino-benzaldehyde) was added to the cells and incubated at37�C for 30 minutes. Solutions were removed, cells werewashed with 20 mL of PBS, and 20 mL of assay buffer wasadded into each well. The fluorometric signal wasdetermined with Envision Multilabel Reader (Perkin-Elmer).
Results
Monensin inhibits prostate cancer cell growthand induces apoptosis
Monensin inhibits prostate cancer cell viability atnanomolar concentrations whereas nonmalignant pros-tate epithelial cells are more resistant (>20-fold) to thecompound (Fig. 1A; ref. 4). The effect of monensin on
cell viabilitywas clearly seen in VCaP (EC50¼ 35 nmol/L)and LNCaP (EC50 ¼ 90 nmol/L) prostate cancer cells,whereas nontumorigenic RWPE-1 (EC50 > 10 mmol/L)and EP156T (EC50 > 1 mmol/L) prostate epithelial cellswere not affected at nanomolar concentrations (Fig. 1A).To determine whether the observed decrease in cellgrowth in response to monensin exposure was becauseof induction of apoptosis, caspase-3 and caspase-7 activ-ities were determined by a quantitative fluorometricassay. Caspase activity was significantly induced inresponse to monensin exposure in VCaP cells (from10 nmol/L) and in LNCaP cells (from 1 mmol/L). Nostatistically significant induction of apoptosis was seenin the nontumorigenic RWPE-1 and EP156T prostatecells, although weak increase in caspase activity wasobserved with 10 mmol/L monensin in EP156T cells(Fig. 1B).
Monensin reduces the expression of androgenreceptor
To get further insights into the growth inhibitorymechanism of monensin, VCaP and LNCaP cells wereexposed to 1 mmol/L of monensin for 3, 6, or 24 hours andAR mRNA expression was measured using quantitativePCR. Monensin reduced AR mRNA levels by 22% (P <0.02) and 48% (P < 0.03) in VCaP cells at 6- and 24-hourtime points (Fig. 2A). In LNCaP cells, AR mRNA levelwas decreased by 45% (P < 0.02) in response to 24-hourmonensin exposure. Accordingly, a significant decreasewas observed in AR protein levels (Fig. 2B). Besides AR,the mRNA expression of androgen-regulated ERG onco-gene was reduced by 30% [P < 0.001 (Fig. 2C)] in VCaPcells, and MYC oncogene was reduced by 60% (P < 0.01)in VCaP cells and 54% (P < 0.03) in LNCaP cells inresponse to 24-hour monensin exposure (Fig. 2D).
Monensin does not affect the subcellularlocalization of androgen receptor
Monensin has been described to alter protein transportand transferrin endocytosis (9, 20). Our results onendocytosis indicated that monensin reduces transferrininternalization in VCaP cells (Supplementary Fig. S1).Because monensin reduced AR expression in the prostatecancer cells, we studied whether monensin affectsnuclear transport of AR in VCaP cells. The results fromimmunofluorescence staining showed slightly reducedAR levels both in the cytoplasm and in the nucleus inresponse to 35-nmol/L monensin exposure (Fig. 3A). Tofurther confirm the result, cytoplasmic and nuclear pro-tein lysates were prepared and separated and run onSDS-PAGE followed by Western blot analysis (Supple-mentary Fig. S2). Flutamide and monensin exposuresalone resulted in significant reduction in AR proteinexpression in the nuclear fractions in comparison tothe control. A combinatorial treatment with monensinand flutamide reduced AR expression even more thaneither of the compounds alone, both in the cytoplasmicand nuclear fractions. In response to flutamide and
A
B
Figure 1.Monensin inhibits cell viability and induces apoptosis in prostatecancer cells. A, the chemical structure of monensin and the cell viabilityresults in response to 48-hour exposure of monensin in VCaP, LNCaP,RWPE-1, and EP156T prostate cells. B, caspace-3 and caspase-7 activityin response to 48-hour monensin treatments (1E�9, 1E�7, and 1E�5 mol/L)in VCaP, LNCaP, RWPE-1, and EP156T cells. Data are presented as mean� SD from 6 independent experiments. Asterisks indicate the statisticalsignificance of the apoptotic induction. ***, P < 0.001.
Ketola et al.
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3178
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

monensin cotreatment, basal PSA protein level wasreduced in both cytoplasmic and nuclear fractions ofthe cells in comparison to flutamide-only–treated cells(Supplementary Fig. S2). A similar combinatorial effecton AR expression was seen in LNCaP cells (Supplemen-tary Fig. S2). Taken together, these results indicate thatmonensin reduces intracellular AR levels especially incombination with flutamide.
Antiandrogens potentiate monensin induced growthinhibition in VCaP and LNCaP cellsBecause monensin exposure reduced AR expression
and a combinatorial treatment with monensin and flu-tamide reduced AR protein expression even more thaneither compound alone, the potential combinatorialeffect of androgen deprivation and monensin on cellviability was studied in VCaP and LNCaP cells. Cellswere treated with 10 mmol/L flutamide or bicalutamidefor 24 hours prior to subsequent exposure to increasingconcentrations of monensin for 48 hours. A synergisticeffect (CI < 0.9) of flutamide and monensin wasobserved with 18–140 nmol/L of monensin in bothVCaP and LNCaP cells (Fig. 3B and C). Synergy wasseen also between bicalutamide and monensin (35–140nmol/L) in VCaP and LNCaP cells (SupplementaryFig. S3).
Monensin induces genes involved in cholesterolsynthesis and oxidative stress defense and showsagonistic effects to terfenadine and antagonisticeffects to progesterone
Genome-wide expression analysis was done in VCaPcells at early time points (3, 6, and 24 hours) to identifypotential early primary effects of monensin as opposed tosecondary alterations. The most significantly altered bio-logical processes in gene ontology analysis (Supplemen-tary Table S2) indicated that cholesterol and lipidmetabolic activity was enriched at 3- and 6-hour timepoints. The 6-hour treatment also induced the expressionof metal binding genes including metallothioneinswhereas 24-hour exposure affected genes involved inprotein transport, cell differentiation, cell cycle, andDNA repair.
Differentially expressed genes at the 6-hour timepoint were compared with the gene expression profilesrepresenting drug responses to more than 1,309 com-pounds using the Connectivity Map approach (Supple-mentary Table S3). Progesterone and pregnenolone,as well as progesterone agonists etynoldiol and dehy-drogesterone, were among the drugs altering geneexpression in an opposite direction than monensin.Progesterone and pregnenolone are precursors ofandrogens and their levels are upregulated in castrate
Figure 2. Monensin reduces theexpression levels of keyoncogenes androgen receptor(AR), ERG and MYC in prostatecancer cells. A, AR mRNAexpression in response to 3-, 6-,and 24-hour monensin exposurein VCaP and LNCaP cells; B, ARprotein expression in response to6- and 24-hour monensinexposure in VCaP and LNCaPcells; C, ERG mRNA expressionin response to 3-, 6-, and 24-hourmonensin exposure in VCaPcells; and D, MYC mRNAexpression in response to 3-, 6-,and 24-hour monensin exposurein VCaP and LNCaP cells.Asterisks indicate statisticalsignificance. *, P < 0.005;**, P < 0.01; ***, P < 0.001.
A
C D
B
Monensin Induces Apoptosis in Prostate Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3179
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

resistant prostate cancers (21, 22). Terfenadine, a hista-mine H1 receptor antagonist, was the most enrichedcompound altering gene expression in the same direc-tion as monensin. Terfenadine induces apoptosis inmelanoma cells (23) and causes massive productionof H2O2 in cultured cerebellar neurons (24). Takentogether, these results suggest that monensin has oppo-site effects to androgen precursors and may induceoxidative stress in prostate cancer cells.
Monensin induces a gene expression signaturecharacteristic of oxidative stress response
Analysis of gene expression profiles also indicatedthat the expression of several previously identifiedoxidative stress–associated markers (25) was altered
(�3 < fold change >3) in monensin exposed VCaP cells.The gene expression changes were validated in VCaP,LNCaP, RWPE-1, and EP156T cells using quantitativePCR (Fig. 4A; Supplementary Figs. S4 and S5). At 3-hourtime point, thioredoxin-interacting protein (TXNIP)was the most downregulated mRNA in VCaP andLNCaP cells (Fig. 4A; Supplementary Fig. S4). TXNIPis an oxidative stress responsive gene regulatingintracellular ROS by inhibiting thioredoxin activity orlimiting its bioavailability (26–29). Metallothioneinswere the most rapidly induced genes in response tomonensin exposure in VCaP and LNCaP cells (Fig. 4A;Supplementary Fig. S4). Metallothioneins are known toprotect cells against oxidative stress by binding to freemetals and H2O2 (30). Interestingly, our previous resultsindicate that VCaP prostate cancer cells are sensitive to
A
B C
Figure 3. Monensin-mediated growth arrest and inhibition of androgen receptor signaling is enhanced by androgen deprivation. A, immunofluorescencestaining of androgen receptor showing the effect of monensin and flutamide in VCaP cells. Cells were androgen deprived by culturing in starvationmediumwith 10% charcoal-stripped serum for 18 hours in absence or presence of 10 mmol/L flutamide prior to subsequent addition of 35 nmol/L monensin for24 hours and stimulation with R1881 for the last 18 hours. For controls, samples without monensin and/or R1881 were prepared. Cells were stainedwith androgen receptor antibody and the results were obtained with confocal microscope and�63 objective. B, Fa-CI plot showing the combinatorial effect ofmonensin and flutamide in VCaP cells. Cells were treated with 10 mmol/L flutamide for 72 hours followed by 18 nmol/L (CI ¼ 0.56), 35 nmol/L(CI ¼ 0.73), 70 nmol/L, (CI ¼ 0.69), or 140 nmol/L (CI ¼ 0.49) monensin exposure for the last 48 hours. C, Fa-CI plot showing the combinatorial effect ofmonensin and flutamide in LNCaP cells. Cells were treated with 10 mmol/L flutamide for 72 hours followed by 18 nmol/L (CI ¼ 0.60), 35 nmol/L(CI ¼ 0.72), 70 nmol/L (CI ¼ 0.54), or 140 nmol/L (CI ¼ 0.33) monensin exposure for the last 48 hours. Fa, fraction affected.
Ketola et al.
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3180
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

oxidative stress and metallothioneins were also inducedin response to disulfiram exposure (4). MT1F andMT1GmRNA expression levels were highly induced inresponse to 3- and 6-hour monensin exposure inVCaP cells (10- to 30-fold). In LNCaP cells the inductionof metallothioneins was seen (2- to 20-fold), at 6- and24-hour time points. In addition, 2 apoptosis-associatedtranscription factors, tumor suppressor KLF6 andATF3 were induced by monensin in a time-dependent manner (Fig. 4A; Supplementary Fig. S5)in VCaP and LNCaP cells. ATF3 is a proapoptoticprotein that is upregulated under stress conditions suchas oxidative stress and DNA damage (31). KLF6has been shown to regulate ATF3 expression in prostatecancer and they are both needed for cancer cellapoptosis in stress conditions (32). Moreover, DNAdamage-inducible transcripts 3 (DDIT3, GADD153,CHOP) and 4 (DDIT4, REDD1) were induced in mon-ensin exposed prostate cancer cells (Fig. 4B; Supple-mentary Fig. S5). DDIT3 and DDIT4 are expressed inendoplasmic reticulum stress–induced apoptosis andDNAdamage (33, 34). gH2AX immunofluorescence stain-ing was also positive in monensin-treated VCaP andLNCaP cells, reflecting DNA damage and onset of apop-tosis (Fig. 4B).
Monensin induces oxidative stress, and the effectof monensin on cell viability is antagonized byantioxidant vitamin C
To validate the oxidative stress induction in response tomonensin exposure, the levels of ROS were detectedusing carboxy-H2DCFDA, a cell-permeant indicatorfor ROS. VCaP, LNCaP, RWPE-1, and EP156T cells wereexposed to monensin or solvent control for 48 hours. As apositive ROS control, cells were exposed to 400 mmol/LH2O2 for 6 hours. Results indicated that monensinsignificantly increases ROS levels in VCaP cells(Fig. 5A). ROS levels were also increased in LNCaP cells,although not as strongly as in the VCaP cells. In contrast,monensin did not induce ROS in nontumorigenicRWPE-1 and EP156T cells.
Vitamin C (L-ascorbic acid) is an antioxidant that acts asa scavenger for a wide range of ROS. Recently, thecytotoxic effects of antineoplastic drugs that producemitochondrial dysfunction, such as loss of mitochondrialmembrane potential and an increase in ROS levels, wereshown to be inhibited by vitamin C in leukemic cells (35).To find out whether monensin-induced cell death can beantagonized by vitamin C treatment, the effect of10 mmol/L vitamin C and 100 nmol/Lmonensin on VCaPand LNCaP cell viability was analyzed after 48 hours of
A
B
Figure 4.Monensin induces oxidative stress and DNA double-strand breaks in prostate cancer cells. A, expression levels of oxidative stress responsiveMT1F,MT1G, ATF3, KLF6, DDIT3, DDIT4, and TXNIP mRNAs in VCaP, LNCaP, RWPE-1, and EP156T cells in response to monensin treatments for 3, 6, and24 hours. B, immunofluorescence staining using DNA damage and apoptosis marker (gH2AX) antibody (red). 40,6-Diamidino-2-phenylindole (blue) wasused as a DNA marker. VCaP and LNCaP cells were treated with 1 mmol/L monensin or solvent control for 3 hours prior to staining. Images were taken withconfocal microscope and �63 objective.
Monensin Induces Apoptosis in Prostate Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3181
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
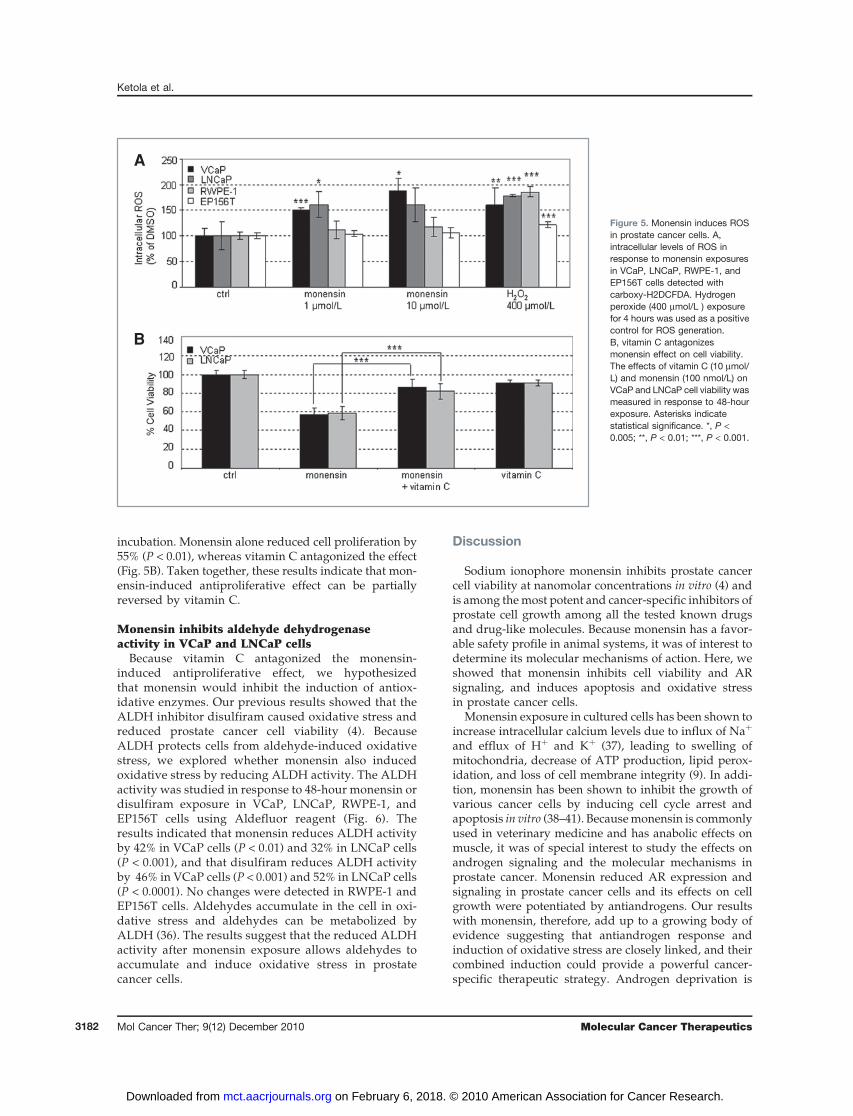
incubation. Monensin alone reduced cell proliferation by55% (P < 0.01), whereas vitamin C antagonized the effect(Fig. 5B). Taken together, these results indicate that mon-ensin-induced antiproliferative effect can be partiallyreversed by vitamin C.
Monensin inhibits aldehyde dehydrogenaseactivity in VCaP and LNCaP cells
Because vitamin C antagonized the monensin-induced antiproliferative effect, we hypothesizedthat monensin would inhibit the induction of antiox-idative enzymes. Our previous results showed that theALDH inhibitor disulfiram caused oxidative stress andreduced prostate cancer cell viability (4). BecauseALDH protects cells from aldehyde-induced oxidativestress, we explored whether monensin also inducedoxidative stress by reducing ALDH activity. The ALDHactivity was studied in response to 48-hour monensin ordisulfiram exposure in VCaP, LNCaP, RWPE-1, andEP156T cells using Aldefluor reagent (Fig. 6). Theresults indicated that monensin reduces ALDH activityby 42% in VCaP cells (P < 0.01) and 32% in LNCaP cells(P < 0.001), and that disulfiram reduces ALDH activityby 46% in VCaP cells (P < 0.001) and 52% in LNCaP cells(P < 0.0001). No changes were detected in RWPE-1 andEP156T cells. Aldehydes accumulate in the cell in oxi-dative stress and aldehydes can be metabolized byALDH (36). The results suggest that the reduced ALDHactivity after monensin exposure allows aldehydes toaccumulate and induce oxidative stress in prostatecancer cells.
Discussion
Sodium ionophore monensin inhibits prostate cancercell viability at nanomolar concentrations in vitro (4) andis among themost potent and cancer-specific inhibitors ofprostate cell growth among all the tested known drugsand drug-like molecules. Because monensin has a favor-able safety profile in animal systems, it was of interest todetermine its molecular mechanisms of action. Here, weshowed that monensin inhibits cell viability and ARsignaling, and induces apoptosis and oxidative stressin prostate cancer cells.
Monensin exposure in cultured cells has been shown toincrease intracellular calcium levels due to influx of Naþ
and efflux of Hþ and Kþ (37), leading to swelling ofmitochondria, decrease of ATP production, lipid perox-idation, and loss of cell membrane integrity (9). In addi-tion, monensin has been shown to inhibit the growth ofvarious cancer cells by inducing cell cycle arrest andapoptosis in vitro (38–41). Becausemonensin is commonlyused in veterinary medicine and has anabolic effects onmuscle, it was of special interest to study the effects onandrogen signaling and the molecular mechanisms inprostate cancer. Monensin reduced AR expression andsignaling in prostate cancer cells and its effects on cellgrowth were potentiated by antiandrogens. Our resultswith monensin, therefore, add up to a growing body ofevidence suggesting that antiandrogen response andinduction of oxidative stress are closely linked, and theircombined induction could provide a powerful cancer-specific therapeutic strategy. Androgen deprivation is
A
B
Figure 5. Monensin induces ROSin prostate cancer cells. A,intracellular levels of ROS inresponse to monensin exposuresin VCaP, LNCaP, RWPE-1, andEP156T cells detected withcarboxy-H2DCFDA. Hydrogenperoxide (400 mmol/L ) exposurefor 4 hours was used as a positivecontrol for ROS generation.B, vitamin C antagonizesmonensin effect on cell viability.The effects of vitamin C (10 mmol/L) and monensin (100 nmol/L) onVCaP and LNCaP cell viability wasmeasured in response to 48-hourexposure. Asterisks indicatestatistical significance. *, P <0.005; **, P < 0.01; ***, P < 0.001.
Ketola et al.
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3182
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

known to decrease antioxidative capacity, elevate ROSanabolism, and sensitize AR-positive prostate cancer tooxidative stress–induced death in vitro and in vivo (42, 43).Because the basal level of oxidative stress is higher inprostate cancer cells than in nonmalignant prostate cells, astrategy that takes advantage of prostate cancer sensitivityto acute exposure of oxidative stress has been proposed(44–47). The results fromthis study indicate thatmonensininduced a gene expression signature characteristic ofoxidative stress response and elevated intracellular levelsof ROS in prostate cancer cells, but not in normal prostateepithelial cells studied. We hypothesize that monensinleads to rapid induction of oxidative stress followed bydownregulation of AR signaling, which then furtherincreases sensitivity of prostate cancer cells to oxidativestress. Moreover, the effect of monensin on cell viabilitywas antagonizedby antioxidant vitaminC, indicating thatthemonensin-induced antiproliferative effect was depen-dent on the elevated level of ROS.Monensin changed the gene expression profile of pros-
tate cancer cells in an opposite direction than progester-one, a precursor of androgens with documentedantioxidant properties (48). Interestingly, progesteronelevels are known to be elevated in hormone refractoryprostate cancers (21, 22). Monensin also reduced theexpression of oncogenes ERG and MYC ERG expressionis regulated by androgens in TMPRSS2-ERG positiveprostate cancers (1, 49) andMYC is a known downstreamtarget of ERG. MYC plays a role in cell cycle progression,apoptosis, and cellular transformation and is frequentlyamplified and overexpressed in prostate cancer (50).MYC is also known to protect cells against oxidativestress (51) and in leukemia cells, the oxidative stress–induced MYC mRNA downregulation has been identi-fied as a mechanism of 2-methoxyestradiol–inducedapoptosis (52). Thus, these results also support the ideathat monensin induces growth inhibition by sensitizingprostate cancer cells to oxidative stress.Furthermore, monensin reduced the ALDH activity in
prostate cancer cells. ALDH is constituted of antioxidantand detoxifying enzymes that metabolize aldehydesaccumulated in the cell under oxidative stress conditions(36). We have previously shown that prostate cancer cellsare sensitive to ALDH inhibitor disulfiram (4). Over-expression of ALDH 1A1 has been shown to reduce
oxidation-induced toxicity in neuroblastoma cells (53).Recently, ALDH 1A1 has also been identified as a markerfor malignant prostate stem cells and predictor of out-come for prostate cancer patients (54, 55). Interestingly,cancer stem cells overexpress ROS detoxifying genes andthus have a stronger antioxidant defense system thantheir nontumorigenic counterparts (56). Moreover,2 structurally highly similar antibiotic ionophores tomonensin—salinomycin and nigericin—have recentlybeen identified as inhibitors of breast cancer stem cellsby the high-throughput screening (57). Inhibition ofALDH activity and the structural similarity to knowncancer stem cell inhibitors suggest that monensin mayhave additional effects on cancer stem cells and theirsensitization to oxidative stress–induced cell death.
We propose monensin as a potential well-tol-erated, in vivo compatible drug with strong proapo-ptotic effects that are specific to prostate cancer cells,and synergistic effects with antiandrogens. Overdosesof monensin have been linked with toxic effects in someanimal species (58) indicating that possible studies withmonensin in humans should be designed carefully.Moreover, our data suggest a general strategy by whichthe effects of antiandrogens could be enhanced bycombinatorial administration with agents that increaseoxidative stress in prostate cancer cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest have been disclosed.
Acknowledgments
We thank Finnish DNA Microarray Centre for doing the Illuminaexperiments and Miro Viitala for excellent technical assistance.
Grant Support
K. Ketola: EU-GENICA project (FP7-HEALTH-2007-A); P. Vainio: TIME(Disseminated Tumour Cells as Targets for Inhibiting Metastasis of EpithelialTumours) project; V. Fey: Academy of Finland; O. Kallioniemi: Academy ofFinland, the Cancer Organizations of Finland and Sigrid Juselius Foundation;and K. Iljin: EU-PRIMA project (contract no. LSHC-CT-204–504587) and Acad-emy of Finland.
The costs of publication of this article were defrayed in part by the pay-ment of page charges. This article must therefore be hereby marked adver-tisement inaccordancewith18U.S.C.Section1734solely to indicate this fact.
Received 04/19/2009; revised 09/21/2010; accepted 09/27/2010;published 12/14/2010.
Figure 6. The inhibition ofaldehyde dehydrogenase (ALDH)activity in prostate cancer cells.ALDH activity was measured withAldefluor assay in responseto 1-mmol/L exposures ofmonensin and disulfiram, andappropriate controls for 48 hoursin VCaP, LNCaP, RWPE-1, andEP156T cells. Asterisks indicatestatistical significance. ***, P <0.001.
Monensin Induces Apoptosis in Prostate Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3183
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

References1. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun
XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factorgenes in prostate cancer. Science 2005;310:644–8.
2. Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, HelgesonBE, et al. TMPRSS2:ETV4 gene fusions define a third molecularsubtype of prostate cancer. Cancer Res 2006;66:3396–4000.
3. Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR,et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 genefusions in prostate cancer. Cancer Res 2008;68:73–80.
4. Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, et al. High-throughput cell-based screening of 4910 known drugs and drug-likesmall molecules identifies disulfiram as an inhibitor of prostate cancercell growth. Clin Cancer Res 2009;15:6070–8.
5. Bjorkman M, Iljin K, Halonen P, Sara H, Kaivanto E, Nees M, et al.Defining the molecular action of HDAC inhibitors and synergism withandrogen deprivation in ERG-positive prostate cancer. Int J Cancer2008;123:2774–81.
6. Westley JW, Evans RH Jr, Sello LH, Troupe N, Liu C, Miller PA.Isolation of novel antibiotics X-14667A and X-14667B from Strepto-myces cinnamonensis subsp. urethanofaciens and their character-ization as 2-phenethylurethanes of monensins B and A. J Antibiot(Tokyo) 1981;34:1248–52.
7. Adovelande J, Schrevel J. Carboxylic ionophores in malaria che-motherapy: the effects of monensin and nigericin on plasmodiumfalciparum in vitro and plasmodium vinckei petteri in vivo. Life Sci1996;59:PL309–15.
8. Otoguro K, Kohana A, Manabe C, Ishiyama A, Ui H, Shiomi K, et al.Potent antimalarial activities of polyether antibiotic, X-206. J Antibiot(Tokyo) 2001;54:658–63.
9. Mollenhauer HH, Morre DJ, Rowe LD. Alteration of intracellular trafficby monensin; mechanism, specificity and relationship to toxicity.Biochim Biophys Acta 1990;1031:225–46.
10. Korenchuk S, Lehr JE, MClean L, Lee YG, Whitney S, Vessella R, et al.VCaP, a cell-based model system of human prostate cancer. In Vivo2001;15:163–8.
11. Harris SE, Rong Z, Harris MA, Lubahn DB. Androgen receptor inhuman prostate carcinoma LNCaP/ADEP cells contains a mutationwhich alters the specificity of the steroid dependent transcriptionalactivation region. Endocrinology 1990;126(Suppl):93–7.
12. Webber MM, Bello D, Kleinman HK, Hoffman MP. Acinar differen-tiation by non-malignant immortalized human prostatic epithelialcells and its loss by malignant cells. Carcinogenesis 1997;18:1225–31.
13. Kogan I, Goldfinger N, Milyavsky M, Cohen M, Shats I, Dobler G, et al.hTERT-immortalized prostate epithelial and stromal-derived cells: anauthentic in vitro model for differentiation and carcinogenesis. CancerRes 2006;66:3531–40.
14. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S,et al. Bioconductor: open software development for computationalbiology and bioinformatics. Genome Biol 2004;5:R80.
15. Smyth GK. Linear models and empirical bayes methods for assessingdifferential expression in microarray experiments. Stat Appl GenetMolBiol 2004;3:1–25.
16. Dennis G, Jr, Sherman BT, Hosack DA, Yang J, GaoW, Lane HC, et al.DAVID: database for annotation, visualization, and integrated discov-ery. Genome Biol 2003;4:P3.
17. Huang da W, Sherman BT, Lempicki RA. Systematic and integrativeanalysis of large gene lists using DAVID bioinformatics resources. NatProtoc 2009;4:44–57.
18. Lamb J. The connectivity map: a new tool for biomedical research. NatRev Cancer 2007;7:54–60.
19. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships:the combined effects of multiple drugs or enzyme inhibitors. AdvEnzyme Regul 1984;22:27–55.
20. Stein BS, Bensch KG, Sussman HH. Complete inhibition of trans-ferrin recycling by monensin in K562 cells. J Biol Chem 1984;259:14762–72.
21. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC,WoodCA, et al.Androgen levels increase by intratumoral de novo steroidogenesisduring progression of castration-resistant prostate cancer. CancerRes 2008;68:6407–15.
22. Leon CG, Locke JA, Adomat HH, Etinger SL, Twiddy AL, NeumannRD, et al. Alterations in cholesterol regulation contribute to theproduction of intratumoral androgens during progression to castra-tion-resistant prostate cancer in a mouse xenograft model. Prostate2010;70:390–400.
23. Jangi SM, Ruiz-Larrea MB, Nicolau-Galmes F, Andollo N, Arroyo-Berdugo Y, Ortega-Martinez I, et al. Terfenadine-induced apoptosis inhuman melanoma cells is mediated through Ca2þ homeostasismodulation and tyrosine kinase activity, independently of H1 hista-mine receptors. Carcinogenesis 2008;29:500–9.
24. Diaz-Trelles R, Novelli A, Vega JA, Marini A, Fernandez-Sanchez MT.Antihistamine terfenadine potentiates NMDA receptor-mediated cal-cium influx, oxygen radical formation, and neuronal death. Brain Res2000;880:17–27.
25. Han ES, Muller FL, Perez VI, Qi W, Liang H, Xi L, et al. The in vivo geneexpression signature of oxidative stress. Physiol Genomics2008;34:112–26.
26. Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H,et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function andexpression. J Biol Chem 1999;274:21645–50.
27. Wang Y, De Keulenaer GW, Lee RT. Vitamin D(3)-up-regulated pro-tein-1 is a stress-responsive gene that regulates cardiomyocyteviability through interaction with thioredoxin. J Biol Chem2002;277:26496–500.
28. Baker AF, Koh MY, Williams RR, James B, Wang H, Tate WR, et al.Identification of thioredoxin-interacting protein 1 as a hypoxia-indu-cible factor 1alpha-induced gene in pancreatic cancer. Pancreas2008;36:178–86.
29. Junn E, Han SH, Im JY, Yang Y, Cho EW, UmHD, et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing thethioredoxin function. J Immunol 2000;164:6287–95.
30. Jimenez I, Gotteland M, Zarzuelo A, Uauy R, Speisky H. Loss of themetal binding properties of metallothionein induced by hydrogenperoxide and free radicals. Toxicology 1997;120:37–46.
31. Hai T, Hartman MG. The molecular biology and nomenclature of theactivating transcription factor/cAMP responsive element bindingfamily of transcription factors: activating transcription factor proteinsand homeostasis. Gene 2001;273:1–11.
32. Huang X, Li X, Guo B. KLF6 induces apoptosis in prostate cancercells through up-regulation of ATF3. J Biol Chem 2008;283:29795–801.
33. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and suffi-cient for ER stress-induced upregulation of REDD1 expression. Bio-chem Biophys Res Commun 2009;379:451–5.
34. Guan L, Han B, Li Z, Hua F, Huang F, Wei W, et al. Sodium seleniteinduces apoptosis by ROS-mediated endoplasmic reticulum stressand mitochondrial dysfunction in human acute promyelocytic leuke-mia NB4 cells. Apoptosis 2009;14:218–25.
35. Heaney ML, Gardner JR, Karasavvas N, Golde DW, Scheinberg DA,Smith EA, et al. Vitamin C antagonizes the cytotoxic effects ofantineoplastic drugs. Cancer Res 2008;68:8031–8.
36. Davydov VV, Dobaeva NM, Bozhkov AI. Possible role of alteration ofaldehyde's scavenger enzymes during aging. Exp Gerontol 2004;39:11–6.
37. Donoho AL. Biochemical studies on the fate of monensin in animalsand in the environment. J Anim Sci 1984;58:1528–39.
38. Park WH, Jung CW, Park JO, Kim K, Kim WS, Im YH, et al. Monensininhibits the growth of renal cell carcinoma cells via cell cycle arrest orapoptosis. Int J Oncol 2003;22:855–60.
39. Park WH, Kim ES, Jung CW, Kim BK, Lee YY. Monensin-mediatedgrowth inhibition of SNU-C1 colon cancer cells via cell cycle arrestand apoptosis. Int J Oncol 2003;22:377–82.
Ketola et al.
Mol Cancer Ther; 9(12) December 2010 Molecular Cancer Therapeutics3184
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

40. Park WH, Kim ES, Kim BK, Lee YY. Monensin-mediated growthinhibition in NCI-H929 myeloma cells via cell cycle arrest and apop-tosis. Int J Oncol 2003;23:197–204.
41. Park WH, Seol JG, Kim ES, Kang WK, Im YH, Jung CW, et al.Monensin-mediated growth inhibition in human lymphoma cellsthrough cell cycle arrest and apoptosis. Br J Haematol2002;119:400–7.
42. Pinthus JH, Bryskin I, Trachtenberg J, Lu JP, Singh G, Fridman E, et al.Androgen induces adaptation to oxidative stress in prostate cancer:implications for treatment with radiation therapy. Neoplasia2007;9:68–80.
43. TamNN, Gao Y, Leung YK, Ho SM. Androgenic regulation of oxidativestress in the rat prostate: involvement of NAD(P)H oxidases andantioxidant defense machinery during prostatic involution andregrowth. Am J Pathol 2003;163:2513–22.
44. Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress inprostate cancer. Cancer Lett 2009;282:125–36.
45. Yossepowitch O, Pinchuk I, Gur U, Neumann A, Lichtenberg D, BanielJ. Advanced but not localized prostate cancer is associated withincreased oxidative stress. J Urol 2007;178:1238-43; discussion1243–4.
46. Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidativestress is inherent in prostate cancer cells and is required for aggres-sive phenotype. Cancer Res 2008;68:1777–85.
47. Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, et al.Global gene expression analysis of reactive stroma in prostate cancer.Clin Cancer Res 2009;15:3979–89.
48. Ozacmak VH, Sayan H. The effects of 17beta estradiol, 17alphaestradiol and progesterone on oxidative stress biomarkers in ovar-iectomized female rat brain subjected to global cerebral ischemia.Physiol Res 2009;58:909–12.
49. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X,Morris DS, et al. Distinct classes of chromosomal rearrangementscreate oncogenic ETS gene fusions in prostate cancer. Nature2007;448:595–9.
50. Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem2006;97:433–47.
51. Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M,et al. c-Myc phosphorylation is required for cellular response tooxidative stress. Mol Cell 2006;21:509–19.
52. Chow JM, Liu CR, Lin CP, Lee CN, Cheng YC, Lin S, et al. Down-regulation of c-myc determines sensitivity to 2-methoxyestradiol-induced apoptosis in human acute myeloid leukemia. Exp Hematol2008;36:140–8.
53. Zhang M, Shoeb M, Goswamy J, Liu P, Xiao TL, Hogan D, et al.Overexpression of aldehyde dehydrogenase 1A1 reduces oxidation-induced toxicity in SH-SY5Y neuroblastoma cells. J Neurosci Res2010;88:686–94.
54. Burger PE, Gupta R, Xiong X, Ontiveros CS, Salm SN, Moscatelli D,et al. High aldehyde dehydrogenase activity: a novel functional markerof murine prostate stem/progenitor cells. Stem Cells 2009;27:2220–8.
55. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker formalignant prostate stem cells and predictor of prostate cancerpatients’ outcome. Lab Invest 2010;90:234–44.
56. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al.Association of reactive oxygen species levels and radioresistance incancer stem cells. Nature 2009;458:780–3.
57. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA,et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009;138:645–59.
58. Langston VC, Galey F, Lovell R, BuckWB. Toxicity and therapeutics ofmonensin: a review. Vet Med 1985;80:75–84.
Monensin Induces Apoptosis in Prostate Cancer Cells
www.aacrjournals.org Mol Cancer Ther; 9(12) December 2010 3185
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

2010;9:3175-3185. Mol Cancer Ther Kirsi Ketola, Paula Vainio, Vidal Fey, et al. Androgen Signaling Leading to Apoptosis in Prostate Cancer CellsMonensin Is a Potent Inducer of Oxidative Stress and Inhibitor of
Updated version
http://mct.aacrjournals.org/content/9/12/3175
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/9/12/3175.full#ref-list-1
This article cites 57 articles, 14 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/9/12/3175.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/9/12/3175To request permission to re-use all or part of this article, use this link
on February 6, 2018. © 2010 American Association for Cancer Research. mct.aacrjournals.org Downloaded from