manchester cancer research centre annual re… · 4 5 welcome to the 2009/10 research report of the...
TRANSCRIPT

Manchester Cancer Research Centre
Research Report 2009/10

Contents
Re
sea
rch
Re
po
rt
200
9/1
0
Chair’s Foreword 4
Introduction 5
The MCRC Partnership 9
Manchester Cancer Research Centre 10Conference - Harnessing Apoptosis
Chris Morrow, Kathryn Simpson, Luke Harrison,
Tetyana Klymenko, Tom Owens and Fiona Foster
Lung Cancer Circulating Tumour Cells as 16
Biomarkers and a Window to Metastasis Biology Tim Ward, Jian-Mei Hou, Matthew Krebs,
Fiona Blackhall and Caroline Dive
DNA Damage Response 20Ivan Ahel
Drug Discovery in the Manchester 24
Cancer Research Centre
Donald Ogilvie and Allan Jordan
Genetic Medicine and the 28
Genesis Prevention CentreD Gareth Evans and Anthony Howell
Cancer Genomics in the Era 34
of High Throughput SequencingCrispin Miller, Jenny Varley and Stuart Pepper
Imaging Research at the Manchester 40Cancer Research Centre
Alan Jackson and Kaye Williams
Obesity and Cancer 46Andrew G Renehan
Role of MKK4 in Ras-Induced Cancers 52Cathy Tournier
The New Patient Treatment Centre 56Malcolm Ranson
Author Biographies 60
Manchester Cancer Research Centre Research Report 2009/10
Manchester Cancer Research CentreThe University of ManchesterWilmslow RoadManchester M20 4BXTel: +44 (0) 161 446 3156www.manchester.ac.uk/mcrc
Founding Partners:
The University of ManchesterOxford Road Manchester M13 9PL Tel: +44 (0) 161 306 6000www.manchester.ac.uk
The Christie NHS Foundation TrustWilmslow RoadManchesterM20 4BX Tel: 0845 226 3000www.christie.nhs.uk
Cancer Research UKAngel Building407 St John StreetLondonEC1V 4ADTel: +44 (0) 20 7242 0200www.cancerresearchuk.org
Copyright © 2010 Manchester Cancer Research Centre

54
Welcome to the 2009/10 Research Report of the Manchester Cancer Research
Centre (MCRC).
The Research Report provides a glimpse into some of the challenges and
achievements over the last two years and emphasises the importance of the
research being undertaken at the MCRC. As a non-scientist and a non-
clinician, what strikes me about the work highlighted within the report is the
diversity of the research undertaken and the innovative approaches being
used to address some of the key outstanding issues in our understanding, and
therefore management, of cancer. This diversity is indicative of the complexity
of cancer as a disease and the need for multifaceted approaches in order to
drive research that leads to improvements in how patients are diagnosed and
treated and improvements in patient outcome.
For the MCRC, 2009/10 has clearly been a successful year, with the fruition of
several long-term strategic initiatives such as the establishment of the MCRC
Drug Discovery Centre and the physical initiation of the new Patient
Treatment Centre at The Christie. These developments provide tangible
evidence of the progress that has been made at the MCRC but also symbolise
the other less visible achievements being made through improving our
understanding of basic biological mechanisms in cancer in order to develop
novel tools and treatment strategies for cancer patients. Importantly, the
research base has been strengthened by attracting leading international
researchers to join the MCRC, giving fresh impetus to research programmes,
adding to the expertise available and bringing opportunities for beneficial
collaborations and the development of holistic research initiatives. This fertile
and nourishing environment is one which fosters and nurtures progress.
Outside of the clinical and scientific world, challenges in the environment we
work in drive innovation and can be motivational; the same is true within
cancer research. The scope and breadth of MCRC research mirrors the
challenge of cancer and highlights the MCRC’s commitment to embrace this
challenge. Ambitious but achievable plans are intended for 2011 and beyond
– in the next Research Report we hope to be able to share with you how some
of these key objectives have been met.
Dr Michael OglesbyChairManchester Cancer Research Centre
Chair’s Foreword
The remit of the MCRC covers the spectrum from basic research through to
translational and clinical research. Our aim is to elucidate the cellular processes
and mechanisms that drive the development and establishment of cancer in
order to identify opportunities for therapeutic intervention and to evaluate
these interventions in clinical trials. This report highlights some key research
areas that exemplify the advantages of this rational and strategic bench to
bedside approach.
Cells have a remarkable capacity to repair DNA damage, a capacity which is
crucial in ensuring accurate replication of the cell’s genomic material.
Aberrant DNA repair mechanisms lead to accumulation of genetic defects
causing disease and the ability to circumvent normal repair mechanisms is
characteristic of many forms of cancer. In order to allow accumulation of
genetic defects, cancer cells may sacrifice specific DNA repair pathways,
making them uniquely susceptible to molecules that impair or inhibit specific
DNA damage response routes. The report by Ivan Ahel, a new recruit to the
MCRC, highlights research being undertaken to better understand the
molecular mechanisms and players in DNA damage response in order to
exploit cancer cell defects in the maintenance of genomic integrity. This
research has led to the identification of novel molecular mediators in DNA
repair processes. Studies on DNA repair mechanisms are particularly exciting
right now since recently developed inhibitors of certain repair pathways are
showing great promise in the clinic.
Despite efficient repair mechanisms, genomic mutations that drive
tumourigenesis do occur leading to cancer development. A major challenge in
maximising the effect of therapeutic interventions is patient stratification
and the identification of those patients whose tumours have a genetic pattern
that makes them more likely to respond to a particular therapy. Since the
publication of the human genome a decade ago spectacular advances in DNA
sequencing technology and techniques have been made and these advances
Introduction
Dr Michael OglesbyChairManchester Cancer Research Centre
Professor Nic JonesDirectorManchester Cancer Research Centre

76
provide great promise for defining the genetic landscape of individual tumours. The report by Miller et al
details the potential and promise of high throughput cancer genomics and the advanced DNA analysis
technology and how they are being used by MCRC researchers to better understand tumour initiation,
progression, prognosis and response to therapies.
The power of defining the genome landscape to match patients to particular treatments depends on
thorough biological knowledge and recognition of the exact mutations that drive tumour growth. A major
focus of MCRC research is to develop gene signature profiles for a range of cancers, the most well
understood currently being breast cancer. The report from Evans and Howell describes the contribution
MCRC researchers have made to a more detailed understanding of breast cancer through rigorous genetic
analysis of samples from thousands of patients. A key goal is identifying genetic profiles that predispose
individuals to tumour development and to use this knowledge to apply preventative interventions to
identified high-risk target populations. MCRC researchers are part of a national initiative that aims to
optimise existing screening programmes by refining risk assessment models and identifying novel
additional risk factors that can improve risk prediction. MCRC researchers were the first to conduct a
comprehensive genetic study on neurofibromatosis type 2, a malignancy that affects skin and nervous
tissue. Based on over a decade of experience, they have secured funding for a national programme on the
management of this disease, a programme which aims to improve patient outcome by identifying and
promoting best practice and a consistent approach to patient management.
Within the MCRC there are substantial efforts to characterise and validate biomarkers that can measure or
predict therapeutic response. A particularly exciting and important area of research involves the
characterisation of circulating tumour cells (CTCs) and evaluating the potential of these cells to act as
biomarkers: to ask whether changes in CTCs can provide prognostic information or an early indication of
therapeutic response, Given the difficulty of accessing biopsy material from certain tumours for genomic
analysis and other studies, for example because of their location, the availability of serum-borne material
that can be routinely monitored before, during and after therapy has significant advantages in terms of
convenience and logistics for both patients and researchers. This approach is described in the report from
Ward et al, and evidence provided for encouraging early results in lung cancer.
The study of molecules as potential biomarkers is not the only method of measuring response to therapy:
advanced imaging approaches are also showing great promise in this regard. The newly formed MCRC
Imaging Group is tasked with promoting the integration of advanced imaging technology across the
breadth of MCRC research in order to fully exploit the range of expertise and world-class facilities available
within the MCRC partnership. Detailed within the report from Jackson and Williams is research aimed at
identification and validation of imaging biomarkers which are directly relevant to tumour behaviour or able
to monitor the impact of therapeutic interventions, preclinical imaging to investigate cell proliferation,
metabolism and cell death, and the use of imaging within clinical research programmes using
multimodality imaging techniques across a range of cancer types.
Understanding and predicting patient response
to therapy including adverse treatment
responses is not limited to studies of genetic
factors but also encompasses research into the
impact of lifestyle and dietary habits on
outcomes. With emerging evidence for obesity
as an adverse prognostic factor in cancer and a
potential marker for increased adverse
treatment effects, MCRC researchers are
conducting studies to better understand and
unravel the complex relationship between
cancer and obesity using both in vitro and in vivo
models, as described in the report from Renehan.
Following groundbreaking research into the
epidemiology of excess weight and cancer,
researchers are now exploring its impact on
treatment response and outcome, the possible
role of molecular mediators such as insulin on
cancer risk and progression and developing
robust statistical methodology for the
evaluation of potential biomarkers in clinical
trials.

98
The University of Manchester
Over the last five years, the partnership has established the MCRC as one of the
largest research centres of its kind in Europe and has strengthened its capability
across the full spectrum of cancer research activity. The MCRC is continuing to
recruit eminent researchers to lead its basic, translational and clinical research
portfolio. The real strength of the MCRC is evident in the closer working relationships
with the partner NHS Trusts. Building on these relationships is vital to expanding our
basic research effort and fostering the translation of the knowledge we gain in to
better patient care.
Professor Dame Nancy Rothwell Professor Rod Coombs
President & Vice-Chancellor Deputy President & Deputy Vice-Chancellor
The Christie NHS Foundation Trust
The MCRC partnership has maximised the impact of activity in cancer research and
is making Manchester on of the largest and most significant centres for cancer
research and treatment.
We are driving forward ambitious plans to provide world class services to our
patients, and developing our clinical research is an important aspect of this. The
new Patient Treatment Centre, which will be the largest early phase clinical trials
unit in the world, provides a fantastic example of the benefits of working in
partnership and brings huge benefits to patients, which is ultimately what our
efforts are all about.
Lord Keith Bradley Caroline Shaw
Chairman Chief Executive
Cancer Research UK
We are proud to have a long association with cancer research in Manchester, and
the MCRC partnership fits exemplifies our vision that ‘Together we will beat cancer’.
Bringing scientists and clinicians closer together will help us to improve our
understanding of cancer and find out how to prevent, diagnose and treat the many
different kinds of the disease. We are committed to building strong partnerships to
maximise the opportunities for discoveries in the laboratory to translate into
benefits for patients. The MCRC is a great example of such a partnership and, with
the development of the Drug Discovery Centre, we have the potential to yield new
treatment options that can have a real impact on cancer patients.
Michael Pragnell Harpal Kumar
Chairman Chief Executive
The MCRC Partnership
Cancer development and progression is a complex process and so also are the mechanisms that control
this process. Cell signalling pathways provide a mechanism by which cells are able to respond and adapt to
changes or stimuli in their environment and play an important role in controlling cell behaviour. Activation
of specific pathways triggers a cascade of events and reactions ‘downstream’ from the initiating activation
point which subsequently impact cell behaviour. Research by Cathy Tournier and colleagues within the
Molecular Cancer Group has provided the first genetic evidence that signalling downstream of MKK-4, a
molecule with potent cell signalling properties, plays an essential role in the formation of skin tumours.
The findings which are featured in this report are particularly noteworthy as until now there has been
conflicting data on the role of MKK-4 in tumour development and therefore its potential as a possible target
for anticancer drug therapy. The team have now been awarded a grant from Cancer Research UK to elucidate
the role of MKK-4 in tumour initiation and progression, under normal conditions and in response to
anticancer therapies.
Improving patient outcome requires innovative approaches to treatment, including the use of existing drugs
in more effective ways such as novel combination strategies and the identification of new target
populations. In parallel, the identification and development of new drugs remains crucially important and
the report from Ogilvie and Jordan focuses on the MCRC Drug Discovery Centre, which is part of a
coordinated policy within Cancer Research UK to increase and prioritise research in this area. In 2009 the
new Centre became a reality with the skills, facilities and partnerships now in place to drive the
development of novel small-molecule based therapies.
Key to our success in improving patient treatment is the ability to thoroughly evaluate promising new
agents in early phase clinical trials. The new £35 million Patient Treatment Centre at The Christie opened in
November 2010, and is described in the report from Ranson. The new Centre will provide for a doubling of
clinical trials activity with an emphasis on increasing capacity for early phase clinical trials. In addition, the
Centre allows for an optimised patient care pathway by bringing together core facilities such as service
chemotherapy and pharmacy under one roof. The expansion and continued commitment to clinical trial
research enhances the international reputation of The Christie and MCRC partners in the conduct of high
quality clinical research with the ultimate goal of improving outcomes for patients with cancer.
We are in a hugely exciting era in cancer research with unrivalled opportunities in terms of increased
knowledge, sophisticated technology and dedicated resources. A recurring theme throughout the highlights
featured in this report is that the challenge to develop a personalised and individual approach to treatment
based on genetic and molecular profiles of individual patients and their disease is being embraced. We aim
to build on the progress already made and to use the advantages that partnership and collaboration within
the MCRC brings to deliver benefits to individuals with cancer.
Professor Nic Jones
Director
Manchester Cancer Research Centre
The Manchester Cancer Research Centre was founded by The University of Manchester, The Christie NHS
Foundation Trust and Cancer Research UK and, with the advent of the Manchester Academic Health Science
Centre (MAHSC), has been expanded to include Central Manchester University Hospitals NHS Foundation
Trust, Salford Royal NHS Foundation Trust, University Hospital of South Manchester NHS Foundation Trust,
Manchester Mental Health and Social Care Trust and NHS Salford (Salford Primary Care Trust).

11
therapeutic targeting of IAPs in cancer cells induce apoptosis. His IAP
antagonists caused Ub-mediated degradation of c-IAP1 and 2, lNF-κB
activation and up-regulation of TNFα. The resulting autocrine/paracrine TNFα
signals induced TNF-RI-dependent apoptosis. In contrast, XIAP neutralisation
by IAP antagonists synergised with anti-DR5 or FasL to induce TNFα
independent apoptosis bypassing the requirement to engage the
mitochondrial apoptosis pathway. Thus, IAP antagonists invoke multiple hits
on apoptotic signals in cancer cells.
Anthony Letai (Dana Faber Cancer Research Institute, Boston) discussed the
molecular mechanisms that set a threshold for drug-induced apoptosis to
explain cancer cell sensitivity and resistance. He described three mechanisms
by which cancers evade pro-apoptotic signals emanating from tumour
microenvironment or from oncogene activation by altering Bcl-2 family
member levels. Significantly, if cancers have evolved to over-express anti-
apoptotic Bcl-2 family proteins they are ‘primed’ for apoptosis and should
readily engage apoptosis after BH-3 mimetic drug treatment. Tony described
his technique, BH-3 profiling, which determines whether a cancer cell is
‘primed’; an approach showing some potential to predict drug response in
lymphoma patients. Saul Rosenberg (Abbott Laboratories, Illinois) described
the development of BH-3 mimetics. ABT-737 was developed using NMR–based
chemical library screening linked to structure-based design, to identify small
molecules that bind with sub-nanomolar affinity to BCL-2, BCL-xL, and BCL-w.
In preclinical studies ABT-737 exhibited single agent activity in almost all
tumour types, but enhanced cytotoxicity of chemotherapy and irradiation
across a wider cancer cell line panel. The orally bioavailable clinical candidate,
ABT-263, is now in phase I/II clinical trials with some early promising results.
The focus switched to clinical utility of drugs targeting direct regulators of
apoptosis and techniques to measure apoptosis in cancer patients. Malcolm
The Manchester Cancer Research Centre Conference: Harnessing Apoptosis
10
Manchester Cancer Research Centre Research Report
The inaugural Manchester Cancer Research Centre Conference entitled
‘Harnessing Apoptosis’ brought together scientists and clinicians from across
Manchester and around the globe to listen to presentations from
international leaders in this field. The conference, which was held at The
Palace Hotel on 17-20 January 2010 attracted 120 delegates, was organised by
Caroline Dive (Paterson Institute for Cancer Research), Charles Streuli
(Wellcome Trust Centre for Cell-Matrix Research, Faculty of Life Sciences, The
University of Manchester) and Esther Walker (MCRC Operations Manager).
Following a warm welcome to Manchester from MCRC Director Nic Jones, the
opening plenary lecture by Doug Green (St. Jude’s, Memphis) gave an
introduction to apoptosis signalling pathways and focused on reconciling an
ongoing controversy regarding two proposed models of regulation of
mitochondrial outer membrane permeabilisation (MOMP) by Bcl-2 family of
proteins. His lab used a BH3-domain-graft method to take advantage of the
differing affinities of Bcl-2 family proteins for each other, to dissect whether
regulation of MOMP occurred by the ‘direct activation/de-repression’ or
‘neutralisation’ model. His new data suggested that both models can occur
dependent upon cellular stress levels. An important ramification may be that
cells are sensitised to death induced by Bcl-2 targeted drugs (BH-3 mimetics)
when ‘primed’ by low levels of cellular stress.
The conference continued with sessions on Inhibitors of Apoptosis Proteins
(IAPs) and the Bcl-2 family. Data presented by Pascal Meier (Institute of Cancer
Research, London), provided further insight into IAP function in vivo. His
group identified a novel mechanism by which IAPs regulate caspases. An
RNAi screen of de-ubiquitinating enzymes (DUBs) in Drosophila identified six
pro-apoptotic DUBs, several of which are associated with the ubiquitin-like
modifier NEDD8. They found that IAP family members act as E3-ligases to
“NEDD8ylate” and inactivate endogenous caspases. Domagoj Vucic
(Genentech, San Francisco) presented mechanisms through which
Manchester Cancer Research Centre Conference:Harnessing Apoptosis By Chris Morrow, Kathryn Simpson, Luke Harrison, Tetyana Klymenko, Tom Owens and Fiona Foster

13
Eileen White (Rutgers University, New Jersey) described a cell model wherein mTOR inhibition promoted
cell cycle arrest and autophagy but when this autophagy was blocked apoptosis ensued. An anti-cancer
therapeutic strategy might therefore be to stress a cell to make it use autophagy as a survival mechanism
and then inhibit autophagy to drive cell death. Marja Jäättelä (Institute of Cancer Biology, Copenhagen)
introduced a different type of cell death, lysosomal cell death, which can be avoided by cancer cells via the
up-regulation of heat shock protein 70 (HSP70) to enhance the activity of lysosomal acidic spingomyelinase
(ASM) and stabilise lysosome membranes. Inhibition of the interaction of HSP70 and ASM by point
mutation of HSP70, or pharmacological inhibition of ASM prevents lysosome stabilisation to promote cell
death and sensitisation to chemotherapeutics. Considering tumour cells frequently show enhanced ASM
activity, targeting of this enzyme may lead to therapeutic benefit.
Using mice with a knock-in mutant of the PI3-kinase isoform p110a that cannot bind RAS-GTP, Julian
Downward (CR-UK London Research Institute) showed that these mice were less tumour-prone in response
to various RAS-dependent tumourigenic assaults. He also reported a RNAi screen for targets that proved
lethal in a mutant RAS background. Of the hits from this screen, MMP7 showed the most therapeutic
promise, with MMP7 inhibitors combined with topotecan demonstrating selective killing of RAS mutant
cell lines. Christine Watson’s (University of Cambridge) presentation concentrated on studies of the
molecular mechanisms that induced apoptotic cell death during mammary gland involution. STAT3, which
is activated during involution, is one of the key drivers of apoptosis, causing increased levels of the PI3-
kinase subunits p55a and p50a, which may inhibit PI3-kinase activity. STAT3 activation reduced levels of
cathepsin inhibitory protein Spi2a, leading to an increase in cathepsin activity and apoptosis, thus
demonstrating two novel molecular mechanisms for STAT3-mediated apoptosis. Bart Vanhaesebroeck
(Queen Mary University, London) described his elegant mouse models where kinase dead PI3-kinase catalytic
isoforms had been knocked-in to predict effects of specific isoform inhibition by small molecule drugs. Cells
in these mice could proliferate and did not undergo apoptosis, although some isotype knock-ins were
embryonic lethal. However, if multiple isoforms were targeted by genetic and chemical means more
The Manchester Cancer Research Centre Conference: Harnessing Apoptosis
12
Manchester Cancer Research Centre Research Report
Ranson (The Christie NHS Foundation Trust) described findings from the first-in-man trial of AEG35156, a
second generation antisense oligonucleotide targeted to XIAP. In this phase I study AEG35156 was well
tolerated and there was some evidence of anti-tumour activity. Discussion followed regarding the need for
pharmacodynamic biomarkers, as well as the need to determine the tumour drug levels to relate to
therapeutic effect. Caroline Dive (Paterson Institute for Cancer Research) continued the biomarker theme,
speaking on circulating biomarkers of cell death and circulating tumour cells (CTCs). In pilot studies, levels
of circulating cytokeratin 18 and nucleosomal DNA correlated with tumour response and/or toxicity and
had potential for predicting response to standard of care chemotherapy. Interest was generated in the
finding that groups of CTCs (microemboli) have increased ability to avoid anoikis, therefore promoting
metastasis. Gerald Cohen (MRC Toxicology Unit, Leicester) discussed BH-3 mimetic treatment of primary
CLL cells where ABT-737/263 was the most efficacious inducer of apoptosis. Culturing primary CLL cells on
CD154 (CD40L)-expressing fibroblasts to model the lymph node environment induced ABT-737 resistance via
up-regulation of Bcl-xL and Bcl-A1. ABT-263 was ~100 fold less potent in whole blood than ABT-737, an effect
attributed to its greater affinity for serum albumin, demonstrating the importance of modelling the cancer
environment using in vitro models.
Elisabeth de Vries (University Medical Centre, Groningen) provided a comprehensive overview of TRAIL
receptor agonists in Phase I/II clinical trials, with emphasis on the use of recombinant TRAIL, the TRAIL-R1
antibody mapatumumab and five TRAIL-R2 antibodies. These agents were generally well tolerated with no
single agent MTD and no significant toxicity. In phase II studies, single agent activity was reported in some
patients with colorectal cancer, non-small cell lung cancer and non-Hodgkin’s lymphoma.
A vigorous debate at the close of day one over whether apoptosis or autophagy regulatory proteins would
prove the best targets for drug development was
masterminded by Henning Walzcak (Imperial College,
London) championing apoptosis and Kevin Ryan
(Beatson Institute for Cancer Research, Glasgow)
arguing for autophagy research. Henning outlined
therapeutic progress with death receptor agonists, BH3
mimetics, and IAP antagonists. Kevin guided the
audience through the complexities and unanswered
questions surrounding autophagy, highlighting the
paradox that reduced autophagy may lead to
tumourigenesis, while autophagy can also be a survival
mechanism for cancer cells in the harsh tumour micro-
environment. The general consensus at the end of the
debate was that apoptosis targeting agents are
showing clinical promise based on decades of basic
research, while the field of autophagy is still very much
in its infancy and only time will tell whether inhibiting
or activating autophagy mechanisms in tumours may
prove clinically beneficial. The convivial atmosphere and
debate format was enjoyed by all.

15
The Manchester Cancer Research Centre Conference: Harnessing Apoptosis
14
Manchester Cancer Research Centre Research Report
pronounced effects were seen. Mice lacking p110δ were resistant to B16 melanoma tumour take and
metastasis indicative of disabling defects in the tumour stroma.
Rakesh Kumar (George Washington University, Washington) gave an overview of p21 activated kinase (PAK)
that plays several roles associated with tumourigenesis. PAK knock-down inhibited tumour cell invasion
and PAK associated with proteins associated with mitosis. PAK was up-regulated in many breast cancers
where its activity associated with tamoxifen sensitivity, raising the possibility that PAK inhibitors might
sensitise resistant breast cancers to tamoxifen.
Paul Workman (Institute of Cancer Research, Sutton) discussed the translation of small molecule PI3-kinase
inhibition to the clinic. Interestingly for inhibitors of a survival pathway, but consistent with data presented
by Bart Vanhaesebroeck, PI3-kinase inhibition did not induce apoptosis, but rather caused cell cycle arrest.
However, when combined with either temozolomide in glioblastoma cells or TRAIL in colorectal cancer cells,
apoptosis was enhanced. A clinical candidate PI3-kinase inhibitor GDC-0941 had good in vivo activity and
was promising results in a phase I clinical trial, with no dose-limiting toxicities reported. Sylvie Guichard
(AstraZeneca, Macclesfield) completed the signalling pathway session discussing the mTOR inhibitor
AZD8055. This has in vitro and in vivo activity, although whether AZD8055 led to cell death or cell cycle
arrest was cell line dependent. Tumour regression was achieved when AZD8055 was combined with the
MEK inhibitor AZD6244 with biomarkers of apoptosis, again demonstrating that while PI3-kinase pathway
inhibitors may not cause apoptosis per se, there is potential for utilising them in combination with other
drugs to great therapeutic benefit.
The second debate of the conference tackled the virtues and problems of in vitro and tumour xenograft
models versus more complex animal models for anti-cancer drug development. Gerard Evan (University of
Cambridge) reported the benefits of mouse models, in particular the use of genetically engineered mouse
models (GEMMs). The use of these mouse models, in which tumours arise from sporadic in situ oncogene
activation or tumour suppressor inactivation, and evolve through acquisition of secondary lesions, allow
for a more accurate modelling of what occurs in human disease. Donald Ogilvie (Paterson Institute for
Cancer Research) pointed out that a better understanding of the causes or possible treatments of cancer
was our aim and whether the information came from GEMMs or tissue culture models, it could all be
informative. As the representative for the use of tissue culture model systems, Donald highlighted the use
of cancer cell line panels to represent the heterogeneity of the disease even within a given cancer subtype.
At the end of an engaging discussion, GEMMs may have just won the vote, but most agreed that multiple
models were useful and none substituted perfectly for the in situ disease in humans.
The final day started with Jos Jonkers (The Netherlands Cancer Institute, Amsterdam) highlighting the use
of GEMMs. His elegant BRCA1 - p53 null model of breast cancer closely mimics the human disease and
drugs inducing double-strand DNA breaks were shown to be particularly effective. Cisplatin and other
platinum drugs decrease tumour volume, but the tumours continually reoccur without developing
resistance. This would suggest that BRCA1 is required for the generation of resistance to cisplatin. The PARP
inhibitor Olaparib inhibited tumour growth and increased survival but prolonged treatment resulted in
resistance mediated by up-regulation of p-glycoprotein. Currently there are no effective treatments for
pancreatic cancer. David Tuveson (CR-UK Cambridge Research Institute) described his GEMM of pancreatic
cancer, which is indistinguishable from the human disease. This model should allow for the identification
of potential new targets in the development of treatments and novel data on the potentially critical role of
ROS in pancreatic cancer was reported.
The conference was concluded by a plenary seminar from Karen Vousden (Beatson Institute for Cancer
Research, Glasgow) who began with an historical overview of tumour suppressor p53 before focusing on
how the accumulated knowledge of p53 is being translated for clinical benefit. She described recent
research which shed light on role of the mutant forms of p53 in regulating of the motility of malignant cells.
She also discussed how under conditions of mild stress, p53 may contribute to cell survival. By example, she
reported how the p53-inducible protein TIGAR is able to modulate the glycolytic pathway, decrease ROS
levels and consequently inhibit autophagy to promote cell survival.
The conference was a great success, spanning the basic research that continues to unravel the control
mechanisms for cell death to the implementation of apoptotic targeted drugs in the clinic. There was much
excitement about the initial clinical trials with drugs such as ABT-263 and IAP inhibitors, even though it
was clear there was still a considerable amount of work to be undertaken, both at the basic and translational
level, before the full benefit of drugs specifically designed to target cell death pathways are realised.

17
The ‘gold-standard’ method for assessing tumour genotype and phenotype is
an invasive tumour biopsy. However, this often challenging for the patient
and difficult to obtain, particularly serially pre and post drug treatment. This
is particularly problematic in lung cancer patients where tumours may be in
high risk or inaccessible regions of the thorax. In many cases, even where
tissue is obtained, there may be insufficient material for molecular analysis.
Here, CTCs/CTM may represent a ‘virtual biopsy’ readily accessible in real-time
from small volume (2-10ml) blood sample. Serial collection and analysis of
CTC/CTM represents an unprecedented opportunity to study the underlying
mechanisms of the metastatic disease process and compare tumour cells
circulating at disease presentation and at disease relapse. If these circulating
cells can be isolated, purified and characterised (using candidate or global
approaches), this could contribute to the goal of personalised medicine.
Detection and enumeration of CTCs
The number of CTCs present in a patient’s bloodstream varies from less than
ten to several thousand per ml in a background of 10 million leukocytes and
5 billion erythrocytes per ml. Efficient and reproducible purification of CTCs is
therefore the proverbial ‘needle in a haystack’ scenario. Until recently very
few, if any, methods for CTC research demonstrated high specificity and
standardised assay protocols preventing their interpretable use in the clinical
setting and especially in multi-site studies. Enrichment techniques decrease
the degree of normal blood cell contamination and are categorised into i)
positive selection by expression of cell surface markers such as EpCam
(epithelial cell adhesion molecule) and ii) size exclusion methods to capture
cells above a certain size threshold chosen to exclude most blood cells.
Positive selection of CTCs poses obvious limitations if there is heterogeneity
in CTC surface marker expression. Eighty percent of human tumours are
epithelial in origin and as such cells within the tumour mass express markers
such as cytokeratins and EpCam. However, the expression of epithelial
markers is predicted to decrease if CTCs have undergone an epithelial to
mesenchymal transition (EMT) as a prelude to cell invasion as part of
metastatic process.
Lung Cancer Circulating Tumour Cells as Biomarkers
16
Lung Cancer Circulating TumourCells as Biomarkers and aWindow to Metastasis Biology
Manchester Cancer Research Centre Research Report
Cancer metastasis is the predominant cause of treatment failure and cancer
fatality. The metastatic process involves migration and invasion of cancer
cells through tissue, degradation of blood vessel walls and intravasation into
the circulation preceding extravasation and clonal growth in environmentally
primed distant ‘niche’ sites according to the ‘seed and soil’ paradigm. The
identification, enumeration and characterisation of tumour cells in the
circulation has become a pre-eminent focus of the Clinical and Experimental
Pharmacology (CEP) Group led by Caroline Dive, based at the Paterson
Institute for Cancer Research where we a) seek to exploit them as biomarkers
in clinical trials and b) gain insights to circulating tumour cell biology that
might uncover innovative therapeutic approaches and novel targets for drug
discovery. Our recent discovery in lung cancer patients of circulating groups
of tumour cells (micro-emboli) suggestive of an endpoint of collective
migration now drives investigations of anoikis suppression in circulating
tumour micro-emboli and the clinical relevance of epithelial to mesenchymal
transition during lung cancer metastasis.
Characterisation of circulating tumour cells (CTCs) and circulating tumour
micro-emboli (CTMs) is challenging and the biology of tumour cells in the
circulation poorly understood due to paucity of validated methods for their
detection and purification. The Veridex CellSearch™ platform, an
immunomagnetic based method for CTC enumeration, has galvanised this
field of research with numerous studies demonstrating utility of CTCs as
prognostic biomarkers. It is widely used clinically, is reproducible and able to
detect CTCs across a range of tumour types inculding breast, colorectal,
prostate, ovarian, bladder and lung. The US Food and Drug Administration
(FDA) approved this technology to monitor progression-free and overall
survival in breast, colorectal and prostate cancers. This and other advances in
technology are now heralding a step-change in our ability to qualify CTC/CTM
biomarkers that predict and/or monitor response to treatment, identify
potential therapeutic targets, and unveil and understand heterogeneity in
tumour cells that survive the circulation to promote metastasis.
By Tim Ward, Jian-Mei Hou, Matthew Krebs, Fiona Blackhall and Caroline Dive

19
Circulating tumour cells and microemboli: collective migration, anoikis and epithelial
mesenchymal transition.
Metastatic spread is postulated to occur via Paget’s ‘seed and soil’ paradigm and via epithelial to
mesenchymal transition (EMT) where down-regulation of epithelial and up-regulation of mesenchymal
cellular characteristics facilitate invasive behaviour of single tumour cells. More recently, the process of
collective migration of cancer cells through tissue has been proposed where clusters or strands of cells
invade through tissues cooperatively,- although the epithelial versus mesenchymal phenotype of these cell
groups has not been reported. In addition, preclinical models suggest a co-operation between epithelial and
mesenchymal cells during the metastatic process that somewhat challenges the strict definition of the
EMT paradigm. In a recent and pilot study of NSCLC and SCLC patients, we identified lung cancer patients
in whom cancer cells circulate as CTM and as single circulating tumour cells (CTCs). CTM were composed
of clusters and strands consistent with the morphologies described for collective migration. Considerable
intra- and inter-patient heterogeneity was observed in EMT markers (vimentin, E-Cadherin, N-Cadherin,
cytokeratin) consistent with incomplete EMT or with co-operation between epithelial and mesenchymal
tumour cells. CTCs, but not cells within CTM, exhibited apoptotic nuclei consistent with the hypothesis
that tumour cells circulating within CTM are more likely to suppress anoikis (the engagement of apoptosis
upon loss of cell-cell contacts and extracellular matrix). Further studies are underway to explore the clinical
significance of these early findings. Venous thromboembolism (VTE) is a well known complication of
metastatic cancer and the presence of tumour cells in the circulation has been associated with increased
risk of VTE in metastatic breast cancer patients. Prospective studies are now underway in the CEP Group to
understand the clinical significance of lung cancer CTM in addition to the molecular mechanisms driving
their behaviour that may provide new insights for therapeutic control.
18
Manchester Cancer Research Centre Research Report
The CEP Group employs the Veridex CellSearch™ approach to enumerate CTCs in clincal trial settings and
the CellScreen ISET platform, a filtration based approach, in research mode to characterise CTCs and CTMs.
The Veridex CellSearch™ platform is a semi-automated system that enriches for CTCs using a ferrofluid
medium of magnetic particles in an EpCAM polymeric coated layer, the EpCAM acting as the capture
antibody. Cells remain intact allowing some morphology assessment and enumeration and CD45
expression is used to identify and exclude leukocytes. CTCs are confirmed by their expression of cytokeratins
(8,18,19) as tumour specific markers. DAPI staining allows assessment of apoptosis via condensed and/or
fragmented nuclear morphology. Enrichment is in the order of 104 to 2x105 fold and sensitivity is one cell
per 0.5ml blood. The fixed and stained cells are washed and dispensed into a cartridge which is inserted into
a ‘MagNest’ device. This attracts cells to a position within a magnetic field and an inbuilt flouresecent
microscope scans this position to produce flourescent images in a gallery format for a manual decision on
what is or is not a CTC. There is an additional spare channel in the standard Veridex CTC kit that allows
examination of a fourth molecule of interest, for example a drug target or pharmacodynamic biomarker. As
examples, the CEP Group is currently examining CD56 (a neuroendocrine marker in small cell lung
carcinoma (SCLC) cells), and Bcl-2 or Mcl-1 (potential biomarkers of senstivity or resistance to the BH-3
mimetic drug ABT 263 respectively). Further analysis is now possible on CTCs recovered from the cartridge
after initial analysis using multi-colour fluorescence in situ hybridisation (FISH). The CEP Group is using this
approach to look for abnormalities in bcl-2 and c-myc gene copy number in SCLC CTCs.
Using the CellScreen ISET platform, blood is filtered through membranes with 8µm pores. Leukocytes are
smaller than CTCs and the vast majority pass through the filter. A blood sample is collected in an EDTA
tube, diluted with buffer to fix the cells and then filtered under vacuum. The system has been shown to
isolate one cell per 1ml blood and successful gene amplification has been a achieved with as few as five
cells per filter. Cytological staining and immunohistochemistry (IHC) can then be performed and the cells
enumerated and characterised. It is also possible, with careful method development, to use the enriched
CTCs for nucleic acid extraction and subsequent genomic analysis and these are ongoing and future
developmental projects in the CEP Group.
Circulating tumour cells in lung cancer patients – a primary focus of the lung cancer disease focus
group in CEP
Lung cancer is a leading cause of cancer related deaths worldwide. Non-small cell lung carcinoma (NSCLC)
lacks validated biomarkers to predict or provide early reporting of treatment response. The CEP Group
investigated whether CTCs are detectable in patients with SCLC and NSCLC and whether they could provide
prognostic information and/or early indication of response to conventional therapy. A high number and
wide range of CTCs were detected using Veridex CellSearch™ in SCLC and the CTC number was both
prognostic and pharmacodynamic. Blood samples were assessed using the Veridex CellSearch™ system for
CTC analysis from 100 patients with previously untreated stage III/IV NSCLC, before and after administration
of one cycle of standard chemotherapy. The CTC number was higher in patients with more advanced stage
IV compared to stage III patients. In multivariate analysis, CTC number was the strongest predictor of overall
survival (OS). This study clearly demonstrated that CTCs are detectable in patients with stage IV NSCLC
where their number is a novel independent prognostic factor.
Lung Cancer Circulating Tumour Cells as Biomarkers
Figure 1. Characterisation ofCTCs. CTCs isolated by eitherthe CellSearch Technology orthe ISET platform were furthermolecularly characterised.Figure A shows a single CTCisolated from SCLC patientwith high nuclear/cytoplasmicratio, bigger cell size (the poreon the membrane is 8 µm indiameter) and irregularnuclear shape. Figure B showsSCLC CTM (black arrow)isolated by ISET and negativelystained for CD45 whereasleukocytes were positivelystained (white arrow). FigureC shows that CTCs detectedfrom SCLC patients can befurther characterisedregarding tumour specificmarker (Ci, CD56), potentialdrug target (Cii, Bcl-2), or drugresistant marker (Ciii, Mcl-1) bythe 4th channel of the VeridexCellSearch™ platform.Similarly SCLC associatedmarkers, NSE and TTF-1 werestained to further identify ISETisolated CTM and CTC (FigureD). Figure E illustrated thatBCL2 FISH analysis can detectBCL2 genetic abnormalitiessuch as amplification andtriploidy from SCLC CTCs.

21
division), transcription (the conversion of DNA into RNA in advance of protein
synthesis by translation) and mitosis (the segregation of chromosomes within
the cell nucleus into two identical daughter nuclei). Poly(ADP-ribose) (PAR) is
a highly negatively-charged polymer that is formed from repeating ADP-ribose
units linked via glycosidic ribose-ribose bonds, and is synthesised by the
poly(ADP-ribose) polymerase (PARP) family of enzymes using a vital cellular
cofactor NAD as a substrate (Figure 1). The recent development of potent PARP
inhibitors has provided powerful tools to study the pathways regulated by
poly(ADP-ribose), as well as providing a very promising class of drugs for
cancer treatment. Specifically, selective inhibition of the single-strand break
repair pathway using permeable PARP inhibitors has been proven to be highly
effective against breast and ovarian cancers. More recent data suggest that
the same inhibitors might be effective against several other cancer types as
well and PARP inhibitors have already entered phase III of clinical trials.
DNA Damage Response
20
DNA Damage Response
Manchester Cancer Research Centre Research Report
DNA is constantly exposed to damage and living organisms have evolved a
variety of DNA repair mechanisms to maintain genome stability. Inadequate
or abnormal DNA repair can cause diseases that in humans are associated
with cancer, neurodegeneration, immunodeficiency or developmental
abnormalities. Therefore, furthering our understanding of the molecular
pathways employed in the mammalian response to DNA damage potentially
provides a basis for the development of new therapies in the treatment of
human disease.
Many cancer therapy procedures, such as radiotherapy and some types of
chemotherapy, work by overwhelming the capacity of the cell to repair DNA
damage, resulting in cell death. Most rapidly dividing cells - cancer cells - are
preferentially affected by such treatments, providing the opportunity to use
DNA damaging agents to selectively kill cancer cells. In addition, the
development of cancer is underpinned by genomic instability and the
accumulation of multiple DNA mutations resulting in the loss of cellular
growth control. In order to accelerate the accumulation of these genetic
changes, cancers often sacrifice specific DNA repair pathways. This can make
cancer cells additionally susceptible to DNA damaging agents and/or to
inhibitors that block alternative repair pathways that allow cancers to thrive
without the full DNA repair repertoire. For these reasons, studying the protein
components involved in repair of damaged DNA has been proven a valuable
strategy in searching for novel approaches and targets in cancer therapy.
There are many pathways and signalling strategies for counteracting the
deleterious effect of DNA damage in humans. In our laboratory we are
particularly interested in studying the DNA repair pathways and protein
functions that are regulated by poly(ADP-ribosyl)ation. Poly(ADP-ribosyl)ation
is a post-translational protein modification employed by several important
nuclear processes including DNA repair, regulation of chromatin structure
(the combination of DNA, RNA, and protein that makes up chromosomes),
cell cycle checkpoint (control mechanisms that ensure the fidelity of cell
By Ivan Ahel
Figure 1. Regulation of DNA damage response by poly(ADP-ribosyl)ation.

22
Manchester Cancer Research Centre Research Report
The principal poly(ADP-ribose) polymerase involved in DNA repair is called PARP1. PARP1 is a DNA damage
sensor protein that specifically recognises breaks in DNA structure, including both single-strand and double-
strand DNA breaks. Upon binding to DNA, PARP1 catalytic activity is activated to produce vast amounts of
poly(ADP-ribose), which makes the DNA damage response a major source of cellular poly(ADP-ribose)
synthesis. The poly(ADP-ribose) produced by PARP1 is covalently attached to many target proteins including
PARP1 itself, histones and tumour suppressor protein p53, which allows the regulation of several different
aspects of the cellular response to DNA damage (Figure 1). Firstly, the local increase of poly(ADP-ribose)
arising at the sites of damaged DNA serves as a platform for the protein complexes involved in trimming
and sealing DNA breaks. In other words, many of the protein factors associated in such DNA repair factories
possess specialised protein modules to specifically bind to poly(ADP-ribose), which allows their timely
recruitment. Another consequence of signalling by PARP1 is the relaxation of chromatin structure via the
displacement of histones at the damaged area. This is particularly important as the chromatinised DNA is
tightly packaged due to it being wrapped around the histones, which in turn makes it refractory to efficient
repair. Finally, excessive poly(ADP-ribosyl)ation as a consequence of DNA damage beyond the cellular repair
capacity is a powerful signal for programmed cell death by apoptosis. Apoptotic signalling is critical for
complex organisms, as it prevents mutations and the development of cancer. Taken altogether, poly(ADP-
ribosyl)ation regulates several key events in DNA damage response and thus is essential for genomic
stability. However, many of the protein components involved and their mechanisms of regulation remain
unknown.
In our laboratory, we are routinely screening for novel proteins that have the ability to respond to DNA
damage in a manner that can be inhibited by treatment with PARP inhibitors. For this, we are using real-
23
time in vivo imaging by confocal microscopy, in combination
with a state-of-the-art laser system, that allows the infliction of
spatially controlled DNA damage to the cell nucleus and
subsequent analysis of the mobilisation of fluorescently
labelled proteins (Figure 2). Utilising this approach we have
recently discovered a novel structural element associated with
several DNA damage response factors which we have named a
poly(ADP-ribose)-binding zinc finger (PBZ) (Figure 3). PBZ is
distinctive of canonical DNA-binding zinc fingers and it is used
by proteins to recognise and bind to poly(ADP-ribose) with a
high affinity. One of the human proteins containing a PBZ
motif is a protein called Checkpoint protein with FHA and RING
domains (CHFR). CHFR is an ubiquitin ligase frequently
inactivated in human epithelial tumours, which acts as a key
modulator of the early mitotic checkpoint (this checkpoint
transiently delays mitosis in response to a variety of stresses). The elucidation of the function of the PBZ
motif gave us a vital clue to discover that the CHFR-dependent checkpoint is regulated by PARPs and that
the PBZ motif in the CHFR protein is critical for checkpoint activation. Another PBZ-regulated protein we are
studying is a protein called Aprataxin-PNK-like factor (APLF). APLF is constitutively associated with the DNA
repair ligases (enzymes responsible for sealing the breaks in DNA) and its down-regulation leads to an
apparent cellular sensitivity to various DNA damaging agents. However, the exact function of APLF in DNA
repair is presently unclear.
Another class of proteins that our research focusses on is macro-domain proteins. The macro-domain is
another module with the capacity to bind poly(ADP-ribose), and the aberrant regulation of several human
macro-domain containing proteins has already been linked to the development of cancer. One of these
proteins is a putative ATP-dependent helicase ALC1 (Amplified in Liver Cancer; also know as CHD1L), which
is frequently over-expressed in human hepatocellular carcinoma (HCC). The role of ALC1 in tumorigenesis
appears to be direct, as uncontrolled expression of ALC1 in mice leads to cancer development. In our recent
work, we provided the molecular evidence that ALC1 acts as a PARP1-regulated chromatin remodelling
enzyme in response to DNA damage and that its histone-repositioning activity is required for efficient DNA
repair. Strikingly, over-expression of ALC1 leads to deregulation of DNA damage signalling pathways and
results in a specific sensitivity to certain types of DNA-damaging drugs. These findings emphasise the
importance of chromatin reorganisation in DNA repair as a significant element in genome stability and
suggest a potential basis for developing a targeted therapy for ALC1-overexpressing tumours.
In conclusion, our research aims to better understand the basis of the response to DNA damage through the
identification of novel molecules that may play a role in this response. Exploring the regulation and function
of these molecules will allow us to establish whether and how they may impact cancer development. We
hope that our fundamental research approach will facilitate translational research via the identification of
potential molecular targets for the development of rational therapeutic interventions.
DNA Damage Response
Figure 2. Recruitment of fluorescently labelled ALC1 protein to the laser-induced DNA break sites in the cellnucleus. The recruitment is blocked by treatment of cells with the specific PARP inhibitor (lower panel).
Figure 3. The structure of the poly(ADP-ribose)-binding zinc finger

25
Projects
In order to make a valuable contribution to cancer drug discovery, we
deliberately work in project areas that are overlooked or currently considered
too risky by the major pharmaceutical companies. This will include for
example, segments of cancer with smaller patient numbers or novel biological
concepts in which the MCRC has world-leading expertise. During late 2009
we presented our cancer drug discovery target “roadshow” to many groups
of scientists and clinicians in the Paterson Institute, The Christie Hospital and
The University of Manchester. These presentations have been followed up
with more detailed discussions and this has allowed us to identify and
prioritise our first drug discovery projects. These projects are focused around
modulation of specific target molecules, in cells or tissues that we believe to
be involved in driving malignant tumour growth and progression. Target
review will be an ongoing activity so that we can keep abreast of new
developments in cancer science and fuel the drug discovery “pipeline” with
the best opportunities.
Once a target is chosen, the aim of drug discovery is to identify and optimise
chemical agents that selectively interfere with the target activity in order to
kill or restrain the tumour cells, without unacceptable effects on normal
tissues. This is an iterative process involving the identification of initial
chemical “hits”, the exploration of their drug potential to create “leads” and
then the optimisation of these leads to create a clinical candidate for testing
in cancer patients. There are several approaches to hit identification which
include screening of existing compound collections and generating and
exploiting detailed structural information on the target. Selection of the final
clinical candidate chemical compound includes optimisation of not just target
modulation but also selectivity, to avoid unwanted side effects, and delivery
to the required site(s) of action in the body. During 2010 we initiated four
MCRC hit identification projects and also engaged in successful high
throughput screening and lead identification collaborative projects with the
CRT Development Laboratory in London
Drug Discovery in the Manchester Cancer Research Centre
24
Drug Discovery in theManchester Cancer Research Centre
Manchester Cancer Research Centre Research Report
In 2009, a £9 million Cancer Research UK five-year programme grant was
awarded to the Paterson Institute for Cancer Research to build a cancer Drug
Discovery Unit in the MCRC. This new initiative arose from a strategy review
in which Cancer Research UK decided to increase significantly their long term
investment in small molecule drug discovery and to align this additional
resource with the core-funded cancer research institutes in Glasgow (Beatson
Institute for Cancer Research) and Manchester (Paterson Institute). The
purpose of co-locating these activities is to maximise the opportunity for
translating the ground-breaking cancer research from these centres of
excellence into novel therapeutic opportunities.
The ultimate aim of the MCRC Drug Discovery Unit is to identify novel drug
therapies to satisfy some of the many unmet clinical needs of cancer patients.
However, drug discovery and clinical development are long and complex
processes and in order to achieve this goal we will need to capitalise on the
outstanding opportunities afforded by the MCRC environment.
During 2009-10, the foundations of the MCRC Drug Discovery Unit, including
strategy, facilities and key recruitment, were laid and the first drug discovery
projects were started.
At the outset, strategic considerations included: 1) What kind of drug
discovery projects should we prosecute in order to make a valuable, unique
contribution to the global war against cancer?, 2) What kind of drug discovery
skills should we focus our limited resources on?, 3) What kind of laboratory
facilities do we need to support these activities? and 4) What kinds of
partnerships, within and beyond the MCRC, do we need to complement our
in-house capabilities in order to achieve our long term objectives?
By Donald Ogilvie and Allan Jordan

27
Partnerships
The first key partner is of course Cancer Research UK who are providing the crucial funding - £9 million for
the first five years. But Cancer Research UK is more than just a source of funding for this new venture. As
well as individual programme grants, Cancer Research UK already supports major Drug Discovery Units in
London, Sutton and Newcastle providing a broad portfolio of projects. The new Units in Manchester and
Glasgow are seeking to complement one another in extending this portfolio into new areas of breaking
cancer science and drug discovery technology. The leaders of these Drug Discovery Units meet regularly to
share expertise, coordinate their activities and identify areas of cooperation and collaboration in order to
maximise the effectiveness of Cancer Research UK drug discovery. As one example of this cooperation, we
are accessing the compound collection and screening technology in the London Unit (CRT-DL) to support our
hit identification projects. Another important part of the Cancer Research UK “family” is Cancer Research
Technology (CRT) which provides us with intellectual property and business development support. These
activities are particularly important for facilitating collaborations, the protection of drug discovery
inventions and, in the longer term, for identification of partners to take our candidate drugs into clinical
trials.
MCRC
Our location in the Paterson Institute, at the heart of the MCRC, is ideal for accessing clinical insight, basic
research expertise and world-leading clinical development technologies and experience. A key component
of the MCRC is the breadth of clinical expertise at The Christie NHS Foundation Trust. This provides direct
insight into the areas of unmet clinical need and the hypotheses to address them but also brings a tangible
connection with our ultimate customer, the cancer patient. At the other end of the MCRC spectrum are the
basic scientists in the Paterson Institute and more broadly in The University of Manchester who provide
insights into the mechanisms of cancer and how to measure these in preclinical models. In the middle are
the translational scientists and clinicians, particularly in the Clinical and Experimental Pharmacology Group
at the Paterson Institute, who provide the roadmap for initial clinical development, particularly in the
validation of novel biomarkers. We are also exploring opportunities to access other key technologies (for
example biophysical chemistry, biochemistry and protein structural analysis) through experts in The
University of Manchester.
Since drug discovery and development takes such a long time (10+ years) and many projects do not progress
to clinical trials we need to need to be able to demonstrate that we are making progress in the shorter term.
In the first five years, this will be primarily through the generation of a unique (within Cancer Research UK)
portfolio of attractive drug discovery projects.
During the first two years of our operation we have laid the foundations of the new MCRC Drug Discovery
Unit and have initiated and advanced our own portfolio of novel drug discovery projects. During the next
period we aim to enhance and progress this portfolio towards the goal of clinical testing of innovative cancer
medicines.
Drug Discovery in the Manchester Cancer Research Centre
26
Manchester Cancer Research Centre Research Report
Skills
Our core expertise is in the areas of cancer drug target selection and drug discovery biology and chemistry
and this focus has been reflected in our recruitment strategy. During the summer of 2009, Allan Jordan
joined the team as Head of Chemistry. Towards the end of the same year, a major campaign was successfully
carried out to recruit a further seven scientists (medicinal/synthetic chemists and biologists) with
pharmaceutical industry experience to kick start the laboratory work from early 2010. All other kinds of
expertise, including clinical insight, biomarkers and other bioscience technologies are being accessed
through collaborations, partnerships and outsourcing as appropriate (see below). Towards the end of 2010
a second wave of recruitment was initiated to expand the team and broaden our skills base to include
computational chemistry and drug metabolism expertise.
Facilities
The major activity during 2009 was the design and construction of a modern drug discovery laboratory.
With the help of external advisers, the Paterson Institute estates team and an excellent building contractor,
the new laboratory was designed and completed on time and handed over in December 2009. An unusual
feature of the design is the co-location of biology and chemistry facilities. This was a deliberate decision to
ensure close interdisciplinary interactions in the design, synthesis and testing of new compounds. Parallel
procurement of key capital equipment ensured that the new laboratory was able to open for work in January
2010. A major project during 2010 has been the construction and optimisation of the informatics
infrastructure including Virtual Screening, Electronic Notebooks and the Dotmatics chemoinformatics
platform. The latter enables tracking of the complete data workflow from novel compound design and
synthesis to biological testing.

29
(discriminatory power). Currently the lifetime risk for a woman in the UK of
developing breast cancer is between 9 and 11%; if she has no family history of
breast cancer the risk is lower at around 8%. Three high risk genes have been
identified to date which predispose to breast cancer in women - TP53, BRCA1
and BRCA2 with the following risks:
• TP53 – 1990, lifetime risk 90%
• BRCA1 – 1994, lifetime risk 60-85%
• BRCA2 – 1995, lifetime risk 50-85%
There are two main types of risk assessment; 1) the chances of developing
breast cancer over a given time-span including lifetime and 2) the chances of
there being a mutation in a known high-risk gene such as BRCA1 or BRCA2.
Whilst some risk assessment models are aimed primarily at solving one of
the questions many also have an output for the other. For instance a
computer algorithm BRCAPRO is primarily aimed at assessing the mutation
probability but can have an output to assess breast cancer risk over time. The
Cuzick-Tyrer model was developed to assess breast cancer risk over time but
does have a read out for BRCA1/2 probability for the individual. In order to
assess breast cancer risks over time as accurately as possible all known risk
factors for breast cancer need to be assessed.
Risk factors
Family history of breast cancer in relatives
• Age at onset of breast cancer
• Bilateral disease
• Degree of relationship (first – for example mother, sister
or greater – for example aunt, cousin)
• Multiple cases in the family (particularly on one side,
for example the maternal lineage)
Genetic Medicine and the Genesis Prevention Centre
28
Genetic Medicine and the Genesis Prevention Centre
Manchester Cancer Research Centre Research Report
The main focus of cancer research in Genetic Medicine has been on genetic
predisposition to breast cancer and neurofibromatosis. Whilst the hunt
continues for a fourth high risk breast cancer gene, genome-wide association
studies, to which we have been a major contributor, continue to find common
genetic variants that enhance risk of breast, colorectal and other cancers. We
have been using these variants to assess variation of risk amongst different
populations. Through an NIHR programme grant we are collecting DNA
samples in the high risk clinic as well as from a group of 60,000 women being
recruited from the National Breast Screening Programme.
Breast Cancer
The authors have published over 200 papers on inherited predisposition to
and aetiology of breast cancer in the last 15 years.
Risk prediction
Breast cancer is diagnosed in 44,000 women and causes 12,000 deaths per
year in the UK. Although breast cancer deaths have decreased in many
Western countries, the incidence of the disease is continuing to rise. In
particular in countries with historically low incidence, breast cancer rates are
rising rapidly making it the world’s most prevalent cancer. The increase in
incidence is almost certainly related to dietary and reproductive patterns
associated with Western lifestyles. Indeed there is evidence from genetic
studies in the US, Iceland and UK of a 3-fold increased incidence in the general
population and also in those at the highest level of risk with BRCA1/2
mutations in the past 80 years. Understandably there is increasing interest
in disease prevention to spare women the trauma of diagnosis and
increasingly aggressive treatment. There is a need, not only to predict which
women will develop the disease, but also to apply drug and lifestyle measures
in order to prevent the disease. Current risk prediction models based on
combinations of risk factors have good overall predictive power, but are still
weak at predicting which particular women will develop the disease
By D Gareth Evans and Anthony Howell
31
Genetic Medicine and the Genesis Prevention Centre
30
Manchester Cancer Research Centre Research Report
• Other related early onset tumours (for example ovary, sarcoma)
• Number of unaffected individuals (large families with many unaffected relatives will be less likely
to harbour a high-risk gene mutation)
Hormonal and reproductive risk factors
Hormonal and reproductive factors have long been recognised to be important in the development of breast
cancer. Prolonged exposure to endogenous oestrogens is an adverse risk factor for breast cancer. Early
menarche and late menopause increase breast cancer risk as they prolong exposure to oestrogen and
progesterone.
Long-term combined HRT treatment (>5 years) after the menopause is associated with a significant increase
in risk. However, shorter treatments may still be associated with risk to those with a family history. The risk
from oestrogen-only HRT appears much less and may be risk-neutral. A meta-analysis also suggested that
both during current use of the Combined Oral Contraceptive and 10 years post use there may be a 24%
increase in risk of breast cancer.
The age at first pregnancy influences the relative risk of breast cancer as pregnancy transforms breast
parenchymal cells into a more stable state, potentially resulting in less proliferation in the second half of the
menstrual cycle. As a result, early first pregnancy offers some protection, whilst women having their first
child over the age of 30 have double the risk of women delivering their first child under the age of 20 years
and these are likely to be similar in those at highest risk from a BRCA1/2 mutation.
Other risk factors
A number of other risk factors for breast cancer are being further validated. Obesity, diet and exercise are
probably interlinked. Mammographic density is perhaps the single largest risk factor that is assessable but
may have a substantial heritable component. Other risk factors such as alcohol intake have a fairly small
effect and protective factors such as breast-feeding are also of small effect unless a number of years of
total feeding have taken place. None of these factors are currently incorporated into available risk
assessment models
Risk factors included in current models
Current risk prediction models are based on combinations of risk factors and have good overall predictive
power, but are still weak at predicting which particular women will develop the disease. New risk prediction
methods are likely to come from examination of a range of high-risk genes as well as single nucleotide
polymorphisms (SNPs) in several genes associated with lower risks. These data would be married in a
prediction programme with other known risk factors to provide a far more accurate individual prediction.
-
Predicting risk of breast cancer at screening (PRoCAS)
PRoCAS is part of a £1.59 million programme grant from NIHR which started in 2009. The overarching aims
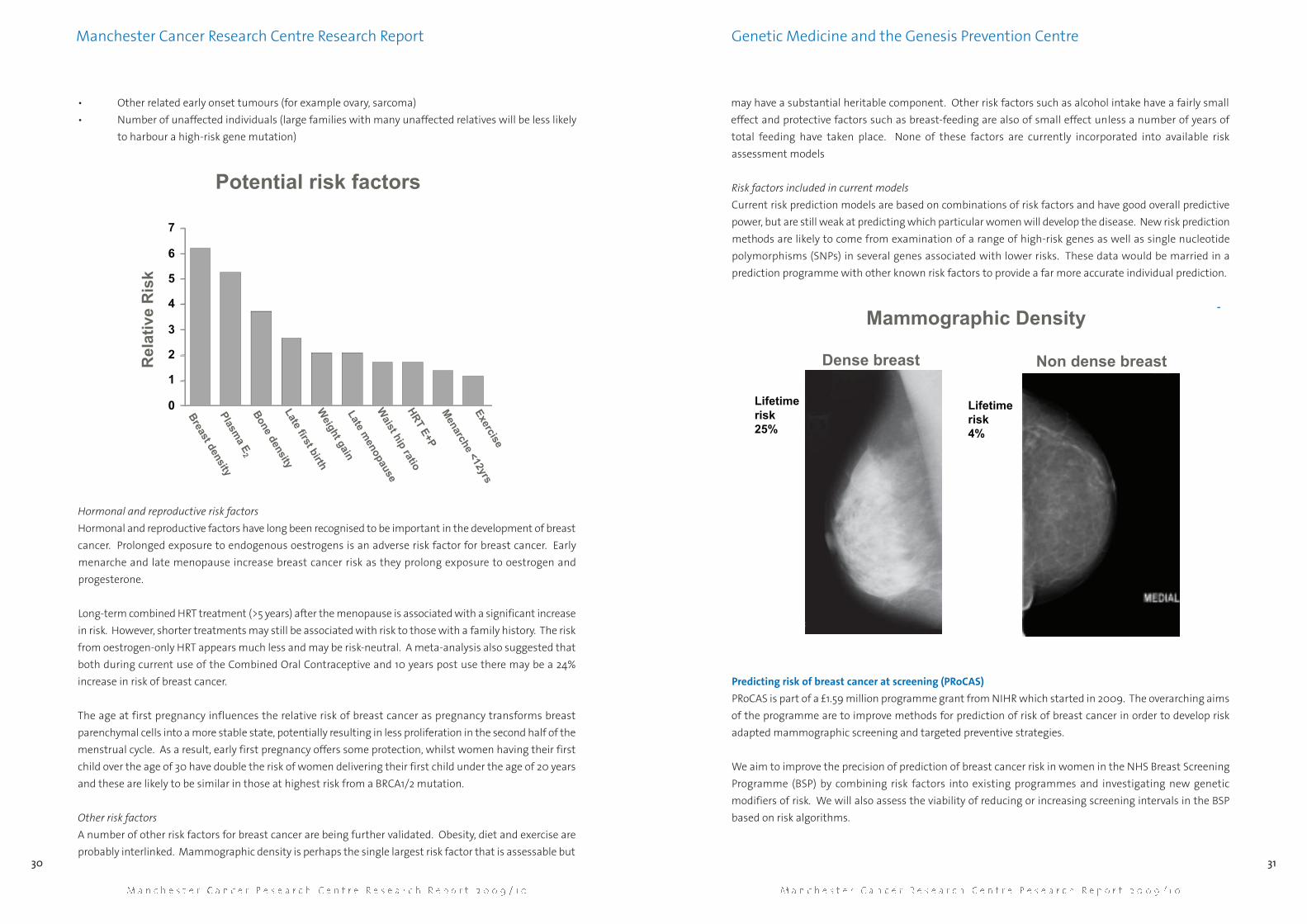
of the programme are to improve methods for prediction of risk of breast cancer in order to develop risk
adapted mammographic screening and targeted preventive strategies.
We aim to improve the precision of prediction of breast cancer risk in women in the NHS Breast Screening
Programme (BSP) by combining risk factors into existing programmes and investigating new genetic
modifiers of risk. We will also assess the viability of reducing or increasing screening intervals in the BSP
based on risk algorithms.
0
1
2
3
4
5
6
7
Breast density
Weight gain
Plasma E
2
Late first birth
Bone density
Late menopause
Waist hip ratio
HRT E+PM
enarche <12yrsExercise
Rel
ativ
e R
isk
Potential risk factors
Lifetimerisk25%
Lifetimerisk4%
Dense breast Non dense breast
Mammographic Density

33
Genetic Medicine and the Genesis Prevention Centre
32
Manchester Cancer Research Centre Research Report
Research plan
New risk prediction methods will be developed from examination of a range of SNPs for high-risk as well
as those associated with lower risks. This will be married in a prediction program with other known risk
factors to provide greatly improved discriminatory power and accuracy of individual risk prediction. We will
include information on mammographic density which is not yet part of a risk prediction model despite
being a greater risk factor than most factors currently included. Risk assessment of women in the BSP will
determine appropriate screening intervals, or if they are at sufficient risk for BSP screening with huge
potential savings to the NHS from more focused use of resources. Importantly for a group of 10,000 women
we are also going to use DNA extracted from a saliva sample to help predict risk. We anticipate the
development of highly predictive algorithms for risk prediction within five years and to have developed
validated prevention programmes appropriately targeted at risk.
Environment
The programme is based in a new £14m breast cancer treatment and prevention centre, The Nightingale
Centre & Genesis Prevention Centre at the University Hospital of South Manchester NHS Foundation Trust,
and builds on development work from ourselves. This is the largest family history clinic and screening
population in the UK. Dissemination to the network will be through our links to Greater Manchester Clinical
Research Network and BSP.
FHrisk protocol
By studying 3,170 women who were unaffected at the time of assessment we previously showed that the
Tyrer-Cuzick model was most accurate at predicting which women would develop the 84 breast cancers
that occurred. This remains the only such validation in the family history clinic (FHC) setting worldwide. We
have now expanded our FHC set to over 8,900 and are already aware of 315 breast cancers that have
occurred. By matching details against the North West Cancer Intelligence Service on 1 December 2009 we
will obtain an accurate assessment of cancer status and predict that a total of 350 breast cancers will have
occurred by that date. Whilst models have good overall predictive power, they are still less good at predicting
which particular women will develop the disease. We will therefore revalidate the original models alongside
a new model - BOADICEA - in this expanded FHC set. We already have risk information on extended family
history, age at menarche, hormonal treatments, age at menopause, and previous breast disease to enter into
the various risk programmes. We will then use information on breast density from mammography and
body mass index (BMI) to improve the predictive value of the best model(s). Data from genetic testing will
also be included with BRCA1/2 information already known on close to 3,200 women. We will obtain DNA
from blood samples on three controls for every breast cancer patient and validate the new genetic variants
being identified. The expected improvements in risk prediction for women will enable more appropriate
targeting of screening and preventive measures.
Neurofibromatosis type 2 (NF2)
NF2 affects the skin and nervous system (including the brain) and is characterised by skin lesions and benign
tumours – neurofibromas – which most commonly occur on the acoustic nerves leading to symptoms
including deafness. Between 1990-92 we carried out the original large clinical and genetic study of NF2. This
research was the definitive description of the epidemiology and genetics of the condition. We were the
first to describe the high level of mosaicism in NF2 and the original evidence for a genotype-phenotype
correlation. We have also published the definitive paper on mortality in NF2, which showed worse survival
in non-specialist units. We have now secured a national bid from the National Commissioning Group for
the management of type 2 neurofibromatosis for £7.1 million annually, which starts in April 2010. We have
published over 80 articles on NF2 that show the importance of multidisciplinary management and these
have shown a survival advantage for patients so managed. This led to a consensus conference at Old Trafford
in 2004, which we organised and chaired, and lead to the publication of national consensus guidelines.
Invitation letter sent
Consent taken & questionnaire completed
Mammogram 1 performed
Initial risk calculation (Tyrer-Cuzick)
Mammogram 2 performed
Breast density results, questionnaire results & DNA results(if applicable) combined to give re-adjusted risk score
OPTIONAL - DNA sample collected (10,000/60,000)
High risk & selection of low risk women informed of risk(if opted to receive risk information)
Flowchart

35
complexity at the DNA level than other organisms. The resolution of this
paradox has come from studies of how the DNA is expressed, and how the
products of gene expression (RNA and proteins) are produced, regulated, and
processed.
The early view of a gene was of a single contiguous piece of DNA that was
transcribed into a messenger RNA (mRNA) intermediate, which in turn was
translated into a functional protein. This simple view has, over the past 40
years, transpired to be immensely complex. A typical gene is now known to
be split into units called exons - sections of the gene which ultimately are
Cancer Genomics in the Era of High Throughput Sequencing
34
Manchester Cancer Research Centre Research Report
Cancer Genomics in the Era ofHigh Throughput Sequencing
Manchester Cancer Research Centre Annual Research Report
When the first working draft sequence of the human genome was published
in 2000, it was the culmination of a massive international effort that some
have likened to that of putting a man on the moon. The Human Genome
Project took thirteen years to complete at a cost of more than of $3billion.
Now, under ten years later, advances in technology are making it possible to
assemble similar amounts of raw sequence in a few weeks using a single
machine. The relatively low cost - currently under $10,000 per genome -
makes it feasible to use the technology for many purposes, ranging from
sequencing the complete genomes of individuals, through characterising
panels of human tumours to detect common mutations, to analysing all the
regions of the genome that are actively transcribed. A variety of competing
technologies exist, each with their own strengths and weaknesses. Within
the Molecular Biology Core Facility (MBCF) at the Paterson Institute for Cancer
Research, we have recently taken delivery of an AB SOLiD™ machine that is
capable of generating over a quarter of a billion residues of high quality
sequence a week. This report will highlight some of the current applications
of this new high throughput sequencing technology (HTS), as well as
describing other platforms that have been established within the Paterson
Institute and which have been widely used within the MCRC.
Each human cell contains 23 pairs of chromosomes comprising 3 billion base
pairs of DNA. Within this DNA are all the determinants for human
development, ageing and, potentially, disease. Until the entire sequence of
the human genome was produced, we had no idea of the exact number of
human genes, and in many cases no clue about their function. Surprisingly,
man was found to have similar numbers of genes to many apparently simpler
organisms such as flies and worms; paradoxically, a very complex and
intelligent organism appeared to have no significantly greater genetic
By Crispin Miller, Jenny Varley and Stuart Pepper
Exon 1 Exon2
Exon 3 Exon 4
Intron 1 Intron 2 Intron 3
Transcription
Alternative Splicing
Translation
Post-translational modification
Promoter
Figure 1. Cartoon showing a simplified structure of a typical gene. At the top of the figure is a stylisedrepresentation of a gene showing the position of a promoter where transcription into the precursor RNA starts,four exons and three introns. After transcription into the pre-RNA introns may be alternatively spliced; in thiscartoon two alternatives are shown. Upon translation two different but related proteins are produced,differing due to the presence or absence of exon 2 material in the transcripts. Finally the proteins may bemodified post-translationally as indicated at the bottom of the figure. In this highly simplified cartoon threedifferent protein products are produced from a single gene.

37
when considering rare types of tumours. The use of FFPE
material, some of which may be many years old, also
allows us to study gene expression in tumours for which
there is information on patient clinical outcome, after
many years’ follow up (Figure 2).
One major use of genomic technologies is the analysis of
DNA from multiple samples, supporting searches for the
polymorphisms and mutations that underpin diseases
such as cancer. There is considerable variability in the
DNA sequence between different individuals
(polymorphisms), and this variability in key regions of the
genome may predispose to diseases such as cancer, as
well as others such as heart disease, diabetes,
neurological and ageing disorders. Not only may normal
variations in our DNA predispose to cancer (for example
by altering the way our cells deal with carcinogenic
agents such as those found in tobacco smoke), but mutations in specific genes within our tissues may be
responsible for the development or progression of a cancer. In addition, many tumours are characterised
by the amplification of some genes and the loss of others, leading to their over- or under-expression. There
is therefore a strong impetus to determine as much as we can about the normal DNA from cancer-prone
individuals as well as investigating, in an unbiased fashion, what genetic changes are occurring in the actual
cancer cells. We can currently carry out this analysis using one of two platforms. Firstly, we can interrogate
Affymetrix SNP GeneChips® in which nearly one million single nucleotide polymorphisms (SNPs) and the
same number of copy number variants (CNVs) are arrayed. Although GeneChips are a very powerful tool for
DNA and expression analysis, the investigations are limited to those regions of the genome represented by
probes on these commercially available arrays. The second platform that can be used to analyse SNPs and
CNVs is the HTS platform which provides unbiased information by analysing the entire genome of individual
tissue or tumour samples.
However, it would be wrong to focus solely on DNA-level analyses when considering the impact of HTS
technology. A consequence of the sheer amount of data it can generate is that it can be used in a wide
variety of assays that require massively parallel DNA or RNA identification. For example, in RNA-Seq studies,
messenger RNA is extracted from a sample, sequenced, and aligned to the genome, before counting the
number of sequences that match to the location of each gene. The result is a global picture of gene
expression.
Cancer Genomics in the Era of High Throughput Sequencing
36
Manchester Cancer Research Centre Research Report
translated and transcribed into the mature protein - separated by introns, which are removed to produce the
mature mRNA transcript, and are therefore not translated. Furthermore, after the gene is translated into a
precursor RNA molecule, there may be multiple exons that can be joined (spliced) in a variety of different
ways, a process known as alternative splicing (Figure 1). Alternative splicing is therefore the mechanism by
which cells can remove different sections of an RNA molecule in a highly coordinated and tightly-regulated
fashion, allowing the expression of a series of distinct, but related, proteins from a single gene. Originally
discovered in the 1970s and initially viewed as a rare event, alternative splicing is now known to affect the
majority of human genes. It allows cells to substantially increase their repertoire of available proteins, and
helps to explain why man has a similar number of genes in comparison to other, apparently simpler,
organisms. Not surprisingly given the genetic basis of cancer, patterns of splicing are markedly different
between tumours and normal cells, and can be indicative of, for example, patient prognosis.
We have recently been using Affymetrix GeneChip® Exon Arrays to identify patterns of alternative splicing.
These arrays feature probesets that target every known and predicted human exon in the genome. One
study that has been carried out as a collaboration between Doctor Crispin Miller (Applied Computational
Biology and Bioinformatics (ACBB) Group at the Paterson Institute) and Professors Catharine West
(Translational Radiobiology Group within the School of Cancer and Enabling Sciences at the University of
Manchester) and Adrian Harris (Weatherall Institute for Mollecullar Medicine at the University of Oxford)
has identified a set of characteristic splicing events in head and neck tumours that could predict for overall
survival.
Since alternative splicing involves removing sections of RNA before joining the remaining fragments
together into a mature mRNA molecule, different, and novel, splice variants can be found in sequencing
data by looking for reads that span the junctions between fragments. The application of direct sequencing
has the potential to give us a completely unbiased picture of all splicing events in a tissue or tumour. This
means that sequencing can be used to track how splicing patterns change between samples, allowing a
systematic exploration of both the functional and predictive consequences of these fine-grained changes
in protein expression.
We have been carrying out gene expression profiling studies in the Paterson Institute for several years now.
This work has been undertaken collaboratively between individual research groups, the MBCF and the ACBB
Group. The MCRC report in 2008 outlined two such studies that demonstrated the power of this technology
for identifying a) gene expression signatures associated with tumour hypoxia and b) for analysis of gene
expression in formalin-fixed paraffin-embedded (FFPE) material. Using the Affymetrix GeneChip® system
we can interrogate samples to determine which of the tens of thousands of human genes are active (or
inactive) in any tissue type. The ability to be able to profile gene expression in FFPE samples is particularly
relevant for clinical studies as large archives of FFPE material are available, and are especially important
Figure 2. Differentially expressed genes identified fromformalin fixed paraffin embedded (FFPE) tissue using
Affymetrix exon arrays. Each row corresponds to a mRNAtarget, each column, a tumour sample. Data are coloured by
expression level. Using this technology we are able to identifystatistically significant changes in expression. Ongoing work is
exploring the potential of these transcripts as biomarkers.

39
Discussion so far has concentrated on the analysis of DNA and RNA, but within the MCRC we have very
powerful biological mass spectrometry capabilities, commonly referred to as proteomics. Although, as
described above, a variety of different protein products can be produced from a single gene due to
alternative splicing; once any protein is produced it can be modified in a wide variety of different ways, all
of which may markedly alter its activity, specificity or function. Within the Biological Mass Spectrometry
Facility at the Paterson Institute we can carry out a variety of different analyses ranging from straightforward
protein identification, global quantitative comparison of protein expression in different tumour or tissue
types through to detailed analysis of post-translational modifications, either of specific proteins or more
global analysis.
Arguably, all these techniques become most powerful when they are used together in repeated rounds of
experimentation, where novel genes and binding sites are discovered, cells are perturbed to disrupt these
loci, and further rounds of sequencing or expression analysis performed in order to investigate the changes
that result. When the ability to be able to investigate protein expression and modification is placed
alongside technologies such as HTS and microarrays, it becomes possible to generate integrated views of
gene expression that encompass DNA, RNA and protein. A major goal therefore is to provide the close
cooperation and experimental integration required to support these complex, multi-platform experiments.
Within the MCRC we have access to a range of cutting edge technologies that will find increasing
application in the analysis of tumour initiation, progression, prognosis and response to therapies. In the
future the application of exomics - the analysis of all expressed sequences in many different tumours - will
provide a valuable picture of potential targets for therapy, and ultimately will help us to target genes and
their products selectively, eventually leading to the Holy Grail of personalised medicine.
Cancer Genomics in the Era of High Throughput Sequencing
38
Manchester Cancer Research Centre Research Report
Exactly how this is done depends on the platform, but with the majority of technologies, a single machine
run can generate many millions of relatively short (10s-100s of nucleotides) sequences, known as reads.
The volume of information that results from this places significant demands on the downstream computing
resources needed to analyse and store HTS experiments, and impacts both on the computing hardware
needed simply to manage the data, and the computational biology required to provide access to
appropriately detailed genome annotation and robust statistics (Figure 3). The ACBB Group at the Paterson
Institute is a highly collaborative mix of scientists from both the numeric- and the biological-sciences. The
majority of the group’s work involves collaborations with both basic and translational scientists within the
MCRC, and it is developing a variety of approaches for analysing HTS data.
Unlike other genome-wide technologies such as microarrays, RNA-Seq has the potential to offer true
nucleotide-level precision whilst generating profiles across the entire genome. This is important because
even with rapid progress in sequencing and analysis, we have yet to fully characterise the human genome.
In part this is because only about 2% of the 3 billion residues in the genome actually code for proteins - the
rest have, until recently, been considered to be relatively inert. However, using technologies including
sequencing and microarrays, many groups have been able to show that the majority of the genome is
expressed, at least at some stage, in some cells. This has led to an increased focus on the 98% of the genome
that does not code for proteins, and has revealed whole new classes of regulatory RNAs that, whilst they do
not originate from protein coding genes, are functional in their own right. RNA-Seq offers a remarkably
straightforward way to identify these genes, and then to explore how their pattern of expression changes
across, for example, normal healthy tissue and tumour cells. The ACBB Group is collaborating with a number
of groups in the search for novel non-coding RNAs with relevance to cancer, much of it driven by the
emerging regulatory role of these molecules.
As well as studying the expression patterns of different genes and splice variants, it is also important to
understand the processes that control these changes. Transcription factors are sets of proteins that bind
to DNA and help regulate the expression of sets of genes. Knowing which transcription factors are bound
to the regulatory region of a given gene can is therefore a key factor when developing an understanding of
the factors that govern its behaviour. ChIP-Seq is a technique in which individual transcription factors are
purified from a cell extract along with the section of DNA to which they are bound. This DNA can then be
released from the protein, sequenced, and aligned to the genome, revealing the locations where the
transcription factor was bound.
Although not yet routine, HTS is already becoming a key platform within the MCRC. Since its strength arises
from its ability to perform a relatively generic task – the efficient sequencing of millions of short reads – it
has a wide variety of applications, and a corresponding potential to benefit the work of many different
groups within the MCRC. Figure 3. RNA-Seq data visualised in the X:Map genome browser developed at the Paterson Institute. In the toppanel, peaks correspond to the number of reads aligning to the genome at that point. Data are associated withannotation representing the location and structure of known genes. The bottom panels provide more detailedannotation, and hyperlinks to other genome annotation resources.

41
four research-dedicated PET cameras: preclinical high-resolution HIDAC PET
and Siemen’s InVeon PET/CT systems for animal studies, a PET/CT whole body
scanner and a high resolution brain camera, which is the only one of its kind
in the UK. Clinical researchers also have access to the University’s research-
dedicated 1.5T MR system at the Wellcome Trust Clinical Research Facility
(WTCRF) and the 3.0T MR system situated at Hope Hospital. In addition, many
research studies are conducted in The Christie clinical imaging facilities. For
animal studies, a 7T preclinical MR is located in the Stopford Building.
Development of the MCRC imaging strategy has been supported by provision
of new imaging equipment and support posts. The Siemen’s InVeon is a state-
of-the-art preclinical PET/CT and was commissioned for use in December 2010.
This will run alongside the HIDAC system, but gives researchers access to CT-
capability, a small animal imaging modality that was previously unavailable
within The University of Manchester. Similarly, the 7T preclinical MR scanner
at the Stopford Building has been upgraded to give internationally
competitive MR capability in preclinical research. For clinical studies a
research dedicated 1.5T whole body MRI scanner has been installed at the
WMIC. These infrastructure changes have occurred in parallel with the
recruitment of three MCRC-funded research associates to specifically drive
forward research in preclinical and clinical MRI and PET. The University has
further supported appointment of two additional research associates to
support development of novel PET-tracers for preclinical application and
analysis of PET data from cancer studies.
Preclinical Imaging Research
Exciting progress has been made with preclinical whole animal imaging. The
key focus of current research programmes is the evaluation of cell population
dynamics (in particular cell proliferation and cell death) and the tumour
microenvironment. PET-based tracers of prolif-eration (18-F-FLT) and
metabolism (18-F-FDG) are being utilised as biomarkers for targeted therapies
(Figures 1 and 2) and also in genetic models where growth arrest is triggered
Imaging Research at the Manchester Cancer Research Centre
40
Imaging Research at the Manchester Cancer Research Centre
Manchester Cancer Research Centre Research Report
Ensuring the MCRC develops and implements a plan that strategically
integrates imaging science across the spectrum of its research is an
important part of establishing the MCRC on an international stage.
Advanced imaging methods play a core role in cancer research, early drug
discovery and increasingly in clinical management. The last 18 months have
seen pivotal changes in Imaging Research at the MCRC with the
implementation of an extensive plan to exploit the expertise in imaging
research and technology of MCRC partner groups within Manchester. These
developments are being coordinated by the newly formed MCRC Imaging
Group.
Under the umbrella of the MCRC Imaging Group, three inter-linked research
themes have been identified and form the basis for strategic developments.
The first focuses on research within imaging science itself, particularly the
development and validation of imaging biomarkers. This grows on the
established strengths and international reputation of Manchester in this area
and is co-ordinated by the MCRC Imaging Group. The second theme is the
strategic and co-ordinated development of preclinical imaging which has
been bolstered by the appointment of Dr Kaye Williams as preclinical lead,
upgrading of dedicated facilities and of the formation of the PReclinical
Imaging Executive (PRIME) to facilitate the development and implementation
of research projects and oversee all modalities of preclinical imaging. The
third theme focuses on translational clinical research with the establishment
of a Translational Research Imaging Group (TRIG). This group will drive
collaborative research and include clinicians and radiobiologists at The
Christie NHS Foundation Trust, the North West Medical Physics team, and
researchers at The University of Manchester.
The imaging research group already has access to high quality, well
established imaging facilities. The Wolfson Molecular Imaging Centre
(WMIC), situated on The Christie campus has its own cyclotron and dedicated
good manufacturing practice (GMP) radiochemistry facilities. The unit has
By Alan Jackson and Kaye Williams
Figure 1. Static PET images of mice with coloniccarcinoma heterografts showing differential
uptake of FDG (reflecting metabolism) and FLT(reflecting cellular proliferation).
Figure 2. Metastatic lung diseasedemonstrated by uptake of FDG (left). Images
on the right show the protective effect of thebioreductive agent NLCQ-1. Metastases can be
demonstrated using PET before anydetrimental effects on the animal are observed
(Cawthorne et al, Br J Cancer, 103, 201-208,2010).

biomarkers can expand. The activity of the TRIG has led to active
projects in brain, lung, breast, ovarian, cervical, colorectal, pancreatic,
renal, prostate, bladder, lymphoproliferative and hepatobiliary
cancers. Many of the studies are multimodality combining
advanced PET and MRI.
One example is an ongoing imaging-based study of Bevacizumab
(Avastin) in patients with metastatic colorectal cancer. This large
prospective clinical study includes over 70 patients each of whom
will have advanced MRI on five separate occasions. The study will
use qualitative biomarkers of the microvascular environment within
the tumour derived from dynamic contrast enhanced MRI. These
methods allow us to produce quantitative images of physiological
parameters including regional blood volume and endothelial
permeability surface area product. We have shown in a previous
study that these biomarkers are sensitive early indicators of
biological effectiveness with this agent (Figure 6). In the present
study we intend to test the hypothesis that early drug-induced
changes in these biomarkers will allow selection of patients who are
likely to show beneficial response. A subgroup of patients will also
have 18-FLT PET studies to measure the effects on tumour cell
proliferation. This powerful multimodality approach allows us to
examine the interactions between the primary anti-angiogenic
effect of the drug and the resultant change, if any, in proliferative
activity.
Another example is the increasing use of advanced PET ligands to
explore tumour biology in glioma. Manchester has a long history of
glioma imaging research based almost entirely around magnetic
resonance (Figure 7). Considerable biomarker development and
validation has occurred in this tumour group with a main focus on
biomarkers of the vascular micro-environment. Several new studies
are combining these advanced MR biomarkers with mechanistically
specific PET studies. These include the use of labelled methionine
(Figure 8) to identify tumour extent and recurrence, PK11195 (a
marker of microglial activation) to study the tumoural and peri-
tumoural inflammatory response in low grade glioma undergoing
dedifferentiation (Figure 9) and the use of 11C-verapamil to measure
the effect of PgP transporter blockade on drug uptake by
glioblastoma (Figure 10).
Imaging Biomarker Research
The development and validation of novel imaging biomarkers is the
core of a successful research imaging strategy. Imaging researchers
must be able to respond to the demands of the clinical and43
Imaging Research at the Manchester Cancer Research Centre
42
Manchester Cancer Research Centre Research Report
upon application of doxycyclin (Figure 3). In addition studies
are underway to validate biomarkers that reflect cell loss and
apoptosis using both PET (Figure 3) and MR imaging
modalities. In the latter case biomarkers derived from diffusion
weighted MRI are being evaluated as robust indicators of cell
death. These have been successfully applied in the context of
radiotherapy response (Figure 4) and are currently being
assessed in genetic models with controllable levels of
apoptosis. Collaborative studies with Dr Andrew Fagan (Centre
for Advance Medical Imaging, St. James’s Hospital Dublin,
Ireland) and Dr Friedrich Wetterling (Computer Assisted
Clinical Medicine, Medical Faculty Mannheim, University of
Heidelberg, Germany) have enabled the initiation of
preliminary multi-nuclear MRI studies (proton and sodium 23)
to complement the diffusion weighted imaging. In the case of
imaging the microenvironment oxygen-enhanced MR has
been applied as a novel MR technique to evaluate tumour
hypoxia (Figure 4) which is also being developed in the clinical
setting (Figure 11). Key to the further development of preclinical imaging in the MCRC is the developmental
of robust tumour models that reflect more accurately clinical disease. To support ongoing translational
research programmes, orthotopic models of glioma and colorectal liver metastases have been generated for
imaging and therapeutic studies within the MCRC (Figure 5).
To support the development of novel PET-tracers for preclinical application, a radiochemistry research
associate has been recruited to the research team. They will contribute to research within cancer and other
disease settings. One particular focus here is the exploitation of a novel technique that has been developed
within the WMIC to allow the 18-F-labelling of macromolecules (for example peptides, nucleic acids,
antibody fragments) for use as PET-tracers. This exciting development has been exemplified by labelling
therapeutic peptides and assessing bio-distribution and metabolism via PET for neuroscience applications.
In addition, the apoptotic tracer ML-10 is at the latter stages of validation for preclinical studies which will
be initiated imminently within the MCRC.
Clinical Imaging Research
Both The Christie NHS Foundation Trust and The University of Manchester have extensive portfolios of
clinical imaging projects and imaging-based clinical trials. The Translational Research Imaging Group (TRIG)
has been formed to coordinate and integrate these research portfolios. The clinical imaging research groups
within the individual MCRC members have strongly synergistic expertise providing considerable added
value to the strategic development of a research imaging portfolio. The TRIG provides access to expertise
not only in clinical and experimental CT, MRI and PET but also in advanced image analysis, systems
modelling, decision support, imaging trial design and implementation and integration of imaging research
into surgical and radiotherapy studies. The role of the TRIG is to ensure strategic development and growth
of clinical imaging research within the MCRC. In order to achieve these goals the TRIG is proactively
interacting with individual clinical research groups within the MCRC to identify areas where imaging
Figure 3. PET-based non-invasive imaging of proliferationand cell loss in genetic “switch” models. Tumour uptake of18F-FLT is an early response biomarker of inhibition ofproliferation (top panels) or activation of apoptosis(bottom panels). Models used express a dominantnegative inhibitor of oncogenic signalling (DN-PI3K; top)or constitutively activate caspase 3 (bottom) in responseto doxycycline treatment. Tracer uptake is maintained incontrol tumours in the same mice. Courtesy ofCawthorne, Smigova, Williams, Morrow, Simpson, Dive.
Figure 4. A) Oxygen enhanced MRI as a potentialbiomarker of hypoxia in tumours. The tumourregions that show a reduction in longitudinal
relaxation rate (R1) during oxygen versus airbreathing (blue regions) correlate with the extent
of tumour hypoxia (brown staining-pimonidazole). Courtesy of Linnik, Waterton and
Parker. B) Apparent diffusion coefficient as anearly response biomarker for radiotherapy (IR). IR
causes a significant increase in ADC within 72h oftherapy. Courtesy of O’Connor, Burke, Williams
and Waterton.
Figure 5. Development of robust preclinicalmodels of colorectal liver metastases and glioma
to support translational imaging andtherapeutic studies within the MCRC. 18F-FLT
uptake into colorectal liver metastases (A) andorthotopic gliomas (C) with comparative T1-weighted coronal MR images (B and D). Also
shown for the glioma is the T1-weighted imagepost-IV gadolinium contrast agent
administration (E). Courtesy of Pathmanaban,Babur, Gieling, Cawthorne, Smigova, Williams
and Bigger.
Figure 6. Changesin endothelial
permeability(images) and
fractional bloodvolume (graph) in a
colorectal livermetastasis treatedwith bevacizumab.

biological research communities by identifying and validating imaging
based biomarkers that are of direct relevance to the biological
mechanisms involved in disease behaviour or therapeutic interventions.
The last 10 years has seen a major focus on the development of imaging
biomarkers that describe the microvascular environment driven, almost
entirely, by the development of anti-angiogenic therapeutic agents. In
the coming years new biomarkers will be required which enable us to
measure novel therapeutic targets and mechanisms. These will include
biomarkers of local invasive behaviour, metastatic behaviour, apoptosis
and cell death. The MCRC Imaging Group is actively involved in the
development and validation of a wide range of novel biomarkers.
Examples include the development of imaging techniques which
measure soluble oxygen concentration for the investigation of hypoxia,
new measures to identify locally invasive behaviour at tumour margins,
novel methods for analysing heterogeneity within tumours from MRI
and PET and analytical techniques which allow the identification and
segmentation of tumour tissue from normal background tissue.
Hypoxia is a major area of research interest in oncology and yet there is
still no totally satisfactory imaging biomarker of tissue hypoxia. In part
this reflects the complexity of the hypoxic mechanism where areas of
well-perfused tissue may show clear cellular hypoxia. An ideal imaging
biomarker would demonstrate cellular hypoxia through a specific
molecular mechanism. Such tracers do exist for PET studies (18F-MISO,
ATSM) but they are not widely available, complex to use and each have
specific disadvantages relating to tracer distribution, severity of hypoxia
required to produce uptake and other factors. Simple measurements of
regional blood flow do not inevitably map to hypoxia and there is
significant effort taking place worldwide to identify a simple, cheap,
widely-applicable imaging biomarker of hypoxia. In Manchester we have
developed a technique to image the concentration of dissolved oxygen
in blood and interstitial tissue using the MR properties of oxygen
dissolved in water. We have already validated this technique in normal
tissues and tumours and are continuing to develop and validate it in a
number of studies (Figure 11).
Many groups of novel agents in development are expected to have a
direct effect on local tumour invasive behaviour and on metastatic
potential. There are remarkably few biomarkers of invasive behaviour
and we are attempting to develop and validate new approaches to
assessing this important hallmark of malignancy. One relatively simple but promising approach has been
to measure the gradients of signal intensity between the tumour and the surrounding normal tissue. We
have applied this technique in patients with glioblastoma where there is believed to be a separate, more
invasive phenotype. Figure 12 shows our technique for mapping the invasive profile of a tumour. This profile
45
Imaging Research at the Manchester Cancer Research Centre
clearly identifies invasive phenotypes associated with worse
prognosis. The analysis must be conducted on appropriate images
including images of free water diffusion, which is affected by tumour
invasion into surrounding white matter and images of fractional blood
volume, which are also specifically affected, by regional tumour
invasion. These methods are now being extended to tumours in
peripheral tissues.
18FLT is a widely used and potentially valuable biomarker of cellular
proliferation. Unfortunately, it is metabolised and cleared by normal
liver tissue so that uptake within the normal liver is high. Traditionally,
this has prevented its use in studies of liver tumours or metastases.
Within the MCRC Imaging Group we have developed a segmentation
method, based on the pharmacokinetic characteristics of tumour and
normal liver tissue which enables us to separate normal from
malignant tissue in FLT studies (Figure 13). This development means
that we can now study proliferative activity within the liver
considerably extending our potential portfolio of techniques.
Advanced cancer imaging research has a clear and exciting future
within the MCRC. PRIME and TRIG provide a new and active forum for
researchers to explore the possibility of imaging based studies in their
area. The imaging research community in Manchester is vibrant and
increasingly successful and we are encouraging all researchers within
the MCRC to take advantage of these facilities.
18FLT is a widely used and potentially valuable biomarker of cellular
proliferation. Unfortunately, it is metabolised and cleared by normal
liver tissue so that uptake within the normal liver is high. Traditionally,
this has prevented its use in studies of liver tumours or metastases.
Within the MCRC Imaging Group we have developed a segmentation
method, based on the pharmacokinetic characteristics of tumour and
normal liver tissue which enables us to separate normal from
malignant tissue in FLT studies (Figure 9). This development means
that we can now study proliferative activity within the liver
considerably extending our potential portfolio of techniques.
Advanced cancer imaging research has a clear and exciting future
within the MCRC. PRIME and TRIG provide a new and active forum for
researchers to explore the possibility of imaging based studies in their
area. The imaging research community in Manchester is vibrant and
increasingly successful and we are encouraging all researchers within
the MCRC to take advantage of these facilities.
44
Manchester Cancer Research Centre Research Report
Figure 7. Quantitative biomarker images of alarge glioblastoma. The image on the left
shows the fractional blood volume within thetumour and in the large feeding vessels. The
image on the right shows a biomarker ofreduced perfusion pressure developed inManchester. This indicates the areas ofreduced blood flow, hypoxia and active
angiogenesis within the tumour.
Figure 8. Images of methionine uptake (top)and contrast enhanced MRI in a patient
with a recurrent low-grade brain tumour.Note the high methionine uptake despite
the relative absence of contrastenhancement on MR.
Figure 9. A fused MR and PK11195 PET imagedemonstrating the distribution of activated
microglial cells within and around a low-grade glioma.
Figure 10. Axial and coronal sectionsthrough region HG1 (a) and HG2 (e) on T1-weighted contrast-enhanced MR imaging
showing tissue segmentation in (b) and (f).GM=red, WM=green, LG=purple, HG1=lightblue, HG2=yellow. VPM K1 maps before andafter TQD are shown centred on HG1 (c and
d), HG2 (g and h).
Figure 11. Two tumours (A and B) imaged usingdissolved oxygen (left), dynamic contrast
enhanced MR (centre) and analysed to show theproportion of perfused tissue (right). Note that
high levels of oxygen transfer are seen in tumourB in areas that are relatively avascular and
poorly perfused. Other areas show extremelylow oxygen transfer despite high perfusion.
Figure 12. The location of a brain tumour (top).The middle two images show the tumour
boundary with measurement lines at every pointallowing the automated calculation of the
tumour invasion profile. The image on the rightshows the distribution of these measurements
demonstrating clear areas of increased (red)invasion and areas of increased infiltration intoperipheral tissues (blue). These measurementscan then be projected on to the source images
(bottom).
Figure 13a & 13b. CT (9A) and FLT (9B) images ina patient with hepatic metastases. On the left-
hand side of B are the source FLT uptake images.Tumour cannot be differentiated from normalliver. On the right are filtered images from the
same dataset using a new technique developedin Manchester to show normal liver (top) and
tumour tissue (bottom).

47
Epidemiology of excess weight and cancer
In collaboration with the University of Bern, Switzerland, the group published
a seminal paper in the Lancet in 2008, quantifying the associations between
increased excess body weight and the risk of several adult cancers. This work
specifically showed that associations are: (i) sex-specific; (ii) common across
populations, including those from Asia-Pacific; and (iii) present for less
common malignancies (such as lymphoma, multiple myeloma, melanoma)
as well as the previously recognised common malignancies (such as post-
menopausal breast, colorectal, and endometrial). In collaboration with
Professor Iain Buchan, at the North West Institute for Bio-Health Informatics
(NIBHI), The University of Manchester, and investigators from the University
of Erasmus, Rotterdam, the group went on to use attributable risk approaches
and probabilistic modelling to show that excess body weight accounts for
Obesity and Cancer
46
Obesity and Cancer
Manchester Cancer Research Centre Research Report
Over the past five years, there has been a fundamental shift in the way the
scientific community investigates lifestyle and dietary aetiological factors for
common adult cancers; away from a ‘reductive approach’, for example dietary
micronutrients, to a more holistic ‘macro approach’ such as obesity. The
Obesity and Cancer Research Group has been at the forefront in quantifying
the association between body mass index (BMI) and risk of several cancer
types, and predicting that obesity may become a greater attributable risk
factor than smoking for new cancer cases in Europe over the next decade.
Evidence is also emerging that obesity is an adverse prognostic factor in
patients with cancer and a predictive lifestyle ‘biomarker’ of adverse
treatment response. To better understand the complex relationships
between obesity and cancer, the group is developing laboratory models along
two lines: chronic insulin exposure in in vitro cell models (hyperinsulinaemia
is a consistent feature in obesity); and in vivo models of human tumour
growth and progression in diet-induced and genetically-induced obese
animals.
Body mass index (BMI: expressed as kg/m2) is the standard method of
approximating body adiposity at a population level. Normal weight is defined
as a BMI between 18.5 and 24.9 kg/m2; overweight as 25.0 to 29.9 kg/m2; and
obesity as a BMI value greater than 30.0 kg/m2. Excess body weight is
overweight and/or obesity. The Obesity and Cancer Research Group
commenced in 2007, led by Andrew Renehan and based in the School of
Cancer & Enabling Sciences at the capital University of Manchester. The
group has parallel clinical and laboratory research activities. An underlying
principle to the research programme is the establishment of robust clinical
observations prior to developing human-mimicking models in the laboratory
(Figure 1). The primary cancer of interest is colorectal. The laboratory activities
are based in the Paterson Institute for Cancer Research, and integrated into
the Clinical and Experimental Pharmacology Group.
By Andrew G Renehan
Large bowel
Intestinal crypt
Obesity in apopulation
Cellularmodels
Figure 1. Cartoon illustrating the principle of taking robust clinical observations tolaboratory models – the ‘population to pipette’ approach.

124,000 new cancer cases in Europe per year. Evidence is also emerging that obesity is an adverse prognostic
factor in patients with cancer, and a predictive lifestyle ‘biomarker’ of adverse treatment response. The
group is currently exploring clinical databases to determine the impact of excess weight on anti-cancer
treatment responses and outcomes.
In vitro experiments – chronic insulin exposure
A variety of candidate biological mechanisms linking obesity with cancer risk and progression have been
suggested. Key among these is the insulin and insulin-like growth factor (IGF) system, and this is a focus in
the laboratory research programme. The acute effects of insulin and IGF-I on cancer cells are well known
as these growth factors increase cell growth, promote cell survival, and increase cell motility, attributes
which favour tumour progression. The effect of chronic insulin exposure on insulin-sensitive tissues, such
as fat and muscle cells, is well characterised in the diabetes and metabolic literature, but there is a paucity
of data on equivalent effects in cancer cells. Chronic insulin exposure is associated with adaptation of the
immediate downstream complex, insulin-receptor substrate (IRS)-1 and -2; specifically, there is decreased
tyrosine phosphorylation (the stimulatory arm) of IRS-1 and increased serine phosphorylation (the inhibitory
arm) of the IRS-1, with ultimate reduction in IRS-1 expression (Figure 2). Our early studies show, for the first
time, that in many cancer cell types, this adaptive mechanism to chronic insulin exposure is lost. In turn,
this led us to hypothesise that this failure to ‘take the breaks off’ insulin and IGF-I signalling may account
for why hyperinsulinaemia and some insulin treatments are associated with increased cancer risk and
progression.
We are currently exploring whether the lack of molecular adaptation among cancer cell types is determined
by mutations in downstream signalling, such as the KRAS and BRAF genes. These are key predictive
biomarkers for new therapies in colorectal cancer and, in turn, obesity and hyperinsulinaemia may be
modifiers of this predictive process.
49
In vivo models of obesity and cancer
Obesity is a complex multi-organ problem and to better understand its influence of cancer risk, progression
and treatment interaction, it is necessary to develop human mimicking animal models. This is not a
straightforward challenge. Thus, for example, the usual manner to develop a diet-induced obese laboratory
animal is through excess fat context in the diet. However, this approach produces only a modest phenotype
of excess insulin and it is not possible to disentangle the effects on tumours of excess dietary fat versus
increased body adiposity per se. Accordingly, we have chosen to use genetically induced obese murine
models where animals are hyperphagic i.e. they eat the same proportions of fat but excess calorie intake
compared with lean counterparts.
Early experiments show that the effects of excess insulin may differ for normal intestinal tissues compared
with neoplastic tissue; an observation consistent with the findings in the in vitro cell systems described
above.
Cancer Prevention Research Network
The Cancer Prevention Research Network (Chairs: Andrew Renehan and Anthony Howell) is a research
network within the Institute for Health Sciences at The University of Manchester. It offers an example of
the Manchester Academic Health Science Centre (MAHSC) in action bringing together disparate disciplines
from across academic teams in Manchester. Two examples in the field of obesity and cancer illustrate the
benefit of this network. First, the obesity and cancer group have supported the prevention team of the
Manchester Breast Centre (leads: Anthony Howell and Michelle Harvie) to optimise the role of surrogate
markers, such as serum IGF-I, in intervention weight-loss trials in women at high risk of breast cancer.
Second, in a collaboration with the Gynaecological Oncology team (lead: Henry Kitchener) and the MRC
Obesity and Cancer
48
Manchester Cancer Research Centre Research Report
InsulinIGF-I
IR
PI3K
AKT
Gene expression/Invasion
BAD
Bcl-2
Survival Gene expression/Proliferation
Hypoxia/ treatment resistanceTranscription
PAK-1 KRAS BRAF
ERK
mTor
IRS-1IRS-1
P302
P307
P612
P636/39
P1101
P 895
P 989
P 1229
tyrosine
serine
Figure 2. Schematic diagram of principle pathways linking insulin and cancer cell biological endpoints. Insert: phosphorylation andadaptation by the IRS-1 complex: insulin resistance is characterised by increased serine phosphorylations.

51
Obesity and Cancer
50
Manchester Cancer Research Centre Research Report
Clinical Trials Unit, the Obesity and Cancer Group are currently undertaking secondary analyses of the effects
of BMI on treatment outcome in women with endometrial cancer treated with the ASPAC trial.
Manchester Birmingham Biostatistics and Biomarkers Collaboration
Biomarkers are increasingly important tools in oncology and, in particular, to facilitate drug development.
The increased integration of these biomarkers into appropriately designed clinical trials is guiding the new
era of personalised therapeutics. For pharmacological and predictive biomarkers, desirable statistical
performance characteristics parallel those for diagnostic tests, such as high sensitivity, specificity and
accuracy. Such qualities are seldom achieved using a single serum biomarker, and thus the combination of
multiple biomarkers to develop a “composite serum biomarker”, with enhanced performance and event
classification is appealing. However, as the number of biomarkers increases, in parallel, there is an increase
in the complexity of statistical handling of data.
To address the needs for robust statistical frameworks to underpin future biomarker-driven trials, the
Clinical and Experimental Pharmacology Group (leads: Caroline Dive and Malcolm Ranson) in collaboration
with the Tumour Angiogenesis Group (lead: Gordon Jayson), jointly one of the largest research groups in
Europe developing biomarker-linked clinical oncology trials, has formed a collaboration with the
Birmingham Cancer Research UK Clinical Trials group (leads: Cindy Billingham and Philip Johnson). The
Birmingham unit has recently become a Medical Research Council (MRC) methodology hub. The statistical
team includes over twenty statisticians and clinical trial researchers. New posts appointed through the
MRC methodology hub scheme, and a two-city bio-statistical post, will contribute dedicated time to the
proposed collaboration.
The collaboration (abbreviated to MBBBC) has been headed by Andrew Renehan and supported by the
Manchester Cancer Research Centre. The first workshop was held in Manchester in February 2010; a second
workshop is planned for Birmingham in June 2010. The first workshop identified that the initial focus of
research will be the statistical handling of multiple blood-borne biomarkers. Future activities will include:
sharing of trial data (Figure 3); development of models to handle longitudinal data, for example, mixed-
effect models, time series modelling; and models to best classify disease events using composite
biomarkers, for example, artificial neural networks.
Time (days)
Figure 3. An example of complex biomarker data sampled longitudinally in a trial of 45 patients undergoing first-line treatment for metastaticcolorectal cancer (The SCOUT trial: data courtesy of Dr Mark Saunders). Each dot represents a sample measurement. Each patient has multiple
repeated measurements, which in turn, are correlated with each other. Some patients died during the time period of the trial (D).

53
The physiological importance of the MAPK cascades has been mostly
demonstrated by embryonic death caused by their functional deletion in mice.
Consistently, studies using cells derived from these genetically modified
animals have provided evidence that MAPK modules regulate numerous
cellular functions in response to distinct extracellular stimuli. For example, in
my previous studies, we have demonstrated that JNK is mainly activated by
cytokines and extracellular stresses. Like other MAPKs, JNK is activated upon
phosphorylation at Thr and Tyr residues by two MAPK kinases, MKK4 and
MKK7. Similar to the early embryonic death caused by the targeted deletion of
the jnk genes, the mkk4-/- and mkk7-/- mice die before birth. The non
redundant function of MKK4 and MKK7 in vivo may be due to their distinct
biochemical properties. Whereas MKK4 can also activate p38 MAPK, MKK7
functions as a specific activator of JNK.
Three genes, jnk1, jnk2, and jnk3 encoding 10 JNK isoforms have been identified
and implicated in promoting neuronal apoptosis during the development of
the central nervous system and in response to brain injury. The characteristic
morphological changes displayed by apoptotic cells reflect the activation of a
tightly regulated intrinsic cell signalling machinery that leads to the activation
of caspases. The ability of JNK to trigger the mitochondrial release of
cytochrome c via post-tanslational modifications of the pro-apoptotic Bcl-2
family members constitutes one mechanism by which JNK activates the
caspase cascade. However, in contrast to other cell types, redistribution of
cytochrome c is not the only regulated step during neuronal apoptosis. De
novo transcription via JNK-dependent regulation of AP-1 activity may
contribute to the additional changes that make a neurone competent to
receive the cytochrome c signal. This is supported by the similarity of the
phenotypic defect displayed by mice carrying a mutated c-jun gene in which
Ser63 and Ser73 residues known to be phosphorylated by JNK are converted
to Ala residues and the jnk3-/- mice.
Role of MKK4 in Ras-induced cancers
52
Role of MKK4 in Ras-inducedcancers
Manchester Cancer Research Centre Research Report
Among the signalling pathways strongly suspected to be involved in
mediating the effect of oncogenes in vitro is the mitogen-activated protein
kinase (MAPK) kinase 4 (MKK4), a non-redundant component of stress
activated MAPK signalling modules. However, the function of MKK4 in
tumourigenesis remains controversial with some studies indicating that
MKK4 is a tumour suppressor, while others have reported a pro-oncogenic
role of MKK4. To clarify these discrepancies, we have examined the role of
MKK4 in the development of skin tumours that occurs in situ in the mouse.
In contrast to the previous studies, this system recapitulates the complex
interactions between tumours and host as well as the cellular heterogeneity
of tumours. This is critical considering that the environment greatly
influences the spatial and temporal regulation of signalling pathways
activated by oncogenes, and consequently determines the biological outcome
of the response. Our results demonstrate that skin-specific MKK4-deficient
mice are resistant to tumourigenesis associated with oncogenic activation
of the ras gene.
Cells in every organism have the ability to respond to signals from their
environment, thanks to clever systems in which external stimuli trigger a
cascade of events which leads to a change in cellular behaviour. These
systems are known as cell signalling pathways and one of them, the mitogen-
activated protein kinase (MAPK) pathway, has been the focus of my group.
The MAPK family comprises three members: extracellular signal-regulated
protein kinase (ERK), c-Jun NH2-terminal protein kinase (JNK, also known as
stress-activated protein kinase, SAPK), and p38 MAPK (also known as cytokine-
suppressive anti-inflammatory drug-binding protein). Two main mechanisms
have been proposed to ensure specific transmission of the signals from
upstream kinases to MAPKs: i) scaffold proteins that assemble the different
components of a cascade; ii) physical interactions between the components
of a cascade. Both mechanisms may operate in parallel and allow different
responses of the same MAPK pathways to different stimuli.
By Cathy Tournier

However, apoptosis does not represent the only functional consequence of JNK activation. For example,
JNK appears to contribute to cell survival, cell proliferation, the immune response, and to mediate the effect
of certain oncogenes, including those associated with over-expression of mutant forms of ras. Ras belongs
to a large family of small GTPases that bind GTP and hydrolyse it to GDP. Constitutive active ras mutants have
been identified in cancers of many different origins, including pancreas (90%), colon (50%), lung (30%),
thyroid (50%), bladder (6%), ovarian (15%), breast, skin, liver, kidney, and some leukaemia. Site directed
mutagenesis have provided evidence that c-Jun is essential for transformation of fibroblasts by activated Ras
proteins. However, despite of its predominant role in controlling the transcriptional activity of c-Jun and c-
Jun expression, the requirement of JNK in Ras-mediated tumour initiation, progression and metastasis is not
rigorously established because of controversial findings.
Like JNK, the role of MKK4 in cancer appears complex with some data suggesting that MKK4 acts as a
tumour and metastasis suppressor, while others pointing to a pro-oncogenic role for MKK4. These
conflicting results have been an obstacle for considering MKK4 a valuable drug targets for cancer therapy.
To clarify these discrepancies, we have examined the effect of mkk4 gene deletion on the development of
skin tumours. The mice were treated with the carcinogen 7, 12-dimethyl-benzanthracene (DMBA) and with
the tumour promoter 12-O-tetradecanoylphorbol
13-acetate (TPA). This simple, versatile and highly
reproducible chemical carcinogenesis protocol
recapitulates the fundamental concept that
tumour development is a multi-step process that
includes: initiation of benign papilloma (mutation
of H-ras following DMBA treatment), promotion (a
clonal expansion of the initated cell triggered by
the repeated treatment with TPA to form a benign
tumour), and progression to malignant carcinoma
(an additional evolutionary step). Using this
model, we have discovered that the number of
tumours per mouse was greatly decreased in mice
lacking MKK4. These results provided the first
genetic demonstration that signalling
downstream of MKK4 is essential for tumour
formation in the skin (Fig. 1).
Based on these data, Cancer Research UK has
awarded us a project grant to specifically define
the role of MKK4 in tumour initiation and
progression, under normal conditions and in
response to anticancer therapies. In particular, we
will determine whether MKK4 is involved in the
development of chemoresistance by promoting
the expression of membrane drug transporters.
Over-expression of membrane drug transporters
such as P-glycoproteins (P-gls) and multidrug
55
resistance proteins (MRPs) constitutes one mechanism by which tumour cells can acquire resistance to
anticancer agents. This represents a substantial obstacle for the successful treatment of certain human
cancers. JNK has been implicated in increasing MRP1 expression. Furthermore, a recent study has shown that
xenografts of MKK4-deficient colon cancer cell lines exhibited greater sensitivity to anticancer agents. This
work is being developed using the chemically induced skin cancer model. We will translate these findings
to another epithelial cancer model: pancreatic cancer. The role of MKK4 in pancreatic cancer is highly
controversial with some studies presenting evidence that MKK4 acts as a tumour promoter while others
suggesting that it is a tumour suppressor. However, none of these studies have used models that accurately
recapitulate the features of premalignant or malignant ductal lesions. We propose to address this issue
using a mouse model which spontaneously develops the full spectrum of human pancreatic intraepithelial
neoplasias (PanINs) following the physiological expression of K-rasG12D in the pancreata. K-rasG12D is a
constitutive active mutant form of K-ras found in human pancreatic cancer. Some of these lesions progress
to invasive and metastatic adenocarcinomas.
However, whether pharmacological inhibition of MKK4 will reproduce the effect of mkk4 gene deletion
remains to be established before the therapeutic implication of inhibiting MKK4 activity for treating human
cancer becomes clear. This key question, namely to explore MKK4 as a potential anticancer drug target, is
being addressed in collaboration with the Drug Discovery Unit at the Paterson Institute for Cancer Research
who has identified a number of compounds that constitute good candidates for inhibiting MKK4 activity.
These compounds will be synthesised and tested in skin and pancreatic cancer cell lines carrying oncogenic
mutation in H-ras or K-ras genes, for their ability to i) inhibit JNK activity, ii) block cell proliferation and iii)
sensitise cells to the apoptotic effect of genotoxic drugs (e.g., doxorubicin). The specificity of the compounds
will be established by comparing their effect to that associated with the down-regulation of MKK4
expression using siRNA technology. The next step will be to test the effect of the compounds on tumour
growth in our mouse models of skin and pancreatic cancer. One potential important outcome of our studies
is the validation of MKK4 as a drug target for the treatment of cancers. In particular, combined with
conventional DNA-damaging chemotherapeutics, inhibitors of MKK4 may provide a new strategy to
overcome resistance of tumours to the current therapies.
Role of MKK4 in Ras-induced cancers
54
Manchester Cancer Research Centre Research Report
DMBA/TPA
MKK4/MKK7
JNK
c-Junp53
p21 EGFR
APOPTOSIS
CELL CYCLEARREST
PROLIFERATION
Ras
Figure 1. Schematic role of MKK4 in skin cancer. Our results show that MKK4has a pro-oncogenic function in the skin. The mechanism by which MKK4promotes tumourigenesis in the skin appears to be mediated via JNK/c-Junsignalling. Downstream targets of c-Jun include the epidermal growth factorreceptor (EGFR). The ability of EGFR to prevent cell differentiation, therebymaintaining keratinocytes in a proliferative state, is one mechanism bywhich c-Jun contributes to tumour formation upon Ras activation. Inaddition, MKK4 may be involved in malignancy by decreasing p53 stability,and thereby down regulating p21 expression, via JNK independently of c-Jun.This model is supported by evidence that the loss of p53 enhances malignantprogression of chemically induced skin tumours.

57
research, the Patient Treatment Centre will provide improved facilities for
service chemotherapy delivery, hospital pharmacy and aseptics and a new
private patient facility.
Phase I trials are experimental studies that seek to define the safety and
pharmacology of new treatments in man and they represent the foundation
stone for future therapeutic developments. Each trial involves the detailed
study of a relatively small number of cancer patients who have usually
received, and have previously failed, established therapies for their tumour.
Each patient is carefully evaluated and detailed clinical observations are
complemented by translational research. Pharmacokinetic studies investigate
how the body distributes and excretes the new agent and pharmacodynamic
studies examine the effects of the new agent on tumour and normal tissues.
The first opportunity to evaluate human pharmacodynamics and to study
proof of mechanism and proof of principle occurs in these Phase I trials.
Although challenging, early phase research helps to define potential
biomarkers to assist in dose optimisation, improve patient selection and
develops the logistics required for future translational research in larger
patient cohorts. Phase I studies are followed by statistically more amendable
phase II trials, where larger cohorts of patients with a single tumour type are
evaluated. The need for biomarkers to allow clinicians to stratify patients and
to optimise treatment has become more important as the molecular
characteristics of tumours has become more clearly defined, and as
mechanism based treatments have been developed. Providing personalised
cancer medicine represents an important objective for improving cancer
patient survival in the future. The recent demonstration of epidermal growth
factor receptor (EGFR) mutations, which define a highly sensitive sub-
population of non small cell lung cancer patients for EGFR inhibitors, the
predictive potential of KRAS mutations in clinical decision making for EGFR
monoclonal antibody therapy in colorectal cancer and the development of
The New Patient Treatment Centre
56
The New Patient Treatment Centre
Manchester Cancer Research Centre Research Report
One of the research areas within the MCRC with an international reputation
is early phase clinical trials and the closely aligned translational research. The
original development and opening of The Christie’s Derek Crowther Unit
(DCU) in 2003 was an important milestone in developing world class early
phase clinical research in Manchester. Since its original opening, the DCU
has flourished, doubling its research activity and patient recruitment, but in
the process becoming too small to support its research portfolio. Over the
last few years Manchester has become one of the most active early phase
clinical trial centres in Europe. During the last three years The Christie has
enrolled over 3,900 patients to 70 Phase I trials, 132 Phase II trials and 44
biomarker studies. During the last five years Manchester’s early phase clinical
research activities have been strengthened by the development of the Clinical
and Experimental Pharmacology Group and of GCLP-compliant research
laboratories in the Paterson Institute for Cancer Research, along with opening
of the Wolfson Medical Imaging Centre (WMIC), the MCRC Biobank and the
Cancer Research UK Drug Discovery Centre. A successful Clinical Research
Infrastructure Award from the CR-UK/Department of Health (DoH)/Wolfson,
coupled with substantial additional investment from The Christie and other
partners, has now enabled the MCRC to enhance the clinical infrastructure
support for early phase clinical research in Manchester.
Detailed proposals for the new Patient Treatment Centre received approval
from The Christie NHS Foundation Trust Board in late 2008, and local
planning approval was granted in December 2008. The original DCU building
was demolished in February 2009 and its research activity relocated to a
temporary building situated on the north side of The Christie site. Building
work on the £35M Patient Treatment Centre commenced in January 2009 and
was opened in November 2010. It will provide space for a doubling of research
activity and make Manchester one of the world’s largest centres for early
phase clinical trials. Alongside improved capacity and facilities for clinical
By Malcolm Ranson

59
The New Patient Treatment Centre
58
Manchester Cancer Research Centre Research Report
BRCA mutation testing in the clinical development of PARP inhibitors in breast and ovarian cancers, are
recent examples of oncology moving towards personalised medicine. In keeping with this it will come as
no surprise that over 80% of the early phase trials conducted by the DCU include biological sample collection
(tissue, blood, urine, etc) and that the translational activities of the DCU have grown steadily since 2004.
The present DCU laboratory processes over 11,000 research samples a year and larger laboratory facilities to
support its growing translational research portfolio have been included in the new Patient Treatment Centre.
Opportunity has also been taken to co-locate this important development in the main treatment area of The
Christie (See Figure 1). This will allow researchers to extend translational science into later phase clinical
trials, an important step since phase II and phase III trials have the necessary statistical power for developing
decision making tools based upon biomarkers. Delivering the necessary high quality clinical science and
conducting it to good clinical practice (GCP) and good clinical laboratory practice (GCLP) involves close
multidisciplinary team working. Support from MCRC partners and from CR UK and DoH to the Manchester
Experimental Cancer Medicine Centre (ECMC) has helped us to respond to the changing research landscape.
This core support has funded key staff for our GCLP research laboratories and provided technical support
staff in pharmacy, radioimmunotherapy, imaging and radiobiology. To support the future growth of
translational research the MCRC has also recently invested in a Laboratory Information Management System
to cover the Paterson and DCU GCLP Laboratories. This will provide powerful linkage and management
tools for large clinical and research laboratory datasets.
As the research portfolios of the DCU and ECMC have grown, the world class biomedical imaging facilities
of the WMIC have become increasingly deployed to provide pharmacodynamic imaging information during
experimental treatment. Recently, a growing trend of cross-cutting research has emerged in which
advanced biomedical imaging is combined and compared with blood and tissue borne biomarkers. These
ground breaking studies should help optimise future patient treatment particularly for anti-angiogenic and
targeted agents. The WMIC currently leads over a dozen imaging protocols within the Manchester ECMC
portfolio and twenty further oncology studies are currently in discussion or set-up.
The pharmaceutical and biotech industry recognises that the UK is a good place to do complex early phase
clinical trials and, although the climate for clinical research in the UK has become more complex and
administratively challenging during the last five years, Manchester has responded to these challenges and
has seen substantial growth in its research activity and in patient recruitment. The Christie’s research and
development department has set itself challenging new targets for trial set-up times and for performance
management to help ensure that Manchester remains internationally competitive.
Historically there has been a dearth of well trained clinical and translational scientists in Europe. Training
of scientists and clinicians with expertise in early phase therapeutic development and in translational
science therefore continues to be an important area of investment. Currently our early phase trial activity
is heavily dependent upon a relatively small number of senior clinical researchers, and two principal
investigators currently account for over half our phase I trial recruitment. To realise the full potential of our
new infrastructure developments, additional senior clinical researchers engaged in early phase clinical
research will be recruited over the next five years. Since 2006, a highly successful Clinical Pharmacology
Fellowship scheme, funded by Cancer Research UK and AstraZeneca, has enabled eight promising young
clinical researchers to develop research skills in early phase clinical trials and experimental pharmacology
and to undertake PhD training. Complemented by training programmes for scientists in GCLP and an MRes
course in Translational Medicine, established by Professor Andrew Hughes, our investment in future talent
remains a key objective.
Figure 1. The new Patient Treatment Centre under construction in March 2010. The main entrance to The Christie is out of frame to theright, and the proximity of the patient treatment centre to many of the wards (to the right of the new build) can be clearly seen.

Chris Morrow, Kathryn Simpson, Luke Harrison and Tetyana Klymenko (Paterson Institute for Cancer Research) and Tom Owens
and Fiona Foster (Faculty of Life Sciences, The University of Manchester). - The Manchester Cancer Research Centre Conference
- Harnessing Apoptosis – page 10
Chris Morrow joined the Clinical and Experimental Pharmacology group at the Paterson
Institute for Cancer Research in April 2004 working as a postdoctoral research associate for
Professor Caroline Dive. Prior to this he read molecular biology at the University of
Edinburgh, gaining a BSc in 2000, before carrying out postgraduate research into cellular
mechanisms ensuring faithful chromosome segregation at The University of Manchester
under the supervision of Professor Stephen Taylor. He now works in the preclinical section
of Professor Dive’s laboratory, focusing on combinations of anti-cancer drugs which induce
cancer cell death and determining the underlying molecular mechanisms behind the drug
interactions.
Kathryn Simpson joined the Clinical and Experimental Pharmacology (CEP) group at the
Paterson Institute for Cancer Research in 2005 as a postdoc to work on small molecule
inhibitors of apoptosis, but also with a focus on developing the clinical and translational
research activities in Professor Caroline Dive’s laboratory. Kathryn became involved in the
implementation of a biomarker research project in CEP, utilising cutting-edge mass
spectrometry technologies, in close collaboration with the expertise in Professor Tony
Whetton’s Stem Cell and Leukaemia Proteomics Research Laboratory. The Clinical
Proteomics Team was soon established, led by Kathryn, in order to help fulfil an unmet
clinical need in cancer care by the discovery of novel biomarkers with the aim of identifying
predictive, prognostic, toxicity and/or response information from the circulating
complement of proteins in patient blood.
Luke Harrison completed a degree in Biochemistry at Newcastle University and
subsequently stayed in Newcastle to undertake a PhD in molecular pharmacology with
Professor Herbie Newell and Dr Mike Tilby. His PhD focused on studying the mechanistic
interactions and therapeutic uses of novel kinase inhibitors in combination with
conventional cytotoxic drugs. After completing his PhD Luke joined Professor Caroline
Dive's Clinical and Experimental Pharmacology group at the Paterson Institute for Cancer
Research in April 2008 as a postdoc where he continues his interest in preclinical
pharmacology. He is currently working on the effect of the tumour microenvironment on
the efficacy of apoptosis-inducing drugs.
61
Author Biographies
60
Author Biographies
Manchester Cancer Research Centre Research Report
Tetyana Klymenko joined the Clinical and Experimental Pharmacology Group in August
2008 as a postdoctoral scientist. She graduated from Kharkov State University in 1999
with her master thesis in biochemistry of viral RNA based on experimental work done at
the Puschino Institute of Protein Research, Russian Academy of Sciences. Following her
work on activation of transcription targets of HIV in Tampere, Finland, as a visiting scientist,
she completed a PhD on regulation of gene expression at the European Molecular Biology
Laboratory (EMBL) in Heidelberg, Germany.
Thomas Owens is in his third year of postdoctoral studies at The University of Manchester.
He gained a BSc in Biochemistry with Industrial Experience from The University of
Manchester in 2002. His placement year was at Evans Vaccines (now Novartis) in Liverpool,
where he worked in the Hepatitis-B-vaccine production facility. Following his degree he
worked on oligonucleotide synthesis at Applied Biosystems, before returning to
Manchester on the Wellcome Trust 4-Year PhD programme in 2003. His postgraduate
studies with Professor Charles Streuli examined the role that IAP proteins had in regulating
apoptosis in mammary epithelial cells. Since completing his PhD he continued to study the
mechanisms of apoptosis regulation with Dr Andrew Gilmore, focusing on the signals that
control the critical point in the life and death decision of a cell.
Fiona Foster is currently undertaking postdoctoral studies at The University of Manchester,
where she is investigating the role of focal adhesion kinase (FAK) in breast cancer initiation.
Fiona undertook a BMedSci at the University of Sydney, Australia, prior to obtaining her
PhD from the same institute in 2001. Fiona began her postdoctoral career in breast cancer
research at the University of Reading in the UK. This involved work on two projects, the first
examining the role of class II PI3 kinases and the second looking at the role of a protein
modification which could potentially make breast cancer cells immuno-suppressive. After
moving to The University of Manchester in 2006, Fiona has continued her research into
breast cancer, looking at the role of apoptosis suppression in breast cancer, first of all that
mediated by the Inhibitor of Apoptosis proteins and now that mediated by FAK.

63
Author Biographies
62
Manchester Cancer Research Centre Research Report
Fiona Blackhall trained in Manchester and for two years in Toronto with Dr Frances
Shepherd. She was appointed Consultant Medical Oncologist at the The Christie NHS
Foundation Trust in 2005, joining the Manchester Lung Group led by Professor Nick
Thatcher. She was awarded Honorary Senior Lecturer in the School of Cancer and Enabling
Sciences at The University of Manchester in 2007. She is an active clinical trialist with a
focus on development of mechanism based therapies and has a major role in biomarker
research, now leading the lung cancer focus group within the Clinical and Experimental
Pharmacology Group at the Paterson Institute for Cancer Research. She is Chair of the
translational subgroup of the recently established European Thoracic Oncology Platform,
a member of the NCRI Lung Clinical Studies Group and leads the MCRC lung cancer
research team.
Professor Caroline Dive is a Senior Group Leader at the Paterson Institute for Cancer
Research and Professor of Pharmacology in the School of Cancer and Imaging Sciences at
The University of Manchester. She is a co-director of the Clinical and Experimental
Pharmacology Group (CEP) within the Paterson Institute for Cancer Research. After
completing her PhD studies in Cambridge, Caroline moved to Aston University's School of
Pharmaceutical Sciences in Birmingham where she started her own group studying
mechanisms of drug induced tumour cell death. She then moved to what became the
Faculty of Life Sciences at The University of Manchester to continue this research and was
awarded a Lister Institute of Preventative Medicine Research Fellowship. Caroline joined
the Paterson Institute for Cancer Research in 2003 to set up the CEP, interfacing with the
Derek Crowther Unit for early clinical trials at The Christie. The CEP aims to drive early
clinical evaluation of novel mechanism-based targeted therapies by providing biomarkers
expertise, analysis of which acts as a readout of drug activity within the body.
Ivan Ahel - DNA Damage Response – page 20
Ivan Ahel obtained his BSc and MSc in Biology at the University of Zagreb, Croatia, in 1997
and 2000 respectively, before undertaking a PhD in Biology with thesis work carried out at
Yale University, USA, between 2000-2003. His undergraduate and PhD research experience
includes the regulation of the DNA damage response in actinomycetes and studies of the
mechanisms ensuring the fidelity of protein biosynthesis. In 2004 Ivan joined the Cancer
Research UK London Research Institute as a postdoctoral fellow investigating the
identification and characterisation of novel DNA repair proteins. In January 2009 he moved
to the Paterson Institute for Cancer Research as Group Leader for the DNA Damage
Response Group.
Tim Ward, Jian-Mei Hou, Matthew Krebs, Fiona Blackhall and Caroline Dive - Lung Cancer Circulating Tumour Cells as
Biomarkers and a Window to Metastasis Biology – page 16
Tim Ward is a translational scientist working within the Clinical and Experimental
Pharmacology Group at the Paterson Institute. He is the Pharmacodynamics Manager
responsible for the development, validation and implementation of all assays used by the
group in early phase clinical trials. He has over 30 years experience in cancer research,
principally anti-cancer drug development specialising in anticancer drug screening and
DNA damaging assays as well as extensive experience in cell culture assays. He currently
sits on the Cancer Research UK New Agents Committee (NAC). His current research
interests are centered on detecting and characterising circulating tumour cells and micro
emboli. He also has a keen interest in utilising circulating DNA as a biomarker of tumour
DNA to detect specific tumour mutations which impact on response to modern targeted
agents.
Jian-Mei Hou gained an MD degree through a seven year training program from West
China University of Medical Science in 2002. She subsequently spent three years studying
towards a PhD that focused on bio-immunotherapy of tumours at the National Key
Laboratory of Biotherapy, West China Hospital, Sichuan University. Jian-Mei spent a year
working on preclinical drug safety evaluation in National Chengdu Centre for Safety
Evaluation of Drugs. In May 2006, she joined the Clinical and Experimental Pharmacology
(CEP) Group at the Paterson Institute as a Cancer Research UK China Fellow. Her research
interests have since been focused on circulating tumour cells as a window into metastasis
biology in small cell lung cancer.
Matthew Krebs is a clinical research fellow within the Clinical and Experimental
Pharmacology Group at the Paterson Institute for Cancer Research. He completed his
degree in Medicine at The University of Leicester in 2001 and took opportunity to spend an
undergraduate elective period at the London Regional Cancer Centre, Ontario, Canada.
Following General Medical training in Manchester he commenced specialist training in
Medical Oncology at The Christie NHS Foundation Trust in 2005. Matthew subsequently
joined the Cancer Research UK/AstraZeneca PhD Clinical Fellowship programme in
November 2007. His research focuses on the isolation and characterisation of Circulating
Tumour Cells from patients with lung cancer with a view to developing these as predictive
and/or pharmacodynamic biomarkers in early phase clinical trials.

64
Manchester Cancer Research Centre Research Report Author Biographies
65
Anthony Howell is Professor of Medical Oncology and is the Director of the Breakthrough
Breast Cancer Research Unit, The University of Manchester. Formerly he was Chairman of
the UKCCCR, the British Breast Group and the Manchester Breast Centre, was the Research
& Development Director of The Christie NHS Foundation Trust and the Cancer Research
Network. His interests are the endocrine therapy and biology of the breast and breast
cancer with a particular interest in prevention. He has published over 500 papers mainly
in these areas.
Crispin Miller, Jenny Varley and Stuart Pepper - Cancer Genomics in the Era of High Throughput Sequencing – page 34
Crispin Miller studied Artificial Intelligence as an undergraduate before completing a PhD
in Computational Biology, which focused on the development of fast pattern matching
tools for nucleotide and protein sequence analysis. This was followed by an MRC training
fellowship in Bioinformatics. Crispin joined the Paterson Institute for Cancer Research in
2002, and currently heads the Applied Computational Biology and Bioinformatics Group.
Jenny Varley obtained her degree and PhD from the University of Leicester before
undertaking postdoctoral work in Geneva funded by a Royal Society Exchange Programme
Fellowship. After further postdoctoral work Jenny was appointed as a non-clinical lecturer
in Pathology at the University of Leicester, and subsequently moved to the Paterson
Institute in 1992. Jenny was head of the Cancer Genetics Group until 2001 and was
awarded the post of Honorary Professor of Experimental Oncology at The University of
Manchester in 2002. Jenny was appointed to the role of Assistant Director (Research) at
the Paterson Institute in 2001, and one of her major roles is developing and managing all
the scientific service units at the Institute.
Donald Ogilvie and Allan Jordan - Drug Discovery in the Manchester Cancer Research Centre – page 24
Donald Ogilvie heads the newly formed Drug Discovery Centre in the MCRC. He joined the
Paterson Institute as a senior group leader in February 2009 after a twenty year career in
the pharmaceutical industry. Donald obtained an MA in Biochemistry at Oxford in 1980
before working at the John Radcliffe Hospital for eight years on the role of proteases in
breast cancer then inherited connective tissue disorders. The latter was the basis of his
D.Phil degree. In 1988 he joined ICI which subsequently became Zeneca then AstraZeneca.
For most of his industrial career, Donald worked on cancer drug discovery and early clinical
development and he was directly responsible for the delivery of ten novel cancer
development compounds, several of which have progressed to Phase II & III clinical trials.
Allan Jordan joined the Drug Discovery Centre in July 2009 as Head of Chemistry. After
gaining a BSc in Chemistry from UMIST in 1993 and a short spell as a teaching assistant in
Arizona, he returned to UMIST to conduct post-graduate research into anticancer natural
products. After post-doctoral work at the University of Reading, he joined RiboTargets in
Cambridge (now Vernalis) where he worked on a number of therapeutic areas at various
stages of the research pipeline. Alongside involvement in a number of oncology
programmes, ultimately leading to the clinical evaluation of Hsp90 inhibitors in
conjunction with Novartis, he became involved in CNS research programmes where he was
a project leader on a GPCR drug discovery programme and was also involved in the
management of Vernalis’ clinical programme for Parkinson’s disease.
D Gareth Evans and Anthony Howell - Genetic Medicine and the Genesis Prevention Centre – page 28
D Gareth Evans has a national and international reputation in clinical and research aspects
of cancer genetics, particularly in neurofibromatosis and breast cancer, and is Chairman of
the NICE (National Institute for Clinical Excellence) Familial Breast Cancer Guideline
Development Group. He has developed a clinical service for cancer genetics in the North
West Region and lecture within the UK and internationally on hereditary breast cancer
and cancer syndromes. He has also developed a regional training programme for clinicians,
nurses and genetic associates in breast cancer genetics, and established a system for risk
assessment and counselling for breast cancer in Calman breast units.

66 6767
Author BiographiesManchester Cancer Research Centre Annual Research Report
Andrew G Renehan – Obesity and cancer – page 46
Andrew G Renehan is a Higher Education Funding Council for England (HEFCE) Senior
Lecturer in the School of Cancer and Imaging Sciences at The University of Manchester,
and Honorary Consultant in Colorectal and Pelvic Surgery at The Christie NHS Foundation
Trust. He heads the Colorectal Cancer and Obesity subgroup within the Clinical and
Experimental Pharmacology Group at the Paterson Institute for Cancer Research, and is
also Chair of the Cancer Prevention Research Network.
Cathy Tournier – Role of MKK4 in Ras-induced cancers – page 52
Cathy Tournier completed her PhD in 1996 and subsequently joined the laboratory of
Professor Roger J Davis (Howard Hughes Medical Institute, University of Massachusetts,
USA) to pursue her training in the field of research on the mitogen-activated protein kinase
(MAPK) signalling pathways. In July 2000, she was appointed as a lecturer in the Faculty of
Life Sciences at the University of Manchester. A year later, she was awarded a Senior
Research Fellowship from the Lister Institute of Preventive Medicine to focus on her
research. Cathy currently leads the Gene Expression and Cell Signalling Group.
Malcolm Ranson – The New Patient Treatment Centre – page 58
Malcolm Ranson completed his PhD in Manchester after obtaining medical and
Pharmacology degrees. Subsequently he was awarded an EORTC/NCI scholarship at the
National Cancer Institute, USA. He is currently Professor of Medical Oncology and
Pharmacology in the School of Cancer and Enabling Sciences at The University of
Manchester and heads the Manchester Experimental Cancer Medicine Centre. He is based
at The Christie where he is the Clinical Director of the Derek Crowther Trials Unit - one of
the largest early phase oncology clinical research units in Europe - and is co-Director of the
Clinical and Experimental Pharmacology Group within the Paterson Institute for Cancer
Research.
Stuart Pepper has run the Molecular Biology Core Facility for the Paterson Institute for
Cancer Research for the last eight years. The Core Facility provides a range of services
including DNA sequencing, DNA extraction and quantitative polymerase chain reaction
(PCR) analysis. The Core Facility also houses the Paterson’s Mass Spectrometry facility,
which provides state-of-the-art protein analysis services. During the last six years, Stuart
has also managed the Cancer Research UK GeneChip Microarray Facility, providing access
to cutting edge microarray technology for all Cancer Research UK grantees. The Core
Facility team have collaborated widely with local and national researchers and have
produced several publications using the latest microarray protocols available.
Alan Jackson and Kaye Williams - Imaging Research at the Manchester Cancer Research Centre – page 40
Alan Jackson joined The University of Manchester in 1993 and was appointed Professor of
Neuroradiology in the Imaging Sciences Group within the Faculty of Human and Medical
Sciences at The University of Manchester in 1995. Prior to this, he was a full-time NHS
clinical neuroradiologist at the Manchester Royal Infirmary. Alan holds honorary chairs in
Neuroimaging at the Universities of Liverpool and Bangor and is also an honorary clinical
consultant at the Salford Hospitals NHS Trust and the Manchester Royal Infirmary.
Kaye Williams received a BSc in Molecular Biology from The University of Manchester and
a PhD from the Paterson Institute for Cancer Research. Following Research Associate and
Research Fellow positions within the Experimental Oncology Group of the School of
Pharmacy and Pharmaceutical Sciences (headed by Professor Ian Stratford), she was
awarded the British Association for Cancer Research/AstraZeneca Frank Rose Young
Scientist Award in 2005. In January 2006 Kaye was appointed Senior Lecturer within the
School of Pharmacy. She currently heads the Hypoxia and Therapeutics Group and is the
MCRC lead for Preclinical Imaging.

Manchester Cancer Research CentreThe University of ManchesterWilmslow RoadManchesterM20 4BX
tel: +44 (0) 161 446 3156fax: +44 (0) 161 446 3109
www.manchester.ac.uk/mcrc