luan van cac chat chong oxy hoa
TRANSCRIPT

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 1/182
I
CONTENTS
ACKNOWLEDGEMENTS VII
SUMMARY VIII
CHAPTER 1 INTRODUCTION
1.1 GENERAL 1
1.2 USES OF COUPLED PHENOLICS 4
1.2.1 Antioxidants 4
1.2.2 Other Uses 6
1.3 METHODS OF PREPARATION OF COUPLED PHENOLICS 6
1.3.1 General Types of Coupling Reaction Mechanisms 8
1.3.2 Chemical and Electrochemical Methods for Oxidatively
Coupling Phenolics 15
1.3.2.1 Chemical oxidative coupling 15
1.3.2.1.1 Vanadium(IV) and vanadium(V) 16
1.3.2.1.2 A (nitrosonaphtholato)metal complex 18
1.3.2.1.3 Activated manganese dioxide 21
1.3.2.1.4 Cupric salts 23
1.3.2.2 Electrochemical oxidative coupling 25 1.3.2.2.1 Direct electrochemical oxidations 25
1.3.2.2.2 Indirect electrochemical oxidations 29
1.4 OBJECTIVES AND MOTIVATION FOR THIS STUDY 32

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 2/182
II
CHAPTER 2 EXPERIMENTAL
2.1 MATERIALS 34
2.1.1 Reagents for Synthesis and Analysis 34
2.2 SYNTHETIC PROCEDURES 36
2.2.1 Reagents for Analysis 36
2.2.1.1 Preparation of 3,3’-di-t -butyl-4,4’-dihydroxybiphenyl 36 2.2.1.2 Preparation of 3,3’,5,5’-tetra-t -butyldiphenoquinone 36
2.2.1.3 Preparation of 3,3’,5,5’-tetra-t -butyl-4,4’-
dihydroxybiphenyl 37
2.2.1.4 Preparation of 3,3’,5,5’-tetra-t -butyl-2,2’-
dihydroxybiphenyl 37
2.2.1.5 Preparation of 3,3’,5,5’-tetramethyl-2,2’-
Dihydroxybiphenyl 38
2.2.2 Preparation of Coupling Agents 38
2.2.2.1 Preparation of silver carbonate/celite 38
2.2.2.2 Preparation of barium manganate 39
2.2.2.3 Preparation of a (nitrosonaphtholato)metal complex
(MnII(1-nnap)2) 39
2.2.2.4 Electrochemical preparation of cerium(IV) from
cerium(III) using a divided cell 40
2.2.2.5 Preparation of silver oxide 42
2.3 EXPERIMENTAL PROCEDURES 43
2.3.1 Oxidative Coupling Reactions 43
2.3.1.1 Oxidation of alkylphenols using silver

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 3/182
III
carbonate/celite 43
2.3.1.2 Oxidation of alkylphenols using copper complexes
of dicarboxylic acids 45 2.3.1.3 Oxidation of alkylphenols using manganese(III)
acetate 44
2.3.1.4 Oxidation of alkylphenols using barium manganate 44
2.3.1.5 Oxidation of alkylphenols using a (nitrosonaphtholato)-
metal complex 45
2.3.1.6 Oxidation of alkylphenols using FeCl3 in an organic
solvent 45
2.3.1.7 Oxidation of alkylphenols using FeCl3 without
solvent 45
2.3.1.8 Oxidation of alkylphenols using Ag2O 46
2.3.1.9 Oxidation of alkylphenols using lead tetra-acetate 46
2.3.1.10 Oxidation of alkylphenols using Ce4+ 46
2.3.1.11 Oxidation of alkylphenols using potassium
ferricyanide 47
2.3.2 Determination of Ce(III) Remaining After the Electrochemical
Oxidation of Ce(III) to Ce(IV) 47
2.3.3 Dealkylation of Dihydroxybiphenyls 48
2.4 ANALYTICAL TECHNIQUES 48
2.4.1 High Performance Liquid Chromatography (HPLC) 48
2.4.2 Nuclear Magnetic Resonance (NMR) Spectroscopy 50 2.4.3 Fourier Transform Infra Red (FTIR) Spectroscopy 50
2.4.4 Gas Liquid Chromatography-Mass Spectrometry (GC-MS) 51
2.4.5 Molecular Orbital Calculations 51
2.5 TERMS AND DEFINITIONS 52

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 4/182
IV
CHAPTER 3 DISCUSSION
3.1 MODES OF PHENOLIC COUPLING 53
3.1.1 Molecular Orbital Calculations for the Coupling of Phenol 56
3.2 THE OXIDATIVE COUPLING OF 2-t -BUTYLPHENOL 58
3.2.1 The Range of Possible Products During the OxidativeCoupling of 2-t -Butylphenol 60
3.2.2 Oxidative Coupling Reactions of 2-t -Butylphenol
using Various Oxidants 63
3.2.2.1 Vanadium(V) oxytrichloride and vanadium(IV)
tetrachloride as coupling agents 65
3.2.2.2 Silver carbonate supported on celite as coupling
Agent 65
3.2.2.3 Copper acetate, in the presence of a dicarboxylic acid,
as coupling agent 70
3.2.2.4 Manganese(III) acetate as coupling agent 71
3.2.2.5 Barium manganate as coupling agent 72
3.2.2.6 Ferric chloride as coupling agent 73
3.2.2.7 Silver oxide as coupling agent 74
3.2.2.8 Potassium ferric cyanide, lead tetra-acetate,
a (nitrosonaphtholato)metal complex and
cerium(IV) sulphate as oxidants 75
3.2.3 Concluding Remarks on the Oxidative Coupling of 2-t -
Butylphenol 75
3.3 THE OXIDATIVE COUPLING OF 2,6-DI-t -BUTYLPHENOL 76

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 5/182
V
3.3.1 Molecular Orbital Calculations for the Oxidative Coupling of
2,6-Di-t -Butylphenol 77
3.3.2 Oxidative Coupling Reactions of 2,6-Di-t -ButylphenolUsing Various Oxidants 81
3.3.2.1 Silver oxide as coupling agent 83
3.3.2.2 Copper(II) acetate/oxalic acid as coupling agent 87
3.3.3 Concluding Remarks on the Oxidative Coupling of 2,6-Di-t -
Butylphenol 88
3.4 THE OXIDATIVE COUPLING OF 2,4-DI-t -BUTYLPHENOL 88
3.4.1 Molecular Orbital Calculations for the Oxidative Coupling
of 2,4-Di-t -Butylphenol 90
3.4.2 Oxidative Coupling Reactions of 2,4-Di-t -Butylphenol Using
Various Oxidants 94
3.4.2.1 Ferric chloride as coupling agent 95
3.4.2.2 Silver oxide as coupling agent 97
3.4.2.3 Potassium ferric cyanide as coupling agent 100
3.4.2.4 Cerium as coupling agent 104
3.4.2.4.1 Identification of Ce(IV) as the preferred
oxidant 104
3.4.2.4.2 Oxidation in MeSO3H mediated by
Ce(IV) ions 107
3.4.2.4.3 Reaction mechanism for the oxidative
coupling of 2,4-di-t -butylphenol usingCe(IV) 119
3.4.3 Concluding Remarks on the Oxidative Coupling of 2,4-Di-t -
Butylphenol 126

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 6/182
VI
3.5 THE OXIDATIVE COUPLING OF 2,4-DIMETHYLPHENOL 127
3.5.1 Oxidative Coupling Reactions of 2,4-Dimethylphenol UsingVarious Oxidants 131
3.5.1.1 Ferric chloride as coupling agent 132
3.5.1.2 Potassium ferric cyanide as coupling agent 136
3.5.1.3 Cerium(IV) as coupling agent 138
3.5.1.3.1 Reaction mechanism for the oxidative coupling
of 2,4-dimethylphenol using Ce(IV) 138
3.5.2 Concluding Remarks on the Oxidative Coupling of 2,4-Dimethylphenol 148
3.6 BUTYLATED PHENOLIC COUPLINGS: COMPARISONS 149
3.6.1 Reactions of 2-t -Butylphenol and 2,6-Di-t -Butylphenol with Ag2O
and Cu(OAc)2/Oxalic Acid 149
3.6.2 Reactions of 2,4-Di-t -Butylphenol and 2,6 -Di-t -Butylphenol with
Ce(IV) in MeSO3H 151
CHAPTER 4 CONCLUSION AND FINAL COMMENTS
REFERENCES 159
APPENDIX 169

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 7/182
VII
ACKNOWLEDGEMENTS
The author wishes to express his sincere appreciation to:
• My promoters, Dr B. Barton and Prof B. Zeelie, for their assistance and
enthusiasm for this work.
• The NRF and Port Elizabeth Technikon for financial support.
• My fellow students, Mteza, Nigel, Daniël, Melissa and Knowledge for their
moral support.
• Dr S. Gouws, Dr G. Rubidge and Prof P. Loyson for their assistance.• The staff and students of the Department of Chemistry at the Port Elizabeth
Technikon for their assistance and moral support.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 8/182
VIII
SUMMARY
The oxidative coupling of 2,6-di-t -butylphenol under mild reaction conditions is well
documented and the subject of many patents. However, the coupling of other mono-
and di- substituted phenols is not as well documented and thus there is scope for
further investigation for providing a convenient, environmentally friendly and
economically viable method for the oxidative coupling of these phenols.
In this study, the oxidative coupling of a variety of alkylated phenolic substrates, 2-t -
butylphenol, 2,6-di-t -butylphenol, 2,4-di-t -butylphenol and 2,4-dimethylphenol, using arange of different oxidizing agents, were investigated by means of experimental
and/or theoretical means. The dibutylated aromatics provided the highest selectivities
to their respective coupled products, with results obtained with the dimethyl analogue
being only satisfactory, and that for 2-t -butylphenol being totally inefficient.
PM3 Molecular orbital (MO) calculations were used to predict the possible modes of
coupling for the substrates 2,6-di-t -butylphenol and 2,4-di-t -butylphenol, and these
results were then compared with those obtained experimentally in the laboratory.
Preliminarily, the coupling of unsubstituted phenolics was also assessed by means of
MO calculations.
Much emphasis was placed on Ce(IV) as the oxidant, and the reaction conditions
under which it was used and the results that were obtained have not been reported
before and are therefore novel. The oxidation of 2,4-di-t -butylphenol using Ce(IV) in
the presence of methanesulphonic acid was optimized to afford high yields andselectivities to the desired ortho C-ortho C coupled product under mild reaction
conditions. Various reaction parameters were also investigated in this case, such as
varying the MeSO3H concentration, the solvent, the reaction temperature, the reaction
time, the substrate loading, the rate of oxidant addition and the substrate to oxidant
ratio. Ce(IV) also gave a high selectivity to the para C-para C coupled product when

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 9/182
IX
using 2,6-di-t -butylphenol as the substrate. However, it was not as effective with 2,4-
dimethylphenol, and even less so with 2- t -butylphenol.
The oxidation reactions of 2-t -butylphenol and 2,4-dimethylphenol with various
coupling agents were also investigated with the intention of obtaining high selectivities
to the respective desired coupled products. In these studies, 2-t -butylphenol afforded
a large number of products, irrespective of the oxidant used. The dimethyl analogue
was more selective, but results were not optimal. It was clear that the number of
substituents on the phenol ring, their nature and their position with regards to the
hydroxyl moiety was of great importance and made a significant impact on thepreferred coupling mode of the substrate. It was observed that steric effects also
played a major role in the outcome of these reactions: 2,6-di-t -butylphenol never
afforded any C-O coupled products whereas 2-t -butylphenol, 2,4-di-t -butylphenol and
2,4-dimethylphenol all appeared to undergo some C-O coupling.
Finally, reaction mechanisms were provided for both the K3Fe(CN)6 and Ce(IV) work,
these reacting in basic and acidic media, respectively. It was proposed that both of
these mechanisms operate through the initial formation of the phenoxyl radical.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 10/182
1
CHAPTER 1
INTRODUCTION
1.1 GENERAL
The chemical industry today is faced with major economic and environmental
challenges. We as scientists have a responsibility towards the efficiency and
profitability of the industry. We thus have to look at developing sustainable processes
that have long-term economic and environmental viability. The chemical industry has
been continually driven by this need for better quality products and much more
effective and efficient production procedures, resulting in an industry that is currentlywell established and one that continues to grow.1
From an initial slow start in the 1850’s, the chemical industry has made tremendous
strides in the field of organic synthesis, this being primarily due to enhanced
competition between the various chemical companies, leading to increased numbers
of products becoming commercially available.2 During the twentieth century, the
industry has experienced exponential growth and this has led to a major improvement
in both our living standards and life expectancy.
Phenol and other phenolics are currently some of the more versatile and important
industrial organic chemicals. Phenol itself was first isolated from coal tar by Runge.3
In 1843, C.F. Gerhardt prepared phenol by heating salicylic acid with lime; the
resulting product was given the name ‘phenol’.4 Until World War II, phenol was
essentially a coal tar extraction product, but due to an increased demand, synthetic
methods replaced extraction from natural resources. Currently, only small amounts of
phenol are obtained from coal tar (SASOL); larger quantities are being formed in
coking or by the low pressure carbonization of wood and brown coal, as well as from
oil cracking. The earlier methods of phenol synthesis via benzenesulphonic acid using
alkali fusion (Scheme 1) and via chlorobenzene (Scheme 2)5 have since been
replaced by more economically and environmentally friendly processes such as the
Hock process, which utilizes cumene as substrate.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 11/182
2
SO3H OH
1. NaOH, 300°C
2. H3O+
Cl OH
1. aq. NaOH, 340°C, 2500 psi
2. H3O++ NaCl
Scheme 1: Preparation of phenol from benzenesulphonic acid (alkali fusion)
Scheme 2: Preparation of phenol from chlorobenzene
The Mitsui group is the world’s second largest producer of phenol through the Hock
process. Acetone is produced as a byproduct in this process, but this is not deemed
a disadvantage of the Hock method since there is also a high demand for acetone
worldwide. The Hock process involves the alkylation of benzene with propene to
afford isopropylbenzene (cumene); cumene is oxidized to the corresponding tert -
hydroperoxide, which is then ultimately cleaved to yield phenol and acetone (Scheme
3).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 12/182
3
OH
[O]
OOH
H3O+
+
O
cumene cumenehydroperoxide
acetone
Scheme 3: The Hock process for the production of phenol
Plants operating the cumene process are found in the USA, Canada, France, Italy,
Japan, Spain, Eastern Europe and Germany, with an overall capacity of 5 000 000
tons per annum.6
By noting the Japanese production output and usage of phenol and phenolic resins
(in tons) through the years 1996 to 2000, merely as an example, as contained in
Table 1.1, one can better comprehend the importance of these compounds in an
industrial capacity (Table 1.1).7
Table 1.1 Japanese production of phenol and phenolic resins (in tons)
Chemical/Year 1996 1997 1998 1999 2000
Increase
1999/2000
Phenol 768 833 851 888 916 3.2%
Phenol Resins 294 303 259 250 262 4.8%
Alkylphenols, such as xylenols, cresols, octylphenols and tert -butylphenols are
generally produced by the alkylation of phenol with methanol or the corresponding
olefins. Alkyphenols can then be reacted further by oxidative coupling to form the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 13/182
4
dihydroxybiphenyls, the focus of this investigation. All of these products have
considerable economic importance because they are used to manufacture
thermosets, insulating foams, adhesives, laminates, impregnating resins, and serveas raw materials for varnishes, herbicides and insecticides.
1.2 USES OF COUPLED PHENOLICS
1.2.1 Antioxidants
One of the more important uses of many phenolic materials is their ability to serve asantioxidants. Antioxidants are merely compounds that are added to, or occur in,
various materials, both living organisms and synthetic organic materials –
antioxidants then readily react with free radicals that would otherwise damage the
materials prematurely. The free radicals are normally the result of autoxidation, a
process that occurs spontaneously all around us all the time due to the oxygen in the
air.
In human blood plasma, α-tocopherol, well known as a component of vitamin E, has
proved to be the most efficient phenol derivative to date to trap damaging phenoxyl
radicals (ROO•),8,9 caused by autoxidation, and therefore acts as an efficient
antioxidant. Uninhibited free radical peroxidation in vivo is implicated in a wide variety
of degenerate diseases such as cancer, heart disease, inflammation and even aging.
Thus, over the last two decades, there has been a tremendous increase in the
research of phenols as antioxidants.10,11
Phenols owe their efficient antioxidant activity to their ability to scavenge radicals by
hydrogen or electron transfer in processes that are much faster than radical attacks
on the substrate. The antioxidant property can be related to the readily abstractable
phenolic hydrogen as a consequence of the relatively low bond dissociation enthalpy
of the phenolic O-H group. Thus phenols and dihydroxybiphenyls are an extremely
important class of antioxidants.12,13

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 14/182
5
To understand the antioxidant strength of phenols and diols, we need to discuss the
reaction of molecular oxygen with organic molecules under mild conditions
(autoxidation). It may be represented by the following chemical reactions (1 – 5).
Initiation: production of RO• (1)
Propagation: R• + O2 → ROO• (2)
ROO• + RH → ROOH + R• (3)
Termination: ROO•
+ RO•
→ products (4)ROO• + PhOH → ROOH + PhO• (5)
While reaction 1 is very fast, having a rate constant of approximately 109 M-1s-1,
reaction 4 is much slower at 101 M-1s-1. Oxidative degradation can therefore occur
readily, and the use of low concentrations of antioxidants is thus important for all
living organisms and for many commercial products in order to reduce the rate of
degradation.
Both phenols and dihydroxybiphenyls behave as antioxidants because of their ability
to undergo reactions such as that shown in reaction 5, thus trapping potentially
damaging peroxyl radicals. This is a much faster reaction than the attack of the
peroxyl radicals on the organic substrate (reaction 3) due to the low bond dissociation
energies for the oxygen-hydrogen bond in the hydroxyaromatic.
The substituents on the aromatic ring have a profound effect on the ability of the
phenol/diol to donate a hydrogen atom. Only those phenols bearing electron-
donating substituents are active as antioxidants, particularly if these are at the ortho
and/or para positions relative to the hydroxyl moiety. This is not unexpected since
electron-donating groups are expected to lower the phenolic O-H bond dissociation
enthalpy and thus increase the reaction rates with peroxyl radicals, implying a more
efficient antioxidant.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 15/182
6
1.2.2 Other Uses
Dihydroxybiphenyls are used in toner resins to increase surface additive adhesionand to optimize cohesion between the toner particles.14 It also acts as a binder resin,
thus eliminating the need for gels to be present in the toner, and enabling the
magnetic brush development system to achieve consistent, high quality copy
images.15
They are also used as inexpensive and simple starting materials for producing
polycarbonate resins,
16
which are used to reinforce rubber vulcanizates.
17
Dihydroxybiphenyls are extensively used in coating agents,18 glass moulding19 and
infrared-reflecting colourants,20 and they are reacted with acid catalysts to form
polymers which are used as a polymer scale deposition preventative agent.21
1.3 METHODS OF PREPARATION OF COUPLED PHENOLICS
The diversity of phenol oxidation products offers interesting synthetic possibilities for
the preparation of simple and polymeric molecules containing phenolic and/or quinoid
structural elements; these can be formed from both like and unlike radical
species.13,22 The successful synthesis of various natural products from phenols has
been well documented from the 1950’s to the present.23-28
Biogenetic oxidative coupling routes were first investigated in 1957,29,30 and the
prevalence of the overall coupling process in the biosynthesis of natural products was
authenticated. Thus the oxidative coupling step has been found to be extremelyimportant in the natural formation of compounds as diverse as lignins,31 lignans,32
tannins,33 plant pigments,22 and an estimated 10% of all known alkaloids.23 (Lignin is
a complex biopolymer that accounts for 20-30% of the dry weight of wood. It is
formed by the free radical polymerization of substituted phenylpropane units to yield
polymers which have a number of functional groups such as aryl ethers, phenols and
benzyl alcohols.34)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 16/182
7
OH
R
OH
R
+ HO OH
R R
The major difficulty with oxidative coupling reactions of phenols is that a large variety
of potential products are possible from a single substrate when carried out in the
presence of various chemical or biological oxidants. This is because the phenolicmolecules are able to undergo both carbon-carbon (Scheme 4 shows para-para
coupling, though ortho-para coupling may also occur) and carbon-oxygen (Scheme 5)
coupling reactions.
Scheme 4: Carbon-carbon oxidative coupling (showingpara-para coupling)
Scheme 5: Carbon-oxygen oxidative coupling
The type of coupled product (whether C-C or C-O coupled) is also dependent on
whether the ortho or para positions bear substituents or not. In addition to these two
potential reaction products, the oxidative coupling of phenols also often allows for the
formation of polymeric materials which, in general, are undesirable (though there are
a few industrial processes where these are of great importance35,36). It has been
reported that when carbon-oxygen coupling occurs, there is a tendency for further
coupling to occur on the resultant substrate, and this leads to the formation of
polymeric products.37
OH
R
OH
R
+ OHO
R
R

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 17/182
8
To understand the effect that both the nature of the reactant and oxidant has on the
type of products that are formed, one must have an understanding of the various
reaction pathways that are possible, from a mechanistic point of view. A summary of literature reports dealing with the various mechanisms is now briefly discussed.
1.3.1 General Types of Coupling Reaction Mechanisms
The reaction pathway for the oxidative coupling of phenols has been extensively
investigated.38,39 There are two main modes of coupling that may be highlighted.
These are an external and an internal oxidation process. In the former, electrons aretransferred from the phenolic compound to an external oxidizing agent, whilst the
internal oxidation process involves an internal oxidation-reduction reaction in which
one substrate molecule is oxidized whilst another is simultaneously reduced. Since
there is no change in the net overall oxidation state, this process may be termed a
“non-oxidative coupling (NOC)” reaction.
In our investigations, only the external oxidative coupling process was studied. For
this reason, literature reports dealing only with this mode are summarized here.
External oxidative coupling reactions may be grouped into two separate classes,
those involving free radical intermediates, and those that are non-radical in nature.
These may further be subdivided into several general mechanistic types.
a) Mechanisms involving free radical intermediates
i) Direct coupling of two phenoxyl radicals (FR1)
ii) Homolytic aromatic substitution (FR2)iii) Heterolytic coupling preceded by two successive one-electron oxidation
steps (FR3)
b) Mechanisms which are non-radical in character
i) Heterolytic coupling preceded by a single two-electron transfer (NR1)
ii) Concerted coupling and electron transfer (NR2)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 18/182
9
It has previously been widely accepted that, in the field of phenol oxidations, the FR1
mechanism is the most viable (without discounting the FR2 mechanism). Most
reviewers have included the FR3 mechanism in their discussions but have attachedlittle importance to it. Until recently, no one has considered the NR1 and NR2
mechanisms as significant enough to warrant a discussion of them in this context.
The para-para (C-C) coupling of a simple 2,6-disubstituted phenol is used to illustrate
the five general types of processes (FR1, FR2, FR3, NR1 and NR2) as listed above.
In all cases, the oxidized phenolic species is written as the neutral phenol molecule,
and only intermediates are shown as unprotonated. The following scheme (Scheme6) highlights the FR1, FR2 and FR3 mechanisms.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 19/182
10
OH
RR
(1)
-e -, -H+RR
O
RR
O
+ (1)
FR2
FR1 coupling of two
FR3-e-
O
RR
+
O
RR
H H
H
OH
RR
-e-
-H+
O
R R
HH
O
R R
(2)
R R
OH
R R
OH
(3)
disproportionation
RR
OH
RR
OH
H
HH
-2e-, -2H+
-H+
pathway (a)
(4)
phenoxy radicals
+ (1)
phenoxy radical
tautomerization
Scheme 6: The FR1, FR2 and FR3 free radical mechanisms

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 20/182
11
The degree of protonation of the phenolic species in each of these mechanisms
depends on various factors, such as the acidity of the species, the nature of the
solvent and the pH of the solution.
The free radical processes are initiated by means of pathway (a) shown in Scheme 6.
The first one-electron transfer from the disubstituted phenol (1) to an oxidant results
in the formation of the phenoxyl radical which is stabilized by resonance, as shown in
the following scheme (Scheme 7).
Scheme 7: Resonance stabilization of the phenoxyl radical
The formation of the phenoxyl radical is well attested, for example by ESR.40,41,42 (It
has been shown9 that the subsequent dimerization thereof fits a diffusion-controlled
model.)
The phenoxyl radical is able to react in one of three ways, each leading to the same
product (Scheme 6).
• Firstly, it may homolytically combine with another phenoxyl radical by mechanism
FR1 to afford compound (2). This dicyclohexadienone rapidly tautomerizes inprotic media to the more stable aromatic biphenol product (3).
• Secondly, the phenoxyl radical may react with the initial substrate (1) via
mechanism FR2 to generate a dimeric radical. Upon loss of an electron and a
proton from this new radical, (2) is formed once again. However, the dimeric
radical may also disproportionate, leading to a dihydro product (4) as well as to (2).
As yet, compounds such as (4), although analogous to similar products produced
R R
O
R R
O
R R
O
RR
O

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 21/182
12
in free radical aromatic substitutions,45 have not yet been observed in oxidative
coupling reactions. This may perhaps be due to the fact that the conversion of (4)
to (3) is a facile one since (3) has enhanced stability due to its aromaticity.
• Thirdly, the phenoxyl radical may be further oxidized by removal of an electron, to
yield a phenoxyl cation, according to mechanism FR3. The initial substrate (1),
with concomitant hydroxyl proton loss, may then heterolytically couple with the
cation to afford (2).
Examples of the NR1 and NR2 non-radical processes are shown in Schemes (8) and
(9), respectively. In both illustrations, the oxidant is represented as a tripositive metalion (M3+), which forms an initial metal-phenolate complex with (1).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 22/182
13
RR
OH
(1)
+ M3+ - H+
O
RR
M2+
O
RR
+ M++ (1), - H+
RR
HH
O
O
RR
RR
OH
RR
OH
(3)
(2)
+
tautomerization
Scheme 8: The NR1 (non-radical) mechanistic pathway

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 23/182
14
OH
RR
(1)
-H+
R R
OM
R R
OH
2+
O
R
R
H
H
O
R
R
RR
OH
R
OH
R
(3)
(2)
- M+
+ M+32
tautomerization
As shown in Scheme 8, the metal complex decomposes into a phenoxyl cation with
concurrent reduction of the metal ion. Subsequently, heterolytic coupling similar to
that shown in Scheme 6 (the FR3 mechanism) affords compound (2) whichundergoes tautomerization, and so the desired product (3) is a result.
Objections, based on energetic grounds, to the formation and stabilization of cationic
intermediates in this mechanism may be obviated by considering the possibility of a
concerted electron transfer, as for the simple NR2 mechanism shown in Scheme 9.
Scheme 9: The NR2 mechanistic pathway

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 24/182
15
1.3.2 Chemical and Electrochemical Methods for Oxidatively Coupling
Phenolics
There has been a tremendous amount of research carried out on the oxidative
coupling of phenols that involves the use of a wide variety of chemical oxidants and/or
catalysts. These include manganese(III) complexes,26,27 silver carbonate/celite,28
molybdenum(VI) and (V),45 cupric salts,46 amongst numerous others.47-55 The
oxidative coupling of phenols through the use of electricity has been documented for
both direct56,57 and indirect58 electrochemical means, but these occur to a much
lesser extent as compared to that of chemical methods.
The wide variety of possible oxidation products that may be obtained under oxidative
coupling conditions is clearly indicated by examples from work done earlier by
scientists such as Barton,29 Thvagarajan59 and Pummerer.60 Subsequent research
has mainly concentrated on the coupling of di- and tri- substituted phenols, and the
literature is virtually devoid of reactions using mono-substituted substrates.
Furthermore, reports suggest that higher selectivities to the carbon-carbon coupled
products are achieved when the substituents on the aromatic ring are large and bulky,
such as the t -butyl moiety, since they prevent carbon-oxygen coupling due to the
steric hindrance that their bulk offers.
In the next sections, research utilizing both the chemical and electrochemical
methods (direct and indirect) for the oxidative coupling of phenols, is summarized.
1.3.2.1 Chemical oxidative coupling
From about as early as the 1920’s, chemists have been researching the oxidative
coupling of phenols using chemical oxidizing systems. It was thought that all
oxidative coupling reactions involved one electron transfers, and therefore that these
oxidations were all free radical reactions. The mechanisms by which the reactions
occurred, and the characteristics of the various oxidizing agents and/or catalysts,

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 25/182
16
were not investigated successfully because they were not well understood; it was
always assumed that coupling occurred through the bonding of two phenoxyl radicals
(FR1) to form the coupled biphenol. However, it has since become clear that thetypes of mechanisms involved are extremely dependent on the nature of the oxidant
and/or catalyst used. Some of these, including vanadium (IV) and (V), a
(nitrosonaphtholato)metal complex, activated manganese dioxide, and cupric salts,
and the reaction pathways they are involved in, will now be discussed further.
1.3.2.1.1 Vanadium(IV) and vanadium(V)
Vanadium(V) oxytrichloride (VOCl3) and vanadium(IV) tetrachloride (VCl4) have beenused to oxidatively couple phenols in aprotic solvents.61 When phenol (5) was used
as the substrate in the presence of VCl4, a dark insoluble residue was initially formed
which was accompanied by the vigorous evolution of HCl gas. This residue was
shown to be a form of vanadium-phenolate species, but when analyzed, the
elemental composition was not consistent with any simple structure. Acid hydrolysis
thereof afforded high yields of the para-para coupled product, identified as 4,4’-
diphenol (6). Also observed were the para-ortho and ortho-ortho coupled products,
identified as 2,4’-diphenol (7) and 2,2’-diphenol (8), as shown in the following scheme
(Scheme 10).
Scheme 10: The oxidative coupling of phenol using VCl4 as oxidant
OH
+ VCl4
OH
OH
OH
OH OH
HO
(5)
(6)
(7)(8)
+ +

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 26/182
17
When the hydrolysis step was carried out in the presence of deuterium oxide, no
carbon-deuterium bonds were formed, indicating that the vanadium is bonded to the
phenolic oxygen. Furthermore, it was found that phenol (5) itself could not becoupled oxidatively using vanadium(V) oxytrichloride but ra ther only those substituted
phenolics, such as the naphthols, that have oxidation potentials lower than (5).
A simple mechanism involving the formation of a vanadium phenolate compound has
previously been proposed, but does not provide explanations for all observations
made. In this proposal, the vanadium-phenolate decomposes to form the phenoxyl
radical and a lower valence vanadium species, whereafter the coupling/dimerizationstep occurs to afford the biphenol. It has been suggested by Carrick61 that phenolic
coupling occurs by a rearrangement of electrons in a complex containing at least two
phenoxide residues and one metal center. Whether vanadium(V) or vanadium(IV)
acts as one or two electron oxidizing agents here is not clear and, furthermore, the
course of the phenolic coupling itself is also not clear. However, the existence of
metal-phenolate compounds has been established, enhancing the possibility that a
non-radical (two electron oxidation) pathway may be involved. The NR2 mechanism
can be used to explain the existence of a metal-phenolate derivative (Scheme 11).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 27/182
18
Scheme 11: The oxidative coupling of a substituted phenol using VOCl3
1.3.2.1.2 A (nitrosonaphtholato)metal complex16
Over the past three decades, the use of a (nitrosonaphtholato)metal complex in these
reactions were investigated both spectroscopically and physically.62,63 However, little
was known about the catalytic ability of these complexes in organic oxidation
reactions,46 and so the coupling reactions of both 2,4- and 2,6- disubstituted phenols,
due to their structural simplicity, were investigated in the presence of this complex.26
+ VOCl3-HCl
O
R R
V
O
Cl
Cl
OH
R R
O
O
HH
RR
R R
+
VOCl3
2VOCl2 + HCl
OH
OH
RR
RR
OH
RR
tautomerization
V
OH
ClCl

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 28/182
19
C(CH3)3(CH3)3C
OH
O2
[Mn(II)
(1-nnap)n]-R3P
O
O
(CH3)3C C(CH3)3
C(CH3)3(CH3)3C
(9)
(10)
Thus when 2,6-di-tert -butylphenol (9) was reacted with
(nitrosonaphtholato)manganese [Mn(II)(1-nnap)2] at 23°C under an oxygen
atmosphere, in the presence of triphenylphosphine, the diphenoquinone (10) was
formed (Scheme 12).
Scheme 12: The oxidative coupling of 2,6-di-t -butylphenol using a
(nitrosonaphtholato)metal complex
Some phosphine compounds are known to activate metal catalysts,64-67 and the
addition of triphenylphosphine as co-ligand to the above reaction increased the yield
of (10) from 5 % (after 10 h) to 93 %. This catalytic activity of [Mn(II)(1-nnap)2] was
demonstrated in a variety of organic solvents such as acetonitrile, tetrahydrofuran,
methanol and ethyl acetate. However, no oxidation products were obtained in
reactions using benzene or acetic acid as solvents. The data obtained from the cyclic
voltammogram of [Mn(II)
(1-nnap)2] showed reversible Mn(II) ? Mn(III) and irreversibleMn(III) → Mn(IV) processes. This indicates that [Mn(II)(1-nnap)2] tends to be oxidized
to a Mn(III) species, implying that it could therefore behave as a one electron transfer
catalyst in these reactions. It was proposed that [Mn(II)(1-nnap)2], after activation by
phosphine, traps molecular oxygen to form complex (11), as shown in Scheme 13.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 29/182
20
Scheme 13: Coupling mechanism using a (nitrosonaphtholato)metal complex
C(CH 3)3(CH3)3C
OH
(9)O
N
N
O
O
O
O
MnIII
Mn-cycle
O
N
N
O
O
O
MnII
Ph3P=O+ HO
.
.
PPh3
+ O2
O
(CH3)3C
(CH3)3C
HO
H
(CH3)3C
(CH3)3C
O
(11) (12)
(13)
-H+[Mn II(1-nnap)2]
(16) [Mn-cycle]
(10)
(14)
PPh3
O
N
N
O
O
O
OH
MnIII
O
PPh3
[MnII(1-nnap)2]
O
(CH3)3C
(CH3)3C
H
H
C(CH 3)3
C(CH 3)3
OH
+(9)
MnIII species
tautomerizationHO
(CH3)3C
(CH3)3C
H
C(CH 3)3
C(CH 3)3
OH
HO
(CH3)3C
(CH3)3C
C(CH 3)3
C(CH 3)3
OH HO
(CH3)3C
(CH3)3C
C(CH 3)3
C(CH 3)3
O
(17)
MnIII species
[MnII(1-nnap)2]
O
(CH3)3C
(CH3)3C
C(CH 3)3
C(CH 3)3
O
(15)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 30/182
21
Complex (11) has manganese in the 3+ oxidation state since this metal ion was found
to be electrochemically stable. It was suggested that complex (11) then abstracts a
hydroxyl hydrogen from (9) to yield the peroxymanganese (12) and the phenoxylradical (13). Complex (12) immediately decomposes to afford phosphine oxide and a
hydroxyl radical. Radical (13) then reacts with (9) to yield the coupled product (14)
which tautomerizes to (15). Thereafter, after a similar oxidation cycle, radical (15)
affords the diphenyl diol (16), which is oxidized by the same catalytic cycle to give
(17). The latter compound is ultimately transformed to the diphenoquinone (10).
1.3.2.1.3 Activated manganese dioxide
37
When activated manganese dioxide was reacted with 2,6-xylenol (18), the analysis of
the product mixture showed the presence of a polyphenylene ether (19), 3,3’,5,5’-
tetramethyl-p ,p ’-biphenol (20) and 3,3’,5,5’-tetramethyldiphenoquinone (21) [Scheme
14].
Scheme 14: Oxidation of 2,6-xylenol using MnO2
The molecular weight of polymer (19) varied substantially, depending on the reactant
ratio and the reaction solvent used, ranging from 2 000 to 20 000. The polymer was
MeMe
OH
MnO2
(19)
n
+
Me Me
Me Me
OH
OH
O
O
Me Me
MeMe
+
(18)
(20) (21)
MeMe
OH
O
MeMe
O
MeMe

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 31/182
22
OH
Me MeO
O
Me Me
O
Me Me
H
(22) + (23) O
Me
O
Me
H
Me
Me
(19) (where n = 0)
(23) + (23)
Me
O
Me
H
Me
Me
O
H
(21)
(23) + O
Me
O
Me Me
Me
(19) (where n = 1)
(23)
(18) (22)
tautomer-
ization
O
OH
Me Me
Me Me
O
MeMe
the major product, with diol (20) and diphenoquinone (21) being formed in much
smaller amounts, when a molar ratio of 3:1 (oxide:xylenol) was used. However, when
(18) was used in molar excess, (21) was the principle product, with a low molecular weight oligomer also being formed. Products (20) and (21) are formed by carbon-
carbon coupling, whilst (19) is formed exclusively by carbon-oxygen coupling
(Scheme 15).
Scheme 15: Reactions showing C-C and C-O coupling using MnO2 as oxidant

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 32/182
23
Scheme 15 shows that the phenoxyl radical may couple with another phenoxyl radical
through either the oxygen (22) or carbon (23) atoms. It was found that when excess
manganese dioxide was used, coupling occurred mainly head to tail (i.e., carbon-oxygen coupling), and thus the main product in this case was (19).
Polymerization may be prevented, if so desired, by using phenolics with large groups
in the 2 and 6 positions, since steric hindrance prevents the phenoxyl oxygen radical
from combining with (23) in such cases. Thus reacting 2,6-di-tert -butylphenol with
manganese dioxide, and having the reactant in excess, results mainly in products
(10) and (16) [see Scheme 13 for structures of (10) and (16)].
1.3.2.1.4 Cupric salts46
Cupric salts of carboxylic acids have been found to oxidize phenols in a manner that
is characteristic of single electron oxidizing agents to yield products coupled at the
ortho or para positions, depending on the substitution of the initial substrate. In this
study, only disubstituted phenols were used. It was with cupric salts that the more
highly oxidized products, like the quinones, were generally not produced (Scheme
16).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 33/182
24
Scheme 16: Oxidation of 2,4-disubstituted phenols using cupric salts
In these reactions, the phenolic compound was in excess and also served as the
solvent. The cupric salt was regenerated by bubbling air through the solution
(Equation 2) and, as a result, could be used in catalytic amounts, with oxygen serving
as the principal oxidizing agent. In the above scheme, when R = H or CH3, it was
found that larger amounts of resinous materials were produced in the presence of
oxygen. Phenol itself gave polymeric products exclusively but, in the absence of oxygen, a light amber oil was produced which consisted mainly of the coupled dimer
(26) [Scheme 17]. Small amounts of other coupled products were also formed such
as the para-para (6) and ortho-para (7) coupled products (Scheme 17).
R
R
OH
R
R R
R
OHOH
+ ...(1)
1/2 O2 + H2O
R
R
OH
2
(24)
(24)
+ 1/2 O2
(25)
R
R R
R
OHOH
(25)
2C
O
R)22Cu(OC
O
R2CuO
C
O
R2HO+
C
O
R2CuO C
O
R2HO+ + C
O
R)22Cu(O ...(2)
+ H2ONett: ...(3)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 34/182
25
OH OH OH
OH
OH
OH
OH
(26)
C
O
R)22Cu(O
(6)
(7)
2 + +
Scheme 17: Oxidative coupling of phenol in the presence of cupric salts
1.3.2.2 Electrochemical oxidative coupling
Electrochemical methods present another useful avenue that may be investigated for
synthesizing organic molecules, particularly for the oxidative coupling of phenols.
Both direct and indirect electrochemical oxidation reactions have been carried out by
other workers in this context, and these are briefly summarized below.
1.3.2.2.1 Direct electrochemical oxidations
Direct electrochemical oxidations involve electron transfer between an organic
reactant and the anode of an electrochemical cell. This results in an intermediate
which then reacts further to form the product. The characteristic features of direct
electrochemical oxidations are as follows:• The redox reagent is the electron itself; electrons are removed either directly or
indirectly from the reactant via an electrode.
• The selectivity of the electrochemical step can be greatly increased by careful
selection of the conditions at the phase boundary, e.g., potential, current densities,
etc.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 35/182
26
• Electrochemical methods can be used to synthesize a wide variety of organic
chemicals: any oxidation that can be carried out using conventional chemical
oxidizing agents can theoretically be carried out in an electrochemical cell.
• Electrochemical syntheses often have a lower environmental impact than
conventional oxidation methods since electrolytic routes often replace toxic
reagents and hazardous process conditions.68
Generally, the phenolic substrate forms an electrogenerated radical species, the
dimerization of which (to afford the desired product) is in competition with a further
one electron oxidation that results in the corresponding cation. In the case of phenolitself, electropolymerization is known to occur at the anode surface resulting in the
formation of a passivating film on the electrode surface.56,57 In addition to polymeric
products, both p -benzoquinone and 4,4’-diphenoquinone are also produced as minor
products (in 20 and 10% yields, respectively) as shown in the following scheme
(Scheme 18).56

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 36/182
27
OH
CPE: +0.9Vvs SCE
25% acetone buffer (pH 5), (C)
OH
+
O
-H+
-e-
O
++H2O
OH
OH
-H+
-2e-, -2H+
O
O
C-C coupling
OH
OH
O
O
(28)
(27)
andtautomerization
-2e-
-2H+
(6)
-e-
Scheme 18: The direct electrochemical oxidation of phenol
The 4,4’-diphenoquinone (28) is formed through biphenol (6). The (27)/(28) ratio may
be controlled to a certain extent: if the electrolysis is carried out at a higher anodic
potential, the amount of (27) may be increased.
In addition to reactions using phenol as substrate, the anodic oxidation of 2,6-
dimethylphenol also leads to the rapid formation of a linear polymer chain. However,

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 37/182
28
phenolic substrates bearing bulkier alkyl substituents afford radicals that are expected
to have enhanced stability. For example, the radical species of 2,6-di-sec -
butylphenol was detected using multiple internal reflection Fourier transform infraredspectroscopy (MIRFTIRS), thus confirming the radical mechanism during the anodic
oxidation of this substrate.
When 2,6-di-tert -butylphenol (9) was reacted under constant current electrolysis
conditions (1.0 mA.cm-2; 2.5 F.mol-1) in MeOH-CH2Cl2, using a divided cell, it was
converted to the corresponding 4,4’-diphenoquinone (10) in 84.7 % yield. A
subsequent electroreduction, achieved by merely altering the current direction,resulted in the formation of biphenol (16) in 92.5 % yield (Scheme 19).53
Scheme 19: Direct electrochemical oxidation of 2,6-di-t -butylphenol
p -Cresol (29) was also electrolyzed at a controlled potential (+0.25 V vs SCE) in a
basic medium to afford Pummerer’s ketone (30) in 74 % yield, as seen in thefollowing scheme (Scheme 20).69
CCE: 1.0 mA cm-1
LiClO4, MeOH-CH 2Cl2
(Pt)
OH
C(CH3)3(CH3)3C
(9)
OH
C(CH3)3(CH3)3C
C(CH3)3(CH3)3C
OH
(16)
O
C(CH3)3(CH3)3C
C(CH3)3(CH3)3C
O
(10)
-2e-, -2H+
+2e-, +2H+

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 38/182
29
Scheme 20: Oxidation of p -cresol to Pummerer’s ketone
1.3.2.2.2 Indirect electrochemical oxidations
In an indirect electrode reaction, a redox couple is used as a catalyst (electron carrier)
for the oxidation or reduction of another species in the system. In such a system, the
electrode can be used to reconvert the redox reagent to an oxidation state where it
can again react with an organic compound. In other words, indirect electrolysis has
distinct advantages over the direct method: firstly, the redox reagent can be recycled,
thus decreasing the problems associated with the use of toxic reagents and,
secondly, the redox catalyst may have increased solubility in water, thus allowing the
reaction to be carried out at high current density in an aqueous electrolyte.
Generally, the most suitable redox couples are inorganic, and include Ce3+/Ce4+,
Mn2+/Mn3+ and Cr 3+/Cr 2O72-. These redox couples are used primarily for oxidations of
organic compounds.
Indirect electrosyntheses may be carried out using one of two methods:
OH
Me
CPE: +0.25 V vs SCE
(1 F mol-1)NaClO4
MeCN-H2O-NaOH
(C)O
Me
O
Me
o-p coupling
O O
HMe
Me
H+
(29)
(30)
Me
OH
O
Me
followed
bytautom.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 39/182
30
• The in-cell method: The reaction between the organic substrate and the redox
reagent, in its active oxidation state, occurs within the cell.
• The ex-cell method: The reaction is carried out in a reactor separate to the cell.
This approach has advantages over the in-cell method in that the conditions for the
electrode reaction and the chemical step may be optimized separately and,
furthermore, that the electrolyte may be purified between the reactor and the cell.
One redox catalyst that has been used successfully for oxidative coupling is the
Ce+3/Ce+4 couple.69 The cerium(IV) ions were generated from cerium(III) in the
presence of perchloric acid. Using 2,6-dimethylphenol as the substrate in aqueous or aqueous-acetonitrile solutions of perchloric acid (0.5 - 1.0 M) at room temperature,
the corresponding 4,4’-diphenoquinone and 1,4-benzoquinone were obtained as the
main products.70
Under similar conditions, the oxidation of 2,6-diisopropylphenol (31a), 2-tert -butyl-6-
methylphenol (31b), 2,6-diphenylphenol (31c) and 2,6-dichlorophenol (31d) resulted
in the formation of the corresponding 4,4’-diphenoquinones (32a-d) in addition to
oligomeric poly(1,4-phenylene) oxides (33a-d) [Scheme 21]. In the case of 2,6-
diphenylphenol (31c), the quantity of carbon-oxygen coupled product was low due to
the steric hindrance associated with the large phenyl groups adjacent to the oxygen
atom.
When 2,6-diisopropylphenol (31a) and 2- tert -butyl-6-methylphenol (31b) were
oxidized by cerium(IV) in a two phase system, namely, in CCl4 and an aqueous
acetonitrile solution of perchloric acid, at a high concentration of perchloric acid (4.0M) in the reacting phase, this afforded the corresponding 1,4-benzoquinones in good
yields. This was not observed with 2,6-diphenylphenol and 2,6-dichlorophenol.
The results obtained from these reactions are summarized in Table 1.2.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 40/182
31
Scheme 21: Indirect oxidation of 2,6-disubstituted phenols
Table 1.2 Oxidation of disubstituted phenols by cerium(IV)
Phenol Procedurea MolarRatiob
Concentrationof HClO4 (M)
Time(min)
Product (yield)(% )
31a A 1:2.15 0.5 0 32a (85), 33a (4), 34a (8)
31a B 1:4.00 4.0 30 32a (3), 34a (90)
31b A 1:2.25 0.5 180 32b (54), 33b (37)
31b B 1:3.75 4.0 90 32b (12), 33b (21), 34b (56)
31c A 1:2.25 0.5 0 32c (70), 33c (23)
31d A 1:1.75 0.5 0 32d (30), 33d (65)
aReactions were either in homogeneous (A) or heterogeneous (B) reaction systems
b Phenolic substrate:cerium(IV) molar ratio
R1
HO
R2
Ce(IV)
H2O-ANHClO4 (0.5-4.0 M)
O
O
R1 R2
R2R1
O O
R1
R2
R1
R2
R1
HO
R2
n33a-d
O O
R1
R2
31a: R1 = R2 = isopropyl31b: R1 =t -Bu; R2 = CH331c: R1 = R2 = Ph31d: R1 = R2 = Cl
+
32a-d
31a or 31b Ce(IV)CCl4/aq. CH 3CN
HClO4 (4.0 M)
34a or 34b

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 41/182
32
1.4 OBJECTIVES AND MOTIVATION FOR THIS STUDY
Dihydroxybiphenyls, as mentioned previously, have many important uses aschemicals in their own right, but also as intermediates in the manufacture of other
materials. Dihydroxybiphenyls are often prepared by means of oxidative coupling
procedures. However, the reaction is only efficient for disubstituted phenols such as
2,6-di-t -butyphenol and 2,4-dimethylphenol. The literature contains many reports on
the successful coupling of these substrate types, but is, however, virtually devoid of
studies carried out on monosubstituted phenols such as 2-t -butylphenol. The reasons
for this are clear: the C-C coupling of 2,4- or 2,6- disubstituted phenols is reallypossible only in the 6- and 4- positions, respectively, leading to reactions that afford
high yields of the desired coupled product. In contrast, a monosubstituted phenol
such as 2-t -butylphenol has two positions available through which C-C coupling may
occur, the 4- and 6- positions.
Hence, in the latter case, complex oxidative coupling reaction mixtures are obtained.
These often contain significant proportions of polymeric materials, and thus low
selectivities to the desired product are a result. This in turn implies tedious and time-
consuming purification steps. There thus appears to be a need to study these
reactions more closely with the view to developing a better understanding as to the
mechanisms at work so that the knowledge base for this reaction type may be
enhanced, and ultimately a better process may be devised.
In addition, it must be mentioned that another factor that has fuelled our interest in
this investigation is the ready availability of the starting materials that are to becoupled. SASOL produces phenol during its petroleum cracking process, and
alkylated phenols may readily be prepared from it. These alkylated phenols serve as
substrates in our coupling reactions.
This study is therefore concerned with the oxidative coupling of various mono- and di-
substituted phenols using chemical and indirect electrochemical oxidation methods.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 42/182
33
The effect of the various substituents already on the aromatic ring on the oxidative
process is also investigated and, furthermore, attention is given to achieving high
conversions and selectivities to specifically carbon-carbon coupled products.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 43/182
34
CHAPTER 2
EXPERIMENTAL
2.1 MATERIALS
2.1.1 Reagents for Synthesis and Analysis
All materials used in the oxidation procedures and syntheses, with their sources and
respective grades, are listed in Tables 2.1 and 2.2, and were used as received.
Table 2.1 Organic reagents for synthesis
CHEMICAL NAME FORMULA SOURCE GRADE
4,4’-Dihydroxybiphenyl C12H10O2 Aldrich CP
Carbon tetrachloride CCl4 Holpro AR
t -Butyl bromide (CH3)3CBr Aldrich CP
Ethyl acetate CH3CO2C2H5 Saarchem CP
Hexane C6H14 BDH Technical
Methanol CH4O BDH HPLC2-Naphthol C10H7OH Saarchem CP
Dichloromethane CH2Cl2 Saarchem AR
Toluene C6H5CH3 Merck Technical
Oxalic acid HO2CCO2H Riedel-de Haen AR
Acetonitrile CH3CN BDH HPLC
Acetic acid CH3CO2H Merck AR
2-t -Butylphenol C10H14O Aldrich AR
2,4-Dimethylphenol C8H10O Riedel-de Haen CP
2,4-Di-t -butylphenol C14H22O Aldrich AR
2,6-Di-t -butylphenol C14H22O Fluka CP
Chloroform CHCl3 Saarchem CP
Succinic acid HO2C(CH2)2CO2H Riedel-de Haen AR

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 44/182
35
Table 2.2 Inorganic and organometallic reagents for synthesis
CHEMICAL NAME FORMULA SOURCE GRADE
Silicon dioxide SiO2 Fluka Technical
Sodium carbonate Na2CO3 Saarchem AR
Celite N/A Hopkin&Williams AR
Potassium ferric cyanide K3Fe(CN)6 Merck AR
Hydrochloric acid HCl Saarchem AR
Silver nitrate AgNO3 Saarchem AR
Barium hydroxide Ba(OH)2 Protea Chemicals CP
Sodium hydroxide NaOH Saarchem CPSodium nitrite NaNO2 Saarchem CP
Sulphuric acid H2SO4 Saarchem Technical
Manganese chloride MnCl2 M&B CP
Cupric acetate Cu(OAc)2 Mallinckcroft AR
Manganese acetate Mn(OAc)3 Merck AR
Triphenylphosphine P(Ph)3 Aldrich AR
Ferric chloride FeCl3 Riedel-de Haen CP
Silver oxide Ag2O Fluka AR
Cerium carbonate Ce2(CO3 )3 Aldrich AR
Methanesulphonic acid CH3SO3H Acros Technical
Ferrous sulphate FeSO4 Unilab Technical
Sodium hydrosulphite Na2S2O4 M&B Technical
Manganese dioxide MnO2 Unilab AR
Ammonium persulphate (NH4)2S2O8 Saarchem CP
Potassium hydroxide KOH Saarchem CP
Ferroin N/A Saarchem AR
Magnesium sulphate MgSO4
Saarchem CP
The reagents used as standard materials for high performance liquid chromatography
(HPLC) are also listed in Table 2.1 (shown before). All standard materials were used
as received. Acetonitrile (Chromasolve), used as mobile phase for HPLC analyses,
was obtained from Merck and also used as received.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 45/182
36
2.2 SYNTHETIC PROCEDURES
2.2.1 Reagents for Analysis
2.2.1.1 Preparation of 3,3’-di-t -butyl-4,4’-dihydroxybiphenyl71
To a mixture of 4,4’-dihydroxybiphenyl (0.3701 g, 1.987 mmol), SiO2 (1.013 g), and
Na2CO3 (1.935 g, 18.25 mmol) in CCl4 (7 mL) was added t -butyl bromide (0.7867 g,
5.741 mmol), and the reaction mixture was stirred vigorously for 24 h at 70°C. The
SiO2 was filtered off and washed with ethyl acetate. The ethyl acetate washings andfiltrate were then combined and the solvent was removed under vacuum. The product
was isolated using thin-layer chromatography with hexane:ethyl acetate (90:10) as
the developing solvent system. The desired product, 3,3’-di-t -butyl-4,4’-
dihydroxybiphenyl, was thus obtained, and had m.p. 182-183°C (lit.71, m.p. 181-
183°C); ?max (CCl4)/cm-1 3600 (OH), 2750-3100 (C-H) and 1583 (C=C); m/z 298 (M+),
283 (M+-15) and 255 (M+-43); dH (CDCl3)/ppm 1.47 (18H, s, CH3), 4.81 (2H, s, OH)
and 6.65-7.50 (6H, m, Ar).
2.2.1.2 Preparation of 3,3’,5,5’-tetra-t -butyldiphenoquinone
2,6-Di-t -butylphenol (0.222 g, 1.075 mmol) was added to silver oxide (0.5147 g, 2.222
mmol) in methanol (25 mL), after which the reaction mixture was stirred for 1 h. The
solids were removed by filtration and washed with hot toluene, the toluene then being
combined with the filtrate. This solution was then concentrated down on the rotary
evaporator to afford crude 3,3’,5,5’-tetra-t -butyldiphenoquinone (99.00 %) as theprimary product, which was further purified by recrystallization using ethyl
acetate:petroleum ether (b.p. 60-80°C); m.p. 247-248°C (lit.28, m.p. 248°C); ?max
(CCl4)/cm-1 2800-3100 (C-H), 1631 (C=O) and 1603 (C=C); m/z 408 (M+), 393 (M+-
15), 366 (M+-42), 351 (M+-57) and 309 (M+-99); dH (CDCl3)/ppm 1.40 (36H, s, CH3)
and 7.73 (4H, s, Ar).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 46/182
37
2.2.1.3 Preparation of 3,3’,5,5’-tetra-t -butyl-4,4’-dihydroxybiphenyl
To a suspension of 3,3’,5,5’-tetra-t -butyldiphenoquinone (1.169 g, 2.861 mmol) inether (50 mL) was added a solution of sodium hydrosulphite (8.030 g, 46.12 mmol) in
aqueous NaOH (1.0 M, 100 mL). After stirring the reaction mixture for 1 h, the
aqueous layer was acidified with concentrated HCl (15 mL). The organic layer was
separated, dried (MgSO4) and concentrated to give 3,3’,5,5’-tetra-t -butyl-4,4’-
dihydroxybiphenyl (99.00 %), which was further purified by recrystallization using
ethyl acetate:petroleum ether (b.p. 60-80°C); m.p. 187-188°C (lit.72, m.p. 185-
186.5°C); ?max (CCl4)/cm
-1
3650 (OH), 2800-3050 (C-H) and 1592 (C=C); m/z 410(M+), 395 (M+-15), 190 (M+-220) and 162 (M+-248); dH (CDCl3)/ppm 1.50 (36H, s,
CH3), 5.20 (2H, s, OH) and 7.30 (4H, s, Ar).
2.2.1.4 Preparation of 3,3’,5,5’-tetra-t -butyl-2,2’-dihydroxybiphenyl
A solution of potassium ferricyanide (6.690 g, 20.32 mmol) and sodium hydroxide
(2.944 g, 73.61 mmol) in water (100 mL) was added drop-wise over 30 min to a
vigorously stirred solution of 2,4-di-t -butylphenol (4.058 g, 19.67 mmol) in methanol
(100 mL). After stirring for a further 90 min, the mixture was poured into water and
extracted with ethyl acetate (3 x 50 mL). The organic layer was then dried (MgSO4)
and concentrated on a rotary evaporator to give 3,3’,5,5’-tetra- t -butyl-2,2’-
dihydroxybiphenyl (83.95 %), which was purified by recrystallization using ethyl
acetate:petroleum ether (b.p. 60-80°C); m.p. 199.5-202.5°C (lit.46, m.p. 200-202°C);
?max (CCl4)/cm-1 3538 (OH), 2800-3050 (C-H) and 1586 (C=C); m/z 410 (M +), 395
(M+
-15), 354 (M+
-56), 339 (M+
-76), 283 (M+
-127), 227 (M+
-183) and 190 (M+
-220); dH (CDCl3)/ppm 1.35 (18H, s, CH3), 1.48 (18H, s, CH3), 5.24 (2H, s, OH) and 7.12-7.43
(4H, m, Ar).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 47/182
38
2.2.1.5 Preparation of 3,3’,5,5’-tetramethyl-2,2’-dihydroxybiphenyl
A Ce4+ solution (20 mL, 2.057 mmol) was added to 2,4-dimethylphenol (0.1366 g,1.118 mmol) in a 50 mL round-bottomed flask and stirred vigorously at 750 rpm for 1
h. The reaction mixture was then extracted using ethyl acetate (3 x 25 mL), and the
organic layer washed with water (3 x 25 mL) and dried (MgSO4). The solvent was
removed under vacuum, and the product was isolated using column chromatography,
with hexane:ethyl acetate (90:10) as the developing solvent system. The desired
product, 3,3’,5,5’-tetramethyl-2,2’-dihydroxybiphenyl, had m.p. 130-134°C (lit.4, m.p.
133-134°C); ?max (CCl4)/cm
-1
3558 (OH), 2858-3050 (C-H) and 1547 (C=C); m/z 242(M+), 227 (M+-15), 199 (M+-43), 165 (M+-77) and 91 (M+-151); dH (CDCl3)/ppm 2.38
(12H, s, CH3), 5.04 (2H, s,OH), 6.63 (2H, s, Ar) and 6.92 (2H, s, Ar).
2.2.2 Preparation of Coupling Agents
2.2.2.1 Preparation of silver carbonate/celite28
Celite was first purified by successively washing with methanol containing 10%
concentrated HCl, and distilled water, until neutral. It was then dried at 120°C for 12
h. This purified celite (30.00 g) was then added to a mechanically stirred solution of
silver nitrate (34.00 g, 200.1 mmol) in distilled water (200 mL). A solution of
Na2CO3·10H2O (30.00 g, 104.9 mmol) in distilled water (300 mL) was then added
slowly to the resulting homogeneous solution. When the addition was complete,
stirring was continued for a further 10 min. The yellow-green precipitate that formed
was filtered off and finally dried on a rotary evaporator over a period of several hours.Every 0.57 g of this silver carbonate/celite reagent contained 1.00 mmol of Ag2CO3.
28

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 48/182
39
2.2.2.2 Preparation of barium manganate73
Preparation of potassium manganate
Potassium hydroxide (5.675 g, 101.1 mmol) was thoroughly mixed with manganese
dioxide (4.345 g, 54.62 mmol) and left in an oven at 350°C for 3 h. The fused green
potassium manganate that so formed was filtered and then used for the preparation
of barium manganate.
Preparation of barium manganate
To a 500 mL flask containing distilled water (100 mL) was added barium hydroxide
(7.698 g, 24.40 mmol), and the pH was adjusted to 7 with dilute hydrochloric acid. To
the resulting warm solution was added potassium manganate (8.236 g, 41.78 mmol)
with stirring. The colour of the reaction mixture immediately changed to dark purple.
The reaction mixture was filtered with suction and the so-obtained dark blue crystals
were washed several times with distilled water, and placed in an oven at 100°C for 24
h to afford active barium manganate.
2.2.2.3 Preparation of a (nitrosonaphtholato)metal complex (Mn II(1-
nnap)2)27
Preparation of 1-nitroso-2-naphthol
After 2-naphthol (14.68 g, 101.8 mmol) was dissolved in hot NaOH (0.6 M, 340 mL),the solution was cooled to 0ºC. NaNO2 (7.054 g, 102.2 mmol) was added, and 6 M
H2SO4 (16 mL) was carefully dropped into the resulting solution during 1.5 h with
stirring. The mixture was stirred for a further 1 h. The brown solid that formed was
filtered, washed with water (250 mL) and dried in a desiccator. The crude material
was recrystallized from petroleum ether (b.p. 60-80ºC) to afford 1-nitroso-2-naphthol
as reddish brown needles; m.p. 107-109ºC (lit.75, m.p. 106-108ºC).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 49/182
40
Preparation of nitrosonaphthol sodium salt
1-Nitroso-2-naphthol (4.015 g, 21.28 mmol) was dissolved in a 10 M NaOH solution(50 mL) at 0ºC during 2 h, and the mixture was stirred at room temperature overnight.
The green solid that formed was filtered, washed with 2 M NaOH solution, and dried
in a desiccator to afford the corresponding sodium salt (3.613 g, 17.11 mmol,
76.25%).
Preparation of MnII(1-nnap)2
Nitrosonaphthol sodium salt (3.182 g, 14.29 mmol) was dissolved in water (200 mL),
and MnCl2 (1.880 g, 9.520 mmol) was added. After stirring for 2 h, the solid that
formed was filtered, thoroughly washed with water and dried in a desiccator. The
solid was recrystallized from CH2Cl2-hexane to give dark brown crystals of MnII(1-
nnap)2 with a m.p. > 300°C (lit.27, m.p. >300°C).
2.2.2.4 Electrochemical preparation of cerium(IV) from cerium(III) using a
divided cell
The required amount of methanesulphonic acid was added to both the anode and
cathode compartments to approximately the same level in each, after which the
required amount of cerium carbonate was slowly added to the anode compartment.
The experimental setup is shown in Figure 2.1.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 50/182
41
A = Anode compartment
B = Cathode compartmentC = Amp meter
D = Power supply
E = Heater/stirrer
Figure 2.1: Experimental setup for the electrochemical generation of Ce(IV)
Both the anode and cathode compartments were heated (60°C) and stirred (500 rpm)
for the designated time period. After completion of this time period, the reactionmixture from the anode compartment was filtered, and a 5 mL sample of the filtrate
was titrated against a ferrous sulphate solution with ferroin as indicator. This was
done in order to determine the Ce4+ concentration. The results obtained for the
oxidation of Ce3+ to Ce4+ in various methanesulphonic acid solutions of varying
concentrations may be observed in Table 2.3, where the data from triplicate titrations
and their averages are listed.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 51/182
42
Table 2.3 Moles of Ce4+ at various MeSO3H concentrations
Methanesulphonic
Acid
Concentration
Moles of Ce 4+
Titration 1 Titration 2 Titration 3 Average
(mmol) (mmol) (mmol) (mmol)
0.5 M 2.975 2.488 2.528 2.664
1.0 M 3.886 3.716 3.454 3.685
2.0 M 3.065 3.564 4.122 3.548
The electrochemical reaction conditions for the oxidation of Ce3+ to Ce4+ in the divided
cell, in various methanesulphonic acid solutions of varying concentrations, are listed
in Table 2.4.
Table 2.4 Conditions for oxidation of Ce3+ to Ce4+
Methanesulphonic Acid
Concentration
Initial Ce3+
Concentration
Volts
(V)
Amperes
(A)
Time
(h)
0.5 M 0.1 M 24.0-26.0 0.4 72
1.0 M 0.1 M 10.0-13.5 0.4 48
2.0 M 0.1 M 5.0-6.8 0.4 24
2.2.2.5 Preparation of silver oxide75
Sodium hydroxide (2.67 g, 66.75 mmol) was added to silver nitrate (10.62 g, 62.52
mmol) in water (100 mL) in a 250 mL round-bottomed flask. The reaction mixture

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 52/182
43
was stirred for 1 h, after which it was filtered. The so-recovered solid was washed
repeatedly with water (200 mL) to result in a brown solid of Ag2O, which was dried in
a vacuum desiccator for 48 h.
2.3 EXPERIMENTAL PROCEDURES
2.3.1 Oxidative Coupling Reactions
2.3.1.1 Oxidation of alkylphenols using silver carbonate/celite73
General procedure
Before use, the silver carbonate/celite reagent (0.2 mmol of Ag2CO3) was freed from
residual water azeotropically by distillation with toluene. The alkylphenol (0.1 mmol)
was then added to the silver carbonate/celite reagent and the reaction mixture was
stirred in toluene (200 mL) for various reaction times. The reaction mixture was then
filtered to remove the solid phase, the solvent evaporated with a rotary evaporator,
and the resulting mixture analyzed by HPLC and GC-MS.
2.3.1.2 Oxidation of alkylphenols using copper complexes of dicarboxylic
acids76
General procedure
Into a 250 mL reaction vessel, which was fitted with a gas addition tube, a condenser,
a thermometer, and a stirrer capable of operating at speeds ranging from
approximately 800 rpm to 10 000 rpm, was added sodium lauryl sulphate (0.10 g,0.35 mmol), deionised water (75 mL) and the alkylphenol (approximately 65 mmol).
To the resulting slurry (which was stirred between 800 and 10 000 rpm depending on
the experiment), was added a mixture of cupric acetate (1.0-50.0 mmol) and a
dicarboxylic acid (1.0-50.0 mmol) in deionised water (50 mL). The resulting mixture
was stirred for 5 min while heating to temperatures ranging from 60 to 80 °C. Sodium
hydroxide (0.4 M, 100 mL) was added during the course of the reaction to maintain

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 53/182
44
the pH of the reaction mixture at 9. The mixture was stirred under oxygen or nitrogen
depending on the experiment. The flow of gas was initially rapid to flush the system.
After approximately 30 min, the gas flow was reduced and maintained at a levelsufficient to cause slow bubbling. The reaction mixture was stirred and maintained
under oxygen or nitrogen for time periods varying from 6 to 30 h. The reaction
mixture was then cooled to room temperature and then acidified to pH 3 with HCl (3
M). The reaction mixture was extracted using ethyl acetate (3 x 50 mL), and the
organic layer washed with water (3 x 50 mL) and dried (MgSO4). The organic layer
was then concentrated on a rotary evaporator and analyzed by HPLC and GC-MS.
2.3.1.3 Oxidation of alkylphenols using manganese(III) acetate26
General procedure
The alkylphenol (7.00 mmol) was added to a mixture containing glacial acetic acid
(130 mL) and manganese (III) acetate (3.753 g, 14.00 mmol). The reaction mixture
was then heated to 100°C for 1 h after which it was cooled down, extracted with
chloroform (3 x 50 mL), and the organic layer washed with water (3 x 50 mL) and
dried (MgSO4). The organic layer was concentrated on a rotary evaporator and
analyzed by HPLC and GC-MS.
2.3.1.4 Oxidation of alkylphenols using barium manganate73
General procedure
The alkylphenol (10.00 mmol) in toluene (50 mL) was added to barium manganate
(12.81 g, 50.00 mmol) in a 100 mL round-bottomed flask. The reaction mixture wasthen stirred at room temperature for 1 h, and then vacuum filtered. The solid was
washed repeatedly with ethyl acetate (total volume of 150 mL), and the combined
organic washings concentrated on a rotary evaporator and analyzed by HPLC and
GC-MS.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 54/182
45
2.3.1.5 Oxidation of alkylphenols using a (nitrosonaphtholato)metal
complex27
General procedure
A mixture of the alkylphenol (1.00 mmol), the (nitrosonaphtholato)manganate
complex (0.0399 g, 0.100 mmol) and triphenylphosphine (0.2885 g, 1.100 mmol), in
dry CHCl3 (30 mL), was stirred for 5 h at 23°C under an oxygen atmosphere (1 atm).
The reaction mixture was then quenched with 2 M HCl (50 mL). The aqueous mixture
was extracted with CHCl3 (3 x 25 mL), and the organic layer washed with water (3 x
25 mL) and dried (MgSO4). The organic layer was then concentrated on a rotaryevaporator and analyzed by HPLC and GC-MS.
2.3.1.6 Oxidation of alkylphenols using FeCl3 in an organic solvent77
General procedure
A mixture of the alkylphenol (7.0 mmol) and FeCl3 (2.271 g, 14.00 mmol), in an
appropriate solvent (20 mL), was stirred in a round-bottomed flask at 50°C for 2 h.
The reaction mixture was then decomposed with dilute HCl (50 mL), and the organic
layer washed with water (3 x 20 mL) and dried (MgSO4). The organic layer was then
concentrated on a rotary evaporator and analyzed by HPLC and GC-MS.
2.3.1.7 Oxidation of alkylphenols using FeCl3 without solvent77
General procedure
The alkylphenol (7.0 mmol) and FeCl3·6H2O (2.271 g, 14.00 mmol) were mixedtogether without any solvent, and the mixture then placed in a test tube and kept at
50°C for 2 h. The reaction mixture was then decomposed with dilute HCl (50 mL),
and the aqueous layer extracted with ethyl acetate (3 x 25 mL). The organic layer
was washed with water (3 x 25 mL), dried (MgSO4), concentrated on a rotary
evaporator and analyzed by HPLC and GC-MS.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 55/182
46
2.3.1.8 Oxidation of alkylphenols using Ag2O78
General procedureThe alkylphenol (10.00 mmol) was added to silver oxide (4.635 g, 20.00 mmol) in 150
mL of methanol, after which the reaction mixture was stirred for 1 h at room
temperature. The solids were removed by filtration and washed with hot toluene, the
toluene then being combined with the filtrate. The resulting organic solution was
concentrated on a rotary evaporator and the reaction mixture analyzed by GC-MS
and HPLC.
2.3.1.9 Oxidation of alkylphenols using lead tetra-acetate79
General procedure
The alkylphenol (7.00 mmol) was dissolved in toluene (100 mL) and stirred while lead
tetra-acetate (6.2058 g, 13.997 mmol) was slowly added to the reaction mixture over
1 h. The reaction mixture was then washed with water (3 x 75 mL) and the organic
layer dried (MgSO4) and concentrated on a rotary evaporator. Analysis was carried
out using GC-MS and HPLC.
2.3.1.10 Oxidation of alkylphenols using Ce4+
General procedure
The required amount of Ce4+ solution and the alkylphenol were added together in a
round-bottomed flask in the required solvent and stirred vigorously at 750 rpm for the
required time period. The reaction mixture was then extracted using ethyl acetate (3x 25 mL), and the organic layer washed with water (3 x 25 mL) and dried (MgSO4).
The organic layer was then concentrated on a rotary evaporator and analyzed by GC-
MS and HPLC.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 56/182
47
2.3.1.11 Oxidation of alkylphenols using potassium ferricyanide
General procedure A solution of potassium ferricyanide (6.585 g, 20.00 mmol) and sodium hydroxide
(2.840 g, 71.00 mmol) in water (100 mL) was added drop-wise during 30 min to a
vigorously stirred solution of the alkylphenol (20.0 mmol) in methanol (100 mL). After
stirring for the required reaction time at room temperature, the mixture was poured
into water and extracted with ethyl acetate (3 x 50 mL), the organic layer washed with
water (3 x 50mL) and then dried (MgSO4). The organic layer was concentrated on a
rotary evaporator and analyzed by GC-MS and HPLC.
2.3.2 Determination of Ce(III) Remaining after the Electrochemical
Oxidation of Ce(III) to Ce(IV)80
Water (140 mL) and concentrated sulphuric acid (1 mL) were added to 20 mL of the
cerium solution obtained after the electrochemical oxidation of cerium carbonate (i.e.,
Ce3+) to Ce4+. (The number of moles of Ce4+ in this amount of solution had already
been determined by titration as described in Section 2.2.2.4, and is denoted as A in
Table 2.5 overleaf.) The reaction mixture was then treated with ammonium
persulphate (1.241 g, 5.438 mmol) and 7 drops of 0.1 M AgNO3, and boiled for 30
min, in order to oxidize any remaining Ce3+ to Ce4+. The reaction mixture was then
cooled down and titrated again against a ferrous sulphate solution (0.0860 M) with
ferroin as the indicator, in order to determine the amount of Ce4+ now present in the
solution (denoted as B in Table 2.5). The difference between the initial Ce4+ and final
Ce4+
values (B-A), therefore, was a measure of the amount of Ce3+
that remainedafter the electrochemical oxidation of the Ce3+ solution.
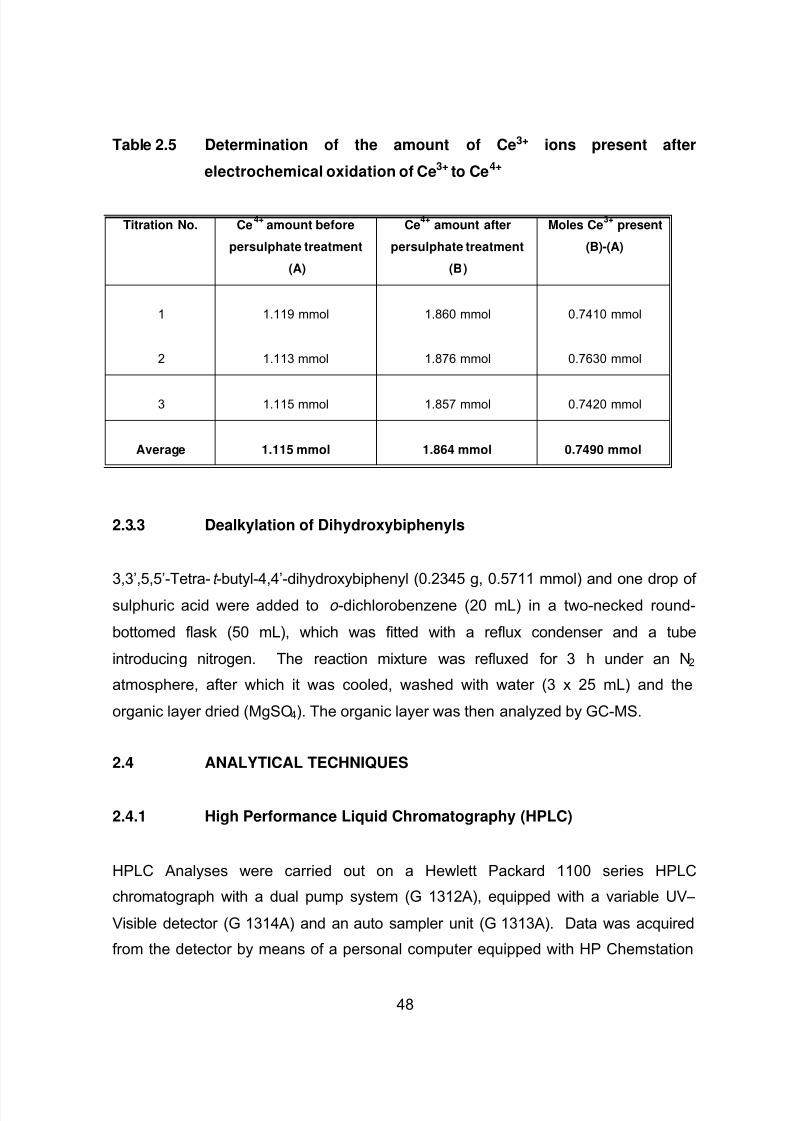
7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 57/182
48
Table 2.5 Determination of the amount of Ce3+ ions present after
electrochemical oxidation of Ce3+ to Ce4+
Titration No. Ce4+
amount before
persulphate treatment
(A)
Ce4+
amount after
persulphate treatment
(B)
Moles Ce3+
present
(B)-(A)
1 1.119 mmol 1.860 mmol 0.7410 mmol
2 1.113 mmol 1.876 mmol 0.7630 mmol
3 1.115 mmol 1.857 mmol 0.7420 mmol
Average 1.115 mmol 1.864 mmol 0.7490 mmol
2.3.3 Dealkylation of Dihydroxybiphenyls
3,3’,5,5’-Tetra- t -butyl-4,4’-dihydroxybiphenyl (0.2345 g, 0.5711 mmol) and one drop of
sulphuric acid were added to o -dichlorobenzene (20 mL) in a two-necked round-
bottomed flask (50 mL), which was fitted with a reflux condenser and a tube
introducing nitrogen. The reaction mixture was refluxed for 3 h under an N2
atmosphere, after which it was cooled, washed with water (3 x 25 mL) and the
organic layer dried (MgSO4). The organic layer was then analyzed by GC-MS.
2.4 ANALYTICAL TECHNIQUES
2.4.1 High Performance Liquid Chromatography (HPLC)
HPLC Analyses were carried out on a Hewlett Packard 1100 series HPLC
chromatograph with a dual pump system (G 1312A), equipped with a variable UV–
Visible detector (G 1314A) and an auto sampler unit (G 1313A). Data was acquired
from the detector by means of a personal computer equipped with HP Chemstation

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 58/182
49
Acquisition software, version A.06.03. All solvents were HPLC grade and were
degassed prior to analysis with a Millipore vacuum-degassing unit. A discovery C8
(serial no. 59354-u) column was used for the analysis of the reaction samples.
As different substrates were used in the reactions, analysis of the mixtures required
different LC conditions. These settings are summarized in Tables 2.6 – 2.9.
Response factors for the compounds of interest were determined by means of a five-
level calibration using standard solutions containing known amounts of the analytes.
Table 2.6 HPLC Conditions for 2-tert -butylphenol reactions
Injector Volume 5 µL
Column µBondpak C18 3.9 mm x 300 mm (Waters)
Wavelength 253 nm
Flow Rate 0.7 cm3 min-1
Mobile Phase 80% MeCN : 20% H2O
Table 2.7 HPLC Conditions for 2,4-dimethylphenol reactions
Injector Volume 1 µL
Column µBondpak C18 3.9 mm x 300 mm (Waters)
Wavelength 289 nm
Flow Rate 0.5 cm3 min-1
Mobile Phase 90% MeCN : 10% H2O

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 59/182
50
Table 2.8 HPLC Conditions for 2,6-di-tert -butylphenol reactions
Injector Volume 5 µL
Column µBondpak C18 3.9 mm x 300 mm (Waters)
Wavelength 267 nm
Flow Rate 0.7 cm3 min-1
Mobile Phase 100% MeCN
Table 2.9 HPLC Conditions for 2,4-di-tert -butylphenol reactions
Injector Volume 1 µL
Column µBondpak C18 3.9 mm x 300 mm (Waters)
Wavelength 289 nm
Flow Rate 0.5 cm3 min-1
Mobile Phase 90% MeCN : 10% H2O
2.4.2 Nuclear Magnetic Resonance (NMR) Spectroscopy
Proton NMR spectra were recorded on a Brücker AX (300 MHz) spectrometer using X
Win NMR software for data analysis. All samples were analyzed using CDCl3 as
solvent.
2.4.3 Fourier Transform Infra Red (FTIR) Spectroscopy
Infra red spectra were recorded on a Brücker Tensor 27 FTIR linked to a Bell
personal computer, equipped with Opus Software version 4.2. All samples were
analyzed using CCl4 as solvent.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 60/182
51
2.4.4 Gas Liquid Chromatography-Mass Spectrometry (GC-MS)
GC-MS Analyses were performed on a Thermo-Finnigan Trace GC coupled to aQuadropole Trace MS+ detector. The GLC was equipped with a Restek-RTX 5 MS
(15 m x 0.25 mm i.d.) column. Data was acquired from the detector by means of a
Bell personal computer equipped with Excaliber version 1.3 software. The
temperature program used is summarized in Table 2.10.
Table 2.10 GLC Temperature program
Initial Temperature 50°C
Initial Hold Time 2.0 min
Program Rate 15 °C min-1
Second Temperature 250°C
Second Hold Time 45.0 min
The mass range capability of the mass spectrometer was from 50 to 1000 atomic
mass units.
2.4.5 Molecular Orbital Calculations
Calculations were carried out using Spartan ’02 (version 119) running under Linux 2.2
on a QuantumStation QS4-1800S machine. Structures were initially partially refined
using the MMFF molecular mechanics facility, where a conformational search was
carried out in order to identify the lowest energy conformer in each case, before these
structures were refined at the PM3 semi-empirical MO level. All geometry
optimisations achieved energy gradient norms of at least 0.01kcal/mol/Å.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 61/182
52
2.5 TERMS AND DEFINITIONS
Terms and definitions like selectivity and conversion need to be clarified. These are:
Selectivity : defined as the ratio of a particular product to the amount of substrate
consumed, and this is expressed as a percentage.
100arg
2 x
remainingsubstratemolesed chsubstratemoles
x product moles ySelectivit
−=
Conversion : defined as the total amount of substrate originally charged that has been
consumed in the formation of the reaction products, expressed as a percentage.
100arg
arg x
ed chsubstrateof moles
remainingsubstrateof molesed chsubstrateof molesConversion
−=

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 62/182
53
CHAPTER 3
DISCUSSION
3.1 MODES OF PHENOLIC COUPLING
There are several mechanisms that may be proposed by which phenolic substrates
may oxidatively couple with one another to form dimers. Naturally, the nature of the
phenolic substrate plays a crucial role in the mode by which it ultimately combines
with another substrate molecule. Of significant importance in this regard is the nature
of substitution of the phenolic ring, not only encompassing the number and type of
substituents, but also the positions they occupy relative to the hydroxyl moiety and
each other.
When one considers, for simplicity sake, the coupling of an unsubstituted phenol with
another such substrate, six modes of coupling may be identified. In five of these, (A-
E) shown in Scheme 22, the immediate precursor to the coupled product is shown as
the phenoxyl radical (which is resonance stabilized, see previous Scheme 7), and theimmediate product upon coupling is the dienone form of the dimer, which then
rearomatizes to form the phenolic analogues.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 63/182
54
O OH
OH
phenoxyl radical dienone dimer phenolic dimer
A
HH
O
O
OH
OH
B
OH
OH
OH
H
OOH
OH
OH
O
OH
O
OH
O
C
D
E
O
OH
Scheme 22: Dienone and phenolic dimers from the coupling of phenoxyl
radicals

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 64/182
55
Scheme 23 is an illustration of the sixth possible mode of coupling (F) in which two
phenoxyl radicals combine through oxygen, resulting in the peroxide as shown.
Scheme 23: Peroxide formation from the coupling of phenoxyl radicals
Thus when two phenoxyl radicals couple with another, they may do so in one of the
following ways:
• Ortho C-ortho C coupling (A): A resonance form of the phenoxyl radical in
which the radical is centered at the ortho position couples with another
identical species;
• Para C-para C coupling (B): A resonance form of the phenoxyl radical in which
the radical is centered at the para position couples with another identical
species;
• Ortho C-para C coupling (C): A resonance form of the phenoxyl radical in
which the radical is centered at the ortho position couples with another
resonance form of the phenoxyl radical in which the radical is centered at the
para position;
• Ortho C-O coupling (D): A resonance form of the phenoxyl radical in which theradical is centered at the ortho position couples with the oxygen-centered
radical of another phenoxyl species;
• Para C-O coupling (E): A resonance form of the phenoxyl radical in which the
radical is centered at the para position couples with the oxygen-centered
radical of another phenoxyl moiety; and
FO O
O
phenoxyl radical peroxide

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 65/182
56
• O-O coupling (F): two phenoxyl moieties combine through their oxygen-
centered radicals.
Because of the numerous pathways through which phenoxyl radicals may react with
one another to form dimers, the oxidative coupling of unsubstituted phenol itself
results in numerous products, and there is no known process in which the yield and
selectivity to one specific product is high enough to term the process a successful
one. In addition to the above six modes of coupling, one must also bear in mind that
dimeric products that form are also capable of reacting further with either the
substrate and/or dimeric products in the reaction mixture, forming polymeric species.Oxidation mixtures of unsubstituted phenols thus result in a complex mixture of
dimeric, polymeric and unreacted compounds, often with poor carbon accountability.
All of these factors make the oxidative coupling of unsubstituted phenol itself a very
unattractive prospect when the desired product is a specific dimeric form, for
example, the industrially useful compound 4,4 ’-dihydroxybiphenyl (6).
3.1.1 Molecular Orbital Calculations for the Coupling of Phenol
In order to ascertain the likelihood that the phenoxyl radicals will couple as shown in
Schemes 22 and 23, it was deemed appropriate to calculate the relative stabilities of
the dienone dimers as well as the phenolic dimers. Since these species are all
isomeric, their relative stabilities can be obtained by comparing their heats of
formation (? f H) directly. Given in Table 3.1 are the theoretical heats of formation for
the coupled dienones (? f Hd) and phenols (? f Hp), calculated at the PM3 semi-empirical
molecular orbital (MO) level. In the final column of the table, the difference betweenthe heats of formation of the phenolic dimers and their corresponding dienones has
also been calculated.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 66/182
57
Table 3.1 PM3 Heats of formation for phenol coupling products
Coupling
mode
? fHd
(kcal/mol)
? fHp
(kcal/mol)
? fHp - ? fHd
(kcal/mol)
E -6.53 -24.52 -17.99
C -5.87 -41.86 -35.99
D -5.61 -23.97 -18.36
B -5.08 -42.19 -37.11
A -4.99 -39.48 -34.49
It is clear from these results that the relative stabilities of the primary coupling
products, the dienones, are very similar. As a result, one would expect to obtain a
distribution of all the possible dienones (C-C and C-O coupled), assuming that their
rates of formation are similar.
These calculations also show that the C-C coupled phenolic products, i.e., coupling
modes A, B and C (where ? f Hp is –39.48, -42.19 and –41.86 kcal/mol, respectively),
are significantly more stable than the C-O coupled products, i.e., coupling modes D
and E (where ? f Hp is –23.97 and –24.52 kcal/mol, respectively), and one would
expect, thus, a predominance of C-C coupled products in the product mixture.
However, the rates at which the dienones are converted to the phenolic dimers are
not known, and so one cannot ultimately make predictions about the final phenolic
dimer product distribution. (It is assumed here that the dienone-phenolic dimer
conversion is irreversible and that no equilibrium exists in this transformation.)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 67/182
58
The peroxide PhOOPh, formed by means of O-O coupling (mode F), can be shown to
be much less stable, having a calculated heat of formation of +34.61 kcal/mol.
From this investigation, it may be concluded that both C-C and C-O dienone products
are likely to form in the oxidative coupling of unsubstituted phenol. Hence their
corresponding phenolic forms are also likely, though it must be reiterated that these
calculations do not convey any information on relative rates of formation, and so the
actual product distribution cannot really be predicted. The peroxide, PhOOPh, on the
other hand, appears unlikely to form due to its low stability relative to the other
products. These calculations thus confirm reports that the oxidative coupling of phenol results in a wide product distribution, both C-C and C-O coupled, and cannot
be used with much success when a single dimer is the desired product.
Experimentally, this work did not involve unsubstituted phenol as a substrate for the
above reasons. However, the above MO study was extrapolated to two other
substrates, 2,4-di-t -butylphenol and 2,6-di-t -butylphenol, the results of which are
discussed in the relevant sections.
3.2 THE OXIDATIVE COUPLING OF 2-t -BUTYLPHENOL
The literature contains many reports that deal with the successful C-C coupling of
disubstituted phenols, such as 2,4- and 2,6- dialkylphenols, but is, however, virtually
devoid of studies carried out on monosubstituted phenols, such as 2- t -butylphenol.
This is certainly because both 2,4- and 2,6- disubstituted phenols can each only
effectively C-C couple at one particular carbon position, namely the 6- and 4-positions, respectively, thus affording high yields and selectivities to the desired
product with these substrate types. (This is obviously assuming that C-C coupling is
less likely to take place at aromatic carbon positions that already bear a substituent,
this assumption having been confirmed by PM3 semi-empirical MO calculations which
are discussed later). However, in the case of the monosubstituted phenols, such as
2-t -butylphenol (35), there are two positions available through which C-C coupling can

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 68/182
59
C(CH3)3
OH
1
2
3
4
5
6
occur, the 4- and 6- positions, and so the number of possible products increases and
therefore results in low yields and selectivities to any one desired product. Thus
these substrate types generally result in reactions that are not significantly successful,and hence their virtual absence of mention in the literature.
(35)
There therefore exists a need to investigate the oxidative coupling of monosubstituted
phenols in more depth in order to, at best, develop a process that leads to higher
yields of the required materials or, at worst, contribute positively towards this field of
chemistry by obtaining further information associated with this reaction, since there
does not appear to be much mention of it in the literature. To this end, a variety of
oxidizing agents were used in the investigation of the oxidative coupling of 2-t -
butylphenol in order to attempt to form the para-para C-C coupled product, 3,3’-di-t -
butyl-4,4’-dihydroxybiphenyl (molecule (39) in Scheme 25), in high yield and
selectivity.
The aim of this section of the work was to determine whether any one particular
oxidizing agent afforded optimal results compared with the other agents used, and
whether the substrate molecule could, in fact, be C-C coupled selectively through itspara position despite the additional availability of its ortho position.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 69/182
60
3.2.1 The Range of Possible Products During the Oxidative Coupling of
2-t -Butylphenol
The reaction mechanisms possible for the oxidative coupling of substituted (mainly
disubstituted) phenols have been discussed at length in the literature.22,23 As
mentioned previously, the number of products possible with 2-t -butylphenol as a
substrate is most likely to be greater than that with 2,4- or 2,6- disubstituted phenols
as substrates. In order to hypothetically predict the types of coupling products
possible with (35), one needs to look at the possible coupling modes of the initial 2-t -
butylphenoxyl radical that is formed. Due to resonance stabilization, the radical maybe centered at either the 6- or the 4- position, or on oxygen itself. In either of the two
cases where the radical is centered on an aromatic carbon atom, a number of
products can form, as is illustrated in Schemes 24 and 25. In these schemes, only
the phenolic forms of the coupled products are given, and not the primary dienone
products, for the sake of brevity.
Scheme 24, in which the radical is centered at position 6, shows how the
monosubstituted phenoxyl radical can couple with either another radical in which the
unpaired electron is centered at the 6- or 4- position, or also on oxygen, affording
phenolic dimers (36), (37) and (38) as products, respectively.
Scheme 25 is similar but the initial phenoxyl radical is centered at position 4, thus
affording compounds (37), (39) and (40) upon combining with the various species as
shown.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 70/182
61
Scheme 24: Modes of coupling of the 2-t -butylphenoxyl radical when the radical
is centered on position 6
OO
O
O
O
OH
OH
(36)
OHOH
OH
O
(37)
(38)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 71/182
62
Scheme 25: Modes of coupling of the 2-t -butylphenoxyl radical when the radical
is centered on position 4
There are therefore three possible modes of C-C coupling when 2-t -butylphenol is
oxidatively coupled, namely para-para , ortho-para and ortho-ortho coupling, as was
the case for unsubstituted phenol. In this study, due to its industrial uses and
importance, the desired product was 3,3’-di-t -butyl-4,4’-dihydroxybiphenyl (39).
Already it is apparent that the number of possible products using 2-di-t -butylphenol as
the substrate are numerous, with, in addition to the three different C-C coupled
OO
O
O
O
OH
OH
(37)
OHOH
(39)
(40)
O
HO

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 72/182

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 73/182
64
determination (where this compared favourably with that given in the literature), as
well as NMR, IR and GC-MS experiments.
The reaction conditions and results for the reaction of 2-t -butylphenol with the various
oxidizing agents are summarized in Table 3.2.
Table 3.2 Reactions of 2-t -butylphenol with various oxidizing agents
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
of (35)
(%)
Selectivity to
(39)
(%)
1 Ag2CO3/Cel ite 1 PhCH3 R.T. 10.98 25.57
2 Ag2CO3/Cel ite 20 PhCH3 R.T. 78.58 3.56
3 Cu(OAc)2/
Oxalic acid
10 H2O 60°C 86.32 1.30
4 Mn(OAc)3 1 CH3CO2H 100°C 85.38 2.14
5 BaMnO4 2 CHCl3 50°C 66.96 4.55
6 FeCl3 2 CH2Cl2 R.T. 100.00 0.00
7 Ag2O 1 MeOH R.T. 96.00 7.29
8 K3Fe(CN)6 1 MeOH R.T. 62.20 0.85
9 Ce(SO4)2 1 H2O R.T. 26.61 25.99

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 74/182
65
3.2.2.1 Vanadium(V) oxytrichloride and vanadium(IV) tetrachloride as
coupling agents
As discussed previously, literature reports show that vanadium(V) oxytrichloride61 and
vanadium(IV) tetrachloride react with phenol to give exclusively para -coupled
products, thus implying that these agents are highly selective and para -directing in
their action. Furthermore, vanadium(V) is reported to follow a non-radical mechanism
in which an intermediate with a considerable cationic character is developed,
ultimately ensuring the exclusive formation of para -coupled products (see previous
Schemes 8 and 9).
82
However, for the purposes of our investigation, due to theprohibitive costs of these oxidants, and due to the fact that a vigorous evolution of
HCl gas is accompanied by their reaction with the substrate, implying both
economical and environmental non-viability, it was decided not to assess their effect
on 2-t -butylphenol as substrate.
3.2.2.2 Silver carbonate supported on celite as coupling agent
Silver carbonate on a celite support is also known28 to be a highly specific and
selective oxidizing agent for C-C coupling when reacted with disubstituted phenols.
For example, when silver carbonate/celite was reacted with 2,6-di-t -butylphenol, the
diphenoquinone (10) was the primary product formed. The redox potential of the
oxidant (Ag+ + e- ? Ag ~0.80 V) is thus high enough to oxidize the initially formed
4,4’-dimer (16) to the corresponding 4,4’-diphenoquinone (10). An attractive prospect
with this agent is that the reactions were performed under very mild conditions.
Though the cost of the agent is rather high, it was deemed plausible that some formof recycle would circumvent this disadvantage, and so it was decided to investigate
silver carbonate/celite as the coupling agent for 2-t -butylphenol, and thus to compare
the results obtained with those achieved with disubstituted phenols.
Thus a water-free silver carbonate/celite oxidant was prepared and the substrate
added to it, and the mixture stirred at room temperature for either 1 h (reaction 1,

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 75/182
66
Table 3.2) or 20 h (reaction 2, Table 3.2). From the results in Table 3.2, it can be
seen that reaction 1 did not afford a high selectivity to the desired coupled product
3,3’-di-t -butyl-4,4’-dihydroxybiphenyl (39). From standard curves using HPLCanalyses, it was determined that in reaction 1, the selectivity to (39) was only 25.57
%.
A GC-MS experiment of the reaction mixture (from reaction 1) showed that there were
four products, at retention times of 12.76, 13.29, 14.18 and 15.27 min, that had the
same molecular ion mass (M+) of 298 mass units (the mass of the desired product).
The GC trace and the associated mass spectra of these four isomeric products maybe observed in Figure 3.1 and Appendices 3.1-3.4, respectively.
Figure 3.1: GC trace of product mixture obtained in reaction 1, Table 3.2
An injection of the standard material for (39) showed that it eluted at 15.27 min,
confirming the presence of the desired product in the reaction mixture. The MS
fragmentation patterns of each of these products (Appendices 3.1-3.4) were found to
be, unsurprisingly, somewhat similar in that many of the mass fragments were
common to all four products. The main difference between these mass spectra was
the relative abundance of the various mass fragments in one spectrum compared to

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 76/182
67
that in another spectrum. It should be noted that products (36), (37), (38) and (40) all
have the same M+ (298 mass units) as the desired product (39). However, to identify
the exact structures of the products from their MS fragmentation patterns alone wasnot possible, and an exhaustive separation procedure would be required followed by
individual characterization. This was not deemed necessary since the objective of
this study was to increase the selectivity to (39).
In an attempt to increase the yield of (39) that was obtained in reaction 1, the reaction
time was extended from 1 h to 20 h, with all other variables remaining constant
(reaction 2, Table 3.2). The effect of this change was a dramatic increase in theconversion of the substrate (from 10.98 to 78.58 %), but with an equally dramatic
decrease in selectivity to (39) [from 25.57 to 3.56 %]. The number of moles of (39)
formed in each of these two reactions was calculated from standard curves using
HPLC analyses, and may be observed in Table 3.3.
Table 3.3 Amount of 3,3’-di-t -butyl-4,4’-dihydroxybiphenyl (39) formed in
reactions 1 and 2
Reaction
No.
Time
(h)
Mass of (35) used
(g)
Moles of (35) used
(mmol)
Moles of (39) formed
(mmol)
1 1 0.1576 1.049 0.0147
2 20 0.1611 1.072 0.0150
It is clear that in both reactions in which the amounts of substrate used was very
similar, irrespective of the reaction time, the same amount of product was formed.
Reaction time did not seem to have an effect on the yield of the desired product (39).
This may imply that the desired reaction is rather fast, and that an increase in time
after the formation of (39) merely resulted in side product formation, and hence the
overall decrease in selectivity to (39). However, it may also be probable that the
increased reaction time allowed the formed product to react further, thus also
accounting for the decrease in selectivity.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 77/182
68
Further scrutiny of the GC trace from reaction 1 (Figure 3.1) revealed the two
products at retention times 20.67 and 21.79 min. MS Data showed both these
products to have an M+ of 446 mass units, and their retention times alone hinted atthe possibility that these were large molecules. When the possible products with m/z
= 446 mass units were investigated, it was found that the C-O coupled product (41) in
Scheme 26 (where n=1), had this required mass. Other workers using 2,6-
dimethylphenol as the substrate and manganese oxide as the coupling agent also
found these types of compounds in their reaction mixtures (see Schemes 14 and
15).37
Scheme 26: The C-O coupling of 2-t -butylphenol to afford (41)
When C-O coupling occurs, the reaction mixture becomes much more complex with
side product formation becoming even more significant. It must also be noted that
when an oxidizing agent is reacted with 2- t -butylphenol, the polyether (41) is only one
OH
(CH3)3C
O
O
(CH3)3C
(CH3)3C
n=1,2,3,.....
OH
(CH3)3C
Ag2CO3/celite
(35)
(41)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 78/182
69
(CH3)3C
OH
C(CH3)3
OH
(CH3)3C
OH
(42)
of the possible products that could have m/z = 446 mass units. Another possibility is
the following C-C coupled product, which is also deemed highly feasible.
Furthermore, a product having both C-O and C-C coupling may also account for the
mass of 446, but mass fragmentation patterns alone do not suffice for the exact
structure determination of such molecules.
Upon completion of reaction 2, i.e., after 20 h reaction time, it was noted that these
two products with m/z = 446 mass units disappeared, an indication that they reacted
further to form longer chain polymers, and which could then not be detected by this
technique. To add credence to the latter statement, reaction mixture 2, upon workup,was very tarry, dark in colour and had a high viscosity, an indication of the presence
of polymers.
It was thus concluded that silver carbonate/celite was not suitable as a coupling agent
for 2-t -butylphenol under the conditions investigated in this study, despite its reported
success with disubstituted phenolics: the reaction was very inefficient and the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 79/182
70
selectivity to (39) was unacceptably low. No further work was thus conducted using
this oxidant and 2-t -butylphenol.
3.2.2.3 Copper acetate, in the presence of a dicarboxylic acid, as coupling
agent
Copper acetate, in the presence of the dicarboxylic acid oxalic acid,46,76 was also
investigated as a coupling agent for 2- t -butylphenol. Cupric salts of dicarboxylic acids
have been reported to couple phenolic substrates, the products of which were
combined at unsubstituted ortho- and para- positions in a manner characteristic of single-electron oxidizing agents. The higher oxidized products such as the
diphenoquinones were generally not produced with this coupling agent. Furthermore,
only reactions of disubstituted phenols have been reported,46 due to the increased
possibility of polymer formation with substrates that are less substituted.
A slurry of the substrate and sodium lauryl sulphate in deionised water was treated
with a mixture of cupric acetate and oxalic acid, also in deionised water, and the
resultant mixture stirred rapidly for 5 min while heating to 60°C. After the addition of
sodium hydroxide (in order to achieve a pH of 9), the mixture was heated and stirred
for 10 h under an oxygen atmosphere, cooled and worked up. Reaction 3 in Table
3.2 summarizes the result of this experiment. Disappointingly, this was not promising:
although the conversion of the substrate was high (86.32 %), the amount of (39)
formed was extremely low, with a selectivity to (39) of 1.30 % being calculated. This
implied that side product formation in this reaction was highly significant. Consider
the HPLC trace obtained upon analysis of the reaction mixture (Figure 3.2):

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 80/182
71
Figure 3.2: HPLC of product mixture obtained in reaction 3, Table 3.2
It is obvious from this trace that the reaction mixture for reaction 3 is highly complex
with many different products visible. It was virtually impossible to isolate/characterize
all of these products due to their great number, and their low occurrence. The
reaction conditions were then varied in order to assess their effect on the reaction:
various other dicarboxylic acids, such as succinic and glutaric acid, were used in
place of oxalic acid, and the reaction was carried out under an N2 rather than O2
atmosphere. Furthermore, the oxidant:substrate molar ratios were varied. None of
these variations provided any significant improvements.
Once again, it was concluded that the cupric salts of dicarboxylic acids were not
suitable as coupling agents for 2-t -butylphenol under the reaction conditions
investigated, despite the positive results reported for 2,6-di-t -butylphenol.46
3.2.2.4 Manganese(III) acetate as coupling agent
It has been reported in the literature 26 that Mn(OAc)3 can be used successfully as a
coupling agent for 2,6-disubstituted phenolic substrates, with quantitative yields to the
desired products being claimed. However, this oxidant, when reacted with
monosubstituted phenols such as p -cresol, p -chlorophenol and 4-t -butylphenol, is

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 81/182
72
also reported to afford mostly polymeric products.26 Despite these reports, an
investigation of this oxidant with 2-t -butylphenol was undertaken.
2-t -Butylphenol was thus treated with a manganese(III) acetate/glacial acetic acid
mixture for 1 h at 100°C. Reaction 4 in Table 3.2 is a summary of the obtained
results. A high conversion (85.38 %) indicated the high reactivity of 2-t -butylphenol
with managanese(III) acetate, but the selectivity to (39) was, once again, extremely
low at 2.14 %. From the GC trace, the products at retention times 12.76, 13.29,
14.18 and 15.27 min (with m/z = 298 mass units) were again prominent (as they were
in the Ag2CO3/celite work), with no trimeric species being observed at retention timesof 20.65 and 21.79 min (with m/z = 446 mass units). The reaction mixture was very
tarry upon work-up, and the presence of long chain polymers was thus a distinct
possibility (these not usually being observable under the GC-MS conditions used).
Thus manganese(III) acetate proved also to be unsuitable for the oxidative coupling
reactions of 2-t -butylphenol under the reaction conditions employed.
3.2.2.5 Barium manganate as coupling agent
Barium manganate is known to effectively couple substituted phenolics,73 and it was
thus decided to investigate its effect on 2-t -butylphenol. This substrate, in CH2Cl2 as
solvent, was treated with excess BaMnO 4 at room temperature for 1 h. The results
obtained are summarized in Table 3.2 (reaction 5). These show, once again, a very
low selectivity to (39) [4.55 %], despite a reasonable conversion of the substrate
(66.96 %). Once again, the GC and HPLC traces indicated that a large number of
products had formed in the reaction. From the GC trace (Appendix 3.5), the isomericproducts at retention times 12.79, 14.21 and 15.30 min were again prominent (as they
were in the silver carbonate/celite work). In addition, products at retention times
20.84, 22.00, 26.41 and 38.04 min all had the same M+ value (446 mass units), and it
has already been speculated that these compounds are isomeric trimeric species
(see the silver carbonate/celite investigation).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 82/182
73
It was concluded that BaMnO4 was therefore not an ideal coupling agent for (35) in
these conditions due to the low selectivity to (39).
3.2.2.6 Ferric chloride as coupling agent
2-Naphthol has been successfully coupled using FeCl3, known as a one-electron
oxidant, as coupling agent.77 2-t -Butylphenol was thus treated with this ferric species
for 2 h at room temperature with chloroform as the solvent. Table 3.2, reaction 6,
illustrates that no p -p coupled product was formed under these conditions, as was
observed from standard HPLC data, and confirmed by GC-MS experiments, despitetotal substrate conversion. The nature of the products obtained in this reaction also
varied substantially from those experiments already discussed, as was apparent from
the lack of common retention times and m/z values when comparing the various
traces. However, due to the poor results achieved in this reaction, these products
were not characterized.
Reports exist that claim that many phenols having steric bulk in the vicinity of the
hydroxyl moiety do not couple successfully in the presence of FeCl3. It has been
stated that this is due to prevention of formation of the phenoxyl–iron complex that is
required to form for an efficient reaction.22 Thus carbon-carbon coupling is only
favoured when the phenoxyl radicals that are produced remain complexed through
oxygen to the respective iron atoms during the coupling step. This may offer a
reasonable explanation for our findings using (35) as the substrate, since this
compound does have steric bulk, in the form of the tert -butyl group, in the ortho
position to the hydroxyl group. FeCl3 was thus not investigated further in thisinstance.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 83/182
74
3.2.2.7 Silver oxide as coupling agent
Silver oxide provided high yields and selectivities to the desired C-C coupled productswhen disubstituted substrates such as 2,6-di-t -butylphenol were employed in its
presence.78 The mechanism of this reaction, however, is possibly the least well
understood of all the oxidative coupling processes, though it is postulated to occur on
the metal surface.22
In our investigation, commercially available silver oxide was used in addition to silver
oxide that had been prepared in our laboratories by treatment of silver nitrate withaqueous sodium hydroxide. The use of this oxidant implies exorbitant costs (unless
some form of recycle may be used for the silver metal so-produced), irrespective of
the results achieved.
The alkylphenol was added to silver oxide in methanol, and the resultant reaction
mixture was stirred at ambient temperature for 1 h. Reaction 7 in Table 3.2 thus
showed a high conversion of the substrate (96.00 %), but with a selectivity of only
7.29 % to (39). The kinds of products obtained were similar to those from the silver
carbonate/celite work, as concluded from GC data. The coupled product 3,3’-di-t -
butyl-4,4’-dihydroxybiphenyl (39) was the most prominent on the GC trace, and no
other products were observed above the retention time of 16 min. It thus initially
appeared as though the selectivity to (39) would be high, but standard calculations
proved otherwise: once again, the conditions under which the GC-MS experiments
were conducted were not conducive to the detection of polymeric materials, as must
have been present in this case to account for the low calculated selectivity.
Silver oxide as coupling agent was thus set aside due to its inefficient action in this
reaction to afford (39).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 84/182
75
3.2.2.8 Potassium ferric cyanide, lead tetra-acetate, a
(nitrosonaphtholato)metal complex and cerium(IV) sulphate as
oxidants
The final oxidants that were investigated for substrate (35) were potassium ferric
cyanide, lead tetra-acetate, a (nitrosonaphtholato)metal complex and cerium(IV)
sulphate. No reactivity between the (nitrosonaphtholato)metal complex27 and 2-t -
butylphenol was observed although the reaction was repeated several times, and so
this reaction was not investigated further. The reaction of lead tetra-acetate79 with 2-
t -butylphenol gave a large variety of products, but the selectivity to the coupledproduct (39) was not quantified.
The use of K3Fe(CN)6 and Ce(SO4)2 as oxidative coupling agents with disubstituted
phenols is well documented.50,83 These reactions were carried out according to
Sections 2.3.1.10 and 2.3.1.11, and the results so-obtained are summarized in Table
3.2, reactions 8 and 9. The first of these was disappointing: potassium ferric cyanide
(reaction 8) allowed the formation of only 0.85 % of (39) of the 62.20 % of substrate
that had been converted. Ce4+, which is generated electrochemically from cerium(III)
carbonate and is thus economically viable and environmentally friendly due to the
potential for regeneration, gave a reasonable selectivity to (39) of 25.99%, but only
26.61 % of the substrate was converted. GC-MS Data showed, once again, that the
range of products was similar to those of the many oxidants discussed previously.
3.2.3 Concluding Remarks on the Oxidative Coupling of 2-t -Butylphenol
From all of the above information, it is clear that the number of modes in which 2-t -
butylphenol can couple is vast, irrespective of the oxidant used, resulting in
experiments that generally produced low selectivities to (39). The highest selectivities
were achieved with silver carbonate on celite, and cerium(IV) sulphate, but these too
were unacceptably low (for purposes of industrial interest). The other oxidants were
virtually totally ineffective at producing the desired result. In most of the cases,

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 85/182
76
OH
C(CH3)3(CH3)3C1
4
(9)
2
35
6
conversion of the substrate was reasonable, and it appeared that the higher
conversions were associated with the lowest selectivities. This was not surprising
since high conversions, in these reactions, generally implied an increased possibilityfor side product and polymer formation. No further work was thus conducted on (35)
as substrate since none of the results obtained showed any promise.
3.3 THE OXIDATIVE COUPLING OF 2,6-DI-t -BUTYLPHENOL
The literature contains many reports that deal with the successful para C-para C
coupling of 2,6-di-t -butylphenol (9).
84-87
In order to understand why this substratereacts so successfully, we may predict that the reasons are due to the fact that (9)
can only effectively couple at one particular carbon position, namely the 4-position,
assuming that coupling at a carbon position already bearing a substituent is less
favoured (as is likely, due to steric implications). This latter assumption, however, will
be investigated further in order to assess its validity.
Furthermore, we may also assume that para C-O coupling will be less likely to occur
than para C-para C coupling because of the increased steric effect that would comeinto play in such a situation, due to the proximity of the t -butyl groups to the hydroxyl
moiety (and thus their proximity to the subsequent O-centered phenoxyl radical
species, effectively hindering its reaction with other radical species). We may thus
predict that side reactions, such as those resulting from para C-O coupling, ortho C-
para C coupling etc., will be less favoured, and that the resultant product mixture in
the oxidative coupling of (9) will show a high yield and selectivity to the desired

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 86/182
77
product, 3,3’,5,5’-tetra-t -butyl-4,4’-dihydroxybiphenyl (16), via the corresponding
dienone form. (Note that, depending on the oxidant, 3,3’,5,5’-tetra-t -
butyldiphenoquinone (10), may also be the isolated product, which is formed by over oxidation of (16), and that this product may readily be reduced back to the phenolic
form (16).)
Previous studies have been conducted to investigate the effect of various substituents
on the aromatic ring in coupling reactions. It has been reported87 that when larger
groups, such as t -butyl, are present, then carbon-carbon coupling predominates.
However, when the phenolic bears smaller substituents, such as a methyl group,carbon-oxygen coupling becomes more predominant. It was further reported that the
relative rate of this competitive reaction depended largely on the reaction conditions.
As confirmation of the above predictions and thus the success with which (9) can be
oxidatively coupled, this substrate has been reported to predominantly form the para
C-para C dimer in very high yields when subjected to electrochemical oxidation, as
shown in Scheme 19 previously.53
3.3.1 Molecular Orbital Calculations for the Oxidative Coupling of 2,6 -Di-
t -Butylphenol
Predictions made in the preceding section required some theoretical backup and
verification, and MO calculations were thus carried out in order to determine the
preferential mode of coupling of (9). All possible modes were taken into account, with
one exception, the oxygen-oxygen coupling mode to afford the peroxide, since thishad been shown in previous calculations to be highly unlikely. Schemes 27a and 27b
are an illustration of the coupling reactions investigated by means of PM3 semi-
empirical MO calculations.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 87/182
78
Scheme 27a: Dienone and phenolic dimers from the coupling of 2,6-di-t -
butylphenoxyl radicals by modes G and H
Of the five modes possible, only two are able to result in a final phenolic form of the
product (where both rings are aromatic), modes G and H (Scheme 27a), with the
other modes (I, J and K, Scheme 27b) forming only the dienone form of the dimers
(where at least one of the rings is not aromatic), due to the nature of their structures.(Dienones from modes I, J and K would require the leaving of one or more t -butyl
groups in order to form phenolic products, which we assume will not be a highly
significant reaction pathway.)
phenoxyl radical dienone dimer phenolic dimer
HH
O
O
t -But -Bu
t -But -Bu
OH
OH
t -Bu
t -Bu t -Bu
t -Bu
H
OH
O
t -Bu t -Bu
t -Bu
t -Bu
O
OH
t -But -Bu
t -Bu
t -Bu
O
t -But -Bu G

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 88/182
79
Scheme 27b: Dienone dimers from the coupling of 2,6-di-t -butylphenoxyl
radicals by modes I, J and K
As done previously for phenol, the relative stabilities of the various dimeric species for
coupling modes G-K were obtained by comparing their heats of formation (? f H)directly. Thus the heats of formation obtained for the coupled dienones (? f Hd) and
phenols (? f Hp) are summarized in Table 3.4.
phenoxyl radical dienone dimer
O t -Bu
Ot -Bu
t -Bu t -Bu
Ot -Bu
H
O
t -Bu
t -Bu t -Bu
O t -Bu
Ot -Bu
t -Bu
t -Bu
I
J
O
t -But -Bu
K

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 89/182
80
Table 3.4 PM3 Heats of formation for 2,6 -di-t -butylphenol coupled products
Coupling
mode
? fHd
(kcal/mol)
? fHp
(kcal/mol)
? fHp - ?fHd
(kcal/mol)
G -84.34 -119.24 -34.90
H -80.29 -94.17 -13.88
I -76.39 N/A N/A
J -59.89 N/A N/A
K -47.35 N/A N/A
It is obvious from the data contained in Table 3.4 that the most stable dienone
intermediate product is the one that results through coupling mode G (where ? f Hd is
-84.34 kcal/mol), i.e., through unsubstituted para C-para C coupling, as predicted
earlier. Approximately 4 kcal/mol less stable is the para C-O coupled dienone
through mode H (with ? f Hd = -80.29 kcal/mol). The relative stabilities of the phenolic
tautomers of these dienone dimers follow the same trend in that mode G affords the
more stable substituted phenolic dimer (? f Hp = -119.24 kcal/mol), with the phenol
from mode H being significantly less stable (by 25.07 kcal/mol with ? f Hp = -94.17
kcal/mol). A plausible reason for these stability differences is the enhanced steric
congestion experienced by products formed through mode H.
Coupling via the substituted ortho C and an unsubstituted para C (mode I) is about 8
kcal/mol less favourable than para C-para C coupling (with ?f Hd = -76.39 kcal/mol).
Steric congestion, once again, is most likely the major contributing factor for this
observation. Similarly, the other two modes of coupling (J and K) that also involve a

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 90/182
81
carbon atom already bearing a substituent are even less favourable, as was predicted
earlier, and for similar reasons.
In conclusion, we may therefore say that the formation of the para C-para C coupled
product will be the most likely when considering calculated heats of formation of the
various species. These MO calculations add credence to previous predictions made
regarding which mode of coupling will be the predominant one when (9) is used as
the substrate of choice.
2,6-Di-t -butylphenol was subjected to the action of a couple of oxidizing agents,namely Ag2O and Cu(OAc)2/oxalic acid, in order to confirm the previous predictions
experimentally in our laboratories, and to compare with the results obtained for other
substrate oxidations. The results of this investigation are reported in the next section.
3.3.2 Oxidative Coupling Reactions of 2,6-Di-t -Butylphenol Using
Various Oxidants
As mentioned previously, the successful oxidative coupling of 2,6-di- t -butylphenol has
been extensively reported in the literature.84-87 In our case, the aim of this
investigation was to determine specifically both conversion and selectivity to the
desired para C-para C coupled products, in order to be able to compare the results
with those obtained when using other substrates, and to confirm data obtained from
MO calculations. Note that, in this case, there are two possible desired products,
3,3’-5,5’-tetra-t -butyl-4,4’-biphenol (16) and 3,3’-5,5’-tetra-t -butyl-4,4’-diphenoquinone
(10). Both of these are obtained by means of coupling mode G, with the quinoneform (10) merely being an oxidized form of the biphenol. Both forms are readily
interchangeable by simple reduction or oxidation (Scheme 28), and hence the
formation of either of these two compounds or a mixture of both is equally desirable,
and a mixture of the two is not necessarily deemed a disadvantage in this reaction.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 91/182
82
Scheme 28: The ready interchangeability of desired products (16) and (10)
The two oxidizing agents that were assessed were Ag2O and Cu(OAc)2/oxalic acid(which were also used previously for 2-t -butylphenol as substrate).
Both standard materials, (16) and (10), required separate preparation so that reaction
mixtures from this investigation could be effectively quantified. The quinone (10) was
prepared by reacting 2,6-di-t -butylphenol with Ag2O resulting in the required product
being virtually quantitatively obtained, and which was further purified by means of
recrystallization. The biphenol (16) was prepared by reducing (10) with sodium
hydrosulphite in the presence of aqueous NaOH, followed by recrystallization. The
structures of both standards were confirmed to be that of (10) and (16) by means of
melting point determinations and the successful comparison of these with reported
values, as well as NMR, IR and GC-MS experiments.
2,6-Di-t -butylphenol (9) was then treated with Ag2O and Cu(OAc)2/oxalic acid, in
separate experiments, and the results so-obtained are summarized in Table 3.5.
HO OHoxidation
reductionO O
(16) (10)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 92/182
83
Table 3.5 Reactions of 2,6-di-t -butylphenol with various oxidizing agents
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
of (9)
(%)
Selectivity
to (10)
(%)
Selectivity
to (16)
(%)
10 Ag2O 1 MeOH R.T. 100 99.00 1.00
11 Cu(OAc)2/
Oxalic acid
10 PhCH3 60°C 100 96.25 3.75
3.3.2.1 Silver oxide as coupling agent
It was previously reported that silver oxide provided high yields and selectivities to the
desired coupled product when disubstituted substrates such as 2,6-di-t -butylphenol
were employed in its presence.78 As described earlier, the mechanism of this reaction
is not well understood, though it is postulated to occur on the metal surface.22
The alkylphenol was added to silver oxide in methanol, and the resultant reaction
mixture was stirred at ambient temperature for 1 h. It was noted in this reaction
(reaction 10, Table 3.5) that 2,6-di-t -butylphenol (9) was highly reactive under the
reaction conditions employed, with a conversion of 100 % being achieved. Upon
analysis by GC, only two products were observed (Figure 3.3).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 93/182
84
Figure 3.3: GC trace of product mixture obtained in reaction 10, Table 3.5
The major product (99 %) at a retention time of 13.77 min was identified as 3,3’,5,5’-
tetra-t -butyldiphenoquinone (10) by using both retention time and mass fragmentation
pattern comparisons with that of the prepared standard material. The molecular ion
had a mass of 408 mass units as is required for this product (see Appendix 3.6). Theother product present in the reaction mixture to a much lesser extent, with a retention
time of 14.11 min, was identified as the biphenol (16), whose retention time and mass
fragmentation pattern corresponded with that of its standard material (with M+ = 410
mass units, Appendix 3.7). The selectivity to (16) was calculated to be only 1.00 %
(Table 3.5). Since both (16) and (10) are desired products, both resulting from the
same coupling mode G (para C-para C), the overall selectivity of this oxidative
coupling reaction may be said to be 100 % (1 % + 99 %).
The predominance of (10) in this reaction is not surprising since the reaction was
carried out with sufficient oxidant for the overall transformation to (10). The
substrate:Ag2O ratio used was 1:2, which is effectively a 1:4 substrate:Ag+ ratio. It is
presumed that one mole equivalent of Ag+ per substrate molecule is required for the
coupling process itself (i.e., two moles of Ag+ are used to form every biphenol product
molecule), while two mole equivalents of Ag+ are then required for the oxidation of the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 94/182
85
so-formed biphenol (16) to the diphenoquinone (10). Thus the amount of oxidant
used was sufficient to ensure the further oxidation of (16) all the way through to (10)
[Scheme 29].
Scheme 29: Oxidation of 2,6-di-t -butylphenol using Ag2O
In addition, silver oxide has a high redox potential (approximately 0.80 V) and thus
has the power to oxidize (16) to (10).
The ratio of the substrate to oxidant was increased to both 1:1 and 1:0.5, and the
results compared with that of 1:2 (Table 3.6).
Table 3.6 The effect of substrate:oxidant ratio variations
Substrate:Oxidant Conversion(%)
Selectivity to (16)(% )
Selectivity to (10)(% )
1:2 100 1.00 99.001:1 98.80 4.05 95.95
1:0.5 71.01 8.75 91.25
OH
O
O
OH
OH
(9)
(16) (10)
2 mol Ag +
22 mol Ag+

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 95/182
86
As the ratio of substrate to oxidant is increased, it is clear that the conversion of
substrate decreases, not unexpectedly. The diphenoquinone (10) remains the
predominant final product, implying that the oxidation of (16) to (10) is possibly amore facile reaction than the coupling of (9) to form (16). In addition, the overall
selectivity topara C-para C coupling remained 100 % in all cases.
The intermediate dienone, i.e., 3,3’,5,5’-tetra-t -butyl-1,1’-dihydro-2,2’,5,5’-
biscyclohexadiene-4,4’-dione (43) [see also Scheme 27a] was never observed in any
of our reaction mixtures. It has been reported88 that the oxidative coupling of 2,6-di-t -
butylphenol with silver oxide, in the absence of air, afforded isolation of this dienoneform of (16), and that this keto tautomer is stable in non-polar solvents,53 but
tautomerizes immediately to (16) in hydroxylic solvents such as methanol. In
addition, Blanchard78 reported that the coupled product (43) was also formed in the
presence of oxygen when 2,6-di-t -butylphenol was oxidized by silver oxide in a non-
polar solvent.
Due to the fact that methanol (a hydroxylic solvent) was our solvent of choice, the
absence of observation of (43) in any of our reaction mixtures (irrespective of
substrate:oxidant ratio) was thus not surprising.
Overall, our findings are thus in agreement with other reports that used silver oxide as
coupling agent, giving para C-para C coupling (mode G) exclusively, with no other
O O
H
H
(43)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 96/182
87
side reactions occurring. No para C-O coupling was ever observed in our
investigations. The previously shown MO calculation data predicted this behaviour.
The bulky t -butyl groups, and thus steric factors, play a significant role in the mode of coupling that (9) preferably undergoes.
3.3.2.2 Copper(II) acetate/oxalic acid as coupling agent
The reaction of copper acetate46,76 in the presence of a dicarboxylic acid, oxalic acid,
with 2,6-di-t -butylphenol was investigated. As reported previously, cupric salts of
dicarboxylic acids have been used to couple phenolic substrates in a manner characteristic of a single-electron oxidizing agent.46
A substrate/sodium lauryl sulphate/deionised water slurry was treated with a cupric
acetate/oxalic acid/deionised water mixture, and stirred rapidly for 5 min while heating
at 60°C. After sodium hydroxide addition (pH 9), the mixture was heated and stirred
for 10 h under an oxygen atmosphere, and then cooled and worked up. Reaction 11
in Table 3.5 summarizes the result of this experiment. (For this reaction, the molar
ratio of the substrate:oxidant was 50:1, the copper salt of oxalic acid thus acting in a
catalytic fashion.) After the required reaction time, no starting material remained, with
the conversion of 2,6-di-t -butylphenol to products being complete. A GC-MS analysis
of the reaction mixture showed that, once again, as with the silver oxide work, only
two products were formed, at retention times of 13.76 and 14.14 min, respectively.
These corresponded with the standards (10) and (16), and the analysis showed again
that (10) was the predominant product (with a selectivity of 96.25 %) as compared to
(16) [with a selectivity of 3.75 %]. Thus the redox potential of the copper salt wasalso high enough to oxidize the initially formed product (16) further to form (10).
These results are thus very similar to those achieved when using Ag2O, reiterating the
ease with which (9) can be oxidatively coupled with high selectivity and yield to the
desired para C-para C coupled product. Once again, the overall selectivity to the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 97/182
88
para C-para C coupled products (16) and (10) is 100 % (3.75 % + 96.25 %), with no
para C-O coupled products being observed.
3.3.3 Concluding Remarks on the Oxidative Coupling of 2,6-Di-t -
Butylphenol
From the above information, it is clear that the number of modes in which 2,6-di- t -
butylphenol can theoretically couple is numerous, though, in practice, this substrate is
highly selective when placed under oxidative coupling conditions. High selectivities to
the desired para C-para C coupled products (16) and (10) were achieved with both Ag2O and Cu(OAc)2/oxalic acid (100 % selective in both instances). This is in
agreement with results obtained in the literature.46,78 No para C-O coupled products
were ever observed. Molecular orbital calculations confirmed these observations. In
addition, it was stated earlier that dealkylation of the coupled product and/or substrate
would not be a significant reaction pathway, and this was found to be so since no
dealkylated products were ever evident. In conclusion, it may thus be said that the
presence of the additional t -butyl group in (9), as compared with that of 2-t -
butylphenol (35), obviously plays a critical role in its choice of mode of coupling, and
steric congestion is also a major consideration in these reactions. No further work
was conducted on (9) as substrate since the results were optimal and well known to
the field, and there was thus no need for further investigation.
3.4 THE OXIDATIVE COUPLING OF 2,4-DI-t -BUTYLPHENOL
The oxidative coupling of 2,4-di- t -butylphenol has not been as well documented asthat of 2,6-di-t -butylphenol. However, as with the 2,6-analogue, it is envisaged that
2,4-di-t -butylphenol (44) will carbon-carbon couple primarily at one particular carbon
position, namely the 6-position, for similar reasons to those discussed earlier for the
2,6-analogue. Thus the oxidative coupling of (44) should lead to the ortho C-ortho C
coupled product (45) with high yield and selectivity.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 98/182
89
OH
C(CH3)3
C(CH3)3
1
6 25 3
4
(44)
Having said this, it must be bourne in mind that although (44) has only one available
unsubstituted carbon position available for coupling, the possibility of unsubstitutedortho C-O coupling occurring may be greater than that for 2,6-di-t -butylphenol since
the hydroxyl moiety (and hence the phenoxyl radical) of the 2,4-analogue has
decreased steric hindrance compared with that of the 2,6-analogue. This is because
there is only one bulky t -butyl group in close proximity to the OH group in the 2,4-
analogue, but two such bulky groups in the 2,6-analogue. One may propose that 2-t -
butylphenol (35) has similar hindrance in the vicinity of the hydroxyl moiety as that of
(44), and thus when one considers that (35), under oxidative coupling conditions,
afforded no less than four products having the same mass as that of the desired
coupled product (39) [see relevant previous section], one may come to the conclusion
that it is highly likely that some unsubstituted C-O coupling did indeed occur with (35)
[though these four isomeric products were not isolated and characterized]. It
therefore appears likely that some unsubstituted C-O coupling should also be likely
with (44). However, in previous reports,89 it was stated that when (44) was oxidized
with di-t -butyl peroxide at 140°C for 24 h, the coupled product 3,3’,5,5’-tetra-t -butyl-
2,2’-dihydroxybiphenyl (45) was solely formed, and no C-O coupled products wereobserved.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 99/182
90
OH OH
(45)
The primary aim of this investigation was to study the oxidative coupling of (44) using
a variety of oxidizing agents. The desired product in these reactions was (45), and an
attempt was made to obtain optimal yields and selectivities to this product. A further
aim was to compare and analyze results obtained here with those obtained for the
other substrates under the same oxidative reaction conditions.
3.4.1 Molecular Orbital Calculations for the Oxidative Coupling of 2,4 -Di-
t -Butylphenol
As before, MO calculations were carried out to determine the preferential mode of
coupling of (44). The heat of formation calculated for 2,4-di-t -butylphenol (-59.90
kcal/mol) suggested that it was marginally more stable than 2,6-di-t -butylphenol
(having a heat of formation of -58.78 kcal/mol), presumably due to the greater steric
crowding associated with the 2,6 -analogue.
The possible coupling modes with respect to available carbon positions available for
coupling were similar to those of 2,6-di-t -butylphenol, with oxygen-oxygen coupling
also being discounted. Schemes 30a and 30b are an illustration of the coupling
reactions investigated by means of PM3 semi-empirical MO calculations.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 100/182
91
O
t -Bu
t -Bu
L
t -But -Bu
t -But -Bu
O
OH H
t -Bu t -Bu
t -Bu t -Bu
HO
OH
M
t -Bu
t -Bu
O
O
t -Bu
t -Bu
Ht -Bu
t -Bu
O
HO
t -Bu
t -Bu
phenoxyl radical dienone dimer phenolic dimer
Scheme 30a: Dienone and phenolic dimers from the coupling of 2,4-di-t -
butylphenoxyl radicals by modes L and M
There are only two possible reaction modes (L and M) that are able to afford phenolic
products with both rings aromatic in nature, via the tautomeric rearrangement of the
corresponding dienone forms. The other modes (N, O, P and Q, Scheme 30b) result
only in the dienone forms of the dimers: as with 2,6-di-t -butylphenol, the formation of
the phenolic products for these latter modes would require the leaving of one or moret -butyl groups and it was assumed that this would not constitute a significant reaction
pathway.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 101/182
92
t -Bu
t -Bu
O
Nt -Bu
t -Bu
H
t -Bu
OO
t -Bu
O
O
t -Bu
t -Bu O
t -Bu
t -Bu
P
t -Bu
t -Bu t -Bu
O
O
t -Bu
phenoxyl radical dienone dimer
t -Bu
t -Bu
t -Bu
O
t -Bu
O
Q
Scheme 30b: Dienone dimers from the coupling of 2,4-di-t -butylphenoxyl
radicals by modes N, O, P and Q

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 102/182
93
The relative stabilities of the various dimeric species for the coupling modes L-Q were
obtained by comparing their heats of formation (? f H), as before. The results thus
obtained for the coupled dienones (? f Hd) and phenols (? f Hp) are summarized in Table3.7.
Table 3.7 PM3 Heats of formation for 2,4 -di-t -butylphenol coupled products
Coupling
mode
? fHd
(kcal/mol)
? fHp
(kcal/mol)
? fHp - ?fHd
(kcal/mol)
L -93.67 -123.31 -29.63
M -90.30 -105.61 -15.31
N -83.61 N/A N/A
O -78.63 N/A N/A
P -59.32 N/A N/A
Q -52.67 N/A N/A
From this data, it can be observed that the most stable dienone intermediate is the
one that results through coupling mode L (where (? f Hd) is -93.67 kcal/mol), formed
through ortho C-ortho C coupling in which both of these ortho positions are
unsubstituted. The unsubstituted ortho C-O coupled dienone (mode M) was
approximately 3.4 kcal/mol less stable than the corresponding ortho C-ortho C
dienone (mode L). The stabilities of the corresponding phenolic tautomers followed
the same trend, with mode L affording the more stable phenolic dimer (? f Hp = -123.31
kcal/mol), followed next by the phenol from mode M. Again, this latter phenol was

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 103/182
94
significantly less stable than that from mode L (by 17.70 kcal/mol with ? f Hp = -105.61
kcal/mol).
Coupling via the unsubstituted ortho C and a substituted para C (mode N) is
approximately 10 kcal/mol less favourable than ortho C-ortho C (with ? f Hd = -83.61
kcal/mol), obviously as a result of steric interactions. Mode O, coupling between a
substituted para C and oxygen is less desired still, having ?f Hd = -78.63 kcal/mol.
Finally, when both coupling carbons are substituted, i.e., modes P and Q, the
coupling reaction is highly disfavoured, as would be expected. In conclusion, we may
therefore say that the formation of the ortho C-ortho C coupled product is most likelyto be the favoured one according to these heats of formation data of the various
species.
2,4-Di-t -butylphenol was then subjected to the action of a variety of oxidizing agents,
and the results of these reported in the next section, and compared with the data
obtained from other substrates in a later section.
3.4.2 Oxidative Coupling Reactions of 2,4-Di-t -Butylphenol Using
Various Oxidants
In the course of this investigation, a number of oxidizing agents were selected for
study. Naturally, agents that were considered both economically and environmentally
advantageous were given priority. Furthermore, the aim here was to obtain optimal
results in terms of the coupling of (44) to (45). The experimental results obtained
were compared to those obtained from MO calculations (with mode L being predictedto be the most favourable), and to results obtained from reactions with other
substrates, where appropriate.
The standard material of the desired product, 3,3’,5,5’-tetra- t -butyl-2,2’-
dihydroxybiphenyl (45), was prepared by reacting 2,4-di-t -butylphenol with K3Fe(CN)3.
The product thus formed required purification by recrystallization. The structure of

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 104/182
95
this compound was confirmed to be that of (45) by means of a melting point
determination, and the successful comparison of this with reported values, as well as
NMR, IR and GC-MS experiments.
2,4-Di-t -butylphenol (44) was then treated with various oxidants, and the optimum
results achieved with each are summarized in Table 3.8.
Table 3.8 Reactions of 2,4-di-t -butylphenol with various oxidizing agents
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (45)
(%)
12 FeCl3 2 PhCH3 50°C 92.00 0
13 Ag2O 1 MeOH R.T. 96.52 5.06
14 K3Fe(CN)6 2 MeOH R.T 96.04 83.95
15 Ce4+ 1 H2O/
MeOH
Reflux 100.00 90.35
3.4.2.1 Ferric chloride as coupling agent
Ferric chloride, known as a one-electron transfer oxidant, has been used extensively
for the oxidative coupling of various phenols.90-92 2,4-Di-t -butylphenol was treated
with this ferric species for 2 h at 50°C in either chloroform or toluene as the solvent.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 105/182
96
Table 3.9 Reactions of 2,4-di-t -butylphenol with FeCl3
Reaction No. Ratio
(oxidant:substrate)
Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (45)
(%)
12 2:1 2 PhCH3 50°C 92.00 0
16 2:1 2 CHCl3 50°C 83.00 0
The results (Table 3.9) showed that 2,4-di-t -butylphenol proved to be highly reactive
with FeCl3 under the reaction conditions employed (reaction 12 and 16), with
conversions of 92.00 and 83.00 % being obtained, respectively. However, in neither
solvent was any desired coupled product (45) formed. A variety of other products
were evident upon analysis of the reaction mixture by GC (Figure 3.4).
Figure 3.4: GC trace of product mixture obtained in reaction 12, Table 3.9

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 106/182
97
Data obtained from MS experiments suggested that, in the case of this substrate, the
products obtained stemmed not only from the oxidative coupling process, but also
from their subsequent dealkylation, as well as that of the starting material. For example, when toluene was used as the reaction solvent, the presence of t -
butyltoluene at the retention time of 7.11 min was very significant, contrary to our
initial assumption that dealkylation would not be a main consideration in these
reactions. In addition, the product at 9.60 min was identified as t -butylphenol, most
likely from the dealkylation of the substrate. Furthermore, products at retention times
15.27 and 13.51 min were identified as the tri- and tetra- debutylated coupled
products, respectively, though these were not isolated and characterized, and so themode of coupling that occurred could not be ascertained.
When CHCl3 was used as the reaction solvent (reaction 16), a large variety of
products were noted, including chlorinated 2,4-di-t -butylphenol, t -butylphenol (due to
dealkylation) and various dealkylated coupled products. It seems that coupling does
indeed occur in these conditions but that the resultant products are further
debutylated.
Once again, the steric bulk afforded by the t -butyl group in the vicinity of the OH
group was not conducive to a clean oxidative coupling process, as verified by results
obtained in the 2-t -butylphenol work and as claimed by other workers in the field.22
Due to the poor results achieved with FeCl3, it was therefore deemed appropriate to
sideline this reaction, and not investigate its use any further with 2,4-di-t -butylphenol
as substrate.
3.4.2.2 Silver oxide as coupling agent
Silver oxide is a known one-electron transfer oxidant that converts phenols to
phenoxyl radicals.12,90 These phenoxyl radicals can then undergo the characteristic
C-C and/or C-O coupling processes. Since silver oxide showed high selectivity to the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 107/182
98
diphenoquinone coupled product (10) when reacted with 2,6-di-t -butylphenol, its
reaction with 2,4-di-t -butylphenol was considered to be of great interest.
The alkylphenol was added to silver oxide in methanol, and the resulting reaction
mixture stirred at ambient temperature for 1 h. Reaction 13 in Table 3.8 indicated the
high reactivity of the substrate in these conditions, with 96.52 % conversion of 2,4-di-
t -butylphenol being observed. However, the reaction was not a selective one, as is
seen by the many products formed according to GC analysis (Figure 3.5).
Figure 3.5: GC trace of product mixture obtained in reaction 13, Table 3.8
The product at retention time 17.51 min had a molecular ion mass (M+) of 438 mass
units and could not be identified, but that at 17.71 min had an M+ of 410 mass units
and was confirmed to be the desired C-C coupled product (45) by comparison with
the standard material. However, the selectivity to biphenol (45) was only 5.06 %.
This was not necessarily surprising since silver oxide, in the case of the 2,6-analogue
(9), afforded mainly the overoxidized diphenoquinone derivative (10), with only a very
small amount of the biphenol (16) being observed. Thus it may appear probable that
(46) could dominate over (45) here also, though its stability (as is known for ortho
versus para quinones) is most likely to be lower than that of (10).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 108/182
99
Two products at retention times 23.84 and 25.70 min had an M+ of 408 mass units,
the required mass for (46). However, because it was not a priority in this
investigation, a separation of these was not carried out, and it is thus not certain
which of the two products was (46), if any. Whatever the case, it is obvious from this
study that the 2,4- and 2,6- di-t -butylphenols behave very differently under identical
conditions when treated with silver oxide. The 2,6-analogue was coupled highly
successfully whilst the 2,4-analogue provided rather disappointing results, with a
number of unwanted side products being formed. Reasons for this are not clear – the
mechanism at work here, as has been stated before, is not well known, but from this
investigation, it may be concluded that the positioning of the alkyl substituents on the
aromatic moiety plays a role in the subsequent reaction of the substrate. It is
plausible that steric hindrance is a factor since one would expect the 4-position of the
2,6-analogue to be less crowded than that of the 6 -position in the 2,4-analogue.
As an aside, and of peripheral interest, was the product at the retention time of 30.74min which MS data showed to have an M+ of 615 mass units (Appendix 3.8). This is
possibly due to a product of multiple coupling such as, for example, the triaryl species
(47) shown in Scheme 31 (where n=1), though the exact nature of the coupling
(whether C-C or C-O) was not verified by further characterization.
O
O
(46)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 109/182
100
Scheme 31: A plausible multi-coupled product (47) with M+ = 615 mass units
Therefore, due to the disappointingly low selectivity to the coupled product (45) and
the subsequent significance of side reactions in these conditions, the reaction of 2,4-
di-t -butylphenol with Ag2O was not investigated further.
3.4.2.3 Potassium ferric cyanide as coupling agent
Ferric cyanide is one of the most widely used oxidizing agents for the generation of
phenoxyl radicals in alkaline solutions.90,92,93 Previous studies94 indicate that the
oxidant must be in excess relative to the substrate, and that K3Fe(CN)6 acts as a one-
electron transfer agent [reaction (A)] involving phenoxide anions (present due to the
basic medium) as the oxidizable substrate. Furthermore, it was found that the rate of oxidation was largely dependent on the basicity of the solution and on the
ferricyanide/ferrocyanide ratio.
ArO-
+ [Fe(CN)6]3- ArO• + [Fe(CN)6]4- (A)
t -Bu
OH
t -Bu
Ag 2O
OH
t -Bu
t -Bu
O
O
t -Bu
t -Bu
t -Bu
t -Bu
n=1,2,3,...
(47)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 110/182
101
2,4-Di-t -butylphenol was thus treated with this ferric species in a basic medium
(NaOH) with the reaction conditions and results summarized in Table 3.10.
Table 3.10 Reactions of 2,4-di-t -butylphenol with K3Fe(CN)6
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (45)
(%)
14 K3Fe(CN)6 2 MeOH/H2O R.T 96.04 83.95
17 K3Fe(CN)6 1 MeOH/H2O R.T 84.53 86.10
It was noted in this reaction that 2,4-di-t -butylphenol (44) was highly reactive under
the reaction conditions employed, with conversions of 96.04 and 84.53 % for reactions 14 and 17, respectively. These reactions were carried out under identical
reaction conditions except that reaction 14 was continued for 2 h, whilst reaction 17
was quenched after only 1 h. The longer reaction (14) afforded the higher conversion
of substrate (not surprisingly), but the selectivity to (45) decreased from 86.10 (after 1
h, reaction 17) to 83.95 % (after 2 h, reaction 14). These results were, however,
significantly superior to those achieved with ferric chloride and silver oxide.
Since potassium ferric cyanide is a known one-electron oxidizing agent, and since the
reaction takes place in a basic medium, the phenoxyl radical is thought to form from
the phenoxide anion (see (A) before). After this, the direct coupling (the FR1
mechanism highlighted earlier, Scheme 6) of two such phenoxyl radicals, in which the
radical is centered at the 6-position (through resonance), takes place to ultimately
afford (45) after tautomerization. Scheme 32 depicts the proposed mechanism at
work in this reaction.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 111/182
102
Scheme 32: Mechanism for the coupling of (44) using potassium ferric cyanide
GC Analysis thus showed a considerable decrease in side product formation when
compared with reactions using Ag2O and FeCl3 (Figure 3.6).
O
anion of
K3Fe(CN)
6
O O
X 2
FR1 mechanism
OH OH
followed by
(45)
_
basic conditions
(44)
tautomerization

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 112/182
103
Figure 3.6: GC trace of product mixture obtained in reaction 14, Table 3.9
The product at a retention time of 17.97 min was the major product and was identified
as the desired ortho C-ortho C coupled product (45) by comparison with a standard.
The product at retention time 17.52 min had the same molecular ion mass (M+) of 410
mass units as (45), though the MS fragmentation pattern differed. This product is
obviously an isomeric form of (45) in which the coupling mode differed, perhaps being
formed by ortho C-O coupling, and affording, as a likely product, dimer (48), though
no isolation was carried out.
O
OH
(48)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 113/182
104
Also notable was the presence of products at retention times of 24.13 and 31.39 min,
their mass fragmentation patterns being indicative, as before, of products (46) and
(47), respectively, which can only form by coupling modes L or M.
3.4.2.4 Cerium(IV) as coupling agent
3.4.2.4.1 Identification of Ce(IV) as the preferred oxidant
In this investigation, two considerations that had to be bourne in mind were the
economic viability and environmental impact of the oxidant of choice. Although the
oxidative coupling reaction of 2,4-di-t -butylphenol (44) with K3Fe(CN)6 gavesatisfactory results, it is not an environmentally acceptable oxidant nor would it be
industrially attractive. The indirect electrochemical oxidation of organic substrates is
becoming more and more economically viable, and there are many compounds that
are known to be capable of acting as indirect oxidants, including transition metal salts,
cobalt, manganese, iron, lead, silver and cerium. Examples of redox couples that
have been studied include Ce3+/Ce4+, Mn2+/Mn3+, and Mn2+/MnO2.95 An alternative
oxidant was thus sought, one that can be regenerated, implying that recycling thereof
would be feasible, and thus being advantageous from an economic point of view.
Furthermore, if the oxidant could be re-used, this would directly have a positive
bearing on the environment since the potential amount of waste requiring
treatment/storage would be minimized. However, the regeneration of many spent
metals to their higher oxidation states is not always effective, since many metal ion
oxidants have certain properties that make this process difficult. However, the
indirect electrochemical oxidation of phenols using a redox couple remains attractive
and, to this end, we investigated it further.
For the purposes of this study, the Ce4+/Ce3+ couple was extensively investigated,
with its recycle (Figure 3.7) being the driving force for our interest. Among the
electron carriers most commonly used for indirect oxidations, cerium salts appear to
be the most suitable when oxidations must be carried out under mild reaction
conditions. The most common electron valencies of cerium salts are three and four,96

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 114/182
105
with cerium(IV) behaving as a one-electron oxidant.83,97 The oxidation potential of the
Ce4+/Ce3+ couple is reported to be dependent on the reaction conditions, such as the
ligand. For example, the oxidation potential of this couple in 1 N perchloric, nitric,sulphuric and hydrochloric acids was observed to be -1.70, -1.61, -1.44 and -1.28
volts, respectively.98-101
Figure 3.7: Electron flow in the indirect electrochemical oxidation process
using the Ce3+/Ce4+ couple
The oxidative coupling of 2,6-disubstituted phenols using Ce(IV) in the presence of
perchloric acid is well documented.69,70 However, an alternative acid would be
preferred since perchloric acid has the potential to lead to the formation of
perchlorates which can be hazardous when in contact with organic chemicals.102 It
was thus decided to investigate the use of W.R. Grace’s technology,103 where Ce(IV)
would be reacted with our substrate in the presence of methanesulphonic acid. There
are a number of advantages of using methanesulphonic acid rather than sulphuric
and perchloric acid. These are:
• It is unreactive with both reactants and products.
• It is stable to anodic and cerium oxidations.
• Ce(III) and Ce(IV) are highly soluble in aqueous methanesulphonic acid.
• It has a high current efficiency (>90 %) at high current density (>400 mA/cm2).
Ce4+
Ce3+
2,4-disubstituted phenol
Coupled product
cathode
anode

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 115/182

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 116/182
107
source of Ce(III). The experimental setup was given previously (Figure 2.1). The
Ce(III) concentration was kept constant (0.1 M) whilst varying the methanesulphonic
acid concentration initially between 0.5 and 2.0 M. An extensive literature surveyfailed to indicate that oxidative coupling of disubstituted phenols, including 2,4-di-t -
butylphenol, had been previously investigated with Ce(IV) in the presence of
methanesulphonic acid. This work is thus entirely novel, and information gathered
from it is deemed to add to the knowledge base of this field of chemistry.
The aim of this investigation was thus to extensively investigate the oxidative coupling
of 2,4-di-t -butylphenol (44) using Ce(IV) as the oxidant in methanesulphonic acid and,in so doing, to determine the effect of the following on this oxidative coupling process:
• Varying the MeSO3H concentration.
• One or two phase systems with or without added co-solvent.
• Varying the reaction temperature.
• Varying the reaction time.
• Substrate loading.
• Varying the substrate:oxidant ratio.
• Varying the rate of oxidant addition to the reaction mixture.
On completion of these studies, it will then be possible to optimize reaction conditions
so as to improve the yield and selectivity to the desired coupled product (45).
3.4.2.4.2 Oxidation in MeSO3H mediated by Ce(IV) ions
For this investigation, the cerium carbonate concentration was kept constant at 0.1 M,
irrespective of the methanesulphonic acid concentration. The minimum concentration
of methanesulphonic acid was set at 0.5 M (MeSO3H is a mono-protic acid). Hence
the methanesulphonic acid concentration was varied between 0.5 and 2.0 M for the
electrochemical oxidation of Ce(III) to Ce(IV). The results obtained are summarized
in Table 2.3 (Experimental section). It was noted that the highest conversion of
Ce(III) to Ce(IV) occurred when the MeSO3H concentration was 1.0 M (when

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 117/182
108
considering the average titration values). The rate of the electrochemical oxidation of
Ce(III) to Ce(IV) also depended on the methanesulphonic acid concentration, with
slower oxidations occurring at lower acid concentrations, as is evident from Table 2.4.
It has been well documented that an increase or decrease in the acid concentration
affects the oxidation strength of the Ce(IV) ions.70,104 The effect of the MeSO3H
concentration on the oxidative coupling of 2,4-di-t -butylphenol (44) to form (45) in the
presence of Ce(IV) ions at room temperature (R.T.) was thus investigated, and the
results summarized in Table 3.11.
Table 3.11 Oxidative coupling of (44) by Ce(IV) at various [MeSO3H] at R.T.
Reactions 18 to 20 were all one hour reactions carried out in aqueous media at
ambient temperature. The ratio of substrate:oxidant was also kept constant at 1:2.
The only variable was the methanesulphonic acid concentration. Initial investigations
under these conditions showed that the highest conversion of (44) and selectivity to
(45) was achieved when the MeSO3H concentration was 1.0 M (reaction 19, Table
3.11), where these values were 43.68 and 42.71 %, respectively. However, due to
the reaction solvent being aqueous, the solubility of the substrate was poor as was
evident from the adherence of the organic substrate to the stirrer bar and the glass
walls of the reaction vessel. This low solubility of the substrate in aqueous media was
presumed to have a deleterious effect on the reactivity of (44) with Ce(IV), and it was
ReactionNo.
[MeSO3H](M)
Time(h)
Solvent Ratio(substrate:oxidant)
Conversionof (44)
(%)
Selectivityto (45)
(%)
18 0.5 1 H2O 1:2 31.06 18.60
19 1.0 1 H2O 1:2 43.68 42.71
20 2.0 1 H2O 1:2 17.05 25.21

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 118/182
109
assumed that this problem required addressing before optimal results would be
achieved.
To decrease this solubility effect, the reaction temperature was increased to 80°C,
whilst keeping all other variables constant (Table 3.12).
Table 3.12 Oxidative coupling of (44) by Ce(IV) at various [MeSO3H] at 80°C
A significant improvement was noted in terms of both the conversion of (44) and the
selectivity to (45) in each of these reactions when compared with those obtained at
R.T. Once again, as at R.T., the reaction using 1.0 M MeSO3H (reaction 22) gave the
best results in terms of conversion of 2,4-di-t -butylphenol and selectivity to the
coupled product (45). In conclusion then, it may be said that an increase in the
reaction temperature from ambient to 80°C increased both conversion and selectivity,
irrespective of the acid concentration. Thus far, the optimal reaction was one carried
out at the elevated temperature using 1.0 M methanesulphonic acid (reaction 22,
Table 3.12).
Not unexpectedly, therefore, these results imply that the solubility of the substrate is
important in the reaction. As above, an increase in reaction temperature is one
method by which the solubility may be increased. Another method is to add a co-
solvent to the aqueous medium, the co-solvent obviously being one in which the
ReactionNo.
[MeSO3H](M)
Time(h)
Solvent Ratio(substrate:oxidant)
Conversionof (44)
(%)
Selectivityto (45)
(%)
21 0.5 1 H2O 1:2 48.47 34.93
22 1.0 1 H2O 1:2 96.11 76.22
23 2.0 1 H2O 1:2 73.43 64.61

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 119/182
110
substrate is very soluble, such as methanol, acetonitrile or dichloromethane. The first
two of these are water-soluble organic solvents and would ultimately afford a one
phase reaction system, whilst dichloromethane is not and would result in a two phasereaction system. To investigate the effects of these co-solvents on the coupling
process, the reactions were initially performed at R.T. for 1 hour. Since previous
results showed that optimal conversions and selectivities were obtained using 1.0 M
methanesulphonic acid concentrations, this was the concentration of choice in the
following reactions. Results so-obtained are summarized in Table 3.13.
Table 3.13 Effect of co-solvents on oxidative coupling of (44) by Ce(IV) at R.T.
The conversion of 2,4-di-t -butylphenol in aqueous solvent with added MeOH or
CH3CN was similar and high at 94.80 and 93.29 %, respectively. However, the
acetonitrile-containing system afforded a lower selectivity to (45) of 62.37 %, whereas
the methanol-containing system was higher at 69.28 %. When CH2Cl2 was used as
the organic co-solvent, there was a significant decrease in the conversion of (44)[reaction 26, 41.87 %] compared with MeOH and CH3CN. The selectivity to the
coupled product (45) was, however, much higher (85.22 %). This low reactivity of
2,4-di-t -butylphenol with Ce(IV) in H2O/CH2Cl2 is most likely due to the presence of
the two phases, where the substrate prefers to reside within the organic solvent,
implying that reaction can only occur effectively at the boundary surface of the two
phases. In such a system, stirring efficiency would be an important consideration.
ReactionNo.
[MeSO3H](M)
Solvent Ratio(substrate:oxidant)
Conversionof (44)
(%)
Selectivityto (45)
(%)
24 1.0 H2O/MeOH 1:2 94.80 69.28
25 1.0 H2O/CH3CN 1:2 93.29 62.37
26 1.0 H2O/CH2Cl2 1:2 41.87 85.22

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 120/182
111
Overall, MeOH as co-solvent thus gave the most promising results at ambient
temperatures.
In the next study, using 1.0 M MeSO3H and MeOH as co-solvent, the reaction was
carried out at various reaction temperatures in order to determine whether an
optimum point could be defined. All other variables were kept constant. The results
obtained are contained in Table 3.14.
Table 3.14 Temperature effect on coupling of (44) by Ce(IV) with MeOH as co-solvent
Reaction No. MeOHvolume
Ratio(substrate:oxidant)
Temperature Conversionof (44)
(%)
Selectivityto (45)
(%)
27 20 mL 1:2 0°C 92.29 73.72
28 20 mL 1:2 R.T. 93.42 78.76
29 20 mL 1:2 45°C 95.09 82.76
30 20 mL 1:2 Reflux 96.96 80.50
The total volume of the MeOH and aqueous MeSO3H was also kept constant (40 mL)
for all of these reactions.
After assessing reactions at 0°C, ambient temperature, 45°C and reflux (65°C), it wasfound that high conversions were obtained throughout, even at 0°C (92.29 %). The
highest conversion occurred at the highest temperature (65°C, 96.96 %). A plot of
conversion versus temperature more clearly summarizes the results obtained (Figure
3.9):

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 121/182
112
Figure 3.9: Plot of conversion versus temperature
Similarly, Figure 3.10 is a graphical summary of the results obtained in terms of
selectivity, where selectivity was plotted against temperature. From this plot, an
optimal selectivity to (45) was achieved at the reaction temperature of 45°C (reaction
29, 82.76 %). However, as seen above, the conversion of (44) at this temperature
(95.09 %) was slightly lower than that obtained at reflux (reaction 30, 96.96 %). Of
interest is the observed decreased selectivity to (45) at the lower temperatures (73.72
% at 0°C and 78.76 % at R.T.). This is a possible indication that the rate of thecoupling reaction to afford (45) increases with increasing temperature, implying that
the lower coupling rates at the lower temperatures provides the reactive species time
to undergo other side reactions, and hence resulting in the observed lower selectivity
at the lower temperatures. In other words, it appears as though the coupling reaction
to form (45) occurs rapidly at the higher temperatures and less so at the lower
temperatures relative to other side reactions: hence, when the reaction temperature
92
93
94
95
96
97
98
0 10 20 30 40 50 60 70
Temperature in Deg. C
C o n v e r s i o n ( % )

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 122/182
113
is decreased, the rate of the desired coupling reaction slows down, therefore
increasing the possibility of side reactions.
Figure 3.10: Plot of selectivity versus temperature
Since the results obtained for reactions 29 and 30 were very similar in terms of both
conversion and selectivity, reflux temperature, due to its ease of application, was
selected as the temperature of choice for further investigations.
A study of the effect of reaction time was then carried out: the substrate (44) was
reacted in 1.0 M MeSO3H for 5 min rather than the usual 1 h, with all other variables
being identical. The comparative result obtained is given in Table 3.15.
73
74
75
76
77
78
79
80
81
82
83
84
0 10 20 30 40 50 60 70
Temperature in Deg. C
S e l e c t i v i t y ( % )

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 123/182
114
Table 3.15 Reaction time effect on the coupling of (44) using Ce(IV)
ReactionNo.
MeOHvolume
Ratio(substrate:oxidant)
Time(min)
Temperature Conversionof (44)
(%)
Selectivityto (45)
(%)
30 20 mL 1:2 60 Reflux 96.96 80.50
31 20 mL 1:2 5 Reflux 94.96 82.97
From this data, it is obvious that there is very little difference between the results
obtained for a reaction of 1 h compared with that of a reaction carried out for 5 min.
Reaction 31 showed only a slight decline in the conversion (from 96.96 to 94.96 %),
but a slight increase in selectivity (from 80.50 to 82.97 %) relative to reaction 30.
Once again, this is probably because the ortho C-ortho C coupling reaction is a rather
rapid one, and so the longer the reaction time, the greater the opportunity for side
product formation.
The next reaction variable that was investigated was the substrate loading by varying
the co-solvent (MeOH) volume, yet keeping the Ce(IV)/MeSO3H volume constant
(i.e., 20 mL) and the amount of 2,4-di-t -butylphenol (44) constant also. Reaction time
and temperature were maintained at the same values throughout. Table 3.16
contains the obtained results. This data was then displayed graphically (Fig. 3.11) by
plotting the percentage selectivity and conversion against the volume of methanol
used (which is then directly related to the substrate loading, with higher methanol
volumes implying lower loadings).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 124/182
115
Table 3.16 Effect of substrate loading on the coupling of (44) using Ce(IV)
Reaction No. MeOHvolume
Ratio(substrate:oxidant)
Temperature Conversionof (44)
(%)
Selectivityto (45)
(%)
32 5 mL 1:2 Reflux 55.98 84.86
33 10 mL 1:2 Reflux 94.47 82.45
30 20 mL 1:2 Reflux 96.96 80.50
34 40 mL 1:2 Reflux 92.93 60.70
Figure 3.11: Plot showing the effect of substrate loading on percentage
conversion and selectivity
0
20
40
60
80
100
120
0 10 20 30 40 50
Volume of MeOH (mL)
P e r c e n t a g e
Conversion Selectivity

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 125/182
116
The best results were obtained in reactions 30 and 33 (Table 3.16) where the volume
of MeOH used was 20 and 10 mL, respectively. It is clear from Figure 3.11 that when
the substrate loading was high (reaction 32, where the MeOH volume used was 5mL), the conversion was a low 55.98 %, while the selectivity was high (84.86 %).
This was surprising since one would have expected that a high substrate loading
would afford high conversions of the starting material. A possible explanation for this
phenomenon is that the substrate solubility in the aqueous medium containing only
small volumes of added co-solvent is low and hence its low conversion. A low
substrate loading, when the volume of MeOH used was 40 mL (reaction 34), showed
a significant decrease in selectivity (60.70 %). This data shows that the substrateloading in these reactions is critical, and should not be too high nor too low for optimal
conversions and selectivities.
The reaction variable investigated next was the oxidant to substrate ratio. In this part
of the investigation, the following reaction variables were kept constant:
• Temperature (reflux).
• Substrate loading (20 mL of MeOH).
• Reaction time (1 hour).
• MeSO3H concentration (1.0 M).
Hence, whilst maintaining a constant substrate amount (moles) in each reaction, the
number of moles of Ce(IV) was varied so that substrate:oxidant molar ratios of 1:0.5,
1:1, 1:1.5, 1:2 and 1:5 were achieved. Refer to Table 3.17 for the results obtained.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 126/182
117
Table 3.17 Effect of substrate:oxidant ratio on the coupling of (44) usingCe(IV)
Reaction No. MeOHvolume
Ratio(substrate:oxidant)
Temperature Conversionof (44)
(%)
Selectivityto (45)
(%)
35 20 mL 1:0.5 Reflux 35.50 94.25
36 20 mL 1:1 Reflux 72.90 91.74
37 20 mL 1:1.5 Reflux 83.35 79.29
30 20 mL 1:2 Reflux 96.96 80.50
38 20 mL 1:5 Reflux 97.12 60.31
The poorest results in terms of conversion were achieved in reaction 35 (35.50 %),
where the substrate to oxidant ratio was 1:0.5. However, this reaction also gave the
greatest selectivity to the coupled product (45) of 94.25 %. The highest conversion of the starting material was achieved in reaction 38 where the substrate to oxidant molar
ratio was the greatest (1:5). This result was to be expected since a larger amount of
oxidant in the reaction mixture increases the availability of the Ce(IV) ions to the
substrate (44), thus increasing the conversion. Furthermore, and as expected, the
selectivity to the coupled product in this reaction was also the lowest (60.31 %):
excess oxidant was obviously available for side reactions or further oxidation of the
formed product.
A graph of both percentage selectivity and conversion versus oxidant:substrate ratio
was then plotted (Fig. 3.12) by using the information in Table 3.17.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 127/182
118
0
20
40
60
80
100
120
0 1 2 3 4 5 6
Molar ratio of Ce(IV) to substrate
P e r c e n t a g e ( % )
conversion selectivity
Figure 3.12: Plot showing the effect of oxidant:substrate ratio on percentage
conversion and selectivity
A molar ratio of 1:2 (substrate:oxidant) thus afforded both high conversion of (44) and
high selectivity to (45), and was thus deemed the optimal ratio in this reaction.
The last variable that was investigated was the rate of addition of the oxidant to the
reaction mixture. In all the previous reactions, the Ce(IV) was added to the reaction
mixture within 30 seconds, except for reaction 15 in Table 3.8 (repeated (Table 3.18)
for convenience), in which the Ce(IV) [in 20 mL MeSO3H] was added to the reactionmixture over a time period of 30 min.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 128/182
119
Table 3.18 Effect of rate of oxidant addition
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (45)
(%)
15 Ce4+
1 MeOH reflux 100.00 90.35
It is clear from this result that these reaction conditions afford superior results in termsof conversion, which is quantitative, and the associated selectivity to the desired
coupled product (45) is still very high at 90.35 %. Thus slow oxidant addition is
clearly favoured over that of its rapid addition.
3.4.2.4.3 Reaction mechanism for the oxidative coupling of 2,4-di-t -butylphenol
using Ce(IV)
To propose a feasible reaction mechanism for the oxidative coupling of 2,4-di-t -
butylphenol (44) using Ce(IV) in the presence of MeSO3H, all the reaction products
need to be identified. The mechanism must then account for these products. Upon a
thorough examination of the reaction mixtures in the various oxidative coupling
reactions using 2,4-di-t -butylphenol (44) as the substrate and Ce(IV) as oxidant, the
following products [(45), (47), (48), (49) and (50)] were identified from their molecular
ion masses and their mass fragmentation patterns. With the exception of (49) and
(50), all of these products were common to those found in reactions using K3Fe(CN)6
as oxidant.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 129/182
120
Since Ce(IV) is reacted with (44) in acidic media, the reaction mechanism probably
differs from that established for potassium ferric cyanide (basic media). (When
potassium ferric cyanide is reacted with (44) in basic medium, the hydrogen of the
phenolic OH is abstracted by base to form the anion (Scheme 32), which is then
oxidized to form the phenoxyl radical.) Since Ce(IV) acts as a one -electron oxidant
and the reaction takes place in acidic medium, the assumption can be made that one
electron is removed from the aromatic ring to form the radical cation (51), as shown inScheme 33. This is most likely a facile process due to the electron rich nature of the
aromatic ring due to the presence of electron-donor substituents such as the hydroxyl
and alkyl groups. This radical cation may then lose a proton to afford the phenoxyl
radical (52). One resonance form of (52) is where the unpaired electron is centered
at carbon position 6. Two of these then couple directly together to afford dimer (53)
O
OH
(48)
OH OH
(45)
OH
t -Bu
t -Bu
O
O
t -Bu
t -Bu
t -Bu
t -Bu(47)
n=1
O
O
(49)
OH
OH
(50)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 130/182
121
which, upon tautomerization, affords (45), the desired product in this reaction, and the
product that was obtained in high yields throughout (relative to any other products).
Scheme 33: Reaction mechanism for the formation of (45)
OH OH
+-H+
O
Ce4+ Ce3+
O
H
(51)
(52)
O
H
OOH H
tautomerization
OHOH
(53)(45)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 131/182
122
Compounds (47) and (48) are readily accounted for: their formation is through C-O
coupling as shown clearly in Scheme 34.
Scheme 34: Reaction mechanism for the formation of (47) and (48)
It is thus plausible that (48) is formed through the direct coupling of radical (52),
where the unpaired electron is centered on oxygen, with another (52) species, but
where the unpaired electron is centered on carbon position 6, thus affording the C-O
coupled product (54). This then tautomerizes to form (48). This product may then be
further oxidized to afford the radical cation (55). Its reaction with (52) yields the cation
O
(52)
O
H
OH
O
O
OH
(48)
tautomerization
Ce4+
Ce3+
O
OH
H
+
O
O
t -Bu
t -Bu
OH
t -Bu
t -Bu
O
t -But -Bu
H
+-H+
O
t -Bu
t -Bu
OH
t -Bu
t -Bu
O
t -But -Bu
(47)
(54)
(55)
(56)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 132/182
123
(56) which may then lose a proton to form trimer (47), the driving force being
rearomatization.
It has been reported that the radical cations are also capable of reacting with water to
ultimately afford dihydroxybenzenes (57) by the pathway shown in Scheme 35.69
Scheme 35: Radical cation reaction with water to form dihydroxybenzenes (57)
Using the same ideology, the formation of products (49) and (50) must certainly be
due to intermediate radical cations reacting with the water that is in the reaction
medium. A possible pathway to explain their formation is presented in Scheme 36.
OH
R
+H2O
OH
OH2
H
R
+
-H+
OH
OH
H
R
-e-
-H+
OH
OH
R(57)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 133/182
124
Scheme 36: Formation of products (49) and (50) from a radical cation
From this scheme, the radical cation (51) produced reacts with water and, after proton
loss, affords the radical (58), which then also undergoes oxidation by losing an
electron. The cation which so forms is then transformed into (50) by the loss of a t -
butyl cation. (Since this is a tertiary carbocation, its stability is reasonably high, and
this step is thus more than plausible. However, since high selectivities to (45) were
observed in these reactions, these debutylated products, though not quantified, werenot formed in amounts significant enough to state that this was a facile process
compared with that of the formation of (45).) Hydroquinone (50) is then readily
oxidized to the quinone (49).
A general trend observed is that an increase in reaction temperature allowed for an
increase in the amount of desired product (45) formed after the same amount of
OH -e -
OH
+
(44)
(51)
+H2O-H+
OH
OH
-e -
OH
OH
(50)
-2e -
-2H+
O
O
(49)
-t -Bu+
(58)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 134/182
125
reaction time (Table 3.14). The desired reaction appears to be a rapid one, and an
increase in reaction temperature plausibly increases it even further, providing less
opportunity for side reactions (such as C-O coupling), and thus possibly accountingfor this general observation. The rate of the desired coupling reaction thus generally
increases with increasing temperature, implying that at lower temperatures, the lower
rate of formation of the desired dimer results in an increased possibility for side
product formation. The statement that the desired coupling reaction is proposed to
occur rather rapidly was also implied by a comparison of the reaction when carried
out for 60 min relative to the reaction when carried out for only 5 min (reaction 30
versus reaction 31, Table 3.15): after only 5 min, a high conversion and selectivity to(45) was obtained, and extending the reaction time to 60 min made no significant
difference to the result.
The effect of substrate loading was also significant in this reaction: the lowest
selectivity to (45) was obtained when the reaction mixture had the lowest substrate
loading (reaction 34, Table 3.16). When the substrate loading was increased, the
selectivity to the desired C-C coupled product also increased. Once again, it may be
concluded that reaction conditions that favour the rapid formation of (45) via the
desired coupling reaction, i.e., by increasing reaction temperature (see above) or
increasing the substrate concentration, disfavour side product formation, and thus an
increase in the selectivity to (45) was observed in these instances. It must be kept in
mind that too high substrate loadings are not ideal since substrate conversions are
rather low in such cases.
Schemes 33-36 therefore account for the products observed in our reaction mixtures,and they hence represent viable pathways by which these products were all formed.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 135/182
126
3.4.3 Concluding Remarks on the Oxidative Coupling of 2,4-Di-t -
Butylphenol
From the results obtained, it is clear that 2,4-di-t -butylphenol coupled primarily by
modes L and M. This substrate was not as selective as 2,6-di-t -butylphenol, but high
selectivities to the ortho C-ortho C coupled product (45) were achieved with both the
K3Fe(CN)6 and Ce(IV) oxidants [86.10 and 96.96 %, respectively]. The use of other
oxidants such as FeCl3 and Ag2O afforded results that were less than satisfactory,
with very little, if any, desired product being formed, despite high conversions. This
was rather surprising, especially in the case of Ag2O, since this oxidant was 100 %selective towards the para C-para C coupled product when the 2,6-analogue was the
substrate. These oxidants are obviously not suitable for the purposes of forming (45),
quite possibly due to the mechanisms by which they react in combination with the
positioning of the substituents on the aromatic rings. Molecular orbital calculations
confirmed the preference for coupling mode L and, to a lesser extent, mode M. The
difference in results obtained for the 2,4- and 2,6-analogues may only be explained in
terms of steric crowding, in which the hydroxyl moiety of the 2,6-analogue is well
“surrounded” by the two bulky t -butyl groups, thus disallowing the formation of C-O
coupled products. The 2,4-analogue, on the other hand, is less crowded in the
vicinity of the OH group, and can thus also form some of the C-O coupled product,
resulting in the observed lower selectivities to the C-C coupled product as compared
with the 2,6-analogue.
The work conducted using Ce(IV) as the oxidant is entirely novel, and the results
obtained in these reactions were very promising indeed, with high selectivities andconversions to the desired coupled dimer (45) being achieved. Optimal reaction
conditions included the use of 1 M aqueous methanesulphonic acid as the medium of
choice with added co-solvent (methanol) such that the resultant solution is a single
phased reaction mixture. Furthermore, the optimal reaction temperature was
approximately 65°C (at reflux, giving high selectivities and conversions and implying
ease of application), and lengthy reaction times were not necessary, probably

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 136/182
127
because the desired coupling reaction takes place rather rapidly. The substrate
loading was an important factor: too high loadings afforded low conversions, and too
low loadings afforded low selectivities. The optimal substrate:oxidant ratio was 1:2,with a slow addition of the oxidant to the reaction medium being slightly favoured over
that of rapid addition. Finally, a mechanism was proposed for this work that
accounted for all the products observed in the reaction mixtures.
3.5 THE OXIDATIVE COUPLING OF 2,4-DIMETHYLPHENOL
From the literature, it was ascertained that the reaction of 2,4-dimethylphenol (59)with various oxidative coupling agents gave complex reaction mixtures.52,105
Furthermore, it was shown that, in the case of the 2,6-dialkylphenols, the bulkiness of
the substituents played an important role in the types of products formed, with the
larger groups, such as t -butyl, giving C-C coupled products almost exclusively.106,107
However, with smaller substituents, such as methyl, C-O coupling has been reported
to occur more readily, and long chain ethers of high molecular weight have been
obtained as products in these cases. When various 2,6-dialkylphenols were oxidizedwith cuprous chloride in nitrobenzene/pyridine, the yield of long chain ethers was
decreased to zero with an increase in bulk of the substituents [-CH3, -CH(CH3)2, -
C(CH3)3].108
Thus the decreased steric effect of the methyl groups of 2,4-dimethylphenol (59),
compared with the t -butyl groups of 2,4-di-t -butylphenol, implies that the course that
OH
Me
Me(59)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 137/182
128
the oxidative coupling process takes by the former substrate may be different to that
taken by the latter substrate, and so resulting in different relative product ranges for
the two substrates. Although 2,4-dimethylphenol (59) has only one availableunsubstituted carbon position available for coupling, the possibility for ortho C-O
coupling occurring may be greater than for 2,4-di-t -butylphenol since the hydroxyl
moiety (and hence the phenoxyl radical) of (59) has decreased steric hindrance.
From the literature, it was also ascertained that the non-bonding interactions between
methyl groups in the transition state for coupling of methyl-substituted phenoxyl
radicals are important.109 It was observed that the oxidation of 2,4-dimethylphenol
gave a much higher yield of C-O coupled products than obtained with p -cresol.
110
Anexamination of the staggered approach (60) for the ortho-ortho coupling of 2,4-
dimethylphenoxyl radicals revealed that there were two sets of non-bonding
interactions between the methyl groups.109
Due to these methyl group interactions, a higher energy pathway for C-C coupling
results, and consequently more C-O coupling occurs because the formation of thelatter bond is much less dependent on efficient SOMO-SOMO interactions between
the two radicals.
However, the possible coupling modes of 2,4-dimethylphenol (59) with respect to
available carbon positions for coupling are similar to those of 2,4-di- t -butylphenol.
Although the oxidative coupling reactions of (59) were not investigated by means of
Me
Me
O
.
MeO
Me
.
(60)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 138/182
129
PM3 semi-empirical MO calculations, the possible coupling modes are illustrated in
Schemes 37a and 37b.
phenoxyl radical phenolic dimer
Scheme 37a: Phenolic dimers from the coupling of 2,4-dimethylphenoxyl
radicals by modes R and S
O
Me
Me
HO
OH
Me Me
Me Me
O
HO
Me
Me
Me
Me
R
S
.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 139/182
130
phenoxyl radical dienone dimer
Scheme 37b: Dienone dimers from the coupling of 2,4-dimethylphenoxyl
radicals by modes T, U, V and W
O
Me
Me
.
H
OO
Me
Me
MeMe
O
O
Me
Me Me
Me
O
O
Me
Me Me
Me
T
U
V
W
Me
Me
Me
Me
OO

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 140/182
131
The assumption is made (as for the 2,4-di-t -butyl analogue) that there are two main
reaction modes that can afford phenolic products, namely modes R and S. The
assumption is also made that, for 2,4-dimethylphenol, the other coupling modes (T, U,V and W, Scheme 37b) result only in the dienone forms of the dimers, with the loss of
methyl groups not being considered to be a pathway that will be favoured by these
dienones, and thus the phenolic forms thereof not being considered significant in
these cases. (The phenolic forms of the dienones can only be formed by methyl
substituents being lost from these dienone substrates.)
The oxidative coupling of (59) was thus assessed by reacting this substrate with avariety of coupling agents, and analyzing the reaction mixtures in order to determine
percentage conversions and yields to the desired product, 3,3’,5,5’-tetramethyl-2,2’-
dihydroxybiphenyl (61).
3.5.1 Oxidative Coupling Reactions of 2,4-Dimethylphenol Using Various
Oxidants
The oxidizing agents considered during the course of this investigation were the same
as those used for 2,4-di-t -butylphenol (with the omission of Ag2O), the aim being to be
able to compare results obtained here with those obtained for the 2,4-di-t -butyl
analogue. This would then provide information on the comparative effect of the
various substituents on the aromatic ring on the coupling process.
OH
Me
Me Me
Me
OH
(61)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 141/182
132
The standard material for the desired product, 3,3’,5,5’-tetramethyl-2,2’-
dihydroxybiphenyl (61), was prepared by reacting 2,4-dimethylphenol with Ce(IV) in
the presence of methanesulphonic acid. The product thus formed was purified bymeans of column chromatography. The structure was confirmed to be that of (61) by
means of a melting point determination, and the successful comparison of this with
reported values, as well as NMR, IR and GC-MS experiments.
The optimum results obtained when 2,4-dimethylphenol (59) was treated with each of
the various oxidants are summarized in Table 3.19.
Table 3.19 Reactions of 2,4-dimethylphenol with various oxidizing agents
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (61)
(%)
39 FeCl3 2 CHCl3 50°C 90.98 49.11
40 K3Fe(CN)6 2 MeOH R.T. 70.86 26.72
41 Ce4+
1 H2O R.T. 76.04 57.58
3.5.1.1 Ferric chloride as coupling agent
The one-electron oxidant, ferric chloride, was reacted with 2,4-dimethylphenol even
though it was not successful at all in coupling the 2,4-di-t -butyl analogue to the
desired coupled product (45). 2,4-Dimethylphenol was treated with this ferric species
for 2 h at 50°C in various solvents (Table 3.20).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 142/182
133
Table 3.20 Reaction of 2,4-dimethylphenol with FeCl3 in various solvents
ReactionNo.
Ratio(substrate:oxidant)
Time(h)
Solvent Temp. Conversion(%)
Selectivityto (61)
(%)
39 1:2 2 CHCl3 50°C 90.98 49.11
42 1:2 2 MeOH 50°C 34.71 11.05
43 1:2 2 PhCH3 50°C 77.88 38.84
44 1:2 2 EtOAc 50°C 36.83 9.59
The results showed that the reactivity of FeCl3 with 2,4-dimethylphenol depended
largely on the solvent employed. The best results for the reaction of FeCl3 with 2,4-
dimethylphenol was achieved when CHCl3 was used as the solvent (reaction 39), with
this affording the highest conversion of (59) and selectivity to (61) of 90.98 and 49.11
%, respectively. Although the selectivity was low and conversion high, no other major
products were detected upon analysis of the reaction mixture by GC (Figure 3.13).
Figure 3.13: GC trace of product mixture from reaction 39, Table 3.20

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 143/182

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 144/182
135
O
Me
Me
O
Me
Me
(64)
OH
Me
Me
O
Me
Me
(65)
Product (63) appears to be one in which one of the methyl substituents has been
oxidized to an aldehydic group. The product at retention time 18.15 min has an
identical m/z value compared with that of (61) (i.e., 242 mass units), and is possibly
either the isomeric Pummerer’s ketone (64) or ortho C-O coupled product (65):
When MeOH and ethyl acetate were used as solvents (reactions 42 and 44) in place
of CHCl3, the results obtained in terms of conversion (34.71 and 36.83 %,respectively) and selectivity (11.05 and 9.59 %, respectively) were very poor, and
these reactions were not further investigated. With toluene as the solvent (reaction
43), a large variety of dealkylated products were obtained, including mono-, di- and
tri- demethylated coupled products at retention times 15.23, 15.17 and 12.86 min
respectively (Figure 3.14). (These dealkylated products were not present in reaction
39 where CHCl3 was the solvent.)
OH
Me
Me
OH
Me
C O
H
(63)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 145/182
136
Figure 3.14: GC trace of product mixture from reaction 43, Table 3.20
The fact that the reaction of FeCl3 with 2,4-dimethylphenol gave at least some of the
desired product, despite being totally ineffective when reacted with 2,4-di-t -
butylphenol (and 2-t -butylphenol), is certainly further evidence that the bulk of the t -
butyl group does indeed prevent formation of the phenoxyl-iron complex,22 whereas
the decreased steric effect associated with the methyl groups allows such an
interaction to some extent.
3.5.1.2 Potassium ferric cyanide as coupling agent
Potassium ferric cyanide gave very promising results when reacted with 2,4-di-t -
butylphenol (44), with a high conversion and selectivity (96.04 and 83.95 %,
respectively) to the desired coupled product (45) being achieved. 2,4-Dimethylphenol was treated with this ferric species under identical reaction
conditions, in basic media (NaOH), and the reaction conditions and results are
summarized in Table 3.21.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 146/182
137
Table 3.21 Reactions of 2,4-dimethylphenol with K3Fe(CN)6
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity
to (45)
(%)
40 K3Fe(CN)6 2 MeOH/H2O R.T 70.86 26.72
The selectivity (26.72 %) to the desired coupled product (61) was thus poor with this
coupling agent. The GC trace, however, shown in Figure 3.15, did not indicate that
many products were formed in this reaction.
Figure 3.15: GC trace of product mixture from reaction 40, Table 3.21
This is indicative again that polymeric materials were formed under these conditions.
GC-MS Experiments suggested that the product eluting at 15.25 min was, once
again, either the ortho C-O coupled product (65) or Pummerer’s ketone (64), since
their M+ values were identical to that of (61) at 242 mass units, but isolation and
further characterization was not considered important here. (The product at retention

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 147/182
138
time 15.54 min was identified as the desired coupled product (61).) Higher elution
times 21.78 and 25.34 min both corresponded to products with an M+ of 360 mass
units, i.e., compounds that were higher than dimeric in nature. The success of thereaction of this ferric species with the 2,4-di-t -butyl analogue could thus not be
mimicked in this case, and all evidence obtained in this reaction hinted at the
significant presence of higher oligomeric species, most likely emanating from C-O
coupling (due to its greater propensity for occurring when the substituents are methyl
groups).
3.5.1.3 Cerium(IV) as coupling agent
For 2,4-di-t -butylphenol (44), coupling mode L (ortho C-ortho C) was found to be the
most dominant, but the dominance of this mode in the oxidation of (44) with Ce(IV)
depended on a number of factors such as temperature, concentration of the oxidant,
the substrate:oxidant ratio, the reaction time and substrate loading. Under the correct
reaction conditions, Ce(IV) in methanesulphonic acid afforded high conversions and
selectivities to the desired product (45) when reacted with 2,4-di-t -butylphenol (44).
However, and as discussed earlier, in the case of the 2,4-dimethyl analogue, it is
highly probable that coupling mode S (ortho C-O) may become more prominent due
to the associated decreased steric effects around the hydroxyl group. In order to
investigate this, 2,4-dimethylphenol was treated with Ce(IV) in methanesulphonic
acid. (This reaction is novel in terms of the reaction conditions used for the coupling
of this substrate by Ce(IV).) The effect of the following reaction parameters on this
oxidative coupling process was investigated:
• Varying the MeSO3H concentration.
• One or two phase systems with or without added co-solvent.
• Varying the reaction temperature.
• Varying the rate of oxidant addition to the reaction mixture.
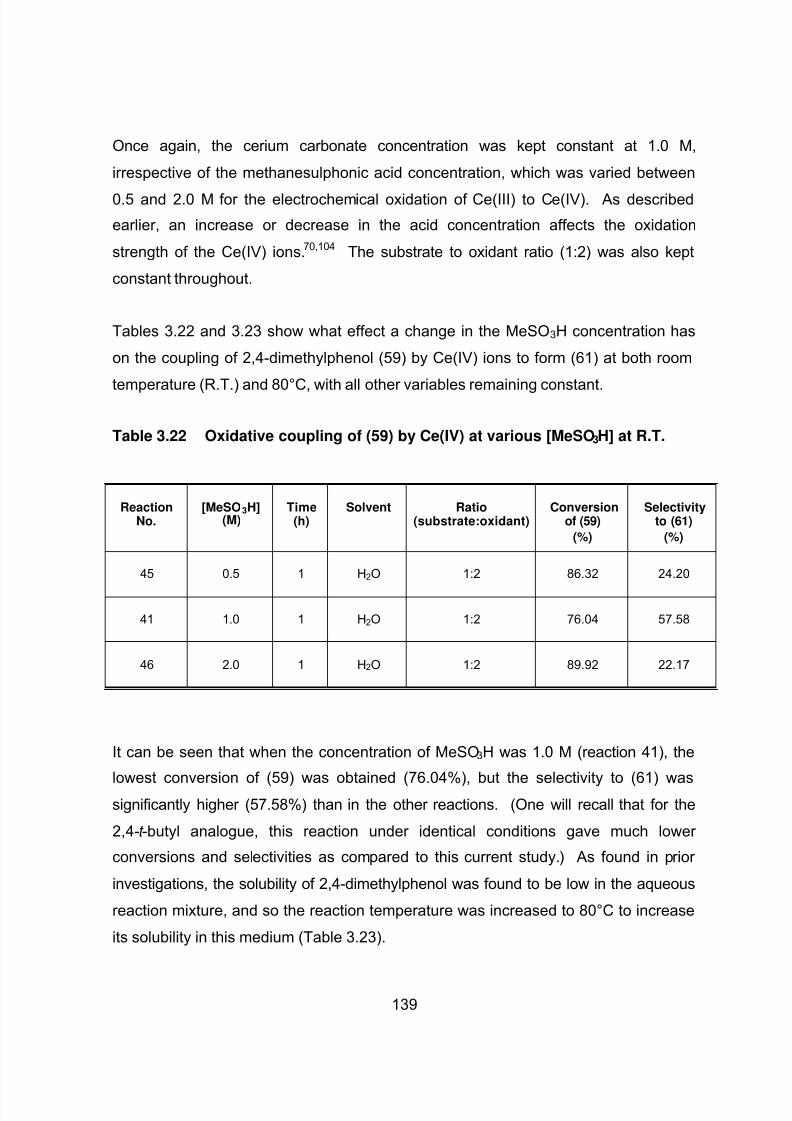
7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 148/182
139
Once again, the cerium carbonate concentration was kept constant at 1.0 M,
irrespective of the methanesulphonic acid concentration, which was varied between
0.5 and 2.0 M for the electrochemical oxidation of Ce(III) to Ce(IV). As describedearlier, an increase or decrease in the acid concentration affects the oxidation
strength of the Ce(IV) ions.70,104 The substrate to oxidant ratio (1:2) was also kept
constant throughout.
Tables 3.22 and 3.23 show what effect a change in the MeSO3H concentration has
on the coupling of 2,4-dimethylphenol (59) by Ce(IV) ions to form (61) at both room
temperature (R.T.) and 80°C, with all other variables remaining constant.
Table 3.22 Oxidative coupling of (59) by Ce(IV) at various [MeSO3H] at R.T.
It can be seen that when the concentration of MeSO3H was 1.0 M (reaction 41), the
lowest conversion of (59) was obtained (76.04%), but the selectivity to (61) wassignificantly higher (57.58%) than in the other reactions. (One will recall that for the
2,4-t -butyl analogue, this reaction under identical conditions gave much lower
conversions and selectivities as compared to this current study.) As found in prior
investigations, the solubility of 2,4-dimethylphenol was found to be low in the aqueous
reaction mixture, and so the reaction temperature was increased to 80°C to increase
its solubility in this medium (Table 3.23).
ReactionNo.
[MeSO3H](M)
Time(h)
Solvent Ratio(substrate:oxidant)
Conversionof (59)
(%)
Selectivityto (61)
(%)
45 0.5 1 H2O 1:2 86.32 24.20
41 1.0 1 H2O 1:2 76.04 57.58
46 2.0 1 H2O 1:2 89.92 22.17

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 149/182
140
Table 3.23 Oxidative coupling of (59) by Ce(IV) at various [MeSO3H] at 80°C
In general, a decrease in terms of both the conversion of (59) and the selectivity to
(61) was noted in each of these reactions when compared to those results at R.T.
This was surprising and in stark contrast to results obtained with the 2,4-di-t -butyl
analogue that afforded much higher conversions and selectivities upon raising the
reaction temperature from ambient to 80°C. Thus, in the case where (59) is the
substrate, an increased reaction temperature is not beneficial to the desired outcome
of the reaction. The reasons for this are not clear, but perhaps the fact that 2,4-
dimethylphenol is a liquid at room temperature whilst 2,4 -di-t -butylphenol is a solid
may affect the reaction in some way. However, it is still very surprising that
conversions dropped upon reaction temperature increase.
As with the 2,4-di-t -butyl analogue, various organic co-solvents were also added to
each of the aqueous mixtures. To this end, MeOH and CH3CN were used in order toafford single-phased reaction systems, whilst CH2Cl2 was used for the biphasic
system. These reactions were performed at R.T. for 1 h while keeping the MeSO3H
concentration constant at 1.0 M (Table 3.24).
Reaction
No.
[MeSO3H](M)
Time
(h)
Solvent Ratio
(substrate:oxidant)
Conversion
of (59)(%)
Selectivity
to (61)(%)
47 0.5 1 H2O 1:2 86.12 17.59
48 1.0 1 H2O 1:2 69.58 57.19
49 2.0 1 H2O 1:2 71.47 16.72

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 150/182
141
Table 3.24 Effect of co-solvents on oxidative coupling of (59) by Ce(IV) at R.T.
The reactivity of 2,4-dimethylphenol towards Ce(IV) ions in the presence of the
organic solvents was high, with reactions 50, 51 and 52 achieving high conversions of
(59) [90.65, 86.17 and 92.46 %, respectively], as expected. These conversions were
much higher than those obtained earlier. However, selectivities dropped significantly.
In the next study, using 1.0 M MeSO3H and CH3CN as co-solvent, the reaction was
carried out at various reaction temperatures with all other variables kept constant.The total volume of the CH3CN and aqueous MeSO3H was also kept constant (40
mL) for all these reactions. It was predicted that increased temperatures in the
presence of this co-solvent would merely decrease selectivities even further. Table
3.25 summarizes the results obtained.
ReactionNo.
[MeSO3H](M)
Solvent Ratio(substrate:oxidant)
Conversionof (59)
(%)
Selectivityto (61)
(%)
50 1.0 H2O/MeOH 1:2 90.65 8.32
51 1.0 H2O/CH3CN 1:2 86.17 23.08
52 1.0 H2O/CH2Cl2 1:2 92.46 6.83

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 151/182
142
Table 3.25 Temperature effect on coupling of (59) by Ce(IV) with CH3CN asco-solvent
Reaction No. CH3CNvolume
Ratio(substrate:oxidant)
Temperature Conversionof (59)
(%)
Selectivityto (61)
(%)
51 20 mL 1:2 R.T. 86.17 23.08
53 20 mL 1:2 80°C 87.03 34.22
54 20 mL 1:2 reflux 78.85 42.81
However, though reasonably high conversions were obtained at all three temperature
settings, the highest selectivity was obtained at the highest temperature (reflux,
reaction 54). Furthermore, the highest conversion was obtained at the lowest
temperature setting (R.T., reaction 51). It is not surprising that increased conversions
are associated with decreased selectivities, but what is surprising is the decreased
conversion at higher reaction temperatures; one would have expected higher
temperatures to be accompanied by higher conversions unless, of course, the
solubility of the substrate decreases with increasing reaction temperature, which
hypothetically appears unlikely. Once again, the reasons for these observations are
thus not clear, and may warrant further investigation in the future.
The optimum reaction conditions for this reaction may therefore be summarized as
follows:• The use of 1.0 M methanesulphonic acid concentration,
• In aqueous solvent with no added co-solvent (i.e., monophasic),
• At room temperature.
When co-solvents were added, reflux temperature afforded the better results.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 152/182
143
The last variable that was investigated was the rate of addition of the oxidant to the
reaction mixture. In all the previous reactions, the Ce(IV) was added to the mixture
within 30 seconds, but in reaction 55 (Table 3.26), the Ce(IV) [in 20 mL MeSO3H] wasadded to the substrate (in 20 mL CH3CN) over a time period of 30 min. Samples
were taken at 15 min and 60 min of reaction time.
Table 3.26 Effect of rate of oxidant addition
Reaction
No.
Oxidant Time
(min.)
Solvent Temp. Conversion
(%)
Selectivity
to (61)
(%)
55 Ce4+
15 CH3CN R.T. 61.10 49.85
55 Ce4+ 60 CH3CN R.T. 82.81 19.24
When one compares the result obtained after 60 min (where the oxidant was added
over 30 min) with that of reaction 51 (where the oxidant was added within 30
seconds), there is no advantage gained with the slower oxidant addition, contrary to
that found for the coupling of the 2,4-di-t -butyl analogue. In fact, slight decreases
were observed in terms of both the conversion and the selectivity to (61). Reaction
51 thus remains the reaction having the optimal results.
3.5.1.3.1 Reaction mechanism for the oxidative coupling of 2,4-dimethylphenolusing Ce(IV)
In order to propose a feasible mechanism by which this reaction occurs, all the
products of reaction 41 (Table 3.22) were identified. The GC trace has the following
appearance (Figure 3.16):

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 153/182
144
Figure 3.16: GC trace of product mixture in reaction 41, Table 3.22
By making use of data obtained in mass spectral experiments and thus by
determining the molecular ion peaks and interpreting their mass fragmentation
patterns, the products (61), (62), (64), (65), (66) and (67) are proposed to have
formed in this reaction. Products eluting at 8.13 and 8.81 min were identified as (66)
and (67), respectively, while those at 14.64, 15.05 and 18.17 min all had the same M+
of 242 mass units. It is obviously not possible to fully characterize and identify theseproducts from their mass fragmentation patterns alone, but it was deemed feasible
that these peaks could be assigned to compounds (64), (65) and some other isomeric
form thereof, such as any of the dienone dimers shown in Scheme 37b. Compounds
eluting at 8.30 and 15.39 min corresponded with the starting material (59) and desired
coupled product (61), respectively.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 154/182
145
The formation of (61) will follow the same mechanism as proposed for the oxidative
coupling of 2,4-di-t -butylphenol (Scheme 33) by Ce(IV) in the presence MeSO3H.
The formation of products (62) and (65) is similar to the mechanism proposed for 2,4-
O
Me
Me
O
MeMe
(64)
OH
Me
Me
O
Me
Me
(65)
OH
Me
Me
OH
Me
Me
(61)
Me
OH
Me
O
O
Me
Me
Me
Me(62)
OH
OHMe
Me
OH
Me
C O
H (67)
(66)

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 155/182
146
di-t -butylphenol to form the products (48) and (47) [Scheme 34]. Pummerer’s ketone
(64) is most likely to be formed by means of the following mechanism (Scheme 38).
Scheme 38: Reaction mechanism for the formation of (64)
The catechol (67) is possibly a result of the reaction of the radical cation with water,
as shown previously in Scheme 35, and it may therefore readily be accounted for.
OH
Me
Me
.Ce4+ Ce3+
-H+
O
Me
Me
Me
O
Me
.
o-p coupling
O
MeH
Me
Me Me
O H+
tautomerization
Me
Me
Me Me
OHOH
(59)
(64)
Me
Me
Me Me
OOH

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 156/182
147
Though the amount of product (66) formed in these reactions was very small, its
formation is of interest. One proposed mechanism that results in this product is
shown in Scheme 39.
Scheme 39: Reaction mechanism for the formation of (66)
OH
Me
Me
Ce4+ Ce3+
-H+
(59)
O
Me
Me
Me
CH2
OH
OH
Me
CH2OH
OH
Me
CHO
(68)(69)
(70)(66)
CH2
OH
Me
+
-e-
H2O -H+
-2e-
-2H+

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 157/182
148
From this scheme, the radical (68) undergoes oxidation by losing an electron to afford
the benzylic cation (69), which then reacts with the water present to form the primary
alcohol (70). This is then further oxidized to form the benzaldehyde (66). (Compound(63), detected in reactions using FeCl3 as oxidant, may thus have been a result of the
coupling of (59) with 4-hydroxy-3-methylbenzaldehyde, formed in a similar fashion to
(66) shown in Scheme 39).
3.5.2 Concluding Remarks on the Oxidative Coupling of 2,4-
Dimethylphenol
From the results obtained, it is clear that 2,4-dimethylphenol coupled primarily by
modes R and S. This substrate was not as selective as 2,6-di-t -butylphenol and 2,4-
di-t -butylphenol. Only moderate selectivities to the ortho C-ortho C coupled product
(61) were achieved with oxidants FeCl3, K3Fe(CN)6 and Ce(IV) [49.11, 26.72 and
57.58 %, respectively]. The difference in the results obtained for 2,4-di-t -butylphenol
and 2,4-dimethylphenol can be explained in terms of steric crowding, in which the
hydroxyl moiety of 2,4-di-t -butylphenol is more sterically hindered by the two bulky t -
butyl groups, thus disallowing the formation of the C-O coupled products which are
more prevalent in the oxidation reactions of 2,4-dimethylphenol. In the case of
K3Fe(CN)6, the major product seems to be that formed by C-O coupling.
Furthermore, the methyl groups themselves are quite plausibly more reactive than the
t -butyl groups of the other substrates, resulting also in the lower observed
selectivities.
Results from the work conducted using Ce(IV) as the oxidant are novel, though theefficiency of the coupling in this reaction was found to be only moderately successful
in terms of selectivity to the coupled product (61). Finally, a mechanism proposed for
this work accounted for all the products observed in the reaction mixtures.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 158/182
149
3.6 BUTYLATED PHENOLIC COUPLINGS: COMPARISONS
In this section, a brief comparison of some of the more important results that have notthus far been discussed and compared, is provided for cases where common
oxidants were used for the various butylated phenol substrates, in order to draw
conclusions from similarities and/or differences observed in the results so-obtained.
(Note that many comparisons have been made in sections prior to this one, but it was
deemed inappropriate to include the discussion that now follows in those self-same
sections.)
3.6.1 Reactions of 2-t -Butylphenol and 2,6-Di-t -Butylphenol with Ag2O
and Cu(OAc)2/Oxalic Acid
Silver oxide was reacted with both 2-t -butylphenol and 2,6-di-t -butylphenol under
identical reaction conditions. The results obtained for these substrates were
significantly different with regards to their selectivity towards the respective desired C-
C coupled products. The results of these reactions are summarized in Table 3.27.
Table 3.27 Comparative data obtained for silver oxide
Reaction
No.
Substrate Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity to p/p
coupled products
(%)
7
2-t -
butylphenol Ag2O 1 MeOH R.T. 96.00 7.29
102,6-di-t -
butylphenol Ag2O 1 MeOH R.T. 100.00 100.00a
aThe diphenoquinone (10) and diol (16) percentages were 96.25 and 3.75 %, respectively.
From these results, it is obvious that when silver oxide is reacted with 2-t -butylphenol
and 2,6-di-t -butylphenol under identical reaction conditions, the selectivity to the
desired para C-para C coupled products is vastly different (7.29 % and 100.00 %,

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 159/182
150
respectively). (Note that with 2,6-di-t -butylphenol, a mixture of the diphenoquinone
(10) and diol (16) were obtained, both resulting from para C-para C coupling, and the
sum of their selectivities equalled 100 %.)
When 2-t -butylphenol and 2,6-di-t -butylphenol were oxidized using a copper(II)
acetate/oxalic acid complex, the results obtained followed the same general trend as
with Ag2O with regards to conversion of starting material and selectivity to the desired
coupled products (Table 3.28).
Table 3.28 Comparative data obtained for copper acetate/oxalic acid
Reaction
No.Substrate Oxidant Solvent Temp. Conversion
(%)
Selectivity to p/p
coupled products
(%)
3 2-t -butylphenol
Cu(OAc)2/
Oxalic acid PhCH3 60°C 86.32 1.30
11
2,6-di-t -
butylphenol
Cu(OAc)2/
Oxalic acid PhCH3 60°C 100.00 100.00
In this case, with 2-t -butylphenol as substrate, the selectivity to the desired coupled
product was extremely low (1.30 %), whereas when the oxidant was reacted with 2,6-
di-t -butylphenol, the desired para C-para C coupled products were formed exclusively
[(16) and (10)].
The fact that 2-t -butylphenol has two unsubstituted carbon positions available (the 4-
and 6-positions) for oxidative coupling plays a significant role in these reactions when
compared with the case when both the 2- and 6- positions are occupied by t -butyl
groups. When the 6-position is blocked by such a bulky group, such as in 2,6-di-t -
butylphenol, it prevents a large variety of C-C and C-O coupled side products from
being formed, therefore accounting for the observed high selectivity. More especially,
C-O coupling would be disfavoured by the proximity of the two bulky groups in the
case of the 2,6-analogue. However, both C-O and C-C coupling (in more than one

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 160/182
151
position) are highly likely for the mono-substituted phenol, thus resulting in the myriad
of products that was observed.
It was further noted that 2-t -butylphenol was less reactive in terms of conversion than
2,6-di-t -butylphenol in the presence of both silver oxide and copper acetate/oxalic
acid. The decreased reactivity of 2-t -butylphenol, as compared to 2,6-di-t -
butylphenol, may be ascribed to the fact that 2-t -butylphenol, with only one electron-
donating alkyl group (by the inductive effect), is most likely less easily oxidized than
2,6-di-t -butylphenol, which has two such alkyl groups, the latter aromatic ring being,
therefore, more electron dense (and thus more readily oxidized) than that of theformer. It was further noted that no diphenoquinone formation was observed in the
oxidation of 2-t -butylphenol, whereas with 2,6-di-t -butylphenol, the diphenoquinone
derivative was the major product (reactions 10 and 11). This is possibly a further
indication of the difference in oxidation potential of the two substrates, as well as of
their resultant coupled products.
3.6.2 Reactions of 2,4-Di-t -Butylphenol and 2,6-Di-t -Butylphenol with
Ce(IV) in MeSO3H
For the sake of this comparative study and in retrospect, it was thought appropriate to
carry out a reaction (reaction 56, Table 3.29) in which 2,6-di-t -butylphenol (9) was
reacted with Ce(IV) under identical reaction conditions to that of reaction 15, in which
the substrate was 2,4-di-t -butylphenol (44).

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 161/182
152
Table 3.29 Comparative data obtained for Ce(IV)
Reaction
No.
Oxidant Time
(h)
Solvent Temp. Conversion
(%)
Selectivity to
desired coupled
products
(%)
15 Ce4+ 1 H2O/ MeOH Reflux 100.00 90.35b
56 Ce4+
1 H2O/ MeOH Reflux 45.01 100.00c
b Selectivity to (45).c
The diphenoquinone (10) and diol (16) percentages were 82.50 and 17.50 %, respectively.
It was observed that 2,6-di-t -butylphenol was significantly less reactive with Ce(IV)
than 2,4-di-t -butylphenol (conversions of 45.01 and 100 %, respectively). However,
2,6-di-t -butylphenol formed para C-para C coupled products exclusively, whereas 2,4-
di-t -butylphenol did not form the desired ortho C-ortho C product (45) exclusively (with
a selectivity of only 90.35 %). This may be due to the increased potential for C-O
coupling in the 2,4-analogue as compared to the 2,6-analogue due to steric
hindrance. In order to investigate possible theoretical reasons for these observations,
MO calculations were carried out on the relevant dienone and phenolic products of
these two reactions.
The relative energy profiles associated with coupling modes G (where the substrate
2,6-di-t -butylphenol undergoes para C-para C coupling [2,6 p,p]), H (where 2,6-di-t -
butylphenol undergoes para C-oxygen coupling [2,6 p,O]), L (where the substrate 2,4-
di-t -butylphenol undergoes ortho C-ortho C coupling [2,4 o,o]) and M (where 2,4-di-t -
butylphenol undergoes ortho C-oxygen coupling [2,4 o,O]) that lead to phenolic
products can be depicted in the following graph (Figure 3.17). (In this figure, dienone
intermediates are denoted as 1 and their corresponding phenolic forms as 2 on the x-
axis. All values plotted are relative to the heat of formation of the phenolic form for

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 162/182
153
0
5
10
1520
25
30
35
40
45
50
0 1 2
R e l a t i v e E
n e r g y ( k c a l / m o l )
2,4 o,o
2,4 o,O
2,6 p,p
2,6 p,O
the ortho C-ortho C coupled product of 2,4-di-t -butylphenol (denoted as 2,4 o,o in the
figure), which was arbitrarily assigned a value of 0 kcal/mol for the purposes of ease
of comparison.)
Figure 3.17: Relative energies of dienones (1) and coupled phenols (2)
From these relative energy profiles, it can be seen that the intermediate dienones,
when coupling the 2,4-analogue, have lower relative energies as compared to the
corresponding 2,6-analogue, implying that the 2,4-analogue more readily forms these
intermediates (because of their greater stability) than the 2,6-analogue. This is in
agreement with experimental findings (Table 3.29) where the conversion of the 2,4-
analogue was much higher than that of the 2,6-analogue in the presence of Ce(IV) as
the oxidant. The driving force for the subsequent tautomerization of these dienones
is their gain in aromaticity, and thus an increase in their stability. These calculationsalso show that the ortho C-O coupling (2,4-di-t -butylphenol) of a pair of the
appropriate phenoxyl radicals is about 10 kcal/mol more favourable than the
corresponding para C-O (2,6-di-t -butylphenol) derivative, as suggested earlier and as
a consequence of the greater steric crowding around oxygen in the 2,6-analogue.
The greater energy difference (4.05 kcal/mol, Table 3.4) between dienones formed by
para C-para C and para C-O coupling reactions of 2,6-di-t -butylphenol as compared

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 163/182
154
with the smaller energy difference (3.37 kcal/mol, Table 3.7) of dienones formed by
ortho C-ortho C and ortho C-O coupling of 2,4-di-t -butylphenol suggests that the 2,6-
analogue favours C-C coupling over C-O coupling to a greater extent than the 2,4-analogue favours C-C coupling over C-O coupling. This may be an explanation for the
observed selectivity difference between these two substrates (when using Ce(IV) and
Ag2O as oxidants), and also for the observation that the 2,4-analogue afforded C-O
coupled product whereas the 2,6-analogue did not. (Note that these MO calculations
do not provide any information on rates of reaction, but only on thermodynamic
aspects thereof.)
Overall, these theoretical considerations thus add credence to experimental findings
obtained in our laboratories.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 164/182
155
CHAPTER 4
CONCLUSION AND FINAL COMMENTS
During the investigation into the oxidative coupling of various mono- and di-
substituted phenols under a variety of reaction conditions using a range of different
coupling agents, a number of conclusions were drawn from the observed results.
The oxidative coupling reactions of 2-t -butylphenol (35) using various oxidants
produced a large number of products, and so the number of coupling modes that 2-t -
butylphenol prefers is numerous under the conditions that were investigated. There
was no observed selectivity to any single product, and both C-C and C-O coupling
appeared to take place in these reactions, amongst others. Although a large variety
of oxidants were assessed, the selectivity to the coupled product 3,3’-di-t -butyl-4,4’-
dihydroxybiphenyl (39) was found to be low irrespective of the oxidant used. The
highest selectivities were achieved with cerium(IV) sulphate (25.99 %) and silver
carbonate on celite (25.57 %), but these selectivities were obtained at low
conversions of 2-t -butylphenol (26.61 and 10.98 %, respectively). (The reactionconditions under which the cerium work was conducted in this case is novel and has
not been reported elsewhere, as indicated by an extensive literature survey.) All the
other oxidants that were used were found to be totally ineffective in producing the
desired coupled product (39). In many of these latter cases, though, the conversion
of the substrate was reasonable. A general trend that was observed was that higher
conversions were usually associated with lower selectivities. It was therefore
concluded that 2-t -butylphenol (35), due to the number of feasible coupling modes
available to this substrate, showed no promise as a substrate for the selective
coupling to afford (39) as the desired product under the reactions conditions that were
investigated. It therefore appears highly unlikely that 2-t -butylphenol may be used as
a substrate in order to form the desired para C-para C coupled product in an
economically viable process due to the non-selectivity displayed by the substrate,
irrespective of the employed oxidant.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 165/182
156
The oxidative coupling reactions of 2,6-di-t -butylphenol (9) can theoretically produce
numerous products through a number of different coupling modes (G-K). Of these
modes, however, G and H were predicted to be more facile, as shown by molecular orbital calculations. When experimentally investigated, it was found that this
substrate was indeed highly selective when placed under oxidative coupling
conditions. It was observed that high selectivities to the desired para C-para C
coupled products (16) and (10) using Ag2O, Cu(OAc)2/oxalic acid and Ce(IV) sulphate
(100 % selectivity in all instances) were achieved. Both Ag2O and Cu(OAc)2/oxalic
acid also gave high conversions of (9) [both 100 %], but Ce(IV) sulphate only
achieved a 45.01 % conversion of (9) after a reaction time of 1 h. These resultsobtained are in agreement with those reported in the literature,46,78 and the molecular
orbital calculations further confirmed these observations. Thus the presence of an
additional t -butyl group in 2,6-di-t -butylphenol (9), as compared with that of 2-t -
butylphenol (35), has a significant effect on the course of the reaction and on the
preferred mode of coupling of (9). The number of feasible coupling modes for (9) is
thus reduced by the additional substituent, and steric congestion also comes into play
when considering the absence of any C-O coupling for (9) [which was present when
the substrate was (35)]. (Note that results obtained from reactions of Ce(IV) with (9)
have not been reported previously.)
When 2,4-di-t -butylphenol (44) was oxidatively coupled using agents potassium ferric
cyanide and cerium(IV) sulphate, a high selectivity to the desired ortho C-ortho C
coupled product (coupling mode L) was observed. The C-O coupling mode also
appears to occur in these reactions (mode M). This substrate was, however, not as
selective as 2,6-di-t -butylphenol (showing 100 % selectivity to para C-para C coupledproducts). The use of other oxidants such as FeCl3 and Ag2O afforded results that
were less than satisfactory in terms of selectivity to the preferred coupled product
(45), despite high conversions of the substrate. Thus both FeCl3 and Ag2O were
found to be unsuitable for the purposes of forming (45), quite possibly due to the
mechanisms by which they react in combination with the positioning of the
substituents on the aromatic rings. Molecular orbital calculations confirmed the

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 166/182
157
preference for coupling mode L and, to a lesser extent, mode M. The difference in
results obtained for the 2,4- and 2,6-analogues may only be explained in terms of
steric crowding, in which the hydroxyl moiety of the 2,6-analogue is well “surrounded”by the two bulky t -butyl groups, thus disallowing the formation of the undesired C-O
coupled products. The 2,4-analogue, on the other hand, is less crowded in the
vicinity of the OH group, and can thus also form some of the C-O coupled product,
resulting in the observed lower selectivities to the C-C coupled product as compared
with the 2,6-analogue. The amount of steric congestion around the OH group in both
2-t -butylphenol and 2,4-di-t- butylphenol is probably somewhat similar, thus explaining
the propensity for both substrates to undergo C-O coupling in these conditions.
Once again, the work conducted with 2,4-di-t -butylphenol using Ce(IV) as the oxidant
is entirely novel, and the results obtained in these reactions were found to be very
promising indeed, with high selectivites and conversions to the desired coupled dimer
(45) being achieved. Investigation of the various reaction conditions then produced
the optimal reaction conditions which included the use of 1 M aqueous
methanesulphonic acid as the medium of choice with added co-solvent (methanol)
such that the resultant solution is a single phased reaction mixture. Furthermore, the
optimal reaction temperature was found to be approximately 65°C (at reflux), and
lengthy reaction times were not necessary, probably because the desired coupling
reaction takes place rather rapidly. The substrate loading was an important factor:
too high loadings afforded low conversions, and too low loadings afforded low
selectivities. The optimal substrate:oxidant ratio was 1:2, with a slow addition of the
oxidant to the reaction medium being favoured over that of rapid addition. This
process can be further investigated in terms of industrial viability since Ce(IV) can beregenerated electrochemically from Ce(III) successfully and since 2,4-di-t -butylphenol
as a substrate gave high selectivities to the desired coupled product.
The oxidative coupling reactions of 2,4-dimethylphenol (59) were not as selective as
that of 2,4-di-t -butylphenol or 2,6-di-t -butylphenol under identical reaction conditions.
From the results obtained, it is clear that 2,4-dimethylphenol coupled primarily by

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 167/182
158
modes R and S. Only moderate selectivities to the desired ortho C-ortho C coupled
product (61) were achieved with oxidants FeCl3, K3Fe(CN)6 and Ce(IV) [49.11, 26.72
and 57.58 %, respectively]. Once again, the difference in the results obtained for 2,4-di-t -butylphenol and 2,4-dimethylphenol can be explained in terms of steric crowding:
the hydroxyl moiety of 2,4-di-t -butylphenol is more sterically hindered by the bulky t -
butyl group, thus decreasing the amount of C-O coupling, which is more prevalent in
the oxidation reactions of 2,4-dimethylphenol (which only has the smaller methyl
substituent in the OH region). (In the case of K3Fe(CN)6, the major product seems to
be that formed by C-O coupling.) Furthermore, the lower selectivities observed for
the dimethyl derivative is also very likely a result of the presence of the benzylic C-Hgroups, which are normally rather activated, especially under radical conditions.
Results from the work conducted using Ce(IV) as the oxidant are again novel, though
the efficiency of the coupling in this reaction was found to be only moderately
successful in terms of selectivity to the coupled product (61).
The aims of this investigation have thus been realized, and the study of the various
coupling reactions, the reaction conditions and the various oxidants and substrates
has led to much new knowledge that may now be added to this field of chemistry.
Many promising results were observed, and feasible reasons given for those
reactions that were not successful. One of the major goals of these investigations
was to find an oxidant that was both environmentally and economically viable, that
afforded high conversions of the substrates and selectivities to the desired coupled
products. From this work, Ce(IV), in the presence of methanesulphonic acid,
appears to be just such a coupling agent, and many promising, novel results were
obtained in oxidative coupling reactions carried out in its presence. The feasibility of its electrochemical regeneration from Ce(III), the fact that substrates such as 2,4-di-t -
butylphenol, when reacted with Ce(IV), gave high conversions and selectivities to the
desired product, and the mild reaction conditions used, makes it an oxidant that may
be feasible for scale-up operations, though scale-up itself will require a separate
intensive study.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 168/182
159
REFERENCES
1. G.C. Gerrans, South Africa – History of the chemical industry ,
http://www.mbendi.co.za/caia.chsahsa01.htm
2. H.A. Nitcoff and B.G. Reuben, “Industrial Organic Chemicals in Perspective,
Part 1: Raw Materials and Manufacture, John Wiley & Sons, Indianapolis,
1996.
3. F.F. Runge, Ann. Phys. Chem., 1834, 31 , 65.
4. C. Gerhardt, Ann. Chim., 1843, vii, 215.
5. J. McMurry, “Organic Chemistry” , 4th Ed., Brooks/Cole Publishing Company,
USA, 1995.
6. H.G. Frank and J.W. Stadelhofer, “Industrial Aromatic Chemistry” , Springer,
New York, 1987, pp. 146-183.
7. J.F. Trembley, Chem. Eng. News , 2000, December 11, 31.
8. J.D. Spikes, H.R. Shen, P. Kopeckova and J. Kopececk, Photochem.
Photobiol., 1999, 70, 130.
9. K.U. Ingold, Spec. Publ. Chem. Soc., 1970, 24, 285.
10. “Natural Antioxidants, Chemistry, Health affects and Applications” , F. Shahidi,
ed., AOCS Press, Champaign, Illonois, 1997.
11. “Antioxidants and Redox Signaling” , C.K. Sen and D.K. Das, eds., M. A.
Liebert, Inc., New York, 1999, Vol. 1.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 169/182
160
12. E.R. Ahwicker, Chem. Rev., 1967, 67, 475.
13. L.R. Mahoney, Angew. Chem., Int. Ed. Engl., 1969, 8, 547.
14. S. Kumar, J.A. Morales-Tirado, D.P. Casalmir, S.M. Silence, Y.S. Molisani,
J.M. Proper, P.L. Jacobs and G. Baer, “Toner with increased surface additive
adhesion” , Xerox Corporation, United States Patent 6,582,866, 2003.
15. E.J. Gutman and B.M. Ferguson, “Toner and developer for magnetic brush
development system” , Xerox Corporation, United States Patent 6,319,647,2001.
16. H. Ito, A. Suwa and J. Kohiruimaki, “Process for producing raw polycarbonate
resin material” , Idemitsu Petrochemical Co., Ltd., United States Patent
6,476,179, 2002.
17. M. Abele, H. Buysch, H. Schrage and H. Vernaleken, “Use of certain phenolic
resins for reinforcing rubber vulcanizates” , Bayer Aktiengesellschaft, United
States Patent 5,214,100, 1993.
18. P. Pier, “Coatings agent and the use thereof” , Bayer Aktiengesellschaft, United
States Patent 6,455,162, 2002.
19. F. Reil, V. Pieper and K. Tonnes, “Production of conductor tracks on plastics
by means of laser energy” , Ticona GmbH, United States Patent 6,417,486,2002.
20. A. Schoenfeld, A. Stohr, “Infrared-reflecting colorants” , Clariant GmbH
(Frankfurt, DE), United States Patent 6,180,025, 2001.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 170/182
161
21. M. Usuki and T. Shimizu, “Polymer scale deposition preventive agent and a
process for producing polymers using the same” , Shin-Etsu Chemical Co., Ltd.
(Tokyo, JP), United States Patent 5,728,781, 1998.
22. A.I. Scott, “Oxidative Coupling of Phenols” , W.I. Taylor and A.R. Battersby,
eds., Dekker, New York, 1967, Chapter 2, p. 95.
23. A.R. Battersby, “Oxidative Coupling of Phenols” , W.I. Taylor and A.R.
Battersby, eds., Dekker, New York, 1967, Chapter 3, p. 119.
24. W.A. Waters, “Mechanisms of Oxidation of Organic Compounds” , Metheun,
London, 1964, pp. 132-149.
25. J.D. Loudon, Progr. Org. Chem ., 1961, 5 , 46.
26. H. Nishimo, N. Itoh, M. Nagashima and K. Kurosawa, Bull. Chem. Soc. Jpn.,
1992, 65, 620.
27. H. Nishimo, H. Satoh, M. Yamashita and K. Kurosawa, J. Chem. Soc., Perkin
Trans. 2 , 1999, 1919.
28. V. Balogh, M. Fetizan and M. Golfier,J. Org. Chem ., 1971, 36, 1339.
29. D.H.R. Barton and T. Cohen, Festschr. Arthur Stoll , 1957,129.
30. H. Erdtman and C.A. Wachtmeister, Festschr. Arthur Stoll , 1957, 144.
31. J.M. Harkin, “Oxidative Coupling of Phenols” , W.I. Taylor and A.R. Battersby,
eds., Dekker, New York, 1967, Chapter 7, p. 323.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 171/182
162
32. K. Weinges and R. Sparig, “Oxidative Coupling of Phenols” , W.I. Taylor and
A.R. Battersby, eds., Dekker, New York, 1967, Chapter 7, p. 323.
33. S.G. Humphries, “Biogenosis of Natural Products” , P. Nufield, ed., Macmillan,
New York, 1963, p. 617.
34. “Photochemistry of Lignocellulosic Materials” , C. Heitner and J.C. Scaiano,
eds., ACS Symposium Series, American Chemical Society, Washington DC,
1993, Vol. 531.
35. G.D. Cooper and A. Katchman, Advan. Chem. Ser., 1969, 91, 660.
36. A.S. Hay, P. Shenian, P.F. Erhardt, W.R. Haaf, and J.E. Theberg, Encycl.
Polym. Sci. Technol ., 1969, 10, 92.
37. E. McNelis, J. Org. Chem., 1966, 31 ,1255.
38. P.D. McDonald and E.A. Hamilton, “Oxidation in Organic Chemistry” , W.S.
Trahanovsky, ed., Academic Press, New York, 1973, Part B, p. 97.
39. D.A. Whiting, “Comprehensive Organic Synthesis” , T.M. Trost and I. Fleming,
eds., Pergamon Press, Oxford, 1993, Vol. 3, p. 659.
40. T.S. Stone and W.A. Waters, J. Chem. Soc., 1964, 213, 4302.
41. E. Muller, H. Eggensperger, A. Rieker, A. Scheffer, H.D. Spanagel, H.B.Stegmann and B. Teisser, Tetrahedron , 1965, 21, 227.
42. E. Muller, A. Rieker, K. Scheffer and A. Moosmayer, Angew. Chem., Int. Ed.
Engl ., 1966, 5, 6.
43. L.R. Mahoney and S.A. Weiner, J. Am. Chem. Soc., 1972, 94, 1412.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 172/182
163
44. G.H. Williams,Spec. Publ. Chem. Soc.,1970, 24, 25.
45. M.L. Larson and F.W. Moore, Inorg. Chem., 1966, 5, 801.
46. W. Kaeding, J. Org. Chem., 1963, 28, 1063.
47. R.G.R. Bacon and D.J. Munro, J. Chem. Soc., 1960, 1339.
48. M.J.S. Dewar and T. Nakay, J.Amer. Chem. Soc., 1968, 90, 7134.
49. T. Kametani, A. Kozuka and K. Fukumato, J. Chem. Soc. (C), 1971, 1021.
50. F.R. Hewgill and G.B. Howie, Aust. J. Chem., 1978, 31, 1061.
51. L. Taimr and J. Pospisil, Tetrahedron Lett., 1971, 30, 2809.
52. G.W.K. Cavill, E.R. Cole, P.T. Gillman and D.J. McHugh, J. Chem. Soc., 1954,
2785.
53. W.T. Reichle, “Oxidative Process for Substituted Phenols” , Union Carbide
Corp., Eur. Pat. Appl., 11 296, 1980.
54. M. Inaba, N. Mitsune and M. Mizutari, “Preparation of 4,4’-Biphenols” ,
Mitsubishi Petrochemicals Co., Ltd., Japanese Patent 04 338 347, 1992.
55. T. Kitamura and M. Yamada, “Preparation of Bisphenols” , Dainippon Ink and
Chemical, Inc., Japanese Patent 04 154 738, 1990.
56. L. Papouchado, R.W. Sandpord, G. Petrie and R.N. Adams, J. Electroanal.
Chem., 1975, 65, 275.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 173/182
164
57. M. Gattrell and D.W. Kirk, J. Electrochem. Soc., 1993, 140, 903.
58. S. Torii, A.L. Dhimane, Y. Araki and T. Inokuchi, Tetrahedron Lett ., 1989, 30,2105.
59. S. Thvagarajan, Chem. Reviews , 1958, 58, 439.
60. R. Pummerer, H. Puttfarcken and P. Schopflocher, Chem. Ber., 1925, 58,
1808.
61. W.L. Carrick, G.L. Karapinka and G.T. Kwaitkowski, J. Org. Chem ., 1969, 34,
2388.
62. J. Charalambous, M.J. Frazer and F.B. Taylor, J. Chem. Soc., 1969, 2787.
63. D. Ray and A. Chakravorty, Inorg. Chem ., 1988, 27, 3292.
64. D. Baluch, J. Charalambous, L. Lan and B. Haines, J. Chem. Soc., Chem.
Commun ., 1988, 1178.
65. R.A. Sheldon and J.K. Kochi, “Metal-Catalyzed Oxidations of Organic
Compounds” , Academic Press, New York, 1981.
66. J.P. Collmann, L.S. Hegedus, J.R. Norton and R.G. Finke, “Principles and
Applications of Organotransition Metal Chemistry” , University Science Books,Mill Valley, California, 1987.
67. E.G. Samsel, K. Srinivasan and J.K. Kochi, J. Am. Chem. Soc., 1985, 107,
7606.
68. F. Walsh and G. Mills, Chemistry and Industry , 1993, 576.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 174/182
165
69. S. Domagala, V. Steglinka and J. Dziegiec, Monatshefte Fur Chemie , 1998,
129, 761.
70. M. Ignaczak and J. Dziegiec, Polish J. Appl. Chem., 1992, 36, 183.
71. Y. Kamitori, M. Hojo, R. Masuda, T. Isumi and S. Tsukamoto, J. Org. Chem.,
1984, 49, 4161.
72. K. Omura, J. Org. Chem., 1998, 63, 10031.
73. H. Firouzabadi and Z. Mostafavipor, Bull. Chem. Soc. Jpn., 1983, 56, 914.
74. C. S. Marvel and P.K. Porter, “Organic Synthesis” , Wiley, New York, 1967,
Coll. Vol. 1, p. 411.
75. W.R. Benson, E.T. McBee and L. Rand, “Organic Synthesis” , Wiley, New York,
1967, Coll. Vol. 5, p. 663.
76. T.F. Rutledge, “Oxidative Coupling of Alkylphenols or 1-Naphthols Catalyzed
by Metal Complexes of Dicarboxylic Acid Compounds ”, ICI Americas Inc., US
Patent 4 098 830.
77. F.Toda, T. Tanaka and S. Iwata, J. Org. Chem., 1989, 54, 3007.
78. H.S. Blanchard, J. Org. Chem ., 1960, 25, 264.
79. F.R. Hewgill and D.G. Hewitt, J. Chem. Soc (C), 1967, 726.
80. A.I Vogel, “Quantitative Inorganic Analysis” , 3rd Ed., Longman Group Limited,
London, 1961, p 325.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 175/182
166
81. H. Musso, Angew. Chem., Int. Ed. Engl., 1963, 2, 723.
82. P.S. Radhakrishnamurti and R.K. Panda, Indian J. Chem., 1970, 8, 946.
83. W.H. Richardson, “Oxidation in Organic Chemistry” , K.B. Wiberg, ed.,
Academic Press, New York, 1965, Chapter 4, Part A, p. 243.
84. A. Nishinaga, H. Tomita, J. Molec. Catalysis , 1980, 7, 179.
85. A. Ramachandraiah, Indian J. Chem., 1989, 28A, 309.
86. V.M. Kathori and J.J. Tazuma, J. Molec. Catalysis , 1976, 14, 180.
87. G.F. Endres, A.S. Hay and J.W. Eustance, J. Org. Chem ., 1963, 28, 1300.
88. M.S. Kharasch and B.S. Joshi, J. Org. Chem., 1957, 22, 1439.
89. D.R. Armstrong, C. Cameron, D.C. Nonhebel and P.G. Perkins, J. Chem. Soc.,
Perkin Trans. 2 , 1983, 588.
90. H. Musso, Angew. Chem., Int. Ed. Engl.,1963, 75, 965.
91. B. Franck and G. Blaschke, Ann. Chem., 1966, 695, 144.
92. A.I. Scott, Quart. Rev. (London), 1965, 19, 1.
93. O. Neunhoeffer and P. Heitmann. Chem. Ber., 1963, 96, 1027.
94. C.G. Haynes, A.H. Turner and W.A. Waters, J. Chem. Soc., 1956, 2823.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 176/182
167
95. D. Pletcher and F.C. Walsh, “Industrial Electrochemistry” , 2nd Ed., Chapman
and Hall, London, 1990, p. 236.
96. R.E. Connick, J. Chem. Soc., 1949, 235.
97. E. Baciochi, C. Rol and L. Mandolini, J. Am. Chem. Soc., 1980, 102, 7597.
98. G.F. Smith and C.A. Getz, Indian Eng. Chem. Anal. Ed., 1938, 10, 191.
99. C.G. King and R.C. Spooner, J. Am. Chem. Soc., 1943, 65, 170.
100. F.B. Baker, T.W. Newton and M. Kahn, J. Phys. Chem., 1960, 64, 109.
101. M. Marrocco and G. Brilmyer, J. Org. Chem., 1983, 48, 1487.
102. L. Leszczyn’ski and J. Dziegiec’, Polish J. Chem., 1994, 68, 1855.
103. W.R. Grace, Cerox Technology , http://www.grace.com.
104. E. Wadsworth, F.R. Duke, and C.A. Goetz, Anal. Chem., 1957, 29, 1824.
105. Nilsson and A. Ronlan, J. Chem. Soc., Perkin Trans 1 , 1973, 2337.
106. A.S. Hay, J. Polymer Sci., 1962, 58, 581.
107. E. Ochiai, Tetrahedron , 1964, 20 , 1831.
108. A.S. Hay, H.S. Blanchard, G.F. Endres and J.W. Eustance, J. Amer. Chem.
Soc., 1959, 81 , 6335.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 177/182
168
109. D.R. Armstrong, C. Cameron, D.C. Nonhebel and P.G. Perkins, J. Chem. Soc.
Perkin Trans. 2 , 1983, 581.
110. P.D. Bartlett and S.F. Nelson, J. Am. Chem. Soc., 1966, 88 , 139.

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 178/182
169
APPENDIX
Appendix 3.1: MS Fragmentation pattern for product with retention time 12.76
min
Appendix 3.2: MS Fragmentation pattern for product with retention time 13.29
min
7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 179/182
170
Appendix 3.3: MS Fragmentation pattern for product with retention time 14.18
min
Appendix 3.4: MS Fragmentation pattern for product with retention time 15.27
min

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 180/182
171
Appendix 3.5: GC trace obtained for BaMnO4 reaction with (35)
Appendix 3.6: MS Fragmentation pattern for product with retention time 13.77
min

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 181/182
172
Appendix 3.7: MS Fragmentation pattern for product with retention time 14.11
min
Appendix 3.8: MS Fragmentation pattern for product with retention time 30.74
min

7/31/2019 Luan Van Cac Chat Chong Oxy Hoa
http://slidepdf.com/reader/full/luan-van-cac-chat-chong-oxy-hoa 182/182
Appendix 3.9: MS Fragmentation pattern for product with retention time 18.07
min