microvillar ca++ signaling: a new view of an old problem
TRANSCRIPT

Microvillar Ca11 Signaling: A New View ofan Old Problem
KLAUS LANGE*Kladower Damm, Berlin, Germany
Proceeding from the recent finding that the main components of the Ca11 signalpathway are located in small membrane protrusions on the surface of differenti-ated cells, called microvilli, a novel concept of cellular Ca11 signaling wasdeveloped. The main features of this concept can be summarized as follows:Microvilli are formed on the cell surface of differentiating or resting cells fromexocytic membrane domains, growing out from the cell surface by elongation ofan internal bundle of actin filaments. The microvillar tip membranes contain allfunctional important proteins synthesized such as ion channels and transportersfor energy-providing substrates and structural components, which are, in rapidlygrowing undifferentiated cells, distributed over the whole cell surface by lateraldiffusion. The microvillar shaft structure, a bundle of actin filaments, forms adense cytoskeletal matrix tightly covered by the microvillar lipid membrane andrepresents an effective diffusion barrier separating the microvillar tip compart-ment (entrance compartment) from the cytoplasm. This diffusion barrier preventsthe passage of low molecular components such as Ca11 glucose and otherrelevant substrates from the entrance compartment into the cytoplasm. Theeffectiveness of the actin-based diffusion barrier is modulated by various signalpathways and effectors, most importantly, by the actin-depolymerizing/reorga-nizing activity of the phospholipase C (PLC)-coupled Ca11 signaling. Moreover,the microvillar bundle of actin filaments plays a dual role in Ca11 signaling. Itcombines the function of a diffusion barrier, preventing Ca11 influx into theresting cell, with that of a high-affinity, ATP-dependent, and IP3-sensitive Ca11
store. Activation of Ca11 signaling via PLC-coupled receptors simultaneouslyempties Ca11 stores and activates the influx of external Ca11. The presentedconcept of Ca11 signaling is compatible with all established data on Ca11
signaling. Properties of Ca11 signaling, that could not be reconciled with thebasic principles of the current hypothesis, are intrinsic properties of the newconcept. Quantal Ca11 release, Ca11-induced Ca11 release (CICR), the cou-pling phenomen between the filling state of the Ca11 store and the activity of theCa11 influx pathway, as well as the various yet unexplained complex kinetics ofCa11 uptake and release can be explained on a common mechanisticbasis. J. Cell. Physiol. 180:19–34, 1999. © 1999 Wiley-Liss, Inc.
One of the most important aspects of cell differenti-ation is the abolishment of cellular autonomy and theorganization of intercellular communication by exter-nal signals recieved at the cell surface. These signalsaffect both metabolic activity and the special functionscharacteristic of the respective type of differentiatedtissue. Signal transduction from the cell surface tointracellular targets occurs via second messengers thatare generated in response to the primary signal.
Of the known second messengers, cytosolic Ca11 isthe most important intracellular signal which hasevolved at an early stage of the evolution. A rise in theintracellular Ca11 concentration triggers a broad spec-trum of functions including motility, contraction, secre-tion, potential generation, and proliferation. In nonex-citable cells like those of the immune system, in glial,endothelial, and epithelial cells, hepatocytes, and oo-cytes, the Ca11 signal is generated by a two-step mech-
anism initiated by the depletion of an intracellularCa11 store. Emptying of this store produces a rapidlocal increase in Ca11 concentration which rapidly de-clines. The prolonged Ca11 signal, necessary to inducemost physiological processes, is generated by openingof a Ca11 influx pathway which was found to be regu-lated by the filling state of the Ca11 store itself (Put-ney, 1986, 1990).
Extensive investigations in this field have yielded avery detail-rich picture of the operational properties ofthis signal system. However, the precise biochemicalmechanisms involved in the main functions of thissystem, Ca11 storage and store-operated influx regu-
*Correspondence to: Klaus Lange, Kladower Damm 25b, 14089Berlin, Germany.
Received 11 November 1998; Accepted 26 January 1999
JOURNAL OF CELLULAR PHYSIOLOGY 180:19–34 (1999)
© 1999 WILEY-LISS, INC.

lation, are still unknown. As it appears, a considerableportion of the existing experimental data is incompat-ible with the classical hypothesis of Ca11 signaling,which has been around for about 20 years. The currentdata do not support the central dogma that the Ca11
store is an intracellular membrane-bounded space, theendoplasmic reticulum, into which Ca11 is activelytransported by an ATP-driven membrane pump. Nei-ther the existence of accumulated calcium in the formof free ions nor the precise morphological substrate ofthese stores has ever been unambiguously demon-strated.
This article presents a new hypothesis of Ca11 sig-naling based on recent experimental data. The mainfeatures of this concept will be discussed as will itscompatibility with the known properties of the Ca11
signal system.
ROLE OF MICROVILLI IN CELLREGULATION: THE CONCEPT OF A
REGULATED ENTRANCE PATHWAY FORCELLULAR SUBSTRATES AND IONS
Localization of integral membrane proteinson microvilli
Some years ago, we presented evidence for the cellsurface regulation of glucose uptake via glucose trans-porters localized in microvilli on the surface of adipo-cytes (Lange and Brandt, 1990a,b; Lange et al., 1989,1990). The glucose transporter isoform, which is spe-cifically expressed in differentiated adipocytes (GLUT-4), was found to be exclusively localized on the tips ofmicrovilli (Lange and Brandt, 1990a,b). Furthermore,stimulation of glucose transport in these cells could becorrelated with typical shape changes of microvillicharacterized by either a conspicuous shortening orballooning of their shaft regions and occasionally thecomplete integration into the plasma membrane(Lange et al., 1990).
Further studies revealed that glucose entering themicrovillar tip compartment via transporters located inthe tip membrane is prevented from reaching the cyto-plasm because the microfilament bundle in the mi-crovillar shaft region represents an effective diffusionbarrier (Lange et al., 1990). Modulation of glucose up-take by various external (e.g., insulin and other growthfactors, glucose starvation, and membrane stress) andinternal (e.g., ATP depletion) signals resulted fromchanges in the effectiveness of the microvillar diffusionbarrier. The morphological correlates of these changesin the barrier function could be demonstrated on theultrastructural level as characteristic shape changes ofmicrovilli.
This work led to the hypothesis that glucose uptakeis regulated by the modulation of the microvillar diffu-sion barrier system. This hypothesis opposes the gen-erally accepted notion that insulin-sensitive glucosetransporters are predominantly localized in a cytoplas-mic storage pool of vesicles from which they were in-serted via exocytosis into the plasma membrane inresponse to insulin and other stimuli. We used a hy-drodynamic shearing technique to remove plasmamembrane extensions from the cell surface. We dem-onstrated that the bulk of the insulin-regulated trans-porter isoform, GluT4, isolated in the microsomal frac-tion using conventional homogenization techniques, is
found in the microvilli-derived vesicle fraction whenshearing is applied before homogenization (Lange andBrandt, 1990a). In a follow-up study (Lange andBrandt, 1990b), the mechanism involved in the segre-gation of transporter proteins to microvillar membranedomains was elucidated. This work not only yielded aplausible explanation for the exclusive localization ofintegral membrane proteins in microvillar membranedomains, but also revealed an important novel aspectof cellular differentiation that points to far-reachingconsequences.
Mechanism of segregation of functionalmembrane proteins to microvilli tips
In rapidly growing tissues such as tumors and em-bryonic cells, exocytosis of membrane vesicles resultsin the immediate integration of newly inserted vesicu-lar membrane patches into the plasma membrane. In-tegral membrane proteins located within these patchesdistribute over the whole cell surface by lateral diffu-sion (Fig. 1a). In contrast, exocytosis in differentiatedcells is not followed by lateral diffusion of the newlyinserted protein components. Instead, the exocyticmembrane domains, presumably stabilized by an in-tact membrane-anchored surface coat of proteoglycans,remain intact and grow out from the membrane surfaceby elongation of an actin filament bundle that formsthe structural scaffold of the microvillar shaft. Thisspecial fate of exocytic membrane domains in differen-tiated cells finally results in an exclusive accumulationof functional membrane proteins in microvillar tip re-gions, whereas the plasma membrane is gradually de-pleted of these components (Fig. 1b) (Lange et al., 1990;Lange and Brandt, 1990b).
The most important consequence following from thisprocess is the functional neutralization of all integralmembrane proteins expressed in differentiated cells.Most of the specialized transporter isoforms and pre-sumably the ion channels of differentiated cells arelocalized beyond a cytoskeletal diffusion barrier. Sub-strate and ion fluxes through these membrane trans-porters or channels terminate within the microvillartip compartment (entrance compartment), sphericaldistensions of various sizes at the ends of the microvilli(Lange et al., 1990) (Fig. 2).
Further diffusion of solutes into the cytoplasm isinhibited by the actin filament bundle of the microvil-lar shafts, which forms a tight cytoskeletal matrix ofhigh negative charge density (Tang and Janmey, 1996)resembling that of a strong cation exchanger. Diffusionof cations along this matrix is strongly attenuated andcritically depends on their charge number (Lin andCantiello, 1993). Due to the higher binding affinity ofthe divalent cations to the external binding sites ofF-actin, the resistance for ionic conduction along thiscytoskeletal structure is much higher for Ca11 andMg11 than for the monovalent cations K1 and Na1.
The peripheral localization of transporters and chan-nels on microvilli may be a necessary precondition forexternal regulation of metabolism and cellular func-tions in differentiated cells. To elucidate the generalsignificance of this new principle of influx regulation,we tried to identify another biochemical system of pu-tative microsomal origin, the ATP-dependent Ca11
store, as the microvillar component.
20 LANGE

Hypothesis of microvillar Ca11 signalingUsing a shearing protocol identical to that used for
the preparation of glucose transporter-containingmembrane fractions from adipocytes, Ca11-storingvesicles were isolated from the surface of a hamsterinsulinoma cell line (HIT; Lange and Brandt, 1993a)and from hepatocytes (Lange et al., 1996). The Ca11
storage properties, the experimental conditions forATP-dependent Ca11 uptake as well as the inhibitorspectrum of these vesicle fractions were identical tothose of the microsomal systems from various cell typesgenerally assigned to the endoplasmic/sarcoplasmic re-ticulum Ca11 ATPases.
Further studies revealed some properties of thesemicrovillar Ca11-storing vesicles that are incompat-ible with the current mechanistic concept of vesicu-lar Ca11 storage (Lange and Brandt, 1993b; Lange etal., 1996):
1. The vesicles were found to be permeable for Ca11
via a La31-sensitive pathway; Ca11 entry and exitwere independent of the presence of IP3.
2. Saponin permeabilization of vesicles, preloaded
with 45-Ca11 in the presence of ATP, did not releasestored 45-Ca11.
3. Various large anions such as ATP and IP3 rapidlyequilibrated the external medium and the vesicle lu-men. This anion pathway was inhibited by the anionchannel inhibitor, 4.49-diisothiocyanostilbene-2.29-dis-ulfonic acid (DIDS).
4. Pretreatment of these vesicles with DIDS alsoinhibited subsequent ATP-dependent Ca11 uptakeinto the vesicles.
5. Treatment of Ca11-preloaded vesicles with DIDSalso inhibited the action of IP3 on the Ca11 store.
These observations suggested that:1. Ca11 storage in these vesicles is not due to an
ATP-driven membrane pump concentrating free Ca11
within the vesicle lumen.2. Consequently, the mechanism of Ca11 storage
can only depend on an ATP-consuming process of Ca11
binding to an intravesicular component of relative highmolecular weight that cannot leave saponin-permeabi-lized vesicles.
3. The IP3-sensitive step of the Ca11 release mech-anism is also located in the vesicle lumen.
The most likely candidate for a microvillar high mo-lecular weight Ca11 store appeared to be the actinsystem, forming the internal cytoskeletal structure ofmicrovilli. F-actin is a well-known ATP-dependenthigh-affinity Ca11-binding system that is abundantlypresent in microvilli. Furthermore, F-actin is the samemicrovillar component proposed to be responsible forthe restricted influx of solutes from the external me-dium. This dual function of F-actin is an intriguingsystemic property with respect to the phenomenon ofcoupling between the Ca11 store and the Ca11 influxpathway. Subsequent studies on the Ca11-bindingproperties of the F-actin system yielded clear evidencefor a possible role of F-actin in cellular Ca11 storage(Lange and Brandt, 1996).
F-actin is an ATP-dependent and IP3-sensitiveCa11 store
Since the work of Gershman et al. (1986) and Carlieret al. (1986), the unusual high affinity of Ca11 to theATP-actin monomer (ATP-G-actin) is well known(Ca11: Kd 5 2–8 nM; Mg11: Kd 5 10 nM). Less knownis the property of actin to effectively shield this high-affinity binding site in the polymerized state (F-actin).Ca11 at the high-affinity binding site of the double-helical F-actin polymers is inaccessible for externalions. Thus, F-actin represents a stable Ca11 storagemolecule exhibiting dissociation rate constants in therange of hours. In contrast, Ca11 bound to actin mono-mers is exchanged with rate constants of seconds, i.e.,about 3,000-fold faster. This property of F-actin, as firstdescribed by Kasai and Oosawa (1968, 1969), is one ofthe main arguments in favor of F-actin as a possibleCa11 storage system.
As yet, no physiological relevance has been attrib-uted to Ca11 storage into F-actin, because the ionicconditions within living cells favor the binding of Mg11
rather than Ca11. However, the localization of theATP-dependent Ca11 store in microvilli and the highpermeability of microvilli-derived vesicles for anionsand cations are new arguments in this discussion, sug-
Fig. 1. a: Insertion of cytoplasmic membrane vesicles into the sur-face of undifferentiated, rapidly growing cells. Due to activated ecto-proteases, the inserted membrane area is destabilized and integratedinto the plasma membrane. b: Microvilli formation on the surfacedifferentiated cells.
Fig. 2. SEM of the surface of an isolated rat hepatocyte. Sphericalextensions at the microvilli tips indicate the location of the entranceor tip compartment.
21MICROVILLAR Ca11 SIGNALING

gesting that Ca11 storage in the tip compartment ofmicrovilli may proceed at much higher Ca11 concen-trations. Because of the presence of open ion channelsin the microvilli tips and the diffusion barrier withinthe microvillar shafts, the ionic composition of the en-trance compartment differs from that of the cytoplasm,reflecting rather that of the external milieu. However,under nonequilibrium conditions, the ionic compositionwithin the tip compartment is largely determined bythe selectivity of its ion channels. According to thereported properties, nonselective cation channels ex-hibit a more than 10-fold higher permeability for Ca11
than for Mg11 (Park and MacKinnon, 1995; Obukhovet al., 1995), giving rise to a preponderance of Ca11
over Mg11 within the entrance compartment undernonequilibrium (storage) conditions.
Compelling evidence for the involvement of the F-actin system came from Ca11-binding experimentswith the microvillar vesicles of HIT cells (Lange andBrandt, 1996). ATP-dependent uptake of 45-Ca11 intothese vesicles was strongly inhibited by pretreatmentphalloidin, a highly specific F-actin–stabilizing toxin(Fig. 3a). As shown in Figure 3b (lower track), thepresence of phalloidin during the loading processlargely reduced the IP3-sensitive Ca11 pool. Vesiclespreloaded with 45-Ca11 and then treated with phalloi-din still released stored 45- Ca11 on the addition of IP3(Fig. 3b, upper track). Because Ca11 uptake into thestore also proceeds in the presence of IP3, the amountof 45-Ca11 released by IP3 in the presence of phalloidinis significantly larger than that in the absence of phal-loidin (upper track). Therefore, IP3-sensitive 45-Ca11
pools stored in the presence and absence of phalloidincan only be compared when the IP3 effect is determinedin the presence of phalloidin in both cases.
Phalloidin specifically inhibits the steady-state dy-namics of depolymerization and polymerization pro-cesses at the ends of actin filaments, also known as“dynamic instability” (Sampath and Pollard, 1991). Ap-parently, phalloidin-mediated suppression of dynamicinstability strongly interferes with the incorporation ofCa11 into the polymer. The phalloidin experimentsyielded the first important hint of the role of F-actin inthe storage process. They also suggested that thesteady-state dynamics of F-actin (dynamic instability)may be critically involved in this process. Further ex-periments on the Ca11-binding and -release propertiesof the actin system were carried out with purified pro-teins in vitro.
Ca11 BINDING AND RELEASE PROPERTIESOF F-ACTIN
Incorporation of Ca11 and Mg11 into F-actinduring in vitro polymerization
Experiments to estimate the relative amounts ofCa11 and Mg11 incorporated into F-actin during poly-merization of G-actin in the presence of ATP and var-ious concentrations of the two ions yielded a surprisingresult. For these experiments, G-actin, dialyzedagainst nominal Ca11-free (.1 mM) Tris buffer con-taining 0.2 mM ATP and 0.1 mM Mg11, was used. Thefinal concentrations of Mg11 and ATP were adjustedimmediately before the polymerization was started byaddition of 50 mM KCl. Regardless of the high concen-tration quotient of Mg11/ Ca11 prior to and during
polymerization, ([Ca11] was between 10 and 30 mMand [Mg11] between 0.1 and 0.4 mM), the proportionsof F-actin-bound Ca11 always amounted to 40–65% ofthe total divalent cation content of the polymer (Langeand Brandt, 1996).
The result of this experiment considerably supportedour idea of a microfilament-based Ca11 store. On theother side, these findings suggested that polymeriza-tion of actin is not an equilibrium process. Obviously,equilibrium parameters such as the dissociation con-stants of the divalent cations from ATP-G-actin (KMg/KCa 5 4 [Estes et al., 1987]) cannot be used to predictthe result of this type of coupled reactions. Instead, the
Fig. 3. a: Inhibition of ATP-dependent 45-Ca11 uptake into mi-crovilli-derived vesicles from HIT cells in the absence (control) andpresence of phalloidin. The lowest line represents the A23187-insen-sitive 45-Ca11 content of loaded vesicles in the presence and absenceof phalloidin. b: Inhibition by phalloidin of the IP3-sensitive Ca11 poolof microvilli-derived vesicles from HIT cells. Upper line (filled sym-bols) shows the 45-Ca content of vesicles loaded in the absence ofphalloidin. At the time point indicated with an arrow, phalloidin (10mM) was added. The points indicated with IP3 represent the 45-Cacontent of the vesicles 1 minute after addition of IP3.
22 LANGE

rate constants for cation binding to ATP-G-actin shouldbe used ( kCa/kMg 5 35 [Estes et al., 1987] to 90 [Gersh-man et al., 1991]). These constants indicate a highrelative preponderance of the addition of Ca-ATP-G-actin to the filaments over that of Mg-ATP-G-actin.Therefore, during nonequilibrium polymerization espe-cially at higher monomer and filament concentrations,when monomer addition is not significantly limited bydiffusion processes, kinetically preferred partial reac-tions such as the exchange of nucleotides and metalions at G-actin may contribute to the preferential ad-dition of Ca11-containing actin monomers and the for-mation of Ca11-enriched actin polymers (Ca/Mg-F-actin).
Ca11 release from Ca-F-actinCa11 release induced by chain breaks. Assembly
of ATP-G-actin occurs at the two ends of actin fila-ments or at a preformed nucleus of at least three sub-units. The two endpieces of actin filaments are knownto exhibit quite different reactivities. The so-calledbarbed end of the filament is the fast-growing site ofthe polymer. At this site, addition (and dissociation)proceeds at a much higher rate than at the oppositeend of the filament (pointed end). Rapid incorporationof ATP-G-actin subunits to this fast-growing end leadsto the formation of a small terminal polymer regioncontaining ATP-actin subunits. Following polymeriza-tion, ATP is rapidly hydrolyzed to ADP, resulting in apolymer chain in which almost all subunits containADP except those at the fast-growing end (for a review,see Carlier, 1991).
In contrast to ATP-G-actin, the respective ADPmonomer exhibits a slow rate of addition and a highrate of dissociation at the barbed end of F-actin. Thedissociation rate constant of ADP-G-actin at this fila-ment end is about 40-fold higher than that of ATP-G-actin (for a review, see Carlier, 1991). Consequently,ADP-actin subunits located at the barbed filamentends dissociate rapidly, giving rise to a sudden short-ening of the filaments which proceeds until the termi-nal region is again stabilized by addition of ATP-con-taining monomers. This interplay between rapiddepolymerization and polymerization processes ac-counts for the steady-state phenomenon of the dynamicinstability of F-actin. Monomer turnover due to dy-namic instability is restricted to only a small region atthe barbed ends of actin filaments, whereas the centralparts including the pointed end regions remain unaf-fected.
On the other hand, the affinity of actin monomers toCa11 decreases about 50-fold when ATP-G-actin ischanged to ADP-G-actin (Selden et al., 1987). Accord-ingly, the actin Ca11 store is characterized by threemolecular forms: 1) ATP-G-actin represents the high-affinity Ca11- binding form of the store; 2) ADP-G-actin is the low-affinity form of the system from whichCa11 can be rapidly and extensively released; and 3)F-actin is the Ca11 storage form.
Proceeding from these well-known facts, some of thein vitro properties of Ca-F-actin could be predicted.Actin polymerization in vitro yields long polymerchains of up to 1,000 subunits (Burlacu et al., 1992).This long-chain F-actin is highly stabilized in the pres-ence of ATP because the concentration of ADP-contain-
ing barbed filament ends is very low. Depolymerizationrates should be moderately higher in the absence ofATP and the presence of ADP, but significantly higherrates of steady-state ADP-actin release can be observedafter increasing the concentration of filament ends bybreaking long filaments into shorter pieces. F-actinfragmentation can be achieved by ultrasonic treat-ment. Increased rates of divalent cation exchange inF-actin during ultrasonic treatment were first de-scribed by Barany and Finkelman (1962) and Kasaiand Oosawa (1968).
Using the Fura-2 fluorescence technique for the de-termination of Ca11 concentrations in the nanomolarrange, release of Ca11 from Ca-F-actin during ultra-sonic treatment could easily be demonstrated (Fig. 4).Following a short ultrasonic pulse of a few seconds, arapid increase of free Ca11 up to a concentration of 500nM was observed. The process is so rapid that thetime-course of Ca11 release could not be resolved byour experimental setup, i.e., Ca11 release was com-plete within seconds during sonication.
Action of profilin as a Ca11 release factor. Stillanother approach was used to demonstrate Ca11 re-lease from F-actin. Profilin is a member of the largefamily of phospholipid-regulated proteins which areknown to affect the dynamics and organization of theactin cytoskeleton. The action of profilin on F- andG-actin comprises different mechanistic aspects:
1. Profilin is able to bind one actin monomer in aCa11- independent way, giving rise to a reduced mono-mer concentration.
2. The profilin-actin complex (profilactin) binds tothe barbed ends of actin filaments, mediating the ad-dition of profilin-bound monomers to this end of thepolymer chain.Therefore, regardless of a reducedmonomer concentration, the resulting rate of monomeraddition at the barbed end remains almost unaffectedby the presence of profilin. In contrast, due to the lowermonomer concentration in the presence of profilin, thenet rate of monomer dissociation at the barbed filamentends is considerably enhanced. By this type of action,profilin promotes a second form of steady-state dynam-ics of F-actin, called “treadmilling” (Perelroizen et al.,1996), characterized by net polymerization at thebarbed ends and concomitant net depolymerization atthe pointed filament ends. This process accelerates therelease of ADP-actin monomers from those parts of thepolymer that are inaccessible to dynamic instability. Inthe absence of profilin, or the analog-acting ADF/cofilinprotein family, treadmilling is slow and does not sig-nificantly contribute to the steady-state monomer turn-over which is then mainly due to dynamic instability(Carlier and Pantaloni, 1997). In the presence of profi-lin, however, massive Ca11 release occurs due to inter-mediary liberation of ADP-G-actin with low affinity toCa11.
3. Profilin accelerates the dissociation rates of diva-lent cations and nucleotides from actin monomers,thereby decreasing the affinity of Ca11 to G-actin byabout 30-fold (Perelroizen et al., 1996).
As shown in Figure 5a, addition of purified bovinespleen profilin to Ca-F-actin induced a sustained re-lease of Ca11 from F-actin without sonication (Langeand Brandt, 1996). Ca11 release from in vitro polymer-ized long-chain F-actin proceeds at a relative large
23MICROVILLAR Ca11 SIGNALING

time scale. Profilin also causes Ca11 release from Ca/Mg-F-actin (Fig. 5b). In this case, however, the profilinaction can only be detected after sonication which con-siderably accelerates the release process. In the ab-sence of profilin, ultrasound does not liberate Ca11
from Ca/Mg-F-actin (not shown).These experiments led to the following three conclu-
sions: 1) Most importantly, one of the PIP/PIP2-regu-lated actin-binding proteins, profilin, was identified asa possible Ca11 release factor. The high relevance ofthis finding results from the fact that activation ofprofilin is a well-known step in the phospholipase C(PLC) pathway of Ca11 signaling. Thus, for the firsttime, the whole chain of biochemical events of Ca11
signaling, from receptor-mediated PLC activationdown to Ca11 release from intracellular Ca11 stores,becomes visible. 2) Not only Mg-free Ca-F-actin can beinduced to release Ca11 but also Ca/Mg-F-actin whichcontains at least 40% Mg11 bound to the high-affinitysites of its subunits. The F-actin storage system is ableto act as a physiological Ca11 source over a wide rangeof its filling state. 3) The rate of profilin-induced Ca11
release from Ca/Mg-F-actin strongly depends on thefilament concentration which is in vivo by more thanone order of magnitude higher than in vitro (Podolskiand Steck, 1990; Burlacu et al., 1992).
Incorporation of Ca11 into Ca/Mg-F-actinIn the presence of Ca11 concentrations below 100
nM, such as those prevailing within unstimulated cells,the Ca/Mg-F-actin copolymer is still able to take upadditional Ca11 in an ATP-dependent manner (Langeand Brandt, 1996). As shown in Figure 6, Ca/Mg-F-actin is a stable Ca11 store even at an ambient Ca11
concentration as low as 40 nM Ca11. By addition ofCaCl2, the concentration of free Ca11 was increased toabout 170 nM. The slow initial Ca11 uptake into thelong-chain polymer could be considerably accelerated
by sonication. Further Ca11 uptake could be initiatedby repeated additions of Ca11. Subsequent addition ofonly 6 mol% profilin induced a significant spontaneousCa11 liberation that was further enhanced by sonica-tion.
Again, sonication-induced chain breaks and the re-sulting high rate of ADP-G-actin release from the gen-erated barbed filament ends is the essential reactiondriving the rapid Ca11 uptake into Ca/Mg-F-actin. Thehigh rate of Ca11 uptake can be explained on the basisof the known rate constants for cation and nucleotideexchange on G-actin. The rate constant for ADP-ATPexchange is 100-fold higher for Ca11-G-actin than forMg-G-actin (Kinosian et al., 1993). The association rateconstant of Ca11 for ATP-G-actin is also 2 orders of
Fig. 4. Ca11 release from Ca-F-actin induced by ultrasonic (us)treatment (from Lange and Brandt, 1996). Ca-F-actin (25 mM) wasrepeatedly (five times for 5 seconds each) treated with ultrasound asindicated with arrows and “us.” The recorder track is interruptedduring the sonication periods.
Fig. 5. a: Profilin-induced Ca11 release from Ca-F-actin (25 mM).Arrows indicate the time of profilin addition giving final concentra-tions of 3 and 6 mM, respectively. b: Profilin-induced Ca11 releasefrom Mg/Ca-F-actin (25 mM). 3 mM profilin was added at the timepoint indicated with an arrow. Ultrasonication (us) of 3–5 secondsduration initiated a rapid Ca11 release.
24 LANGE
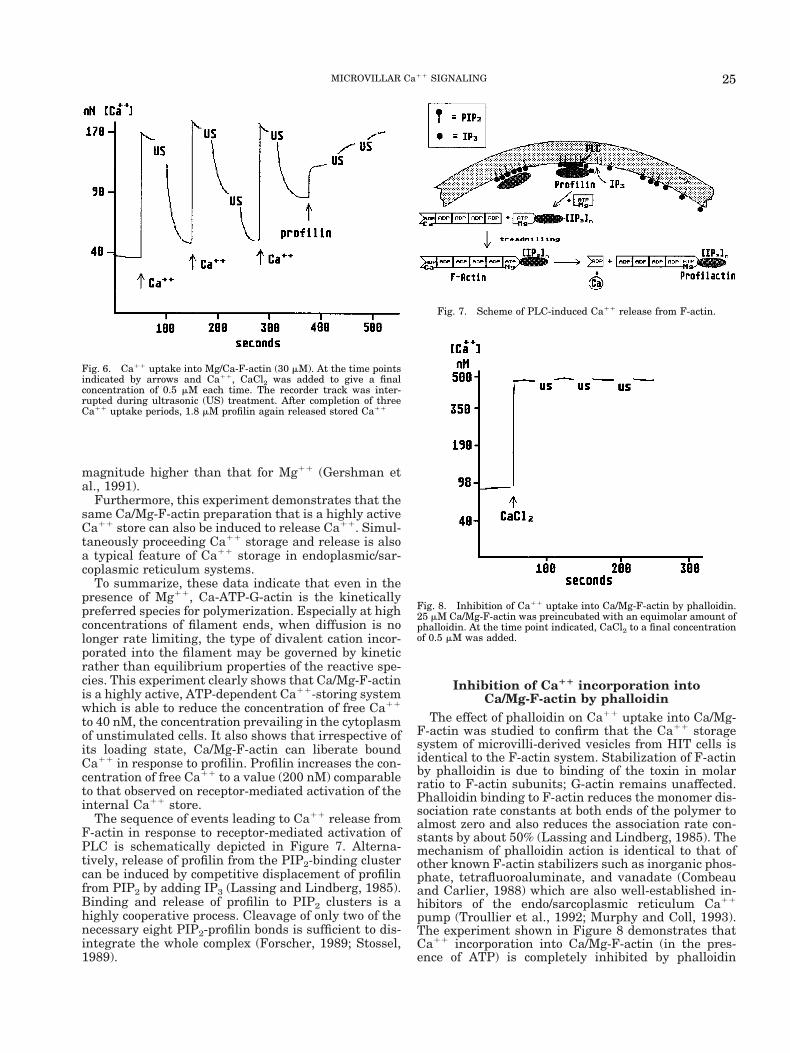
magnitude higher than that for Mg11 (Gershman etal., 1991).
Furthermore, this experiment demonstrates that thesame Ca/Mg-F-actin preparation that is a highly activeCa11 store can also be induced to release Ca11. Simul-taneously proceeding Ca11 storage and release is alsoa typical feature of Ca11 storage in endoplasmic/sar-coplasmic reticulum systems.
To summarize, these data indicate that even in thepresence of Mg11, Ca-ATP-G-actin is the kineticallypreferred species for polymerization. Especially at highconcentrations of filament ends, when diffusion is nolonger rate limiting, the type of divalent cation incor-porated into the filament may be governed by kineticrather than equilibrium properties of the reactive spe-cies. This experiment clearly shows that Ca/Mg-F-actinis a highly active, ATP-dependent Ca11-storing systemwhich is able to reduce the concentration of free Ca11
to 40 nM, the concentration prevailing in the cytoplasmof unstimulated cells. It also shows that irrespective ofits loading state, Ca/Mg-F-actin can liberate boundCa11 in response to profilin. Profilin increases the con-centration of free Ca11 to a value (200 nM) comparableto that observed on receptor-mediated activation of theinternal Ca11 store.
The sequence of events leading to Ca11 release fromF-actin in response to receptor-mediated activation ofPLC is schematically depicted in Figure 7. Alterna-tively, release of profilin from the PIP2-binding clustercan be induced by competitive displacement of profilinfrom PIP2 by adding IP3 (Lassing and Lindberg, 1985).Binding and release of profilin to PIP2 clusters is ahighly cooperative process. Cleavage of only two of thenecessary eight PIP2-profilin bonds is sufficient to dis-integrate the whole complex (Forscher, 1989; Stossel,1989).
Inhibition of Ca11 incorporation intoCa/Mg-F-actin by phalloidin
The effect of phalloidin on Ca11 uptake into Ca/Mg-F-actin was studied to confirm that the Ca11 storagesystem of microvilli-derived vesicles from HIT cells isidentical to the F-actin system. Stabilization of F-actinby phalloidin is due to binding of the toxin in molarratio to F-actin subunits; G-actin remains unaffected.Phalloidin binding to F-actin reduces the monomer dis-sociation rate constants at both ends of the polymer toalmost zero and also reduces the association rate con-stants by about 50% (Lassing and Lindberg, 1985). Themechanism of phalloidin action is identical to that ofother known F-actin stabilizers such as inorganic phos-phate, tetrafluoroaluminate, and vanadate (Combeauand Carlier, 1988) which are also well-established in-hibitors of the endo/sarcoplasmic reticulum Ca11
pump (Troullier et al., 1992; Murphy and Coll, 1993).The experiment shown in Figure 8 demonstrates thatCa11 incorporation into Ca/Mg-F-actin (in the pres-ence of ATP) is completely inhibited by phalloidin
Fig. 6. Ca11 uptake into Mg/Ca-F-actin (30 mM). At the time pointsindicated by arrows and Ca11, CaCl2 was added to give a finalconcentration of 0.5 mM each time. The recorder track was inter-rupted during ultrasonic (US) treatment. After completion of threeCa11 uptake periods, 1.8 mM profilin again released stored Ca11
Fig. 7. Scheme of PLC-induced Ca11 release from F-actin.
Fig. 8. Inhibition of Ca11 uptake into Ca/Mg-F-actin by phalloidin.25 mM Ca/Mg-F-actin was preincubated with an equimolar amount ofphalloidin. At the time point indicated, CaCl2 to a final concentrationof 0.5 mM was added.
25MICROVILLAR Ca11 SIGNALING

(Lange and Brandt, 1996). Even repeated sonicationdid not induce Ca11 uptake.
Because of the high specificity of the phalloidin ac-tion on F-actin, this experiment provides compellingevidence that Ca11 binding to F-actin is involved in theATP-dependent Ca11 uptake of vesicular systems.Moreover, it strongly indicates that monomer dissoci-ation is a critical step for Ca11 incorporation into Ca/Mg-F-actin because monomer dissociation from F-actinis almost completely blocked by phalloidin (Sampathand Pollard, 1991).
CONCEPT OF MICROVILLARCa11 SIGNALING
Proceeding from the above findings, the followingconception of the Ca11 signaling system can be postu-lated (Fig. 9):
1. The surfaces of differentiated cells are coveredwith microvilli such as observed on freshly isolatedhepatocytes and other cell types.
2. Cation and anion channels are located at the tipsof microvilli.
3. The shaft regions of microvilli contain a densematrix of microfilaments arranged as bundles. In theabsence of stimulating signals, Ca11 can enter the tipcompartments of microvilli but is hindered from enter-ing the cytoplasm by the underlying microfilamentbundle which forms a diffusion barrier for ions, espe-cially for divalent cations.
4. Ca11 entering the microvillar tip compartmentcan be stored into the adjacent portions of the F-actinbundles.
5. ATP, which is used for this process, is supplied byglucose influx via microvillar glucose transporters andglycolysis. Glycolytic enzymes are tightly associatedwith filamentous actin (Nanhua and Masters, 1987;Mejean et al., 1989; Bereiter-Hahn et al., 1995).
6. Following stimulation of the Ca11 signal pathwayvia PLC-coupled receptors, the phosphatidylinositolphosphates, PIP and PIP2, were cleaved by activated
PLC. As a consequence, actin-binding and severingproteins such as profilin, gelsolin, and ADF/cofilin,which were originally bound to these phospholipids,were released and liberated Ca11 from F-actin. Liber-ated IP3 remains bound to profilin/gelsolin, stabilizingtheir active state.
7. F-actin-severing proteins of the gelsolin/cofilinfamily act on the bundle structure of the microvillarshaft regions, impairing its function as a diffusion bar-rier and increasing the entry of external Ca11 into thecytoplasm.
Morphological correlate of the activated Ca11
signal pathwayActivation of the Ca11 signal pathway is always
accompanied by morphological shape changes of mi-crovilli, indicating the loss of their internal structuralorganization, especially of the tight connection betweenthe microfilament bundles and the covering lipid mem-brane. This morphological correlate of the activatedmicrovillar influx systems has been demonstrated fordifferent cell types including C6 glioma cells (Lange etal., 1989), 3T3-L1 adipocytes (Lange et al., 1990), HITcells (Lange and Brandt, 1993a), and isolated rat hepa-tocytes (Lange et al., 1997).
As an example, morphological changes occurring onthe surface of vasopressin-treated hepatocytes areshown in Figure 10. Scanning electron microscopy(SEM) reveals that the small and regularly-formed mi-crovilli of the unstimulated hepatocyte surface are rap-idly transformed by vasopressin into large irregularprotrusions. As shown by additional experiments, theloss of the normal shape of microvilli is always corre-lated with activation of Ca11 influx regardless of theway by which this activation was caused (Lange et al.,1997). Receptor-mediated activation of the PLC/ Ca11
signal pathway acts on the microvillar structure viadepolymerization/reorganization of the microfilamentbundle. Mechanical stress (Lange et al., 1996), action ofproteases on the cell surface (Lange et al., 1997), thap-
Fig. 9. Receptor-operated Ca11 signal pathway. Dual function ofmicrofilament bundles of the microvillar shaft as diffusion barrier andCa11 store. PLC-induced liberation of actin-binding and severing
proteins initiates the reorganization of the cytoskeletal diffusion bar-rier, simultaneously releasing Ca11 from F-actin and increasing theinflux of external Ca11 via cation channels in the tip membrane.
26 LANGE

sigargin-induced store depletion (Lange et al., 1997),and alteration of membrane properties by lipophilicchemicals also accelerate Ca11 influx into the cell. Theactions of all these effectors are accompanied by essen-tially the same type of shape changes of microvilli(Gartzke et al., 1997).
Organization of the vesicular Ca11 storeAs pointed out above, conventional homogenization
of cells generates a small vesicle fraction derived fromcell surface protrusions such as microvilli, which be-comes part of the microsomal fraction after sedimenta-tion. Although this vesicle fraction is not derived fromthe intracellular endoplasmic reticulum or Golgi vesi-cle system, a biochemical distinction from intracellularmembranes is almost impossible because all proteinsthat have passed through the membrane systems of theendoplasmic reticulum and Golgi apparatus finally be-come components of the microvillar system. It is of nouse to search for marker proteins differentiating mi-crovilli-derived vesicles from endoplasmic reticulumand Golgi vesicles. Until now, the most compellingevidence for the shear stress-induced formation of mi-crovillar vesicles comes from a SEM study of hepato-cytes treated with a low-force hydrodynamic shearingtechnique. In this study, the surface of hepatocytes wasshown at an intermediate stage of shearing when partof the microvilli showed a vesicular appearance butstill adhered to the cell surface. The hepatocyte surfacewas also shown at a final stage with a smooth surfacedevoid of microvilli. Furthermore, the isolated mem-brane fraction was shown to form a homogenous pop-ulation of spherical vesicles with uniform size (Langeet al., 1996).
Since most of our knowledge on Ca11 storage wasacquired using vesicle systems from different celltypes, all known properties of vesicular Ca11 storageshould also be reflected by the new concept of Ca11
signaling. The main features of the current concept ofvesicular Ca11 storage are schematically depicted inFigure 11a. Experiments with microvilli-derived vesi-cles from HIT cells and hepatocytes have suggested anentirely different model of vesicular Ca11 storage (Fig.11b). First, an IP3-independent, La31--sensitive Ca11-
permeable pathway of the vesicle membrane equili-brates the vesicular lumen with external Ca11 withinseconds (Lange et al., 1996; Lange and Brandt, 1993b).Second, the vesicles also contain an anion-conducting(DIDS-inhibitable) pathway that is permeable for IP3and ATP (Lange et al., 1996; Lange and Brandt,1993b). Third, actin filaments within the vesicles storeCa11 in an ATP-dependent way. Fourth, external IP3enters the vesicle lumen via the anion pathway andinduces Ca11 release by activating PIP/PIP2-regulatedactin-binding/severing proteins (Lange and Brandt,1993b). In microvilli lacking profilin/gelsolin, Ca11
storage and IP3 sensitivity can dissociate. For instance,vesicles derived from the basal surface of hepatocyteswere found to be IP3 insensitive (Lange et al., 1996).
Although the organization of this storage model iscompletely different from that of the current concept,the properties of this model are in accord with allknown characteristics of the Ca11 signaling system.Some of these characteristics are even more consistentwith the novel hypothesis than with the current con-cept of Ca11 storage. For instance:
1. Ca11 storage in permeabilized cells is more con-sistently explained by Ca11 binding to a high molecu-lar structure than by a concentrated mechanism be-cause it is not easy to reconcile that saponin (or otherdetergents) readily perforates the plasma membranebut does not affect the permeability of cytoplasmic ves-
Fig. 10. SEM of the surface of a vasopressin-stimulated hepatocyte(2 minutes). Control see Figure 2.
Fig. 11. a: Current concept of vesicular Ca11 storage. CytoplasmicCa11 is concentrated by an ATP-driven membrane-located Ca11
pump in the vesicle lumen. Delivery of Ca11 occurs via an IP3-sensitive Ca11 channel (IP3 receptor). b: Ca11 storage into microvilli-derived vesicles.
27MICROVILLAR Ca11 SIGNALING

icles. Detergent treatment of Ca11-storing vesiclesonly causes a small portion of stored Ca11 to leave thevesicle lumen but does not affect the thapsigargin-sensitive portion of stored Ca11(Lange et al., 1996).F-actin-bound Ca11 is retained within permeabilizedvesicles and cells unless the detergent-induced poresare large enough to allow the exit of actin monomers(40 kDa).
2. Up to now, the IP3-sensitive Ca11 channel aspostulated by the current hypothesis could not be es-tablished in microsomal preparations. Instead, micro-somal Ca11 channels that have been detected withpatch clamp techniques are IP3 independent (Schmidet al., 1990; Zweifach and Lewis, 1993; Duszynski etal., 1995).
3. Inhibition of ATP entry via the vesicular anionchannel by DIDS was shown to inhibit Ca11 uptakeinto microvilli-derived vesicles (Lange and Brandt,1993b; Lange et al., 1996) and cerebellar microsomes(Michelangeli, 1993). These findings demonstrate thatATP entry into vesicles, but not external ATP, is es-sential for Ca11 storage, clearly contradicting the as-sumption of a membrane-located Ca11 pumping AT-Pase.
4. Similarly, blockade of IP3 entry into Ca11-loadedvesicles by DIDS also inhibits Ca11 release (Mi-chelangeli, 1993; Lange and Brandt, 1993b).
As discussed in the following section, the F-actin-based model of Ca11 storage readily explains all essen-tial properties of Ca11 signaling.
Essential properties of cellular Ca11 signalingare compatible with the new concept
The best recommendation for a new hypothesis is itsability to account for even those problems that re-mained unresolved by the current concept. Several ofthe essential properties of cellular Ca11 signalingcould hardly be reconciled with the idea of Ca11 pump-ing into intracellular vesicles, but are intrinsic proper-ties of the concept of microvillar Ca11 signaling. Espe-cially three of the most extensively studied features ofthe Ca11 signal system appear to be “exotic” to thecurrent concept: 1) The functional coupling betweenthe filling state of the Ca11 store and the activity of theCa11 influx pathway; 2) the “quantal” kinetics of Ca11
release from the store; and 3) Ca11 induced Ca11
release from the store.The discovery of these special properties has led to a
variety of supplementary postulates in order to adaptthe Ca11 pump hypothesis to the changed state ofknowledge. However, most of these auxiliary assump-tions could not be verified by experiments.
Functional coupling between the Ca11 storeand the Ca11 influx pathway. Putney (1990) wasthe first to show that receptor stimulation mobilized anintracellular Ca11 store which is subsequently refilledin the presence of extracellular Ca11. Later on, usingthe IP3-independent effector thapsigargin, it was dem-onstrated that store depletion alone was sufficient tostimulate Ca11 influx (Thastrup et al., 1989). A wealthof publications on this subject have appeared since,however, the mechanism of this coupling phenomenonis still unknown (for a review, see Parekh and Penner,1997). The most compelling explanation for store-oper-ated Ca11 fluxes has been provided by Perdue (1971),
Dunlop and Larkins (1988), Guillemette et al. (1988),Rossier et al. (1991), and Lievremont et al. (1994). Theypropose that Ca11-storing organelles, distinct from theendoreticulum, are localized near or at the cytoplasmicface of the plasma membrane where they, either di-rectly or via second messengers, regulate Ca11 channelactivity in the plasma membrane in a Ca11 content-dependent manner. However, neither biochemical normorphological data confirming this notion could be pre-sented.
As depicted in Figure 9, the concept of microvillarsignaling comprises store-coupled Ca11 influx as anintrinsic property, resulting from the unique arrange-ment of Ca11 store and Ca11 channels within mi-crovilli. Ultimately, this coupling phenomenon is due tothe identity of the Ca11 store with the Ca11 influxregulator, the cytoskeletal diffusion barrier. The samemechanisms that stimulate Ca11 release from thestore also affect the diffusion barrier system (Fig. 9). Inaddition, microvillar signaling also offers an explana-tion for some forms of store-independent activation ofCa11 influx, the most important of which are the vol-ume- and membrane stretch-activated Ca11 pathways,also termed mechanoactivated signaling. Mechanoacti-vation can be explained by mechanical disruption ofthe relative weak interactions between microvillar fil-ament bundles and the covering lipid membrane, ex-erted by either shear stress at the cell surface such asacoustic stimulation of hair cell microvilli or by shearstress induced by blood flow over vascular endothelialcells or by osmotically induced changes in cell volume(volume regulation).
Origin of the quantal Ca11 release. Another se-vere shortcoming of the current concept of Ca11 sig-naling is its inability to provide a systematic explana-tion for the observed quantal kinetics of Ca11 releasefrom the internal stores. Although much experimentalwork has been done in this field, no satisfying expla-nation could be found. The main problem results fromthe postulate of an IP3-regulated Ca11 channel. If anIP3-regulated channel is involved, it is hardly conceiv-able that only a small portion of the vesicle contentshould be liberated by subsaturating IP3 concentra-tions. Also, the observed initial high rate of Ca11 re-lease at subsaturating IP3 concentrations contradictsthe channel hypothesis. None of the presently knownchannel types exhibit properties of this type. Even theassumption of stores with different IP3 receptor sensi-tivities cannot adequately explain the staircase-likekinetics of Ca11 release.
The concept of microvillar Ca11 signaling presents asystematic solution of this problem without additionalpostulates and is based exclusively on establishedknowledge. The well-known functional link betweenthe PLC action on membrane lipids and the actin cy-toskeleton represents a step that imparts quantal orincremental properties to the whole signal pathway. Asshown by several laboratories, the interaction of actin-binding and -severing proteins to membrane phospho-lipids such as PIP and PIP2 follows highly cooperativekinetics (Lassing and Lindberg, 1985; Janmey et al.,1987; Goldschmidt-Clermont et al., 1990, 1991). Mem-brane binding of proteins of the profilin/gelsolin typeonly occurs on clusters of PIP/ PIP2 containing at leastsix to eight molecules of the phospholipid. The tight
28 LANGE

membrane interaction of profilin/gelsolin is destabi-lized by removal of only two to three of the six to eightbinding sites per PIP/ PIP2-protein complex (Fig. 7) (fora review, see Forscher, 1989; Stossel, 1989).
Liberation of actin-binding proteins of the profilin/gelsolin family from the inositol phosphate headgroupsof PIP/ PIP2 can be achieved in two ways. The first wayis by receptor-activated PLC which is able to hydrolyzePIP/ PIP2 even when these lipids are shielded by boundprofilin/gelsolin (Janmey et al., 1987; Goldschmidt-Clermont, 1991). The second way, by IP3 either liber-ated during PLC action or externally added, competeswith PIP/ PIP2 for binding to profilin (Lassing andLindberg, 1985). In either case, the loss of only a fewbonds between the proteins and the phospholipid clus-ters breaks down the whole complex. Whereas the lib-erated proteins activate a partial Ca11 release fromF-actin, the unchanged residual PIP or PIP2 lipids ofthe original binding clusters redistribute within theplane of the membrane by lateral diffusion. Therefore,the concentration of PIP/ PIP2 within the area of theactivated membrane domain is increased and the re-maining PIP/ PIP2 -profilin/gelsolin complexes are sta-bilized to a level exceeding that of the original pre-stimulation state. Additional Ca11 release can only beinitiated by a further increase of the ambient IP3 con-centration. The cooperativity of this process is identicalto that of IP3-induced Ca11 release (Hill factor of 2–3;Hiro et al., 1995).
Temperature dependence is a characteristic featureof the quantal nature of Ca11 release. As shown byKindman and Meyer (1993), the quantal effect is abol-ished at 11°C. At this temperature, addition of a sub-saturating concentration of IP3 released the whole con-tent of the IP3 -sensitive Ca11 store. This finding is inaccord with the notion of a binding equilibrium be-tween PIP/ PIP2 binding clusters and free PIP/ PIP2molecules moving within the plane of the inner bilayerleaflet of the plasma membrane by lateral diffusion. Atlow temperatures, lipid layers undergo a phase transi-tion to a relative rigid state which impairs the lateraldiffusion of lipids. Consequently, gelsolin-PIP/ PIP2complexes on the membrane surface can no longer bestabilized by lateral redistribution of PIP/ PIP2 mole-cules from decaying complexes. Thus, at low tempera-tures, all gelsolin/lipid complexes can be cleaved at aconstant IP3 concentration.
The interaction system of phospholipids with actin-binding proteins represents an excellent example forthe generation of incremental signaling properties inbiochemical mechanisms. It provides a qualitative sat-isfying explanation for the observed incremental (orquantal) IP3 effects, the staircase kinetics, which en-ables the cell to leave the all-or-nothing principle offixed threshold activation in favor of a graded responseon changing signal levels. This type of activation prin-ciple resembles physiological perceptive systems with agliding activation threshold to eliminate long-lastingbasal signal levels.
Ca11-induced Ca11 release. A third essentialproperty of the cellular Ca11 store that cannot be ex-plained on the basis of the current hypothesis of Ca11
storage is Ca11-induced Ca11 release (CICR). CICR issupposed to be involved in the mechanism of intracel-lular Ca11 oscillations and wave propagation of muscle
cells. The activation of this type of Ca11 release frominternal stores can be observed at submicromolar cyto-solic Ca11 levels. One possible mechanism for CICRfrom F-actin may be the Ca11-induced activation of thenumerous Ca11-regulated F-actin-binding proteinssuch as the vertebrate capping and severing proteins,gelsolin, villin, and adseverin. Activation of these pro-teins also occurs at or below micromolar cytosolic Ca11
levels that can be generated by stimulated entry ofexternal Ca11 via store-independent pathways(Stossel, 1989).
Complex Ca11 uptake and release kinetics areintrinsic properties of Ca11 uptake into F-actin
Kinetics of Ca11 uptake into F-actin. Accordingto Favre et al. (1996), the biphasic kinetics of ATP-dependent Ca11 uptake into intracellular stores ex-hibit two characteristic properties. First, the rapid ini-tial Ca11 uptake is limited to a constant time span.Regardless of the used external Ca11 concentration[Ca11]ex), the rapid initial uptake process declines to alow sustained level within 20 minutes. Second, initialrates of rapid Ca11 uptake depend on [Ca11]ex anddisplay saturation kinetics with an EC50 of about 400nM Ca11.
In order to reconcile these kinetic properties with thecurrent Ca storage concept, Favre et al. (1996) pos-tulated a feedback inhibition of the Ca11 pumpingATPase by the Ca11 concentration within the storagevesicles. However, in order to explain the rapid cessa-tion of the initial uptake process and the independenceof its termination of [Ca11]ex, the concentration-inhi-bition relationship between the Ca11 load and the ini-tial uptake rate was postulated to be “highly supralin-ear,” as indicated by an exponential slope factor of 5-6in their mathematical model. These findings clearlyshow that normal (linear) negative feedback Ca11, asassumed for the lumenal site of the pumping ATPase,is completely insufficient to account for the observeduptake kinetics.
In contrast to the ATPase pumping hypothesis, Ca11
storage into F-actin exactly mirrors these Ca11 uptakeproperties which essentially reflect the existence of twodifferent Ca11 uptake pathways with different affini-ties and velocities.
High-rate, low-affinity Ca11 uptake. Due to the dy-namic instability of F-actin, discrete events of rapiddissociation of ADP-actin-Mg monomers occur at thebarbed ends of filaments (k2
B 5 6 in Fig. 12). Afterexchange of the bound ADP for ATP, actin monomersrapidly reassociate with other filaments that bear ATP-monomer caps at their barbed ends. According to thedegree of exchange of Mg11 for Ca11, they contributeto the uptake of Ca11 into F-actin. As indicated by thelow dissociation rate constant (k2
P 5 0.4 in Fig. 12),monomer dissociation from the pointed filament ends isslow compared with that at the barbed ends. Conse-quently, the main contribution to Ca11 uptake comesfrom ADP-G-actin-Mg released during barbed-end dy-namic instability.
Exchange of Mg11 for Ca11 in Mg-ADP-actin is avery slow process (kex
Mg 5 0.04) compared with barbed-end dissociation which is 150-fold faster. Still slower isthe exchange of ADP for ATP (kex
N 5 0.01). Thus,exchange of Mg11 for Ca11 on Mg-ADP-actin is the
29MICROVILLAR Ca11 SIGNALING

rate-limiting step of barbed-end monomer cycling. Con-sequently, the main stream of monomers, recruited forreassociation at the barbed end, flows from Mg-ADP-actin via Ca-ADP-actin to Ca-ATP-actin which is themost rapidly polymerizing species (k1
B 5 1.4) (Fig. 12).As indicated by the rate constants, Mg-ADP-actin rep-resents the most abundant intermediate monomerform to which binding of Ca11 occurs. Once Ca-ADP-actin is formed, the remaining steps of the pathway arerapid and their pool sizes are small. Ca11 incorpora-tion into F-actin proceeds via:
Mg-ADP-G-actin 1 Ca11 3 Ca-ADP-G-actin
1ATP 3 Ca-ATP-G-actin 3 Ca-F-actin.
According to this scheme, the initial rate of Ca11
uptake into F-actin mainly depends on the rate of Ca11
incorporation into the large ADP-monomer pool. Atconstant ambient [Ca11], this rate is proportional to[Ca11] and to the proportion of Mg-ADP-actin/Ca-ADP-actin which is always highest at the beginning ofthe Ca11 incorporation but rapidly declines as F-actinsubunits increasingly contain Ca11. At the final stagesof the loading process, liberated ADP-actin monomerscan no longer bind additional Ca11 and the uptakeprocess slows down.
The time period until cessation of the rapid Ca11
uptake largely depends on the kinetics of dynamic in-stability such as the number of filaments, the initiationrate of breakdown periods, and the length of the fila-ment region which is subject to dynamic instability.Since these factors are nearly independent of free[Ca11], uptake termination is also invariant to [Ca11].
As shown by Favre et al. (1996), the initial rate ofCa11 uptake into F-actin depends on the applied[Ca11] and follows saturation kinetics (Kd about 400nM). Below this concentration, initial rates are propor-tional to [Ca11]; at higher concentrations, the incre-ments on initial uptake rates rapidly decrease (Favreet al., 1996). Evidently, the EC50 for the dependence of
initial Ca11 uptake rates on [Ca11] is identical withthe Kd of Ca11 for ADP-G-actin. At 400 nM Ca11, theKd of Ca11 for ADP-G-actin, the ADP-G-actin pool ishalf saturated with Ca11. At lower concentrations,Ca11 binding declines linearly, at higher concentra-tions binding becomes saturated.
Low-rate, high-affinity Ca11 uptake. F-actin storagealso accounts for the second type of Ca11 uptake, pro-ceeding with a much lower rate but a higher affinity,which becomes visible after the first low-affinity pro-cess has slowed down. This second component of Ca11
storage is conducted by the intermediary pool of ATP-G-actin with high affinity for Ca11 (Kd 5 2–8 nM inFig. 12). Exchange of Ca11 for Mg11 in ATP-G-actinproceeds with only one third of the rate as with ADP-G-actin (k2
Mg5 0.013 sec-1). The rate of this uptakeprocess is also slower because the pool size of ATP-G-actin is much smaller than that of ADP-G-actin (Fig.12). Thus, the slow high-affinity uptake process is con-ducted via:
Mg-ADP-G-actin 1 ATP 3 Mg-ATP-G-actin
1 Ca11 3 Ca-ATP-G-actin 3 Ca-F-actin.
This type of Ca11 uptake into F-actin has been visual-ized in vitro using free [Ca11] between 400 and 40 nMas described above (Lange and Brandt, 1996).
Kinetics of Ca11 release from F-actin. Manypublications have reported the dependence of the Ca11
release process on the Ca11 concentration either in thecytoplasm or in the store itself. The accumulated datagive a puzzling picture when projected on the currentmodel of Ca11 storage, but are easily to be reconciledwith Ca11 storage into F-actin.
Using permeabilized smooth muscle cells (A7r5),Missiaen et al. (1992) and Parys et al. (1993) demon-strated that the extent of IP3-induced 45-Ca11 releasefrom 45-Ca11-loaded stores is a function of the concen-tration of external 40-Ca11(added together with IP3).In the presence of 300 nM 40-Ca11, IP3 released almosttwofold the amount of 45-Ca11 than in the absence of40-Ca11. The same dependence of label efflux on ex-ternal [40-Ca11] was observed for thapsigargin-stimu-lated Ca11 efflux. Furthermore, the relative portion of45- Ca11 released by IP3 plus 300 nM Ca11 was in-creased at lower filling states of the store. The abovementioned authors suggested an activation of the IP3receptor by cytoplasmic Ca11 and a complex depen-dence of the open state of the IP3 receptor channel onthe external and luminal Ca11 and IP3 concentrations.
When Ca11 dissociation from actin monomers repre-sents the true release step, the accelerated and en-hanced 45-Ca11 release merely results from exchangeof G-actin-bound 45-Ca11 for added 40-Ca11. This typeof 45- Ca11 liberation does not represent enhanced andenlarged net liberation of bound Ca11.
At lower loading states, a higher amount of 45-Ca11
originally bound to actin is cycled back into F-actinafter IP3-induced 45-Ca11 release. Therefore, additionof IP3 without 40- Ca11 results in a strongly reducednet amount of released 45- Ca11. Release of 45-Ca11
due to isotope exchange, however, proceeds to an al-most identical level as with the filled store: down to afinal level of 10% of the maximum load which is also
Fig. 12. Flow diagram of uptake of Ca11 into F-actin under initialrate conditions. Rate constants (k) and equilibrium dissociation con-stants (K) are taken from Parys et al. (1993) and Horne and Meyer(1995). Because association rate constants of cations are very largecompared to their dissociation rate constants, they do not influencethe general flow of intermediates under non-steady-state conditions.
30 LANGE

reached when (IP3 plus 40-Ca11) is added to the fullstore.
As the Kd for Ca11 dissociation from ADP-G-actin is380 nM, the 40-Ca11 concentration of 300 nM used forthese experiments represents an almost optimal con-centration. Addition of 1 mM 40-Ca11 was shown toincrease 45-Ca11 release only by additional 25% (Mis-siaen et al., 1992). Essentially the same results werereported by Horne and Meyer (1995) and Combettes etal. (1994), both applying the 45- Ca11 technique fortheir studies.
In contrast, Shuttleworth (1992), using the directCa11 measurement by indo-1 fluorescence, concludedfrom his experiments that “Ca11 release from IP3-sensitive stores is not modulated by intraluminalCa11.”
Identity, localization, and propertiesof the IP3 receptor
As depicted in Figures 7 and 9, the molecular targetsfor the IP3 action on Ca11 release from F-actin areactin-binding and severing proteins of the gelsolin/pro-filin family, some of which (gelsolin, villin, adseverin)are Ca11 regulated. In this model, IP3 receptors areproteins of this family which are deactivated by bind-ing to PIP/PIP2 clusters. The action of IP3 on thesecomplexes is due to competitive displacement of theinositol phosphate moieties of the PIP/PIP2 bindingclusters from their binding sites on actin-binding pro-teins (Lassing and Lindberg, 1985; Janmey et al.,1987). On the other side, Ca11-regulated actin-bindingproteins including gelsolin and cofilin change their af-finity to F-actin in a Ca11-dependent manner. In thepresence of micromolar Ca11, proteins of the gelsolin/cofilin family dissociate from the lipid membrane toform complexes with the ADP-containing subunits ofF-actin (Coue and Korn, 1986). F-actin-bound gelsolinmolecules exhibit free PIP/PIP2-binding sites to whichIP3 can associate with higher affinity than to the lipid-bound proteins (Lassing and Lindberg, 1985) (Fig. 13).
According to this scheme, only the membrane-boundlow-affinity form of gelsolin is the functional active IP3receptor. The F-actin-bound high-affinity form, how-ever, cannot contribute to additional Ca11 release be-cause it is the receptor form “just in action.” Ca11-dependent transformation of the functional low-affinityIP3 receptors to nonfunctional high-affinity receptorshas been reported for various cell models, in a mostdetailed form for permeabilized hepatocytes (Pietri etal., 1990a,b; Rouxel et al., 1992; Dufour et al., 1997).
Interestingly, the process of IP3 receptor transforma-tion exhibits a specific temperature sensitivity, whichis identical to that of the quantal Ca11 release (Rouxelet al., 1992). Once IP3 receptors have been transformedinto the high-affinity state, reversal to the low-affinitystate is completely blocked at low temperature (4°C).At 37°C, however, IP3 receptors are rapidly reverted tothe low-affinity state (t1/2 , 1 s) (Rouxel et al., 1992).This “locking” of IP3 receptors in the high-affinity stateis identical to the inhibition of quantal Ca11 release atlow temperatures. It is due to the phase transition inlipid membranes at low temperatures which lowers thefluidity of the membrane components, thereby inhibit-ing the formation of PIP/PIP2 clusters necessary forbinding of proteins of the gelsolin/profilin family.
Cytoplasmic IP3 receptors. IP3 receptors of thegelsolin type, as discussed in the previous section, dif-fer in their properties from the classical receptor typeof an integral membrane protein. Instead, this effectorsystem consists of a membrane-bound receptor that isreleased to the cytoplasm when activated by IP3. Thistype of receptor, although originally located at the in-ner face of the plasma membrane, can also exist as acytoplasmic component.
Using heparin-Sepharose as well as IP3 affinity chro-matography, two recent publications reported the iso-lation of cytoplasmic protein complexes which may rep-resent cytoplasmic forms of the high-affinity IP3receptor. First, Banno et al.(1992) prepared by PIP/PIP2 elution of a heparin column loaded with plateletcytosol a complex of PLC (g and d) with two otherproteins forming a tight complex of 130 kDa. This wasidentified as the very stable 1:1 actin/gelsolin complexcontaining 1 mol Ca11. In contrast to gelsolin, thiscomplex was ineffective in inhibiting PIP2 hydrolysisby PLC (g and d). Second, Kanematsu et al. (1992),using an IP3 affinity column, isolated a quite similarprotein complex from rabbit brain cytosol. This com-plex also consisted of PLCd and an as yet unidentified130 kDa protein with high IP3-binding activity. Fur-thermore, a specific association of gelsolin and actinwith the PLCg1 was observed in human platelets (Bal-dassare et al., 1997). A similar association of PLC withthe Ca11-regulated F-actin-severing protein, villin,was observed in brush border microvilli (Khurana etal., 1997).
IP3-independent, receptor-operatedCa11 channels
One of the most important implications followingfrom the present concept of Ca11 signaling is the in-volvement of IP3-independent Ca11 channels in recep-tor-operated Ca11 signal pathways. As yet, two differ-ent cation channel types have been postulated forcellular Ca11 signaling. A microsomal IP3-sensitivechannel and a plasma membrane channel regulated byan unknown putative signal assumed to account for itsstore-dependent regulation. However, all detected mi-crosomal cation channels were found to be independent
Fig. 13. Gelsolin as IP3 receptor.
31MICROVILLAR Ca11 SIGNALING

of IP3 (Schmid et al., 1990; Filipovic and Sackin, 1991;Zweifach and Lewis, 1993; Lange and Brandt, 1993b;Duszynski et al., 1995; Lumpkin and Hudspeth, 1995;Lange et al., 1996).
The concept of microvillar Ca11 signaling needs onlyone IP3-independent microvillar cation channel to ex-plain all known functions of the Ca11 signal pathway.This channel, located in the microvillar tip membrane,provides a constitutionally open cation pathway allow-ing Ca11 influx into the microvillar tip region of intactmicrovilli as well as Ca11 entry into the cytoplasmafter receptor-mediated breakdown of the microvillardiffusion barrier. One of the most direct studies offer-ing experimental proof of the existence of an IP3-inde-pendent, constitutionally open Ca11 channel on mi-crovilli was a study on hair cells published by Lumpkinand Hudspeth (1995).
Direct visualization of Ca11 signaling on thecell surface
Hair cell model of microvillar Ca11signaling.Lumpkin and Hudspeth’s (1995) study of Ca11 signal-ing in audioreceptor cells (hair cells) provided completeexperimental confirmation for the proposed microvillararrangement of Ca11 channels and the diffusion bar-rier system. Hair cells represent the only known cellu-lar model on which microvilli are large enough fordirect light microscopic observation of internal Ca11
movements. The authors examined the site of Ca11
entry into the hair cell microvilli (also called stereo-cilia) during acoustic stimulation using laser scanningconfocal microscopy coupled with the fura-2 fluores-cence technique.
The results of this study were described in the fol-lowing way: “An unstimulated hair cell showed a ‘tipblush’ of enhanced fluorescence at the hair bundle’stop, which we attribute to Ca11 permeation throughtransduction channels at rest. Upon mechanical stim-ulation, individual stereocilia displayed increased flu-orescence that originated near the tips and spread to-wards their bases.” The authors further conclude: “Ourresults confirm that mechanoelectrical transduction oc-curs near stereociliary tips.” These findings confirm themain features of the presented concept of Ca11 signal-ing: localization of IP3-insensitive, open Ca11 channelsin the tip membrane of microvilli; existence of a mi-crovillar tip compartment (entrance compartment) towhich external Ca11 has free access; function of themicrovillar actin filament bundle as diffusion barrierfor Ca11 in the unstimulated state; and modulation ofthe cytoskeletal diffusion barrier by a specific stimulus,here mechanical stimulation by acoustic waves.
Mechanosensitive regulation of Ca11 entry into haircells strongly suggests that mechanical activation ofcation permeability may be an intrinsic property of theF-actin diffusion barrier system. Consequently, othertypes of mechanoactivated channels may also functionin a similar manner such as volume regulation andblood pressure regulation.
Ca11 signaling in Xenopus oocytes. The idea ofmicrovillar Ca11 signaling is further supported by re-cent studies on Ca11 signaling in Xenopus oocytes us-ing confocal microscopy. Unfortunately, the resolutionof light microscopy is insufficient to resolve small mem-brane details such as the microvilli of normal cells.
Even the microvilli of the large Xenopus oocyte are ofthe same dimension as those of other epithelial cellsand, therefore, are not detectable by light microscopy.However, as shown by Eidne et al. (1994), receptor-operated Ca11 increases initiated in the immediatevicinity of the plasma membrane and spread out intothe inner cytoplasm: “An initial transient planar waveof calcium propagating just below the surface of themembrane is followed by a second slower phase.” Re-lease of Ca11 from internal stores was responsible forthe initial response while influx of external Ca11 wasinvolved in the second phase. Almost the same obser-vation was reported by Nathanson et al. (1992) forvasopressin receptor-expressing Xenopus oocytes: “Va-sopressin induced a single concentric wave of increasedCa11 that radiated inward from the plasma mem-brane.” In a recent paper, Peterson and Berridge (1996)noted: “Depletion of intracellular calcium stores in alocalized region of Xenopus oocytes was found to evokecapacitative calcium entry exclusively colocalizedacross the stimulated area of the plasma membrane,arguing against the involvement of a highly diffusiblecalcium influx factor.” These findings suggest the im-mediate physical association of the Ca11 store andinflux pathway as postulated for microvillar Ca11 sig-naling and demonstrate that the whole system istightly associated with the plasma membrane.
CONCLUSIONSA novel conception of Ca11 signaling is presented
that attributes essential cellular control functions tomicrovilli on the surface of differentiated or restingcells. Integral membrane proteins such as transportersand ion channels have been identified as microvillarcomponents. Furthermore, the microvillar core proteinF-actin was shown to function as an ATP-dependent,IP3-sensitive Ca11 store. It also represents the diffu-sion barrier between the microvilli tip compartmentand the cytoplasm regulating the rate of Ca11 influxinto the cytoplasm. Most convincingly, this new con-cept implicates as systematic properties all the as yetunexplained complex features of Ca11 signaling suchas store-coupled Ca11 influx, quantal Ca11 release,and CICR as well as the complicated Ca11 uptake andrelease kinetics of the Ca11 store. Furthermore, it pro-vides a clear mechanistic idea of all components in-volved in Ca11 storage and release as well as theircellular arrangement.
Control of essential cell functions via microvillar sys-tems is a novel aspect of cellular differentiation. Itappears to represent an old evolutionary principlehighly conserved from early forms of life such as cili-ates and slime molds up to highly organized multicel-lular organisms. According to the evolutionary rule notto leave a successful principle, microvilli have beenadapted for a multitude of functions during the evolu-tion of higher life. The number of possible functions ofthis system in cell physiology may be much higher thanwe can see at the moment. Further work will be nec-essary to elucidate the whole physiological relevance ofthis important regulatory principle.
LITERATURE CITEDBaldassare JJ, Henderson PA, Tarver A, Fisher GJ. 1997. Thrombin
activation of human platelets dissociates a complex containing gel-
32 LANGE

solin and actin from phosphatidylinositide-specific phospholipaseCgamma1. Biochem J 324:283–287.
Banno Y, Nakashima T, Kumada T, Ebisawa K, Nonomura Y, NozawaY. 1992. Effects of gelsolin on human platelets cytosolic phospho-inositide phospholipase C isoenzymes. J Biol Chem 267:6488–6494.
Barany M, Finkelman F. 1962. The lability of the F-actin-boundcalcium under sonic vibration. Biochim Biophys Acta 63:98–105.
Bereiter-Hahn J, Stubig C, Heymann V. 1995. Cell cycle-relatedchanges in F-actin distribution are correlated with glycolytic activ-ity. Exp Cell Res 218:551– 560.
Burlacu S, Janmey PA, Borejdo J. 1992. Distribution of actin filamentlengths measured by fluorescence microscopy. Am J Physiol 262:C569–C577.
Carlier MF. 1991. Actin: protein structure and filament dynamics.J Biol Chem 266:1–4.
Carlier M-F, Pantaloni D. 1997. Control of actindynamics in cellmotility. J Mol Biol 269:459–467.
Carlier M-F, Pantaloni D, Korn ED. 1986. The exchangeability ofactin-bound calcium with various divalent cations to high-affinityand low-affinity binding sites on ATP-G-actin. J Biol Chem 261:10778–10784.
Combeau C, Carlier M-F. 1988. Probing the mechanism of ATP hy-drolysis on F-actin using vanadate and the structural analogs ofphosphate BeF3
- and AlF4-. J Biol Chem 263:17429–17436.
Combettes L, Hannaert-Merah Z, Coquil J-F, Rousseau C, Claret M,Swillens S, Champeil P. 1994. Rapid filtration studies of the effectof cytosolic Ca11 on inositol 1,4,5-trisphosphate-induced 45- Ca11
release from cerebellar microsomes. J Biol Chem 269:17561–17571.Coue M, Korn ED. 1986. Interaction of plasma gelsolin with ADP-
actin. J Biol Chem 261:3628–3631.Dufour J-F, Arias IM, Turner TJ. 1997. Inositol 1,4,5-trisphosphate
and calcium regulate the calcium channel function of the hepaticinositol 1,4,5-trisphosphate receptor. J Biol Chem 272:2675–2681.
Dunlop ME, Larkins RG. 1988. GTP- and inositol 1,4,5-trisphosphate-induced release of calcium-45 ion from a membrane store colocal-ized with pancreatic islet cell plasma membrane. Biochem J 253:67–72.
Duszynski J, Elenski M, Tillotson DL, LaNoue KF. 1995. Hormone-regulated Ca11 channel in rat hepatocytes revealed by whole cellpatch clamp. Cell Calcium 18:19–29.
Eidne KA, Zabavnik J, Allan WT, Trewavas AJ, Read ND, AndersonL. 1994. Calcium waves and dynamics visualized by confocal mi-croscopy in Xenopus oocytes expressing cloned TRH receptors.J Neuroendocrinol 6:173–178.
Estes JE, Selden LA, Gershman LG. 1987. Tight binding of divalentcations to monomeric actin. Binding kinetics support a simplifiedmodel. J Biol Chem 262:4952–4957.
Favre CF, Schrenzel J, Jacquet J, Lew DP, Krause K-H. 1996. Highlysupralinear feedback inhibition of Ca11 uptake by the Ca11 load ofintracellular stores. J Biol Chem 271:14925–14930.
Filipovic D, Sackin H. 1991. A calcium-permeable stretch-activatedcation channel in renal proximal tubule. Am J Physiol 260:F119-F129.
Forscher P. 1989. Calcium and polyphosphosinositide control of cy-toskeletal dynamics. TINS 12:468–474.
Gartzke J, Lange K. 1996. Biochemische Grundlagen der Wirkungvon Tumorpromotoren und Testung von Schadstoffen an isoliertenZellen. Ergo Med 20:30–43.
Gershman LC, Selden LA, Estes JE. 1986. High affinity binding ofdivalent cations to actin monomers is much stronger than previ-ously reported. Biochem Biophys Res Commun 135:607–614.
Gershman LC, Selden LA, Estes JE. 1991. High affinity divalentcation exchange on actin. Association rate measurements supportthe simple competitive model. J Biol Chem 266:76–82.
Goldschmidt-Clermont PJ, Machesky LM, Baldassare JJ, Pollard TD.1990. The actin-binding protein profilin binds to PIP2 and inhibitsits hydrolysis by phospholipase C. Science 247:1575–1578.
Goldschmidt-Clermont PJ, Kim JW, Machewsky LM, Rhee SG, Pol-lard TD. 1991. Regulation of phospholipase C-c1 by profilin andtyrosine phosphorylation. Science 251:1231–1233.
Guillemette G, Balla T, Baukal AJ, Catt KJ. 1988. Characterization ofinositol 1,4,5-trisphosphate receptors and calcium mobilization in ahepatic plasma membrane fraction. J Biol Chem 263:4541–4548.
Hiro J, Michikawa T, Miyawaki A, Furuichi T, Okura I, Mikoshiba K.1995. Kinetics of calcium release by immunoaffinity-purified inosi-tol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles.J Biol Chem 270:19046–19051.
Horne JH, Meyer T. 1995. Luminal Ca11 regulates the inositoltrisphosphate receptor of rat basophilic leukemia cells at a cytosolicsite. Biochemistry 34:12738–12746.
Janmey PA, Iida K, Yin HL, Stossel TP. 1987. Polyphosphoinositide
micelles and polyphosphoinositide-containing vesicles dissociateendogenous gelsolin-actin complexes and promote actin assemblyfrom the fast growing end of actin filaments blocked by gelsolin.J Biol Chem 262:12228– 12236.
Kanematsu T, Takeya H, Watanabe Y, Ozaki S, Yoshida M, Koga T,Iwanaga S, Hirata M.1992. Putative inositol 1,4,5-trisphosphatebinding protein in rat brain cytosol. J Biol Chem 267:6518–6525.
Kasai M, Oosawa F. 1968. The exchangeability of actin-bound calciumwith various divalent cations. Biochim Biophys Acta 154:520–528.
Kasai M, Oosawa F. 1969. Behavior of divalent cations and nucleo-tides bound to F-actin. Biochim Biophys Acta 172:300–310.
Khurana S, Arpin M, Patterson R, Donowitz M. 1997. Ileal microvillarprotein villin is tyrosine-phosphorylated and associates with PLC-gamma1. Role of cytoskeletal rearrangement in the carbachol-in-duced inhibition of ileal Na1 absorption. J Biol Chem 272:30115–30121.
Kindman LA, Meyer T. 1993. Use of intracellular Ca11 stores fromrat basophilic leukemia cells to study the molecular mechanismleading to quantal Ca11 release by inositol 1,4,5-trisphosphate.Biochemistry 32:1270–1277.
Kinosian HJ, Selden LA, Estes JE, Gershman LC. 1993. Nucleotidebinding to actin, Cation dependence of nucleotide dissociation andexchange rates. J Biol Chem 268:8683–8691.
Korn ED, Carlier M-F, Pantaloni D. 1987. Actin polymerization andATP hydrolysis. Science 238:638–644.
Lange K, Brandt U. 1990a. Insulin-responsive glucose transportersare concentrated in a cell surface-derived membrane fraction of3T3-L1 adipocytes. FEBS Lett 261:459–463.
Lange K, Brandt U. 1990b. Restricted localization of the adipocyte/muscle glucose transporter species to a cell surface-derived vesiclefraction of 3T3-L1 adipocytes. Inhibited lateral mobility of integralplasma membrane proteins in newly inserted membrane areas ofdifferent iated 3T3-L1 cells. FEBS Lett 276:39–41.
Lange K, Brandt U. 1993a. The IP3-sensitive calcium store of HITcells is located in a surface-derived vesicle fraction. FEBS Lett320:183–188.
Lange K, Brandt U. 1993b. Rapid uptake of calcium, ATP, and inositol1,4,5-trisphosphate via cation and anion channels into surface-derived vesicles from HIT cells containing the inositol 1,4,5-trisphosphate-sensitive calcium store. FEBS Lett 325:205–209.
Lange K, Brandt U. 1996. Calcium storage and release properties ofF-actin: evidence for the involvement of F-actin in cellular calciumsignaling. FEBS Lett 395:137–142.
Lange K, Brandt U, Keller K, Zimmermann B. 1989. Endogenousregulation of 2-deoxyglucose uptake in C6 glioma cells correlateswith cytoskeleton-mediated changes of surface morphology. J CellPhysiol 140:29–43.
Lange K, Brandt U, Zimmermann B.1990. Relationship between in-sulin stimulation and endogenous regulation of 2-deoxyglucose up-take in 3T3-L1 adipocytes. J Cell Physiol 142:1–14.
Lange J, Schlieps K, Lange K, Brandt U, Knoll-Kohler E. 1996.Evidence for the involvement of cell surface microvilli in the cal-cium signaling pathway of isolated rat hepatocytes. Detection of theATP-dependent calcium store in a cell surface-derived vesicle frac-tion. Exp Cell Res 228:189–196.
Lange J, Schlieps K, Lange K, Brandt U, Knoll-Kohler E. 1997.Activation of calcium signaling in isolated rat hepatocytes is accom-panied by shape changes of microvilli. Exp Cell Res 234:486–497.
Lassing I, Lindberg U. 1985. Specific interaction between phosphati-dylinositol 4,5-bisphosphate and profilactin. Nature 314:472–474.
Lievremont J-P, Hill A-M, Hilly M, Mauger J-P. 1994. The inositol1,4,5-trisphosphate receptor is localized on specialized sub-regionsof the endoplasmic reticulum in rat liver. Biochem J 300:419–427.
Lin EC, Cantiello HF. 1993. A novel method to study the electrody-namic behavior of actin filaments: evidence for cable-like propertiesof actin. Biophys J 65:1371– 1378.
Lumpkin EA, Hudspeth AJ. 1995. Detection of Ca11 entry throughmechanosensitive channels localizes the site of mechanoelectricaltransduction in hair cells. Proc Natl Acad Sci USA 92:10297–10301.
Mejean C, Pons F, Benyamin Y, Roustan C. 1989. Antigenic probeslocate binding sites for the glycolytic enzymes glyceraldehyde-3-phosphate dehydrogenase, aldolase, and phosphofructokinase onthe actin monomer in microfilaments. Biochem J 264:671–677.
Michelangeli F. 1993. The effects of amino acid-reactive reagents onthe functioning of the inositol 1,4,5-trisphosphate-sensitive calciumchannel from rat cerebellum. Cell Signal 5:33–39.
Missiaen L, De Smedt H, Droogmans G, Casteels R. 1992. LuminalCa11 controls the activation of the inositol 1,4,5-trisphosphate re-ceptor by cytosolic Ca11. J Biol Chem 267:22961–22966.
Murphy AJ, Coll RJ. 1993. Formation of a stable inactive complex of
33MICROVILLAR Ca11 SIGNALING

the sarcoplasmic reticulum calcium ATPase with magnesium, be-ryllium, and fluoride. J Biol Chem 268:23307–23310.
Nanhua C, Masters C. 1987. The influence of calcium ions on adsorp-tion of glycolytic enzymes to cellular structure. Biochem Int 15:835–842.
Nathanson MH, Moyer MS, Burgstahler AD, O’Carroll A-M, Brown-stein MJ, Lolait SJ. 1992. Mechanism of subcellular Ca11 signalingevoked by stimulation of the vasopressin V1a receptor. J Biol Chem267:23282–23289.
Obukhov AG, Jones SVP, Degtiar VE, Lockhoff A, Schultz G, Hes-cheler J. 1995. Ca11 permeable large-conductance nonselective cat-ion channels in rat basophilic leukemia cells. Am J Physiol 269:C1119–C1125.
Parekh AB, Penner R. 1997. Store depletion and calcium influx.Physiol Rev 77:901–930.
Park C-S, MacKinnon R. 1995. Divalent cation selectivity in a cyclicnucleotide-gated ion channel. Biochemistry 34:13328–13333.
Parys JB, Missiaen L, De Smedt H, Casteels R. 1993. Loading depen-dence of inositol 1,4,5-trisphosphate-induced Ca11 release in theclonal line A7r5. Implications for the mechanism of quantal release.J Biol Chem 268:25206 – 25212.
Perdue J. 1971. The isolation and characterization of plasma mem-branes from cultured cells. III. The adenosine triphosphate-depen-dent accumulation of Ca11 by chick embryo fibroblasts. J BiolChem 246:6750–6759.
Perelroizen I, Didry D, Christensen H, Chua N-H, Carlier M-F. 1996.Role of nucleotide exchange and hydrolysis in the function of pro-filin in actin assembly. J Biol Chem 271:12302–12309.
Petersen CC, Berridge MJ. 1996. Capacitive calcium entry is colocal-ized with calcium in Xenopus oocytes: evidence against a highlydiffusible calcium influx factor. Pluegers Arch 432:286–292.
Pietri F, Hilly M, Mauger JP. 1990a. Calcium mediates the intercon-version between two states of the liver inositol 1,4,5-trisphosphatereceptor. J Biol Chem 265:17478–17485.
Pietri F, Hilly M, Claret M, Mauger JP. 1990b. Characterisation of 2reversible states of the inositol 1,4,5-trisphosphate receptor in rathepatocytes. Gastroenterol Clin Biol 14:710–714.
Podolski JL, Steck TL. 1990. Length distribution of F-actin in Dictyo-stelium discoideum. J Biol Chem 265:1312–1318.
Putney JW, Jr. 1986. A model for receptor-regulated calcium entry.Cell Calcium 7:1–12.
Putney JW, Jr. 1990. Capacitative calcium entry revisited. Cell Cal-cium 11:611–624.
Rossier MF, Bird GSJ, Putney JW. 1991. Subcellular distribution ofthe calcium-storing inositol 1,4,5-trisphosphate-sensitive or-ganelles in the rat liver. Biochem J 274:643–650.
Rouxel FP, Hilly M, Mauger J-P. 1992. Characterization of a rapidlydissociating inositol 1,4,5-trisphosphate-binding site in liver mem-branes. J Biol Chem 267:20017–20023.
Sampath P, Pollard TD. 1991. Effects of cytochalasin, phalloidin, and pHon the elongation of actin filaments. Biochemistry 30:1973–1980.
Schmid A, Dehlinger-Kremer M, Schulz I, Gogelein H. 1990. Volt-age-dependent InsP3-insensitive calcium channels in membranesof pancreatic endoplasmic reticulum vesicles. Nature 346:374 –376.
Selden LA, Gershman LC, Kinosian HJ, Estes JE. 1987. Conversion ofATP-actin to ADP-actin reverses the affinity of monomeric actin forCa11 vs Mg11. FEBS Lett 217:89–93.
Shuttleworth TJ. 1992. Ca11 release from inositol trisphosphate-sensitive stores is not modulated by intraluminal (Ca11). J BiolChem 267:3573–3576.
Stossel TP. 1989. From signal to pseudopod. How cells control cyto-plasmic actin assembly. J Biol Chem 264:18261–18264.
Tang JX, Janmey PA. 1996. The polyelectrolyte nature of F-actin andthe mechanism of actin bundle formation. J Biol Chem 271:8556–8563.
Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK,Bjerrum PJ, Christensen SB, Hanley MR. 1989. Thapsigargin, anovel molecular probe for studying intracellular calcium releaseand storage. Agents Actions 27:17–23.
Troullier A, Girardet J-L, Dupont Y. 1992. Fluoroaluminate com-plexes are bifunctional analogues of phosphate in sarcoplasmicreticulum Ca11 ATPase. J Biol Chem 267:22821–22829.
Zweifach A, Lewis RS. 1993. Mitogen-regulated Ca11 current of Tlymphocytes is activated by depletion of intracellular stores. ProcNatl Acad Sci USA 90:6295–6299.
34 LANGE