molecular cell article · molecular cell article ... (clb2) in cim3-1 cells at 37 c. clb2...
TRANSCRIPT

Molecular Cell
Article
Failure of Amino Acid HomeostasisCauses Cell Death following Proteasome InhibitionAmila Suraweera,1 Christian Munch,2 Ariane Hanssum,1 and Anne Bertolotti1,*1MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH, UK2Department of Cell Biology, Harvard Medical School, Boston, MA 02115, USA
*Correspondence: [email protected]://dx.doi.org/10.1016/j.molcel.2012.08.003
SUMMARY
The ubiquitin-proteasome system targets manycellular proteins for degradation and thereby con-trols most cellular processes. Although it is wellestablished that proteasome inhibition is lethal, theunderlying mechanism is unknown. Here, we showthat proteasome inhibition results in a lethal aminoacid shortage. In yeast, mammalian cells, and flies,the deleterious consequences of proteasome inhibi-tion are rescued by amino acid supplementation. Inall three systems, this rescuing effect occurs withoutnoticeable changes in the levels of proteasome sub-strates. In mammalian cells, the amino acid scarcityresulting from proteasome inhibition is the signalthat causes induction of both the integrated stressresponse and autophagy, in an unsuccessful attemptto replenish the pool of intracellular amino acids.These results reveal that cells can tolerate proteinwaste, but not the amino acid scarcity resultingfrom proteasome inhibition.
INTRODUCTION
The controlled degradation of proteins by the proteasome is
essential in all cells and impairment of the ubiquitin-proteasome
system contributes to many pathological conditions (Hershko
and Ciechanover, 1998; Finley, 2009). Although the proteasome
inhibitor Bortezomib is used for the treatment of multiple
myeloma (Hideshima et al., 2001; Goldberg, 2007; Navon and
Ciechanover, 2009), it remains unclear how proteasome failure
kills cells.
Because the ubiquitin-proteasome system controls the degra-
dation of a large number of cellular proteins including short-lived,
regulatory, and damaged or misfolded proteins (Hershko and
Ciechanover, 1998; Schwartz and Ciechanover, 2009), it has
been assumed that accumulation of no-longer needed proteins
underlies the toxicity of proteasome inhibition. However, how
such proteins become harmful to cells and organisms remains
unclear. Failure to degrade key regulatory proteins could also
perturb cellular function. The transcription regulator NF-kB is
one such regulatory protein activated by proteasome cleavage
(Palombella et al., 1994). NF-kB is constitutively activated in
multiple myeloma and thus was thought to be a prominent
242 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc
target of proteasome inhibitors (Schwartz and Ciechanover,
2009). However, as selective inhibition of NF-kB does not
recapitulate the cytotoxic effects of proteasome inhibition
(Hideshima et al., 2002), alternative mechanisms need to be
considered.
The function of the proteasome in protein waste disposal
has been extensively studied (Hershko and Ciechanover, 1998;
Finley, 2009). In addition to this well-described function, the
proteasome also recycles amino acids. The importance of this
aspect of proteasomal degradation for normal cell and organ-
ismal function is unclear. In contrast, recycling amino acids is
a well-established function of autophagy in starving cells (Naka-
togawa et al., 2009). Under conditions of amino acid starvation,
both autophagy and proteasomal degradation are required to
maintain adequate amino acid levels for sustaining protein
synthesis (Onodera and Ohsumi, 2005; Vabulas and Hartl,
2005). Despite the established importance of protein recycling
under conditions of severe nutrient deficiency, it is unclear
whether protein degradation contributes an important fraction
of the supply of amino acids under physiological conditions.
Because the mechanisms underlying the toxicity resulting
from proteasome inhibition are unclear, and as it is unknown
whether protein degradation contributes an essential fraction
of the intracellular amino acid pool under normal conditions,
we examined whether the lethality resulting from proteasome
inhibition is mediated by perturbation of amino acid homeo-
stasis. In three model systems—yeast, mammalian cells, and
Drosophila—we find that proteasome inhibition causes a lethal
shortage of amino acids and the lethality resulting from protea-
some inhibition is rescued upon amino acid supplementation,
without any detectable decrease in the levels of proteasome
substrates in proteasome-inhibited cells.
RESULTS
Failure of Amino Acid Homeostasis Causes Cell Deathfollowing Proteasome Inhibition in YeastWe first assessed whether amino acid homeostasis was altered
following proteasome impairment in yeast. As expected (Ghislain
et al., 1993), exposure to 37�C for 20 hr was lethal to the yeast
proteasome mutant cim3-1, harboring a thermosensitive muta-
tion in the regulatory subunit Rpt6 (Figure S1A available online).
We next monitored the levels of free amino acids in yeast cells
following proteasome inhibition. Exponentially growing yeast
cells were inoculated at an optical density of 0.2 and grown at
30�C in the standard rich medium yeast peptone dextrose
.
H
Ura3-3(GFP)
Ponceau S
cim3-1
25 °
C
37 °
C
37 °
C 0
.5 h
37 °
C 0
.5 h
+ A
A
37 °
C 1
h
37 °
C 1
h +
AA
B
% o
f ini
tial
A
I
100
97
37
54
35S-methionine
Coomassie
AA - + - + - + - +WT cim3-1WT cim3-1
30 °C 37 °C
label WT/ cim3-1 100 19 88 %
C
dead cells
Fluorescence intensity Propidium iodide
Cel
l num
ber
G
4 hours 6 hours
WT
cim
3-1
Ponceau S
0 hours
WT cim3-1 WT cim3-1
30 °
C
30 °
C30
°C
37 °
C
37 °
C +
AA
30
°C
37 °
C37
°C
+ A
A
30 °
C37
°C
37 °
C +
AA
30 °
C37
°C
37 °
C +
AA
Ubn10483
49
37 °C + AA
4 °C
37 °C
cim3-1
21 h at 4 °C or
37 °C +/- AA
2 d at 30 °C
plate
1
2
3
+ AA
cim3-1
23 h at 38 °C24 h at 30 °C
48 h at 30 °C
+ AA
pre1-1
23 h at 37 °C25 h at 30 °C
48 h at 30 °C
E FD
CLB2
Ponceau
30 °
C W
T
30 °
Ccim3-1
30 °
C
37 °
C
37 °
C +
AA
30 °
C
37 °
C
37 °
C +
AA
0 hours 4 hours
WT cim3-1
WT
25 °
C
37 °
C
37 °
C 0
.5 h
37 °
C 0
.5 h
+ A
A
37 °
C 1
h
37 °
C 1
h +
AA
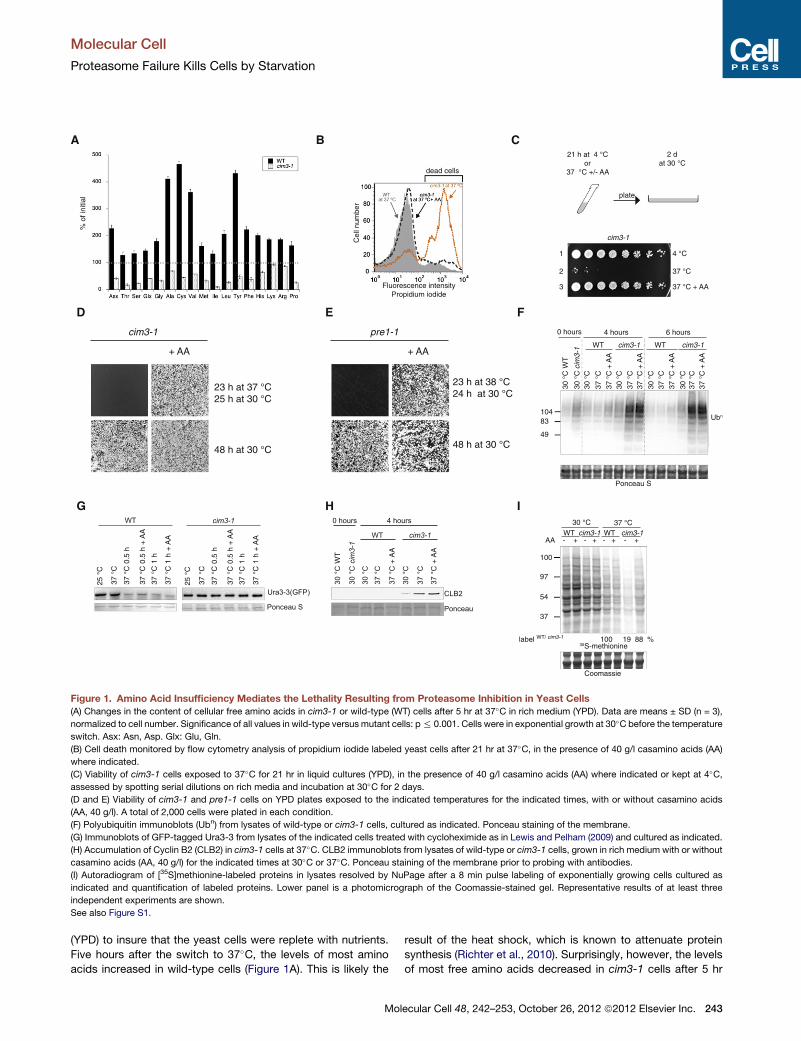
Figure 1. Amino Acid Insufficiency Mediates the Lethality Resulting from Proteasome Inhibition in Yeast Cells
(A) Changes in the content of cellular free amino acids in cim3-1 or wild-type (WT) cells after 5 hr at 37�C in rich medium (YPD). Data are means ± SD (n = 3),
normalized to cell number. Significance of all values in wild-type versus mutant cells: p% 0.001. Cells were in exponential growth at 30�C before the temperature
switch. Asx: Asn, Asp. Glx: Glu, Gln.
(B) Cell death monitored by flow cytometry analysis of propidium iodide labeled yeast cells after 21 hr at 37�C, in the presence of 40 g/l casamino acids (AA)
where indicated.
(C) Viability of cim3-1 cells exposed to 37�C for 21 hr in liquid cultures (YPD), in the presence of 40 g/l casamino acids (AA) where indicated or kept at 4�C,assessed by spotting serial dilutions on rich media and incubation at 30�C for 2 days.
(D and E) Viability of cim3-1 and pre1-1 cells on YPD plates exposed to the indicated temperatures for the indicated times, with or without casamino acids
(AA, 40 g/l). A total of 2,000 cells were plated in each condition.
(F) Polyubiquitin immunoblots (Ubn) from lysates of wild-type or cim3-1 cells, cultured as indicated. Ponceau staining of the membrane.
(G) Immunoblots of GFP-tagged Ura3-3 from lysates of the indicated cells treated with cycloheximide as in Lewis and Pelham (2009) and cultured as indicated.
(H) Accumulation of Cyclin B2 (CLB2) in cim3-1 cells at 37�C. CLB2 immunoblots from lysates of wild-type or cim3-1 cells, grown in rich medium with or without
casamino acids (AA, 40 g/l) for the indicated times at 30�C or 37�C. Ponceau staining of the membrane prior to probing with antibodies.
(I) Autoradiogram of [35S]methionine-labeled proteins in lysates resolved by NuPage after a 8 min pulse labeling of exponentially growing cells cultured as
indicated and quantification of labeled proteins. Lower panel is a photomicrograph of the Coomassie-stained gel. Representative results of at least three
independent experiments are shown.
See also Figure S1.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
(YPD) to insure that the yeast cells were replete with nutrients.
Five hours after the switch to 37�C, the levels of most amino
acids increased in wild-type cells (Figure 1A). This is likely the
Mol
result of the heat shock, which is known to attenuate protein
synthesis (Richter et al., 2010). Surprisingly, however, the levels
of most free amino acids decreased in cim3-1 cells after 5 hr
ecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc. 243

Molecular Cell
Proteasome Failure Kills Cells by Starvation
at 37�C (Figure 1A). Unlike wild-type cells, cim3-1 cells did not
grow at 37�C (Figure S1B), indicating that the decrease in intra-
cellular amino acids in cim3-1 cells was not due to an exhaustion
of the nutrient supply from the media as a result of cell growth.
Thus, inhibition of proteasomal degradation, under conditions
of nutrient sufficiency, decreases the levels of intracellular amino
acid in yeast cells.
We then investigated whether increasing the supply of amino
acids could rescue cim3-1 cells. Exponentially growing wild-
type or cim3-1 yeast cells were inoculated at an optical density
of 0.2 and cultured in the standard rich medium YPD for 21 hr,
labeled with propidium iodide to reveal dead cells, and analyzed
by flow cytometry. Unlike wild-type cells, the vast majority of
cim3-1 cells died after 21 hr of culture at 37�C in rich media
(Figure 1B). supplementation with amino acids prevented the
death of the cim3-1 cells exposed to 37�C (Figure 1B). To deter-
mine whether such cells were viable, they were spotted on rich
plates and grown at the permissive temperature. Unlike the non-
supplemented cim3-1 cells, cim3-1 cells that had been exposed
to 37�C in the presence of additional amino acids were viable
and gave rise to the same number of colonies as wild-type cells
at 30�C (Figure 1C). This revealed that amino acid supplementa-
tion prevents death of proteasome-inhibited yeast cells. This
rescuing effect occurs without promoting cell growth because
the number of viable cells exposed to 37�C in the presence of
amino acids equaled the number of cells prior exposure to
37�C (Figure 1C, compare lane 1 to lane 3). Supplementation
with amino acids also protected cim3-1 cells exposed to 37�Con plates (Figure 1D), as well as the proteasome mutant pre1-1
cells, harboring a thermosensitive mutation in the b4 subunit of
the 20S proteasome (Heinemeyer et al., 1991) (Figure 1E), but
not the ndc80-1mutant cells (Figure S1C), which harbors a ther-
mosensitive mutation unrelated to the proteasome, in a spindle
body component (Wigge et al., 1998).
Although rescuing viability, addition of amino acids did not
noticeably decrease the levels of polyubiquitinated proteins
(Figure 1F) or the unstable GFP-tagged Ura3-3 (Lewis and
Pelham, 2009) that accumulated in cim3-1 cells at 37�C (Fig-
ure 1G). Thus, amino acid supplementation rescues from the
lethality resulting from proteasome inhibition without promoting
a recovery of proteasome function.
Proteasomal degradation is required for progression through
the cell cycle (Hershko, 2005) and at nonpermissive temperature,
cim3-1 cells stop dividing (Ghislain et al., 1993) and (Figure S1B)
and accumulate cyclins, such as CLB2 (Ghislain et al., 1993).
Although rescuing the viability of proteasome-inhibited cells,
amino acids supplementation did not alleviate the growth arrest
resulting from proteasome inhibition (Figure S1B). Consistently,
the levels of CLB2 were indistinguishable in cim3-1 cells at
37�C with or without amino acids supplementation (Figure 1H).
This confirmed that amino acids did not rescue proteasomal
degradation or the resulting growth impairment in proteasome
inhibited cells, while rescuing viability.
We next examined if the marked decrease in amino acid noted
in cim3-1 cells impacted on protein synthesis. Note that, as ex-
pected (Richter et al., 2010), heat shock reduced protein
synthesis in wild-type cells (Figure 1I). Strikingly, we found that
in cim3-1 cells, protein synthesis at 37�C was reduced to 19%
244 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc
of the wild-type cells exposed to the same temperature (Fig-
ure 1I). Supplementation with amino acids rescued translation
rates in the cim3-1 cells to nearly wild-type levels (Figure 1I).
Amino Acid Supplementation Rescues theDeleterious Consequences of Proteasome Inhibitionin Mammalian CellsBecause proteasome function is conserved in evolution, we next
examined whether inhibition of the proteasome altered amino
acid homeostasis in mammalian cells. Treatment of mammalian
NIH 3T3 cells with the proteasome inhibitorsMG-132 or Bortezo-
mib decreased the levels of the amino acids asparagine/aspar-
tate and cysteine, whereas the levels of other amino acids
remained largely unchanged or only marginally increased (Fig-
ure S2A). To address whether the decrease in cysteine and
asparagine/aspartate were detrimental to cells following protea-
some inhibition, we examined whether supplementation with
these amino acids affected cell viability of proteasome-inhibited
cells. Amino acids were added after a transient treatment with
the proteasome inhibitor to ensure that the amino acids did not
interfere with the proteasome inhibitors. Addition of cysteine
following treatment with MG-132 for 8 hr dramatically increased
cell survival and markedly reduced apoptosis in a dose-depen-
dent manner, in contrast to other amino acids, the reducing
agent b-mercaptoethanol, or the antioxidant ascorbic acid
(Figures S2B and S2C). Cysteine also markedly increased cell
viability following Bortezomib treatment (Figure S2D). Aspara-
gine further increased viability, when added together with
cysteine following an 8 hr MG-132 treatment, whereas it only
had a minor effect on its own (Figure 2A). It may not be a coinci-
dence that both cysteine and asparagine are conditionally
essential amino acids (Reeds, 2000), absent in the Dulbecco’s
modified Eagle’s medium (DMEM). Although rescuing the
viability of proteasome-inhibited cells, amino acid supplementa-
tion had no significant effect on the proliferation of untreated
cells (Figure S2E) and neither cysteine nor asparagine detectably
altered proteasome activity (Figure S2F). Thus, supplementation
with the critical amino acids depleted upon proteasome inhibi-
tion rescued survival of �90% of proteasome-impaired cells,
without rescuing proteasome activity.
To dissect the mechanism underlying the rescued viability of
proteasome-inhibited cells by amino acid supplementation, we
next focused on cysteine addition as it had a potent effect. We
monitored the abundance of polyubiquitinated proteins as well
as the proteasome reporter substrates UbG76V-GFP, ZsGreen-
ODC, and GFP-CL1 (Dantuma et al., 2000; Hoyt et al., 2005;
Gilon et al., 1998; Bence et al., 2001). Although markedly
increasing the survival of three different cell lines following
proteasome inhibition (Figure S2G), addition of cysteine did not
reduce the abundance of either polyubiquitinated proteins (Fig-
ure 2B) or the three different rapidly degraded, proteasome
reporter-substrates UbG76V-GFP, ZsGreen-ODC, and GFP-CL1
(Figure 2C). Similarly, cells accumulated indistinguishable levels
of the proteasome reporter substrate ZsGreen-ODC when
treated with MG-132 alone or together with 1 mM cysteine
(Figure 2D). Addition of cysteine together with MG-132 also
rescued the viability of proteasome-inhibited cells (Figure S2G).
These results demonstrate that cysteine did not antagonize
.

A
Asn (mM) - 10 - 2.5 5 10
Cys 1 mM - - + + + +
*
**V
iabi
lity
(%)
MG-132 8 h
+/- Cys, Asn 20 h
2 days
***
B
Tubulin
100
97
54
Wash - - 0.5 1 2 4 6 - - 0.5 1 2 4 6 (h)
- Cys + Cys
Ubn
MG-132 - + + + + + + - + + + + + +
D E
MG-132 +/- Cysco-treatment
Cys - + - + - + - + - + - + MG-132 0 1 2 4 6 8 (h)
ZsGreen-ODC 293T
Flu
ores
cenc
e in
tens
ity
n.s.
n.s.
n.s.
Tubulin
Cys - - - +
MG-132 - + - +
p105
p50
100
97
37
54
Ubn
MG-132 6 h +/- Cys 6 h
UbG76V-GFP NIH-3T3
Flu
ores
cenc
e in
tens
ity
MG-132 ��� ����� ����������Cys ��� ����� �
****
n.s.
ZsGreen-ODC 293T
****
MG-132 ��� ����� ����������Cys ��� ����� �
Flu
ores
cenc
e in
tens
ity
n.s.
GFP-CL1 293T
****
MG-132 ��� ����� ����������Cys ��� ����� �
Flu
ores
cenc
e in
tens
ity
n.s.
C
Figure 2. Failure of Amino Acid Homeostasis Causes Cell Death following Proteasome Inhibition
(A) Viability of NIH 3T3 cells, assessed by the reduction of WST-8 into formazan, after 2 days of growth, following treatment with 10 mM MG-132 for 8 hr and a
20 hr washout in regular medium, with or without cysteine (Cys) and/or asparagine (Asn). Data are means ± SD (n = 4). *p% 0.01, **p% 0.001, and ***p% 0.0001.
n.s., not significant.
(B) Polyubiquitin (Ubn) and tubulin immunoblots of lysates from NIH 3T3 cells exposed to 10 mM MG-132 treatment for 6 hr, followed by a washout in regular
medium, with or without 1 mM Cys supplementation, for the indicated time.
(C) Fluorescence of proteasome reporter-substrates UbG76V-GFP (Dantuma et al., 2000), ZsGreen-ODC (Hoyt et al., 2005), and GFP-CL1 (Gilon et al., 1998;
Bence et al., 2001) in NIH 3T3 or 293T cells, following treatment with 10 mM MG-132 and a washout for the indicated time, with or without Cys. Data are
means ± SD (n = 3). *p % 0.01, **p % 0.001, and ***p % 0.0001. n.s., not significant.
(D) Fluorescence of proteasome reporter-substrates ZsGreen-ODC (Hoyt et al., 2005) following treatment with 10 mMMG-132 either alone or together with 1 mM
Cys where indicated.
(E) Polyubiquitin (Ubn), tubulin, and NF-kB (p105 and p50) immunoblots of lysates of 293T cells overexpressing p105 and treated with 10 mM MG-132 for 8 hr,
either alone or together with 1 mM Cys, where indicated. Representative results of at least three independent experiments are shown.
See also Figure S2.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc. 245

Molecular Cell
Proteasome Failure Kills Cells by Starvation
MG-132, while rescuing viability of cells following proteasome
inhibition.
The proteasome activates the transcription regulator NF-kB
by proteolytic cleavage of the precursor p105 into p50
(Palombella et al., 1994). Although rescuing cell viability (Fig-
ure 2A), cysteine supplementation did not reduce the impairment
of the proteasome-mediated processing of the transcription
regulator NF-kB, p105 into p50, in cells treated with MG-132
for 8 hr (Figure 2E). These results reveal that while rescuing
from the lethality resulting from proteasome inhibition, supple-
mentation with an amino acid selectively depleted following
proteasome inhibition did not decrease the levels of polyubiqui-
tinated proteins, proteasome reporter substrates and the impair-
ment of NF-kB processing in proteasome-inhibited cells. Thus,
cells can tolerate proteasome inhibition but not the resulting
amino acid insufficiency.
The Integrated StressResponse Is Inducedby theAminoAcid Shortage Resulting from Proteasome InhibitionProteasome inhibition, like many different stresses, induces the
integrated stress response (ISR) (Jiang and Wek, 2005).
However, the signal responsible for the ISR induction in protea-
some-inhibited cells is unknown. The ISR is an adaptive
response to many forms of stresses, which converge into phos-
phorylating the a subunit of translation initiation factor 2 (eIF2a)
thereby reprogramming protein synthesis. When eIF2a is phos-
phorylated, general protein synthesis is attenuated, whereas
ATF4 is selectively translated (Harding et al., 2003). ATF4 is a
transcription factor, which controls expression of genes
involved in amino acid import and biosynthesis (Harding et al.,
2003), as well as the transcription factor CHOP, which in turn
induces GADD34 (Figure 3A). GADD34 is a regulatory subunit
of the serine/threonine phosphatase PP1, which recruits PP1
in stressed cells to dephosphorylate eIF2a and terminates stress
signaling (Novoa et al., 2003). Having found that proteasome
inhibition caused a lethal shortage in both cysteine and aspara-
gine, and because genes involved in the biosynthesis of cysteine
and asparagine are prominent targets of the ISR (Harding et al.,
2003), we examined whether amino acid scarcity may be the
signal that induces the ISR in proteasome inhibited cells (Fig-
ure 3A). If so, amino acid supplementation should attenuate
ISR signaling in response to proteasome inhibition. In the
absence of MG-132, the ISR was not induced, confirming that
untreated cells were not stressed (Figure 3B). As expected
(Jiang and Wek, 2005), MG-132 induced the ISR, and this is
manifested by the phosphorylation of eIF2a, expression of
ATF4 and the pro-death proteins CHOP and GADD34 (Fig-
ure 3B). Strikingly, the levels of CHOP and GADD34 remained
elevated in the MG-132 cells 6 hr after the removal of the protea-
some inhibitor, in agreement with the finding that such cells
were committed to die (Figure 2A). In contrast, in cells supple-
mented with cysteine, a marked decrease in the levels of
CHOP and GADD34 was detected 4–6 hr following the cysteine
washout (Figure 3B), in good agreement with the cytoprotective
effect of cysteine upon proteasome inhibition (Figure 2A). When
added together with MG-132, cysteine virtually abrogated the
induction of the ISR resulting from proteasome inhibition,
without altering the levels of polyubiquitinated proteins (Fig-
246 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc
ure S3). This reveals that the amino acid shortage resulting
from proteasome inhibition is the signal that induces the ISR in
proteasome-inhibited cells.
To confirm that CHOP-mediated cell death following protea-
some inhibition (Jiang andWek, 2005), wemonitored the survival
of wild-type and CHOP�/� cells (Marciniak et al., 2004)
following proteasome inhibition. Genetic ablation of CHOP
markedly enhanced survival of proteasome-inhibited cells (Fig-
ure 3C). The decreased CHOP levels in NIH 3T3 cells supple-
mented with cysteine following proteasome inhibition could be
due to an enhanced degradation or a decreased synthesis. We
found that addition of cysteine did not rescue proteasomal
degradation (Figures 2B–2E) and the stability of both GADD34
and CHOP were indistinguishable in cells that had been supple-
mented or not with cysteine, following proteasome inhibition
(Figure 3D). Thus, the low levels of CHOP in proteasome-
inhibited cells supplemented with cysteine did not reflect an
increased degradation but rather, a decreased synthesis of this
stress protein, as a result of the dephosphorylation of eIF2a
following cysteine supplementation (Figure 2B).
The marked attenuation of eIF2a phosphorylation by cysteine
supplementation in proteasome-inhibited cells suggested that
the supply of cysteine might be rate-limiting for global protein
synthesis in proteasome-inhibited cells. If so, cysteine supple-
mentation may rescue global protein synthesis while attenuating
translation of stressed proteins such as CHOP. We next tested
this hypothesis. As expected (Jiang andWek, 2005), proteasome
inhibition caused a pronounced inhibition of protein synthesis
(Figure 3E). Remarkably, addition of 1 mM cysteine largely pre-
vented the translation attenuation resulting form proteasome
inhibition (Figure 3E). GCN2 is the eIF2a kinase that mediates
the ISR induction in response to amino acid deprivation (Harding
et al., 2000; Zhang et al., 2002). Genetic ablation of GCN2 virtu-
ally abolished eIF2a phosphorylation upon proteasome inhibition
(Figure 3F). Furthermore, activated and phosphorylated GCN2
was detected in cells deprived of leucine or MG-132-treated
cells, and addition of cysteine markedly reduced GCN2 phos-
phorylation upon proteasome inhibition (Figure 3G). This estab-
lishes that inhibition of proteasomal degradation causes a
shortage of critical amino acids that induce the ISR via GCN2.
To confirm that the attenuation of eIF2a phosphorylation by
cysteine in proteasome inhibited cells is due to the replenish-
ment of this otherwise limiting amino acid, as opposed to a
suppression of ISR signaling by an indirect effect, we tested
whether cysteine affected eIF2a phosphorylation induced by
ultraviolet (UV) irradiation, a different stress than amino acid limi-
tation (Deng et al., 2002). We found that the induction of eIF2a
phosphorylation by UV was not reduced by supplementation
with cysteine (Figure 3H). Thus, cysteine supplementation selec-
tively alleviates eIF2a phosphorylation in proteasome-inhibited
cells, but not in UV-irradiated cells.
Amino Acid Scarcity in Proteasome-Inhibited CellsSignals the Induction of AutophagyAlthough polyubiquitination is the canonical signal to target
proteins for proteasomal degradation, recent studies have
revealed that polyubiquitinated proteins can also be degraded
by autophagy (Kirkin et al., 2009). Autophagy is also induced
.

A B
C D
E F
G H
Figure 3. Amino Acid Supplementation Alle-
viates the Induction of the Integrated Stress
Response upon Proteasome Inhibition
(A) Scheme depicting the integrated stress
response induction upon proteasome inhibition.
The signal responsible for eIF2a phosphorylation
(P-eIF2a) upon proteasome inhibition is unknown.
(B) Polyubiquitin (Ubn), GADD34, ATF4, phos-
phorylated eIF2a (P-eIF2a), eIF2a, and CHOP
immunoblots of lysates from NIH 3T3 cells treated
with 10 mMMG-132 for 6 hr followed by a washout
in the presence of 1 mM cysteine (Cys) for the
indicated time.
(C) Viability of wild-type and CHOP�/� mouse
embryonic fibroblasts, assessed by the reduction
of WST-8 into formazan, after 2 days of growth,
following treatment with 10 mM MG-132 for the
indicated time and a washout in regular medium.
Data are means ± SD (n = 4), normalized to cell
number. *p % 0.01, ***p % 0.0001.
(D) GADD34, CHOP, and tubulin immunoblots of
lysates from NIH 3T3 cells treated with 10 mMMG-
132 for 4 hr followed by a washout in the presence
of cycloheximide (CHX, 100 mg/ml), with or without
1 mM Cys for the indicated time.
(E) Autoradiogram of [35S]methionine-labeled
proteins in lysates resolved by NuPage after a
8 min pulse labeling of exponentially growing cells
cultured as indicated. Quantification are presented
below each lane. Lower panel is a photomicro-
graph of the Coomassie-stained gel.
(F) eIF2a immunoblots of lysates from GCN2�/�or wild-type mouse embryonic fibroblasts treated
with 10 mM MG-132 for the indicated time.
(G) Immunoblots of GCN2 immunoprecipitated
from lysates of cells either left untreated, leucine-
deprived (-Leu), or treated with 10 mM MG-132,
with or without 1 mM Cys. The top panel was
probed with antisera against the phosphorylated
GCN2 and the bottom panel with an antibody
recognizing both the phosphorylated and the
nonphosphorylated GCN2.
(H) Immunoblots of lysates from NIH 3T3 cells
following a dose response analysis of UV irradia-
tion, in the presence or in absence of 1 mM
cysteine (Cys). Representative results of at least
three independent experiments are shown.
See also Figure S3.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
when proteasomal degradation is inhibited (Rideout et al., 2004;
Iwata et al., 2005) and alleviates the toxicity resulting from pro-
teasome inhibition (Korolchuk et al., 2010; Pandey et al., 2007).
However, the mechanisms underlying autophagy induction
when proteasomal degradation is compromised are unknown.
As expected (Korolchuk et al., 2010), MG-132 increased the
number of GFP-LC3 labeled autophagosomes in HeLa cells
stably expressing GFP-LC3 (Figures 4A and 4B). Strikingly,
cysteine supplementation markedly decreased the number of
fluorescent puncta in proteasome-inhibited cells (Figures 4A
and 4B). In contrast, supplementation with other amino acids,
reducing agents or antioxidants did not noticeably reduced the
number of GFP-LC3 puncta in proteasome inhibited cells (Fig-
Mol
ure S4). Cysteine is not a generic suppressor of autophagosome
accumulation because cysteine did not decrease the number of
GFP-LC3 puncta that accumulated in cells treated with Bafilo-
mycin A1 (Figures 4A and 4B), an inhibitor of autophagosome-
lysosome fusion (Mizushima et al., 2010). MG-132 treatment
also increased the levels of LC3-II (Figures 4C and 4D) but this
was markedly decreased in the presence of cysteine (Figures
4C and 4D). These results establish that the decrease in a critical
amino acid is the signal that induces autophagy when proteaso-
mal degradation is inhibited. To assess whether induction of
autophagy contributed to protect cells from proteasome-inhibi-
tion, we compared the survival of wild-type or Atg5�/� cells
(Kuma et al., 2004) following proteasome inhibition. We found
ecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc. 247

A B
C D
E
Figure 4. Amino Acid Supplementation Suppresses Autophagy Resulting from Proteasome Inhibition
(A) Confocal micrographs of HeLa cells stably expressing GFP-LC3 (Thurston et al., 2009) either untreated or following 10 mMMG-132 treatment, with or without
1 mM Cys, where indicated. Nuclei were stained with H33258.
(B) Quantification of GFP-LC3 dots in at least 100 cells exposed to the indicated treatment. Data are means ± SEM. ***p % 0.0001.
(C) LC3 and tubulin immunoblots of lysates from cells treated with 100 nMBafilomycin A1 (Baf A1) for 15 hr, or 10 mMMG-132 alone or together with 1mMCys for
the indicated time.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
248 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc.

Molecular Cell
Proteasome Failure Kills Cells by Starvation
that Atg5�/� cells were significantly more sensitive to MG-132
treatment and this was manifest already following 1 hr of protea-
some inhibition (Figure 4E).
Themammalian target of rapamycin (mTOR) signalingpathway
senses and responds to amino acid availability to modulate
protein synthesis via the phosphorylation of substrates such as
S6 kinase 1 (S6K1) (Ma and Blenis, 2009) (Figure 5A). To evaluate
whether autophagy contributed to replenish the pool of intracel-
lular amino acids upon proteasome inhibition, we measured the
levels of phosphorylation of threonine 389 of the mTORC1
substrate S6K1 (Ma and Blenis, 2009). In agreement with the
decrease in critical amino acids following proteasome inhibition,
S6K1 phosphorylation decreased 1 hr after MG-132 addition
(Figure 5B). Four hours following MG-132 addition, S6K1 phos-
phorylation increased (Figure 5B). This increase was abrogated
by Bafilomycin A1, indicating that it was dependent on autopha-
gic degradation (Figure 5B). Amino acid sensing by mTOR is
mediated by the Rag GTPases and dominant negative mutants
of the Rag GTPases abrogate mTOR stimulation by amino acids
(Sancak et al., 2008). Strikingly, overexpression of dominant
negative mutants of the Rag GTPases that prevent amino acid
sensing by mTORC1 (Figure 5A) also abolished the increased
phosphorylation of S6K1 that followed proteasome inhibition
(Figure 5C). These results reveal that amino acid scarcity is the
signal that induces autophagy when the proteasome is inhibited,
in an attempt to rescue amino acid homeostasis.
This model predicts that blocking amino acid consumption by
inhibiting protein synthesis should recapitulate the rescuing
effects of amino acid supplementation in proteasome inhibited
cells. Indeed, we found that addition of cycloheximide together
with MG-132 prevented the decreased S6K1 phosphorylation
resulting from treatment with MG-132 for 1 hr (Figure 5D). Con-
sistently, the decrease in cysteine and asparagine/aspartate re-
sulting from proteasome inhibition was markedly attenuated in
cells cotreated with cycloheximide (Figure 5E). Strikingly, cyclo-
heximide protected cells from the lethality resulting from protea-
some inhibition (Figure 5F). The rescuing effect of cycloheximide
in proteasome-inhibited cells was selective because cyclohexi-
mide increased the lethality of cells exposed to UV irradiation
(data not shown). Thus, inhibition of protein synthesis markedly
attenuates the depletion of critical amino acid following protea-
some inhibition as well as the lethality resulting from proteasome
inhibition.
An Amino Acid Imbalance Underlies the LethalityResulting from Proteasome Inhibition in Drosophila
We next assessed whether proteasome inhibition had detri-
mental consequences on amino acid homeostasis in an organ-
ismal context. We exposed Drosophila to proteasome inhibitors
and monitored amino acid levels. Bortezomib significantly
reduced the levels of asparagine/aspartate, threonine/serine,
glutamine/glutamate, phenylalanine, and proline in Drosophila
(D) Quantification of the LC3-II/tubulin ratio in three independent experiments. V
(E) Viability of wild-type and Atg5�/� mouse embryonic fibroblasts, assessed
treatment with 10 mMMG-132 for the indicated time and a washout in regular med
not significant.
See also Figure S4.
Mol
(Figure 6A). Bortezomib also increased the levels of glycine (Fig-
ure 6A), similar to prolonged starvation in humans (Felig et al.,
1969). As expected (Vernace et al., 2007), exposure of flies to
Bortezomib was lethal (Figure 6B). Amino acid supplementation
markedly increased survival of proteasome-inhibited flies (Fig-
ure 6B), without detectable changes in the levels of proteasome
substrates (Figure 6C). Thus, proteasome inhibition caused an
intolerable amino acid imbalance in flies and the resulting
lethality was rescued upon supplementation with amino acids.
DISCUSSION
Here, we show that the detrimental consequences of protea-
some inhibition are largely abrogated by supplementation with
amino acids in yeast,mammalian cells, andDrosophila (Figure 7).
Strikingly, the rescuing effect of amino acids occurs without
reducing the accumulation of proteasome substrates. These
findings reveal that cells can survive with the protein waste
they accumulate when the proteasome is inhibited but the result-
ing amino acid deficiency is lethal. Thus, the proteasome
substrates accumulating upon proteasome inhibition appear
detrimental to cells as they sequester a pool of critical amino
acids that would otherwise be recycled.
Although abrogating death of proteasome-inhibited cells,
amino acid supplementation does not abrogate the requirement
for proteasomal degradation for regulatory functions such as cell
division. The amino acid scarcity resulting from proteasome
inhibition induces the classical set of responses to amino acid
starvation, in an vain attempt to sustain amino acid homeostasis.
Although it had been previously reported that proteasome inhibi-
tion, like many different stresses, induces eIF2a phosphorylation
and that the resulting translation attenuation is abrogated by
genetic impairment of eIF2a phosphorylation (Jiang and Wek,
2005), the underlyingmechanismwas unknown.We have shown
here that the amino acid decrease following proteasome
inhibition is the signal that activates GCN2 and induces eIF2a
phosphorylation, leading to attenuation of protein synthesis (Fig-
ure 3). Cysteine is the least abundant cellular amino acid and we
found that cysteine scarcity following proteasome inhibition
impairs protein synthesis. Upon persistent inhibition of the pro-
teasome and the subsequent amino acid insufficiency, the ISR
is chronically activated, thereby leading to the expression of
the pro-apoptotic protein CHOP (Figure 3B). Supplementation
with a critical amino acid, depleted upon proteasome inhibition,
virtually suppresses ISR signaling and cell death (Figure 3B),
similar to genetic ablation of CHOP (Figure 3C) or impairment
of eIF2a phosphorylation, which reduces death following protea-
some inhibition (Jiang and Wek, 2005).
The mechanisms underlying the crosstalk between autopha-
gic and proteasomal degradation have remained elusive. In
this study, we provide the missing link between autophagy
induction and proteasome inhibition. We show that the signal
alues were normalized to untreated cells. Data are means ± SEM. *p % 0.05.
by the reduction of WST-8 into formazan, after 2 days of growth, following
ium. Data are means ± SD (n = 4), normalized to cell number. **p% 0.001. n.s.,
ecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc. 249

A
C
MG-132 0 1 2 3 4 8 0 1 2 3 4 8 (h) Mock transfected RagB-DN transfected
S6K1
FLAG
P -T389-S6K1
Baf A1
MG-132 0 1 2 3 4 8 0 1 2 3 4 8 (h)
P -T389-S6K1
S6K1
B
Amino acids
RagA/B
RagC/D
GTP
GDP
mTOR
RagA/B
RagC/D
GDP
GTP
P -T389-S6K1
1 h
MG
-132
D
S6K1
P -T389-S6K1
E
% s
urvi
val
CHX - +
F
MG-132 10 µM
Cys AsxUT 100 100MG-132 73.3 ±1.9 64 ±2.7MG-132 + CHX 88.6 ±1.6 97.7 ±3
1 h
MG
-132
+
CH
X
***
Figure 5. Autophagy and Rag GTPases-Dependent Amino Acid
Signaling to mTOR upon Proteasome Inhibition
(A) Model for amino acid signaling to mTOR by the Rag GTPases (RagA/BGTP-
RagC/DGDP). Amino acid sensing by the Rag GTPases is blocked by dominant
negative mutants (RagBGDP-RagCGTP) (Sancak et al., 2008).
Molecular Cell
Proteasome Failure Kills Cells by Starvation
250 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc
responsible for autophagy induction upon proteasome inhibition
is the resulting amino acid scarcity, the canonical inducer of
autophagy. Autophagy aims at protecting cells from the delete-
rious consequences of proteasome inhibition, as cells lacking
Atg5, a key autophagy component are more sensitive to protea-
some inhibition. Both the ISR and autophagy are involved as
early as 1 hr following proteasome inhibition to adapt cells to
the resulting changes. Our work shows that autophagy and
proteasomal degradation act in a concerted manner to adjust
the supply of amino acids to the cellular needs. Following pro-
teasome inhibition, autophagy attempts to restore amino acid
homeostasis, as manifested by an increased S6K1 phosphoryla-
tion via the canonical, Rag GTPases-dependent amino acid
signaling pathway to mTOR. The findings reported here reveal
how changes in amino acid homeostasis are integrated with
protein metabolism and cell death (Figure 7).
We found here that the deleterious consequences of protea-
some inhibition are rescued upon amino acid supplementation
in yeast, mammalian cells, andDrosophila. These findings estab-
lish the importance of proteasomal degradation in recycling
amino acids for normal cell and organismal function. There is
a qualitative difference in the required amino acids in the three
different systems that is not surprising as yeast, mammalian
cells, andDrosophila have different properties and requirements.
Yeast cells required a broad range of amino acids to survive pro-
teasome inhibition, perhaps because they have higher metabolic
needs, as they divide more rapidly than mammalian cells. Upon
proteasome inhibition, mammalian cells had a prominent need
for cysteine and asparagine and we show that the decrease in
cysteine and asparagine/aspartate were largely prevented by
blocking protein synthesis in proteasome-inhibited cells (Fig-
ure 5E). Notably, enzymes required for the metabolism of
cysteine and asparagine, two conditionally essential amino
acids, are under the control of the ISR, thus underscoring an
evolutionary pressure to maintain the levels of asparagine and
cysteine in the cell. In addition, the importance of asparagine
starvation has been previously recognized and is the basis for
the use of L-asparaginase for the treatment of acute lympho-
blastic leukemia (Muller and Boos, 1998).
The proteasome inhibitor Bortezomib is used in cancer
therapy but it is limited to the treatment of multiple myeloma
(B) Immunoblots of lysates from 293T cells exposed to 10 mM MG-132 treat-
ment revealed with phospho-T398-S6K1 or S6K1 antibodies. Cells were either
left untreated or treated with 100 nM Baf A1 for the indicated time.
(C) Immunoblots with phospho-T398-S6K1 or S6K1 antibodies of lysates of
cells mock transfected or transfected with dominant negative forms of RagB
(RagB-DN, FLAG-tagged).
(D) Immunoblots with phospho-T398-S6K1 or S6K1 antibodies of lysates of
293T cells either left untreated or treated with 10 mM MG-132 with or without
100 mM cycloheximide (CHX) for 1 hr.
(E) Changes in the content of cellular free cysteine (Cys) or asparagine/
aspartate (Asx) in cells treated with 10 mM MG-132 with or without 50 mM
cycloheximide (CHX) for 4 hr.
(F) Viability of NIH 3T3 cells, assessed by the reduction of WST-8 into
formazan, after 2 days of growth, following treatment with 10 mMMG-132 with
or without 50 mM cycloheximide (CHX) for 8 hr. Data are means ± SD (n = 4),
normalized to cell number. ***p % 0.0001. Representative results of at least
three independent experiments are shown.
.

A
Am
ino
acid
(nm
ol)
**
**
***
***
B
0
20
40
60
80
100
0 1 2 3 4
Untreatedno AA+ AA
Bortezomib treated
***
**
**
C
Ponceau
Unt
reat
ed
-
Bortezomib
+ A
A
Ubn
***
Sur
viva
l (%
)
Days
**
Figure 6. Amino Acid Imbalance Mediates
the Lethality Resulting from Proteasome
Inhibition in Drosophila
(A) Changes in the content of free amino acids
in Drosophila treated with or without 50 mM
Bortezomib for 3 days. Asx, asparagine/aspartate;
Glx, glutamine/glutamate. Data are means ± SEM
(30 flies per conditions, from six independent
experiments). **p % 0.005; ***p % 0.001.
(B) Survival of Drosophila either untreated or
treated with 50 mM Bortezomib, with or without
additional amino acids (AA), at the concentration
used in Grandison et al. (2009). Data are means ±
SEM (400 flies per condition, from four indepen-
dent experiments). **p % 0.005; ***p % 0.001.
(C) Polyubiquitin (Ubn) immunoblots from lysates
of flies treated with 50 mM Bortezomib and
additional amino acids, where indicated. Ponceau
staining of the membrane. Representative results
of at least three independent experiments are
shown.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
and restricted by adverse effects, such as peripheral neuropathy
(Richardson et al., 2003). So far, the mechanisms by which
proteasome inhibitors kill cells have remained elusive. Failure
of amino acid homeostasis not only explains the lethality result-
ing from proteasome inhibition but also provides a foundation for
the development of novel cancer therapies.
EXPERIMENTAL PROCEDURES
Mammalian Cell Culture
NIH 3T3, 293T (ATCC), 293T cells stably expressing ZsGreen-ODC
(ZsProsensor-1 cells, Clontech), and HeLa cells were maintained at 37�C in
DMEM supplemented with 10% fetal bovine serum (FBS), penicillin, and
streptomycin. MEFs cells were cultured in DMEM supplemented with
Molecular Cell 48, 242–253,
penicillin, streptomycin, glutamine, 13 nonessen-
tial amino acids (Sigma), sodium pyruvate, and
10% FBS.
Assessment of Cell Viability
Cells were plated (day 0) in DMEM supplemented
with 10% FBS bovine serum in 24-well plates
(Nunc), at a density of 8,000 cells/ml. The next
day (day 1), media were replaced with DMEM
supplemented with 0.5% FBS. On day 2, cells
were treated with proteasome inhibitors for the
indicated period of time. Media containing pro-
teasome inhibitors were replaced with DMEM
supplemented with 0.5% FBS and the indicated
additives. Four wells per condition were used.
On day 3, media were replaced with DMEM
supplemented with 10% FBS. Cells were kept
for 2–3 days as indicated. Cell viability was
assessed by measuring the reduction of WST-8
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-
(2,4-disulfophenyl)-2H-tetrazolium] into formazan
using Cell Viability Counting Kit-8 (Dojindo) ac-
cording to the supplier’s recommendation.
Flow Cytometry and Confocal Microscopy
Flow cytometry analyses were performed on
a FACSCalibur flow cytometer (BD Biosciences)
and analyzed with FlowJo and confocal microscopy was carried out as
described in Munch et al. (2011).
Immunoblot Analyses and Assessment of Translation Rates
Cell lysis, immunoblots, and measurement of translation rates were performed
as described in Tsaytler et al. (2011). Note that cells were not starved in methi-
onine-free media before labeling with 35S-methionine for monitoring protein
synthesis.
Yeast Strains, Media, and Culture
Cim3-1 or pre1-1 and their respective isogenic wild-type strains (Ghislain
et al., 1993; Heinemeyer et al., 1991) and ndc80-1 (Wigge et al., 1998) were
grown in YPD medium using standard techniques, as in Dehay and Bertolotti
(2006).
October 26, 2012 ª2012 Elsevier Inc. 251

Figure 7. Proteasome Inhibition Causes an Intolerable Amino Acid Imbalance and the Resulting Lethality Is Rescued upon Supplementation
with Amino Acids
The amino acid shortage resulting from proteasome inhibition is the signal that induces eIF2a phosphorylation and autophagy, in a vain attempt to replenish
to pool of amino acid and sustain vital protein synthesis rates. In yeast, mammalian cells, and Drosophila, the lethality resulting from proteasome inhibition is
rescued upon amino acid supplementation. This rescuing effect occurs without a detectable decrease in the levels of proteasome substrates in proteasome-
inhibited cells.
Molecular Cell
Proteasome Failure Kills Cells by Starvation
Amino Acid Analyses
Amino acid analysis from yeast cells and mammalian cells were performed
as described, respectively (Onodera and Ohsumi, 2005; Vabulas and Hartl,
2005).
Flies
Wild-type Oregon R flies were reared on standard food. Amino acids were
extracted in 600 ml methanol from 5 frozen female flies per condition, as in
Hayward et al. (1993). The suspension was centrifuged and the supernatant
collected, vacuum-dried, and analyzed. Some amino acids were below detec-
tion levels in such analyses. For survival experiments, 20 flies, both females
and males, were kept in vials lined with 3 mm filter paper and fed a solution
of 5% sucrose with either vehicle (0.5% DMSO, control) or with Bortezomib
(50 mM in DMSO), for the indicated time, as in Vernace et al. (2007). Under
the same conditions MG-132 (50 mM in DMSO) was not toxic. In Figure 6B,
1.5 ml of 5% sucrose with or without amino acids at the concentration used
in Grandison et al. (2009) was added together with the proteasome inhibitor.
Proteins were extracted from five frozen female flies per conditions in 250 ml
boiling Laemmli buffer supplemented with 10 mM NEM and 50 mM DTT.
Statistical Analysis
Data are presented as means and SD or SEM, as indicated. Where indicated,
two groups were compared with a two-tailed nonpaired Student’s t test.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and Supplemental Experi-
mental Procedures and can be found with this article online at http://dx.doi.
org/10.1016/j.molcel.2012.08.003.
ACKNOWLEDGMENTS
We are grateful to D. Owen for amino acid analysis; C. Mann, D.H. Wolf, J. Kil-
martin, M. Lewis, N. Mizushima, B. McGrath, D. Cavener, H. Harding, D. Ron,
W. Greene, M. Narita, and F. Randow for reagents; M. Freeman, S. Aldaz
252 Molecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc
Casanova, and M. Zettl for help and advice on the work with Drosophila;
Z. Zhong for help with the revision; G. Lingley and P. Margiotta for help with
figures and illustrations; and M.M. Babu, M. Goedert, M.H. Hastings, D.
Komander, and S. Munro for discussions. This work was supported by the
UK Medical Research Council.
Received: February 2, 2012
Revised: June 4, 2012
Accepted: August 3, 2012
Published online: September 6, 2012
REFERENCES
Bence, N.F., Sampat, R.M., and Kopito, R.R. (2001). Impairment of the ubiqui-
tin-proteasome system by protein aggregation. Science 292, 1552–1555.
Dantuma, N.P., Lindsten, K., Glas, R., Jellne, M., and Masucci, M.G. (2000).
Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-
dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543.
Dehay, B., and Bertolotti, A. (2006). Critical role of the proline-rich region in
Huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 281,
35608–35615.
Deng, J., Harding, H.P., Raught, B., Gingras, A.C., Berlanga, J.J., Scheuner,
D., Kaufman, R.J., Ron, D., and Sonenberg, N. (2002). Activation of GCN2 in
UV-irradiated cells inhibits translation. Curr. Biol. 12, 1279–1286.
Felig, P., Owen, O.E., Wahren, J., and Cahill, G.F.J., Jr. (1969). Amino acid
metabolism during prolonged starvation. J. Clin. Invest. 48, 584–594.
Finley, D. (2009). Recognition and processing of ubiquitin-protein conjugates
by the proteasome. Annu. Rev. Biochem. 78, 477–513.
Ghislain, M., Udvardy, A., and Mann, C. (1993). S. cerevisiae 26S protease
mutants arrest cell division in G2/metaphase. Nature 366, 358–362.
Gilon, T., Chomsky, O., and Kulka, R.G. (1998). Degradation signals for
ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 17,
2759–2766.
.

Molecular Cell
Proteasome Failure Kills Cells by Starvation
Goldberg, A.L. (2007). Functions of the proteasome: from protein degradation
and immune surveillance to cancer therapy. Biochem. Soc. Trans. 35, 12–17.
Grandison, R.C., Piper, M.D., and Partridge, L. (2009). Amino-acid imbalance
explains extension of lifespan by dietary restriction in Drosophila. Nature 462,
1061–1064.
Harding, H.P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron,
D. (2000). Regulated translation initiation controls stress-induced gene
expression in mammalian cells. Mol. Cell 6, 1099–1108.
Harding, H.P., Zhang, Y., Zeng, H., Novoa, I., Lu, P.D., Calfon, M., Sadri, N.,
Yun, C., Popko, B., Paules, R., et al. (2003). An integrated stress response
regulates amino acid metabolism and resistance to oxidative stress. Mol.
Cell 11, 619–633.
Hayward, D.C., Delaney, S.J., Campbell, H.D., Ghysen, A., Benzer, S.,
Kasprzak, A.B., Cotsell, J.N., Young, I.G., and Miklos, G.L. (1993). The slug-
gish-A gene of Drosophila melanogaster is expressed in the nervous system
and encodes proline oxidase, a mitochondrial enzyme involved in glutamate
biosynthesis. Proc. Natl. Acad. Sci. USA 90, 2979–2983.
Heinemeyer, W., Kleinschmidt, J.A., Saidowsky, J., Escher, C., and Wolf, D.H.
(1991). Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional
proteinase: mutants unravel its function in stress induced proteolysis and
uncover its necessity for cell survival. EMBO J. 10, 555–562.
Hershko, A. (2005). The ubiquitin system for protein degradation and some of
its roles in the control of the cell division cycle. Cell DeathDiffer. 12, 1191–1197.
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev.
Biochem. 67, 425–479.
Hideshima, T., Richardson, P., Chauhan, D., Palombella, V.J., Elliott, P.J.,
Adams, J., and Anderson, K.C. (2001). The proteasome inhibitor PS-341
inhibits growth, induces apoptosis, and overcomes drug resistance in human
multiple myeloma cells. Cancer Res. 61, 3071–3076.
Hideshima, T., Chauhan, D., Richardson, P., Mitsiades, C., Mitsiades, N.,
Hayashi, T., Munshi, N., Dang, L., Castro, A., Palombella, V., et al. (2002).
NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 277,
16639–16647.
Hoyt, M.A., Zhang, M., and Coffino, P. (2005). Probing the ubiquitin/protea-
some system with ornithine decarboxylase, a ubiquitin-independent
substrate. Methods Enzymol. 398, 399–413.
Iwata, A., Riley, B.E., Johnston, J.A., and Kopito, R.R. (2005). HDAC6 and
microtubules are required for autophagic degradation of aggregated hunting-
tin. J. Biol. Chem. 280, 40282–40292.
Jiang, H.Y., and Wek, R.C. (2005). Phosphorylation of the alpha-subunit of the
eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and
enhances apoptosis in response to proteasome inhibition. J. Biol. Chem.
280, 14189–14202.
Kirkin, V., McEwan, D.G., Novak, I., and Dikic, I. (2009). A role for ubiquitin in
selective autophagy. Mol. Cell 34, 259–269.
Korolchuk, V.I., Menzies, F.M., and Rubinsztein, D.C. (2010). Mechanisms of
cross-talk between the ubiquitin-proteasome and autophagy-lysosome
systems. FEBS Lett. 584, 1393–1398.
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T.,
Ohsumi, Y., Tokuhisa, T., and Mizushima, N. (2004). The role of autophagy
during the early neonatal starvation period. Nature 432, 1032–1036.
Lewis, M.J., and Pelham, H.R. (2009). Inefficient quality control of thermosen-
sitive proteins on the plasma membrane. PLoS ONE 4, e5038.
Ma, X.M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated
translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318.
Marciniak, S.J., Yun, C.Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R.,
Nagata, K., Harding, H.P., and Ron, D. (2004). CHOP induces death by
promoting protein synthesis and oxidation in the stressed endoplasmic
reticulum. Genes Dev. 18, 3066–3077.
Mizushima, N., Yoshimori, T., and Levine, B. (2010). Methods in mammalian
autophagy research. Cell 140, 313–326.
Mol
Muller, H.J., and Boos, J. (1998). Use of L-asparaginase in childhood ALL. Crit.
Rev. Oncol. Hematol. 28, 97–113.
Munch, C., O’Brien, J., and Bertolotti, A. (2011). Prion-like propagation of
mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl.
Acad. Sci. USA 108, 3548–3553.
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics
and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol.
Cell Biol. 10, 458–467.
Navon, A., and Ciechanover, A. (2009). The 26 S proteasome: from basic
mechanisms to drug targeting. J. Biol. Chem. 284, 33713–33718.
Novoa, I., Zhang, Y., Zeng, H., Jungreis, R., Harding, H.P., and Ron, D. (2003).
Stress-induced gene expression requires programmed recovery from transla-
tional repression. EMBO J. 22, 1180–1187.
Onodera, J., and Ohsumi, Y. (2005). Autophagy is required for maintenance of
amino acid levels and protein synthesis under nitrogen starvation. J. Biol.
Chem. 280, 31582–31586.
Palombella, V.J., Rando, O.J., Goldberg, A.L., and Maniatis, T. (1994). The
ubiquitin-proteasome pathway is required for processing the NF-kappa B1
precursor protein and the activation of NF-kappa B. Cell 78, 773–785.
Pandey, U.B., Nie, Z., Batlevi, Y., McCray, B.A., Ritson, G.P., Nedelsky, N.B.,
Schwartz, S.L., DiProspero, N.A., Knight, M.A., Schuldiner, O., et al. (2007).
HDAC6 rescues neurodegeneration and provides an essential link between
autophagy and the UPS. Nature 447, 859–863.
Reeds, P.J. (2000). Dispensable and indispensable amino acids for humans.
J. Nutr. 130, 1835S–1840S.
Richardson, P.G., Barlogie, B., Berenson, J., Singhal, S., Jagannath, S., Irwin,
D., Rajkumar, S.V., Srkalovic, G., Alsina, M., Alexanian, R., et al. (2003). A
phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J.
Med. 348, 2609–2617.
Richter, K., Haslbeck, M., and Buchner, J. (2010). The heat shock response:
life on the verge of death. Mol. Cell 40, 253–266.
Rideout, H.J., Lang-Rollin, I., and Stefanis, L. (2004). Involvement of macroau-
tophagy in the dissolution of neuronal inclusions. Int. J. Biochem. Cell Biol. 36,
2551–2562.
Sancak, Y., Peterson, T.R., Shaul, Y.D., Lindquist, R.A., Thoreen, C.C.,
Bar-Peled, L., and Sabatini, D.M. (2008). The Rag GTPases bind raptor and
mediate amino acid signaling to mTORC1. Science 320, 1496–1501.
Schwartz, A.L., and Ciechanover, A. (2009). Targeting proteins for destruction
by the ubiquitin system: implications for human pathobiology. Annu. Rev.
Pharmacol. Toxicol. 49, 73–96.
Thurston, T.L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F.
(2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the
proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Tsaytler, P., Harding, H.P., Ron, D., and Bertolotti, A. (2011). Selective inhibi-
tion of a regulatory subunit of protein phosphatase 1 restores proteostasis.
Science 332, 91–94.
Vabulas, R.M., and Hartl, F.U. (2005). Protein synthesis upon acute nutrient
restriction relies on proteasome function. Science 310, 1960–1963.
Vernace, V.A., Arnaud, L., Schmidt-Glenewinkel, T., and Figueiredo-Pereira,
M.E. (2007). Aging perturbs 26S proteasome assembly in Drosophila mela-
nogaster. FASEB J. 21, 2672–2682.
Wigge, P.A., Jensen, O.N., Holmes, S., Soues, S., Mann,M., and Kilmartin, J.V.
(1998). Analysis of the Saccharomyces spindle pole by matrix-assisted laser
desorption/ionization (MALDI) mass spectrometry. J. Cell Biol. 141, 967–977.
Zhang, P., McGrath, B.C., Reinert, J., Olsen, D.S., Lei, L., Gill, S., Wek, S.A.,
Vattem, K.M., Wek, R.C., Kimball, S.R., et al. (2002). The GCN2 eIF2alpha
kinase is required for adaptation to amino acid deprivation in mice. Mol.
Cell. Biol. 22, 6681–6688.
ecular Cell 48, 242–253, October 26, 2012 ª2012 Elsevier Inc. 253