methodological studies of α
TRANSCRIPT

METHODOLOGICAL STUDIES OF α-HALOGENATED CARBONYLS AND THE
SYNTHETIC INVESTIGATION OF DIHYDRORESORCYLIDE
by
KRISTINA CLAIRE PROBASCO
MICHAEL P. JENNINGS, COMMITTEE CHAIR
KEVIN H. SHAUGHNESSY
TIMOTHY S. SNOWDEN
JOHN M. RIMOLDI
PAUL A. RUPAR
A DISSERTATION
Submitted in partial fulfillment of the requirements
for the Doctor of Philosophy
in the Department of Chemistry
and Biochemistry
in the Graduate School of
The University of Alabama
TUSCALOOSA, ALABAMA
2020

Copyright Kristina Claire Probasco 2020
ALL RIGHTS RESERVED

ii
ABSTRACT
The research presented herein consisted of projects with focuses on metal enolates and
silane chemistry, and their uses in methodology and total synthesis. The projects were divided
into three distinct chapters.
The first chapter covers the development of highly functionalized pyran motifs that are
commonly found in classes of natural products such as Bryostatins via an intramolecular Heck
reaction with a novel palladium enolate transfer. A bromoethoxy pentanoate compound was
synthesized through several steps and was then subjected to catalytic reactions with conditions
found in literature where many variations were changed in attempts to obtain the desired six-
membered ring.
The second chapter consists of the total synthesis of Dihydroresorcylide via a novel
palladium enolate ring closure. This structure has been synthesized twice before, however both
syntheses undergo a ring closing metathesis to create the macrocycle. The macrocyclization
attempts were based on literature published by Buchwald and Hartwig.
The third project studies the halogenation of a trialkylsilyl bond through what is believed
to be a bromonium ion intermediate followed by an SN2 like elimination according to work
published by Tamao and company. Majority of the halogenations proceeded in good yields and
with complete inversion of stereochemistry.

iii
LIST OF ABBREVIATIONS AND SYMBOLS
9-BBN 9-borabicyclo[3.3.1.]nonane
Ac2O acetic anhydride
BF3•OEt2 boron trifluoride diethyl etherate
Bn benzyl
CSA camphorsufonic acid
DBA dibenzylideneacetone
DCM dichloromethane
DIBAL-H diisobutyl aluminum hydride
DIAD diisopropyl azodicarboxylate
DIPEA diisopropyl ethyl amine
DMAP 4-dimethyl amino pyridine
DME dimethoxyethane
DMF N,N-dimethylformamide
DMP Dess-Martin periodinane
DMSO dimethyl sulfoxide
dr diastereomeric ratio
E- entgegen (opposite, trans-)
equiv equivalents
EWG electron withdrawing group

iv
GC(II) Grubbs’ generation (II) catalyst
HMPA hexamethylphosphoramide
HRMS high resolution mass spectroscopy
Hz hertz
IR infrared
Ipc2BOCH3 (−)-diisopinocampheylmethoxy borane
J coupling constant
KHMDS potassium hexamethyl disilazideLDA
LDA lithium diisopropylamide
LiHMDS or
LHMDS
lithium bis(trimethylsilyl)amide
M molar
mCPBA meta-chloroperoxybenzoic acid
MHz megahertz
mmol millimole
mol mole
MEM methoxy ethoxy methyl
MOM methoxy methyl
MTBE methyl tert-butyl ether
NA not applicable
nBuLi n-butyllithium
ND not determined
NEt3 triethylamine

v
NMR nuclear magnetic resonance
NOE nuclear Overhauser enhancement
NR no reaction
o- ortho-
-OTf trifluoromethane sulfonate (triflate)
p- para-
PTSA (TsOH) p-toluenesulfonic acid
Py (pyr) pyridine
(R)- rectus (clockwise)
rt room temperature
(S)- sinister (counterclockwise)
TBAF tetra-n-butyl ammonium fluoride
TBS tert-butyldimethylsilyl
TEA Triethylamine
TEMPO 2,2,6,6-tetramethyl-1-piperidinyloxy free radical
TES triethylsilyl
TFA trifluoroacetic acid
THF tetrahydrofuran
THP tetrahydropyran
TMS trimethylsilyl
TMSOTf trimethylsilyl trifluoromethanesulfonate
TPS triphenylsilyl
Z- zuzammen (together, cis-)

vi
ACKNOWLEDGEMENTS
The first person I want to thank is my wonderful husband, Michael Probasco. We have
had a lot of the craziness of life happen, but you are always there being steady and helping me to
take everything in stride. You have helped me grow as a person and as a chemist over these last
few years and I cannot express my gratitude enough. Without you my days would be sullen. You
keep me laughing and help me to remain motivated. I love you more than I could accurately
express and I am so proud to be your wife.
My growth as a synthetic chemist comes from the help of several people. I wish to show
appreciation to my doctoral advisor, Dr. Michael P. Jennings, for giving me encouragement and
advice to help me improve my laboratory skills. My committee members also deserve thanks for
their investment in me along with their knowledgeable research suggestions. Thank you to Dr.
Michael P. Jennings, Dr. Timothy S. Snowden, Dr. Kevin H. Shaughnessy, Dr. Paul A. Rupar,
and John M. Rimoldi. Gratitude should also be given to Dr. Ken Belmore for assistance with
NMR experiments and Qiaoli Liang for completing mass spec analysis in a timely manner. Also,
a special thank you to Dr. Douglas Masterson, Dr. David Rankin, and Robin Wilson for seeing
something in me that I didn’t see in myself and encouraging me to persevere.
I want to state my upmost appreciation to my family, Karen Morris, Otis Morris, Jesse
Mejia, and the Lee Morris family. Thank you for supporting me and never giving up on me. I
appreciate the countless vent sessions and the understanding when I don’t call. I would not be
where I am without your love.

vii
Finally, I want to express my gratitude to the people I call friends. Shelby Dickerson and
Megan Roark, you both have been there for me from undergraduate studies to now. Both of you
are incredible and I will never be able to convey how much you have kept me calm and centered
these last several years. Thank you to Cameron Massey for the long random lab talks and
keeping my husband occupied with video games when I have been too busy.

viii
CONTENTS
ABSTRACT ....................................................................................................................... ii
LIST OF ABBREVIATIONS AND SYMBOLS .............................................................. iii
ACKNOWLEDGEMENTS ............................................................................................... vi
LIST OF TABLES ............................................................................................................. xi
LIST OF FIGURES ......................................................................................................... xiii
LIST OF SCHEMES..........................................................................................................xv
LIST OF NMRS .............................................................................................................. xvii
CHAPTER 1: PALLADIUM ENOLATE TRANSFER VIA AN
INTRAMOLECULAR HECK REACTION .......................................................................1
1.1 Introduction ....................................................................................................................1
1.2 Palladium Enolates.........................................................................................................2
1.3 The Heck Reaction .........................................................................................................4
1.4 Synthesis of Ethyl (E)-5-(2-bromoacetoxy)-5-phenylpent-2-enoate and the
Attempts to Undergo an Intramolecular Heck Reaction ....................................................11
1.5 Conclusion ...................................................................................................................22
1.6 Supporting Information for Chapter 1 .........................................................................22

ix
1.7 References for Chapter 1 .............................................................................................27
CHAPTER 2: TOTAL SYNTHESIS OF DIHYDRORESORCYLIDE ...........................42
2.1 Introduction ..................................................................................................................42
2.2 Isolation and Structural Elucidation, Biological Properties and Reactions, and
Synthesis of Dihydroresorcylide ........................................................................................43
2.3 Retrosynthetic Analysis ...............................................................................................48
2.4 Total Synthesis of Dihydroresorcylide: Aromatic Synthon .........................................49
2.5 Total Synthesis of Dihydroresorcylide: Aliphatic Synthon .........................................54
2.6 Total Synthesis of Dihydroresorcylide: Combining Synthons ....................................56
2.7 Future Works ...............................................................................................................59
2.8 Conclusion ...................................................................................................................60
2.9 Supporting Information for Chapter 2 .........................................................................61
2.10 References for Chapter 2 ...........................................................................................69
CHAPTER 3: STEREOSELECTIVE HALO-SUCCINIMIDE FACILITATED α-
HALOGENATIONS OF SUBSTITUTED α-TRIALKYLSILYL-ß-SUBSTITUTED
-α,ß-UNSATURATED ESTERS ......................................................................................92
3.1 Introduction ..................................................................................................................92
3.2 The Generation of α-Trialkylsilyl-α, β-Unsaturated Esters .........................................94
3.3 The Halogenation Reaction ..........................................................................................96
3.4 (Z)-α-Halogen-α, β-Unsaturated Esters Compound Library ......................................100
3.5 Future Works .............................................................................................................105

x
3.6 Conclusion .................................................................................................................105
3.7 Supporting Information for Chapter 3 .......................................................................105
3.8 References for Chapter 3 ...........................................................................................112

xi
LIST OF TABLES
Table 1.1 Base Study with Pd(OAc)2 ................................................................................14
Table 1.2 Base study with PdCl2 and PPh3 .......................................................................16
Table 1.3 Base Study with PdCl2 and P(o-tolyl)3..............................................................16
Table 1.4 Solvent Study ....................................................................................................17
Table 1.5 Pd2(dba)3 Studies ...............................................................................................18
Table 1.6 Frank Glorius and Ionic Heck Conditions ........................................................19
Table 1.7 Chloroacetoxy Intramolecular Heck Attempts ..................................................20
Table 1.8 Studies with Compound 1.19 ............................................................................22
Table 1.9 Studies with Compounds 1.20 ..........................................................................22
Table 2.1 Triflate Coupling Experiments ..........................................................................50
Table 2.2 Triflate Coupling with Silyl Enol Ether ............................................................52
Table 2.3 Bromobenzene and Ethyl Bromobenzoate Coupling ........................................52
Table 2.4 Pd Enolate Macrocycle Formation ....................................................................59
Table 2.5 Silyl Enol Ether Conversion..............................................................................59
Table 3.1 Vinyl Silane Compound Library .......................................................................96

xii
Table 3.2 Solvent Dependence Studies .............................................................................97
Table 3.3 Bromination Compound Library .....................................................................101
Table 3.4 Chlorination Compound Library .....................................................................104

xiv
LIST OF FIGURES
Figure 1.1 Pyran Motif .................................................................................................................. 1
Figure 1.2 Bryostatin and Exiguolide ........................................................................................... 1
Figure 1.3 General Pd Enolate ...................................................................................................... 2
Figure 1.4 Formation of Metal Enolate ......................................................................................... 3
Figure 1.5 Metal Enolate Coordination......................................................................................... 3
Figure 1.6 Relative Stability of Pd Enolates ................................................................................. 4
Figure 1.7 Genaralized Mizoroki-Heck Reaction ......................................................................... 5
Figure 1.8 Cis/Trans Isomerization ............................................................................................... 8
Figure 1.9 Example of 6-Endo Cylization .................................................................................. 10
Figure 1.10 6-Exo Cyclization .................................................................................................... 10
Figure 1.11 Products Isolated Under Base Study With Pd(OAc)2 .............................................. 15
Figure 1.12 Product Isolated with Stoichiometric PdCl2 Catalyst .............................................. 18
Figure 1.13 Reference Heck Reaction ........................................................................................ 19
Figure 2.1 Resorcylic Acid Backbone of RALs ......................................................................... 42
Figure 2.2 Dihydroresorcylide .................................................................................................... 43

xiv
Figure 2.3 Culvularin .................................................................................................................. 44
Figure 2.4 Retrosynthetic Analysis of 2013 Synthesis ............................................................... 45
Figure 2.5 Weinreb Amides Utilized for Synthesis .................................................................... 46
Figure 2.6 Hydrogen Bound 2.24................................................................................................ 50
Figure 2.7 Side Product Formed From 2.26................................................................................ 51
Figure 2.8 Silyl Enol Ether Formation ........................................................................................ 51
Figure 2.9 Coupling with Silyl Enol Ether ................................................................................. 52
Figure 3.1 Example Reaction Studied by Tamao ....................................................................... 93
Figure 3.2 Versatility of Vinyl Silanes ....................................................................................... 94
Figure 3.3 General Halogenation Reaction ................................................................................. 94
Figure 3.4 Copper Facilitated Silyl Ketene Acetal Formation .................................................... 95
Figure 3.5 Sample Halogenation for Optimization ..................................................................... 97
Figure 3.6 Key NOE Interactions for 3.3a ................................................................................ 101
Figure 3.7 Key NOE Interactions for 3.3d................................................................................ 103
Figure 3.8 TES Bromination ..................................................................................................... 105

xv
LIST OF SCHEMES
Scheme 1.1 Generalized Catalytic Cycle of Heck Reaction ......................................................... 6
Scheme 1.2 Preactivation of Pd Catalyst ....................................................................................... 6
Scheme 1.3 Synthesis of ethyl (Z)-2-(2-oxo-6-phenylthetrahydro-4H-pyran-4-ylidene)acetate 12
Scheme 1.4 Proposed Catalytic Cycle for Intramolecular Heck Cyclization .............................. 13
Scheme 1.5 Synthesis of 4-methylene-6-phenyltetrahydro-2H-pyran-2-one .............................. 21
Scheme 2.1 Total Synthesis of Dihydroresorcylide Published in 2013 ...................................... 46
Scheme 2.2 Total Synthesis of Dihydroresorcylide Published in 2017 ...................................... 47
Scheme 2.3 Retrosynthetic Analysis of 2.1 ................................................................................. 49
Scheme 2.4 Synthetic Pathway 1 of Aromatic Synthon .............................................................. 50
Scheme 2.5 Synthetic Pathway 2 of Aromatic Synthon .............................................................. 53
Scheme 2.6 Synthetic Pathway 3 of Aromatic Synthon .............................................................. 54
Scheme 2.7 Grignard Method of Synthetic Pathway 1 for Aliphatic Formation ........................ 55
Scheme 2.8 Oxidation Method of Synthetic Pathway 1 for Aliphatic Formation....................... 55
Scheme 2.9 Combination Method of Synthetic Pathway 1 for Aliphatic Formation .................. 55
Scheme 2.10 Synthetic Pathway 2 for Aliphatic Formation ....................................................... 56

xvi
Scheme 2.11 Synthesis of Dihydroresorcylide Via Pathway 2 ................................................... 57
Scheme 2.12 Synthesis of Dihydroresocylide Via Pathway 3 .................................................... 60
Scheme 3.1 Hypothesized Mechanism of Bromination .............................................................. 99
Scheme 3.2 Possible Mechanism of 3.3d .................................................................................. 103

xvii
LIST OF NMRS
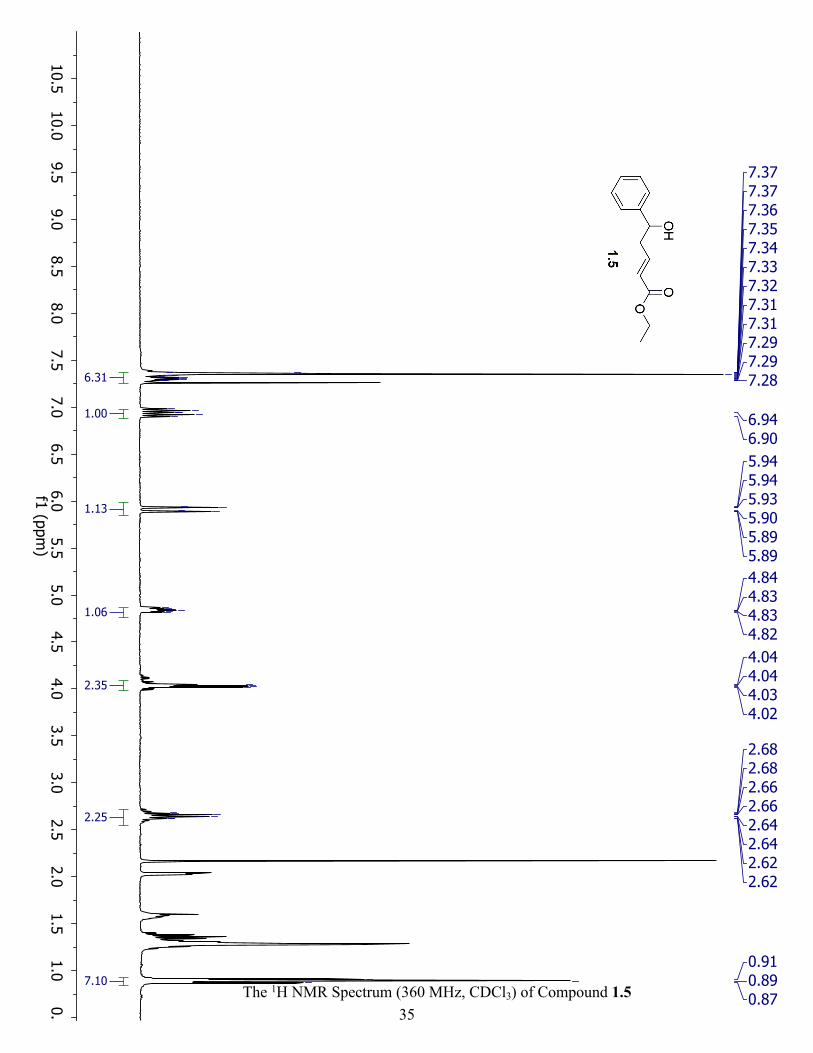
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.3 ................................................... 34
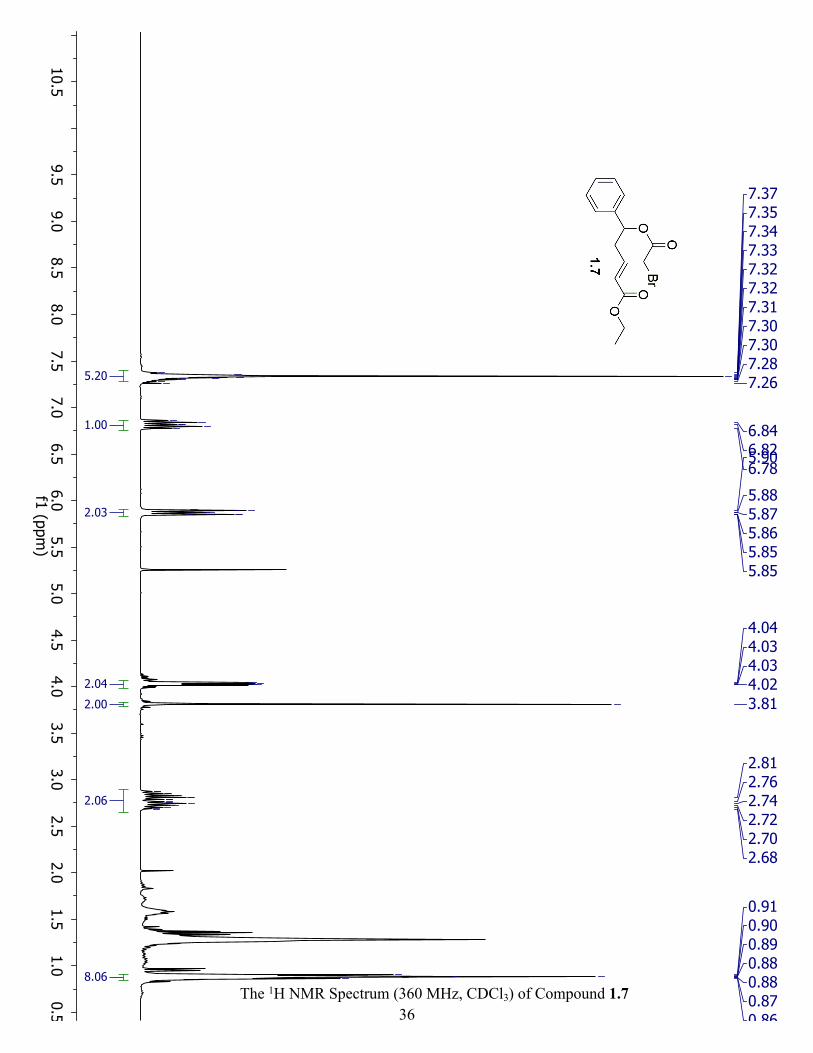
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.5 ................................................... 35
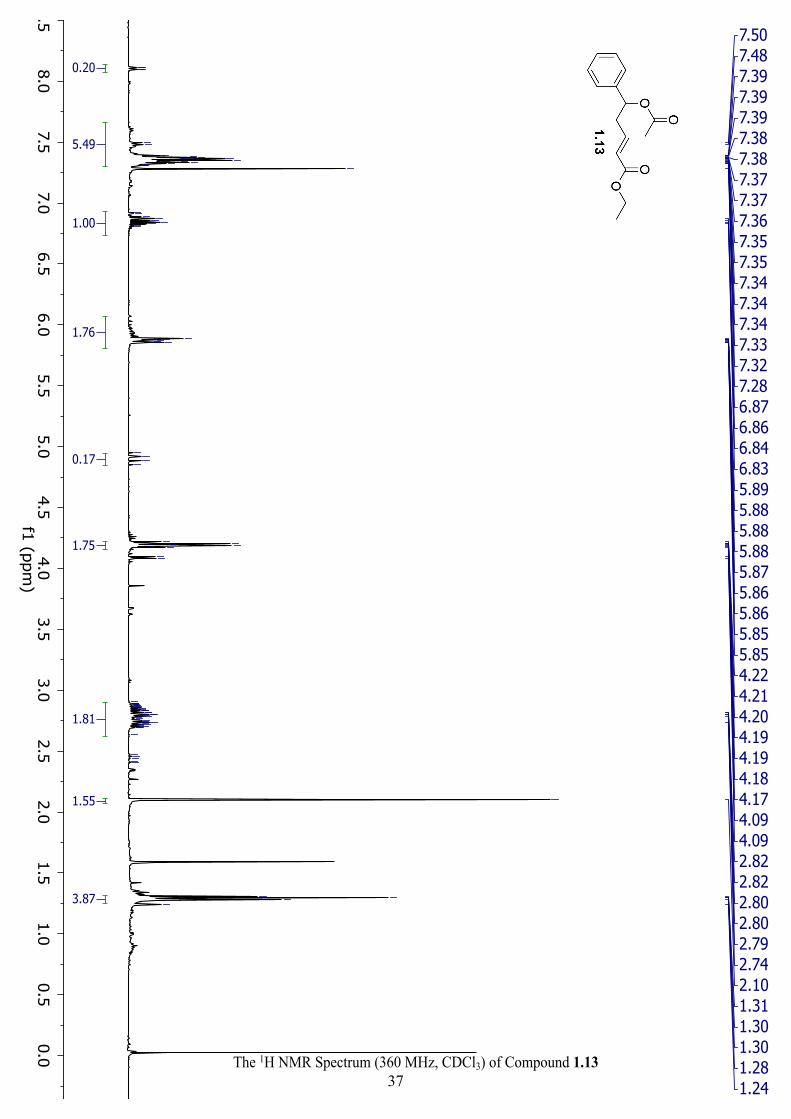
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.7 ................................................... 36
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.13 ................................................. 37
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.17 ................................................. 38
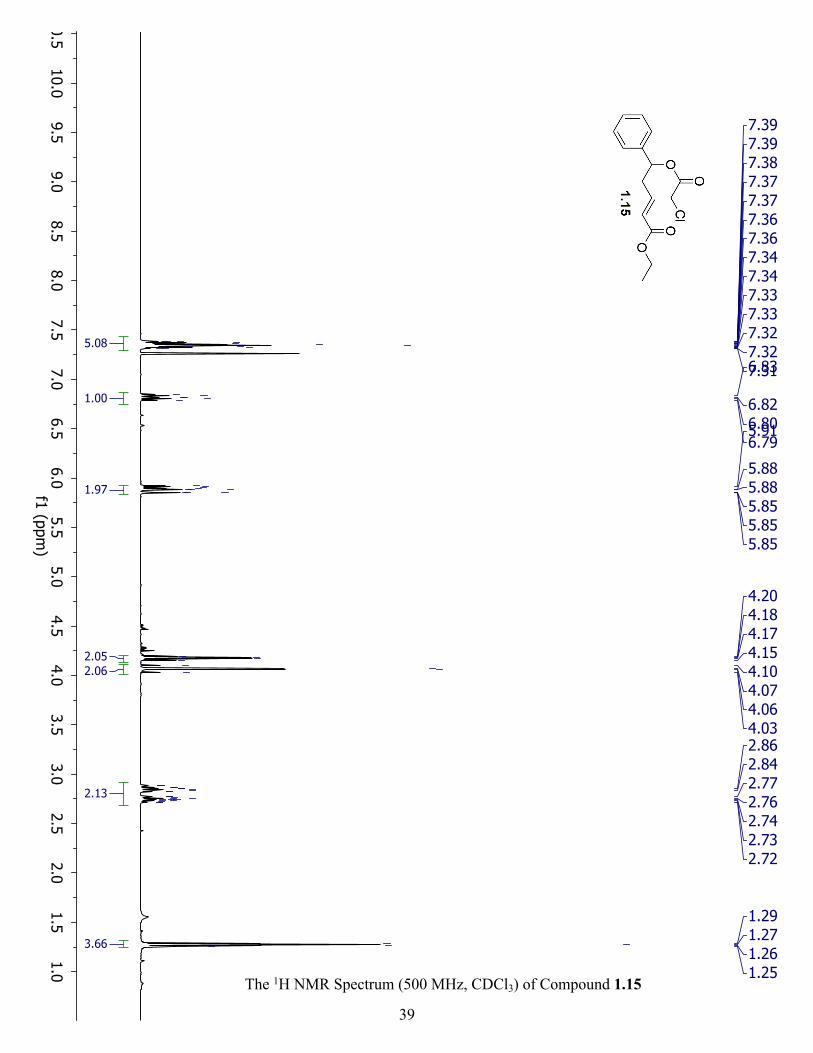
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.15 ................................................. 39
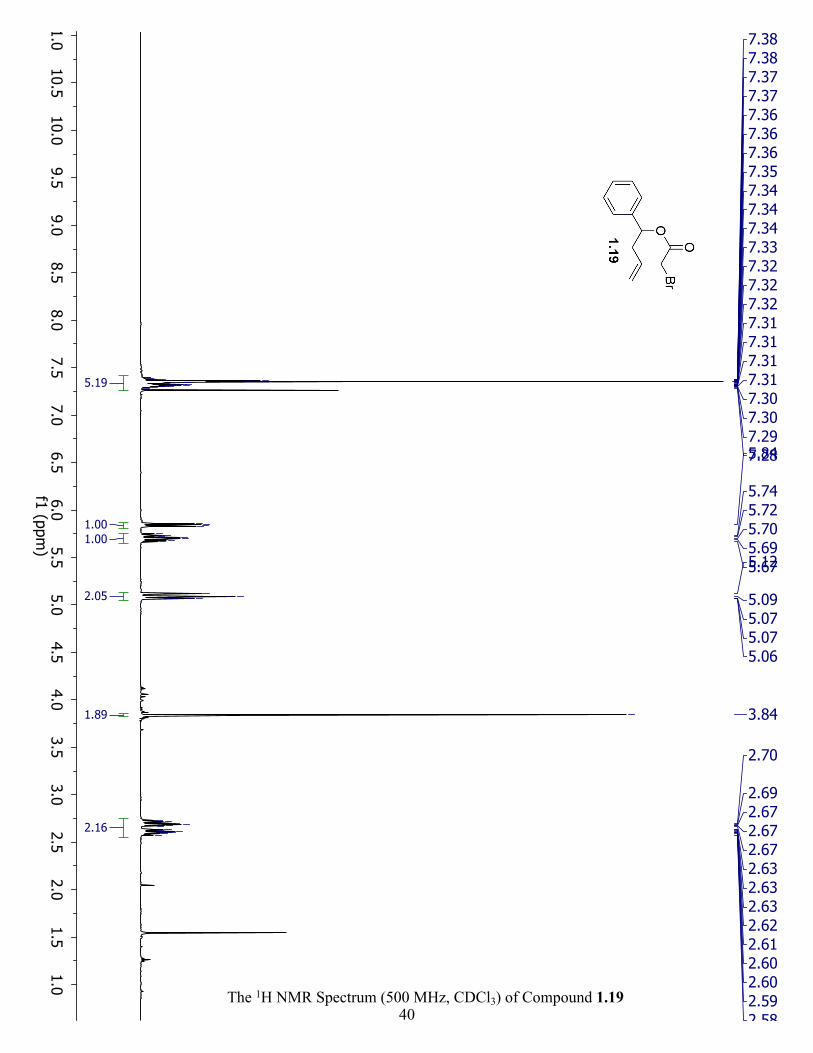
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.19 ................................................. 40
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.20 ................................................. 41
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.25 ................................................. 73
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.26 ................................................. 74
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.27 ................................................. 75
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.28 ................................................. 76
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.31 ................................................. 77
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.33 ................................................. 78
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.34 ................................................. 79

xviii
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.35 ................................................. 80
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 2.35 ................................................ 81
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.20 ................................................. 82
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 2.20 ................................................ 83
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.20, A ............................................ 84
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.22 ................................................. 85
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.39 ................................................. 86
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.40 ................................................. 87
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.23 ................................................. 88
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.41 ................................................. 89
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.21 ................................................. 90
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.42 ................................................. 91
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2a ............................................... 115
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2b ............................................... 116
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2c ............................................... 117
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2d ............................................... 118
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2e ............................................... 119
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2f ................................................ 120

xix
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2g ............................................... 121
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.2g .............................................. 122
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2h ............................................... 123
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2i ................................................ 124
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.2i ............................................... 125
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3a ............................................... 126
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3a .............................................. 127
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3b ............................................... 128
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3c ............................................... 129
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3c .............................................. 130
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3d ............................................... 131
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3d .............................................. 132
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3e ............................................... 133
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3f ................................................ 134
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3g ............................................... 135
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3g .............................................. 136
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3h ............................................... 137
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3h .............................................. 138

xx
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4a ............................................... 139
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4b ............................................... 140
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.4b .............................................. 141
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4c ............................................... 142
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.4c .............................................. 143
1
CHAPTER 1: PALLADIUM ENOLATE TRANSFER VIA AN INTRAMOLECULAR HECK
REACTION
1.1 Introduction
Development of highly functionalized small molecules to serve as intermediates in synthesis
of natural products, or other biologically active macromolecules, is desirable. The pyran motif
shown in Figure 1.1 is exhibited in the class of biological compounds called Bryostatin and (-)
Exiguolide (Figure 1.2) and will be the focus of this research.1, 2
Pd enolates (Figure 1.3) have been utilized to undergo various transformations3, 4 and often
an enolate intermediate is proposed with both intermolecular and intramolecular Heck reaction

2
takes place.5, 6 However, a Pd enolate transfer is novel when introduced into an intramolecular
Heck reaction.
The following research examines the ability to perform a Pd enolate transfer via an
intramolecular Heck reaction with halogenated acetoxy α, β unsaturated esters and their olefin
analogs.
1.2 Palladium Enolates
Pd enolates have found their way into use for several decades. It has been reported that there
are three known ways to create a metal enolate as shown in Figure 1.4.7 The first way, A,
involves a metal anion displacement which is frequently shown through a transmetallation of a
variety of transition metals such as silver, or copper.8, 9 In part B, allyl coordination is visible by
removal of a halide on the Pd complex, for example, a PdCl2 has shown to undergo this
transformation.10 The final way, C, focuses on silyl enolates. Silyl enolate, similar to transition
metals, experience alteration through transmetallation.11 Upon formation of the metal enolate
complex, there are now four ways in which the Pd would coordinate.

3
Figure 1.5 details the ways in which Pd enolate coordination can occur.12 Carbon
coordination as shown in A, is the first method of coordination.13 2. B, exhibits the formation of
an oxoallyl species.8, 14 3. The formation of the oxygen coordinated enolate is shown in C.15 D
exhibits bridging as the final potential species in enolate coordination.16 The bonding to the
metal center can be influenced by a variety of factors.
According to Culkin and Hartwig, the coordination of the metal enolate is dependent on a
variety of factors from electronics to ligands. After performing NMR studies, the ketones with α-

4
methyl or methylene hydrogens and a bidentate phosphine ligand were all bound to the carbon
whereas α-methine protons were oxygen bound. Figure 1.6 summarizes the bonding and stability
of these interactions. Electronically, the carbon is favored when there is a phosphine trans on the
metal complex while oxygen is favored when there is an aryl ligand in the trans position.17
Pd enolates have been studied and are utilized in many different processes from initiation of
homocoupling,18 enantioselective Michael reactions,19 intramolecular ring formations,20, 21
formation of quaternary centers,22, 23 and synthesis.3, 4 The usefulness of these groups has
expanded organometallic coupling like that of the Heck reaction.
Olefins containing carbonyls have been found to produce a Pd enolate during a Heck
reaction under various conditions.6, 24, 25 However, despite all the research over these topics, a Pd
enolate has not been generated first and then treated to form a second enolate from the olefin.
The development of the intermolecular and intramolecular Heck reaction will be discussed in the
following section.
1.3 The Heck Reaction
The Mizoroki-Heck reaction, or Heck reaction, was founded by Tsutomu Mizoroki and
Richard Heck. The catalytic process involves cross coupling of an unsaturated halide and various
substituted olefins as shown in Figure 1.7.26

5
The proposed catalytic cycle goes through preactivation, oxidative addition, migratory
insertion, and hydride elimination shown in Scheme 1.1. Preactivation (Scheme 1.1, A) is the
reduction of the Pd, typically a Pd(II), into an active species, Pd(0), via removal of a ligand like
phosphine with assistance of a hard nucleophile like water.27-29 Phosphines are common ligands
utilized in cross coupling reactions and it will reduce to yield a phosphine oxide, while dba,
another ligand frequented in literature, will not undergo an oxide formation.30, 31 It is believed
that the nucleophile can either perform a nucleophilic substitution with the ligand on the metal
complex (outer shell mechanism), or the species will coordinate to the metal complex and go
through a reductive elimination (inner shell mechanism). The following scheme (Scheme 1.2)
exhibits a basic example of the activation. However, it was found that an excess of ligand can
hinder this step as it will not allow for the catalyst to remain active, which has shown to halt the
coupling from occurring.32

6
Oxidative addition (Scheme 1.1, B) follows preactivation and involves the active Pd catalyst
inserting itself in a carbon-halogen bond via a concerted process where all bonds that are broken
and formed happen at the same time ultimately changing the oxidation state of the Pd.33 The
halogen reactivity is as follows: I>>OTf>Br>>Cl showing that iodine is significantly more
reactive than other halogens and chlorine is the least likely to have bond insertion transpire.34
Cis/trans isomerization will occur and although it was thought to be a simpler process, studies

7
were conducted by Arturo and workers finding that there are in fact several pathways this can go
through.
These extensive studies showed that there are multiple associative and dissociative means to
go from the less stable cis isomer to the more stable trans. Figure 1.8 illustrates the findings of
this research. It should be noted that the solvent coordination pathway shown on the right side of
the figure can occur in the same manner with a metal halide complex. After insertion cis/trans
isomerization takes place oxidative addition is complete and the rate determining migratory
insertion takes place.35
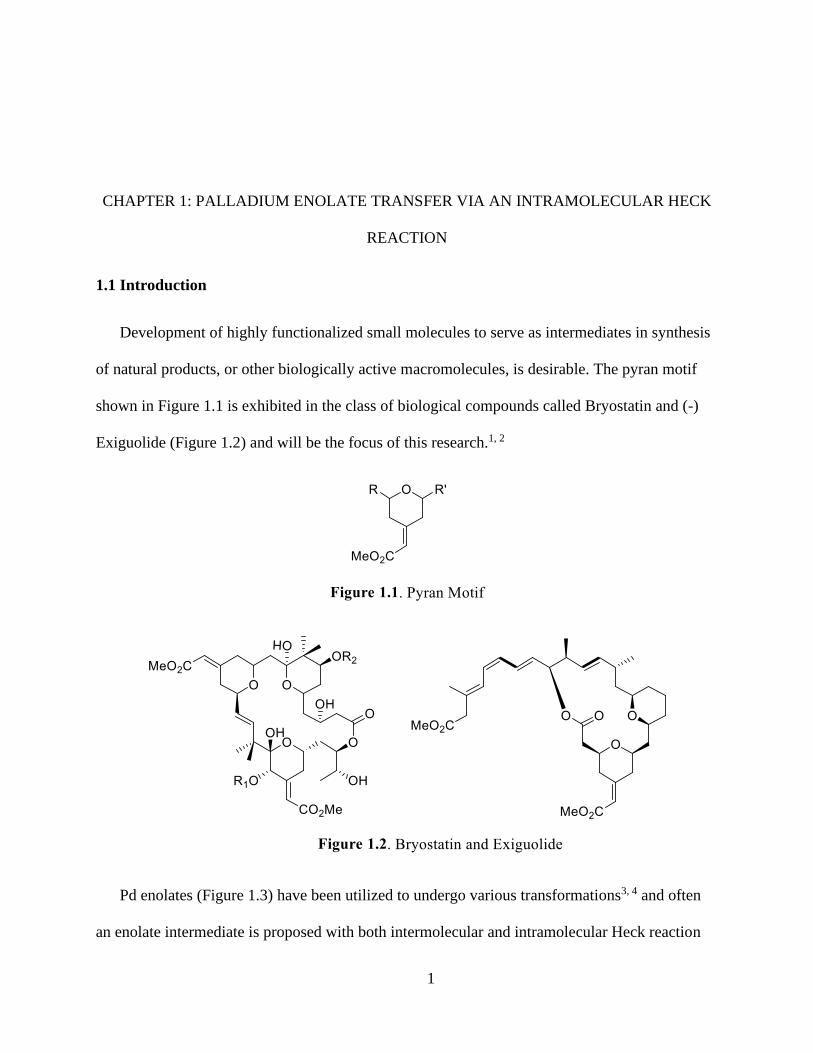
8
Unlike other cross couplings, migratory insertion is the bond forming step in the Heck
reaction shown in Scheme 1.1, C. This step can often exhibit the generation of a Pd enolate and
as discussed earlier olefin type, ligands, and cone angles all influence the connectivity at this
stage.17 There are three different pathways by way this can proceed; 1. The Pd halide complex

9
will act as a carbanion where the insertion is similar to a vinylic nucleophilic substitution.33 2.
The attack can happen with neutral or cationic catalyst systems and proceeds through a classic
electrophilic addition which is most supported by literature. 3. The insertion happens via a
concerted SN2 addition.36-38 In order for the attack and bond formation to occur, a ligand needs to
be lost and can be either neutral or ionic. Monodenate ligands tend to follow the neutral pathway
while multidentate ligands tend to be ionic.39-41
Electronics also play a role in migratory insertion. An electron rich olefin can go through
both neutral and ionic intermediates for the Pd attack. The Pd will bind to the atom with the
highest electron density. While steric effects can override the outcome of couplings, electronics
will still dominate in intermolecular Heck reactions.42-47
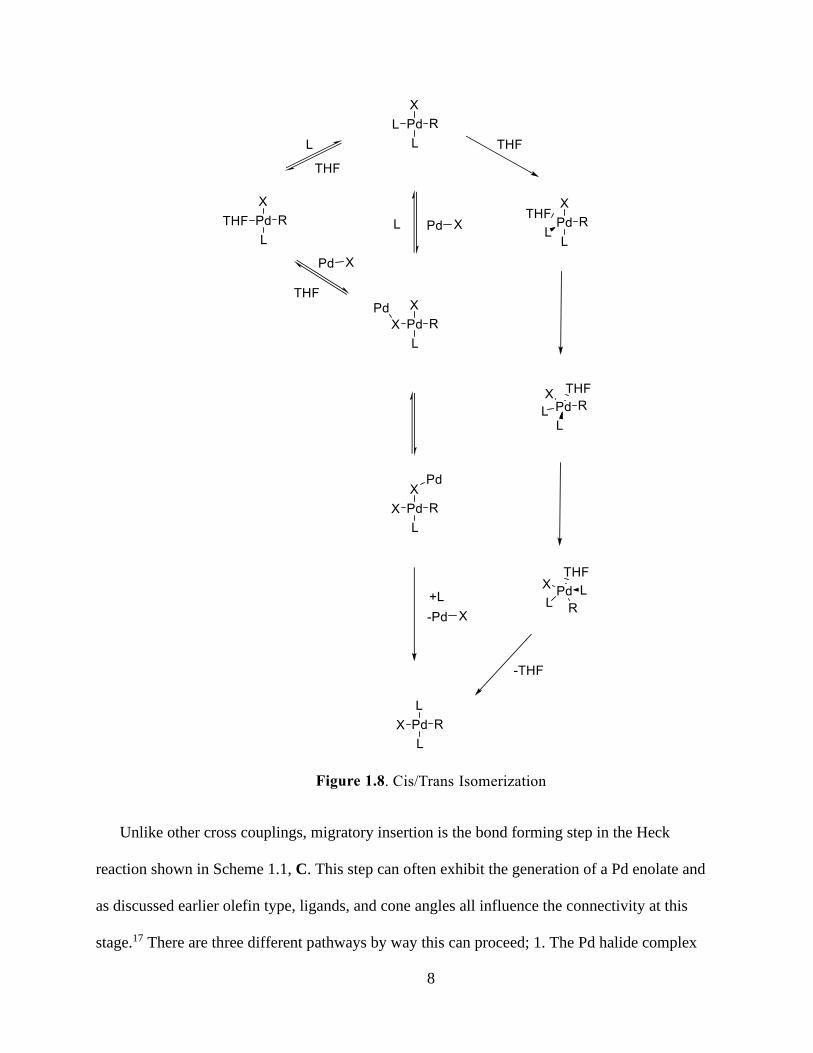
Intramolecular Heck reactions follow the same catalytic cycle as the previously shown in the
intermolecular cycle (Scheme 1.1), however, steric effects will be the dominating factors of
migratory insertion. During intramolecular Heck reactions, the endo cyclization is typically not
present for smaller rings (5, 6, and 7) due to sites being sterically hindered. There have been
specific examples of a 6-endo cyclization transpiring under specific circumstances; however the
process did not occur via the 6-endo ring closure, but through a sequence of 5-exo trig followed
by 3-exo trig cyclizations (Figure 1.9).48-50 6-exo trig (Figure 1.10) intramolecular Heck
reactions are most common cyclizations found.51-54 This cyclization is desirable in the formation
of the 6-exo ring crucial to the research presented herein.
10
Hydride elimination is the final step of the catalytic cycle which removes the Pd via a
concerted syn elimination to afford the final alkene.55, 56 The newly formed Pd hydride complex
must be quickly scavenged by a base to avoid readdition to the olefin which would ultimately
give the wrong stereochemistry. Bases can also influence where the elimination will occur
whether internal or terminal due to availability of proton sources.57
As shown in Scheme 1.1, the E isomer predominates as the final alkene. Again, electronics
can also influence this outcome. The more electron rich the olefin, the more likely only the E
configuration will be isolated.17

11
While the Heck reaction has been widely studied, and Pd enolates can occur throughout this
process, there is no current literature presence of a Pd enolate transfer taking place to form a six
membered exo cycle. The research conducted regarding this novel venture follows.
1.4 Synthesis of Ethyl (E)-5-(2-bromoacetoxy)-5-phenylpent-2-enoate and the Attempts to
Undergo an Intramolecular Heck Reaction
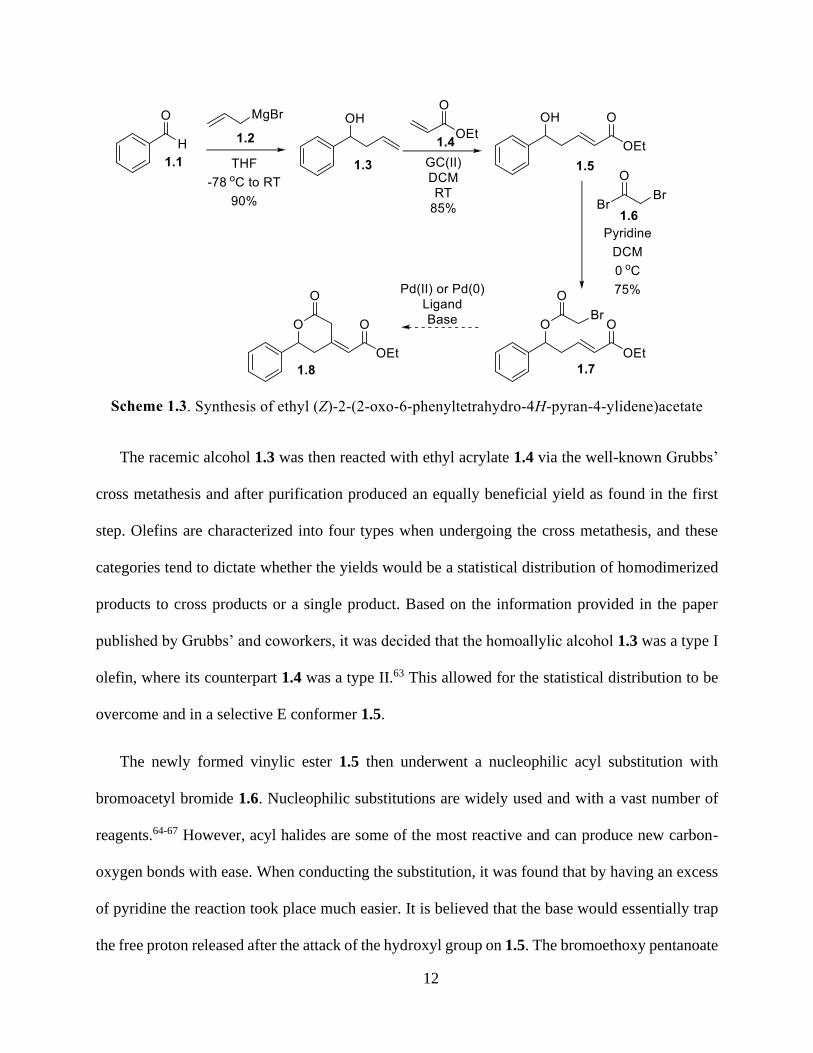
Development of the targeted ring product 1.8 required a four step, linear synthetic pathway as
shown in Scheme 1.3. In the first step benzaldehyde 1.1 underwent a Grignard reaction with an
allylmagnesium bromide 1.2 to afford the homoallylic alcohol 1.3 in high yields. Grignard
reactions undergo nucleophilic attack of the carbonyl species present and are frequently utilized in
organic synthesis due to the high yields ultimately produced.58-62 It was found that when
conducting the aforementioned reaction, if the temperature was not maintained for a certain period
of time or if the Grignard reagent was not added dropwise, there would be a decrease of yield.
However, the product did not require any purification other than longer time on the vacuum to
remove excess solvent which also proved desirable.
12
The racemic alcohol 1.3 was then reacted with ethyl acrylate 1.4 via the well-known Grubbs’
cross metathesis and after purification produced an equally beneficial yield as found in the first
step. Olefins are characterized into four types when undergoing the cross metathesis, and these
categories tend to dictate whether the yields would be a statistical distribution of homodimerized
products to cross products or a single product. Based on the information provided in the paper
published by Grubbs’ and coworkers, it was decided that the homoallylic alcohol 1.3 was a type I
olefin, where its counterpart 1.4 was a type II.63 This allowed for the statistical distribution to be
overcome and in a selective E conformer 1.5.
The newly formed vinylic ester 1.5 then underwent a nucleophilic acyl substitution with
bromoacetyl bromide 1.6. Nucleophilic substitutions are widely used and with a vast number of
reagents.64-67 However, acyl halides are some of the most reactive and can produce new carbon-
oxygen bonds with ease. When conducting the substitution, it was found that by having an excess
of pyridine the reaction took place much easier. It is believed that the base would essentially trap
the free proton released after the attack of the hydroxyl group on 1.5. The bromoethoxy pentanoate
13
1.7 was formed in moderate yields.68 Upon purification it was found that there was starting material
still present which could have potentially been avoided by increasing the equivalents of pyridine;
however, the yield consistently given was proficient enough to begin development of the novel
intramolecular Heck reaction via Pd enolates by formation of the desired exocyclic alkene 1.8.
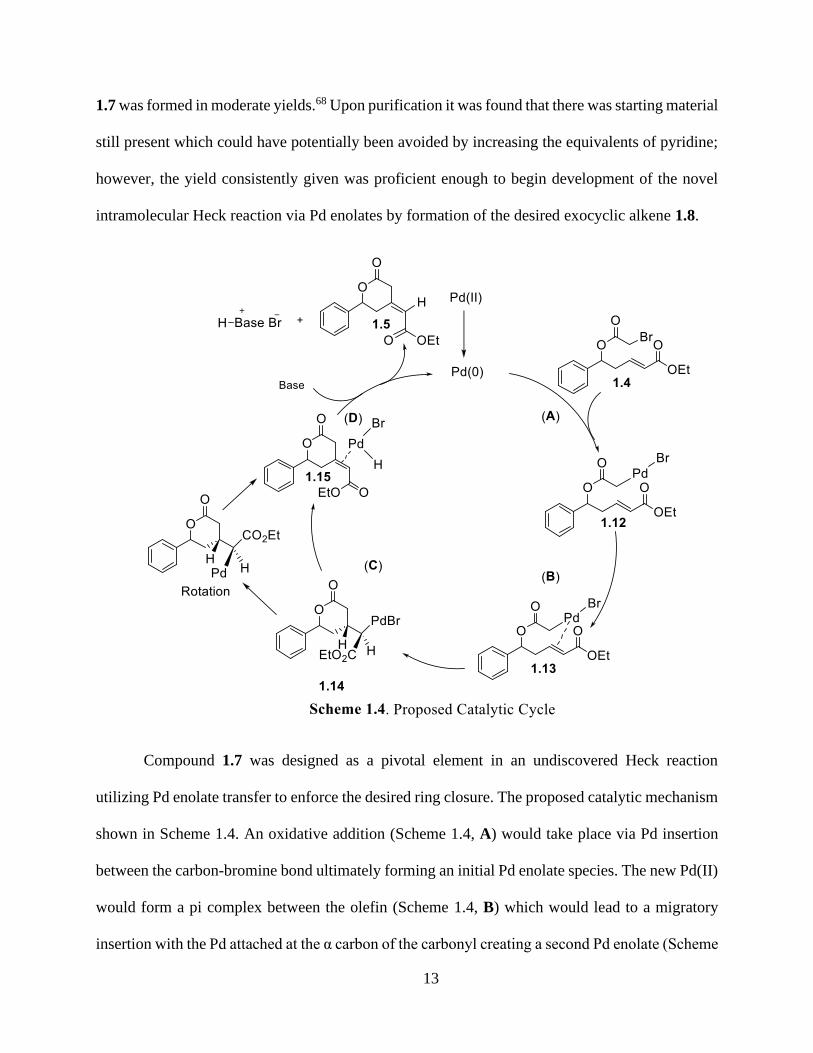
Compound 1.7 was designed as a pivotal element in an undiscovered Heck reaction
utilizing Pd enolate transfer to enforce the desired ring closure. The proposed catalytic mechanism
shown in Scheme 1.4. An oxidative addition (Scheme 1.4, A) would take place via Pd insertion
between the carbon-bromine bond ultimately forming an initial Pd enolate species. The new Pd(II)
would form a pi complex between the olefin (Scheme 1.4, B) which would lead to a migratory
insertion with the Pd attached at the α carbon of the carbonyl creating a second Pd enolate (Scheme

14
1.4, C). This species would undergo the highly important β-hydride and reductive elimination
(Scheme 1.4, D) to form the desired alkene 1.8. This theoretical cycle would continue in this
manner until the starting material was used up. Due to the ring closure generating a compound that
is six membered, it can be assumed that the Pd would not react with the structure any further as it
would be stable.
The bromoethoxy pentanoate 1.7 underwent reactions with conditions that were similar to
that found in literature for Heck reactions both inter and intramolecularly.6, 69-71 Table 1.1 shows
that 1.8 was subjected to a series of reactions under specific conditions, where the catalyst, ligand,
and solvent were Pd(OAc)2 at 0.03 equivalents, P(o-tolyl)3 at 0.09 equivalents, and toluene
respectively. The base varied because of relative strength of the base, size, and frequency of the
material found in literature. In entry 1-3, K2CO3, K3PO4, and NaHCO3 were added into the reaction
system because these are shown to be utilized a great deal when researching desirable conditions
and they are noncoordinating with the catalyst. Both entries 4 and 5 exploited the bulkiness of the
two amines TEA and Hunig’s base. Despite the use of literature-based circumstances, a series of
unwanted products were isolated.
Entry Catalyst (0.03 eq) Ligand (0.09 eq) Base (5.0 eq) Solvent Product
1 Pd(OAc)2 P(o-tol)3 K2CO3 Toluene 1.7, 1.14
2 Pd(OAc)2 P(o-tol)3 K3PO4 Toluene 1.7, 1.14
3 Pd(OAc)2 P(o-tol)3 NaHCO3 Toluene 1.7, 1.14
4 Pd(OAc)2 P(o-tol)3 TEA Toluene 1.13, 1.14
5 Pd(OAc)2 P(o-tol)3 Hunig’s Toluene 1.13, 1.14
Table 1.1 Base Study with Pd(OAc)2
Compound 1.13 (Figure 1.10) was the material collected when the amine bases were
introduced. The acetyl group formation was caused by the fact that there are β hydrogens present
on the base which allowed for a β hydrogen elimination to occur readily. Therefore, it was decided

15
to steer away from these types of hydrogen sources. While the salt like bases in entries 1-3 did not
show any adverse results, it was found that an SN2 type reaction between the acetate group of
Pd(OAc)2 and the bromoethoxy pentanoate 1.7 giving rise to 1.14 (Figure 1.11). Due to the side
reactions taking place with the catalyst and bases, it was decided to change the catalyst to Pd(Cl)2.
Table 1.2 shows a second series of base studies conducted and a change of ligand to PPh3
occurred. It was thought that potentially the methyl group on P(o-tolyl)3 pushed the cone angle
larger which could have affected coordination to the catalyst. The bases in entries 1-3 were chosen
because of the lack of additional hydrogens available to cause unfavorable side reactions.
NaH2PO4 was chosen in entry 4 despite having an available hydrogen because the hydride source
is not on the ß-phosphine thus rendering the ß-hydride elimination not possible. Entry 5 utilized
imidazole, which would be an amine base source that would not cause the β-hydride elimination
previously illustrated because of the hydride sources already being on sp2 carbon centers. The
material recovered was starting material 1.7 with a negligible amount of what appeared to be
desired product. However, recovery of the compound proved futile and it was reasoned that an
increase in catalyst loading may help improve the yield and allow for isolation of the questionable
species.

16
Entry Catalyst (0.03 eq) Ligand (0.09 eq) Base (5.0 eq) Solvent Product
1 Pd(Cl)2 PPh3 K3PO4 Toluene 1.7
2 Pd(Cl)2 PPh3 K2PO4 Toluene 1.7
3 Pd(Cl)2 PPh3 K2CO3 Toluene 1.7
4 Pd(Cl)2 PPh3 NaH2PO4 Toluene 1.7
5 Pd(Cl)2 PPh3 Imidazole Toluene 1.7
Table 1.2. Base Study with PdCl2 and PPh3
In Table 1.3 the catalyst remained Pd(Cl)2, however the equivalents was increased to 0.09
and the ligand was changed back to P(o-tolyl)3 since there was so significant improvements
concerning PPh3. Entries 1-3 were potassium and sodium species chosen yet again for the inability
to coordinate to the catalyst or give up hydrogens for unfavorable reactions. NaOH, as shown in
entry 4, was utilized because of the strong basicity exhibited by the compound in solution. It was
thought that the system could benefit from a stronger base in order to help regenerate the Pd
catalyst drive the reaction to completion. Entry 5 lists pyridine as a base of choice despite having
β hydrogens. However, these hydrogens are not likely to be removed due an unfavorable formation
of an alkyne species. All entries, except for 4, produced arguably similar results as the previous
study where potential product was formed but in incredibly small amounts. Entry 4 underwent a
nucleophilic substitution and was reduced back to compound 1.5. It was then decided to set base
studies aside and focus on solvent effects.
Entry Catalyst (0.09 eq) Ligand (0.09 eq) Base (5.0 eq) Solvent Product
1 Pd(Cl)2 P(o-tol)3 K2CO3 Toluene 1.7
2 Pd(Cl)2 P(o-tol)3 K3PO4 Toluene 1.7
3 Pd(Cl)2 P(o-tol)3 NaHCO3 Toluene 1.7
4 Pd(Cl)2 P(o-tol)3 NaOH Toluene 1.7
5 Pd(Cl)2 P(o-tol)3 Pyridine Toluene 1.7
Table 1.3. Base Study with PdCl2 and P(o-tolyl)3
17
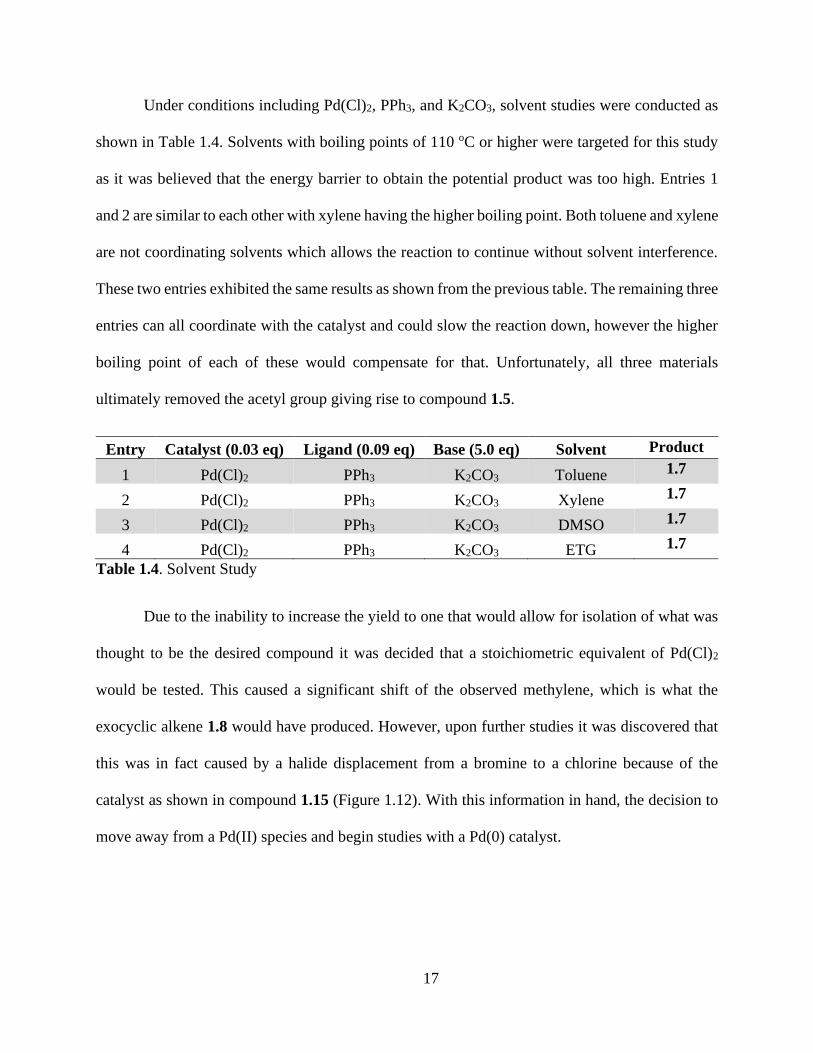
Under conditions including Pd(Cl)2, PPh3, and K2CO3, solvent studies were conducted as
shown in Table 1.4. Solvents with boiling points of 110 oC or higher were targeted for this study
as it was believed that the energy barrier to obtain the potential product was too high. Entries 1
and 2 are similar to each other with xylene having the higher boiling point. Both toluene and xylene
are not coordinating solvents which allows the reaction to continue without solvent interference.
These two entries exhibited the same results as shown from the previous table. The remaining three
entries can all coordinate with the catalyst and could slow the reaction down, however the higher
boiling point of each of these would compensate for that. Unfortunately, all three materials
ultimately removed the acetyl group giving rise to compound 1.5.
Entry Catalyst (0.03 eq) Ligand (0.09 eq) Base (5.0 eq) Solvent Product
1 Pd(Cl)2 PPh3 K2CO3 Toluene 1.7
2 Pd(Cl)2 PPh3 K2CO3 Xylene 1.7
3 Pd(Cl)2 PPh3 K2CO3 DMSO 1.7
4 Pd(Cl)2 PPh3 K2CO3 ETG 1.7
Table 1.4. Solvent Study
Due to the inability to increase the yield to one that would allow for isolation of what was
thought to be the desired compound it was decided that a stoichiometric equivalent of Pd(Cl)2
would be tested. This caused a significant shift of the observed methylene, which is what the
exocyclic alkene 1.8 would have produced. However, upon further studies it was discovered that
this was in fact caused by a halide displacement from a bromine to a chlorine because of the
catalyst as shown in compound 1.15 (Figure 1.12). With this information in hand, the decision to
move away from a Pd(II) species and begin studies with a Pd(0) catalyst.

18
Shown in Table 1.5, the Pd(0) catalyst that was employed was Pd2(dba)3. This was a bulkier
catalyst and was already in the active state making the compound air sensitive which meant great
care was taken to complete set up. The ligand of choice was for entries 1-3 was PPh3 and toluene
was the solvent system due to lack of decomposition on the starting material. K2CO3, DIPEA, and
pyridine were the bases that were studied. Due to the change in the catalyst, the β-hydride
elimination to form an acetyl side product did not occur as it had previously. All three entries
provided either starting material, compound 1.5, or a mixture of both. In entry 4, P(o-tolyl)3 was
exchanged with PPh3 to see if any positive results could be discerned and unfortunately this was
not the case.
Entry Catalyst (0.03 eq) Ligand (0.09 eq) Base (5.0 eq) Solvent Product
1 Pd2(dba)3 PPh3 K2CO3 Toluene 1.7
2 Pd2(dba)3 PPh3 Hunig’s Toluene 1.7
3 Pd2(dba)3 PPh3 Pyridine Toluene 1.7
4 Pd2(dba)3 P(o-Tol)3 Hunig’s Toluene 1.7
Table 1.5. Pd2(dba)3 Studies
Frank Glorius and coworkers published work concerning similar compounds. Due to this
literature material, the conditions listed were followed exactly (see Table 1.6) in an attempt to
obtain any usable products.72 Again, only compound 1.5 was recovered. Entry 2 shows the
conditions found in literature for conducting an ionic Heck reaction, which follows the same
pathway as that of a neutral Heck reaction but tends to be more reactive with pi complexes.73 The

19
only material recovered after experiment was starting material 1.5 and pure silver pellets. At this
point, the technique of setting up these reactions came into question.
Catalyst Equiv. Ligand Equiv. Base Equiv. Solvent Product
1 Pd(PPh3)2Cl2 0.05 P(o-Tol)3 0.09 Hunig’s 1.5 Acetonitrile 1.7
2 Pd(TFA)2 0.1 P(o-Tol)3 0.09 Ag2CO3 2 1,4-dioxane 1.7
Table 1.6. Frank Glorius and Ionic Heck Conditions
A reference Heck reaction shown in Figure 1.13 was conducted using iodobenzene 1.16
and ethyl acrylate 1.4. After purification the iodoester 1.17 was successfully isolated and in
moderate yields. Because of the positive outcome, it was concluded that technique was not the
hindering the formation of the exocyclic alkene 1.8. The question arose if the Pd insertion between
the carbon-bromine bond was occurring due to the inability to recover any material other than that
of starting material or compound 1.5.
With that question in mind, the chloroethoxy pentanoate analog was synthesized. The
theory is that potentially the bromine atom was too large to allow the insertion, therefore with a
smaller atom the Pd catalyst could successfully insert for the oxidative addition. The synthetic
pathway remained the same with the exchange of bromoacetyl bromide 1.6 for chloroacetyl
chloride. Table 1.7 shows ligand studied conducted where monodentate phosphine ligands were
compared to a bidentate phosphine ligand. Bidentate ligands tend to be useful as they force the

20
bite angle of a catalyst to be set parameters, often times driving the cycle forward. Unfortunately,
none of entries 1-3 yielded any material other than starting material or compound 1.5. Entry 4
shows the conditions for the published article by Frank Glorius and coworkers, which also proved
to be futile.
Catalyst Equiv. Ligand Equiv. Base Equiv. Solvent Product
1 Pd2(dba)3 0.03 PPh3 0.09 Hunig’s 5 Toluene 1.5, 1.15
2 Pd2(dba)3 0.03 P(o-tol)3 0.09 Hunig’s 5 Toluene 1.5, 1.15
3 Pd2(dba)3 0.03 DPPE 0.09 Hunig’s 5 Toluene 1.5, 1.15
4 Pd(PPh3)2Cl2 0.05 P(o-tol)3 0.09 DIPEA 1.5 Acetonitrile 1.5, 1.15
Table 1.7. Chloroacetoxy Intramolecular Heck Attempts
Another theory arose believing that the ester group may not be allowing the reaction to
progress as desired either due to size, location, or possible coordination of the Pd. Further analogs
1.19 and 1.20 were developed and tested with this in mind.
The synthetic pathway to create analogs 1.19 and 1.20 is shown in Scheme 1.5. This
method is very similar to the generation of the original bromoethoxy penanoate (1.7) compound.
In the first step, benzaldehyde 1.1 underwent a Grignard reaction with allylmagnesium bromide
1.2 which again was successful with high yields and did not need purification. In the previous
synthetic strategy (Scheme 3), the second portion would be to conduct a cross metathesis; however,
the formation of the ester 1.5 would not be necessary for either analog. Therefore, the second step
consisted of reacting either bromoacetyl bromide or chloroacetyl chloride with 1.3 via a
nucleophilic substitution to afford both analogs. The yields of the substitutions were comparable
to the previous experiments.

21
The proposed catalytic cycle of derivatives 1.19 and 1.20 would be similar to that of the
original except the only Pd enolate to be formed would be during the oxidative addition when the
Pd insertion of the carbon-bromine bond occurred. While this would change the scope of the
project because the Pd enolate transfer would not be present, the information gathered could give
insight to assist in the understanding of the difficulties presented with compound 1.7.
Analog 1.19 underwent a series of experiments similar to that of the bromo and
chloroethoxy pentanoate shown in Table 1.8. In entries 1-2 monodentate ligands were compared
to determine if the cone angle would have a substantial effect on the products recovered. Bidentate
ligand, DPPE, was utilized shown in entry 3. All three ligand choices did not have any desirable
impact and removal of the acetyl group to afford compound 3 was the only recoverable product.
Due to those results, it was decided to attempt the reaction with a lower amount of base. Hunig’s
base was decreased from a total of five equivalents to two. However, compound 1.3 continued to
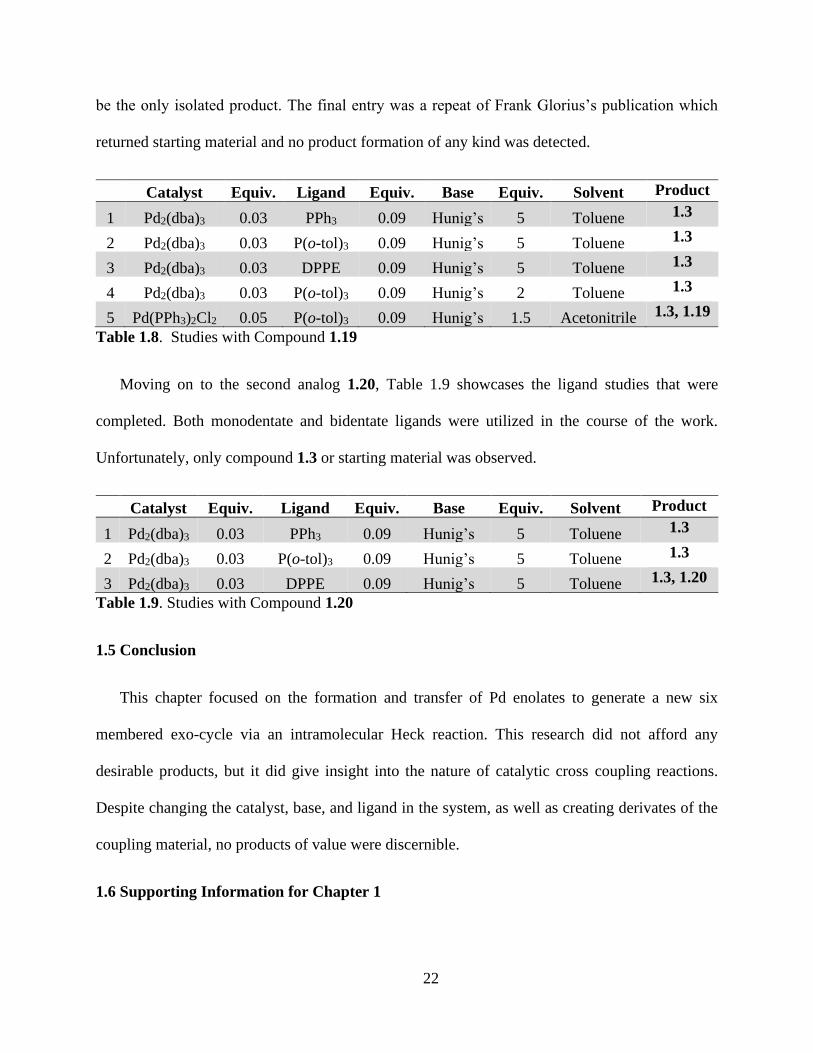
22
be the only isolated product. The final entry was a repeat of Frank Glorius’s publication which
returned starting material and no product formation of any kind was detected.
Catalyst Equiv. Ligand Equiv. Base Equiv. Solvent Product
1 Pd2(dba)3 0.03 PPh3 0.09 Hunig’s 5 Toluene 1.3
2 Pd2(dba)3 0.03 P(o-tol)3 0.09 Hunig’s 5 Toluene 1.3
3 Pd2(dba)3 0.03 DPPE 0.09 Hunig’s 5 Toluene 1.3
4 Pd2(dba)3 0.03 P(o-tol)3 0.09 Hunig’s 2 Toluene 1.3
5 Pd(PPh3)2Cl2 0.05 P(o-tol)3 0.09 Hunig’s 1.5 Acetonitrile 1.3, 1.19
Table 1.8. Studies with Compound 1.19
Moving on to the second analog 1.20, Table 1.9 showcases the ligand studies that were
completed. Both monodentate and bidentate ligands were utilized in the course of the work.
Unfortunately, only compound 1.3 or starting material was observed.
Catalyst Equiv. Ligand Equiv. Base Equiv. Solvent Product
1 Pd2(dba)3 0.03 PPh3 0.09 Hunig’s 5 Toluene 1.3
2 Pd2(dba)3 0.03 P(o-tol)3 0.09 Hunig’s 5 Toluene 1.3
3 Pd2(dba)3 0.03 DPPE 0.09 Hunig’s 5 Toluene 1.3, 1.20
Table 1.9. Studies with Compound 1.20
1.5 Conclusion
This chapter focused on the formation and transfer of Pd enolates to generate a new six
membered exo-cycle via an intramolecular Heck reaction. This research did not afford any
desirable products, but it did give insight into the nature of catalytic cross coupling reactions.
Despite changing the catalyst, base, and ligand in the system, as well as creating derivates of the
coupling material, no products of value were discernible.
1.6 Supporting Information for Chapter 1

23
General Procedure: All of the reactions were performed under Ar in flame-dried glassware. All
starting materials, solvents, reagents, and catalysts were commercially available and used
without further purification. The NMR spectra were recorded with either a 360 or 500 MHz
Bruker spectrometer. 1H and 13C NMR spectra were obtained using CDCl3 as the solvent with
chloroform (CHCl3 1H: δ = 7.26 ppm, CDCl3
13C: δ = 77.0 ppm) as the internal standard.
Column chromatography was performed using 60-200 µm silica gel. Analytical thin layer
chromatography was performed on silica coated glass plates with F-254 indicator. Visualization
was accomplished by UV light (254 nm) and KMnO4.
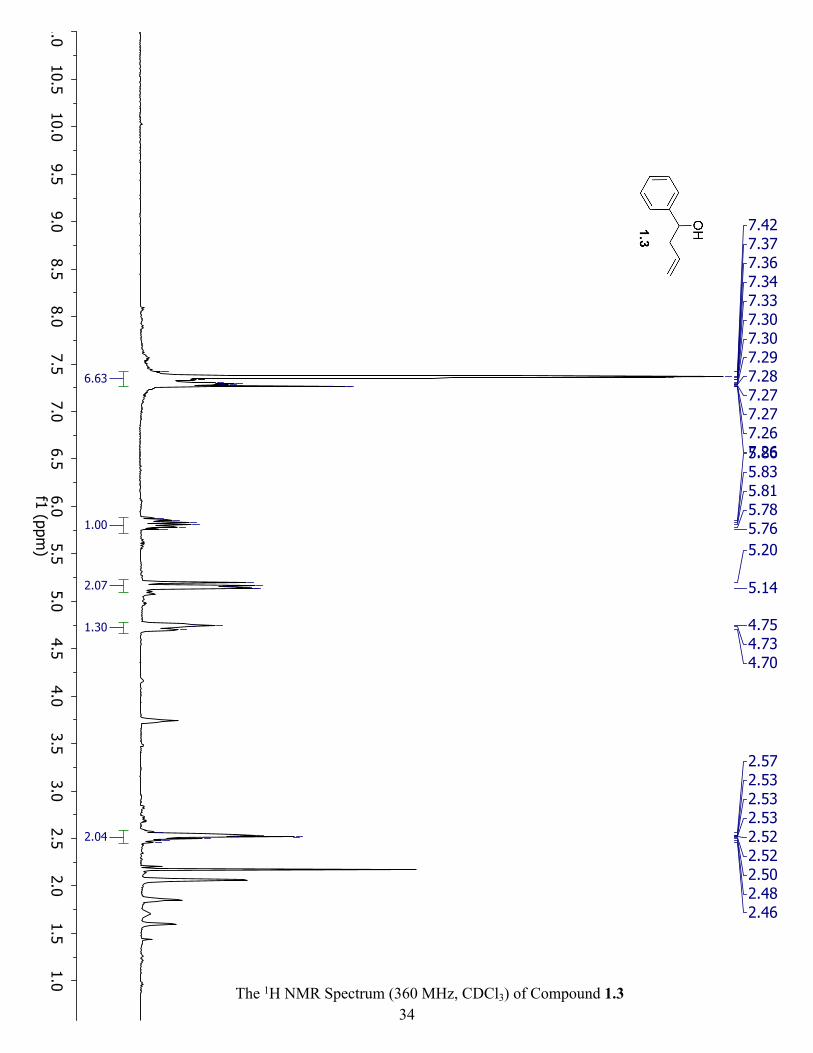
Synthesis of 1-phenylbut-3-en-1-ol (1.3): To a flame dried and purged RBF, a solution of 1.1
(9.4 mmol, 0.96 mL, 1.0 eq.) in anhydrous THF (1.0 M, 10 mL) was added. The mixture was
cooled to -78 oC where 1.2 (1.0 M, 10.4 mL, 1.1 eq.) was added dropwise. The reaction stirred
overnight allowing the temperature to raise to RT. It was then carefully quenched with DI water
and extracted three times with 10 mL of diethyl ether. All the organic layers were combined and
dried with MgSO4 and concentrated. There was no purification needed. Yield: 1.3 g, 90% as
light-yellow oil. 1H NMR (360 MHz, CDCl3) δ 7.34 (m, 5H), 5.82 (td, J = 17.2, 7.3 Hz, 1H),
5.17 (m, 2H), 4.73 (m, 1H), 2.52 (m, 2H).
Synthesis of ethyl (E)-5-hydroxy-5-phenylpent-2-enoate (1.5): Homoallylic alcohol 1.3 (0.68
mmol, 0.10 g, 1.0 eq.) was dissolved in DCM (0.2 M, 2.3 mL) and added to a flame dried and
purged flask. The acrylate 1.4 (3.4 mmol, 0.36 mL, 5.0 eq.) was added dropwise followed by
addition of the Grubbs’ (II) catalyst (0.03 mmol, 0.03 g, 0.05 eq.). The mixture was stirred
overnight at RT and then concentrated. Purification took place via column chromatography in
20% EtOAc and hexanes to afford the desired 1.5. Rf: 0.7; Yield: 0.11 g, 74% as dark brown oil.

24
1H NMR (360 MHz, CDCl3) δ 7.33 (m, 6H), 6.94 (m, 1H), 5.91 (dt, J = 15.7, 1.4 Hz, 1H), 4.83
(m, 1H), 4.03 (dd, J = 5.8, 2.8 Hz, 2H), 2.66 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H).
Synthesis of ethyl (E)-5-(2-bromoacetoxy)-5-phenylpent-2-enoate (1.7): To a flame dried and
purged flask, a solution of 1.5 (0.45 mmol, 0.10 g, 1.0 eq) and DCM (0.3 M, 1.5 mL) was added.
The reaction was cooled to 0 oC where pyridine (0.91 mmol, 0.07 mL, 2.0 eq.) was added,
followed by compound 1.6 (0.91 mmol, 0.07 mL, 2.0 eq) dropwise. The mixture stirred
overnight raising the temperature to RT where is was then quenched with saturated NH4Cl and
extracted three times with 10 mL of DCM. The organic layers were combined and washed with
10 mL of saturated CuSO4. The solution was then dried with MgSO4 and concentrated.
Purification took place via column chromatography in 5% EtOAc in hexanes to afford the
desired 1.7. Rf: 0.2; Yield: 0.09 g, 57% as dark brown oil. 1H NMR (360 MHz, CDCl3) δ 7.33
(m, 5H), 6.82 (dt, J = 15.5, 7.3 Hz, 1H), 5.87 (ddd, 2H), 4.03 (dd, J = 5.8, 2.9 Hz, 2H), 3.81 (s,
2H), 2.74 (m, 2H), 0.88 (m, 3H).
Ethyl (E)-5-acetoxy-5-phenylpent-2-enoate (1.13) (Table 1.1, Entry 4): Purified from 5%
EtOAc in hexanes. Rf: 0.2; Yield: 0.008 g, 20% as a yellow oil.
Ethyl (E)-5-(2-acetoxyacetoxy)-5-phenylpent-2-enoate (1.14) (Table 1.1, Entry 1): Purified
from 5% EtOAc in hexanes. Rf: 0.2; Yield: 0.01 g, 20% as a yellow oil.
Ethyl (E)-5-(2-chloroacetoxy)-5-phenylpent-2-enoate (1.15) (Table 1.4, Entry 1): Purified
from 20% EtOAc in hexanes. Rf: 0.2; Yield: 0.03 g, 50% as a yellow oil.
Synthesis of ethyl cinnamate (1.17): To a flame dried and purged flask, acrylate 1.4 (1.0 mmol,
0.11 mL, 1.0 eq.) and iodobenzene 1.16 (1.2 mmol, 0.14 mL, 1.2 eq.) were added to anhydrous
toluene (0.05 M, 20 mL). Pd2(dba)3 (0.03 mmol, 0.03 g, 0.03 eq.) and PPh3 (0.09 mmol, 0.02 g,
0.09 eq.) were sequentially added followed by Hunig’s base (5.0 mmol, 0.87 mL, 5.0 eq.). The

25
reaction was refluxed overnight where the material was filtered and concentrated. The desired
1.17 was visible by 1H NMR. 1H NMR (500 MHz, CDCl3) δ 7.53 (m, 6H), 6.45 (d, J = 16.0 Hz,
1H), 4.28 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H).
Synthesis of ethyl (E)-5-(2-chloroacetoxy)-5-phenylpent-2-enoate (1.15): To a flame dried
and purged flask, a solution of 1.5 (0.45 mmol, 0.10 g, 1.0 eq) and DCM (0.3 M 1.5 mL) was
added. The reaction was cooled to 0 oC where pyridine (0.91 mmol, 0.07 mL, 2.0 eq.) was added,
followed by compound chloroacetyl chloride (0.91 mmol, 2.0 eq) dropwise. The mixture stirred
overnight raising the temperature to RT where is was then quenched with saturated NH4Cl and
extracted three times with 10 mL of DCM. The organic layers were combined and washed with
10 mL of saturated CuSO4. The solution was then dried with MgSO4 and concentrated.
Purification took place via column chromatography in 20% EtOAc in hexanes to afford the
desired 1.15. Rf: 0.6: Yield: 0.05 g, 39% as light brown oil. 1H NMR (500 MHz, CDCl3) δ 7.36
(m, 5H), 6.82 (dt, J = 15.5, 7.3 Hz, 1H), 5.88 (m, 2H), 4.18 (q, 2H), 4.06 (m, 2H), 2.76 (m, 2H),
1.27 (dd, J = 12.2, 5.1 Hz, 3H).
Synthesis of 1-phenylbut-3-en-1-yl 2-bromoacetate (1.19) and 1-phenylbut-3-en-1-yl 2-
chloroacetate (1.20): To a flame dried and purged flask, a solution of 1.3 (00.68 mmol, 0.10 g,
1.0 eq) and DCM (0.3 M, 2.3 mL) was added. The reaction was cooled to 0 oC where pyridine
(1.3 mmol, 0.11 mL, 2.0 eq.) was added, followed by bromoacetyl bromide or chloroacetyl
chloride (1.3 mmol, 0.10 mL, 2.0 eq) dropwise. The mixture stirred overnight raising the
temperature to RT where is was then quenched with saturated NH4Cl and extracted three times
with 10 mL of DCM. The organic layers were combined and washed with 10 mL of saturated
CuSO4. The solution was then dried with MgSO4 and concentrated. Purification took place via
column chromatography in 20% EtOAc in hexanes to afford the desired 1.19 and 1.20. Rf: 0.6

26
and 0.7 respectively: Yield: 0.07g, 39% as light brown oil. 1H NMR (500 MHz, CDCl3) 1.19 δ
7.34 (m, 5H), 5.84 (dd, J = 7.8, 5.9 Hz, 1H), 5.71 (ddt, J = 17.1, 10.2, 7.0 Hz, 1H), 5.07 (m, 2H),
3.84 (s, 2H), 2.63 (m, 2H). 1H NMR (500 MHz, CDCl3) 1.20 δ 7.34 (m, 5H), 5.88 (dd, J = 7.8,
5.9 Hz, 1H), 5.70 (ddt, J = 17.2, 10.2, 7.0 Hz, 1H), 5.09 (ddd, J = 9.5, 5.5, 1.2 Hz, 2H), 4.07 (dd,
2H), 2.63 (m, 2H).

27
1.7 References for Chapter 1
1. Pettit, G. R.; Herald, C. L.; Doubek, D. L.; Herald, D. L.; Arnold, E.; Clardy, J.,
Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846-6848.
2. Zhang, Z.; Xie, H.; Li, H.; Gao, L.; Song, Z., Total Synthesis of (-)-Exiguolide. Org.
Lett. 2015, 17, 4706-4709.
3. Schuppe, A. W.; Huang, D.; Chen, Y.; Newhouse, T. R., Total Synthesis of (-)-
Xylogranatopyridine B via a Palladium-Catalyzed Oxidative Stannylation of Enones. J. Am.
Chem. Soc. 2018, 140, 2062-2066.
4. Sole, D.; Urbaneja, X.; Bonjoch, J., Synthesis of the 4-Azatricyclo[5.2.2.04,8]undecan-
10-one Core of Daphniphyllum Alkaloid Calyciphylline A Using a Pd-Catalyzed Enolate
Alkenylation. Org. Lett. 2005, 7, 5461-5464.
5. Lee, D.-H.; Taher, A.; Hossain, S.; Jin, M.-J., An efficient and general method for the
Heck and Buchwald-Hartwig coupling reactions of aryl chlorides. Org. Lett. 2011, 13, 5540-
5543.
6. Xu, H.-J.; Zhao, Y.-Q.; Zhou, X.-F., Palladium-Catalyzed Heck Reaction of Aryl
Chlorides under Mild Conditions Promoted by Organic Ionic Bases. J. Org. Chem. 2011, 76,
8036-8041.
7. Burkhardt, E. R.; Bergman, R. G.; Heathcock, C. H., Synthesis and reactions of nickel
and palladium carbon-bound enolate complexes. Organometallics 1990, 9, 30-44.
8. Ito, Y.; Aoyama, H.; Hirao, T.; Mochizuki, A.; Saegusa, T., Cyclization reactions via
oxo-π-allylpalladium(II) intermediates. J. Am. Chem. Soc. 1979, 101, 494-496.
9. Ito, Y.; Hirao, T.; Saegusa, T., Synthesis of α,β-unsaturated carbonyl compounds by
palladium(II)-catalyzed dehydrosilylation of silyl enol ethers. J. Org. Chem. 1978, 43, 1011-
1013.
10. Kurosawa, H.; Majima, T.; Asada, N., Synthesis, structures, stabilities, and reactions of
cationic olefin complexes of palladium(II) containing the η5-cyclopentadienyl ligand. J. Am.
Chem. Soc. 1980, 102, 6996-7003.
11. Guo, Y.; Tao, G.-H.; Blumenfeld, A.; Shreeve, J. n. M., Impact of Silyl Enol Ether
Stability on Palladium-Catalyzed Arylations. Organometallics 2010, 29, 1818-1823.

28
12. Albeniz, A. C.; Catalina, N. M.; Espinet, P.; Redon, R., Bonding Modes in Palladium(II)
Enolates: Consequences for Dynamic Behavior and Reactivity. Organometallics 1999, 18, 5571-
5576.
13. Vicente, J.; Abad, J. A.; Chicote, M.-T.; Abrisqueta, M.-D.; Lorca, J.-A.; Ramirez de
Arellano, M. C., Synthesis of New Ketonyl Palladium(II) and Platinum(II) Complexes with
Nitrogen-Donor Ligands. Crystal Structure of [Pt{CH2C(O)Me}2(bpy)]. Organometallics 1998,
17, 1564-1568.
14. Sodeoka, M.; Ohrai, K.; Shibasaki, M., Catalytic Asymmetric Aldol Reaction via Chiral
Pd(II) Enolate in Wet DMF. J. Org. Chem. 1995, 60, 2648-2649.
15. Sodeoka, M.; Tokunoh, R.; Miyazaki, F.; Hagiwara, E.; Shibasaki, M., Stable diaqua
palladium(II) complexes of BINAP and Tol-BINAP as highly efficient catalysts for asymmetric
aldol reactions. Synlett 1997, 463-466.
16. Veya, P.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C., Terminal and bridging bonding
modes of the acetophenone enolate to palladium(II): the structural evidence and the insertion of
isocyanides. Organometallics 1993, 12, 4899-4907.
17. Culkin, D. A.; Hartwig, J. F., C-C Bond-Forming Reductive Elimination of Ketones,
Esters, and Amides from Isolated Arylpalladium(II) Enolates. J. Am. Chem. Soc. 2001, 123,
5816-5817.
18. Lei, A.; Zhang, X., A novel palladium-catalyzed homocoupling reaction initiated by
transmetalation of palladium enolates. Tetrahedron Lett. 2002, 43, 2525-2528.
19. Hamashima, Y.; Hotta, D.; Sodeoka, M., Direct Generation of Nucleophilic Chiral
Palladium Enolate from 1,3-Dicarbonyl Compounds: Catalytic Enantioselective Michael
Reaction with Enones. J. Am. Chem. Soc. 2002, 124, 11240-11241.
20. Qian, H.; Widenhoefer, R. A., Mechanism of the Palladium-Catalyzed Intramolecular
Hydroalkylation of 7-Octene-2,4-dione. J. Am. Chem. Soc. 2003, 125, 2056-2057.
21. Widenhoefer, R. A., Palladium-catalyzed alkylation of unactivated olefins. Pure Appl.
Chem. 2004, 76, 671-678.
22. Hamashima, Y.; Sodeoka, M., Development of catalytic asymmetric reactions via chiral
palladium enolates. Chem. Rec. 2004, 4, 231-242.
23. Streuff, J.; White, D. E.; Virgil, S. C.; Stoltz, B. M., A palladium-catalyzed enolate
alkylation cascade for the formation of adjacent quaternary and tertiary stereocenters. Nat. Chem.
2010, 2, 192-196.

29
24. Kantam, M. L.; Srinivas, P.; Yadav, J.; Likhar, P. R.; Bhargava, S., Trifunctional
N,N,O-Terdentate Amido/Pyridyl Carboxylate Ligated Pd(II) Complexes for Heck and Suzuki
Reactions. J. Org. Chem. 2009, 74, 4882-4885.
25. Wang, A.-E.; Xie, J.-H.; Wang, L.-X.; Zhou, Q.-L., Triaryl phosphine-functionalized N-
heterocyclic carbene ligands for Heck reaction. Tetrahedron 2005, 61, 259-266.
26. Heck, R. F., Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem.
Res. 1979, 12, 146-151.
27. Ioele, M.; Ortaggi, G.; Scarsella, M.; Sleiter, G., A rapid and convenient synthesis of
tetrakis(triphenylphosphine)palladium(0) and -platinum(0) complexes by phase-transfer
catalysis. Polyhedron 1991, 10, 2475-2476.
28. Grushin, V. V.; Alper, H., Alkali-induced disproportionation of palladium(II) tertiary
phosphine complexes, [L2PdCl2], to LO and palladium(O). Key intermediates in the biphasic
carbonylation of ArX catalyzed by [L2PdCl2]. Organometallics 1993, 12, 1890-1901.
29. Grushin, V. V., Catalysis for Catalysis: Synthesis of Mixed Phosphine-Phosphine Oxide
Ligands via Highly Selective, Pd-Catalyzed Monooxidation of Bidentate Phosphines. J. Am.
Chem. Soc. 1999, 121, 5831-5832.
30. Roffia, P.; Gregorio, G.; Conti, F.; Pregaglia, G. F.; Ugo, R., Catalysis by palladium
salts. VIII: An easy synthesis of zerovalent palladium and platinum triphenylphosphine catalysts
by reduction with alkaline alkoxides. J. Mol. Catal. 1977, 2, 191-201.
31. Amatore, C.; Jutand, A.; Medeiros, M. J., Formation of zerovalent palladium from the
cationic complex Pd(PPh3)2(BF4)2 in the presence of PPh3 and water in DMF. New J. Chem.
1996, 20, 1143-1148.
32. Amatore, C.; Jutand, A.; Khalil, F.; M'Barki, M. A.; Mottier, L., Rates and mechanisms
of oxidative addition to zerovalent palladium complexes generated in situ from mixtures of
Pd0(dba)2 and triphenylphosphine. Organometallics 1993, 12, 3168-3178.
33. Rappoport, Z., Nucleophilic vinylic substitution. A single- or a multi-step process? Acc.
Chem. Res. 1981, 14, 7-15.
34. Jutand, A.; Mosleh, A., Rate and Mechanism of Oxidative Addition of Aryl Triflates to
Zerovalent Palladium Complexes. Evidence for the Formation of Cationic (σ-Aryl)palladium
Complexes. Organometallics 1995, 14, 1810-1817.
35. Casado, A. L.; Espinet, P., On the Configuration Resulting from Oxidative Addition of
RX to Pd(PPh3)4 and the Mechanism of the cis-to-trans Isomerization of [PdRX(PPh3)2]
Complexes (R = Aryl, X = Halide). Organometallics 1998, 17, 954-959.

30
36. Ozawa, F.; Kubo, A.; Hayashi, T., Catalytic asymmetric arylation of 2,3-dihydrofuran
with aryl triflates. J. Am. Chem. Soc. 1991, 113, 1417-1419.
37. Cabri, W.; Candiani, I.; DeBernardinis, S.; Francalanci, F.; Penco, S.; Santo, R., Heck
reaction on anthraquinone derivatives: ligand, solvent and salt effects. J. Org. Chem. 1991, 56,
5796-5800.
38. Sato, Y.; Nukui, S.; Sodeoka, M.; Shibasaki, M., Asymmetric heck reaction of alkenyl
iodides in the presence of silver salts. Catalytic asymmetric synthesis of decalin and
functionalized indolizidine derivatives. Tetrahedron 1994, 50, 371-382.
39. Sato, Y.; Sodeoka, M.; Shibasaki, M., On the role of silver salts in asymmetric Heck-
type reaction. A greatly improved catalytic asymmetric synthesis of cis-decalin derivatives.
Chem. Lett. 1990, 1953-1954.
40. Cabri, W.; Candiani, I.; DeBernardinis, S.; Francalanci, F.; Penco, S.; Santo, R., Heck
reaction on anthraquinone derivatives: ligand, solvent and salt effects. J. Org. Chem. 1991, 56,
5796-5800.
41. Ozawa, F.; Kubo, A.; Hayashi, T., Catalytic asymmetric arylation of 2,3-dihydrofuran
with aryl triflates. J. Am. Chem. Soc. 1991, 113, 1417-1419.
42. Grigg, R.; Sridharan, V.; Sukirthalingam, S., Alkylpalladium(II) species. Reactive
intermediates in a bis-cyclization route to strained polyfused ring systems. Tetrahedron Lett.
1991, 32, 3855-3858.
43. Meyer, F. E.; Parsons, P. J.; De Meijere, A., Palladium-catalyzed polycyclization of
dienynes: surprisingly facile formation of tetracyclic systems containing a three-membered ring.
J. Org. Chem. 1991, 56, 6487-6488.
44. Grigg, R.; Dorrity, M. J.; Malone, J. F.; Sridharan, V.; Sukirthalingam, S., Palladium-
catalyzed polycyclization-anion capture processes. Tetrahedron Lett. 1990, 31, 1343-1346.
45. Zhang, Y.; Negishi, E., Metal-promoted cyclization. 25. Palladium-catalyzed cascade
carbometalation of alkynes and alkenes as an efficient route to cyclic and polycyclic structures.
J. Am. Chem. Soc. 1989, 111, 3454-3456.
46. Carpenter, N. E.; Kucera, D. J.; Overman, L. E., Palladium-catalyzed polyene
cyclizations of trienyl triflates. J. Org. Chem. 1989, 54, 5846-5848.
47. Liu, C.-H.; Cheng, C.-H.; Cheng, M.-C.; Peng, S.-M., Palladium-Catalyzed Addition of
Alkyne to Norbornene Derivatives. Unusual Ring Formation and Expansion Reactions.
Organometallics 1994, 13, 1832-1839.

31
48. Rawal, V. H.; Michoud, C., An unexpected Heck reaction. Inversion of olefin geometry
facilitated by the apparent intramolecular carbamate chelation of the σ-palladium intermediate. J.
Org. Chem. 1993, 58, 5583-5584.
49. Owczarczyk, Z.; Lamaty, F.; Vawter, E. J.; Negishi, E., Apparent endo-mode cyclic
carbopalladation with inversion of alkene configuration via exo-mode cyclization-
cyclopropanation rearrangement. J. Am. Chem. Soc. 1992, 114, 10091-10092.
50. Albeniz, A. C.; Espinet, P.; Lin, Y.-S., Cyclization versus Pd-H Elimination-Readdition:
Skeletal Rearrangement of the Products of Pd-C6F5 Addition to 1,4-Pentadienes. J. Am. Chem.
Soc. 1996, 118, 7145-7152.
51. Tietze, L. F.; Modi, A., Regioselective silane-terminated intramolecular Heck reaction
with alkenyl triflates and alkenyl iodides. Eur. J. Org. Chem. 2000, 1959-1964.
52. Nagasawa, K.; Zako, Y.; Ishihara, H.; Shimizu, I., Stereoselective synthesis of 1α-
hydroxyvitamin D3 A-ring synthons by palladium-catalyzed cyclization. Tetrahedron Lett. 1991,
32, 4937-4940.
53. Maruyama, O.; Yoshidomi, M.; Fujiwara, Y.; Taniguchi, H., Palladium(II)-copper(II)-
catalyzed synthesis of mono- and dialkenyl-substituted five-membered aromatic heterocycles.
Chem. Lett. 1979, 1229-1230.
54. Shue, R. S., Catalytic coupling of aromatics and olefins by homogeneous palladium(II)
compounds under oxygen. J. Chem. Soc. D. 1971, 1510-1511.
55. Albert, K.; Gisdakis, P.; Roesch, N., On C-C Coupling by Carbene-Stabilized Palladium
Catalysts: A Density Functional Study of the Heck Reaction. Organometallics 1998, 17, 1608-
1616.
56. Deeth, R. J.; Smith, A.; Hii, K. K.; Brown, J. M., The Heck olefination reaction; a DFT
study of the elimination pathway. Tetrahedron Lett. 1998, 39, 3229-3232.
57. Spencer, A., Stereochemical course of the palladium-catalyzed arylation of disubstituted
activated alkenes with benzoyl chloride. J. Organomet. Chem. 1982, 240, 209-216.
58. De Joarder, D.; Jennings, M. P., Convergent synthesis of (+)-xestodecalactone A via a
Pd-catalyzed α-arylation reaction. Tetrahedron Lett. 2013, 54, 3990-3992.
59. De Joarder, D.; Jennings, M. P., Umpolung Pd-Catalyzed α-Arylation Reactions in
Natural Product Synthesis: Syntheses of (+)-Xestodecalactone A, (-)-Curvularin, (+)-12-
Oxocurvularin and (-)-Citreofuran. Eur. J. Org. Chem. 2015, 2015, 3303-3313.
60. Hu, N.; Dong, C.; Zhang, C.; Liang, G., Total Synthesis of (-)-Indoxamycins A and B.
Angew. Chem., Int. Ed. 2019, 58, 6659-6662.

32
61. Carrick, J. D.; Jennings, M. P., An Efficient Formal Synthesis of (-)-Clavosolide A
Featuring a "Mismatched" Stereoselective Oxocarbenium Reduction. Org. Lett. 2009, 11, 769-
772.
62. Lee, S.-M.; Lee, W.-G.; Kim, Y.-C.; Ko, H., Synthesis and biological evaluation of α,β-
unsaturated lactones as potent immunosuppressive agents. Bioorg. Med. Chem. Lett. 2011, 21,
5726-5729.
63. Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H., A General Model for
Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360-11370.
64. Kim, D. W.; Song, C. E.; Chi, D. Y., Significantly Enhanced Reactivities of the
Nucleophilic Substitution Reactions in Ionic Liquid. J. Org. Chem. 2003, 68, 4281-4285.
65. Ju, Y.; Kumar, D.; Varma, R. S., Revisiting Nucleophilic Substitution Reactions:
Microwave-Assisted Synthesis of Azides, Thiocyanates, and Sulfones in an Aqueous Medium. J.
Org. Chem. 2006, 71, 6697-6700.
66. Jadhav, V. H.; Kim, J. G.; Jeong, H. J.; Kim, D. W., Nucleophilic Hydroxylation in
Water Media Promoted by a Hexa-Ethylene Glycol-Bridged Dicationic Ionic Liquid. J. Org.
Chem. 2015, 80, 7275-7280.
67. Zhao, X.; Zhuang, W.; Fang, D.; Xue, X.; Zhou, J., A Highly Efficient Conversion of
Primary or Secondary Alcohols into Fluorides with n-Perfluorobutanesulfonyl Fluoride-
Tetrabutylammonium Triphenyldifluorosilicate. Synlett 2009, 2009, 779-782.
68. Sawant, K. B.; Jennings, M. P., Efficient Total Syntheses and Structural Verification of
Both Diospongins A and B via a Common δ-Lactone Intermediate. J. Org. Chem. 2006, 71,
7911-7914.
69. Samanta, S.; Mohapatra, H.; Jana, R.; Ray, J. K., Pd(0) catalyzed intramolecular Heck
reaction: a versatile route for the synthesis of 2-aryl substituted 5-, 6-, and 7-membered O-
containing heterocycles. Tetrahedron Lett. 2008, 49, 7153-7156.
70. Jimenez, F.; Fernandez, A.; Boulifa, E.; Mansour, A. I.; Alvarez-Manzaneda, R.;
Chahboun, R.; Alvarez-Manzaneda, E., Diastereoselective Intramolecular Heck Reaction
Assisted by an Acetate Group: Synthesis of the Decahydrobenzofluorene Derivative
Dasyscyphin E. J. Org. Chem. 2017, 82, 9550-9559.
71. Zhou, W.; An, G.; Zhang, G.; Han, J.; Pan, Y., Ligand-free palladium-catalyzed
intramolecular Heck reaction of secondary benzylic bromides. Org. Biomol. Chem. 2011, 9,
5833-5837.
72. Glorius, F., Palladium-catalyzed Heck-type reaction of 2-chloro acetamides with olefins.
Tetrahedron Lett. 2003, 44, 5751-5754.

33
73. Rousee, K.; Bouillon, J.-P.; Couve-Bonnaire, S.; Pannecoucke, X., Stereospecific
Synthesis of Tri- and Tetrasubstituted α-Fluoroacrylates by Mizoroki-Heck Reaction. Org. Lett.
2016, 18, 540-543.
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.010.5
11.0f1 (ppm
)
2.04
1.30
2.07
1.00
6.63
2.462.482.502.522.522.532.532.532.57
4.704.734.75
5.14
5.205.765.785.815.835.867.267.267.277.277.287.297.307.307.337.347.367.377.42
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.334
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
10.5f1 (ppm
)
7.10
2.25
2.35
1.06
1.13
1.00
6.31
0.870.890.91
2.622.622.642.642.662.662.682.68
4.024.034.044.044.824.834.834.845.895.895.905.935.945.946.906.94
7.287.297.297.317.317.327.337.347.357.367.377.37
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.535
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
f1 (ppm)
8.06
2.06
2.002.04
2.03
1.00
5.20
0.860.870.880.880.890.900.91
2.682.702.722.742.762.81
3.814.024.034.034.04
5.855.855.865.875.88
5.906.786.826.84
7.267.287.307.307.317.327.327.337.347.357.37
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.736
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
f1 (ppm)
3.87
1.55
1.81
1.75
0.17
1.76
1.00
5.49
0.20
1.241.281.301.301.312.102.742.792.802.802.822.824.094.094.174.184.194.194.204.214.225.855.855.865.865.875.885.885.885.896.836.846.866.877.287.327.337.347.347.347.357.357.367.377.377.387.387.397.397.397.487.50
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 1.1337

0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
10.5f1 (ppm
)
3.10
2.06
1.00
7.40
1.341.361.37
4.264.284.294.30
6.446.47
7.397.397.407.407.537.537.547.557.697.72
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.17
38
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.010.5
f1 (ppm)
3.66
2.13
2.062.05
1.97
1.00
5.08
1.251.261.271.29
2.722.732.742.762.772.842.864.034.064.074.104.154.174.184.20
5.855.855.855.885.88
5.916.796.806.82
6.837.317.327.327.337.337.347.347.367.367.377.377.387.397.39
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.15
39
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.010.5
11.0f1 (ppm
)
2.16
1.89
2.05
1.001.00
5.19
2.582.592.602.602.612.622.632.632.632.672.672.672.69
2.70
3.84
5.065.075.075.09
5.125.675.695.705.725.74
5.847.287.297.307.307.317.317.317.317.327.327.327.337.347.347.347.357.367.367.367.377.377.387.38
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.1940
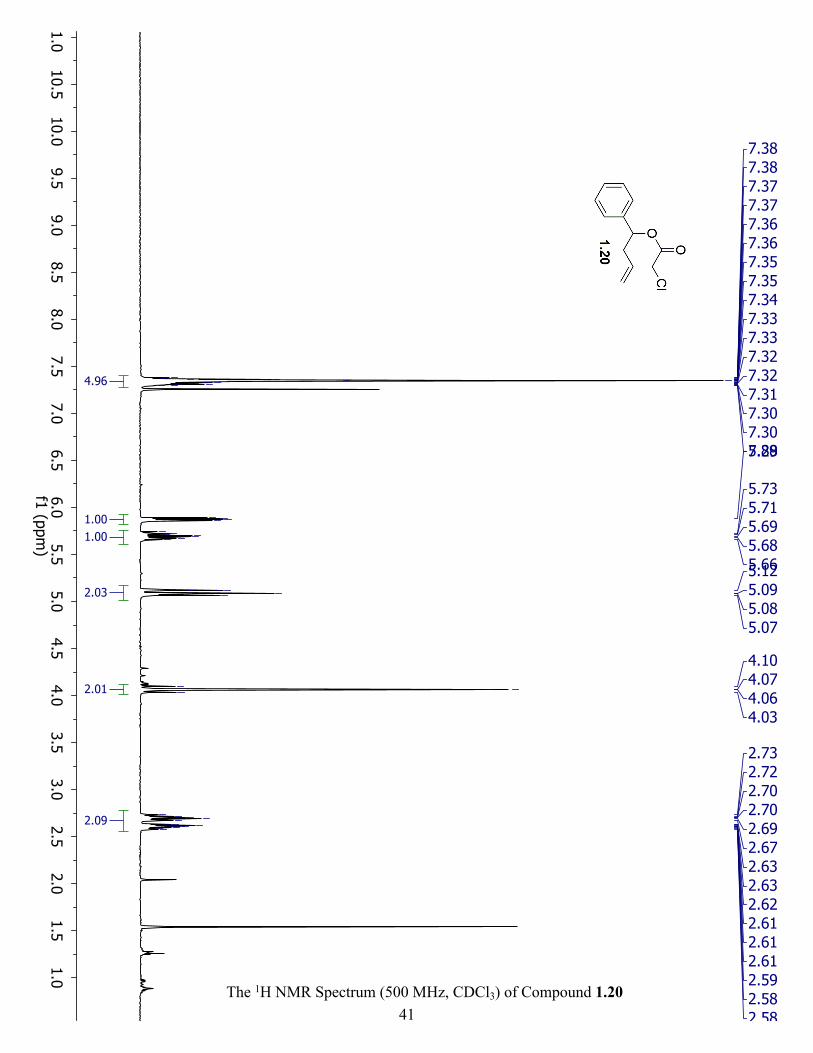
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.010.5
11.0f1 (ppm
)
2.09
2.01
2.03
1.001.00
4.96
2.582.582.592.612.612.612.622.632.632.672.692.702.702.722.73
4.034.064.074.10
5.075.085.095.125.665.685.695.715.73
5.887.297.307.307.317.327.327.337.337.347.357.357.367.367.377.377.387.38
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 1.2041

42
CHAPTER 2: TOTAL SYNTHESIS OF DIHYDRORESORCYLIDE
2.1. Introduction
As discussed in a previous chapter, Pd enolates have been extensively studied and are a
focus of this research. These intermediates allow for various reactions such as, Michael
reactions,1 homocoupling,2 and intramolecular ring formations,3 to occur and are even found in a
couple of examples of total synthesis.4, 5 Despite being widely utilized, the ring closure of natural
product macrocycles via Pd enolates remains an unutilized method in synthesis especially in
resorcylic acid lactones (RALs).
A class of compounds called RALs are divided into three types based on the backbone
present as shown in Figure 2.1 and are predominately found in strains of fungi. The first RAL,
Radicicol, was isolated in 1953 by Delmotte and company.6 Since then, a large quantity of these
compounds has been discovered and been found to have various biological properties ranging
from cancer cell inhibitors to antiviral activity.7, 8

43
RAL synthesis utilizes a variety of methods to obtain the desired compounds, but
formation of the macrocycle moiety is widely conducted through RCM.9-11 For example, the
Jennings’ group has previously created one of these compounds called Ponchonin J with a key
reaction being an oxocarbenium allylation.12 The purpose of this research is to synthesize
Dihydroresorclide, 2.1, shown in (Figure 2.2) via a Pd enolate ring closure rather than RCM.
2.2 Isolation and Structural Elucidation, Biological Properties and Reactions, and
Synthesis of Dihydroresorcylide
Dihydroresorcylide 2.1 has been isolated twice from two different fungal strains. The first
was in 2008, Poling and company examine Acremonium zeae, a protective endophyte found in
maize, and several new compounds were found including 2.1.13 After a series of extractions with
EtOAc and MeOH, a final extraction with acetonitrile afforded the new material. At first, the
thought was that the recovered species was that of Culvularin, 2.2, (Figure 2.3), but upon closer
inspection of the 1H NMR data, chemical shifts not consistent with Culvularin 2.2 were present.
Based on this information, more spectroscopic techniques like 13C NMR, HMBC, and mass
spectrometry were performed. The stereochemistry was also determined to be (S), though this
has come under scrutiny and will be discussed further. The material was isolated a second time
in 2016 from fungus Gliomastix sp found in a sponge giving rise to the mindset that 2.1 could be

44
common among fungal strains.14, 15 The compound was then subjected to biological testing and a
variety of biotransformations.
Poling subjected 2.1 to antifungal and leaf-wound puncture assays to determine if the
material was part of a protective endophyte relationship. Based on results of the assays,
Dihydroresorcylide 2.1 did not exhibit any fungistatic behavior against at least two samples, but
it did prove to be phytotoxic in the leaf-puncture wound assay producing “elongated lesions”
averaging 1.37 mm. This led to the thought that the product was facilitating the spread of the
endophyte into the maize tissues.15 Work conducted by Zhan and coworkers, published in 2010
and 2011, examined if the natural product could undergo biotransformations with a halogenase
and a glycolytransferase. These experiments were successful as the aromatic ring was
chlorinated at the green positions, and a glucose moiety was coupled with the hydroxy at the red
position shown in Figure 2.2.16, 17 In 2017, Dihydroresorcylide was tested as a PTP1B inhibitor.18
A PTP1B inhibitor changes the sensitivity to insulin and is a promising method of treating type 2
diabetes.19 Based on the bioassays conducted, the (R) isomer of 2.1 was a highly selective
inhibitors compared to the (S) counterpart. This means that the stereochemistry does matter.
Molecular docking studies were conducted to attempt to understand why there is selectivity.
Based on the modeling, the (R) isomer has the ability to have hydrogen-bonding, pi-pi stacking,
and hydrophobic interactions that the (S) does not. In addition to the biological testing, the
natural product has been synthesized twice by other groups.

45
The first total synthesis occurred in 2013 and the retrosynthesis published had three main
cuts being an esterification, carbonylation, and an RCM (Figure 2.4).20 Based on Scheme 2.1, the
synthesis was conducted via nine steps. The first step began with orcinol monohydrate 2.3 which
underwent a series of three steps to give 2.4. 2.4 was then saponified to yield 2.5 which was then
subjected to successful Mitsunobu esterification with (R)-hept-6-en-2-ol. The new ester 2.6 then
underwent a carbonylation. The carbonylation with a Weinreb amide (Figure 2.5, 2.10) proved
difficult so the amide was changed to 2.11 which overcame the difficulty to afford 2.7. The
material was diluted significantly in DCM and reacted with GC(II) for an efficacious RCM to
give the macrocycle 2.8. The macrocycle 2.8 was subjected to a hydrogenation and with the
removal of the alkene, the demethylation of both methoxy groups was attempted. It was found
that reagents such as BBr3 would not properly remove the groups, and after several attempts AlI3
in benzene worked the best to give the desired 2.1. The second synthesis was not conducted
until 2017.
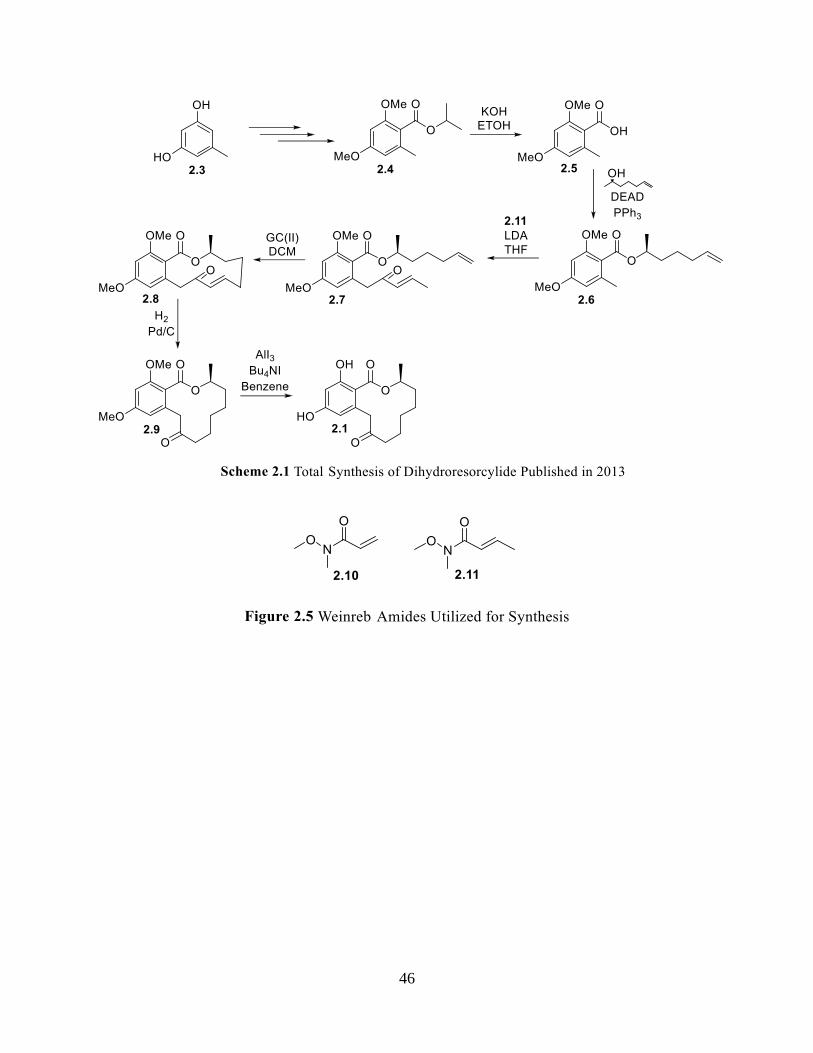
46
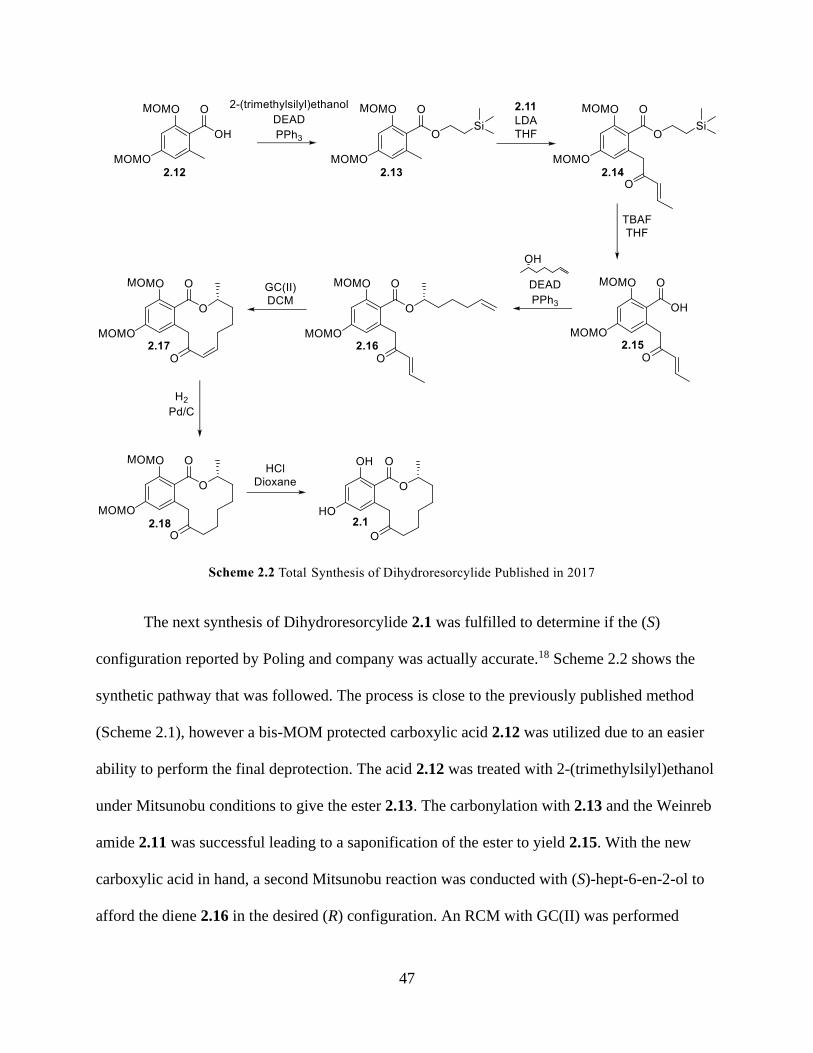
47
The next synthesis of Dihydroresorcylide 2.1 was fulfilled to determine if the (S)
configuration reported by Poling and company was actually accurate.18 Scheme 2.2 shows the
synthetic pathway that was followed. The process is close to the previously published method
(Scheme 2.1), however a bis-MOM protected carboxylic acid 2.12 was utilized due to an easier
ability to perform the final deprotection. The acid 2.12 was treated with 2-(trimethylsilyl)ethanol
under Mitsunobu conditions to give the ester 2.13. The carbonylation with 2.13 and the Weinreb
amide 2.11 was successful leading to a saponification of the ester to yield 2.15. With the new
carboxylic acid in hand, a second Mitsunobu reaction was conducted with (S)-hept-6-en-2-ol to
afford the diene 2.16 in the desired (R) configuration. An RCM with GC(II) was performed

48
followed by subsequent hydrogenation to give 2.18. The MOM deprotection was fruitful and
yielded (R)-Dihydrorescorcylide 2.1. With the material synthesized it was now time to discover
which configuration was the correct one.
It was noted that both (R) and (S) have identical NMR data so the difference came down
to the optical rotation of the two. Upon previous synthesis, the rotation was opposite what was
published in 2008 for the (S), After generating the (R) isomer, optical rotation was examined and
determined to be identical to the literature value thus confirming the stereochemistry to be the
(R) conformer which aligns with other work published by Poling and coworkers in 2008.13
Dihydroresorcylide 2.1 has been isolated, extensively studied, and synthesized twice. This
research discussed within, aims at obtaining either isomer of the RAL through a Pd enolate ring
closure.
2.3 Retrosynthetic Analysis
Dihydroresorcylide 2.1 can be broken down into several substructures as exhibited in the
retrosynthesis in Scheme 2.3. The first of two major cuts could occur at the benzyl methylene to
break open the macrocycle. The second major cut could occur at the oxygen of the ester breaking
the piece into two components that would be the focus of the synthesis; an aromatic synthon A
and an aliphatic synthon B. Both A and B would undergo their own transformations before being
joined together. For instance, the two hydroxy groups attached to the phenyl ring would need to
be protected over the course of the synthetic approach and it is feasible to do this with methoxy
groups, or the aliphatic chain would be created through a cross metathesis of two olefins with
one containing an alcohol and the other a ketone. Once the formation of the two synthons are
complete, they could be combined through an esterification reaction and the macrocycle could be
created by way of a Pd enolate ring closure. This type of ring closure is novel to total synthesis
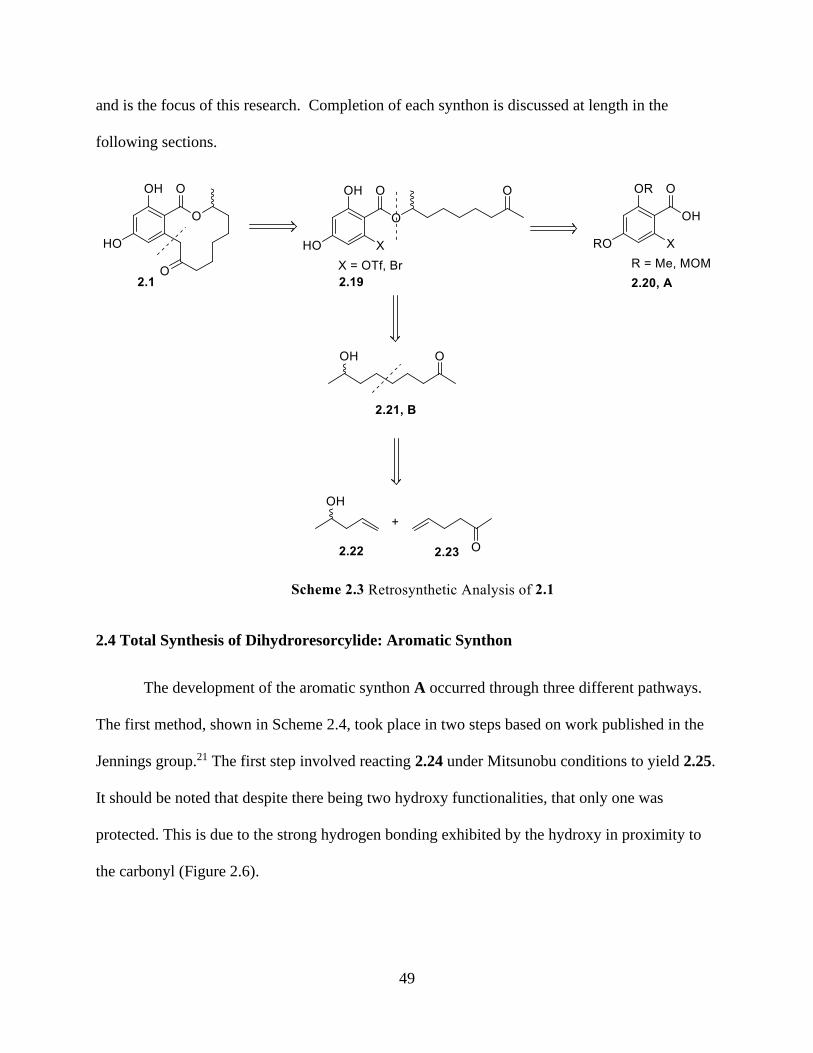
49
and is the focus of this research. Completion of each synthon is discussed at length in the
following sections.
2.4 Total Synthesis of Dihydroresorcylide: Aromatic Synthon
The development of the aromatic synthon A occurred through three different pathways.
The first method, shown in Scheme 2.4, took place in two steps based on work published in the
Jennings group.21 The first step involved reacting 2.24 under Mitsunobu conditions to yield 2.25.
It should be noted that despite there being two hydroxy functionalities, that only one was
protected. This is due to the strong hydrogen bonding exhibited by the hydroxy in proximity to
the carbonyl (Figure 2.6).
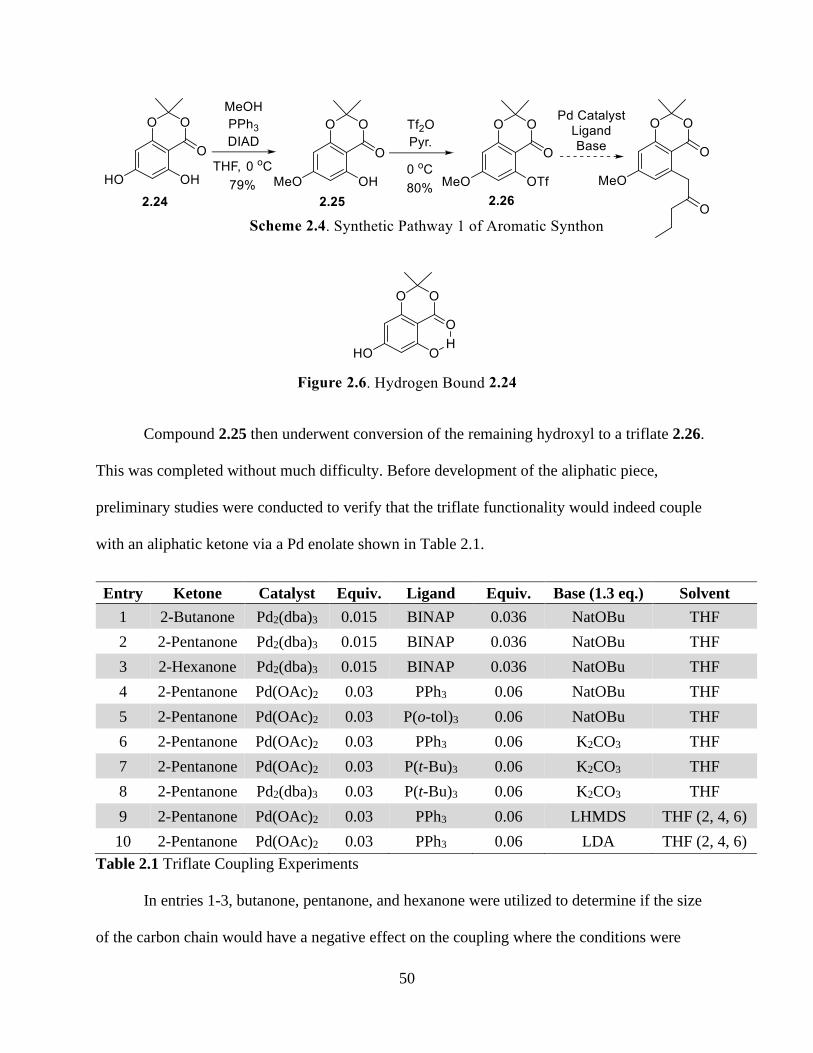
50
Compound 2.25 then underwent conversion of the remaining hydroxyl to a triflate 2.26.
This was completed without much difficulty. Before development of the aliphatic piece,
preliminary studies were conducted to verify that the triflate functionality would indeed couple
with an aliphatic ketone via a Pd enolate shown in Table 2.1.
Entry Ketone Catalyst Equiv. Ligand Equiv. Base (1.3 eq.) Solvent
1 2-Butanone Pd2(dba)3 0.015 BINAP 0.036 NatOBu THF
2 2-Pentanone Pd2(dba)3 0.015 BINAP 0.036 NatOBu THF
3 2-Hexanone Pd2(dba)3 0.015 BINAP 0.036 NatOBu THF
4 2-Pentanone Pd(OAc)2 0.03 PPh3 0.06 NatOBu THF
5 2-Pentanone Pd(OAc)2 0.03 P(o-tol)3 0.06 NatOBu THF
6 2-Pentanone Pd(OAc)2 0.03 PPh3 0.06 K2CO3 THF
7 2-Pentanone Pd(OAc)2 0.03 P(t-Bu)3 0.06 K2CO3 THF
8 2-Pentanone Pd2(dba)3 0.03 P(t-Bu)3 0.06 K2CO3 THF
9 2-Pentanone Pd(OAc)2 0.03 PPh3 0.06 LHMDS THF (2, 4, 6)
10 2-Pentanone Pd(OAc)2 0.03 PPh3 0.06 LDA THF (2, 4, 6)
Table 2.1 Triflate Coupling Experiments
In entries 1-3, butanone, pentanone, and hexanone were utilized to determine if the size
of the carbon chain would have a negative effect on the coupling where the conditions were

51
based on literature published by Buchwald.22 However, the only material recovered was starting
material 2.26. Entries 4-7 change the catalyst from a Pd(0) to a Pd(II) and examined
monodentate ligands with various salt bases.23-25 Unfortunately, only starting material 2.26 was
only recovered again. Entry 8 utilized a Pd(0) catalyst with a monodentate ligand, and again
there were no promising results. Entries 9-10 experimented with stronger lithium bases like LDA
and LHMDS where the reaction time was varied between two, four, and six hours. There was
only starting material 2.26 isolated. However, after repeating the experiments it was found that
the base was causing the ring to open forming 2.27 shown in Figure 2.7. With that information in
hand, it was decided to attempt coupling with the aliphatic ketone as a silyl enol ether.
The formation of the silyl enol ether was based on work completed by Tanabe with an
example shown in Figure 2.8.26 With the conversion complete, Table 2.2 exhibits the conditions
found in literature to attempt the coupling.27, 28 All three entries only provided starting material
2.28. Due to the inability to create a new carbon-carbon bond, it was decided that the triflate
would be replaced with a bromine.

52
Entry Catalyst Ligand Additive Solvent Product
1 Pd2(dba)3 DPPF LiOAc THF 2.26
2 Pd2(dba)3 P(t-Bu)3 Bu3SnF Toluene 2.26
3 Pd(PPh3)2Cl2 NA Bu3SnF THF 2.26
Table 2.2 Triflate Coupling with Silyl Enol Ether
Bromobenzene 2.29 and ethyl 2-bromobenzoate 2.30 with the silyl enol ether 2.28
(Figure 2.9) were chosen as pseudo compounds to attempt the new bond formation. In Table 2.3,
the conditions for these attempts were laid out. After several attempts, the ethyl 2-
bromobenzoate 2.30 did show some coupling product in low yields of about 10-20%. Despite the
low yield, this gave indication that the target reaction would work so it was concluded to change
the pathway of the aromatic portion from a triflate to a bromine.
Entry Catalyst Ligand Additive Solvent Product
1 Pd2(dba)3 P(t-Bu)3 ZnCl2 THF 2.30
2 Pd(PPh3)2Cl2 NA Bu3SnF THF 2.31
Table 2.3 Bromobenzene and Ethyl Bromobenzoate Coupling
In Scheme 2.5, 1-bromo-3,5-dimethoxybenzene 2.32 underwent a Vielsmeire-Haack
reaction to yield compound 2.33.29 It was found that purification would need to occur

53
immediately following work-up in order to avoid decomposition and gave a white powder.
Compound 2.33 was subjected to a selective demethylation using BBr3 to afford 2-bromo-6-
hydroxy-4-methoxybenzaldehyde 2.34. There was no demethylation product of the hydroxy at
the 4 position or any dihydroxy material isolated due to the ability of the hydroxy at the 6
position to hydrogen bond with the aldehyde. Compound 2.34 underwent a protection of the
hydroxy group with MOM-Cl.30 There were difficulties initially getting this reaction to proceed,
but it was found that the addition of the phase transfer catalyst TBAI allowed for the protection
to be completed in a moderate yield.12 The final step of the second aromatic synthon pathway is a
Pinnick oxidation with 2.35 to generate 2.20, A.
This final compound 2.20, A was designed to join with the aliphatic piece via an
esterification reaction. This will be discussed in a following section. After several attempts the
esterification was completed however there were difficulties getting the material completely
pure. Upon no longer having any more material to perform the esterification or subsequent
reactions, it was decided to change the synthetic pathway yet again to remove the MOM-Cl
protection all together.

54
Scheme 2.6 shows the final pathway for synthesis of the aromatic portion. The first step
involves conducting a Veilsmeire-Haack reaction on 2.32 same as the previous pathway.
However, the second step goes directly to the Pinnick oxidation to form 2.20, A. With this
material in hand, more attempts to perform the esterification are underway and development of
the aliphatic synthon will be discussed in the following section.
2.5 Total Synthesis of Dihydroresorcylide: Aliphatic Synthon
The synthesis of the aliphatic synthon B was developed through two pathways. Scheme
2.7 through 2.9 goes through the development of the first method of generation of compound
2.41. Scheme 2.7 illustrates reacting (S)-propylene oxide 2.37 with vinylmagnesium bromide to
form 2.22 in moderate to high yields with no need for purification.31
Scheme 2.8 utilizes pent-4-en-1-ol 2.38 to undergo a oxidation to form 2.39.
Unfortunately, a completely accurate yield was never obtained due to the inability to get the
material purified, however the material recovered was approximately 60-75%. With aldehyde
2.39 in hand, a Grignard reaction with methylmagnesium bromide was conducted to afford the
resulting alcohol 2.40 in an 85% yield. A final oxidation was completed with 2.40 to give hex-5-
en-2-one 2.23, however an accurate yield could not be calculated due to the inability to remove
the remaining DMP.

55
Scheme 2.9 shows the combination of compounds 2.22 and 2.23 via a cross metathesis.
Unfortunately, even though this reaction is widely known the conversion would not take place.
Both compounds tend to homodimerize based on what is known about Grubbs cross metathesis
and would need an additive to obtain the desired species 2.41.32 At this time the amount of hex-
5-en-2-one 2.23 had diminished and it was decided to purchase 2.23 as it was more cost effective
and avoided the issues with purification ultimately reducing the overall number of steps in the
synthon synthesis.
Now that commercially available material 2.23 was obtained, the synthetic pathway was
altered as shown in Scheme 2.10. The Grignard creating homoallylic alcohol 2.22 was still
conducted and was then coupled with 2.23 after an efficient additive was found. Based on
literature, phenol was the best additive because of its ability to coordinate to the metal complex

56
and slow down homodimerization.33 After several attempts with no positive results it was
decided to distill the phenol, which turned the material from brown to clear. After the distillation,
the metathesis was successful with an overall yield of 67% which lead to the final step of
generation of the aliphatic synthon.
Hydrogenation of 2.41 concluded the aliphatic piece. The hydrogenation was conducted
with Pd/C in ethanol under 1 atm of H2.34 This reaction while successful was only able to
produce a 60% yield. The reasons are not clear, but this reaction is being optimized. Synthon
piece A and B were coupled together as mentioned previously and will be discussed in the
following section.
2.6 Total Synthesis of Dihydroresorcylide: Combining Synthons
As shown in Scheme 2.11, with the aromatic synthon 2.20 in hand, it would be reacted
with the aliphatic synthon 2.21 via an esterification reaction to form the new ester 2.42. The ester
would then undergo a direct Pd enolate ring closure to generate macrocycle 2.43. The
macrocycle would be treated with a strong acid to complete the deprotection to give compound
2.44 which would be followed by a demethylation with BBr3 to give the final Dihydroresorcylide
2.1.
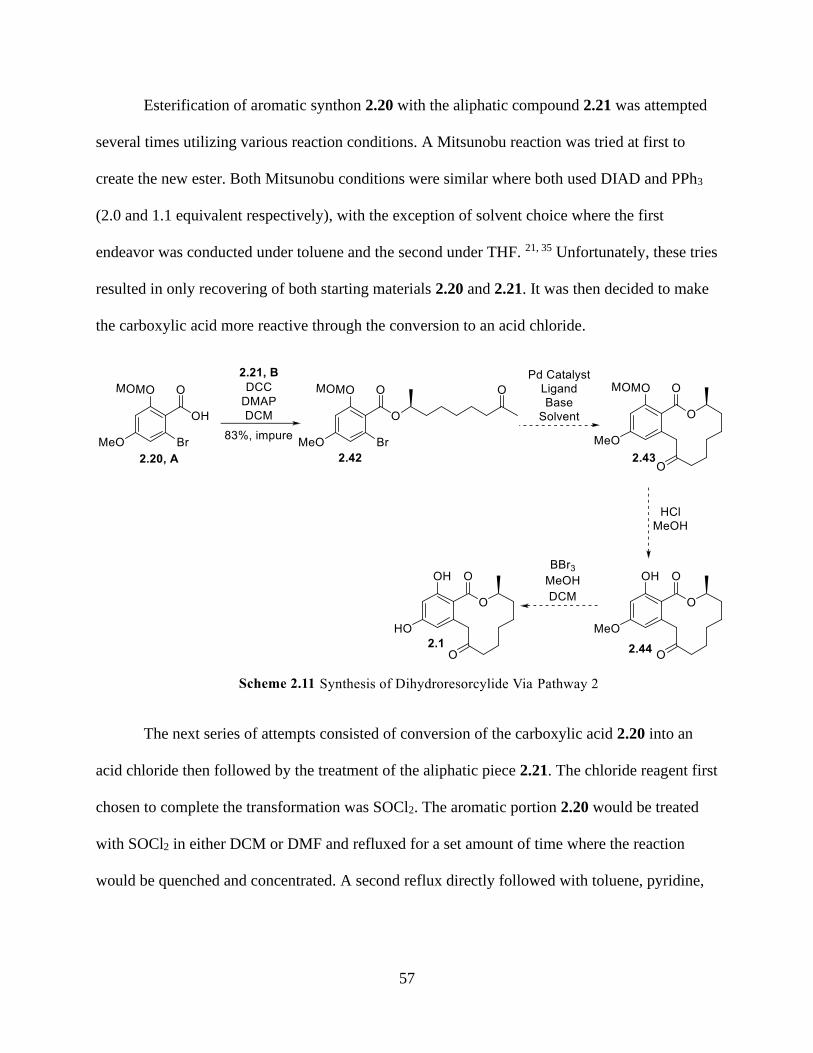
57
Esterification of aromatic synthon 2.20 with the aliphatic compound 2.21 was attempted
several times utilizing various reaction conditions. A Mitsunobu reaction was tried at first to
create the new ester. Both Mitsunobu conditions were similar where both used DIAD and PPh3
(2.0 and 1.1 equivalent respectively), with the exception of solvent choice where the first
endeavor was conducted under toluene and the second under THF. 21, 35 Unfortunately, these tries
resulted in only recovering of both starting materials 2.20 and 2.21. It was then decided to make
the carboxylic acid more reactive through the conversion to an acid chloride.
The next series of attempts consisted of conversion of the carboxylic acid 2.20 into an
acid chloride then followed by the treatment of the aliphatic piece 2.21. The chloride reagent first
chosen to complete the transformation was SOCl2. The aromatic portion 2.20 would be treated
with SOCl2 in either DCM or DMF and refluxed for a set amount of time where the reaction
would be quenched and concentrated. A second reflux directly followed with toluene, pyridine,

58
and 2.21.36 Again, there was no discernible ester formation detected which prompted the change
from SOCl2 to oxalyl chloride.
Two different reactions with oxalyl chloride were conducted.10, 37 The first employed
TPPO in acetonitrile, oxalyl chloride, and TEA to combine the two pieces while the second used
both DMF and DMAP as catalysts with oxalyl chloride in TEA and DCM. While both of these
processes were different, they both yielded the same results which was lack of esterified product
and only starting materials 2.20 and 2.21. Due to the inability to recover the desired compound
2.42, it was decided to forgo the acid chloride conversion for reactions with the carboxylic acid
as is.
Fischer esterification was utilized as the next attempt to join 2.20 and 2.21.38 The
materials were refluxed in THF and catalytic H2SO4 and again did not yield and promising
results other than the recovery of starting material. Therefore, the final method of creating the
new ester bond was to try a Steiglich esterification.39 The reaction was conducted several times
based on the work published by Santandrea in 2014, however the recrystallization was not
successful. With this in mind the work up was changed to allow for column purification which
led to the completion of the new ester bond to form compound 2.42. There were, however, some
difficulties with removing leftover DMAP even after column purification. Despite the slight
impurity, it was decided to take the material 2.42 and try to perform the desirable ring closure.
Generating the macrocycle 2.43 via a Pd enolate ring closure was attempted three times
based on work published by both Buchwald and Hartwig shown in Table 2.4.22, 40 All three
entries utilized Pd2(dba)3 as the catalyst of choice and LDA as a base. In entry 2, the ligand was
switched from DPPF to another bidentate ligand, BINAP, and in entry 3 the number equivalents
of base changed from 1.1 to 3.0. The only recoverable material from the reactions was starting
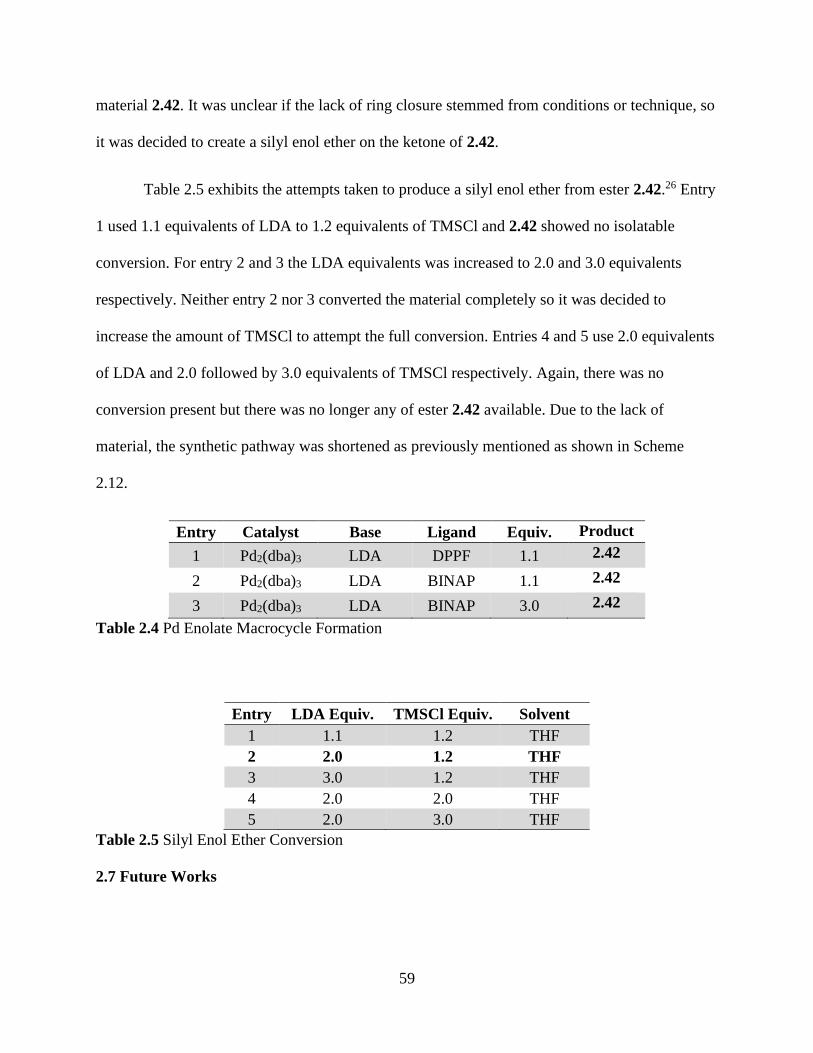
59
material 2.42. It was unclear if the lack of ring closure stemmed from conditions or technique, so
it was decided to create a silyl enol ether on the ketone of 2.42.
Table 2.5 exhibits the attempts taken to produce a silyl enol ether from ester 2.42.26 Entry
1 used 1.1 equivalents of LDA to 1.2 equivalents of TMSCl and 2.42 showed no isolatable
conversion. For entry 2 and 3 the LDA equivalents was increased to 2.0 and 3.0 equivalents
respectively. Neither entry 2 nor 3 converted the material completely so it was decided to
increase the amount of TMSCl to attempt the full conversion. Entries 4 and 5 use 2.0 equivalents
of LDA and 2.0 followed by 3.0 equivalents of TMSCl respectively. Again, there was no
conversion present but there was no longer any of ester 2.42 available. Due to the lack of
material, the synthetic pathway was shortened as previously mentioned as shown in Scheme
2.12.
Entry Catalyst Base Ligand Equiv. Product
1 Pd2(dba)3 LDA DPPF 1.1 2.42
2 Pd2(dba)3 LDA BINAP 1.1 2.42
3 Pd2(dba)3 LDA BINAP 3.0 2.42
Table 2.4 Pd Enolate Macrocycle Formation
Entry LDA Equiv. TMSCl Equiv. Solvent
1 1.1 1.2 THF
2 2.0 1.2 THF
3 3.0 1.2 THF
4 2.0 2.0 THF
5 2.0 3.0 THF
Table 2.5 Silyl Enol Ether Conversion
2.7 Future Works

60
Compound 2.45 and 2.21 would be joined via an esterification shown in Scheme 2.12.
This method will potentially include EDC to replace DCC in another Steiglich esterification
attempt, as well as, Mitsunobu and Yamaguchi reactions will be experimented with. Once ester
2.46 is obtained, the material will undergo the macrocyclization followed by a demethylation of
both methoxy groups to afford Dihydroresorcylide 2.1. This synthetic pathway is currently being
conducted, but it is hopeful that the work will yield positive results.
2.8 Conclusion
Dihydroresorcylide was isolated from maize and has been synthesized two times before.
However, the macrocycle formation has not gone through a Pd enolate ring closure as this is
novel to the research presented herein. The synthetic pathway of Dihydroresorcylide has changed
multiple times throughout the course of this work. The attempt to generate the natural product is
currently underway and the previous pathways will give valuable insight into the future
endeavors.

61
2.9 Supporting Information for Chapter 2
General Procedure: All of the reactions were performed under Ar in flame-dried glassware. All
starting materials, solvents, reagents, and catalysts were commercially available and used
without further purification. The NMR spectra were recorded with either a 360 or 500 MHz
Bruker spectrometer. 1H and 13C NMR spectra were obtained using CDCl3 as the solvent with
chloroform (CHCl3 1H: δ = 7.26 ppm, CDCl3 13C: δ = 77.0 ppm) as the internal standard.
Column chromatography was performed using 60-200 µm silica gel. Analytical thin layer
chromatography was performed on silica coated glass plates with F-254 indicator. Visualization
was accomplished by UV light (254 nm) and KMnO4.
Synthesis of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one (2.25): A
solution of 2.24 (24.0 mmol, 5.0 g, 1.0 eq.) and THF (0.2 M, 120 mL) was added to a flame
dried and purged flask and then cooled to 0 oC. MeOH (42.0 mmol, 1.82 mL, 1.75 eq.) and PPh3
(26.0 mmol, 6.87 g, 1.1 eq.) were added sequentially and the mixture stirred for five minutes.
DIAD (26.0 mmol, 5.14 mL, 1.1 eq.) was added dropwise and the reaction stirred at 0 oC for four
hours. The solution was then concentrated and purified via column chromatography in 10%
EtOAc and hexanes to afford 2.25. Rf: 0.2; Yield: 4.21 g, 79% as white solid. 1H NMR (360
MHz, CDCl3) δ 10.45 (s, 1H), 6.15 (d, J = 2.3 Hz, 1H), 6.00 (d, J = 2.3 Hz, 1H), 3.82 (s, 3H),
1.73 (s, 6H).21
Synthesis of 7-methoxy-2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-5-yl
trifluoromethanesulfonate (2.26): To a flame dried and purged flask, a solution of 2.25 (11.2
mmol, 2.5 g, 1.0 eq.) in pyridine (0.15 M. 74.4 mL) was added and then the reaction was cooled
to 0 oC. Tf2O (16.7 mmol, 2.81 mL, 1.5 eq.) was then added in one portion and the mixture
stirred at 0 oC for 24 hours. The contents were then diluted with 50 mL of EtOAc where the

62
organic layer was washed with 50 mL of saturated CuSO4, followed by 50 mL of DI water, and
then underwent a final wash with 50 mL of brine. The organic material was dried with MgSO4
and the concentrated. Purification took place via column chromatography in 10% EtOAc in
hexanes to afford 2.26. Rf: 0.17; Yield: 3.72 g, 80% as a white solid. 1H NMR (500 MHz,
CDCl3) δ 6.53 (d, J = 2.3 Hz, 1H), 6.48 (d, J = 2.4 Hz, 1H), 3.88 (s, 3H), 1.74 (s, 6H).21
Tert-butyl 2-hydroxy-4-methoxy-6-(((trifluoromethyl)sulfonyl)oxy)benzoate (2.27) (Table
2.1, Entry 4): Purified via column chromatography in 10% EtOAc in hexanes. Rf: 0.20; Yield:
0.063g, 60% as a white solid. 1H NMR (500 MHz, CDCl3) δ 11.87 (s, 1H), 6.47 (d, J = 2.5 Hz,
1H), 6.32 (d, J = 2.2 Hz, 1H), 3.83 (s, 3H), 1.63 (s, 9H).21
Synthesis of trimethyl(non-1-en-2-yloxy)silane (2.28): nBuLi in hexanes (0.77 mmol, 0.70
mL, 1.1 eq.) and DIPA (0.77 mmol, 0.11 mL, 1.1 eq.) in THF (0.17 M, 4.0 mL) were added to a
flame dried and purged flask and cooled to -78 oC. The reaction stirred for 30 minutes
maintaining temperature and then ketone 2.28 (0.70 mmol, 0.10 g, 1.0 eq.) was added dropwise.
The mixture stirred for another 30 minutes still at -78 oC where TMSCl (10.5 mmol, 1.34 mL,
1.5 eq.) was added dropwise. The solution stirred for one and a half hours maintaining the
temperature and it was then poured over a mixture of ice and hexanes. Extraction then occurred
three times with 10 mL of hexanes. The combined organic layers were washed with brine and
dried with MgSO4 where the final solution was concentrated to afford the desired silyl enol ether
(2.28). There was no purification necessary. 1H NMR (500 MHz, CDCl3) δ 4.05 (d, J = 1.5 Hz,
2H), 2.02 (m, 2H), 1.44 (m, 2H), 1.30 (m, 10H), 0.90 (t, J = 7.0 Hz, 3H), 0.22 (s, 9H).
Ethyl 2-(2-oxohexyl)benzoate (2.31) (Table 2.3, Entry 2): Material did not undergo
purification and still contained residual solvent. Yield: 0.025 g, 20% as a yellow oil. 1H NMR
(500 MHz, CDCl3) δ 7.46 (d, J = 1.4 Hz, 1H), 7.34 (dd, J = 7.6, 1.2 Hz, 1H), 7.17 (d, J = 7.6 Hz,

63
1H), 4.30 (d, J = 7.1 Hz, 1H), 4.09 (s, 1H), 2.53 (t, J = 7.5 Hz, 2H), 1.58 (m, 2H), 1.35 (td, J =
7.2, 2.5 Hz, 5H), 0.92 (t, J = 7.3 Hz, 3H).
Synthesis of 2-bromo-4,6-dimethoxybenzaldehyde (2.33): 2.32 (23.2 mmol, 5.0 g, 1.0 eq.) was
dissolved in DMF (0.08 M, 28.9 mL) and placed into a flame dried and purged flask. The flask
was then cooled to 0 oC where POCl3 (57.8 mmol, 5.41 mL, 2.5 eq.) was added dropwise and
allowed to stir at 0 oC for ten minutes. The reaction then stirred at RT for 30 minutes followed by
heating to 100 oC for four hours. The mixture was cooled and diluted with 50 mL of DI water
and then extracted three times with 50 mL of EtOAc. The combined organic layers were dried
with MgSO4 and concentrated. Purification via column chromatography in 30% EtOAC in
hexanes was conducted to afford 2.33. Rf: 0.2; Yield: 1.86 g, 33% as a white solid. 1H NMR
(500 MHz, CDCl3) δ 10.32 (s, 1H), 6.79 (d, J = 2.2 Hz, 1H), 6.44 (d, J = 2.2 Hz, 1H), 3.88 (d, J
= 12.8 Hz, 6H).29
Synthesis of 2-bromo-6-hydroxy-4-methoxybenzaldehyde (2.34): A solution of 2.33 (0.41
mmol, 0.10 g, 1.0 eq.) in DCM (0.20 M, 1.0 mL) was added to a flame dried and purged flask
that was then cooled to 0 oC. BBr3 (0.20 mmol, 0.02 mL, 0.50 eq.) was added dropwise and the
mixture stirred for 30 minutes at 0 oC. MeOH was added dropwise and the mixture was
concentrated. The material was extracted three times with 5 mL of EtOAc and the combined
organic layers were dried with MgSO4 and concentrated to afford 2.34. There was no purification
needed. Yield: 0.58 g, 64% as a white solid. 1H NMR (500 MHz, CDCl3) δ 12.47 (s, 1H), 10.11
(s, 1H), 6.75 (d, J = 2.4 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 3.85 (s, 3H).29
Synthesis of 2-bromo-4-methoxy-6-(methoxymethoxy)benzaldehyde 2.35: A flame dried and
purged flask was cooled to 0 oC and to it, 2.34 (0.43 mmol, 0.10 g, 1.0 eq.), TBAI (0.09 mmol,
0.03 g, 0.2 eq.), DIPEA (3.0 eq.), and MOMCl (0.87 mmol, 0.06 mL, 2.0 eq.) in DCM (0.2 M,

64
1.0 mL), were added. The reaction temperature was raised to RT and the mixture stirred for 24
hours. The flask was then cooled to 0 oC and quenched with 5 mL of saturated NaHCO3.and 5
mL of DI water. The aqueous was extracted with DCM and the combined organic layers were
dried with MgSO4 and concentrated. Purification took place via column chromatography in 25%
EtOAc in hexanes to afford (2.35). Rf: 0.3; Yield: 0.13 g, 79% as a white solid. 1H NMR (500
MHz, CDCl3) δ 10.33 (s, 1H), 6.85 (d, J = 2.3 Hz, 1H), 6.71 (d, J = 2.3 Hz, 1H), 5.26 (s, 2H),
3.85 (s, 3H), 3.51 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 188.96, 164.32, 161.47, 126.56,
117.72, 113.53, 101.34, 95.13, 56.68, 55.93.
Synthesis of 2-bromo-4-methoxy-6-(methoxymethoxy)benzoic acid (2.20): A round bottom
flask was flame dried, purged, and then cooled to 0 oC. A solution of 2.35 (0.36 mmol, 0.10 g,
1.0 eq.) dissolved in t-BuOH and H2O (3:1 ratio, 2.2 mL and 0.92 mL) was added to the flask
followed by 2-methyl-2-butene (2.2 mmol, 0.23 mL, 6.0 eq.) in one portion. Two more mixtures
of t-BuOH and H2O were made (1:1 ratio, 0.92 mL) and in one NaClO2 (1.5 mmol, 0.13 g, 4.0
eq.) was dissolved and in the other NaH2PO4 (0.55 mmol, 0.07 g, 1.5 eq.). The solution of
NaClO2 was added to the system dropwise followed by the solution of NaH2PO4. The reaction
was raised to room temperature and stirred for five hours where it was then quenched with 10
mL of saturated NH4Cl. The material was then extracted with 10 mL of EtOAc three times. The
combined organic layers were then dried with MgSO4 and concentrated. Purification took place
via column chromatography in 5% MeOH in DCM to afford 2.20. Rf: 0.09; Yield 0.08 g, 80% as
a white solid. 1H NMR (500 MHz, CDCl3) δ 6.79 (d, J = 2.1 Hz, 1H), 6.70 (d, J = 2.1 Hz, 1H),
5.20 (s, 2H), 3.80 (s, 3H), 3.49 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 162.07, 156.41, 120.75,
111.67, 101.51, 95.34, 56.76, 56.06.

65
Synthesis of 2-bromo-4,6-dimethoxybenzoic acid (2.20, A): A round bottom flask was flame
dried, purged, and then cooled to 0 oC. A solution of 2.33 (0.41 mmol, 0.10 g, 1.0 eq.) dissolved
in t-BuOH and H2O (3:1 ratio, 2.2 mL and 0.92 mL) was added to the flask followed by 2-
methyl-2-butene (2.4 mmol, 0.26 mL, 6.0 eq.) in one portion. Two more mixtures of t-BuOH and
H2O were made (1:1 ratio, 0.92 mL) and in one NaClO2 (1.6 mmol, 0.15 g, 4.0 eq.) was
dissolved and in the other NaH2PO4 (0.62 mmol, 0.07 g, 1.5 eq.). The solution of NaClO2 was
added to the system dropwise followed by the solution of NaH2PO4. The reaction was raised to
room temperature and stirred for five hours where it was then quenched with 10 mL of saturated
NH4Cl. The material was then extracted with 10 mL of EtOAc three times. The combined
organic layers were then dried with MgSO4 and concentrated. Purification took place via column
chromatography in 5% MeOH in DCM to afford 2.20, A. Rf: 0.12; Yield 0.09 g, 80% as a white
solid. 1H NMR (360 MHz, CDCl3) δ 6.74 (d, J = 2.2 Hz, 1H), 6.43 (d, J = 2.1 Hz, 1H), 3.84 (d, J
= 11.9 Hz, 6H).
Synthesis of (S)-pent-4-en-2-ol (2.22): CuI (0.87 mmol, 0.16 g, 0.5 eq.) was added to a flame
dried and purged flask followed by THF (0.18 M, 10 mL). The flask was cooled to -78 oC and
vinylmagnesium bromide (1.0 M, 3.4 mL, 2.0 eq.) was added dropwise. The reaction stirred for
fifteen minutes and then (S)-propylene oxide (1.7 mmol, 0.10 g, 1.0 eq.) was added. The mixture
stirred overnight where the temperature gradually rose to RT. The temperature was lowered to 0
oC and the reaction was quenched with saturated NH4Cl. An extraction took place three time
with 10 mL of diethyl ether and the combined organic layers were dried with MgSO4 and
concentrated. Purification was not needed. Yield: 0.13 g, 90% as a yellow oil. 1H NMR (360
MHz, CDCl3) δ 5.81 (m, 1H), 5.12 (m, 2H), 3.85 (dd, J = 12.0, 6.0 Hz, 1H), 2.18 (m, 2H), 1.21
(d, J = 6.2 Hz, 3H).

66
Synthesis of pent-4-enal (2.39): To a flame dried and purged flask, 2.38 (1.2 mmol, 0.10 g, 1.0
eq.) and DMP (2.3 mmol, 0.99 g, 2.25 eq.) in DCM (0.17 M, 6.8 mL) were added at RT. The
reaction stirred for three hours and was then quenched with 3 mL of saturated NaHCO3 and 3
mL of Na2S2O3. The mixture stirred for an additional 30 minutes and was then extracted three
times with 10 mL of DCM. The combined organic layers were dried with MgSO4 and
concentrated. Purification could not be conducted and was taken on to the next step. Yield: 0.07
g, 75% as yellow oil. 1H NMR (360 MHz, CDCl3) δ 9.73 (t, J = 1.6 Hz, 1H), 5.72 (m, 2H), 4.95
(m, 4H), 2.04 (ddd, J = 8.4, 7.0, 4.2 Hz, 4H).
Synthesis of hex-5-en-2-ol (2.40): To a flame dried and purged flask, THF (0.24 M, 5.0 mL)
was added. The flask was cooled to -78 oC and methylmagnesium bromide (3.0 M, 0.80 mL, 2.0
eq.) was added dropwise. The reaction stirred for fifteen minutes and then (2.39) (1.2 mmol, 0.10
g, 1.0 eq.) was added. The mixture stirred overnight where the temperature gradually rose to RT.
The temperature was lowered to 0 oC and the reaction was quenched with saturated NH4Cl. An
extraction took place three time with 10 mL of diethyl ether and the combined organic layers
were dried with MgSO4 and concentrated. Purification was not needed. Yield: 0.10 g, 85% as a
yellow oil. 1H NMR (360 MHz, CDCl3) δ 5.79 (ddt, J = 16.9, 10.1, 6.7 Hz, 1H), 4.96 (m, 2H),
3.63 (t, J = 6.5 Hz, 1H), 2.04 (m, 2H), 1.45 (m, 3H), 1.18 (m, 2H).
Synthesis of hex-5-en-2-one (2.23): To a flame dried and purged flask, 2.40 (1.0 mmol, 0.10 g,
1.0 eq.) and DMP (1.2 mmol, 0.51 g, 1.2 eq.) in DCM (0.17 M, 6.0 mL) were added at RT. The
reaction stirred for three hours and was then quenched with 3 mL of saturated NaHCO3 and 3
mL of Na2S2O3. The mixture stirred for an additional 30 minutes and was then extracted three
times with 10 mL of DCM. The combined organic layers were dried with MgSO4 and
concentrated. Purification could not be conducted, and an accurate yield could not be obtained.

67
1H NMR (360 MHz, CDCl3) δ 5.78 (m, 1H), 5.02 (m, 2H), 2.80 (dt, J = 38.1, 7.3 Hz, 1H), 2.36
(m, 4H), 1.25 (s, 3H).
Synthesis of (R,E)-8-hydroxynon-5-en-2-one (2.41): A solution of 2.22 (1.2 mmol, 0.10 g, 1.0
eq.), 2.23 (3.5 mmol, 0.41 mL, 3.0 eq.), PhOH (0.58 mmol, 0.06 g, 0.5 eq.), and GC(II) (0.02
mmol, 0.02 g, 0.015 eq.) in DCM (0.2 M, 6.0 mL) was added to a flame dried and purged flask at
RT. The reaction stirred overnight where it was then concentrated and purified via column
chromatography in 25% EtOAc in hexanes. Rf: 0.1; Yield: 0.12 g, 66% as brown oil. 1H NMR
(360 MHz, CDCl3) δ 5.47 (m, 2H), 3.78 (ddd, J = 9.3, 7.4, 5.5 Hz, 1H), 2.51 (t, J = 7.2 Hz, 2H),
2.26 (m, 2H), 2.12 (m, 6H), 1.18 (m, 3H).
Synthesis of (R)-8-hydroxynonan-2-one (2.21): A solution of 2.41 (0.64 mmol, 0.10 g, 1.0 eq.)
in EtOH (0.06 M, 10 mL) was added to a flame dried and purged flask. The flask was then
placed under 1 atm of H2 overnight. The resulting mixture was carefully opened to air and
filtered through celite and washed with copious amounts of DCM. The material was concentrated
and purified via column chromatography in 25% EtOAc in hexanes. Rf: 0.2; Yield: 0.04 g, 42%
as yellow oil. 1H NMR (360 MHz, CDCl3) δ 3.76 (m, 1H), 2.39 (d, J = 7.4 Hz, 1H), 2.10 (s, 3H),
1.54 (dd, J = 14.7, 7.4 Hz, 2H), 1.35 (m, 6H), 1.15 (d, J = 6.2 Hz, 3H).
Synthesis of (S)-8-oxononan-2-yl 2-bromo-4-methoxy-6-(methoxymethoxy)benzoate (2.42):
To a flame dried and purged flask, 2.20 (0.34 mmol, 0.10 g, 1.0 eq.) and 2.21 (0.69 mmol, 0.11
g, 2.0 eq.) were added to DCM (0.05 M, 7.0 mL). DCC (069 mmol, 0.14 g, 2.0 eq.) and DMAP
(1.0 mmol, 0.13 g, 3.0 eq.) were sequentially added to the reaction and it stirred at RT for 18
hours. The mixture was then placed in the freezer for 5 hours and the filtrate was filtered and
purified. Purification took place via column chromatography in 30% EtOAc in hexanes, there
was residual solvent left over. Rf: 0.3; Yield: 0.12 g, 83% as white solid. 1H NMR (360 MHz,

68
CDCl3) δ 7.37 (s, 1H), 7.08 (s, 1H), 5.15 (dd, J = 12.7, 6.1 Hz, 1H), 3.91 (d, J = 3.7 Hz, 6H),
2.42 (td, J = 7.4, 2.5 Hz, 2H), 2.12 (m, 3H), 1.78 (m, 4H), 1.30 (m, 7H).

69
2.10 References for Chapter 2
1. Hamashima, Y.; Hotta, D.; Sodeoka, M., Direct Generation of Nucleophilic Chiral
Palladium Enolate from 1,3-Dicarbonyl Compounds: Catalytic Enantioselective Michael
Reaction with Enones. J. Am. Chem. Soc. 2002, 124, 11240-11241.
2. Lei, A.; Zhang, X., A novel palladium-catalyzed homocoupling reaction initiated by
transmetalation of palladium enolates. Tetrahedron Lett. 2002, 43, 2525-2528.
3. Qian, H.; Widenhoefer, R. A., Mechanism of the Palladium-Catalyzed Intramolecular
Hydroalkylation of 7-Octene-2,4-dione. J. Am. Chem. Soc. 2003, 125, 2056-2057.
4. Schuppe, A. W.; Huang, D.; Chen, Y.; Newhouse, T. R., Total Synthesis of (-)-
Xylogranatopyridine B via a Palladium-Catalyzed Oxidative Stannylation of Enones. J. Am.
Chem. Soc. 2018, 140, 2062-2066.
5. Sole, D.; Urbaneja, X.; Bonjoch, J., Synthesis of the 4-Azatricyclo[5.2.2.04,8]undecan-
10-one Core of Daphniphyllum Alkaloid Calyciphylline A Using a Pd-Catalyzed Enolate
Alkenylation. Org. Lett. 2005, 7, 5461-5464.
6. Delmotte, P.; Delmotte-Plaquee, J., A new antifungal substance of fungal origin. Nature.
1953, 171, 344.
7. Jana, N.; Nanda, S., Resorcylic acid lactones (RALs) and their structural congeners:
recent advances in their biosynthesis, chemical synthesis and biology. New J. Chem. 2018, 42,
17803-17873.
8. Brase, S.; Glaser, F.; Framer, C., Chemistry of Mycotoxins. Springer: 2012; p 280 pp.
9. Garbaccio, R. M.; Stachel, S. J.; Baeschlin, D. K.; Danishefsky, S. J., Concise
Asymmetric Syntheses of Radicicol and Monocillin I. J. Am. Chem. Soc. 2001, 123, 10903-
10908.
10. Moulin, E.; Barluenga, S.; Winssinger, N., Concise Synthesis of Pochonin A, an HSP90
Inhibitor. Org. Lett. 2005, 7, 5637-5639.
11. Geng, X.; Danishefsky, S. J., Total Synthesis of Aigialomycin D. Org. Lett. 2004, 6, 413-
416.

70
12. Martinez-Solorio, D.; Belmore, K. A.; Jennings, M. P., Synthesis of the Purported ent-
Pochonin J Structure Featuring a Stereoselective Oxocarbenium Allylation. J. Org. Chem. 2011,
76, 3898-3908.
13. Poling, S. M.; Wicklow, D. T.; Rogers, K. D.; Gloer, J. B., Acremonium zeae, a
Protective Endophyte of Maize, Produces Dihydroresorcylide and 7-
Hydroxydihydroresorcylides. J. Agric. Food Chem. 2008, 56, 3006-3009.
14. He, W.-J.; Zhou, X.-J.; Qin, X.-C.; Mai, Y.-X.; Lin, X.-P.; Liao, S.-R.; Yang, B.;
Zhang, T.; Tu, Z.-C.; Wang, J.-F.; Liu, Y., Quinone/hydroquinone meroterpenoids with
antitubercular and cytotoxic activities produced by the sponge-derived fungus Gliomastix sp.
ZSDS1-F7. Nat. Prod. Res. 2017, 31, 604-609.
15. Wicklow, D. T.; Poling, S. M.; Summerbell, R. C., Occurrence of pyrrocidine and
dihydroresorcylide production among Acremonium zeae populations from maize grown in
different regions. Can. J. Plant Pathol. 2008, 30, 425-433.
16. Zeng, J.; Zhan, J., A novel fungal flavin-dependent halogenase for natural product
biosynthesis. Chembiochem 2010, 11, 2119-2123.
17. Zeng, J.; Valiente, J.; Zhan, J., Generation of two new macrolactones through sequential
biotransformation of dihydroresorcylide. Nat. Prod. Commun. 2011, 6, 223-226.
18. Jiang, C.-S.; Zhang, L.; Gong, J.-X.; Li, J.-Y.; Yao, L.-G.; Li, J.; Guo, Y.-W., Concise
synthesis and PTP1B inhibitory activity of (R)- and (S)-dihydroresorcylide. J. Asian Nat. Prod.
Res. 2017, 19, 1204-1213.
19. Combs, A. P., Recent Advances in the Discovery of Competitive Protein Tyrosine
Phosphatase 1B Inhibitors for the Treatment of Diabetes, Obesity, and Cancer. J. Med. Chem.
2010, 53, 2333-2344.
20. Zhang, L.; Ma, W.; Xu, L.; Deng, F.; Guo, Y., Efficient total synthesis of (S)-
dihydroresorcylide, a bioactive twelve-membered macrolide. Chin. J. Chem. 2013, 31, 339-343.
21. Trotter, T. N.; Albury, A. M. M.; Jennings, M. P., Total Synthesis of 7-Deoxy-6-O-
methylfusarentin Featuring a Chelation-Controlled 1,3-Reetz-Keck-Type Allylation. J. Org.
Chem. 2012, 77, 7688-7692.
22. Palucki, M.; Buchwald, S. L., Palladium-Catalyzed α-Arylation of Ketones. J. Am. Chem.
Soc. 1997, 119, 11108-11109.
23. Barnard, C. F. J., Palladium-Catalyzed Carbonylation-A Reaction Come of Age.
Organometallics 2008, 27, 5402-5422.

71
24. Cacchi, S.; Morera, E.; Ortar, G., Palladium-catalyzed carbonylation of enol triflates. A
novel method for one-carbon homologation of ketones to α,β-unsaturated carboxylic acid
derivatives. Tetrahedron Lett. 1985, 26, 1109-1112.
25. Kawatsura, M.; Hartwig, J. F., Simple, Highly Active Palladium Catalysts for Ketone and
Malonate Arylation: Dissecting the Importance of Chelation and Steric Hindrance. J. Am. Chem.
Soc. 1999, 121, 1473-1478.
26. Okabayashi, T.; Iida, A.; Takai, K.; Nawate, Y.; Misaki, T.; Tanabe, Y., Practical and
Robust Method for Regio- and Stereoselective Preparation of (E)-Ketene tert-Butyl TMS Acetals
and β-Ketoester-derived tert-Butyl (1Z,3E)-1,3-Bis(TMS)dienol Ethers. J. Org. Chem. 2007, 72,
8142-8145.
27. Carfagna, C.; Musco, A.; Sallese, G.; Santi, R.; Fiorani, T., Palladium-catalyzed
coupling reactions of aryl triflates or halides with ketene trimethylsilyl acetals. A new route to
alkyl 2-arylalkanoates. J. Org. Chem. 1991, 56, 261-263.
28. Iwama, T.; Rawal, V. H., Palladium-Catalyzed Regiocontrolled α-Arylation of
Trimethylsilyl Enol Ethers with Aryl Halides. Org. Lett. 2006, 8, 5725-5728.
29. Won, M.; Kwon, S.; Kim, T.-H., An efficient synthesis of Alternariol. J. Korean Chem.
Soc. 2015, 59, 471-474.
30. Kluge, A. F.; Untch, K. G.; Fried, J. H., Prostaglandins. X. Synthesis of prostaglandin
models and prostaglandins by conjugage addition of a functionalized organocopper reagent. J.
Amer. Chem. Soc. 1972, 94, 7827-7832.
31. De Joarder, D.; Jennings, M. P., Convergent synthesis of (+)-xestodecalactone A via a
Pd-catalyzed α-arylation reaction. Tetrahedron Lett. 2013, 54, 3990-3992.
32. Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H., A General Model for
Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360-11370.
33. Forman, G. S.; McConnell, A. E.; Tooze, R. P.; Van Rensburg, W. J.; Meyer, W. H.;
Kirk, M. M.; Dwyer, C. L.; Serfontein, D. W., A Convenient System for Improving the
Efficiency of First-Generation Ruthenium Olefin Metathesis Catalysts. Organometallics 2005,
24, 4528-4542.
34. De Joarder, D.; Jennings, M. P., Umpolung Pd-Catalyzed α-Arylation Reactions in
Natural Product Synthesis: Syntheses of (+)-Xestodecalactone A, (-)-Curvularin, (+)-12-
Oxocurvularin and (-)-Citreofuran. Eur. J. Org. Chem. 2015, 2015, 3303-3313.
35. Avuluri, S.; Bujaranipalli, S.; Das, S.; Yadav, J. S., Stereoselective synthesis of 5'-
hydroxyzearalenone. Tetrahedron Lett. 2018, 59, 3547-3549.

72
36. Bugarin, A.; Connell, B. T., Chiral Nickel(II) and Palladium(II) NCN-Pincer Complexes
Based on Substituted Benzene: Synthesis, Structure, and Lewis Acidity. Organometallics 2008,
27, 4357-4369.
37. Jia, M.; Jiang, L.; Niu, F.; Zhang, Y.; Sun, X., A novel and highly efficient
esterification process using triphenylphosphine oxide with oxalyl chloride. R. Soc. Open Sci.
2018, 5, 171988/1-88/12.
38. Lehman, J. W., Operational Organic Chemistry: A Problem-solving Approach to the
Laboratory Course. Pearson Prentice Hall: 2009.
39. Santandrea, J.; Bedard, A.-C.; Collins, S. K., Cu(I)-Catalyzed Macrocyclic Sonogashira-
Type Cross-Coupling. Org. Lett. 2014, 16, 3892-3895.
40. Culkin, D. A.; Hartwig, J. F., Palladium-Catalyzed α-Arylation of Carbonyl Compounds
and Nitriles. Acc. Chem. Res. 2003, 36, 234-245.
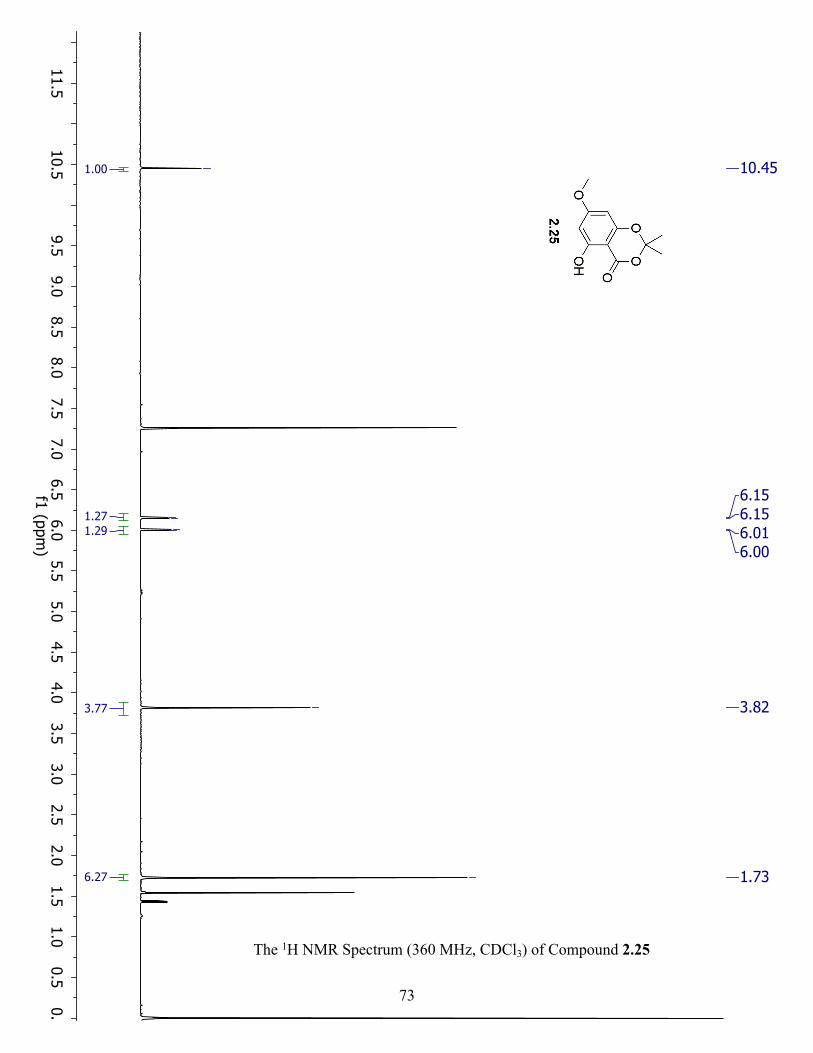
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
6.27
3.77
1.291.27
1.00
1.73
3.82
6.006.016.156.15
10.45
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.25
73
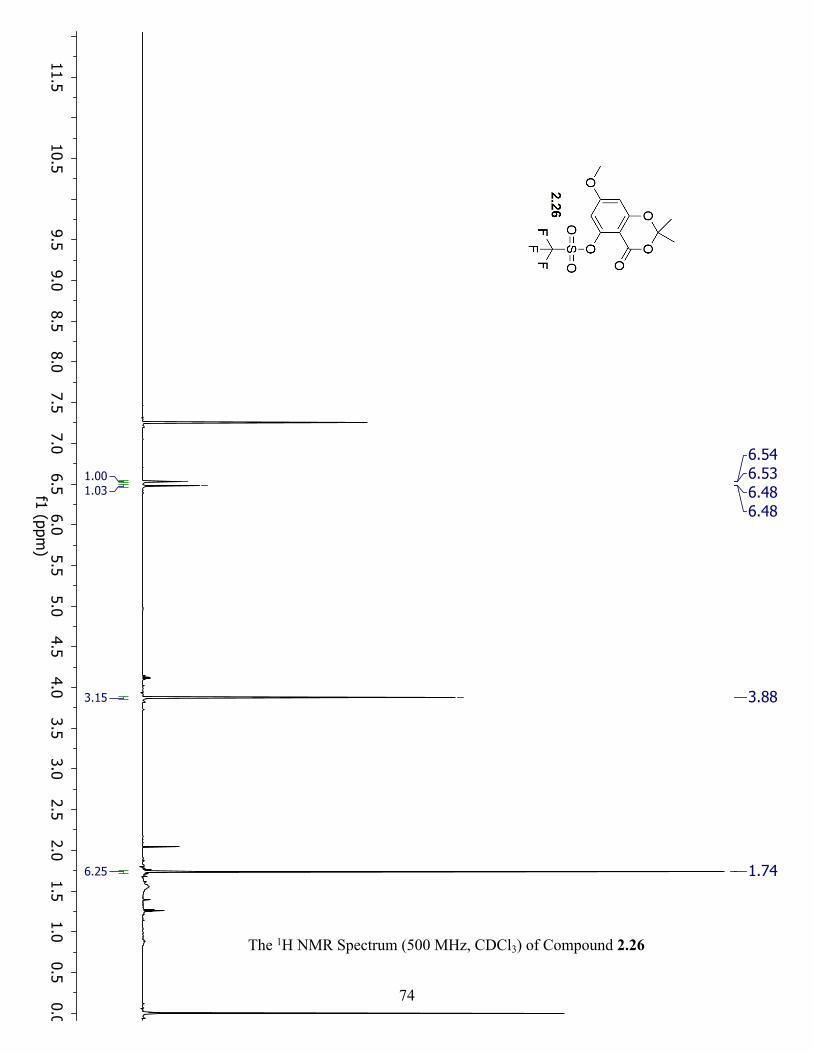
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
6.25
3.15
1.031.00
1.74
3.88
6.486.486.536.54
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.26
74
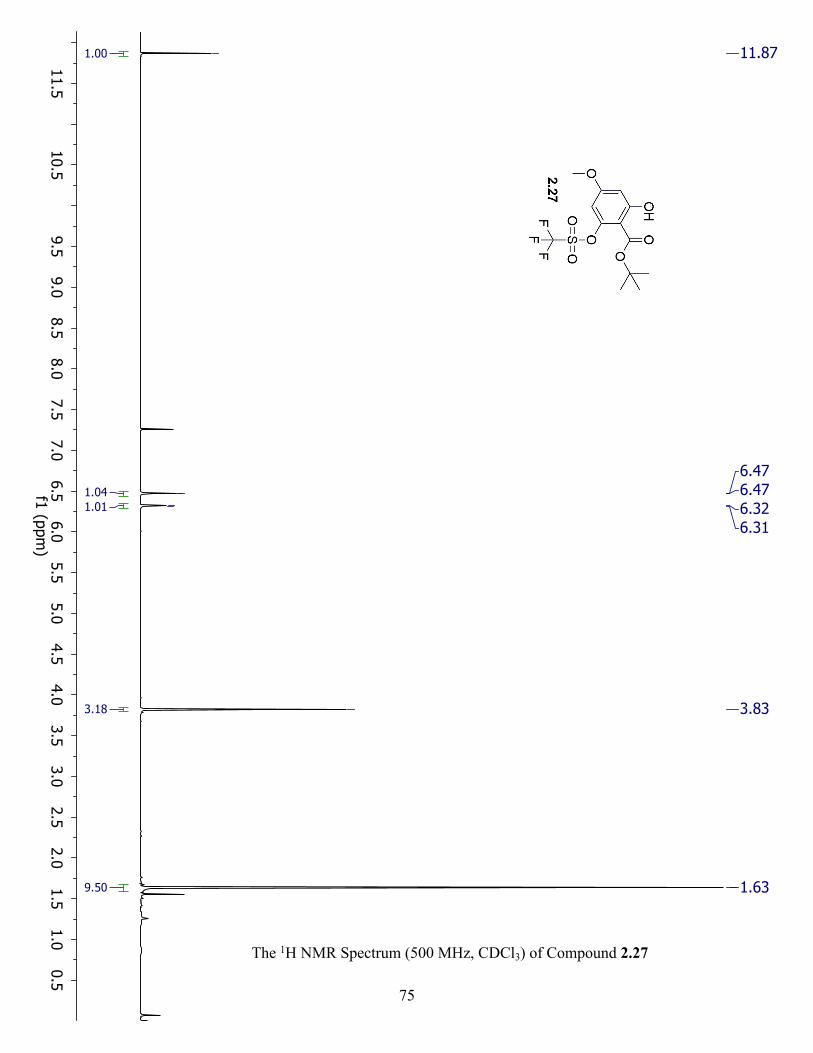
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
9.50
3.18
1.011.04
1.00
1.63
3.83
6.316.326.476.47
11.87
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.27
75

0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
9.14
3.63
9.722.08
2.10
2.00
0.220.880.90
1.301.442.002.022.03
4.054.05
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.28
76
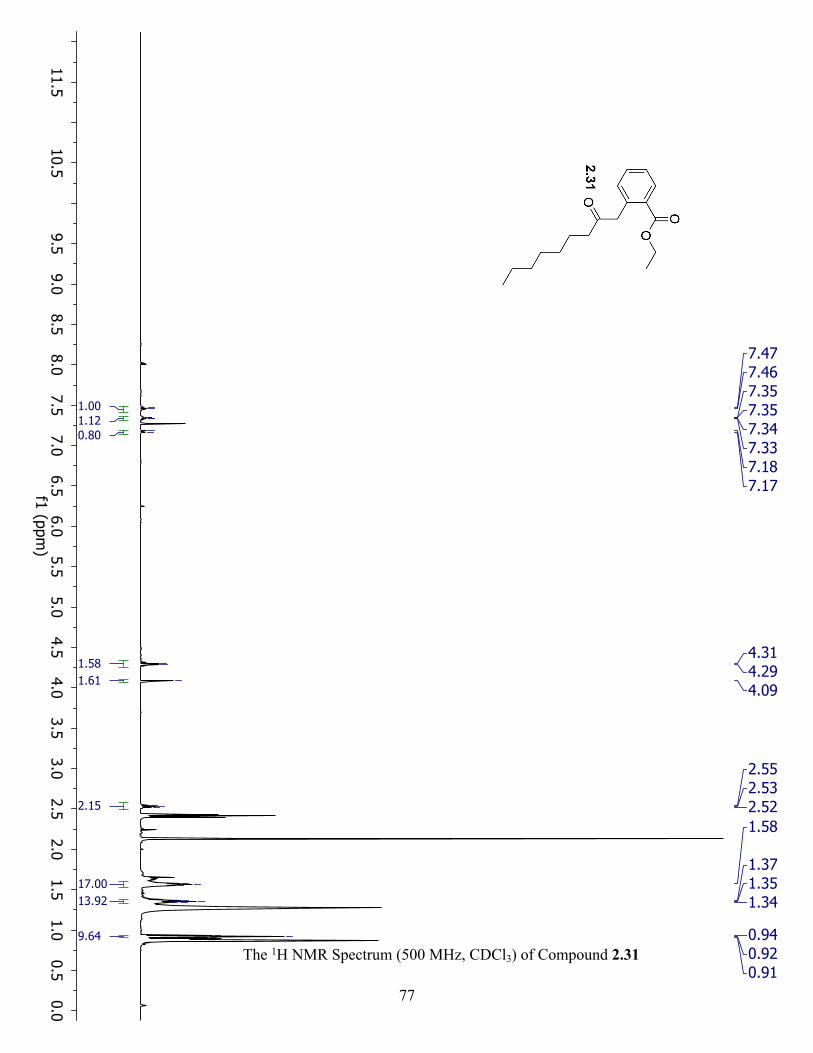
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
9.64
13.9217.00
2.15
1.611.58
0.801.121.00
0.910.920.94
1.341.351.37
1.582.522.532.55
4.094.294.31
7.177.187.337.347.357.357.467.47
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.31
77
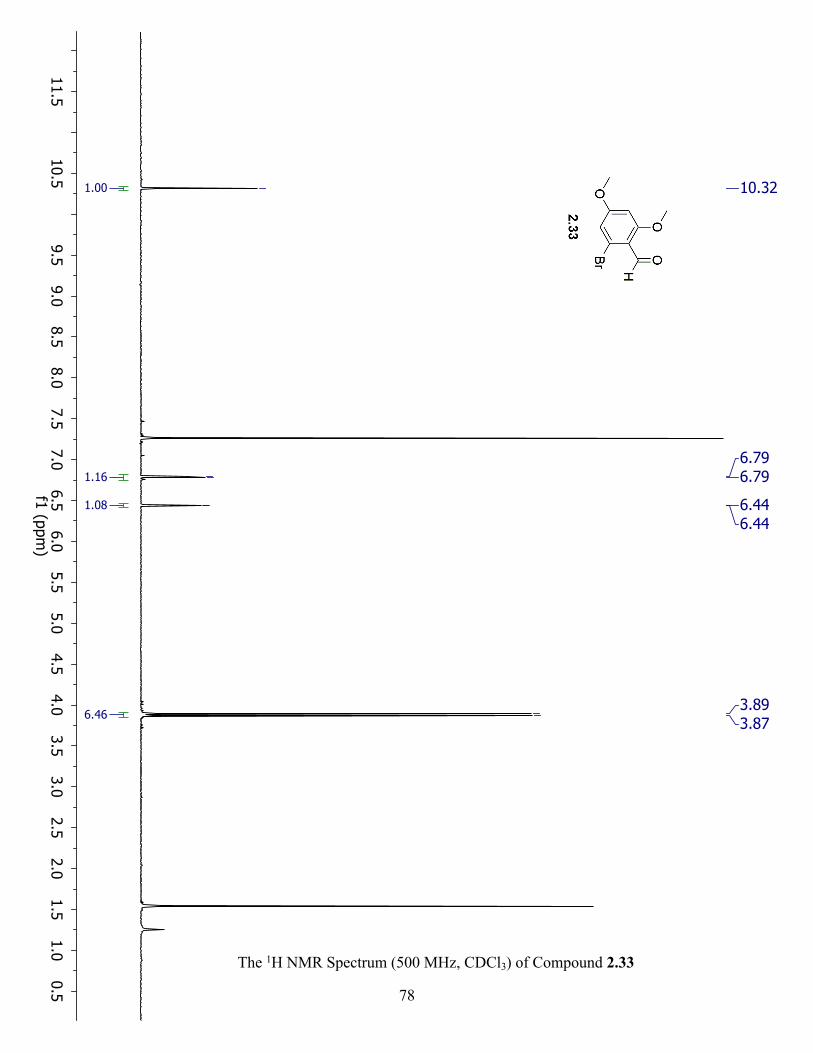
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
6.46
1.08
1.16
1.00
3.873.89
6.446.44
6.796.79
10.32
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.33
78
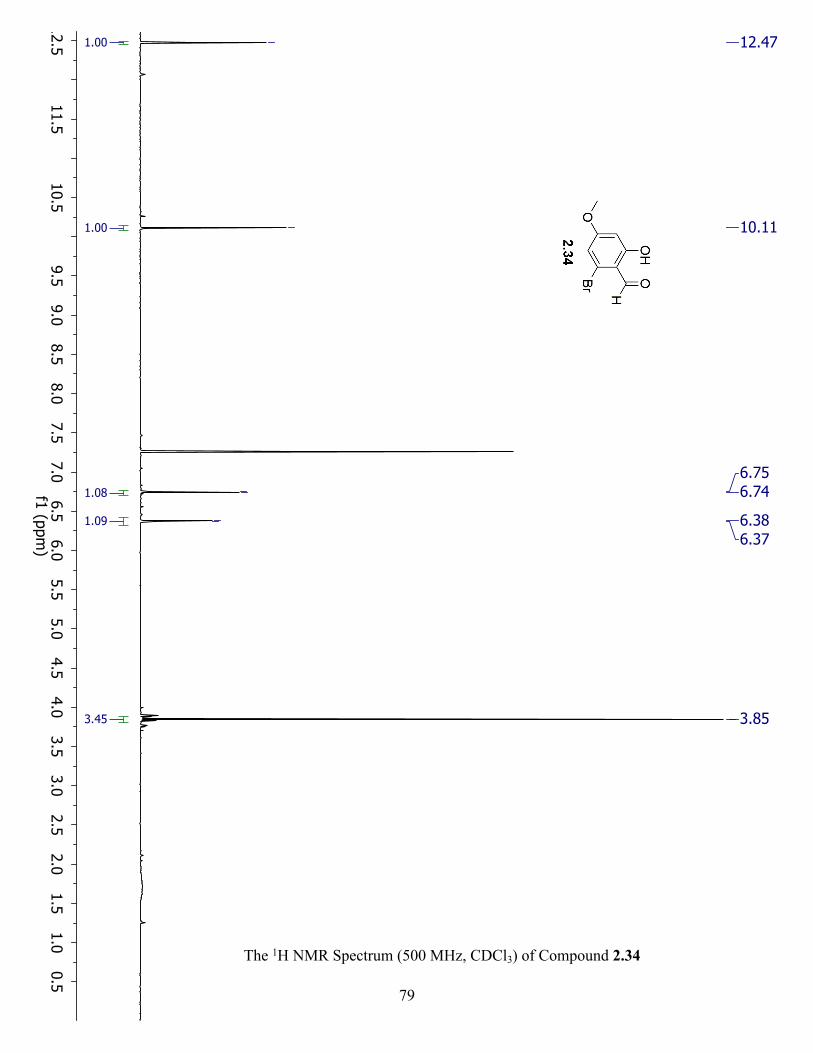
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.512.5
f1 (ppm)
3.45
1.09
1.08
1.00
1.00
3.85
6.376.38
6.746.75
10.11
12.47
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.34
79
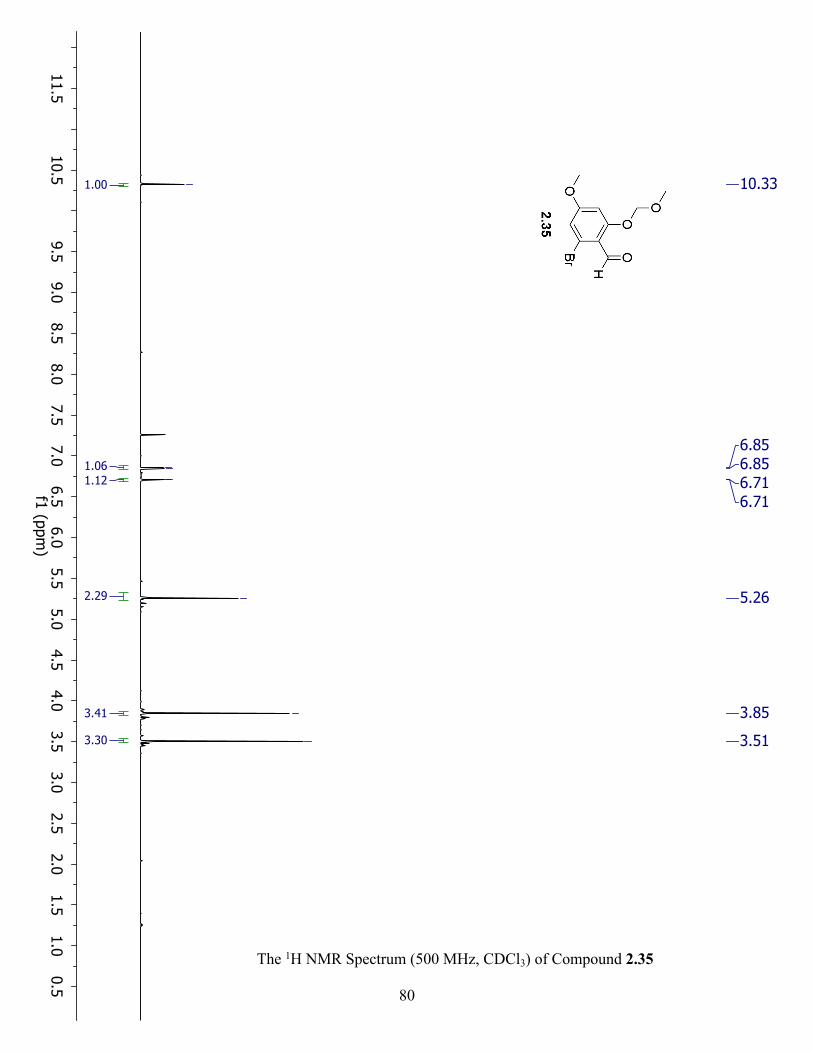
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
3.30
3.41
2.29
1.121.06
1.00
3.51
3.85
5.26
6.716.716.856.85
10.33
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.35
80
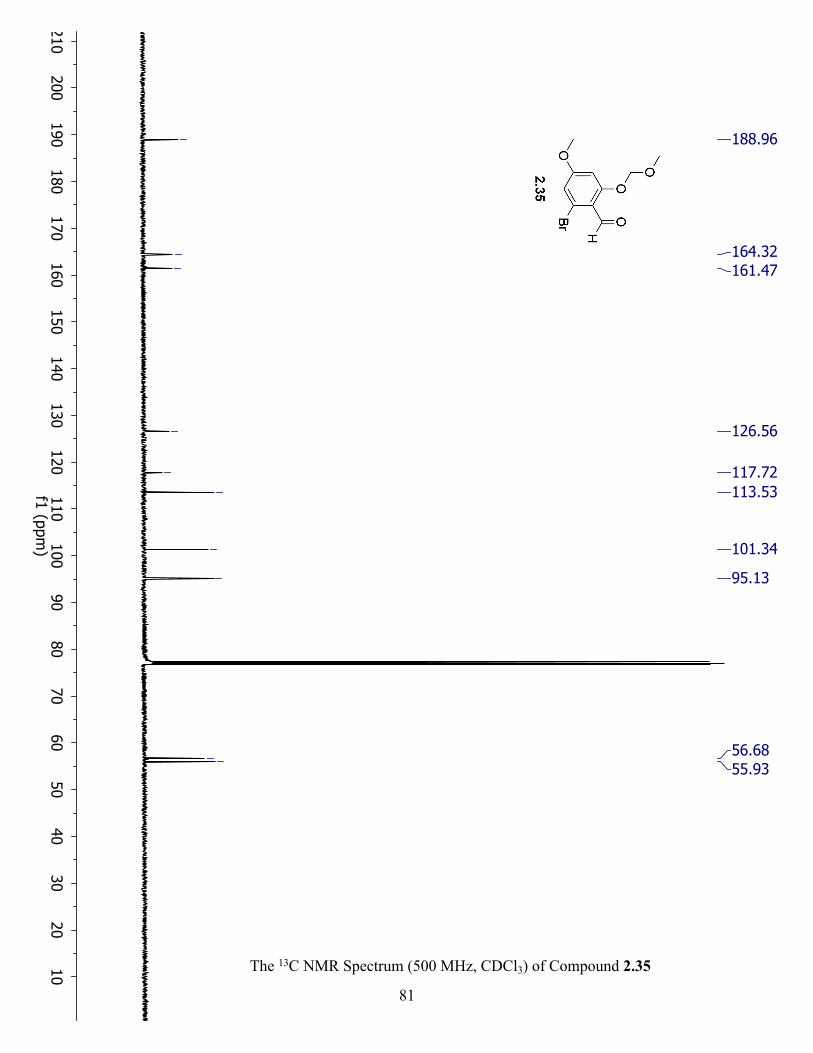
1020
3040
5060
7080
90100
110120
130140
150160
170180
190200
210f1 (ppm
)55.9356.68
95.13
101.34
113.53117.72
126.56
161.47164.32
188.96
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 2.35
81
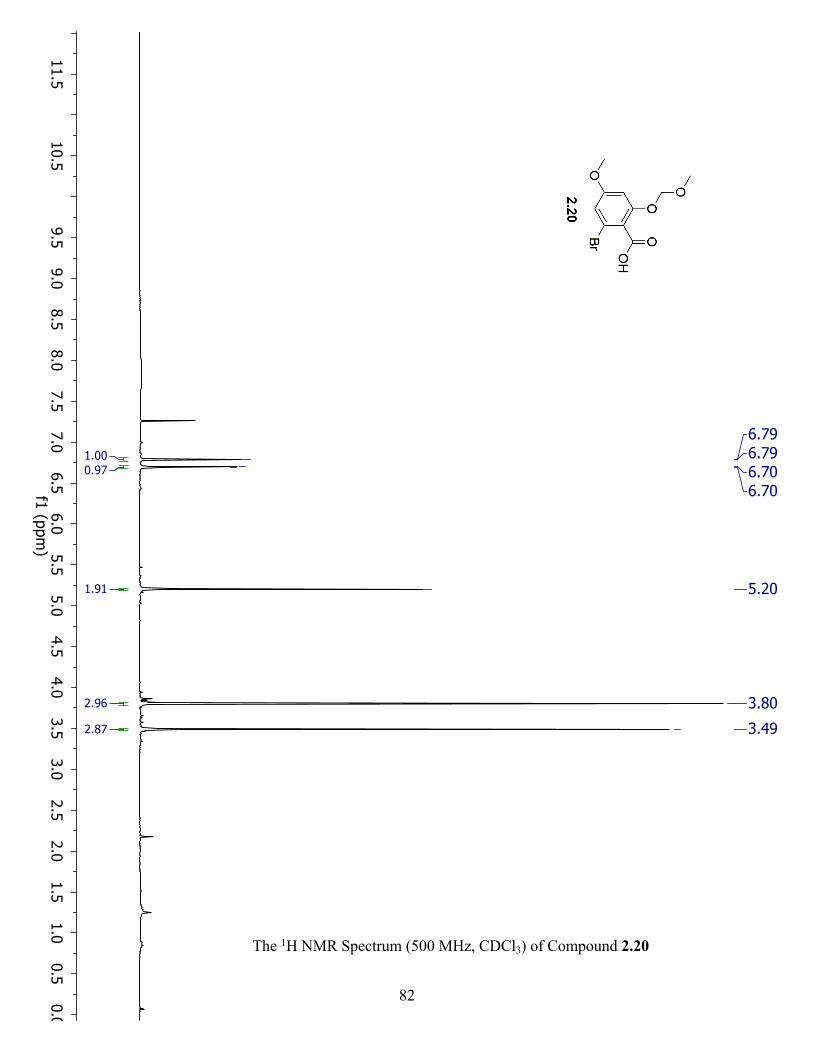
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
2.87
2.96
1.91
0.971.00
3.493.80
5.20
6.706.706.796.79
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 2.20
82
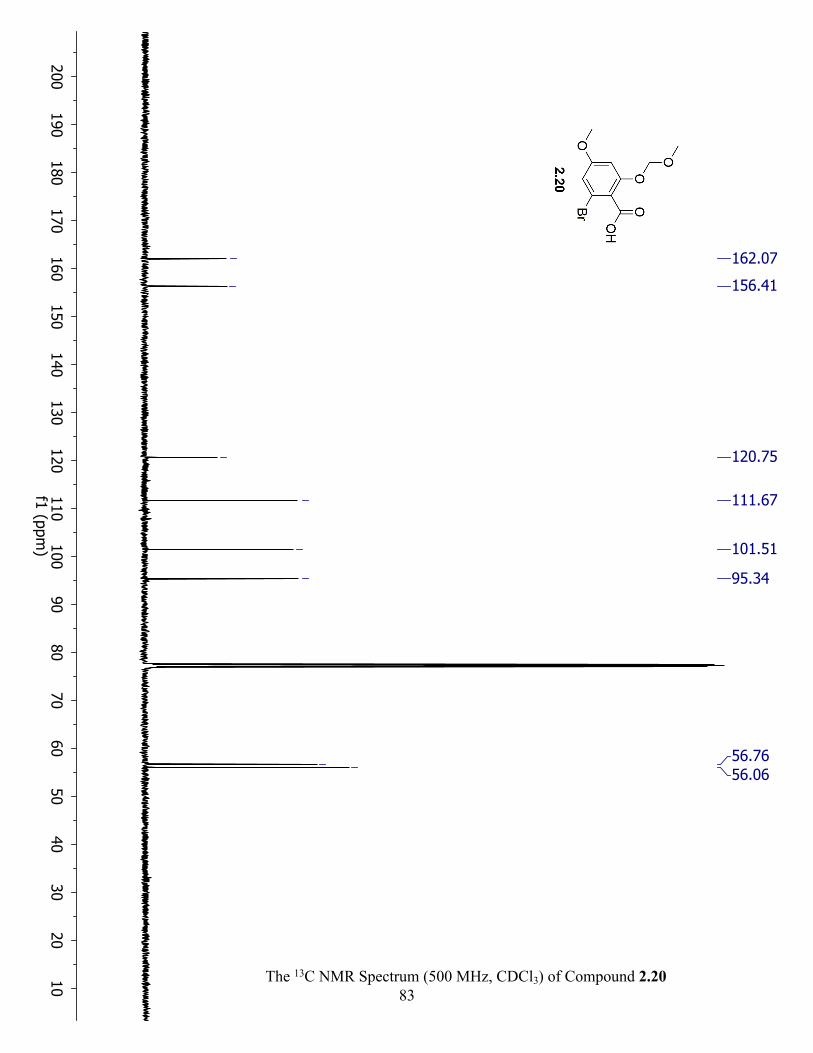
1020
3040
5060
7080
90100
110120
130140
150160
170180
190200
f1 (ppm)
56.0656.76
95.34
101.51
111.67
120.75
156.41162.07
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 2.2083
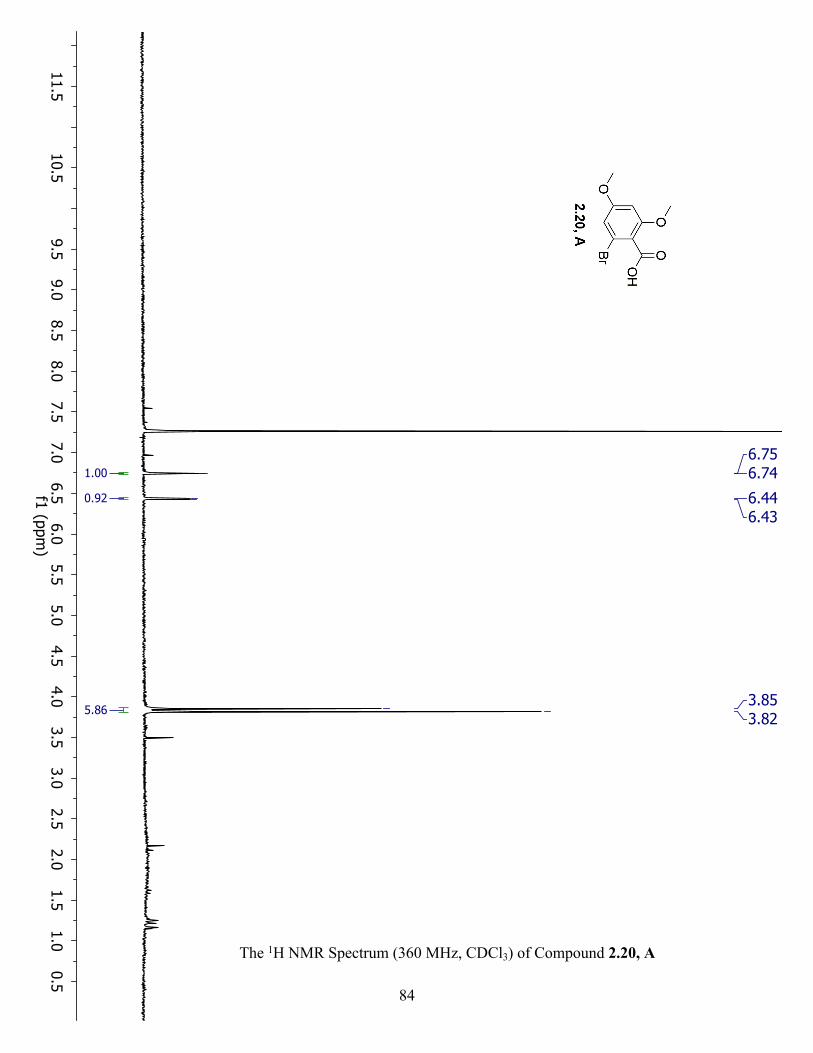
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
5.86
0.92
1.00
3.823.85
6.436.446.746.75
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.20, A
84
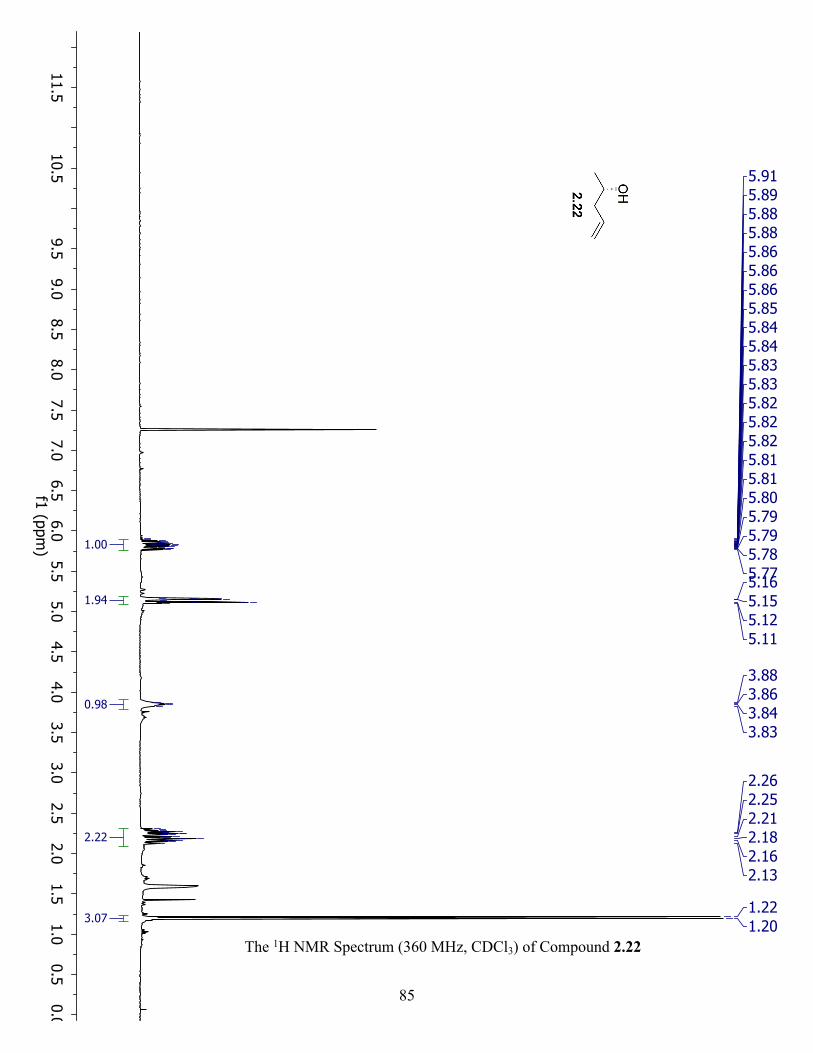
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
3.07
2.22
0.98
1.94
1.00
1.201.22
2.132.162.182.212.252.26
3.833.843.863.88
5.115.125.155.165.775.785.795.795.805.815.815.825.825.825.835.835.845.845.855.865.865.865.885.885.895.91
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.22
85
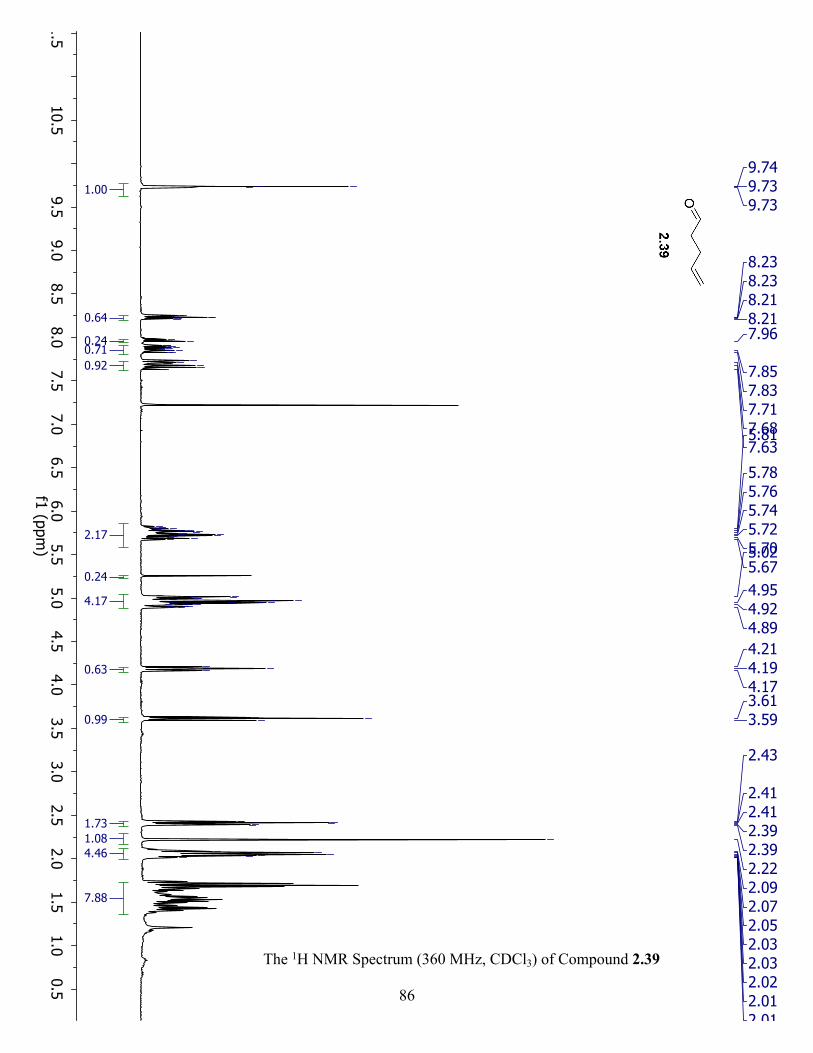
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
7.88
4.461.081.73
0.99
0.63
4.17
0.24
2.17
0.920.710.24
0.64
1.00
2.012.012.022.032.032.052.072.092.222.392.392.412.41
2.43
3.593.614.174.194.214.894.924.95
5.025.675.705.725.745.765.78
5.817.637.687.717.837.85
7.968.218.218.238.23
9.739.739.74
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.39
86
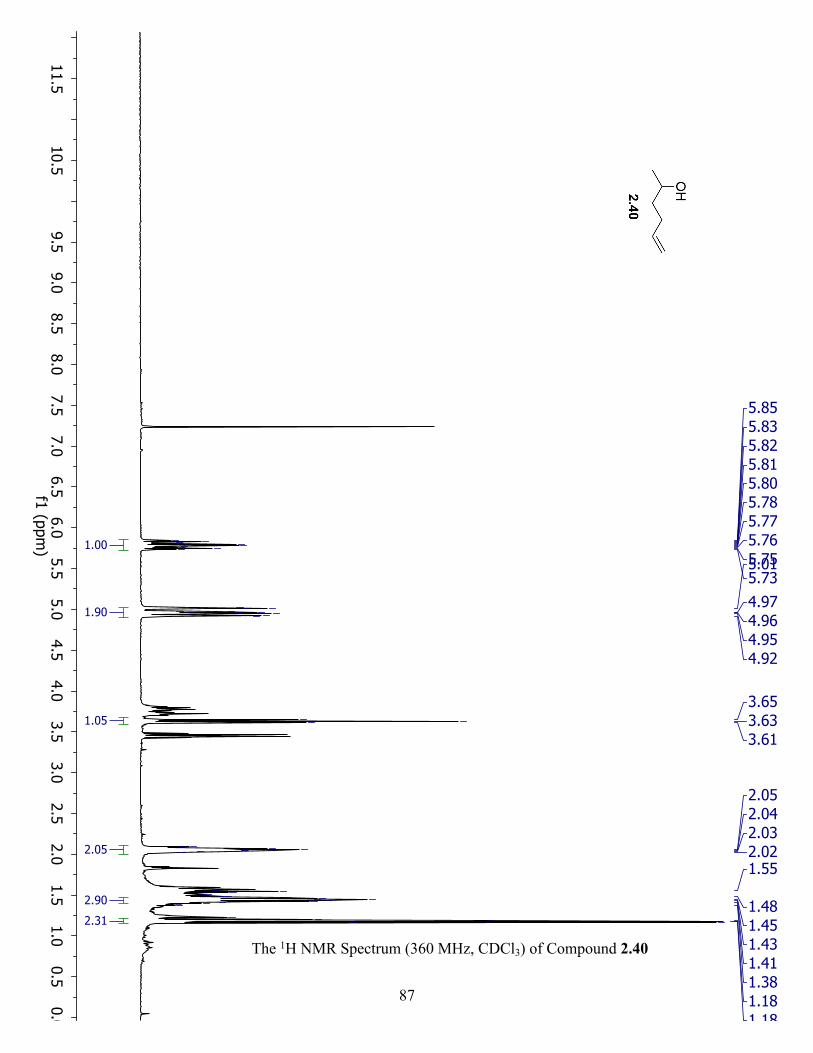
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
2.312.90
2.05
1.05
1.90
1.00
1.181.181.381.411.431.451.48
1.552.022.032.042.05
3.613.633.65
4.924.954.964.97
5.015.735.755.765.775.785.805.815.825.835.85
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.40
87
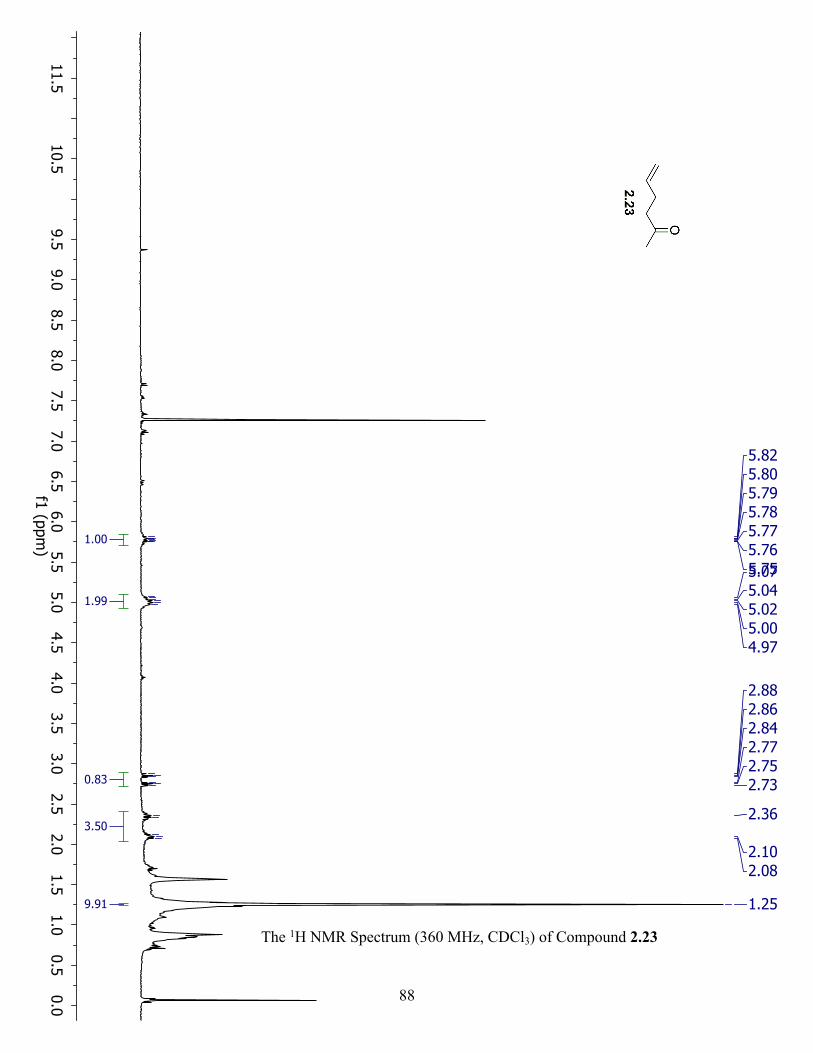
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.511.5
f1 (ppm)
9.91
3.50
0.83
1.99
1.00
1.25
2.082.10
2.36
2.732.752.772.842.862.88
4.975.005.025.045.075.755.765.775.785.795.805.82
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.23
88
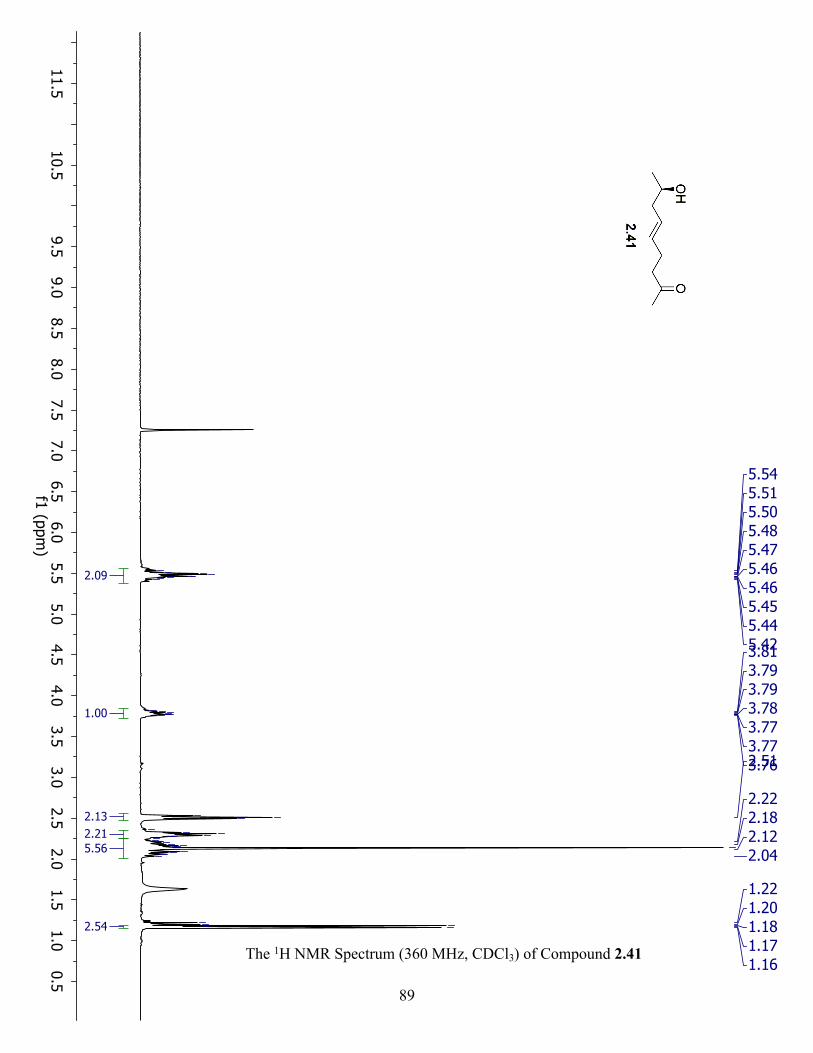
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
2.54
5.562.212.13
1.00
2.09
1.161.171.181.201.22
2.042.122.182.22
2.513.763.773.773.783.793.793.815.425.445.455.465.465.475.485.505.515.54
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.41
89
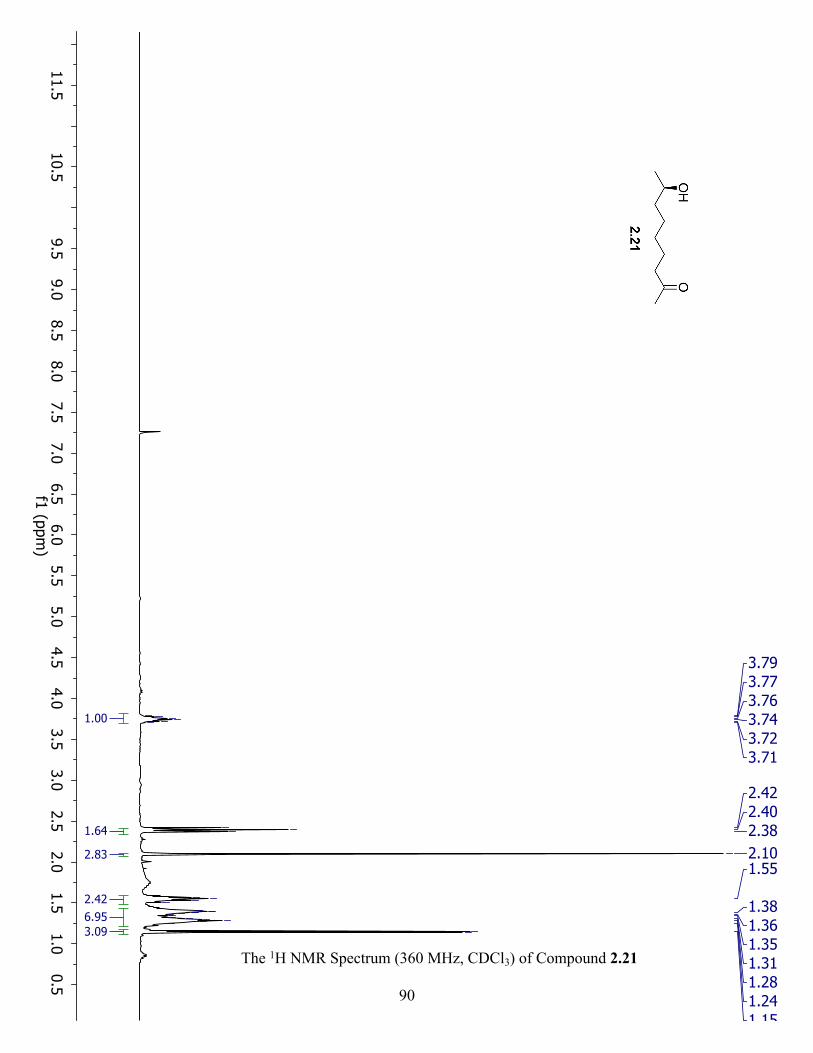
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
3.096.952.42
2.83
1.64
1.00
1.151.241.281.311.351.361.38
1.552.102.382.402.42
3.713.723.743.763.773.79
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.21
90
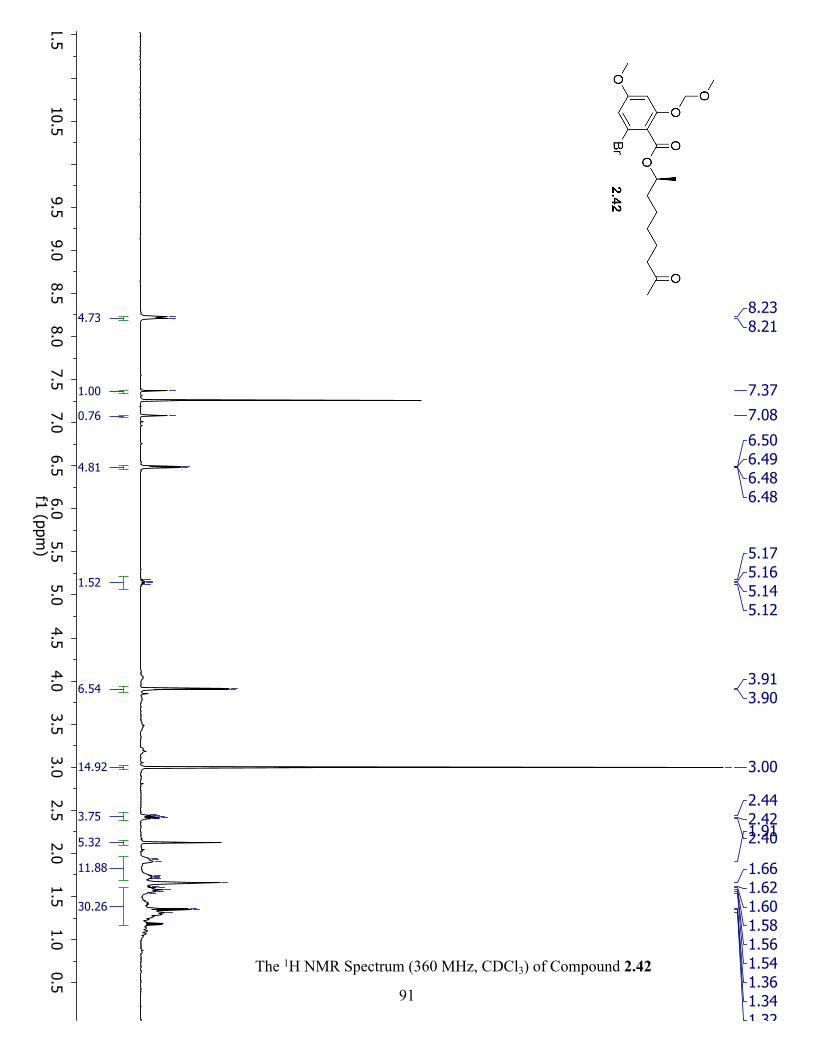
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.5
11.5f1 (ppm
)
30.26
11.88
5.32
3.75
14.92
6.54
1.52
4.81
0.76
1.00
4.73
1.321.341.361.541.561.581.601.621.66
1.912.402.422.44
3.00
3.903.91
5.125.145.165.17
6.486.486.496.507.087.37
8.218.23
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 2.42
91

92
CHAPTER 3: Stereoselective Halo-Succinimide Facilitated α-Halogenations of Substituted α-
Trialkylsilyl-ß-Substituted-α,ß-Unsaturated Esters
3.1 Introduction
Halogenations, specifically brominations, have been widely utilized in chemical reactions
because these atoms act as good leaving groups and help facilitate the formation of a variety of
new bonds. These take place across alkanes and benzylic carbons through a free radical
mechanism1 while alkenes and alkynes go through an electrophilic substitution2 and aromatics
undergo substitution via electrophilic aromatic substitution (EAS).3 There are several reagents
commonly used to conduct the brominations.
HBr and Br2 are some of the chemicals readily used to complete the halogenations.
However, Br2 is incredibly toxic leading to the study of other halogen sources. Transition metals
like, Fe, Pd, and Ru, have proven to be highly compatible for this addition or substitution on
complex materials.4-6 Another reagent that has proven to be useful when conducting
brominations is N-bromosuccinimide (NBS).7, 8
NBS is most known for its usefulness in the Wohl-Ziegler reaction where the halogen is
added at an allylic or benzylic position.9 It has two known mechanisms of bromine addition. The
first is a radical pathway and the second is the introduction of a bromonium ion complex.7 NBS
has been utilized with different types of compounds like vinyl silanes.

93
In 1987, Tamao et al. published work looking at the ability to substitute a vinyl trimethyl
silyl functional group with a halogen (Figure 3.1) and how that mechanism would work. Various
halogen sources such as, ICl, Br2, and NBS were utilized along with solvents like, DMF, CCl4,
and THF, which were successful in producing high yields of the desired halogenated compounds.
The mechanism was then studied to determine that the stereochemistry of the resulting alkene,
whether Z or E, and if it could be manipulated based on reaction conditions.10
In 1996, there was a compound library published that conducted halogenations under the
conditions set forth by Tamao, except these were completed with N-iodosuccinimide (NIS).11
These conditions were also implemented during the synthesis of several pharmaceutical or
natural products like Leukotriene B3, Haterumalide NA, and (S)-Jamaicamide C.12-14 There is
also material published concerning the bromination of styrene and related analogs and
dibrominations of arylethylenes.15, 16 However, there are not any current reports of brominations
concerning α-trialkylsilyl-α,ß-unsaturated esters.
Figure 3.2 shows the general structure of the aforementioned silanes and how versatile
these compounds are. Over the past several years these motifs have undergone different
transformations, often in a stereoselective manner, to increase the number of reactive sites.17-19
These small molecules have been studied concerning halogenations, however these studies
concerned addition of the halogen at the γ position and not the α shown in the research presented
within.20

94
Figure 3.3 shows the general reaction that will be discussed further. The halogenated
compounds could be used in Sonagashira, Sukuki, or Negishi coupling, all of which have had
their uses in total synthesis.21-23 The following work will discuss the novel results of this study
by using Tamao’s conditions as a guide.
3.2 The Generation of α-Trialkylsilyl-α, β-Unsaturated Esters
The formation of these highly functionalized vinyl silanes through ethyl propiolate (3.1)
have been extensively studied by the Jennings’ research group.18 They are believed to go through
a complex copper mediated cycle with the Grignard reagent to give a silyl ketene acetal
intermediate that upon work-up will selectively give the formation of the (E) silane (Figure 3.4).
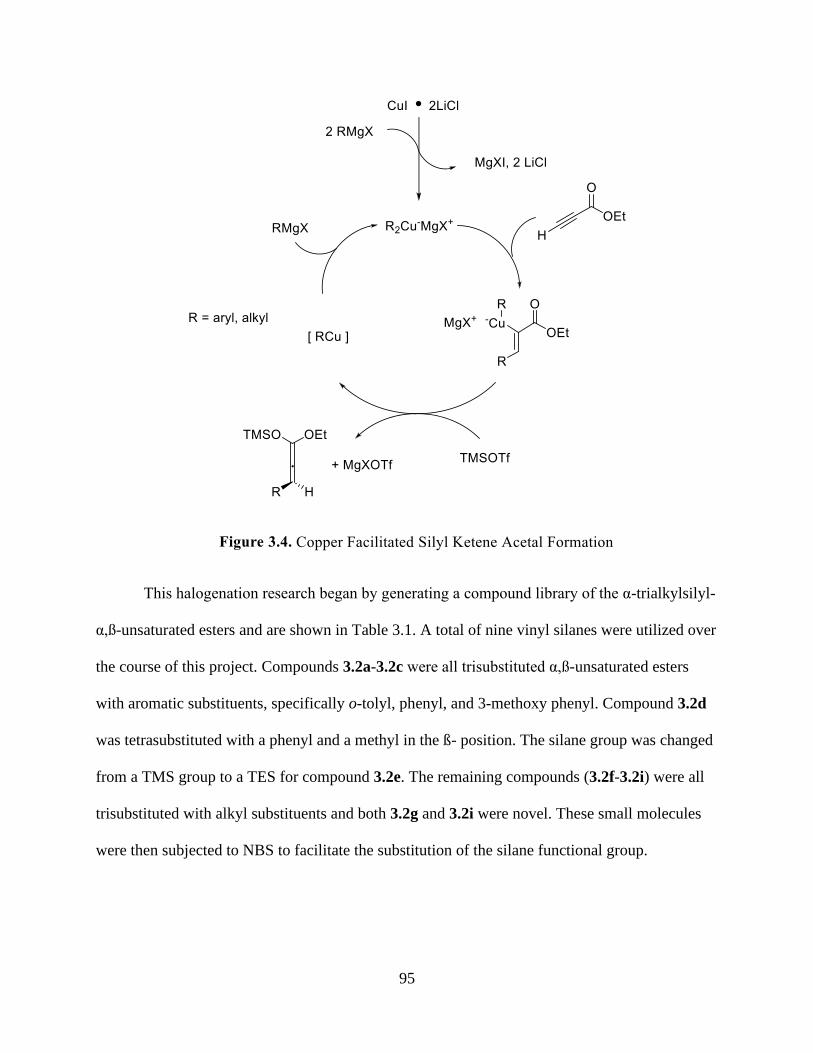
95
This halogenation research began by generating a compound library of the α-trialkylsilyl-
α,ß-unsaturated esters and are shown in Table 3.1. A total of nine vinyl silanes were utilized over
the course of this project. Compounds 3.2a-3.2c were all trisubstituted α,ß-unsaturated esters
with aromatic substituents, specifically o-tolyl, phenyl, and 3-methoxy phenyl. Compound 3.2d
was tetrasubstituted with a phenyl and a methyl in the ß- position. The silane group was changed
from a TMS group to a TES for compound 3.2e. The remaining compounds (3.2f-3.2i) were all
trisubstituted with alkyl substituents and both 3.2g and 3.2i were novel. These small molecules
were then subjected to NBS to facilitate the substitution of the silane functional group.
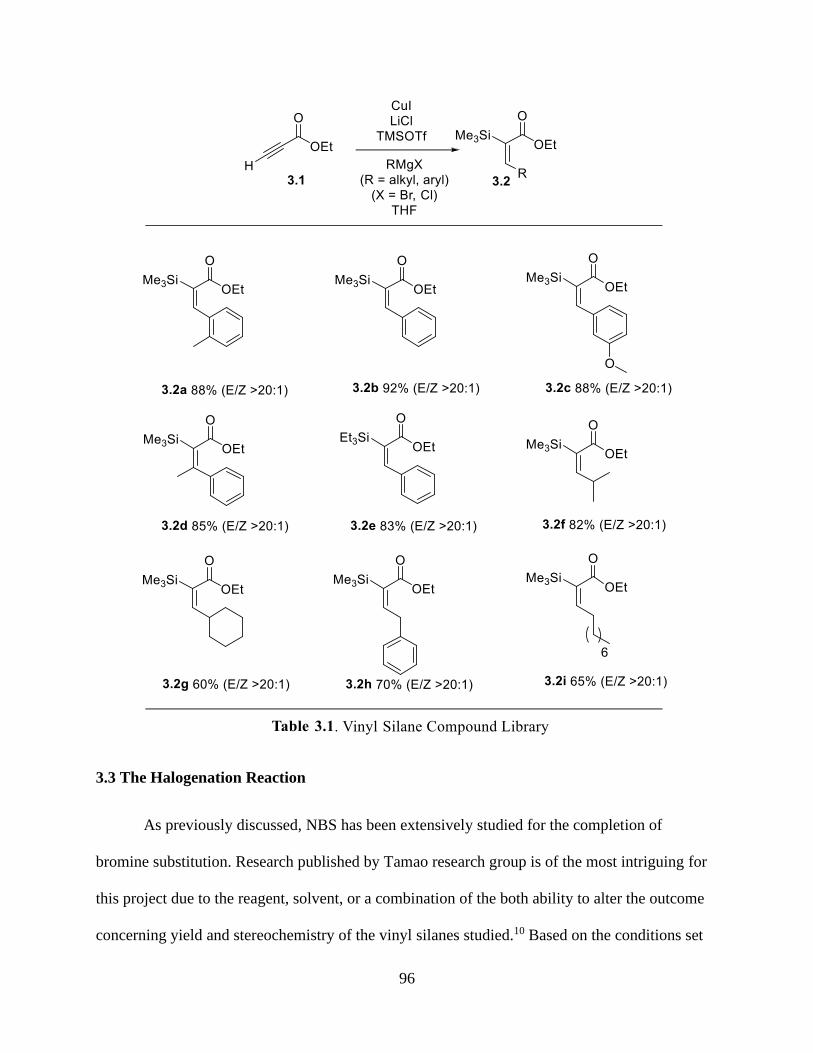
96
3.3 The Halogenation Reaction
As previously discussed, NBS has been extensively studied for the completion of
bromine substitution. Research published by Tamao research group is of the most intriguing for
this project due to the reagent, solvent, or a combination of the both ability to alter the outcome
concerning yield and stereochemistry of the vinyl silanes studied.10 Based on the conditions set

97
forth, a sample study was conducted to optimize reaction conditions concerning the unsaturated
ester silanes. Figure 3.5 shows the model reaction which was carried out under Ar at room
temperature while stirring for 18-24 hours with vinyl silane 3.2a, 2.0 equivalents of NBS, and
the solvent of choice. The conversion percent was established by crude NMR due to the
substantial shift of the ß proton seen when comparing starting material and products. Table 3.2
lists the findings of the solvent dependence study.
Entrya Solvent Conversion* %
1b DMF 100
2c DMF 63
3 DME 75
4 THF 53
5 Ether 26
6 DCM 10
7 Toluene 10
Table 3.2. Solvent Dependence Studies. *conversion calculated via crude 1H NMR. a. All entries
utilized 3.2a due to availability. 1b was conducted with 2.0 equivalents of NBS. 2c was
conducted with 4.0 equivalents of NBS.
All entries utilized 3.2a as the vinyl silane of choice due to the availability of the
compound. Entries 1 and 2 were both conducted with the aprotic solvent DMF, however, the
equivalents of NBS was altered 2.0 equivalents and 4.0 equivalents for entry 1 and 2

98
respectively. It was interesting to discover that double the equivalents had significant impact on
the conversion which is possibly due to the increase of Br2 formation in the system. Entries 3 and
4 explore other aprotic solvents such as DME and THF, and these two entries exhibited a
decreased amount of conversion that took place in the reaction. Entries 5-7 implemented other
organic solvents of varying polarity such as diethyl ether, DCM, and toluene, all of which also
showed a substantial decrease of silane conversion. It should be noted that the conversion
percent for these entries is significantly lowered than the previous donor solvents and cannot be
determined to be accurate. This is consistent with the work published by Tamao showing that as
the polarity decreases, so did silane bond cleavage. The less electronegative or nucleophilic a
solvent, the less chance there is for it to attack the ß-carbon and ring open the bromonium ion
intermediate. All the solvents appeared to give solely the (Z) isomer of the newly formed
brominated compounds leading to the belief that the reaction was not solvent dependent
regarding stereochemistry but was dependent regarding conversion. Based on the studies by
Tamao and ourselves, we can theorize a possible mechanism (Scheme 3.1).10
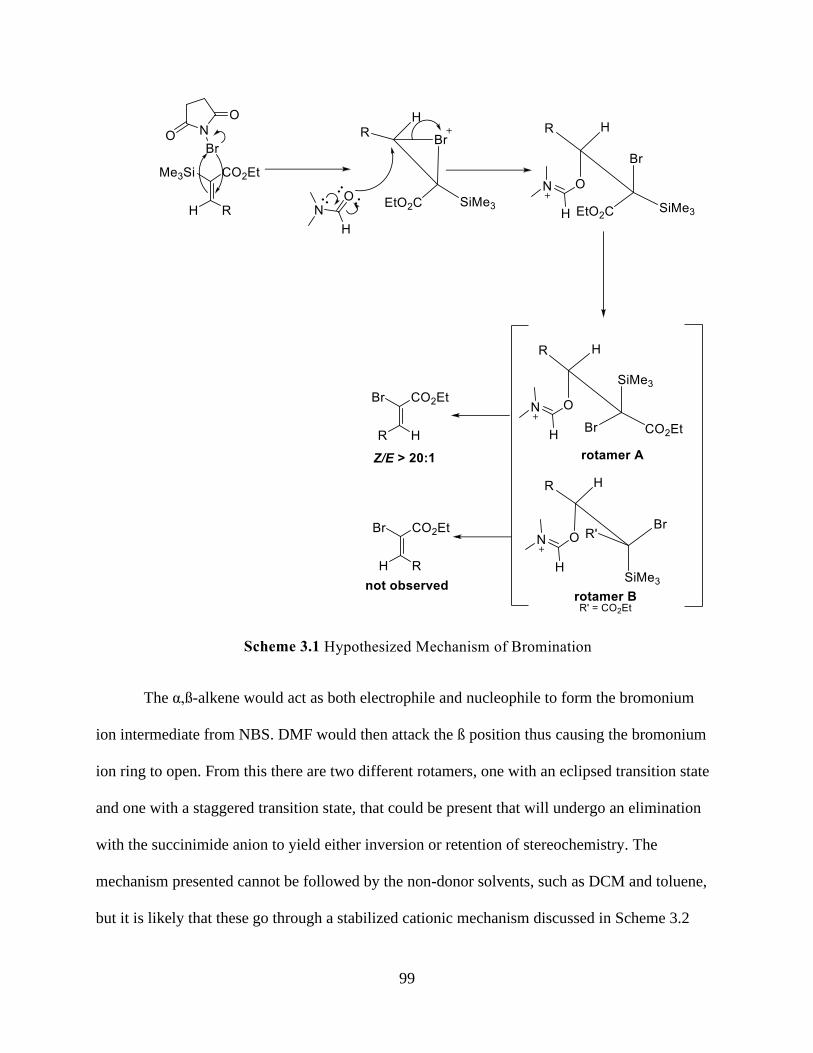
99
The α,ß-alkene would act as both electrophile and nucleophile to form the bromonium
ion intermediate from NBS. DMF would then attack the ß position thus causing the bromonium
ion ring to open. From this there are two different rotamers, one with an eclipsed transition state
and one with a staggered transition state, that could be present that will undergo an elimination
with the succinimide anion to yield either inversion or retention of stereochemistry. The
mechanism presented cannot be followed by the non-donor solvents, such as DCM and toluene,
but it is likely that these go through a stabilized cationic mechanism discussed in Scheme 3.2

100
later in the chapter. A compound library of α-bromo-α,ß-unsaturated esters was generated and
discussed in the following section.
3.4 (Z)-α-Halogen-α, β-Unsaturated Esters Compound Library
The transformation of α-trialkylsilyl-α,ß-unsaturated esters into α-halogen-α,ß-
unsaturated esters took place via 2.0 equivalents of NBS in DMF. Table 3.3 shows the molecules
that were generated with the previously mentioned vinyl silanes. The yields for the compounds
ranged from 58%-90% and while most gave complete inversion of stereochemistry, which was
verified by NOE studies shown in Figure 3.6, there were a few unexpected results.
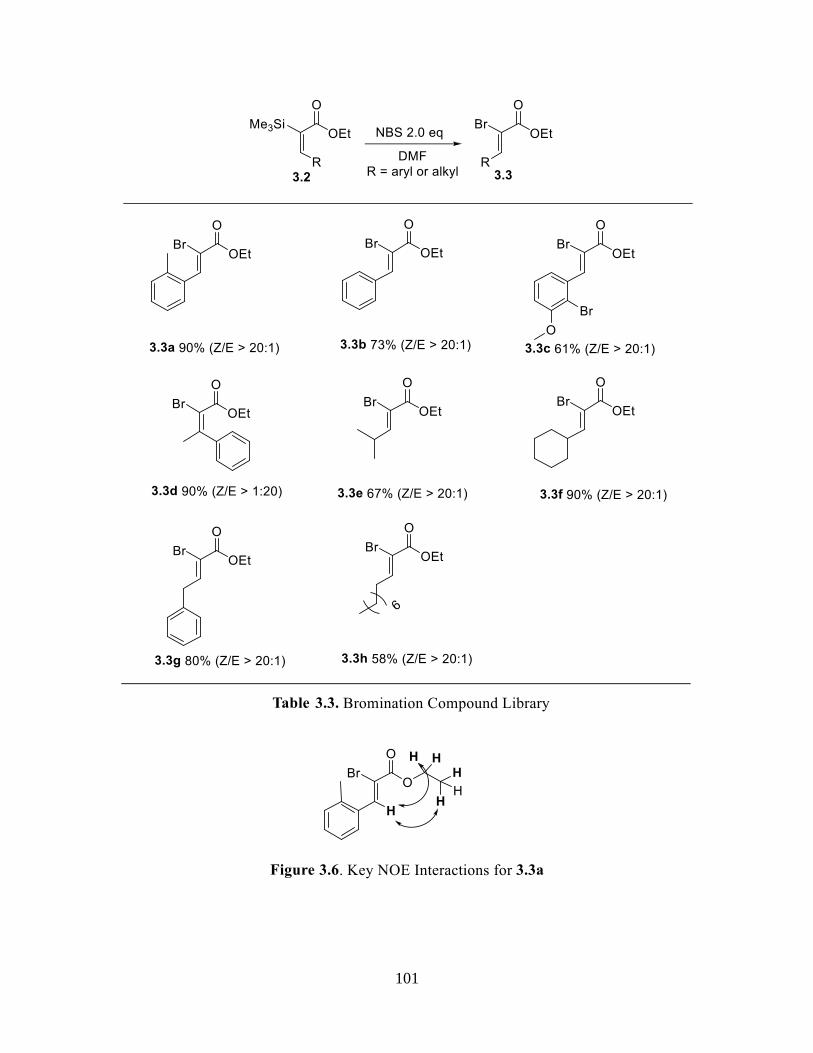
101

102
Compounds 3.3a and 3.3b both proceeded without any incident to give a yellow oil in
yields of 90% and 73% respectively. Compound 3.3c was the only material to have a
dibromination occur and it is theorized that the vinyl silane is an anisole analog which is a strong
ortho/para director so while the substitution of the silane is occurring so is an aromatic
substitution. It should also be noted that 3.3c was isolated in a 61% yield as a yellow powder and
was the only material to be a solid. Compounds 3.3e-3.3h proceeded to be isolated all as oils and
with inversion of stereochemistry. 3.3d gave intriguing results compared to the others.
The tetrasubstituted 3.3d was the only compound to give complete retention of
stereochemistry which was confirmed by NOE studies of both the vinyl silane, 3.2d, and the
brominated product, 3.3d, shown in Figure 3.7. 3.2d (E isomer) was confirmed to be the product
instead of 3.2d (Z isomer) by the lack of interactions between the ß-methyl and the ester. The
only interactions that occurred with the ester of 3.3d (E isomer) were from the aromatic group.
When considering the mechanism proposed, in order to have retention of stereochemistry, an
eclipsed rotation would take place which is a higher energy transition state than its staggered
counterpart. Due to this, it can be assumed that another mechanism (Scheme 3.2) took place
likely involving a hyperconjugated carbocation. This mechanism shows the silane activating the
double bond to allow the attack of the bromine on the NBS but not the subsequent attack by the
bromine as shown in Scheme 3.1. The C-Br bond would form in the α-position leaving a
stabilized tertiary carbocation in the ß-position. The carbocation would be further stabilized due
to σ orbital overlap of the silane group which is also known as the ß-silicon effect.24 Elimination
would subsequently follow leaving the halogenation to have retention of stereochemistry other
than the expected inversion.
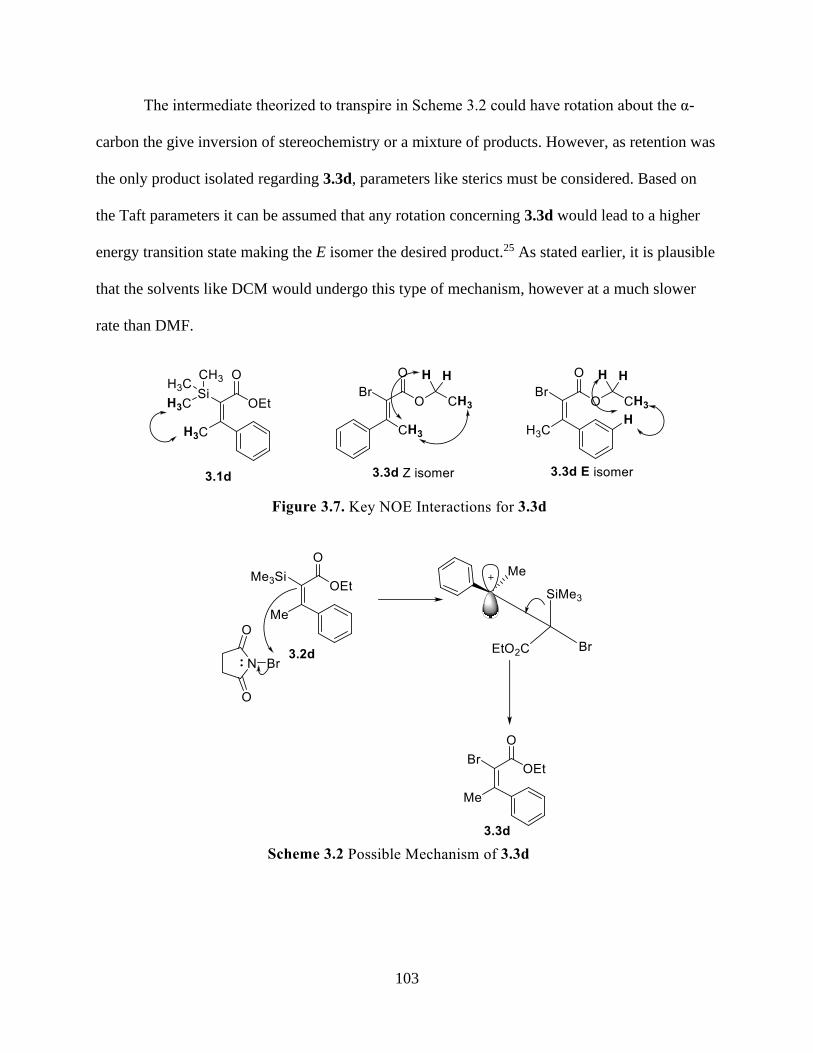
103
The intermediate theorized to transpire in Scheme 3.2 could have rotation about the α-
carbon the give inversion of stereochemistry or a mixture of products. However, as retention was
the only product isolated regarding 3.3d, parameters like sterics must be considered. Based on
the Taft parameters it can be assumed that any rotation concerning 3.3d would lead to a higher
energy transition state making the E isomer the desired product.25 As stated earlier, it is plausible
that the solvents like DCM would undergo this type of mechanism, however at a much slower
rate than DMF.

104
Table 3.4 shows the isolation of three chlorinations (3.4a-3.4c) using NCS instead of
NBS. These reactions were also heated to 60 oC for 18-24 hours in lieu of just stirring at room
temperature. Compound 3.4a was the only one of the three to give a 1:1 ratio of the (Z) and (E)
isomers where the other two produced complete inversion. It is a possibility that the mechanisms
in Scheme 3.1 and 3.2 are competing.
Another reaction that was conducted is shown in Figure 3.8. The vinyl silane, 3.2e, was
subjected to standard conditions to give rise to 3.3b. The vinyl silane was similar to that of 3.2b
with the TMS group being changed to a TES group. Instead of giving complete inversion of
stereochemistry, it was found that a 3:1 (Z)/(E) mixture was generated. It is theorized that the
TES group was more sterically hindering so as to not allow for rotation. All of the work
conducted in this project furthers the versatility of these α-trialkylsilyl-α,ß-unsaturated esters.

105
3.5 Future Works
It is hopeful that in the future the (Z)-ß-substituted-α-trialkylsilyl-α,ß-unsaturated esters
could be studied to determine if the halogenation is both stereoselective and stereospecific. It
would also be desirable to conduct full mechanistic studies of the halogenations, especially those
that gave mixtures of isomers or complete retention of stereochemistry.
3.6 Conclusion
The formation of vinyl silanes was successful and were utilized to create α-bromo-α,ß-
unsaturated esters with both aryl and alkyl substituents in a stereoselective manner with good
yields. These small molecules have multiple reactive sites making them useful in the synthesis of
natural products and other pharmaceuticals.
3.7 Supporting Information for Chapter 3
All of the reactions were performed under Ar in flame-dried glassware. All starting materials,
solvents, reagents, and catalysts were commercially available and used without further
purification, with the exception of N-Bromosuccinimide, which was recrystallized in H2O. The
NMR spectra were recorded with either a 360 or 500 MHz Bruker spectrometer. 1H and 13C
NMR spectra were obtained using CDCl3 as the solvent with chloroform (CHCl3 1H: δ = 7.26
ppm, CDCl3 13C: δ = 77.0 ppm) as the internal standard. Column chromatography was performed

106
using 60-200 µm silica gel. Analytical thin layer chromatography was performed on silica coated
glass plates with F-254 indicator. Visualization was accomplished by UV light (254 nm) and
KMnO4.
General experimental procedure for the formation of -trialkylsilyl--unsaturated
esters: CuI (0.029 g, 0.15 mmol) and LiCl (0.013 g, 0.30 mmol) was placed in a 100 mL round
bottom flask (flame dried under vacuum) under Ar. Dry THF (20 mL) was added and the
mixture was stirred at rt for a period of 0.5 h until complete dissolution had occurred. The clear,
light yellow homogeneous solution was cooled to –78 C, and ethyl propiolate (0.294 g, 3.0
mmol) was added, followed by TMSOTf (3.3 eq., 1.8 mL, 9.9 mmol). After 5 minutes at –78
C, the aryl or alkyl Grignard reagent (1.2 eq., 3.6 mmol) was added dropwise via syringe, and
the solution was stirred at –78 C for 1 h and allowed to warm to rt. The reaction was quenched
with H2O and the product extracted with Et2O (3 x 25 mL), and the combined organic layers
were washed with deionized H2O followed by saturated NH4Cl. The organic layer was
separated, dried with MgSO4, and concentrated in vacuo to give the crude product, which was
then analyzed by 1H NMR spectroscopy to determine diastereoselectivity. Column
chromatography of the crude material (3% ethyl acetate in hexanes) afforded the pure vinyl
silane products.
Ethyl (E)-3-(o-tolyl)-2-(trimethylsilyl)acrylate (3.2a): Yield: 0.692 g, 88%: 1H NMR (500
MHz, CDCl3) 7.15 (m, 4H), 7.03 (s, 1H), 4.08 (q, J = 7.2 Hz, 2H), 2.31 (s, 3H), 1.09 (t, J = 7.2
Hz, 3H), 0.25 (s, 9H).18
Ethyl (E)-3-phenyl-2-(trimethylsilyl)acrylate (3.2b): Yield: 0.684 g, 92%: 1H NMR (500 MHz,
CDCl3) 7.25 (m, 5H), 6.77 (s, 1H), 4.15 (q, J = 7.2 Hz, 2H), 1.16 (t, J = 7.2 Hz, 3H), 0.19 (s,
9H).18

107
Ethyl-(E)-3-(3-methoxyphenyl)-2-(trimethylsilyl)acrylate (3.2c): Yield: 0.734 g, 88%: 1H
NMR (500 MHz, CDCl3) 7.23 (t, J = 7.8 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.89 (s, 1H), 6.83
(dd, J = 8.3, 3.0 Hz, 1H), 6.78 (s, 1H), 4.21 (q, J = 7.2 Hz, 2H), 3.79 (s, 3H), 1.23 (t, J = 7.2 Hz,
3H), 0.24 (s, 9H).18
Ethyl-(E)-3-phenyl-2-(trimethylsilyl)but-2-enoate (3.2d): Yield: 0.668 g, 85%: 1H NMR (500
MHz, CDCl3) 7.26 (m, 5H), 3.86 (q, J = 7.2 Hz, 2H), 2.19 (s, 3H), 0.92 (t, J = 7.2 Hz, 3H), 0.28
(s, 9H).18
Ethyl-(E)-3-phenyl-2-(triethylsilyl)acrylate (3.2e): Yield: 0.722 g, 83%: 1H NMR (360 MHz,
CDCl3) 7.31 (m, 5H), 6.79 (s, 1H), 4.18 (q, J = 7.2 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H), 1.01 (t, J
= 7.6 Hz, 9H), 0.75 (q, J = 7.6 Hz, 6H).18
Ethyl-(E)-4-methyl-2-(trimethylsilyl)pent-2-enoate (3.2f): Yield: 0.702 g, 82%: 1H NMR (360
MHz, CDCl3) 5.89 (d, J = 9.3 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.92 (m, 1H), 1.29 (t, J = 7.0
Hz, 3H), 1.00 (d, J = 6.6 Hz, 6H), 0.13 (s, 9H).18
Ethyl (E)-3-cyclohexyl-2-(trimethylsilyl)acrylate (3.2g): Yield: 0.46 g, 60%, light yellow oil:
1H NMR (500 MHz, CDCl3) δ 5.93 (d, J = 9.3 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 2.63 (m, 1H),
1.68 (m, 5H), 1.30 (t, J = 7.1 Hz, 2H), 1.26 (m, 2H), 1.12 (m, 3H), 0.12 (s, 9H). 13C NMR (125
MHz, CDCl3) δ 170.6, 156.4, 133.8, 59.8, 40.3, 32.5, 25.9, 25.6, 14.4, -1.3. IR (NaCl): 2977, 2927,
1713, 1606, 1448, 1263, 1192, 855 cm-1. HRMS (EI-EBE Sector) m/z: [M]+ Calcd for C14H26O2Si
254.1696; found 254.1702. Rf = 0.50, 3% EtOAc in hexanes.
Ethyl-(E)-4-phenyl-2-(trimethylsilyl)but-2-enoate (3.2h): Yield: 0.734 g, 70%: 1H NMR (360
MHz, CDCl3) 7.27 (m, 5H), 6.30 (t, J = 7.0 Hz, 1H), 4.27 (q, J = 7.0 Hz, 2H), 3.74 (d, J = 7.0
Hz, 2H), 1.35 (t, J = 7.2 Hz, 3H), 0.18 (s, 9H).18

108
Ethyl (E)-2-(trimethylsilyl)undec-2-enoate (3.2i): Yield: 0.56 g, 65%, yellow oil: 1H NMR (500
MHz, CDCl3) δ 6.15 (t, J = 7.3 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 2.35 (m, 2H), 1.43(m, 2H), 1.29
(m, 10H), 1.27 (t, J = 7.1 Hz, 3H), 0.88 (t, J = 6.9 Hz, 3H), 0.13 (s, 9H). 13C NMR (125 MHz,
CDCl3) δ 170.6, 151.8, 135.9, 59.9, 31.9, 31.7, 29.4, 29.3, 29.2, 29.1, 22.7, 14.4, 14.1, -1.3. IR
(NaCl): 2956, 2925, 2871, 2855, 1715, 1464, 1247,1191, 1033, 840, 754. HRMS (EI-EBE Sector)
m/z: [M]+ Calcd for C16H32O2Si 284.2160; found 284.2172. Rf = 0.62, 4% EtOAc in hexanes.
General experimental procedure for the -halogenated--unsaturated esters: In a dark
room, NBS or NCS (0.80 mmol, 140 mg, 2.0 equiv.) was added to a (flame-dried under vacuum)
round-bottom flask under Ar. The flask was covered in aluminum foil and then a solution of vinyl
silane (3.2a-3.2i) (0.40 mmol, 0.10 g, 1.0 equiv.) in anhydrous DMF (1.00 mL) was added in one
portion. The mixture was stirred at rt for a minimum of 24 hours where it was then quenched with
saturated Na2CO3 (1.00 mL) and the product was extracted with CH2Cl2 (3 x 10 mL), and the
combined organic layers were washed with deionized H2O (3 x 10 mL). The organic layer was
dried with MgSO4 and concentrated in vacuo to give the crude product. Column chromatography
of the crude material (3% Et2O in pentane) afforded the pure halogenated products 3.3a-3.3h and
3.4a-3.4c in yields ranging from 58%-91%.
Ethyl (Z)-2-bromo-3-(o-tolyl)acrylate (3.3a): Yield: 0.092 g, 90%, yellow oil. 1H NMR (500
MHz, CDCl3) δ 8.29 (s, 1H), 7.66 (d, J = 7.5 Hz, 1H), 7.27 (m, 4H), 4.35 (q, J = 7.1 Hz, 2H), 2.32
(s, 3H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 163.1, 140.6, 137.0, 133.7, 130.1,
129.4, 128.6, 125.6, 115.7, 62.8, 19.9, 14.2. IR (NaCl): 1720, 1481, 1257, 865, 482 cm -1. HRMS
(EI-EBE Sector) m/z: [M]+ Calcd for C12H13BrO2 268.0099; found 238.0105. Rf = 0.39, 3% Et2O
in pentane.

109
Ethyl (Z)-2-bromo-3-phenylacrylate (3.3b): Yield: 0.074 g, 73%, yellow oil. 1H NMR (500
MHz, CDCl3) δ 8.22 (s, 1H), 7.86 (m, 2H), 7.43 (m, 3H), 4.36 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1
Hz, 3H).26
Ethyl (Z)-2-bromo-3-(2-bromo-3-methoxyphenyl)acrylate (3.3c): Yield: 0.060 g, 61%, yellow
solid. 1H NMR (500 MHz, CDCl3) δ 8.27 (s, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 3.0 Hz,
1H), 6.83 (dd, J = 8.8, 3.0 Hz, 1H), 4.38 (q, J = 7.1 Hz, 2H), 3.82 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H).
13C NMR (125 MHz, CDCl3) δ 162.8, 158.4, 140.4, 135.2, 133.6, 117.1, 116.7, 115.8, 114.8, 63.0,
55.6, 14.2. IR (NaCl): 1720, 1463, 1280, 1234, 908, 729. HRMS (EI-EBE Sector) m/z: [M]+ Calcd
for C12H12Br2O3 361.9153; found 361.9164. Rf = 0.29, 3% Et2O in pentane.
Ethyl (E)-2-bromo-3-phenylbut-2-enoate (3.3d): Yield: 0.075 g, 70%, clear oil. 1H NMR (500
MHz, CDCl3) δ 7.32 (m, 3H), 7.18 (m, 2H), 3.95 (q, J = 7.1 Hz, 2H), 2.32 (s, 3H), 0.91 (t, J = 7.1
Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 164.9, 147.0, 141.2, 128.3, 128.0, 126.8, 111.1, 61.7,
25.9, 13.4. IR (NaCl): 1288, 1277, 1042, 700. HRMS (EI-EBE Sector) m/z: [M]+ Calcd for
C12H13BrO2 268.0099; found 268.0090. Rf = 0.18, 3% Et2O in pentane.
Ethyl (Z) and (E)-2-chloro-3-phenylacrylate (3.4a): Yield: 0.040 g, 89%, yellow oil. (Z)-
product: 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 7.63 (d, J = 7.5, 1H), 7.43 (m, 4H), 4.36 (q,
J = 7.1 Hz, 2H), 1.39 (t, J = 7.0 Hz, 3H); (E)-product: 1H NMR (500 MHz, CDCl3) δ 7.30(m, 5H),
7.21 (s, 1H), 4.21 (q, J = 7.2, 2H), 1.18 (t, J = 7.1, Hz, 3H).27, 28
Ethyl (Z)-2-bromo-4-methylpent-2-enoate (3.3e): Yield: 0.069 g, 67%, yellow oil. 1H NMR
(500 MHz, CDCl3) δ 7.08 (d, J = 9.3 Hz, 1H), 4.27 (q, J = 7.1 Hz, 1H), 2.86 (m, 1H), 1.33 (t, J =
7.1 Hz, 2H), 1.09 (d, J = 6.7 Hz, 3H).29

110
Ethyl (Z)-2-bromo-3-cyclohexylacrylate (3.3f): Yield: 0.083 g, 90%, yellow oil. 1H NMR (500
MHz, CDCl3) δ 7.10 (d, J = 9.2 Hz, 1H), 4.27 (q, J = 7.1 Hz, 1H), 2.57 (m, 1H), 1.72 (m, 5H),
1.33 (m, 2H), 1.33 (t, J = 7.1 Hz, 2H), 1.19 (m, 3H).29
Ethyl (Z)-2-bromo-4-phenylbut-2-enoate (3.3g): Yield: 0.082 g, 80%, yellow oil. 1H NMR (500
MHz, CDCl3) δ 7.43 (t, J = 7.3 Hz, 1H), 7.33 (m, 2H), 7.25 (m, 3H), 4.28 (q, J = 7.1 Hz, 2H), 3.70
(d, J = 7.3 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 162.4, 144.0, 137. 2,
128.8, 128.6, 126.9, 117.0, 62.5, 38.4, 14.1. IR (NaCl): 2360, 2341, 1652, 1255, 992, 799. HRMS
(EI-EBE Sector) m/z: [M]+ Calcd for C12H13BrO2 268.0099; found 268.0096. Rf = 0.42, 3% Et2O
in pentane.
Ethyl (Z)-2-bromoundec-2-enoate (3.3h): Yield: 0.059 g, 58%, yellow oil. 1H NMR (500 MHz,
CDCl3) δ 7.29 (t, J = 7.2 Hz, 1H), 4.28 (t, J = 7.1 Hz, 2H), 2.34 (m, 2H), 1.51 (m, 2H), 1.33 (t, J
= 7.1 Hz, 3H), 1.29 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 162.6,
146.3, 116.3, 62.3, 32.1, 31.8, 29.3, 29.3, 29.2, 27.5, 22.6, 14.2, 14.1. IR (NaCl): 2926, 1730, 1718,
1235. HRMS (EI-EBE Sector) m/z: [M]+ Calcd for C13H23BrO2 290.0881; found 290.0882. Rf =
0.45, 3% Et2O in pentane.
Ethyl (Z)-2-chloro-4-methylpent-2-enoate (3.4b): Yield: 0.068 g, 82%, yellow oil. 1H NMR
(500 MHz, CDCl3) δ 6.88 (d, J = 9.4 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 2.90 (m, 1H), 1.34 (t, J =
7.1 Hz, 3H), 1.08 (d, J = 6.7 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 162.8, 148.3, 122.8, 62.2,
29.1, 21.1, 14.2. IR (NaCl): 1734, 1721, 1251, 1145, 1041, 754 cm -1. HRMS (EI-EBE Sector)
m/z: [M]+ Calcd for C8H13ClO2 176.0604; found 176.0601. Rf = 0.40, 3% Et2O in pentane.
Ethyl (Z)-2-chloro-4-phenylbut-2-enoate (3.4c): Yield: 0.052 g, 80%, yellow oil. 1H NMR (500
MHz, CDCl3) δ 7.33 (m, 2H), 7.23 (m, 3H), 7.22 (t, J = 7.4 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 3.71
(d, J = 7.4 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 162.4, 140.2, 137.4,

111
128.8, 128.6, 126.8, 125.2, 62.30, 35.6, 14.1. IR (NaCl): 1730, 1268, 1043 cm -1. HRMS (EI-EBE
Sector) m/z: [M]+ Calcd for C12H13ClO2 224.0604; found 224.0605. Rf = 0.27, 3% Et2O in pentane.

112
3.8 References for Chapter 3
1. Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J., Free radical bromination by the H2O2–
HBr system on water. Tetrahedron Lett. 2006, 47, 7245-7247.
2. Roberts, I.; Kimball, G. E., The Halogenation of Ethylenes. J. Am. Chem. Soc. 1937, 59,
947-948.
3. Galabov, B.; Nalbantova, D.; Schleyer, P. v. R.; Schaefer, H. F., Electrophilic Aromatic
Substitution: New Insights into an Old Class of Reactions. Acc. Chem. Res. 2016, 49, 1191-1199.
4. Brown, H. C.; Stock, L. M., Relative Rates of Bromination of Benzene and the
Methylbenzenes. Partial Rate Factors for the Bromination Reaction1-3. J. Am. Chem. Soc. 1957,
79, 1421-1425.
5. Kong, W.; Che, C.; Wu, J.; Ma, L.; Zhu, G., Pd-Catalyzed Regio- and Stereoselective
Addition of Boronic Acids to Silylacetylenes: A Stereodivergent Assembly of β,β-Disubstituted
Alkenylsilanes and Alkenyl Halides. J. Org. Chem. 2014, 79, 5799-5805.
6. Imazaki, Y.; Shirakawa, E.; Ueno, R.; Hayashi, T., Ruthenium-Catalyzed
Transformation of Aryl and Alkenyl Triflates to Halides. J. Am. Chem. Soc. 2012, 134, 14760-
14763.
7. Incremona, J. H.; Martin, J. C., N-bromosuccinimide. Mechanisms of allylic bromination
and related reactions. J. Am. Chem. Soc. 1970, 92, 627-634.
8. Jyothi, D.; HariPrasad, S., A remarkably simple one-step procedure for the preparation of
α-bromo-α,β-unsaturated carbonyl compounds. Synlett 2009, 2309-2311.
9. Djerassi, C., Brominations with N-Bromosuccinimide and Related Compounds. The
Wohl-Ziegler Reaction. Chem. Rev. 1948, 43, 271-317.
10. Tamao, K.; Akita, M.; Maeda, K.; Kumada, M., Silafunctional compounds in organic
synthesis. 32. Stereoselective halogenolysis of alkenylsilanes: stereochemical dependence on the
coordination state of the leaving silyl groups. J. Org. Chem. 1987, 52, 1100-1106.
11. Stamos, D. P.; Taylor, A. G.; Kishi, Y., A mild preparation of vinyliodides from
vinylsilanes. Tetrahedron Lett. 1996, 37, 8647-8650.

113
12. Babudri, F.; Fiandanese, V.; Hassan, O.; Punzi, A.; Naso, F., New synthesis of
leukotriene B3 methyl ester from bis(trimethylsilyl) unsaturated derivatives. Tetrahedron 1998,
54, 4327-4336.
13. Hayakawa, I.; Ueda, M.; Yamaura, M.; Ikeda, Y.; Suzuki, Y.; Yoshizato, K.; Kigoshi,
H., Second-Generation Total Synthesis of Haterumalide NA Using B-Alkyl Suzuki–Miyaura
Coupling. Org. Lett. 2008, 10, 1859-1862.
14. Graf, K. M.; Tabor, M. G.; Brown, M. L.; Paige, M., Synthesis of (S)-Jamaicamide C
Carboxylic Acid. Org. Lett. 2009, 11, 5382-5385.
15. Pawluc, P.; Hreczycho, G.; Szudkowska, J.; Kubicki, M.; Marciniec, B., New One-Pot
Synthesis of (E)-β-Aryl Vinyl Halides from Styrenes. Org. Lett. 2009, 11, 3390-3393.
16. Pawluc, P.; Hreczycho, G.; Walkowiak, J.; Marciniec, B., A new facile synthesis of 1,1-
dibromo-2-arylethenes. Synlett 2007, 2061-2064.
17. Johnson, D. A.; Jennings, M. P., Tandem Copper-Catalyzed Conjugate Addition-
Diastereoselective Protonation of (E)-α-Trialkylsilyl-β-Alkyl(Aryl)-α,β-Unsaturated Esters. Org.
Lett. 2018, 20, 6099-6103.
18. Johnson, D. A.; Mueller Hendrix, A. J.; Jennings, M. P., Diastereoselective Syntheses of
(E)-α-Trialkylsilyl α,β-Unsaturated Esters, α-Silane-Substituted Conjugated Silyl Ketene
Acetals, and α,γ-Substituted Allylsilanes. J. Org. Chem. 2018, 83, 9914-9928.
19. Probasco, M. S.; Johnson, D. A.; Jennings, M. P., Stereoselective One-Pot
Deconjugation, Aldol, and Stabilized Peterson Olefination of α-Trialkylsilyl-β-alkyl-α,β-
Unsaturated Esters. Org. Lett. 2019, 21, 1379-1383.
20. Fealy, L. M.; Jennings, M. P., Stereoselective halo-succinimide mediated γ-halogenations
of substituted α-trialkylsilyl-β,γ-unsaturated esters. Tetrahedron Lett. 2020, 61, 151384.
21. Mi, X.; Huang, M.; Feng, Y.; Wu, Y., Discovery of A Novel Palladium Catalyst for the
Preparation of Enynes with a Copper- and Ligand-Free Sonogashira Reaction. Synlett 2012,
2012, 1257-1261.
22. Kasai, Y.; Ito, T.; Sasaki, M., Total Synthesis of (−)-Polycavernoside A: Suzuki–
Miyaura Coupling Approach. Org. Lett. 2012, 14, 3186-3189.
23. Haut, F.-L.; Speck, K.; Wildermuth, R.; Möller, K.; Mayer, P.; Magauer, T., A Negishi
cross-coupling reaction enables the total synthesis of (+)-stachyflin. Tetrahedron 2018, 74, 3348-
3357.

114
24. Sommer, L. H.; Dorfman, E.; Goldberg, G. M.; Whitmore, F. C., The Reactivity with
Alkali of Chlorine—Carbon Bonds Alpha, Beta and Gamma to Silicon1,2. J. Am. Chem. Soc.
1946, 68, 488-489.
25. Taft, R. W., Linear Free Energy Relationships from Rates of Esterification and Hydrolysis of
Aliphatic and Ortho-substituted Benzoate Esters. J. Am. Chem. Soc. 1952, 74, 2729-2732.
26. Ganesh, M.; Namboothiri, I. N. N., Stereospecific approach to α,β-disubstituted
nitroalkenes via coupling of α-bromonitroalkenes with boronic acids and terminal acetylenes.
Tetrahedron 2007, 63, 11973-11983.
27. Arthuis, M.; Lecup, A.; Roulland, E., Pd0-Catalyzed carbonylation of 1,1-dichloro-1-
alkenes, a new selective access to Z-α-chloroacrylates. Chem. Commun. 2010, 46, 7810-7812.
28. Hosseini-Sarvari, M.; Sharghi, H.; Etemad, S., Solvent-free Knoevenagel condensations
over TiO2. Chin. J. Chem. 2007, 25, 1563-1567.
29. Olpp, T.; Brueckner, R., Stereoselective preparation of (E)-α-bromo acrylates from
mixtures of brominated Ando phosphonates. Synthesis 2004, 2135-2152.
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
9.31
3.13
3.16
2.11
1.004.28
0.25
1.081.091.11
2.31
4.054.074.084.10
7.037.087.087.107.107.117.117.137.157.157.167.177.177.187.197.217.22
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2a
115

-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
9.18
3.14
2.05
1.00
5.22
0.19
1.141.161.18
4.124.144.164.18
6.777.207.217.227.227.237.237.247.257.257.267.267.277.287.297.297.30
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2b
116
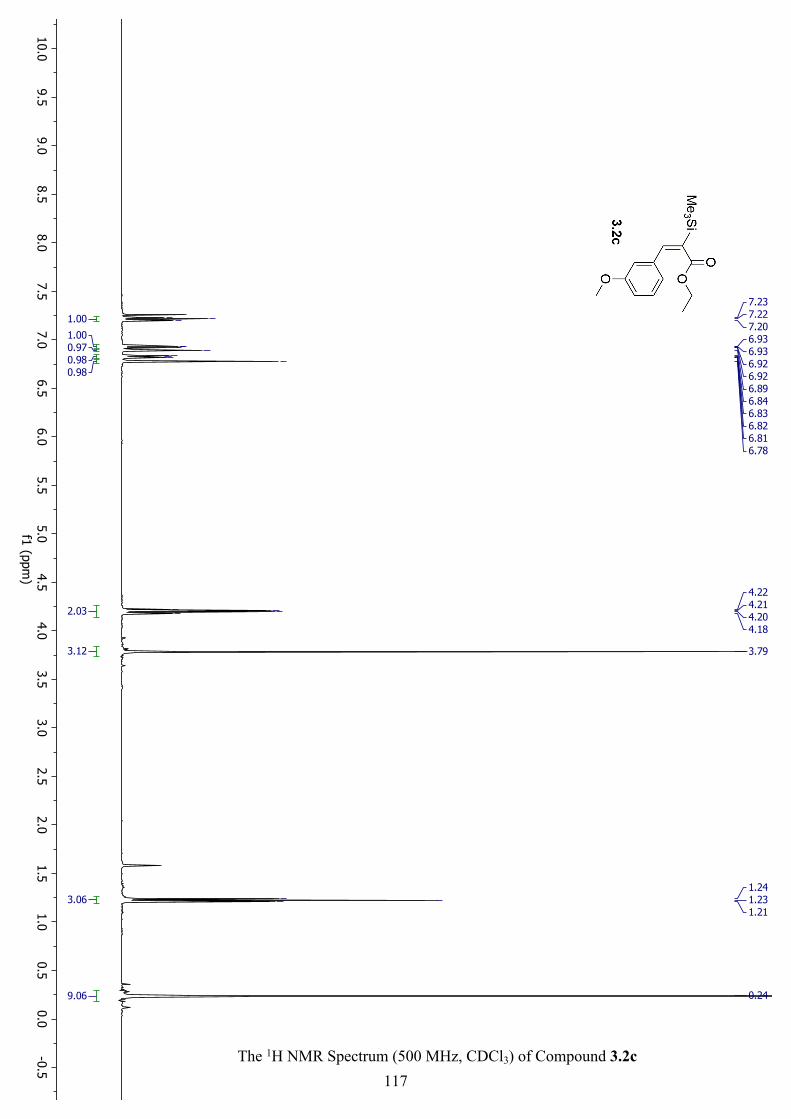
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
9.06
3.06
3.12
2.03
0.980.980.971.00
1.00
0.24
1.211.231.24
3.79
4.184.204.214.22
6.786.816.826.836.846.896.926.926.936.937.207.227.23
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2c117
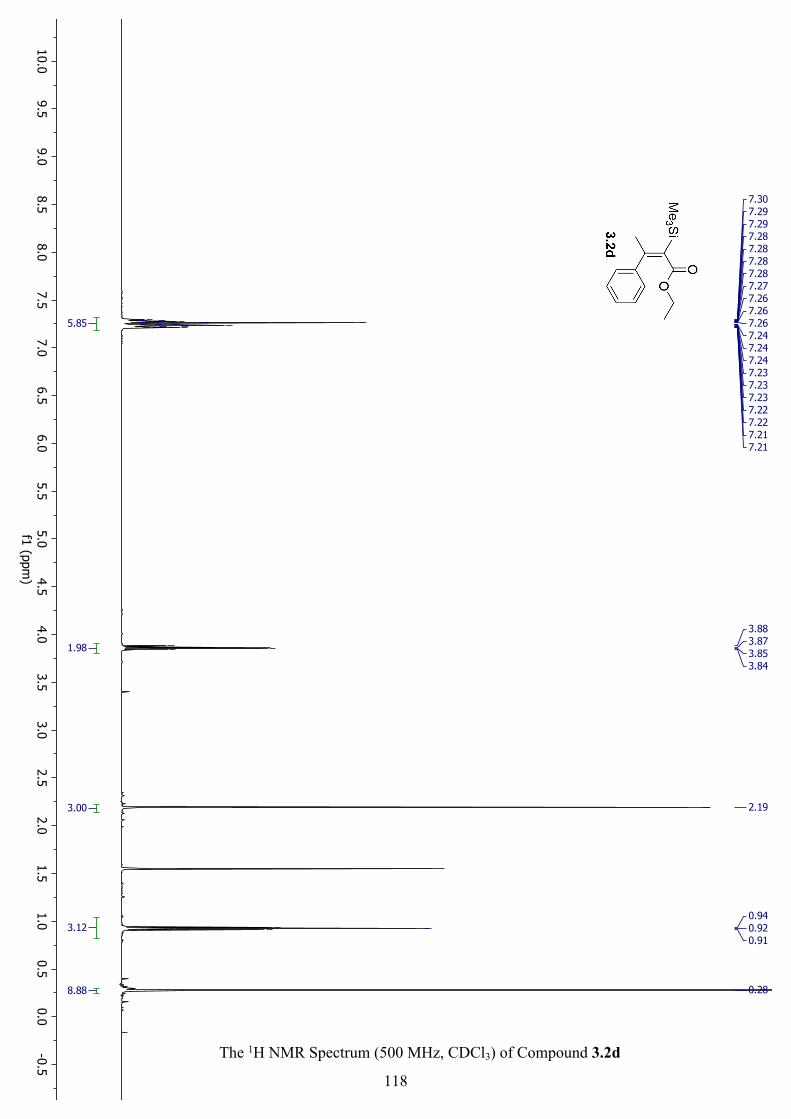
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
8.88
3.12
3.00
1.98
5.85
0.28
0.910.920.94
2.19
3.843.853.873.88
7.217.217.227.227.237.237.237.247.247.247.267.267.267.277.287.287.287.287.297.297.30
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2d
118
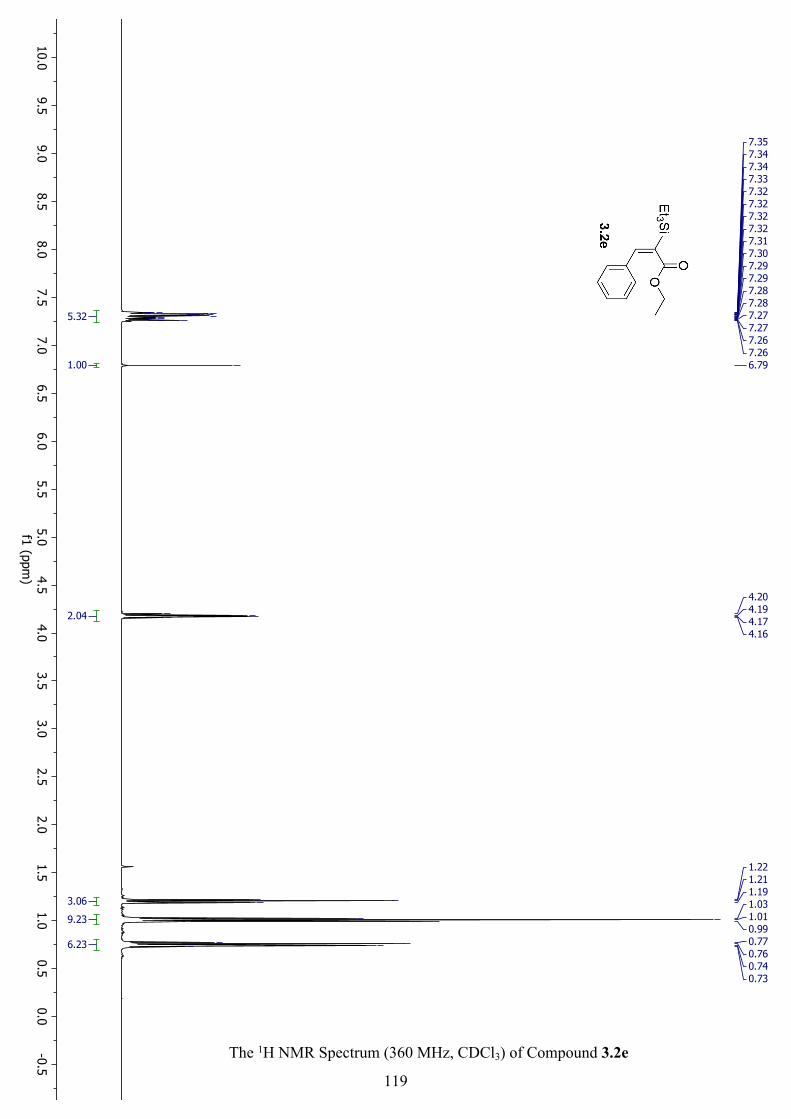
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
6.23
9.23
3.06
2.04
1.00
5.32
0.730.740.760.770.991.011.031.191.211.22
4.164.174.194.20
6.797.267.267.277.277.287.287.297.297.307.317.327.327.327.327.337.347.347.35
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2e
119
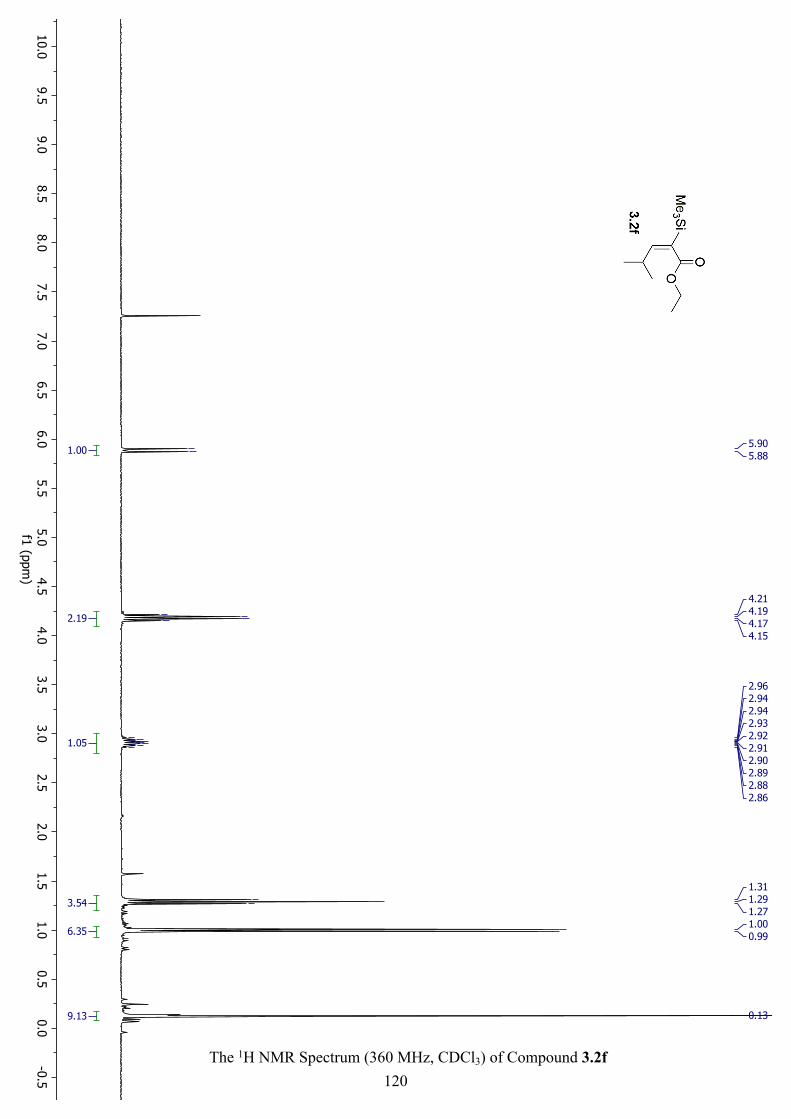
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
9.13
6.35
3.54
1.05
2.19
1.00
0.13
0.991.001.271.291.31
2.862.882.892.902.912.922.932.942.942.96
4.154.174.194.21
5.885.90
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2f120
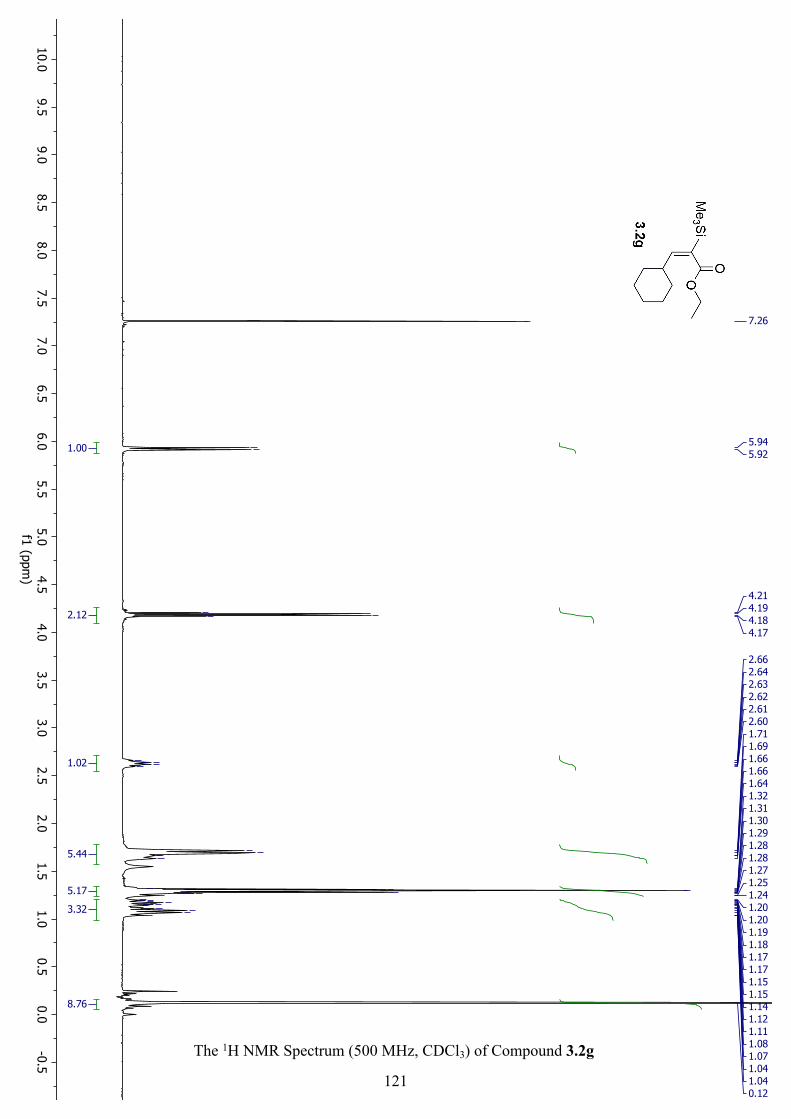
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
8.76
3.32
5.17
5.44
1.02
2.12
1.00
0.121.041.041.071.081.111.121.141.151.151.171.171.181.191.201.201.241.251.271.281.281.291.301.311.321.641.661.661.691.712.602.612.622.632.642.66
4.174.184.194.21
5.925.94
7.26
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2g
121
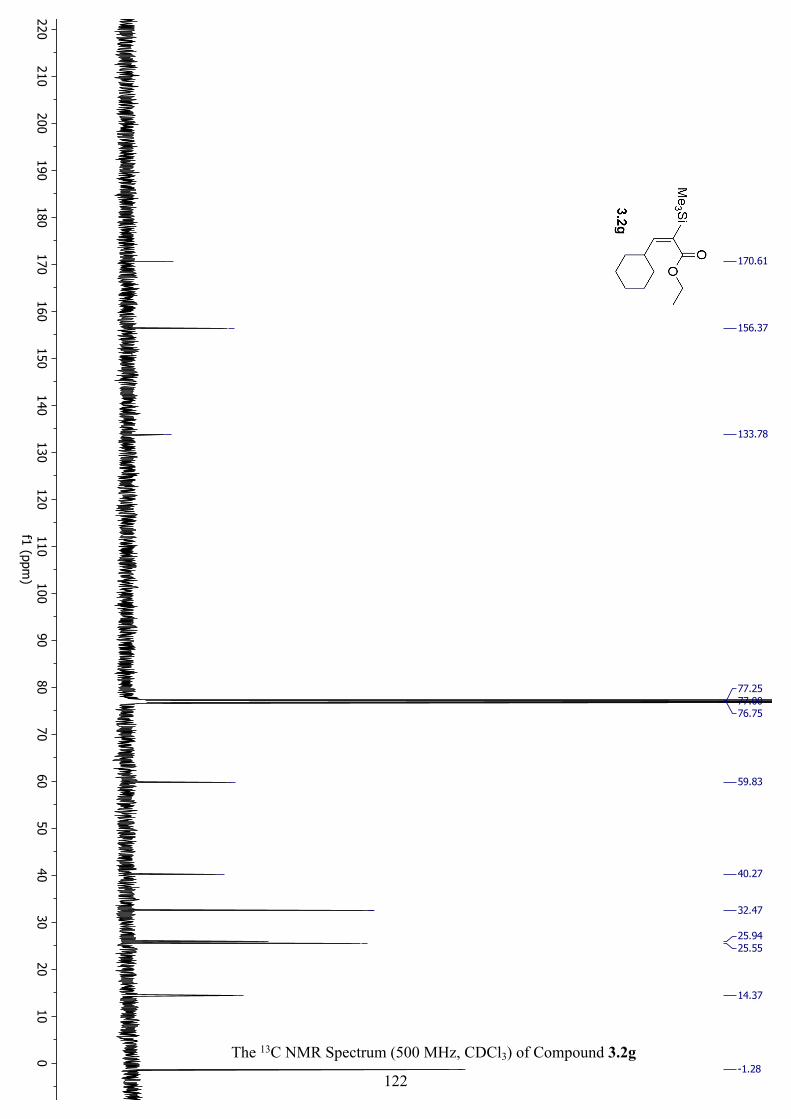
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
-1.28
14.37
25.5525.94
32.47
40.27
59.83
76.7577.0077.25
133.78
156.37
170.61
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.2g
122
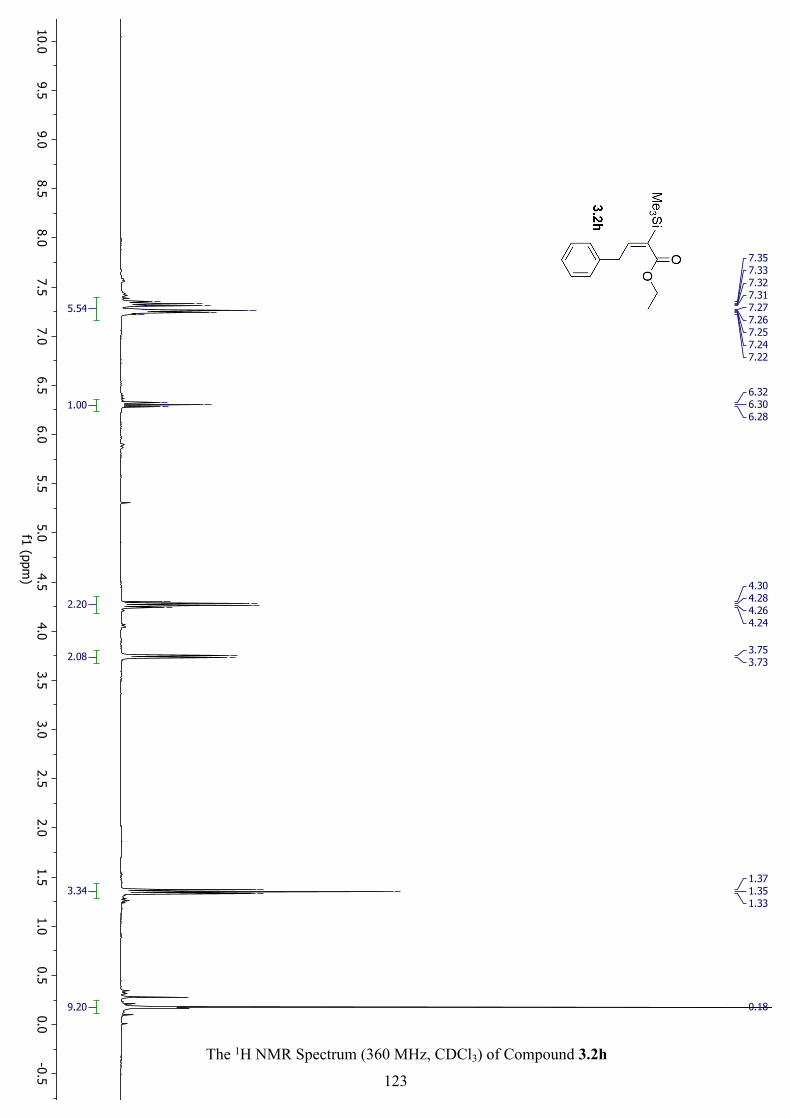
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
9.20
3.34
2.08
2.20
1.00
5.54
0.18
1.331.351.37
3.733.75
4.244.264.284.30
6.286.306.32
7.227.247.257.267.277.317.327.337.35
The 1H NMR Spectrum (360 MHz, CDCl3) of Compound 3.2h
123
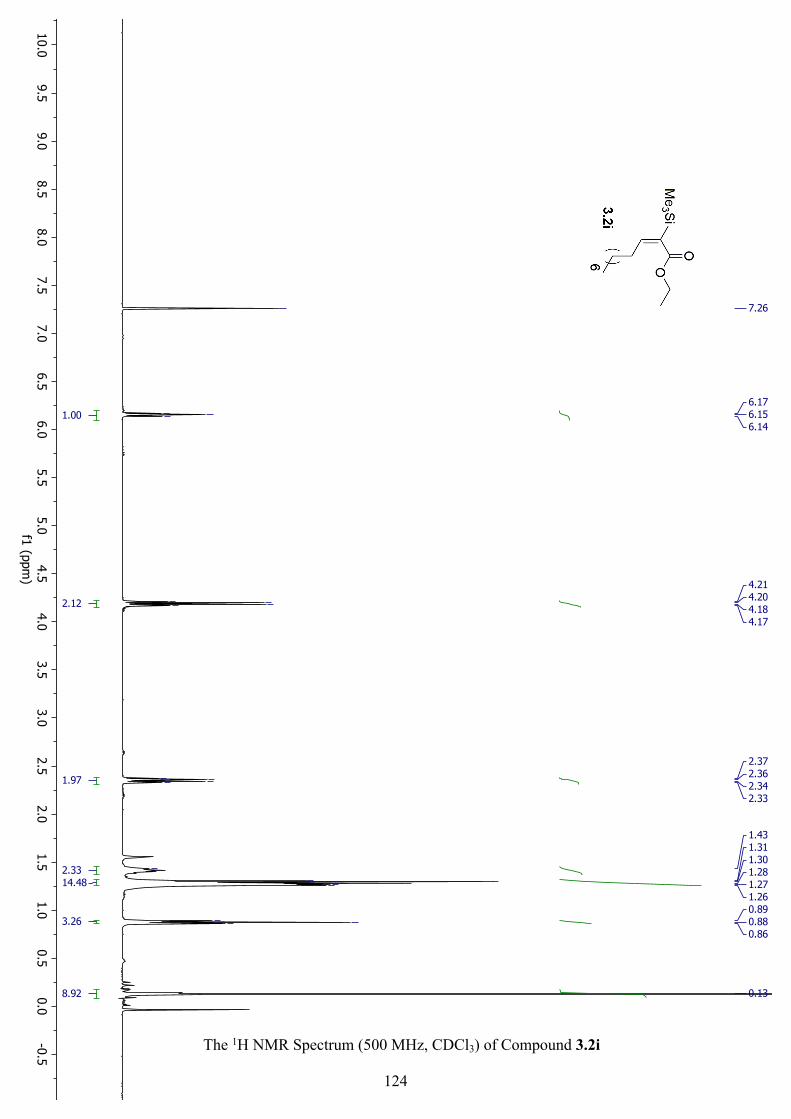
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
8.92
3.26
14.482.33
1.97
2.12
1.00
0.13
0.860.880.891.261.271.281.301.311.43
2.332.342.362.37
4.174.184.204.21
6.146.156.17
7.26
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.2i
124
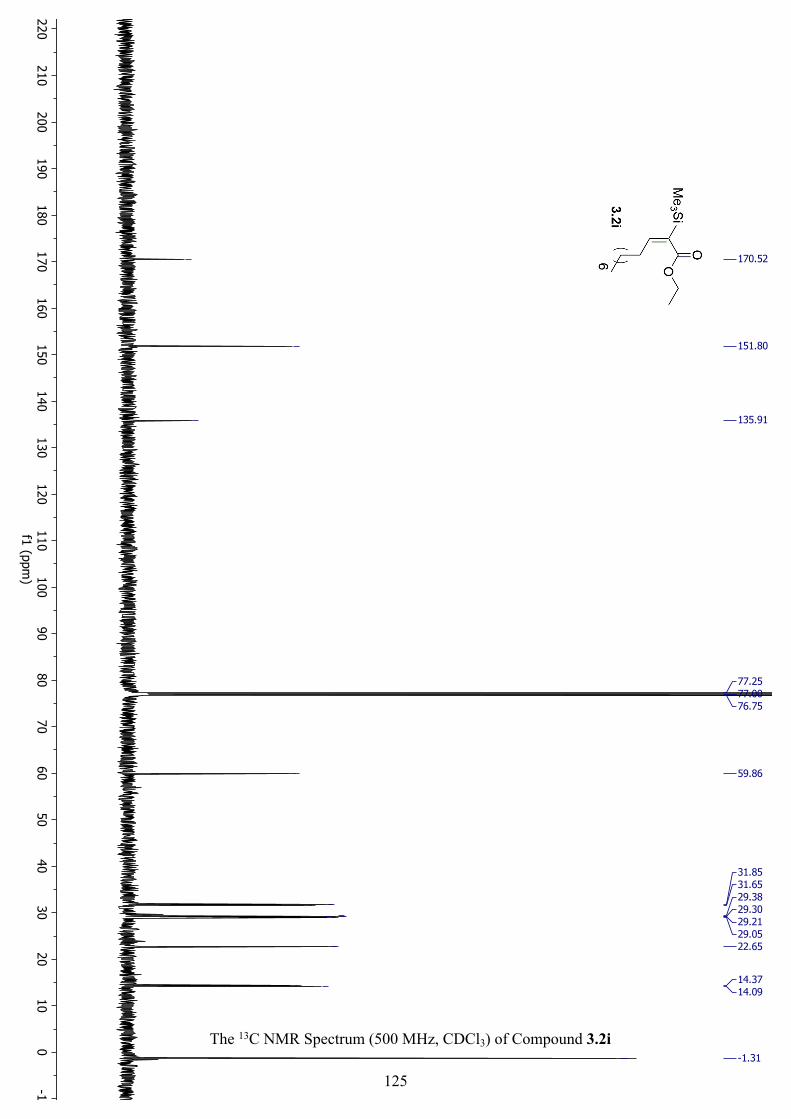
-100
1020
3040
5060
7080
90100
110120
130140
150160
170180
190200
210220
f1 (ppm)
-1.31
14.0914.37
22.6529.0529.2129.3029.3831.6531.85
59.86
76.7577.0077.25
135.91
151.80
170.52
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.2i
125
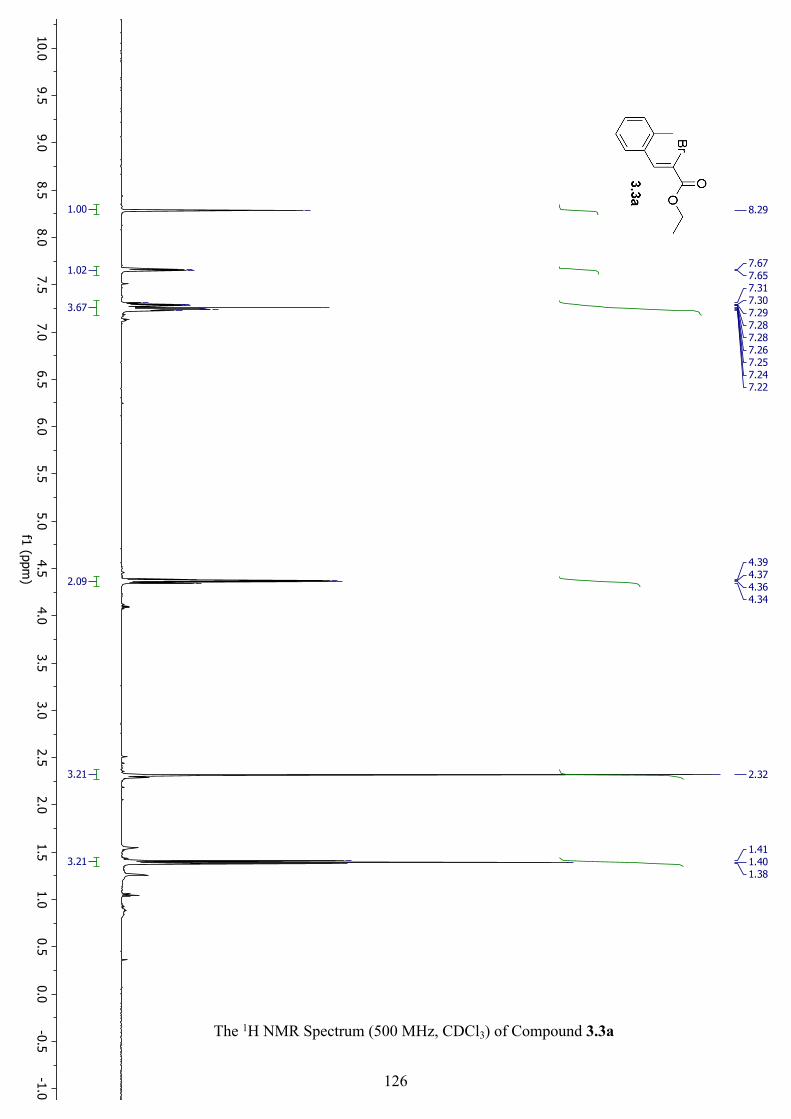
-1.0-0.5
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.0f1 (ppm
)
3.21
3.21
2.09
3.67
1.02
1.00
1.381.401.41
2.32
4.344.364.374.39
7.227.247.257.267.287.287.297.307.317.657.67
8.29
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3a
126
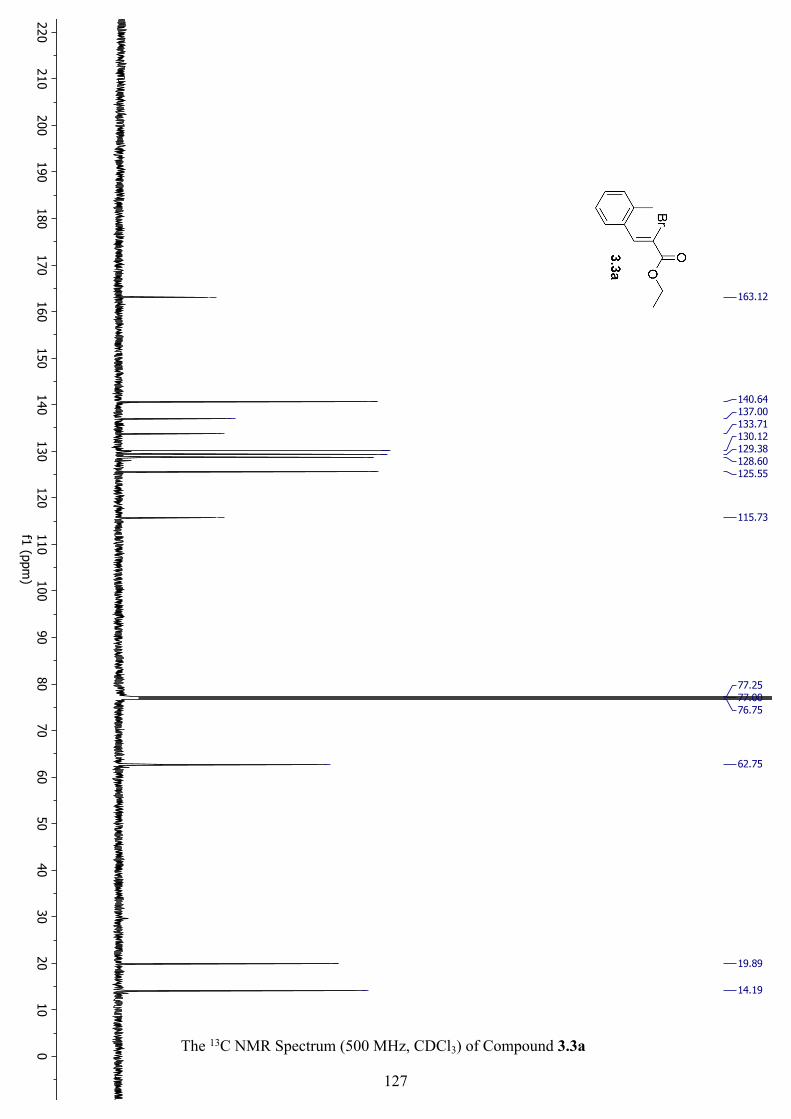
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.19
19.89
62.75
76.7577.0077.25
115.73
125.55128.60129.38130.12133.71137.00140.64
163.12
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3a
127
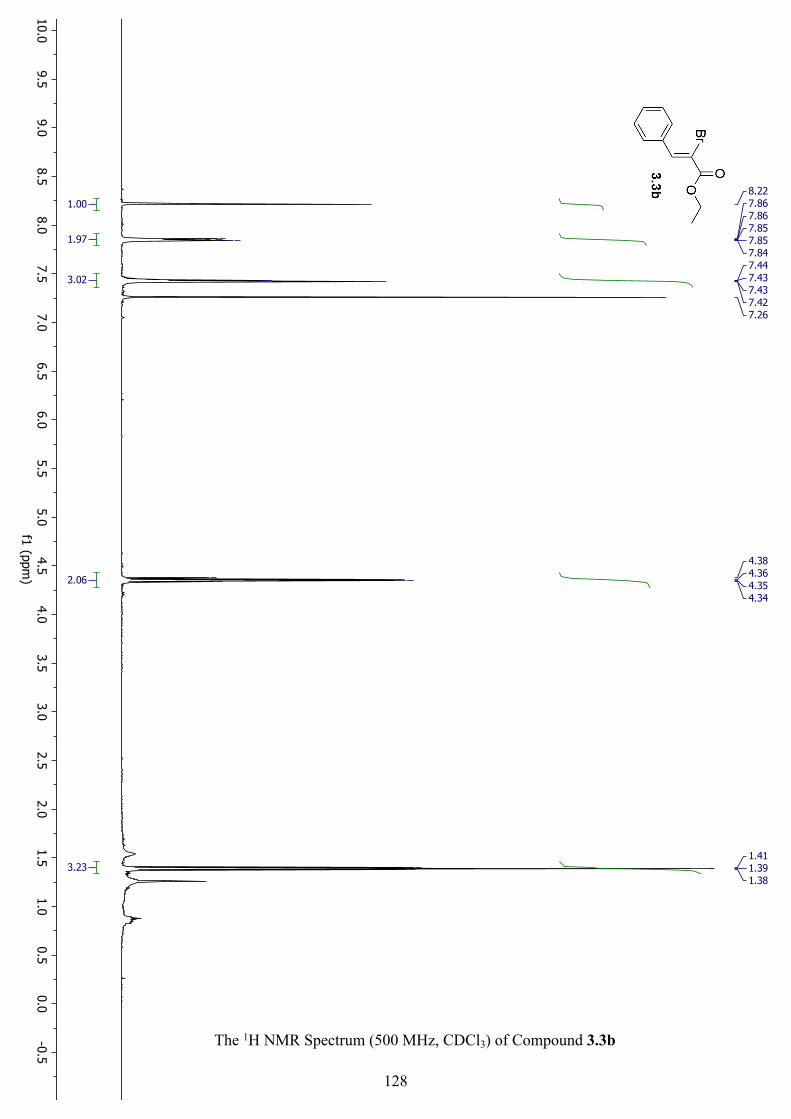
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
3.23
2.06
3.02
1.97
1.00
1.381.391.41
4.344.354.364.38
7.267.427.437.437.447.847.857.857.867.868.22
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3b
128
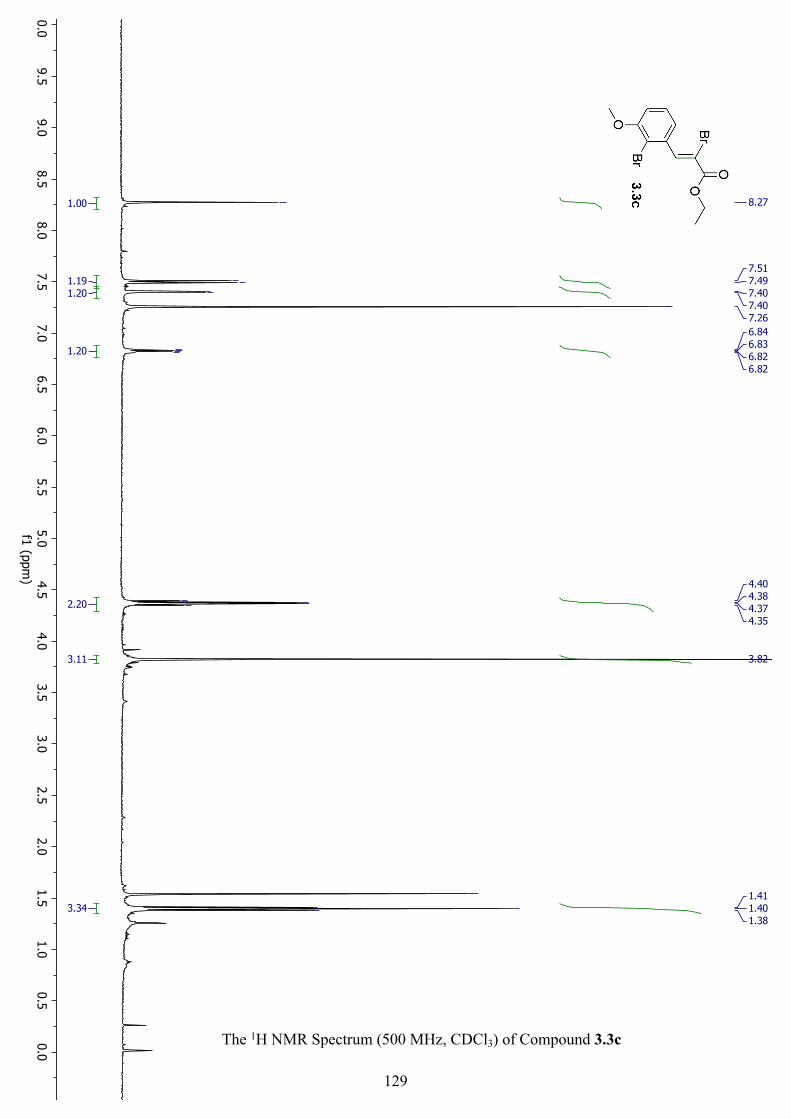
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.0f1 (ppm
)
3.34
3.11
2.20
1.20
1.201.19
1.00
1.381.401.41
3.82
4.354.374.384.40
6.826.826.836.847.267.407.407.497.51
8.27
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3c
129
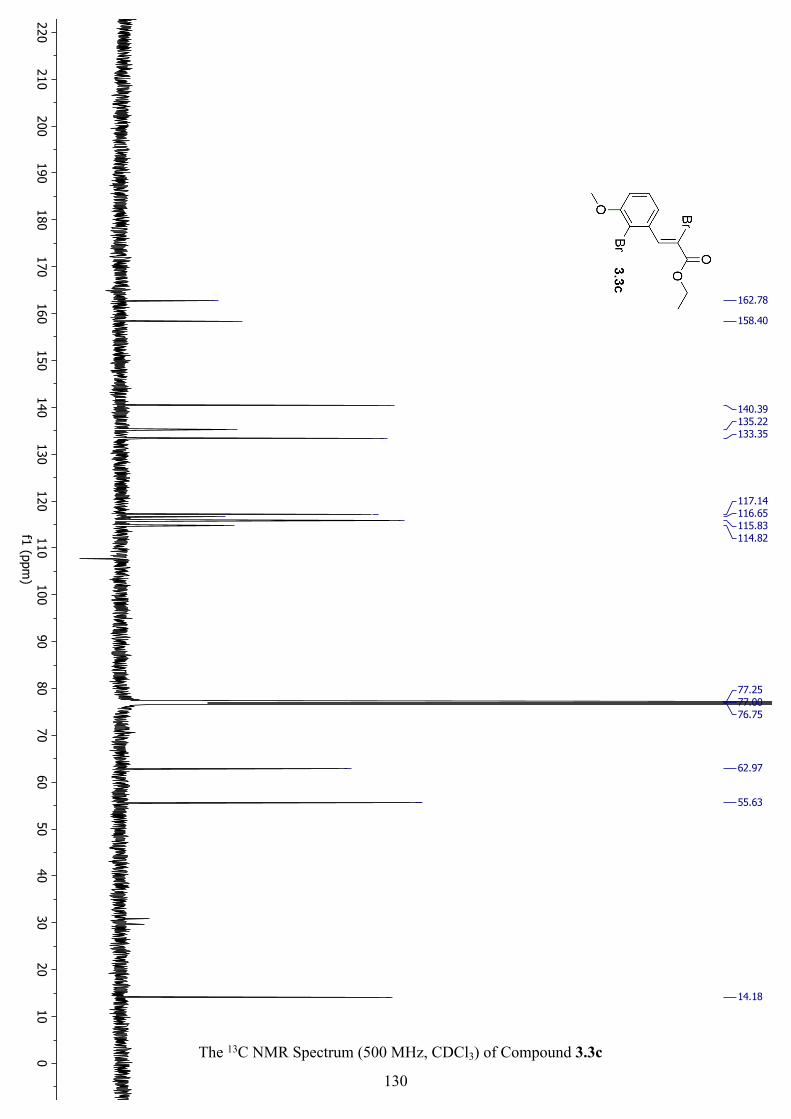
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.18
55.63
62.97
76.7577.0077.25
114.82115.83116.65117.14
133.35135.22140.39
158.40
162.78
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3c
130
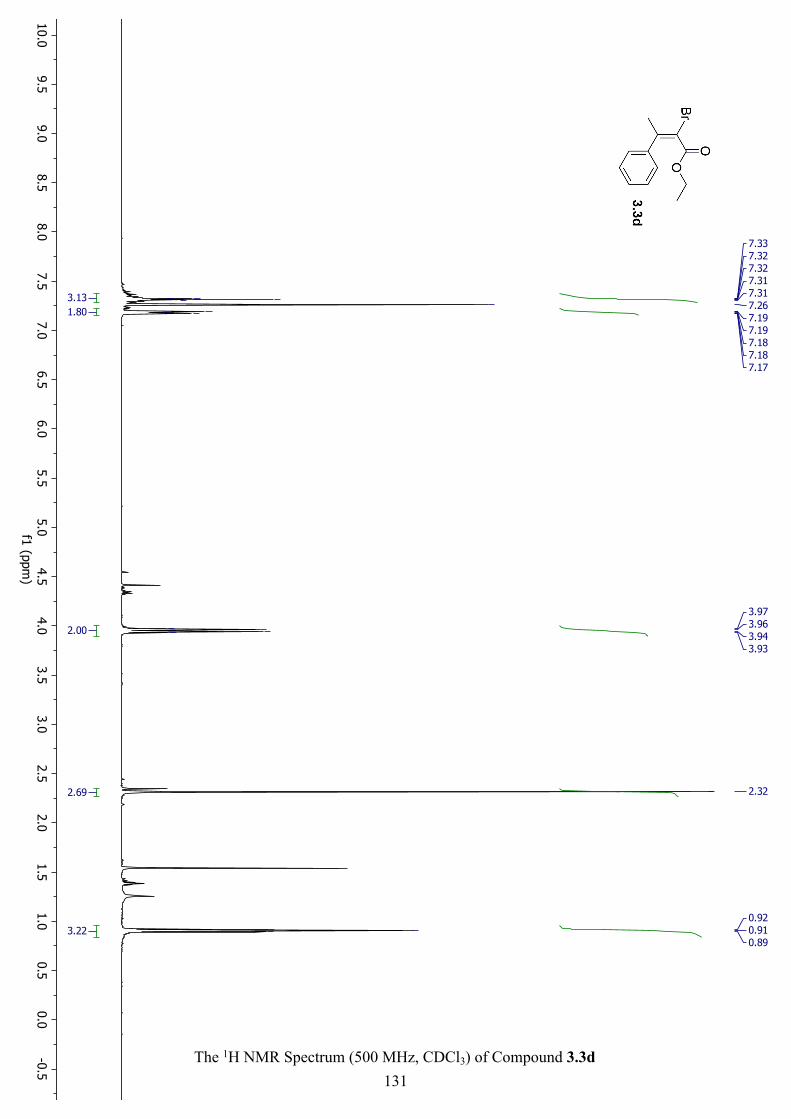
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
3.22
2.69
2.00
1.803.13
0.890.910.92
2.32
3.933.943.963.97
7.177.187.187.197.197.267.317.317.327.327.33
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3d131
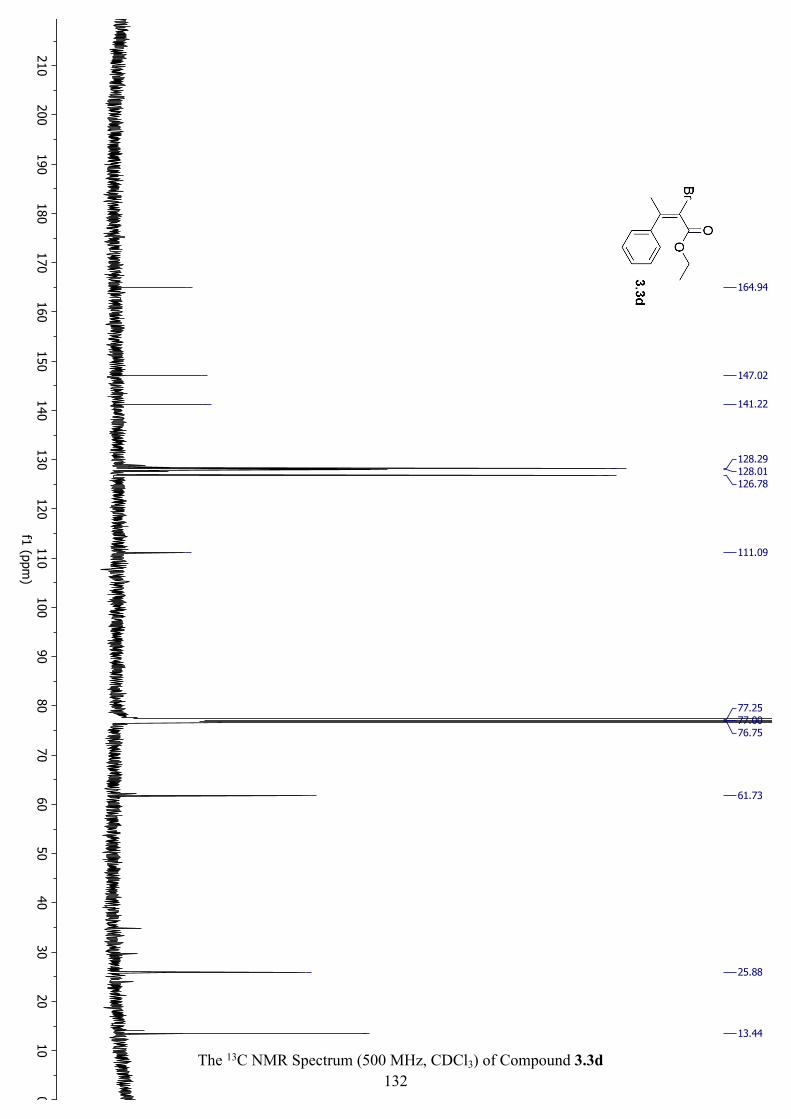
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
f1 (ppm)
13.44
25.88
61.73
76.7577.0077.25
111.09
126.78128.01128.29
141.22
147.02
164.94
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3d132
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
6.34
3.17
1.02
2.04
1.00
1.081.091.321.331.35
2.832.842.842.852.862.862.872.88
4.254.264.274.29
7.077.097.26
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3e
133
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.0f1 (ppm
)
3.72
5.54
5.47
1.04
2.06
1.00
1.161.191.211.321.331.341.671.671.681.701.701.701.711.741.741.762.542.552.562.562.572.572.582.592.60
4.244.264.274.29
7.097.107.26
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3f134
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
3.29
2.05
2.14
3.532.161.00
1.311.321.34
3.693.70
4.254.274.284.30
7.237.247.247.267.267.277.317.337.347.417.437.44
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3g
135
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.14
38.35
62.50
76.7577.0077.25
116.98
126.85128.63128.80
137.22
144.02
162.39
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3g
136
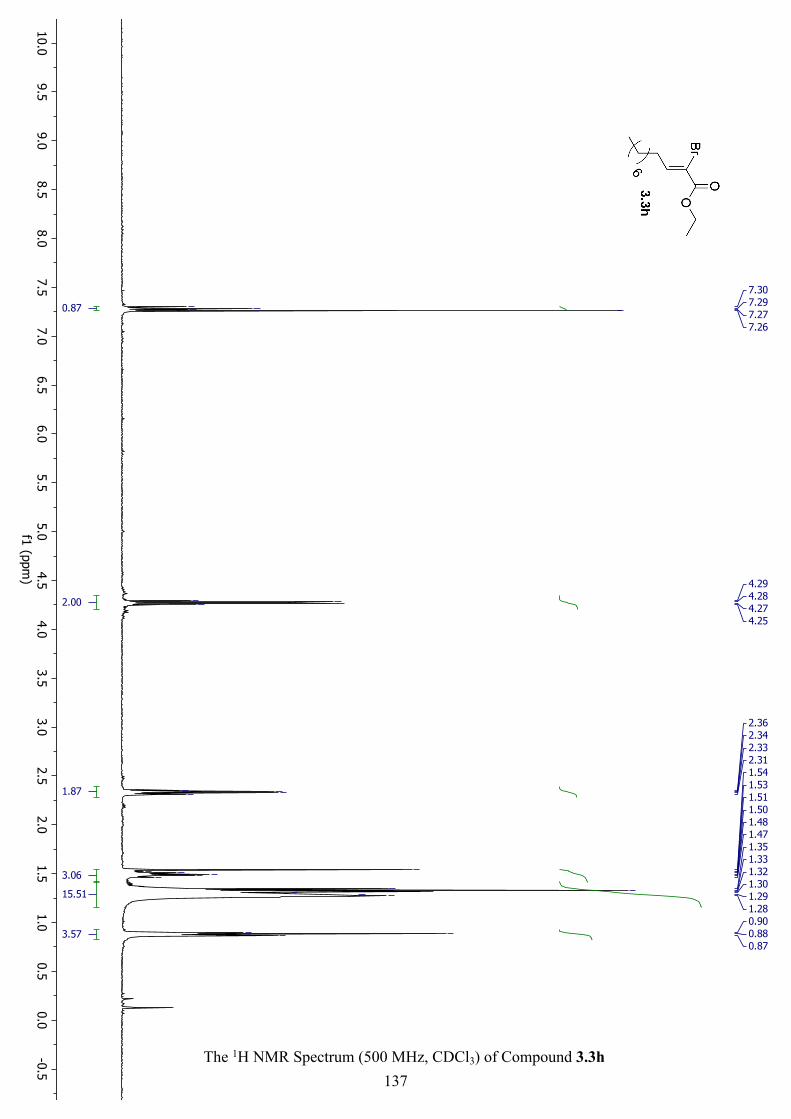
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
3.57
15.51
3.06
1.87
2.00
0.87
0.870.880.901.281.291.301.321.331.351.471.481.501.511.531.542.312.332.342.36
4.254.274.284.29
7.267.277.297.30
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.3h137
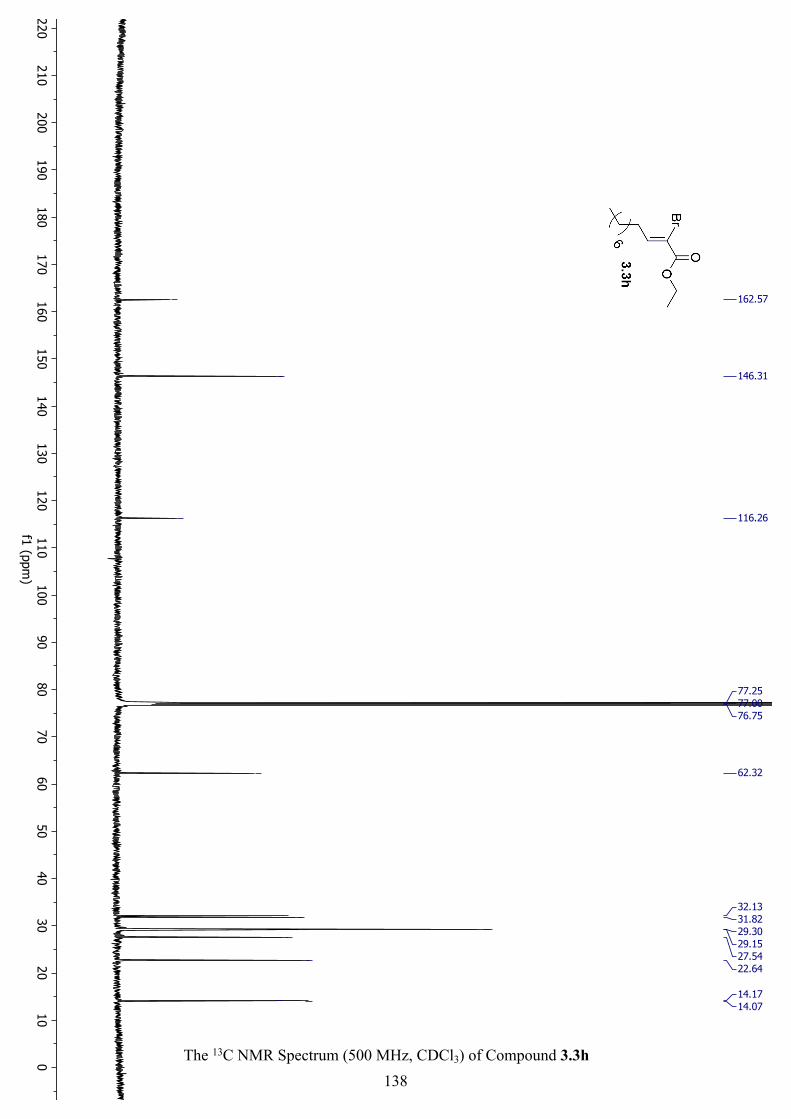
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.0714.17
22.6427.5429.1529.3031.8232.13
62.32
76.7577.0077.25
116.26
146.31
162.57
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.3h138
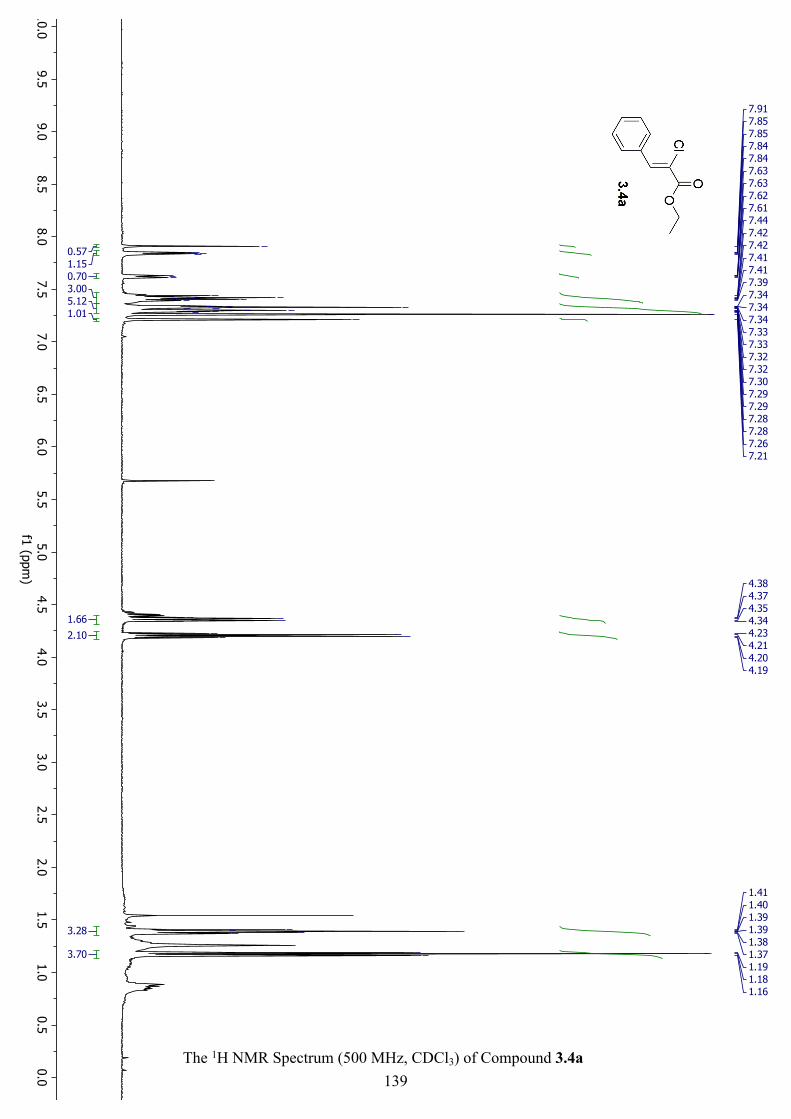
0.00.5
1.01.5
2.02.5
3.03.5
4.04.5
5.05.5
6.06.5
7.07.5
8.08.5
9.09.5
10.0f1 (ppm
)
3.70
3.28
2.10
1.66
1.015.123.000.701.150.57
1.161.181.191.371.381.391.391.401.41
4.194.204.214.234.344.354.374.38
7.217.267.287.287.297.297.307.327.327.337.337.347.347.347.397.417.417.427.427.447.617.627.637.637.847.847.857.857.91
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4a139
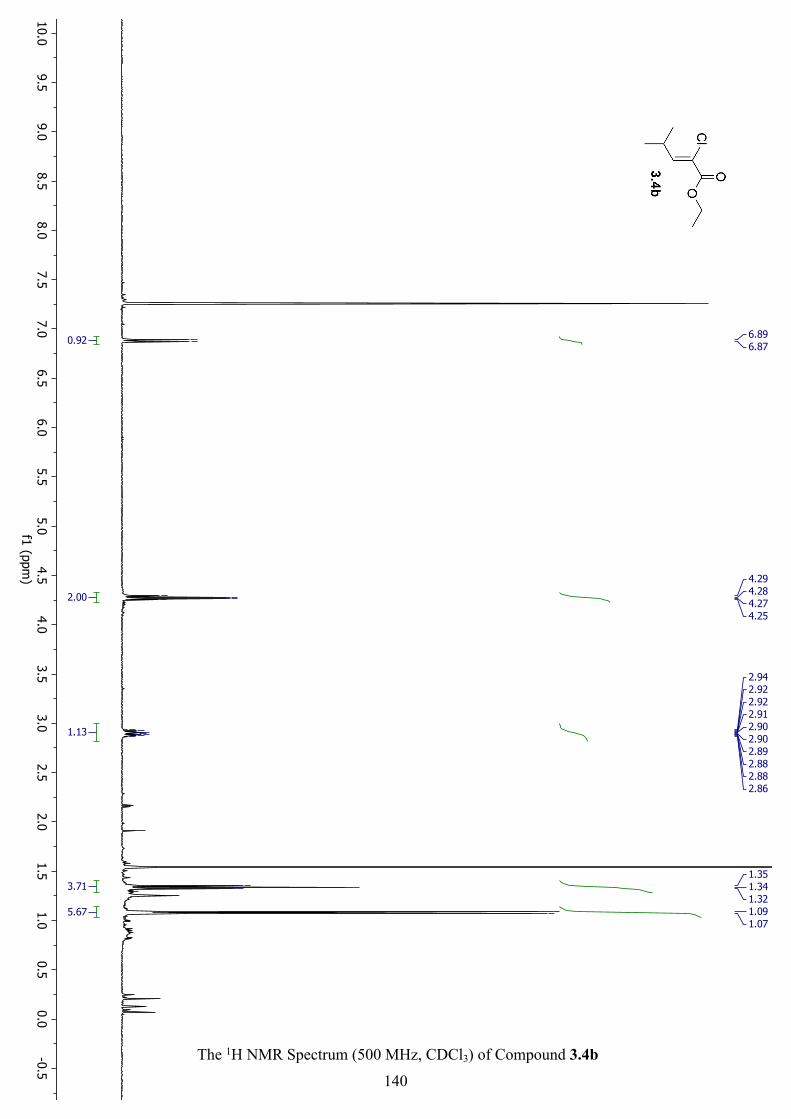
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
5.67
3.71
1.13
2.00
0.92
1.071.091.321.341.35
2.862.882.882.892.902.902.912.922.922.94
4.254.274.284.29
6.876.89
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4b
140
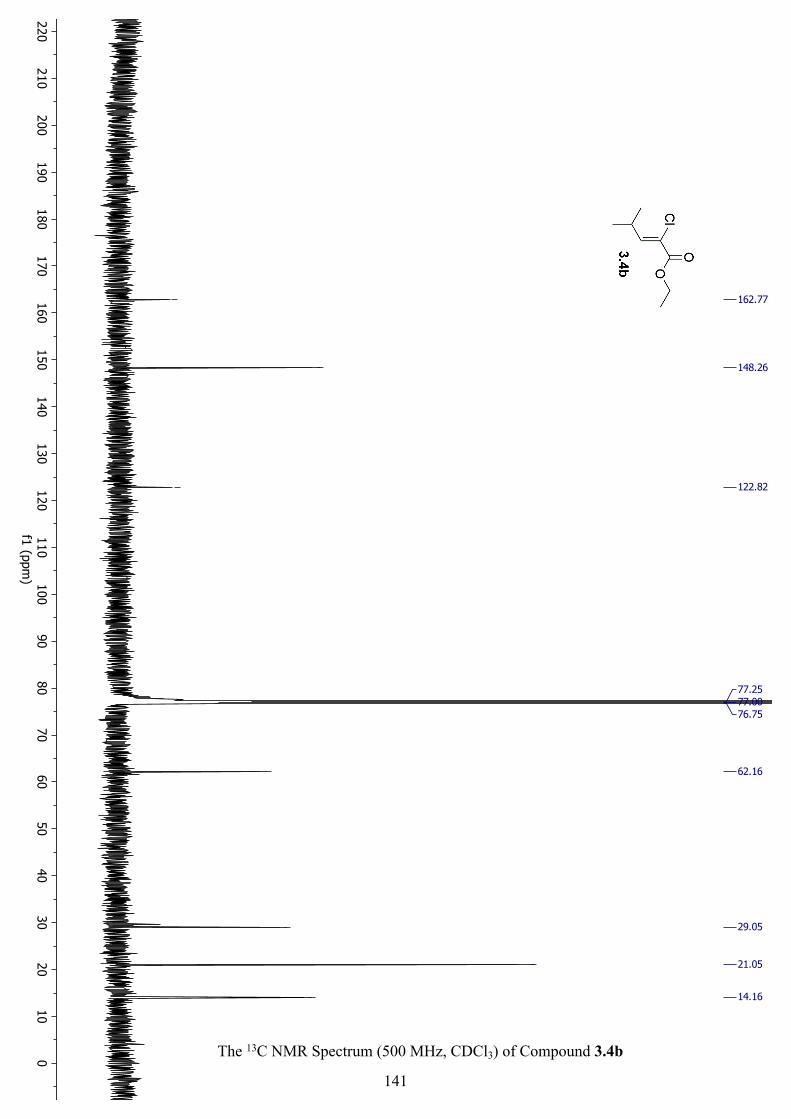
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.16
21.05
29.05
62.16
76.7577.0077.25
122.82
148.26
162.77
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.4b
141
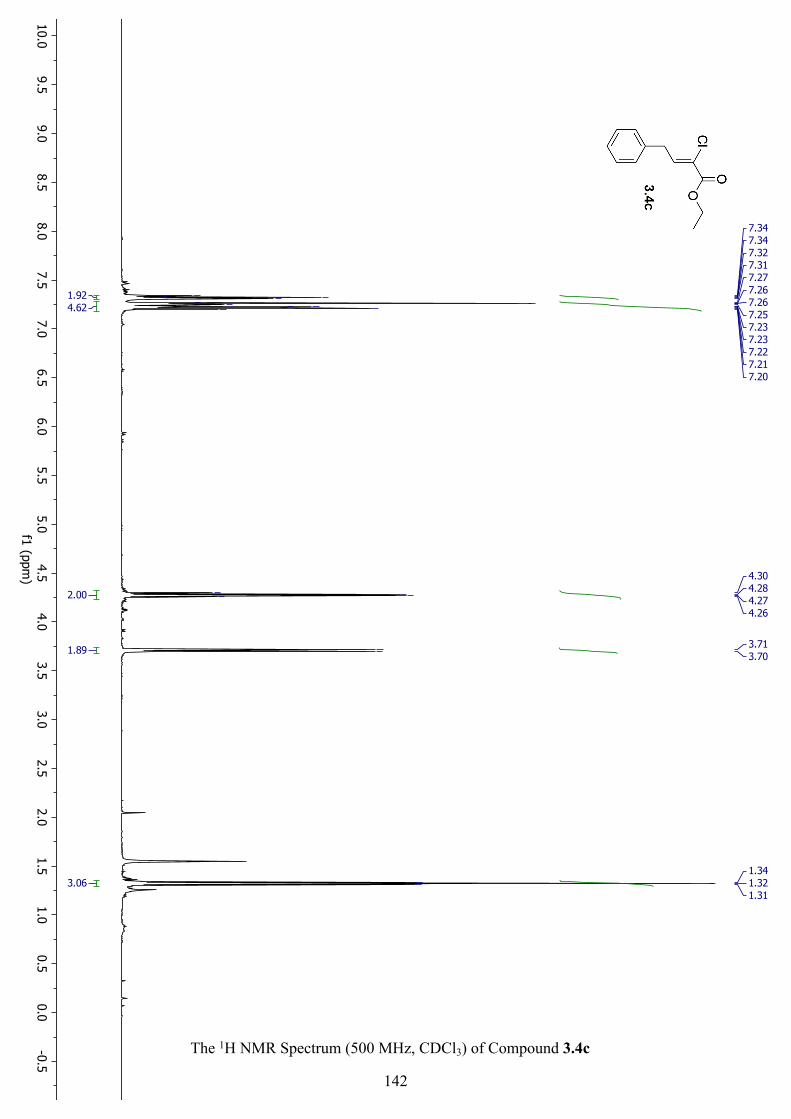
-0.50.0
0.51.0
1.52.0
2.53.0
3.54.0
4.55.0
5.56.0
6.57.0
7.58.0
8.59.0
9.510.0
f1 (ppm)
3.06
1.89
2.00
4.621.92
1.311.321.34
3.703.71
4.264.274.284.30
7.207.217.227.237.237.257.267.267.277.317.327.347.34
The 1H NMR Spectrum (500 MHz, CDCl3) of Compound 3.4c
142
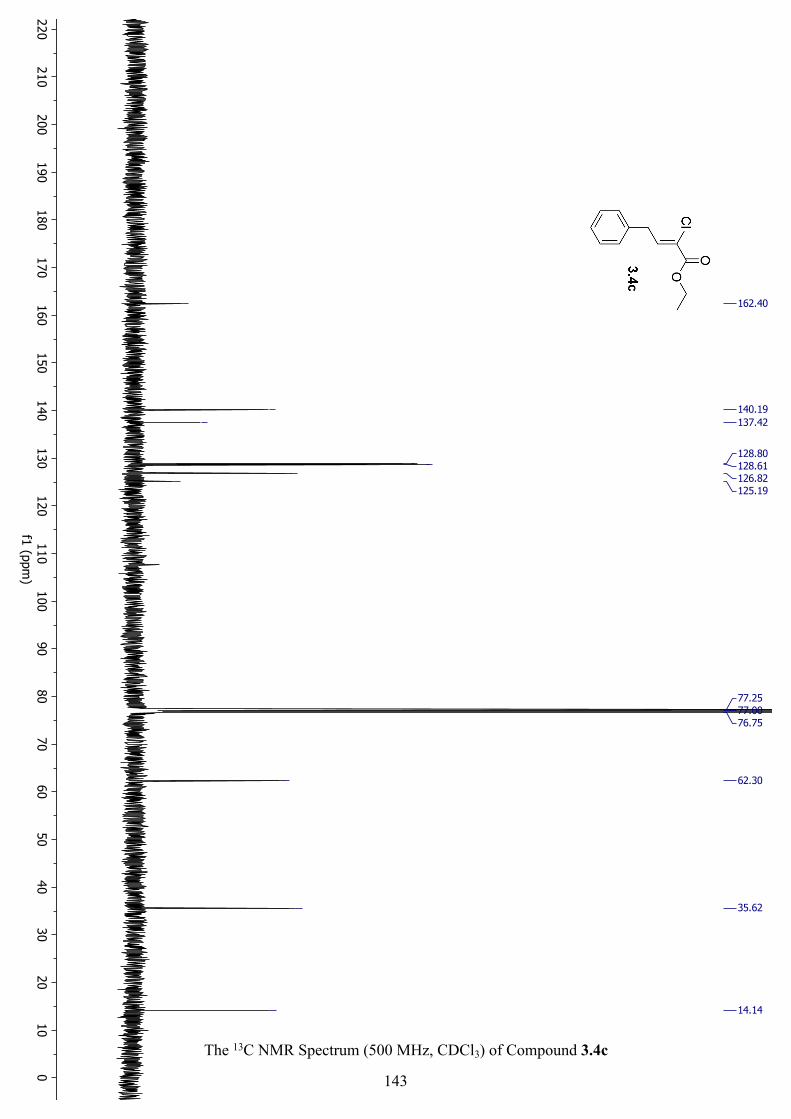
010
2030
4050
6070
8090
100110
120130
140150
160170
180190
200210
220f1 (ppm
)
14.14
35.62
62.30
76.7577.0077.25
125.19126.82128.61128.80
137.42140.19
162.40
The 13C NMR Spectrum (500 MHz, CDCl3) of Compound 3.4c
143