mcbl documentation
TRANSCRIPT

MCBL DocumentationRelease 10
Saranga
Jul 07 2020
About The MCBL
1 MCIC Computational Biology Lab (MCBL) 3
2 MCBL membership 5
3 MCBL servers and computing resources 7
4 Download from BaseSpace 11
5 Share runs and projects in BaseSpace 17
6 Using BaseMount 23
7 Download from hudsonalphaorg 31
8 Filter a CASAVA-generated fastq file 33
9 Fastq adapter removal and QC 35
10 DESeq2 with phyloseq 39
11 GBS 43
12 A basic microbiome analysis 47
13 Indices and tables 51
Python Module Index 53
Index 55
i
ii
MCBL Documentation Release 10
About The MCBL 1
MCBL Documentation Release 10
2 About The MCBL
CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

About The MCBL
1 MCIC Computational Biology Lab (MCBL) 3
2 MCBL membership 5
3 MCBL servers and computing resources 7
4 Download from BaseSpace 11
5 Share runs and projects in BaseSpace 17
6 Using BaseMount 23
7 Download from hudsonalphaorg 31
8 Filter a CASAVA-generated fastq file 33
9 Fastq adapter removal and QC 35
10 DESeq2 with phyloseq 39
11 GBS 43
12 A basic microbiome analysis 47
13 Indices and tables 51
Python Module Index 53
Index 55
i
ii
MCBL Documentation Release 10
About The MCBL 1
MCBL Documentation Release 10
2 About The MCBL
CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

ii
MCBL Documentation Release 10
About The MCBL 1
MCBL Documentation Release 10
2 About The MCBL
CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
About The MCBL 1
MCBL Documentation Release 10
2 About The MCBL
CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
2 About The MCBL
CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 1
MCIC Computational Biology Lab (MCBL)
11 Our goal
Our mission is to build core support and intellectual leadership in the area of bioinformatics to support research atthe OARDC by providing an engaging work environment space infrastructure and training for performing researchinvolving biological data analysis
We aspire for the MCBL to become the place to be for learning and performing bioinformatics research at the OARDCthe place where ideas are discussed and exchanged students and users learn from each other and get help and supportfrom our experienced staff when needed and we as a community move our bioinformatics knowledge forward
We specialize in working with High-throughput Sequencing (HTS) data and providing training in reproducible scienceusing modern tools
We are happy to help you to carry out your own analysis This will include helping with experimental design dis-cussing the most effective way to carry out your data analysis providing the computational infrastructure (softwarescripts and computers) interpreting results and answering questions you might come across along the way We canalso process and analyze HTS data for you using either standardized or custom pipelines
12 MCBL services
Data analysis Contacts
bull Processing analyzing and interpreting HTS databull WGS RADseqGBS RNAseq metagenomics microbiomics bull Quality controlbull Genotypingbull Genome assembly and annotationbull Differential expression analysesbull Population genetic and genomic analysesbull GWAS (e)QTL analyses
Jelmer Poelstra
3
MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
Workshops amp training Contacts
bull UNIX command line and bash scriptingbull Running analyses at the Ohio Supercomputer Center (OSC)bull Running analyses in the cloudbull R - general bioinformatics R markdown dashboardingbull Python - general bioinformaticsbull Reproducible compute environments with containers and condabull Reproducible analysis pipelines with Snakemakebull Version control with git and Githubbull Data management and deposition in public repositories
Jelmer Poelstra
High-throughput Sequencing services Contacts
bull Illumina Miseq bull Tea Meulia (di-rector)
bull Fiorella Cis-neros Carter
See also
Main MCIC page for more details on sequencing services
4 Chapter 1 MCIC Computational Biology Lab (MCBL)
CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 2
MCBL membership
21 MCBL membership benefits
bull Access to the MCBL and MCBL computers 247
bull Free access to all MCBL activities workshops user group meeting etc
bull Access to 1 TB data storage space for the duration of the membership
bull Assistance with experimental design bioinformatic analyses and interpretation of your data
22 How to apply for an MCBL membership
Step 1 Fill out and submit the MCBL application form MCBL Application
Step 2 Submit your membership fee to MCIC
Step 3 Contact Jelmer Poelstra for login credentials
Note Access to MCBL resources will be granted till we receive the payment Once the form is completed andsubmitted a notification e-mail will be sent to the membership applicant and the PI
23 MCBL membership duration
Membership is offered for a 6 month or 1 year period at a time
5
MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
24 MCBL membership termination
MCBL membership will be terminated after the membership period ends or upon a written request from user or PI toterminate the membership
25 Contacts
Person InformationTea Meulia (direc-tor)
Questions regarding membership
Jelmer Poelstra MCBL server access and remote accessJody Whittier MCBL payments
6 Chapter 2 MCBL membership
CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 3
MCBL servers and computing resources
31 Servers
Server Processors Cores MemoryLocalDisk
mcic-ender-svr four 240GHz ten-core Intelreg Xeon processors E7-4870 40 10TB
16TB
mcic-ender-svr2 four 200GHz ten-core Intelreg Xeon processors E7-4850 40 12TB
10TB
mcic-ent-srvr two 267GHz six-core Intelreg Xeon processors X7542 12 250GB 20TB
32 Workstations
Workstation Processors Cores MemoryLocalDisk
mcic-galaxy-srvr two 347GHz six-core Intelreg Xeon processors X5690 12 94GB
26TB
mcic-mac-srvr two 293GHz six-core Intelreg Xeon processors X5670 12 64GB
40TB
7
MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
33 Desktops
Desktop Processors Cores MemoryLocalDisk
mcic-sel019-d1 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d2 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d3 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d4 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d5 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d6 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
mcic-sel019-d7 one 300GHz four-core Intelreg Xeon processors i7-4578U 7 32GB
10TB
34 Software
The following bioinformatics software are availabe through the MCBL Please contact Jelmer Poelstra for details onavailability of the software
341 Commercial Software
Application Version DescriptionCLCBio Work-bench
851 A comprehensive and user-friendly analysis package for analyzing com-paring and visualizing next generation sequencing data
Blast2GO Pro Pro A complete framework for functional annotation and analysis
8 Chapter 3 MCBL servers and computing resources
MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
342 Open Source Software
Application Version Description ModuleName
Bowtie 1102-223 An ultrafast and memory-efficient tool for aligning sequencingreads to long reference sequences
Bowtie-ltversiongt
Cd-hit 461 A very widely used program for clustering and comparing pro-tein or nucleotide sequences
cd-hit-vltversiongt
Cutadapt 142181 Removes adapter sequences from high-throughput sequencingdata
Cutadaptltversiongt
Exonerate 220 A generic tool for pairwise sequence comparison ExonerateltversiongtExpress 151 eXpress is a streaming tool for quantifying the abundances of
a set of target sequences from sampled subsequencesexpress-ltversiongt
Fastqc 151 Aims to provide a simple way to do some quality controlchecks on raw sequence data coming from high throughputsequencing pipelines
Fastqc-ltversiongt
GenomeAnalysisTK32-2 GATK tools for error modeling data compression and variantcalling
GenomeAnalysisTK-ltversiongt
Maker 2318 MAKER is a portable and easily configurable genome annota-tion pipeline
Makerltversiongt
Mothur 133135 Tool for analyzing 16S rRNA gene sequences Mothur-ltversiongt
Mummer 323 A system for rapidly aligning entire genomes whether in com-plete or draft form
Mummerltversiongt
Pandaseq 28 A program to align Illumina reads optionally with PCRprimers embedded in the sequence and reconstruct an over-lapping sequence
Pandaseqltversiongt
Qiime 18 A program for comparison and analysis of microbial commu-nities primarily based on high-throughput amplicon sequenc-ing data
Qiime-ltversiongt
Rsem 1216 A software package for estimating gene and isoform expres-sion levels from RNA-Seq data
rsem-ltversiongt
Samtools 0119 Provides various utilities for manipulating alignments in theSAM format including sorting merging indexing and generat-ing alignments in a per-position format
Samtools-ltversiongt
SNAP 0119 A new sequence aligner that is 3-20x faster and just as accurateas existing tools like BWA-mem Bowtie2 and Novoalign
SNAPltversiongt
Trim-fastq 122 A Fastq quality trimmer Trim-fastq-ltversiongt
Trinity r20140717 a novel method for the efficient and robust de novo reconstruc-tion of transcriptomes from RNA-seq data
Trinity
34 Software 9
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
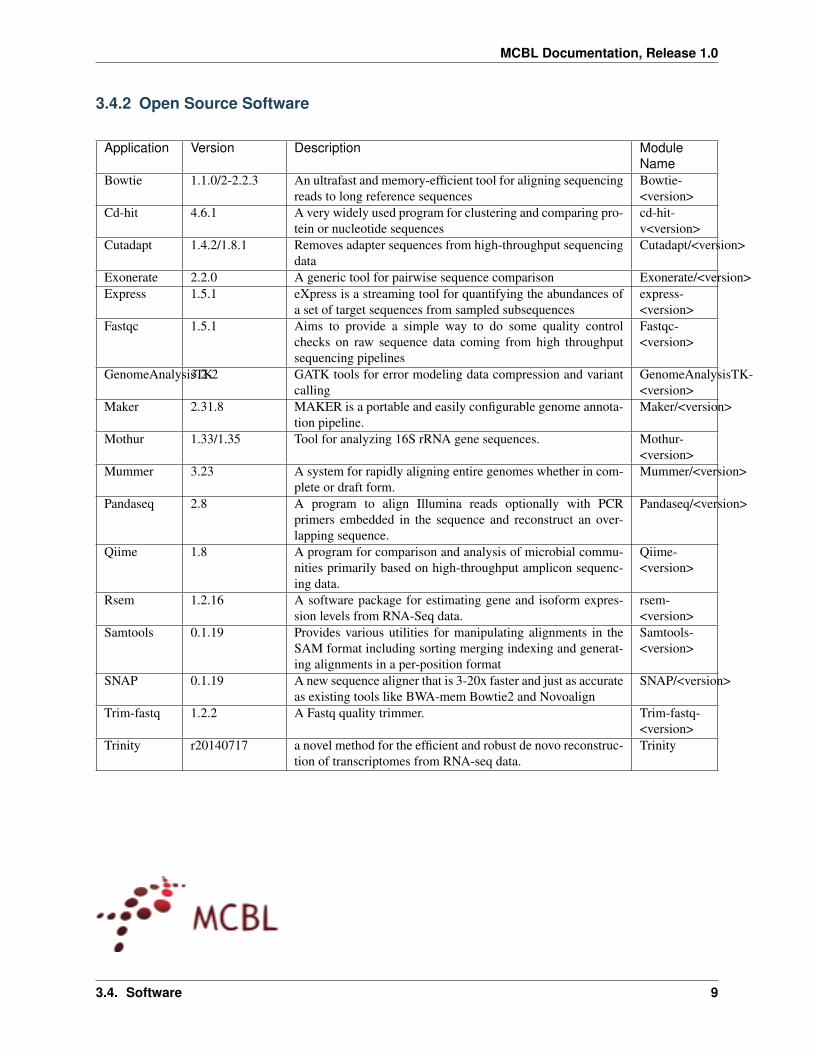
MCBL Documentation Release 10
10 Chapter 3 MCBL servers and computing resources
CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 4
Download from BaseSpace
Note
Purpose This short tutorial will show you how to download MiSeq sequencing data from Illumina BaseS-pace
Author Wirat Pipatpongpinyo
Date July 6 2020
41 Step-by-step guide to download your sequence data from BaseS-pace
1 You will receive two invitation emails frombasespace-noreplyilluminacom for the trans-fer of ownership to you one for the sequencing runand another for the project To be able to fullyaccess the data you must accept both invitations
11
MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
2 Once you click the link (Click here to acceptthis transfer of ownership) in one of the invi-tation emails you will be taken to the Illumina BaseSpacewebsite where you can log in to your account
3 After you have logged in BaseSpace will bring you tothe DASHBOARD tab Click Accept in both boxes to takethe ownership of both the run and project
4 Once transfer is acceptedclick on the name of the run
12 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
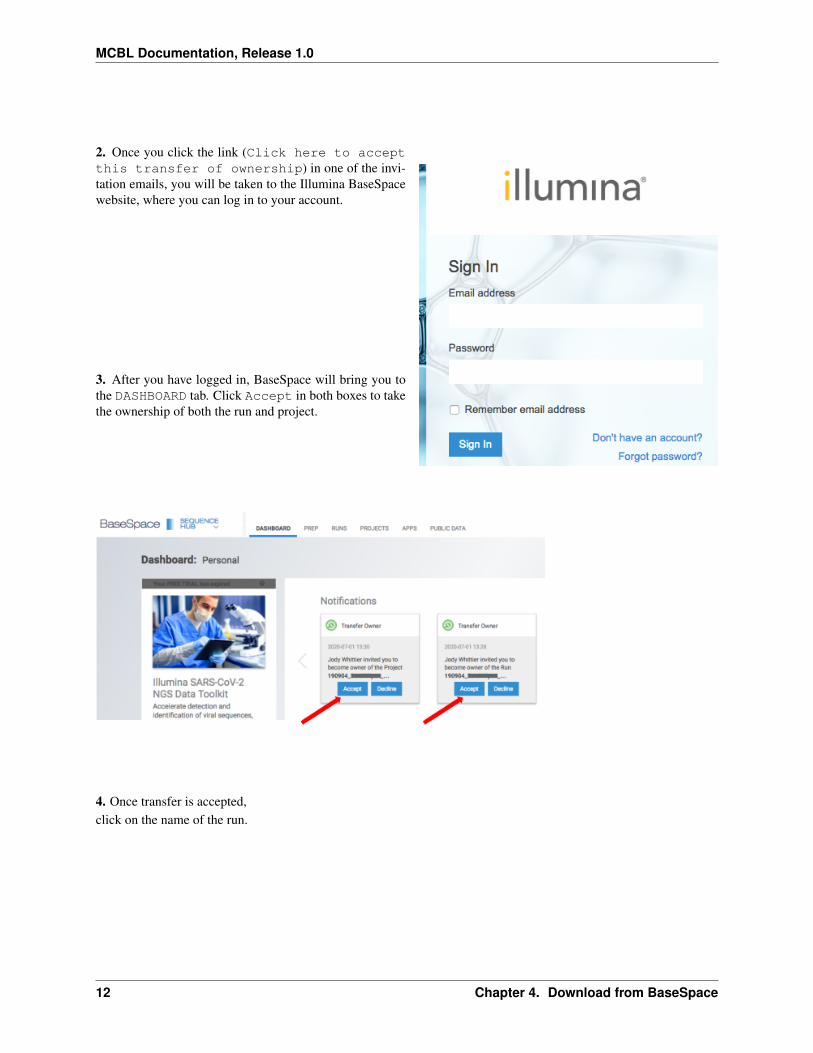
MCBL Documentation Release 10
5 Under the SUMMARY tab click Download
6 The Download Run screen will pop upIf thisis the first time you download from BaseSpace you willneed to install the downloader software click Installthe BaseSpace Sequence Hub Downloader
41 Step-by-step guide to download your sequence data from BaseSpace 13
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
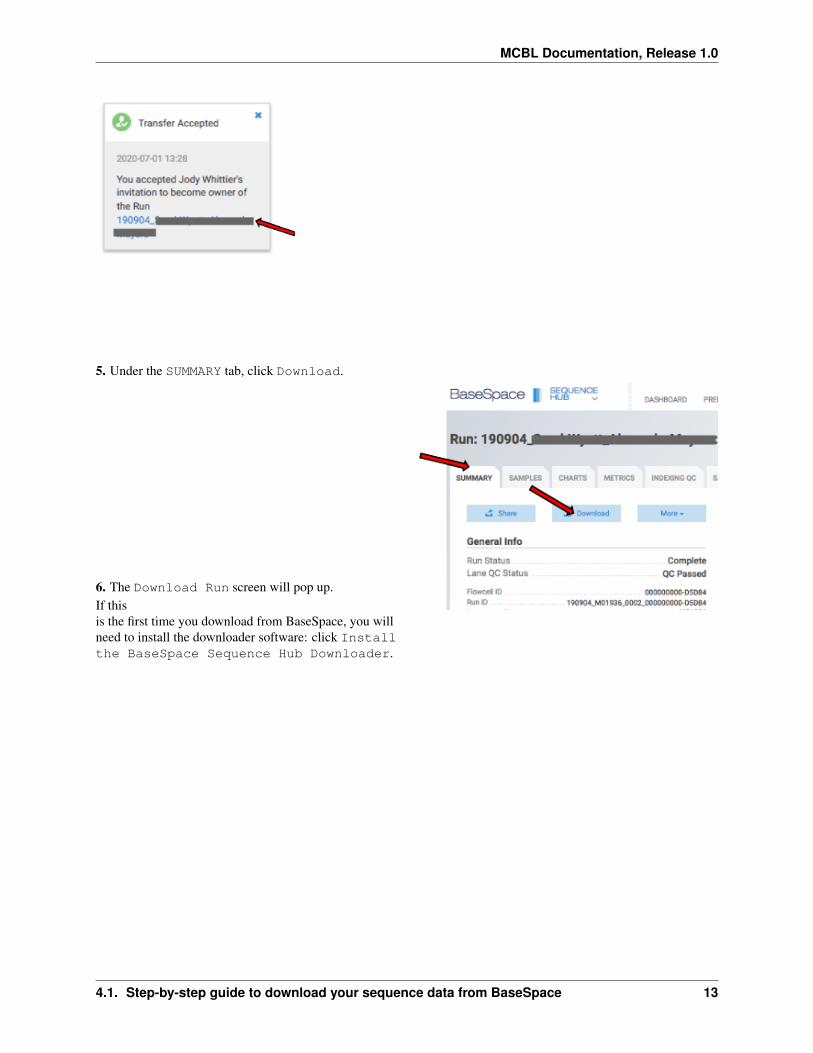
MCBL Documentation Release 10
7 After the BaseSpace downloader has beeninstalled select FASTQ as the file typeand click Download
14 Chapter 4 Download from BaseSpace
MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
8 The Confirm Download screen will pop up whereyou can select a directory to download the filesinto In this case the files will be stored atCBaseSpace Click START DOWNLOAD
41 Step-by-step guide to download your sequence data from BaseSpace 15
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
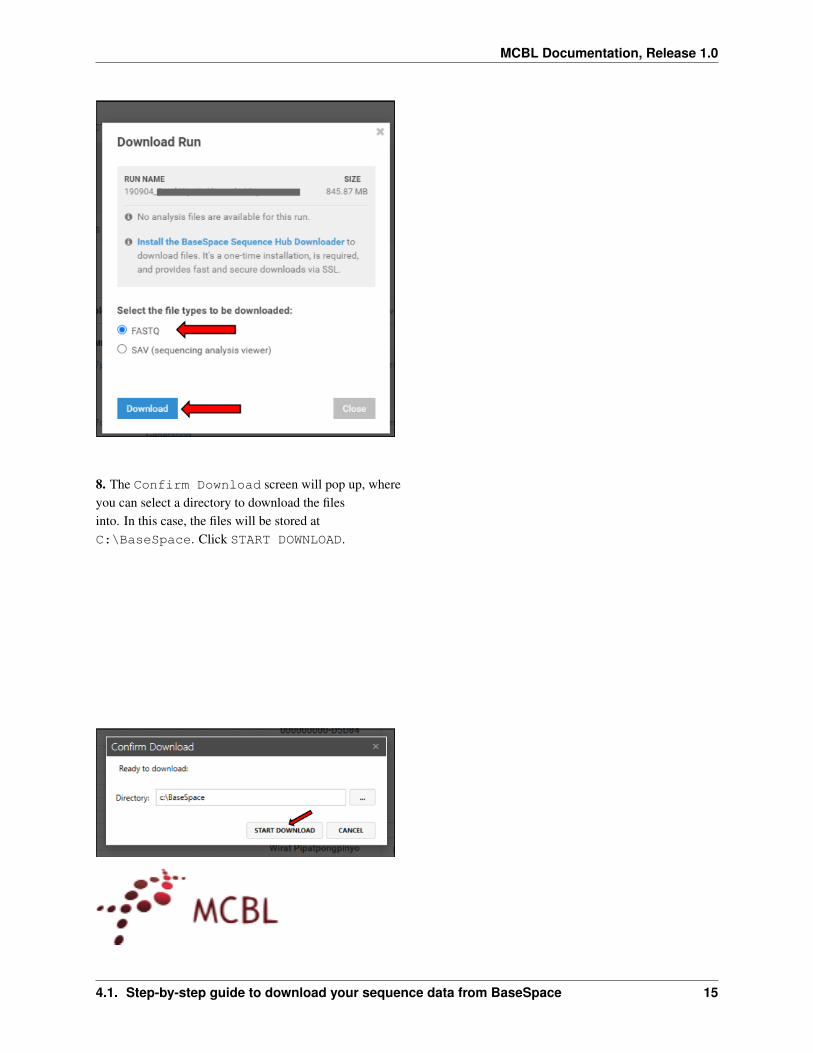
MCBL Documentation Release 10
16 Chapter 4 Download from BaseSpace
CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 5
Share runs and projects in BaseSpace
Note
Purpose These two short tutorials will show you how to share sequencing runs and projects respectivelyon Illumina BaseSpace
Author Wirat Pipatpongpinyo
Date July 6 2020
bull Sharing runs
bull Sharing projects
51 Sharing runs
17
MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 Go to the Illumina BaseSpace website and sign in
2 Click on the RUNS tab
3 Select the name of the runyou want to share
18 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
4 Go to the SUMMARYtab
5 Click the SHAREbutton
6 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator
51 Sharing runs 19
MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
8 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects
1 Got to the Illumina BaseSpace website and sign in
2 Click on the PROJECTS tab
3 Select the Project that youwould like to share
20 Chapter 5 Share runs and projects in BaseSpace
MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
4 Click the Share Project button
5 Enter the email address (linked to aBaseSpace account) of yourcollaborator
6 Click Add Collaborator(Repeat Steps 6 and 7 if you want toshare with multiple collaborators)
7 Click Save Settings Yourcollaborator will receive an email frombasespace-noreplyilluminacom
52 Sharing projects 21
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
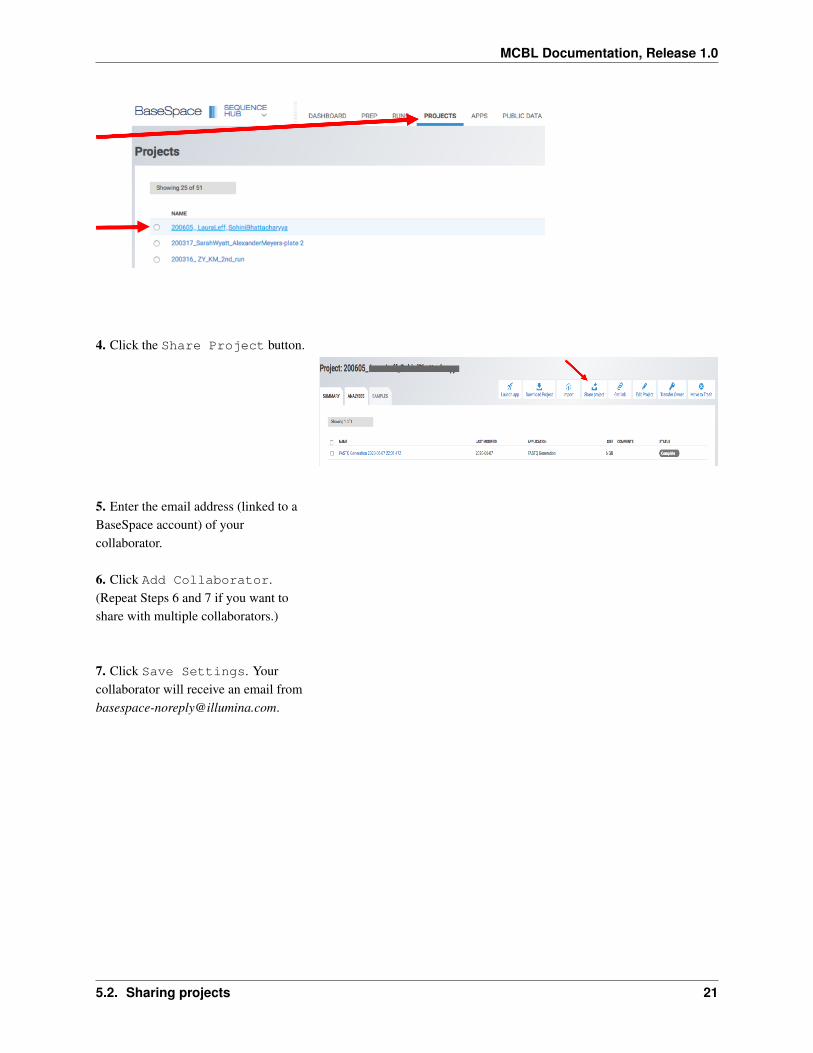
MCBL Documentation Release 10
22 Chapter 5 Share runs and projects in BaseSpace
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-
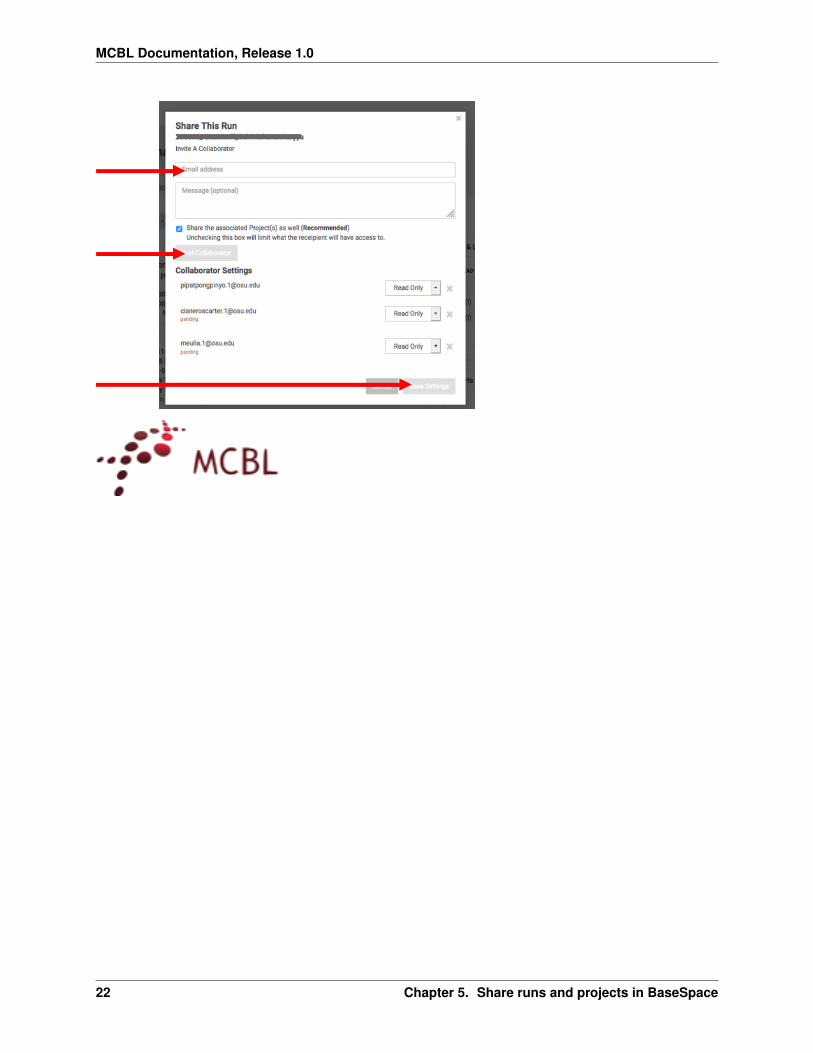
CHAPTER 6
Using BaseMount
bull Introduction to BaseMount
bull Getting Started with BaseMount
ndash Example
bull Basic analysis of fastq files
bull Basic analysis on Alignment files (BAM)
bull Metadata
bull Limitations of BaseMount
61 Introduction to BaseMount
What is BaseMount
BaseMount is Illumina software that enables access to your BaseSpace storage as a Linux file system on the commandline
Advantages of BaseMount
bull Have access to your Projects Runs and App results as your local files
bull Can run local apps on basespace data without downloading data to your local computer
bull Can save your local storage space
ldquoAlthough BaseMount does facilitate file download we would recommend that since BaseMountallows convenient fast cached access to your BaseSpace metadata and files you may find that many
23
MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
operations can be carried out without the need to download locally During our testing we have usedBaseMount to grep through fastq files extract blocks of reads from bam files and even use IGV on thebam files directly all without downloading files locally This can be more convenient than includinga download step and saves on the overheads of local storagerdquo -Illumina
Official page
62 Getting Started with BaseMount
Quick Install
1 sudo bash -c $(curl -L httpsbasemountbasespaceilluminacominstall)
Manual install
Supported Operating Systems Ubuntu CentOS
Ubuntu 14 amp 15
1 wget httpsbintraycomartifactdownloadbasespaceBaseSpaceFS-DEBbsfs_11631-1_rarr˓amd64deb
2 wget httpsbintraycomartifactdownloadbasespaceBaseMount-DEBbasemount_012rarr˓463-20150714_amd64deb
3 sudo dpkg -i --force-confmiss bsfs_11631-1_amd64deb4 sudo dpkg -i basemount_012463-20150714_amd64deb
Mounting Your BaseSpace Account
1 basemount --config config_file_prefix mount-point folder2 basemount --config user1 ~BaseSpace_Mount
621 Example
1 mkdir exportNFSSarangaBaseSpace2 mkdir exportNFSMariaBaseSpace3 basemount --config Maria exportNFSMariaBaseSpace
Thenopenyourin-ter-netbrowser
AfteryouclickldquoAc-ceptrdquo
24 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
Toac-cessthefoldertype
1 cdrarr˓rarr˓export
rarr˓NFSrarr˓Mariararr˓BaseSpacerarr˓
2 ls
Projectsrarr˓
rarr˓Runs
Toseethe
contents in the Projects
1 ls -lsh exportNFSSarangaBaseSpaceProjectsHiSeq 2500 - v4 reagentsrarr˓TruSeq PCR Free (4 replicates of NA12877)SamplesNA12877_Files
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain10Filestotal 85M85M -r--r--r-- 1 root root 85M Oct 5 1409 Brain10_S22_L001_R1_001fastqgz
exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNA Expression (NFkBrarr˓amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain11Filestotal 62M62M -r--r--r-- 1 root root 62M Oct 5 1409 Brain11_S23_L001_R1_001fastqgz
63 Basic analysis of fastq files
You can get basic information about your fastq files without having to download them For instance
bull View sequences inside Fastq files
bull Get the number of reads for each fastq file
bull Get basic statistics and read length distribution
Example View your data
63 Basic analysis of fastq files 25
MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | head -n 4
M0343848000000000-AGGNU11101117921006 1N013NTCAATCCCCAGCAGTGGAATAAGGCCTGTTGTTCCTTGCAGTGGATCCTG+88ABFFGCFEEGltFFltFDFFEGGFGGFCFGFFGGEGGGGGGFGGFGGGGG
Example Count the number of sequences using native Linux commands
1 zcat exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)SamplesBrain1rarr˓FilesBrain1_S13_L001_R1_001fastqgz | grep -c M03438
838876
FILE SIZE 85M
TIME TAKEN 1327s
Example Count the number of sequneces using using fastqutils
1 fastqutils names exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz | wc -l
838876
FILE SIZE 85M
TIME TAKEN 2300s
Example Get the Length distribution and other statistics
1 fastqutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓SamplesBrain1FilesBrain1_S13_L001_R1_001fastqgz
Space basespacePairing FragmentedQuality scale IlluminaNumber of reads 838876
Length distributionMean 510StdDev 00Min 5125 percentile 51Median 5175 percentile 51Max 51
(continues on next page)
26 Chapter 6 Using BaseMount
MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(continued from previous page)
Total 838876
Quality distributionpos mean stdev min 25pct 50pct 75pct max count1 335948650337 215033983948 2 34 34 34 34 8388762 336285434319 201137931152 12 34 34 34 34 8388763 336910246568 181306178651 11 34 34 34 34 8388764 336861812711 184720681841 11 34 34 34 34 8388765 336998292954 173881480815 12 34 34 34 34 8388766 372788564698 249830369426 2 38 38 38 38 8388767 373219820331 239576158974 2 38 38 38 38 8388768 371795640834 272499009837 2 38 38 38 38 8388769 37158373824 276769991544 10 38 38 38 38 83887610 370991517221 293041515545 10 38 38 38 38 83887611 37080357526 300286915879 10 38 38 38 38 83887612 370752506926 293530617165 10 38 38 38 38 83887613 371372705859 285104291311 10 38 38 38 38 83887614 370559641711 300820427859 10 38 38 38 38 83887615 370753877808 299577677236 10 38 38 38 38 83887616 370950426523 292821324956 10 38 38 38 38 83887617 37204973083 264727500649 10 38 38 38 38 83887618 371618308308 275627448135 10 38 38 38 38 83887619 370932426247 292692822638 10 38 38 38 38 83887620 371103548081 289001817524 10 38 38 38 38 83887621 37117659821 290476888945 10 38 38 38 38 83887622 371280034236 280218109494 10 38 38 38 38 83887623 369527546383 318334971719 9 37 38 38 38 83887624 371825096915 270092644993 10 38 38 38 38 83887625 372478948021 258021368225 10 38 38 38 38 83887626 371056830807 304029966339 10 38 38 38 38 83887627 370417129588 321755695865 9 38 38 38 38 83887628 369245287742 346922882082 9 38 38 38 38 83887629 370233538687 322132395112 9 38 38 38 38 83887630 369768440151 336846192959 9 38 38 38 38 83887631 369019378311 351733823479 10 38 38 38 38 83887632 370442532627 315497307231 10 38 38 38 38 83887633 370763462061 305842992907 10 38 38 38 38 83887634 369112836701 351048896466 10 38 38 38 38 83887635 36871104907 350954115324 10 37 38 38 38 83887636 369835911386 329022069717 9 38 38 38 38 83887637 369526103977 340894509478 9 38 38 38 38 83887638 369730198504 335198104312 10 38 38 38 38 83887639 36925962836 342925499742 9 38 38 38 38 83887640 369641508399 33749237703 10 38 38 38 38 83887641 369701290775 337108719426 9 38 38 38 38 83887642 36925310773 346541098262 9 38 38 38 38 83887643 364323821399 437591782904 9 37 38 38 38 83887644 366849510536 384804558398 10 37 38 38 38 83887645 368000574578 371757395575 10 37 38 38 38 83887646 36743696327 389392229051 9 37 38 38 38 83887647 366721195981 406597933483 10 37 38 38 38 83887648 367998965282 373484635736 9 37 38 38 38 83887649 369068074423 346537970313 10 38 38 38 38 83887650 36828634983 367970695764 10 37 38 38 38 83887651 367344208202 375309659394 9 37 38 38 38 838876
Average quality string(continues on next page)
63 Basic analysis of fastq files 27
MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(continued from previous page)
BBBBBFFFFFFFFFFFFFFFFFEFFFFEFEEFFEEEEEEEEEEEEEEEEEE
FILE SIZE 85M
TIME TAKEN 110m
64 Basic analysis on Alignment files (BAM)
Example Check bam headers
1 samtools view -H exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeqrarr˓Targeted RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)rarr˓AppSessionsTopHat Alignment No cSNP or RNA Editing found (36 Samples)rarr˓AppResults26970091Brain1FilesalignmentsBrain1alignmentsbam
HD VN10 SOcoordinateRG ID19 SMBrain1SQ SNchrM LN16571SQ SNchr1 LN249250621SQ SNchr2 LN243199373SQ SNchr3 LN198022430SQ SNchr4 LN191154276SQ SNchr5 LN180915260SQ SNchr6 LN171115067SQ SNchr7 LN159138663SQ SNchr8 LN146364022SQ SNchr9 LN141213431SQ SNchr10 LN135534747SQ SNchr11 LN135006516SQ SNchr12 LN133851895SQ SNchr13 LN115169878SQ SNchr14 LN107349540SQ SNchr15 LN102531392SQ SNchr16 LN90354753SQ SNchr17 LN81195210SQ SNchr18 LN78077248SQ SNchr19 LN59128983SQ SNchr20 LN63025520SQ SNchr21 LN48129895SQ SNchr22 LN51304566SQ SNchrX LN155270560SQ SNchrY LN59373566PG IDTopHat VN207 CLilluminadevelopmentIsisRNA24195IsisRNA_Toolsrarr˓bintophat --bowtie1 --read-realign-edit-dist 1 --segment-length 24 -o datainputrarr˓AlignmentsamplesBrain1replicatesBrain1tophat_main -p 1 --GTF illuminararr˓developmentGenomesHomo_sapiensUCSChg19AnnotationGenesgenesgtf --rg-id 19 --rarr˓rg-sample Brain1 --library-type fr-firststrand --no-coverage-search --keep-fasta-rarr˓order illuminadevelopmentGenomesHomo_sapiensUCSChg19SequenceBowtieIndexrarr˓genome datainputAlignmentsamplesBrain1replicatesBrain1filteredBrain1_S20_rarr˓L001_R1_001fastqgz
FILE SIZE 17M
TIME TAKEN 0006s
28 Chapter 6 Using BaseMount
MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
Example Check basic statistics on a Bam file
1 bamutils stats exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targetedrarr˓RNA Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)AppSessionsrarr˓TopHat Alignment No cSNP or RNA Editing found (36 Samples)AppResultsrarr˓26970091Brain1FilesalignmentsBrain1alignmentsbam
Reads 766531Mapped 766531Unmapped 0
Flag distribution[0x010] Reverse complimented 383242 5000[0x100] Secondary alignment 47 001
Reference counts countchr1 32252chr10 6829chr11 45127chr12 74524chr13 24336chr14 81664chr15 254chr16 5704chr17 46662chr18 9922chr19 25644chr2 32527chr20 10708chr21 22chr22 7805chr3 83585chr4 42487chr5 47865chr6 106512chr7 18024chr8 6921chr9 25334chrM 0chrX 31823chrY 0
65 Metadata
In each directory BaseMount provides a number of hidden files with extra BaseSpace metadata These are hiddenfiles and their names start with a ldquordquo
1 cd exportNFSSarangaBaseSpaceProjectsMiSeq v3 TruSeq Targeted RNArarr˓Expression (NFkB amp Cell Cycle Human Brain-Liver-UHRR)Samples
2 ls -l List only the hidden files
-r-------- 1 swijeratne swijeratne 149 Nov 16 1129 curl
(continues on next page)
65 Metadata 29
MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(continued from previous page)
-r--r--r-- 1 swijeratne swijeratne 40674 Nov 16 1129 json
Display content of the json file
1 cat json
Query through a json file qith ldquojqrdquo
1 cat json | jq ResponseItems[] | select(NumReadsPF ) | Name Name PF rarr˓NumReadsPF
2 cat json | jq ResponseItems[] | select(NumReadsPF ) | ( Name)t(NumReadsPF)rarr˓
3 cat json | jq ResponseItems[] | select(NumReadsPF gt 747912) | ( Name)t(rarr˓NumReadsPF)
66 Limitations of BaseMount
According to Illumina
Every new directory access made by BaseMount relies on FUSE the BaseSpace API and the userrsquos credentials Thismechanism means that as currently available BaseMount does not support the following types of access
bull Cluster access where many compute nodes can access the files FUSE mounted file systems are per-host andcannot be accessed from many hosts without additional infrastructure
bull BaseMount also doesnrsquot refresh files or directories In order to reflect changes done via the Web GUI in yourcommand line tree you currently need to unmount (basemount ndashunmount ) and restart BaseMount
bull The Runs Files directory is not mounted automatically for you as there can be 100k + files available in thatmount which can take a couple minutes to load for really large runs You can still mount this directory manuallyif needed
bull In general lots of concurrent requests can cause stability issues on a resource constrained system Keep in mindthis is an early release and stability will increase
30 Chapter 6 Using BaseMount
CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 7
Download from hudsonalphaorg
Note
Required OS OS x or Linux Windows users please contact Maria Elena Hernandez-Gonzalez
Software wget curl
Terminal emulator
bull Terminal (OS x)
bull Genome Terminal or Other Emulator (Linux)
Author This document was created by Saranga Wijeratne
71 Download
1 Create a Samplestxt file with your sample links (the links are provided in the Excelsheet) as follows
Content of the Samplestxthttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzhttpmysampledownloadorgdld4Meuliamyprojectnumberdata_150522C6V7FANXX_rarr˓s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Use the Terminal and navigate to the location where Samplestxt is saved
1 If your Samplestxt is saved under ~Downloads2 $ cd ~Downloads
31
MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
3 On OS x issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do curl --progress-bar -O $f done
4 On Linux issue the following command to download your files
1 $ for f in $(cat Samplestxt ) do wget -v $f done
72 Check checksum
To detect errors which may have been introduced during the downloading you have to run checksum on your down-loaded files
1 Navigate to the location where you have downloaded your files
1 If your files are saved under ~Downloads2 $ cd ~Downloads
2 Then if yoursquore on OS x Terminal type in the following command
1 $ md5
MD5 (C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgz) =rarr˓d41d8cd428f00b204e9800998ecf8427eMD5 (C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgz) =rarr˓d49d8cdf00j204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz) =rarr˓d47d8cd98dfds0b204e9800998ecf8427eMD5 (C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgz) =rarr˓d42d8cd98f00bdfse9800998ecf8427e
If yoursquore on Linux terminal type in the following commmand
1 $ md5sum
d41d8cd428f00b204e9800998ecf8427e C6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_rarr˓D501_SL104549fastqgzd49d8cdf00j204e9800998ecf8427ed56 C6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_rarr˓D506_SL104602fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_rarr˓D501_SL104565fastqgzd47d8cd98dfds0b204e9800998ecf8427e C6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_rarr˓D508_SL104628fastqgz
Tip Match these checksum values with the values provided in the Excelsheet For any samples with mismatchingchecksum you have to re-download the samples
32 Chapter 7 Download from hudsonalphaorg
CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 8
Filter a CASAVA-generated fastq file
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Illumina CASAVA-18 FASTQ Filter
Purpose This document provides instructions about how to remove Passing Filter (PF) failed reads froma Fastq file
More Read more about PF here and here
Author This document was created by Saranga Wijeratne
81 Software installation
Note If you are runing this on MCBL mcic-ender-svr please skip the installation Following command will load thesoftware module to your environment
1 $ module load fasq_filter01
On your own server
Warning If you donrsquot have administrator privileges on the machine you wouldnrsquot be able run sudo (lastcommand in the following code block) commands
33
MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 $ wget httpcancancshledulabmembersgordonfastq_illumina_filterfastq_illumina_rarr˓filter-01targz
2 $ tar -xzf fastq_illumina_filter-01targz3 $ cd fastq_illumina_filter-014 $ make5 $ sudo cp fastq_illumina_filter usrlocalbin
Tip Put your executables in ~bin or full-path to executables in $PATH in the absence of sudo privilages
82 Filter a fastq file
Input File C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz
Output File C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
1 $ zcat C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz | fastq_illumina_filter -rarr˓vvN | gzip gt C8EC8ANXX_s2_1_illumina12index_1_SL143785filteredfastqgz
83 Filter multiple fastq files
Input File Fastq_filenamestxt
Output Files Individual Fastq files
1 Create a Fastq_filenamestxt file with your Fastq filenames in seperate lines as follows
Content of SamplestxtC6V7FANXX_s8_0_TruseqHTDual_D712-TruseqHTDual_D508_SL104628fastqgzC6V7FANXX_s3_0_TruseqHTDual_D703-TruseqHTDual_D501_SL104549fastqgzC6V7FANXX_s5_0_TruseqHTDual_D709-TruseqHTDual_D506_SL104602fastqgzC6V7FANXX_s8_0_TruseqHTDual_D705-TruseqHTDual_D501_SL104565fastqgz
2 Save the above file in the same folder with your Fastq files
3 Use the Terminal and navigate to the location where Fastq_filenamestxt is saved
1 If your Fastq_filenamestxt is saved under ~Downloads2 $ cd ~Downloads
4 Type in the following command to filter Fastqs in the Fastq_filenamestxt
1 $ for f in $(cat Fastq_filenamestxt) do zcat $f | fastq_illumina_filter -vvN |rarr˓gzip gt $ffastqgzfilteredfastqgzdone
34 Chapter 8 Filter a CASAVA-generated fastq file
CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 9
Fastq adapter removal and QC
Note
Required OS OS x or Linux Windows users please contact Saranga Wijeratne
Software Trimmomatic
Purpose This document provides instructions about how to remove adapters and filter low quality basesfrom a Fastq file
More Read more about Read trimming adapter removing here
Author This document is created by Saranga Wijeratne
91 Load the software
Note If you are runing this on MCBL mcic-ender-svr following command will load the software module to yourenvironment
1 $ module load Trimmomatic322
then you can get the help how to run Trimmomatic
1 $ java -jar $TRIMHOMEtrimmomatic-033jar
92 Files needed
Input Files In put files should be in fastq formatcompressed fastq( fq fastq fqgzfastqgz) Read Introduction eg C8EC8ANXX_s2_1_illumina12index_1_SL143785fastq
35
MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
C8EC8ANXX_s2_1_illumina12index_1_SL143785fastqgz s_1_1_sequencetxtgzlane1_forwardfqgz
Adapter File Currently following Adapter sequence files are hosted in MCBL server
bull TruSeq2-PEfa
bull TruSeq2-SEfa
bull TruSeq3-PEfa
bull TruSeq3-SEfa
bull NexteraPE-PEfa
Warning If you want to make your own adapter sequence file please read the The Adapter Fasta section andMaking cutome clipping files here before you make your Adapter sequence file
93 Code examples
Single-end fastq files
1 $java -jar $TRIMHOMEtrimmomatic-033jar SE -threads 12 s_1_1_sequencetxtgz lane1_rarr˓forwardfqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-SEfa23010 LEADING3rarr˓TRAILING3 SLIDINGWINDOW415 MINLEN36
Paired End Fastq Files
1 $java -jar $TRIMHOMEtrimmomatic-033jar PE -threads 12 C8EC8ANXX_s2_1_rarr˓illumina12index_1_SL143785fastqgz C8EC8ANXX_s2_2_illumina12index_1_SL143785fastqrarr˓gz C8EC8ANXX_s2_1_Trimmed_1Pfastqgz C8EC8ANXX_s2_1_Trimmed_1Ufastqgz C8EC8ANXX_rarr˓s2_2_Trimmed_1Pfastqgz C8EC8ANXX_s2_2_Trimmed_1Ufastqgz ILLUMINACLIP$TRIMHOMErarr˓adaptersTruSeq3-PEfa23010 LEADING3 TRAILING3 SLIDINGWINDOW415 MINLEN36
Multiple fastq files
Tip Assumption has been made that your data in ldquoRaw_Datardquo folder
Input Files C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzC6EF7ANXX_s3_1_illumina12index_19_SL100997fastqgz C6EF7ANXX_s3_1_illumina12index_22_SL100998fastqgzC6EF7ANXX_s3_1_illumina12index_25_SL100999fastqgz C6EF7ANXX_s3_1_illumina12index_27_SL101000fastqgzC6EF7ANXX_s3_1_illumina12index_3_SL100994fastqgz C6EF7ANXX_s3_1_illumina12index_5_SL100995fastqgzC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz C6EF7ANXX_s3_2_illumina12index_19_SL100997fastqgzC6EF7ANXX_s3_2_illumina12index_22_SL100998fastqgz C6EF7ANXX_s3_2_illumina12index_25_SL100999fastqgzC6EF7ANXX_s3_2_illumina12index_27_SL101000fastqgz C6EF7ANXX_s3_2_illumina12index_3_SL100994fastqgzC6EF7ANXX_s3_2_illumina12index_5_SL100995fastqgz
These are paired-end fastq files eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgz andC6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz belongs to single sample
Adapter File $TRIMHOMEadaptersTruSeq3-PEfa (Make sure you change this accordingly)
Output Files Each paired-end read (eg C6EF7ANXX_s3_1_illumina12index_10_SL100996fastqgzand C6EF7ANXX_s3_2_illumina12index_10_SL100996fastqgz) will give 4 outputs
36 Chapter 9 Fastq adapter removal and QC
MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Pfastqgz - for paired for-wad reads
bull Q_trimmed_6EF7ANXX_s3_1_illumina12index_10_SL100996_1Ufastqgz - for unpairedforward reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Pfastqgz - for paired re-verse reads
bull Q_trimmed_6EF7ANXX_s3_2_illumina12index_10_SL100996_1Ufastqgz - for unpaired re-verse reads
1 $cd Raw_Data make sure you change the folder name accordingly2 $mkdir Trimmed_Data Output will be saved here3 $files_1=(_s3_1_fastqgz)files_2=(_s3_2_fastqgz)sorted_files_1=($(printf
rarr˓sn $files_1[] | sort -u))sorted_files_2=($(printf sn $files_2[] |rarr˓sort -u))for ((i=0 ilt$sorted_files_1[] i+=1))do java -jar $TRIMHOMErarr˓trimmomatic-033jar PE -threads 12 -trimlog Trimmed_Datalog-j3stat -phred33 $rarr˓sorted_files_1[i] $sorted_files_2[i] Trimmed_DataQ_trimmed_$sorted_files_1[i]rarr˓_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_files_1[i]-Ufastqgz Trimmed_rarr˓DataQ_trimmed_$sorted_files_2[i]_1Pfastqgz Trimmed_DataQ_trimmed_$sorted_rarr˓files_1[i]-Ufastqgz ILLUMINACLIP$TRIMHOMEadaptersTruSeq3-PE23010rarr˓LEADING3 TRAILING3 SLIDINGWINDOW420 MINLEN40 ampgtTrimmed_Datastattxt done
93 Code examples 37
MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
38 Chapter 9 Fastq adapter removal and QC
CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 10
DESeq2 with phyloseq
Note
Required OS OS x or Linux
Software R phyloseq R library
Purpose This document provides instructions about how to find differentially abundant OTUs for micro-biome data
More Read more about phyloseq DEseq2 here and here
Author This document was created by Saranga Wijeratne
101 Software installation
Note If you are runing this on MCBL mcic-sel019-d please skip the installation The following command inR-Studio will load the software module to your environment
1 gt library(phyloseq) packageVersion(phyloseq)2 [1] lsquo1162rsquo version
Install the phyloseq package as follows
1 gt source(httpbioconductororgbiocLiteR)2 gt biocLite(phyloseq)
39
MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
102 Import data with phyloseq
For this step you need Biom and mapping files generated by the Qiime pipeline
Input biom file otu_table_mc10_w_taxbiom
Qiime mapping file mappingtxt
Output file DESeq2_Out
Copy all the input files to your ldquoWorking Directoryrdquo before you execute the following commands
1 Filenames2 biom_file lt- otu_table_mc10_w_taxbiom3 mapping_file lt- mappingtxt4
5 Import the biom file with phyloseq6 biom_otu_tax lt- import_biom(biom_file)7
8 Import the mapping file with phyloseq9 mapping_file lt- import_qiime_sample_data(mapping_file)
10
11 Merge biom and mapping files with phyloseq12 merged_mapping_biom lt- merge_phyloseq(biom_otu_taxmapping_file)13
14 Set column names in the taxa table15 colnames(tax_table(merged_mapping_biom)) lt- c(kingdom Phylum Class Order
rarr˓Family Genus species)
Now your merged mapping and Biom output should look as follows
1 merged_mapping_biom2
3 phyloseq-class experiment-level object4 otu_table() OTU Table [ 315 taxa and 9 samples ]5 sample_data() Sample Data [ 9 samples by 8 sample variables ]6 tax_table() Taxonomy Table [ 315 taxa by 7 taxonomic ranks ]
The mapping file should look like this
1 head(mapping_file)2
3 Sample Data [40 samples by 7 sample variables]4 XSampleID BarcodeSequence LinkerPrimerSequence InputFileName IncubationDate
rarr˓Treatment Description5 S1 S1 NA NA S1fasta 0
rarr˓ CO CO16 S2 S2 NA NA S2fasta 0
rarr˓ CO CO27 S3 S3 NA NA S3fasta 0
rarr˓ CO CO38 S4 S4 NA NA S4fasta 15
rarr˓ CO CO49 S5 S5 NA NA S5fasta 15
rarr˓ CO CO5
To remove taxonomy level tags assigned to each level (k__ p__ etc) issue the following commands
40 Chapter 10 DESeq2 with phyloseq
MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 tax_table(merged_mapping_biom) lt- gsub(k__([[alpha]]) 1 tax_table( merged_rarr˓mapping_biom))
2 tax_table(merged_mapping_biom) lt- gsub(p__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
3 tax_table(merged_mapping_biom) lt- gsub(c__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
4 tax_table(merged_mapping_biom) lt- gsub(o__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
5 tax_table(merged_mapping_biom) lt- gsub(f__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
6 tax_table(merged_mapping_biom) lt- gsub(g__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
7 tax_table(merged_mapping_biom) lt- gsub(s__([[alpha]]) 1 tax_table(merged_rarr˓mapping_biom))
8 tax_table(merged_mapping_biom) lt- gsub(p__([)1 tax_table(merged_mapping_rarr˓biom))
9 tax_table(merged_mapping_biom) lt- gsub(c__([)1 tax_table(merged_mapping_rarr˓biom))
10 tax_table(merged_mapping_biom) lt- gsub(o__([)1 tax_table(merged_mapping_rarr˓biom))
11 tax_table(merged_mapping_biom) lt- gsub(f__([)1 tax_table(merged_mapping_rarr˓biom))
12 tax_table(merged_mapping_biom) lt- gsub(g__([)1 tax_table(merged_mapping_rarr˓biom))
13 tax_table(merged_mapping_biom) lt- gsub(s__([)1 tax_table(merged_mapping_rarr˓biom))
103 Testing for differential abundance among OTUs
Input file merged_mapping_biom
Output files DESeq2_Outtxt
1 Load the DESeq2 package into your R environment
1 library(DESeq2)2 packageVersion(DESeq2)3 [1] lsquo1124rsquo
2 Assign DESeq2 output name and padj-cutoff
1 filename_out lt- DESeq2_Outtxt2 alpha lt- 001
3 Convert to DESeqDataSet format The phyloseq_to_deseq2() function converts the phyloseq-format mi-crobiome data (ie merged_mapping_biom) to a DESeqDataSet with dispersion estimated using the experi-mental design formula (ie ~ Treatment)
1 diagdds lt- phyloseq_to_deseq2(merged_mapping_biom ~ Treatment)
4 Run DESeq
1 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)2
3 estimating size factors
(continues on next page)
103 Testing for differential abundance among OTUs 41
MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(continued from previous page)
4 estimating dispersions5 gene-wise dispersion estimates6 mean-dispersion relationship7 final dispersion estimates8 fitting model and testing
Warning If you are getting the following error
Error in estimateSizeFactorsForMatrix(counts(object) locfunc geoMeans =rarr˓geoMeans) every gene contains at least one zero cannot compute lograrr˓geometric meansCalls estimateSizeFactors estimateSizeFactors -gt local -gtrarr˓estimateSizeFactorsForMatrix
Then please execute the following code (see here for more info)
1 gm_mean lt- function(x narm=TRUE) exp(sum(log(x[x gt 0]) narm=narm) rarr˓length(x))
2 geoMeans lt- apply(counts(diagdds) 1 gm_mean)3 diagdds lt- estimateSizeFactors(diagdds geoMeans = geoMeans)4 diagdds lt- DESeq(diagdds test=Wald fitType=parametric)
5 Process the results
The results function creates a table of results Then the res table is filtered by padj lt alpha
1 res lt- results(diagdds cooksCutoff = FALSE)2 sigtab lt- res[which(res$padj lt alpha) ]3 sigtab lt- cbind(as(sigtab dataframe) as(tax_table(merged_mapping_
rarr˓biom)[rownames(sigtab) ] matrix)) Bind taxonomic info to final resultsrarr˓table
4 writecsv(sigtab ascharacter(filename_out)) Write `sigtab` to file
42 Chapter 10 DESeq2 with phyloseq
CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 11
GBS
This documents a pipeline for the analysis of GBS (Genotyping-By-Sequencing) data
Note
Required OS OS x or Linux
Software Tassel 5
Documentation Tassel 50 Wiki
Author This document is created by Saranga Wijeratne
111 File formats
1 File formats that will be using in this analysis
bull HDF5
bull VCF
bull Hapmap
bull Plink
bull Projection Alignment
bull Phylip
bull FASTA more
bull Fastq
bull Numerical Data
ndash Phenotype Format
43
MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
ndash Trait Format
ndash Covariate Format
ndash Marker Values as Numerical Co-variates
bull Square Numerical Matrix
bull Table Report
bull TOPM (Tags on Physical Map)
112 Files you need to have
The following files need to be present before you start the pipeline
1 Sequencing data files (fastq or fastqgz)
Note Fastq files should follow this naming convention (more on page 7 here) - FLOWCELL_LANE_fastqgz(eg AL2P1XXX_2_fastqgz) - FLOWCELL_s_LANE_fastqgz (eg AL2P1XXX_s_2_fastqgz) -code_FLOWCELL_s_LANE_fastqgz (eg 00000000_AL2P1XXX_s_2_fastqgz)
1 To rename a fastqgz file2 $ mv AE_S1_L001_R1_001fastqgz AL2P1XXX_1_fastqgz
2 GBSv2 key file (example key file more information)
3 A reference genome
113 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
114 GBSv2 pipeline
1 Load Tassel 50 module
44 Chapter 11 GBS
MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 $ module load Tassel50
2 Useful commands
To check all the plugins available type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for given Plugin Ex GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options in here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Execute the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
114 GBSv2 pipeline 45
MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
46 Chapter 11 GBS
CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 12
A basic microbiome analysis
121 QIIME
Note
Required OS OS x or Linux
Software Qiime 19
Documentation Qiime tutorial
Author This document was created by Saranga Wijeratne
1211 File formats
This section includes descrption of varies file formats including Qiime scripts and parameters files Read more here
Qiime Script index Index of all the scripts used in Qiime
Metadata mapping files Metadata mapping files provide per-sample metadata
Tip A metadata mapping file example is given here Read the section carefully If you are planning to create themapping file by hand read this section
Biom file OTU observation file Read more here
1212 Download the files
For this tutorial we will use Mothur tutorial data from Schloss Wiki These data are 16s rRRNA Amplicons sequencedwith Illumina MiSeq
47
MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
1 Create folders
Make a new directory MCICQiime and then cd to move into the dirctory
1 $ mkdir MCICQiime2 $ cd MCICQiime
2 Download data
Download data from Schloss Wiki
For this tutorial download only dataset shown in the image below (ie Example data from Scholoss lab)
Inside the MCICQiime dir issue the following command to get the data The data has been zipped and we will useunzip -j to extract all the files to same directory we are in right now
1 $ wget httpwwwmothurorgwimagesdd6MiSeqSOPDatazip2 $ unzip -j MiSeqSOPDatazip
Rename the files for downstream analyses
1 $ for f in fastq do mv $f $f_Lfastqdone
And explanation of the preceding commands
bull for f in fastq reads any file that ends with fastq one at a time
bull do starts the body of the for loop
bull mv $f do mv $f $f_Lfastq rename $f (ie F3D0_S188_L001_R1_001fastq) to$f_Lfastq (ie F3D0_S188fastq)
bull done finishes the loop
3 An explanation of the data
The files and experiment are described in the Schloss Wiki
Because of the large size of the original dataset (39 GB) we are giving you 21 of the 362 pairsof fastq files For example you will see two files F3D0_S188_L001_R1_001fastq andF3D0_S188_L001_R2_001fastq These two files correspond to Female 3 on Day 0 (ie theday of weaning) The first and all those with R1 correspond to read 1 while the second and all those withR2 correspond to the second or reverse read These sequences are 250 bp and overlap in the V4 region of
48 Chapter 12 A basic microbiome analysis
MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
the 16S rRNA gene this region is about 253 bp long So looking at the files in the MiSeq_SOP folderthat yoursquove downloaded you will see 22 fastq files representing 10 time points from Female 3 and 1 mockcommunity You will also see HMP_MOCKv35fasta which contains the sequences used in the mockcommunity that we sequenced in fasta format
1213 GBSv2 pipeline plugins
Plugin DescriptionGBSSeqToTagDBPluginExecuted to pull distinct tags from the database and export them in the fastq format MoreTagExportToFastqPluginRetrieves distinct tags stored in the database and reformats them to a FASTQ file MoreSAMToGBSdbPlugin Used to identify SNPs from aligned tags using the GBS DB MoreDiscoverySNPCallerPluginV2Takes a GBSv2 database file as input and identifies SNPs from the aligned tags MoreSNPQualityProfilerPluginScores all discovered SNPs for various coverage depth and genotypic statistics for a given
set of taxa MoreUpdateSNPPositionQualityPluginReads a quality score file to obtain quality score data for positions stored in the snpposition
table MoreSNPCutPosTagVerificationPluginAllows a user to specify a Cut or SNP position for which they would like data printed MoreGetTagSequenceFromDBPluginTakes an existing GBSv2 SQLite database file as input and returns a tab-delimited file
containing a list of Tag Sequences stored in the specified database file MoreProductionSNPCallerPluginV2Converts data from fastq and keyfile to genotypes then adds these to a genotype file in VCF
or HDF5 format More
1214 GBSv2 pipeline
1 Load the Tassel 50 module
1 $ module load Tassel50
2 Useful commands
To check all the available plugins type
1 $ run_pipelinepl -Xmx200g -ListPlugins
To check all the parameters for a given plugin eg GBSSeqToTagDBPlugin type
1 $ run_pipelinepl -fork1 -GBSSeqToTagDBPlugin -endPlugin -runfork1
Tip Users are recommended to read more about GBS command line options here Page 1-2
3 File preparation
Create necessary folders and copy your raw data (fastqs) reference file and key file to appropriate folder
1 $ mkdir fastq ref key db tagsForAlign hd5
4 Commands for the pipeline
1 $ run_pipelinepl -Xmx200g -fork1 -GBSSeqToTagDBPlugin -i fastq -k keyTomato_keyrarr˓txt -e ApeKI -db dbTomatodb -kmerLength 85 -mnQS 20 -endPlugin -runfork1
2 $ run_pipelinepl -fork1 -TagExportToFastqPlugin -db dbTomatodb -o tagsForAlignrarr˓tagsForAlignfagz -c 5 -endPlugin -runfork1
(continues on next page)
121 QIIME 49
MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
(continued from previous page)
3 $ cd ref4 $ bwa index -a is S_lycopersicum_chromosomes250fa5 $ cd 6 $ bwa samse refS_lycopersicum_chromosomes250fa tagsForAligntagsForAlignsai
rarr˓tagsForAligntagsForAlignfagz gt tagsForAligntagsForAlignsam7 $ run_pipelinepl -fork1 -SAMToGBSdbPlugin -i tagsForAligntagsForAlignsam -db db
rarr˓Tomatodb -aProp 00 -aLen 0 -endPlugin -runfork18 $ run_pipelinepl -fork1 -DiscoverySNPCallerPluginV2 -db dbTomatodb -sC chr00 -
rarr˓eC chr12 -mnLCov 01 -mnMAF 001 -endPlugin -runfork19 $ run_pipelinepl -fork1 -ProductionSNPCallerPluginV2 -db dbTomatodb -e ApeKI -i
rarr˓fastq -k keyTomato_key2txt -kmerLength 85 -mnQS 20 -o hd5HapMap_tomatoh5 -rarr˓endPlugin -runfork1
50 Chapter 12 A basic microbiome analysis
CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

CHAPTER 13
Indices and tables
bull genindex
bull modindex
bull search
51
MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
52 Chapter 13 Indices and tables
Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

Python Module Index
aAbout 1
bbasemount 21basespace 9basespace-share 15
cComputingResources 6
dDataDownloading 30DESeq2-phyloseq 37
ffastq_qc 34FilterFastq 32
gGBS 42
hHome 1
mMembership 4Microbiome 45
53
MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

MCBL Documentation Release 10
54 Python Module Index
Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-

Index
AAbout (module) 1
Bbasemount (module) 21basespace (module) 9basespace-share (module) 15
CComputingResources (module) 6
DDataDownloading (module) 30DESeq2-phyloseq (module) 37
Ffastq_qc (module) 34FilterFastq (module) 32
GGBS (module) 42
HHome (module) 1
MMembership (module) 4Microbiome (module) 45
55
- MCIC Computational Biology Lab (MCBL)
- MCBL membership
- MCBL servers and computing resources
- Download from BaseSpace
- Share runs and projects in BaseSpace
- Using BaseMount
- Download from hudsonalphaorg
- Filter a CASAVA-generated fastq file
- Fastq adapter removal and QC
- DESeq2 with phyloseq
- GBS
- A basic microbiome analysis
- Indices and tables
- Python Module Index
- Index
-