módulo i: moléculas pequeñas . módulo ii:...
TRANSCRIPT

CARRERA DE ESPECIALIZACION EN BIOTECNOLOGIA INDUSTRIAL
FCEyN-INTIMateria de Especialización CEBI_E1
Técnicas de análisis en biotecnologíaMódulo I: Moléculas pequeñas.
Módulo II: Macromoléculas.
Docente a cargo: SANTAGAPITA, Patricio
CEBI_E1_1 : Electroforesis

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
La electroforesis es una técnica para separar o resolver moléculas en una mezcla, bajo la influencia de un campo eléctrico.
Las moléculas en solución, bajo la influencia de un campo eléctrico, migran a una velocidad determinada por la relación carga:masa, observando para cada una de ellas una determinada movilidad electroforética

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Fuerza impulsora: V aplicado.
La velocidad de la molécula será proporcional al gradiente de voltaje que la molécula perciba a su alrededor.
Leyes: Ley de Ohm
Ley de Joule (calor producido):
La fuente aplicará el V y la I. La R estará dada por el gel, buffer, electrodos.
La velocidad de migración (v) dependerá de la carga efectiva (q) y del coeficiente de fricción f (relacionado con el tamaño y la forma).
V=voltaje; I: intensidad; R: resistencia; P:potencia (W);

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
PAGE: polyacrilamide gel electrophoresis
2 condiciones: nativa y desnaturalizante
SDS-PAGE: método para separar proteínas sólo por tamaño
SDS: sodium dodecyl sulphate
El detergente “linealiza” las proteínas. Así, poseen la misma carga por masa
INDEPENDENCIA CONFORMACIÓN ESPACIAL

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
SDS: estructura y función
- Detergente aniónico desnaturalizante
- Rompe interacciones hidrofóbicas y cubre la superficie de la proteína
- Enmascara las cargas positivas con sus cargas negativas
- No rompe puentes disulfuro inter o intra- catenarios
Se agrega aprox. 1,5 g / g de proteína

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Tratamiento de la muestra:
Además del agregado del SDS, la muestra puede ser calentada a 95-98°C durante 3-4 minutos, en presencia de DTT (ditiotreitol) o 2-mercaptoetanol, que contribuyen a desnaturalizar las proteínas reduciendo enlaces disulfuro y rompiendo estructuras terciarias (plegamientos) y cuaternarias (asociaciones de subunidades).
Recordatorio: este procedimiento no se utiliza cuando es necesario mantener la estructura nativa de la proteína para otros análisis

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Por lo tanto, las proteínas en la muestra:
Sólo presentan estructura primaria (linealizadas). Serán más largas cuanto mayor sea su masa.
Tienen carga neta negativa, por lo que migrarán al polo positivo cuando se las someta a un campo eléctrico.
Las proteínas no presentan diferencias en cargas (todas tienen la misma tendencia a acercarse al polo positivo con la misma fuerza) y no presentan diferencias espaciales en su conformación.
Resta poner las proteínas en un ambiente que las separe por tamaño, una estructura tridimensional en forma de red o esponja con un determinado tamaño de poro.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
s-s SDS, calor
Proteínas con SDS
+
–
Entonces, un esquema de lo que sucederá ...

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Pasos:
1) Preparación del gel y de la muestra (en paralelo)
2) Corrida del gel
3) Revelado de las proteínas en el gel, mediante el método de tinción
Requiere conocimiento
previo
Dependerádel uso

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Técnica: enlaces interesantes:
Como preparar un gel (How to Make an SDS-PAGE gel)http://www.youtube.com/watch?v=EDi_n_0NiF4
Como correr un gel (How to Run an SDS-PAGE gel)http://www.youtube.com/watch?v=XUjLO-ek2C8&feature=related
Como realizar la tinción (How to Stain an SDS-PAGE gel)http://www.youtube.com/watch?v=b-1dXzU4iOw&feature=related

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Poliacrilamida: polímero de acrilamida.
Se produce un gel cuyo tamaño de poro puede regularse cambiando las proporciones de acrilamida y bisacrilamida utilizadas en el momento de la preparación de la mezcla y la polimerización.
El gel no es sólido, sino que presenta túneles, poros y una forma de redtridimensional de fibras.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Estructuras involucradas en la polimerización:
t
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
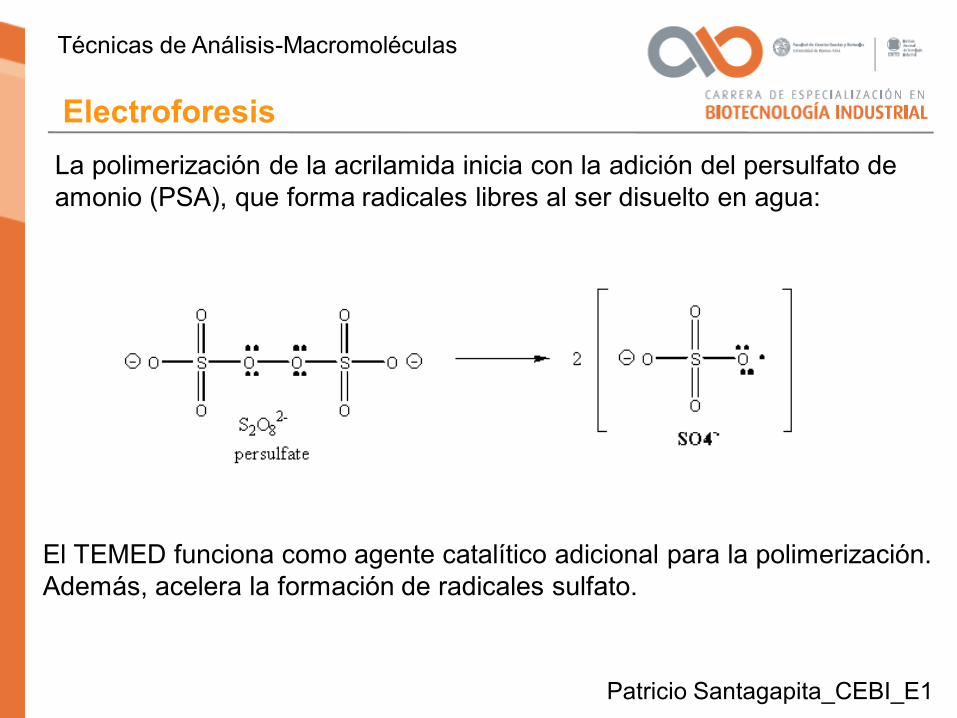
La polimerización de la acrilamida inicia con la adición del persulfato de amonio (PSA), que forma radicales libres al ser disuelto en agua:
El TEMED funciona como agente catalítico adicional para la polimerización.Además, acelera la formación de radicales sulfato.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
El radical inicia al polimerización de la acrilamida.
Sólo se forma un gel rígido cuando se adiciona la bis-acrilamida a la mezcla, uniendose así cadenas de poliacrilamida adyacentes.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
La cantidad de bisacrilamida adicionada es la que controla el grado de entrecruzamiento, y por lo tanto, el tamaño de poro.
IMPORTANTE: el efecto del tamaño de poro es OPUESTO al que normalmente se verifica en cromatografía. Si bien en ambos casos las proteínas grandes presentan dificultad para entrar por los poros, en cromatografía eluyen rápidamente. En electroforesis, eluyen lentamenteya que al no entrar facilmente en el gel, el transporte producido por la corriente eléctrica es dificultoso.
El tamaño de poro no es simple de controlar como suele serlo en las resinas utilizadas en cromatografía.
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
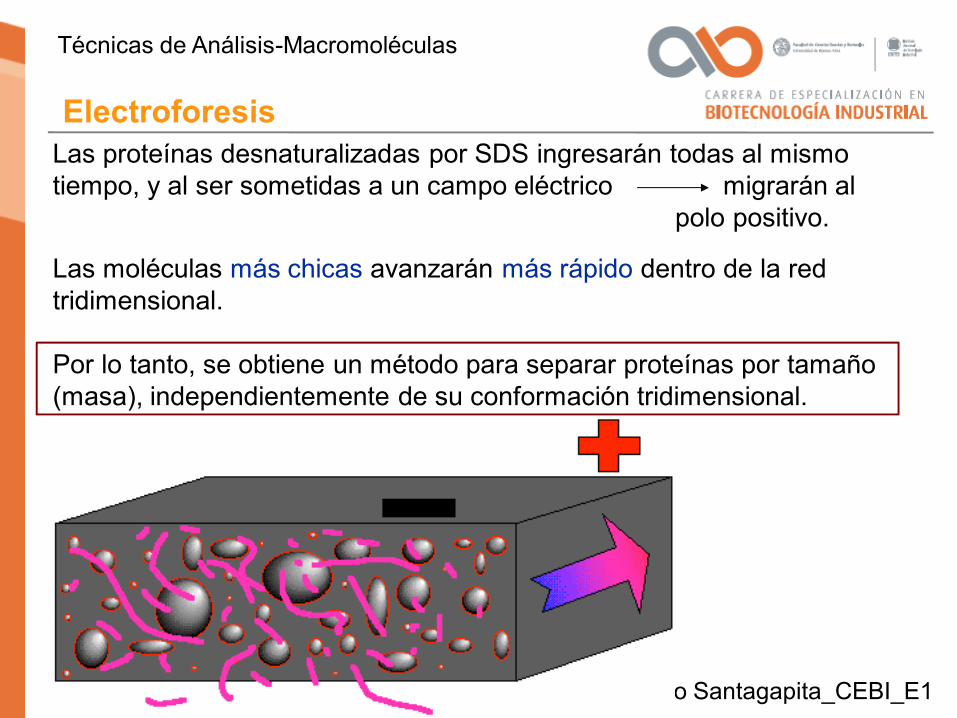
Las proteínas desnaturalizadas por SDS ingresarán todas al mismo tiempo, y al ser sometidas a un campo eléctrico migrarán al
polo positivo.
Las moléculas más chicas avanzarán más rápido dentro de la red tridimensional.
Por lo tanto, se obtiene un método para separar proteínas por tamaño (masa), independientemente de su conformación tridimensional.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Resultado de la corrida: ¿líneas o bandas?
En una muestra que contiene sólo una proteína de una secuencia y tamaño determinados, tendremos miles de copias de la misma molécula.
Cada una de estas copias recorrerá el gel por diferentes caminos, siendo algunas retenidas durante un poco más de tiempo. Esto genera una banda (y no una línea definida) en el gel.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Resultado de la corrida: ¿líneas o bandas?
De la misma manera, la técnica SDS-PAGE sólo separa proteínas del mismo tamaño. Por lo tanto, proteínas con diferente secuencia de aminoácidos pero de la misma longitud, correrán juntas en la misma banda, sin poder ser identificadas.
Como regla general (aunque depende ENORMEMENTE del gel preparado), esta técnica permite separar moléculas cuyos tamaños difieren hasta en un 10-5 %, y se utilizan markers o ladders con bandas de peso molecular conocidas como referencia en los geles.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Varios diseños de aparatos posibles

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Los geles de acrilamida se preparan entre dos placas de vidriotransparente.
BIORAD ©

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Técnica:Requiere preparar los vidrios en posición vertical, utilizando un spacerstandard (1 mm por ej). Esto minimiza el contacto del gel con el aire, ya que el O2 inhibe la polimerización por reacción con los sulfatos.
Preparar las mezclas de ambos geles (stack y de corrida o resolutivo), sin agregar PSA ni TEMED.
BIORAD ©

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Técnica (cont.)Agregar TEMED y PSA, en ese orden, sólo al gel de corrida, mezclar porinversión varias veces y volcar entre los vidrios.
Antes de que termine de polimerizar el gel de corrida, agregar TEMED y PSA al gel stacking, y también volcarlo entre los vidrios, sobre el gel de corrida.
Poner el peine y dejar polimerizar.
Antes de cargar las muestras, sacar los peines con cuidado y lavar los wells con buffer de corrida.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
La resolución de los geles depende de las cantidades agregadas para la preparación de la mezcla, y se expresa en % de acrilamida (geles de 10, 12, 15%, etc).
Para geles de 10% o menos, se usa un gel de stacking de 4%, mientras que para geles mayores al 10%, es recomendable un stacking de 6%.
Los geles stacking son laxos para permitir que todas las proteínas corran libremente, hasta encontrarse con el gel de corrida, que es mucho más denso. Esto hace que todaslas proteínas de la muestra se encuentren en la misma línea de partida cuando comienzan a correr por el gel de corrida o resolutivo.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
PAGE: polyacrilamide gel electrophoresis % de gel
%T: contenido total A+B.A > %T, menor tamaño de poroSe elegirá el %T de acuerdo a la mezcla que se debe separar.El %C también modifica el tamaño. Al 5% de bis se obtienen los geles de menor tamaño. > o < %C hace que el gel sea de mayor tamaño de poro.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Se presentan mezclas standard para geles de corrida o resolutivos. El pH del buffer es 8,8.
7.5 %
17.5 %
7.5 %
13.5 %

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Mezcla standard para gel destacking de 4,5%. El pH del bufferes 6,8
Solución para la muestra (5X): tiene azul de bromofenol para indicar el frente de la corrida y como indicador de pH, y glicerol para darle mayor densidad que el buffer al momento de la siembra en la calle.
El azul de bromofenol tiene carga positiva en pH > 4,6, por lo que corredelante de todas las proteínas.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Buffer de corrida: es el buffer con el que se llena la cuba y en el que sesumerge el gel.
Para una cuba standard de Bio Rad (Protean II) son necesarios 500 ml de una solución del buffer anterior 1X, por lo que deberán diluirse 50 ml del Buffer 10X en agua destilada o milliQ.
Se corren los geles a 200 volts constantes, hasta que el azul del frente de corrida llegue al borde inferior del gel. Son unos 50-60 minutos. La progresión de la corrida puede monitorearse también utilizando markerscoloreados.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Luego de la electroforesis, el gel debe ser teñido para detectar las proteínas separadas, o las proteínas transferidas a una membrana para su detección específica posterior (Western blot).
Las tinciones mas comunes son Coomassie Blue, PageBlu, o tinción Argéntica (con plata).
Recordar que las proteínas se observan como bandas diferentes en el gel, y se utiliza un marcador con proteínas de peso molecular conocido como referencia en una de las calles del gel.
La electroforesis en gel es el primer método utilizado para la determinación de pureza en proteínas.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
El colorante Coomassie Blue es cuantitativo. Puede utilizarse paracuantificar especies diferentes en un gel. Sin embargo, se adhiere conmenor afinidad a las proteínas con alto grado de glicosilación. Límite dedetección: 100 ng.
Se une a las histidinas y aminoácidos aromáticos de las proteínas a detectar.
Colorantes comúnmenteutilizados: Coomassie BlueR250 (se obtiene mejorresolución), Coomassie BlueG250 (es más sensible),Coomassie Violet R150.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Existe también una tinción coloidal de Coomassie Blue, en la cual se usaCoomassie Blue G250 (en lugar de R250) en una solución que tambiéncontiene acido fosfórico, sulfato de amonio, y metanol al 20%.
Esta tinción es mucho más sensible, aunque con mayores errores a la hora de cuantificar respecto a la que utiliza R250.
Se recomienda el uso con proteínas de bajo peso molecular, o cuando es necesaria la detección de especies en muy baja concentración.
En estos casos también es recomendable aumentar el porcentaje demetanol de los buffers, incluyendo el buffer de transferencia en los Western Blots.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
La tinción argéntica es muy sensible (detecta impurezas que no se ven por otros métodos) pero no es cuantitativa (tiene diferente afinidad por lasdistintas proteínas). Límite de detección: 1 ng.
El mecanismo químico de la tinción argéntica todavía no se conoce. Es unmétodo muy sensible a suciedad en los vidrios, materiales e impurezas en los reactivos.
Hay que trabajar con mucho cuidado, evitar el contacto con el gel y utilizar buffers recién preparados y fríos.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
PageBlu: solución teñidora de proteínas.
Protocolo mucho más simple
Más sensible que Coomasie Blue R-250
Única solución. No contiene ác. Acético o metanol
Más rápido que CB
Útil para Western Blot
de Thermo Scientific

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Tinción PAS específica para glicoproteínas tinción del ácido periódico-Schiff-PAS para glicoproteínas
Concentraciones de proteínas entre 1 y 6 mg/ml
(Glicoprotein Detection Kit, Sigma)

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Tinción PAS
“Las bandas de proteínas se fijaron al gel de poliacrilamida con ácido tricloroacético al 12,5% (p/v) durante 30 minutos, el exceso de ácido se eliminó por lavado con agua y a continuación los glicanos unidos a la estructura de las glicoproteínas se hicieron reaccionar con ácido periódico al 1% (p/v) en ácido acético al 3% (v/v). El exceso de los iones periodato e iodato se eliminaron lavando con agua (6 veces durante 10 minutos) y a continuación se desarrolló el color por inmersión del gel en el reactivo de Schiff durante 1 h en oscuridad. El gel se decoloró con una solución de Na2S2O5 al 0,5% (p/v) y lavado con agua bidestilada.”
Benavent, 2007, Tesis Facultad de Ciencias de Madrid

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Marcadores: muchas opciones en el mercado.
Debe cubrir todo el rango de interés.
Puede poseer doble utilidad: PAGE y Western blot.
de Invitrogen

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
de Thermo Scientific

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Los geles finalmente son secados, para extraerles la mayor parte de sucontenido acuoso.
El secado puede hacerse en estufas cerradas, o en secadoras con planchas calientes (80°C) y la aplicación de vacío.
El secado por aire caliente se realiza en geles contenidos entre dos planchas de celofan, y es ideal para densitometría, escaneado y almacenamiento. La tendencia al quebrado es mucho menor.
BIORAD ©
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Algunos ejemplos de tinciones...

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Mas ejemplos...
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
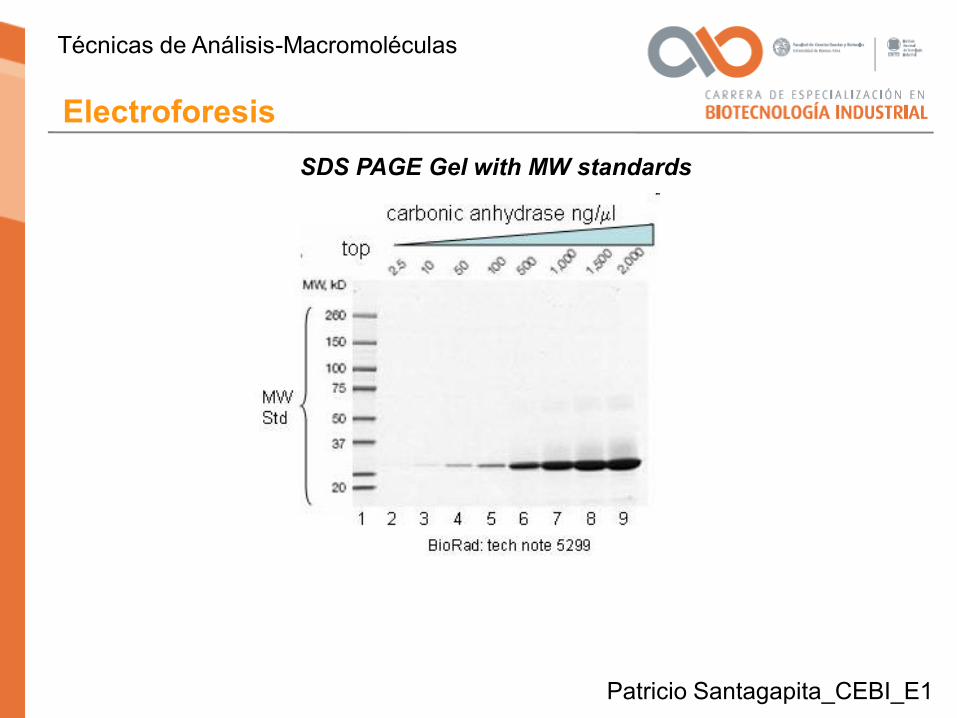
SDS PAGE Gel with MW standards

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Estimación del peso molecular
10 (36 mm)
15 (27.5 mm)
20 (22 mm)25 (17 mm)
37 (12 mm)
0
5
10
15
20
25
30
35
40
45
50
0 10 20 30 40
Distance migrated (mm)
BIORAD ©

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
10
100
0 10 20 30 40Distance migrated (mm)
Size
(kD
)Analisis del peso
molecular con papel semi-log
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
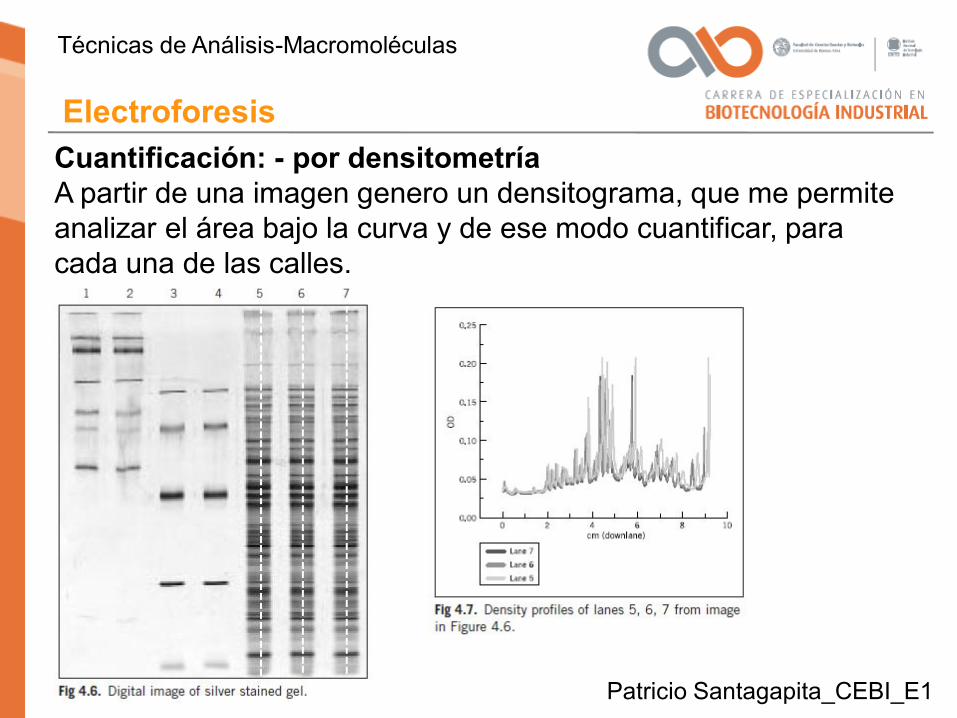
Cuantificación: - por densitometríaA partir de una imagen genero un densitograma, que me permite analizar el área bajo la curva y de ese modo cuantificar, para cada una de las calles.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Cuantificación: - por análisis de imagenA partir de una imagen adquirida por un escaner o cámara, se analiza la imagen. Softs libres (ej. ImageJ, http://imagej.nih.gov/ij/).
Tutorial: https://www.youtube.com/watch?v=JlR5v-DsTdsUsing ImageJ to quantify protein bands on a PAGE gel.
http://openwetware.org/wiki/Protein_Quantification_Using_ImageJ

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Existen sistemas automatizados para hacer electroforesis de proteínas y ácidos nucleicos, con circuitos capilares microvolumétricos conteniendo una matriz similar a la poliacrilamida.
Estos sistemas son automáticos, rápidos, y generan un perfil de absorbancia versus tiempo de cada muestra, similar los picos del HPLC analítico.
Luego estos picos son transformados en bandas con la densidad apropiada en un gel virtual emitido por el equipo.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Recomendaciones:
Utilizar geles SDS-PAGE para determinar pureza en muestras, cuando sea necesaria una cuantificación.
Optar por métodos coloidales cuando se busque una proteína de bajo PM.
Aumentar el porcentaje de metanol en los buffers de tinción cuando hayaque detectar proteínas de bajo PM, para fijarlas mejor.
Tricina: su uso es necesario con proteínas pequeñas o péptidos. Muy altaresolución. Más complejos de preparar. Geles mas duros y difíciles dealmacenar. Ver protocolo adjunto (Schägger, Nat. Protocols, 2006,1:16)

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Recomendaciones:
Utilizar geles de plata para detectar impurezas en mínima concentración. Se recomienda utilizarlos para el seguimiento y desarrollo de métodos depurificación.
No analizar muestras con alta concentración de proteínas.
Las cantidades que mejor se observan en geles de Coomassie Blueestán entre 200 y 2000 ng.
Para geles de plata, entre 25 y 200 ng.
No comparar cuantitativamente proteínas con diferente grado deglicosilación.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Por lo tanto, el paso n° 1 antes de realizar el gel:
No analizar muestras con alta concentración de proteínas
Determinación de concentración de proteínas
Existen varios métodos (Bradford, Lowry, Biuret, etc). Se entrará en detalle más adelante en el curso
Conocer el tipo de proteínas para realizar tinciones diferenciales

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
PAGE: polyacrilamide gel electrophoresis
2 condiciones: nativa y desnaturalizante
La condición NATIVA permite estudiar moléculas que retienen su estructura, actividad (enzimática, x ej.), e interacciones (complejos).
La migración dependerá de TAMAÑO, FORMA y CARGA.
Suelen ser de menor resolución que los SDS-PAGE.
El protocolo es similar al descripto, pero se evita el SDS, DTT, calor, etc.
Útil para todos los casos que se deba retener funcionalidad, interacciones, conformación. Para determinar el PM: SDS-PAGE.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Condición NATIVA: La migración de las proteínas dependerá de su carga neta, que dependerá del pH (dependiendo de su punto isoeléctrico). Sólo las proteínas que estén cargadas negativamente migrarán al polo +.
El punto isoeléctrico (pI) de una proteína es el pH donde la carga neta de la misma es nula. No es ni positiva ni negativa, por lo que al someterla a un campo eléctrico, no tiende a moverse hacia ninguno de los dos polos.
Los pI pueden variar entre 2 y 13, pero la mayoría de las proteínas tiene un pI entre 3 y 7.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Aminoácidos cargados
(+): Lys, Arg, His(-): Asp, Glu
¿Y si mi proteína tiene un pI > 9? Hay que invertir la polaridad

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Ejemplo de marcadores (alto peso molecular)

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
El método Ferguson (1964)
Los autores usaron 7 T% diferentes

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Geles de agarosa
Uso más general: separar ADN, o ARN por tamaño
Migran hacia el polo positivo porque ya cargadas eléctricamente
Proteínas: sii alto peso molecular
Aplicaciones: - luego de digestión con enzimas específicas- análisis de productos de PCR
De esta forma, separo fragmento linealizados, y la conformación de doble hélice no es un problema
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
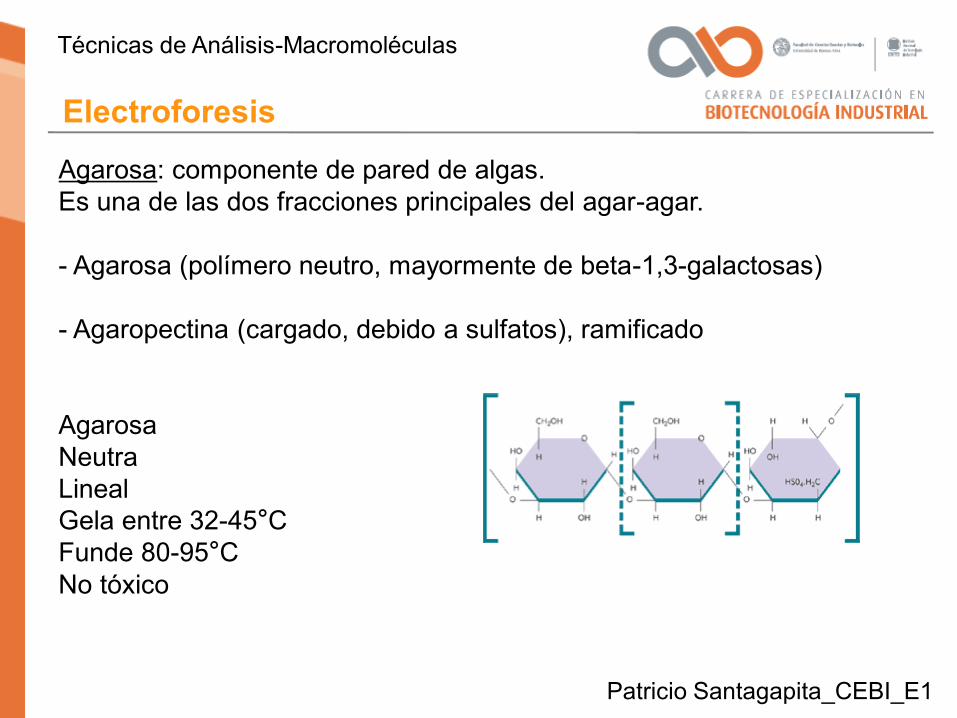
Agarosa: componente de pared de algas. Es una de las dos fracciones principales del agar-agar.
- Agarosa (polímero neutro, mayormente de beta-1,3-galactosas)
- Agaropectina (cargado, debido a sulfatos), ramificado
AgarosaNeutraLinealGela entre 32-45°CFunde 80-95°CNo tóxico
Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
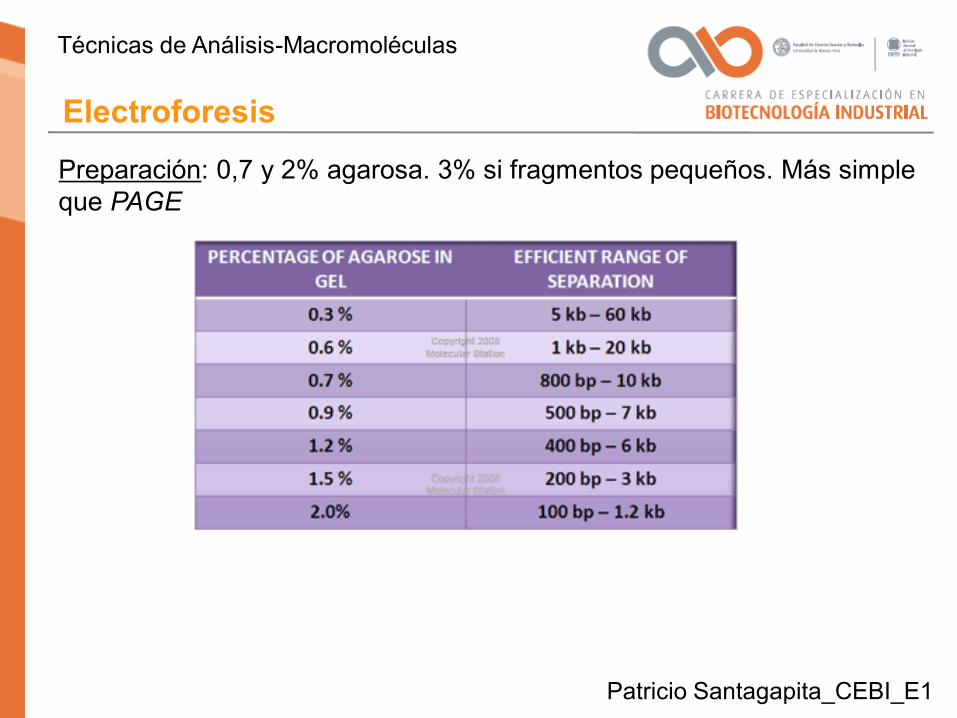
Preparación: 0,7 y 2% agarosa. 3% si fragmentos pequeños. Más simple que PAGE

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Tinción: bromuro de etidio (se intercala entre las bases), SYBR Green (Sigma), plata.
Revelado: al UV.
Comparando intensidad y posición respecto al marcador, puedo cuantificar y conocer el PM, respectivamente
Buffers de corrida: - TAE (Tris/ acetate/ EDTA)- TBE (Tris/ borate/ EDTA)- SB (Sodim borate)

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Geles de agarosa
Protocolo útil: http://www.methodbook.net/dna/agarogel.html
Electroforesis gel: desarrollo de la técnicahttps://www.youtube.com/watch?v=2UQIoYhOowMHow to Make and Run an Agarose Gel (DNA Electrophoresis)

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Electroforesis en geles de agarosa. Esquema

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Fragmentos de DNA separados en gel de agarosa
Ladders (en kb), a) por Enz. Restricc, b) marcadores

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Geles de agarosa
Desarrollos específicos:
Sistema de electroforesis por microchip para análisis de ADN/ARNMCE-202 MultiNA (Shimadzu, Biotech- Jenck S.A.)
https://www.jenck.com/productos/producto/mce-202-multina
Alta reproducibilidad, detecciones de hasta 6 pb, automatización

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
¿Se pueden separar proteínas en geles de agarosa?
Poliacrilamida Agarosa
Myosin Heavy Chain
ActinTropomyosin
10
15
2025
375075
100150250
Myosin Light Chains 20
25375075
100150250
Myosin Heavy Chain
ActinTropomyosin
Myosin Light Chains
BIORAD ©

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Algunas empresas que comercializan equipos:
BIO RAD, www.bio-rad.com
Labnet International Inc., http://www.labnetlink.com/
Scie-Plas (UK), www.scie-plas.co.uk
Cole-parmer instrument company, www.colepalmer.com

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas

Ready Gel®Precast Gel Assembly
Step 1 Step 2
Step 3 Step 4Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
BIORAD ©

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
10
100
0 10 20 30 40Distance migrated (mm)
Size
(kD
)PROBLEMAS

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Problema 1
Una enzima presenta varias isoformas posibles. La electroforesis en condiciones nativas de una muestra conteniendo una mezcla de isoformas de una enzima X muestra 4 bandas definidas, a 70, 90, 120 y 140 kDa.Cuando se trata a la muestra con SDS y β-mercaptoetanol, por SDS-PAGE se obtienen bandas con una movilidad relativa que pudo ser asignada a dos bandas de 20 y 50 kDa.Dibuje los geles obtenidos, señalando los polos de la corrida, e indique la estructura cuaternaria de las posibles isoformas a partir de los datos.

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Problema 2
Se ha determinado la movilidad relativa de la lactato deshidrogenasa y de varias proteínas patrón en electroforesis en gel de poliacrilamida al 7,5% en presencia de SDS, obteniéndose los valores de la tabla. Calcule la masa molecular estimada de la LDH.
Curso Lic. Biología, Universidad de Alcalà, España

Patricio Santagapita_CEBI_E1
Electroforesis
Técnicas de Análisis-Macromoléculas
Problema 3
La electroforesis de una enzima a la que previamente se ha tratado con SDS y β-mercaptoetanol, por SDS-PAGE da 2 bandas con una movilidad relativa de 0,51 y 0,56. La masa molecular de la enzima nativa, determinada mediante Sephadex G-200 y ultracentrifugación, es de 130 000. Indique la estructura cuaternaria de la enzima y la masa molecular de sus subunidades. La movilidad relativa de proteínas patrón en las mismas condiciones es:
Curso Lic. Biología, Universidad de Alcalà, España