modern drug discovery and development - …...361 modern drug discovery and development john c....
TRANSCRIPT

361
Modern Drug Discovery and Development
John C. Alexander and Daniel E. Salazar
Chapter 25
of America ( PhRMA, 2005 ), for every 10 000 compounds synthesized, only one will be approved by the FDA. Of these approvals, only one in three will cover the cost of development ( PhRMA, 2005 ). A study from Tufts University ( DiMasi et al. , 2003 ) estimated the cost of the development of one compound reaching the market was over $800 million and was increasing at a rate of 7.4% over inflation.
This chapter presents an overview of the drug discovery process from the laboratory bench through post-marketing approval. A brief history of drug discovery through the ages is included to provide perspective on the current situ-ation and anticipated changes in the near future. Aspects of drug discovery include finding drug targets, identifying and optimizing lead compounds, preclinical studies, and phase I through phase IV clinical studies. New paradigms such as pharmacokinetic modeling and simulations are also dis-cussed. Numerous examples are included to illustrate vari-ous processes and principles.
DRUG DISCOVERY THROUGH THE TWENTIETH CENTURY
Drug discovery began with observations that various natural products produced a desirable response in the patient, e.g.,
BACKGROUND: THE IRONY OF INNOVATION
In 2006 global pharmaceutical sales were estimated to be $643 billion, with the majority of sales in North America, Europe and Japan, according to Intercontinental Marketing Services ( IMS Health, 2007 ). The Centre for Medicines Research (CMR) reported that the global investment in pharmaceutical R & D exceeds $55 billion and has grown more than 58% during the past 10 years (CMR, 2006). The irony of this investment in innovation is that although there are numerous targets, greater investment and better technology, the number of compound approvals has been declining. In 2006, only 18 new molecular entities and 4 new biologic license applications were approved in the United States (US), a new low that was reported in Nature Reviews Drug Discovery ( Owens, 2007 ).
The research and development of new medicines is a lengthy process, taking over 11.5 years from the identifica-tion of a drug candidate to its introduction to the market ( Fig. 25.1 ). The development times are surprisingly similar between the US, Europe and Japan and also have not changed much over the past decades (CMR, 2006). Not only is time for new drug development very long but attrition is also severe. According to the Pharmaceutical Research and Manufacturers
Stage 1DRUG DISCOVERY
Stage 2PRE-CLINICAL
Stage 3CLINICAL TRIALS
Stage 4FDA REVIEW
10 000Compounds
1FDA Approved
Drug250 Compounds 5 Compounds
PHASE I20–100 Volunteers PHASE III
1000–5000 Volunteers
PHASE II100–500 Volunteers
1.5 YEARS6 YEARS6.5 YEARS
IND
SU
BM
ITT
ED
ND
A S
UB
MIT
TE
D
FIGURE 25.1 Process of drug development (Reproduced from PhRMA, 2005 ). A color version of this figure is available on the Clinical and Translational Science companion website which can be accessed at www.elsevierdirect.com/companions/9780123736390
Clinical and Translational ScienceCopyright © 2009 Elsevier Inc. All rights reserved

PART | VII Human Pharmacology362
the use of the foxglove plant in the mid-1700s for the treat-ment of heart failure ( Goldman, 2001 ). Beginning in the nineteenth century, advances in chemistry enabled isolation of the active substance and de novo synthesis of active com-pounds such as aspirin by Bayer ( Vane, 2000 ). Subsequently, chemical modifications of key functionalities to enhance effi-cacy were achieved. For example, the discovery of penicillin in 1928 and its introduction into clinical use in the 1940s was followed by the introduction of semi-synthetic penicil-lins over the next 40 years ( Rolinson, 1998 ).
Importantly, these same advances in chemistry also allowed the discernment of distinct enzymes and cellu-lar receptors by pharmacologists and biochemists, thereby providing the basis for rational drug discovery and devel-opment that evolved during the mid-twentieth century and is still generally in use today ( Fig. 25.2 ). Once a target was identified, a screening method is developed, based on knowledge of the disease, and validated. Libraries of natu-ral product extracts or pure chemicals are ‘ screened ’ against a cellular, tissue, or even whole animal model to discover a ‘ hit ’ that is a chemical or mixture with recognizable activity. Once a hit was discovered, analogue synthesis proceeded to improve activity in the screen and hopefully develop a structure activity relationship. This is known as the lead identification stage. Once a lead or several lead compounds were identified that had sufficient pharmacological activity, preliminary animal toxicology and pharmacokinetic studies as well as studies characterizing the pharmaceutical prop-erties of the compounds were conducted to support lead
compound optimization. Usually, additional analogues have to be synthesized to obtain a compound deemed sufficient in all these aspects and thus suitable for clinical develop-ment. The clinical candidate would then undergo the studies required by regulatory authorities to proceed to clinical test-ing and hopefully make it to the market. Most of the time, the initial clinical candidate failed due to poor pharmacoki-netics, safety, or efficacy.
An example of the twentieth-century drug discovery process was the discovery of the histamine 2 (H 2 ) receptor antagonists. In 1956, Code discovered that histamine could stimulate gastric acid secretion ( Code, 1956 ). In addition, several different investigators postulated that there were subtypes of histamine receptors ( Folkow et al. , 1948 ; Ash and Schild, 1966 ). Based on this foundation, James Black and his colleagues began studying the ability of histamine analogues to block histamine-induced gastric acid secre-tion and identified several lead compounds ( Molinder, 1994 ). As Molinder (1994) describes, the initial lead H 2receptor antagonists had no oral bioavailability and several had very poor pharmaceutical characteristics. In addition, the first H 2 receptor antagonist tested clinically, metiamide, produced severe agranulocytosis in several patients and was discontinued from development. Eventually, improve-ments in pharmacokinetics and drug safety were made through exploring additional molecules. This research led to the discovery, development and marketing of cimetidine,the first selective H 2 receptor antagonist. Following clinical studies demonstrating the efficacy of cimetidine in gastric
Lead optimization
Analoguesynthesis
PharmacologySafety,
Pharmacokinetics,Pharmaceutics
Clinical development
Leadidentification
Compoundscreening
Analoguesynthesis
Hit identification
Target discovery
Assay validation
FIGURE 25.2 General process of drug discovery. A color version of this figure is available on the Clinical and Translational Science companion web-site which can be accessed at www.elsevierdirect.com/companions/9780123736390

Chapter | 25 Modern Drug Discovery and Development363
and duodenal ulcer, it would go on to revolutionize the treatment of these ailments, becoming the first drug to achieve ‘ blockbuster ’ status as defined by more than $1 bil-lion in global annual sales and is available today over-the-counter without a prescription. Finally, while cimetidine is a very good drug, it has enough affinity for histamine 1 (H 1 ) receptors that, at high concentrations, it can produce adverse effects ( Freston, 1987 ); it has a fairly short plasma half-life and thus must be dosed at least twice per day; and it can significantly inhibit the metabolism of other medi-cations ( Ostro, 1987 ). Thus, subsequently discovered and marketed H 2 receptor antagonists were more selective for the H 2 receptor, had somewhat longer half-lives and were less susceptible to metabolic drug interactions ( Ostro,1987 ), illustrating that clinical feedback to drug discovery is essential for new drugs to provide meaningful advances in therapeutics.
DRUG DISCOVERY IN THE TWENTY-FIRST CENTURY
In the twenty-first century, the overall process of drug dis-covery is largely unchanged from the previous century; however, the biotechnology and informatics revolutions of the late 1980s and 1990s have changed the specifics of the stages of drug discovery in manifold ways. Furthermore, due to these advances, many activities that were formerly conducted as part of lead optimization, such as absorption,
pharmacokinetic and toxicology profiling, are now con-ducted as high throughput screening assays closely associ-ated with lead identification.
Drug target discovery
Contemporary drug target discovery employs techniques such as genomics, proteomics, genetic association and reverse genetics as well as clinical and in vivo studies to identify potential targets ( Lindsay, 2003 ) ( Fig. 25.3 ). During valida-tion, the demonstration of the role of the target in disease phenotype, modulation of gene expression and/or protein function in both cell and animal models is used to sup-port the target selection before using the target in the drug discovery process.
The elucidation of the human genome is having a fun-damental impact on drug target discovery. For example, researchers now have characterized the molecular targets of all FDA-approved drugs ( Imming et al. , 2007 ; Overington et al. , 2006 ) showing that there are only 324 different molecular targets for approved drugs and that most of these targets belong to only 10 gene families. Furthermore, while an exact determination of the potential number of drug tar-gets is not possible, estimates based on the human genome suggest there may be somewhere between 600 and 1500 drug targets ( Hopkins and Groom, 2002 ).
Although the effect of highly penetrant genes on diseases has long enabled drug discovery, e.g., familial hypercho-lesterolemia and drugs for the treatment of atherosclerosis
Disease model Target identification Target validation
Molecular approach(A)
(B)
Clinical samples
Cell models
Systems approach
Patients
Animal models
Genomics, proteomics,genetic association
Forward genetics
Clinical sciences
Forward geneticsReverse genetics
Disease tissueexpression
Modulation in cell models(messenger RNA knockout,
protein overexpression)
Drugdiscovery
Modulation in animal models(KO/transgenic mice)
FIGURE 25.3 An overview of molecular- and system-based approaches to target discovery. Target discovery is composed of three steps: the provi-sion of disease models/tissues, target identification and target validation. The ‘ molecular ’ approach (A) uses techniques such as genomics, proteomics, genetic association and reverse genetics, whereas the ‘ systems ’ approach (B) uses clinical and in vivo studies to identify potential targets. During validation, modulation of gene expression and/or protein function in both cell and animal models is used to confirm the role of the target prior to passing into the discovery pipeline (Adapted from Lindsay, 2003 , with permission)

PART | VII Human Pharmacology364
( Lusis et al. , 2004 ), there are now newer techniques that allow for the discovery of the association of low penetrance genes with human disease. For example, a recent study employing whole genome scanning of subjects with diabe-tes and matched controls by the Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University and Novartis Institutes for BioMedical Research (2007) has found several new genes associated with diabetes and trig-lyceride levels. Certainly, the new genetic targets uncovered from this study are already the focus of new drug discovery efforts. Furthermore, the emphasis on gene-based target dis-covery is highlighted by the fact, that at least one biophar-maceutical company, deCODE, states on its website that it is ‘ applying its discoveries in human genetics to develop drugs and diagnostics for common diseases ’ (see http://www.decode.com/ ).
Lead identification
Accompanying the advancements in the ability to discov-ery new targets for drugs, the fields of high throughput screening (HTS) and combinatorial chemistry have dra-matically altered the lead identification process. HTS is currently conducted with protein, cell and even organism-based assays ( Nicholson et al. , 2007 ). Based on a survey of HTS laboratories and suppliers, it was estimated that HTS laboratories could perform an average of 55 000 screens per week in 1998 ( Fox et al. , 1999 ). A more recent sur-vey has found that many drug discovery laboratories can now perform over 100 000 screening assays per week ( Fox et al. , 2006 ). In addition, it was estimated that in 2004, the 54 HTS laboratories surveyed generated over 700 lead com-pounds and 100 compounds tested in humans ( Fig. 25.4 ).
The ability to rapidly screen compounds was accompa-nied in parallel by the ability to synthesize new molecules. During the 1980s it was recognized that creation of new molecules for screening against targets and for lead optimi-zation was rate-limiting in the discovery of new drugs. In the late 1980s and 1990 many pharmaceutical companies built on the work of Merrifield in solid phase peptide syn-thesis ( Krieger et al. , 1976 ) and formed groups devoted to combinatorial synthesis, the simultaneous preparation of all possible combinations of two or more mutually reactive sets of chemical monomers, either as individual compounds or mixtures ( Rabinowitz and Shankley, 2006 ). These groups were able to increase the output of compounds per chem-ist and thereby create large diverse chemical libraries for screening against many targets and, most importantly, rap-idly generate libraries based on a “ hit ” to obtain a lead or for subsequent lead optimization. Depending on the meth-ods used, the libraries produced by combinatorial chemistry can contain mixtures of compounds and sometimes impu-rities that compromised the ability to interpret screening results. However, using the technique of parallel synthesis,
libraries of 10–1000 compounds of good purity can be pre-pared at one time ( Rabinowitz and Shankley, 2006 ). These so-called focused libraries are now widely employed to go from hit to lead and lead optimization. While certainly combinatorial chemistry is being used to discover drugs, natural product extracts are still yielding drug candidates ( Newman and Cragg, 2007 ) and combinatorial biosynthesis is an area that may further improve the ability to generate novel therapeutic compounds ( Floss, 2006 ). Finally, virtual screening, the process of using computer-based methods to discover new ligands on the basis of biological structures, is becoming more widely used in the pharmaceutical industry ( Shoichet, 2004 ). Recently, Pang (2007) has demonstrated that virtual screening was able to identify chemicals that penetrate and rescue cells from viral infection. Importantly, in this process, the target structure was predicted by com-puters solely from the protein’s gene sequence and thus may be termed virtual ‘ genome to lead ’ discovery.
Lead optimization
The lead optimization stage typically includes further study in various pharmacological models intended to predict the compound’s efficacy in humans. Since these are typically
328 326
662
746
2000(from 45 labs)
2002(from 44 labs)
2003(from 38 labs)
2004(from 43 labs)
0
100
200
300
400
500
600
700
800
Nu
mb
er o
f le
ads
46
62
74
104120
100
80
60
40
20
02000
(from 18 labs)2002
(from 23 labs)2003
(from 28 labs)2004
(from 26 labs)
Nu
mb
er o
f d
rug
can
did
ates
(A)
(B)
FIGURE 25.4 Historical comparison of (A) the number of leads and (B) the number of drug candidates being tested in humans emerging from high throughput screening as reported by survey participants (Reproduced from Fox et al. , 2006 , with permission)
Chapter | 25 Modern Drug Discovery and Development365
specific to the intended indication for the compound they are beyond the scope of this chapter. Instead, the common elements of the lead optimization phase of drug discov-ery are discussed in this section. First, in order for a lead compound to progress to the clinic, it must be able to be formulated for delivery by the intended route. If a drug is to be given orally, it should have adequate solubility and permeability as well as negligible luminal transport by P-glycoprotein for absorption to take place. Second, once absorbed it should have pharmacokinetics consistent with the intended use. Finally, the candidate compound should have a safety profile such that risk does not outweigh the projected benefit of the compound. Lead candidates that do not pass criteria set for these lead optimization elements by drug discovery teams are rejected, resulting in additional synthetic work to find an optimized lead.
The solubility and permeability of lead candidates were traditionally determined empirically with a good deal of effort. By examining the computed physical chemical prop-erties of drugs that entered phase II clinical development, and thus assumed to have adequate pharmaceutical prop-erties, Lipinski and colleagues (2001) were able to derive the ‘ rule of 5 ’ used to identify compounds that may have absorption or permeability issues. The rule of 5 states that poor absorption and/or permeation are more likely if one or more of the following conditions are met:
● there are more than 5 H-bond donors (expressed as the sum of OHs and NHs);
● the MWT is over 500; ● the Log P is over 5 (or MLogP is over 4.15); ● there are more than 10 H-bond acceptors (expressed as
the sum of Ns and Os).
Importantly, compound classes that are substrates for biological transporters are exceptions to the rule. To deter-mine if a potential lead compound may be a substrate of intestinal efflux transporters, many companies now assess compound permeability across Caco-2 cell monolayers ( Balimane et al. , 2006 ). Caco-2 cells are cultured human adenocarcinoma cells that undergo spontaneous enterocytic differentiation. It has been demonstrated that the extent of drug absorption correlates well with permeability across Caco-2 monolayers. Between the rule of 5 and Caco-2 permeability, most lead compounds now have good absorp-tion capability.
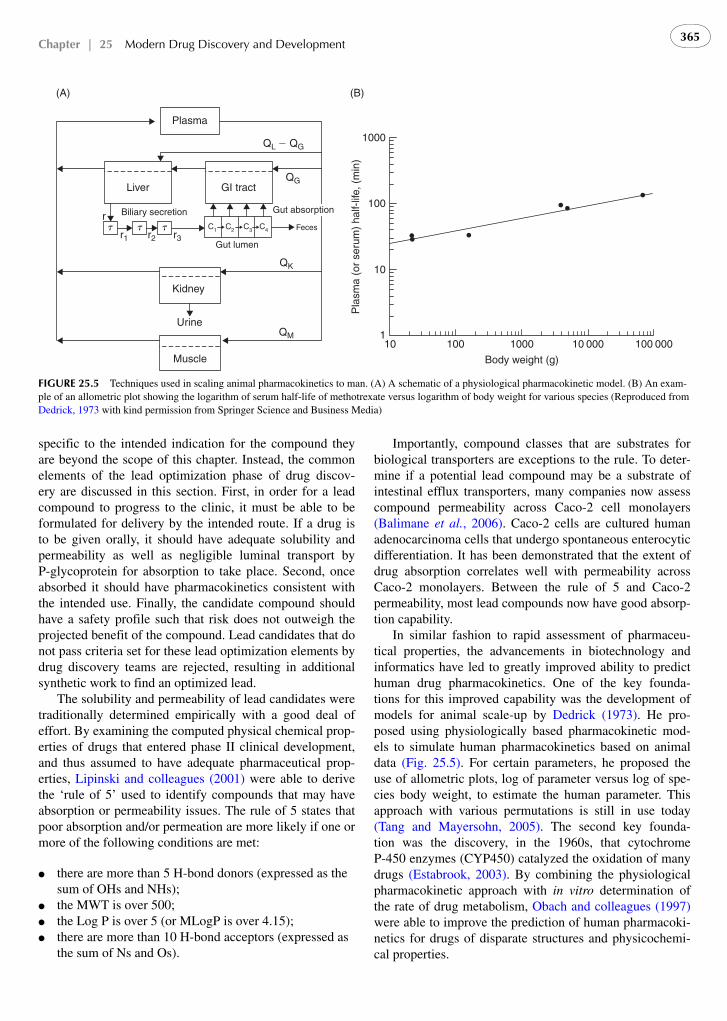
In similar fashion to rapid assessment of pharmaceu-tical properties, the advancements in biotechnology and informatics have led to greatly improved ability to predict human drug pharmacokinetics. One of the key founda-tions for this improved capability was the development of models for animal scale-up by Dedrick (1973) . He pro-posed using physiologically based pharmacokinetic mod-els to simulate human pharmacokinetics based on animal data ( Fig. 25.5 ). For certain parameters, he proposed the use of allometric plots, log of parameter versus log of spe-cies body weight, to estimate the human parameter. This approach with various permutations is still in use today ( Tang and Mayersohn, 2005 ). The second key founda-tion was the discovery, in the 1960s, that cytochrome P-450 enzymes (CYP450) catalyzed the oxidation of many drugs ( Estabrook, 2003 ). By combining the physiological pharmacokinetic approach with in vitro determination of the rate of drug metabolism, Obach and colleagues (1997) were able to improve the prediction of human pharmacoki-netics for drugs of disparate structures and physicochemi-cal properties.
Liver GI tract
Plasma
QL � QG
QG
Feces
Gut lumen
QK
QM
Gut absorption
C1 C2 C3 C4t t tr1 r2 r3
r Biliary secretion
Kidney
Urine
Muscle10 100 1000 10 000 100 000
1
10
100
1000
Pla
sma
(or
seru
m)
half-
life,
(m
in)
Body weight (g)
(A) (B)
FIGURE 25.5 Techniques used in scaling animal pharmacokinetics to man. (A) A schematic of a physiological pharmacokinetic model. (B) An exam-ple of an allometric plot showing the logarithm of serum half-life of methotrexate versus logarithm of body weight for various species (Reproduced from Dedrick, 1973 with kind permission from Springer Science and Business Media)

PART | VII Human Pharmacology366
Today, most large pharmaceutical companies have high throughput and well validated in vitro assays for deter-mining compound metabolism by recombinant CYP450 enzymes. High throughput assays are now used at the lead identification stage to eliminate compounds that might have too rapid a metabolism or too great a potential for drug interactions ( Zlokarnik et al. , 2005 ). Highly validated in vitro assays are used at the lead optimization stage to provide more accurate predictions of human pharmacoki-netics and to better predict clinical drug – drug interactions ( Walsky and Obach, 2004 ; Obach et al. , 2006 ).
The impact of greater ability to predict the human phar-macokinetics of new compounds is highlighted by data showing that the attrition rate for new drugs due to poor pharmacokinetics or bioavailability appears to have dropped from about 40% in 1991 to about 10% in 2000 ( Kola and Landis, 2004 ). Importantly, pharmaceutical manufacturers have worked together to encourage standardization of the in vitro CYP450 drug interaction studies to allow for better assessment and comparison of different drugs ( Bjornsson et al. , 2003 ). This has helped the FDA to formulate guid-ances for industry that support not only the design of in vitroand clinical drug interaction studies but also allows for drug interaction labeling based on in vitro CYP450 studies ( US Department of Health and Human Services, 1997a, 2006c ).
An essential element of modern lead compound optimi-zation for safety includes profiling of compounds in vitrofor ‘ off target ’ activity in assays selected to predict major adverse drug events ( Whitebread et al. , 2005 ). Many of these assays are intended to predict cardiovascular risk, such as activity in the hERG assay as predictive of the risk of the potentially lethal torsade de pointe arrhythmia, although they can also be predictive of adverse impact on the endocrine, central nervous, or other systems. While these assays are useful to gauge risk for pharmacologi-cally induced toxicity, compounds may also induce toxicity through direct or indirect interactions with DNA or through reactive metabolites that form covalent adducts with pro-teins or DNA (Liebler and Guengrich, 2005). During the lead optimization stage, companies will conduct in vitroand at times in vivo genotoxicity studies to eliminate com-pounds with carcinogenic potential as per the FDA guid-ance ( US Department of Health and Human Services, 1997c ). In addition, several companies are conducting in vitro and in vivo studies that determine the covalent bind-ing of compounds to microsomal or hepatic proteins ( Evans et al. , 2004 ), with the aim of assessing the potential for hepatotoxicity and idiosyncratic toxicities. While these gen-otoxicity studies and covalent binding studies are not com-pletely predictive of human risk, in many cases any potential risk can outweigh the benefit of the candidate compound especially if there is already a marketed pharmacotherapy for the indication of interest. Toxicogenomics, the integra-tion of ‘ -omic ’ technologies, bioinformatics and toxicol-ogy, has seen significant investment in the pharmaceutical
industry for both predictive and mechanism-based toxicol-ogy in an effort to identify candidate drugs more quickly and economically ( Boverhof and Zacharewski, 2006 ).While this approach may hold future promise for support-ing lead optimization efforts and generating new biomark-ers for clinical safety evaluation, the key proof of principle experiments have yet to emerge. Finally, as part of the lead optimization stage, compounds will undergo short-term in vivo toxicology and safety pharmacology studies. The duration of study, the species that are selected, and the doses chosen for study vary from compound to compound largely depending on the intended indication, the compound’s met-abolic profile, and the standards of the organization that is developing the compound. These studies are usually the last tests that a compound must pass before becoming a clini-cal candidate and proceeding on to the studies required by regulatory guidance before clinical testing.
PRECLINICAL DEVELOPMENT
Once a clinical candidate has been identified, preclinical safety, pharmacokinetic, pharmacodynamic and pharmaceu-tical studies are conducted to support initiation of clinical trials. While there are some differences in the specific pre-clinical studies required for initiating clinical trials in various regions, e.g., between US and EU, the International Conference on Harmonisation (ICH) has produced guide-lines that are relatively similar between the regions. The ICH guidelines for the US can be found on the internet at http://www.fda.gov/cder/guidance/index.htm .
The elements of preclinical development are driven by the clinical plans to evaluate the compound and the regu-latory requirements to support those clinical trials. For example, Table 25.1 shows the duration of repeated dose toxicity studies in rodent and non-rodent studies required by the ICH M3 guideline for phase I, II and III stud-ies of various durations in various regions of the world ( US Department of Health and Human Services, 1997b ).Typically, most phase I studies have treatment durations of no longer than 2 weeks; thus, toxicology studies to sup-port an Investigational New Drug (IND) filing usually have treatment durations of 2 weeks. Toxicology studies with longer treatment durations will be conducted subsequently to support clinical trials of longer duration. The species selection and design for these non-clinical safety studies should be tailored to the compound and the indication that is being pursued ( Greaves et al. , 2004 ). It is also required to measure the blood or plasma concentrations as part of these studies or in satellite groups of animals in order to enhance the value of the toxicological data generated, both in terms of understanding the toxicity tests and eventually in comparison with clinical data as part of the assessment of risk and safety in humans. Importantly, these toxicology and toxicokinetic studies must be conducted under ‘ Good

Chapter | 25 Modern Drug Discovery and Development367
Laboratory Practice ’ (GLP) conditions to ensure the qual-ity of the data.
The results of the toxicology studies are used to determine the maximum recommended safe starting dose for initial clin-ical trials ( US Department of Health and Human Services, 2005 ). The approach is to use a fraction of the no observable adverse effect dose (NOAEL), as determined by the toxicol-ogy studies, and convert this to the human equivalent dose by using body surface area ( Fig. 25.6 ). Alternatively, one can perform simulations of the human pharmacokinetics as discussed under ‘ Lead Optimization ’ above and then select doses for initial studies based on comparison with the phar-macokinetics in animals at doses that produce pharmaco-logical effects and concentrations observed in the toxicology study at the NOAEL. In this approach one should consider that there could be large differences in plasma protein bind-ing between species and take this into account.
This alternative approach is particularly useful if animal pharmacokinetic – pharmacodynamic (PK/PD) studies have been performed. PK/PD models describe the relationship between drug plasma concentrations and pharmacologic effects by mathematical equations. PK/PD models were first described in the 1960s ( Levy 1964a, 1964b ) and are now
available for a wide diversity of pharmacological responses ( Mager et al. , 2003 ). By combining pharmacokinetic models that predict human pharmacokinetics and PK/PD models developed in animals, it is possible to simulate the dose and time course of pharmacological activity before conducting a clinical study.
An example of this approach that was performed at our company for a recent factor Xa (FXa) inhibitor is shown in Fig. 25.7 . Pharmacokinetic studies of this compound were performed in several species and because the compound was nearly completely eliminated by renal excretion, sim-ple allometry was considered adequate to simulate the human pharmacokinetics ( Fig. 25.7A ). Since it was found that the absolute oral bioavailability differed between spe-cies, simulations of the human pharmacokinetics were per-formed for various doses and bioavailabilities. A PK/PD study conducted in monkeys found a simple linear relation-ship between plasma concentration of the compound and anti-FXa activity; this allowed simulations of the expected time course of anti-FXa activity at various doses and abso-lute bioavailabilities ( Fig. 25.7B ).
With sufficient clinical data linking the pharmacologi-cal activity to the clinical endpoint, it is possible to simu-late a dose response as well. Enoxaparin, a low molecular weight heparin whose principle pharmacological activity is inhibition of FXa, is indicated for the prophylaxis of deep vein thromboembolism in several postoperative situations (Lovenox package insert, 2005). Using published data on the safety and efficacy of enoxaparin ( Colwell and Spiro, 1995 ; Colwell et al. , 1995 ), its pharmacokinetics, and the relative potency for anti-factor Xa activity, we were able to simulate the dose response for the probability of venous thromboembolism and major bleeding for our FXa inhibi-tor ( Fig. 25.8 ) without conducting a clinical trial.
While clearly these simulations had many assumptions, they are helpful to optimize the design of early clinical tri-als. For example, if there is a great deal of uncertainty in the predicted human pharmacokinetics or pharmacody-namics one might consider phase I studies of a more lim-ited nature to determine these properties at a limited dose and/or duration and thereby save time and money. This approach is now supported by a recent guideline US FDA Guidance on Exploratory IND studies ( US Department of Health and Human Services, 2006a ). This guidance allows for single dose toxicology studies to support single doses less than 1/100 th of the estimated pharmacologically active dose up to 100 μ m. This type of human ‘ microdose ’ study permits limited characterization of the human pharmacoki-netics with modern analytical methodology or imaging studies via positron emission tomography ( Lappin et al. , 2006 ). Whether the pharmacokinetics at these very low, non-pharmacological doses will predict those at higher doses remains uncertain ( Boyd and Lalonde, 2007 ), the value for use of such low doses in positron emission tom-ography imaging studies is clear ( Fowler et al. , 1999 ). The
TABLE 25.1 Duration of repeated dose toxicity studies to support phase I and II trials in the European Union and phase I, II and III trials in the United States and Japana
Duration of clinical trials
Minimal duration of repeated dose toxicity studies
Rodents Nonrodents
Single dose 2–4 weeks b 2 weeks
Up to 2 weeks 2–4 weeks b 2 weeks
Up to 1 month 1 month 1 month
Up to 3 months 3 months 3 months
Up to 6 months 6 months 6 months c
� 6 months 6 months Chronicc
a In Japan, if there are no phase II clinical trials of equivalent duration to the planned phase III trials, conduct of longer duration toxicity studies should be considered.
b In the European Union and the United States, 2-week toxicity studies are the minimum duration. In Japan, 2-week nonrodent and 4-week rodent studies are needed. In the United States, as an alternative to 2-week studies, single dose toxicity studies with extended examinations can support single dose human trials.
c Data from 6 months of administration in nonrodents should be available before the initiation of clinical trials longer than 3 months. Alternatively, if applicable, data from a 9-month nonrodent study should be available before the treatment duration exceeds that which is supported by the available toxicity studies.

PART | VII Human Pharmacology368
exploratory IND guidance also provides information on the preclinical studies needed to support clinical studies up to 7 days ’ duration with the objective of determining the phar-macologically active human dose. The preclinical toxi-cology testing strategy from the guidance is displayed in Fig. 25.9 . This strategy reduces the amount of preclinical toxicology required to initiate a clinical study; however, as can be seen in Fig. 25.9 , the criteria for stopping dose escalation in the study does not permit determination of the human maximum tolerated dose. The maximum tolerated
dose can be important in subsequent clinical development so the full possible dose range can be explored for safety and efficacy and ultimately the risk/ benefit of the compound.
CLINICAL DEVELOPMENT
Traditionally, clinical development has been divided into different phases from I to IV. The process is described in Fig. 25.1 . The development of new drugs is very highly regulated and the development requirements in the US are
Determine NOAELs(mg/kg) in toxicity
studies
Is there justification for extrapolatinganimal NOAELs to human equivalent dose
(HED) based on mg/kg (or otherappropriate normalization)?
No
Yes
Convert each animal NOAELto HED (based on body
surface area; see Table 25.1)
Select HED from most appropriate species
Choose safety factor anddivide HED by that factor
HED (mg/kg) � NOAEL (mg/kg)(or other appropriate
normalization)
Maximum recommendedstarting dose (MRSD)
Consider lowering dose based on avariety of factors, e.g., PAD
Step 1
Step 2
Step 3
Step 4
Step 5
FIGURE 25.6 Algorithm for determining the maximum recommended starting dose for drugs administered systemically to normal volunteers (Reproduced from US Department of Health and Human Services, 2005 )

Chapter | 25 Modern Drug Discovery and Development369
defined specifically in the Code of Federal Regulations, Title 21 ( FDA, 1998 ).
Phase I studies
After extensive preclinical toxicology testing in several ani-mal species, ‘ first-in-human studies ’ are initiated in a small number of human volunteers who are exposed initially to
very small single doses that are estimated to have no toxic effects. The goal is to obtain pharmacologic information and dose response but always with the understanding that safety of the volunteer is of paramount importance. Since there can be no therapeutic benefit for the volunteer, the risk must be very low. After safety is established with the initial exposure, the dose of the medicine may be gradually increased and be given more frequently.
80 160 320
0 8 16 24 32 40 48
0 8 16 24 32 40 48
F � 0.18 F � 0.5F � 0.09
F � 0.18 F � 0.5F � 0.09
F � 0.045
F � 0.045
8000
6000
4000
2000
0402010
80 160 320
402010
Cp
(ng/
ml)
8000
6000
4000
2000
0
0 8 16 24 32 40 48 0 8 16 24 32 40 48
0 8 16 24 32 40 48 0 8 16 24 32 40 48
Time (hr)
Time (hr)
4
2
0
�2
�4
4
2
0
�2
�4
Log
(ant
i-FX
a) (
IU/m
l)
(A)
(B)
FIGURE 25.7 An example of the simulation of (A) human pharmacokinetics and (B) pharmacodynamics for a range of doses and bioavailabilities for a factor Xa inhibitor from preclinical data. The dose in mg is shown above each panel in the simulation. Cp is the plasma concentration of the drug, F is the absolute oral bioavailability, and FXa is the anti-factor Xa activity in plasma

PART | VII Human Pharmacology370
While the safety experience in ‘ first-in-human ’ studies has been excellent ( Stein, 2003 ), almost all of the agents pre-viously tested had been small molecules with known phar-macologic mechanisms that were quickly reversible. More recently drug development has begun to shift to biological
molecules targeted mainly for oncology and immunologic disorders. According to CMR data (2006), 11% of all the drugs in development over the past five years were biotech-derived, along with 20% of the product approvals. These agents often have novel mechanisms of action, a high degree of species specificity and/or new immune system targets. Because of these changes, pharmacologic and safety testing in animals may be less predictive than in the past ( Kenter and Cohen, 2006 ).
Suntharalingam et al . (2006) recently reported a ‘ first-in-human ’ study where six volunteers became seriously ill after receiving injections of TGN1412, a novel human-ized monoclonal antibody targeted as a CD28-specific T-lymphocyte receptor agonist for autoimmune diseases or B-cell chronic lymphocytic leukemia. In preclinical studies in Rhesus monkeys, large doses were well tolerated. While 100% homology was thought to exist between the bind-ing sites in humans and monkeys, this has now been dis-puted ( Kenter and Cohen, 2006 ). This was the first human trial with this novel antibody but important information was available from trials with antibodies for similar targets such as CTLA-4 (cytotoxic T lymphocyte-associated anti-gen). This unfortunate study has been extensively studied by European regulatory authorities and a final report and rec-ommendation is now available from the UK Medicine and
SafetypharmacologyCNSPulmonary
2-week toxicologystudy in rodentspecies
Establishment of NOAEL
In vivomicronucleus
Repeat dose toxicologystudy in nonrodent
Nonrodentequivalent or lesssensitive
Calculation of clinicalstop dose (whichever islowest) Observation of
adverse clinicalresponse
Clinical equivalent of ½ ofAUC in rat or the nonrodentAUC – whichever is lower
Clinical equivalent
to ¼ rat NOAEL
Achievement of pharmacologicaleffect or targetmodulation
Candidateexcluded fromexp IND
2-week toxstudy innonrodent
Nonrodent moresensitive
SafetypharmacologyCV
Calculation of clinical startdose, of rat NOAEL50
FIGURE 25.9 A preclinical toxicology testing strategy for exploratory Investigational New Drug applications (eINDs) designed to administer pharmacologically active doses (Reproduced from US Department of Health and Human Services, 2006a )
Hip VTEKnee VTEMajor bleeds
100
80
60
40
20
403020100
0
20
40
60
80
100
0
DV
T (
%)
Dose (mg)
Maj
or b
leed
s (%
)
FIGURE 25.8 Simulated dose response for the probability of venous thromboembolism (VTE) postoperatively in patients undergoing hip replacement (Hip VTE) and knee replacement (Knee VTE). The prob-ability of experiencing a major bleeding event (Major Bleeds) was also simulated

Chapter | 25 Modern Drug Discovery and Development371
Healthcare Products Regulatory Agency [MHRA] report ( Expert Scientific Group, 2006 ). Also the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency [EMEA] has issued new draft guide-lines for first-in-man clinical trials for potentially high-riskmedicinal products (CHMP, 2007). Many additional efforts are under way as reported by Steinbrook (2002a, 2002b) to further protect research subjects and volunteers after deaths of volunteers at the University of Rochester and Johns Hopkins, as well as after the death of 18-year-old Jesse Gelsinger in a gene-transfer trial at the University of Pennsylvania.
Not only safety and pharmacology but potential efficacy can also sometimes be projected even from phase I studies. For example, atorvastatin (Lipitor) (80 mg) produced major reduction of over 50% in LDL cholesterol in volunteers after only 14 days of treatment ( Cilla et al. , 1996 ), predict-ing superior efficacy in patients ( Fig. 25.10 ). Subsequently, atorvastatin became the best selling drug in the world ( IMSHealth, 2007 ).
For drugs developed to block the renin – angiotensin – aldosterone pathway, innovative studies ( Brunner et al. , 1980 ; Ferguson et al. , 1977 ) of the first angiotensin-converting-enzyme inhibitor, captopril, in volunteers uti-lized infusions with angiotensin I to raise blood pressure
to determine the optimal dosage. The efficacy measure was the dose of the experimental drug required to block the rise in the angiotensin-induced blood pressure increase. Results from this study predicted very closely the doses that were eventually approved ( Fig. 25.11 ).
For the development of the cyclooxygenase type 2 (COX-2) inhibitors, an important early question was whether they would have the analgesic benefit of non-steroidal anti-inflammatory drug (NSAID) class because animal studies may be not so predictive for analgesic drugs. An excellent model to assess this is a dental pain model where patients receive the analgesic agent(s) or placebo after third molar extraction and the pain intensity can be measured quite accurately ( Mehlisch, 2002 ). The COX-2 inhibitor, celecoxib, was included in a dental pain study and total pain relief at 4 hours after the dose was significantly better for subjects treated with celecoxib compared with placebo-treated subjects ( Moberly et al. , 2007 ). Also, potential NSAID gastrointestinal (GI) toxicity can be assessed in healthy subjects. Celecoxib was studied in an upper GI endoscopy study and no ulcers occurred in subjects receiv-ing seven days of treatment with celecoxib or placebo, compared with 19% of subject receiving naproxen ( Simonet al. , 1998 ). These results supported the hypothesis that selective COX-2 inhibitors would have a better GI adverse reaction profile than conventional NSAIDs.
Traditionally, in oncology, the toxicity of the drug can-didates has precluded healthy volunteer studies and only patients refractory to previous treatment have been included. While the objective of these studies has been to determine safety and pharmacokinetic properties, suggestions of efficacy can sometimes be obtained. It has been estimated that between 10 to 15% of patients may show some benefit. In oncology, the distinction between the early phases of drug development is not as clear ( Khandekar and Khandekar, 2006 ).
Total cholesterol
b.i.d.
q.d.
10 20 40 80�50
�40
�30
�20
�10
0
% R
educ
tion
from
bas
elin
e
Atorvastatin dose (mg/day)
LDL cholesterol
b.i.d.
q.d.
10 20 40 80�60
�50
�40
�30
�20
�10
0
% R
educ
tion
from
bas
elin
e
Atorvastatin dose (mg/day)
FIGURE 25.10 Effect of regimen and total daily atorvastatin dose on total cholesterol and LDL cholesterol levels. Values are least-squares mean percentage reduction from baseline after 14 days of atorvastatin (Reproduced from Cilla et al. , 1996 , with permission)
100
90
80
70
60
50
40
30
20
10
00 1 2
% C
ontr
ol r
espo
nse
3 4
1 mg2.5 mg
5 mg10 mg
20 mg
Hours
FIGURE 25.11 Inhibition of systolic pressor responses to angiotensin I in 14 healthy men after incremental doses of oral captopril. Mean responses of three subjects are shown for each dose of captopril except for 20 mg where data are derived from two subjects only (Reproduced from Ferguson et al. , 1977 , with permission)

PART | VII Human Pharmacology372
Proof of concept (phase II)
After safety is established in phase I studies of single and multiple doses of the new medicine in up to 100 volunteers treated for about two weeks, the next step is to test the new medicine in patients with the targeted disease. The objective is to confirm safety in patients and to determine potential efficacy and dose response. Different dosage regi-mens are employed over varying periods of time to obtain a suggestion of therapeutic benefit and the optimal dosage regimen. These data are quite predictive for diseases such as hypertension and diabetes where the blood pressure or fasting blood glucose levels are accepted as evidence of
efficacy. Temple (1999) has presented the FDA view on the use of surrogate markers, which are acceptable for cardio-vascular drug approval. For other conditions, biomarkers are now actively being developed to guide discovery, early development and possibly earlier approval for serious con-ditions ( Wagner et al. , 2007 ).
For heart failure, an interesting phase II study with spironolactone utilizing atrial naturetic factor (ANF) levels showed that very low doses were efficacious ( Fig. 25.12 ) in heart failure when used in addition to conventional therapy with digitalis, loop diuretics and ACE inhibitors without producing significant hyperkalemia (The RALES Investigators, 1996). Previously, the concomitant use of ACE inhibitors and spironolactone had been contraindi-cated because of the hyperkalemia risk. Changes in ANF levels used to demonstrate potential efficacy were later confirmed in a severe heart failure study where the risk of both mortality and morbidity were substantially reduced by the addition of spironolactone ( Pitt et al ., 1999 ).
In radiology, the development of non-ionic contrast agents was an important advance. By reducing the osmolarity, adverse effects such as patient discomfort during arteriog-raphy could be alleviated. This was investigated by Dotteret al . (1985) in a unique study design where patient verbal responses were recorded and compared during a cross-over study of iopamidol and a traditional ionic contrast agent. Use of iopamidol was associated with a substantial reduc-tion in painful symptoms during arteriography ( Fig. 25.13 ).
For agents without established biomarkers, novel end-points may be needed for proof of concept. For athero-sclerosis when development is initiated with a new agent without an effect on a known risk factor or with a reli-able biomarker, an imaging endpoint such as carotid artery ultrasound measurement of carotid intima-media thick-ness (CIMT) may be helpful since it is viewed as a con-firmed surrogate at least for the statin class. Crouse (2006) recently reviewed approaches with CIMT and other meth-ods to image atherosclerosis.
100
20
15
10
5
0
�100
�200
�300
�400
0
N-T
erm
inal
AN
F (
pmol
/l)P
RA
(ng
/ml.h
�1 )
12.5 mg 25 mg 50 mg 75 mg
SpironolactonePlacebo
FIGURE 25.12 With spironolactone therapy, significant changes from baseline were seen in plasma renin activity (PRA) (p � 0.002) and N-terminal pro-atrial natriuretic factor (ANF) (p � 0.022) at Week 12 (Reproduced from RALES Investigators, 1996, p. 906, with permission from Elsevier)
1 2 3 4 5 6 7 8 9 10 1 2 3 4
10
15
10
55
Num
ber
of p
atie
nts
Num
ber
of p
atie
nts
Iopamidol 300
Hypaque 60
Patient’s subjective scoring Observer’s scoring
(A) (B)
FIGURE 25.13 Comparison of discomfort and pain with iopamidol 300 and Hypaque 60 during iliofemoral runoff arteriography in 22 subjects. (A) Subject’s subjective assessment of discomfort and pain on a scale of 1 to 10, with 1 being a mild heat sensation and 10 the worst pain experienced. (B) Angiographer’s objective assessment of subjects ’ discomfort and pain on a scale of 1 to 4, with 1 being the least and 4 being the most severe pain. (Reproduced from Dotter et al. , 1985 , with permission)

Chapter | 25 Modern Drug Discovery and Development373
A study by Brousseau et al. (2004) with a novel cholesterol ester transfer protein (CETP) inhibitor, torcetrapib, showed very impressive increase in HDL and reduction in LDL cho-lesterol alone or in combination with a statin in patients with low HDL cholesterol levels. Reduction in LDL but not HDL is a very well established surrogate or biomarker for athero-sclerosis. However, a long-term outcome study called the Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events (ILLUMINATE) had to be terminated after a recommendation by the data safety moni-toring board ‘ because of an imbalance of morbidity and car-diovascular events ’ ( Pfizer, 2006 ). Following the termination of development, there were two reports from cardiovascular imaging studies with torcetrapib. An intravascular ultrasound (IVUS) study of the coronary arteries by Nissen et al . (2007) showed no significant decrease in the progression of athero-sclerosis in patients receiving torcetrapib plus atorvastatin versus atorvastatin alone. A study by Kastelein et al . (2007) with carotid ultrasound not only showed that torcetrapib failed to reduce the progression of atherosclerosis but there was evi-dence of progression in the segment of the common carotid. In both studies, torcetrapib was also associated with an eleva-tion of blood pressure of unknown cause that may have had an adverse cardiovascular effect. If these imaging studies were conducted first, there would have been no rationale for conducting an endpoint trial.
Phase III trials
The final stage before product registration is called phase III. During this phase, larger numbers of patients are treated with one or more doses for longer periods of time in con-trolled studies versus placebo or active agents. The goal is to confirm the suggestion of efficacy in proof-of-concept stud-ies but more importantly to evaluate the safety in terms of adverse events during long-term treatment. It is recognized that the statistical power is limited to detect rare adverse reactions with exposure of usually only months with limited numbers treated for longer than 6 months and one year. The ICH has produced guidelines for drugs intended for long-term treatment of non-life threatening conditions (1994). It is suggested that the numbers of volunteers and patients exposed to the new agent before registration be about 1500 with 300 – 600 treated for six months and 100 treated for a minimum of one year. Depending on the safety observed in animal studies, earlier human studies, and information from related compounds, much greater exposure may be neces-sary for an adequate assessment of risk and benefit.
Phase IV trials
The final postmarketing phase of drug testing is becom-ing more and more important to explore the safety in larger number of patients after longer-term treatment and also to confirm the efficacy in terms of clinical endpoints.
Statins
One of the best examples is the development of the statin class of drugs. These drugs were designed to inhibit HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. The class was first discovered in the 1970s at the Sankyo Co. in Japan ( Endo et al. , 1976 ). The develop-ment of this class of drug was also added by the work of Brown and Goldstein (1986) , who won the Nobel Prize for Medicine in1985 for their work on understanding the role of the LDL receptor and regulation of cholesterol homeostasis.
It is difficult now to understand that when this class was developed there were doubts about the ‘ lipid hypoth-esis ’ of the benefit of cholesterol reduction by diet or drugs on coronary disease mortality ( Tobert, 2003 ). This began to change with the publication of the results of the LipidResearch Clinics Coronary Primary Prevention Trial (1984) that found a reduction in the incidence of coronary heart disease (CHD) with cholestyramine treatment. Based on these results, an NIH Consensus Conference (1985) recom-mended that the risk of CHD could be reduced by lowering LDL cholesterol by diet and drugs.
Statins were approved by regulatory agencies on the basis of studies in thousands of patients that demonstrated favorable effects on LDL cholesterol, HDL cholesterol and triglycerides. The only safety issues were occasional eleva-tions of serum transaminases without evidence of hepatic disease, increases in CPK levels and myopathy as well as rare cases of rhabdomyolysis. However, findings of tumors in preclinical animal studies and the absence of benefit on total mortality in earlier lipid-lowering trials raised the question of a possible cancer risk ( Oliver, 1991 ).
Before this class of drugs could be widely prescribed, long-term safety and mortality studies had to be conducted. The first of these was the placebo-controlled Scandinavian Simvastatin Survival Study (4S) in over 4000 patients with CHD and elevated cholesterol studied over five years on a low fat diet. For subjects treated with simvastatin, there was a 30% reduction in all-cause mortality and a 34% reduction in the combination of CHD death and non-fatal myocardial infarction along with few adverse effects com-pared with subjects treated with placebo ( 4S Study Group, 1994 ). Many subsequent secondary prevention trials in high-risk patients, such as the CARE trial ( Sacks et al. , 1996 ) in patients with average cholesterol levels after myo-cardial infarction, the LIPID study (1998) and the study by the Heart Protection Study Collaborative Group (2002) have confirmed and extended these findings.
The question of primary prevention was first addressed in the West of Scotland Prevention Study (WOSCOPS study) by Shepherd et al . (1995) in males with elevated cholesterol (mean cholesterol 272 mg/dl) without a history of CHD, conducted in the greater Glasgow, Scotland, area. This geo-graphic area was selected because it had one of the highest incidences of coronary artery disease in the world related to a high rate of risk factors such as smoking, poor diet and

PART | VII Human Pharmacology374
genetic risk factors. In this trial of 6595 men treated with pravastatin or placebo for 4.9 years, there was a significant reduction in the incidence of myocardial infarction and death from cardiovascular causes for pravastatin-treated subjects compared with placebo-treated subjects.
Following the results of the 4S and WOSCOPS trials as well as previous trials with other lipid lowering agents, there was a marked change in medical opinion about the value of cholesterol lowering. A former critic wrote an editorial entitled ‘ Statins prevent coronary disease ’ in TheLancet that strongly recommended lowering of cholesterol in patients with elevated cholesterol and at high risk of car-diovascular disease ( Oliver, 1995 ). A recent analysis by the Cholesterol Treatment Trialists ’ (CTT) Collaborators
(2005) of data from long-term trials in more than 90 000 patients with ischemic heart disease confirmed that after a mean of 5 years of statin treatment, there was a reduction in the incidence of major coronary events, coronary revas-cularizations and stroke by about 20% per mmol/l LDL cholesterol ( Fig. 25.14 ) without evidence of an increase in cancer risk or other significant toxicity.
Drugs that block the renin – angiotensin – aldosterone (RAA) system
It was proposed in the past that angiotensin II had a del-eterious effect on the cardiovascular system beyond that caused by blood pressure elevation ( Brunner and Gavras,
Cause of death
Vascular causes
cHD
Stroke
Other vascular
Any non-cHD vascular
Any vascular
Non-vascular causes:
Cancer
Respiratory
Trauma
Other/unknown
Any non-vascular
Any death
Events (%) RR(cI)Treatment(45054)
Control(45002)
1548 (3.4%) 1960 (4.4%)
291 (0.6%)
302 (0.7%)
593 (1.3%)
265 (0.6%)
289 (0.6%)
554 (1.2%)
2102 (4.7%)
1094 (2.4%) 1069 (2.4%)
125 (0.3%)
51 (0.1%)
98 (0.2%)
487 (1.1%)
3832 (8.5%)
1730 (3.8%) 1801 (4.0%)
4354 (9.7%)
57 (0.1%)
550 (1.2%)
2553 (5.7%)
0.81 (0.76–0.85)
0.91 (0.74–1.11)
0.93 (0.83–1.03)
0.83 (0.79–0.87)
1.01 (0.91–1.12)
0.82 (0.62–1.08)
0.89 (0.59–1.34)
0.95 (0.90–1.01)
0.88 (0.84–0.91)
0.87 (0.73–1.03)
0.95 (0.78–1.16)
0.5 1.0 1.5
Treatmentbetter
Controlbetter
Effect p < 0.0001
FIGURE 25.14 Proportional effects on cause-specific mortality per mmol/l LDL cholesterol reduction. Diamonds � totals and subtotals (95% CI); squares � individual categories (horizontal lines are 99% CIs); area of square is proportional to amount of statistical information in that category. Relative risks (RRs) are weighted to represent reduction in rate per mmol/l LDL cholesterol reduction achieved by treatment at 1 year after randomiza-tion. The 26 deaths on active treatment vs. 31 deaths on control treatment in a post-CABG trial could not be subclassified into vascular and non-vascular causes, but were known not to be due to CHD and were assigned to other non-vascular deaths (Reproduced from Cholesterol Treatment Trialists ’ Collaborators, 2005, p. 1269, with permission from Elsevier)

Chapter | 25 Modern Drug Discovery and Development375
2002 ). In postmarketing trials, it was shown that drugs that block the renin – angiotensin – aldosterone (RAA) system had many benefits beyond their initial use as antihypertensives. These additional benefits included prevention and treatment of heart failure, reduction in ventricular remodeling after myocardial infarction, reduction in the rate of progression of diabetic nephropathy, and possibly even reduction of the increased incidence of diabetes in hypertensive patients.
Heart failure and myocardial infarction
Captopril, an angiotensin-enzyme-converting (ACE) inhibi-tor, was first approved for hypertension but then developed for heart failure patients with New York Heart Association (NYHA) Class III and IV heart failure refractory to digitalis and diuretics. This was the first new treatment approved for heart failure since digitalis and the diuretics. Improved survival was first shown with ACE inhibitors by the effect of enalapril on mortality in subjects with severe congestive heart failure ( CONSENSUS Trial Study Group, 1987 ). A subsequent trial by the SOLVD investigators (1992) showed that treatment with enalapril prevented the development of overt heart failure in patients without heart failure but with left ventricular dysfunction.
Pfeffer and Frohlich (2006) described some good examples of ‘ translational research ’ , including the innova-tive studies in remodeling after myocardial infarction by Pfeffer et al. (1985) , first in a coronary ligation model in rats followed by a pilot ventricular enlargement clinical study ( Pfeffer et al ., 1988 ) and finally in the survival and ventricular enlargement (SAVE) trial ( Pfeffer et al. , 1992 ). In the SAVE study, asymptomatic left ventricular dysfunc-tion patients treated with captopril for 3 to 16 days after myocardial infarction had a 19% reduction in mortality compared with subjects treated with placebo. Subsequent trials in almost 100 000 patients confirmed the prolongation of survival for subjects treated with ACE inhibitors after myocardial infarction ( Pfeffer, 1995 ). Similar benefits after myocardial infarction were shown by Pfeffer et al. (2003) with angiotensin receptor blockers (ARBs).
Clinical trials with mineralocorticoid antagonists have also shown benefit on survival in patients with heart dis-ease. An example of such a mineralocorticoid antagonist is spironolactone ( Pitt et al. , 1999 ), mentioned in the section on ‘ Proof of Concept ’ and in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) ( Pitt et al ., 2003 ). Dluhy and Williams (2004) made the important observation that aldosterone only becomes toxic to the cardiovascular system when produced in excessive amounts relative to the level of sodium intake.
Diabetes
Another area of innovation for the RAA blockers was treat-ment of patients with type 2 diabetes. A meta-analysis by Abuissa et al. (2005) of pooled data from 12 randomized
controlled trails showed that treatment of patients with hypertension with ACE inhibitors or ARBs was associated with a 25% reduction in the incidence of newly diagnosed diabetes.
RAA system blockage has also been valuable in slowing the progression of diabetic nephropathy. Studies in animal models of diabetes showed that ACE inhibitors were the only antihypertensives that could reduce increased glomeru-lar capillary pressure by blocking the effects of angiotensin II on efferent arteriolar resistance and slow the progression of renal disease, as summarized in an editorial by Remuzziand Ruggenenti (1993) .
Lewis et al. (1993) conducted a randomized placebo-controlled trial with captopril in 409 patients with type 1 diabetes and elevated urinary protein excretion. There was a 50% reduction in the composite endpoint of death, dial-ysis and transplantation in the captopril-treated patients. They also concluded that captopril was more effective than blood pressure lowering alone with other classes of antihy-pertensive agents.
Studies in type 2 diabetes with the ARBs including irbesartan ( Lewis et al ., 2001 ) and losartan ( Brenner et al. , 2001 ) have extended the evidence for the value of RAA system blockade for renoprotection in patients with hyper-tension and diabetes. Parving et al. (2001) found that irbe-sartan was also renoprotective in patients with diabetes and microalbuminuria without renal impairment.
NEW DRUG DEVELOPMENT PARADIGMS
Given the high costs of clinical trials as well as the very high attrition rate for drug in development, in 2004 the US Food and Drug Administration (FDA) proposed a ‘ critical path ’ initiative for new ways and opportunities to improve drug development ( US Department of Health and Human Services, 2004 ) ( Fig. 25.15 ). This ‘ critical path ’ report was updated in 2006 ( US Department of Health and Human Services, 2006b ) with the lessons that have been found since 2004, including a clear consensus that biomar-ker development, streamlining of clinical trials, and use of modeling and simulation were the most important areas for improving medical product development. A summary and status of these initiatives can be found at www.fda.gov/oc/initiatives/criticalpath/ . An example of such a new paradigm for development of rationally targeted oncol-ogy drugs is the development of imatinib (GLEEVEC), a protein – tyrosine kinase inhibitor for chronic myelogenous leukemia ( Mauro et al. , 2002 ). This program incorporated the use of biomarker development and streamlined clinical trials as promoted by the critical path initiative.
As with preclinical development, PK/PD modeling and simulation can be used extensively to support clinical drug development at all phases ( Chien et al. , 2005 ). In order to use modeling and simulation effectively, the work must be

PART | VII Human Pharmacology376
planned in conjunction with clinical trials. Figure 25.16 shows a schematic of a PK/PD modeling and simulation plan that was designed to support development of CS-706, a novel COX-2 inhibitor ( Kastrissios et al. , 2006 ; Rohatagi et al. , 2007 ; Moberly et al. , 2007 ). In the early develop-ment of this compound, five studies were conducted, two single ascending dose studies in healthy subjects, a mul-tiple ascending dose study in healthy subjects, a study in
healthy subjects to determine the frequency of GI erosions and ulcers by endoscopy and a dose ranging efficacy trial in acute postoperative dental pain. The primary objectives of the modeling plan were to characterize the effects of CYP450 genotypes on pharmacokinetics; correlate plasma concentrations with ex vivo determined inhibition of COX-1 and COX-2 activity; and correlate the ex vivo inhibition of COX-1 with gastrointestinal erosion frequency and COX-2
Basicresearch
Prototypedesign ordiscovery
Preclinicaldevelopment Clinical development
FDA filing,approval,launchpreparation
Translational research
Critical path research
FIGURE 25.15 Relationship between the different types of research and how they support the product development process. Basic research is directed toward fundamental understanding of biology and disease processes. Basic research provides the foundation for product development as well as trans-lational and critical path research. Translational research is concerned with moving basic discoveries from concept into clinical evaluation and is often focused on specific disease entities or therapeutic concepts. Critical path research is directed toward improving the product development process itself by establishing new evaluation tools (Adapted from US Department of Health and Human Services, 2006b )
Phase 1 GI safety
Phase 2 Dental pain
Phase 1 SD/MD
Phase 1 high dose
PD 1
PD 2
PKCp
Cox-2inhibition
Cox-1inhibition
Tooth extraction
Osteoarthritis Pain andsymptom relief
Pain relief
GI ulcers orerosions
Literature Vioxx, Celebrex, NSAIDs
Literature Vioxx, Celebrex
Tooth extraction and osteoarthritis
Tooth extraction andosteoarthritis
Competitor doses
CS-706regimen
Projectionsin
Japanese
PK in otherpopulations:Japanese
GenotypeEthnicity other
covariates
FIGURE 25.16 A schematic of a PK/PD modeling and simulation plan for a COX-2 inhibitor CS-706. The plan incorporated data from five clinical studies with CS-706 and from the literature (shaded boxes) The PK model incorporated demographic variables and CYP450 genotypes and was used to simulate PK for various dose regimens in Western and Japanese subjects. The PK/PD model incorporated results of ex vivo COX-1 and COX-2 inhibition as well as gastrointestinal erosion and analgesia subsequent to tooth extraction

Chapter | 25 Modern Drug Discovery and Development377
inhibition with postoperative pain relief. The model was then used to simulate PK/PD responses across different dose regimens and populations by using the different demo-graphics and frequency distributions of CYP450 genotypes ( Kastrissios et al. , 2006 ; Rohatagi et al. , 2007 ). An additional important objective was to understand the PK/PD profile and potential safety and efficacy compared to existing marketed compounds such as celecoxib or rofecoxib. Figure 25.17 shows the relationship between COX-1 inhibition and GI erosion and ulceration based on the modeling. For compari-son, the historical GI erosion rate of celecoxib and rofecoxib of about 5% and for aspirin, naproxen and ibuprofen of about 65% ( Simon et al. , 1998 ; Lanza et al. , 1999 ). Based on the PK-PD model for COX-1 inhibition, it was estimated that daily doses of 50 mg CS-706 would not have significantly greater frequency of ulcers than celecoxib or rofecoxib. Clearly, the model-based predictions would need to be vali-dated in well-designed clinical trials; however, this approach can be helpful to optimize the design of those trials as well as support appropriate decision making.
REFERENCES
Abuissa , H. , Jones , P.G. , Marso , S.P. and O’Keefe , J.H. Jr. ( 2005 ) Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers for prevention of type 2 diabetes . J. Am. Coll. Cardiol. 46 , 821 – 826 .
Baigent , C. , Keech , A. , Kearney , P.M. , Blackwell , L. , Buck , G. et al. (2005) Cholesterol Treatment Trialists ’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trails of statins . Lancet 366 , 1267 – 1278 .
Ash , A.S.F. and Schild , H.O. ( 1966 ) Receptors mediating some actions of histamine . Br. J. Pharmacol. Chemother. 27 , 427 – 439 .
Balimane , P.V. , Han , Y.-H. and Chong , S. ( 2006 ) Current industrial prac-tices of assessing permeability and P-glycoprotein interaction . AAPSJournal 8 , E1 – E13 .
Bjornsson , T.D. , Callaghan , J.T. , Einolf , H.J. , Fischer , V. , Gan , L. et al. ( 2003 ) The conduct of in vitro and in vivo drug – drug interaction studies: a pharmaceutical research and manufacturers of America (PhRMA) perspective . Drug Metabol. Dispos. 31 , 815 – 832 .
Boverhof , D.R. and Zacharewski , T.R. ( 2006 ) Toxicogenomics in risk assessment: applications and needs . Toxicol. Sci. 89 , 352 – 360 .
Boyd , R.A. and Lalonde , R.L. ( 2007 ) Nontraditional approaches to first-in-human studies to increase efficiency of drug development: will microdose studies make a significant impact? . Clin. Pharmacol. Ther. 81 , 24 – 26 .
Brenner , B.M. , Cooper , M.E. , De Zeeuw , D. , Keane , W.F. , Mitch , W.E. et al. ( 2001 ) Effects of Losartan on renal and cardiovascular out-comes in patients with type 2 diabetes and nephropathy . N. Engl. J. Med. 345 , 861 – 869 .
Brousseau , M.E. , Schaefer , E.J. , Wolfe , M.L. , Bloedon , L.T. , Digenio , A.G. et al. ( 2004 ) Effects of an inhibitor of cholesteryl ester transfer pro-tein on HDL cholesterol . N. Engl. J. Med. 350 , 1505 – 1515 .
Brown , M.S. and Goldstein , J.L. ( 1986 ) A receptor-mediated pathway for cholesterol homeostasis . Science 232 , 34 – 47 .
Brunner , H.R. and Gavras , H. ( 2002 ) Angiotensin blockade for hyperten-sion: a promise fulfilled . Lancet 359 , 990 – 992 .
Brunner , H.R. , Gavras , H. , Waever , B. , Textor , S.C. , Turini , G.A. et al. ( 1980 ) Clinical use of an orally acting converting enzyme inhibitor: Captopril . Hypertension 2 , 558 – 566 .
Chien , J.Y. , Friedrich , S. , Heathman , M.A. , de Alwis , D.P. and Sinha , V. ( 2005 ) Pharmacokinetics/pharmacodynamics and the stages of drug development: role of modeling and simulation . AAPS Journal 7 , E544 – E559 .
CHMP (Committee for Medicinal Products for Human Use) ( 2007 ) Guideline on Requirements for First-in-Man Clinical Trials for Potential High-Risk Medicinal Products , 1-11. Doc. Ref.EMEA/CHMP/SWP/28367/2007 Corr . London : European Medicines Agency .
Cilla , D.D. Jr. , Whitfield , L.R. , Gibson , D.M. , Sedman , A.J. and Posvar , E.L. ( 1996 ) Multiple-dose pharmacokinetics, pharmacodynamics, and
Data
Logistic fit
Asprin/naproxen/ibuprofen
100
�100 �50 0 50 100 150
80
60
40
20
0
Gas
tric
ero
sion
/ulc
er (
%)
COX-1 inhibition (%)
VIOXX/celebrex
Naproxen
CS-706/200 mg
CS-706/100 mg
Placebo
FIGURE 25.17 The relationship between ex vivo inhibition of COX-1 activity and the probability of endoscopically determined GI erosion or ulcers. For comparison, the graph shows data on the frequency of erosions observed with aspirin, naproxen, ibuprofen, Celebrex (celecoxib) and Vioxx (rofecoxib) as well as CS-706. A color version of this figure is available on the Clinical and Translational Science companion website which can be accessed at www.elsevierdirect.com/companions/9780123736390

PART | VII Human Pharmacology378
safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects . Clin. Pharmacol. Ther. 60 , 687 – 695 .
CMR (Centre for Medicines Research International Ltd) ( 2006 ) CMR International 2006/2007 Pharmaceutical R & D Factbook . Surrey, UK : CMR .
Code , C.F. ( 1956 ) Histamine and gastric secretion . In: Wolstenholme GEW( C.M. O’Connor , ed.) , pp. 189 – 219 . London : Churchill .
Collins , R. , Armitage , J. , Parish , S. , Sleight , P. , Peto , R. Heart Protection Study Collaborative Group et al. ( 2002 ) MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebo-controlled trial . Lancet 360 , 7 – 22 .
Colwell , C.W. and Spiro , T.E. ( 1995 ) Efficacy and safety of enoxaparin to prevent deep vein thrombosis after hip arthroplasty . Clin. Orthop. Rel. Res. 319 , 215 – 222 .
Colwell , C.W. , Spiro , T.E. , Trowbridge , A.A. , Stephens , J.W. , Gardiner , G.A. et al. ( 1995 ) Efficacy and safety of enoxaparin versus unfractionated heparin for prevention of deep venous thrombosis after elective knee arthroplasty. Enoxaparin Clinical Trial Group . Clin. Orthop. Rel. Res. 321 , 19 – 27 .
CONSENSUS Trial Study Group ( 1987 ) Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) . N. Engl. J. Med. 316 , 1429 – 1435 .
Crouse , J.R. III. ( 2006 ) Imaging atherosclerosis: state of the art . J. Lipid Res. 47 , 1677 – 1699 .
Dedrick , R.L. ( 1973 ) Animal scale-up . J. Pharmacokinet. Biopharm, 1 , 435 – 461 .
DiMasi , J.A. , Hansen , R.W. and Grabowski , H.G. ( 2003 ) The price of innovation: new estimates of drug development costs . J. Health Econ. 22 , 151 – 185 .
Dluhy , R.G. and Williams , G.H. ( 2004 ) Aldosterone – villain or bystander? N Engl. J. Med. 351 , 8 – 10 .
Dotter , C.T. , R ö sch , J. , Erlandson , M. , Buschman , R.W. and Ogilvie , R. ( 1985 ) Iopamidol arteriography: discomfort and pain . Radiology 155 , 819 – 821 .
Endo , A. , Kuroda , M. and Tsujita , Y. ( 1976 ) ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium cit-rinium . J. Antibiotics (Tokyo) 29 , 1346 – 1348 .
Estabrook , R.W. ( 2003 ) A passion for P450s (remembrances of the early history of research on cytochrome P450) . Drug Metabol. Dispos. 31 , 1461 – 1473 .
Evans , D.C. , Watt , A.P. , Nicoll-Griffith , D.A. and Baillie , T.A. ( 2004 ) Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development . Chem Res. Toxicol. 17 , 3 – 16 .
Expert Scientific Group ( 2006 ) Expert Scientific Group on Phase One Clinical Trials, Final Report . Norwich, UK : The Stationery Office .
FDA (1998) CFR Title 21: Food and Drugs. Part 312: Investigational New Drug Application. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart � 312 .
Ferguson , R.K. , Turini , G.A. , Brunner , H.R. and Gavras , H. ( 1977 ) A spe-cific orally active inhibitor of angiotensin-converting enzyme in man . Lancet 1 , 775 – 778 .
Floss , H.G. ( 2006 ) Combinatorial biosynthesis – potential and problems . J. Biotechnol. 124 , 242 – 257 .
Folkow , B. , Haeger , K. and Kahlson , G. ( 1948 ) Observations on reactive hyperaemia as related to histamine on drugs antagonizing vasodilata-tion induced by histamine and on vasodilator properties of adenosin-etriphosphate . Acta Physiol. Scand. 15 , 264 – 278 .
Fowler , J.S. , Volkow , N.D. , Wang , G.-J. , Ding , Y.-S. and Dewey , S.L. ( 1999 ) PET and drug research and development . J. Nucl. Med. 40 , 1154 – 1163 .
Fox , S. , Farr-Jones , S. , Sopchak , L. , Boggs , A. , Nicely , H.W. et al. ( 2006 ) High-throughput screening: update on practices and success . J. Biomol. Screen. 11 , 864 – 869 .
Fox , S. , Farr-Jones , S. and Yund , M.A. ( 1999 ) High-throughput screen-ing for drug discovery: continually transitioning into new technology . J. Biomol Screen. 4 , 183 – 186 .
Freston , J.W. ( 1987 ) Safety perspectives on parenteral H 2 -receptor antago-nists . Am. J. Med. 83 , 58 – 67 .
Goldman , P. ( 2001 ) Herbal medicines today and the roots of modern phar-macology . Ann. Intern. Med. 135 , 594 – 600 .
Greaves , P. , Williams , A. and Eve , M. ( 2004 ) First dose of potential new medicines to humans: how animals help . Nature Rev. Drug Discov. 3 , 226 – 236 .
Hopkins , A.L. and Groom , C.R. ( 2002 ) The druggable genome . Nature Rev. Drug Discov. 1 , 727 – 730 .
ICH (1994) ICH Harmonised Tripartite Guideline, The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions E1 . ICH. 27 October 1994. 1–3 Current Step 4 version.
Imming , P. , Sinning , C. and Meyer , A. ( 2007 ) Drugs, their targets and thenature and number of drug targets . Nature Rev. Drug Discov. 5 , 821 – 834 , [published correction appears in Nature Rev. Drug Discov. 6, 126] .
IMS Health (2007) IMS Health reports global pharmaceutical market grew 7.0 percent in 2006, to $643 billion [press release]. IMS Health, 20 March 2007.
Kastelein , J.J.P. , van Leuven , S.I. , Burgess , L. , Evans , G.W. , Kuivenhoven , J.A. et al. ( 2007 ) Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia . N. Engl. J. Med 356 , 1620 – 1630 .
Kastrissios , H. , Rohatagi , S. , Moberly , J. , Truitt , K. , Gao , Y. et al. ( 2006 ) Development of a predictive pharmacokinetic model for a novel cyclooxygenase-2 inhibitor . J. Clin. Pharmacol 46 , 537 – 548 .
Kenter , M.J.H. and Cohen , A.F. ( 2006 ) Establishing risk of human experimentation with drugs: lessons from TGM1412 . Lancet 368 , 1387 – 1391 .
Khandekar , J. and Khandekar , M. ( 2006 ) Phase 1 clinical trials . Arch. Intern. Med 166 , 1440 – 1441 .
Kola , I. and Landis , J. ( 2004 ) Can the pharmaceutical industry reduce attrition rates? Nature Rev. Drug Discov 3 , 711 – 715 .
Krieger , D.E. , Erickson , B.W. and Merrifield , R.B. ( 1976 ) Affinity purification of synthetic peptides . Proc. Natl Acad. Sci. USA 73 , 3160 – 3164 .
Lanza , F.L. , Rack , M.F. , Simon , T.J. , Quan , H. , Bolognese , J.A. et al. ( 1999 ) Specific inhibition of cyclooxygenase-2 with MK-0966 is associated with less gastroduodenal damage than either aspirin or ibu-profen . Aliment. Pharmacol. Ther. 13 , 761 – 767 .
Lappin , G. , Kuhnz , W. , Jochemsen , R. , Kneer , J. , Chaudhary , A. et al. ( 2006 ) Use of microdosing to predict pharmacokinetics at the therapeu-tic dose: experience with 5 drugs . Clin. Pharmacol. Ther. 80 , 203 – 215 .
Levy , G. ( 1964 a ) Relationship between Elimination Rate of Drugs and Rate of Decline of Their Pharmacologic Effects . Journal of Pharmaceutical Sciences 53 , 342 – 343 .
Levy , G. ( 1964 b ) Relationship between rate of elimination of tubocurarine and rate of decline of its pharmacological activity . Br. J. Anaesth. 36 , 694 – 695 .
Lewis , E.J. , Hunsicker , L.G. , Bain , R.P. and Rohde , R.D. ( 1993 ) Theeffect of angiotensin-converting-enzyme inhibition on diabetic neph-ropathy . N. Engl. J. Med. 329 , 1456 – 1462 .

Chapter | 25 Modern Drug Discovery and Development379
Lewis , E.J. , Hunsicker , L.G. , Clarke , W.R. , Berl , T. , Pohl , M.A. et al. ( 2001 ) Renoprotective effect of the angiotensin-receptor antago-nist irbesartan in patients with nephropathy due to type 2 diabetes . N. Engl. J. Med . 345 , 851 – 860 .
Liebler , D.C. and Guengerich , F.P. ( 2005 ) Elucidating mechanisms of drug-induced toxicity . Nature Rev. Drug Discov. 4 , 410 – 420 .
Lindsay , M.A. ( 2003 ) Target discovery . Nature Rev. Drug Discov. 2 , 831 – 838 .
Lipid Research Clinics Coronary Primary Prevention Trial ( 1984 ) The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease . JAMA 251, 351 – 364 .
Lipinski , C.A. , Lombardo , F. , Dominy , B.W. and Feeney , P.J. ( 2001 ) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings . Adv. Drug Deliv. Rev. 46 , 3 – 26 .
Lovenox package insert. Bridgewater, NJ: Aventis Pharmaceuticals Inc. Lusis , A.J. , Fogelman , A.M. and Fonarow , G.C. ( 2004 ) Genetic basis of
atherosclerosis. Part I: New genes and pathways . Circulation 110 , 1868 – 1873 .
Mager , D.E. , Wyska , E. and Jusko , W.J. ( 2003 ) Diversity of mechanism-based pharmacodynamic models . Drug Metabol. Dispos. 31 , 510 – 519 .
Mauro , M.J. , O’Dwyer , M. , Heinrich , M.C. and Druker , B.J. ( 2002 ) STI571: A paradigm of new agents for cancer therapeutics . J. Clin. Oncol. 20 , 325 – 334 .
Mehlisch , D.R. ( 2002 ) The efficacy of combination analgesic therapy in relieving dental pain . J. Am. Dent. Assoc. 133 , 861 – 871 .
Moberly , J.B. , Xu , J. , Desjardins , P.J. , Daniels , S.E. , Bandy , D.P. et al. ( 2007 ) A randomized, double-blind, celecoxib- and placebo-controlled study of the effectiveness of CS-706 in acute postoperative dental pain . Clin. Ther. 29 , 399 – 412 .
Molinder , H.K.M. ( 1994 ) The development of cimetidine: 1964 – 1976 . J. Clin. Gastroenterol. 19 , 248 – 254 .
Newman , D.J. and Cragg , G.M. ( 2007 ) Natural products as sources of new drugs over the last 25 years . J. Nat. Products 70 , 461 – 477 .
Nicholson , R.L. , Welch , M. , Ladlow , M. and Spring , D.R. ( 2007 ) Small-molecule screening: advances in microarraying and cell-imaging technologies . ACS Chem. Biol. 2 , 24 – 30 .
NIH Consensus Conference (1985) Lowering blood cholesterol to prevent heart disease. JAMA 253, 2080–2086.
Nicklas , J.M. , Pitt , B. , Timmis , G. , Breneman , G. , Jafri , S.M. SOLVD Investigators et al. ( 1992 ) Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions . N. Engl. J. Med. 327 , 685 – 691 .
Nissen , S.E. , Tardif , J.-C. , Nicholls , S.J. , Revkin , J.H. , Shear , C.L. et al. ( 2007 ) Effect of torcetrapib on the progression of coronary athero-sclerosis . N. Engl. J. Med. 356 , 1304 – 1316 .
Obach , R.S. , Baxter , J.G. , Liston , T.E. , Silber , B.M. , Jones , B.C. et al. ( 1997 ) The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data . J Pharmacol. Exper. Ther. 283 , 46 – 58 .
Obach , R.S. , Walsky , R.L. , Venkatakrishnan , K. , Gaman , E.A. , Houston , J.B. et al. ( 2006 ) The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug interactions . J. Pharmacol. Exper. Ther. 316 , 336 – 348 .
Oliver , M.F. ( 1991 ) Might treatment of hypercholesterolemia increase non-cardiac mortality? Lancet 337 , 1529 – 1531 .
Oliver , M.F. ( 1995 ) Statins prevent coronary heart disease . Lancet 346 , 1378 – 1379 .
Ostro , M.J. ( 1987 ) Pharmacodynamics and pharmacokinetics of parenteral histamine (H 2 )-receptor antagonists . Am. J. Med. 83 , 15 – 22 .
Overington , J.P. , Al-Lazikani , B. and Hopkins , A.L. ( 2006 ) How many drug targets are there? Nature Rev. Drug Discov. 5 , 993 – 996 .
Owens , J. ( 2007 ) 2006 drug approvals: finding the niche . Nature Rev. Drug Discov. 6 , 99 – 101 .
Pang , Y.P. ( 2007 ) In Silico drug discovery: solving the ‘ target-rich and lead-poor ’ imbalance using the genome-to-drug-lead paradigm . Clin.Pharmacol. Ther. 81 , 30 – 34 .
Parving , H.-H. , Lehnert , H. , Br ö chner-Mortensen , J. , Gomis , R. , Andersen , S. et al. ( 2001 ) The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes . N. Engl. J. Med. 345 , 870 – 878 .
Pfeffer , M.A. ( 1995 ) ACE inhibition in acute myocardial infarction . N.Engl. J. Med. 332 , 118 – 120 .
Pfeffer , M.A. and Frohlich , E.D. ( 2006 ) Improvements in clinical out-comes with the use of anigiotensin-converting enzyme inhibitors: cross-fertilization between clinical and basic investigation . Am. J. Physiology Heart Circ. Physiol. 291 , H2021 – H2025 .
Pfeffer , M.A. , Braunwald , E. , Moye , L.A. , Basta , L. , Brown , E.J. et al. ( 1992 ) Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators . N. Engl. J. Med. 327 , 669 – 677 .
Pfeffer , M.A. , Lamas , G.A. , Vaughan , D.E. , Parisi , A.F. and Braunwald , E. ( 1988 ) Effect of captopril on progressive ventricular dilation after anterior myocardial infarction . N. Engl. J. Med. 319 , 80 – 86 .
Pfeffer , M.A. , McMurray , J.J.V. , Velazquez , E.J. , Rouleau , J.-L. , K ø ber , L. et al. ( 2003 ) Valsartan, captoril, or both in myocardial infarction com-plicated by heart failure, left ventricular dysfunction . N. Engl. J. Med. 349 , 1893 – 1906 or both [published correction appears in N. Engl. J. Med . 2004; 350, 203] .
Pfeffer , J.M. , Pfeffer , M.A. and Braunwald , E. ( 1985 ) Influence of chronic captopril therapy on the infarcted left ventricle of the rat . Circ. Res. 57 , 84 – 95 .
Pfizer ( 2006 ) In interests of patient safety, Pfizer stops all torcetrapib clinical trials; company has notified FDA and is in the Process of notifying all clinical investigators and other regulatory authorities [press release] , 2 December 2006 . Pfizer Inc.
PhRMA ( 2005 ) What goes into the cost of prescription drugs? , June 2005 . Washington, DC : Pharmaceutical Research and Manufacturers of America .
Pitt , B. , Remme , W. , Zannad , F. , Neaton , J. , Martinez , F. et al. ( 2003 ) Eplerenone, a selective aldosterone blocker, in patients with left ventricu-lar dysfunction after myocardial infarction . N. Engl. J. Med. 348 , 1309 – 1321 . [published erratum appears in N. Engl. J. Med. 2003; 348, 2271] .
Pitt , B. , Zannad , F. , Remme , W.J. , Cody , R. , Castaigne , A. et al. ( 1999 ) The effect of spironolactone on morbidity and mortality in patients with severe heart failure . N. Engl. J. Med. 341 , 709 – 717 .
Pitt , B. , Pierard , L.A. , Bilge , A. , Bourassa , M.G. , White , M. RALES Investigators et al. ( 1996 ) Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (The Randomized Aldactone Evaluation Study [RALES]) Am. J. Cardiol. 78 , 902 – 907 .
Rabinowitz , M. and Shankley , N. ( 2006 ) The impact of combinatorial chemistry on drug discovery . In: The Process of New Drug Discovery and Development , 2nd edn ( C.G. Smith and J.T. O’Donnell , eds) , pp. 56 – 77 . New York : Informa Healthcare .
Remuzzi , G. and Ruggenenti , P. ( 1993 ) Slowing the progression of dia-betic nephropathy . N. Engl. J. Med. 329 , 1496 – 1497 .

PART | VII Human Pharmacology380
Rohatagi , S. , Kastrissios , H. , Gao , Y. , Zhang , N. , Xu , J. et al. ( 2007 ) Predictive population pharmacokinetic/pharmacodynamic model for a novel COX-2 inhibitor . J. Clin. Pharmacol. 47 , 358 – 370 .
Rolinson , G.N. ( 1998 ) Forty years of β -lactam research . J. Antimicrob. Chemother. 41 , 589 – 603 .
4S Study Group ( 1994 ) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) . Lancet 344 , 1383 – 1389 .
Sacks , F.M. , Pfeffer , M.A. , Moye , L.A. , Rouleau , J.L. , Rutherford , J.D. et al. ( 1996 ) The effect of pravastatin on coronary events after myo-cardial infarction in patients with average cholesterol levels . N. Engl. J. Med. 335 , 1001 – 1009 .
Saxena , R. , Voight , B.F. , Lyssenko , V. , Burtt , N.P. , de Bakker , P.I.W. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research et al. ( 2007 ) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels . Science 316 , pp. 1331 – 1336 .
Shepherd , J. , Cobbe , S.M. , Ford , I. , Isles , C.G. , Lorimer , A.R. et al. ( 1995 ) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia . N. Engl. J. Med. 333 , 1301 – 1307 .
Shoichet , B.K. ( 2004 ) Virtual screening of chemical libraries . Nature 432 , 862 – 865 .
Simon , L.S. , Lanza , F.L. , Lipsky , P.E. , Hubbard , R.C. , Talwalker , S. et al. ( 1998 ) Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor . Arthritis Rheum. 41 , 1597 – 1602 .
Stein , C.M. ( 2003 ) Managing risk in healthy subjects participating in clin-ical research . Clin. Pharmacol. Ther. 74 , 511 – 512 .
Steinbrook , R. ( 2002 a ) Protecting research subjects – the crisis at Johns Hopkins . N. Engl. J. Med. 346 , 716 – 720 . [published correction appears in N. Engl. J. Med. 2002; 346,1678-a] .
Steinbrook , R. ( 2002 b ) Improving protection for research subjects . N. Engl. J. Med. 346 , 1425 – 1430 . [published correction appears in N. Engl. J. Med. 2002; 346, 1838-a] .
Suntharalingam , G. , Perry , M.R. , Ward , S. , Brett , S.J. , Castello-Cortes , A. et al. ( 2006 ) Cytokine storm in a phase 1 trial of the anti-CD28 mon-oclonal antibody TGN1412 . N. Engl. J. Med. 355 , 1018 – 1028 .
Tang , H. and Mayersohn , M. ( 2005 ) A novel model for prediction of human drug clearance by allometric scaling . Drug Metabol. Dispos. 33 , 1297 – 1303 .
Temple , R. ( 1999 ) Are surrogate markers adequate to assess cardiovascu-lar disease drugs? JAMA 282 , 790 – 795 .
Tobert , J.A. ( 2003 ) Lovastatin and beyond: The history of the HMG-COA reductase inhibitors . Nature Rev. Drug Discov. 2 , 517 – 526 .
Tonkin , A. , Aylward , P. , Colquhoun , D. , Glasziou , P. , Harris , P. LIPID(Long-Term Intervention with Pravastatin in Ischaemic Disease) Study Group et al. ( 1998 ) Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels . N. Engl. J. Med. 339 , 1349 – 1357 .
US Department of Health and Human Services (1997a) Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) Guidance for Industry: Drug Metabolism/Drug Interaction Studies in the Drug
Development Process: Studies In Vitro . Rockville, MD: US Department of Health and Human Services. April 1997.
US Department of Health and Human Services (1997b) Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) Guidance for Industry: M3 Nonclinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals . Rockville, MD: US Department of Health and Human Services. July 1997.
US Department of Health and Human Services (1997c) Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) Guidancefor Industry: S2B Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals. Rockville, MD: US Department of Health and Human Services. July 1997.
US Department of Health and Human Services (2004) Food and Drug Administration. Challenge and Opportunity on the Critical Path to New Medical Products . Rockville, MD: US Department of Health and Human Services. March 2004.
US Department of Health and Human Services (2005) Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers . Rockville, MD: US Department of Health and Human Services. July 2005.
US Department of Health and Human Services (2006a) Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Guidance for Industry: Investigators, and Reviewers: Exploratory IND Studies . Rockville, MD: US Department of Health and Human Services. January 2006.
US Department of Health and Human Services (2006b) Food and Drug Administration. Critical Path Opportunities Report . Rockville, MD: US Department of Health and Human Services. March 2006.
US Department of Health and Human Services (2006c) Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) Guidance for Industry: Drug Interaction Studies – Study Design, Data Analysis, and Implications for Dosing and Labeling: Draft Guidance. Rockville, MD: US Department of Health and Human Services. September 2006.
Vane , J. ( 2000 ) Aspirin and other anti-inflammatory drugs . Thorax 55 , S3 – S9 .
Wagner , J.A. , Williams , S.A. and Webster , C.J. ( 2007 ) Biomarkers and surrogate endpoints for fit-for purpose development and regulatory evaluation of new drugs . Clin. Pharmacol. Ther. 81 , 104 – 107 .
Walsky , R.L. and Obach , R.S. ( 2004 ) Validated assays for human cyto-chrome P450 activities . Drug Metabol. Dispos. 32 , 647 – 660 .
Whitebread , S. , Hamon , J. , Bojanic , D. and Urban , L. ( 2005 ) In vitrosafety pharmacology profiling: an essential tool for successful drug development . Drug Discov. Today 10 , 1421 – 1433 .
Zlokarnik , G. , Grootenhuis , P.D.J. and Watson , J.B. ( 2005 ) High through-put P450 inhibition screens in early drug discovery . Drug Discov. Today 10 , 1443 – 1450 .