mining shifting-and-scaling co-regulation patterns on gene expression profiles jin chen sep 2012
TRANSCRIPT

Mining Shifting-and-Scaling Co-Regulation Patterns on Gene Expression Profiles
Jin ChenSep 2012
Motivation
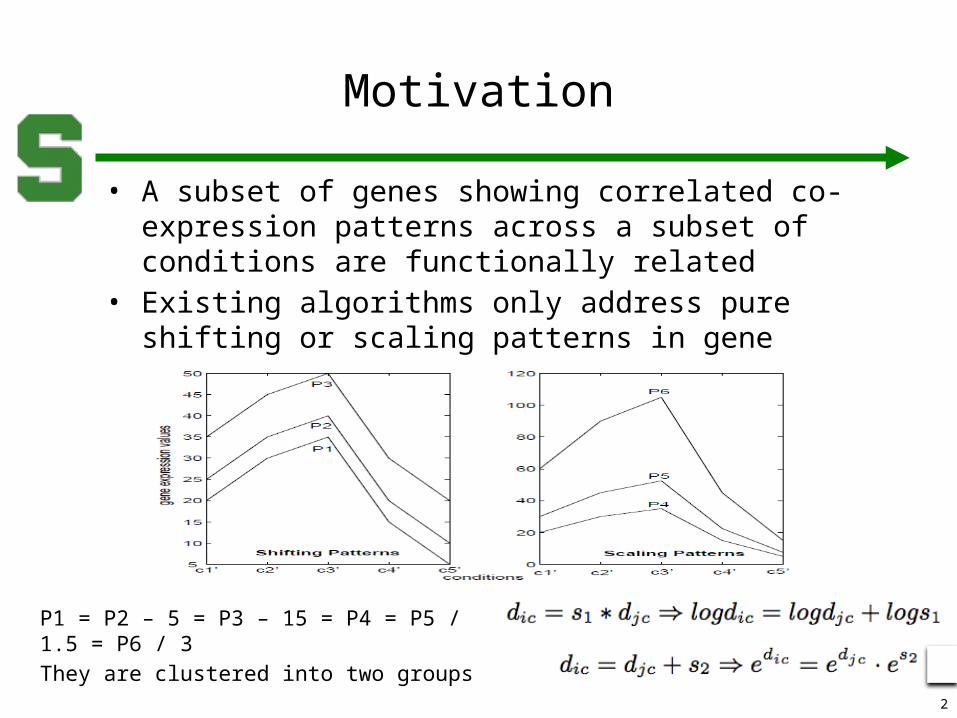
• A subset of genes showing correlated co-expression patterns across a subset of conditions are functionally related
• Existing algorithms only address pure shifting or scaling patterns in gene expression profiles.
2
P1 = P2 – 5 = P3 – 15 = P4 = P5 / 1.5 = P6 / 3
They are clustered into two groups

Motivation
How to group the previous genes into one cluster?We need to handle shifting and scaling
patterns simultaneously!
Three genes g1, g2 and g3 are correlated under all the above conditions:
g2 = -2.5 * g3 + 35 = -g1 + 30

Definition of Correlation
• Correlation means any of a broad class of statistically relationships between random variables and data values.
• In this paper, we only focus on linear correlation, including shifting and scaling.
• Positive and negative correlation correspond to positive and negative scaling factors respectively.

Definition of Bi-clustering
5
• Simultaneous clustering of the rows and columns of a matrix, e.g. group genes which have similar expression patterns under a subset of conditions.

Clustering
Definition: the assignment of a set of observations into subsets (called clusters) so that observations in the same cluster are similar in some sense.
6
The result of a cluster analysis shown as the coloring of the squares into three clusters.
K-means

Density-based Subspace Clustering
• Discover arbitrary-shaped clusters under a subspace. A cluster is regarded as a region, in which the density of data objects exceeds a threshold
• Suffer from the problem that each data object can only be assigned to one cluster only

Hierarchical Clustering
• Use previous established clusters to find successive clusters.
• It has two categories: agglomerative ("bottom-up") or divisive ("top-down")
• Only applicable to full space clustering.

Pattern-based and Tendency-based Biclustering
• Pattern-based biclustering measures similarities between objects based on the coherent pattern they exhibit. It only identifies pure shifting or scaling patterns
• Tendency-based biclustering focuses on linear ordering of gene expression levels without coherent guarantee
• Both methods fail to address the issue of negative correlation in subspace
• Both methods disregard the fact that patterns with smaller variations in expression values are probably of little biological meaning
• Both methods miss the co-regulated genes that have shifting-and-scaling patterns due to varying individual sensitivities

10
New algorithm: reg-cluster• A reg-cluster exhibits the following characteristics which
are suitable for expression data analysis:
– Increase or decrease of gene expression levels across any two conditions of a reg-cluster is in proportion, allowing small variations defined by the coherence threshold
– Increase or decrease of gene expression levels across any two conditions of a reg-cluster is significant with regard to the regulation threshold
– Genes of a reg-cluster can be either positively correlated or negatively correlated

11
Challenges
• The biggest challenge is the need of a novel coherent cluster model that can capture the more general shifting-and-scaling co-regulation patterns
• Another challenge is how to apply a non-negative regulation threshold. Tendency-based models of are not suitable for adopting a regulation threshold

Regulation Measurement
• Notations: dica and dicb are expression levels of gene gi under condition ca and cb respectively; γ is a user-defined gene expression threshold.
• gi is up-regulated from condition cb to ca if dica – dicb > γ • gi is down-regulated from condition cb to ca if dica – dicb < -γ• We represent them as:
We call cb the regulation predecessor of ca ( ) and ca the regulation successor of cb ( ).

RWave Model
• To effectively find the regulation chains by keeping a record of the bordering regulation relationships
order and find minimum pairs which exceeds threshold (γ1 = γ2 = 4.5 and γ3 = 1.8)

Coherent Measurement
The shifting and scaling correlation between gene expression data di and dj under condition set Y can be expressed as a linear equation:
The correlation between di and dj can equally be expressed in the following condition:
where dick+1 and dick are neighboring expression levels of gene gi after all the levels are sorted non-descending order; and djck+1 and djck are neighboring expression levels of gene gj. Here ic2 and ic1 are baseline condition pairs

Coherent Measurement (cont’d)
• A coherent score for gene gi on conditions ck and ck+1 given baseline condition-pair c1 and c2 is defined as:
• Genes share the same coherent scores under a subset of conditions are shifting-and-scaling patterns.
• In practice, a coherent threshold є is applied to flexibly control the coherence of the clusters.

Reg-Cluster Model Definition
• In order to decide whether a subset of genes are shfiting-and-scaling patterns, the reg-cluster model proposed in this paper requires that both regulation and coherence requirements be satisfied:– All the genes should form a regulation chain under the subset of
conditions, either up-regulation or down-regulation, i.e.
– Any pair of genes should have a difference of coherence score smaller than the given coherence threshold є, i.e.

Algorithm and pruning
• The basic idea of the algorithm is to systematically identify the representative regulation chain for each validated reg-cluster.
• The algorithm performs a bi-directional depth-first search on the RWave model for representative regulation chains.
• 4 pruning strategies are applied: minimum gene number, minimum condition number, regulation threshold and coherent threshold.

Algorithm and pruning (cont’d)
• To avoid redundancy due to opposite chains with the same members, they also prune regulation chains which have fewer than half positive correlated gene members.
• They called positive correlated gene members p-members and negative correlated gene members n-members.
• Representative chains which survive the pruning steps and have with maximal gene set will be the output reg-clusters.
18

Algorithm

Efficiency
The running time of reg-cluster is evaluated on synthetic datasets
Effectiveness
21
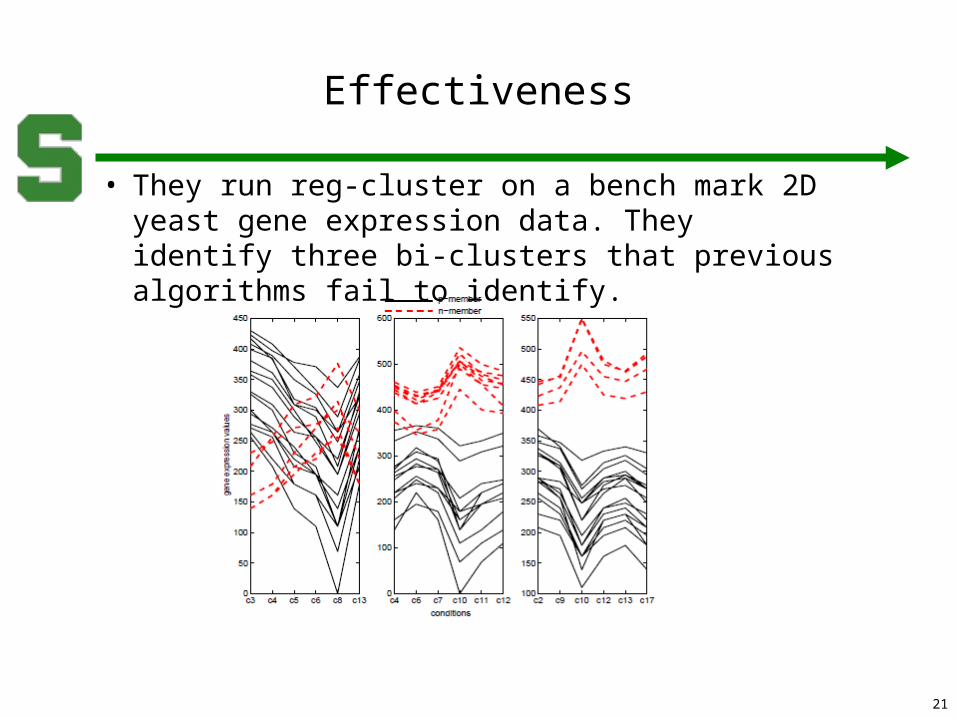
• They run reg-cluster on a bench mark 2D yeast gene expression data. They identify three bi-clusters that previous algorithms fail to identify.

Biological Significance Evaluation
22
Yeast genome gene ontology term finder is used to evaluate the biological significance of the bi-clusters in three categories.

23
• Cons of Reg-cluster:• Identify arbitrary shifting-and-scaling co-regulation
patterns
• Address both positive and negative correlation in the subspace
• Allow flexible regulation threshold to quantify up or down regulation
• Experiments proved that the bi-clusters found are of biological significance in a variety of biological process
Conclusions

24
• How to choose proper regulation and coherence thresholds to have a satisfactory tradeoff between sensitivity and specificity?
• The model propose can only handle linear correlation between co-regulated genes. This will still miss a lot of cases where co-regulated genes have non-linear patterns.
• Do we need a measurement of similarity between bi-clusters to combine those which engage in similar biological processes?
Discussion