mastering regulatory approval in new orphan drug markets
TRANSCRIPT

1
Mastering Regulatory Approval in New
Orphan Drug Markets
Lewis Lau, RAC
Independent Regulatory Science Researcher
Humber-RAC Work Group
Toronto, Canada

2
The views and opinions expressed in the following PowerPoint slides are those of the individual presenter and should not be attributed to Drug Information Association, Inc. (“DIA”), its directors, officers, employees, volunteers, members, chapters, councils, Communities or affiliates, or any organization with which the presenter is employed or affiliated.
These PowerPoint slides are the intellectual property of the individual presenter and are protected under the copyright laws of the United States of America and other countries. Used by permission. All rights reserved. Drug Information Association, Drug Information Association Inc., DIA and DIA logo are registered trademarks. All other trademarks are the property of their respective owners.
Disclaimer

3
While many regulators continue to provide
strong incentives to drug development for rare
diseases (e.g. PRV program), obtaining prompt
regulatory approval of orphan drug in
jurisdictions where orphan drug framework is
not yet formalized OR still in development can
be challenging
What would be the best regulatory strategy for
pharma in these jurisdictions? Canada presents
a good case study
Overview

4
In 2012, Health Canada announced its determination to establish orphan drug (OD) framework.
In August 2014, Health Canada has announced pilot project targeting patient input from Canadians with rare diseases to help inform future reviews of orphan drugs
In late 2014, the parliament of Canada has passed the Protecting Canadians from Unsafe Drugs Act (Vanessa's Law). Further efforts and incentives to formalize the orphan drug regulatory framework are expected in 2015.
Background
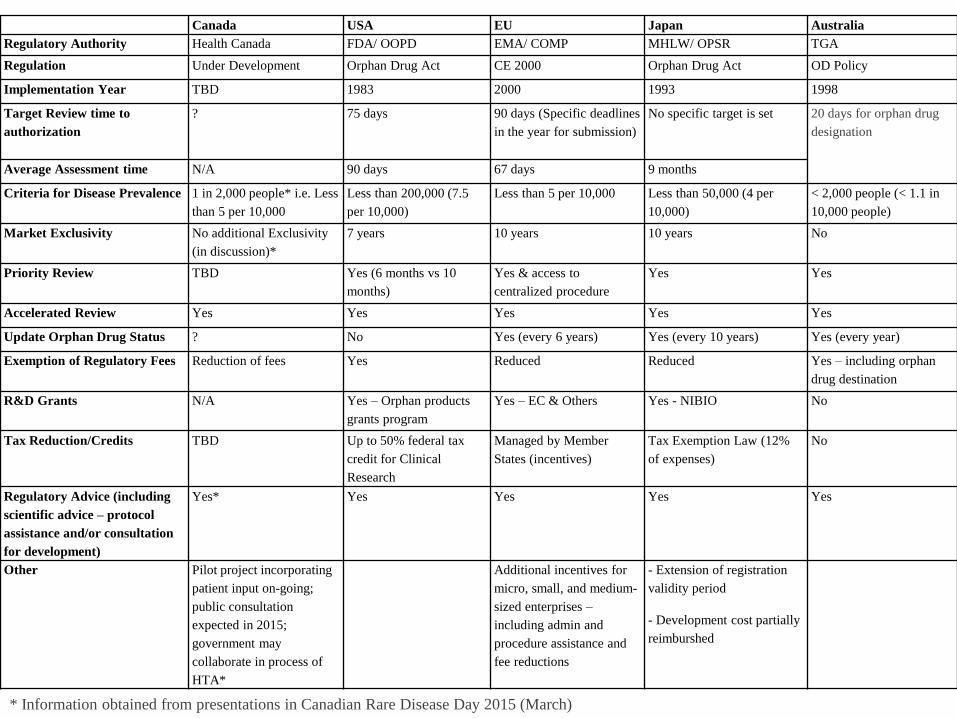
* Information obtained from presentations in Canadian Rare Disease Day 2015 (March)
Canada USA EU Japan Australia
Regulatory Authority Health Canada FDA/ OOPD EMA/ COMP MHLW/ OPSR TGA
Regulation Under Development Orphan Drug Act CE 2000 Orphan Drug Act OD Policy
Implementation Year TBD 1983 2000 1993 1998
Target Review time to
authorization
? 75 days 90 days (Specific deadlines
in the year for submission)
No specific target is set 20 days for orphan drug
designation
Average Assessment time N/A 90 days 67 days 9 months
Criteria for Disease Prevalence 1 in 2,000 people* i.e. Less
than 5 per 10,000
Less than 200,000 (7.5
per 10,000)
Less than 5 per 10,000 Less than 50,000 (4 per
10,000)
< 2,000 people (< 1.1 in
10,000 people)
Market Exclusivity No additional Exclusivity
(in discussion)*
7 years 10 years 10 years No
Priority Review TBD Yes (6 months vs 10
months)
Yes & access to
centralized procedure
Yes Yes
Accelerated Review Yes Yes Yes Yes Yes
Update Orphan Drug Status ? No Yes (every 6 years) Yes (every 10 years) Yes (every year)
Exemption of Regulatory Fees Reduction of fees Yes Reduced Reduced Yes – including orphan
drug destination
R&D Grants N/A Yes – Orphan products
grants program
Yes – EC & Others Yes - NIBIO No
Tax Reduction/Credits TBD Up to 50% federal tax
credit for Clinical
Research
Managed by Member
States (incentives)
Tax Exemption Law (12%
of expenses)
No
Regulatory Advice (including
scientific advice – protocol
assistance and/or consultation
for development)
Yes* Yes Yes Yes Yes
Other Pilot project incorporating
patient input on-going;
public consultation
expected in 2015;
government may
collaborate in process of
HTA*
Additional incentives for
micro, small, and medium-
sized enterprises –
including admin and
procedure assistance and
fee reductions
- Extension of registration
validity period
- Development cost partially
reimburshed

6
Problem: Orphan drug development often implies smaller and shorter clinical trials, how did these drugs obtain prompt approval in Canada without the aid of a proper OD framework?
Method: Our team screened the database of OrphaNet (~180 orphan drugs) and gathered 27 drugs approved and recognized as OD by both FDA and EMA as OD. Within the list of the 27 drugs, 20 of them have also been approved by Health Canada (note: there is no OD regulation in Health Canada jurisdiction). Further analysis has been conducted by analyzing the Summary Basis of Decisions (SDB) published by Health Canada.
What we did?
7
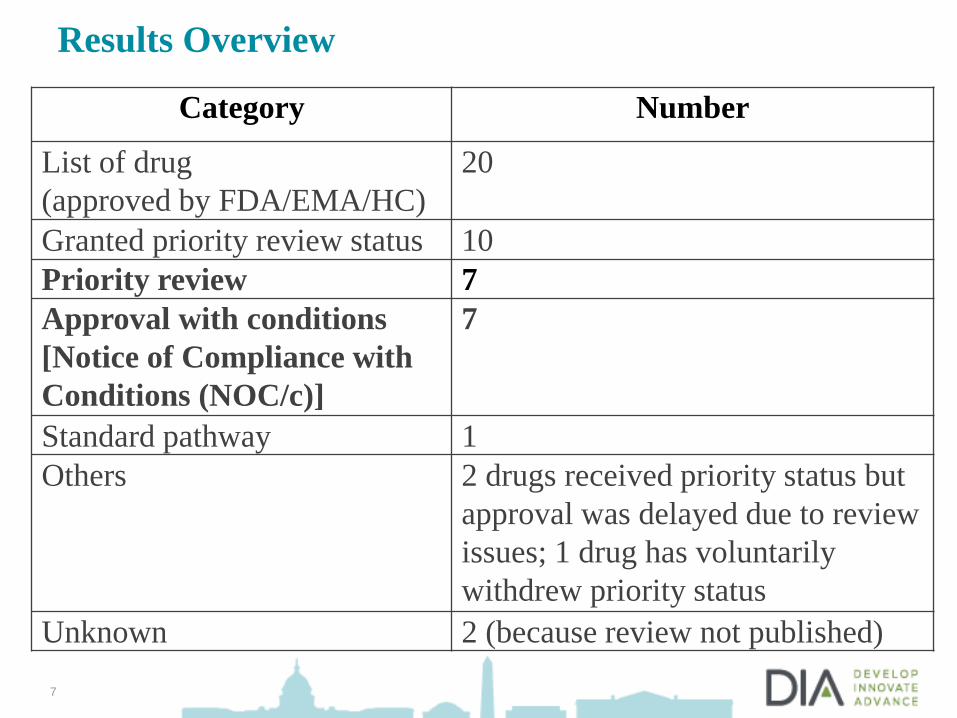
Results Overview
Category Number
List of drug
(approved by FDA/EMA/HC)
20
Granted priority review status 10
Priority review 7
Approval with conditions
[Notice of Compliance with
Conditions (NOC/c)]
7
Standard pathway 1
Others 2 drugs received priority status but
approval was delayed due to review
issues; 1 drug has voluntarily
withdrew priority status
Unknown 2 (because review not published)
8
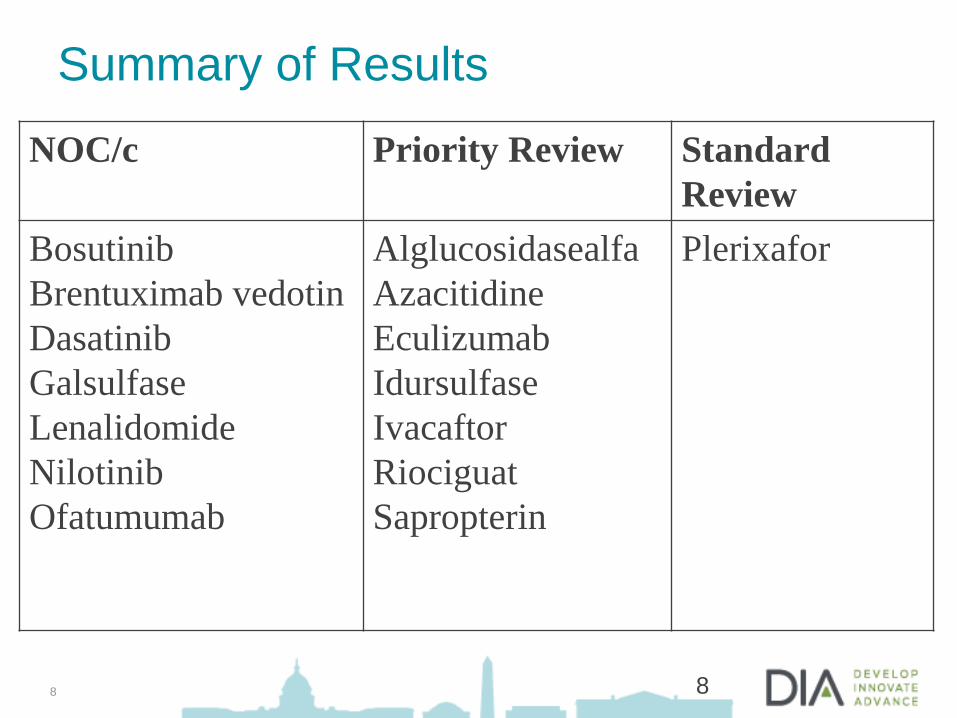
Summary of Results
8
NOC/c Priority Review Standard
Review
Bosutinib
Brentuximab vedotin
Dasatinib
Galsulfase
Lenalidomide
Nilotinib
Ofatumumab
Alglucosidasealfa
Azacitidine
Eculizumab
Idursulfase
Ivacaftor
Riociguat
Sapropterin
Plerixafor
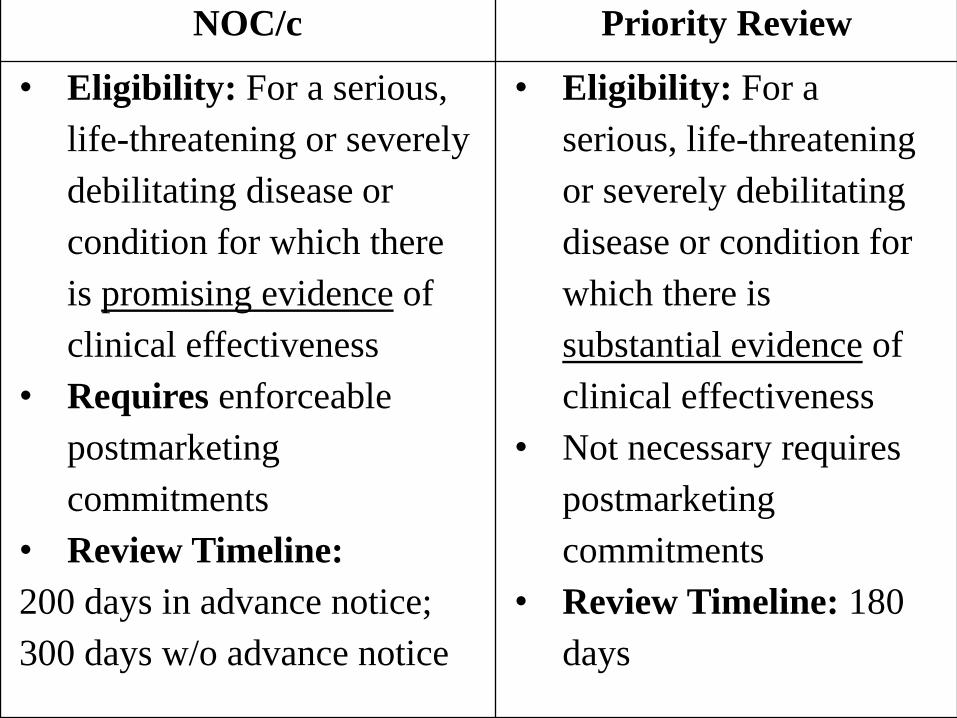
NOC/c Priority Review
• Eligibility: For a serious,
life-threatening or severely
debilitating disease or
condition for which there
is promising evidence of
clinical effectiveness
• Requires enforceable
postmarketing
commitments
• Review Timeline:
200 days in advance notice;
300 days w/o advance notice
• Eligibility: For a
serious, life-threatening
or severely debilitating
disease or condition for
which there is
substantial evidence of
clinical effectiveness
• Not necessary requires
postmarketing
commitments
• Review Timeline: 180
days

10
Clinical Evidences: NOC/c VS Priority Reivew
Promising Clinical Evidence: evidence based
on well-controlled and well-conducted clinical
trials establishing that the drug product has an
effect on a surrogate or clinical endpoint that is
reasonably likely to predict clinical benefit
Substantial Clinical Evidence: evidence
consisting of at least two adequate and well
controlled clinical studies, each convincing on
its own to establish effectiveness of the drug
involved
10

11
Highlights on NOC/c Drug (approval with Conditions)
11
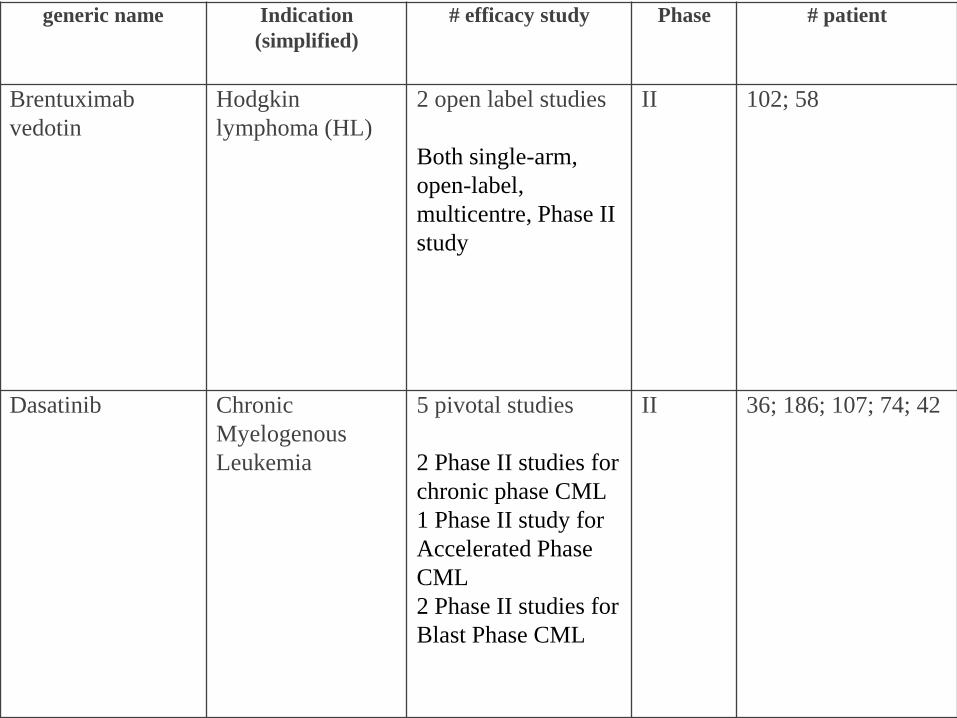
generic name Indication
(simplified)
# efficacy study Phase # patient
Brentuximab
vedotin
Hodgkin
lymphoma (HL)
2 open label studies
Both single-arm,
open-label,
multicentre, Phase II
study
II 102; 58
Dasatinib Chronic
Myelogenous
Leukemia
5 pivotal studies
2 Phase II studies for
chronic phase CML
1 Phase II study for
Accelerated Phase
CML
2 Phase II studies for
Blast Phase CML
II 36; 186; 107; 74; 42

generic name Indication (simplified) # efficacy study Phase # patient
Nilotinib Chronic Myelogenous
Leukemia
1 pivotal study:
Approval based on
interim analysis of the
Phase II study
II 105
Ofatumumab Chronic lymphocytic
leukaemia
1 pivotal study:
Phase II, single-arm,
multicentre study
II 154

Postmarketing Commitments Brentuximab
vedotin
• Submit final data of on-going clinical studies - if unsuccessful,
indications for may be withdrawn
• Several final clinical study reports, safety updates of on-going clinical
trials;
• other post-marketing analyses requested by other regulators
Dasatinib
• Periodic and final efficacy analysis
• Submit 2-year follow-up data from the clinical studies supporting
efficacy;
• Post-market reports of safety and PK studies
Nilotinib
• Final efficacy data
• Study report specific to assess the use of nilotinib in patients with
hepatic impairment.
• Commit to nonclinical Study on drug interaction study with a substrate
of CYP2C9;
• Addition assessment of cardiac safety
• Non-clinical Study: A two-year, nilotinib long-term carcinogenicity
study in rats.
Ofatumumab
• Final study data
• Additional assessment including cardiac safety, anti-drug antibody
response
• Safety updates; Risk Management Plan

15
Highlights on Priority Review Drug
15

generic name Indication
(simplified)
# efficacy study Phase # patient
Azacitidine Myelodysplastic
Syndrome
1 pivatol controlled Phase
III efficacy study
(international,
multicentre, controlled
open-label, randomized,
parallel-group)
2 uncontrolled phase II
studies
1 controlled phase III
supporting study
II &
III
358 (in pivotal
Phase III study)
Eculizumab Paroxysmal
nocturnal
haemoglobinuria
2 placebo-controlled
studies
III 184 (from all
efficacy
studies)
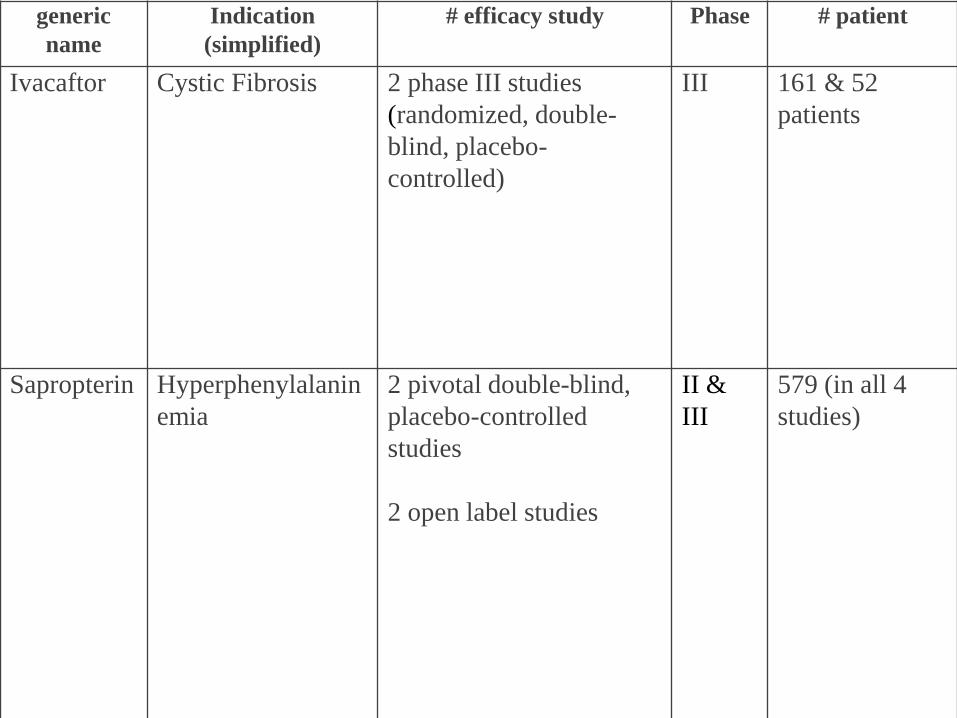
generic
name
Indication
(simplified)
# efficacy study Phase # patient
Ivacaftor Cystic Fibrosis 2 phase III studies
(randomized, double-
blind, placebo-
controlled)
III 161 & 52
patients
Sapropterin Hyperphenylalanin
emia
2 pivotal double-blind,
placebo-controlled
studies
2 open label studies
II &
III
579 (in all 4
studies)
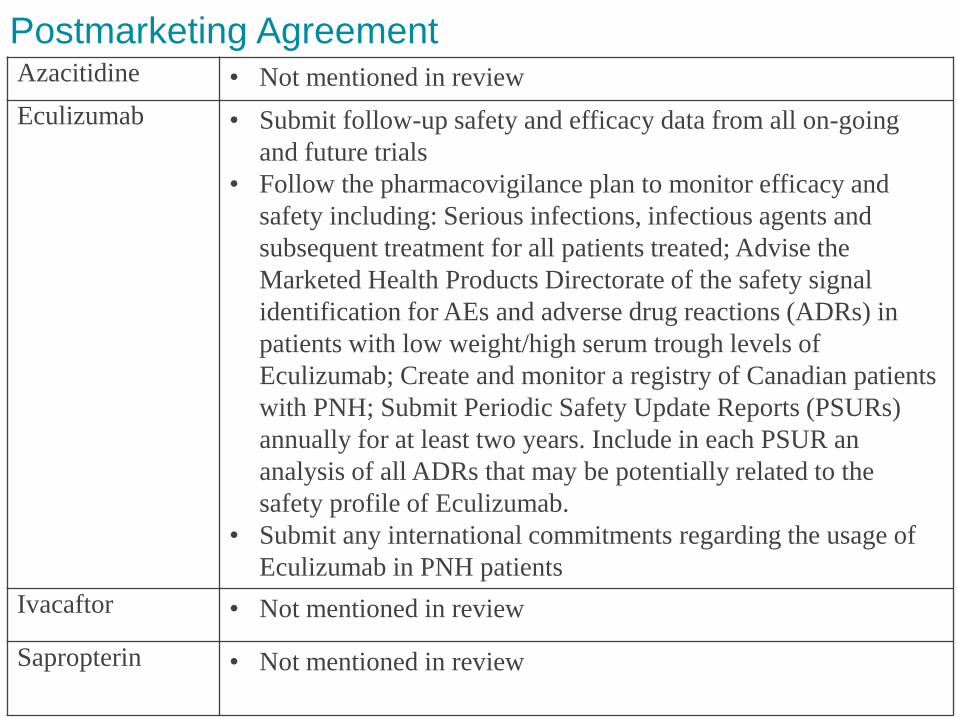
Postmarketing Agreement Azacitidine • Not mentioned in review
Eculizumab • Submit follow-up safety and efficacy data from all on-going
and future trials
• Follow the pharmacovigilance plan to monitor efficacy and
safety including: Serious infections, infectious agents and
subsequent treatment for all patients treated; Advise the
Marketed Health Products Directorate of the safety signal
identification for AEs and adverse drug reactions (ADRs) in
patients with low weight/high serum trough levels of
Eculizumab; Create and monitor a registry of Canadian patients
with PNH; Submit Periodic Safety Update Reports (PSURs)
annually for at least two years. Include in each PSUR an
analysis of all ADRs that may be potentially related to the
safety profile of Eculizumab.
• Submit any international commitments regarding the usage of
Eculizumab in PNH patients
Ivacaftor • Not mentioned in review
Sapropterin • Not mentioned in review

19
Lessons Learned
Based on the efficacy data summaries from SBD,
the approval pathway will fall into the mainly
following:
NOC/c: marketing approval supported by Phase II
clinical trials (with smaller # of patients), requires
commitment to provide new/updated data
Priority Review: indication + supporting data
clearly meets priority review eligibilities. A robust
clinical package backed by Phase III data. No new
commitment is required for the approval.

20
Lessons Learned (Continue)
Sponsor should determine and obtain
agreement with regulators to determine
regulatory pathway applicable to support
marketing approval.
Other recommendations: understanding of the
local treatment paradigm, patient’s
epidemiology; collaboration with local patient
advocacy group.
20

21
Public Health Impact
Current pathway for Canadian patient to obtain
unapproved drug is Special Access Programme
(SAP) – insufficient and inefficient
If the drug is not being commercialized, what is
the incentive for physicians to prescribe?
Reimbursement will remain challenging
No framework = No incentives (while many
orphan drugs are developed by small
companies with low start-up budget or “garage
science”)

Canadian HTA/Reimbursement
pan-Canadian Oncology
Drug Review
(pCORDR)
Common Drug Review
(CDR)
Health Canada Approval
pan-Canadian Pricing Alliance Negotiations
Provincial Review and Evaluation for Funding
Non-CDR product/
non-pCODR Products

23
Conclusion
Use scientific judgments to determine approval
pathway applicable to the data package in
support of the approval of orphan indication(s)
Increased influence in patient input: pharma
must identify treatment paradigm, patient
epidemiology; and other important key
stakeholders
Reimbursement will remain to be challenging
Health agencies, sponsors and patient groups
must collaborate to enable patients to have
prompt access to drug treatments

24
Lewis Lau, RAC
Kindly contributed by:
Giri Venkiteela, MSc, RAC; Mukesh Kumar, RAC;
Daniel Lee, MSc; Wojciech Kulacz, MSc, RAC;
Raje Devanathan, MSc; Vadim Lysenko, RAC;
Bhaskar Anand, PhD; Carla Maxemous, PharmD,
Anchalee Srisombun
Special Thanks:
Sameer Thapar, PharmD; Stephen Li, MBA;
Judy Chapman; Abhiram Pushparaj, Ryan Thompson
Thank You