management of the bleeding patient dr. alan tinmouth director, adult regional hemophilia and...
TRANSCRIPT

Management of the Bleeding Patient
Dr. Alan TinmouthDirector, Adult Regional Hemophilia and Bleeding
Disorders ClinicFebruary 1st, 2005

Outline
Review the mechanism of blood coagulation Understand the tests used in the investigation
of bleeding disorders Understand the therapeutic options in the
management of bleeding patients “Hemostasis” Poker

Overview of Hemostasis

Primary Hemostasis: Platelets
I. Adhesion Platelet glycoprotein Ib/V/IX adheres to
subendothelium binding is primarily to vWFII. Activation
Shape change formation of pseudopods Anionic phospholipids (phospatidylserine and
phosphoethanolamine) exposed on external membrane
Glycoprotein IIb/IIIa exposed on surface Secretion of platelet granules
III. Aggregation Platelets bind to each other via Gp IIb/IIIa receptor
and soluble fibrinogen and vWF
FORMATION OF HEMOSTATIC PLUG
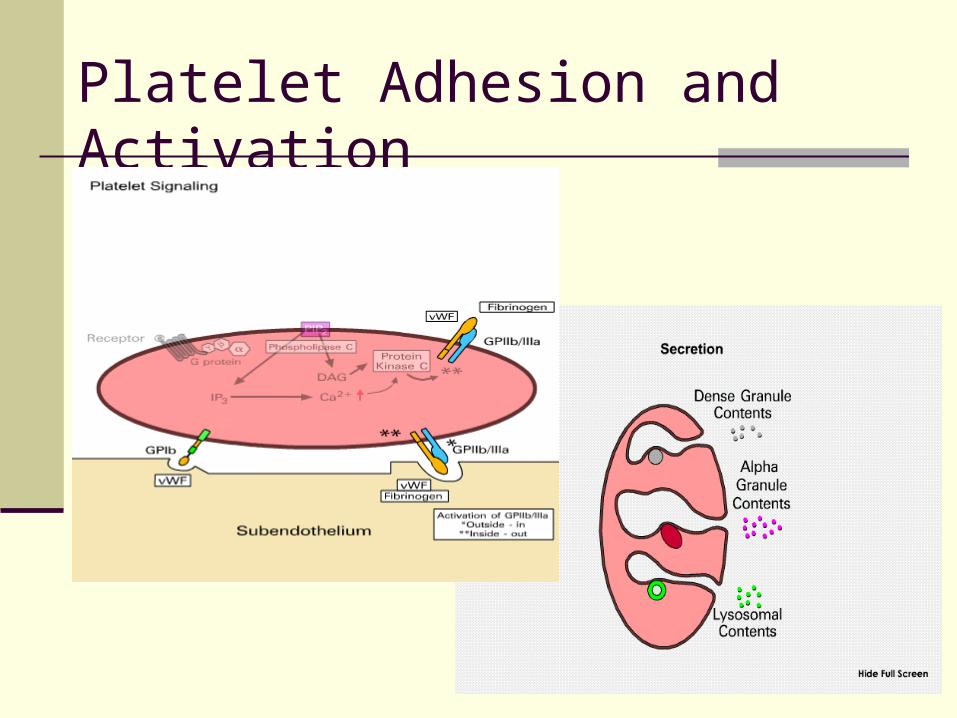
Platelet Adhesion and Activation
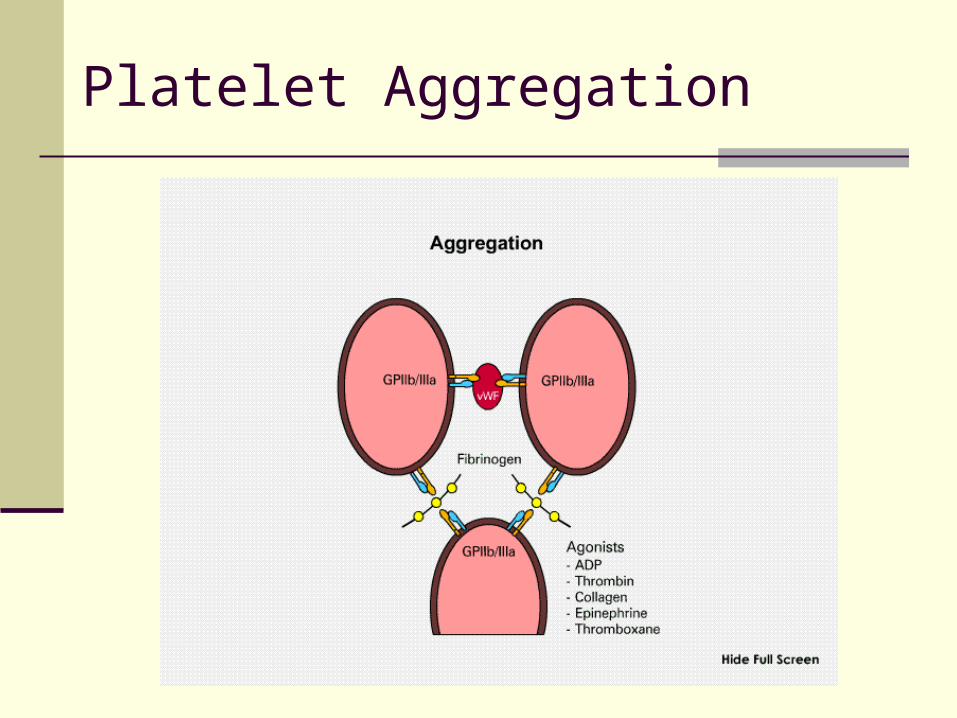
Platelet Aggregation

Secondary Hemostasis: Coagulation Factors
Coagulation factors are proteins that circulate primarily in inactive state (proenzyme), which are converted to active enzyme
Activation of coagulation factors is a stepwise process of one activated factor activating subsequent factor(s)
Historically separated into the intrinsic pathway and extrinsic pathway Intrinsic and extrinsic pathways believed to act
independently and lead to formation of fibrin clot through separate “coagulation cascades”
Model fails to explain many coagulation abnormalities seen clinically

Coagulation in a test tube
XII XIIa
XI XIa
IX IXa
X Xa +VIIIa
II IIa
VIIa VII +TF
Fibrinogen Fibrin
INT
RIN
SIC
EX
TR
INS
IC
COMMON
+Va
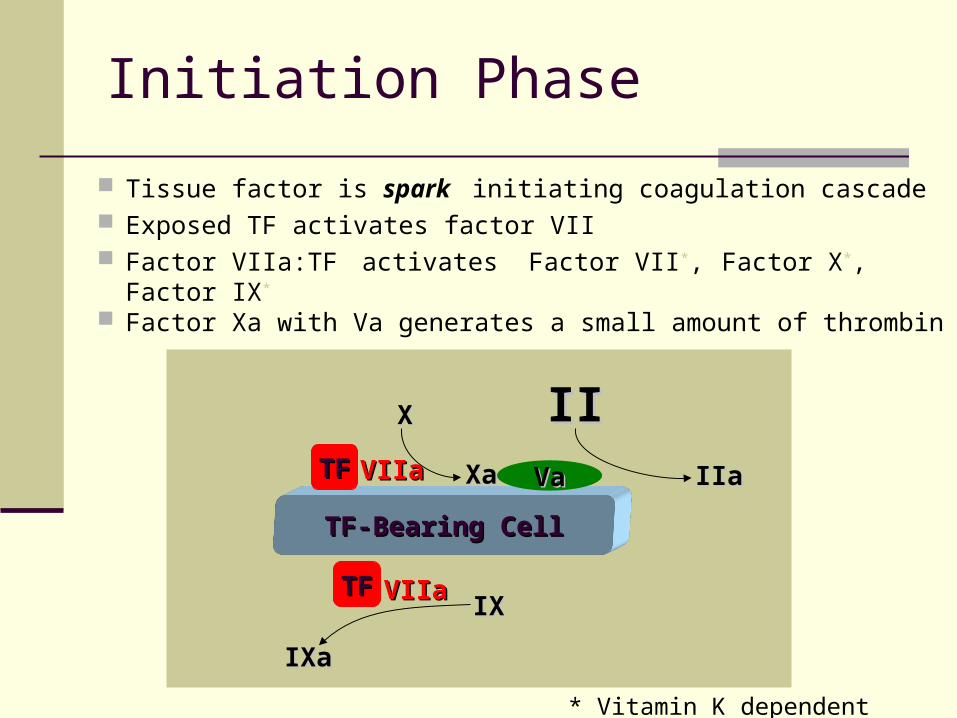
Initiation Phase
Tissue factor is spark initiating coagulation cascade Exposed TF activates factor VII Factor VIIa:TF activates Factor VII*, Factor X*, Factor IX*
Factor Xa with Va generates a small amount of thrombin
TF-Bearing CellTF-Bearing Cell
TFTF
VaVa
VIIaVIIa
TFTF VIIaVIIa
XX
XaXa
IIIIIIaIIa
IXIX
IXaIXa
* Vitamin K dependent clotting factors
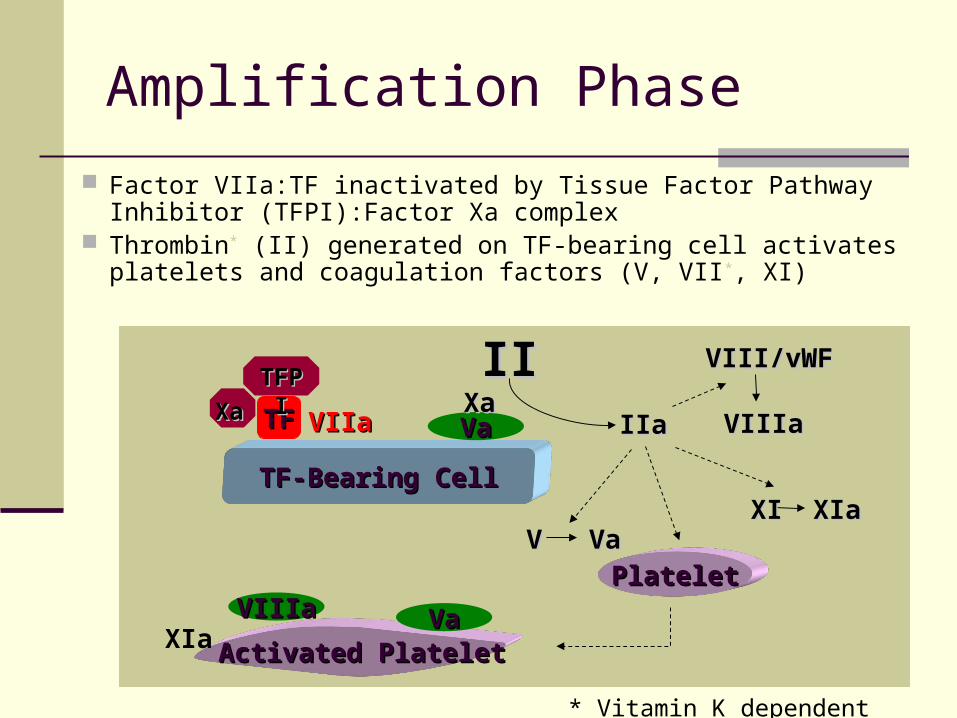
Amplification Phase
Factor VIIa:TF inactivated by Tissue Factor Pathway Inhibitor (TFPI):Factor Xa complex
Thrombin* (II) generated on TF-bearing cell activates platelets and coagulation factors (V, VII*, XI)
TF-Bearing CellTF-Bearing Cell
Activated PlateletActivated Platelet
PlateletPlateletVIIIaVIIIa VaVa
VaVaTFTF VIIaVIIaXaXa
VV VaVa
VIII/vWFVIII/vWF
VIIIaVIIIa
TFPITFPI
XaXa
IIIIIIaIIa
XIa
* Vitamin K dependent clotting factors
XIXI XIaXIa

Propagation Phase
Activated platelets serve as phospholipid platform for generation of large amounts of thrombin
Factor IXa with VIIIa (+ Ca2+) [tenase] activates factor X
Factor Xa with Va (+ Ca2+) [pro-thrombinase] produces large thrombin burst
Thrombin then generates fibrin and fibrin clot
Activated PlateletActivated Platelet
TF-Bearing CellTF-Bearing Cell
TFTF
VIIIaVIIIa VaVa
VIIaVIIa
IIIIIXaIXa XX
IXaIXa IIaIIaXaXa
IXIX
IXIX
XIaXIa
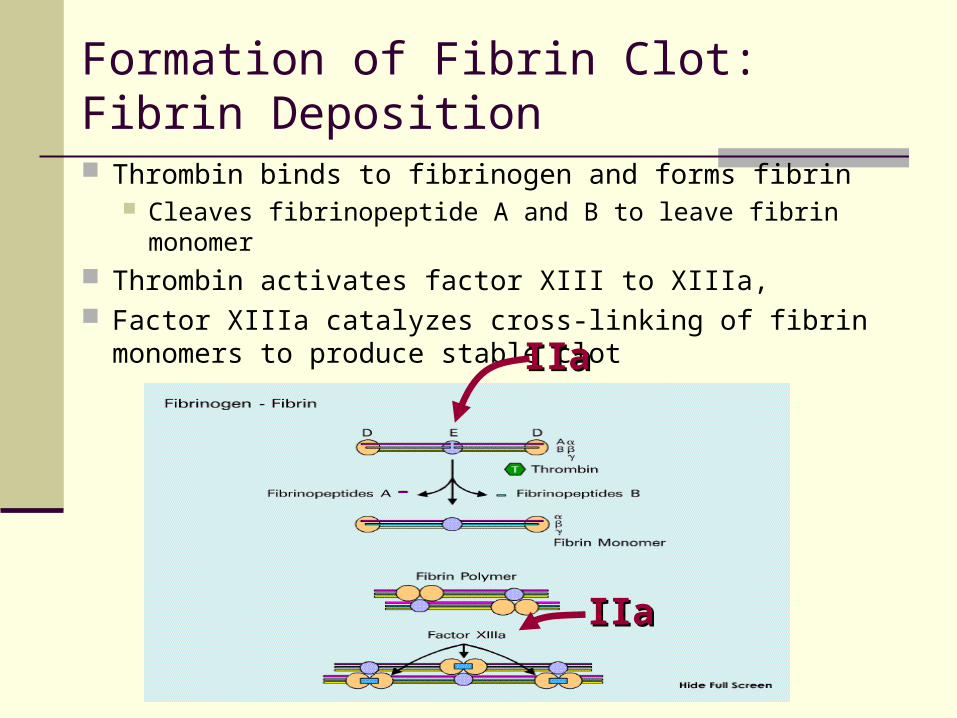
Formation of Fibrin Clot: Fibrin Deposition
Thrombin binds to fibrinogen and forms fibrin Cleaves fibrinopeptide A and B to leave fibrin monomer
Thrombin activates factor XIII to XIIIa, Factor XIIIa catalyzes cross-linking of fibrin monomers
to produce stable clot IIaIIa
IIaIIa

Coagulation Factor Pathway in Vivo

Fibrinolysis
Fibrin clots are temporary scaffolding that allow for cellular wound healing Dissolution of clots needed to maintain vessel
patency Plasmin (converted from plasminogen) is primary agent
of fibrinolysis Fibrinolysis is controlled by
Activators of plasminogen activation Inhibitors of plasminogen activation Inhibitors of plasmin

Fibrinolysis and Plasmin Inhibition
Fibrinolysis Plasminogen and t-PA bind to fibrin lysine sites
Activated plasmin protected from degradation by alpha2-antiplasmin
Plasmin optimally positioned to degrade fibrin
Plasmin Inhibition Circulating plasmin is rapidly inactivated by alpha2-
antiplasmin Prevents systemic (widespread) fibrinolysis
Thrombin activated fibrinolytic inhibitor (TAFI) removes lysine sites from fibrin to downregulates fibrinolysis during initial clot formation
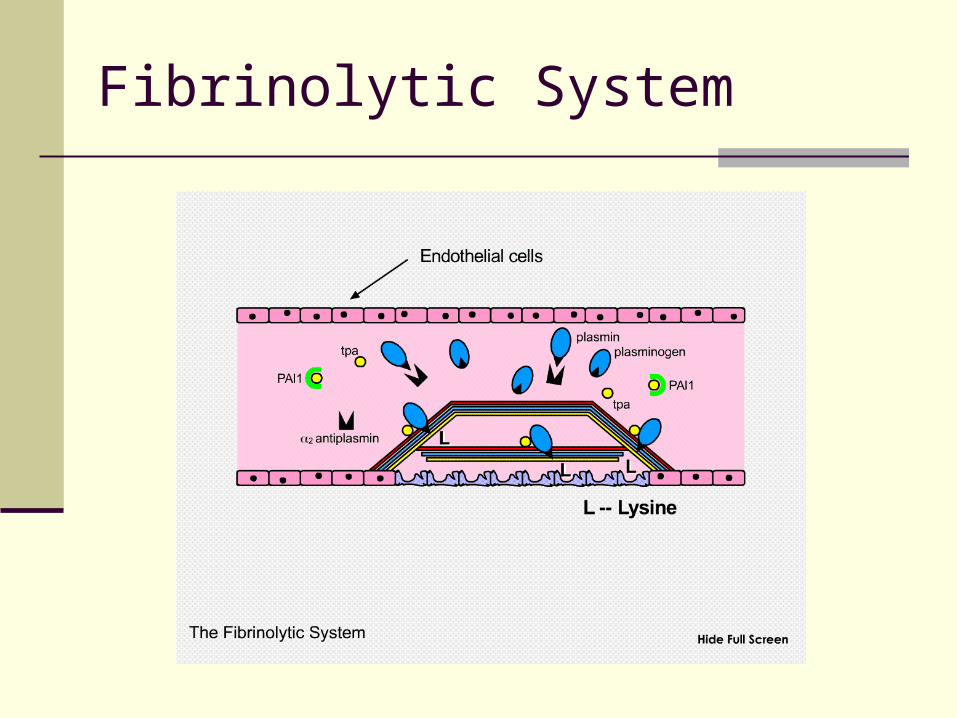
Fibrinolytic System

Laboratory Investigations

Platelet Disorders
Platelet Count Platelet Morphology (blood film) Platelet Aggregation / Release von Willebrand Studies
von Willebrand Antigen Ristocetin Cofactor von Willebrand Multimers
Bleeding Time

Coagulation Factor Disorders
Prothrombin Time / International Normalized Ratio
Activated Partial Thromboplastin Time Thrombin Time 50:50 Mixing Studies Coagulation Factor Assays

Activated Partial Thromboplastin Time
XII XIIa
XI XIa
IX IXa
X Xa +VIIIa
II IIa
VIIa VII +TF
Fibrinogen Fibrin
INT
RIN
SIC
EX
TR
INS
IC
COMMON
+Va

Activated Partial Thromboplastin Time
XII XIIa
XI XIa
IX IXa
X Xa +VIIIa
II IIa
VIIa VII +TF
Fibrinogen Fibrin
INT
RIN
SIC
EX
TR
INS
IC
COMMON
+Va

XII XIIa
XI XIa
IX IXa
X Xa +VIIIa
II IIa
VIIa VII +TF
Fibrinogen Fibrin
INT
RIN
SIC
EX
TR
INS
IC
COMMON
+Va
Thrombin Time (TT)

Fibrinolytic System
Euglobulin Lysis Alpha 2 Antiplasmin Factor XIII levels

Therapy

Platelet Transfusions - Products
Random Donor PlateletsRandom Donor Platelets Separated from whole blood donations Dose = 5 units (250-300 mls) ABO matched preferable but not mandatory Increases platelet ct by 5-10 x 109/L per unit
Single Donor PlateletsSingle Donor Platelets Collected from 1 donor by apheresis Equivalent to 5-6 random donor platelets Decrease donor exposure Can be matched if patient has identified antibodies to
platelets (alloimmune platelet refractoriness)

Indications for Platelet Transfusions
ThrombocytopeniaThrombocytopenia
Platelet DysfunctionPlatelet Dysfunction Congenital Acquired
TherapeuticTherapeutic Plat ct < 50x109/L Plat dysfunction
ProphylacticProphylactic Prior to invasive
procedures Plat ct < 50-100x109/L
Prevent spontaneous bleeding
Plat ct 10x109/L

Fresh Frozen Plasma
Made from whole blood within 8 hrs of collection Contains all coagulation factors
Minimum factor VIII level of 0.7 IU/ml 200-250 mls / unit Effect may only last 4 hrs (t1/2 of factor VII)
Indications:Indications:
INR / PTT > 1.5 x normal
andand
1. Bleeding or
2. Emergency procedure or operation

Fresh Frozen Plasma
Dose:Dose: 10-15 ml/kg for bleeding patients Sufficient to increase all individual coagulation factors
by 30% (minimum hemostatic level) 5-7 ml/kg may be sufficient for warfarin reversal
AlternativesAlternatives Vitamin K
2 mg will correct INR in 12 -24 hrs No effect on PTT (factors VIII, IX, XI) Oral dose more effective than subcutaneous Intravenous associated with anaphylactic reactions

Cryoprecipitate
Precipitate collected from plasma thawed at 40C 10-15 mls/unit Contains specific clotting factors from plasma
Factor VIII Fibrinogen von Willebrand’s Factor Factor XIII
IndicationsIndications1. Fibrinogen < 1.0 g/L2. Dysfibrinogenemia

Cryoprecipitate
DoseDose 1 unit / 5-10 kg body weight (total 8-10 units) t ½ of 3-5 days
AlternativesAlternatives Factor VIII or IX Deficiency
recombinant factor VIII or IX Von Willebrand’s Disease
virally inactivated plasma derived factor concentrates Other factor deficiencies
recombinant factor concentrates virally inactivated plasma derived factor concentrates

DDAVP
Synthetic vasopressin Release of VIII and vWF from endothelium May also help with platelet dysfunction 2-3 fold rise in levels Dose – 0.3 ug/kg q 12-24 hours Tachyphylaxis may occur after 2-3 doses

Antifibrinolytics
Tranexamic acid (Cyclokapron) 20-25 mg/kg po or 10 mg/kg IV q8h
Epsilon aminocaproic acid (Amicar) 50-60 mg/kg po q6h
Competitive inhibition with plaminogen activator (t-PA) Prevents fibrinolysis (clot breakdown) Promotes thrombosis Relative contraindication in
renal bleeding

Recombinant Factor VIIa
Initiates coagulation by interaction with TF Only approved for treatment of hemophilia with
inhibitors Treat/prevent bleeding in other patient groups?
Efficacy not proven Risk of thrombosis? Primary use is bleeding patients refractory to other
treatments Dose for hemophilia 90 ug/kg q2-3h
Lower dose often effective in other patients 30-40 ug/kg Vials of 1.2 mg, 2.4mg, 4.8mg
Cost ~ $1000/mg

“Hemostasis” Poker

One-of-a-Kind
DisordersINR aPTT TT/Fib Platelets
N N N
N N N
N N N
N N N
Liver diseaseVit K defCoumadinFactor VII
Heparin Antiphospholipid AbFactor VIII, IX, XIvon Willebrand’s(Factor XII)
HypofibrinogenemiaDysfibrinogenemiaThrombin InhibitorsHeparin
ITPTTP/HUSDrugsBone MarrowSplenomegaly

Pair
DisordersINR aPTT TT/Fib Platelets
N N
Factor II, V, X, Coumadin Overdose

Three-of-a-Kind
DisordersINR aPTT TT/Fib Platelets
N
Liver disease (acute)

Full House
DisordersINR aPTT TT/Fib Platelets
Liver disease (chronic)DIC

Nothing
Von Willebrand’s Disease Mild Hemophilia A or B Mild Factor XI deficiency Platelet Function Disorder Factor XIII deficiency Alpha 2 antiplasmin deficiency Plasminogen Activator Inhibitor deficiency