liver cirrhosis – epidemiological and clinical aspects · pdf file2 abstract liver...
TRANSCRIPT

Section of Gastroenterology and Hepatology Department of Internal Medicine Sahlgrenska University Hospital
Göteborg University Göteborg, Sweden
Liver cirrhosis – Epidemiological and
Clinical Aspects
Steingerður Anna Gunnarsdóttir
Göteborg 2008

2
ABSTRACT
Liver cirrhosis – Epidemiological and Clinical Aspects Steingerður Anna Gunnarsdóttir Department of Internal Medicine
Sahlgrenska University Hospital, Göteborg University, Sweden
Liver cirrhosis is the end-stage of many different chronic liver diseases. Limited data exists on the epidemiology, natural history and complications of liver cirrhosis such as esophageal varices and malignancies in the Nordic countries after the discovery of hepatitis C (HCV). Most hepatocellular carcinomas (HCC) develop in patients with liver cirrhosis but data on the occurrence of other malignancies than HCCs in these patients are scarce. Gastrointestinal (GI) symptoms such as nausea, vomiting, abdominal pain, and diarrhea are common in patients with advanced liver disease but the importance of portal hypertension for these symptoms is unexplored.
The aims of the present study were to evaluate the incidence, outcome and complications of liver cirrhosis in a Swedish population and in Iceland and the effects of portal hypertension on small bowel motility and small intestinal bacterial overgrowth (SIBO) in patients with liver cirrhosis.
The annual incidence of liver cirrhosis in Gothenburg was 15.3 ±2.4/100.000 compared to 3.3 ±1.2/100.000 in Iceland, p<0.0001. In Gothenburg 50% of the patients had alcoholic cirrhosis compared to 29% in Iceland (p<0.0001).
Only 9% of patients died in their first variceal bleeding, that is within one week of their first bleeding episode. Of the patients diagnosed with esophageal varices after a bleeding episode, 55% had a bleeding episode during follow-up compared to only 13% of the patients diagnosed without a bleeding episode. Variables predicting mortality in a multivariate analysis were: Child-Pugh class, bleeding before diagnosis, age and bilirubin levels. Causes of death were in 26% of cases liver failure, 19% variceal bleeding and the rest other causes.
Patients with liver cirrhosis had 267 times increased risk of hepatocellular cancer. Among patients with HCV cirrhosis, 19% developed HCC and 20% of those with HCV and alcoholic liver disease (ALD). We observed 13 times increased risk of cholangiocarcinoma and also increased risk for esophageal, pancreatic, pulmonary and colorectal cancer than in the general population.
Patients with liver cirrhosis and portal hypertension had more motility disturbances in the small intestine compared to those without portal hypertension and seemed to have a higher risk of small intestinal bacterial overgrowth.
Conclusions: The incidence of liver cirrhosis is low in Iceland, 24% of the incidence in Gothenburg. The difference is due to lower incidence of alcoholic liver disease and HCV cirrhosis in Iceland. In patients with liver cirrhosis and esophageal varices a bleeding is still a strong risk factor for recurrent bleeding. The mortality is high but mainly from other causes than variceal bleeding and few die in the first bleeding. The risk of HCC in cirrhosis is mostly associated with HCV and is the same in HCV patients with and without alcohol overconsumption. Other malignancies than HCC are more common in patients with cirrhosis than in the general population. Abnormal small bowel motility and SIBO is common in patients with liver cirrhosis with concomitant portal hypertension. Portal hypertension per se might be significantly related to small bowel abnormalities observed in patients with liver cirrhosis. Keywords: Liver cirrhosis, etiology, alcoholic liver disease, mortality, portal hypertension, esophageal varices, variceal bleeding, hepatitis C, hepatocellular cancer, malignancies, small intestinal motility, small intestinal bacterial overgrowth. ISBN: 978-91-628-7460-5

3
In memory of my excellent tutor, Professor Rolf Olsson (1936-2008)
The Basics of Life Eldur er bestur A man needs warmth, með ýta sonum the warmth of fire og sólar sýn. and of the shining sun. Heilindi sitt A healthy man ef maður hafa náir is a happy man án við löst að lifa. who´s neither ill nor injured. (Hávamál) (Translated by Björn Jónasson)

4
Liver cirrhosis – Epidemiological and Clinical Aspects Copyright© Steingerður Anna Gunnarsdóttir [email protected] ISBN: 978-91-628-7460-5 Published by: Department of Internal Medicine The Sahlgrenska Academy at Göteborg University, Sweden Printed in Sweden Chalmers Tekniska Högskola AB, Chalmers reproservice, Göteborg 2008

5
LIST OF PAPERS
This thesis is based on the following papers, which will be referred to in the text by their Roman numerals: I. Liver cirrhosis in Iceland and Sweden: incidence, etiology and outcomes. Gunnarsdottir SA, Olsson R, Ólafsson S, Cariglia N, Westin J, Thódleifsson B, Björnsson ES. In manuscript II. Characteristics, prognosis and outcome of patients with oesophageal varices in a university hospital in Sweden 1994-1999. Gunnarsdottir SA, Olsson R, Björnsson ES. Scand J Gastroenterol 2005;40:1462-1468 III. Development of different malignancies in patients with liver cirrhosis. Gunnarsdottir SA, Kalaitzakis E, Björnsson ES. In manuscript IV. Small intestinal motility disturbances and bacterial overgrowth in patients with liver cirrhosis and portal hypertension. Gunnarsdottir SA, Sadik R, Shev S, Simrén M, Sjövall H, Stotzer PO, Abrahamsson H, Olsson R, Björnsson ES. Am J Gastroenterol 2003;98:1362-1370

6
CONTENTS
Page
ABBREVIATIONS 8 1. INTRODUCTION 1.1. Background 9 1.2. Epidemiology 10 1.3. Alcoholic liver disease 11 1.4. Natural history and complications 12 1.5. Portal hypertension 14 1.6. Variceal bleeding 16 1.7. Small intestinal disturbances 18 1.8. Hepatocellular cancer and other malignancies 20 2. AIMS OF THE PRESENT STUDIES 22 3. METHODS 3.1. Paper I 23 3.1.1. Subjects 23 3.1.2. Data collection 23 3.1.3. Information obtained from medical records 24 3.2. Paper II 24 3.2.1. Subjects 24 3.2.2. Data collection 24 3.2.3. Information obtained from medical records 25 3.3. Paper III 25 3.3.1. Subjects 25 3.3.2. Data collection 25 3.3.3. Follow-up 26 3.3.4. End-points 26 3.4 Paper IV 26 3.4.1. Subjects 26 3.4.2. Manometry 27 3.4.3. Analysis of manometric data 28 3.4.4. Diagnosis of bacterial overgrowth 29 3.4.5. Variceal pressure measurements 30 3.5. Statistical methods 31 3.5.1. In papers I and II 31

7
3.5.2. In paper III 32 3.5.3. In paper IV 32 4. RESULTS 4.1. Liver cirrhosis in Gothenburg and Iceland, epidemiology and natural history 33 4.1.1. Patients characteristics 33 4.1.2. Incidence 33 4.1.3. Survival analysis 34 4.1.4. Variables associated with overall mortality 36 4.2. Esophageal varices, prognosis and outcome 37 4.2.1. Patients characteristics 37 4.2.2. Outcome 37 4.2.3. Variables associated with overall mortality 39 4.2.4. Use of prophylactic therapy 40 4.3. Hepatocellular carcinoma and other malignancies 40 4.3.1. Patients characteristics 40 4.3.2. Mortality 41 4.3.3. Overall risk of cancer 42 4.4. Small intestinal motility disturbances and bacterial overgrowth 44 4.4.1. Conventional manometric evaluation 44 4.4.2. Evaluation with high temporospatial resolution 47 4.4.3. Small intestinal bacterial overgrowth 47 4.4.4. Variceal pressure 48 5. DISCUSSION 5.1. Epidemiology and of liver cirrhosis in Sweden and Iceland 49 5.2. Characteristics, prognosis and outcome of patients with esophageal varices 52 5.3. Liver cirrhosis and cancer risk 55 5.4. Small intestinal motility disturbances and bacterial overgrowth 58 6. CONCLUSIONS 61 7. ACKNOWLEDGEMENTS 62 8. REFERENCES 63 PAPERS I-IV

8
ABBREVIATIONS
AIH Autoimmune hepatitis ALD Alcoholic liver disease B Bleeding CC Cholangiocarcinoma CI Confidence interval CP Child-Pugh CRC Colorectal cancer DD Distal duodenum FHVP Free hepatic venous pressure GI Gastrointestinal GO Gothenburg HBV Hepatitis B virus infection HC Healthy controls HCC Hepatocellular carcinoma HCV Hepatitis C virus infection HVPG Hepatic venous pressure gradient IC Iceland J Jejunum KM Kaplan Meier LC Liver cirrhosis MELD Model of end-stage liver disease MI Motility index MMC Migrating motor complex NALD Non alcoholic liver disease NASH Non-alcoholic steato-hepatitis NS Non-significant OV Oesophageal varices OLT Orthotopic liver transplantation P Prebleeding PBC Primary biliary cirrhosis PD Proximal duodenum PH Portal hypertension PHG Portal hypertensive gastropathy PPG Portal pressure gradient PSC Primary sclerosing cholangitis SBP Spontaneous bacterial peritonitis SD Standard deviation SEM Standard error of the mean SIBO Small intestinal bacterial overgrowth SIR Standard incidence ratio SW Sweden T Portal vein thrombosis WHVP Wedged hepatic venous pressure

9
1. INTRODUCION 1.1 BACKGROUND The word cirrhosis comes from the Greek word kirrhos, which means orange yellow (1). Laennec gave cirrhosis its name kirrhos in 1819 in a brief footnote to his treatise De l’auscultation mediate (2). The definition of cirrhosis remains morphological, described by a working party for the World Health Organization (WHO) in 1978 as: “a diffuse process characterized by fibrosis and the conversion of normal liver architectures into structurally abnormal nodules” (3).
Cirrhosis is a chronic disease of the liver in which diffuse destruction and regeneration of hepatic parenchymal cells has occurred, in which diffuse increase in connective tissue has resulted in disorganization of the lobular architecture. The triad of parenchymal necrosis, regeneration and scarring is always present regardless of individual clinical manifestations (4). In the evolution of many chronic liver diseases cirrhosis is a stage that is considered to be irreversible. Cirrhosis can be stabilized by controlling the primary disease but its presence implies consequences such as portal hypertension, intrahepatic shunting of blood, impaired parenchymal function affecting protein synthesis, hormone metabolism and excretion of bile and bile salts. The most common complications are: gastrointestinal hemorrhage, ascites, encephalopathy, bacterial infections, renal failure, hepatocellular carcinoma and hepatic failure (5). Certain reversible components of cirrhosis have been indicated where significant histological improvement have occurred with regression of cirrhosis but complete resolution with a return to normal architecture seems unlikely (6). The underlying immunological response has usually been acting for months or years where inflammation and tissue repairing are in progress simultaneously which leads in the end to fibrosis and cirrhosis (7).
The main causes of cirrhosis are: alcoholic liver disease (ALD), hepatitis B (HBV), hepatitis C (HCV), non-alcoholic steatohepatitis (NASH),

10
haemochromatosis, auto-immune hepatitis (AIH), primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC) (8). The natural history of cirrhosis can be divided into a preclinical and a subsequent clinical phase. The preclinical phase is usually prolonged over several years; once clinical events occur, such as, ascites, encephalopathy, variceal bleeding or the development of hepatocellular carcinoma the remaining course of the disease is much shorter and usually fatal (9). For liver cirrhosis there still is no curable treatment available except for liver transplantation. 1.2 EPIDEMIOLOGY In developing countries viral hepatitis is the leading cause of cirrhosis and in the developed countries ALD, HCV and NASH are the most significant causes of cirrhosis (7). In many developed countries the death rates from liver cirrhosis have been declining in the recent years with some exceptions (10). World wide death rates from alcohol related liver cirrhosis has been decreasing but an increase has been observed in a few Eastern European countries and England (11). In the United States (USA) there has been an increase in the proportion of patients with HCV compared to ALD in the recent years (12). Studies on patients characteristics at diagnosis show that the mean age is around 60 years and majority of the patients are males with the male/female ratio ranging from 1.3-4 (13-17). The highest mortality from liver cirrhosis is in the age group 60-70 years (14). There has been a reduction in hospitalization and mortality from liver diseases in Sweden from 1969-2001, which has been shown to be associated with reduced sales of spirits (18). Figures on the incidence of liver cirrhosis have been largely lacking in many countries such as Sweden, as well as Finland among the Nordic countries. The mortality and incidence of liver cirrhosis per 100.000 inhabitants in the Nordic countries are shown in Table 1.

11
Country Incidence Mortality Years Method/source Sweden (19) 6.7 1995 Official mortality from LC Finland (19) 10.6 1995 Official mortality from LC Denmark (19) 16.7 1995 Official mortality from LC Denmark Men (20) Women (20)
26.5 11.8
2001-2005 National wide hospital registry
Norway (21) 13.4 1999-2004 Retrospective confirmation of diagnosis from hospital registry
Iceland (22) 4.8 2.8
1950-1990 1970-1990
Retrospective confirmation of diagnosis. Death certificates data.
Table 1. Previously published studies on the incidence and mortality of liver cirrhosis per 100.000 inhabitants in the Nordic countries. There are some difficulties in comparing the numbers in the different countries because in many of these studies the mortality from liver cirrhosis is presented from national wide databases without evaluation of the grounds of the diagnosis and risk of underreporting, which has been estimated in Norway to be 30% to the Norwegian Death Registry (21). There have been changes in alcohol consumption in many of these countries in the last decades and it is therefore important to estimate the influence of these changes on the epidemiology of liver cirrhosis. 1.3 ALCOHOLIC LIVER DISEASE ALD is one of the leading causes of liver cirrhosis in the Western World. The risk of acquiring alcohol cirrhosis has been suggested to increase when alcohol ingestion increases (23) and in a meta-analysis it has been demonstrated that even a low consumption increases the risk, for 25g/day the estimated relative risk was 1.5-3.6 (24). Epidemiological studies from around the world have documented a direct correlation between per capita ethanol consumption and liver related mortality rates (25-27).
Mortality from alcoholic and even non-alcoholic liver cirrhosis has been shown to follow trends in total alcohol consumption per inhabitant showing that alcohol related deaths were underreported (28).

12
Whether there is a threshold of consumption where alcoholic liver disease develops has been studied and it has been estimated that the risk of liver injuries increases when the consumption exceeds 40 g/day (29), 20-40 g/day (30), 30 g/day (26), 12-24 g/day for women and 24-36 g/day for men (31), 40-60 g/day for women (32) and 40-80 g/day (33). According to these studies the threshold is near 40 g/day, which is 1.1 L of bear, 0.44 L of wine or 0.11 L of spirits (33). Another interesting issue is for how long time the high alcohol intake has to exist to develop alcoholic liver disease and it has been estimated to take 16-20 years for men and 10-17 for women (34).
Even though investigators have tried to find a threshold for alcohol consumption and liver injury, only a small proportion of people who have a high alcohol intake will develop liver cirrhosis. In a study from Italy where the average consumption was 60 g/day or more, only 4.2 % had alcoholic liver disease (26). In another cohort of individuals drinking 5 drinks a day the risk of liver cirrhosis was 5.9 % (35). The total mortality from liver cirrhosis is often used to estimate changes in the prevalence of alcoholic liver diseases over time and comparing different nations. In the last decades there has been a decrease in the mortality from liver cirrhosis in most European countries and in the United States (27, 36). The average death rates from chronic liver diseases have been declining in the European Union but an increase has been observed in England (10, 37, 38).
1.4 NATURAL HISTORY AND COMPLICATIONS OF LIVER
CIRRHOSIS Clinical features of cirrhosis derive from morphological alterations and usually reflect severity of hepatic damage rather then the etiology of the underlying liver disease. The loss of hepatocellular mass may lead to jaundice, edema, coagulopathy, and a variety of metabolic abnormalities. Fibrosis and distorted vasculature lead to portal hypertension and its sequelae, including gastroesophageal varices and splenomegaly. Ascites and hepatic encephalopathy result from both portal hypertension and hepatocellular insufficiency. Other major complications of liver cirrhosis and portal hypertension are spontaneous bacterial peritonitis, hepatorenal

13
syndrome and heptocellular carcinoma. The sequelae are also thrombocytopenia as a result of hypersplenism and in the alcoholic patients a direct bone marrow suppression by ethanol. Diminished protein synthesis can lead to reduced production of factors I, II, V, VII, IX and X. This may also be worsened by concomitant malabsorption of the fat-soluble vitamin K due to cholestasis (39).
It has been observed that liver cirrhosis also affects other organs and organ-systems such as the gastrointestinal tract (40) and nutrition (41, 42) as well as the kidneys (43, 44) cardiovascular (45), respiratory (46-48) and the skeletal system (49-51).
The prognosis of liver cirrhosis ranges according to the severity of the liver disease and is significantly reduced after the development of decompensation (52-54).
A new scheme for the analysis of the natural history of cirrhosis and the identification of prognostic factors was proposed and agreed upon at the International Consensus Workshop of Baveno IV (55, 56). The scheme identifies four clinical stages of cirrhosis: stage 1 which is characterized by the absence of esophageal varices and of ascites; stage 2 which is characterized by the presence of esophageal varices without ascites and without bleeding. The development of ascites, with or without varices, in a patient who has never bled is the hallmark of stage 3. Gastrointestinal bleeding with or without ascites characterizes stage 4. Cirrhosis is defined as compensated when the patient is in stages 1 and 2, decompensated in stages 3 and 4 (55, 56). Dividing the patients in to these 2 groups of compensated and decompensated cirrhosis is very useful in terms of prognosis as these are two distinct stages of cirrhosis with different predictors of survival (55, 56). The yearly mortality rates have been calculated based on a large natural history study and the yearly risk of death according to the clinical stage is: in stage 1: 1.0%, in stage 2: 3.4%, in stage 3: 20% and in stage 4: 57% (57). Even among patients with cirrhosis and mild portal hypertension the survival rate is significantly decreased compared to the general population (58).

14
To assess the severity of the liver disease, scoring models have been used. The oldest model is the Child-Pugh (CP) classification (59). The CP classification is based on bilirubin level, prothrombin time, albumin and the presence and/or severity of ascites and encephalopathy (Table 2). Table 2. The Child-Pugh classification
Points 1 2 3 Encephalopathy (grade) none 1 and 2 3 and 4 Ascites absent slight moderate S-bilirubin (mmol/L) <35 35-50 >50 S-albumin (g/L) >35 28-35 <28 INR <1.3 1.3-1.8 >1.8 The points for all five parameters involved are summed up and patients classified into one of the three classes A, B or C. Class A: 5-6 points, Class B: 7-9 points and Class C: >10 points. As liver transplantation is the only curable treatment for end-stage liver diseases it has become even more important to predict patients survival for prioritization on waiting lists for liver transplantations. One of the drawbacks of the CP classification is subjective assessment (encephalopathy and ascites) and another model has been developed for the assessment of prognosis which is the Model of End-stage Liver Disease (MELD) (60). In this model there are only objective parameters, bilirubin, prothrombin time and creatinine. It has been shown to be reliable to estimate mortality risk for patients with end-stage liver disease of various etiologies (61, 62). 1.5 PORTAL HYPERTENSION Portal hypertension is the most common and lethal complication of chronic liver disease and its consequences are: gastroesophageal varices, variceal hemorrhage, ascites, renal dysfunction, encephalopathy, hypersplenism and hepatopulmonary syndrome. Portal hypertension is defined as an increase in portal pressure in which the pressure gradient between the portal vein and inferior vena cava (the portal pressure gradient) is increased above the upper

15
normal limit of 5 mmHg. The clinical significance increases above 10 mmHg (formations of varices) and above 12 mmHg (variceal bleedings and ascites) (63-66). This value of 10-12 therefore represents the threshold for defining portal hypertension as clinically significant. The portal pressure gradient depends on the blood flow and vascular resistance in the portal venous system. It begins with increased resistance to portal blood flow and is aggravated by an increase in portal venous inflow. The increased resistance can be prehepatic (as in portal vein thrombosis), intrahepatic (as in cirrhosis) or posthepatic (as in hepatic vein thrombosis or heart disease). Increased hepatic vascular tone is the basis for the use of vasodilators to treat portal hypertension in cirrhosis. Portal inflow is increased by splanchnic vasodilatation which can be counteracted with vasoconstrictors and betablockers which is the prerequisite for use of these drugs in portal hypertension. Clinically portal pressure is assessed by measuring the hepatic venous pressure gradient (HVPG), which is the difference between the wedged hepatic venous pressure (WHVP) and free hepatic venous pressure (FHVP), at hepatic vein catheterization (67).
The complications of portal hypertension are: upper gastrointestinal bleeding from ruptured gastroesophageal varices and portal hypertensive gastropathy (PHG), ascites, renal dysfunction hepatic encephalopathy, arterial hypoxemia, disorder in the metabolism of drugs or endogenous substances that are normally eliminated by the liver, bacteremia, and hypersplenism (68). These complications are major causes of death and the main indications for liver transplantation in patients with liver cirrhosis.
Factors contributing to the increase in hepatic vascular resistance in cirrhosis are: reduction in the release of endogenous vasodilators (such as nitric oxide) (69, 70) and an increase in endogenous vasoconstrictor activity (such as angiotensin, norepinephrine and vasopressin) (68).
Splanchnic vasodilatation is usually associated with peripheral vasodilatation and a systemic hyperkinetic syndrome, characterized by reduced arterial pressure and peripheral resistance and increased plasma volume and cardiac output (68). Peripheral vasodilatation plays a major role

16
in the activation of endogenous neurohumoral systems that cause sodium retention and expansion of the plasma volume, followed by an increase in the cardiac index (71). The pressure in esophageal varices can be measured at endoscopy by direct puncture of the varices (72) or with an endoscopic pressure-sensitive gauge attached to the tip of the endoscope (72, 73, 74). This is non-invasive technique, Varipress®. It is based on the assumption that because of their thin walls and lack of external tissue support, varices behave as elastic structures, thus, the pressure needed to compress a varix (which can be sensed by a pressure gauge) equals the pressure inside the varix. High sensitivity pressure transducer is used to record the measurements (75). The variceal pressure is calculated as the difference in pressure when the capsule is free in the esophageal lumen and when it is applied over a varix. Several recordings are usually obtained for each patient and the result given is the mean of at least three satisfactory tracings. Several studies have shown a good correlation between this technique and the more invasive variceal puncture technique (72, 73). 1.6 VARICEAL BLEEDING The most important clinical consequences of portal hypertension are related to the formation of portal-systemic collaterals, including gastroesophageal varices, which are responsible for one of the main complications of portal hypertension, upper gastrointestinal bleeding (67).
The main cause of variceal bleeding is thought to be excessive hydrostatic pressure inside the varices, which is a consequence of increased portal pressure. Many studies have shown that variceal bleeding does not occur before the HVPG reaches a threshold value of 12 mmHg (63, 65, 66). Variceal bleeding occurs when the tension exerted by the thin wall of a varix is beyond a critical value, determined by the elastic limit of the vessel, then the variceal wall cannot resist further dilation and variceal rupture occurs.

17
According to Frank’s modification of Laplace’s law, variceal wall tension (WT) can be defined by the following equation: WT = (Pi – Pe) x r/w. In which Pi is the intravariceal pressure, Pe the pressure in the esophageal lumen, r the radius of the varix and w the thickness of its wall (68, 76, 77). This equation explains the prognostic value of the red color signs indicating areas where the wall of a varix is especially thin (77, 78). At the diagnosis of cirrhosis, varices are present in about 40% of compensated patients and in 60% of those who present with ascites (79). The HPVG usually increases with worsening liver function and continued alcohol abuse and may decrease when liver function improves and alcohol is discontinued (80). Prospective studies have demonstrated that variceal hemorrhage does not occur if the HVPG is reduced, spontaneously or pharmacologically to levels below 12 mmHg (66, 80, 81), or more then 20% from baseline (82). The incidence of esophageal varices in patients with cirrhosis increases about 5%-8% per year (67, 83). In a meta-analytic review it is recommended that patients with liver cirrhosis should undergo endoscopic screening regularly. Those without varices every other year and those with small varices every year (84). Patients with medium-sized to large varices are considered to be at considerable risk for bleeding, and they should receive therapy to prevent variceal bleeding (85, 86). The risk for both rebleeding and mortality peaks during the first days after bleeding, it declines slowly thereafter and after 6 weeks becomes constant and virtually equal to that before bleeding (87). It has been demonstrated that changes in management and therapy of variceal bleeding has led to improvement in survival over the past decades (88, 89), but information on the prognosis of patients with esophageal varices in Sweden is limited.

18
1.7 GASTROINTESTINAL MOTILITY DISORDERS AND SMALL INTESTINAL BACTERIAL OVERGROWTH IN LIVER CIRRHOSIS
The liver and the biliary tree arise from the foregut during fetal development. This common embryologic origin causes the close relation between the liver and the gastrointestinal tract (40). The venous blood from the gastrointestinal tract passes the liver through the portal venous system before it joins the systemic circulation. Gastrointestinal symptoms and disturbances in gastrointestinal secretory, digestive, and motor functions appear to be common among patients with liver disease (90). Small intestinal dysmotility, bacterial overgrowth, changes in mucosal permeability can enhance bacterial translocation and therefore increase the risk of systemic infections and sepsis in patients with liver cirrhosis (91). Patients with liver disease have high prevalence of gastrointestinal symptoms such as nausea, vomiting, abdominal pain and diarrhea (90-92). It has been demonstrated that portal hypertension can cause morphological alterations in the intestinal mucosa (93). A protein loosing enteropathy has been described in cirrhosis (94, 95), and the severity correlates with clinical stage and the presence of portal hypertension and hypoalbuminemia (96). Studies of small-intestinal motility, in both an animal model of portal hypertension and in patients with chronic liver disease and portal hypertension have shown delayed transit through the small intestine (97-99). Studies have also shown small-intestinal dysmotility and in particular abnormalities in the frequency and propagation of phase III of the migrating motor complex (98-102). Several studies have shown a high prevalence of small-intestinal bacterial overgrowth (SIBO) in patients with chronic liver disease (100,101,105-107).

19
The pathophysiology of intestinal dysfunction is not fully understood. One of the many hypotheses is autonomic dysfunction which has been detected in high proportion of patients with cirrhosis and also in patients with extrahepatic portal hypertension, indicating that the portal hypertension per se plays an important role in the pathogenesis of autonomic dysfunction (108). Gastric and autonomic motor dysfunctions have been shown to correlate in patients with cirrhosis (109). The clinical significance of gastrointestinal dysfunction in patients with liver cirrhosis for the development of SIBO and risk of spontaneous bacterial peritonitis (SBP) is somewhat obscure. Edema of the intestinal wall, a consequence of hypoalbuminemia and portal hypertension are factors that can contribute to a disruption of mucosal function and facilitate bacterial translocation into the portal circulation (110). It has been shown that translocation occurs in experimental models of cirrhosis, portal hypertension and ascites (111-115). Conflicting results exist on the permeability of the intestinal mucosa in patients with liver cirrhosis with some studies demonstrating increased permeability (116-118) whereas others have not (119-121). Abnormal intestinal permeability has been postulated to be of limited importance in the pathophysiology of bacterial infections in patients with liver cirrhosis (122).
Recent gastrointestinal hemorrhage is a known risk factor for SBP and sepsis in humans (123,124). It is of interest that in an animal model translocation was exacerbated by the induction of hemorrhage (112). The consequences of SIBO and bacterial translocation could lead to activation of tumor necrosis factor (TNF) and other cytokines that affect homeostatic functions and it has been suggested that activation of TNF could potentiate the hyperdynamic circulation that is the hallmark of portal hypertension (125). The effect of portal hypertension per se on small bowel motility and small intestinal bacterial overgrowth is not fully understood. Changes in intestinal motility and bacterial overgrowth have not been compared in patients with liver cirrhosis with and without portal hypertension although a correlation

20
between changes in small bowel motility, bacterial overgrowth and liver cirrhosis have been demonstrated. 1.8 HEPATOCELLULAR CARCINOMA (HCC) AND OTHER
MALIGNANCIES IN PATIENTS WITH LIVER CIRRHOSIS Hepatocellular carcinoma (HCC) is the fifth most common malignancy globally in men and the eight most common in women (126). The geographic areas with the lowest incidence rates include northern Europe, Australia, and North America (126). In the past decades the number of cases of HCC has increased in the United States (127), with a shift in the incidence rates in the younger age groups as well as in Australia, Central Europe, the United Kingdom and Japan (126). Rates for liver cancers in men are typically 2 to 4 times higher than in women (126). It is unclear whether the higher risk of HCC in males reflects an increased susceptibility of men to the tumor or a greater exposure to such environmental risk factors as hepatitis viruses and alcohol. The majority of patients worldwide with HCC have underlying liver cirrhosis, supported by the fact that in 80% of autopsies of patients with HCC, cirrhosis is found (128). The annual incidence of HCC in patients with compensated cirrhosis is about 3%, and HCC has been identified as a relevant cause of death in these patients (129,130). Studies have shown a strong correlation between hepatitis C virus (HCV) infection and HCC (127,131). A meta-analysis of 32 case-control studies the estimated risk for the development of HCC was 17.5 fold greater in HCV carriers than in non-carriers (131). The severity of the hepatitis is the crucial factor for increased risk for HCC in carriers of HCV, the strongest predictor of HCC was cirrhosis (132-138). The increased risk of hepatocellular carcinoma (HCC) in patients with liver cirrhosis is well established and liver cirrhosis can therefore be considered a premalignant condition (139). Among patients with alcoholic liver cirrhosis who died during a 15 years follow-up, 20% died from malignancies, 10% from HCC and 10% from

21
other malignancies (140). Studies both from Denmark and Japan have reported an increased risk for the development of cholangiocarcinoma (CC) in patients with liver cirrhosis (141,142). Pulmonary and pancreatic cancers were also found to be associated with cirrhosis with 4-5 times increased risk. Furthermore a 1.5 times increased risk for colorectal cancer (CRC) was observed (141). In a French study the prevalence of colon adenomatous polyps, was over two times higher in patients with cirrhosis than in those without cirrhosis after controlling for alcoholism (143). There are limited data on the occurrence of other malignancies than HCCs in patients with liver cirrhosis. Changes in hormonal levels, impaired metabolism or alterations in immune function could influence the cancer risk in patients with liver cirrhosis. Information of the occurrence of hepatic and extrahepatic malignancies in patients with liver cirrhosis in Sweden is scarce and it has not been compared previously with that in the general population.

22
2. AIMS OF THE PRESENT STUDIES
To evaluate the incidence, etiology and prognosis of patients with liver cirrhosis both in Sweden and Iceland. To investigate prognosis in relation to different etiologies, and the relationship between alcohol consumption in these countries and the incidence of alcoholic cirrhosis in the last decades. To evaluate the outcome of patients with esophageal varices of cirrhotic and non-cirrhotic causes. Also to evaluate relationship with the etiology of cirrhosis and treatment used, and to which extent different treatment for esophageal varices have been applied in a University Hospital in Sweden in recent years. To assess the risk of hepatic and extrahepatic malignancies in a Swedish cohort of patients with liver cirrhosis compared to that in the general Swedish population, and to identify the most important predictors for the development of different forms of malignancies in patients with cirrhosis. To investigate the effects of portal hypertension on small bowel motility and small intestinal bacterial overgrowth in patients with liver cirrhosis, and whether portal hypertension could have a pathogenic role in altering small bowel motility and small intestinal bacterial contents in cirrhotic patients.

23
3. METHODS The studies were approved by the Ethics Committee at the University of Gothenburg the Sahlgrenska Academy. In study I all the participants gave informed consents. Studies II, III, and IV are record studies the cohort of patients have been studied retrospectively. Study III was also approved by the Ethics Committee at the Icelandic University Hospital, Landspitali. 3.1 Paper I 3.1.1. Subjects A retrospective analysis was performed on all patients with liver cirrhosis diagnosed for the first time in Gothenburg, Sweden serving a population of approximately 600.000 inhabitants during the 10-year period 1994-2003. In comparison we analyzed all patients with the diagnosis liver cirrhosis during the same time period in Iceland (approximately 300.000 inhabitants). Study outcomes were overall mortality during follow-up and causes of death. Patients were followed until death or, if alive, until data were collected from their medical records. The end-points were death, transplantation or follow-up if the patient was still alive. 3.1.2. Data collection All patients diagnosed with liver cirrhosis during hospitalization or as outpatients were retrieved with a search in a computerized diagnoses database at the Sahlgrenska University Hospital Gothenburg Sweden and also at Landspitali University Hospital and Akureyri District Hospital in Iceland, serving the whole population of Iceland. Medical records were reviewed and only patients in whom the cirrhosis was diagnosed for the first time during these years were included. The medical records of eligible patients were reviewed to obtain necessary clinical, laboratory, imaging and follow-up data. Diagnosis and etiology of the cirrhosis was based on a liver biopsy or on compatible clinical, laboratory and imaging findings, based on the presence of at least two of the following: characteristic imaging features,

24
esophageal or gastric varices, ascites, increased INR that could not be attributed to any other cause. 3.1.3. Information obtained from medical records The following information was obtained from medical records: gender, age at diagnosis, etiology of the liver cirrhosis and the diagnostic work-up of the etiology, presence of ascites and esophageal varices, laboratory results at diagnosis: hemoglobin, platelets count, prothrombinkomplex (PTK) or international normalization ratio (INR), albumin, bilirubin, AST, ALT, ALP, time of death and cause of death. If the information on the cause of death if this information was lacking in the medical records the causes of death were retrieved from the Swedish Epidemiological Center (the Swedish registry of Causes of Death from the National board of Health and Welfare), and from the Icelandic Registry of Causes of Death. 3.2 Paper II 3.2.1. Subjects This study was a retrospective analysis of all patients with esophageal varices diagnosed for the first time at Sahlgrenska University Hospital (serving a population of approximately 600 000 inhabitants) during the 6-year period 1994-1999; and thus there was no selection. Patients with liver cirrhosis as well as those with non-cirrhotic portal hypertension were included. Death in variceal bleeding was defined as death within 1 week after a variceal bleeding. Study outcomes were frequency of bleeding during follow-up, death in variceal bleeding and overall mortality during follow-up. Patients were followed until death or, if alive, until the day data were collected from their medical records. The end-points were death, transplantation or follow-up if the patient was still alive. 3.2.2. Data collection All patients diagnosed with esophageal varices during hospitalization, or as outpatients in the endoscopy unit, were retrieved from the hospital diagnoses register. A long list of patients with this diagnosis was provided from

25
different departments. All these records were reviewed and only patients in whom the varices were diagnosed for the first time during these years were included. The medical records of eligible patients were reviewed to obtain necessary endoscopic, intensive care unit (ICU), clinical, laboratory, imaging and follow-up data. Diagnosis and etiology of the cirrhosis was based on a liver biopsy or on compatible clinical, laboratory and imaging findings, based on the presence of at least two of the following: characteristic imaging features, esophageal or gastric varices, ascites, increased INR that could not be attributed to any other cause. 3.2.3. Information obtained from medical records The information obtained was apart from gender: age at diagnosis, etiology of portal hypertension and cirrhosis, presence of ascites, laboratory results at diagnosis: haemoglobin, platelets count, coagulation factors II, VII, X, (PTK), albumin, bilirubin, AST, ALT, ALP, hepatitis serological tests, recorded bleeding episodes, acute therapy to stop bleeding, indication for gastroscopy, primary and secondary prophylactic therapy, time of death and diagnosis (those that we could not find in the medical records were retrieved from the Causes of Death Register). 3.3 Paper III 3.3.1. Subjects The study cohort comprised all liver cirrhosis patients registered over the 12 year period 1994-2005 during hospitalization or as outpatients at the Sahlgrenska University Hospital Gothenburg Sweden serving a population of approximately 600.000 inhabitants. 3.3.2. Data collection The patients were identified through search of computerized diagnoses database at the Sahlgrenska University Hospital. Medical records were reviewed and only patients in whom the cirrhosis was diagnosed for the first time during these years were included. Diagnosis of the cirrhosis was based on a liver biopsy or on compatible clinical, laboratory and imaging findings,

26
based on the presence of at least two of the following: characteristic imaging features, esophageal or gastric varices, ascites, increased INR that could not be attributed to any other cause. All patients’ records were scrutinized to confirm the diagnosis of liver cirrhosis and to obtain necessary clinical, laboratory, imaging and follow-up data. 3.3.3. Follow-up Two strategies were used for follow-up: 1. through the National Swedish Cancer Registry which provided follow-up of all patients to 20 November, 2007 and 2. follow-up to latest clinical visit, at the latest to 27 November, 2007. Linkage between registries was possible through the individually unique ten-digit national registration number assigned to all Swedish residents. The National Cancer Registry was founded in 1985 and the reporting is close to 98% complete (144,145). From the Death Registry we ascertained date of death during follow-up and also checked if any death due to cancer was reported that was not registered in the National Swedish Cancer Registry. 3.3.4. End-points Endpoints were overall mortality during follow-up and causes of death, or last date of clinical follow-up. Patients were also censored at time for liver transplantation. 3.4. Paper IV 3.4.1. Subjects Twenty-four patients with liver cirrhosis were included: 12 had esophageal varices and thus portal hypertension (PH group), and 12 had no clinical signs of portal hypertension, with no esophageal varices on endoscopy and no history of ascites (LC group).

27
The manometry results were compared with the findings from 32 healthy controls (15 women, 17 men; mean age 35 yr, age range 23–54 yr) (HC group). 3.4.2. Manometry Antroduodenojejunal pressure was recorded after an overnight fast with an eight-channel catheter as previously described (146). The catheter was introduced nasally and placed, under fluoroscopic guidance, with three side ports in the gastric antrum 1.5 cm apart, three in the proximal descending part of the duodenum 2 cm apart (to allow analysis of the propagation of individual pressure waves) (147), one in the distal duodenum close to the ligament of Treitz, and one in the proximal jejunum. The water-perfused pressure catheter had an outer diameter of 4.8 mm, a central lumen of 1.8 mm for the guidewire, and eight lumens with a diameter of 0.8 mm for pressure recording, the placement of the side ports is shown in Fig 1.
Figure 1. The placement of the side ports on the catheter used for antroduodenojejunal pressure recordings. The pressure recording side ports were located 2, 17, 30, 32, 34, 45.5, 47, and 48.5 cm from the tip of the catheter. The catheter was connected to pressure transducers, and recordings were made by a PC polygraph (Synectics Medical, Stockholm, Sweden), which converted the pressure data into digital information at 4 Hz and transferred it to an IBM-compatible computer. The individual recordings were displayed on the computer screen

28
during the recording and were stored for later analysis. The patients were in a semireclining position during the registration. Fasting motility was recorded for 5 h and another 1 h after a standard meal, which consisted of porridge made from about 200 ml of water and 50 g rolled oats, 150 ml milk, white bread (50 g), butter (10 g), and about 13 g of cheese (16% fat); total energy content of the meal was 500 kcal). A condensed tracing was used for conventional evaluation of motility disorders in the small intestine (148). 3.4.3. Analysis of manometric data Phase III of the gastric antrum was defined as a sequence of three pressure waves per min for at least 1 min in temporal relationship with a duodenal phase III and followed by motor quiescence (phase I). In the small intestine the phase III was defined as a sequence of regular pressure waves, 10.5–14/min for at least 2 min, migrating in aboral direction and followed by motor quiescence (phase I). Phase II was defined as motor activity below the phase III frequency but more than 2 contractions/10 min. Phase I was defined as a period with contractions not more than 2/10 min and preceded by a phase III. The duration of MMC was defined as the time passing between two consecutive phases III. For calculation of the motility index in the four segments studied, the following channels were used: channel 3 in the antrum, channel 5 in the proximal duodenum, channel 7 in the distal duodenum, and channel 8 in the jejunum. A long cluster of intestinal activity was defined as a sequence of pressure waves at the maximal contractile activity (10–14/min), lasting ≥30 sec and preceded and followed by motor quiescence for >30 sec but not followed by a motor phase I–like quiescence. The channels used were numbers 4, 5, 6, 7, and 8. Analysis of interdigestive motility and individual pressure waves was performed by inspection on the computer screen. Motility variables measured were the duration of MMC, the number of phases I, II, and III and their duration, phase III motility index (area under the curve mm Hg × sec),

29
and the migration velocity of phase III from the proximal duodenum to the distal duodenum and from the distal duodenum to the jejunum. The motility index for the last 30 min of phase II (late phase II), the number of propagated single contractions during phase II, and the total number of long clusters of contractions during phase II were measured. Postprandial recordings were analyzed for motility index and propagated single contractions. The MMC cycles were analyzed qualitatively and quantitatively with a commercially available program (Polygram, version 5.06 X1, Synectics Medical). The basic menu of this program was used for calculations. The area under the curve was used as motility index, expressed as mm Hg × sec. Evaluation with high temporospatial resolution was performed to analyze the propagation pattern of individual pressure waves in the proximal duodenum, (channels 4–6) in late phase II and postprandially (the first 30 min after the test meal). The direction of propagation was determined visually on the computer screen. Conventional evaluation of the condensed manometry data were performed by one of the investigators, who was blinded to patient group. Motor abnormalities were divided into neuropathic- and myopathic-like patterns. Patterns were defined as neuropathic-like if: 1) phase III started earlier in the more distal segments of the small bowel studied (retrograde activity front) or were interrupted for ≥1 min; or 2) there were bursts of regular pressure waves in phase II with higher contraction frequency than normal (i.e., ≥15/min); or 3) a phase III appeared 30 min after the start of the meal (no fed pattern); or 4) a steady elevation above baseline after the meal was observed. Recordings with persistently low amplitude of contractions but with normal coordination patterns were considered to be myopathic-like. 3.4.4. Diagnosis of bacterial overgrowth Aspirate from the jejunum was obtained at the manometry via the central lumen of the catheter and collected in a sterile glass tube. Specimens were cultured (within 2 h) for aerobic and anaerobic bacteria on blood agar plates

30
with 4% defibrinated horse blood in aerobic and anaerobic atmosphere of 10% CO2 and N2, and for selective cultivation of gram-negative strains on Drigalski agar under aerobic conditions. Yeast fungus was cultured on Sabouraud's agar. The incubation time of the plates was at least 48 h. Identification of the microorganisms was based on colony characteristics, Gram staining, and biochemical and gas chromatographic tests. Cultures from jejunal aspirate were considered positive if the total count of colonic bacteria was >105 colony-forming units/ml (104,149,150), whereas growth of upper respiratory tract flora was not regarded as positive. Biopsies were taken from the stomach during gastroscopy to verify whether the patients had atrophic gastritis or not. 3.4.5. Variceal pressure measurements Variceal pressure measurements were performed in all 12 patients with esophageal varices with a noninvasive technique that employs a pressure-sensitive capsule connected to an endoscope (Varipress®, Labotron, Barcelona, Spain) (72-74). The capsule is perfused with a constant flow of nitrogen; when applying it over the esophageal varix the pressure needed to perfuse the capsule equals the variceal pressure. A high sensitivity pressure transducer was used for recording. Variceal pressure was calculated as the difference in pressure when the capsule was free in the esophageal lumen and when it was applied over a varix. Several recordings were obtained in each patient, and the results given represent the mean of at least three satisfactory tracings obtained in each patient. A schematic figure of the Varipress method is shown in Fig 2 and a typical registration is shown in Fig 3.

31
Figure 2. Schematic figure of the Varipress method for variceal pressure measurements.
Figure 3. A Varipress registration from one patient, showing 6 tracings. 3.5. Statistical methods 3.5.1. In papers I and II Mean, SD, range, medians and percentages were calculated for descriptive purposes. When comparing two groups, Fisher’s exact test was used for dichotomous variables; for non-ordered categorical variables, the chi-square test was performed and for ordered categorical variables Mantel-Haenszel’s test was used. When comparing three groups, Kruskal-Wallis ANOVA was performed for continuous variables.

32
For comparison of the incidence in Gothenburg and Iceland the optimal test (151) for comparison of two poisson distributions has been used. For survival analysis, Kaplan-Meier estimates were calculated and formally tested with the log-rank test. In order to select independent predictors to survival stepwise Cox proportional hazard regression analysis were performed. Only variables that affected survival time at univariate test (p<0.1) were included as possible predictors in the multivariate analysis. Hazard ratios with 95% confidence intervals were calculated with Cox models for descriptive purpose. All tests were two-tailed and conducted at 5% significance level. 3.5.2. In paper III Mean, standard deviation (SD), medians and percentages were calculated for descriptive purposes. When comparing two groups, Fisher’s exact test was used for dichotomous variables. For comparison of continuous variables the t-test was used. All tests were two-tailed and were conducted at a 5% significance level. The expected number of cases used to calculate SIRs was obtained by multiplying age- and calendar specific person-years in the cohort with the corresponding incidence in the entire Swedish population. Confidence intervals of SIRs were calculated assuming Poisson-distributed number of observed cancer cases. 3.5.3. In paper IV Results are presented as mean ± SEM. For comparison between two groups, Fisher nonparametric permutation test was used. Pitman nonparametric permutation test was used for all correlation analyses. In addition, Pearson correlation coefficient was calculated for descriptive purposes. All significance tests were performed two-sided and conducted at a 5% significance level.

33
4. RESULTS 4.1. Liver cirrhosis in Gothenburg and Iceland, epidemiology and natural history 4.1.1. Patients characteristics A total of 1016 patients were eligible to be included in the study 918 in Gothenburg Sweden (SW) and 98 patients in Iceland (IC). A gender difference was found between the countries with 69.3% of the patients were of male gender but in Iceland 52% were males (p=0.001). Higher age at diagnosis was observed in Gothenburg (60.0 ± 12.4) years vs. in Iceland (63.7 ± 13.4) years (p=0.0004). In Gothenburg there was a significant difference when comparing the age of the patients with ALD (58.5 ± 10.5 years) vs. NALD (62.3 ± 14.7 years), (p<0.0001), but no difference in the Icelandic group. Only 9% in GO and 5% in IC had HCV (p=NS). There were a higher proportion of patients with PBC, AIH and heamochromatosis in IC than in GO (p<0.001). The etiology was unknown that is cryptogenic cirrhosis in GO 16% and IC 20 % (p=NS). Concerning the etiology of the liver cirrhosis, alcohol abuse was much more common in SW than in IC 50% and 29% respectively and when those who also were infected with hepatitis C were included this difference was, 62% and 32%, respectively (p<0.0001). A higher proportion of patients in GO than in IC had ascites, 61% vs 34% (p<0.0001). The mean Child-Pugh score was higher in Gothenburg 9.0±2.5 vs in Iceland 7.3±2.7 (p<0.0001). 4.1.2. Incidence The incidence per 100.000 inhabitants for each year as well as proportion of alcoholic etiology is shown in figure 6. The mean annual incidence per 100.000 inhabitants in Gothenburg was 15.3 ± 2.4 compared to Iceland 3.3 ± 1.2 p<0.00001. In SW the incidence and proportion of alcohol etiology has been fairly constant over the study period but in IC the proportion of alcoholic etiology varied more between the years (Figure 4).
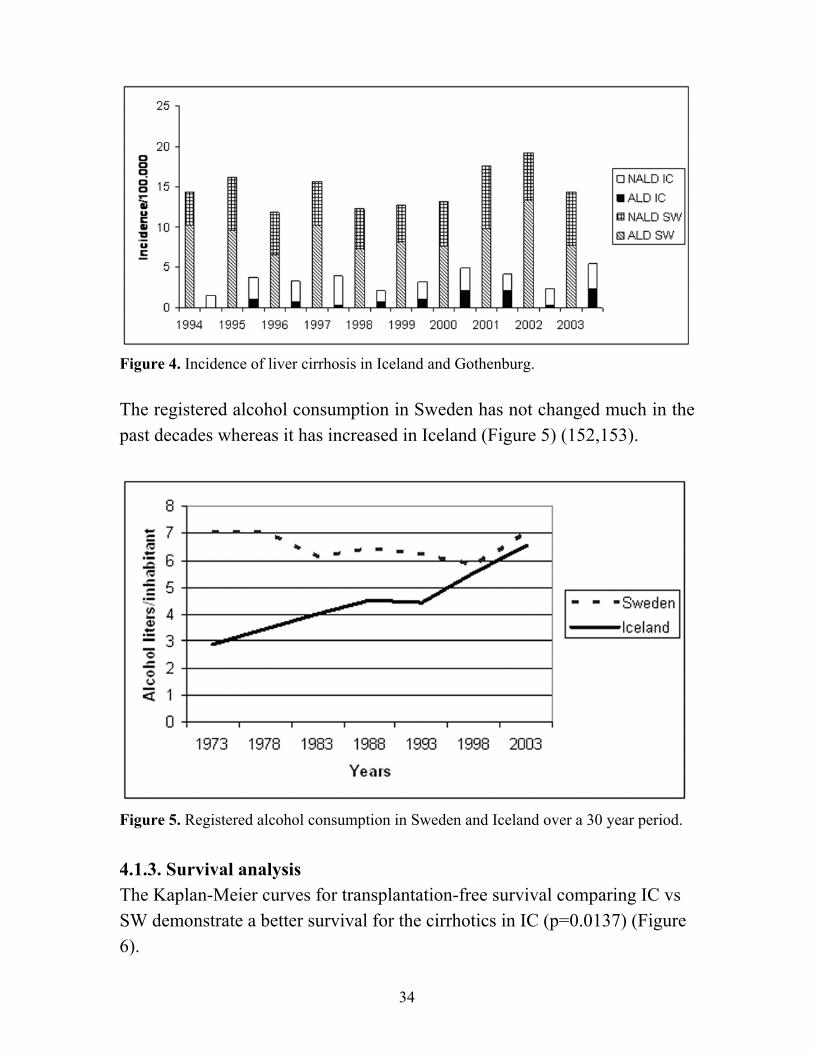
34
Figure 4. Incidence of liver cirrhosis in Iceland and Gothenburg. The registered alcohol consumption in Sweden has not changed much in the past decades whereas it has increased in Iceland (Figure 5) (152,153).
Figure 5. Registered alcohol consumption in Sweden and Iceland over a 30 year period. 4.1.3. Survival analysis The Kaplan-Meier curves for transplantation-free survival comparing IC vs SW demonstrate a better survival for the cirrhotics in IC (p=0.0137) (Figure 6).

35
Figure 6. KM-curve comparing survival of patients in Iceland and Gothenburg. However, for the same Child-Pugh (CP) groups in the different countries there was no significant difference in survival (Figure 7a CP-A, 7b CP-B, 7c CP-C).
Figure 7a Figure 7b
Figure 7c No significant differences were observed in survival for ALD vs NALD in neither Gothenburg nor Iceland (p=NS) Figure 8 a and b.

36
Figure 8 a Gothenburg Figure 8 b Iceland 4.1.4. Variables associated with overall mortality
Table 3. Univariate and multivariate survival analysis for Sweden Univariate analysis Multivariate analysis Variables HR (95% CI) p HR (95% CI) p for death for death C-P score 1.220 (1.181-1.260) <0.0001 1.180 (1.135-1.227) <0.0001 Age (year) 1.016 (1.009-1.022) <0.0001 1.023 (1.016-1.030) <0.0001 Bilirubin 1.002 (1.002-1.003) <0.0001 1.001 (1.001-1.002) 0.0002 Gender Woman 1 1 Man 1.205 (1.008-1.440) 0.0348 1.295 (1.073-1.563) 0.0071 AST 1.013 (1.006-1.020) 0.0004 1.011 (1.002-1.019) 0.0153 HR = Hazard ratios, CI = Confidence interval Table 4. Univariate and multivariate survival analysis for Iceland. Univariate analysis Multivariate analysis ________________________________________________________________________ Variables HR (CI) p HR (CI) p for death for death Etiology unknown 3.091 (1.746-5.471) <0.0001 4.059 (2.097-7.860) <0.0001 C-P score 1.125 (1.021-1.238) 0.0171 1.209 (1.087-1.344) 0.0005 Gender Woman 1 1 Man 2.005 (1.168-3.441) 0.0090 2.220 (1.252-3.937) 0.0064 Age (year) 1.029 (1.003-1.055) 0.0257 1.027 (1.005-1.050) 0.0154
37
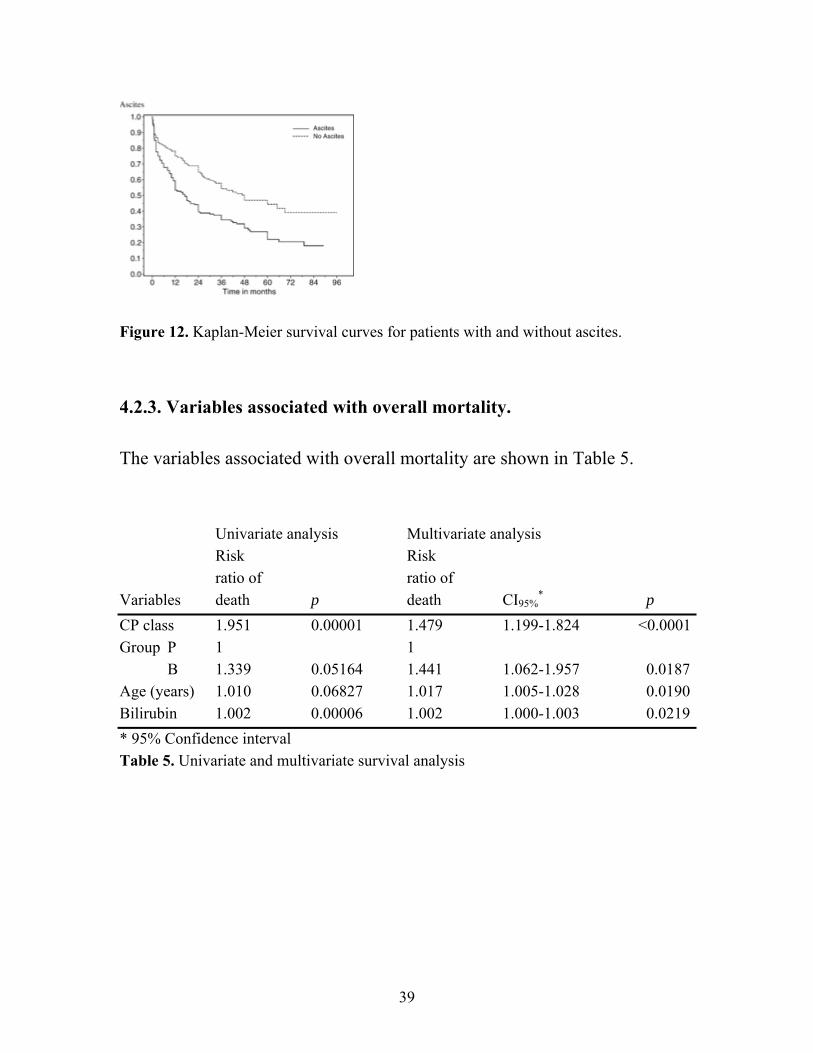
4.2 Esophageal varices, prognosis and outcome 4.2.1. Patients characteristics A total of 312 patients were eligible to be included in this study. Two-hundred-and-ninety-seven patients had liver cirrhosis, 197 with a diagnosis of OV obtained before the first bleeding (P), 92 with OV diagnosed after bleeding (B). Eight (3%) of the patients were diagnosed at autopsy (A) and were excluded from further analysis. Fifteen (5%) of the patients had portal vein thrombosis (T), without liver cirrhosis (7/15, 47% of these were diagnosed in association with a bleeding episode). In groups B and P the etiology of the cirrhosis was alcohol abuse in 54% of the patients (including those who were also infected with the hepatitis C virus. There was no significant difference between groups B and P in the frequency of alcohol-related etiology when combining the etiology groups alcohol abuse and alcohol abuse + HCV (58% versus 53%; p=NS). Thirty-eight patients (38/289, 13%) had other identified etiology of the cirrhosis than alcohol or HCV: primary biliary cirrhosis (PBC) 16; primary sclerosing cholangitis (PSC) 5; hepatitis B 4; auto-immune hepatitis (AIH) 4; haemochromatosis 3; non-alcoholic steatohepatitis (NASH) 3; congenital fibrosis 3. In 53 patients (17%) the etiology was unknown. The incidence as well as the proportion of alcoholic etiology was fairly constant over time (Fig.9).
Figure 9. Number of patients diagnosed for each year and the proportion of alcohol etiology. 4.2.2. Outcome In the B group, 8/92 (9%) died in their first bleeding. In 4/8 the etiology was alcoholic liver disease. During follow-up, 46/84 (55%) group B patients

38
experienced at least one bleeding episode as compared to 26/197 (13%) of those in group P (p<0.001), in spite of a much higher rate of prophylactic treatment in the B patients (96% versus 56%). When comparing patients in Child-Pugh class A + B versus C, there was no difference in rebleeding frequency (p=NS). The median follow-up was 24 months (range 0-96); 20 months in group B, 25 in group P and 36 in group T (p=NS). Transplantation-free survival for the three groups is shown in fig 10. There was a tendency towards a worse prognosis in group B vs. group P (p=0.0516).
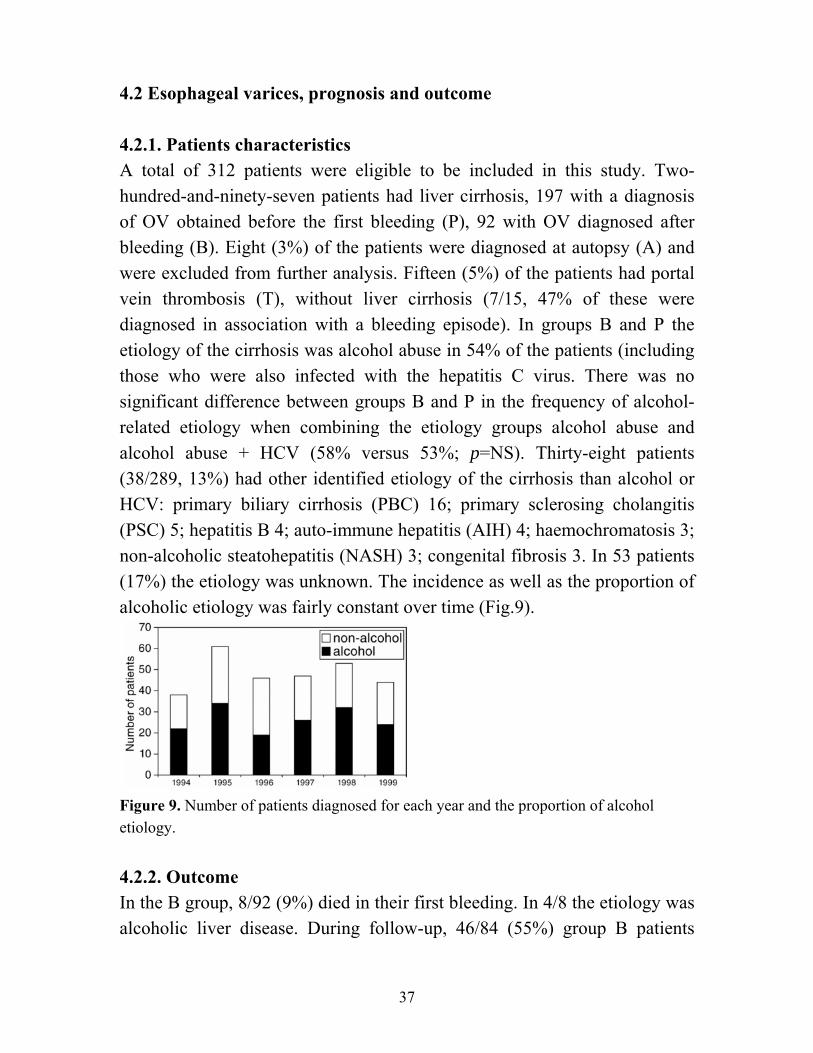
Figure 10. Kaplan-Meier survival curves for the three patient groups B, P and T. Because of the different etiology of the portal hypertension compared to groups B and P, the T group was excluded from the further survival analysis. There was a significant difference in survival related to CP classes (p<0.001) (Fig.11), and to the presence or absence of ascites (p<0.001) (Fig.12).
Figure 11. Kaplan-Meier survival curves for the different Child-Pugh classes A, B and C.
39
Figure 12. Kaplan-Meier survival curves for patients with and without ascites. 4.2.3. Variables associated with overall mortality. The variables associated with overall mortality are shown in Table 5.
Univariate analysis Multivariate analysis Risk Risk ratio of ratio of
Variables death p death CI95%* p
CP class 1.951 0.00001 1.479 1.199-1.824 <0.0001 Group P 1 1 B 1.339 0.05164 1.441 1.062-1.957 0.0187 Age (years) 1.010 0.06827 1.017 1.005-1.028 0.0190 Bilirubin 1.002 0.00006 1.002 1.000-1.003 0.0219 * 95% Confidence interval Table 5. Univariate and multivariate survival analysis

40
4.2.4. Use of prophylactic therapy In group P, only 128/197 (65%) were treated with primary prophylactic therapy. The majority 41/69 (60%) of the patients who did not receive primary prevention had small varices and were therefore judged as having a low risk of bleeding, or they were lost during follow-up due to alcohol or drug abuse. During follow-up, 26/197 (13%) patients in group P had a bleeding episode; 20/128 (16%) of the patients who received primary preventive therapy experienced variceal bleeding during follow-up. Four patients in group P died in the first bleeding and all had received primary preventive therapy, two of them pharmacological and two both pharmacological and endoscopic therapies. In group B, 8 of the 92 patients died in the first bleeding and 81 of the surviving 84 (96%) had secondary prophylactic therapy. Forty-six (55%) of these patients had a bleeding during follow-up. There was no significant difference in bleeding rate during follow-up when comparing different primary or secondary preventive therapies when this was grouped into pharmacological, endoscopic and combined therapy, or when comparing those with alcoholic liver disease versus other etiologies. 4.3 Hepatocellular carcinoma and other malignancies 4.3.1. Patients characteristics Clinical characteristics of 1019 patients with liver cirrhosis are shown in Table 6.

41
Table 6 Sex (male/female) 68%/32% (696/323) Mean Child-Pugh score (±SD) 9.1 ± 2.5 Mean age at diagnosis of liver cirrhosis in years (±SD) 59.5 ± 12.4 Child-Pugh class A 19% (198/1019) B 36% (367/1019) C 45% (454/1019) Etiology: Alcoholic liver disease (ALD) 48% (492/1019) Hepatitis C (HCV) 10% (103/1019) ALD +HCV 12% (123/1019) Primary Biliary Cirrhosis 5% (45/1019) Hepatitis B 3% (28/1019) Cryptogenic 15% (157/1019) Other 7% (71/1019) Mean time of follow (months) up to end point (±SD) 39 ± 37 Years of follow-up to death, OLT or latest clinical control* 3290 person-years Patients treated with liver transplantation 11% (109/1019) Rate of death to the latest clinical control 64% (654/1019) * Endpoint 27 November, 2007 OLT, orthotopic liver transplantation
4.3.2. Mortality The frequency and cause of death, 1019 liver cirrhosis patients are shown in Table 7. Table 7 Mortality rate 64% (654/1019) Primary cause of death (n = 654) Liver disease (non-malignant) 53% (349/654) Liver failure 37% (242/654 Variceal bleeding 8% (49/654) Infections 8% (53/654)
Cancer 22% (141/654) HCC 14% (87/654) Other cancers 8% (54/654) Other 25% (164/654) Reasons for death due to causes other than cancer/chronic liver disease (n = 164) GI-bleeding (other than variceal) 11% (17/164) Various others 66% (109/164) Heart disease 9% (14/164) Alcohol dependency 7% (12/164) Other diseases and accidents 50% (83/164) Unknown 23% (38/164)
42
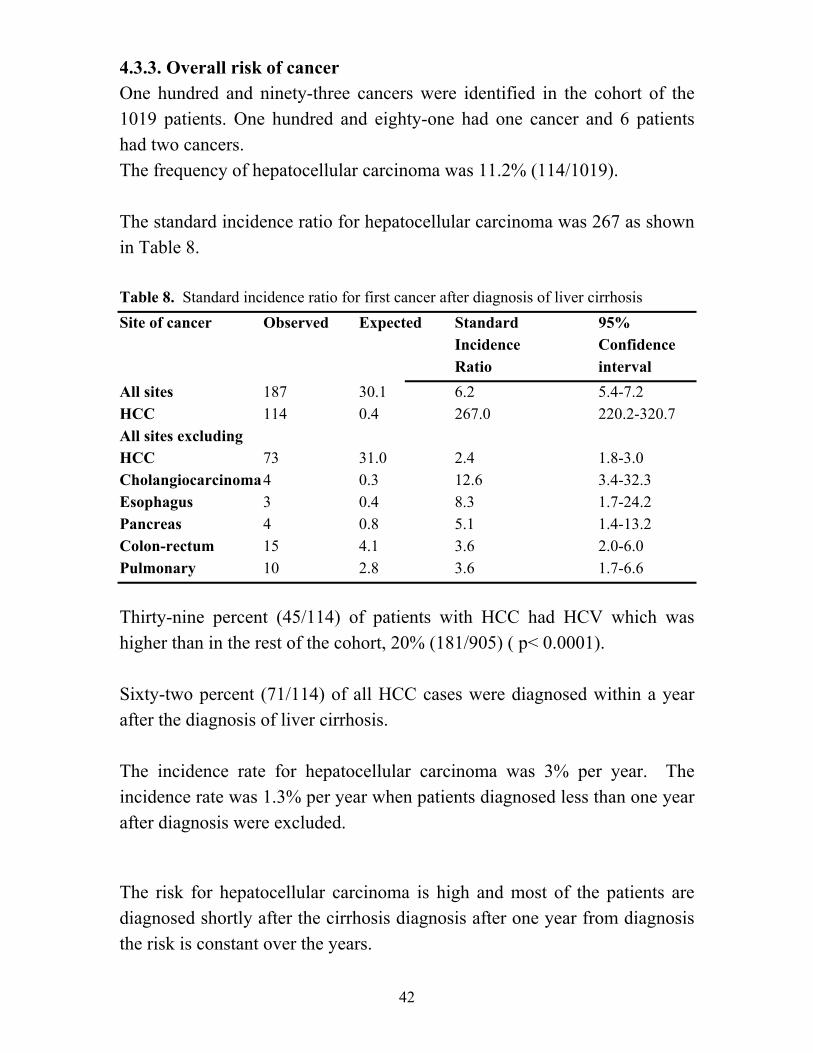
4.3.3. Overall risk of cancer One hundred and ninety-three cancers were identified in the cohort of the 1019 patients. One hundred and eighty-one had one cancer and 6 patients had two cancers. The frequency of hepatocellular carcinoma was 11.2% (114/1019). The standard incidence ratio for hepatocellular carcinoma was 267 as shown in Table 8. Table 8. Standard incidence ratio for first cancer after diagnosis of liver cirrhosis Site of cancer Observed Expected Standard 95% Incidence Confidence
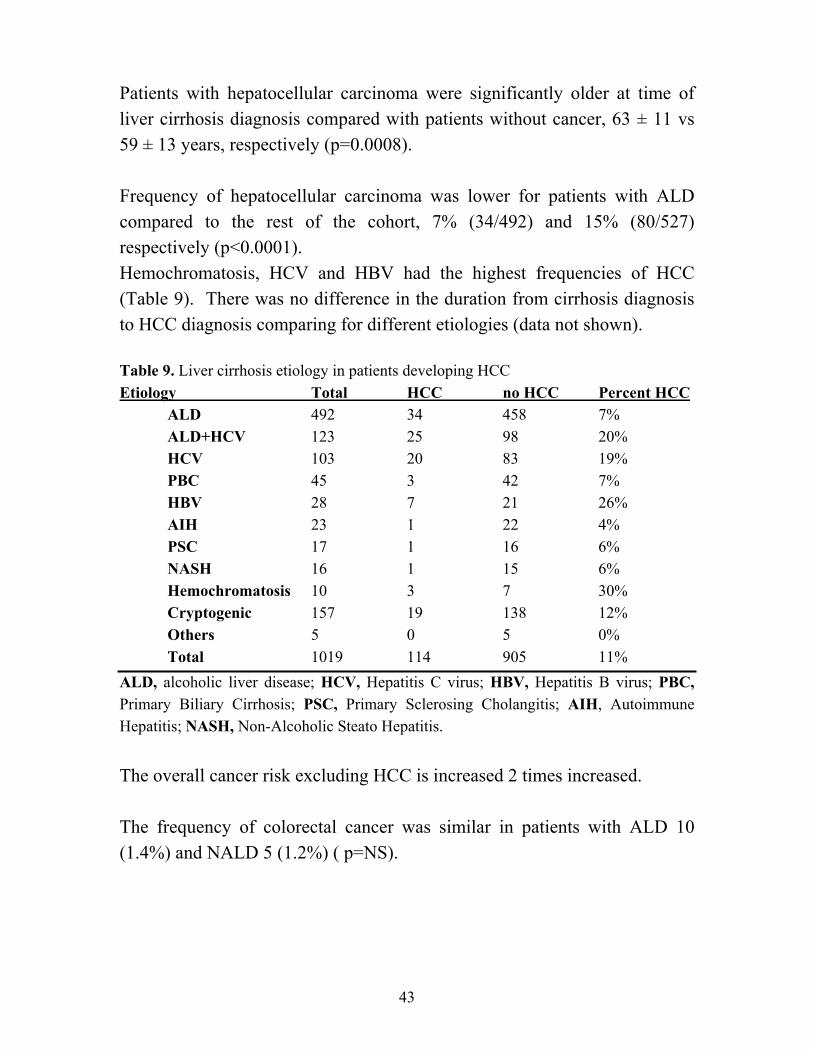
Ratio interval All sites 187 30.1 6.2 5.4-7.2 HCC 114 0.4 267.0 220.2-320.7 All sites excluding HCC 73 31.0 2.4 1.8-3.0 Cholangiocarcinoma 4 0.3 12.6 3.4-32.3 Esophagus 3 0.4 8.3 1.7-24.2 Pancreas 4 0.8 5.1 1.4-13.2 Colon-rectum 15 4.1 3.6 2.0-6.0 Pulmonary 10 2.8 3.6 1.7-6.6 Thirty-nine percent (45/114) of patients with HCC had HCV which was higher than in the rest of the cohort, 20% (181/905) ( p< 0.0001). Sixty-two percent (71/114) of all HCC cases were diagnosed within a year after the diagnosis of liver cirrhosis. The incidence rate for hepatocellular carcinoma was 3% per year. The incidence rate was 1.3% per year when patients diagnosed less than one year after diagnosis were excluded. The risk for hepatocellular carcinoma is high and most of the patients are diagnosed shortly after the cirrhosis diagnosis after one year from diagnosis the risk is constant over the years.
43
Patients with hepatocellular carcinoma were significantly older at time of liver cirrhosis diagnosis compared with patients without cancer, 63 ± 11 vs 59 ± 13 years, respectively (p=0.0008). Frequency of hepatocellular carcinoma was lower for patients with ALD compared to the rest of the cohort, 7% (34/492) and 15% (80/527) respectively (p<0.0001). Hemochromatosis, HCV and HBV had the highest frequencies of HCC (Table 9). There was no difference in the duration from cirrhosis diagnosis to HCC diagnosis comparing for different etiologies (data not shown). Table 9. Liver cirrhosis etiology in patients developing HCC Etiology Total HCC no HCC Percent HCC ALD 492 34 458 7% ALD+HCV 123 25 98 20%
HCV 103 20 83 19% PBC 45 3 42 7%
HBV 28 7 21 26% AIH 23 1 22 4%
PSC 17 1 16 6% NASH 16 1 15 6%
Hemochromatosis 10 3 7 30% Cryptogenic 157 19 138 12% Others 5 0 5 0% Total 1019 114 905 11% ALD, alcoholic liver disease; HCV, Hepatitis C virus; HBV, Hepatitis B virus; PBC, Primary Biliary Cirrhosis; PSC, Primary Sclerosing Cholangitis; AIH, Autoimmune Hepatitis; NASH, Non-Alcoholic Steato Hepatitis. The overall cancer risk excluding HCC is increased 2 times increased. The frequency of colorectal cancer was similar in patients with ALD 10 (1.4%) and NALD 5 (1.2%) ( p=NS).

44
4.4. Small intestinal motility disturbances and bacterial overgrowth 4.4.1. Conventional manometric evaluation In the 24 patients 10 (42%) demonstrated motor abnormalities. Four of these patients had portal hypertension (one of these four had SIBO), and six patients had liver cirrhosis without portal hypertension ( p=NS).). All of these alterations were neuropathic-like, and no myopathic-like manometry pattern was identified. A registration from one of the patients is shown in Fig 13.
Figure 13. Condensed manometry recording in a patient with liver cirrhosis and portal hypertension, exhibiting a high number of long clusters. Three of the patients (two PH and one LC) had no phase III during the 5-h fasting recording as compared with two HC subjects (NS). In 30% of the PH patients, 55% of the LC patients, and 53% of the HC subjects an antral phase III was recorded (NS for all comparisons). Both patient groups and the control group had 0–4 phases III/5 h (PH 1.8 ± 0.4 vs LC 2.4 ± 0.4 vs HC 1.9 ± 0.1; NS).

45
Cycle length of the MMC in the proximal duodenum was significantly shorter in the LC patients compared with the HC subjects (83 ± 7 vs 125 ± 11 min; p < 0.05), but there was no significant difference between the patient groups (PH 117 ± 22 min vs LC vs HC; NS). Cycle length of the MMC in the distal duodenum was similar in all the groups (PH 120 ± 18 vs LC 85 ± 8 vs HC 110 ± 10 min; NS). There were no significant differences in the duration of phase I or phase II in the distal duodenum (channel 7) when comparing the three different groups (data not shown). Duration of phase III in the proximal duodenum was significantly longer in the PH group 6.00 ± 0.56 minutes, compared to the LC group 3.64 ± 0.34, p<0.01. In the jejunum it was also significantly longer in the PH 8.00 ± 0.80 minutes compared to both the LC group 5.27 ± 0.76 and the HC group 6.14 ± 0.32, (p<0.05). The duration of phase III did not differ between the patients with and without SIBO (data not shown). The motility index (MI) in phase III in the proximal and distal duodenum was higher in the PH group compared with the other groups. The migration velocity of phase III was similar in the groups. The PH group had a higher number of long clusters in phase II, 9.1 ±2.1 compared with HC subjects 4.9 ± 0.8, (p<0.05). The MI in late phase II in the antrum and distal duodenum was similar in the three groups, but in the proximal duodenum PH patients had a significantly higher MI than did LC patients, and in the jejunum, LC patients had a significantly higher MI than did HC subjects (Fig. 14).

46
Figure 14. Motility index in late phase II (30 min). The PH group is shown with black bars, the LC group with grey bars, and the HC with white bars. *p < 0.05. PD = proximal duodenum; DD = distal duodenum; J = jejunum. Postprandially, there was a significant difference in the MI in the proximal and distal duodenum between the PH and HC groups, but no difference between the patient subgroups. In the jejunum, the MI was significantly higher in the patients groups compared with controls, but no difference was observed between the patient subgroups (Fig 15).
Figure 15. Motility index postprandially, PH-group in black, LC-group in grey and HC-group in white. PD is porximal duodenum, DD is distal duodenum and J is jejunum. *p<0.05 compared with HC, **p<0.01 compared with HC.

47
4.4.2. Evaluation with high temporospatial resolution The direction of propagation of individual pressure waves was analyzed for the last 30 min of the phase II in the proximal duodenum, and marked changes were observed in patients with portal hypertension compared with the other two groups. A significantly higher proportion of individual pressure waves were retrograde in the PH group compared with the HC group (p < 0.001) and LC group (p < 0.001) (Fig 2). Similar propagation patterns were observed postprandially in the proximal duodenum, with a higher proportion of retrograde pressure waves in the PH compared with the HC group (p < 0.001) and LC group (p = 0.003) (Fig 16).
Figure 16. Percentage of retrograde pressure waves out of all propagated individual pressure waves in the duodenal segment, in late phase II and postprandially. The PH group is shown with black bars, the LC group with grey bars, and the HC with white bars. *p < 0.01 compared with LC and HC. There was no significant difference between patients with Child-Pugh score A or B when comparing any of the motility variables (data not shown). 4.4.3. Small intestinal bacterial overgrowth SIBO was observed in 4 of the 12 patients in the PH group (33%) but in none of the 12 patients in the LC group (p = 0.09). The following bacteria were cultured: Citerobacter, Escherichia coli, Enterobacter, Enterococci, and Klebsiella; one patient had two different bacterial strains (both Klebsiella and Enterococci). Three of the patients had biopsy-verified atrophic gastritis, and one of them had SIBO. Only one patient was treated with acid

48
suppressive therapy (omeprazole), and this patient did not have SIBO. The only significant difference in motility parameters between patients with and without SIBO was the cycle length of the MMC (SIBO 156.5 ± 63.5 min vs without SIBO 87.8 ± 6.2 min; p < 0.05). There was no difference in the number of clusters or retrograde pressure waves in patients with and without SIBO (data not shown). 4.4.4. Variceal pressure There was no significant difference in the variceal pressures in patients with different etiologies for liver disease. There was a significantly higher variceal pressure in patients with SIBO than without SIBO (25.5 ± 0.5 vs 18.9 ± 1.5 mm Hg; p = 0.015). There was a negative correlation between variceal pressure and motility index in the proximal duodenum postprandially (r = −0.84; p < 0.01), but no other correlation between variceal pressure and motility variables were found (data not shown).

49
5. DISCUSSION 5.1 Epidemiology of liver cirrhosis in Sweden and Iceland The results of the current study show a higher incidence of liver cirrhosis in Gothenburg, Sweden, 15.3/100.000 inhabitants compared to only 3.3 per 100.000 inhabitants in Iceland. There was no time trend in the incidence for neither of the countries during the study period and the incidence in Iceland has decreased by 27% compared to the period 1970-1990 (22). The lower incidence in Iceland is predominantly explained by lower prevalence of alcohol and hepatitis C cirrhosis than in Sweden. The mortality from liver cirrhosis is declining in most western countries in concert with reduction in alcohol consumption (11). We observed a declining incidence of liver cirrhosis in Iceland in spite the fact that alcohol consumption has been increasing in the last decades. In the last 28 years the alcohol consumption has increased by 51% (from 4.33 L/inhabitant 15 years and older in 1980 to 6.52 L in 2003 (153), the increase is 27% from 1991 5.14 L to 2003 6.52 L in Iceland (153) and the increase has continued from 2003-2007 when it was 7.53 (15% in 4 year) but in Sweden the increase has only been 11% from 1991 6.3 L to 2003 7.0 L and then decreased until 2006 when it was 6.8 L (3% decrease in 3 years) (152). Estimation of the total consumption indicates 17% increase from 8.80 L in 1976 to 10.31 in 2003 and then a 5% decrease to 2007 when it was 9.83 L in 2007 (154,155). In Iceland estimates of the total alcohol consumption in the 70s was 4.15 L (156) and in the 80s it was estimated to be 6.22 L (157). Recent estimates of the total alcohol consumption that is the unregistered alcohol consumption in Iceland are not available. Comparisons with other epidemiological studies can be difficult because of different methods used. A few incidence studies from the Nordic countries have been published. A study from Denmark demonstrated an incidence of liver cirrhosis over the period 2001-2005 to be 26.5 in men and 11.8 in women, a high number compared to our results especially because they

50
included only patients with alcoholic liver cirrhosis. The alcohol consumption in Denmark has been stable over the past 30 year around 12 L per inhabitant which is also higher than in Sweden and Iceland as mentioned above (20). In Norway the incidence was in the period 1999-2004 13.4 per 100.000 inhabitants and 53% was due to ALD (21). The results from Sweden in the current study are very similar to these figures, 50% of the Swedish cohort had alcoholic liver disease. The alcohol consumption is also similar in these countries, although somewhat lower in Norway, the average consumption in Norway was 5.8 L per inhabitant over the period 1999-2004 (21). Explanations for the low incidence of liver cirrhosis in Iceland in spite of increase in alcohol consumption could be several. One is that the effect of overconsumption of alcohol on the liver for the development of cirrhosis takes years, it has been estimated to take 16-20 years for men and 10-17 for women (34). Because of these large changes in consumption in Iceland in the last decades and the fact that it is continuing to increase it would be interesting in the future to follow the alcoholic liver cirrhosis incidence in correlation to changes in alcohol consumption over a long period of time. A long follow-time would be needed taking into account the delay from overconsumption to development of cirrhosis. Other explanation could be genetic factors. It has been demonstrated that genetic factors can influence the predisposition to alcoholism and alcoholic liver fibrosis (158). Genetic studies have indicated that the majority of females in the Icelandic founding population had Gaelic ancestry (mostly from Ireland) but the majority of males had Scandinavian ancestry (159). Linkage studies have not demonstrated that chromosomes related to alcoholism in the Irish population are different from Scandinavian or Caucasian chromosomes (160). Genes that are related to alcohol liver damage and fibrosis can affect the development of liver cirrhosis (158,161), no information is available on the prevalence of these genes in the Swedish or Icelandic populations. Analysis of cirrhosis mortality in Western Europe has revealed that in Ireland the prevalence of liver cirrhosis is lower than expected from per capita alcohol consumption (162).

51
Another possible explanation for this low incidence of alcoholic cirrhosis in Iceland is an extensive secondary prevention of alcoholism. In 2002 there were 204/100.000 inhabitants inpatients beds for detoxication, rehabilitation and after-care (163). Alcoholics Anonymous is very strong in Iceland, in the 1990s the number of AA meetings/week /100.000 inhabitants 15 years and older increased from 11 to 14, and it corresponded to the increase in alcohol consumption (163). In Sweden the number of AA meetings/week /10.000 inhabitants 15 years and older is estimated to be 1.5 (164) and another self helping organization Länkarna held 0.3 meetings/week/15 years and older inhabitant (165). It can not be excluded that the more active AA in Iceland compared to AA and Länkarna in Sweden contribute to the difference in alcohol cirrhosis incidence in these countries. It has been demonstrated that the efficacy of AA membership and meeting attendance for the population prevention of consequences of alcoholism is important (166). Another explanation could be a change in alcohol consumption from heavy to moderate drinkers as there were radical changes in Icelandic drinking habits after 1989 when beer was available for the first time since 1915. The proportion of spirits went down in the decade after this change and has been low during the period of our study (167). Mortality form liver diseases has decreased in Sweden over the last decades (18, 168). Increased treatment for alcoholism may also play role here for the reduction observed in alcohol-related harm in Sweden in recent years (169). It is interesting that in the study of liver cirrhosis mortality in Sweden over the period 1969-2001 the decrease in liver-related mortality were associated with reduction in sales of spirits, but there were no change in sales of beer or wine in this period (18). Iceland is a low alcohol consumption country and according to a study on the relationship of alcohol restrictive measures and per capita alcohol consumption there was a strong inverse correlation between alcohol consumption and restrictive measures. Iceland had the second highest restrictive indicator and the lowest consumption which probably contributes to the low incidence of alcoholic cirrhosis in Iceland in spite of increase in consumption in the last decades (168). A study on the drinking habits and

52
beverages consumed in correlation to liver cirrhosis development is difficult to do because the lack of objectivity but could give important information for public health and health care and restrictive measures planning. The proportion of alcoholic liver disease of the whole cohort was 50% in Sweden and that is similar to what was found in a Swedish study from the 1960s where 54% of the cirrhosis were probably of alcoholic etiology (171). Other studies from western countries have demonstrated much higher proportion of alcoholic liver disease. A study from Texas from the 1980s, 90% had ALD (87) and in a study from Stockholm in the same period, 80% were considered to have ALD but this was before the identification of hepatitis C (172). In a Spanish study 85% of the patients were thought to be of alcoholic etiology (173). In southern Europe the etiology is different, over 50% of patients with liver cirrhosis the etiology is hepatitis C (174). 5.2. Characteristics, prognosis and outcome of patients with esophageal varices. In the present study we observed that esophageal varices are more common in males than females (which is the case in patients with liver cirrhosis in general) and that the mean age at diagnosis is around 60 years which is slightly older than in other studies but were the males are also in majority (88, 175,176). When combining the etiology groups ALD and those with ALD and hepatitis C, 52% of the patients had ALD +/- HCV. The majority of patients that were diagnosed with varices under the study period had not had a bleeding episode at diagnosis and only 13% of them had a variceal bleeding during follow-up. It is recommended that patients with liver cirrhosis are screened regularly, every or every other year to find those in need of primary prophylactic therapy (84). In our study every patient underwent an endoscopy but non-invasive methods for the prediction of esophageal varices have been under evaluation, one of the parameters that have been tested are platelet count and spleen size which were promising for

53
hepatitis C cirrhosis but less useful for ALD cirrhosis (177). In a newly published longitudinal study on the predictive value of platelet count on the development of gastroesophageal varices the results indicated that platelet count was inadequate noninvasive marker for predicting the presence or development of varices in cirrhosis, although they found a decreased risk of varices in patients with mild portal hypertension (<10 mmHg) it can not be used as a surrogate marker for changes in portal pressure (178). In our study platelet counts were not a predicting factor for survival but we did not have accurate figures about the spleen size and could therefore not evaluate the combination of these parameters in the current study. It would be useful in the future if there was a reliable and less invasive method than, endoscopy to screen for esophageal varices in patients with cirrhosis. In the present study 65% of the patients received primary prophylactic therapy which is the same as observed in a French study were similar proportion of patient had a bleeding episode (173). After the first variceal bleeding the risk for rebleeding without secondary prophylactic treatment has been observed to be up to approximately 70% (179). In the current study 55% of the patients had a recurrent bleeding although 96% received secondary prophylaxis. The results of our study indicate that it is an independent negative predictor of overall survival to present with a variceal bleeding, and also a strong risk factor for recurrent bleeding The high rebleeding risk of 55% in the current study during a 20 months of follow-up is higher than has been observed in RCT studies of secondary prevention of variceal bleeding. It has been reported to be 29% to 45% after follow-up of 20-25 months in patients receiving beta-blockers, sclerotherapy or ligation (180-182). However, our figures were similar to those found in a meta-analysis of beta-blockers alone as a secondary prophylactic therapy where 61% had a rebleeding during a 25 months follow-up (181). In these RCT and meta-analysis the proportion of patients with CP-class C was lower than in the present study with 40% belonging to CP-class C, and in one of the studies the patients were also younger (182). Our results indicate that in practical clinical medicine the endoscopic follow-up and treatment is less rigorous than in randomized controlled trials.

54
In a prospective study on the prognosis of patients with acute variceal bleeding independent predictors of failure to control bleeding were: CP-class, shock at admission and non-alcoholic cirrhosis (183). In the current study patients who presented with their first variceal bleeding, 9% died in the first bleeding (defined as death within one week from admission to hospital) and 4 of the 8 patients (50%) had ALD. The very limited number of patients makes it difficult to draw any firm conclusions about different risk in different etiologies for unfavorable outcome. But the proportion of 9% is lower than was observed in a French study were the mortality from variceal bleeding 2000 was 14.5% and interestingly in this study the mortality in CP-class A and B was zero for the year 2000 but 32% in CP-class C patients (89). Again our number of patients is probably too small to compare the mortality risk for different CP-classes. The mortality in patients who were diagnosed with varices during their first variceal bleeding was high, 59/92 (64%) died during follow-up and 23/92 (25%) died from variceal bleeding during the mean follow-up of 20 months. Mortality from variceal bleeding has been declining in the last decades, from 42% mortality in 1981 (87,89) to 15-20% in recent years (88,89,176,184,185). According to our results the mortality from variceal bleeding was low for this period 1994-1999 probably due to the improvements in the acute therapy over the last decades including endoscopic sclerotherapy, band ligation, frequent use of vasoactive drugs such as terlipressin, somatostatin and perhaps better treatment in the intensive care units. More frequent use of antibiotics in patients with variceal bleeding has also found to be a contributing factor of a better outcome (89) but we did not systematically study use of antibiotics in the current study. However, the current standard of care is use of antibiotic in association with variceal bleeding. It is interesting that the mortality for patients with liver cirrhosis and esophageal varices is still very high. According to the scheme suggested by the International Consensus Workshop of Baveno IV the yearly mortality

55
risk for group B would be 57% (stage 4) and for group P 3.4% - 20% (stage 2 and 3) (55, 56). In the present study the mortality was 64% for group B and 49% for group P and the mean follow-up time 20 and 25 months respectively. The difference was not as much as would be expected according to the Baveno IV scheme although belonging to group B was one of the negative predictors for survival in the multivariate analysis. In our study the mean CP-score of 8.9 was exactly the same for group B and P and therefore the proportion of patients in each CP-class very similar and that was observed to be one of the independent predictors of survival in the multivariate analysis. It seems that the risk of death is very high once a patient with liver cirrhosis has been diagnosed with esophageal varices. Although the risk of dying from variceal bleeding is much higher for those that have survived a bleeding episode, the risk of dying from liver failure, infections, hepatocellular carcinoma and other malignancies is still very high both for group B and P patients. 5.3. Liver cirrhosis and cancer risk We assessed the risk of HCC and other cancers in patients with liver cirrhosis of different etiologies. We used a retrospective/prospective approach. The patients were identified retrospectively but then followed prospectively. The strength of the study is the quality of the Swedish cancer registry to which the patients data were linked to. It has been evaluated that the registry is almost complete (144,145). As expected we observed an increased risk of HCC in patients with liver cirrhosis, the frequency was 11% under the mean follow-up of 39 months. It is interesting that in a prospective Spanish study on HCC in decompensated liver cirrhosis the follow-up was also 39 months and the frequency was also 11% (186). Most of the patients (62%) were diagnosed within a year from the cirrhosis diagnosis. Probably these patients have had compensated liver cirrhosis for a

56
long time although it is unclear, we only identified patients with the diagnosis in the hospital files. The annual incidence was 6% for patients with hepatitis C cirrhosis and in the rest of the cohort the incidence was 2.7% per year. This high incidence in HCV-induced cirrhosis was although lower than have been observed in a study where the incidence rate was 9% per year for hepatitis C cirrhosis (187). In another study on HCV cirrhosis the incidence was only 3.5 per year (188). The cumulative risk of HCC over 3 years has been reported to be approximately 10.4 for HCV cirrhosis (189). Hepatocellular cancer was significantly more common in patients with HCV compared to other etiologies and that is in agreement with other studies (186,190). There was even a lower risk for patients with ALD than the rest of the cohort to develop HCC similar to that observed in some other studies (186,187,190). Although in a Danish study there was no risk difference for HCC development when comparing different etiologies for liver cirrhosis, but they lacked information about hepatitis status (140). Conflicting results exist concerning the influence of the combination of alcoholic and hepatitis C liver cirrhosis on the development of HCC (187, 191-193). We did no observe any difference in the incidence of HCC in patients with HCV with or without alcohol overconsumptions (20% and 19% respectively) and that is in agreement with some other studies (186,193). These results suggest a direct role of the virus as a carcinogenic agent in HCC development and when a patients already has this very strong risk factor the impact of alcohol overconsumption does not seem to increase the risk. In the present study development of HCC in cirrhosis of other etiologies was 30% for hemochromatosis, 26% in patients with HBV in 20 % with HCV and only in 7% of patients with ALD. There are limited data on HCC in cirrhosis of other etiologies but a high risk has been observed for hemochromatosis and HCV and a lower risk for ALD (186,190). Patients with HCC were significantly older at the time of diagnosis than the rest of the cohort which is in line with previous studies (133, 194, 195).

57
Our results indicate that the overall risk of other malignancies than HCC in patients with liver cirrhosis was 2-3 times higher than that in the general Swedish population. We observed a 13 times increased risk for cholangiocarcinoma and 4-5 times the risk for pancreatic and pulmonary cancers and 3.6 times increased risk for colorectal cancer (CRC). This is somewhat higher risk than was reported in a Danish study although they observed increased risk in patients with cirrhosis for all of the above mentioned cancer types (141). We found no difference in the risk for CRC when comparing patients with ALD vs NALD cirrhosis and that is in agreement with a French study where colonic adenomas were more prevalent in patients with cirrhosis than in those without after controlling for alcoholism (143). Other studies have indicated an increased risk of CRC and high intake of alcohol (196,197) but other increased risk of rectal cancer but not of colon cancer in association with high alcohol intake (198). Studies on the benefit of preventive care in patients with liver cirrhosis have concentrated on liver related complications (199). Our results indicate that it is important to think of malignancies in other organs than the liver when a patient is newly diagnosed with liver cirrhosis. The bases for this increased risk of extrahepatic malignancies in patients with liver cirrhosis is unclear but hypotheses about the reasons for colorectal cancer include differences in the bile acid metabolism, genetic predisposition or immunological disturbances (142). It is also possible that a malignancy that has not yet been diagnosed could have such an impact on the body immunology or homeostasis that it could in some way lead to an earlier presentation of the complications of cirrhosis than would be otherwise. It would be interesting to prospectively study patients with liver cirrhosis and collect data about the risk factor for different types of malignancies unfortunately we lacked these data in the current study because information

58
about these factors such as smoking and other environmental factors were often missing in the medical records. An interesting meta-analysis was published recently and the investigators wanted to answer the question whether a lifestyle modification in the form of reduction in coffee consumption could change the course of liver cirrhosis by influencing the risk of HCC development. According to this meta-analysis results an increased coffee consumption may reduce the risk of liver cancer (200). Further prospective studies to evaluate the optimal dose and comparisons of risk reduction for different preparation of the beans and the brewing method before any concrete recommendations can be done, but the question has been raised: “Does a latte a day keep the hepatologist away”? (201). 5.4 Small intestinal motility disturbances and bacterial overgrowth. Patients with cirrhosis and portal hypertension were found to have abnormal propagation pattern of duodenal individual pressure waves. They had higher frequency of retrograde pressure waves in the proximal duodenum both in phase II of the MMC and postprandially both in comparison with patients with liver cirrhosis without clinical signs of portal hypertension and with healthy subjects. A higher number of long clusters were observed in patients with portal hypertension than in healthy controls. The increased retroperistalsis both in phase II and postprandially could be a restricting factor for the aboral flow of contents in the small intestine as it has been shown that the motility and flow of contents are closely related in the small intestine (202). These motility abnormalities might influence the flow of contents and cause gastrointestinal symptoms. Patients with liver cirrhosis often suffer from malnutrition and they have high frequency of gastrointestinal symptoms and that it is correlated to recent weight loss (90). Patients with encephalopathy have prolonged orocecal transit in comparison with patients with advanced liver disease

59
without encephalopathy (97), the increased retroperistalsis might therefore increase the risk of encephalopathy in these patients. Studies have shown contradictory results concerning the importance of portal hypertension on small intestinal motility disturbances. Portal hypertension in animal model has been shown to change intestinal myoelectrical activity without influencing intestinal propulsion (203). Other animal models have demonstrated delayed orocecal transit in portal hypertension (98) and in human no changes in small intestinal transit in patients with portal hypertension (204). Another study showed delayed small intestinal transit in males but no changes in females in comparison to healthy controls (99). The higher number of long clusters in the portal hypertension group compared to healthy controls is in agreement with results from previous studies on patients with cirrhosis (102-104). A correlation between the severity of the liver disease and clusters in the small intestine has been demonstrated (103) but we found no difference in the number of long clusters comparing patients with CP-A and CP-B. Both that and the high difference of the retrograde pressure waves comparing patients with liver cirrhosis with and without portal hypertension indicates that the portal hypertension per se plays an important role in these motility disturbances. Increased severity of gastrointestinal symptoms in patients with liver cirrhosis has been shown to be associated to impaired health related quality of live (90) and that cluster activity could be a nonspecific response to stress (205). Thus, it could be of interest to investigate further the relationship of motility disturbances, gastrointestinal symptoms and quality of live in patients with liver cirrhosis and portal hypertension. There was a higher prevalence of SIBO in the patients with portal hypertension, 33% of those patients had SIBO whereas none of the patients with liver cirrhosis without portal hypertension.

60
In previous studies on SIBO in patients with liver cirrhosis breath test has been used for the diagnosis of SIBO. In some of them SIBO has been demonstrated (100,101,105) but another study did not demonstrate SIBO in patients with liver cirrhosis and portal hypertension (206). The sensitivity of glucose hydrogen breath test has been shown to be only 62% and the specificity 83% (207). Glucose breath hydrogen test was shown to correlate poorly with microbiological culture of jejunal secretions in patients with liver cirrhosis (208). Thus, the method used in the current study to diagnose SIBO was based on cultures which can be considered the gold standard in the diagnosis of SIBO. The patients with SIBO had higher variceal pressure than those without SIBO which could indicate that portal hypertension directly and/or by causing motility disturbances have a significant role in causing SIBO in these patients. The patients with SIBO had increased cycle length of MMC, theoretically it could predispose to SIBO. Other motility variables were similar in patients with and without SIBO. A study with a higher power is needed to evaluate further this relationship of liver cirrhosis and portal hypertension and SIBO. Although we observed an indication for the increased risk of SIBO in patients with liver cirrhosis and portal hypertension (33%) compared to those without portal hypertension where none of the patients had SIBO it was not a significant difference. Further studies are needed to evaluate the importance of SIBO for the prognosis of patients with liver cirrhosis and portal hypertension. The strong relationship between retrograde pressure waves and portal hypertension should also be investigated further to explore the role of portal hypertension per se on the motility disturbances, probably most accurately by studying intestinal motility before and after transjugular intrahepatic portosystemic shunt.

61
6. CONCLUSIONS ♦ The incidence of liver cirrhosis is still very low in Iceland, and only
24% of that in Gothenburg. Alcoholic liver cirrhosis is uncommon in Iceland. ALD accounts for only 29%-32% of the cases in Iceland but 50%-62% of those in the Swedish population. No trends for increase in the incidence of cirrhosis were observed in neither of these countries despite increase in alcohol consumption during the study period.
♦ ALD accounts for 50% of cases of liver cirrhosis and esophageal
varices in Gothenburg, Sweden. In these patients the mortality is high but etiology does not seem to influence mortality. Majority of patients dies of other complications than variceal bleeding. Most of the patients received primary and secondary prophylactic therapy of proven efficacy.
♦ The risk of HCC is 267 times increased in patients with liver cirrhosis
and the overall risk for other cancers is 2.4 times increased, of those the highest for cholangiocarcinoma but also for esophageal, pancreatic, pulmonary and colorectal cancer. There is an increased risk for HCC in patients with HCV but it is not increased further by excessive alcohol consumption.
♦ Patients with portal hypertension have abnormal propagation pattern
of individual pressure waves, with a high proportion of the pressure waves propagating in a retrograde direction. This pattern was only found in those with portal hypertension but not those with cirrhosis without portal hypertension. This might indicate that portal hypertension per se is related to small bowel abnormalities observed in patients with liver cirrhosis.

62
7. ACKNOWLEDGEMENTS I wish to express my sincere gratitude to those who supported me and helped and made this work possible. In particular I would like to thank: Einar Björnsson, my tutor for excellent supervising. For introducing me to the world of research and sharing with me his extensive knowledge in hepatology. Especially for his enthusiasm in making this work possible and always very fast feedback on my work and for believing in me. Rolf Olsson, my co-tutor, for excellent supervising too. For supporting my and encouraging me in my work and for sharing with me his extensive knowledge in hepatology and for his helpfulness when ever needed. Magnus Simrén, Hasse Abrahamsson, Per-Ove Stotzer, Henrik Sjövall, Evangelos Kalaitzakis and Bjarni Thodleifsson my co-authors for their excellent collaboration and interesting scientific discussions. My other co-authors, Riadh Sadik, Sigurður Ólafsson, Nick Cariglia, Johan Westin and Steven Shev for their collaboration. The excellent staff at the gastrointestinal motility lab Gisela Ringström, Pia Agerforz, Pernilla Jerlstad and Anette Lindh for excellent technical assisstence and helpfulness. My colleagues at the Department of Gastroenterology Anders Kilander, Rolf Gilberg, Inga-Lill Friis-Liby, Hans Strid, Antal Bajor, Anreas Pischel and Iris Posserud. All the patients and the healthy volunteers who participated in the studies. All my friends for their support and encouragement and for making life more fun and easier to live. To my family, my parents, my sisters and their families. They have all of them helped my enormously all my life, encouraged, loved and always been there. Thank you all from all my heart.

63
8. REFERENCES 1. Arey LB, Burrows W, Greenhill JP, Hewitt RM, editors board. Dorland’s
illustrated medical dictionary 23rd edition. Philadlphia: Press of W.B Saunders Companty;1962:286.
2. Duffin JM. Why does cirrhosis belong to Laennec? CMAJ 1987;137:393-396 3. Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The
morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J Clin Pathol 1978;31:395-414.
4. Conn HO. Cirrhosis. In: Diseases of the liver 4th edition. Edited by Schiff L. Philadelphia: J.B. Lippincott Company;1975:833.
5. Millward-Sadler GH, Hahn EG, Wright R. Cirrhosis: An appraisal. In:Liver and biliary disease. Pathophysiology, diagnosis, management. 2nd edition. Edited by Wright R, Millward-Sadler GH, Alverti KGMM, Karran S. London, Bailliére Tindall W.B. Saunders Company1985:821.
6. Iredale JP, Guha IN. The evolution of cirrhosis. In: Textbook of hepatology from basic science to clinical practice. 3rd edition. Edited by Rodés J, Benhamou J-P, Blei A, Reichen J, Rizzetto M. Oxford, Blackwell Publishing 2007:583-589..
7. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199-210.
8. Guha NI, Iredale JP. Clinical and diagnostic aspects of cirrhosis. In: Textbook of hepatology from basic science to clinical practice. 3rd edition. Edited by: Rodés J, Benhamou J-P, Blei A, Reichen J, Rizzetto M. Oxford, Blackwell Publishing 2007:604-622.
9. de Franchis R, Dell’Era A. Non-invasive diagnosis of cirrhosis and the natural history of its complications. Best Pract Res Clin Gastroenterol 2007;21:3-18.
10. Vass A. Rates of liver cirrhosis rise in England, fall in Europe. BMJ 2001;323:1388.
11. Besetti C, Levi F, Lucchini F, Zatonski WA, Negri E, La Vecchia C. Worldwide mortality from cirrhosis: an update to 2002. J Hepatol 2007;46:827-839.
12. Nguyen GC, Segev DL, Thuluvath PJ. Nationwide increase in hospitalizations and hepatitis C among inpatients with cirrhosis and sequelae of portal hypertension. Clin Gastroenterol Hepatol 2007;5:1092-1099.
13. Sørensen HT, Thulstrup AM, Mellemkjar L, Jepsen P, Christensen E, Olsen JH, Vilstrup H. Long-term survival and cause-specific mortality in patients with cirrhosis of the liver: a nationwide cohort study in Denmark. J Clin Epidemiol 2003;56:88-93
14. Haliday ML, Coates RA, Rankin JG. Changing trends of cirrhosis mortality in Ontario, Canada 1911-1986. Int J Epidemiol 1991;20:199-208.
15. Corrao G, Zambon A, Torchio P, Arico S, La Vecchia C, di Orio F and Collaborative Groups for the Study of Liver Diseases in Italy. Attributable risk for symptomatic liver cirrhosis in Italy. J Hepatol 1998;28:608-614.
16. Stroffolini T, Sagnelli E, Almasio P, Ferringno L, Craxi A, Mele A, on behalf of the Italian Hospitals Collaborating Groups. Characteristics of liver cirrhosis in Italy: results from a multicenter national study. Dig Liver Dis 2004;36:56-60.

64
17. Moreau R, DelegueP, Pessione F, Hillarire S, Durand F, Lebrec D, Valla D-C. Clinical characteristics and outcome of patients with cirrhosis and refractory ascites. Liver Int 2004;24:457-464.
18. Stokkeland K, Brandt L, Ekbom A, Ösby U, Hultcrantz R. Morbidity and mortality in liver diseases in Sweden 1969-2001 in relation to alcohol consumption. Scan J Gastroenterol 2006;41:463-468.
19. Simpura J, Tigerstedt C, Hanhinen S, Lagerspetz M, Leifman H, Moskalewicz J, Törrönen J. Alcohol misuse as a health and social issue in the Baltic Sea region. A summary of findings from the Baltic Study. Alcohol Alcohol 1999;34:805-823.
20. Jepsen P, Vilstrup H, Sørensen HT. Alcoholic cirrhosis in Denmark population-based incidence, prevalence and hospitalization rates between 1988 and 2005: a descriptive cohort study. BMC Gastroenterol 2008;8:3.
21. Heukeland JW, Lorgen I, Schreiner LT, Frigstad, S-O, Brandsæter B, Bjøro K, Bang C, Raknerud N, Konopski Z. Incidence rates and causes of cirrhosis in a Norwegian population. Scand J Gastroenterol 2007;42:1501-1508.
22. Ludviksdottir D, Skulason H, Jakobsson F, Thorisdottir A, Cariglia N, Magnusson B, Thjodleifsson B. Epidemiology of liver cirrhosis morbidity and mortality in Iceland. Eur J Gastroenterol Hepatol 1997;9:61-66.
23. Klatsky AL, Armstrong MA. Alcohol, smoking, coffee, and cirrhosis. Am J Epidemiol 1992;136:1246-1257.
24. Corrao G, Bagnardi V, Zambon A, Torchio P. Meta-analysis of alcohol intake in relation to risk of liver cirrhosis. Alcohol Alcohol 1998;33:381-392.
25. Kerr WC, Fillmore KM, Marvy P. Beverage specific alcohol consumption and cirrhosis mortality in a group of English-speaking beer-drinking countries. Addiction 2000;95:339-346.
26. Beeentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Crose L, Sasso F, Pozzato G, Cristianini G, Brandi G, and the Dionysos Study Group. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997;41:845-850.
27. Ramstedt M. Per capita alcohol consumption and liver cirrhosis mortality in 14 European countries. Addiction 2001;96:19-34.
28. Ramstedt M. Alcohol consumption and liver cirrhosis mortality with and without mention of alcohol – the case of Canada. Addiction 2003;98:1267-1276.
29. Batey RG, Burns T, Benson RJ, Byth K. Alcohol consumption and the risk of cirrhosis. Med J Aust 1992;156:413-416.
30. Tuyns AJ, Pequignot G. Greater risk of ascitic cirrhosis in females in relation to alcohol consumption. Int J Epidemiol 1984;13:53-57.
31. Becker U, Deis A, Sørensen TIA, Grønbæk M, Borch-Johnsen K, Müller CF, Schnohr P. Prediction of risk of liver disease by alcohol intake, sex and age: a prospective population study. Hepatology 1996;23:1025-1029.
32. Norton R, Baetey R, Dwyer T, MacMahon S. Alcohol consumption and the risk of alcohol related cirrhosis in women. Br Med J (Clin Res Ed) 1987;295:80-82.
33. Savolainen VT, Liesto K, Männikkö A, Penttilä A, Karhunen PJ. Alcohol consumption and alcoholic liver disease: evidence of a threshold level of effects of ethanol. Alcohol Clin Exp Res 1993;17:1112-1117.
34. Saunders JB, Davis M, Williams R. Do women develop alcoholic liver disease more readily than men? Br Med J 1981;282:1140-1143.

65
35. Becker U, Grønbæk M, Johansen D, Sørensen TI. Lower risk for alcohol induced cirrhosis in wine drinkers. Hepatology 2002;35:868-875.
36. Roizen R, Kerr WC, Fillmore KM. Cirrhosis mortality and per capita consumption of distilled spirits. United States 1947-94.: trend analysis. Br Med J 1999;319:666-670.
37. Leon DA, McCambridge J. Liver cirrhosis mortality rates in Britain from 1950 to 2002: an analysis of routine data. Lancet 2006:367:52-56.
38. Leon DA, McCambridge J. Liver cirrhosis mortality rates in Britain, 1950-2002. Lancet 2006;367:645.
39. Podolsky DK, Isselbacher KJ. Cirrhosis and alcoholic liver disease and major complications of cirrhosis. In: Harrison’s principles of Internal Meidcine 14th edition. Edited by Fauce AS, Braunwald E, Isselbacher KJ, Wilson JD, Martin JV, Kasper DL, Hause SL, Longo DL. Published by McGraw-Hill 1998. 1704-1717.
40. Quigley EMM. Gastrointestinal dysfunction in liver disease and portal hypertension: gut-liver interactions revisited. Dig Dis Sci 1996:41:557-563.
41. Alberino F, Gatta A Amodio P, Merkel C, Di Pascoli L, Boffo G, Caregaro L. Nutrition and survival in patients with liver cirrhosis. Nutrition 2001;17:445-450.
42. matos C, Porayko MK, Francisco-Ziller N, Dicecco S. Nutrition and chronic live disease. J Clin Gastroenterol 2002:35:391-397.
43. Cárdenas A. Hepatorenal syndrome: A dreaded complication of end-stage liver disease. Am J Gastroenterol 2005;100:460-467.
44. Dagher L, Moore K. The hepatorenal syndrome. Gut 2001;49:729-737. 45. Wong F, Girgrah N, Graba J, Allidina Y, Liu P, Blendis L. The cardiac response
to exercise in cirrhosis. Gut 2001;49:268-275. 46. Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and
hepatopulmonary syndrome. Lancet 2004:363:1461-1468. 47. Huffmyer JL, Nemergut EC. Respiratory dysfunction and pulmonary disease
in cirrhosis and other hepatic disorders. Respir Care 2007;52:1030-1036. 48. Møller S, Krag A, Henriksen JH, Bendtsen F. Pathophysiological aspects of
pulmonary complications of cirrhosis. Scand J Gastroenterol 2007:42:419-427. 49. Collier J. Bone disorders in chronic liver disease. Hepatology 2007;46:1271-
1278. 50. Crawford BAL, Kam C, Donaghy AJ, McCaughan GW. The heterogeneity of
bone disease in cirrhosis: a multivariate analysis. Osteoporos Int 2003;14:987-994.
51. Monegal A, Navasa M, Guañabens N, Peris P, Pons F, Martinez de Osaba MJ, Rimola A, Rodés J, Muños-Goméz J. Osteoporosis and bone mineral metabolism disorders in cirrhotic patients referred for orthotopic liver transplantation. Calcif Tissue Int 1997;60:148-154.
52. Porpst A, Propst T, Zangerl G, Ofner D, Judmaier G, Vogel W. Prognosis and life expectancy in chronic liver disease. Dig Dis Sci 1995;40:1805-1815.
53. Gines P, Quintero E, Arroyo V, TEres J, Bruguera M, Rimola A, Caballeria J, Rodes J, Rozman C. Compensated cirrhosis: natural history and prognostic factors. Hepatology 1987;7:122-128.
54. Gattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almsio O, Nevens F, Solinas A, Mura D, Brouwer JT, Thomas H, Njapoum C, Casrin C, Bonetti P, Fuschi P, Basho J, Tocco A, Bhalla A, Galassini R, Noventa F, Schalm SW, Realdi G. Morbidity and mortality in compensated cirrhosis type C: A

66
retrospective follow-up study of 384 patients. Gastroenterology 1997;112:463-472.
55. de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Heptol 2005;43:167-176.
56. D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systemic review of 118 studies. J Hepatol 2006;44:217-231.
57. D’Amico G, Morabito A, Pagliaro L, Marubini E, and the liver study group of “V. Cervello” Hospital. Survival and prognostic indicators in compensated and decompensated cirrhosis. Dig Dis Sci 1986;31:468-475.
58. Ytting H, Møller S, Henriksen JH, Larsen K, Bendtsen F. Prognosis in patients with cirrhosis and mild portal hypertension. Scand J Gastroenterol 2006;41:1446-1453.
59 Pugh RN, Marray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973;60:646-649.
60. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, D’Amico G, Dickson ER, Kim WR. A model to predict survival in patients with end-stage liver disease. Hepatology 2001;33:464-470.
61. Said A, Williams J, Holden J, Remington P, Gangnon R, Musat A, Lucey MR. Model for end-stage liver disease score predicts mortality across a broad spectrum of liver disease. J Hepatol 2004;40:897-903.
62. Kamath PS, Kim WR. The model for end-stage liver disease (MELD). Hepatology 2007;45:797-805.
63. Gacia-Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology 1985;5:419-424.
64. Vallet A, MArleau D, Huet M, Martin F, Farley A, Villeneuve JP, Lavoie P. Hemodynamic evaluation of patients with intrahepatic portal hypertension. Relationship between bleeding varices and the portohepatic gradient. Gastroenterology 1975;69:1297-1300.
65. Rigau J, Bosch J, Bordas JM, Navasa M, Mastai R, Kravetz D, Bruix J, Feu F, Rodés J. Endoscopic measurement of variceal pressure in cirrhosis correlation with portal pressure and variceal hemorrhage. Gastroenterology 1989;96:873-880
66. Casado M, Bosch J, Garcia-Pagan JC, Bru C, Bañares R, Bandi JC, AScorsell A, Rodríguez-Laiz JM, Gilabert R, Fue F, Shorlemer C, Echenagusia A, Rodés J. Clinical events after transjugular intrahepatic portosystemic shunt: correlation with hemodynamic findings. Gastroenterology 1998;114:1296-1303.
67. Bosch J, D’Amico G, Garcia-Pagan JC. Portal hypertension. In: Diseases of the liver 9th edition. Edited by Schiff ER, Sorrell MF, Maddrey WC. Lippincott Williams 6 Wilkins, Philadelphia 2003:428-485.
68. Bosch J, Garcia-Pagan JC. Complications of cirrhosis. 1. Portal hypertension. J Hepatol 2000;32:141-156.
69. González-Abraldes J, García-Pagan JC, Bosch J. Nitric oxide and portal hypertension. Metab Brain Dis 2002;17:311-324.

67
70. Gupta Tk, Tourner M, Ching MK, Groszmann RJ. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation of cirrhotic rats. Hepatology 1998;28:926-931.
71. Colombato LA, Albillos A, Groszmann RJ. Temporal relationship of peripheral vasodilatation, plasma volume, expansion and the hyperdynamic circulatory state in portal-hypertensive rats. Hepatology 1992;15:323-328.
72. Bosch J, Bordas JM, Rigau J, Viola C, Mastai R, Kravetz D, Navasa M, Rodés J. Noninvasive measurement of the pressure of esophageal varices using an endoscopic gauge: the comparison with measurements by variceal puncture in patients undergoing endoscopic sclerotherapy. Hepatology 1986;6:667-672.
73. Nevens F, Sprengers D, Feu F, Bosch J, Fevery J. Measurements of variceal pressure with an endoscopic pressure-sensitive gauge: validation and effect of propranolol therapy in chronic conditions. J epatol 1996;24:66-73.
74. Escorsell A, Bordas JM, Feu F, Garcia-Pagan JC, Gines A, Bosch J, Rodés J. Endoscopic assessment of variceal volume and wall tension in cirrhotic patients: Effects of pharmacological therapy. Gastroenterology 1997;113:1640-1646.
75. Nevens F, Fevery J. Measurements of variceal pressure and its clinical implications. Scand J Gastroenterol 1994;29:6-10.
76. Dell’Era A, Bosch J. Review article: The relevance of portal pressure and other risk factors in acute gastro-oesophageal variceal bleeding. Aliment Pharmacol Ther 2004;20:8-15.
77. Sharara A, Rockey DC. Gastroesophageal variceal hemorrhage. N Engl J Med 2001;345:669-681.
78. Merkel C, Zoli M, Siringo S, van Buuren H, Magalotti D, Angeli P, Sacerdoti D, Bolondi L, Gatta A. Prognostic indicators of risk for first variceal bleeding in cirrhosis: a multicenter study in 711 patients to validate and improve the North Italian Endoscopic Club (NIEC) index. Am J Gastroenterol 2000;95:2915-2920.
79. Schepis F, Camma C, Niceforo D, Magnano A, Pallio S, Cinquegrani M, D’Amico g, Pasta L, Craxi A, Saitta A, Raimondo G. Which patients with cirrhosis should undergo endoscopic screening for esophageal varices detection? Hepatology 2001;33:333-338.
80. Vorobioff J, Groszmann RJ, Picabea E, Gamen M, Villavicencio R, Bordato J, Morel I, Audano M, Tanno H, Lerner E, Passamonti M. Prognostic value of hepatic venous pressure gradient measurements in alcoholic cirrhosis: a 10-year prospective study. Gastroenterology 1996;111:701-709.
81. D’Amico G, Garcia-Pagan JC, Luca A, Bosch J. Hepatic vein pressure gradient reduction and prevention of variceal bleeding in cirrhosis: a systematic review. Gastroenterology 2006;131:1611-1624.
82. Feu F, Garcia-Pagan JC, Bosch J, Luca A, Escorsell A, Rodés J, Terés J. Relation between portal pressure response to pharmacotherapy and risk of recurrent variceal haemorrhage in patients with cirrhosis. Lancet 1995;346:1056-1059.
83. Christensen E, Fauerholdt L, Schilichting P, Juhl P, Poulsen H, Tygstrup N, CLS. Aspects of natural history of gastrointestinal bleeding in cirrhosis and the effect of prednisone. Gastroenterology 1981;81:944-952.
84. D’Amico G, Pagliaro L, Bosch J. The treatment of portal hypertension: a meta-analytic review. Hepatology 1995;22:332-354.

68
85. Poynard T, Cales P, Pasta L, Ideo G, Pascal JP, Pagliaro L, Lebrec D. Beta-adrenergic-antagonist drugs in the prevention of gastrointestinal bleeding in patients with cirrhosis and esophageal varices. An analysis of data and prognostic factors in 589 patients form four randomized clinical trials. Franco-Italian Multicenter Study Group. N Eng J Med 1991;324:1532-1538.
86. Pagliaro L, D’Amico G, Sørensen TI, Lebrec D, Burroughs AK, Morabito A, Tiné F, Politi F, Traina M. Prevention of first bleeding in cirrhosis. A meta-analysisof randomized trials of non-surgical treatment. Ann Intern Med 1992;117:59-70.
87. Graham D, Smith J. The course of patients after variceal hemorrhage. Gastroenterology 1981;80:800-809.
88. Chalasani N, Kahi C, Francois F, Pinto A, Marathe A, Bini EJ, Pandya P, Sitaraman S, Shen J. Improved patient survival after acute variceal bleeding: a multicenter, cohort study. Am J Gastroenterol 2003;98:653-659.
89. Carbonell, N, Pauwels A, serfaty L, Fourdan O, Levey VG, Poupon R. Improved survival after variceal bleeding in patients with cirrhosis over the past two decades. Hepatology 2004;40:652-659.
90. Kalaitzakis E, Simrén M, Olsson R, Henfridsson P, Hugosson I, Bengtsson M, Björnsson E. Gastrointestinal symptoms in patients with liver cirrhosis. Associations with nutritional status and health-related quality of life. Scan J Gastroenterol 2006;41:1464-1472.
91. Quigley EMM. The liver and gastrointestinal disorders. In: Diseases of the liver 9th edition. Edited by Schiff ER, Sorrell MF, Maddrey WC. Published by Lippincott Wliiliams & Wilkons, Philadelphia 2003: 511-527.
92. Zetterman RK. Altered gastrointestinal motility in chronic liver disease. In: Gut and the liver. Edited by Blum HE, Bode CH. UK Kluwer Academic Publishers, 1998:307-315.
93. Norman GA, Atkins JM, Seelig, Gomez-Sanchez C, Krejs GJ. Water and electrolyte movement and mucosal morphology in the jejunum of patients with portal hypertension. Gastroenterology 1980;79:707-715.
94. Stanley AJ, Gilmour HM, Ghosh S, Ferguson A, McGilchrist AJ. Transjugular intrahepatic portosystemic shunt as a treatment for protein-losing enteropathy caused by portal hypertension. Gastroenterology 1996;111:1679-1682.
95. Conn HO. Is protein-losing enteropathy a significant complication of portal hypertension. Am J Gastroenterol 1998;93:127-128.
96. Iber FL. Protein loss into the gastrointestinal tract in cirrhosis of the liver. Am J Clin Nutr 1966;19:219-222.
97. Van Thiel DH, Fagiuoli S, Wright HI, Chien M-C, Gavaler JS. Gastrointestinal transit in cirrhotic patients: effect of hepatic encephalopathy and its treatment. Hepatology 1994;19:67-71.
98. Reilly JA, Quigley EMM, Forts CF, Rikkers LF. Small intestinal transit in the portal hypertensive rat. Gastroenterology 1991;100:670674.
99. Sadik R, Abrahamsson H, Björnsson E, Gunnarsdottir A, STotzer P-O. Etiology of portal hypertension may influence gastrointestinal transit. Scand J Gastroenterol 2003;38:1039-1044.
100. Chang CS, Chen GH, Lien HC, Yeh H-Z. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology 1998;28:1187-1190.

69
101. Madrid AM, Hurtado C, Venegas M, Cumsille F, Defilippi C. Long-term treatment with cisapride and antibiotics in live cirrhosis: effect on small intestinal motility, bacterial overgrowth, and liver function. Am J Gastroenterol 2001;96:1251-1255.
102. Chesta J, Defilippi CL, Defilippi C. Abnormalities in proximal bowel motility in patients with cirrhosis. Hepatology 1993;17:828-832.
103. Madrid AM, Cumsille F, Defilippi C. Altered small bowel motility in patients with liver cirrhosis depends on severity of liver disease. Dig Dis Sci 1997;42:738-742.
104. Madrid AM, brahm J, Buckel E, Silva G, Defilippi C. Orthotopic liver transplantation improves small bowel motility disorders in cirrhotic patients. Am J Gastroenterol 1997;92:1044-1045.
105. Morencos FC, Castano GH, Ramos LM, Arias MJL, Ledesma F, Romero FP. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig Dis Sci 1996;41:552-556.
106. Bauer TM, Steinbrückner B, Brinkmann FE, Ditzen AK, Schwacha H, Aponte JJ, Pelz K, Kist M, Blum HE. Small intestinal bacterial overgrowth in patients with cirrhosis: prevalence and relation with spontaneous bacterial peritonitis. Am J Gastroenterol 2001;96:2962-2967.
107. Pardo A, Bartoli R, Lorezo-Zuniga V, Planas R, Vinado B, Riba J, Cabré E, Santos J, Luque T, Ausina V, Gassull MA. Effect of cisapride on intestinal bacterial overgrowth and bacterial translocation in cirrhosis. Hepatology 2000;31:858-863.
108. Rangari M, Sinha S, Kapoor D, Mohan JC, Sarin SK. Prevalence of autonomic dysfunction in cirrhotic and noncirrhotic portal hypertension. Am J Gastroenterol 2002;97:707-713.
109. Miyajima H, Nomura M, Muguruma N, Okahisa T, Shibata H, Okamura S, Honda H, Shimizu I, Harada M, Saito K, Nakaya Y, Ito S. Relationship among gastric motility, autonomic activity, and portal hemodynamics in patients with liver cirrhosis. J Gastroenterol Hepatol 2001;16:646-659.
110. Ramachandran A, Balasubramanian KA. Intestinal dysfunction in liver cirrhosis: its role in spontaneous bacterial peritonitis. J Gastroenterol Hepatol 2001;16:607-612.
111. Llovet JM, Bartoli R, Planas R, Cabré E, Jimenez M, Urban A, Ojanguren I, Arnal J, Gassull MA. Bacterial translocation in cirrhotic rats: its role in the development of spontaneous bacterial peritonitis. Gut 1994;35:1648-1652.
112. Sorell WT, Quigley EMM, Jin G, Johnson TJ, Rikkers LF. Bacterial translocation in the portal hypertensive rat: studies on basal conditions and on exposure to hemorrhagic shock. Gastroenterology 1993;104:1722-1726.
113. Garcia-Tsao G, Lee FY, Barden GE, Cartun R, West AB. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology 1995;108:1835-1841.
114. Garcia-Tsao G, Albillos A, Barden GE, West AB. Bacterial translocation in acute and chronic portal hypertension. Hepatology 1993;17:1081-1085.
115. Llovet JM, Bartoli R, March F, Planas R, Vinado B, Cabré E, Arnal J, Coll P, Ausina V, Gassull MA. Translocated intestinal bacteria cause spontaneous bacterial peritonitis in cirrhotic patients molecular biological evidence. J Hepatol 1998;28:307-313.

70
116. Parlesak A, Schafer C, Schutz T, Bode C, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 2000;32:742-747.
117. Campillo B, Pernet P, Baries PN, Richardet JP, Devanlay M, Aussel C. Intestinal permeability in liver cirrhosis: relationship with severe septic complications. Eur J Gastroenterol Hepatol 1999;11:755-759.
118. Pascual S, Sch J eseban Á, Zapater P, Casellas JA, Aparicio JR, Girona E, Gutiérrez A, Carnices F, Plazón JM, Sola-Vera J, Pérz-Mteo M. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 2003;50:1482-1486.
119. Ersöz G, Aydin A, Erdem S Yuksel D, Akarca U, Kumanlioglu K. Intestinal permeability in liver cirrhosis. Eur J Gastroenterol Hepatol 1999;11:409-412.
120. Fujii T, Seki T, Maruoka M, Tanaka J, Kawashima Y, Watanabe T, Sawamura T, Inoue K. Lactulose-L-rhamnose intestinal permeability test in patients with liver cirrhosis. Hepatol Res 2001;19:158-169.
121. Huglo D, De Botton S, Canva-Delcambre V, Colombel JF, Wallaert B, Steinling M, Marchandise X. Simultaneous determination of pulmonary and intestinal permeability in patients with alcoholic liver cirrhosis. Eur J Nucl Med 2001;28:1505-1511.
122. Kalaitzakis E, Johansson J-E, Bjarnason I, Björnsson E. Intestinal permeability in cirrhotic patients with and without ascites. Scand J Gastroenterol 2006;41:326-330.
123. Deschenes M, Villeneuve JP. Risk factors for the development of bacterial infections in hospitalized patients with cirrhosis. Am J Gastroenterol 1999;94:2193-2197.
124. Bernard B, Cadraner JF, Valla D, Escolano S, Jrlier V, Opolon P. Prognostic significance of bacterial infection in bleeding cirrhotic patients: a prospective study. Gastroetnerolgy 1995;108:1828-1834.
125. Lopez-Talavera JC, Merrill WW, Groszmann RJ. Tumor necrosis Factor-α: a major contributor to the hyperdynamic circulation in pre-hepatic portal hypertensive rats. Gastroenterology 1995;108:761-767.
126. Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology 2004;127:S5-S16.
127. El-Serag HB. Hepatocellular carcinoma: Recent trends in the united states. Gastroenterology 2004;127:S27-S34.
128. Simonetti RG, Cammá C, Fiorello F Politi F, D’Amico G, Pagliaro L. Hepatocellular carcinoma. A worldwide problem and the major risk factors. Dig Dis Sci 1991;36:962-72.
129. Colombo M, de Franchis R, Del Ninno E, Sangiovanni A, De Fazio C, Tommasini M, Donato MF, Piva A, Di Carlo V, Dioguardi N. Hepatocellular carcinoma in Italian patients with cirrhosis. N Engl J Med 1991;325:675-680.
130. Donato MF, Arosio A, Del Ninno E, Ronchi G, Lampertico P, Morabito A, Balestrieri MR, Colombo M. High rates of hepatocellular carcinoma in cirrhotic patients with high liver cell proliferative activity. Hepatology 2001;34:523-528.
131. Donato F, Bofetta P, Puoti M. A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int J Cancer 1998;75:347-354.

71
132. Tradati F, Colombo M, Mannucci PM, Rumi MG, De Fazio C, Gamba G, Ciavarella N, Rocino A, Morfini M, Scaraggi A, Taioli E. A prospective multicenter study of hepatocellular carcinoma in Italian hemophiliacs with chronic hepatitis C. Blood 1998;91:1173-1177.
133. Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H, Kawashima T. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993;328:1797-1801.
134 Takano S, Yokosuka O, Imazeki F, Tagawa M, Omata M. Incidence of heptocellular carcinoma in chronic hepatitis B and C: a prospective study of 251 patients. Hepatology 1995;21:650-655.
135 Shibata M, Morizane T, Uchida T, Yamagami T, Onozuka Y, Nakano M, Mitamura K, Ueno Y. Irregular regeneration of hepatocytes and risk of hepatocellular carcinoma in chronic hepatitis and cirrhosis with hepatitis C virus infection. Lancet 1998;351:1773-1777.
136 Benvegnú L, Fattovich G, Noventa V, Tremolada F, Chemello L, Cecchetto A, Alberti A. Concurrent hepatitis B and C virus infection and risk of hepatocellular carcinoma in cirrhosis: a prospective study. Cancer 1994;74:2442-2448.
137 Bruno S, Silini E, Crosignani A, Borzio F, Leandro G, Bono F, Asti M, Rossi S, Larghi A, Cerino A, Podda M, Mondelli MU. Hepatitis C virus genotypes and risk of hepatocellular carcinoma in cirrhosis: a prospective study. Hepatology 1997;25:754-758.
138 Romeo R, Rumi MG, Del Ninno E, Colombo M. Hepatitis C virus genotype 1b and risk of hepatocellular carcinoma. Hepatology 1997;26:1077.
139. Bell H, Jahnsen J, Kittang E, Raknerud N, Sandvik L. Long-term Prognosis of Patients with Alcoholic Liver Cirrhosis: a 15-Year Follow-up Study of 100 Norwegian Patients Admitted to One Unit. Scand J Gastroenterol 2004;39:858-863.
140. Sørensen HT, Friis S, Olsen JH, Thulstrup AM, Mellemkjaer L, Linet M, Trichopoulos D, Vilstrup H, Olsen J. Risk of liver and other types of cancer in patients with cirrhosis – a nation wide cohort study in Denmark. Hepatology 1998;28:921-925.
141. Kobayashi M, Ikeda K, Saitoh S, Suzuki F, Tsubota A, Suzuki Y, Arase Y, Murashima N, Chayama K, Kumada H. Incidence of primary cholangiocellular carcinoma of the liver in Japanese patients with hepatitis C virus-related cirrhosis. Cancer 2000;88:2471-2477.
142. Naveau S, Chaput JC, Bedossa P, Poynard T, Pauphilet C, Ink O, Houdayer C, Aubert A. Cirrhosis as an independent risk factor for colonic adenomas. Gut 1992;33:535-540.
143. El-Serag HB, Rudolph KL. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007;132:2557-2576.
144. Mattsson B, Rutqvist L, Wallgren A. Undernotification of diagnosed cancer cases to the Stockholm Cancer Registry. Int Epidemiology 1985;14:64-69.
145. Helgesson O, Bengsson C, Lapidus L, Marck C, Sparén P. Malignant disease observed in a cohort of women. A validation of Swedish Cancer Registry data. Scand J Soc Med 1994;22:46-49.

72
146. Björnsson ES, Abrahamsson H. Interdigestive gastroduodenal manometry in humans. Indication of duodenal phase III as a retroperistaltic pump. Acta Physiol Scand 1995;153:221-230.
147. Björnsson ES, Abrahamsson H. MMC-related duodenojejunal antegrade and retrograde peristalsis in humans. Neurogastroenterol Motil 1994;6:303-309.
148. Camilleri M. Study of human gastroduodenojejunal motility: Applied physiology in clinical practice. Dig Dis Sci 1993;38:785-794.
149. Stotzer PO, Björnsson ES, Abrahamsson H. Interdigestive and postprandial motility in small intestinal bacterial overgrowth. Scand J Gastroenterol 1996;31:875-880.
150. Stotzer PO, Brandberg A, Kilander A. Diagnosis of small intestinal bacterial overgrowth in clinical praxis: A comparison of the culture of small bowel aspirate, duodenal biopsies and gastric aspirate. Hepatogastroenterolgy 1998;45:1018-1022.
151. Lehmann EL. Testing statistical hypotheses. Wiley, New York 1959:140-143. 152. Statens Folkhälsoinstitut. Swedish National Institute of Public Health (SNIPH).
http://www.fhi.se/statistik Försäljningsstatistik för alkoholdrycker, page 13. 153. The National Statistical Institute of Iceland (Hagstofa Íslands).
http://www.statice.is price and consumption: sales of alcohol 1980-2005. 154. The Swedish Council for information on Alcohol and other Drugs.
Centralförbundet for alcohol- och narkotikaupplysning. www.can.se 155. Centre for Social Research on Alcohol and Drugs, Stockhom University, Sweden.
www.sorad.su.se/alcohol_statisics 156. Thjodleifsson B. Áfengisneysla Íslandinga og áhrif hennar á heislufar.
Manneldismál 1980;2:51-55. 157. Gudmundsdóttir A. Ikke-registrerad alcoholconsumption på Island.
Alcoholpolitik – Tidskrift för nordisk alcoholforskning 1990;7:155-164. 158. Bataller R, North KE, Brenner DA. Genetic polymorphisms and the
progression of liver fibrosis: a critical appraisal. Hepatology 2003;37:493-503. 159. Helgason A, Sigurdardottir S, Nicholson J, Sykes B, Hill EW, Bradley DG,
Bosnes V, Gulcher JR, Ward R, Stefánsson K. Estimating Scandinavian and Gaelic ancestry in the male settlers of Iceland. Am J Hum Genet 2000;67:697-717.
160. Kuo P, Neale MC, Riley BP, Webb BT, Sullivan PF, Vittum J, Patterson DG, Thiselton DL, van den Oord EJ, Walsh D, Kendler KS, Prescott CA. Identification of susceptibility loci for alcohol-related traits in the Irish Affected Sib Pair Study of Alcohol Dependence. Alcohol Clin Exp Res 2006;30:1807-1816.
161. Utsunomiya T, Okamoto M, Wakiyama S, Hashimoto M, Fukuzawa K, Ezaki T, Aishima S, Yoshikawa Y, Hanai T, Inoue H, Barnard GF, Mori M. A specific gene-expression signature quantifies the degree of hepatic fibrosis in patients with chronic liver disease. World J Gastroenterol 2007 21;13:383-390.
162. Ramstedt M. Alcohol-related mortality in 15 European countries in the postwar period. Eur J Popul 2002;18:307-323.
163. Olafsdottir H. Trends in alcohol consumption and alcohol-related harms in Iceland. Nordic studies on alcohol and drugs 2007;24 (suppl.):47-60.
164. Bergmark K, Imgard Eisenbach-Stangl E, Rosequist P. The links and alcoholics anonymous: two AA movements in Sweden. Diversity in unity studies of alcoholics anonymous in eight societies. NAD Publication 1998;33:83.

73
165. Kurube N. Självhjälp och överlevnad. En studie av Länkarna. Stockholm. Stockholms universitet, Institutionen för social arbete – Socialhögskolan. 1997.
166. Smart RG, Mann RE. The impact of programs for high-risk drinkers on population levels of alcohol problems. Addiction 2000;95:37-51.
167. Olafsdottir H. The dynamics of shifts in alcoholic beverage preference: effects of the legalization of been Iceland. J Stud Lcohol 1998;59:107-114.
168. Ramstedt M. Has the impact of population drinking on harm become weaker in Sweden? An analysis of the development oin alcohol consumption and alcohol-related harm in Sweden 1990-2005. Nordic studies on alcohol and drugs 2007;24: (suppl.) 73-83.
169. Romelsjö A. Can an improvement and increase in treatment explain unexpected trends in alcohol-related harm in Sweden? Addiction 2007;102:669-671.
170. Brand DA, Saisana M, Rynn LA, Pennoni F, Lowenfels AB. Comparative analysis of alcohol control polices in 30 countries. PLoS Med 2007;4:e151.
171. Olsson R. The natural history of esophageal varices. Digestion 1972;6:65-74. 172. Söderlund C. Long-term survivors after variceal haemorrhage. Scand J
Gastroenterol 1987;22:665-671. 173. Nidegger D, Ragot S, Berthelémy P, Masliah C, Pilette C, Martin T, Bianchi A,
Paupard T, Silvain C, Beauchant M. Cirrhosis and bleeding: the need for very early management. J Hepatol 2003;39:509-514.
174. D’Amico G, De Franchis R and a cooperative study group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003;38:599-612.
175. Nevens F, Broeckaert L, Rutgeerts P, Van Steenbergen W, Fevery J. The long-term morbidity and mortality rate in a cohort of patients with liver cirrhosis and oesophageal varices. Hepato-Gastroenterol 1995;42:979-984.
176. Arguedas MR, McGuire BM, Fallon MB, Abrams GA. The use of screening and preventive therapies for gastro-esophageal varices in patients referred for evaluation of orthotopic liver transplantation. Am J Gastroenterol 2001;96:833-837.
177. Sen S, Griffiths WJH. Non-invasive prediction of oesophageal varices in cirrhosis. World J Gastroenterol. 2008;14:2454-5
178. Qamar AA, Grace ND, Groszmann RJ, Guadalupe G-T, Jaime B, Burroughs AK, Maurer R, Planas R, Escorsell A, Garcia-Pagna JC, Patch D, Matloff DS, Makuch R for the Portal Hypertension Collaborative Group. Platelet count is not a predictor of the presence or development of gastroesophageal varices in cirrhosis. Hepatology 2008;47:153-159.
179. Burroughs AK, McCormick PA. Natural history and prognosis of variceal bleeding. Baillieres Clin Gastroenterol 1992;6:437-450.
180. D’Amico G, Pragliaro L, Bosch J. Pharmacological treatment of portal hypertension: an evidence-based approach. Semin Liver Dis 1999;19:475-505.
181. Bernard B, Lebrec D, Matthurin P, Opolon P, Poynard T. Propranolol and sclerotherapy in the prevention of gastro-intestinal rebleeding in patients with cirrhosis: a meta-analysis. J Hepatol 1997;26:312-324.
182. Lo GH, Lai KH Cheng JS, Chin MH, Huang HC Hsu PI, Lin C-K. Endoscopic variceal ligation plus nadolol and sucralfate compared with ligation alone for the prevention of variceal rebleeding: a prospective, randomized trial. Hepaotlogy 2000.;32:461-465.

74
183. Abraldes JG, Villanueva C, Bañares R, Aracil C, Catalina MV, Garci A-Pagán JC, Bosch J; Spanish Cooperative Group for Portal Hypertension and Variceal Bleeding. Hepatic venous pressure gradient and prognosis in patients with acute variceal bleeding treated with pharmacologic and endoscopic therapy. J Hepatol 2008;48:229-236.
184. Stokkeland K, Brandt L, Ekbom A, Hultcrantz R. Improved prognosis for patients hospitalized with esophageal varices in Sweden 1969-2002. Hepatology 2006;43:500-505.
185. McCormick PA, O’Keefe C. Improving prognosis following a first variceal haemorrhage over four decades. Gut 2001;49:682-685.
186. Sola R, Alvarez MA, Balleste B, Montoliu S, Rivera M, Miquel M, Crera I, Morillas RM, Coll S, Planas R. Probability of liver cancer and survival in HCV-related or alcoholic decompensated cirrhosis. A study of 377 patients. Liver International 2006;26:62-72.
187. Miyakawa H, Iwumi N, Marumo F, Sato C. Roles of alcohol, hepatitis virus infection, and gender in the development of hepatocellular carcinoma in patients with liver cirrhosis. Alcohol Clin Exp Res 1996;20: (suppl.1):91A-94A.
188. Hutchinson S, Bird SM, Goldberg D. Modeling the current and future disease burden of hepatitis C among injection drug users in Scotland. Hepatology 2005;42:711-723.
189. Ikeda K, Saitoh S, Koida K, Arase Y, Tsubota A, Chayama K, Kumada H, Kaanishi M. A multivariate analysis of risk factors for hepatocellular carcinogenesis: a prospective observation of 795 patients with viral and alcoholic cirrhosis. Hepatology 1993:18:47-53.
190. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004;127:S35-50.
191. Chiba T, Matsuzaki Y, abei M, Shoda J, Aikawa T, Tanaka N, Osuga T. Multivariate analysis of risk factors for hepatocellular carcinoma in patients with hepatitis C virus-related liver cirrhosis. J Gastroenterol 1996;31:255-258.
192. Simonetti, rG, Camma C, Fiorello F, Cottone M, Rapicetta M, Marion L, Fiorention G, Crexi A, Ciccaglione A, Giuseppetti R, Stroffolini T, Pagliaro L. Hepatitis C virus infection as a risk factor for hepatocellular carcinoma in patients with cirrhosis. A case control study. Ann Intern Med 1992;116:97-102.
193. Kuper H, Ye W, Broomé U, Romelsjö A, Mucci LA, Ekbom A, Adami H-O, Trichopoulos D, Nyrén O. The risk of liver and bile duct cancer in patients with chronic viral hepatitis, alcoholism or cirrhosis. Hepatology 2001;34:714-718.
194. Velázquez FR, Rodríguez M, Navascués CA, Linares A, Pérez R, Sotorríos NG, Marínez I, Rodrigo L. Prospective analysis of risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Hepatology 2003;37:520-527.
195. Degos F, Christidis C, Ganne-Carrie N, Farmachidi J-P, Degott C, Guettier C, Trinchet J-C, Bearugrand M, Chevret S. Hepatitis C virus related cirrhosis. Time to occurrence of hepatocellular carcinoma and death. Gut 2000;47:131-136.
196. Thygesen LC, Grønbæk M, Johansen C. Colorectal cancer in Denmark 1943-1997. Dis Colon Rectum 2004;47:1232-1241.
197. Bagnardi V, Blangiardo M, La vecchia C, Corrao G. A meta-analysis of alcohol drinking and cancer risk. Br J Cancer 2991;85:1700-1705.

75
198. Pedersen A, Johansen C, Grønbæk M. Relations between amount and type of alcohol and colon and rectal cancer in a Danish population based cohort study. Gut 2003;52:861-867.
199. Riley TR, Smith JP. Preventive care in chronic liver disease. J Gen Intern Med 1999;14:699-704.
200. Larsson SC, Wolk A. Coffee consumption and risk of liver cancer: a meta-analysis. Gastroenterology 2007;132:1740-1745.
201. Fix OK. Does a latte a day keep the hepatologist away? Hepatology 2008;47:348-451.
202. Kerlin P, Zinsmeister A, Philips S. Relationship of motility to flow of contents in the human small intestine. Gastroenterology 1982;82:701-706.
203. Stewart JJ, Battarbee HD, Farrar GE, et al. Intestinal myo-eletrical activity and transit time in chronic portal hypertension. Am J Physiol 1992;263:G474-479.
204. Madsen JL, Brinch K, Hansen EF, Fuglsang S. Gastrointestinal motor function in patients with portal hypertension. Scand J Gastroenterol 2000;35:490-493.
205. Quigley EMM. Intestinal manometry-technical advances, clinical limitations. Dig Dis Sci 1992;37:10-13.
206. Bac D-J, Swart GR, van den Berg JWO, Silson JHP. Small bowel wall function in patients with advanced liver cirrhosis and portal hypertension: Studies on permeability and luminal bacterial overgrowth. Eur J Gastroenterol Hepatol 1993;5:383-387.
207. Corazza GR, Menozzi MG, Strocchi A, Rasciti L, Vaira D, Lecchini R, Avanzini P, Chezzi C, Gasbarrini G. The diagnosis of small bowel bacterial overgrowth. Reliability of jejunal culture and inadequacy of breath hydrogen testing. Gastroenterology 1990;98:302-309.
208. Bauer T, Schawacha H, Steinbrückner B, Brinkmann FE, Ditzen AK, Kist M, Blum HE. Diagnosis of small intestinal overgrowth in patients with cirrhosis of the liver: Poor performance of the glucose breath hydrogen test. J Hepatol 2000;33:382-386.