medicinal chemistry introduction
TRANSCRIPT

Medicinal Chemistry Introduction
13 Dic. 2019University Tor Vergata
Prof. Vincenzo Summa PhD

What is medicinal chemistry?
• Simple idealistic answer: using organic chemistry to synthesise new drugs
• More realistic answer:
Medicinal chemistry is an infinitely complicated multi-disciplinary
science that uses organic chemistry, biochemistry, physics, biology,
computational chemistry and many other scientific disciplines to
drive the discovery and development of new drug therapies
• Medicinal chemistry can be exciting and rewarding
– the decision making engine that brings forward new drugs
– opportunity to do both applied science and be involved in creative/innovative research
• Medicinal chemistry is always demanding
– developing medicines is a long, expensive and far from easy process
Good: Bad:
Isentress Thalidomide
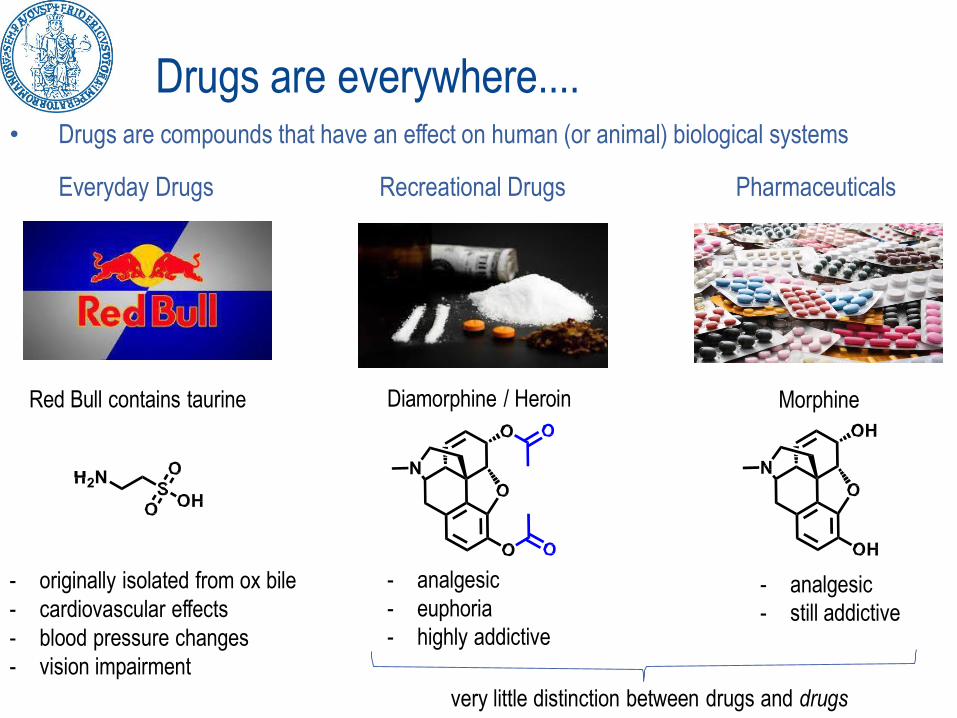
Drugs are everywhere....• Drugs are compounds that have an effect on human (or animal) biological systems
Everyday Drugs Recreational Drugs Pharmaceuticals
Red Bull contains taurine
- originally isolated from ox bile
- cardiovascular effects
- blood pressure changes
- vision impairment
Diamorphine / Heroin
- analgesic
- euphoria
- highly addictive
Morphine
- analgesic
- still addictive
very little distinction between drugs and drugs

Drug discovery through history – ‘pre-scientific’ drugs
• Nature was the original source of drugs and has been making them for a long time
– no need for medicinal chemistry if you can find the finished drug in nature
– lots of drugs have come in full or in part from nature either pre-scientific or post-scientific
- chew willow bark to ease fever and
reduce inflammation
- white willow = Salix alba
- contains salicylic acid (aspirin)
Salicylic acid Aspirin (Prodrug)
- sweet wormwood extracts from
Chinese folklore dramatically blocked
plasmodium parasite growth
- sweet wormwood = Artemisa annua
- active species identified in 1972
Artemisinin – 2015 Nobel Prize in Medicine
Hippocrates (c.400BC) Tu Youyou (1930 to date)

Drug discovery through history – serendipitous discoveries
Alexander Fleming 1928
Penicillin G
- Staphylococci bacterial cultures left
beside open window during Fleming’s
vacation grew moldy
- mold appeared to kill some of the
bacteria
- penicillin antibiotics isolated by Florey &
Chain in time to save thousands of lives
in 2’nd world war
Pfizer (1998)
Sildenafil
- compound discovered in 1991 and reached
clinical trials for chest pain / angina
- found to be fairly ineffective treatment for
heart disease
- some male patients in the trials reported
unexpected side effects (and some refused
to give back their left over pills!)
- eventually marketed as Viagra
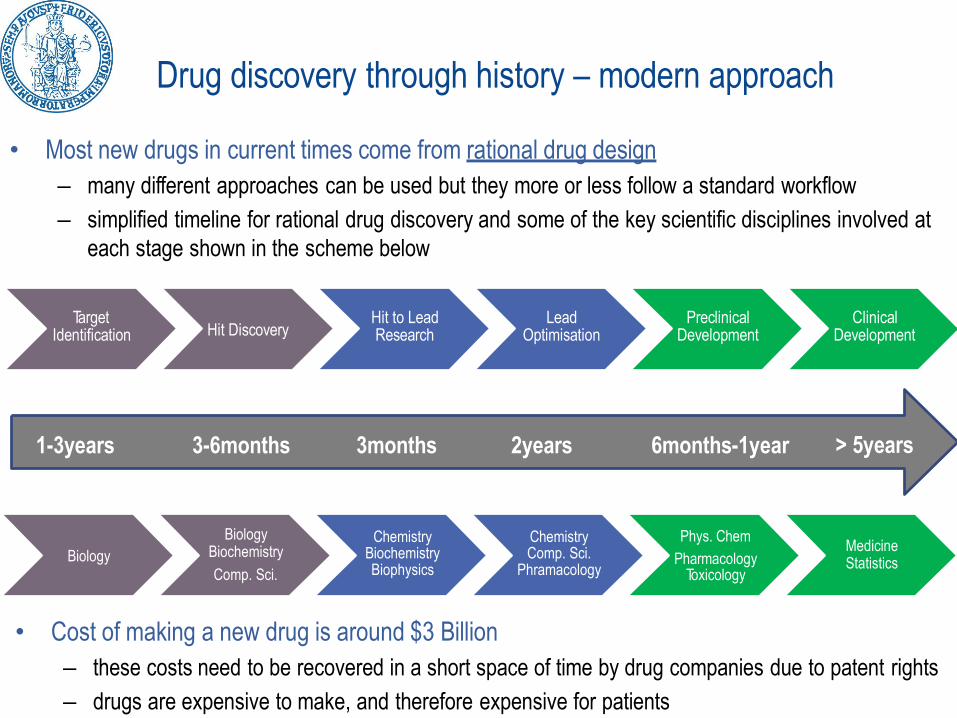
Drug discovery through history – modern approach
• Most new drugs in current times come from rational drug design
– many different approaches can be used but they more or less follow a standard workflow
– simplified timeline for rational drug discovery and some of the key scientific disciplines involved at
each stage shown in the scheme below
TargetIdentification Hit Discovery
Hit to LeadResearch
LeadOptimisation
PreclinicalDevelopment
ClinicalDevelopment
Biology
BiologyBiochemistry
Comp. Sci.
ChemistryBiochemistryBiophysics
ChemistryComp. Sci.
Phramacology
Phys. Chem
PharmacologyToxicology
MedicineStatistics
1-3years 3-6months 3months 2years 6months-1year > 5years
• Cost of making a new drug is around $3 Billion
– these costs need to be recovered in a short space of time by drug companies due to patent rights
– drugs are expensive to make, and therefore expensive for patients
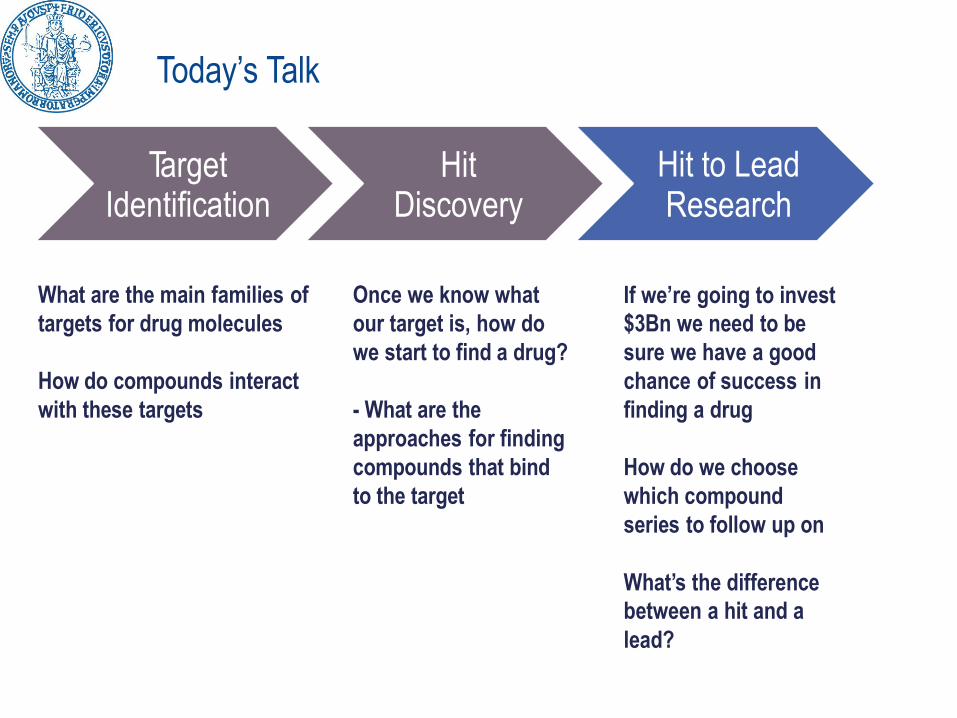
Today’s Talk
TargetIdentification
HitDiscovery
Hit to LeadResearch
What are the main families of
targets for drug molecules
How do compounds interact
with these targets
Once we know what
our target is, how do
we start to find a drug?
- What are the
approaches for finding
compounds that bind
to the target
If we’re going to invest
$3Bn we need to be
sure we have a good
chance of success in
finding a drug
How do we choose
which compound
series to follow up on
What’s the difference
between a hit and a
lead?

Target ID : knowing the target is key for drug discovery
• Biological targets where drugs interact are usually (but not always) natural polymers:
proteins, DNA, RNA etcetc
• 3 main protein target types:
– Enzymes : catalyse specific chemical processes that influence biological events and pathways
› Proteases – hydrolysis reactions, usually protein hydrolysis
› Kinases – phosphorylation reactions
› Polymerases – facilitate DNA / RNA synthesis
– Receptors : bind naturally occuring molecules and the binding triggers a physiological event
› Neurotransmitter receptor – neuronal signalling
› Growth hormone receptors – switch on and off the pathways that lead tissues to undergo growth
– Protein-protein interactions : two (or more) proteins that bind together to influence a downstream
biochemical event in a physiological pathway
› Protein-protein interactions are extremely challenging targets for medicinal chemistry
TargetIdentification Target identification/validation timeline : many months - many years

Target ID – Enzymes
• Enzymes convert substrates (S) to products (P) at an ACTIVE SITE that usually mimics the
reaction transition state (TS)
– enzymes essentially lower the barrier to reactions by making the transition state more viable
Active Site Inhibitors - Inhibitors that bind in the active site
instead of the enzymes natural
substrate
-competitive inhibitors
- inhibitor binding blocks access to the substrate
Suicide Inhibitors - Inhibitors that covalently bind to the
enzyme to inactive its mechanism of
action
- also known as irreversible inhibitors
- inhibitor binding chemically inactivates the enzyme
preventing it from turning over any further substrate
Fre
eE
nerg
y
TS
S
P
Progress of Reaction

Target ID – Enzymes
• Enzymes convert substrates (S) to products (P) usually at an ACTIVE SITE that mimics the
reaction transition state (TS)
– enzyme essentially lower the barrier to reactions by making the transition state more viable
Fre
eE
nerg
y
Allosteric Inhibitors - Inhibitors that bind at sites distal to
the active site.
-non-competitive inhibitors
- can be many allosteric sites on a single enzyme
-inhibitor binding often blocks a conformation of the
protein that prevents productive processing of the
substrate
TS
S
P
Progress of Reaction
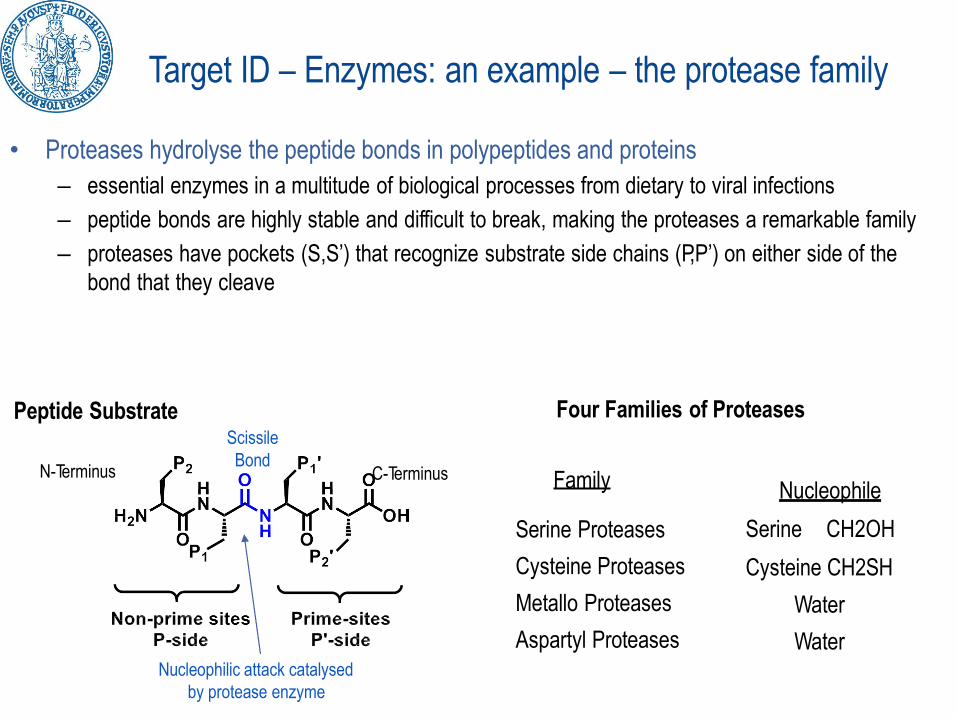
Target ID – Enzymes: an example – the protease family
• Proteases hydrolyse the peptide bonds in polypeptides and proteins
– essential enzymes in a multitude of biological processes from dietary to viral infections
– peptide bonds are highly stable and difficult to break, making the proteases a remarkable family
– proteases have pockets (S,S’) that recognize substrate side chains (P,P’) on either side of the
bond that they cleave
Peptide Substrate Four Families of Proteases
Serine Proteases
Cysteine Proteases
Metallo Proteases
Aspartyl Proteases
Scissile
BondFamily Nucleophile
Serine CH2OH
Cysteine CH2SH
Water
WaterNucleophilic attack catalysed
by protease enzyme
N-Terminus C-Terminus
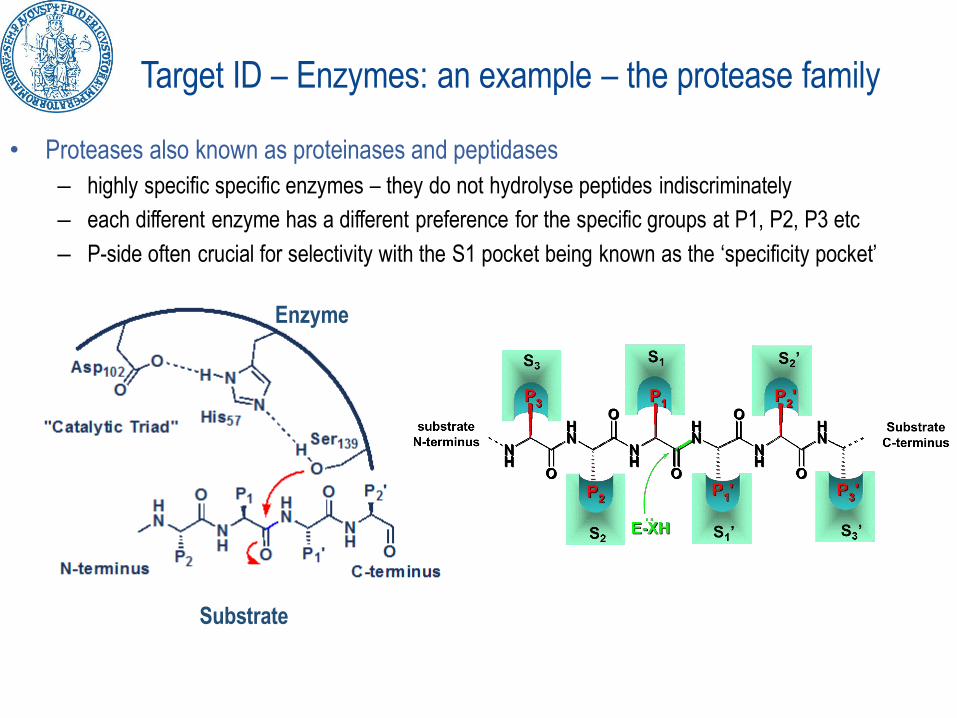
Target ID – Enzymes: an example – the protease family
• Proteases also known as proteinases and peptidases
– highly specific specific enzymes – they do not hydrolyse peptides indiscriminately
– each different enzyme has a different preference for the specific groups at P1, P2, P3 etc
– P-side often crucial for selectivity with the S1 pocket being known as the ‘specificity pocket’
Enzyme
Substrate

Target ID : Receptors
• Receptors are usually extracellular / cell-surface protein bundles
– Binding of an endogenous small molecule to the receptor can set off
a chain of complicated events INSIDE the cell
• Medicinal chemists can design small molecules that interact at the receptor
– binding of an exogenous small molecule to the receptor can achieve different outcomes
Antagonist
Exogenous
Small Molecule
Intracellular
SignalDescription
Agonist
(Partial Agonist)
Inverse Agonist
Antagonists are the receptor equivalent of enzyme inhibitors
- bind to receptor but do not produce intramolecular events
- block the binding of endogenous ligand
Off
Agonists are molecules that bind and switch on the receptor
- produce same effect as natural ligand, but usually stronger
On
Partly On
ReversedInverse agonists bind to the same receptor but produce
the OPPOSITE effect inside the cell
(Partial Inverse Agonist) Partly reversed

Hit discovery: what is a hit?
HitDiscovery
• A hit is a molecule that binds to the target protein at the site of interest
– classic model of interaction is Emil Fischer’s ‘lock and key’ theory
– protein binding sites are the locks, small molecule ligands are the keys
– early compounds in drug development and only need to bind & have a nice ‘tractable’ structure
– cell-based activity can be optimised later
Keap-1 protein with published ligand

Hit discovery: what is a hit?
• A hit is a molecule that binds to the target protein at the site of interest
– small molecules (ligands) bind to proteins through a range of potential interactions
– overall binding follows the laws of thermodynamics
Direct binding involves enthalpy-driven
interactions between protein/ligand
Flexible structures can be less
complementary to the protein ‘lock’ but are
beneficial to entropy
- can ‘pre-pay’ entropy penalty needed to
get the binding conformation by making a
constrained inhibitor
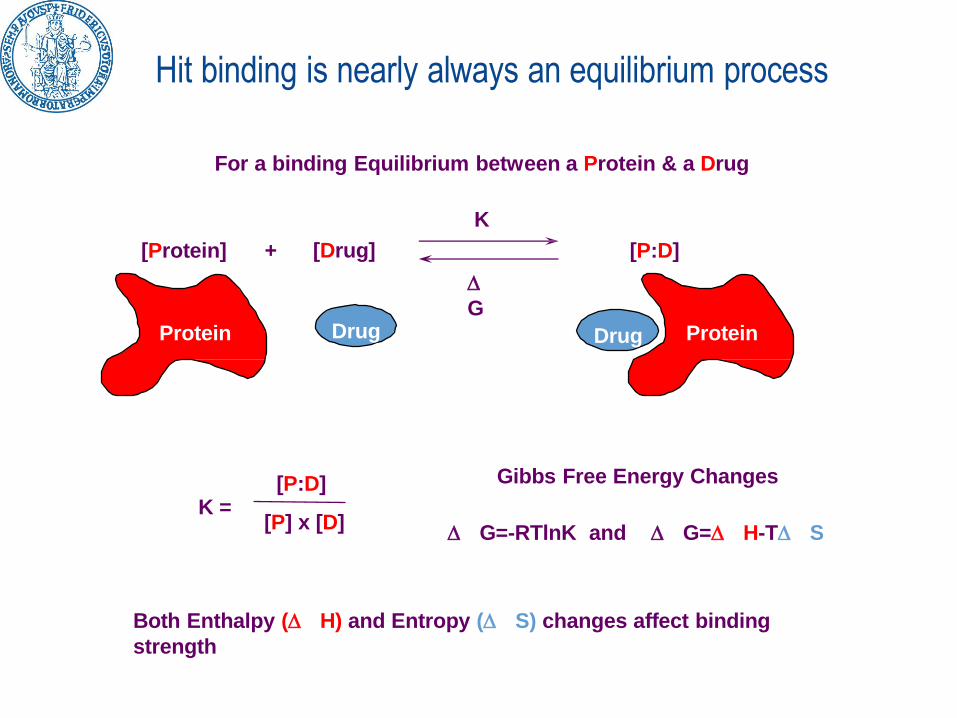
Hit binding is nearly always an equilibrium process
[P:D]
[P] x [D]
Both Enthalpy ( H) and Entropy ( S) changes affect binding
strength
Gibbs Free Energy Changes
G=-RTlnK and G= H-T S
[Protein] + [Drug] [P:D]
For a binding Equilibrium between a Protein & a Drug
K
G
Drug ProteinDrugProtein
K =
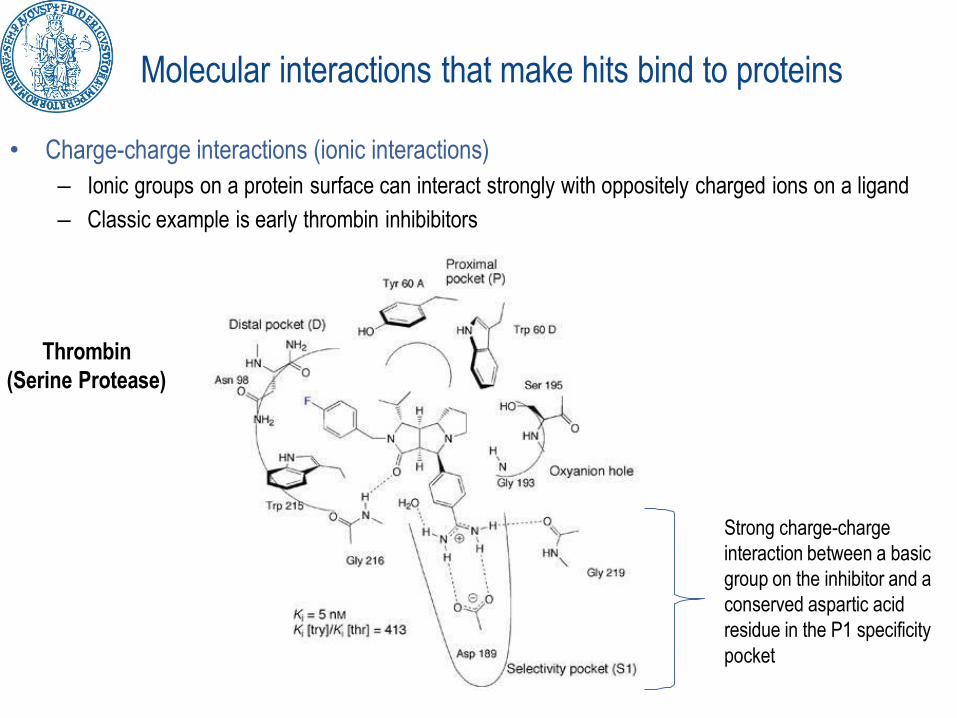
Molecular interactions that make hits bind to proteins
• Charge-charge interactions (ionic interactions)
– Ionic groups on a protein surface can interact strongly with oppositely charged ions on a ligand
– Classic example is early thrombin inhibibitors
Thrombin
(Serine Protease)
Strong charge-charge
interaction between a basic
group on the inhibitor and a
conserved aspartic acid
residue in the P1 specificity

NB. When a drug moves from the aqueous medium into the ‘Binding Site’ it has to
break H-Bonds with water, de-solvate etc. These processes require energy, so the
net energy available for binding is only a fraction of the above bond energies.
Drug-Protein Interactions
Bond Example kJ/mol
Van der Waal
Hydrophobic
Dipole - Dipole
Hydrogen
Xe…Xe, alkyl groups 2
Ph…Ph ( -stacking) 5
C=O…HN-R ( +/ -)...( +/ -) 5
35H2O…H2O (X-H) …(Y-R)
Ion - Dipole F-…H2O (+/-ve)…(+/-) 170
Ion - Ion H+…Cl- (+ve)…(-ve) 450
Covalent C-O 350
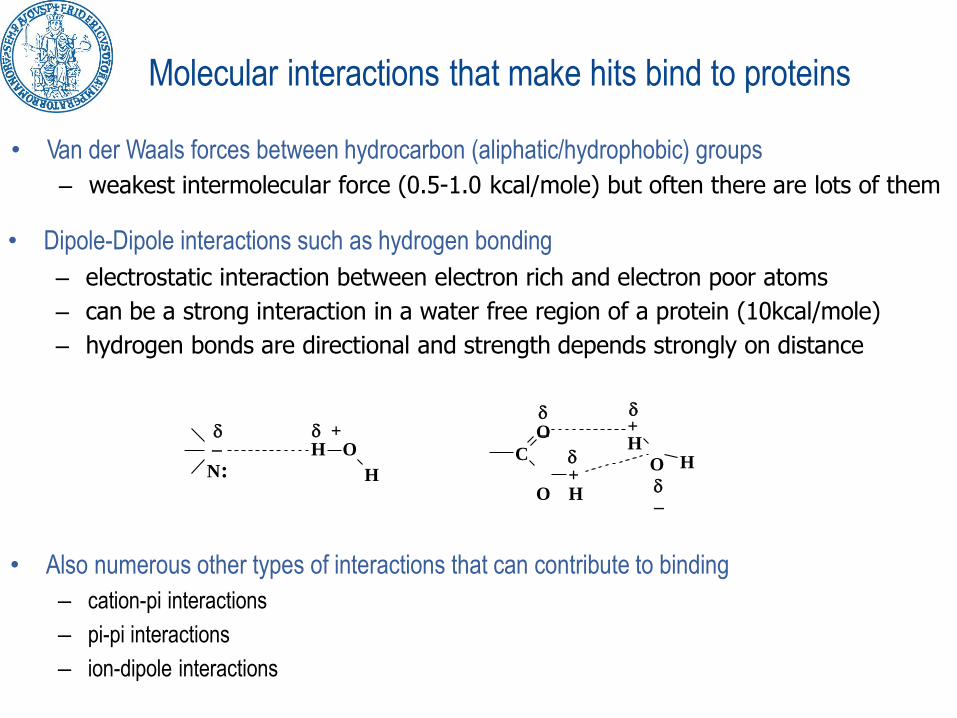
• Van der Waals forces between hydrocarbon (aliphatic/hydrophobic) groups
– weakest intermolecular force (0.5-1.0 kcal/mole) but often there are lots of them
• Dipole-Dipole interactions such as hydrogen bonding
– electrostatic interaction between electron rich and electron poor atoms
– can be a strong interaction in a water free region of a protein (10kcal/mole)
– hydrogen bonds are directional and strength depends strongly on distance
• Also numerous other types of interactions that can contribute to binding
– cation-pi interactions
– pi-pi interactions
– ion-dipole interactions
N:
+H O
H
C
O
+
O H
+H
O
H
Molecular interactions that make hits bind to proteins

Entropy – hard to understand but important
• Based on the properties of the drug molecule, need to think about entropic effects involved in
dissolution – drugs need to be free from solvent to undergo enthalpic interactions with protein
When a hydrophobic drug is placed into water, the structure of
the water around the drug is more ordered.
This creates more ordered H2O-H2O H-bonding
This is lower entropy and is not favoured
Water molecules are preferably in a highly
disordered state.
Each molecule maximises H-bonds to other
water molecules
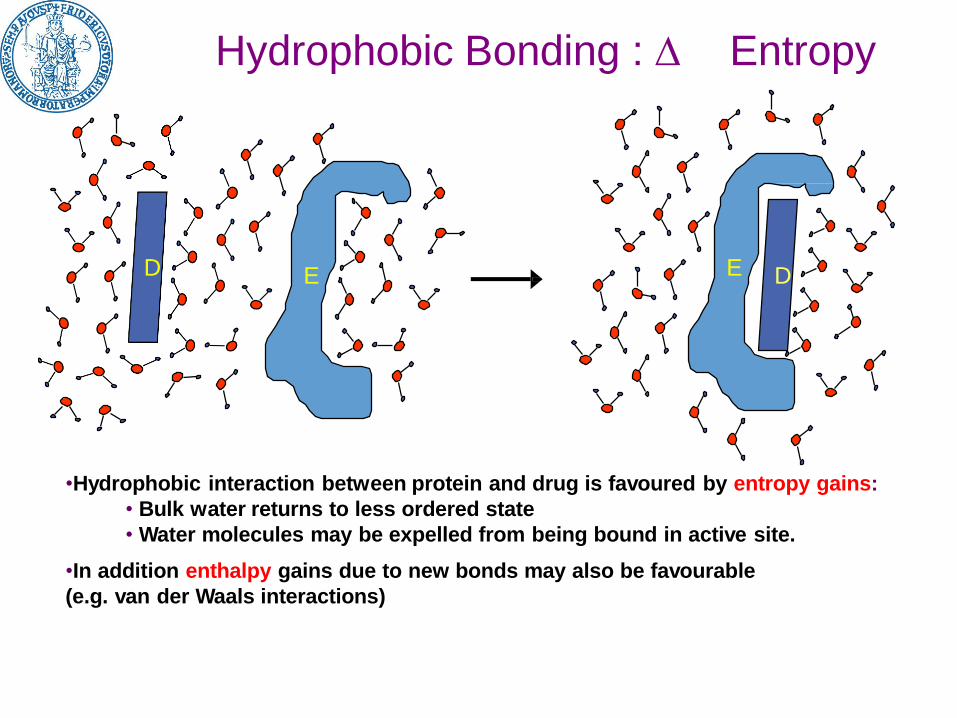
Hydrophobic Bonding : Entropy
D E
•Hydrophobic interaction between protein and drug is favoured by entropy gains:
• Bulk water returns to less ordered state
• Water molecules may be expelled from being bound in active site.
•In addition enthalpy gains due to new bonds may also be favourable
(e.g. van der Waals interactions)
DE

How are hits found? – High Throughput Screening
• High Throughput Screening (HTS)
– most companies have collections of potential hits from 50,000 – 3 million compounds!
– test ALL of these compounds at one or two different concentrations to see if they bind
– select only the compounds that bind and then measure them at 10-20 concentrations to determine
IC50
IC50 – concentration of drug that
causes 50% inhibition of enzyme or
receptor activity
EC50 – effective concentration of
drug that causes 50% inhibition of
enzyme or receptor activity
- e.g. cell based assays
CC50 – concentration of drug that
causes 50% cytotoxicity (cell death)
in a cell based assay

• Therapeutic window / therapeutic index can be important even for hits (selectivity index)
– therapeutic index is the ratio of toxic dose (TD50) / effective dose (ED50)
- usually needs to be >100
- higher TI means safer medicines
- two ways to improve the TI
➢ make cpds more effective
➢ make cpds less toxic
• For hits a selectivity index can be used to prioritize compounds
– selectivity index is the ratio of CC50 / EC50 or even IC50
How are hits found? – High Throughput Screening

What properties do compound collections target
• IRBM’s compound collection is 350000 compouds with carefully monitored properties
Structural diversity is a key to covering
‘Chemical Space’
- Redundancy needed to avoid false
negatives but should not be too high
- Murcko scaffolds often used to
compare molecules computationally
Mwt influences many things %Csp3 impacts solubility PSA influences absorption
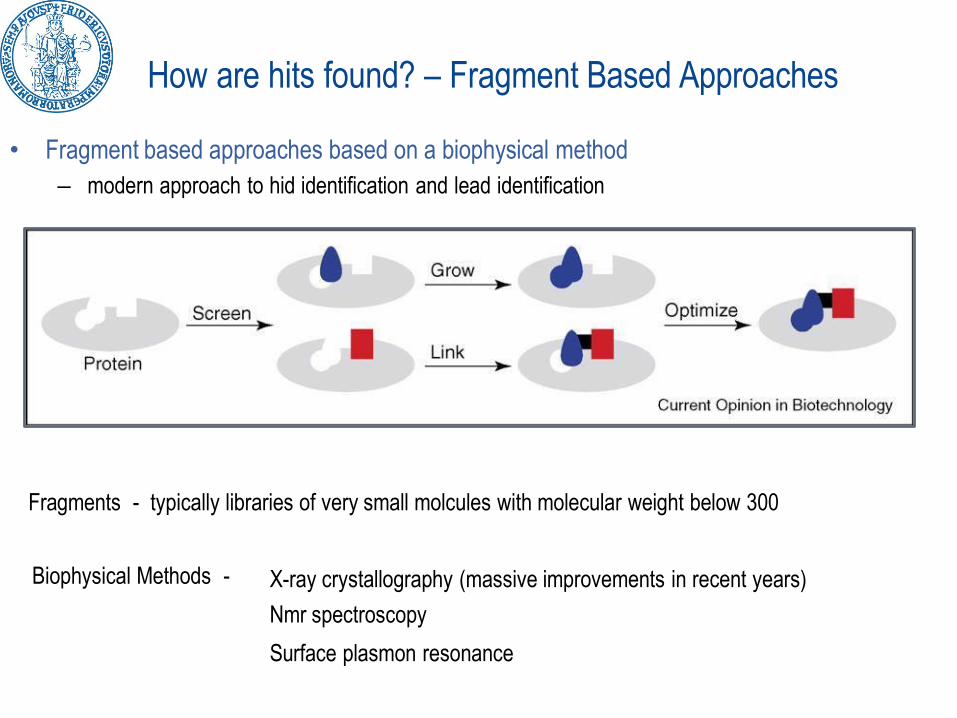
• Fragment based approaches based on a biophysical method
– modern approach to hid identification and lead identification
Fragments - typically libraries of very small molcules with molecular weight below 300
Biophysical Methods - X-ray crystallography (massive improvements in recent years)
Nmr spectroscopy
Surface plasmon resonance
How are hits found? – Fragment Based Approaches

• Design from a natural product or endogenous ligand
– many drugs have come from natural receptor ligands as the original hits
How are hits found? – Other Approaches
Adrenalin Salbutamol (GlaxoSmithKline for asthma)
• ‘Me too!’ approaches
– copying other companies drugs or patent literature is an art
– not always easy to copy, but can be a very productive way to get to new starting points
– competition in pharmaceutical industry is huge, and highlights the importance of patents
› need to cover broad range of patent space to block me-too approaches from competitors
Viagra (Pfizer) Levitra (Bayer)

Hit to lead efforts – where medicinal chemistry starts to get
interesting
Hit to LeadResearch
• Hits are compounds that inhibit the target protein with a measurable IC50
Hit to Lead Medicinal Chemistry
Structure Activity Relationships
• Leads are compounds that show improved potency with respect to the hit AND have some
other key properties
– the specific properties that medicinal chemists look for in a lead compound vary
– some key features are:
› chemical stability
› lead-likeness (tractable structure, suitable physicochemical properties)
› suitable early DMPK (Drug Metabolism & Pharmacokinetic) profile

What properties do drugs need to have?
• Whole range of properties and behaviours that are needed for them to be successful
– needs to be soluble in WATER
› all chemical reactions / interactions in humans occur in an aqueous solvent
› apart from bones, humans are well over 80% water
– needs to be stable in acidic environment of the stomach
– needs to get into the human bloodstream (absorption)
– needs to remain in circulation (metabolism / elimination as the body tries to get rid of the drug)
– needs to get to the site of action (distribution)
– needs to be non-toxic
– needs to be novel / patentable
• The fewer weak points (liabilities) a lead has in terms of these long term targets the higher
the probability of success of evolving the lead compound to a drug
– hit to lead work aims to get rid of any obvious issues in the hit compound
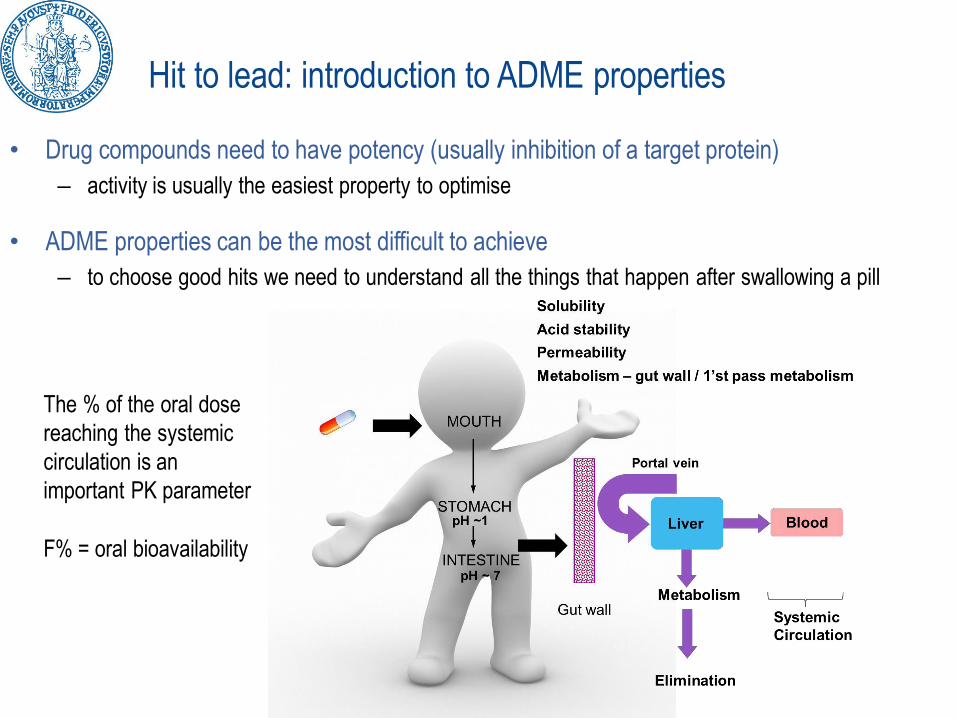
Hit to lead: introduction to ADME properties
• Drug compounds need to have potency (usually inhibition of a target protein)
– activity is usually the easiest property to optimise
• ADME properties can be the most difficult to achieve
– to choose good hits we need to understand all the things that happen after swallowing a pill
The % of the oral dose
reaching the systemic
circulation is an
important PK parameter
F% = oral bioavailability

Hit to Lead – example of a hit-to-lead process
HTS Screening Hit
IC50 19uM
Solubility >3mg/mL
Not patentable
Medicinal Chemistry Analysis
R = Me: IC50 200 uM
R = H: IC50 2 uM
Acid group
is needed
but better at
6-position
SAR at the Acid Group
R = CH3 IC50 90 uM
R = Ph IC50 120 uM
R = Chex IC50 0.7 uM
R = Cbut IC50 5 uM
SAR at the N-Alkyl Group
A cycloalkyl
group is
needed
SAR at the C2 Aromatic Group
R = CH3 IC50 Inactive
R = 4-Cl-Ph IC50 0.4 uM
R = Chex IC50 Inactive
An aryl
group is
needed at
C2
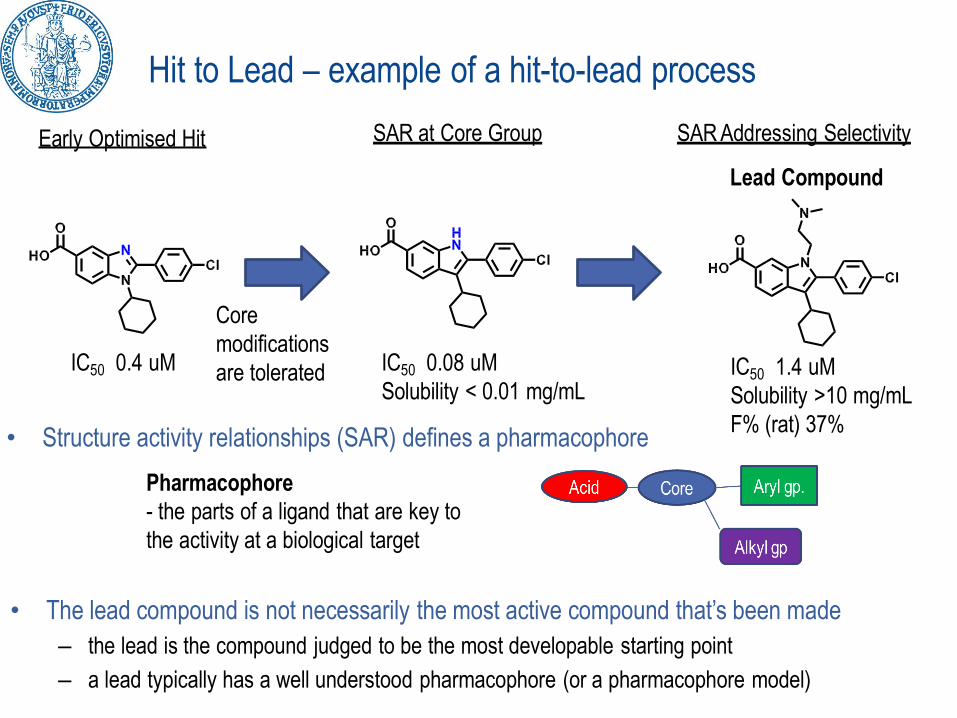
Hit to Lead – example of a hit-to-lead process
• Structure activity relationships (SAR) defines a pharmacophore
Pharmacophore
- the parts of a ligand that are key to
the activity at a biological target
IC50 0.4 uM IC50 0.08 uM
Solubility < 0.01 mg/mLIC50 1.4 uM
Solubility >10 mg/mL
F% (rat) 37%
SAR Addressing Selectivity
Lead Compound
SAR at Core Group
• The lead compound is not necessarily the most active compound that’s been made
– the lead is the compound judged to be the most developable starting point
– a lead typically has a well understood pharmacophore (or a pharmacophore model)
Early Optimised Hit
Core
modifications
are tolerated
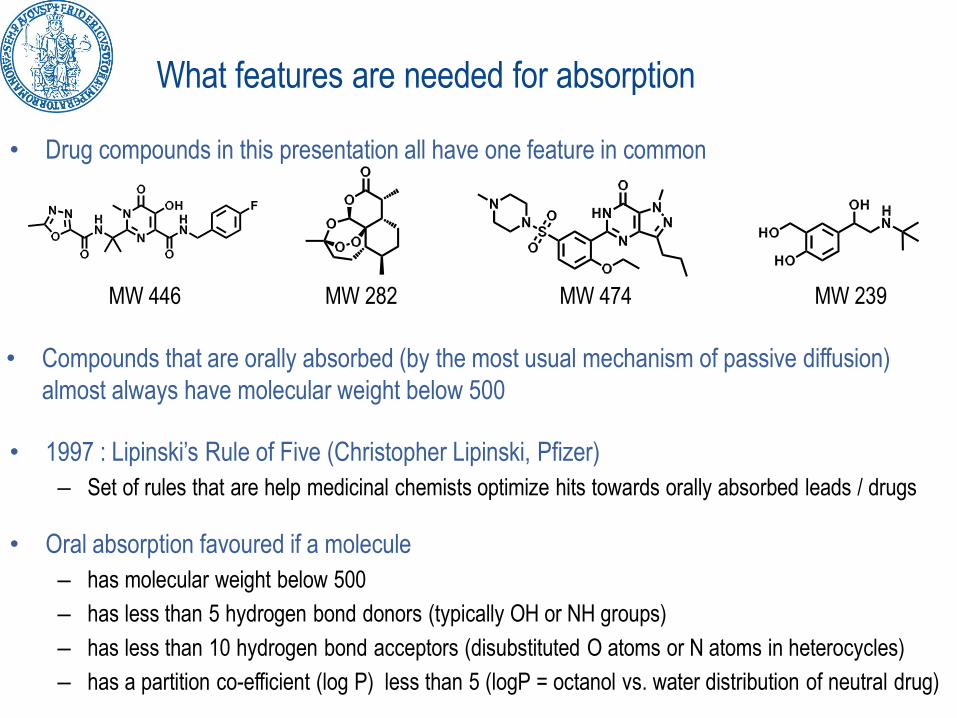
What features are needed for absorption
• Drug compounds in this presentation all have one feature in common
MW 446 MW 282 MW 474 MW 239
• Compounds that are orally absorbed (by the most usual mechanism of passive diffusion)
almost always have molecular weight below 500
• 1997 : Lipinski’s Rule of Five (Christopher Lipinski, Pfizer)
– Set of rules that are help medicinal chemists optimize hits towards orally absorbed leads / drugs
• Oral absorption favoured if a molecule
– has molecular weight below 500
– has less than 5 hydrogen bond donors (typically OH or NH groups)
– has less than 10 hydrogen bond acceptors (disubstituted O atoms or N atoms in heterocycles)
– has a partition co-efficient (log P) less than 5 (logP = octanol vs. water distribution of neutral drug)

Mechanisms of drug absorption
Transcellular absorption
– Main route for most oral drugs
– Drug must be in solution at cell surface
– pKa important - drug must be unionised
– Lipophilicity important
– Lipinski’s ‘Rule of 5’
Paracellular absorption
– drug passes through gaps between cells
– inefficient – pores have << surface area than cellular surface so hard for drugs to find the pores
– restricted to small polar molecules
Active Transport
– drugs carried through membrane by a transporter – requires energy
– many transporters exist for nutrient molecules, eg glucose, amino acids
– SAR specific – few drugs absorbed by this route

What you learnt about the Drug Discovery Process…..
1) The HITs are never the best & most active molecules2) You need to confirm the HITs, by synthesis or by acquisition of
cmpds3) Primary exploratory SAR 4) Identify a lead, the lead is never the Drug5) Led optimization phase: long, difficult, multiparameters needs to
be taken in consideration6) Identification of Preclicnial Candidate.... It shoold have the
potential to became a drug
HIT to Lead - Lead to preclinical candidate:
Each of these point implies different chemistry stretegies to obain the maximun results in shorter time
Today we will talk about point 3 and 4
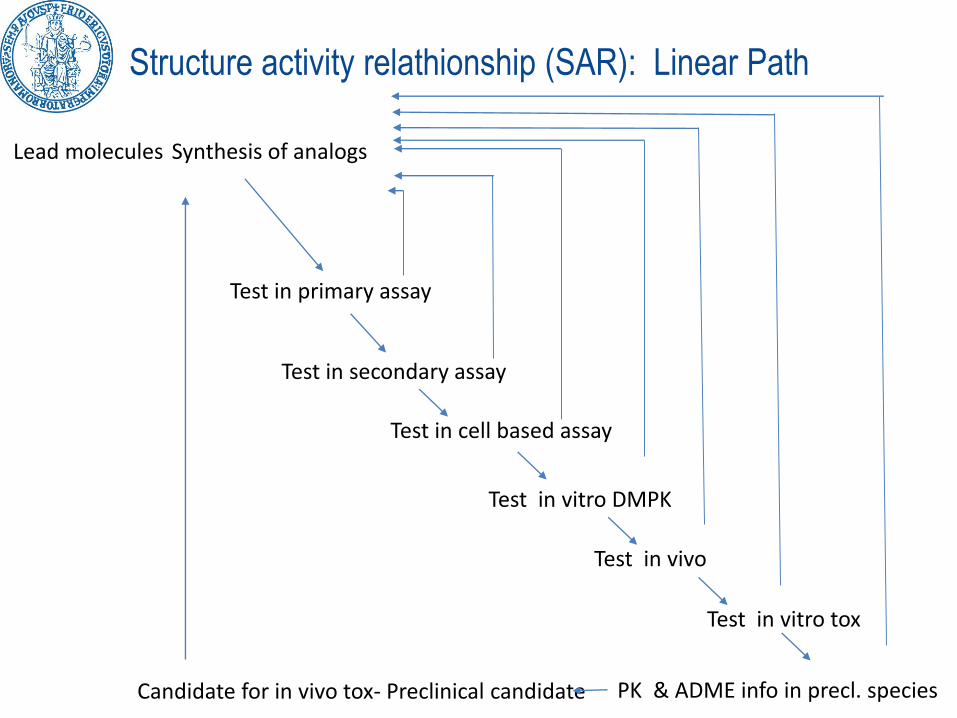
Structure activity relathionship (SAR): Linear Path
Lead molecules Synthesis of analogs
Test in primary assay
Test in secondary assay
Test in cell based assay
Test in vitro DMPK
Test in vivo
Test in vitro tox
PK & ADME info in precl. speciesCandidate for in vivo tox- Preclinical candidate
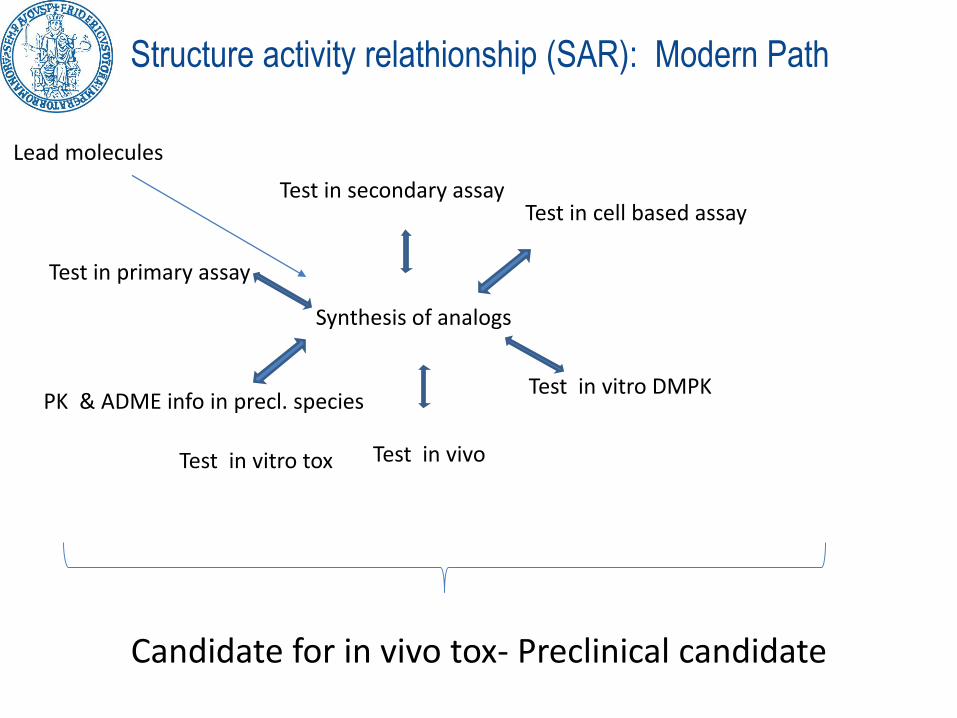
Structure activity relathionship (SAR): Modern Path
Lead molecules
Synthesis of analogs
Test in primary assay
Test in secondary assayTest in cell based assay
Test in vitro DMPK
Test in vivoTest in vitro tox
PK & ADME info in precl. species
Candidate for in vivo tox- Preclinical candidate

Analogs Synthesis .... A big dilemma
First: define the strategy guided by the level of knowledge on the program
Assumtion: HIT validated!
Small SAR information available: explore the scaffold to identify minimal active template ( small number of analogs, acquisiton of commercial cmpds)
Minimal template known: esploration of the SAR of side chains to improve activity against the targets ( large set of compunds needed to explore the chemical space around the core)
SAR in conjuntions in vitro DMPK acitivities to identifiy liabilities (a good number of compounds needed)
SAR to fix DMPK issues (focus synthesis to answer specific question or hypothesis)
Scale up to fully characterize a small set of compounds both in vitro and in vivo (optimization of the synthesis)

Scaffold exploration definition of minimal requirements
Hit Lead investigation: Goal identify the minimal active template – define pharmacofore
1 case: HIV integrase program
The HITs
Inactive compounds
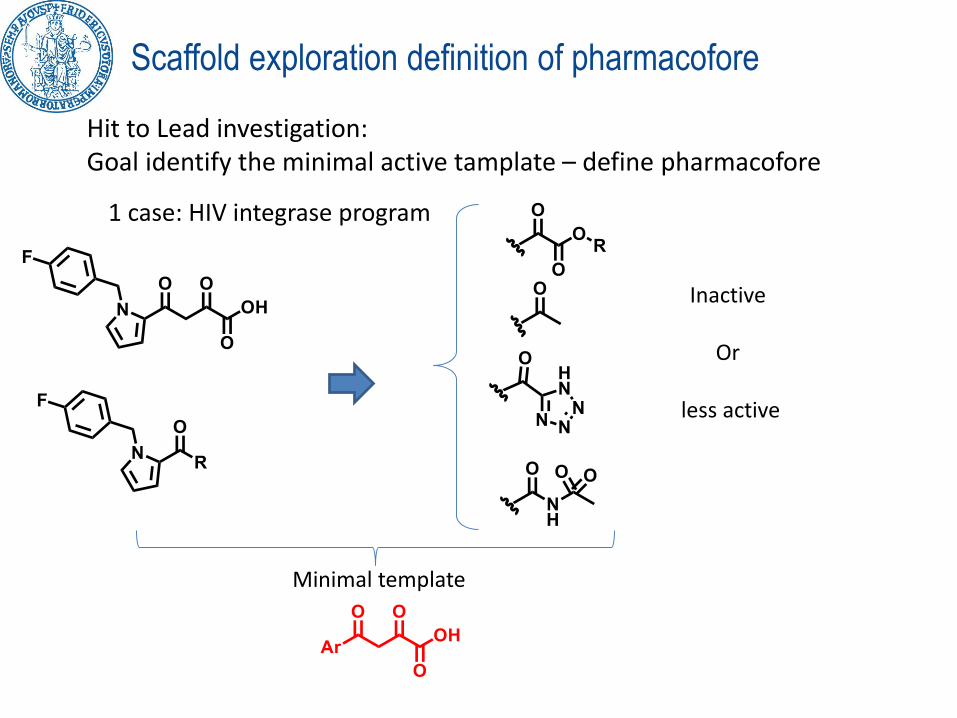
Scaffold exploration definition of pharmacofore
Hit to Lead investigation: Goal identify the minimal active tamplate – define pharmacofore
1 case: HIV integrase program
Inactive
Or
less active
Minimal template
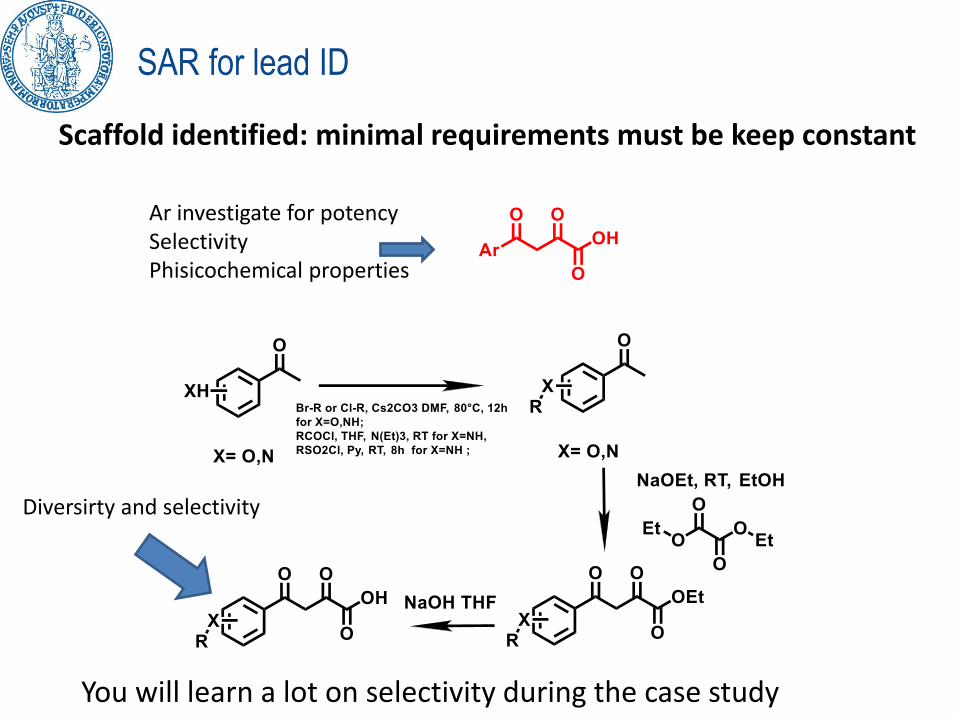
SAR for lead ID
Scaffold identified: minimal requirements must be keep constant
Ar investigate for potencySelectivityPhisicochemical properties
Diversirty and selectivity
You will learn a lot on selectivity during the case study

Quinolone antibacterial Pharmacophore - Excercise
Active Compounds
Inactive Compounds
Minimal template
?
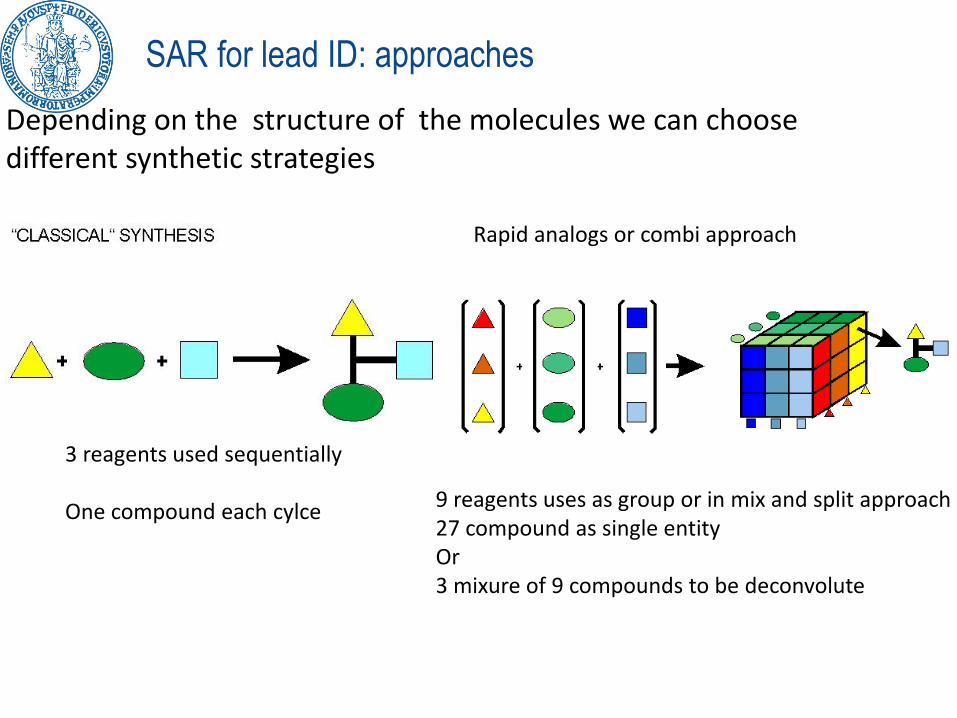
SAR for lead ID: approaches
3 reagents used sequentially
One compound each cylce
Rapid analogs or combi approach
9 reagents uses as group or in mix and split approach27 compound as single entityOr3 mixure of 9 compounds to be deconvolute
Depending on the structure of the molecules we can choose different synthetic strategies
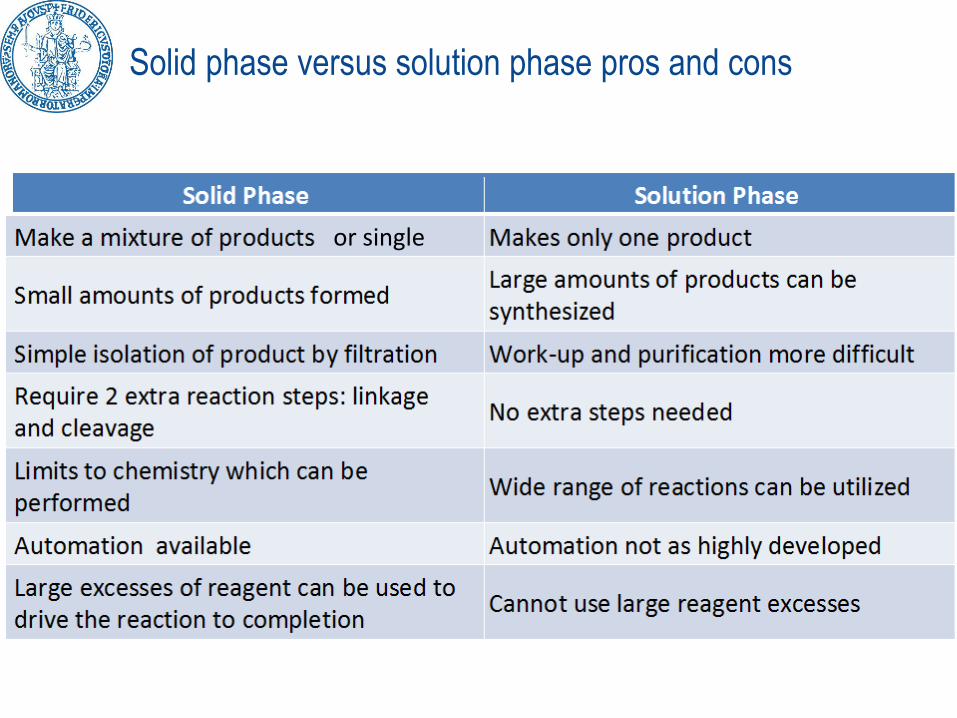
Solid phase versus solution phase pros and cons
or single

MIX and split approach
When to use this approach?1) Chemistry must be very efficient and modular2) Easy work up of the reactionExample 3 reagents available X,Y,Z to be combined to form XYZ molecules
At the end we have 3 pools of nine compounds each, ready for testing

YXY
Deconvolution of the mixtures
Active mixture resynthesis of the pool
YXY
Active mixture resynthesis of the single cmpds

Practical aspects

Parallel synthesis
Most preferred method to create very robust data You must try to apply the methods any time you have a good chemistry in handsUse as much as possible supported reagents and scavangers to run reaction in solution
Common intermediate

Example 1: reactions carried out by supported reagents
Example 1:Library of 4000 compounds prepared by 400 mixture of 10 compounds

Example 1: Deconvolution of the active pool

Example 1: Comparison standard chemistry vs supported
reagent
The biological results are comparable between the two synthetic metodologies
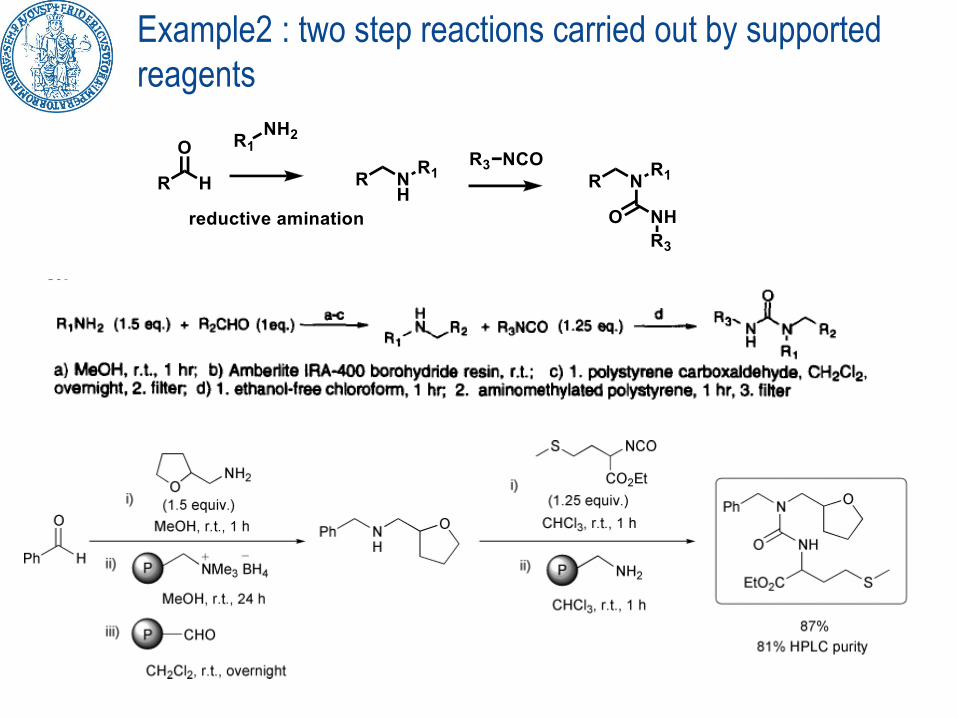
Example2 : two step reactions carried out by supported
reagents

Example 3: multistep synthesis carried out by supported
reagents

Consideration about polymer supported reagents
Very convinient for sigle step and standard reactions
Needs to have good number of testing reactions to optimize the procedure per single step
Allow to set up a parallel synthesis reducing the work up time drasticaly (dedicated tools are needed)
cost effective methodology
Reccomanded for screening compounds
Not applicable for scale up work or gram scale synthesis

Overview – take home messages
• Drugs and where they often originate
– Nature, good fortune or hard work
• Should know some of the key interactions that are involved in protein-ligand intearactions
– Different energy contributions from VdW, electrostatic interactions
• Seen the 3 main types of protein targets that drugs interact with
– Enzymes, receptors, protein protein interactions
• Broad overview of the three early stages of drug discovery from a medicinal chemistry point
of view
TargetIdentification
HitDiscovery
Hit to LeadResearch

Thank you for your attention