m b g 8 6 8 0
DESCRIPTION
Rui Pires Martins PhD Candidate, CMMG. M B G 8 6 8 0. computer applications in molecular genetics. before we start…. changes to .login file? created two directories in genetics “traces” “class” (or something??) transferred a copy of files to “class” by wsFTP or through Windows. - PowerPoint PPT PresentationTRANSCRIPT

M B G 8 6 8 0Rui Pires Martins PhD Candidate, CMMG
computer applications in molecular genetics

before we start…
• changes to .login file?
• created two directories in genetics• “traces”• “class” (or something??)
• transferred a copy of files to “class”• by wsFTP or through Windows
Mgb8680 | DNA sequencing

• DNA sequencing• chromatogram/trace data• chromas v.1.45• staden suite
• preGAP4• GAP4• trev• spin
Mgb8680 | DNA sequencing
outline

Mgb8680 | DNA sequencing
DNA sequencing jargon
• read a DNA sequence
• trace a chromatographic representation of DNA sequencing data
• contiguous sequence several reads with common spans joined together
• consensus the resulting sequence from several contigs that overlap
• template “sense” strand
• complement “anti-sense” strand
5’ ATTGGAGATCCGACTAATCCA 3’3’ TAACCTCTAGGCTGATTAGGT 5’
Mgb8680 | DNA sequencing
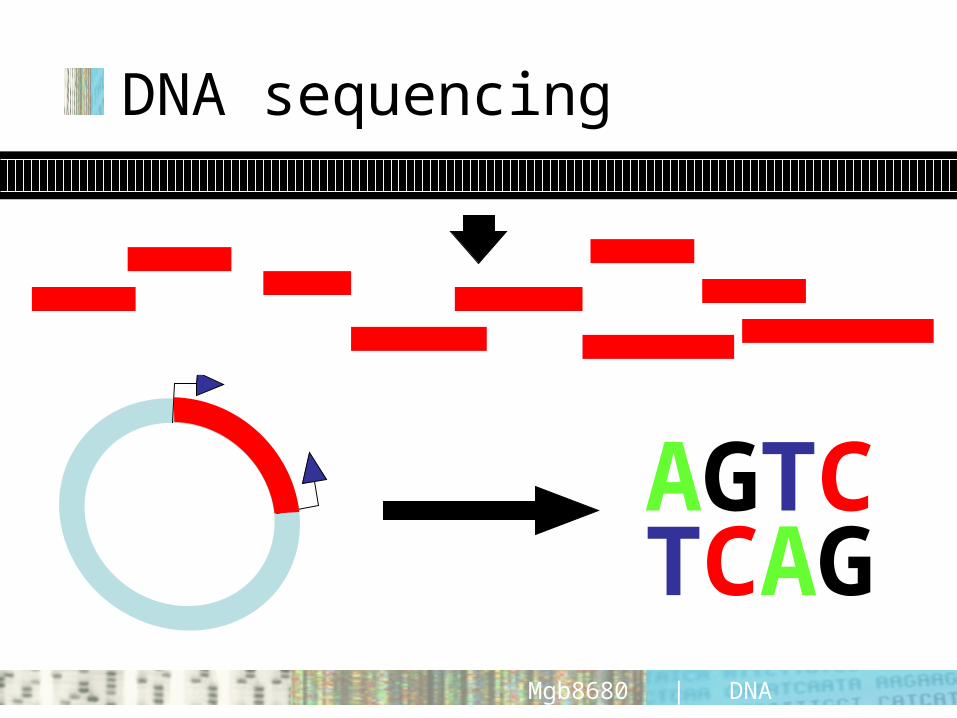
DNA sequencing
AGTCTCAG
Mgb8680 | DNA sequencing
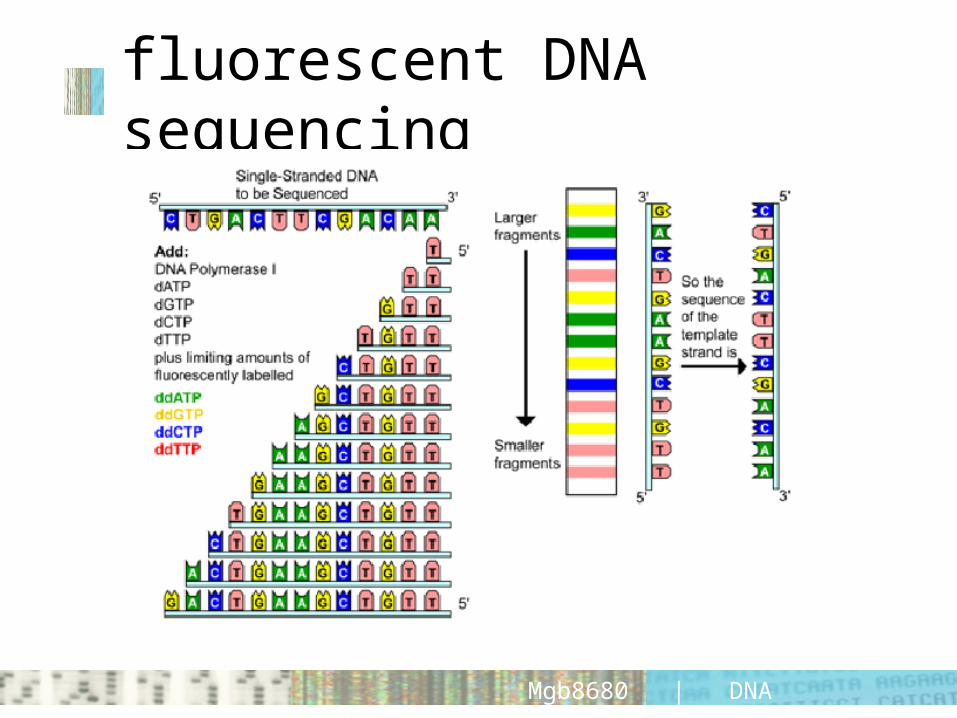
fluorescent DNA sequencing

• each nucleotide is colour coded• “good” sequence reads have well-defined peaks
Mgb8680 | DNA sequencing
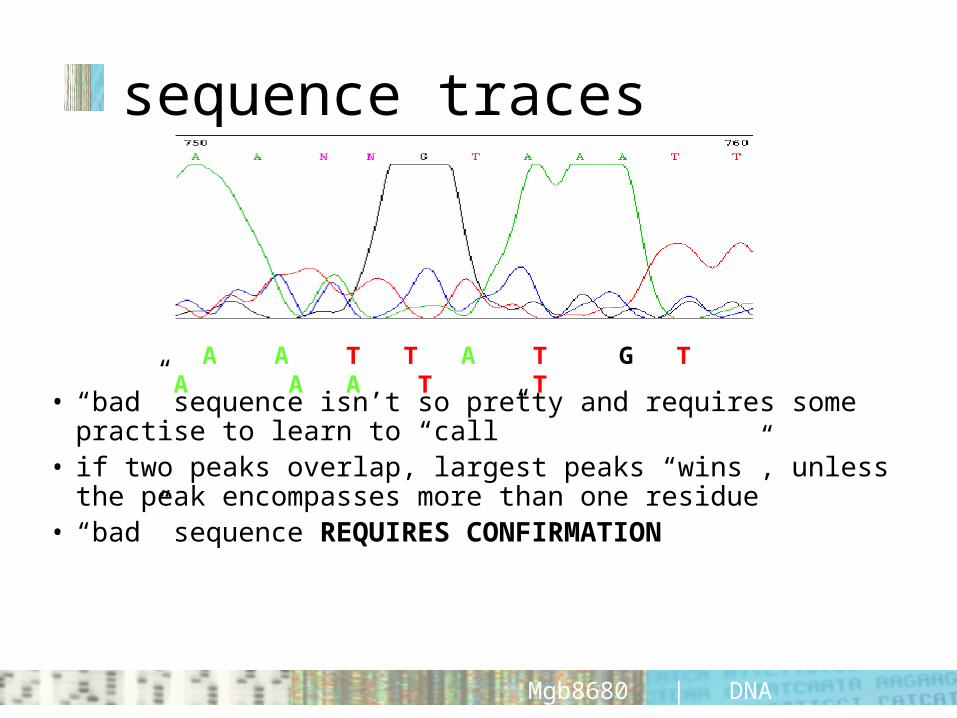
sequence traces
Mgb8680 | DNA sequencing
sequence traces
• “bad” sequence isn’t so pretty and requires some practise to learn to “call”
• if two peaks overlap, largest peaks “wins”, unless the peak encompasses more than one residue
• “bad” sequence REQUIRES CONFIRMATION
A A T T A T G T A A A T T

Mgb8680 | DNA sequencing
Chromas v.1.45• basic chromatogram/trace reading/viewing
programme for ab1 and scf files• freeware, works in Windows environments• some limited editing capabilities
• examples: forward.ab1 & reverse.ab1• Compare to forward.seq and reverse.seq
Mgb8680 | DNA sequencing
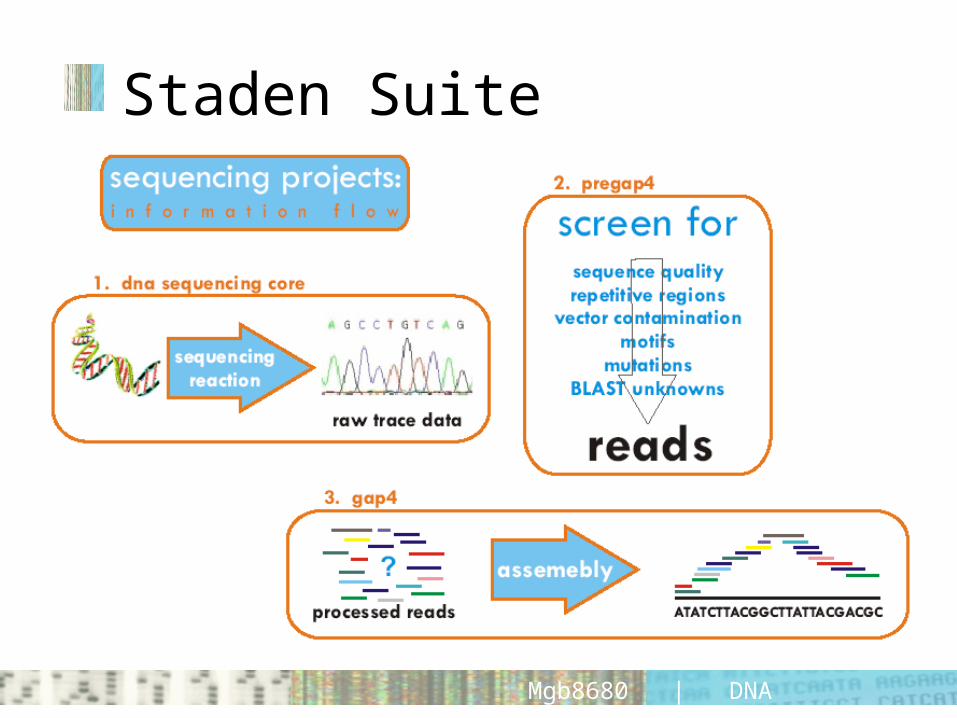
Staden Suite

Mgb8680 | DNA sequencing
Staden suite• very comprehensive suite of programmes for
sequence analysis, manipulation and assembly• (was?) free to academics• preGAP4 processes/manipulates raw data prior
to assembly• GAP4 (genome assembly) assembles/
manipulates processed reads into contigs; analyzes sequence integrity; organizes sequencing projects
• trev trace viewing programme; can be used along GAP or on its own.

Mgb8680 | DNA sequencing
Staden suite• examples: lb3.ab1, lb4.ab1, ub3l.ab1,
ub3lup.ab1, ub4.ab1, ubml.ab1, ubmup.ab1• vector file: pBSK+antisense5to3
• you will learn to read these files into preGAP4; process them; then assemble the files into a contig using GAP4.
• trev will be used to edit the sequence reads• you will also learn to produce a finished
sequence file that could be submitted to GenBank

Mgb8680 | DNA sequencing
assignment1. Finish the assembly of the 7 files into as long a contig as you can generate. Be
sure to edit any sequence ambiguities as you go. Submit a final text file (fastA format) with this sequence.
2. Repeat the assembly. Only this time, shotgun all 7 files at once. What happened? Are there any advantages to the manual process? (HINT: you’ll have to create a new database in Staden to do this)
3. Use one of the trace readers/editors to edit the following residues from reverse.ab1
270 280 290 300
GCCCCTACACTCGNNNGCCTGCCCGCCTCTCAA4. Assemble the forward.ab1 and reverse.ab1 files into a staden reads database.
What is different this time (i.e. do you notice any annotations or tagged regions in the contigs; and if so what?) What advantages can you see to tagging these regions before you try to assemble them?
email answers as text to [email protected] by Sunday night
help/questions can also be directed to [email protected] or through
MSN messenger ([email protected])