lights, camera, actin: divergent roles of β and γ
TRANSCRIPT

i
Lights, Camera, Actin:
Divergent roles of β- and γ-
cytoplasmic actin in vaccinia
virus infection
NOORUL BISHARA MARZOOK
A thesis submitted in fulfillment of requirements for the degree of Doctor of Philosophy
FACULTY OF SCIENCE
SCHOOL OF MOLECULAR BIOSCIENCE
UNIVERSITY OF SYDNEY
2017

ii
TABLE OF CONTENTS
Table of Contents ........................................................................................................... ii
Acknowledgements ....................................................................................................... v
Declaration ................................................................................................................... vii
Abstract ....................................................................................................................... viii
List of Figures ................................................................................................................ x
List of Publications Arising From This Work.............................................................. xi
Abbreviations Used ..................................................................................................... xii
Chapter 1: Introduction ............................................................................................... 1 1.1 The Cytoskeleton ............................................................................................................ 2
1.1.1 The Eukaryotic Cytoskeleton ...................................................................................... 3 1.1.1.1 The Actin Cytoskeleton......................................................................................................... 5 1.1.1.2 Actin Dynamics ..................................................................................................................... 5
1.2 Host-Pathogen Interactions At The Cytoskeleton ......................................................... 9 1.2.1 Knocking On Actin’s Door – Cell Entry ...................................................................... 11
1.2.1.1 Virus-cell surfing ................................................................................................................. 11 1.2.1.2 Clathrin-mediated entry ...................................................................................................... 11 1.2.1.3 Macropinocytosis ................................................................................................................ 13
1.2.2 Viral Revolution – Seizing the Means of Cellular Transportation ............................... 15 1.2.2.1 Intracellular transport .......................................................................................................... 15 1.2.2.2 Intracellular replication........................................................................................................ 16 1.2.2.3 Post-replicative transport and assembly ............................................................................ 17
1.2.3 Pathogen Exit ........................................................................................................... 18 1.2.4 Pathogens Are Doing It For Themselves – Hijacking Actin-Based Motility ................ 19
1.3 Poxviruses ..................................................................................................................... 24 1.3.1 Vaccinia Virus and its Life Cycle ............................................................................... 27
1.3.1.1 Vaccinia virus and the actin cytoskeleton........................................................................... 30 1.4 Project Aims .................................................................................................................. 37
Chapter 2: Materials and Methods ........................................................................... 38 2.1 Building blocks .............................................................................................................. 39
2.1.1 Reagents .................................................................................................................. 39 2.1.2 Cell lines ................................................................................................................... 40 2.1.3 Viruses ..................................................................................................................... 41 2.1.4 Buffers and solutions ................................................................................................ 42 2.1.5 Primary antibodies used for immunoblots ................................................................. 43 2.1.6 Secondary antibodies used for immunoblots ............................................................ 43 2.1.7 Reagents for immunofluorescent staining ................................................................. 44 2.1.8 Primers ..................................................................................................................... 45 2.1.9 Vector constructs made and/or used ........................................................................ 46
2.2 Fantastic viruses and how we use them ...................................................................... 48 2.2.1 Viral infection ............................................................................................................ 48 2.2.2 Transfection .............................................................................................................. 48 2.2.3 Plaque assays .......................................................................................................... 48 2.2.3.1 Plaque picking for virus purification ........................................................................ 48 2.2.3.2 Plaque visualisation ............................................................................................... 48 2.2.3.3 Plaque size measurement ..................................................................................... 49 2.2.4 EEV release assays ................................................................................................. 49 2.2.5 Virus DNA extraction ................................................................................................ 49
2.3 Under the Microscope ................................................................................................... 51 2.3.1 Immunofluorescence assays .................................................................................... 51 2.3.2 Image acquisition ...................................................................................................... 51 2.3.2.1 Wide-field microscopy ............................................................................................ 51 2.3.2.2 Confocal microscopy ............................................................................................. 51

iii
2.3.2.3 Live-cell wide-field microscopy .............................................................................. 52 2.3.3 Image analysis ......................................................................................................... 52 2.3.3.1 Actin tail measurements ........................................................................................ 52 2.3.3.2 Virus particles at the cell surface ........................................................................... 52 2.3.3.3 Measuring virus speed ........................................................................................... 52
2.4 DNA ................................................................................................................................ 53 2.4.1 Polymerase chain reaction (PCR) and cloning .......................................................... 53 2.4.2 Plasmid vector construction ...................................................................................... 53
2.5 Proteins .......................................................................................................................... 55 2.5.1 Bacterial expression of proteins ................................................................................ 55 2.5.2 Protein purification using GST-pull-down assays ...................................................... 55 2.5.3 SDS-PAGE gel electrophoresis ................................................................................ 55 2.5.4 Immunoblot assays for proteins of interest ................................................................ 56
2.6 The Silent Treatment ..................................................................................................... 57 2.6.1 siRNA ....................................................................................................................... 57 2.6.2 siRNA protocol ......................................................................................................... 57
Chapter 3: Developing an optimised VACV gene-tagging method ....................... 58 3.1 Introduction ................................................................................................................... 59
3.1.1 Fluorescent Markers: The Highlights ........................................................................ 61 3.1.2 Fluorescent Labelling Goes Viral: Applications for Virology ...................................... 65 3.1.3 Creating Recombinant VACV ................................................................................... 66 3.1.4 Dominant Selection and Fluorescent Markers – With Their Powers Combined ......... 68 3.1.5 VACV Genes Of Interest ........................................................................................... 71
3.1.5.1 F12L.................................................................................................................................... 71 3.1.5.2 A36R ................................................................................................................................... 71 3.1.5.3 A3L ..................................................................................................................................... 72 3.1.5.4 F1L...................................................................................................................................... 72
3.2 Results ........................................................................................................................... 74 3.2.1 Minimal homology length required for homologous recombination in VACV ............. 74 3.2.2 Designing the recombination vector .......................................................................... 76 3.2.3 TDS vectors containing synthetically designed oligonucleotides provide a rapid and efficient method for recombinant VACV generation ........................................................... 80 3.2.4 Successful creation of recombinant VACV ................................................................ 83 3.2.5 Characterisation of recombinant VACV .................................................................... 86 3.2.6 Recombinant viruses carrying more than one fluorescent tag can be created .......... 88
3.3 Disccussion ................................................................................................................... 90
Chapter 4: Understanding virus-induced cell migration in a natural host ........... 95 4.1 Introduction ................................................................................................................... 96
4.1.1 VACV-Induced Cell Motility ....................................................................................... 96 4.1.2 VACV Protein F11L .................................................................................................. 97 4.1.3 ECTV and Cell Motility .............................................................................................. 98
4.2 Results ......................................................................................................................... 100 4.2.1 ECTV encodes a homolog of VACV protein F11..................................................... 100 4.2.2 Design of TDS vector to create ECTV- ΔF11 .......................................................... 102 4.2.3 Creation of ECTV- ΔF11 ......................................................................................... 104
4.3 Discussion ................................................................................................................... 106
Chapter 5: Divergent roles of β- and γ-actin in VACV-induced actin comet
formation 109 5.1 Section Heading .......................................................................................................... 110
5.1.1 The Role of Actin in VACV Infection ....................................................................... 110 5.1.2 VACV actin-based motility as a model to study actin dynamics .............................. 110 5.1.3 Features of VACV-induced actin comets ................................................................ 112 5.1.4 Cytoplasmic Actin: A Tale of Two Isoforms ............................................................. 115 5.1.5 Actin Isoforms and Intracellular Pathogens ............................................................. 118
5.2 Results ......................................................................................................................... 119 5.2.1 VACV actin comets contain both β- and γ-actin ...................................................... 119

iv
5.2.2 β- and γ-actin are abundant in VACV-induced actin comets in apical and basal regions of the cell ............................................................................................................ 121 5.2.3 Composition of VACV-induced actin comets under cytoplasmic actin knockdown .. 124 5.2.4 Apical-basal location of VACV-induced actin comets does not affect their cytoplasmic actin composition under knockdown ................................................................................ 127 5.2.5 Extent of cytoplasmic actin knockdown is dependent on cell type ........................... 129 5.2.6 Characterising cytoplasmic actin knockdown levels in selected cell types .............. 131 5.2.7 Silencing β-actin attenuates VACV-induced actin comet formation in cells ............. 134 5.2.8 Loss of β-actin reduces VACV-induced actin comet length ..................................... 136 5.2.9 VACV-induced actin comets exhibit greater speed under γ-actin knockdown ......... 138
5.3 Discussion ................................................................................................................... 141
Chapter 6: Divergent roles of β- and γ-actin in VACV spread ............................. 145 6.1 Introduction ................................................................................................................. 146
6.1.1 Actin and VACV Spread ......................................................................................... 146 6.2 Results ......................................................................................................................... 149
6.2.1 Extracellular virus release is reduced under β-actin knockdown ............................. 149 6.2.2 VACV motility to the cell surface is not actin isoform-dependent ............................. 151 6.2.3 Src is recruited to CEV even under β-actin knockdown .......................................... 153 6.2.4 VACV plaque size is significantly larger in cells under β-actin knockdown .............. 155 6.2.5 Expression of GST-bound VCA domain and its non-actin-binding mutant .............. 157 6.2.6 The VCA domain of N-WASP does not show specificity for one actin isoform ........ 160
6.3 Discussion ................................................................................................................... 162
Chapter 7: Conclusions and Future Directions .................................................... 165 7.1 VACV AS A FLUORESCENT CELL BIOLOGICAL MARKER ..................................... 166 7.2 BETA-ACTIN IN VACV INFECTION AND BEYOND .................................................... 168 7.3 INVESTIGATING THE BIOCHEMICAL BASIS FOR BETA-ACTIN DEPENDENCE ON VACV-MOTILITY – A CASE FOR ENA/VASP .................................................................... 170 7.4 CELL MIGRATION IN ORTHOPOXVIRUS INFECTION ............................................... 175
Chapter 8: References ............................................................................................ 177

v
ACKNOWLEDGEMENTS
It would be impossible for me to list, on one page, everyone that deserves to be immeasurably thanked for getting me to this point with my PhD. It may take a village to raise a child, but it definitely requires a sizeable city of excellent people to see someone (in particular, someone like myself) through a PhD. They are the real heroes of this story. Therefore I’ll limit my mentions to those who I think might actually give this a read. Rest assured that everyone else will be thanked in person, in real life, unlike the people mentioned here who will have to merely content themselves with making it into my thesis acknowledgements. I’m kidding, the cheques are in the mail. Firstly to Tim, thank you for accepting me into your eclectic menagerie, I mean…lab. I thought I knew what patience and prescience looked like before I met you, but I guess I was wrong. Your faith in me gave me faith in myself, and my grasp of science is so much greater thanks to you, even though I know you look at my data using your Apple watch. I did not appreciate just how much viruses danced until now (although the fancy new microscope helped), so I thank you for that. Thank you to Dean, for being a genius and making all of this seem so easy. Thank you to Helena, for understanding that it wasn’t, for always making sure I was ok, and for getting me through those countless times I showed up at your desk/house. Thank you to Chris, for all the coerced pep talks I thought I wanted, and the refreshing sass I actually needed. You all make me want to be a better scientist and I am grateful to have followed in your footsteps. To the rest of the Nous Sommes: vous étes pretty rad. Thank you to Anjali for being my constant blackup, Mel for that teaspoon of cement that’s still working its way through my veins, and Caitlin for making it to my second tier… jk you’re my bae for life. Andrew, your fried food addiction kept me going. Thank you for making this lab the second dysfunctional family I always wanted. Thank you to Marj, for always giving me strength and leaving me in stitches, often simultaneously. You will forever remain an inspiration to me. Thanks to Alice for being my amazing gym buddy (and regular buddy!) and thank you Mario for your 11th-hour PyMol magic. To Sharissa for introducing me to your friends β and γ; your help and expertise were invaluable. Thank you to Jaime, for taking the calls that saved my life (Skype and otherwise). Thanks to Kara for making me a better writer, and to James for teaching me it’s ok to be a shit one. To Imran and Asmara, thank you for opening up your arms and homes, for giving me a space to write, and for putting food in my stomach. I will always be grateful for your years of support. Thank you to Wapa for making me stubborn enough to see this thing through. Many many many thank yous to Byron, for being foolish enough to love someone who’s finishing their PhD. For all your late night visits to the lab, your meals-on-heels, and for being the buffest little baby, I am forever obliged to share my chicken bones with you, I suppose. I really don’t think I could’ve done this without you. And finally, thank you to ‘science’, for giving me a reason to keep pushing, and to ‘comedy’, for giving me the tools to.

vi
Ok, I lied, this is going to take two pages. There is no way I could hope to articulate the
thanks my mother deserves for every single thing she’s done for me. I literally and
metaphorically would not be here if it weren’t for her relentless determination, kindness,
generosity, and love. I would like to dedicate this work to her.
For Umma, Forever Ago

vii
DECLARATION
The work presented in this thesis is, to the best of my knowledge and belief, original except as acknowledged in the text. All assistance, particularly in the published work, is acknowledged in the appropriate chapters within the text.
I hereby declare that I have not submitted this material, either in full or in part, for a degree at this or any other institution.
N. B. Marzook

viii
ABSTRACT
Viruses and other intracellular pathogens require access to host cells for their replication
and spread. The actin cytoskeleton represents a physical barrier to these organisms,
although many have evolved various ways to circumvent, or even hijack, this system to their
advantage. Vaccinia virus (VACV) is one such organism that is capable of manipulating the
host actin cytoskeleton to facilitate virus dissemination. It is capable of expediting its own
movement out of cells by nucleating actin beneath virus particles, creating F-actin ‘tails’ or
comets that propel virions across the cell surface.
VACV is a dsDNA virus of the Poxviridae family, and was the live vaccine used in the
eradication of smallpox. It is often used as a model organism for studying virus-host
interactions; its large genome and virion size render it highly amenable to genetic
manipulation and fluorescent live-cell microscopy, respectively. The tagging of VACV
proteins with fluorescent proteins has been an indispensable approach to further
understanding of not only virus-host interactions, but also for teasing apart host molecular
mechanisms, particularly within pathways of actin dynamics.
To this end, we developed a novel, optimised protocol for generating recombinant VACV.
After determining the minimal requirements for targeted homologous recombination during
VACV replication, recombinant vector generation was simplified. We coupled this with the
method of Transient Dominant Selection (TDS) using metabolic selection and fluorescent
reporter screening, to streamline the rapid and modular generation of poxviruses expressing
fluorescently labelled virus and/or host proteins. In particular, we used this method to
generate a recombinant VACV capable of expressing Lifeact-GFP, a fluorescent marker
capable of highlighting F-actin on infection, thus enabling the live tracking of VACV comets
using real-time fluorescence microscopy.
VACV can also induce motility of infected cells to enhance viral spread. We attempted to
create a recombinant ectromelia virus (ECTV, the causative agent of mousepox) lacking
F11, the viral protein responsible for virus-induced cell motility, while also expressing
Lifeact-GFP. VACV with an F11 truncation was found to fare poorly in infectious mouse
models, and we therefore aimed to re-create this experiment with ECTV in its natural host.
Unfortunately the F11L gene proved to be reticent to easy genetic manipulation.

ix
Finally, we undertook a closer examination of F-actin in VACV-induced actin-based motility.
F-actin is composed of two isoforms in the cytoplasm: β- and γ- cytoplasmic actin. Despite
differing only by four amino acids at the N-terminus, recent studies have outlined distinct
distributions and functions for both isoforms in normal cellular processes. We employed
recently developed monoclonal antibodies to β- and γ-actin, as well as specific siRNA
knockdown techniques to examine the distribution and role of the two isoforms in VACV-
induced actin comets. Initiation of actin comet formation appears to have an essential
requirement for β-actin, the knockdown of which results in reduced length and number of
actin comets, as well as reduced virus release. Conversely, speed of virus movement was
enhanced when γ-actin was silenced, indicating a moderating effect on the rate of actin
polymerisation by this isoform. We aimed to narrow down the cause of the dependency on
β-actin for VACV actin-based motility by specific pull-down assays, however a clear answer
was not forthcoming.
This study represents the first investigation into the role of individual actin isoforms in actin-
based motility, and implicates the importance of the relative distribution of these two
isoforms in initiating VACV-induced actin comet formation. Further study may underpin the
importance of β-actin over γ-actin in other pathogens that also employ actin-based motility,
and may provide an answer for limiting actin-assisted spread of intracellular pathogens.

x
LIST OF FIGURES
Three biopolymers make up the eukaryotic cytoskeleton. ................................................... 4 Modes of actin nucleation. ................................................................................................... 7 HIV particles move along filopodia towards T-cells .......................................................... 10 SGIV engages with actin-rich protrusions on the cell surface during entry. ...................... 14 Pathogens exploiting actin-based motility. ......................................................................... 21 The life cycle of VACV ..................................................................................................... 28 Signaling pathways used by VACV to initiate microtubule- (left) and actin-based (right)
motility. ................................................................................................................................................. 32 Plasmid vector restriction maps. ......................................................................................... 54 Range of available monomeric fluorescent proteins .......................................................... 62 Method of transient dominant selection ............................................................................. 69 Quantitative analysis of recombination efficiencies between recombinant vectors and the
VACV genome ...................................................................................................................................... 75 Creating the Transient Dominant Selection (TDS) recombination vector ......................... 77 Map of synthetic oligonucleotide carrying homology regions for fluorescent gene
insertion. ................................................................................................................................................ 79 Outline of the experimental procedure to create recombinant VACV using TDS. ............ 82 Recombinant viruses created using modified TDS recombination. ................................... 85 Characterisation of recombinant VACV. ........................................................................... 87 Creation of recombinant Lifeact-GFP/RFP-A3 VACV. .................................................... 89 Comparison of F11 orthologs in VACV and ECTV. ....................................................... 101 Creation of the TDS vector to make ECTV-ΔF11 ........................................................... 103 Creation of an ECTV-ΔF11 virus. .................................................................................... 105 Comparison of truncated sequences in ECTV-ΔF11 and VACV-ΔF11. ......................... 107 Incorporation of G-actin into VACV-induced actin comets occurs at the virus surface. . 113 Differences in cytoplasmic actin isoforms. ...................................................................... 115 VACV actin comets contain both β- and γ-actin. ............................................................. 120 Composition of VACV actin comets created throughout a cell. ...................................... 121 Distribution of β- & γ-actin in VACV comets under actin knockdown. .......................... 126 Composition of VACV actin comets under actin knockdown throughout a cell. ............ 128 β- and γ-actin knockdown efficiency differs with cell type. ............................................ 130 Effect of actin knockdown on chosen cell lines. .............................................................. 133 Production of VACV-induced actin comets during actin knockdown. ............................ 135 VACV actin comet lengths under actin knockdown. ..................................................... 137 Live-cell analysis of actin comet speed under actin knockdown. .................................. 139
Actin nucleation cascade inititated by A36. ..................................................................... 148 EEV release under actin knockdown. ............................................................................... 150 Effect of actin knockdown on VACV motility to the cell surface. .................................. 152 Src is recruited to CEV irrespective of actin knockdown. ............................................... 154 VACV plaque size under actin knockdown. .................................................................... 156 Production and purification of GST-tagged VCA and VCA RA/RA mutant in bacteria. 159 GST-VCA pull-down assays to determine binding preferences for β- or γ-actin. ........... 161 VASP is important for VACV actin comet formation. .................................................... 171 Alignment of β-actin:profilin:VASP-GAB. ..................................................................... 174 Opposing forces acting on the RhoA signalling pathway can influence the integrity of the
cortical actin cytoskeleton and cell migration. .................................................................................... 176

xi
LIST OF PUBLICATIONS ARISING FROM THIS WORK
Marzook N.B., Latham S., Lynn H., McKenzie, C., Chaponnier, C., Grau G., Newsome T.P. (2017) The divergent roles of beta and gamma actin in vaccinia virus infection. Cytoskeleton 74 (4) pp. 170-183. Marzook, N.B., Newsome, T. P. (2016) Viruses That Exploit Actin-Based Motility for Their Replication and Spread. Handbook of Experimental Pharmacology. Berlin, Heidelberg, Springer Berlin Heidelberg: 1-25. Newsome, T.P. and Marzook N.B. (2015) Viruses that ride on the coat-tails of actin nucleation. Semin Cell Dev Bio (46) pp. 155-63. Marzook N.B., Procter D.J., Lynn H., Yamamoto Y., Horsington J., Newsome T.P. (2014) Methodology for the efficient generation of fluorescently-tagged vaccinia viruses. Journal of Visualised Experiments (83), e51151, doi:10.3791/51151.

xii
ABBREVIATIONS USED
AcMNPV – Autographa californica multiple nucleopolyhedrovirus
Arp2/3 – actin-related protein-2/3 complex
ATCC – American Type Culture Collection
ATP – adenosine triphosphate
CEV – cell-associated enveloped virus
DNA – deoxyribonucleic acid
dpi – days post-infection
ECTV – ectromelia virus
EEV – extracellular enveloped virus
EV – enveloped virus
EVH2 – Ena/VASP homology 2 domain
F-actin – filamentous actin
FBS – foetal bovine serum
FP – fluorescent protein
G-actin – globular/monomeric actin
GAB – G-actin binding domain
GFP – green fluorescent protein
gpt – guanine phosphoribosyl transferase gene
Grb2 – growth factor receptor-bound protein 2
GST – glutathione S-transferase
hpi – hours post infection
HRP – Horse Radish Peroxidase
IFA – immunofluorescence assay
IMV – intracellular mature virus
kDa – kiloDalton
LB – Luria Broth
MOI – multiplicity of infection
MPA – mycophenolic acid
N-WASP – Neural Wiskott-Aldrich syndrome protein
NLS – nuclear localisation sequence
NPF – nucleation promoting factor
PFU – plaque forming unit(s)

xiii
RFP – red fluorescent protein
RhoA – Ras homolog gene family, member A
RNA – ribonucleic acid
SD – standard deviation
SDS – sodium dodecyl sulphate
SDS-PAGE – sodium dodecyl sulphate polyacrylamide gel electrophoresis
SFM – serum-free media
TDS – transient dominant selection
VACV – vaccinia virus
VARV – variola virus
VASP – vasodilator-stimulated phosphoprotein
WH2 – WASP homology 2 domain
WR – Western Reserve strain of VACV

CHAPTER 1: Introduction
The University of Sydney 2016
1
Chapter 1: INTRODUCTION

CHAPTER 1: Introduction
The University of Sydney 2016
2
1.1 THE CYTOSKELETON
Author’s note: Sections of this chapter have been previously
published under two reviews:
Newsome, T.P. and Marzook N.B. (2015). Viruses that ride on the coat-
tails of actin nucleation. Semin Cell Dev Bio (46) pp. 155-63.
Marzook N.B. and Newsome T.P. (2016). Viruses that exploit actin-based
motility for their replication and spread. Chapter in The Actin Cytoskeleton;
Handbook of Experimental Pharmacology, ed. Brigitte Jockusch, Springer
Publishing.
“Nothing happens until something moves” – A. Einstein
The cytoskeleton is a dynamic network of biopolymers tasked with giving a cell its shape
and connecting it with its external environment, enabling it to move, and providing a
scaffold that anchors everything else within. To study the cytoskeleton is to study its
flexibility, as it is predisposed by its very organisation to be manipulated in many ways
based on a cell’s most pressing task(s) at hand [1].
While the presence of a cytoskeleton was initially thought to be exclusive to eukaryotic
cells, studies over the past 15 years have identified many bacterial and archaeal
proteins homologous to those that comprise the eukaryotic cytoskeleton [2, 3], starting
with the discovery of actin-like filaments in Bacillus subtilis [4]. Since then, bacterial
homologues of almost every class of eukaryotic cytoskeletal proteins have been
discovered, except for the presence of cytoskeletal-associated motor proteins [5]. These
homologues function to maintain cell shape and length, aid in cell division and anchor
other organelle-like structures within [6]. No doubt there is much we have to learn about
the bacterial and archaeal cytoskeletons, however this study will focus on new frontiers
that are as yet unchartered within the eukaryotic cytoskeleton itself.

CHAPTER 1: Introduction
The University of Sydney 2016
3
1.1.1 The Eukaryotic Cytoskeleton
The role of the eukaryotic cytoskeleton is varied and essential to almost all aspects of
cellular function and can only be understood as the sum of a number of different, yet
interconnected and interacting, parts. These parts can be divided into 3 broad
categories, each comprised of different biopolymers (Figure 1.1).
First, there is the microtubule (MT) network, consisting of a tubular polymer made up by
a heterodimer of two isotypes of the protein tubulin (α- and β-tubulin). MTs are primarily
responsible for cargo transport within the cell, although they can also affect cell shape,
motility and mitosis [7-9]. MTs originate under the control of nucleators such as γ-tubulin,
and this is generally considered to occur at a perinuclear microtubule organising centre
(or MTOC) called the centrosome. However more recent studies have discovered the
existence of secondary MTOCs such as the nuclear envelope, the Golgi complex or
even the cell cortex [10]. β-tubulin is capable of hydrolysing GTP during polymerisation
[11], lending itself to dynamic polymerisation events known as ‘dynamic instability’, a
property of microtubules whereby stochastic switching between prolonged phases of
polymerisation and depolymerisation are possible [12, 13]. These stochastic movements
are usually isolated to the growing (or ‘plus’) ends [14] of the microtubule and enables
associations with cell organelles and the cortex [15]. MTs are controlled by several
microtubule associated proteins (MAPs) which serve to stabilise or destabilise the MT
network and/or promote MT function at the plus ends [16]. Additionally, the microtubule
motors kinesin and dynein travel along MTs, carrying cargo to and from its plus ends
respectively [17].
Next are the intermediate filaments (IF), a large and highly diverse protein family [18].
This sets them apart from the tubulins and actins of the microtubule and actin
cytoskeletal networks, where sequence diversity is not as rampant [19]. Structurally,
they consist of α-helical coiled-coil filaments that constitute the major structural element
of eukaryotic cells. IFs are divided into two kinds: cytoplasmic IFs that play a major role
in stabilising cell shape [20], and the nuclear IFs comprised of lamins which are attached
to the inner nuclear membrane and constitute the nuclear lamina [21].

CHAPTER 1: Introduction
The University of Sydney 2016
4
Finally, there is the actin cytoskeleton, whose prominence for this study warrants a more
detailed explanation, which can be found in the section following.
Three biopolymers make up the eukaryotic cytoskeleton.
Each polymer consists of distinct subunits and are controlled by different motor proteins,
however they interact to collectively to determine cell shape, structure and transport. From
[22].

CHAPTER 1: Introduction
The University of Sydney 2016
5
1.1.1.1 The Actin Cytoskeleton
Actin plays an essential role in the function of eukaryotic cells. For example, the cortical
actin network forms a structural and protective barrier to extracellular stresses. In
addition, force-generation by actin polymerisation promotes a variety of processes from
vesicle motility to the deformation of membranes as macromolecule complexes are
passed between the cytoplasm and the outside of the cell [23, 24].
Actin filaments are composed of actin monomers that are expressed from multiple loci
that give rise to six highly conserved actin isoforms: two striated muscle (α-skeletal actin
and α-cardiac actin), two smooth muscle (α- smooth actin and γ-smooth actin), and two
cytoplasmic actins (β- and γ-cytoplasmic actin) [25, 26]. The muscle isoforms exhibit
tissue specific expression, while β- and γ-cytoplasmic actins (henceforth referred to as
β- or γ-actin) are the most abundant in non-muscle cells [27] and often exist in 2:1 ratio
in epithelial cell lines like HeLa and chicken embryo fibroblasts [28]. Recently, increasing
interest has surrounded these two actin isoforms since the discovery of their differing
roles in cell attachment and contraction (β-actin) and cell motility (γ-actin) [29]. These
concepts will be further expanded upon in section 5.1.
1.1.1.2 Actin Dynamics
Briefly, actin exists as G-actin (globular actin, a 43-kDa ATP-ase), or soluble actin
monomers, which can undergo polymerisation — promoted by accessory factors — to
form F-actin (filamentous actin), the insoluble polymer form of actin [30, 31]. The
spontaneous polymerisation of actin is inefficient, as the formation of actin ‘nuclei’
consisting of actin dimers or trimers is kinetically unfavourable [32]. Actin assembly is
initiated by the creation of free ‘barbed’ (or growing) ends on existing filaments by
filament uncapping or severing proteins, or by the de novo nucleation of new actin
filaments. G-actin monomers are sequentially added to the growing barbed end, while
the other end of the filament is referred to as the ‘pointed’ end, from which disassembly
of actin monomers takes place in a process referred to as actin ‘treadmilling’ [33].
Proteins or protein complexes that increase the number of actin filaments are called
actin nucleators, which in turn promote overall polymerisation, after the creation of more

CHAPTER 1: Introduction
The University of Sydney 2016
6
filaments that are available to extend [34]. Figure 1.2 provides an overview of some of
the major actin nucleators. The first class are the formins, a highly conserved family of
proteins which are capable of nucleating and promoting the polymerisation of
unbranched actin filaments [35]. The formin homology 2 (FH2) domain initiates actin
assembly by binding to and stabilising actin dimers and trimers, and remains associated
with the growing barbed end of the actin filament [36]. In addition to stimulating actin
polymerisation, formins such as mDia2 are also implicated in stabilising the microtubule
network [37].
Next, we have the Arp2/3 complex, a stable assembly of 7 polypeptides, two of which
are actin-related proteins Arp2 and Arp3 [38]. Unlike the formins, Arp2/3 binds to the
side of an existing actin filament, nucleating a daughter filament at a 70o angle to the
original in a Y-branch shape [39, 40]. The Arp2/3 complex possesses minimal
biochemical activity on its own, and must be activated by nucleation promoting factors
(NPFs). There are two classes of NPFs: Class I NPFs that are capable of binding to both
Arp2/3 through a central (C) and acidic (A) domain, and G-actin through a WASP-
homology-2 (WH2) domain [41]; and the Class II NPFs which contain an Arp2/3-binding
region but an F-actin- binding domain instead of a G-actin-binding one [38]. WASP (or
N-WASP) and WAVE proteins are examples of class I NPFs, which localise the Arp2/3
complex and G-actin to the site of actin branch formation [42, 43]. These protein families
are sensitive to signalling molecules involved in actin remodelling, such as the Rho
family of GTPases (including Rac, Cdc42 and RhoA) [44-46]. (See section 1.3.1.1.3 for
more details.)
Finally, the third class of actin nucleators includes the Spire proteins. Spire contains four
WH2 domains which binds an actin monomer each, resulting in the formation of an
elongated stable nucleus for the formation of unbranched actin filaments [47]. Like the
Arp2/3 complex, Spire proteins remain attached to the pointed negative end of the
growing actin filament [48].

CHAPTER 1: Introduction
The University of Sydney 2016
7
Modes of actin nucleation.
The spontaneous nucleation of actin monomers to polymerise into filaments is kinetically
unfavourable and hence requires several actin nucleators to enhance this process. Formins
and Spire proteins promote the formation of unbranched actin filaments, while Arp2/3 binds to
existing filaments from which it creates branching daughter filaments at 70o angles. Both
Arp2/3 and Spire remain at the pointed end of newly formed filaments. Figure from [36].
In addition to facilitating movement by force generation, elements can traverse actin
filaments as cargo, similar to the microtubule network. Myosins are a class of motor
proteins that associate with actin filaments and mediate transport along them [49, 50].
There are 18 different classes of myosins known to date, and their functions range from
intracellular transport and endocytosis to cell adhesion and migration [51-53]. Other
players in the actin polymerisation process include actin depolymerisers, actin bundlers,
and filament severing and capping proteins [34]. Therefore, many classes of protein
interact with, or are implicated in, actin-based motility.

CHAPTER 1: Introduction
The University of Sydney 2016
8
Actin polymerisation can be affected at different stages: actin monomers can be
sequestered by the drug latrunculin A (LatA), preventing the formation of actin filaments
by binding G-actin in a 1:1 ratio [54, 55], or growth can be halted by capping the growing
end of actin filaments using cytochalasins (A-E and H), which prevent both the addition
of new monomers and the disassembly of the actin filament at that end [56, 57].
Additionally, drugs such as jasplakinolide specifically block actin filament disassembly,
essentially fixing existing filaments within a cell by halting actin treadmilling [58]. The
various ways pathogens utilise actin in its many forms can be understood by studying
the effects of inhibitors of actin dynamics on virus replication.

CHAPTER 1: Introduction
The University of Sydney 2016
9
1.2 HOST-PATHOGEN INTERACTIONS AT THE CYTOSKELETON
Viruses require entry into, and exit from, host cells for their replication, and hence the
ability to interface with actin is an opportunity to facilitate this process. Many pathogens
have developed both unique and sometimes convergent mechanisms of manipulating
the host actin cytoskeleton and associated machinery [59-64]. This section will highlight
different stages in the replication cycle of several viruses that utilise the actin network to
promote infection, replication and spread.
While some viruses require interactions with actin for a particular stage of their
replication cycle, others rely on actin for multiple events including entry, intracellular
transport and exit. For example, HIV-1 subverts actin remodelling at the cell surface
prior to entry, which concentrates co-receptors CD4 and CXCR4 that are required for
virus entry, while treatment of cells with cytochalasin D prevents the same [65, 66].
Binding of viral gp120 receptors induces localised F-actin rearrangements through a
RhoA-, Rac1-, Arp2/3- and moesin- (a protein that links the plasma membrane to the
actin cytoskeleton)-dependent mechanism [67-69]. While transport of internalised virus
particles towards the nucleus is microtubule-based, this switches to an actin-based
mechanism at the perinuclear region, prior to nuclear entry [70]. Treatment of cells with
latrunculin prior to infection reduces virus cytoplasmic transport leading to an
accumulation of particles in proximity to the plasma membrane. On the other hand,
treatment 1 hour post-infection (hpi) results in an accumulation of particles adjacent to
the nucleus [70]. This indicates a requirement for actin in both cell and nuclear entry.
Other HIV proteins including Gag and Nef also interact with the actin cytoskeleton during
later stages of infection, which is important for viral assembly and/or budding [71-73].
Finally, cell-to-cell transmission of HIV is facilitated by the actin-dependent formation of
virological synapses and/or filopodia [74, 75]. High resolution imaging of budding HIV
particles by cryo-electron tomography reveals a directed arrangement of cortical actin
filaments around budding sites, half of which are associated with F-actin-rich filopodia
[76]. This use of filopodia for viral transport can be followed by the live imaging of HIV-
infected dendritic cells, where virus particles hijack the dendritic–CD4 T cell contacts. As
illustrated in Figure 1.3, newly-formed virus particles are moved along filopodial
trajectories that are pivoted from the dendritic cell surface towards T cells [77].

CHAPTER 1: Introduction
The University of Sydney 2016
10
HIV particles move along filopodia towards T-cells
HIV particles (in white) are present on the tips of filopodia (F-actin in red) produced by
infected dendritic cells. Scale bars are 5 μm. Figure adapted from [77].

CHAPTER 1: Introduction
The University of Sydney 2016
11
1.2.1 Knocking On Actin’s Door – Cell Entry
1.2.1.1 Virus-cell surfing
Actin-rich protrusions called filopodia, which are structures used by cells to interact with
their environment, are exploited by viruses to infect cells [78]. Filopodia exhibit
retrograde actin flow [79, 80] that can be harnessed by viruses to traverse or ‘surf’ the
cell surface prior to internalisation to seek endocytic hotspots [81]. Herpes simplex virus-
1 (HSV-1) induces dendritic filopodia formation in neuronal cells upon infection, which
virus particles bind to, and traverse to reach the cell body [82-84]. This process is actin-
dependent and virus infection induces RhoA and Cdc42 activation [83, 85]. In addition,
treatment of cells with cytochalasin D prior to infection leads to a reduction in cell entry
[84], highlighting the importance of underlying actin dynamics for this process. Similarly,
the Murine leukaemia virus (MLV), the Avian leukosis virus (ALV) and the Human
Papillomavirus type 16 all show similar filopodial ‘surfing’ prior to internalisation [81, 86].
Therefore, for many viruses, this is their first encounter with the actin cytoskeleton and
engaging with filopodia aids in their movement towards the cell body and favourable
centers of endocytosis. Here viruses face further challenges before they access the
intracellular space. These subsequent steps may also be actin-dependent and are
outlined below.
1.2.1.2 Clathrin-mediated entry
Clathrin-mediated endocytosis (CME) occurs via clathrin-coated pits (CCP), specialised
plasma membrane invaginations typically up to 0.2 μm in size [87, 88]. This process is
mediated by adaptor proteins such as AP-2, allowing the CCP to pinch off from the
plasma membrane into the cytosol with the aid of dynamin [89]. Dynamin in turn can
interact with the actin cytoskeleton through its ability to recruit cortactin, a promoter of
actin nucleation and an actin bundler [90]. CME is a major pathway by which the cell
shuttles molecular cargo across the membrane, and a site targeted by many viral (and
some bacterial) pathogens [91]. Movement of clathrin-coated structures towards the
cytosol is accompanied by the recruitment of actin at the site of budding, and actin
polymerisation may provide the mechanical force required to detach and propel these
structures away from the membrane [92, 93]. Myosin VI, an actin-based molecular

CHAPTER 1: Introduction
The University of Sydney 2016
12
motor, localises to CCPs further supporting a role of actin in this process [52]. Although
analysis of CCP formation in the presence of cytochalasin D or latrunculin A reveals that
an intact actin cytoskeleton is required for the sustained assembly of new CCPs [94, 95],
it does not divulge a direct role in the specific events leading to regular CCP creation,
such as their initiation or subsequent endocytosis [94]. However, actin polymerisation is
required for the formation and internalisation of what are known as ‘clathrin coated
plaques’, or more stable clathrin-coated structures which may carry viruses or bacterial
particles [96, 97]. Therefore, actin may only be recruited when the size of the CCP
needs to accommodate large objects (greater than 0.2 μm) and the force-generating
properties of actin polymerisation are then required for vesicle budding and scission [88].
Viruses such as influenza A [98] and Vesicular Stomatitis Virus (VSV) [96] induce CCP
formation following virus-receptor binding. Single particle tracking of lipophilic dye-
labeled influenza viruses and enhanced yellow fluorescent protein (EYFP)-labeled
clathrin enabled the visualisation of clathrin-mediated endocytosis of 65% of internalised
influenza virus particles. The appearance of EYFP-clathrin on the cell surface after viral
binding suggests the de novo formation of CCP at influenza virus particles [98]. Physical
forces exerted by the acto-myosin and microtubule dynamics are required for uncoating
of influenza A virus post-entry [99], highlighting the importance of both in this process.
Finally, eGFP (Green Fluorescent Protein)-tagged actin, Arp3 and cortactin were found
to localise to virus-containing CCPs and the inhibition of actin polymerisation results in
reduced internalisation of VSV [96].
Kaposi’s sarcoma-associated herpesvirus (KSHV), African swine fever virus (ASFV) and
dengue virus (DENV-1) utilise a dynamin-dependent, clathrin-mediated cell entry
pathway, as inhibitors of CCP assembly such as dextrose and chlorpromazine reduce
virus entry and trafficking [100-104]. KSHV also induces a rearrangement of the actin
cytoskeleton almost immediately following infection, with distinct actin filaments or
spikes appearing on the cell surface at 15 minutes post-infection (mpi) in association
with the majority of KSHV particles. In addition, chemically disrupting the actin
cytoskeleton, or regulators of actin nucleation like Rho GTPases, N-WASP and Arp2/3,
reduces the entry and trafficking of virus particles to the nucleus, supporting the
importance of de novo actin nucleation in this process [101].

CHAPTER 1: Introduction
The University of Sydney 2016
13
1.2.1.3 Macropinocytosis
Macropinocytosis is an actin-dependent, growth factor-induced endocytic process that
enables the uptake of extracellular macromolecules and fluid [105, 106]. Unlike CME,
macropinocytosis requires actin cytoskeleton remodelling, as treatment with
cytochalasin D reduces membrane ruffling [107]. Actin-mediated cell surface projections
such as lamellipodia- and filopodia-related membrane ruffling initiates macropinocytosis,
although they do not always result in an endocytic event. In addition, PI3-kinase activity
[108], Na+/H+ exchange pumps and Rac1 and Cdc42 signalling [109] are all involved in
macropinocytosis. Macropinocytosis is able to non-selectively accommodate
endocytosis of large macromolecular complexes (0.2-5 μm) and fluids [110]. As a result,
many larger pathogens exploit this non-receptor mediated process to enter host cells.
Orthopoxviruses such as vaccinia and variola viruses are large, enveloped DNA viruses
that exploit macropinocytosis to gain access to the host cytoplasm. Vaccinia virus
(VACV) produces two morphological distinct infective forms following a replication cycle:
intracellular mature virus and extracellular enveloped virus, both of which enter cells in a
macropinocytic, actin-, PAK1- and Na+/H+ exchange-dependent manner [111-113].
Both forms of the virus induce the formation of cell-wide membrane blebs (containing
Rac1, RhoA, ezrin and cortactin) during entry, which in the case of mature VACV entry,
is triggered by exposed phosphatidylserine in the virus envelope [112]. Uptake by cells
of extracellular fluid marked by Alexa 488-labeled dextran following exposure to virus is
indicative of induction of macropinocytosis activity in infected cells.
Viruses may engage multiple cell entry pathways, possibly to widen their host range or
cell-type tropism. A novel marine Iridovirus, the Singapore Grouper Iridovirus (SGIV)
uses both clathrin-mediated endocytosis and macropinocytosis to enter cells, as
inhibitors of both are capable of reducing the entry of fluorescently labeled virus particles
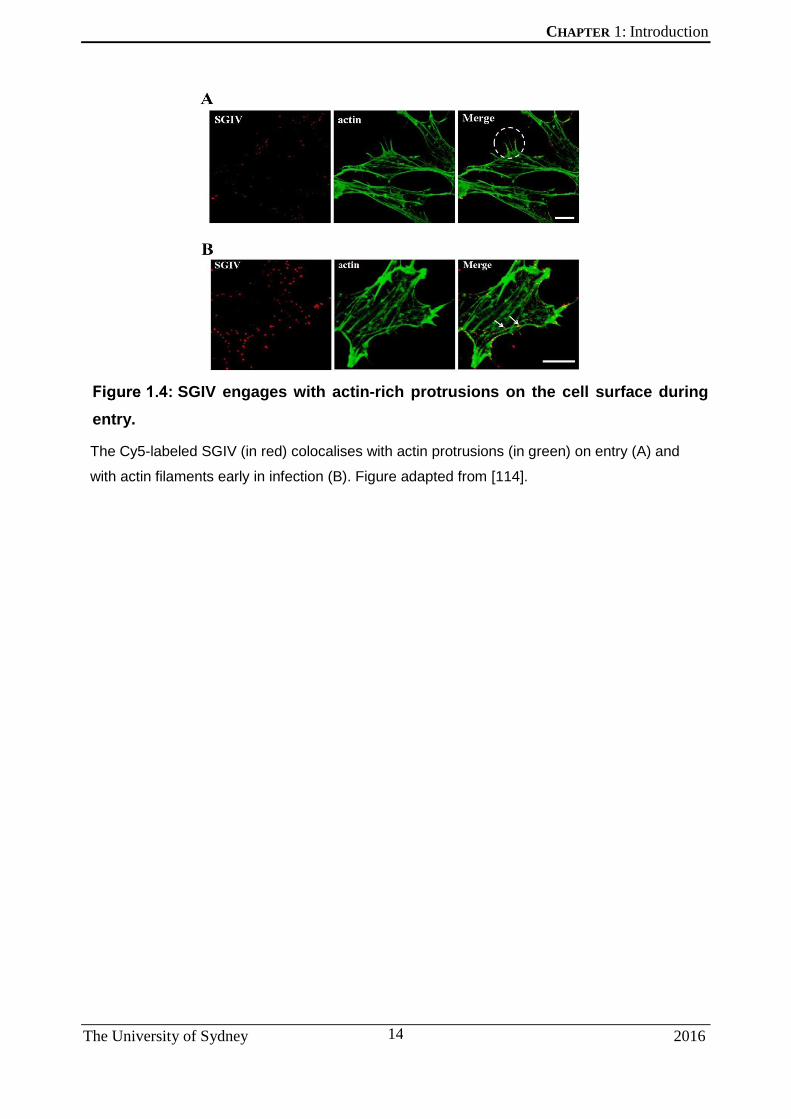
[114]. Interestingly, virus particles were also observed engaging with actin-rich
protrusions on the cell surface during the early stages of viral entry (Figure 1.4).
CHAPTER 1: Introduction
The University of Sydney 2016
14
SGIV engages with actin-rich protrusions on the cell surface during
entry.
The Cy5-labeled SGIV (in red) colocalises with actin protrusions (in green) on entry (A) and
with actin filaments early in infection (B). Figure adapted from [114].

CHAPTER 1: Introduction
The University of Sydney 2016
15
1.2.2 Viral Revolution – Seizing the Means of Cellular Transportation
In addition to microtubules, actin also plays a role in the transport of endocytosed
vesicles away from the cell periphery [115-117]. There are two forms of actin-based
transport within cells. One is based on the acto-myosin network where cargo travels
along actin microfilaments aided by the myosin motor proteins. The other form is based
on highly localised actin polymerisation occurring on the surface of the cargo itself [118].
Following entry via endocytosis, many pathogens, both bacterial and viral exploit the
force-generating reaction of actin polymerisation to aid movement within host cells [119].
Actin-myosin dynamics can also influence various stages of the viral replication cycle,
not only from its movement away from sites of entry but to (and the creation of) regions
of genome replication, progeny assembly, and subsequent return to the plasma
membrane for release. Here we highlight several viruses that exploit both mechanisms
for the completion of their intracellular life cycles.
1.2.2.1 Intracellular transport
Influenza virus displays actin-dependent transport of virus following endocytosis in the
cell periphery (distances within 2 μm from the point of initial virus binding), however this
is superseded by a burst of microtubule-based movement towards the nucleus (the site
of viral RNA synthesis) [120]. On the other hand, intracellular movement of HBV as
imaged by single-particle tracking of labeled surface antigen HBsAg reveals rapidly
moving virus particles that rely on actin- but not microtubule-based motility [121]. This
was revealed by comparing virus movement in cells treated with either cytochalasin D or
nocodazole, inhibitors of the actin- or microtubule-network respectively. In addition,
labeled HBsAg-infected cells transfected with GFP-tagged actin revealed their
colocalisation, confirming the intracellular association of HBV and actin.

CHAPTER 1: Introduction
The University of Sydney 2016
16
1.2.2.2 Intracellular replication
Following delivery of incoming virus to their site of replication, engagement with the actin
cytoskeleton can be used to promote the replication and assembly of progeny virus.
Respiratory syncytial virus (RSV) relies on both actin and profilin (an actin monomer
binding protein) to stimulate the transcriptional activity of RSV polymerase [122]. During
a measles virus infection (MV), the creation of viral replication centers close to the
nucleus is dependent on cofilin, an actin-severing factor [123]. RNA-mediated
knockdown of cofilin decreases ribonucleoprotein (RNP) complex formation and MV
RNA synthesis. Interestingly, the phosphorylation of cofilin, which renders it
enzymatically inactive [124], increases during MV infection suggesting a tight temporal
regulation of activity. The role of cofilin in actin dynamics is a multi-factorial one, as the
severing of actin filaments can both suppress the elongation of existing F-actin
structures but also create sites for branching of new actin filaments via the Arp2/3
complex [125]. Actin severing increases the G-actin pool and cofilin also possesses
actin-nucleating activity. HSV-1 replication in neuronal cells relies on F-actin dynamics,
although this occurs via a bi-phasic process: the cofilin-1-mediated assembly of F-actin
during early stages of entry, followed by the disassembly of F-actin during later stages of
replication [126]. HIV-1 also induces cofilin-mediated actin dynamics to aid in entry and
nuclear localisation of the virus [127]. Therefore cofilin may act as a sensitive regulator
of F-actin dynamics that is targeted by several viruses to aid in various stages of their
replication, and hence shows potential as a novel anti-viral target.
Many viruses replicate, transcribe their genomes and assemble progeny in the nucleus
of host cells. Several viruses engage with actin in the nucleus for successful replication
[128]. In addition to AcMNPV being able to manipulate intracellular actin for its own
motility in the cytoplasm (see section 1.2.4), nuclear F-actin is also essential for
AcMNPV nucleocapsid morphogenesis [129]. P78/83 is a viral WASP-like protein that
interacts with Arp2/3, which translocates into the nucleus following infection [130], along
with monomeric G-actin [131], to induce nuclear actin polymerisation. P78/83 is
stabilised by a further AcMNPV-nucleocapsid protein C42, which is essential for viral-
induced actin polymerisation in the nucleus [132]. AcMNPV VP80 also interacts with
actin in the nucleus and may play a myosin-like role in transport of nucleocapsids to the
nuclear surface [133].

CHAPTER 1: Introduction
The University of Sydney 2016
17
1.2.2.3 Post-replicative transport and assembly
Transport of retroviral RNA such as HIV-1 gag mRNA out of the nucleus is actin-
dependent [134, 135] and β-actin colocalises with nuclear viral RNA ‘tracks’ (curvilinear
structures observed by fluorescence microscopy) [135]. Marburg virus (MARV)
nucleocapsids also travel along, and between, F-actin filaments through the cytosol from
viral replication centers to the plasma membrane [136]. This is facilitated by an actin
cytoskeletal regulator IQGAP1, whose suppression reduces MARV release [137]. Actin-
dependent host motor protein myosin 10 is also co-transported along with mature MARV
nucleocapsids to filopodia, which serve as sites of MARV budding and release [136].
While alpha-herpesviruses such as pseudorabies virus (PRV) and HSV-1 were thought
to rely on nuclear F-actin for transport of nucleocapsids [138], more recent studies refute
this hypothesis [139]. While it is clear that treatment of neuronal or mouse embryonic
fibroblast (MEF) cells with latrunculin A reduces intranuclear capsid motility, Bosse et al
discovered that treating cells with other actin inhibitors such as jasplakinolide (stabilises
actin and stops actin treadmilling) did not replicate phenotype [139]. Infecting MEFs that
stably expressing Lifeact, a live F-actin-binding probe bound to GFP, with capsid-tagged
PRV in the presence of LatA revealed the formation of thick actin rods that also bound to
nucleocapsids in an immunoprecipitation assay, thus preventing capsid motility. This
finding calls into question the use of broad-acting drugs that disturb actin dynamics to
understand the role of actin in viral replication, as it appears that the modes of viral
manipulation of host actin may be more nuanced (both spatially and temporally
controlled, and/or dependent on delicate actin homeostasis) than initially thought.

CHAPTER 1: Introduction
The University of Sydney 2016
18
1.2.3 Pathogen Exit
The final stage in the viral replication cycle is release from the infected host cell. As with
entry, viral egress requires a reckoning of the many barriers to cell exit, particularly in
the case of non-lytic viruses. Actin is necessary for the budding of measles virus (MV)
and respiratory syncytial virus (RSV) particles as the inhibition of actin dynamics
reduces cell-free virus titres, although viral protein synthesis is unaffected [140, 141].
The role of actin in MV release was determined by the use of different actin inhibitors;
cytochalasin D reduced transport of viral capsids (complexes of the MV M protein and
newly formed nucleocapsids) from the nucleus to the plasma membrane, confirming the
requirement for intact actin filaments for this process. Jasplakinolide treatment reduced
virus release but not viral synthesis, supporting a role for actin dynamics in MV particle
budding and release [142]. Here the authors propose an interaction between the M
protein of the measles virus and F-actin, which was subsequently confirmed by
Wakimoto et al by immunoprecipitation of the viral M protein and actin following MV
infection [143]. Interactions between the M protein of other Paramyxoviruses such as
Sendai and Newcastle disease viruses and actin have also been observed [144].
Virus infection can also induce the creation of intercellular connections that facilitate
virus spread. Infectious influenza A virus cores can travel along actin-containing
connections between cells in the absence of budding or release of cell-free virions [145].
Retroviruses such as MLV and HIV-1 also spread by establishing cell–cell filopodial
bridges or conduits, which can be inhibited by disrupting actin dynamics [75, 146],
however the role of actin in this process is distinct from that involved in budding or entry
[75]. Interestingly, prions have also been shown to utilise this actin-dependent method
for spread in neuronal cells [147].

CHAPTER 1: Introduction
The University of Sydney 2016
19
1.2.4 Pathogens Are Doing It For Themselves – Hijacking Actin-Based Motility
We have seen how many viruses are reliant on actin dynamics for their entry, replication
and spread, however a few pathogens have the ability to direct and control actin
dynamics themselves for the purposes of actin-based motility. While many pathogens
have developed several mechanisms of affecting the cytoskeleton (from mimicking
formins, Spire proteins and NPFs, to exploiting tyrosine kinases [148-150]), the use (or
abuse) of the Arp2/3 complex for actin-driven motility has proven particularly useful for
the elucidation of the intricacies of actin dynamics at the molecular level. The propulsion
of pathogens by the localised stimulation of actin nucleation at the pathogen/host
interface has been a powerful research model, leading to significant insights into the
regulation of actin dynamics, as well as deepening our understanding of novel
pathogenesis mechanisms. During normal cellular functioning, actin nucleation is a
highly dynamic and seemingly capricious process. In contrast, the assembly of actin
filaments by bacteria species such as Listeria and Shigella, and viruses like vaccinia
virus (VACV) and the baculovirus Autographa californica multiple nucleopolyhedrovirus
(AcMNPV), is robust and highly localised, while also being amenable to genetic
manipulation. Recent studies have begun to shed light on the role of actin-based motility
as a virulence mechanism in the replication cycle of these pathogens. In fact, the ability
to perturb actin has been proposed as a ‘pattern of pathogenesis’ employed by
infectious microbes that may be recognised by the immune system as a hallmark of
infection [151].
Several intracellular pathogenic bacteria gain access to non-phagocytic cells by
manipulating the actin cytoskeleton. They utilise the Arp2/3 complex to move in an intra-
and inter-cellular manner via actin-based motility on so-called actin comets or tails,
oriented such that their fast-growing ends are directed toward the pathogen, enabling
the rapid spread of infection between cells [119, 152]. Examples of such bacteria that
travel on actin-derived comets include Listeria, Shigella and Rickettsia species. Some of
these organisms encode proteins that interact directly with the Arp2/3 complex, while
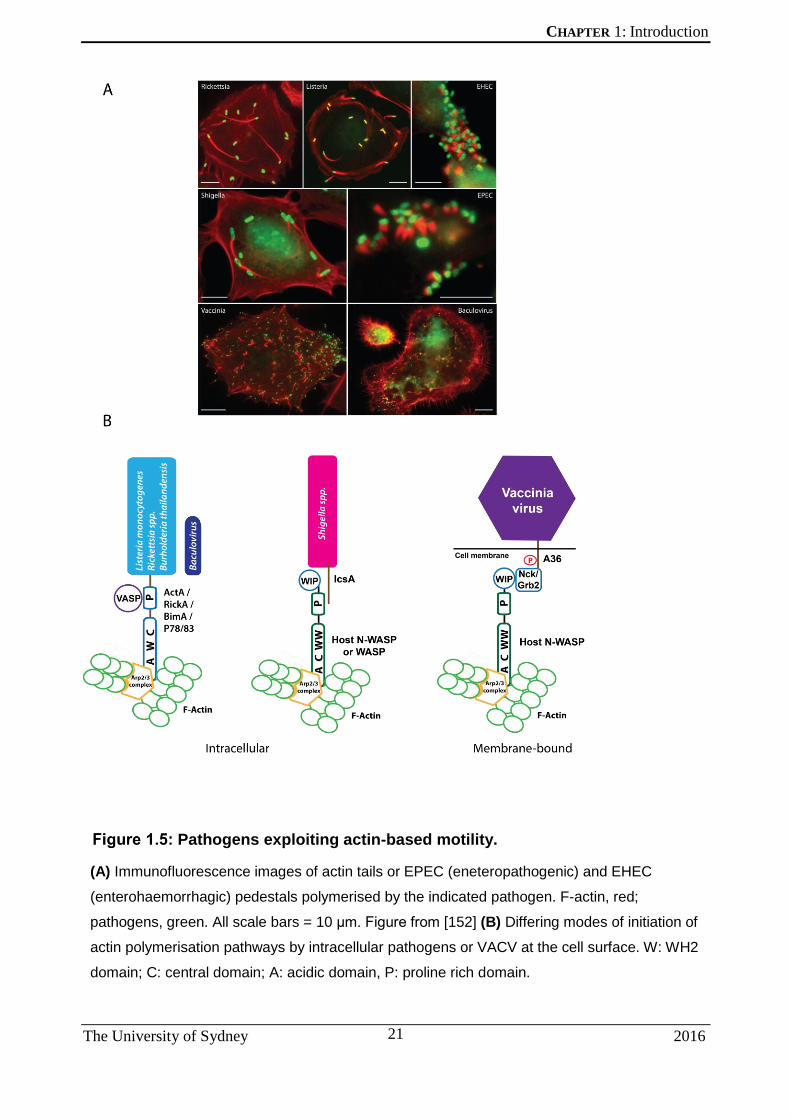
others encode proteins that recruit various host NPFs first. Figure 1.5 depicts examples
of pathogens undergoing actin-based motility, along with providing a brief overview of
the actin-nucleating machinery in some of these organisms. ActA is produced by the

CHAPTER 1: Introduction
The University of Sydney 2016
20
gram-positive Listeria monocytogenes, and was indeed the first ever NPF of Arp2/3 to
be identified [153]. The C-terminal end of ActA possesses a transmembrane domain that
is inserted into the cell membrane, while the N-terminal end has C and A regions
(described previously in section 1.1.1.2), as well as a WH2 domain similar to other
WASP proteins [152]. A proline-rich region (P) enhances actin assembly of actin
filaments beneath the bacterium [154]. ActA paved the way for the discovery of the
Arp2/3 complex, through expression of ActA in fractionated cytoplasmic cell extracts and
identifying the minimum requirements for motility [155, 156]. The actin adaptor protein
Ena/VASP also binds to the P region on ActA and recruits the actin monomer-binding
protein profilin, which enhances bacterial motility [152]. Bacteria that produce similar
NPF mimics containing WH2 homologies capable of activating Arp2/3 include Rickettsia
spp. that produce RickA [157] and Burkholderia thailandensis which produces BimA
[158]. In contrast, the IcsA protein, which is on the outer membrane of gram-negative
Shigella spp. [159], cannot activate the Arp2/3 complex directly, but instead relies on the
recruitment of the cellular NPF N-WASP [160], which then activates the Arp2/3 complex
[161]. IcsA also requires the activity of other host cell-signalling proteins such as Abl
kinase [162] and Toca-1, an activator of N-WASP [163]. On the other hand, actin-based
motility of Listeria using ActA is independent of any regulation by host signalling
pathways [152]. Therefore, it appears as if pathogens developed two methods of Arp2/3
activation: by mimicking NPFs such as activated N-WASP or by recruiting cellular N-
WASP.
CHAPTER 1: Introduction
The University of Sydney 2016
21
Pathogens exploiting actin-based motility.
(A) Immunofluorescence images of actin tails or EPEC (eneteropathogenic) and EHEC
(enterohaemorrhagic) pedestals polymerised by the indicated pathogen. F-actin, red;
pathogens, green. All scale bars = 10 μm. Figure from [152] (B) Differing modes of initiation of
actin polymerisation pathways by intracellular pathogens or VACV at the cell surface. W: WH2
domain; C: central domain; A: acidic domain, P: proline rich domain.

CHAPTER 1: Introduction
The University of Sydney 2016
22
Apart from bacterial pathogens, only one example of intracellular transport mediated by
virally stimulated actin nucleation has been characterised. Autographa californica
multiple nucleopolyhedrovirus (AcMNPV) is a Baculovirus of lepidopterans that initiates
actin polymerisation 5–30 mpi after endocytosis of virus particles [164, 165]. Actin
nucleation by the AcMNPV is akin to that of bacterial intracellular pathogens, in that
motility promotes the exploration of intracellular space and dispersal of progeny [152].
Viral nucleocapsid protein P78/83 is a viral NPF located on one end of the viral particle
and activates the Arp2/3 complex, inducing localised actin nucleation at the virus surface
[130, 166]. On entering a host cell, AcMNPV particles use their actin-driven motility to
either navigate to the nucleus, where uncoating and gene expression can occur, or to
proceed to neighbouring uninfected cells via cell surface spikes. These spikes appear 2
hpi – prior to the creation of virus progeny – and hence the AcMNPV that are present in
these cell spikes must derive from the infecting inoculum [165]. The addition of the
myosin inhibitor butanedione monoxime reduced transport of AcMNPV to the nucleus,
suggesting a role for the actin-myosin network in complementing intracellular transport
of the virus [167]. Thus incoming virus are presented with two alternative routes: the
nucleus for the initiation of replication or the seeking out of cell surface spikes to
facilitate the infection of surrounding cells. The second route is restricted by the onset of
the early expression of the envelope protein GP64, an entry receptor that is incorporated
into nucleocapsids by budding at the plasma membrane. Thus the spread of virus may
be enhanced when cells become infected with a high dose of virus, such as that derived
from an occlusion body. A subset of virus would translocate to the nucleus and initiate
early gene expression, including that of GP64, while a portion of the inoculum would
traverse the infected cell and be passed to adjacent cells.
Finally, the orthopoxvirus vaccinia was found to move by the power of actin
polymerisation on the tips of actin tails, as a means of being projected from the surface
of an infected cell [168, 169]. Infected cells typically exhibit virus-tipped membrane
protrusions that are rich in F-actin and are visible by scanning electron microscopy [170].
The viral protein A36 was implicated in the initiation of this process, however, like IcsA,
required the recruitment of several host signaling molecules and the NPF N-WASP for
the eventual activation of Arp2/3 and initiation of actin polymerisation. The details of this
process are expanded upon in section 1.3.1.1.

CHAPTER 1: Introduction
The University of Sydney 2016
23
While pathogens have developed varying mechanisms for initiating actin nucleation,
methods of actin filament depolymerisation appear to be conserved [119] and reliant on
host accessory proteins. Actin Depolymerising Factor (ADF, or cofilin) and capping
proteins are involved in actin depolymerisation [171] and are also essential for actin-
based motility of Listeria and Shigella, by the maintaining the pool of G-actin available
for incorporation into filaments [155, 172, 173]. Cofilin is also responsible for the
depolymerisation of VACV comets, the RNAi-mediated knockdown of which produces
comets of greater lengths [174].

CHAPTER 1: Introduction
The University of Sydney 2016
24
1.3 POXVIRUSES
Poxviruses (family Poxviridae) are double-stranded DNA viruses that replicate in the
cytoplasm of host cells [175]. The poxvirus family in divided into two subfamilies: the
Entomopoxvirinae and Chordopoxvirinae (which infect insects and chordates
respectively). Each subfamily contains several genera each, which are outlined along
with some examples in Table 1.1.
Table 1.1 Members of the Poxviridae family
Subfamily Genus Examples
Chordopoxvirinae Avipoxvirus
Fowlpox virus
Capripoxvirus Sheeppox virus
Leporipoxvirus
Myxoma virus
Molluscipoxvirus
Molluscum contagiosum virus
Orthopoxvirus Variola virus, cowpox virus, vaccinia virus
Parapoxvirus Orf virus
Suipoxvirus Swinepox virus
Yatapoxvirus Yaba monkey tumour virus
Entomopoxvirinae Alphaentomopoxvirus Melolontha melolontha virus
Betaentomopoxvirus Amsacta moorei virus
Gammaentomopoxvirus Chironomus luridus virus

CHAPTER 1: Introduction
The University of Sydney 2016
25
Poxviruses, along with asfarviruses, iridoviruses and phycodnaviruses, are part of the
large nuclear and cytoplasmic DNA viruses of eukaryotes (NCLDV) [176]. While
considered to be one of the largest of animal viruses [177], the recent discovery of giant
protist viruses such as Mimivirus and other Pandoraviruses has called their relative
magnitudes into question [178-180]. Nevertheless, their large size of 200 – 400 nm
enables them to be visualised by light microscopy, while analysis by electron microscopy
reveals not an icosahedral or helical shape enjoyed by other viruses, but an oval or
brick-shaped virion consisting of a walled biconcave core surrounded by lateral bodies
[181]. This core contains a very large genome, which can vary from 135 to 360 kb based
on all currently sequenced poxviruses, similar to other large DNA viruses [175]. These
genomes are relatively compact with an approximate rate of one gene per 1 kb [182]. Of
these, 49 genes are present in all sequenced poxviruses, while 90 are common to all
chordopoxviruses [182]. These essential genes, involved in replication, transcription,
and assembly are clustered at the centre of the genome, while those genes that provide
host-specificity commonly reside at the flanking regions at either end of the viral genome
[183, 184]. Chordopoxviruses exhibit diverse host ranges and virulence. For example,
variola virus (VARV) only infects, and is highly virulent to, humans, while cowpox
(CPXV) and monkeypox (MPXV) viruses infect a wide range of mammal species [175].
While specific genes known as ‘host range genes’ are necessary for the ability of a
poxvirus to replicate in certain host cells, they exhibit less specificity when it comes to in
vitro entry of cells in tissue culture [185].
Poxviruses are so named for the characteristic feature of the disease produced by the
best known members of the group [186], of which smallpox is the most infamous. VARV
is causative agent of smallpox and is the only human disease to have been successfully
eradicated [187]. Initial attempts to control the spread of smallpox used variolation,
which was the practice of introducing a small amount of infectious material from a
smallpox-infected individual to a healthy one to prevent disease. Variolation was widely
practiced in the East, from where it spread to Europe and finally the U.K. [188]. In 1798,
Edward Jenner popularised the safer practice of using the cowpox virus (CPXV) to
immunise individuals instead (this is where we obtained the term ‘vacciniation’ — ‘vacca’
being the Latin word for cow). Although this was thought to have been subsequently
replaced with the use of vaccinia virus (VACV), whose natural host remains unknown.
Several theories on the origin of VACV exist, including that it may have somehow

CHAPTER 1: Introduction
The University of Sydney 2016
26
derived from co-cultivation of VARV and CPXV by repetitive virus production [188], or
that it originated from the horsepox virus (HSPV) since an infection of horses with VACV
reproduces the clinical signs of HSPV [189]. However since the horsepox virus is
believed to be all but extinct [190], this mystery remains unresolved until now. Still,
poxviruses provide an intriguing case study for the evolutionary origins of not only
double-stranded DNA viruses, but all viruses [191].
Since its eradication, all known VARV stocks were centralised to two maximum-security
laboratories in the US and Russia, although the possibility of the existence of rogue
stocks remains. In addition, with the advent of increasingly accessible and feasible
methods of oligonucleotide synthesis, the assembly of a VARV-like virus is more and
more feasible. Fears of a smallpox recurrence have been stoked with the rise of the
ubiquitous threat of terrorism [192]. Despite the apparent abolition of smallpox, efforts to
completely destroy VARV stocks have been postponed, citing the need for more live
virus experimentation and the development of newer and even safer vaccines in the
event of a smallpox resurgence [193-195]. Indeed if a smallpox outbreak were to occur
now, it would be 25 years since the cessation of worldwide vaccination programs,
leaving whole generations susceptible to this deadly disease. Therefore VACV remains
a widely studied virus, spawning several generations of safer and more stable vaccine
candidates [188]. Currently VACV is studied not just for its use in our immunisation
against smallpox but also as a tool for understanding the fundamentals of cell and
molecular biology [196, 197] and increasingly as a vector for cancer treatment [198,
199]. Additionally, orthopoxviruses pose an increasing risk both in terms of zoonotic
infections as well as transmissible infections in non-immunised humans [200, 201].

CHAPTER 1: Introduction
The University of Sydney 2016
27
1.3.1 Vaccinia Virus and its Life Cycle
VACV is a member of the Orthopoxviridae genus of the Poxviridae family, possessing
the ability to infect a broad range of organisms including humans, cows and rodents
[202]. Recent outbreaks of VACV have identified its presence in dogs and opossums,
raising questions as to whether several animal species are capable of acting as
reservoirs of this virus [203].
Widely used VACV strains that have been sequenced include the Copenhagen (VACV-
Cop) and Western Reserve (VACV-WR) strains (and can be accessed at
www.viprbrc.org). These strains house a 200 kb genome encoding over 200 proteins
[204, 205]. VACV genes were traditionally annotated based on the DNA fragments
produced by a HindIII digest of the entire genome, ranging from A (the largest) to P (the
smallest) [204, 206]. These fragments are subdivided using numbers that denote the
position of the ORF in that fragment in the 5’-3’ direction. Since VACV replicates in the
cytoplasm, its mRNAs are not spliced and the genome does not contain any introns
[181], making this numbering system ideal. Finally, gene names are suffixed with L or R,
to signify if they are read in the left or right direction for transcription and hence protein
names do not contain the L or R suffix. Therefore, for example, the VACV gene A3L is
the third gene located on the largest HindIII restriction fragment, is transcribed from left
to right, and produces the protein A3.
A productive replication cycle of the prototypal VACV strain Western Reserve takes a
minimum of 6-8 hpi to produce two morphologically distinct but mature infectious forms
of the virus [207] (see Figure 1.6 for an overview). Briefly, the two forms of the virus
produced are mature virions (MV), and WV (wrapped virions) or EV (enveloped virions).
The enveloped virions can be further subdivided based on their position relative to the
cell: IEV (intracellular enveloped virions), CEV (cell-associated enveloped virions) and
EEV (extracellular enveloped virions).

CHAPTER 1: Introduction
The University of Sydney 2016
28
The life cycle of VACV
From right to left: virions enter the host by fusion with the cell membrane. Virus cores travel to
the centre of the cell where perinuclear viral factories are set up. VACV DNA is replicated and
rudimentary packages containing viral core components called immature virus (IV) are created.
IV gradually develop into intracellular mature virus (IMV) as VACV DNA is packaged inside and
proteolytic cleavage of core proteins transforms the virions into the characteristic brick-shaped
mature virus (MV). A subset of MV travel along microtubules to attain a secondary membrane
derived from the trans-Golgi network (TGN) or endosomes to produce wrapped or intracellular
enveloped virus (WV/IEV). These travel along microtubules again to fuse with the cell
membrane creating cell-associated extracellular virus (CEV), at which point the nucleation of
actin polymerisation can eject the virus from the cell, creating extracellular enveloped virus or
EEV. Adapted from [207].
The infectious cycle begins with the entry of virus particles by fusion with the cell
membrane [208]. Viral core contents are released into the cytoplasm and travel along
microtubules towards the centre of the cells where viral replication centres or ‘virus
factories’ are established [209]. These factories are perinuclear and create mature virus
(MV) particles, which are the ‘simplest’ infectious particles produced, and are only
released by lysis of the infected cell [181, 208]. These particles have a single
membrane, derived from the endoplasmic reticulum, and comprise over 100 viral

CHAPTER 1: Introduction
The University of Sydney 2016
29
proteins with a range of post-translational modifications [181, 210-212]. A subset of MV
travel from virus factories along microtubules to acquire two additional membrane layers
from the early endosome/trans-Golgi network compartment [213, 214]. These are
referred to as wrapped virus (WV). In acquiring additional membranes of a different
origin to MV, WV possess an additional complement of viral proteins that are integral to,
or associated with, these membranes; these are referred to as WV-specific proteins.
Three WV-specific proteins A36, F12 and E2 recruit and stabilise the microtubule motor
complex kinesin-1 at the cytoplasmic virus surface. This interaction acts to haul virus
cargo from the site of WV wrapping, typically located between the host nucleus and virus
factory, to the cell periphery [215-222]. A36 is a type Ib integral membrane protein of
221 amino acids that lies at the heart of WV transport events, mediating interactions with
both microtubule and actin cytoskeletons [215, 223, 224]. From the N-terminus, a short
transmembrane domain anchors the protein to the WV outer envelope, with the
remainder of the protein protruding into the cytoplasm. Although lacking recognised
domains, two WD/WE motifs associate with the tetratricopeptide repeats (TPR) of
kinesin light chain (KLC), a component of kinesin-1 [221]. Efficient anterograde virus
transport also requires a second pathway, involving a complex of F12 and E2 that also
binds KLC (specifically the KLC-2 isoform) [222]. How the cytoplasmic proteins F12 and
E2 are tethered to the virus is not yet fully understood, but part of the answer may be an
interaction between F12 and A36 [225]. Anterograde transport mediated by kinesin-1
translocates WV to the vicinity of the cell surface. Figure 1.7 (on the left) provides an
overview of the host and viral proteins involved in VACV microtubule-based motility.
Access to the plasma membrane is granted by the cytoplasmic viral protein F11 that
globally downregulates RhoA GTPase signalling, thereby clearing a path for the virus
through the dense cortical F-actin [226]. This occurs via a PDZ domain (a commonly
occurring protein-binding domain, although the first to be discovered in a viral protein) in
F11, which binds to Myosin-9A, an inhibitor of RhoA signalling [227]. F11 also promotes
the migration of infected cells by inhibiting RhoA activity [228, 229]. Therefore, VACV
has developed a method to manipulate the regular functioning of the cortical actin
cytoskeleton, which obstructs virus access to the cell surface, to facilitate its release.
Upon reaching the cell periphery, the outer WV membrane fuses with the plasma
membrane leaving a cell-associated extracellular virus (CEV). Here, virus particles
switch to actin-based transport at the cell periphery [170, 215, 216].

CHAPTER 1: Introduction
The University of Sydney 2016
30
1.3.1.1 Vaccinia virus and the actin cytoskeleton
Several viruses have developed methods of both manipulating existing actin polymers
and promoting actin nucleation for their movement, and VACV is one such virus that is
capable of both [63]. It is the virus with the best-characterised molecular mechanism for
how cellular actin nucleation pathways are repurposed for the promotion of virus
transport. While treatment of cells with a low concentration of latrunculin B stimulates
virus movement to the cell periphery and does not affect virus release, latrunculin B at
higher concentrations and cytochalasin D reduce virus release, indicating the
importance of actin dynamics for VACV exit [226].
1.3.1.1.1 Initiation of actin nucleation at the cell surface
After traversing the cortical cytoskeleton, VACV particles that reach the cell periphery
fuse with the plasma membrane but remain attached to it. Following this fusion event,
there is an abrupt rearrangement to the complement of virus-associated proteins. Figure
1.7 (on the right) depicts some of the viral and host proteins involved in VACV egress
from the cell. Clathrin and the clathrin adaptor AP-2 accumulate on the cytoplasmic
surface of extracellular virus [62, 91]. In parallel to clathrin accumulation there is an
abrupt disassociation of F12, E2 and kinesin-1 from WV [62, 219, 230]. Viral epitopes
present between the two WV membranes (the periplasmic space) are now accessible on
the cell surface, and A36 polarises to the side of the virus particle remaining in contact
with the infected cell [62, 231]. Polarisation of A36 is a product of this protein localizing
exclusively to the outermost viral membrane, a unique attribute among integral WV
membrane proteins [224]. Exposure of WV at the surface of the cell triggers tyrosine
phosphorylation at two residues on A36, Y112 and Y132 (A36Y112, A36Y132), by Src and
Abl cytoplasmic kinases [232-236]. The extracellular SCR (Short Consensus Repeat)
domains of WV envelope protein B5 (specifically SCR4) that now reside on the cell
surface are required for the localisation of active kinases to the virus but exactly how this
phosphorylation event is so tightly restricted to the cell surface is not fully understood
[234]. It may be that other WV proteins obstruct kinases from accessing A36, access
that is granted by the modification of associated proteins. Furthermore, the
serine/threonine kinase, casein kinase 2 (CK2) is also necessary for the association of

CHAPTER 1: Introduction
The University of Sydney 2016
31
active Src with motile viruses [237]. Currently, CK2 has not been localised to virus
particles so it is unclear if it acts directly at the site of actin nucleation [237]. A36 is
heavily phosphorylated at serine residues [238] and represents a potential target for
CK2, but the role of A36 serine phosphorylation is yet to be determined.
At least five Src and Abl kinases (Src, Fyn, Yes, Abl, Arg) localise to and phosphorylate
A36 [233-236], with in vitro kinase assays demonstrating some specificity for individual
kinases at each of the two sites: Yes, for example, exclusively targets A36Y112 [236].
Loss-of-function experiments support the notion that substantial redundancy of function
operates between these kinases [235]. Finally, it is a platform of tyrosine-phosphorylated
A36 on the cytoplasmic surface of an extracellular virus that is essential to recruit the
cellular orchestrators of actin nucleation.

CHAPTER 1: Introduction
The University of Sydney 2016
32
Signaling pathways used by VACV to initiate microtubule- (left) and actin-based
(right) motility.
Actin nucleation is utilised by viral pathogens to mediate viral egress at the cell surface (VACV). WV particles
of VACV recruit the microtubule motor kinesin-1 through a WE and a WD motif on the envelope protein A36
and then activate actin nucleation initiated by the phosphorylation of A36 at two tyrosine residues. Viral
proteins are shown in orange and cellular proteins are shown in green; arrows indicate pathways and
interactions, and when mapped to specific domains, they are shown by connecting lines. Adapted from [239].

CHAPTER 1: Introduction
The University of Sydney 2016
33
1.3.1.1.2 Cellular components involved in the core actin nucleation cascade
Actin nucleation by extracellular WV is executed by the cellular actin nucleator, the
Arp2/3 complex, which is activated by the Type I NPF N-WASP, to promote de novo
seeding of actin filaments at 70° branch points on existing actin filaments [43, 130, 233,
240, 241]. When VACV-infected cells are observed by live-cell microscopy at 6–8 hpi,
activity of the Arp2/3 complex propels WV laterally across apical and basal membranes
at speeds of 18–24 μm/min with F-actin localizing adjacent to virus particles. Following
nucleation, actin polymerisation is in a constant state of flux; as rapidly as actin is
nucleated at the cytoplasmic/virus interface and filaments extend, actin polymers are
disassembled, giving rise to a characteristic comet morphology (also referred to as actin
‘tails’). Those with an interest in parsing how N-WASP is able to co-ordinate multiple
signals have been successful by utilising characteristics of virus motility as a proxy for
Arp2/3 complex activity. For example, it is possible to quantify the speed of virus motility,
the frequency of comet initiation and the length of actin comets. These criteria reflect the
magnitude and quality of N-WASP activation of the Arp2/3 complex. Typically, cells
infected with VACV display 5–50 virus-associated actin comets of about 3.5 μm in
length, although there is great variation between cell types [219, 241]. At any one point
in time, 5–30% of CEV will be adjacent to an actin comet [62, 168, 242].
Recruitment of N-WASP to the cytoplasmic surface beneath extracellular WV is initiated
by the phosphorylation of residues A36Y112 and A36Y132 that, with the surrounding amino
acid residues, form binding sites for the SH2 domains of the cellular adaptor proteins
Nck1/Nck2 and Grb2, respectively [233, 243]. N-terminal SH3 domains of Nck bind a
poly-proline tract in WASP Interacting Protein (WIP), which itself binds N-WASP through
a WASP Binding Domain (WBD) [43, 243-245]. WIP function can be replaced by a WIP
homologue, WIRE [245]. Further stabilizing N-WASP at the virus, Grb2 is likely to bind
N-WASP via its own SH3 domains [241, 243]. Disruption of either arm that acts to
stabilise N-WASP at virus particles (A36Y112/Nck and A36Y132/Grb2) has quite distinct
consequences. Loss of the A36Y112/Nck arm abolishes actin comet formation while loss of
the A36Y132/Grb2 arm results in reduced frequency of comet initiation and shorter comets
but faster motility of virus particles [241].

CHAPTER 1: Introduction
The University of Sydney 2016
34
The contribution of both arms to actin-based motility is apparent when one considers the
turnover of the core cascade during actin nucleation. This can be studied by using
Fluorescence Recovery After Photobleaching (FRAP) to examine the recovery of GFP-
tagged transgenes at motile WV. Using this approach, N-WASP that was associated
with virus particles was found to have a turnover rate of 2.68 ± 0.12 s [241]. Abrogating
Grb2 function results in increased turnover of N-WASP, confirming that Nck and Grb2
combine to stabilise N-WASP at the virus surface. In the absence of the A36Y132/Grb2
arm, fewer viruses initiate actin nucleation, N-WASP turns over at a higher rate but virus
particles are propelled at a faster speed [241]. Increased speed might come at a cost to
the robustness of motility, as loss of AP-2 also results in faster virus motility concomitant
with a reduction in the duration of transport [62]. These findings allude to a fine balance
between stable recruitment of actin nucleation machinery, actin nucleation activity, and
robust and efficient virus transport. We might consider A36-mediated recruitment of Nck,
WIP, Grb2, N-WASP and the Arp2/3 complex the core cascade that leads to actin-based
motility of WV, but this is really the tip of the iceberg regarding how nucleation is
regulated by VACV.
1.3.1.1.3 Regulating the core cascade
The recruitment and activation of N-WASP at the WV interface are inextricably linked, as
interactions with N-WASP will inevitably act to relieve auto-inhibitory associations.
However, virus-induced actin nucleation is subject to higher orders of regulation. For
example, the density and clustering of A36 at WV plays a role in how actin is nucleated.
The clathrin adapter AP-2 is recruited transiently to WV via an interaction with A36,
which also leads to the recruitment of clathrin [62]. Under conditions of AP-2 knockdown,
A36 fails to coalesce to a discrete platform at extracellular virus particles. These viruses
initiate comets but they take longer to do so, they move faster but travel for shorter
durations, and N-WASP turnover is reduced. Structured Illumination Microscopy (SIM)
reveals the coalescence of A36 beneath WV but lacks the resolution necessary to
confirm the model that local density of A36 impacts virus motility. Support for this
mechanism was derived from expressing a combination of functional and non-functional
(for actin nucleation) versions of A36 in different ratios. Decreasing the number of active
A36 proteins at the virus increased comet length and increased speed, mimicking the
phenotype of loss of AP-2 and thereby supporting the model [62]. Very recent studies

CHAPTER 1: Introduction
The University of Sydney 2016
35
have found NPF-like motifs in the VACV A36 protein, which recruit AP-2 and clathrin to
the site of actin polymerisation [246]. Loss of these C-terminal motifs on A36 reduces
actin-based motility, and thus, despite the association of AP-2 and clathrin being
transient, the consequences on the capacity for actin-based transport are longer term.
Rho-family GTPases such as Rho, Cdc42 and Rac play a central role in actin dynamics
in many contexts. It should not be surprising that during VACV replication their
regulation is complex and their roles are multiple. How RhoA modulates cortical actin
facilitating virus release has already been described, but RhoA function is far more
pleiotropic with additional roles in microtubule dynamics, cell detachment and cell
migration [227-229, 247, 248]. Rho GTPases also participate in actin-based motility
through interactions with the nucleation machinery. N-WASP possesses a GTPase
binding domain that binds active Cdc42 (GTP-bound) relieving autoinhibition of N-
WASP, often in synergy with Nck [249]. Active Cdc42 is locally generated at virus
particles by the Rho guanine-nucleotide exchange factor intersectin-1 (ITSN1), further
stabilizing N-WASP, enhancing Arp2/3 complex activation and facilitating virus motility.
The simplicity of a linear pathway leading to actin-based virus motility is further
challenged by the revelation that another class of actin nucleator is recruited to actin
comets and facilitates their formation: the formins [242]. Unlike the Arp2/3 complex
where activity leads to highly branched actin networks, formins nucleate and extend
actin polymers resulting in long, bundled filaments of actin [250]. That these two modes
of nucleation could act together during VACV infection has precedence in many cellular
functions and even in the motility of another pathogen, Shigella flexneri [149]. The
localisation of the formin FHOD1 to VACV-induced actin comets requires the active form
of Rac1 and prior recruitment of N-WASP, so FHOD1 activity is downstream of Arp2/3
complex activity [242]. Until now, no fine-scale analysis of F-actin beneath WV has been
conducted in the absence of FHOD1 activity that might reveal structural differences in
the actin network formed at virus particles. Loss of FHOD1 or Rac1 decreases the
efficiency of comet initiation and those that do form travel at a reduced velocity [251]. It
is instructive that both active Cdc42 and Rac1 directly participate in VACV-induced actin
nucleation despite being globally inactivated by infection at the time point of actin-based
motility [247]; clearly, high-resolution spatial analysis is needed for a comprehensive
appreciation of their roles.

CHAPTER 1: Introduction
The University of Sydney 2016
36
It is quite clear that actin is targeted for manipulation by a number of viruses as a result
of the fundamental roles it plays in a cell. While many of their techniques may be unique,
trends in how actin is repurposed during virus replication can be observed. Viruses
require a rearrangement of the cortical actin cytoskeleton to gain entry to cells, however
the size of the virus plays a role here. While smaller viruses such as HIV and DENV-1
can enter by actin-assisted clathrin-mediated pathways, larger adenoviruses and
orthopoxviruses harness the more flexible macropinocytic entry mechanism. Following
entry, actin-mediated cellular transport pathways present an efficient means for invading
pathogens to travel to sites of replication an/or exit. More complex viruses with larger
genomes such as VACV and AcMNPV have evolved to encode proteins that specifically
interact with actin cytoskeleton signalling pathways to initiate their movement.
While the use of various actin destabilizing drugs to study the role of actin in virus
infection has been invaluable, care must be taken in their interpretation as these drugs
often induce broad or off-target effects in a cell. A more precise understanding of the
specific function of these drugs and their use in combination may be useful to narrow
down the roles of actin at various stages of the virus replication cycle. Additionally,
caution must be observed when using different viral strains to answer broad questions
on viral-actin interactions as we have seen that different viral strains have evolved
different relationships to actin depending on their specific host cell targets in vivo.
Signalling cascades initiated by VACV and AcMNPV result in activity of the Arp2/3
complex at virus particles. This provides a compelling opportunity to dissect the
dynamics of actin filament assembly and elongation with a minimal toolbox both in vitro
and in vivo, and understand how force is generated. For example, a recent study used
electron tomography to reveal that AcMNPV particles in vivo were trailed by a fishbone-
like array of filaments with 4-5 filaments in close proximity to virus particles [252]. Thus
the extension of few actin polymers is sufficient to push nucleocapsids through the
cytoplasm. Induction of actin comets by VACV is subject to far greater regulation and is
mediated and fine-tuned by a multitude of host factors before culminating in Arp2/3
complex activity. This powerful pathogen model has afforded the opportunity to study
how these host pathways are sequentially assembled and to correlate real-time cell
biology with actin nucleation activity.

CHAPTER 1: Introduction
The University of Sydney 2016
37
1.4 PROJECT AIMS
The actin cytoskeleton plays a vital role in VACV infection. Any attempt to understand
VACV pathogenesis and spread will require a close following of its association with
actin. Advancements both in the field of fluorescent microscopy and oligonucleotide
synthesis provided a unique opportunity for us to develop a novel mechanism for the
rapid generation of fluorescently tagged viruses. We hope to use this method to create a
recombinant VACV that would be capable of fluorescently highlighting the actin
cytoskeleton once it infects a host cell. This tool would be invaluable for the study of
VACV actin-based motility in tandem with live-cell microscopy (see Chapter 3).
We also hope to use this tool to elucidate a lesser-understood manipulation of VACV on
the actin cytoskeleton: that of viral-induced cell motility. Currently, VACV is known to
induce cell motility in infected cells, and this ability is beneficial for the infection of VACV
in mice. We hope to utilise the aforementioned recombinant poxvirus creation
techniques to create a recombinant ectromelia virus (ECTV), whose natural host is the
mouse, to examine this process in a true smallpox-like infection in vivo (see Chapter 4).
Finally, current research trends have been gradually teasing apart the differing roles of
the two cytoplasmic actin isoforms: β-actin and γ-actin. While we know that several
pathogens induce actin-based motility as part of their infectious cycles, no study so far
has looked at the role of these two actin isoforms in this process. Through the use of
siRNA and novel highly specific antibody staining techniques, we aimed to discern the
specific roles and requirements of the two cytoplasmic actins using VACV as a model of
actin-based motility (See Chapters 5 and 6).

CHAPTER 2: Materials and Methods
The University of Sydney 2016
38
Chapter 2: MATERIALS AND METHODS

CHAPTER 2: Materials and Methods
The University of Sydney 2016
39
2.1 BUILDING BLOCKS
2.1.1 Reagents
Chemicals, reagents and kits used and/or mentioned in this thesis are listed below,
along with their suppliers and product numbers in brackets:
• 0.25% Trypsin-EDTA 1X (Invitrogen) (Cat: # 25200114)
• 30% Acrylamide/Bis Solution, 37.5:1 (2.6% C) (Bio-Rad Laboraroties) (Cat: # 161-0158)
• 3',3",5',5"-tetrabromophenolsulfonphthalein (bromophenol blue) (Sigma-Aldrich) (Cat: # B8026)
• 6-Well Flat-Bottom Plate with Lid (Corning-Falcon) (Cat: # 353046)
• 12-Well Flat-Bottom Plate with Lid (Corning-Falcon) (Cat: #353043)
• 24-Well Flat-Bottom Plate with Lid (Corning-Falcon) (Cat: #353047)
• μ-Dishes 3cm No. 1.5 glass (Ibidi) (Cat: #81151)
• Acrylamide/Bis solution 30% 37.5:1 (Bio-Rad) (Cat: #1610158)
• Agarose (Bioline) (Cat: # Bio41025)
• Alexa Fluor® 568 Phalloidin (Invitrogen) (Cat: # A12380)
• Amersham ECL Westertn Blotting Detection Reagent (GE Health)(Cat: # RPN3243)
• Amersham Hyperfilm ECL (GE Health)(Cat: # 28-9068-37)
• Ammonium Persulphate for electrophoresis (APS) (Sigma-Aldrich) (Cat: # A3678)
• Ampicillin (Astral Scientific) (Cat: # AM0339)
• Boric Acid (Astral Scientific) (Cat: # AM0588)
• Bovine Serum Albumin, Nuclease free (Fisher Biotec) (Cat: # BSA-50)
• Carboxymethylcellulose sodium salts, medium viscosity (Sigma-Aldrich) (Cat: # C9481)
• 4', 6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich) (Cat: # D9542)
• D-Glucose (Astral Scientific) (Cat: # AM0188)
• Dimethylsulfoxide (DMSO) (Sigma-Aldrich) (Cat: # D2650)
• dNTP Set (Bioline) (Cat: # Bio39026)
• Dulbecco's Modified Eagle Medium (D-MEM) (1X), liquid (High Glucose) (Invitrogen) (Cat: #11995073)
• ECL Western Blotting Reagent (GE Health) (Cat: # RPN2106)
• Ethidium Bromide (Amresco) (Cat: # X328)
• Ethylenediaminetetraacetic Acid (EDTA) Disodium Salt Dihydrate (Astral Scientific) (Cat: # AM0105)
• Fetal Bovine Serum (FBS) (Diethelm Keller Siber Hegner, DKSH) (Cat: # SFBS)
• Frosted l End 1 Side, 1.0-1.2mm (Livingstone) (Cat: # 7105-1A)
• GelRedTM Nucleic Acid Gel Stain (Biotium) (Cat: # 41003)
• Glutathione Sepharose® 4B (GE Healthcare) (Cat: # 17-0756-01)
• Glycerol, minimum 99% GC (Sigma-Aldrich) (Cat: # G5150)
• Glycine (Astral Scientific) (Cat: # AM0167)
• Hybond-C Extra (Amersham Biosciences, GE) (Cat: # RPN203E)
• Hyperladder I (Bioline) (Cat: # Bio33026)
• Immersion Oil (Olympus) (Cat: # AV9602)
• Lens Paper (Olympus) (Cat: # AX6476)
• Lipofectamine 2000 Transfection Reagent (Invitrogen) (Cat: # 11668027)
• Magnesium Acetate Tetrahydrate (Sigma-Aldrich) (Cat: # M5661)
• Magnesium Chloride (APS Chemical) (Cat: # 296)
• β-Mercaptoethanol (Sigma-Aldrich) (Cat: # M3148)
• 2-(N-Morpholino)ethanesulfonic acid hydrate (MES) (Sigma-Aldrich) (Cat: # M8250)
• Microscope Coverslips No.1 Thickness Circular, 12mm (Livingstone) (Cat: # CS12RD)

CHAPTER 2: Materials and Methods
The University of Sydney 2016
40
• Minimum Essential Medium Eagle, with EAR (MEM) (Sigma-Aldrich) (Cat: # M2279)
• Modified Eagle Medium (MEM) (2X), liquid (Invitrogen) (Cat: # 11935046)
• Mycophenolic Acid (Sigma-Aldrich) (Cat: # M5255)
• Opti-MEM Reduced Serum Medium (Gibco) (Cat: # 51985034)
• Paraformaldehyde (PFA) (Sigma-Aldrich) (Cat: # P6148)
• Penicillin-Streptomycin-Glutamine (100X) (Cat: # 10378-016)
• Phenol Buffer Saturated (pH 6.7/8.0) (Astral Scientific) (Cat: # 0945)
• Phenol:Chloroform (pH6.7/8.0) premixed with isoamyl (25:24:1) (Astral Scientific) (Cat: # 0883)
• Phosphate Buffered Saline Tablet (Astral Scientific) (Cat: # AME404)
• Polyvinyl Alcohol 4-88 (Mowiol) (Sigma-Aldrich) (Cat: # 81381)
• Polyoxyethylene Sorbitan Monolaurate (Tween-20) (Sigma-Aldrich) (Cat: # p2287)
• P-Phenylenediamine Free Base (Sigma-Aldrich) (Cat: # P6001)
• QiaexII Gel Extraction Kit (Qiagen) (Cat: # P20021)
• Qiaprep Spin Minikit (Qiagen) (Cat: # P27106)
• Rubidium Chloride (Sigma-Aldrich) (Cat: # 215260)
• 5mL Serological Pipets (Becton Dickinson) (Cat: # 357543)
• 10mL Serological Pipets (Becton Dickinson) (Cat: # 357551)
• 25mL Serological Pipets (Becton Dickinson) (Cat: # 357525)
• Snap Strip II PCR tubes 8-Strip Standard Tube & with Individual Attached Flat Caps (Astral Scientific) (Cat: # I324500)
• Sodium Chloride (Astral Scientific) (Cat: # AMX190)
• Sodium Dodecyl Sulfate (SDS) (Astral Scientific) (Cat: # AM0227)
• Syringe Filter (Diethelm Keller Siber Hegner, DKSH) (Cat: # 431227)
• Tetracycline Hydrochloride Crystalline (Sigma-Aldrich) (Cat: # T3383)
• 175 cm2 Tissue Culture Flask with Vented Cap (Corning-Falcon) (Cat: #353112)
• 75 cm2 Tissue Culture Flask with Vented Cap (Corning-Falcon) (Cat: #353136)
• Tris-hydroxymethyl-aminomethane (Tris Base) (Astral Scientific) (Cat: # AM0479)
• Wizard® SV Gel and PCR Clean-Up System (250 preps) (Promega) (Cat: # A9282)
• Wizard® Plus SV Miniprep DNA Purification System + Vaccum Adaptors (250 preps) (Promega) (Cat: # A1470)
• Xanthine (Sigma-Aldrich) (Cat: # X4002)
2.1.2 Cell lines
Cell lines used in this study include BSC-1 (monkey kidney epithelial cell line; ATCC
CCL-26, CRUK strain; kind gift from M. Way), HeLa (human cervical cancer cell line;
ATCC CCL-2, CRUK strain; kind gift from M. Way), GBM A-172 (glioblastoma cell line;
ATCC CRL-1620; kind gift from Prof. R. Christopherson) and hCMEC-D3 (human
cerebral microvascular endothelial cell line; kind gift from Prof C.O. Couraud). Cells were
grown in Gibco Dulbecco’s modified Eagle Medium (DMEM; Invitrogen) which was
supplemented with 5% foetal bovine serum (FBS), 292 μg/ml L-glutamine, 100 units/ml
penicillin and 100 μg/ml streptomycin, and incubated at 37°C in a 5% CO2-enriched
atmosphere.

CHAPTER 2: Materials and Methods
The University of Sydney 2016
41
2.1.3 Viruses
The VACV-WR strain was a gift from Michael Way, Cancer Research UK, and was the
parent strain used for several of the recombinant viruses used in this study. These
strains and their origins are listed in Table 2.1. ECTV strain Moscow was a gift from
Professor RM Buller, St. Louis University School of Medicine.
Table 2.1 Viruses used and generated
VIRUS DESCRIPTION GENERATED BY
VACV-WR VACV strain Western Reserve (ATCC
VR-1354) ATCC VR 1354
VACV-WR A36 YdF VACV strain WR with two point
mutatuins in the A36R gene J. Horsington
ECTV-Mos ECTV strain Moscow ATCC VR 1374
VACV-WR Lifeact-
GFP
VACV strain WR constitutively
expressing Lifeact-GFP C. McKenzie
VACV-WR GFP-A3L VACV strain WR with A3L N-
terminally tagged with GFP N. B. Marzook
VACV-WR GFP-F1L VACV strain WR with F1L N-
terminally tagged with GFP N. B. Marzook
VACV-WR A3-RFP VACV strain WR with A3L N-
terminally tagged with RFP T. Newsome
VACV-WR Lifeact-
GFP/A3-RFP
VACV strain WR with A3L N-
terminally-tagged with RFP and
constitutively expressing Lifeact-GFP
N. B. Marzook

CHAPTER 2: Materials and Methods
The University of Sydney 2016
42
2.1.4 Buffers and solutions
Buffers and solutions used in this study, including sources or compositions (where
available) are as follows:
BUFFER/SOLUTION COMPOSITION/SOURCE
Blocking buffer (for IFA) 1% bovine serum albumin (BSA) and 2% foetal bovine serum in cytoskeletal buffer (CB)
Blocking buffer (for immunoblots)
5% (w/v) skim milk in PBS with 0.1% Tween-20
Cell lysis buffer (bacterial and mammalian)
1% Triton X-100 (v/v), 200 μM phenylmethylsulfonyl fluoride (PMSF) in PBS
Crystal violet solution 0.5% (w/v) in 20% methanol solution (Sigma-Aldrich)
Cytoskeletal buffer (CB) 10 mM 2-(N-morpholino) ethanesulfonic acid (MES) buffer, 0.15 M NaCl, 5 mM EGTA, 5 mM MgCl2, 50 mM glucose, pH 6.1
Luria-Bertani (LB) broth 10 g/L NaCl, 10 g/L tryptone, 5g/L yeast extract; in MilliQ water
Luria-Bertani (LB) agar 10 g/L NaCl, 10 g/L tryptone, 5g/L yeast extract, 15 g/L bacteriological agar
MOWIOL mounting solution 10% (w/l) polyvinyl alcohol 4-88 (Sigma-Aldrich), 25% (w/v) glycerol, 0.1 M Tris, pH 8.5
Mycophenolic acid (MPA) Sigma-Aldrich (M3536-50MG); dissolved in 0.1 N NaOH
Phosphate Buffered Saline (PBS) PBS tablets; Astral Scientific, Cat # AME404
Phosphate Buffered Saline – Tween 20 (PBS-T)
PBS with 0.1% Tween-20
Phosphate Buffered Saline – Tween 20 and milk (PBS-T milk)
PBS with 0.1% Tween-20 and 5% w/v skim milk powder
SDS-PAGE sample buffer 62.5 mM Tris-HCl, 0.25 M glycerol, 2% SDS, 0.01% (w/v) bromophenol blue, 12.5% (v/v) β-mercaptoethanol

CHAPTER 2: Materials and Methods
The University of Sydney 2016
43
Xanthine Sigma-Aldrich (X0626-5G); dissolved in 0.1 N NaOH
2.1.5 Primary antibodies used for immunoblots
All antibodies were diluted in PBS-T milk unless stated otherwise.
2.1.6 Secondary antibodies used for immunoblots
ANTIBODY SPECIES DILUTION SOURCE
α-Rabbit-HRP Goat 1:2000 EMD Millipore
α-Mouse-HRP Goat 1:2000 EMD Millipore
α-Rat-HRP Goat 1:2000 EMD Millipore
ANTIBODY SPECIES DILUTION SOURCE
α-A36 Rabbit 1:2000 [253]
α-beta-actin (loading control)
Mouse 1:2000 Sigma-Aldrich (AC-74)
α-GFP Mouse 1:2000 Thermo Fisher Scientific (MA5-15349)
α-human β-actin
Mouse 1:500 (diluted in 5% BSA in PBS)
Specifically raised against β-actin; Courtesy Prof. C. Chaponnier
α-human γ-actin
Mouse 1:10,000 (diluted in 5% BSA in PBS)
Specifically raised against γ-actin; Courtesy Prof. C. Chaponnier
α-actin (α-pan-actin) Mouse 1:5000 EMD Millipore (MAB1501)
α-GST Rat 1:2000 Sigma-Aldrich (SAB4200055)

CHAPTER 2: Materials and Methods
The University of Sydney 2016
44
2.1.7 Reagents for immunofluorescent staining
All antibodies and reagents were diluted in IFA blocking buffer, unless stated otherwise.
NAME SPECIES / LABEL DILUTION SOURCE
Primary Antibodies
α-B5 Rat 1:300 19C2, [168]
α-Src Mouse 1:200 Clone 327, M. Way
α-human β-actin Mouse; IgG1 only 1:50 (diluted in 2% BSA in PBS)
C. Chaponnier
α-human γ-actin Mouse; IgG2b only 1:100 (diluted in 2% BSA in PBS)
C. Chaponnier
Secondary antibodies
α-Rat Goat; Alexa Fluor 350 1:200 Invitrogen
α-Rat Goat; Alexa Fluor 568 1:200 Invitrogen
α-Mouse Goat; Alexa Fluor 488 1:200 Invitrogen
α-Mouse (IgG1) Goat, IgG1-specific; CY2
1:200 Jackson Immunotech
α-Mouse (IgG2b) Goat, IgG2b-specific; CY3
1:200 Jackson Immunotech
Other reagents
Phalloidin Alexa Fluor 488 1:2000 Invitrogen
Phalloidin Alexa Fluor 568 1:2000 Invitrogen
MitoTracker Red CMXRos
N/A (binds to mitochondria)
1:10,000 Thermo Fisher Scientific
DAPI N/A (binds to dsDNA) 1 μg/mL (in CB) Sigma-Aldrich

CHAPTER 2: Materials and Methods
The University of Sydney 2016
45
2.1.8 Primers
All primers used in this study are outlined below. Restriction sites are in bold, bases
added to maintain the frame are in green and stop codons are in red.
NAME TARGETING REGION DNA SEQUENCE
In Chapter 3
GFP.BamHI For. GFP sequence from pE/L GFP, for the
creation of the GFP tag for insertion into
TDS vector; see Figure 3.4 (restriction sites
are inverted since it is an N-terminal tag)
GGATCCAAGGGCGAGGA
GCTGTTC
GFP.NotI Rev. GCGGCCGCCCTTGTACA
GCTCGTC
A4 seq. For. Amplifying area encompassing the end of
A4L and the start of A3L
GATGCAAGGGAGTATAC
G
A3 seq. Rev. GACAATGAATTGCATACA
F2 seq. For. Amplifying area encompassing the end of
F2L and the start of F1L
CTGGAGATAGAATAGCTC
F1 seq. Rev. ATTGCTAGCCTCATCTTC
In Chapter 4
ECTV F12L LA
NotI For. 3’ end of ECTV F12L; for creation of LA for
ECTV F11L deletion; see Figure 4.2
AAGCGGCCGCACTTGAA
CGCAGCCACAAC
ECTV F12L LA
NheI Rev.
AAACTAGTGCTAGCCGAT
AATTAATAATATTGTTTTT
CAC
ECTV ΔF11L RA
NheI For. 3’ end of ECTV F11L; for creation of ECTV
ΔF11L RA containing likely promoter
sequence of F10L; see Figure 4.2
AAGCTAGCAAGCTTTCCT
GTATGTTAACCGAG
ECTV ΔF11L RA
BamHI Rev.
AAGGATCCGTCGACTGA
ATCATTGGCAACACC
pE/L NheI. For For the creation of a pE/L Lifeact-GFP insert AAGCTAGCCCCCTCGAG
AAAAATTG

CHAPTER 2: Materials and Methods
The University of Sydney 2016
46
GFP.NheI Rev to replace ECTV F11; pE/L Lifeact-GFP
used as template; see Figure 4.2
AAGCTAGCTTACTTGTAC
AGCTCGTCCATG
In Chapter 6
VCA NotI For. To amplify the rat N-WASP (O08816) VCA
domain to be inserted into the pMW-GST
vector; see Figure 2.1B
GGCGGCCGCGACCATCA
AGTTCCAGCT
VCA EcoRI Rev. GAATTCTCAGTCTTCCCA
CTCATC
2.1.9 Vector constructs made and/or used
All plasmids made and/or used in this study are described below. All possessed
Ampicillin resistance genes for selection after bacterial transformation.
NAME DESCRIPTION CREATED BY
In Chapter 3
Synthetic oligonucleotide 1 – multi-gene cassette
De novo synthesised oligonucleotide containing 5 cassettes of 300 bp each, corresponding to the LA/RA of chosen viral genes; see Figure 3.5
GenScript®
TDS recombination vector (empty)
TDS recombination vector with gpt and mCherry genes under the VACV pE/L promoter [254]; see Figure 3.2
T. P. Newsome
A3L LA/RA TDS recombination vector
Synthesised 300bp cassette (corresponding to A3L LA/RA) inserted into TDS recombination vector
N. B. Marzook
F1L LA/RA TDS recombination vector
Synthesised 300bp cassette (corresponding to F1L LA/RA) inserted into TDS recombination vector
N. B. Marzook
A3L LA/GFP/RA TDS recombination vector
GFP [255] inserted in between LA and RA of A3L LA/RA TDS recombination vector; see Figure 3.4
N. B. Marzook
F1L LA/GFP/RA TDS recombination
GFP [255] inserted in between LA and RA of F1L LA/RA TDS recombination vector;
N. B. Marzook

CHAPTER 2: Materials and Methods
The University of Sydney 2016
47
vector see Figure 3.4
pE/L GFP For synthesis of the GFP fragment to be inserted into the TDS vector; see Figure 3.4 and 4.2, and the expression of GFP controlled by the VACV pE/L promoter; see Figure 3.7
T. P. Newsome
In Chapter 4
ECTV ΔF11L LA/RA Intermediate TDS vector containing the ECTV ΔF11L LA and RA; see Figure 4.2. step 1
J. Horsington
ECTV ΔF11L LA/pEL Lifeact-GFP/RA
Final TDS vector containing ECTV ΔF11L LA and RA with pE/L Lifeact-GFP sequence inserted in between; see Figure 4.2, step 2
N. B. Marzook
pE/L Lifeact-GFP Plasmid vector for transient expression of Lifeact-GFP in VACV-infected cells; used to amplify pE/L Lifeact-GFP with NheI restriction sites on each end for insertion into ECTV ΔF11L LA/RA; see Figure 4.2
H. Lynn
In Chapter 6
Synthetic oligonucleotide 2 – VCA-RA/RA
De novo synthesised oligonucleotide containing the VCA domain with 2 point mutations (R410A and R438A)
DNA 2.0 Inc.
GST-VCA For the expression of GST-VCA in bacteria; VCA domain obtained by PCR from rat N-WASP
N. B. Marzook
GST-VCA-RA/RA For the expression of GST-VCA-RA/RA in bacteria; VCA-RA/RA domain was synthesised de novo
N. B. Marzook

CHAPTER 2: Materials and Methods
The University of Sydney 2016
48
2.2 FANTASTIC VIRUSES AND HOW WE USE THEM
2.2.1 Viral infection
For infection, virus stock was diluted (to the desired multiplicity of infection (MOI)) in
DMEM not supplemented with foetal bovine serum (FBS), called serum-free media
(SFM), and applied to phosphate-buffered saline (PBS)-washed cells. Cells were
incubated at 37°C with a 5% CO2 atmosphere for 1 hour before being recovered with
fresh growth medium supplemented with 5% or 10% FBS (depending on cell type), 292
μg/ml L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin.
2.2.2 Transfection
For the creation of recombinant VACV, TDS plasmids were transfected into cells 1 hour
post infection (hpi) using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. Cells were scraped after 24 hours and lysed using three
rounds of freeze-thaw cycles with liquid nitrogen to release virus particles.
2.2.3 Plaque assays
A monolayer of cells (BSC-1, unless stated otherwise) were infected as above, but were
rescued instead with a mixture of GIBCO Modified Eagle Medium (MEM; Invitrogen)
similarly supplemented as the growth medium described above, as well as 0.45% Ultra
Pure Agarose (Invitrogen) for purification of individual plaques, or 1.5% carboxy-methyl
cellulose (CMC) for plaque visualization. Cells for fixed 3 days post-infection (dpi)
2.2.3.1 Plaque picking for virus purification
Virus plaques were picked using a P1000 pipette tip such that an agarose plug,
along with the cells containing virus beneath that plug, were contained in the tip.
The contents of the tip were discharged into 100 μL of SFM, subjected to three
freeze-thaw cycles with liquid nitrogen, and used to infect a monolayer of BSC-1
cells for amplification or further rounds of purification, as appropriate. Cells were
rescued 1 hpi with DMEM containing 5% FBS, or the agarose solution as above.
2.2.3.2 Plaque visualisation
The CMC overlay on cells was aspirated 3 dpi, followed by washing with PBS at
least three times. At this point, plaques were visualised in two ways. They could

CHAPTER 2: Materials and Methods
The University of Sydney 2016
49
be visualised using an Olympus BX51 microscope (see section 2.3.2.1 for
details), either with prior immunofluorescent staining (see section 2.3.1 for
details), or directly in the case of fluorescent VACV. Alternatively, plaques were
fixed and stained with crystal violet (0.5% (w/v) in 20% methanol solution) for 15
min, followed by three rounds of washing with PBS. Regions of clearing in the cell
monolayer caused by the lysis of infected cells were unstained, and these regions
were visualized by scanning with a high-resolution gel scanner (BioRad GS-800).
2.2.3.3 Plaque size measurement
Size of plaques (visualised either by fluorescence or scanning post-crystal violet
staining) was measured using the program FIJI (an open source image
processing software based on ImageJ, ver. 2.0.0-rc-43/1.51g). A horizontal line
was drawn across each plaque, giving a measurement in pixels, which was then
converted to mm using a fixed scale measurement.
2.2.4 EEV release assays
Cells in a 12-well plate, at a confluency of 70-80%, were infected by the VACV of choice
at an MOI of 0.1 for 1 hour. Cells were then washed twice with PBS and overlaid with an
exact amount of DMEM containing FBS (5-10% depending on cell type). Precise and
consistent volumes of the supernatant were collected at 16 hpi. Plaque assays using 10-
fold serial dilutions of the supernatant were conducted on BSC-1 cells, as described
above. Plaques were enumerated from three experimental replicates and statistical
analysis was carried out with GraphPad PRISM software (ver 6.0h).
2.2.5 Virus DNA extraction
Virus genomic DNA was extracted in order to confirm recombinant genotypes by PCR.
This was done by two methods. The first involved scraping virus-infected cells into 1 mL
of SFM, followed by centrifugation at 16100 rcf for 10 min at 4oC (Eppendorf
Microcentrifuge 5415R). The supernatant was removed and the cell pellet was
resuspended in 500 μL TE, 0.1% SDS by vortexing to lyse cells. 500 μL of phenol :
chloroform : isoamyl alcohol (25:24:1) was added to the cell lysate and mixed by
inversion. This was centrifuged at 16100 rcf for 4 min at 4oC, following which the top
aqueous later was transferred to a new tube. This step was repeated once more,
followed by the addition of 1 mL 100% chilled ethanol and 50 μL 3M NaAcetate to the
aqueous layer. This was cooled to -80oC for 1 hour to precipitate viral DNA, followed by

CHAPTER 2: Materials and Methods
The University of Sydney 2016
50
centrifugation again at 16100 rcf for 30 min at 4oC. The supernatant was removed and
the DNA was allowed to dry at 50oC for 10 minutes. The DNA was resuspended in MilliQ
water and used for subsequent sequencing or PCR analysis.

CHAPTER 2: Materials and Methods
The University of Sydney 2016
51
2.3 UNDER THE MICROSCOPE
2.3.1 Immunofluorescence assays
Cells were grown on glass coverslips, treated with siRNA and/or infected with viruses as
appropriate, and fixed using 3% paraformaldehyde (PFA) in cytoskeletal buffer for 15
minutes at room temperature. Cells were then washed three times in PBS and stored at
4oC until staining.
Cells were then permeabilised (unless stated otherwise) in 0.1% Triton X-100 in CB for 5
minutes. The only exception to this was in the case of cells being stained with α-β- or α-
γ-actin, which were permeabilised with ice-cold methanol (-20oC) for 5 minutes.
Permeabilisation was followed by washing three times in PBS, and blocking in IFA
blocking buffer for 20 minutes. Cells were then incubated in the primary antibody diluted
in blocking buffer for at least 40 minutes, followed by three more rounds of washing in
PBS. Similarly, cells were incubated in the respective secondary antibody, followed by
Alexa Fluor-conjugated phalloidin where required. Finally, cells were incubated in DAPI
for 1 minute, washed twice in PBS and once in MilliQ water, and mounted onto glass
slides with MOWIOL mounting media containing 1% (w/v) P-phenylenediamine (Sigma-
Aldrich). Slides were incubated at 37oC for 10 minutes, and stored at 4oC prior to
imaging.
2.3.2 Image acquisition
2.3.2.1 Wide-field microscopy
An Olympus BX51 Microscope with a reflected fluorescence system was used to image
cells both by phase-contrast and fluorescent microscopy. Other components included a
Mercury Burner (U-RFL-T), F- view monochrome fluorescence camera and DAPI (347
nm/442 nm [#31013v2]), eCFP (436 nm/480 nm [#49001]), FITC (495 nm/515 nm
[#31001]) and TxRed (584 nm/610 nm [#31004]) Chroma filters. Micrographs were
captured using AnalySIS LS Starter (Olympus Soft Imaging Systems, ver. 2.8), and
edited using Photoshop CS5.1 (Adobe, ver. 16.04) and FIJI (ver. 2.0.0-rc-43/1.51g).
2.3.2.2 Confocal microscopy
Where indicated, images of dual-labeled actin comets were captured on a ZEISS LSM
510 confocal microscope, at 63x magnification with 1.4 NA at room temperature. Z-

CHAPTER 2: Materials and Methods
The University of Sydney 2016
52
stacks were also obtained in this way, using the Zen software package (Carl Zeiss
MicroImaging), and analysed using the FIJI image analysis package.
2.3.2.3 Live-cell wide-field microscopy
Where indicated, fixed and live images were captured using the Nikon Eclipse Ti-E
inverted microscope system, equipped with an Andor Ultra 888 EMCCD camera, a
Lumencor Spectra X fluorescent light source, and Semrock standard DAPI, FITC and
TxRED filter sets. For live cell images, temperature was maintained at 37oC in a 5%
CO2-enriched atmosphere.
2.3.3 Image analysis
2.3.3.1 Actin tail measurements
Length of tails was measured using FIJI image analysis software (ver. 2.0.0-rc-
43/1.51g). A freehand line was drawn with the *Straight* tool, and it’s length was
measured with the Measure function in pixels, later converted to μm using a scale bar.
2.3.3.2 Virus particles at the cell surface
The number of VACV particles on the surface of infected cells was counted by
visualising non-permeabilised cells stained for the envelope protein B5. Particles were
counted using the Cell Counter tool on FIJI (ver. 2.0.0-rc-43).
2.3.3.3 Measuring virus speed
Cells infected with VACV-WR Lifeact-GFP were imaged using the Nikon Eclipse Ti-E
inverted microscope system at 40x magnification in a chamber maintained at 37oC in a
5% CO2-enriched atmosphere. Images were captured every 4 seconds over a period of
5 minutes using NIS-Elements AR (v4.51.00) image capture software. Maximal intensity
projections for 1 min intervals over the 5 min time course were created, and lengths of
actin comets in these projections were measured using FIJI (ver. 2.0.0-rc-43). Speed
was calculated as length of comets over the time interval of that projection (1 min in this
case).

CHAPTER 2: Materials and Methods
The University of Sydney 2016
53
2.4 DNA
2.4.1 Polymerase chain reaction (PCR) and cloning
PCR was carried out using standard protocols with primers listed above (section 2.1.7),
either using plasmids (listed in section 2.1.8) or viral genomic DNA as the template.
PCR products were cleaned using the QIAquick PCR Purification Kit (QIAGEN).
Plasmids were cut using 5U of restriction enzymes (NEB) in the appropriate buffer at
37oC for 10 minutes. Products of digests were separated on a 1% agarose gel made in
TBE buffer (10.781g/L Tris-base, 0.744g/L EDTA and 5.5g/L Boric acid) and desired
vector backbones or inserts were extracted using the QIAquick Gel Extraction Kit
(QIAGEN).
DNA ligations were performed according to standard protocols using T4 DNA ligase
(NEB) overnight at 4oC. This was then transformed into XL 10-Gold Ultracompetent
Cells (Stratagene, La Jolla, CA, USA, Cat: # 200314), and plated onto Luria Broth (LB)
agar plates supplemented with ampicillin (50 μg/mL). Successful colonies were amplified
and plasmids extracted using the Qiaprep Spin Miniprep kit (QIAGEN) before being
verified by diagnostic digests and sequencing. All created vectors were sequenced at
Australian Genome Research Facility Ltd.
2.4.2 Plasmid vector construction
Plasmid vector backbones used in this study are described here. Promoters used were
either the synthetic pE/L viral promoter [254] or the bacterial T7 promoter [256] used in
the pMW-GST vector [236].

CHAPTER 2: Materials and Methods
The University of Sydney 2016
54
Plasmid vector restriction maps.
The plasmid vectors used in this thesis are described here. (A) pE/L GFP vector for expression
of GFP in VACV-infected cells, (B) pMW-GST bacterial expression vector, and (C) GPT
selection vector.

CHAPTER 2: Materials and Methods
The University of Sydney 2016
55
2.5 PROTEINS
2.5.1 Bacterial expression of proteins
Plasmids expressing either GST-VCA or GST-VCA-RA/RA (see Figure 2.1 for vector
backbone) were transformed into bacterial BL-21 cells and grown overnight at 37oC.
Single colonies were selected and used to incubate starter cultures in LB broth for a
maximum of four hours. 10 μL of this was used to inoculate conical flasks containing 1 L
of LB broth, and cells were monitored until they reached an OD of about 0.5. Bacterial
cells were pelleted at 4000 g, washed and lysed in bacterial lysis buffer (section 2.1.3)
by sonication to release expressed protein.
2.5.2 Protein purification using GST-pull-down assays
Expressed protein was purified by passing the bacterial lysate over Glutathione-
containing Sepharose beads (GE Healthcare). This was followed by a few rounds of
washing in lysis buffer, and purified protein was then denatured by boiling the beads in
SDS-PAGE sample buffer.
For the purification of actin using the GST-VCA and GST-VCA-RA/RA constructs,
mammalian cell lysates (lysed in the same lysis buffer as above) were passed over
Glutathione Sepharose beads containing bound GST-VCA or GST-VCA-RA/RA protein.
After three rounds of washing in lysis buffer, the beads containing bound GST constructs
and actin were added to SDS-PAGE sample buffer.
2.5.3 SDS-PAGE gel electrophoresis
Mammalian cells (either infected or uninfected) or protein expressed from bacterial cells
were harvested and lysed in sodium dodecyl sulphate (SDS)- polyacrylamide gel
electrophoresis (PAGE) sample buffer by heating at 95oC for 5 minutes. Proteins were
separated by SDS-PAGE, using a resolving gel (10% acrylamide-Bis solution [37.5:1],
0.375 M Tris-HCl, pH 8.8, 0.1% [wt/ vol] SDS, 0.1% ammonium persulfate (APS), and
0.1% N,N,N,N-tetramethylethylenediamine [TEMED]), after a stacking gel layer (4% to
30% acryl- amide-Bis solution [37.5:1], 0.375 M Tris-HCl, pH 6.8, 0.1% [wt/vol] SDS,
0.1% APS, and 0.1% TEMED). The gel was run for 1.5 hours at 100v in a Mini-Protean
Tetra Cell (BioRad) and either fixed in 0.5 % Coomassie Blue G-250 (Sigma; prepared
in 50% methanol with 10% acetic acid) for visualization of proteins, or transferred to a
membrane for immunoblotting.

CHAPTER 2: Materials and Methods
The University of Sydney 2016
56
2.5.4 Immunoblot assays for proteins of interest
Following electrophoresis, proteins were transferred to a nitrocellulose membrane
(Amersham ProTran, GE Healthcare) using a Mini Trans-Blot (Bio-Rad) system and
buffers according to manufacturer’s instructions. Membranes were then blocked
overnight at 4oC in PBST-milk (5% [w/v] skim milk and 0.1% Tween 20 in PBS).
Membranes were probed with primary antibodies diluted in PBST-milk (table 2.3) for
approximately 1 h. Membranes were then washed three times in PBST-milk before
probing for 30 min with secondary antibodies conjugated to horseradish peroxidase
(HRP), also diluted in PBST-milk (table 2.3). After at least three further washes in PBST
and PBS, protein bands were visualised using enhanced chemiluminescence reagent
(ECL) (Amersham ECL Prime, GE Healthcare) applied on top of the membrane.
Chemiluminescence was detected by exposure on Amersham Hyperfilm photographic
film (GE Healthcare) and development using a CP1000 photographic film developer.

CHAPTER 2: Materials and Methods
The University of Sydney 2016
57
2.6 THE SILENT TREATMENT
2.6.1 siRNA
β- and γ-actin were targeted for silencing using a 1:1 (or 1:1:1 for γ-actin) mixture of 2
(or 3 for γ-actin) siRNAs respectively.
NAME SOURCE
Hs_ACTB_8; SI04205306 QIAGEN
Hs_ACTB_9; SI04287759 QIAGEN
Hs_ACTG1_8; SI04155480 QIAGEN
Hs_ACTG1_9; SI04361007 QIAGEN
Hs_ACTG1_10; SI04364871 QIAGEN
2.6.2 siRNA protocol
Cells were washed twice in SFM and incubated in Opti-MEM (Gibco) for 1 hour. Cells
were then transfected with the respective siRNA (50 μM final concentration) with
Lipofectamine® 2000 (1 μL/mL) (Thermo Fisher Scientific) in Opti-MEM for six hours,
followed by replacement with DMEM supplemented with 10% FBS. Knockdown was
allowed to proceed for 72 hours prior to infection, or analysis by immunoblot or
immunofluorescence.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
58
Chapter 3: DEVELOPING AN
OPTIMISED VACV GENE-TAGGING
METHOD

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
59
3.1 INTRODUCTION
Author’s note: Sections of the following chapter have been published
in the Journal of Visualised Experiments in 2014 as: Marzook N.B., Procter
D.J., Lynn H., Yamamoto Y., Horsington J., Newsome T.P. (2014)
Methodology for the efficient generation of fluorescently-tagged vaccinia
viruses. Journal of Visualised Experiments (83), e51151, doi:
10.3791/51151.
Viruses F13L-GFP and Lifeact-GFP were created by H. Lynn and C.
McKenzie respectively. Figure 3.3 was generated by Y. Yamamoto, and the
cassette described in Figure 3.5 was designed by J. Horsington. All
remaining work described was carried out by N.B. Marzook.
Orthopoxviruses have large double-stranded DNA genomes (180-220 kb) that encode
upwards of 200 predicted open reading frames (Goebel 1990, Smith 1991). Replication
of these viruses occurs in the cytoplasm and involves the formation of a perinuclear
virus factory, where mature viruses (MV) are made. A subset of MV acquire two
additional membranes in the trans-Golgi network, to generate wrapped viruses (WV),
which are the only morphological form capable of initiating actin nucleation (reviewed by
Roberts and Smith 2008, Newsome and Marzook 2015, and see Introduction section
1.3.1).
Orthopox genomes are amenable to genetic manipulation due to their aforementioned
replication in the cytoplasm (allowing efficient delivery of recombination templates) and
their high degree of homologous genetic recombination with great accuracy [257, 258],
which is a feature of VACV replication. Generating recombinant viruses relies on
homologous recombination mediated by a VACV-encoded DNA polymerase [259], and
linear DNA molecules with homologies as little as 12 bp are sufficient to mediate
recombination in VACV-infected cells [260]. These principles were the foundation
underpinning our goal to optimise VACV gene tagging, utilising a minimal amount of
gene homology and selection techniques for the fast and efficient production of
fluorescent VACV. Optimally, a new methodology would also enable the excision of any
extraneous genes or selection markers, thus enabling the creation of recombinant VACV
carrying more than one fluorescence gene, through sequential genetic modifications.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
60
The ability to quickly and efficiently create recombinant viruses has proven to be key to
expanding our knowledge of poxvirology, from understanding the viruses themselves, to
interactions with their hosts. Viruses expressing fluorescent proteins, which may not only
tag viral proteins, but also be capable of highlighting specific structures in infected cells,
would facilitate future studies of virus-host interactions. The re-modelling of the host
actin cytoskeleton by VACV, as outlined earlier (see section 1.3.1.1), has led to several
key insights of not only virus-based actin motility, but also of the more transient, yet
fundamental, machinery and regulation of actin nucleation within a cell with the help of
fluorescently-tagged proteins.
For example, several GFP-tagged constructs expressing proteins (or specific domains
thereof) involved in the VACV actin polymerisation signalling cascade were used to
tease apart their recruitment at the point of actin comet formation [233, 234, 236, 241,
243]. This defined the role of the N-WASP-WIP complex in actin polymerisation both in
VACV-induced, as well as cellular, actin polymerisation [43]. The dynamics of signalling
proteins, such as their turnover rates at the site of actin polymerisation, can also be
monitored by observing fluorescently tagged proteins like N-WASP, WIP, Grb2 and Nck
at VACV-induced actin comets [241]. Even turnover of actin itself in actin comets can be
monitored by photoactivation techniques (discussed below) [62].
It is with these applications in mind that we set out to create a rapid and efficient method
of creating recombinant VACV that labels the actin cytoskeleton and virus particles
during a live infection, which could then be applied to further study of the role of actin in
VACV infection and spread.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
61
3.1.1 Fluorescent Markers: The Highlights
The discovery of green fluorescent protein (GFP) as an accessory protein to
bioluminescence in the jellyfish Aequorea victoria in 1962 (Shimomura 1962), followed
by its eventual cloning (Prasher 1992) and its expression in several model systems [261-
263], have opened up a new avenue of scientific research into the visualisation of
proteins within cells, tissues or whole systems (Tsien, 1998, Giepmans 2006, Rizzo
2009, Chudakov 2010, Kremers 2011). The fluorescent labelling of proteins is one of the
powerful tools available to us in the quest to understand a protein’s localisation, and
hence function, in a cell (Crivat 2012). In-frame fusions of intrinsically fluorescent tags to
proteins are usually minimally disruptive to the protein (Crivat 2012, Modesti 2011) and
enable tracking localisation over time and space.
Fluorescent proteins (FPs) range in size with monomers typically approximately 25 kDa
(compared to organic fluorophores such as TexasRed which are around 1 kDa (Kremers
2011)) and possess a characteristic central helix surrounded by a β-barrel composed of
11 β-sheets (Ormo 1996). The light-emitting region of the protein, or the chromophore, is
located at the centre of the β-barrel (formed by residues 65-67 in the Aequora victoria
GFP protein (Chudakov 2010)), and it is believed that this structure is similar for all FPs
(Remington 2006). Although the GFP protein sequence is quite resistant to truncation
[261] a few mutations introduced in the amino acid region surrounding the chromophore
increased its intensity of fluorescence when excited at 488 nm, folding efficiency, and
maturation at 37oC [264], creating what is known as enhanced GFP or EGFP. A number
of other mutations have been identified that improve particular characteristics of GFP,
such as the stability and aggregation tendencies of the protein (see [265] for a review).
A. victoria GFP could also be mutated to shift its emission spectrum to blue, violet, cyan
and yellow [266-268]. FPs with emission spectra beyond this are not GFP-derived,
rather they are the result of the discovery of DsRed and other FPs [269-271] in
Anthozoa species of coral. The work of others to extend its emission spectra to the
orange and yellow range [272, 273], and transform it from being an obligate tetramer to
a monomer [274], has generated a palette of FPs to select from, allowing for the imaging
of several tagged components within a complex biological system [265] (Figure 3.1).

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
62
Range of available monomeric fluorescent proteins
Range of available monomeric FPs arranged by emission maxima on the visible spectrum, with
columns depicting their relative brightness. Figure obtained from [265].
These diverse fluorophores are suitable for a range of applications, the most prominent
one being the imaging of fusion proteins, both in fixed and live cells or systems. This
ability to express FP-protein fusions enables a deeper understanding of the localisation,
and hence function, of proteins of interest, as was described by the first instance of such
a fusion construct used to study mRNA transport complexes in Drosophila oocytes [275].
The rainbow of available options when it comes to FPs also allows multi-colour imaging
of several proteins, or structures they may localise to, at the same time. Multi-channel
imaging is possible as long as the excitation/emission spectra of each FP do not
overlap, or if they do, such as in the case of FPs with increased Stokes shifts (where
their emission and excitation spectra are at least 100 nm apart), this can be applied to a
form of dual-colour imaging whereby we may excite two visibly different FPs with the
same laser [276]. This has seen the simultaneous imaging of at least six different
subcellular structures with the same laser line [277], although the possibility of imaging
ten different channels in a single system exists [265].
While the number of available FPs with non-overlapping absorption/emission spectra is
technically the limit for multi-colour imaging of cells [278], other factors to consider are

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
63
their expression systems. The simultaneous transient overexpression of several fusion
proteins in a cell may be detrimental to the cell itself, as well as to the individual
expression profiles of the proteins. Abundance, and hence brightness, of all proteins
should be comparable to minimise spectral bleed-through [265], which may prove
difficult when transfecting cells with multiple FP-expression plasmids. Therefore stable
cell lines expressing fusion proteins, or more tightly regulated systems such as
recombinant viruses capable of expressing fusion proteins in infected cells from their
genomes, are a more desirable option.
Finally, there is always the chance that a fusion protein may disrupt the structure and/or
function of the protein target, by causing misfolding, decrease in expression, or a
reduction in protein activity. Identifying the correct terminus for tagging is also important,
and can be achieved by examining the functional domains of the protein to be tagged,
and their role in protein structure and/or localisation [265]. Sometimes it may even be
necessary to place the tag in between the target protein to achieve functionality [279,
280]. The use of linker regions 6-10 amino acids long in between the tag and target
protein, based on the structure and function of both the tag and protein, is also
recommended [281]. Even so, observations made by fluorescently tagged proteins may
not always be accurate, as evidenced by the contentious study of bacterial cytoskeletal
protein MreB [282]. YFP-tagged MreB was found to organise into helical structures [283,
284], and hence dictated our understanding of this protein. However, cryo-EM studies
instead revealed a more punctate and patchy localisation of the same protein in its
native state, as well as when internally tagged by mCherry [285]. Therefore, it appears
that N-terminal tagging of MreB with YFP specifically caused a helical-folding artefact
that is not observed in its untagged, or alternatively tagged state. As a result, care must
be taken to assess and validate the function of fusion proteins by other methods where
possible.
Another major use of fusion FPs, particularly multi-colour labelling, is the ability to
dissect protein-protein interactions. The simplest way to infer an interaction between two
proteins is to assay for their colocalisation, usually to other larger subcellular structures,
using distinct fluorescing proteins, and is still a powerful tool that is widely used [286-
288]. However, increasingly sensitive instrumentation and continuous refining of the
available palette of FPs has paved the way for more complex studies of protein
interaction. The principle of Förster resonance energy transfer (FRET), which relies on

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
64
the non-radioactive transfer of energy from one molecule (or donor) to another (the
acceptor) if they are within 10 nm of each other [289] has become a popular choice for
understanding protein-protein interactions in cells with greater spatial resolution [290].
FRET analysis measures the intensity of fluorescence emission from the acceptor
molecule after excitation of the donor molecule. This is subject to a number of controls
due to cross-talk between spectrally similar FRET pairs [291]. A more sensitive form of
FRET imaging is FRET-FLIM (fluorescence lifetime imaging microscopy), which relies
on measuring the lifetime of fluorescence emitted by different FPs, and their subsequent
reduction when in proximity to acceptor FPs [292, 293]. It is also much more sensitive,
and independent of emission intensity, requiring fewer controls and is also capable of
monitoring changes to the local protein environment [294, 295]. Unfortunately, FRET-
FLIM also relies on highly specialised and expensive instrumentation and hence is not
viable for mainstream use [294]. FRET can also be applied to the field of biosensors
[296], such as the creation of biosensors to study oncogenic signalling molecules [297].
In addition to protein-protein interactions, advanced fluorescence microscopy techniques
can be employed to study molecular dynamics within a cell. Fluorescence recovery after
photobleaching (FRAP) is one such technique that relies on the fact that high intensity
excitation of FPs can cause them to photobleach, i.e. reduce their emission signal, thus
allowing the monitoring of particles into, and out of, a particular area of a cell that has
been bleached [295, 298]. The scope of a FRAP experiment can be expanded from a
photobleached region to the whole cell by measuring fluorescence loss in
photobleaching (FLIP), whereby one area is subjected to repetitive photobleaching,
while the rest of the cell is monitored for a decrease in fluorescence intensity as a result
of movement of bleached particles out of that area [294, 299]. Photobleaching can also
be coupled to FRET and is divided into two categories: donor or acceptor
photobleaching. Donor photobleaching measures the bleaching rate of the donor FP
with and without the presence of the acceptor and generally takes longer timeframes,
while acceptor photobleaching measures changes in the emission intensity of donor FPs
before and after the acceptor FP is photobleached. As a result, acceptor photobleaching
is relatively faster and can also be carried out in live cell systems [294]. In addition,
photoactivatable or photoswitchable fluorescent proteins such as PA-GFP, PS-CFP2
and Dendra2 [300, 301], whose fluorescence intensities or emission wavelengths can be

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
65
modified by specific intense irradiation, can be used to track a specific subset of tagged
proteins within a larger system in time and space.
Advanced microscopy techniques enabling super-resolution of cellular structures that
overcome the diffraction limit of traditional light microscopy combined with fluorescence
tagging that offers several advantages over higher-resolving tools like electron
microscopy, which is generally time-consuming, requires greater technical skills and can
only be performed in fixed samples [302]. Super-resolution microscopy has enabled us
to image multiple labelled proteins simultaneously, with enough resolution to be able to
discern cellular structures and dynamics in fixed and live samples [303].
3.1.2 Fluorescent Labelling Goes Viral: Applications for Virology
The applications of fluorescent markers are only limited by the nature of the tags (pliable
to a point by exploratory mutations or rational design), the technology of our
fluorescence detection tools, and to the properties of the protein being tagged.
Fluorescent tagging of viral proteins has proven invaluable to the study of host-pathogen
interactions [304-307]. Studies can range from the use of fluorescently tagged HIV-1 to
track the uncoating of single virus particles in time and space [308], to the creation of
replication competent fluorescent viruses for use in anti-viral screening assays [309,
310].
Fluorescent labelling of VACV can yield extremely bright virus particles due to the large
size of orthopox particles, which allows the incorporation of many fluorescent proteins
per virion [304]. Vaccinia virus has the capacity to carry large fragments of foreign DNA
[311] and furthermore, the lack of rigid capsid symmetry may permit a degree of
flexibility when expressing viral protein gene fusions from their endogenous loci [312].
Since the creation of a C-terminal fusion of VACV envelope protein B5 to the enhanced
green fluorescent protein (GFP), and the discovery that this fusion protein still localised
to the Golgi and was capable of restoring a B5R deletion mutant virus [217],
fluorescently-tagged VACV proteins have been employed to study various aspects of the
replication cycle at the subcellular level. The same B5-GFP tagged VACV, as well as

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
66
VACV expressing GFP fused to another IEV protein F13, were used to describe the
microtubule-based motility to the cell surface, before switching to actin-based cell egress
[170, 215]. GFP tagged F13 was found to not overtly disrupt virus assembly or actin
comet formation [215]. Similarly, fluorescently tagged wrapped VACV have been used to
further elucidate intracellular morphogenesis and movement of viruses [209, 214, 304].
By labelling distinct morphological components of VACV particles - such as the core and
envelope proteins - with complementary tags, more complex questions of virus entry and
uncoating can be understood. For example Schmidt et al [112] created a doubly tagged
VACV, where core protein A5 was fused to mCherry and envelope protein F13 to GFP,
enabling the tracking of both wrapped and unwrapped virions during entry and
morphogenesis in a single infected cell.
3.1.3 Creating Recombinant VACV
A number of methodologies have been employed for the creation of recombinant VACV
[313]. Initially, inactivating insertions of foreign DNA into the VACV thymidine kinase
(TK) locus were selected for by plaque assay in TK- cell lines, with the addition of 5-
bromodeoxyuridine as a thymidine substitute [314, 315]. TK- VACV mutants could also
be rescued by insertion of the herpesvirus TK gene [316].
Selectable markers such as the Escherichia coli beta-galactosidase (beta-gal) gene can
also be introduced into the TK gene, which allows the selection of blue plaques in the
presence of a beta-gal indicator to the overlay media [317]. Beta-gal alone my be
introduced, along with a foreign gene, into VACV and recombinant viruses can be
selected for by picking blue plaques when grown in an agarose overlay containing X-gal
[318, 319]. The use of fluorescent proteins as screen-able markers, enabling isolation of
viral plaques based on fluorescence is another popular technique [217, 320], which
forms part of the foundation of our method.
The dominant selectable marker gene gpt (the E. coli xanthine-guanine phosphoribosyl
transferase) is widely used to efficiently create and select recombinant VACV. When
myocophenolic acid (an inhibitor of purine metabolism which normally blocks VACV

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
67
replication), xanthine, and hypoxanthine are added to the media of infected cells, only
recombinant viruses carrying the gpt transgene are selected for [321, 322]. Therefore
recombination cassettes that embed gpt, as well as the desired exogenous DNA
bounded by regions of homology, into the VACV genome, can be used to create
recombinant VACV. Finally, selection based on rescue of an attenuated growth
phenotype has proven to be quite popular [323-325]. For example, a deletion mutant of
VACV missing envelope protein A27 presents with a small plaque phenotype, thus
providing easy pickings of recombinant viruses when infected cells are also transfected
with a plasmid carrying the rescue A27L gene as well as the desired exogenous DNA
[326]. The advantage of such a method is that it does not leave behind superfluous
selection DNA in the VACV genome, however it does not usually allow site-directed
tagging of specific VACV genes, say with FPs, since the site of homologous
recombination is normally directed to the gene being rescued, or to a non-essential site
in the VACV genome.
As most methods of creating recombinant VACV involve the use of selection markers
that often remain in the VACV genome, the inability to modify VACV genes themselves,
as well as numerous selection steps often requiring complementary cell lines, we aimed
to create a simple and efficient method of creating recombinant VACV that did not result
in any extraneous genes in the final product, while also providing the option of site-
specific tagging of VACV genes. A major benefit of a recombinant virus that does not
retain its selection markers is our continued ability to add exogenous DNA to the same
virus using the same selection methods.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
68
3.1.4 Dominant Selection and Fluorescent Markers – With Their Powers Combined
We chose to develop a method for the selection of fluorescent viruses using a
combination of fluorescent screening and metabolic selection. Following on from their
use of the gpt gene for metabolic selection [322], Falkner and Moss expanded its use in
1990 for the creation of marker-free VACV by transient dominant selection (TDS) [327].
A vector containing the gpt gene, along with the desired exogenous DNA flanked by
regions of homology to the VACV genome, was created. When VACV-infected cells are
transfected with this plasmid, a single recombination event causes the entire TDS
plasmid to integrate into the genome. When these recombinant VACV are grown in cells
under metabolic selective pressure with the use of mycophenolic acid (MPA; an inhibitor
of purine metabolism) and xanthine (a purine precursor that can be converted to guanine
by gpt), only those carrying the gpt gene will be able to survive. The removal of MPA
from growth media in successive rounds of plaque purification will cause a second
recombination event where the gpt gene is excised; either reverting the virus to its
original sequence, or producing a recombinant virus only containing the desired DNA
addition (Figure 3.2).

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
69
Method of transient dominant selection
First, a single cross-over event introduces the entire plasmid into the VACV genome. Only
VACV expressing the gpt gene will survive by metabolic selection. Once selection pressure is
removed, a second cross-over event occurs within the VACV genome, excising the gpt gene
while leaving the added DNA sequence at the target site. The other possible recombination
event will revert the virus to the wild-type genome. Not pictured: antibiotic resistance genes and
origin of replication sites on the TDS plasmid. Image adapted from [327].

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
70
A further selection step can be incorporated into the TDS vector, by adding a further
mode of screening for primary recombinant plaques using a constitutively expressed
fluorescent marker gene, as described by Cordeiro et al [229]. In this case, the gpt
gene is accompanied by red fluorescent protein mCherry (Figure 3.3), thus enabling
both metabolic selection as well as fluorescent screening by eye when picking
recombinants after the initial crossover step. Once metabolic selection pressure is
removed, both gpt and mCherry genes are excised, leaving behind two possible
genomic outcomes: the original virus, or a recombinant one containing only the desired
gene addition. For our method, we aimed to develop an efficient method to rapidly tag
VACV genes with fluorescent genes. Designing a plasmid containing exogenous DNA
flanked by homologous sites usually involves several rounds of PCR and cloning
techniques. The increasingly affordable economics of DNA synthesis [328, 329] has
meant that we can reduce the steps involved in creating such a vector by simply
designing and synthesising oligonucleotide cassettes of minimal homology lengths –
making sure it will allow for efficient recombination while still keeping down costs of
DNA synthesis. These designed cassettes contain restriction sites both within the
regions of homology (such that any fluorescent gene of choice can be inserted for use
as a tag), as well as flanking it (such that it can be cloned into the TDS vector). As a
result, plaques exhibiting both mCherry and the desired tag fluorescence are selected
after the initial recombination step, while once selection pressure is removed, correctly
resolved viruses that have lost the mCherry gene, leaving behind only the desired
fluorescent tag, can be picked. The excision of the selection markers allows the
possibility to combine multiple fluorescent tags through sequential modifications,
enabling us to create viruses with several fluorescently labelled viral proteins
simultaneously. A step-by-step description of the technique developed follows.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
71
3.1.5 VACV Genes Of Interest
For this pilot study, we chose five VACV genes of interest to be tagged, and hence 150
bp regions of homology, corresponding to the left and right arms from the point of tag
insertion for each gene, were designed into the synthesised cassette. The five genes
were A36R, A3L, F1L, F12L and F13L. The F13L-GFP virus was created and
described by H. Lynn [330]. A brief description of the other selected VACV genes
follows:
3.1.5.1 F12L
F12L encodes a 65 kDa protein which enables the microtubule-mediated egress of IEV
particles to the cell surface [230]. The loss of this protein causes a reduction in
virulence, produces a small plaque phenotype [331] and results in the absence of CEV
on the infected cell surface [230]. More recent studies have shown that F12 associates
with viral protein E2, which is essential for IMV morphogenesis [219], as well as with
another IEV-associated protein A36, which is necessary for viral egress [225]. It also
shows structural similarities to the cellular kinesin light chain [220] and like A36, also
interacts with the kinesin-1 motor complex during virus egress [222]. There is much to
be understood about the interactions between F12, A36 and E2 at the point of IEV-CEV
transformation, which a recombinant VACV expressing fluorescently tagged F12 may
be able to address.
3.1.5.2 A36R
A36R encodes a 45 kDa type Ib transmembrane protein and is exclusively present on
the outer of the two IEV membranes [224]. In addition to aiding in microtubule-based
transport of mature virions to the cell surface [215, 218], A36 is also crucial for actin-
based motility of VACV [233, 234, 253, 332], as its phosphorylation by host proteins
begins a signal cascade that ultimately results in Arp-2/3 mediated polymerisation of
actin beneath virus particles [43, 236, 241], where A36 localises before the virion is

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
72
released [332]. A36 is also expressed and presented on the cell surface early in
infection, enabling what is known as ‘super repulsion; where infectious virus particles
are repelled from already infected cells by actin projections as a result of A36
expression, thereby enabling VACV to leap-frog over cells until reaching an uninfected
target [333]. More recently, research has shown that A36 itself may itself possess
nucleation-promoting factors (NPFs) that recruit N-WASP and associated proteins
implicated in VACV release [246]. Although recombinant VACV expressing tagged A36
such as A36-YFP exist [332], we wanted to be able to develop a system to easily tag
this crucial protein with different proteins to answer varying questions, especially when
creating double-tagged viruses.
3.1.5.3 A3L
The A3 protein is expressed as a 72.5 kDa precursor, and cleaved during virus
maturation into one of about 65 kDa [334, 335]. Mutations in, or the loss of A3
altogether, respectively results in either the production of defective cores [336], or their
complete loss [337]. This is because it forms the inner layer of the VACV core [338,
339], and hence is present in both IMV (the most abundant infectious VACV particle) as
well as IEV. A3 is the fourth most abundant protein in VACV [210], and hence any
recombinant VACV expressing fluorescently tagged A3 would produce relatively bright
particles capable of highlighting the virus factory (where virus cores are created) [231].
3.1.5.4 F1L
F1L encodes F1, a comparatively smaller protein at 26 kDa. Additionally, unlike the
VACV mentioned so far, F1 is neither a structural protein, nor is it involved in the VACV
transport. Instead, F1 is responsible (along with a few others [340]) for the ability of
VACV to inhibit host cell apoptosis. This occurs through its association with (and
inhibition of) Bcl-2-like proteins Bim and Bak, pro-apoptotic proteins responsible for the
ultimate release of cytochrome c from mitochondria [341-344]. F1 also localises to
mitochondria, into which its C-terminal transmembrane domain is inserted [345]. The
sequence of F1 bears no resemblance to eukaryotic proteins involved in apoptosis, and

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
73
yet its structure is capable of attaining similar folds as those that are involved in the
apoptotic pathway [346]. Structural studies into its interaction with Bcl-2-like proteins is
only just starting to be understood [346], and hence advanced microscopy techniques,
such as super-resolution microscopy, which are capable of revealing detailed structural
information of tagged proteins such as F1 and its interacting partners and/or
organelles, would be extremely useful in learning more about this important viral
survival technique.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
74
3.2 RESULTS
3.2.1 Minimal homology length required for homologous recombination in VACV
Previous studies have examined the minimal homology requirements for VACV-
mediated recombination of linear and circular DNA molecules [347]. Recombination
between three different types of DNA molecules was examined: linear-linear, circular-
circular, and linear-circular, with decreasing efficiencies respectively. In linear molecules,
16bp of homology was found to be sufficient for a 4% recombination efficiency, which
reduced by up to 50 times for circular molecules. DNA molecules tested for
recombination encoded overlapping regions of the luciferase gene, and recombination
frequencies were assessed by luciferase assays. We wanted to determine the minimum
homology length required for recombination within the VACV genome itself. For this, we
used a series of plasmids containing gpt and mCherry genes, along with varying regions
of homology to the VACV genome. BSC-1 monolayers were infected with VACV and
transfected 1 hour post-infection (hpi) with three recombination vectors containing
regions of homology of 500 bp, 100 bp, or 70 bp to the VACV genome. Cells were
recovered 24 hpi and lysed to release the recombinant viruses formed. Plaque assays
were performed on cell lysates with GPT selection media and plaques showing mCherry
fluorescence were counted as successful recombinants. It was determined that
homologous regions of 70 bp in the TDS vector are sufficient to allow the insertion of
exogenous DNA into the VACV genome by homologous recombination (Figure 3.3),
with the number of successful recombination events (as determined by mCherry-positive
plaques) increasing with respect to homology length.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
75
Quantitative analysis of recombination efficiencies between
recombinant vectors and the VACV genome
BSC-1 monolayers were infected with VACV and transfected 1 hpi with three recombination
vectors containing regions of homology of varying lengths. Cells were recovered 24 hr post-
infection and lysed to release the recombinant viruses formed. Plaque assays were performed
under GPT selection and plaques showing mCherry fluorescence were counted as successful
recombinants (n=3 replicate experiments). Figure generated by Y. Yamamoto.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
76
3.2.2 Designing the recombination vector
The plasmid backbone used to develop our methodology has been described previously
[229], however it has been expanded upon in this case to rapidly and efficiently tag
multiple VACV genes of choice (Figure 3.4). Although it was determined that 70 bp was
sufficient for homologous recombination to occur using this system, we opted to use 150
bp to increase the efficiency of recombination while still keeping down the costs of
oligonucleotide synthesis. Firstly, 150 bp long flanking regions of homology (referred to
as the left and right arms) were identified, based on whether the viral gene of interest
was to be N- or C-terminally tagged (Figure 3.4b). An oligonucleotide sequence
comprising the 150 bp left and right arms, separated by a pair of restriction sites of
choice (NotI and BamHI), was designed for synthesis. These restriction sites matched
those flanking the open reading frame of our fluorescent tags (Figure 3.4c). It is
possible to use a NotI restriction site as a three amino acid linker between the left arm
and the start of the fluorescent tag. Primers incorporating NotI and BamHI into GFP
were created (as described in Table 2.1.8) and used to create a GFP sequence
containing the matching restriction sites, for incorporation into the TDS vector in
between the left and right arms of homology. A second, different pair of restriction sites
(HindIII and SalI) was also designed such that they flanked the entire sequence,
allowing incorporation into the TDS vector once synthesised. This second pair of
restriction sites corresponds to those present on the TDS vector (which has had its NotI-
BamHI sites blunt-ended first, for later incorporation of the GFP tag; see Figure 2.1C).
After obtaining the synthesised fragment in a commercial vector (see Table 2.1.9),
digestion using the restriction enzymes corresponding to the flanking restriction sties
was performed and resulting fragments cloned into the TDS vector as regions of
homology. This resulting vector was then cut by restriction enzymes corresponding to
sites in between the left and right homology arms, enabling insertion of the fluorescent
tag, also cut by the same restriction enzymes (Figure 3.4d). Since we aimed to create
recombinant VACV with multiple fluorescently tagged viral proteins, we created an
oligonucleotide containing several homology cassettes corresponding to five viral genes
of interest (Figure 3.5A). Each homology cassette consisted of the 150 bp left and right
homology arms separated by NotI-BamHI restriction sites (which also served as the
fusion protein linker – see Figure 3.5B), and is also bound by HindIII and SalI restriction
sites. Once synthesised, the five cassettes were separated by a HindIII-SalI restriction

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
77
digest, followed by cloning into the TDS vector also cut by the same restriction enzymes.
The identity of each vector was determined by testing with a further restriction digest
based on ‘kill-cut’ sites (specific sites that will linearise the plasmid) were also
incorporated into the cassettes, either at the site of the linker or within the arms
themselves (Figure 3.5B) This enabled the easy identification of plasmids containing the
LA/RA sites for each gene.
Creating the Transient Dominant Selection (TDS) recombination
vector
The (a) TDS vector with gpt and mCherry selection markers. (b) Left and right arms (LA and
RA) of homology are designed with specific restriction sites in between and flanking the arms of
homology. Restriction sites in between the left and right arms used in this method were NotI
and BamHI, the NotI site also being used as a linker between the gene and fluorescent tag. (c)
Fluorescent tags compatible with this method are flanked by corresponding restriction sites.
Some tags explored were eGFP (enhanced green fluorescent protein), RFP (red fluorescent
protein), Cerulean (an improvement on ECFP, a cyan fluorescent protein, by site-directed
mutagenesis [348] and mini-SOG, a fluorescent protein engineered from GFP, which creates a
product resolvable by EM on illumination [349]. (d) Cloning steps involved in the generation of
the final TDS recombination vector. The synthesized oligonucleotide containing the left and
right flanking arms was first cloned into the TDS vector. This provides a recombination vector
into which any tag of choice can be shuttled in and out by cloning into the restriction sites
incorporated in between the left and right arms.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
78

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
79
Map of synthetic oligonucleotide carrying homology regions for fluorescent gene insertion.
(A) Five VACV genes were chosen for fluorescent tagging. 150 bp-long left and right homology arms corresponding to each gene were selected,
depending on whether the tag was to be N- or C-terminal. All arm pairs were flanked by HindIII-SalI restriction sites. (B) The left and right arms
were separated by NotI-BamHI restriction sites for tag insertion, and also contained a unique restriction ‘kill-cut’ site, enabling their identification
once cut out of the cassette and cloned into the TDS vector. These kill-cut sites were present within the linker regions for A, C and E, while they
were located in the right arms for B (*; XbaI) and D (**; SpeI).

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
80
3.2.3 TDS vectors containing synthetically designed oligonucleotides provide a
rapid and efficient method for recombinant VACV generation
Figure 3.6 provides a step-by-step outline for the procedure described below, with
representative fluorescent plaque images of an A3L-GFP recombinant VACV
depicted for each step of the selection process.
A monolayer of BS-C-1 cells were infected with VACV in serum-free media at an
MOI > 1, and transfected with the TDS recombination vector of choice (in the case of
Figure 3.6, a recombination vector aimed at N-terminally tagging the VACV A3L
gene with GFP, see Table 2.1.9 for vectors used) 1 hpi. Cells were recovered after
24 hours, freeze-thawed to release virus particles and a plaque assay with a liquid
overlay of 10% FBS-DMEM and GPT selection reagents mycophenolic acid (25
µg/ml) and xanthine (250 µg/ml) was performed. After a 24-hour incubation, the
liquid overlay was removed and virus plaques exhibiting diffuse red fluorescence
corresponding to the incorporation of mCherry from the TDS vector into the virus
were picked. Since the recombination vector was aimed at creating an N-terminal
tag to the VACV core protein A3, green fluorescence was also observed in these
plaques. Picked plaques were amplified with GPT selection reagents and a plaque
assay was repeated, but with an agarose overlay under GPT selection. 2-3 dpi,
plaques exhibiting both red and green fluorescence were picked and amplified, but
with no GPT selection this time. A plaque assay of amplified plaques was performed
with an agarose overlay, again with no selection. Plaques that have lost their diffuse
red fluorescence but retained the localised fluorescence corresponding to the A3-
GFP tag were picked, amplified and subjected to another plaque assay under no
selection. At this point, all resulting plaques had lost their red fluorescence
corresponding to mCherry, but retained the green fluorescence corresponding to A3-
GFP. Thus, within four rounds of plaque purification, pure recombinant VACV
containing only the desired fluorescent tag can be obtained.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
81

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
82
Outline of the experimental procedure to create recombinant VACV using TDS.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
83
Figure 3.6 Description: Events occurring at the genetic and cellular levels are depicted, along with
representative plaque images outlining the steps following the creation of recombinant VACV GFP-
A3L. (A) Cells infected with vaccinia virus were transfected with the TDS recombination vector. (B) In
this figure, only the result of left-hand recombination is depicted and the example uses GFP as the
fluorescent tag of choice. Right-hand recombination would result in the entire TDS plasmid being
incorporated into the genome in a similar way, except the tag would be fused to the entire target gene
in the intermediate step, i.e. step C. A plaque assay was performed on a cell monolayer with the
recombination mix and subjected to GPT selection. (C) Plaques exhibiting both red and green
fluorescence, corresponding to mCherry and GFP expression respectively, were picked and amplified.
Loss of red fluorescence corresponding to the loss of the gpt and mCherry genes occurs after removal
of GPT selection (D), and plaques exhibiting exclusively green fluorescence are picked and amplified
(E).
3.2.4 Successful creation of recombinant VACV
Of the homology arms corresponding to the five VACV genes designed, three were
successfully used to create recombinant VACV: GFP-A3L (GFP N-terminally tagged to
the VACV core protein-encoding gene A3L (Jensen 1996)), GFP-F1L (GFP N-terminally
tagged to viral protein-encoding gene F1L which localises to the mitochondria and
inhibits apoptosis (Wasilenko 2005)) (depicted in Figure 3.7), and F13L-GFP (described
by Lynn, H. [330]). Additionally, a Lifeact-GFP VACV, a virus constitutively expressing
Lifeact (Reidl 2008) fused to GFP and capable of highlighting the actin cytoskeleton of
an infected cell in real-time, was also created using the same method (described by
McKenzie, C [350]). The two oligonucleotides that did not produce recombinant VACV
were those corresponding to A36R and F12L. While attempts to create a GFP-A36R
virus reached the final stages of plaque purification, the intensity of green fluorescence
required to be able to pick successful recombinants following removal of GPT selection
reagents was not achieved, due to the low abundance of A36 protein produced by
VACV within a cell, compared to the expression levels of core protein A3 or envelope
protein F13. The creation of an F12-GFP virus by this method was not attempted due to
time constraints.
Confirmation of the creation of successful VACV recombinants GFP-A3L and GFP-F1L
was done by three methods. Firstly, confirmation of the site of insertion of the GFP gene

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
84
into the VACV genome was done by PCR. Primers spanning the site of insertion were
used in both cases. Genomes of both GFP-A3L and GFP-F1L viruses showed an
increase in size by about 700 bp when compared to the parental VACV strain (Figure
3.7A). This corresponds to the predicted increase in size by 741 bp for both
recombinants (accounting for the 717 bp of Bright Human GFP and 24 bp of the linker
region – see Figure 3.5B). Secondly, a western blot was conducted to determine GFP
expression by both recombinants. HeLa cells were infected with the recombinant viruses
and scraped 24 hpi. A vector expressing GFP under a VACV pE/L promoter [254] was
also transfected into cells infected by VACV-WR as a positive control. GFP is around 27
kDa in size, while the expected sizes for GFP-A3 and GFP-F1 are 92 kDa (65 kDa + 27
kDa) and 53 kDa (26 kDa + 27 kDa) respectively, all of which roughly correspond with
our observations (Figure 3.7B). A re-blot of the same membrane confirms expression of
the VACV protein A36, and actin as a loading control. Finally, the localisation of the
tagged proteins was observed by immunofluorescence (Figure 3.7C). A3 is a core
VACV protein, and hance GFP-A3 appeared as distinct points, localising particularly to
distinct regions around the nucleus, which we can assume is the virus factory. F1, being
a protein that localises to the mitochondria, was found to essentially highlight
mitochondrial-like structures in the cell. A3 is a much more abundant protein than F1
and hence, cells expressing GFP-A3 were much more readily visible than those
expressing GFP-F1.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
85
Recombinant viruses created using modified TDS recombination.
Agarose gel image of PCR results showing successful inclusion of the GFP gene at the desired
locus in both GFP-A3L and GFP-F1L viruses. Genomic DNA from each recombinant virus as
well as the parent VACV-WR strain were as templates. (B) Western blot of GFP expression in
lysates of HeLa cells infected with the recombinant viruses depicted. A vector expressing GFP
under the control of a VACV pE/L promoter was also included. The same blot was stripped and
re-probed for A36 and actin. (C) Fluorescence images of plaques and individual cells infected
by the respective recombinant viruses. A3 is a core protein of VACV and F1 localises to
mitochondria.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
86
3.2.5 Characterisation of recombinant VACV
Having confirmed that our recombinant VACV contain the fluorescent gene insert at the
right location and that it is being expressed, our next step was to characterise these
viruses to compare its replication dynamics and known functions (and/or localisations)
within an infected cell. A plaque assay comparing the parental strain WR to GFP-A3L
and GFP-F1L did not reveal any significant differences in plaque sizes between them
(Figure 3.8A). Wide-field live microscopy of cells infected with GFP-A3L was
conducted, using which GFP-tagged particles were observed in peri-nuclear virus
factories and engaging in microtubule-based transport towards the cell periphery over a
period of 10 minutes (Figure 3.8B). MitoTracker Red, which stains live mitochondria,
was used to confirm the localisation of GFP-F1 to mitochondria. Interestingly, F1 was
found to localise only to the mitochondrial membrane, while MitoTracker Red stained
the entire organelle (Figure 3.8C, inset). This agrees with F1 possessing a C-terminal
transmembrane domain, which facilitates its tight binding to the mitochondrial
membrane [345].

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
87
Characterisation of recombinant VACV.
(A) Quantitative analysis of plaque diameters, along with representative images of plaques
created by WR, GFP-A3L and GFP-F1L viruses in a monolayer of BSC-1 cells 5 dpi (n=12;
14 for WR; statistical analysis performed using PRISM v6 by Student’s t-test). (B) Real-time
tracking of GFP-A3 positive virus particles in a HeLa cell over a 10-min period 8 hpi. Images
were captured with in the FITC channel with a Nikon Eclipse Ti-E inverted microscope (see
section 2.3.2.3 for microscope details). Magnified regions outlined in the original image are
depicted as a time course following a single GFP+ particle. (C) HeLa cell infected with GFP-
F1L and stained with MitoTracker Red 10 hpi. Magnified regions outlined in the original
image depicting labelled mitochondria are indicated below. Scale bar is 10 μm.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
88
3.2.6 Recombinant viruses carrying more than one fluorescent tag can be created
Once pure stocks of recombinant VACV are generate, cells can be co-infected with two
(or more) VACV carrying different tags to create recombinant VACV carrying more than
one fluorescent tag, broadening their applicability in studying VACV infection in real-
time (provided their emission spectra do not overlap). We used a previously created
RFP-A3 VACV to co-infect cells containing our TDS-made recombinants, to generate
double- and triple-tagged VACV. Lifeact-GFP VACV and RFP-A3 created a VACV that
allowed us to visualise the formation of actin tails by virus particles at the cell surface in
real-time (Figure 3.9A). A maximal intensity projection of a live movie taken over 5
minutes reveals the total path lengths of the actin comets (Figure 3.9B), which may be
used to calculate speed of virus movement. A plaque assay was conducted to compare
the double-tagged virus to its original parent strains. The plaques produced by the
Lifeact-GFP/RFP-A3L virus were significantly smaller than those created by the
individual Lifeact-GFP and RFP-A3L viruses, as well as the parental VACV-WR strain.
There was no difference between the individually tagged recombinant VACV and the
parental strain. Therefore expressing both fluorescent tags at once may be additively
taxing on the replication dynamics of this double-tagged virus.

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
89
Creation of recombinant Lifeact-GFP/RFP-A3 VACV.
(A) Wide-field microscopic images of a HeLa cell infected with the Lifeact-GFP/RFP-A3L virus
over time. Scale bar is 10 μm. (B) Maximal projection of a 5-minute video of the infected HeLa
cell. (C) Quantitative analysis of plaque diameters, created by the viruses indicated in a
monolayer of BSC-1 cells 5 dpi (statistical analysis performed using PRISM v6 by Student’s t-
test; * p<0.05, *** p<0.001).

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
90
3.3 DISCCUSSION
This technique describes a novel protocol for an efficient and modular method to tag
specific genes in the VACV genome. This method also ensures that the only change to
the viral genome is the addition of the tag, leaving behind no extraneous DNA in the
form of selection markers. The uses of tagged viral proteins are many and varied,
ranging from understanding virus morphogenesis and trafficking, discerning colocalising
proteins and hence inferring possible functions, to discovering the purposes of as yet
uncharacterized VACV proteins based on visualising their localisation and behaviour
within an infected cell.
This technique takes advantage of the increasingly accessible and affordable ability to
synthesise custom oligonucleotides. The short arm length required for homologous
recombination enables its direct synthesis, eliminating several time-consuming rounds of
PCR and cloning. While smaller homology lengths would also enable recombination,
100 bp homology lengths provided sufficient recombination frequency such that viruses
that could be readily generated and identified with metabolic selection and screening by
fluorescence. DNA fragments of this size can be commercially synthesized at relatively
low cost greatly facilitating the production of multiple vectors for the creation of
recombinant viruses. Although 70 bp was found to be sufficient to create recombinants
by this method, we opted to increase the homology length to 150 bp to provide greater
recombination frequency while keeping down costs for synthesis of
the oligonucleotide sequence of flanking regions.
The other aspects of the TDS vector are the fluorescence of mCherry and metabolic
GPT selection, which are used to isolate viral recombination intermediates. A similar
method involving the use of both fluorescent and metabolic selection has been
described previously [351], although this was done by inserting transgenes in between
two essential VACV genes to promote their stability, instead of being targeted to specific
genes of interest. In our case, virus intermediates can be resolved, following the removal
of selection, to a virus with a tagged gene or back to the parental type, allowing the
selection of the desired recombinant virus by imaging the fluorescence of the tagged
gene of interest. An advantage of this is that fluorescently tagged proteins are expressed

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
91
at endogenous levels in the cell, since tags are fused to the gene of interest, and hence
the viral promoter controls its expression.
However, this secondary selection is only applicable for tagging highly expressed viral
genes that produce sufficient fluorescence to be detected in a plaque assay. Therefore
VACV proteins that are comparatively less abundant, like A36 and F12, proved harder to
isolate via this method, and is one of the limitations of this technique. Without this, it may
be possible to pick recombinant viruses based on mCherry fluorescence under
metabolic selection, followed by picking of several non-fluorescent plaques after
selection is removed, of which at least 50% would contain the desired recombinant
viruses. Those possible recombinants could then be identified by molecular strategies
such as PCR. Alternatively, one could envisage the insertion of a complete expression
cassette, for example a fluorescent protein under a strong viral promoter. In this case
the left and right arms would define the point of insertion rather than the viral gene to be
tagged.
Another advantage of this technique is the ability to create recombinant VACV
containing more than one tagged gene, since the selection markers are excised during
the process. By excising selectable markers, the TDS method allows for the serial
addition of various fluorescent proteins or the combination of TDS-based tagging with
TDS-based gene deletions for phenotypic analyses [352]. While double-tagged viruses
can be made by co-infection of two single-tagged parents [231], and as we have done,
recent studies have shown that co-infection of two VACV strains produces genomes with
a patchwork or crossover events from each parent, at a rate of one crossover/12 kbp in
the case of one study [353]. Therefore, while this may not be problematic if both viruses
came from the same parent, the modular addition of tags to one virus may prove more
faithful to the original VACV strain.
Nevertheless, multiple-tagged viruses can prove very beneficial for understanding the
more complex processes involved in VACV morphogenesis, especially with the advent
of sensitive live cell imaging. Imaging studies with this virus could be used to study
movement, morphogenesis and wrapping of virus during virus replication.
There are some key steps that proved helpful during the experimental procedure. The
liquid overlay proved crucial for the detection and isolation of red/green fluorescent

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
92
plaques. We believe that the combination of the GPT selection reagents and agarose
overlay deterred the growth of recombinant viruses, and therefore switched to a liquid
overlay for the first step of amplifying viruses following transfection. It is also important to
pick fluorescent plaques showing localized tag colour fluorescence for enrichment and
purification, as intermediates resulting from left-arm recombination may result in diffuse
fluorescence observed in plaques if the left arm also contains a promoter sequence.
The mCherry marker gene in the TDS vector may also be replaced by gfp, for example,
to allow for the easy incorporation and selection of mCherry as a fluorescent tag.
Some techniques described above vary slightly from established methods of creating
recombinant vaccinia virus. For example, the MOI of virus used to create recombinants
is normally less than 1 (Broder 1997), however the use of higher MOIs has been
sufficient for the creation of recombinant vaccinia virus by this method. The pre-
incubation of cells with GPT selection reagents (mycophenolic acid, xanthine and
hypoxanthine) for recombinant VACV selection was recommended in the first iteration of
TDS [322], and repeated by some since [354], but not by others [355, 356]. The purpose
of pre-incubation of cells has never been expressly stated, but one might guess that it
depletes cells of purines (especially guanine monophosphate, whose production MPA
specifically inhibits), which further enhances selection of gpt+ recombinant VACV in
infected cells. The first description of the use of the E. coli gpt gene as a selection
marker does not mention pre-incubation, since it involved selecting for transformed cells
themselves, and also described the use of both MPA and aminopterin to completely
block purine synthesis [357]. We opted to forego pre-incubation of our cells, since MPA
also slows down the growth of mammalian cells in general [357], and this method was
still sufficient to detect gpt+ VACV, particularly since we possessed the added
advantage of mCherry+ selection. Furthermore, we opted to only use MPA and xanthine
in the selection reagents, as others have [358], since hypoxanthine is only a necessary
supplement if both MPA and aminopterin (which blocks the de novo synthesis of all
purines [359]) are used as inhibitors.
As mentioned previously, the use of protein tags may also disrupt the properties of the
original protein. We attempted to address these issues by comparing phenotypes such
as plaque size with respect to the WR strain, live-cell microscopy to track tagged VACV
in the case of GFP-A3, and the use of alternative staining methods such as MitoTracker
Red in the case of GFP-F1 (Figure 3.8). We did not observe statistical differences in the

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
93
plaque sizes of single-tagged viruses GFP-A3L and GFP-F1L, however GFP-A3L
exhibited a trend towards smaller plaques similar to that observed previously for a YFP-
A3L virus (although it remained statistically insignificant) [226] and a Dendra2-A3L virus
[330]. A3 is an abundant 65 kDa coat protein and hence tagging it with the 27 kDa-large
GFP might hamper efficient viral assembly. GFP-F1 could serve as a valuable
alternative to dye-based labelling of mitochondria, along with mitochondrial markers
such as mitoGFP/YFP/RFP, since they enable imaging for longer periods of time (24
hours or longer), compared to fluorescent dyes which usually cause the disintegration of
mitochondria within an hour of labelling [360]. The significantly smaller plaque sizes
exhibited by the double-tagged Lifeact-GFP/RFP-A3L virus resulted in our preference for
use of the single-tagged Lifeact-GFP virus for the study of actin comet formation in
further studies (see Chapters 4-6).
Recombinant VACV are generally stable over time [311, 361-363], although recombinant
MVA (Modified Vaccinia Ankara – an attenuated strain of VACV) carrying HIV env and
gag-pol genes were found to have lost transgene expression due to silencing mutations
and/or deletions following several passages [364]. However this was likely due to their
detrimental effects on virus replication, and recombinants were stabilised once more
stable versions of the HIV proteins were inserted instead. More recent studies have
shown that VACV accumulates around 1 x 10-8 mutations per replication cycle, and
larger deletions may occur after around 70 passage events [353]. Hence, care must be
taken to maintain a stock of VACV at early passage time points, not only for new
recombinants, but also for parental strains. VACV can also undergo both inter- and
intramolecular recombination between regions of homology in its genome that are as
close as 2.4 kb [257]. Therefore recombinant viruses containing multiple similar
fluorescent tags – such as GFP and CFP which are 97% homologous [365] – may
undergo undesirable permutations of recombination resulting in either the swapping of
tags or their complete loss, based on their relative positions on the VACV genome.
Finally, there may be a limit to the number of fluorescently tagged proteins that can be
incorporated into a single virus. One aspect of this is the ability to visualize them all
simultaneously; given the overlapping nature of emission spectra of available fluorescent
tags, it is important to select them carefully to ensure minimal spectral bleed-through.
Bleed-through is caused when two fluorophores have overlapping excitation or emission
spectra such that fluorescence emission from one protein is detected in a channel meant

CHAPTER 3: Developing an optimised VACV gene-tagging method
The University of Sydney 2016
94
for the other [287, 366]. This can be overcome by choosing tags with minimally
overlapping spectra, such as those that might be detected by DAPI, FITC and TxRED
channels. Additionally, the use of appropriate light filter sets and sensitive detection
methods with the ability to apply spectral unmixing [367-369], which can correct for
some bleed-through, may also be beneficial. Fortunately, the modular nature of this
technique enables the simple substitution of fluorescent tags in the TDS vector, based
on compatibility with other staining and/or tag choices.
VACV has been extensively used in imaging studies owing to many characteristics of
the virus that are favourable to live-cell microscopy. Fluorescent tags are expressed
from the viral genome, eliminating the need for transfection, enabling primary cells
derived from infected animals or non-transfectable cells to be easily analysed. Initially,
fluorescent VACVs were used for simple subcellular tracking of virus movement
(reviewed in [370]), but more recent approaches have expanded their utility to include
FRET studies [371], FRAP at single virus particles [241], promoter reporters [372],
intravital imaging [373], and structural studies [62, 231, 332]. Fluorescent VACV pave
the way for in vivo imaging experiments, which will be the final step in understanding the
true nature of a viral infection of its host. All these techniques could be within easier and
closer reach coupled with this method of creating recombinant VACV with fluorescently
tagged genes.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
95
Chapter 4: UNDERSTANDING VIRUS-
INDUCED CELL MIGRATION IN A
NATURAL HOST

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
96
4.1 INTRODUCTION
Author’s note: All experimental work described below was carried out
by the author, except for the creation of the intermediate TDS vector
described by Step 1 in Figure 4.2, which was carried out by J.
Horsington.
4.1.1 VACV-Induced Cell Motility
In addition to the disruption of the actin cytoskeleton during entry and exit, and the
induction of actin-based motility of its own particles, VACV holds sway over the
cytoskeleton by another manner: that of cell motility. Cells grown in a monolayer were
found to migrate 8-12 hpi, followed by the induction of projections in the cytoskeleton
[374]. Since then, VACV has become a model for the study of cell migration, mimicking
the transition of cell morphologies and phenotypes during cancer metastasis [375, 376].
Few other viruses have been recorded inducing a similar phenomenon – these include
the human T-cell Leukemia Virus type I (HTLV-1), which promotes migration of infected
cells [377], or the Rous sarcoma virus (RSV), whose transformation of infected cells with
the oncogene v-Src can induce metastasis of cells [378].
The VACV gene responsible for inducing cell motility was discovered by comparison of
strain WR – capable of causing this effect – with the attenuated strain Modified Virus
Ankara (MVA), which is not. By introducing regions of the genome missing from MVA
into MVA-infected cells, and assaying the resulting cellular morphology, vaccinia gene
F11 was identified to be responsible [228, 379]. The VACV replication cycle consists of
differing changes of cell migration, from detachment to migration and eventual
resettlement of cells and re-establishment of cell-cell contacts [248], and hence it is
important to determine which stages are influenced by F11. Live cell microscopy of
VACV expressing truncated versions of F11 revealed that it is responsible for the
rearrangement of the actin cytoskeleton early on in infection, followed by detachment
and migration of cells. The subsequent re-establishment of cell-cell contacts however,
was F11-independent [248]. F11 expression can also augment the spread of related
Poxvirus family: myxoma virus (MYXV), which does not express an F11 ortholog.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
97
Although MYXV carries orthologs of many Orthopoxvirus genes, it lacks one
corresponding to the F11L gene [380]. An F11-expressing MYXV was found to have
significantly improved rates of infection and dissemination in a wide range of cancer
cells [381], further supporting its importance in virus spread.
4.1.2 VACV Protein F11L
The viral gene F11L is conserved among orthopoxviruses, is expressed as early as 2
hpi, and is believed to act by binding to the Rho GTPase [229, 247], a key regulator of
actin dynamics [382], thus preventing it from binding to downstream signalling partners
Rho-associated kinase (ROCK) and mDia [228, 247]. A more recent study has found
that F11 acts as a scaffolding protein inhibiting RhoA signalling by binding to Myosin 9A,
a GTPase-activating protein [227]. The loss of F11 also induces observable changes in
cell morphology. Cells infected with VACV either lacking F11 or expressing an F11
dominant negative mutant exhibit prominent stress fibres 8 hpi [229], whereas cells
infected with wild-type virus characteristically undergo the loss of visible stress fibres
due to its inhibition of RhoA signalling [228, 383].
The inhibition of RhoA signalling by VACV was also found to be vital for virus particles to
access the cell cortex, following microtubule-based transport, but prior to the induction of
actin-based motility [226]. The expression of a dominant negative form of F11 reduces
the presence of CEV on the cell surface, as well the number of virus particles released
into the supernatant [226]. Thus, F11 enhances the release of VACV by modulating
cortical actin dynamics through RhoA signalling.
F11 enhances the cell-to-cell spread of VACV in a cell monolayer; this can be visualised
by the initial loss of cell-cell contacts, followed by the migration of infected cells away
from the plaque centre [229]. The loss of F11 not only attenuates the cell-to-cell spread
of VACV in a monolayer, but also adversely affects the spread of infection from the
primary site of inoculation in mice. Given the remarkable ability of this protein to
influence the actin cytoskeleton, and promote cell migration following virus infection, we
sought to improve upon this in vivo study by using a more species-appropriate
orthopoxvirus: ectromelia virus (ECTV).

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
98
4.1.3 ECTV and Cell Motility
Discovered in the 1930s after the introduction of mice as a live laboratory model, ECTV
is the causative agent of mousepox [384]. ECTV shares around 90% genetic similarity to
VACV and VARV strains [183, 385], and a typical ECTV infection of mice presents very
similarly to that of smallpox in humans [386-388]. As a result, ECTV-infected mice
provide a model for studying orthopoxvirus infection in vivo, given the potentially lethal
nature of VARV and the unknown host origins of VACV. Its infectivity at low doses,
restriction to a particular host, and high mortality rate make ECTV a credible model for a
smallpox-like infection in a natural host [177, 387, 389]. However, this has meant that
most studies involving ECTV mainly focus on immune responses to an infection [387,
390-393]. Given the remarkable ability of orthopoxviruses to influence the actin
cytoskeleton it is surprising that, to date, only a handful of studies have examined the
special relationship between the two [394-396]. Like VACV, ECTV infection also causes
the loss of stress fibres in infected cells [396], and it also relies on actin-based motility,
including the generation of actin comets for the spread of infection [394].
Despite these similarities between ECTV and VACV, the nature of their respective
infections differ in a few ways: the replication cycle of ECTV is about 1.5 times slower
than that of VACV in BSC-1 cells [394], and is more attenuated compared to VACV in
rabbit RK13 cells [397]. Most importantly, infection of mice with VACV is non-lethal at
similar doses resulting in minimal pathologies when compared with ECTV [398, 399].
Therefore, care must be taken when studying VACV infection in mice since, possessing
an unknown host, it is not clear what a normal course of VACV infection might resemble.
We aimed to expand upon the study by Cordeiro et al [229], whereby mice infected with
VACV lacking F11, the gene responsible for cell motility, were found to experience
attenuated infections. However given the differing natures of VACV and ECTV infection
in mice, this study can only be extrapolated so far.
Little is known about the ability of ECTV to induce cell motility. A study by Roberts in
1962 believed to have found evidence for the migration of infected dermal cells from the
scarification site in mice [400], although whether these cells were migratory tissue
macrophages, or infected by EEV released from the initial site of infection is unknown.
Despite the ability of VACV-infected cells to enhance BSC-1 cell motility [228, 374], a

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
99
very recent study found that ECTV infection impeded motility of murine fibroblasts
compared to mock-infected cells [395]. While mock-infected fibroblast cells migrated into
a wound in a scratch assay, ECTV-infected cells impeded migration of cells by around
1.8-fold [395]. This goes against observations of VACV inducing migration of infected
cells, however different cell types undergo different migratory events [401]. BSC-1 and
HeLa cells migrate differently in response to VACV infection (unpublished data) and
hence, murine fibroblast cells may represent another variant along this trend. Even if
ECTV-infected cells do not undergo infection-induced migration, the question as to why
the ECTV F11L gene has remained highly conserved remains pertinent, if not more so.
Since we know that an in vivo infection of mice with VACV lacking F11L impedes its
spread compared to the parent VACV, this provides an opportunity to compare those
findings with ECTV infection in mice. Therefore a system capable of tracking an ECTV
infection, and assessing the role of F11 (or an ECTV homolog) therein, would prove very
beneficial in understanding the importance of this unique trait to the spread of a poxvirus
infection in its natural host.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
100
4.2 RESULTS
4.2.1 ECTV encodes a homolog of VACV protein F11
A genome comparison of VACV-WR and ECTV-Mos (Moscow strain) revealed ECTV
gene 034 to be an ortholog of the F11L gene, a possibility confirmed by previous
comparisons of ECTV-Mos to VACV-COP (Copenhagen strain) [385]. The two
sequences are 96% identical (Figure 4.1A) and are flanked by orthologous genes as
well, with VACV genes F12L and F10L corresponding to ECTV genes 035 and 033
respectively. Henceforth, genes will be referred to as ECTV-F11L, VACV-F11L, ECTV-
F10L and so on. VACV-F11L binds to RhoA in a manner similar to ROCK, and indeed
mutation of the (partially) homologous region between VACV-F11L and ROCK (depicted
by F11-VK in Figure 4.2B) abrogates binding of VACV-F11 to RhoA [228]. ECTV-F11L
is 100% homologous to VACV-F11L at this region (Figure 4.2B) and hence we may
infer that ECTV also possesses similar RhoA binding abilities. Indeed, ECTV infection
has already been shown to display other hallmarks of VACV-F11 expression, such as
the loss of stress fibres, a general upheaval of the actin cytoskeleton, and the formation
of actin projectiles [394-396]. Since truncation of VACV-F11L proved greatly beneficial in
discerning its importance to the spread of VACV [229], we hypothesised that targeting
the ECTV-F11L gene for truncation would prove similarly illustrative of the functions of
this gene in an ECTV infection of mice.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
101
Comparison of F11 orthologs in VACV and ECTV.
(A) Alignment of F11L and F12L genes in VACV-WR (top) and ECTV-Mos (bottom). Regions used as left and right homology arms, in vectors
designed for the construction of ΔF11 VACV and ECTV viruses, are highlighted in grey. (B) Alignment of homologous protein sequences found in
the F11L genes of VACV and ECTV, as well as ROCKI and the F11-VK virus described in [229], from which this image was adapted. Matching
amino acids between all four sequences are highlighted in red and, the mutations in F11L that abrogate RhoA binding are highlighted in green.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
102
4.2.2 Design of TDS vector to create ECTV- ΔF11
In order to generate a recombinant ECTV with an F11L deletion, we first examined the
steps involved in the creation of the VACV-ΔF11 virus by Cordeiro et al [229]. The TDS
vector carrying gpt and mCherry genes was also employed in that case, however the left
and right homology arms for recombination were much longer (depicted in grey in
Figure 4.1). The entire VACV-F12L gene was incorporated into the vector as the left
arm, while the last 386 bp of VACV-F11L was cloned and inserted into the vector as the
right homology arm. This region of F11L is believed to contain the promoter sequence
for F10L, and hence was retained in the final VACV ΔF11 virus [229]. A VACV-ΔF12
virus was used as the parent virus for the creation of VACV-ΔF11, since this vector also
functioned as an F12L rescue vector, in addition to creating an F11L truncation.
We also opted to use the TDS system (described in Chapter 3) for the creation of an
ECTV-ΔF11 virus, however since we also needed to incorporate more than 150 bp of
the right arm (to retain any possible ECTV-F10L promoter element), we created the left
and right arms by PCR (see Table 2.1.8) – whose sizes were 263 kb and 337 kb
respectively. These were cloned into the TDS vector, after its (Figure 4.2A; step 1),
followed by a Lifeact-GFP sequence under the control of the pE/L VACV promoter [254],
which was also cloned in between the left and right arms, effectively replacing the region
of F11L to be deleted (Figure 4.2A; step 2). As mentioned previously, Lifeact is a 17 aa
peptide that binds to filamentous actin [402], which when bound to a fluorescent protein,
can effectively highlight the actin cytoskeleton when expressed in a cell. The Lifeact-
GFP sequence was created by PCR using primers containing the same restriction site
on both forward and reverse primers, such that the sequence could be inserted using
only one site (see Table 2.1.8 for primer sequences). Figures 4.2B and 4.2C
respectively describe the process of homologous recombination that should occur
between the TDS vector and the ECTV genome, and the resulting successful ECTV-
ΔF11 virus genome once TDS selection is removed. Therefore, cells that are transfected
with this TDS vector and have undergone homologous recombination would appear both
red (due to mCherry fluorescence) and green (due to Lifeact-GFP). Once GPT selection
is removed, ECTV plaques only expressing Lifeact-GFP would by necessity also have a
truncated F11 gene, and hence can be picked for further purification.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
103
Creation of the TDS vector to make ECTV-ΔF11
(A) The left and right arms of homology, corresponding the 3’ end of ECTV-F12L and the last
337 bp of ECTV-F11L were cloned by PCR and inserted into the TDS vector containing gpt and
mCherry genes. A Lifeact-GFP sequence was then inserted in between the two homology
arms. (B) Homologous recombination between the vector and the ECTV genome. (C) Following
removal of GPT selection and resolving out of the TDS vector from the ECTV genome, the
sequence of the desired recombinant ECTV-ΔF11 population is depicted here.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
104
4.2.3 Creation of ECTV- ΔF11
Several attempts were made to create the ECTV-ΔF11 virus using transient dominant
selection. Despite initially successful first or second rounds of transfection and plaque
purification, we were unable to isolate a stable, recombinant ECTV-ΔF11. The
advantage of having an ECTV-ΔF11 virus that also expresses Lifeact-GFP under a
VACV promoter is that one can immediately identify a successful deletion mutant by the
presence of stress fibres, since loss of F11 function leads to the re-appearance of stress
fibres in a VACV infection [229]. Cells infected by ECTV expressing both mCherry and
Lifeact-GFP that still retained stress fibres were present after initial transfection-infection
steps (Figure 4.3A), and even persisted to create mCherry+ and Lifeact-GFP+ plaques
in a second round of purification under GPT selection (Figure 4.3B). However, Lifeact-
GFP+ cells failed to endure beyond this step, when cultured with or without GPT
selection reagents.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
105
Creation of an ECTV-ΔF11 virus.
(A) HeLa cells were infected with ECTV and then transfected with the ΔF11 TDS vector 1 hpi. Cells were fixed 36 hpi and stained for DAPI.
Lifeact-GFP was imaged through the FITC channel and mCherry through TxRED. Stress fibres can be seen in Lifeact-GFP+ cells. Scale bar = 20
μm. (B) Following transfection, cells were also scraped 36 hpi, lysed by 3x freeze-thaw cycles, and used to infect BSC-1 cells under GPT
selection. Plaques of successful ECTV-ΔF11 recombinants expressing both Lifeact-GFP and mCherry were imaged 3 dpi.

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
106
4.3 DISCUSSION
The creation of F11 deletion viruses has long-proven to be a difficult process. Several
attempts were made to create a VACV-ΔF11 virus by homologous recombination, but
were met with failure [228, 403]. F11 was initially thought to be essential for viral
replication [228], however further attempts were able to create recombinant VACV
containing a nonsense mutation in F11 (which introduced a stop codon halfway through
the F11L gene) such that it was not expressed by replicating virus, thus proving that F11
may not be essential after all [403]. Eventually, Cordeiro et al were successful in
creating a VACV-ΔF11 virus by the TDS method using both GPT and mCherry selection
[229].
Our efforts were similarly designed, except in two aspects: first, we did not opt to create
the ECTV-ΔF11 virus by rescue of an entire gene (as Cordeiro et al did, by employing
the whole VACV F12L gene as the right homology arm and using VACV-ΔF12, a virus
severely deficient in microtubule transport and actin-based motility [331], as the parent),
and secondly, our left homology arm was around 110 bp shorter (Fig. 4.4). The left
homology arm used for the creation of VACV-ΔF11 was cited by Cordeiro as being 386
bp long in order to retain the F10L promoter sequence, which is supposedly located at
the 3’ end of the F11L gene. Unfortunately, no further evidence or explanation is
provided in this regard. F10 is an essential protein involved in VACV morphogenesis,
required for the proper formation of viral membranes [404]. It is expressed late during
infection [405], and the consensus vaccinia late promoter sequence TAAATG [406] is
located right before its start codon in both ECTV and VACV genomes (depicted by
asterisks in Fig. 4.4). Since the length of the right homology arms in the ΔF11 plasmids
used to create VACV and ECTV differed by around 110 bp, we wanted to ensure that no
other promoter sequences were lost in this process – which would be responsible for
the instability of ECTV-ΔF11 due to the loss of F10 expression. Therefore we aligned
the sequences surrounding F11L in both ECTV and VACV, and highlighted all putative
promoter sequences (Fig. 4.4). These included TAAATG, as well as TAAAT and TAAA,
other common late promoters in VACV [407, 408]. Although it is unusual for promoter
sequences to exist beyond position -30 from the site of transcription [408], the citing of
promoter sequences existing within the last 386 bp of VACV-F11L [229] prompted this

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
107
investigation. However, promoter sequences were not identified within this 110 bp
region, and therefore, our inability to isolate an ECTV-ΔF11 virus was likely not due to
any disruption of F10 expression. In order to ensure that some other likely promoter
sequence was not overlooked, a TDS vector containing the entire 386 bp end of ECTV-
F11L could be created and used to repeat this experiment. Alternatively, a pE/L
promoter sequence [254] could be incorporated into the very end of the right homology
arm, to ensure transcription of F10L.
Comparison of truncated sequences in ECTV-ΔF11 and VACV-ΔF11.
F11L sequences in ECTV-Mos (top) and VACV-WR (bottom) are aligned, with the truncated
regions in their respective ΔF11 recombinants highlighted in yellow, and the retained regions
(by virtue of them being the right homology arms in the TDS vector) highlighted in grey. The
ECTV-ΔF11 right homology arm extends into the first 43 bp of F10L, which also contains its
promoter elements.
It is possible that the success of Cordeiro et al in creating a VACV-ΔF11 virus was due
to their use of a VACV-ΔF12L virus as the parent strain, and a TDS vector which
simultaneously rescued F12L and incorporated the F11L truncation (see Fig. 4.1A). In
essence, the virus might ‘prefer’ being rescued from the severely attenuated ΔF12L
phenotype over the loss of F11L (Cordeiro, J. V., PhD thesis [409]), thus increasing the
chances of recovering a VACV-ΔF11 virus. Therefore, the experiment could be
repeated by creating an ECTV-ΔF12L virus first, followed by using a TDS rescue vector,
which both rescued ECTV F12L and incorporated an ECTV F11L truncation.
Alternatively, a method similar to Kato et al, whereby a nonsense mutation was

CHAPTER 4: Understanding virus-induced cell migration in a natural host
The University of Sydney 2016
108
introduced midway through VACV F11L [403] could be attempted to minimise any flow-
on effects of ECTV F11L gene truncation.
It is clear that the study of the F11L gene is a contentious one, both in terms of its
necessity for viral morphogenesis and its position within the orthopoxvirus genome.
Nevertheless, the potential insights to be gained from the creation of an ECTV-ΔF11L
virus, and a study thereof in a natural host setting, would greatly contribute to the role of
this gene – not only in poxvirus infection and spread but also of the cytoskeletal
mechanisms involved in virus-induced cell migration.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
109
Chapter 5: DIVERGENT ROLES OF
Β- AND Γ-ACTIN IN VACV-INDUCED
ACTIN COMET FORMATION

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
110
5.1 SECTION HEADING
Author’s note: Sections of this chapter have been published in the
journal Cytoskeleton, under the title “Divergent roles of β- and γ-actin
isoforms during spread of vaccinia virus”. Its authors are N Bishara
Marzook, Sharissa L Latham, Helena Lynn, Christopher McKenzie,
Christine Chaponnier, Georges E Grau & Timothy P Newsome. Images
depicted in Figures 5.1-5.4 were taken by S. Latham. All other
experimental and analytical work presented here was carried out by N.B.
Marzook.
5.1.1 The Role of Actin in VACV Infection
Remodelling of the host actin cytoskeleton by vaccinia virus (VACV) occurs at multiple
stages during the replication cycle and facilitates virus spread via a number of distinct
mechanisms [410]. VACV has the uncommon ability for an intracellular pathogen to
induce actin polymerisation at its surface. The nucleation of actin in the cytoplasm
beneath extracellular virus, or wrapped virions (WV) results in comet-like structures of F-
actin in the underlying cytoplasm (also referred to as actin tails or comets) that propel
virus particles across the surface (apical or basal) of infected cells [43, 168, 233, 411].
5.1.2 VACV actin-based motility as a model to study actin dynamics
VACV-induced actin comet formation constitutes one of the best-characterised
pathways of actin nucleation, leading to a number of key insights in not only the
mechanism of actin manipulation by VACV, but in also understanding the fundamentals
of actin nucleation in general. Elucidation of the mechanisms of actin nucleation at the
biochemical level typically requires the setting up of highly technical assays [412].
Briefly, this consists of setting up an in vitro system whereby actin is extracted from
rabbit muscle acetone powder using a cocktail of several buffers and dialysing
equipment over lengthy periods of time [412, 413]. Actin polymerisation is induced by
altering the pH or the addition of salts, and can be monitored by tracking the increase in

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
111
light scatter or viscosity of the solution as the actin filaments grow. Alternatively, purified
actin is labelled with a fluorophore called pyrene, so that its increase in signal intensity
once incorporated into an actin polymer can be quantitatively measured to provide
kinetic data on actin polymerisation dynamics [414, 415]. Other accessory proteins can
be studied by their individual purification for later reconstitution into the same in vitro
assay. Alternatively, cytoplasmic extracts containing all cellular proteins involved in actin
polymerisation can be isolated from cell extracts [416]. These extracts have been used
to study the motility of bacteria such as E. coli, which don’t normally enter mammalian
cells but have been engineered to express certain proteins involved in actin-based
motility [417]. In addition, cytosolic extracts can be used to perform biomimetic motility
assays in which polystyrene beads are coated with an accessory protein of interest and
actin-based movement can be tracked [418, 419].
Observations made using in vitro systems do not always align perfectly with complex
and highly regulated in vivo pathways (many of which remain uncharacterised) [420].
While observations can be made on whole cells to understand functions of accessory
actin proteins, or actin itself, observable phenotypes are often restricted to effects far
downstream of the event of actin polymerisation itself, such as cell shape or motility
[421, 422].
As pathogens utilise cellular actin nucleation pathways, studying these pathways not
only helps further our understanding of microbial actin-based motility, but also the
mechanisms by which cytoskeletal systems are regulated in non-disease states. The
pathogen-induced actin comet provides us with a metaphorical light in the dark
stochastic depths of global actin dynamics within a cell – in the form of an observable,
measurable phenotype – which can then be evaluated in response to varying stimuli.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
112
5.1.3 Features of VACV-induced actin comets
The fundamentals of VACV-initiated actin nucleation have been described previously
(see section 1.3.1.1). Briefly, during the VACV replication cycle EV particles travel to
the cell periphery on microtubules where the viral protein F11 clears a path through the
cortical actin for the virion to gain access to the cell surface [226]. Exocytosed CEV
remain attached to the surface of the host cell. The plasma membrane in contact with
extracellular virus expresses a number of integral and membrane-associated viral
proteins, including A36 [224, 231]. Following a signalling cascade leading to the
recruitment and activation of N-WASP and the Arp2/3 complex, actin filaments are
nucleated in the underlying cytoplasm beneath virus particles giving rise to F-actin
comets, which usually appear 6-8 hpi [43, 168, 241].
Analysis of actin comets reveals that unlike bacterial comets, VACV-generated actin
filaments branch out at 45o angles from the central axis of the comet. However, they are
similar to Listeria comets with their barbed or fast-growing filament ends pointed toward
the virus particle [169]. Incubating VACV-infected cells in G-actin labelled with
rhodamine (a fluorescent dye) reveals the recruitment of G-actin, and not pre-formed F-
actin, to VACV particles specifically at the virus surface and not along internal sites
along the length of the actin comet [169] (Figure 5.1). This incorporation occurs
regardless of whether labelled G-actin is supplied at levels below or above the critical
concentration of actin required for incorporation into the pointed end of actin filaments in
vitro. Therefore, actin polymerisation only occurs at the free barbed ends close to the
virus surface.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
113
Incorporation of G-actin into VACV-induced actin comets occurs at the
virus surface.
VACV-infected HeLa cells were fixed after a 10 second incubation with rhodamine-labelled G-
actin. Phalloidin signal (which only labels F-actin) is viewed in green (A) and rhodamine in red
(B), with both merged in (C). Figure adapted from [169].
As actin filaments extend, the force generated propels the CEV across the surface of
the cell [168, 416]. Rapid disassembly of newly formed filaments leads to the
characteristic comet morphology [119, 152, 173]. Measuring aspects of VACV-induced
actin comets, such as the length of actin comets in fixed cell samples or the speed of
actin comets in live-cell movies captured of infected cells, can provide insights into roles
of proteins involved in the actin polymerisation/depolymerisation cycle. A few factors
have been found to influence the properties of VACV-induced actin comets. The stability
of N-WASP at the virus surface can effect the speed of VACV actin-based movements
[241]. Removal of the stabilising effect of the protein Grb2, or expression of an N-WASP
mutant lacking the ability to bind to the barbed end of actin filaments (N-WASP-RA/RA)
results in a faster rate of virus movement, while also reducing the length of actin comets
[241]. In contrast, loss of the clathrin adaptor AP-2, normally recruited to the virus
particle prior to actin nucleation, produces actin comets of longer lengths while also
increasing the speed of virus movement [62]. However, VACV particles take longer to
initiate actin nucleation, and also have a slower rate of actin filament disassembly once
formed, which contributes to the longer comet morphology. Finally, different isoforms of
the proteins comprising the Arp2/3 complex are less (in the case of ARPC1A and

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
114
ARPC5) or more (with ARPC1B and ARPC5L) efficient at promoting actin
polymerisation. Depletion of the more efficient isoforms by siRNA produces shorter
comets, while knockdown of the less efficient isoforms produces longer comets [174].
This difference in Arp2/3 complex isoform functions also plays out in actin filament
disassembly: those filaments nucleated by ARPC1B- or ARPC5L-containing Arp2/3 are
more resistant to F-actin depolymerisation [174].
Therefore, it is clear that various proteins recruited to the actin polymerisation cascade
at the virus surface can affect the rate of virus particle movement. But how might the
nature of the G-actin monomer itself, incorporated into the growing actin comet with the
help of these accessory proteins, impact VACV actin comet dynamics? A closer
examination of cytoplasmic actin would be the first step in answering this question.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
115
5.1.4 Cytoplasmic Actin: A Tale of Two Isoforms
“Two actins, both alike in sequence
In the cytoplasm, where we lay our scene”
As mentioned earlier (in section 1.1.1.1), actin is composed of 6 isoforms with very
similar amino acid sequences [25]. Of those, the cytoplasmic actin isoforms β-actin and
γ-actin are the most abundant in non-muscle cells [27]. The two cytoplasmic actin
isoforms are completely conserved from birds to mammals, only differing by four
biochemically similar amino acids [26] (Figure 5.2).
Differences in cytoplasmic actin isoforms.
(A) Crystal structure of β-actin monomer highlighting the positions of the 4 amino acids
differing between it and γ-actin with D1-D3 in green and V10 in pink. ATP is depicted as an
orange stick and bound Ca2+ or Mg2+ ion is depicted as a red circle. (B) Model arrangement of
the actin trimer in F-actin. Image adapted from [423]. (C) Differences between β- and γ-actin
exist only in the first 10 amino acid positions.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
116
Despite their amino acid differences being conservative, β-actin is the more acidic of the
two isoforms [423]. β-actin and γ-actin usually exist in a 2:1 ratio in cells [28], however
this ratio is inverted in cell types like auditory hair cells [424]. Studies into the functional
basis for the presence of two conserved cytoplasmic actin isoforms have been largely
based on two methods: examining the global and local effects of siRNA knockdown on
the cell and/or organism, or by inferring functions from their differential localisations
within a cell. Mice that homozygous for mutant β-actin alleles are embryonic lethal [425,
426]. On the other hand, while mice with homozygous knockouts of the γ-actin gene
experience no developmental issues, they often fail to thrive and experience
progressive hearing loss in adulthood [427, 428]. Therefore it appears that while γ-actin
is not essential in the early development of the cytoskeleton, it is required for the
maintenance and long-term stability of F-actin structures, especially in ear stereocilia
where its role is vital [427].
Inferring functions based on the differential localisation of the two isoforms has yielded
conflicting results. β-actin was repeatedly observed localising to migratory, or more
dynamic regions, of a cell such as lamellipodia by staining of either β- or γ-actin-specific
mRNA [429, 430] or isoform-specific antibodies [431, 432]. At the same time, γ-actin
was found to be more uniformly distributed throughout the cell [429]. A more recent
study by Dugina et al [29] used antibodies raised against N-terminal nanopeptides of β-
and γ-actin. They discovered that under resting conditions, β-actin localised to ventral
stress fibres, and cell-cell contacts, and were more baso-laterally present in general,
while γ-actin were more apically abundant in dorsal stress fibres. Upon the induction of
cell migration by creating a scratch in a cell monolayer, γ-actin was enriched in
lamellipodia [29, 433] while β-actin was present in bundles close to the substrate [29].
Isoform-specific functions of cytoplasmic actin were also examined by the specific
depletion of either isoform, either by siRNA-induced gene silencing or by knocking out
the gene entirely [29, 427, 434-436]. Despite incomplete knockdown achieved by the
siRNA technique, drastic changes in cell morphology and motility were observed under
both β- and γ-actin depletion in epithelial cells and fibroblasts [29]. β-actin-depleted cells
lost stress fibres and exhibited broad protrusions at the leading edge of cells, while loss
of γ-actin reduced the presence of lamellipodia, assuming a more contractile phenotype.
Cell motility can be used to assay proper functioning of the actin cytoskeleton.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
117
Knockdown of γ-actin also reduces directional migration of cells [29, 435], while the
speed of motility is reduced during depletion of either isoform [29]. Thus, it can be
concluded that while γ-actin is important for directional migration, β-actin may be
important for short-term cell motility. Complete ablation of the β-actin gene also causes
migration defects in primary mouse embryonic fibroblasts [436], while overexpression of
β-actin in myoblasts drastically increases cell motility [437]. However this increase in
migration was found to not correlate with an increase in the rate of actin polymerisation
(since over-expression of a β-actin mutant defective in polymerisation also increased
cell motility), but to depend on myosin function [437]. Indeed overexpression of both
isoforms was found to increase cell motility in human colon cancer cells [438],
suggesting the overall importance for a maintenance of a precise balance between
ratios of β- and γ-actin for proper control of cell migration.
Finally, the differing functions of the cytoplasmic actin isoforms can be analysed by
understanding the molecular basis behind their differences. Actin accessory proteins, for
example, may show preferential binding to one isoform over the other. L-plastin was
found to preferentially bind to β-actin, although a mechanism for this remains unclear
[439]. Additionally, β-actin can be post-translationally modified by N-terminal
arginylation, which has been proposed to regulate actin polymerisation and lamella
formation in motile cells [440]. A major hurdle in assessing isoform differences at the
biochemical level has been the inability to study them in isolation as pure isoform
preparations. Bergeron et al [423] were able to express individual cytoplasmic actin
isoforms, either as single or mixed populations, in insect cells using a baculovirus-driven
expression vector. Using this system, they were able to deduce that, in the presence of
Ca2+ or Mg2+ ions, while both isoforms are completely co-polymerisable, pure β-actin
polymerises at a much faster rate than γ-actin. Phosphate release during actin
treadmilling was also found to be twice as fast compared with γ-actin. In addition, the
slower polymerisation rate of γ-actin appears to result from slower nucleation and
elongation rates, as well as greater stability of the pure γ-isoform filament [423].
Therefore it is likely that any imbalance in actin isoform concentrations that leans
towards an increase in γ-actin monomers would create an energy barrier to F-actin
formation, and also increase the stability of any filaments that are formed.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
118
5.1.5 Actin Isoforms and Intracellular Pathogens
Cells may rely on the delicate balance of actin isoforms for their proper functioning, but
so do the intracellular pathogens that infect them. The E2 glycoprotein of classical swine
fever virus interacts with β-actin, the loss of which adversely affects early virus
replication [441]. The coronavirus M protein, a transmembrane protein that sits in the
viral envelope also interacts with β-actin [442]. Disruption of actin filament assembly by
cytochalasin D causes a reduction in virus assembly and budding. With regard to
pathogens that rely on actin-based motility, a recent study found that siRNA-mediated
knockdown of β-actin, but not γ-actin, impairs Listeria infection of HeLa cells [443].
Given the demonstration of efficient individual silencing of the cytoplasmic actin
isoforms, as well as the availability of antibodies specific for each isoform as described
earlier, we decided to investigate the roles, if any, of β-actin and γ-actin in VACV-
induced formation of actin comets. One virus particle initiating an actin comet in
essence represents a functional unit of actin nucleation. VACV-induced actin-based
motility provides us with a unique ability to evaluate actin dynamics at fixed loci in space
and time, and hence serves as a unique model to further our understanding of the roles
of β- or γ-actin.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
119
5.2 RESULTS
5.2.1 VACV actin comets contain both β- and γ-actin
Since β- and γ-actin are differently localised within in a cell (as outlined in section
5.1.4), our first aim was to determine the actin isoform composition of VACV-induced
actin comets. Actin comets generated by a sub-population of CEV are evident in VACV
infected cells at 7-9 hpi [168, 444]. hCMEC/D3 cells were infected with VACV-WR, fixed
8 hpi and probed with β- or γ-actin-specific antibodies . Antibodies were created by
exposing mice to synthetic N-terminal nanopeptides containing the four differing amino
acids between the two isoforms and screened by triple ELISA [29]. Although antibodies
against β-actin and γ-actin were raised in the same species, they belong to different IgG
subclasses (IgG1 and IgG2b respectively), such that they can be probed simultaneously
with secondary antibodies specific to those subclasses. Immunofluorescence
micrographs revealed the presence of both actin isoforms in hCMEC/D3 cells (Figure
5.3A), and actin comets (Figure 5.3B). All VACV-induced actin comets observed
comprised both β- and γ-actin, although β-actin staining appeared stronger closer
towards the VACV particle, while γ-actin staining showed this isoform trailed further
behind (Figure 5.3B, 5.4B). However, no comets containing specifically one actin
isoform were seen. This is in agreement with the two actins being co-polymerisable. β-
and γ-actin comets are produced exclusively in VACV-infected cells (Figure 5.3C);
VACV-infected cells can be identified by the presence of a DAPI-stained peri-nuclear
virus factory. This represents the first examination of VACV-induced actin comets for
cytoplasmic actin isoform composition.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
120
VACV actin comets contain both β- and γ-actin.
(A) Fluorescent micrographs of hCMEC/D3 cells infected with VACV-WR and fixed 8 hpi. Cells
were stained with anti- β-actin (green), anti-γ-actin (red) and DAPI (blue). (B) Close-ups of
outlined sections in (A). (C) Micrograph showing an infected hCMEC/D3 cell producing actin
comets (note the presence of the DAPI-stained peri-nuclear viral factory) next to an uninfected
one. Scale bar is 10 μm. Images captured by S. Latham.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
121
5.2.2 β- and γ-actin are abundant in VACV-induced actin comets in apical and
basal regions of the cell
VACV-induced actin comets are readily imaged at the level of the substrate due to the
low z-axis profile of adherent cells. Additionally, β-actin preferentially localises to the
basal region, while γ-actin preferentially localises to apical regions of HSCF cells
(Dugina 2009). We therefore aimed to determine if the preference of actin isoforms
along the apical-basal axis of a cell was reflected in their localisation to actin comets on
the apical or basal membrane. hCMEC/D3 cells were infected with VACV-WR and fixed
8 hpi. Following staining for β - and γ-actin, confocal microscopy was used (see section
2.3.2.2) to obtain z-stack images of infected cells (Figure 5.4A).
Closer inspection of actin comets (representative images shown in Figure 5.4B) reveals
the presence of both β- and γ-actin in comets produced throughout the cell. It was noted
that virus-associated actin comets displayed decreasing length towards the apical
surface (results not shown). This led us to conclude that VACV-associated actin comets
are composed of both cytoplasmic actin isoforms irrespective of their basal or apical
localisation.
Composition of VACV actin comets created throughout a cell.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
122
Fluorescent micrographs of hCMEC/D3 cells infected with VACV-WR and fixed 8 hpi. Cells were
fixed in 1% PFA, permeabilised with ice-cold methanol, and stained with anti-β-actin (green),
anti-γ-actin (red) and DAPI (blue). Individual z-stack planes of a single field of view for VACV-
infected cells are shown in (A), along with close-ups of actin comets from three z-planes (B).
Scale bar is 10 μm. Images captured by S. Latham

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
123

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
124
5.2.3 Composition of VACV-induced actin comets under cytoplasmic actin
knockdown
Since previous studies reported not only different localisations, but also different
functions for β- and γ-actin within a cell, we wanted to determine if there was a distinct
role for either isoform in VACV-induced actin polymerisation, despite their co-
localisation at virus particles. A cocktail of isoform-specific siRNA sequences employed
previously [29] (described in section 2.6) was used to specifically deplete β- or γ-actin
expression in cells. These cells were subsequently infected with VACV-WR in order to
determine a role, if any, for the two isoforms in VACV actin-based motility.
hCMEC/D3 cells were treated with control siRNA (referred to as the ‘scrambled siRNA’),
β-, or γ-actin-specific siRNA for 72 hours before being infected with VACV-WR and fixed
8 hpi. Immunofluorescence assays of siRNA-treated cells stained with anti-β-actin and
anti-γ-actin-specific antibodies again showed localisation of both actin isoforms in the
cytoskeleton of scrambled siRNA-treated cells. Levels of β-actin and γ-actin were
unaffected by scrambled siRNA, however both were significantly reduced in the
presence of their respective siRNA, as shown by IFA (Figure 5.5A). Cells treated for
knockdown of either actin were readily identified by IFA (by appearing almost
completely red or green during β-actin or γ-actin knockdown respectively) through
labelling with isoform-specific antibodies (described in section 2.1.7). Thus we were
able to confirm their specificity as well as the efficiency of knockdown at the resolution
of single cells.
As with the control cells from Figure 5.3, both β-actin and γ-actin were detected in virus-
associated actin comets in the scrambled siRNA-treated cells (Figure 5.5B). γ-actin,
which generally stains abundantly throughout the cell and the cell periphery, was greatly
reduced in γ-actin knockdown cells. Stress fibres, which comprise mostly of β-actin [29],
were readily visible under γ-actin knockdown. This aligns with findings (by other studies
and ours) of compensatory expression of cytoplasmic actins when either one is depleted
[29, 427, 428]. The presence of stress fibres during VACV infection is also indicative of
the increased expression of β-actin, as VACV infection is known to reduce actin stress
fibres frequency [229]. Cells experiencing γ-actin knockdown that were infected with

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
125
VACV displayed morphologically normal actin comets. These comets were composed
almost exclusively of β-actin although there was some residual γ-actin-positive staining
when compared to scrambled controls. Cells experiencing β-actin knockdown showed
greatly reduced staining of β-actin by and a corresponding increase in γ-actin staining at
the cell periphery. Stress fibres were not visible in these cells, and most notably, the
presence of actin comets was greatly reduced. Cells that exhibited the most efficient β-
actin knockdown displayed the greatest attenuation in comets. In these cells, the few
comets that were observed were almost completely bereft of β-actin, with small
accumulations of β-actin at the virus interface. It is difficult to discern if this observation
reflects a distinct localisation of β-actin to the proximal region of the virus-associated
actin comet, or is simply reflective of the 3D topography of the comet structure. The
thickest region of the comet is adjacent to the virus, which is where weak localisations
are most likely to be observed. Even under knockdown, actin comets observed
possessed both β- and γ-actin, since knockdown is not 100% effective. Although
reduced β-actin expression appeared to attenuate actin comets, the presence of a β-
actin ‘seed’ was detected for all instances of VACV-induced actin comet formation.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
126
Distribution of β- & γ-actin in VACV comets under actin knockdown.
(A) Fluorescent micrographs of hCMEC/D3 cells treated with the isoform-specific siRNA
indicated, infected with VACV-WR and fixed at 8 hpi. Cells were stained with anti-β-actin
(green), anti-γ-actin (red) and DAPI (blue). Scale bar is 10 μm. (B) Close-ups of sections from
each treatment, outlined in A. Original images captured by S. Latham

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
127
5.2.4 Apical-basal location of VACV-induced actin comets does not affect their
cytoplasmic actin composition under knockdown
To further characterise our observations of both actin isoforms being present in actin
comets even under actin knockdown, we examined comets produced throughout the z-
axis of a VACV-infected cell treated with actin isoform-specific siRNA. hCMEC/D3 cells
undergoing knockdown were infected with VACV-WR and fixed 8 hpi. Cells were
stained with β- and γ-specific antibodies, and Z-stack images of infected cells were
taken by confocal microscopy (described in section 2.3.2.2).
In both scrambled siRNA- (Figure 5.6A) and γ-actin-targeting siRNA-treated cells
(Figure 5.6C), both γ- and β-actin isoforms comprise the VACV-induced actin comets,
irrespective of whether they were present at the basal or apical membrane (Figure
5.6B, 5.6D). γ-actin is still present in cells treated with γ-actin-targeting siRNA as
knockdown never reaches 100%. These data led us to conclude that VACV-associated
actin comets are composed of both actin isoforms irrespective of their basal or apical
localisation. Cells treated with β-actin-targeting siRNA did not produce abundant virus-
associated comets (Figure 5.6E). However, in the instances where comets were
present residual β-actin was present at basal and near-apical locations (Figure 5.6F).
Thus, the absence of a requirement for γ-actin was not due to the examination of solely
basal actin comets. In addition, apical comets, where γ-actin is present, are not
specifically disrupted by γ-actin knockdown.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
128
Composition of VACV actin comets under actin knockdown
throughout a cell.
hCMEC/D3 cells treated with scrambled siRNA (A,B), γ-actin-targeting siRNA (C, D), and β-
actin-targeting siRNA (E, F) were infected with VACV-WR and fixed 8 hpi. Cells were stained
with anti-β-actin (green) and anti-γ-actin (red) antibodies, along with DAPI (blue). Individual z-
stack planes of a single field of view for each treatment are pictured (A, C, E), along with close-
ups of actin comets from specific z-planes in each treatment (B, D, F). Scale bar is 10 μm.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
129
5.2.5 Extent of cytoplasmic actin knockdown is dependent on cell type
Our results until now have been restricted to examining the effects of actin knockdown
on actin comets in a single cell line: hCMEC/D3, an endothelial cell line (Weksler 2013).
While VACV infection of endothelial cells has been previously studied, they were mainly
restricted to human umbilical cord vascular endothelial cells (HUVECs) in the context of
immune responses to VACV infection [445, 446], or of endothelial cells in vivo in the
context of oncolytic therapies using various recombinant VACV strains [447, 448]. The
knockdown efficiency of β- and γ-actin in this endothelial cell line has been studied
previously, and was assessed at 44.6% and 63.2% respectively, compared to the
scrambled negative control [449]. Having established a phenotypic difference in VACV-
induced actin comets under β-actin knockdown in hCMEC/D3 cells, we aimed to extend
our analysis to other cell types, to establish whether the β-actin requirement was a
peculiarity that was restricted to hCMEC/D3-infected cells, or a general phenomenon
that may be observable in other cell types more relevant to VACV infection studies.
Cell lines chosen for testing were HeLa (a human epithelial cell line), GBM
(gliobastoma; a human neural tumour cell line) and BSC-1 (a monkey kidney epithelial
cell line) (see section 2.1.2 for sources). These cells were treated with either a cocktail
of two β-actin-targeting siRNAs or three γ-actin-targeting siRNAs, and a scrambled
siRNA as a negative control for 48 hours in conditions identical to those used for actin
knockdown in hCMEC/D3 cells [449]. Immunoblots specifically targeting β- or γ-actin
were carried out on cell lysates (Figure 5.7A). HeLa and GBM cell lines showed a
reduction in expression of the targeted actin at levels that were comparable, if not more
efficient, when compared to those achieved in hCMEC/D3 cells (Figure 5.7B). However
all three cell lines showed greater efficiencies for β-actin knockdown than γ-actin. BSC-1
cells, however, were not as receptive to knockdown as the other cell types, possibly
owing to the specificity of the siRNA to the human β- and γ-actin isoforms. While other
techniques to measure actin knockdown, such as quantitation of mRNA levels, could
have been carried out, the efficacy of these particular siRNA and their phenotypic
effects have been verified and published previously [29].
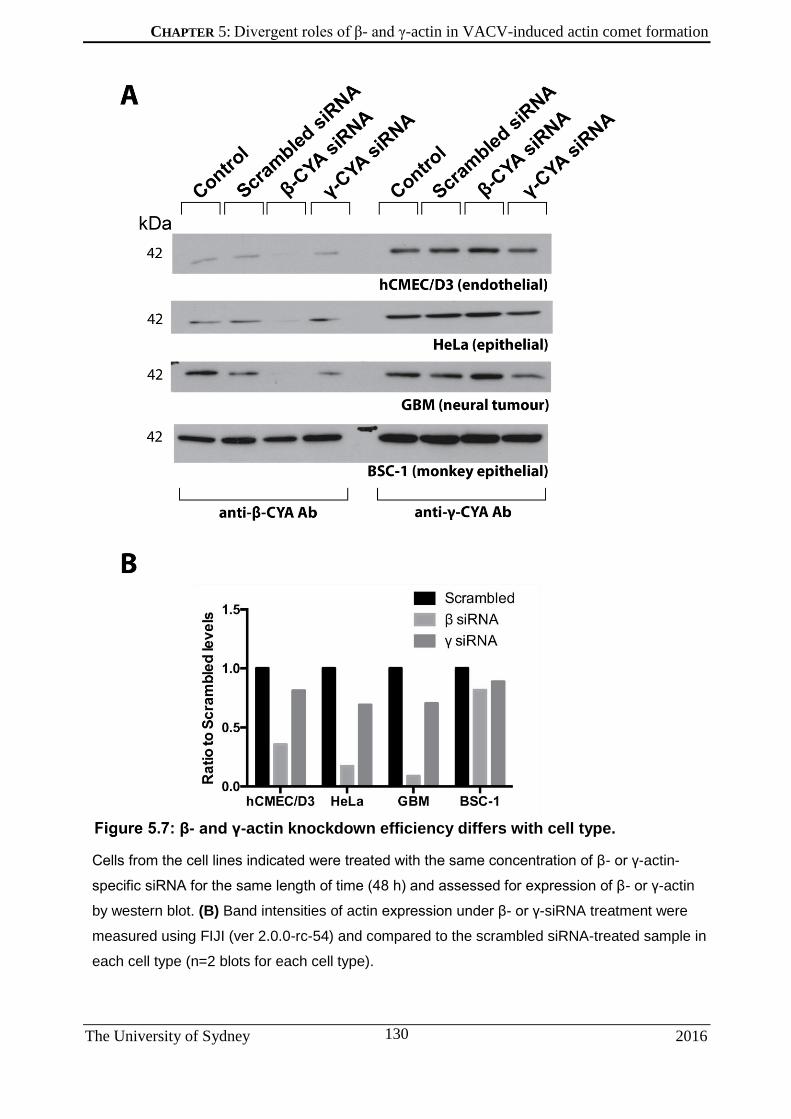
CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
130
β- and γ-actin knockdown efficiency differs with cell type.
Cells from the cell lines indicated were treated with the same concentration of β- or γ-actin-
specific siRNA for the same length of time (48 h) and assessed for expression of β- or γ-actin
by western blot. (B) Band intensities of actin expression under β- or γ-siRNA treatment were
measured using FIJI (ver 2.0.0-rc-54) and compared to the scrambled siRNA-treated sample in
each cell type (n=2 blots for each cell type).

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
131
5.2.6 Characterising cytoplasmic actin knockdown levels in selected cell types
Since BSC-1 cells did not exhibit efficient actin knockdown, HeLa and GBM cell lines,
along with hCMEC/D3 cells, were selected for ongoing experiments. Although GBM
cells are not commonly used for the study of VACV, glioblastoma cells have been tested
with VACV in its capacity as an oncolytic agent [450], and the mouse glioma cell line
GL261 was found to support high levels of virus replication in cell culture [451]. Our
ability to identify prominent VACV-induced actin comets in infected GBM cells, as well
as their ability to form adequate cell monolayers (unlike HeLa cells) – which was
required for ensuing experiments such as plaque assays – resulted in their use for
further study. Additionally, we increased the duration of actin knockdown from 48 h to 72
h, as this appeared to enhance knockdown of γ-actin in HeLa and GBM cells.
Like previous studies conducted on HSCF and HaCaT cells (Dugina 2009), cytoplasmic
actin knockdown caused distinct morphological changes to HeLa cells (Figure 5.8A). β-
actin silencing produced an increase in circularity and protrusions at the leading edge of
cells, as well as a greater number of multinucleated cells compared to the scrambled
control. γ-actin silencing produced an elongated, contractile phenotype in HeLa cells.
Immunoblots of HeLa and GBM cell lysates under actin silencing were also conducted,
with a pan-actin antibody as a control (Figure 5.8B). Densitometry analysis showed that
β-actin knockdown caused a significant reduction of β-actin to about 31% and 8%
compared to levels in the scrambled siRNA control in HeLa and GBM cells respectively
(P<0.05, n=2) (Figure 5.8C1). γ-actin siRNA treatment produced a comparatively less
efficient knockdown of γ-actin to about 52% and 35% in HeLa and GBM cells
respectively (P<0.05, n=2) (Figure 5.8C2). Interestingly, knockdown of β-actin led to a
corresponding significant increase in γ-actin in both cell types. Similar results were
observed in the hCMEC/D3 endothelial cell line [449] This suggests that under
knockdown conditions, cells may overexpress one isoform to compensate for loss of the
other. This has not been observed for actin isoforms in other studies, either using the
same cocktail of isoform-specific siRNA [29] or using only one of those sequences
[435]. This could be due to their use of different cell types, varying doses of siRNA or

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
132
incubation times, and/or the use of only a single actin isoform-specific siRNA in one of
the studies.
The use of a pan-actin antibody which detects both cytoplasmic isoforms revealed that
there is a compensatory effect of actin isoform expression during either β-actin or γ-
actin knockdown in HeLa cells, as there is no significant difference in total actin levels
under either knockdown condition (Figure 5.8D). This corresponds to observations by
other studies where neither isoform knockdown significantly affected the total level of
actin in HSCF cells (Dugina 2009) and A549 human lung epithelial cells [452]. GBM
cells, however, show significantly reduced levels of total actin expression during γ-actin
knockdown (Figure 5.8D).

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
133
Effect of actin knockdown on chosen cell lines.
(A) Phase-contrast images of HeLa cells 72 h after treatment with respective siRNA. Scale bar is
100 μm. (B) Immunoblot of HeLa and GBM cells treated with respective siRNA for 72 hours. Cell
lysates were probed with either mouse anti-β-actin (left) or anti-γ-actin (left) antibodies with a pan-
actin antibody as a control. (C) Ratio of densitometry measurements of β-actin (C1) or γ-actin (C2)
levels under specified siRNA treatments compared to a scrambled siRNA control are shown for
HeLa and GBM cells. (D) Total actin levels in cells under siRNA treatment. Ratio of densitometry
measurements from the immunoblot of total actin (in B) under specified siRNA treatments
compared to a scrambled siRNA control are shown for HeLa and GBM cells.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
134
5.2.7 Silencing β-actin attenuates VACV-induced actin comet formation in cells
To further characterise the effect of β-actin depletion on actin comet formation,
hCMEC/D3 cells were infected with VACV-WR following treatment with respective
siRNAs, and visualised by immunofluorescence assay to reveal the morphology of the
actin cytoskeleton and VACV-induced actin comets (Figure 5.9A). We then assayed the
efficiency of actin comet nucleation by VACV by picking infected cells at random
(identifiable by their characteristic peri-nuclear virus factories) and of those, cells with 10
or more actin comets were noted. Treatment of cells with siRNA targeting β-actin
resulted in a significant reduction in the percentage of infected cells containing at least
10 actin comets compared to the scrambled negative control (Figure 5.9B).
There was no significant difference in actin comet production between the γ-actin-
depleted and scrambled siRNA-treated cells. Since it was not possible to stain for β-
and γ-actin specifically, in addition to phalloidin, DAPI and viral envelope staining, the
level of actin knockdown achieved in the individual cells chosen could not be
ascertained. As a result, our results for the number of cells with more than 10 actin
comets produced under β-actin depletion may be conservative. Nevertheless, it appears
as if the presence of β-actin is necessary for the initiation of VACV-induced actin
comets.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
135
Production of VACV-induced actin comets during actin knockdown.
hCMEC/D3 cells were treated with the respective siRNA for 72 hours, followed by VACV-WR
infection at an MOI > 5. Cells were fixed 8 hpi and stained for F-actin (green), envelope protein
B5 (red) and DAPI (blue). Scale bar is 10 μm. (B) The number of cells with more than 10 actin
comets was enumerated for each condition (‘***’: p<0.001; n=40 cells for each treatment, with
3 replicate experiments performed).

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
136
5.2.8 Loss of β-actin reduces VACV-induced actin comet length
As knockdown of β-actin reduced the formation of VACV-induced actin comets (Figure
5.9), we next examined the effect of β-actin knockdown on comet length, which is
related to the speed of virus motility and the stability of N-WASP and its activation of the
Arp2/3 complex [241, 332]. We aimed to use the VACV Lifeact-GFP virus created via
the method described in Chapter 3 to efficiently label VACV-induced actin comets in
real-time as they are produced, removing the need for the use of plasmid-based
expression of Lifeact-GFP. HeLa cells treated with the respective siRNA were infected
with both the VACV Lifeact-GFP and VACV WR viruses and fixed 8 hpi. Following
staining for envelope protein B5 and DAPI (and F-actin in the case of VACV WR
infection), cells were visualised by fluorescence microscopy. On comparing the two
viruses, we found that while both provided adequate highlighting of F-actin, post-
staining with phalloidin provided clearer and more abundant actin comets for
measurement (Figure 5.10A). Cell boundaries were also more clearly defined with
phalloidin. While the use of VACV Lifeact-GFP has obvious advantages in other areas
of analysis including live-cell microscopy, we opted to use post-staining with phalloidin
for actin comet measurements.
VACV-WR was used to infect hCMEC/D3 and HeLa cells treated with siRNA specific for
either β-actin or γ-actin, which were then fixed 8 hpi. Actin comets were visualised by
fluorescence microscopy and lengths of comets were measured using FIJI image
analysis software (ver 2.0.0-rc-43/1.51g) (Figure 5.10B1, B2). In both cell types
examined, VACV-induced actin comets produced in β-actin knockdown cells were
significantly shorter compared to those produced in the γ-actin knockdown or scrambled
siRNA-treated cells. There was no significant difference in comet length between the γ-
actin knockdown and scrambled siRNA-treated cells. In HeLa cells, the length of comets
in scrambled siRNA-treated and γ-actin knockdown cells was slightly greater than those
in control cells, however, just as in hCMEC/D3 cells, there was no significant difference
in comet length between the scramble siRNA-treated and γ-actin knockdown cells.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
137
VACV actin comet lengths under actin knockdown.
(A) HeLa cells were treated with siRNA indicated and infected with either VACV Lifeact-GFP or
VACV-WR to observe actin comet formation. Infected cells were fixed 8 hpi and stained for
envelope protein B5 (red), DAPI (blue) and phalloidin (green) in the case of VACV-WR
infection only. White arrows indicate actin comets. Scale bar is 10 μm. (B) hCMEC/D3 (B1) and
HeLa (B2) cell lines were treated with the siRNA as indicated for 72 hours and infected with
VACV-WR at an MOI > 3. Cells were fixed 8 hpi, followed by staining for F-actin (green). Actin
comet lengths were measured using FIJI (ver. 2.0.0) image-analysing software and statistical
analyses were carried out using GraphPad PRISM (ver. 6 for Mac OSX), with non-parametric t-
tests used to determine significance in differences between parameters (‘ns’: p > 0.05, ‘*’: p ≤
0.05, ‘****’: p ≤ 0.0001, n=60 comets each, with 2 experimental replicates). Means and
standard deviations (SD) for each group are provided below.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
138
5.2.9 VACV-induced actin comets exhibit greater speed under γ-actin knockdown
Since we revealed a reduction in actin comet length during silencing of β-actin
expression, we next tested if depletion of either actin had an effect on comet speed.
HeLa cells subjected to β- or γ-actin knockdown were infected with VACV Lifeact-GFP
and imaged live 7-9 hpi. Speeds were calculated by measuring actin comet lengths in
maximal projections of 1 min intervals from a 5 min video (Figure 5.11A). Maximum
intensity projections of frames captured over the entire 5 min period (at 4 sec intervals)
reveals the distance covered by the actin comet over time. Comets initiated by VACV in
HeLa cells with γ-actin knockdown spanned greater distances over the same period of
time (Figure 5.11A; right-most column). This measurement was used to calculate
speeds of actin comets under the different conditions.
Interestingly, while there was no significant difference in speed of comets produced
under β-actin depletion, scrambled siRNA-treated, and control cells, those produced
under γ-actin depletion were significantly faster compared to all other conditions by
more than 2-fold: 0.14 μm/sec in γ-actin-depleted cells compared to an average of 0.05
μm/sec in the other three conditions, including β-actin knockdown (Figure 5.11B).
Therefore, despite the finding that actin comets produced under β-actin knockdown are
shorter, their speeds remain the same relative to the controls. However, since β-actin
knockdown reduces the number of actin comets produced in general, the number of
comets available for analysis of speed was much lower compared to the other
conditions. There was no significant difference in length of actin comets produced by γ-
actin knockdown and scrambled siRNA-treated HeLa cells (Figure 5.11B2). This implies
that while β-actin is necessary for the initiation of VACV comets, the ratio of β-actin to γ-
actin in a cell may determine the speed of VACV motility, whereby the presence of γ-
actin in the actin comet has a moderating effect on virus speed.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
139
Live-cell analysis of actin comet speed under actin knockdown.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
140
Figure 5.11 description: HeLa cells were treated with respective siRNA for 72 h, followed by
infection with VACV Lifeact-GFP. Live cell microscopy was carried out 7-9 hpi (see methods
section 2.x for details) where images of actin comets were captured at 4 sec intervals over 5
mins. (A) Maximum intensity projections for 1 min intervals (15 consecutive frames) and the
entire 5 min interval (75 consecutive frames) were obtained. Scale bar is 10 μm. (B) Lengths of
actin comets over the 1 min time intervals were measured using FIJI (ver. 2.0.0) image-
analysing software and statistical analyses were carried out using GraphPad PRISM (ver. 6 for
Mac OSX), with one-way ANOVA and Tukey’s multiple comparison tests (n= at least 10
comets in 3 cells each, except for β-actin-targeting siRNA treated cells); ‘**’: p ≤ 0.01, ‘****’: p ≤
0.0001). Means and standard deviations (SD) for each group are provided below.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
141
5.3 DISCUSSION
The studies described in this chapter represent the first foray into examining the actin
isoform composition of not only VACV-induced actin comets, but of any actin comets
induced by a pathogen within a host cell. While both isoforms were found to be present
in actin comets, the exact ratios or spatial distribution of β- and γ-actin within a single
comet could not be adequately determined. More advanced super-resolving
fluorescence microscopy techniques such as structured-illumination microscopy might
enable a closer look at their fine-scale distribution [453].
We showed that actin comets produced by VACV during the late stages of an infection
have a reliance on the β-actin isoform. This was achieved by specific depletion of either
β-actin or γ-actin using siRNA, and observing the effects of this knockdown at the late
stages of a VACV infection. Depletion of β-actin quelled VACV-induced actin nucleation,
indicating a specific requirement for this isoform rather than it being due to a general
reduction in actin expression.
IFA images with specific actin-isoform staining reveals individual cells that were easily
identifiable as having undergone either β- or γ-actin knockdown, i.e., they were either
strongly red or green, as opposed to the co-localisation seen in scrambled-siRNA
treated cells. However, actin comets comprising both actins were still recorded under
both conditions, albeit less so under β-actin knockdown. Hence, an IFA of a cell treated
with anti-β-actin siRNA that appears red cannot be quantified as completely deficient of
β-actin. β- or γ-actin-specific siRNA were previously shown to cause incomplete
knockdown of their respective targets by immunoblotting methods [29, 435], and we
have observed similar results (Figure 5.8). Therefore, the immunoblot results may either
reflect a mixed population of cells, consisting of those experiencing actin knockdown to
a certain maximum extent and those that are not, or all cells experiencing the same
extent of incomplete actin knockdown. Incomplete knockdown was nonetheless
sufficient to cause significant changes to cell morphology and behaviour, as seen by
both Dugina [29] and our observations recorded here.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
142
Due to this variability of actin knockdown at the cellular level, we observed cells with
reduced β-actin expression that gave rise to few virus-associated comets. Even in these
cells, however, small accumulations of β-actin were evident at the virus surface (Figure
5.5B). These results are consistent with an absolute, albeit dosage-sensitive,
requirement for β-actin in VACV actin nucleation and actin-based motility. This further
suggests that the phenotype of reduced actin comets is due to the loss of β-actin and
not due to off-target effects of the siRNA. Indeed the residual expression of β-actin at
the virus surface bears some resemblance to the rhodamine-labelled G-actin monomers
that are incorporated into growing actin comets at the virus surface (Figure 5.1).
Whether β-actin monomers are preferentially recruited to points of VACV-induced actin
nucleation could be tested by incubating VACV-infected cells with differentially labelled
populations of β- or γ-actin. However this would require the expression and isolation of
pure populations of each actin isoform.
Previous studies have demonstrated significant morphological and functional changes in
various cell types with incomplete knockdown of actin isoforms, such as taking on a less
or a more contractile phenotype, or an increase/decrease in lamellipodial structures,
stress fibres, and cell motility [29, 435, 454]. We also observed morphological
differences in HeLa cells treated with actin-targeting siRNA for 72 hours. HeLa cells
undergoing β-actin silencing were more circular and showed an increased tendency to
form larger multinucleated cells. As a result, analysis of the effect of VACV infection with
β-actin knockdown was more difficult, compared to scrambled or γ-actin silenced cells.
β-actin-silenced cells with single nuclei had to be sought first before assessments (such
as the number of actin comets observed) could be made. Clearly the siRNA cocktails
used to silence β- and γ-actin were effective and the knockdown efficiencies obtained
were sufficient to cause the attenuation of actin comet production observed under β-
actin knockdown.
It was unfortunate that the VACV Lifeact-GFP virus was inferior in terms of adequately
highlighting actin comets in infected cells, when compared to an IFA of a phalloidin-
stained cell (Figure 5.10). As the expression of Lifeact-GFP is controlled by the VACV
pE/L promoter, the amount of protein expressed may be insufficient to effectively bind to
all F-actin in the cell. This is why we opted to use phalloidin-staining for our actin comet
length measurements. However, VACV Lifeact-GFP was viable for our live-cell

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
143
measurements (Figure 5.11), which was used to track VACV comets over time. Using
this technique, we observed that the speed of VACV comets in γ-actin-depleted cells
was greater than those in control, scrambled- or β-actin siRNA-treated cells. The
knockdown of γ-actin in HeLa cells causes a compensatory increase in the levels of β-
actin (Figure 5.8C1). This indicates that comets with greater speeds observed in γ-
depleted cells may be caused by a shift in the ratio of cytoplasmic actins in favour of β-
actin, which is in agreement with the previously shown ability of β-actin to polymerise
faster than γ-actin [423]. So far, two factors are known to increase VACV actin-based
speed, and are related to actin NPFs or accessory proteins. They are the reduced
stability of N-WASP (which also reduces comet length) [241] and the loss of recruitment
of AP-2 to the virus during initiation of the polymerisation cascade (which also increases
comet length) [62]. While comet speeds were increased under γ-actin knockdown, we
did not see a corresponding change in comet length compared to the scrambled siRNA
control, indicating an increase in the rate of actin polymerisation as well as
depolymerisation in these comets. As opposed to actin accessory proteins having an
effect on the rate of actin dynamics, a change in the ratio of actin isoforms may be
sufficient to influence the rate of polymerisation of VACV-induced actin comets.
We can test the role of γ-actin as a moderator of comet speed by performing the same
assay on cells under γ-actin knockdown for a shorter period of time (48 h as opposed to
72 h), which will result in reduced efficiencies of γ-actin depletion. If comet speeds are
slower, we would be able to further confirm the role of γ-actin as a regulator of actin
polymerisation speeds. Additionally, a Rho kinase inhibitor was found to selectively
disorganise β-actin bundles, without disturbing γ-actin [29]. Treatment of γ-actin-
depleted cells with this inhibitor could be monitored for its effect on VACV-induced
comet formation and/or speed, to confirm the role of β-actin in initiating actin comet
nucleation and promoting polymerisation. Finally, myosin II A is known to localise to β-
actin [29] while β-actin gene knockout cells show increased expression of genes with
myosin activity [436]. Moreover, overexpression of β-actin and a mutant β-actin that is
defective in polymerisation both increase cell motility, which can be retarded by addition
of a myosin inhibitor [437]. Therefore it is clear that β-actin is regulated by myosin
activity, a feature that can be tested by observing VACV comet speeds under β-actin
overexpression with/without myosin inhibitors.

CHAPTER 5: Divergent roles of β- and γ-actin in VACV-induced actin comet formation
The University of Sydney 2016
144
VACV-associated actin comets were fewer and shorter when levels of β-actin were
reduced, with no corresponding change in comet speed. Reductions in actin comet
length have been observed previously, either due to mutations in the viral A36 envelope
protein [332], or mutations in cellular N-WASP, which abrogates its ability to bind to
actin monomers and reduce its stability during actin polymerisation [241]. As we also
observed a significant reduction in the length of actin comets during β-actin depletion,
we hypothesised that altered binding efficiencies, such as a preference for β-actin by N-
WASP, may contribute to the β-actin-dependent actin nucleation phenotype. This
possibility was explored and is described in the next chapter. In addition, the
contribution of β- and γ-actin to VACV infectivity and spread in general will also be
examined.
Until now, studies of actin isoforms involved inferring function from localisation, or
observing whole-cell changes in movement and/or morphology under knockdown or
gene ablation. Work described here provides insights into the function of both isoforms
in different aspects of VACV-induced cell motility, serving as an observable and
traceable functional unit of actin polymerisation. Localisation studies alone would only
have given us part of the picture – that VACV comets comprise both actin isoforms. This
is unsurprising, since both actins readily copolymerise [423]. More mechanistic
differences in the cytoplasmic actins arise once siRNA-mediated knockdown is
achieved. Here we found that while β-actin is necessary for the initiation of VACV-
induced actin polymerisation, γ-actin is required to regulate the speed of comet
movement. This contributes a novel facet of differing actin roles to the small, but
growing, body of information we possess on these isoforms.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
145
Chapter 6: DIVERGENT ROLES OF
Β- AND Γ-ACTIN IN VACV SPREAD

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
146
6.1 INTRODUCTION
Author’s note: Sections of this chapter have been published in the
journal Cytoskeleton, under the title “Divergent roles of β- and γ-actin
isoforms during spread of vaccinia virus”. Its authors are N Bishara
Marzook, Sharissa L Latham, Helena Lynn, Christopher McKenzie,
Christine Chaponnier, Georges E Grau & Timothy P Newsome.
So far, we have analysed the functions of β-actin and γ-actin in VACV-induced actin
comets. However, the role of actin in VACV spread can be measured through
phenotypes other than the morphology of the actin comet (more details provided below).
Therefore, we aimed to explore these actin isoforms in the larger context of VACV
infection and spread, the results of which are outlined in this chapter. In addition we
attempted to address the dependence on β-actin for initiation of actin nucleation by
VACV.
6.1.1 Actin and VACV Spread
Efficient VACV spread is not only reliant on proper actin-based motility [62, 168, 207,
239, 455] as described in Chapters 1 and 5, but also by microtubule-based motility to
the cell surface [215, 216, 247], release of EEV [235, 332, 352, 456], VACV-infected cell
motility [228, 229] and cell-to-cell spread by plaque formation [331, 457].
The intercellular dissemination of VACV depends on its ability to form actin comets.
However, IEV particles need to gain access to the cell periphery before they can initiate
actin-based motility. Disruption of viral proteins involved in microtubule-based transport
such as F12 [331] and A27 [214, 458] can attenuate virus plaque size (i.e. the zone of
clearing in a cell monolayer created by lysis of infected cells originating from a
theoretical single viral ‘plaque-forming unit’), which can be used as a measurement of
virus fitness and infectivity [324]. Once at the cell surface, CEV initiate a complex
cascade of events ending in the polymerisation of actin beneath virus particles,
generating a force that propels them out of the cell (see section 1.3.1.1.1). Mutant
strains unable to undergo actin-based motility are attenuated in whole-animal mouse

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
147
infection models and deficient in cell-to-cell spread in vitro [223, 411, 456, 457, 459-
461]. Disruptions to viral membrane proteins such as A36, which is involved both in
microtubule- and actin-based motility, severely attenuates orthopoxvirus plaque
formation and spread in vivo [224, 332, 394, 411].
A36 is critical for the release of CEV from the cell surface, and therefore the generation
of EEV. While CEV are important for spread of VACV between adjacent cells, EEV
mediate long-range dissemination of virus [216, 462-464]. Despite making up only 1% of
virus progeny, EEV is the morphological variant against which protective immune
responses are directed [465]. The release of CEV particles as EEV requires an
untethering of the virus particle from the plasma membrane; A36-mediated actin
nucleation at the virus surface provides the force required for this process [332]. Briefly,
the phosphorylation of A36 generates binding sites for Nck and Grb2 adaptor proteins,
which then stabilise the actin NPF N-WASP (see section 1.3.1.1.1 for a detailed
explanation). The C-terminal VCA domain of N-WASP possesses two domains with
WH2 homology (‘V’ referring to the verprolin homology segment consisting of a single,
or both, WH2 domains), a central (‘C’, central or connector domain) and a short acidic
(‘A’) domain. N-WASP activates the Arp2/3 complex (bound to the side of an existing
actin filament) via the CA domain [117], while the V region containing the WH2 domains
binds to monomeric actin and activates polymerisation of a new actin filament under the
virus particle [466, 467] (see Figure 6.1A). Other envelope proteins such as A33 and
A34 are also responsible for regulating release of EEV, mutations in which can also
enhance EEV release [468, 469]. Localised actin nucleation by A36 and B5 expressed
on the surface of cells may allow for the ‘super-repulsion’ or leap-frogging of CEV or
EEV over infected cells, until an uninfected one is reached [333, 470]. Finally, VACV
can also induce infected cell motility, which is mediated by the viral protein F11L and its
influence on the cortical actin cytoskeleton (described in detail in section 4.1.1). The
ability of VACV to induce cell motility is vital for proper plaque formation and efficient
spread of infection of mice in vivo [229].

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
148
Actin nucleation cascade inititated by A36.
(A) Transmembrane protein A36 is present below CEV, sticking into the cytoplasm, where it is
phosphorylated by Src or Abl kinases. This creates binding sites for Nck and Grb2, which
stabilise WIP and N-WASP below the virus. Arp2/3 is recruited to the VCA domain, as it also
binds to an existing F-actin filament. Actin monomers also bind to the WH2 (V) domain on N-
WASP, which initiates the polymerisation of a new actin filament, creating a force that points
towards the VACV particle. (B) Describes what we know so far about the nature of the actin
composition of the VACV-induced comet: that it consists of both actin isoforms. However, the
requirement for β-actin to initiate actin nucleation may be reflected in a preference for β-actin
by the VCA WH2 domains.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
149
6.2 RESULTS
6.2.1 Extracellular virus release is reduced under β-actin knockdown
Since a clear attenuation of VACV-induced actin nucleation during β-actin knockdown
was evident, we next determined its effect on virus release, which is dependent on actin
nucleation [332]. HeLa cells under β-actin or γ-actin knockdown were infected with
either VACV-WR, or the mutant strain VACV-A36YdF. VACV-A36YdF contains point
mutations of the two tyrosine residues in A36R that, upon phosphorylation, are required
for the recruitment of the actin polymerisation machinery. Phenylalanine substitutions at
these two sites renders A36 incapable of being tyrosine-phosphorylated. VACV-A36YdF
is therefore unable to induce actin comets [170, 215] and a subsequent reduction in
release of extracellular enveloped virus (EEV) from the cell is observed [332].
Supernatants from infected cells were collected and plaque assays were performed to
determine the infectious EEV titre of each condition (Figure 6.2). In cells infected by
VACV-WR, EEV release into the supernatant (measured in plaque forming units or
pfu/mL) was significantly reduced under β-actin knockdown. These results are
consistent with our previous observations that disrupting actin-based motility leads to a
corresponding reduction in EEV release. Cells infected by VACV-A36YdF showed greatly
reduced EEV release when compared to VACV-WR as has been previously reported
[332]. However, no difference in EEV release of VACV-A36YdF was observed under
either actin knockdown condition. All phenotypes associated with actin-based motility
are ablated in VACV- A36YdF, and β-actin knockdown does not exacerbate defects in
EEV release in a VACV- A36YdF. This suggests that β-actin also disrupts EEV release
through actin-based motility rather than other WV protein interactions known to affect
EEV release [471].

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
150
EEV release under actin knockdown.
HeLa cells were treated with the specified siRNA for 72 hours and then infected with the
specified VACV (WR strain or VACV-A36YdF, depicted as YdF), at an MOI of 0.1. Supernatants
were collected at 16 hpi and used to perform plaque assays on BSC-1 cells to estimate viral
titre (‘ns’: p>0.05, ‘*’: p≤ 0.05, n=3).

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
151
6.2.2 VACV motility to the cell surface is not actin isoform-dependent
To rule out the possibility that depleting β-actin would affect motility of virus particles to
the cell surface and indirectly perturb EEV release, we assessed the number of WV
reaching the cell surface, or CEV (cell-associated extracellular virion).
HeLa cells under knockdown were infected with VACV-WR and fixed 8 hpi. Cells were
stained with an anti-B5 antibody (against viral envelope protein B5), prior to
permeabilising the cells. Since protein antibodies cannot pass through the lipid cell
membrane, only enveloped virus particles at the cell surface would be labelled (Figure
6.3A). The number of CEV was counted for each treatment using the FIJI cell counter
plugin tool (v 2.0.0-rc-54/1.51h).
As previously described, β-actin knockdown resulted in a higher percentage of
multinucleated cells compared to scrambled siRNA and γ-actin knockdown in uninfected
(Figure 5.8A) as well as infected (data not shown) HeLa cells. This phenomenon has
also been previously observed in uninfected HaCaT cells [29]. The presence of greater
numbers of multinucleated cells in the β-actin siRNA-treated samples made it difficult to
identify single infected cells for counting the number of CEV. Although single infected
cells were preferentially selected for ease of counting, the likelihood of those cells not
having their β-actin silenced is possible. There was no significant difference in the
number of anti-B5-stained virus particles at the cell surface between all three siRNA
treatment conditions (Figure 6.3B). This suggests that microtubule-based VACV motility
to the cell surface is not affected by the silencing of either actin isoform.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
152
Effect of actin knockdown on VACV motility to the cell surface.
HeLa cells were treated with the siRNA as indicated for 72 hours and infected with VACV-WR
at an MOI > 3. Cells were fixed 8 hpi, followed by staining with anti-B5 prior to permeabilisation
(A). The number of B5-positive virus particles at the cell surface was counted in each case (B)
(n=10 cells each, in 2 repeat experiments; scale bar is 10 μm).

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
153
6.2.3 Src is recruited to CEV even under β-actin knockdown
Src activity is required for VACV actin comet formation and localises to virus particles
undergoing actin-based motility [233, 235]. This is mediated by a signalling cascade that
is triggered once enveloped VACV reach the plasma membrane of an infected cell. EV
that travelled via microtubules to the cell membrane with the help of recruited kinesin,
switch to actin-based motility at the cell surface after the recruitment of Src kinase [215,
234]. The co-localisation of Src with CEV only occurs after virus particles no longer
associate with kinesin, indicating a transition away from microtubule-based motility,
which is followed by the Src-induced phosphorylation of viral protein A36 (see section
1.3.1.1.1), the first step in the actin-polymerisation pathway resulting in the formation of
virus-associated actin comets.
HeLa cells subjected to actin depletion for 72 hours were infected with WR and fixed 8
hpi. Cells were stained for viral envelope protein B5 and phalloidin prior to
permeabilisation, followed by staining for Src. This was to only allow for visualisation of
virus particles at the cell surface. Src localised to non-permeabilised B5 in all instances
of actin knockdown (Figure 6.4). In the case of scrambled siRNA- and γ-actin siRNA-
treated cells, B5 positive particles at the heads of actin comets localised to Src as
expected. Under β-actin knockdown, even though the number of actin comets produced
was greatly attenuated, B5 positive particles on the cell surface still localised to Src.
Therefore, not only is microtubule-based transport unaffected by β-actin depletion,
neither is recruitment of Src to CEV, the first step in the switch to actin-based motility of
VACV.
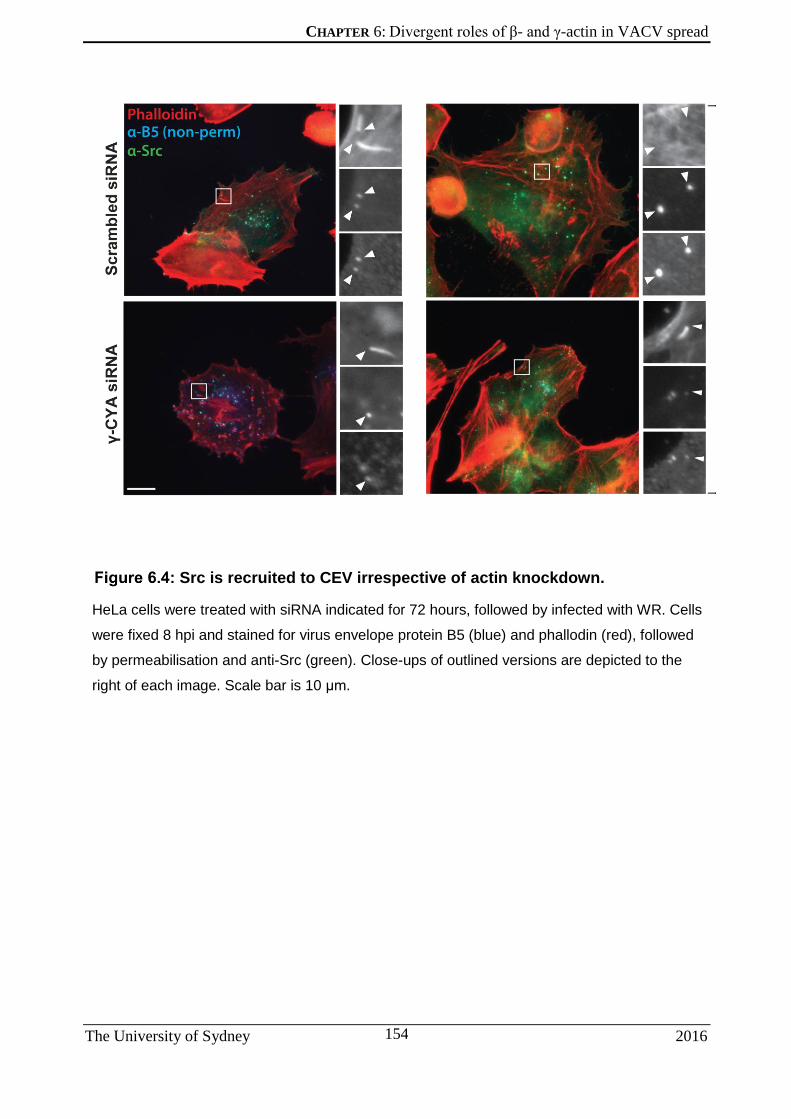
CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
154
Src is recruited to CEV irrespective of actin knockdown.
HeLa cells were treated with siRNA indicated for 72 hours, followed by infected with WR. Cells
were fixed 8 hpi and stained for virus envelope protein B5 (blue) and phallodin (red), followed
by permeabilisation and anti-Src (green). Close-ups of outlined versions are depicted to the
right of each image. Scale bar is 10 μm.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
155
6.2.4 VACV plaque size is significantly larger in cells under β-actin knockdown
Since actin-based motility in VACV enhances cell-to-cell spread, with VACV-A36YdF
producing a reduced plaque phenotype compared with parental strains [331, 332, 457],
we aimed to test how β-actin knockdown in cells might affect VACV plaque size or
morphology.
GBM cells (selected over HeLa due to their amenability to forming monolayers) were
subjected to actin knockdown and allowed to near confluence, at which point cells were
infected with VACV-WR at an MOI of 0.1. Three days post infection, viral plaques were
fixed and their diameters measured (Figure 6.5A). Confounding expectations, plaques
produced by VACV-WR treated with β-actin knockdown were significantly larger than
those compared to plaques produced in cells under γ-actin knockdown or treated with
scrambled siRNA (Figure 6.5B). These results indicate that knockdown of β-actin
produces a complex phenotype and that other mechanisms may compensate for the
expected reduction in plaque size elicited by the reduction in actin-based motility. The
tendency for cells undergoing β-actin knockdown to form aggregates of multinucleated
cells may also a play role in this greater plaque phenotype.
One issue encountered with the plaque assay was the inability to get perfect
monolayers of GBM cells, although they were more readily formed by GBM than HeLa
cells. The confluency of cells at the time of siRNA transfection had to be optimised over
a few attempts, to ensure an adequate monolayer would be formed 72 hours later, such
that a VACV plaque assay could be performed over another 72 hours. Often the
monolayer would be too sparse (especially at the centre of the well, as can be seen for
all four cases in Figure 6.5A) or too dense after 72 hours of siRNA treatment. A
compromise in cell seeding concentration had to be reached, to achieve sufficient
monolayer surface area to perform a plaque assay 72 hours post siRNA treatment,
before GBM cells became overcrowded or actin knockdown effects were sub-optimal.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
156
VACV plaque size under actin knockdown.
GBM cells were treated with the specified siRNA for 72 hours and infected with VACV-WR at
an MOI of 0.1. Cells were fixed and stained with crystal violet 3 dpi (A) to measure plaque
diameters (B). (‘****’: p ≤0.0001, n=30 plaques each, with 2 experimental replicates).

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
157
6.2.5 Expression of GST-bound VCA domain and its non-actin-binding mutant
Having uncovered a strict requirement for β-actin for the efficient formation of VACV-
induced actin comets, we next aimed to further define this mechanism. N-WASP is an
actin NPF that is recruited to the site of actin comet formation during VACV egress [43,
233] (see section 1.3.1.1.2). The VCA domain of N-WASP recruits the Arp2/3 complex
and two actin monomers, branching off from an existing actin filament. If the VCA
domain were to have a preference for the β-actin isoform, it would explain the
dependence on β-actin for actin-based virus transport (see Figure 6.1B).
A bacterial expression plasmid containing glutathione S-transferase (GST) tagged to the
VCA domain of rat N-WASP (generated by PCR; see section 2.1.8 and Figure 2.1C)
was created. Initially, GST-tagged VCA proteins were expressed in bacteria at two
different temperatures – 37oC or 23oC – and purified using Sepharose beads bound to
glutathione (Figure 6.6A). Cells grown at 23oC exhibited lower expression levels but
also lower presence of breakdown products (size of GST bound to the VCA domain
protein should be around 37 kDa). Therefore all future bacterial protein expression
cultures were carried out at 23oC.
Another plasmid expressing a GST-tagged VCA domain containing two arginine-alanine
substitutions in its actin-binding region, VCA R410A/R438A (called VCA RA/RA; see
Table 2.1.9 for details), which effectively abrogate actin binding [472] was created
(Figure 6.6B). The two GST-VCA constructs along with a control GST-expressing
plasmid were expressed in bacteria and purified by glutathione bound to Sepharose
beads (Figure 6.6C). This method was deemed efficient enough for the next step, which
was to testing the actin-binding capabilities of the respective VCA constructs.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
158

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
159
Production and purification of GST-tagged VCA and VCA RA/RA
mutant in bacteria.
(A) SDS-PAGE of GST-pulldown purification steps of bacterial lysate from cells expressing the
GST-VCA plasmid. Cells were grown at 37oC (left) or 23oC (right). (B) Sequence alignment of
original VCA domain from rat N-WASP and the VCA RA/RA mutant. Image made using
Geneious Pro v5.5.3. (C) SDS-PAGE of GST-pulldown purifications of bacterial lysate from
cells grown at 23oC, expressing the GST-VCA, GST-VCA RA/RA and GST control plasmids.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
160
6.2.6 The VCA domain of N-WASP does not show specificity for one actin isoform
Having successfully expressed and purified both GST-tagged VCA domains from
bacterial cells, we next had to test their ability to bind (or not bind, as in the case of
GST-VCA RA/RA) actin.
GST-tagged VCA proteins were expressed in bacteria, purified and used to enrich for
actin by passing HeLa cell lysates over the bound, immobilized protein and probing for
actin isoforms. As expected, actin bound to GST-VCA and did not bind to the GST-VCA
RA/RA (Figure 6.7A). We then examined whether the actin that bound to the VCA
domain was specifically one actin isoform. Both anti-β-actin and anti-γ-actin antibodies
bound to an immunoblot of cell lysates passed over GST-VCA protein (Figure 6.7B),
suggesting that the VCA domain does not specifically bind to one actin isoform over the
other.
While we were unable to conclude that the N-WASP VCA domain has an absolute
preference for β-actin over γ-actin, it may still have a binding preference for one over the
other. Since the antibodies for the two actins are different, with different binding
efficiencies, a direct comparison of the β- and γ-actin band intensities from Figure 6.7B
is not possible, and hence this experiment cannot be quantitative. Additionally, the
presence of salts favours stabilised F-actin over individual actin monomers [413]. Salts
in the cell lysis buffer used to obtain cellular actin, which was passed over bound GST,
may have favoured the presence of F-actin over β- and γ-actin monomers. Since we
have seen that F-actin usually consists of both cytoplasmic actin isoforms, our ability to
precisely study binding preferences of β- and γ-actin to N-WASP as individual
monomers using this method may be compromised.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
161
GST-VCA pull-down assays to determine binding preferences for β-
or γ-actin.
Immunoblots of cell lysate passed over GST-VCA or GST-VCA RA/RA bound to glutathione-
containing Sepharose beads. Immunoblots were probed with either anti-actin and anti-GST
antibodies (A) or antibodies specific to β-actin or γ-actin (B).

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
162
6.3 DISCUSSION
The results presented in this chapter provide an overview of insights into the effect of β-
actin depletion on the phenotype of VACV infection and spread.
While our data support a role for β-actin knockdown in the release of EEV through actin
nucleation (Figure 6.2), we also found that it did not disrupt the intracellular movement
of enveloped virus particles to the cell periphery (Figure 6.3). This is as expected, as
both the generation of intracellular enveloped VACV, as well as their transport to the cell
periphery, is independent from release of EEV particles [170, 332, 394, 473]. While the
depletion of γ-actin by siRNA can suppress microtubule dynamics in SH-EP cells
expressing GFP-labelled tubulin [474], we did not observe any difference in the number
of virus particles either being released or being transported to the cell periphery under γ-
actin depletion. As the major effects observed by Po’uha et al (2013) were a decrease in
microtubule shortening rates and a delayed metaphase-anaphase transition, this may
have been insufficient to grossly perturb the microtubule-based transport of VACV.
Any doubts on the efficiency of our CEV detection method, owing to the multinucleated
nature of β-actin-depleted cells, and the potentially differing penetrability of cell
membranes under actin knockdown (thus affecting CEV staining) may be addressed by
performing live-cell tracking experiments of VACV with fluorescently-tagged envelope
proteins (such as the F13L-GFP virus) in actin depleted cells. This was beyond the
scope of the time frame of this current project, but is an avenue we hope to explore in
the near future. However, the inability to discern cells undergoing actin depletion from
those that haven’t from within a population of siRNA-transfected cells, while also
visualising EV, remains a challenge. Additionally, single step growth curve assays to
determine VACV replication during actin knockdown may be pertinent, given the
detrimental effects of β-actin depletion on replication of the coronavirus [442] and the
classical swine fever virus [441]. However, previous studies have shown that
cytochalasin D (an inhibitor of actin polymerisation) does not affect the formation of CEV
[216] and hence VACV replication is likely to be independent of actin dynamics.

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
163
Not only have we shown that viral transport to the cell surface is unaffected by β-actin
knockdown, but also that Src-mediated switching of microtubule-based transport as
virus particles reach the cell periphery is unaffected by loss of β-actin (Figure 6.4).
Hence, the attenuation of VACV-induced actin comets, and the reduced release of EEV
into the culture medium under β-actin knockdown must be caused at a point
downstream of this, in the N-WASP-mediated actin polymerisation process. As we also
observed a significant reduction in the length of actin comets during β-actin depletion in
the previous chapter (Figure 5.10), we hypothesized that altered binding, such as a
preference for β-actin by N-WASP, may contribute to the actin nucleation phenotype. To
test this model, we expressed GST-tagged fusions of the VCA domain of N-WASP (its
actin-binding region of N-WASP; see [475] for a review), or a mutated version of the
domain with abrogated actin-binding ability (VCA-RA/RA, [472]). The ability of these
tagged proteins to bind to actin was examined, and no preference for either actin
isoform was observed by immunoblot. Additionally, phosphorylation of the N-WASP
VCA domain has been shown to enhance actin polymerisation activity [476], a post-
translational modification that may not be present in bacterially-expressed GST-VCA
domains. Nonetheless, our results do not support a specific binding affinity of the WH2
domains of N-WASP for β-actin as the underlying mechanism that results in a
requirement for this isoform in VACV actin-based motility. A recent genome-wide siRNA
screen performed during infection of HeLa cells with the bacterial pathogen Listeria,
also known to employ actin-based motility for infection [477], showed that siRNA-
mediated knockdown of β-actin, but not γ-actin, impaired infection [443]. These findings
suggest that specificity for actin isoforms for actin nucleation might be at the level of the
Arp2/3 complex, rather than with its activator N-WASP.
Our most surprising observation was the increase in VACV plaque size during β-actin
knockdown in GBM cells. γ-actin is a known regulator of Rho-associated kinase
(ROCK)-mediated cell migration, and the knockdown of γ-actin has been shown to
reduce cell migration [29, 454]. Additionally, we observed a significant increase in γ-
actin expression during β-actin knockdown in GBM cells (Figure 5.8C2), which may
have had an additive effect, contributing to increased viral plaque size. An enhancement
of directional cell migration during β-actin depletion, as well as a reduction in migration
during γ-actin depletion, has also been observed previously [29]. This increase in
motility during β-actin knockdown could also be playing a role in VACV virus-induced

CHAPTER 6: Divergent roles of β- and γ-actin in VACV spread
The University of Sydney 2016
164
cell motility, contributing to an increased plaque size. The VACV protein F11 is also
responsible for virus-induced cell migration, through its inhibition of RhoA signalling
[228, 229]. This may explain why we have not observed a corresponding reduction in
plaque size during γ-actin knockdown. An analysis of cell migration during actin
depletion with and without VACV infection would go a long way to address some of
these questions.
Interestingly, β-actin synthesis has also been associated with Rho signalling [478] and
the use of a Rho kinase inhibitor can cause the selective disorganisation of β-actin
bundles while leaving γ-actin undisturbed [29]. Clearly there is a strong link between β-
actin and Rho signalling, such that the depletion of β-actin may have a significant effect
on Rho and its downstream signalling targets. The use of ROCK inhibitors under
conditions of β- and γ-actin knockdown may, in future studies, clarify the complex
networks involved in this process. It is evident, however, that cell motility is an important
determinant of VACV plaque size.

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
165
Chapter 7: CONCLUSIONS AND
FUTURE DIRECTIONS

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
166
7.1 VACV AS A FLUORESCENT CELL BIOLOGICAL MARKER
We have shown that VACV continues to serve as a valuable vector for the fluorescence-
based study of protein function. Its relative ease of undergoing site-directed homologous
recombination, and ability to carry exogenous genes with little attenuation make it a
great tool in this regard. We have taken advantage of increasingly affordable
oligonucleotide synthesis tools to determine the minimal homology lengths required —
both by VACV for efficient and detectable levels of recombination events, as well as that
required by labs in search of time — and cost-effective methods for recombinant VACV
creation. While advances are being made for the use of the CRISPR-Cas9 system for
efficient editing of VACV [479, 480], we are still a while away from this becoming an
easily reproducible and time-sensitive tool.
Under this system, regions surrounding the desired locus for gene insertion can be
quickly designed in silico and manufactured, even as part of a cassette carrying multiple
oligonucleotides for the tagging of several genes, which can easily be inserted into a
recombination vector (as described in Figure 3.5). This frees up valuable time and
resources that would previously have been spent on several PCR and cloning steps
required to assemble the molecular biological tools.
This method offers recombinant VACV selection based on two levels: metabolic
selection and fluorescence screening, providing an extra tier of assessment for the
correct identification of recombinants. The fluorescence-screening step itself contains
two layers of selection: that of the constitutively expressed mCherry, and the promoter-
driven expression of the GFP (or other fluorescent gene of choice)-tagged viral protein
of interest. Approximately localised expression of the fluorescent protein of interest in a
plaque assay provides an initial confirmation of the success of fluorescent tag insertion
at the chosen locus.
We have used this method to engineer VACV that are capable of labelling both viral and
cellular structures during infection (see Figure 3.8). We are currently working on
purifying triple-labelled VACV capable of highlighting multiple cellular structures, such
as the nucleus or mitochondria, simultaneously during an infection. This will hopefully
eliminate the need for the complicated (and often eventually cytotoxic, as is the case for

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
167
many live dyes [481, 482]) lengths imaging cell biologists have to go to in order to image
multiple compartments within a cell in real-time. The success of the GFP-F1 protein is
especially promising for the study of this viral protein and its localisation to mitochondria
within a cell.
Live-cell imaging for the study of protein function is a valuable and necessary
complement to the more traditional practices of immunolabeling, as fixation and labelling
procedures often induce artefacts and distortions in protein structure and localisation
[483]. With the rapid advancement of imaging technology, capable of analysing
structures and protein-protein interactions at the molecular level [303, 484], the ability to
easily create fluorescently tagged viral proteins will only become more relevant. We
believe this technique is a valuable addition to the tools currently available to this end.

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
168
7.2 BETA-ACTIN IN VACV INFECTION AND BEYOND
This study represents the first cytoplasmic actin isoform-specific analysis of pathogen-
induced actin-based motility, as well as the first evidence found for a particular role
played by one isoform over the other in this context. We have confirmed a reliance on
the β-actin isoform for the initiation of VACV-induced actin comets, as well as EEV
release.
While previous studies have found β-actin to be essential for embryonic development of
mice [436], it is evidently also required for facilitating VACV infection and spread.
Additionally, the speed of actin comets showed an increase under γ-actin knockdown
(and hence, under increased presence of β-actin – as observed by western blot analysis
from Figure 5.8C2) in HeLa cells. Actin comets in these cells are comprised of more β-
than γ-actin (see Figure 5.5B), and hence this increase in comet speed is consistent
with the previous discovery that β-actin enjoys a much faster rate of polymerisation (and
depolymerisation) that its γ-actin counterpart [423]. Therefore, while VACV is dependent
on β-actin for initiation of actin-based motility, γ-actin plays a role in the regulation of
comet speed.
To determine the mechanism underpinning the β-actin requirement for VACV-induced
comets, we focused on N-WASP, the nucleation-promoting factor responsible for
activating the Arp2/3 complex and seeding the nucleation of a new branched F-actin
filament below the virus particle [232, 233, 241]. Our hypothesis that the actin-binding
WH2 domain on N-WASP would show a preference for β-actin over γ-actin was not
supported, although further improvements in experimental design may allow the role of
N-WASP to be revisited. Specifically, the use of pure isolated monomers of actin
isoforms in similar pull-down assays (and reverse pull-down assays with actin isoforms
as bait), as opposed to a cell lysate that likely comprised mixed populations of the two
isoforms in F-actin form.
Many intracellular pathogens undergo actin-based motility and a common thread tying
them all together is their dependence on the Arp2/3 complex for actin nucleation [152,
239], although a new study has discovered that certain species of virulent Burkholderia
express Ena/VASP mimics for their actin-nucleating activity instead [485]. These

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
169
species were found to not require any Arp2/3 activity for F-actin nucleation, and were
able to nucleate actin polymerisation in a manner similar to host Ena/VASP NPFs.
Nevertheless, since Arp2/3 activity is linked to actin-based motility of many pathogens
including VACV, it may be that the requirement for β-actin lies at the level of the Arp2/3
complex instead of N-WASP. Pyrene actin polymerisation reactions with Arp2/3 and
individual actin isoforms would help clear up some questions on this matter. However, it
would be pertinent to first determine the reliance on β-actin for actin-based motility of all
pathogens known to do so. For if those species of Burkholderia that do not rely on
Arp2/3 complex-mediated actin nucleation were found to also require β-actin for motility,
it would follow that the β-actin specificity lies elsewhere.

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
170
7.3 INVESTIGATING THE BIOCHEMICAL BASIS FOR BETA-ACTIN
DEPENDENCE ON VACV-MOTILITY – A CASE FOR ENA/VASP
We have shown that the dependence on β-actin for VACV-induced actin-based motility
does not lie within N-WASP-actin interactions. N-WASP is an NPF that is recruited to
the site of actin nucleation below virus particles, which activates the actin nucleator
Arp2/3. However, it is not the only actin accessory protein recruited to VACV or other
pathogens that produce actin comets. Ena/VASP proteins are a family of actin
regulatory proteins implicated in actin assembly and cell motility [486] that are also
recruited to actin comets in VACV [232, 487, 488], Listeria and Shigella [155, 489, 490]
and baculovirus [252]. The vasodilator-stimulated phosphoprotein (VASP) can bind to
both F- and G-actin and regulate actin dynamics, especially at the barbed end of
growing actin filaments [486, 491, 492]. VASP contains a WH2-like domain known as
Ena/VASP homology domain 2 (EVH2) situated at the C-terminal region which is
required for G-actin binding and actin nucleation activity [493, 494].
Not only does VASP localise to pathogen-induced actin comets, but the expression of
its inactive mutant form also inhibits comet formation in VACV and Shigella [488].
Expression of the dominant interfering VASPΔB in VACV-infected cells reduces both the
proportion of cells with actin tails, as well as the number of tails per cell [488]. Therefore
VASP is important for actin-based motility of these intracellular pathogens. The
presence of VASP also enhances the speed of protrusion in lamellipodia [495], and
propulsion of Listeria [155] and baculovirus [252], as well as that of beads in
reconstituted actin polymerisation assays [496, 497]. Analysis by in vitro TIRF
microscopy has revealed the ability of VASP to directly accelerate filament elongation
by delivering monomeric actin to the growing barbed end [498] Interestingly, VASP
appears to antagonise the formation of Arp2/3 complex-based actin filament branches
[495-497, 499].
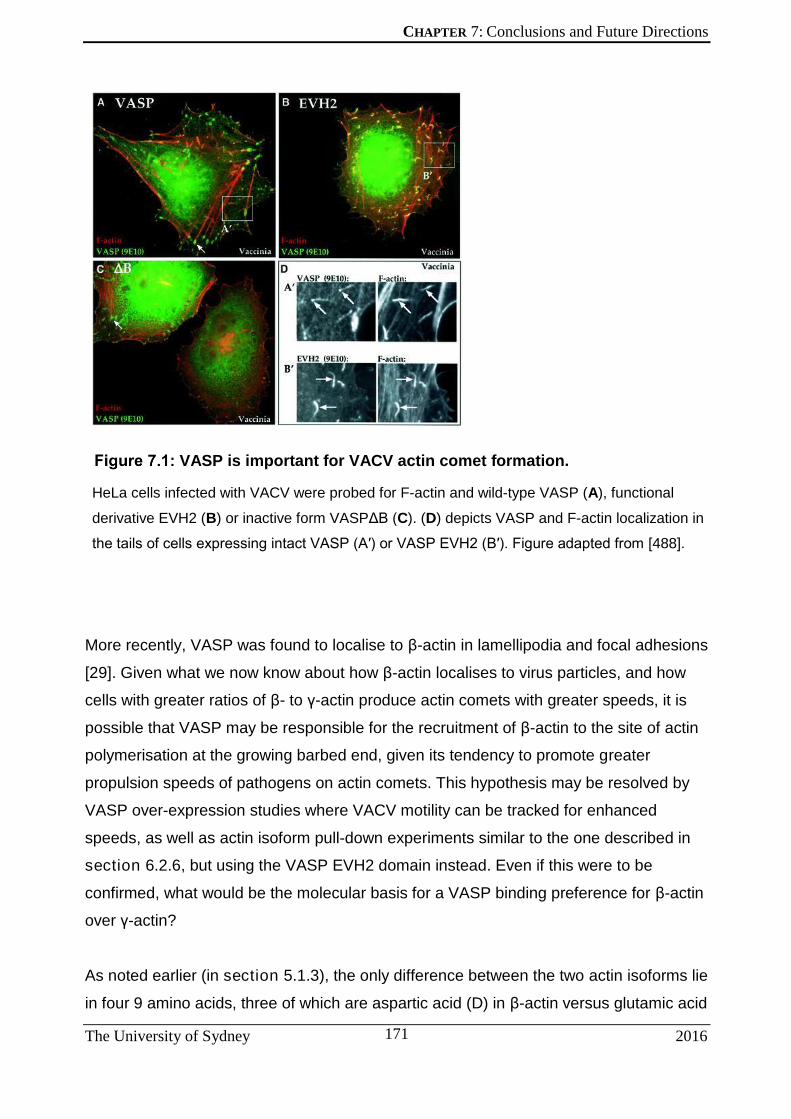
CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
171
VASP is important for VACV actin comet formation.
HeLa cells infected with VACV were probed for F-actin and wild-type VASP (A), functional
derivative EVH2 (B) or inactive form VASPΔB (C). (D) depicts VASP and F-actin localization in
the tails of cells expressing intact VASP (A′) or VASP EVH2 (B′). Figure adapted from [488].
More recently, VASP was found to localise to β-actin in lamellipodia and focal adhesions
[29]. Given what we now know about how β-actin localises to virus particles, and how
cells with greater ratios of β- to γ-actin produce actin comets with greater speeds, it is
possible that VASP may be responsible for the recruitment of β-actin to the site of actin
polymerisation at the growing barbed end, given its tendency to promote greater
propulsion speeds of pathogens on actin comets. This hypothesis may be resolved by
VASP over-expression studies where VACV motility can be tracked for enhanced
speeds, as well as actin isoform pull-down experiments similar to the one described in
section 6.2.6, but using the VASP EVH2 domain instead. Even if this were to be
confirmed, what would be the molecular basis for a VASP binding preference for β-actin
over γ-actin?
As noted earlier (in section 5.1.3), the only difference between the two actin isoforms lie
in four 9 amino acids, three of which are aspartic acid (D) in β-actin versus glutamic acid

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
172
(E) in γ-actin. Aspartic acid has a lower pKa than glutamic acid, and hence is more
acidic, and is reflected by the fact that β-actin is the more acidic isoform [423]. A
proposed mechanism for WH2 domain binding to actin monomers and initiating
nucleation involves the possession of sequence elements, which engage in electrostatic
interactions with an actin monomer. These interactions decrease the electronegativity of
G-actin to lower the free energy of nucleation associated with elementary steps of the
regular actin nucleation pathway [500], in a method known as ‘facilitated spontaneous
nucleation’. Interestingly, unlike WH2 domains in other NPFs, the N-WASP VCA region
(containing the WH2 domains) does not possess this ability, and is therefore unable to
nucleate actin polymerisation in the absence of the Arp2/3 complex, although binding of
Arp2/3 without a mother filament still does not confer nucleating activity to VCA [500]. In
contrast, VASP binding to G-actin is salt-dependent, and hence is based on electrostatic
interactions [501]. Therefore, the stronger electrostatic interactions afforded by β-actin
compared to γ-actin due to its more acidic amino acids may create a greater interaction
with VASP compared to N-WASP.
Structural analysis of the actin monomer crystal does not reveal a significant location for
these acidic residues near binding sites to actin-binding proteins, since the extreme N-
terminus of the monomer is part of an unstructured finger that reaches out into solution
from the protein surface [423, 502]. Ferron et al [503] have attempted to analyse the
interaction between VASP and monomeric actin at a structural level. While no
interactions between the N-terminal domain of profilin-bound actin and the G-actin-
binding domain (GAB) of VASP were recorded, a closer inspection of the crystal
structure analysed by the group (PDB code: 2PBD) is missing the first four amino acids
of actin (which is where the acidic D residues on β-actin lie – see Figure 5.2). When we
aligned the crystal structure of β-actin:profilin (PDB code: 1HLU) to the
profilin:actin:VASP structure instead, we observed the extreme N-terminal region
appear as a protrusion previously unseen by Ferron et al (Figure 7.2A, highlighted in
yellow). This arm comes into relatively close contact with the VASP GAB domain (at a
distance of 8.4 Å at their closest – see Figure 7.2B,D). Residues are thought to interact
at distances from about 5-7 Å [504, 505], so whether there is potential for the N-terminal
β-actin residues and GAB domain to interact is yet to be determined. Obtaining a crystal
structure of full-length β-actin:profilin:VASP GAB could help to address this question. In
addition, the Valine at position 9 (highlighted in orange in Figure 7.2C) is at the centre

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
173
of the structural core in subdomain 1 of actin [423] – which is also responsible for
creating the target-binding cleft along with subdomain 3, where the majority of VASP
GAB binds to actin [503]. Whether the difference between residues V or I (present in γ-
actin) at position 9 results in a slight conformational shift in this cleft, shifting a binding
preference for β-actin to the VASP GAB over γ-actin also remains to be seen. Finally,
an alignment of the VASP GAB to the actin-binding WH2 domain of WASP shows that
they both bind to the same actin-binding cleft on actin, although the VASP GAB is
rotated by 45o and sits forward by half a helical turn compared to WASP-WH2 [503]
(Figure 7.2E). Therefore while VASP and WH2 bind to the same regions on actin, their
differing conformations in this bound state could dictate an underlying preference for
one actin isoform over the other.
Many questions still persist regarding the pre-requisition for β-actin for VACV actin-
based motility. It is clear however that the particular actin dynamics possessed by β-
actin align with those offered by VASP, which warrants a closer examination of the link
between the two.

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
174
Alignment of β-actin:profilin:VASP-GAB.
The crystal structure of the actin:profilin:VASP-GAB complex (2PBD) was modified by aligning
β-actin (1HLU) to the structure instead. (A) Front-on view of the structure (β-actin in grey; 3 N-
terminal AAs in yellow; profilin in cyan; ATP in dark blue; VASP-GAB domain in magenta and
stick residue outlines). (B, D) Top-down and close-up front view showing the proximity of
VASP-GAB and N-terminal β-actin residues. (C) Top-down and through view showing the
location of Valine (orange) at position 9 within the actin-binding cleft created by subdomains 1
and 3. Figures created using MacPyMol v 1.7.4.5. (E) Figure adapted from [503], showing
alignment of VASP-GAB (magenta) and WASP-WH2 (green) to profilin:actin (cyan:grey).

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
175
7.4 CELL MIGRATION IN ORTHOPOXVIRUS INFECTION
Finally, we have reported the surprising finding of VACV producing bigger plaques
under β-actin knockdown. This was unexpected due to the established correlation
between actin-based motility and plaque size [331, 332]. On the other hand, the ability
of VACV to induce cell motility is also required for proper plaque formation [229]. In the
event of β-actin depletion, we find a compensatory increase in γ-actin expression in both
HeLa and GBM cells (Figure 5.8C1), and γ-actin has been linked to a role in cell motility
and migration [29, 435, 438, 454]. However, we do not see a corresponding decrease in
plaque size under γ-actin depletion, which may be explained by the role of viral protein
F11 in promoting cell migration and VACV release, presumably winning out over the
negative effects of γ-actin loss, or insufficient levels of γ-actin knockdown altogether.
Therefore, VACV plaque formation under β-actin knockdown presents a juxtaposition of
two potentially opposing forces: the viral protein F11, which promotes cell migration
through inhibition of RhoA signalling, and the increase in γ-actin, itself a potential
regulator of RhoA signalling [454] (see Figure 7.3). An analysis of cell migration under
actin knockdown, with or without VACV and Rho kinase inhibitors, would help gain
greater insight into the complex forces at play in such a system.
Yet other studies have linked β-actin with a role in cell migration [434, 506], however
this may be due to the use of different cell lines, varying methods of achieving
knockdown or gene ablation, and varying methods of measuring cell motility and/or
migration. Still, the degree of contribution of either actin isoform to cell motility is not yet
resolved.
The study of cell motility is significant not only in the context of orthopoxvirus infection,
but also for the study of tumour cell metastasis in the development of cancers [507].
Regulation of actin polymerisation is essential for the control of cancer cell migration,
and many studies have proven a correlation between the ability of cancer cells to
metastasise and the disruption of their actin polymerisation dynamics [508-510]. Given
what we now know about how differing β- and γ-actin levels may influence cell migration
in the context of VACV infection and plaque formation, elucidating the role of F11 in this

CHAPTER 7: Conclusions and Future Directions
The University of Sydney 2016
176
context is more vital than ever.
Opposing forces acting on the RhoA signalling pathway can
influence the integrity of the cortical actin cytoskeleton and cell migration.
The challenge often posed by intracellular pathogens is their reliance on essential
pathways for their replication and spread, making the design of antimicrobial targets all
the more arduous. The actin cytoskeleton presents the invading pathogen with a
number of hurdles in its pursuit of establishing an infection. Often these pathogens end
up using our systems against us, as is the case of pathogen exploiting actin-based
motility. In the case of VACV actin-based motility, we have discovered a requirement for
just one of the two ubiquitous cytoplasmic actin isoforms to this end. Soon we will be
able to tell whether this dependence is shared by all other pathogens capable of
abusing host actin dynamics for their motility. While β-actin is initially required for the
proper development of cells, γ-actin is more important for their long-term survival,
making β-actin a promising future target against intracellular pathogens.

CHAPTER 8: References
The University of Sydney 2016
177
Chapter 8: REFERENCES

CHAPTER 8: References
The University of Sydney 2016
178
References
1. Fletcher, D.A. and R.D. Mullins, Cell mechanics and the cytoskeleton. Nature, 2010. 463(7280): p. 485-92.
2. Amos, L.A., F. van den Ent, and J. Lowe, Structural/functional homology between the bacterial and eukaryotic cytoskeletons. Curr Opin Cell Biol, 2004. 16(1): p. 24-31.
3. Wickstead, B. and K. Gull, The evolution of the cytoskeleton. J Cell Biol, 2011. 194(4): p. 513-25.
4. Jones, L.J., R. Carballido-Lopez, and J. Errington, Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell, 2001. 104(6): p. 913-22.
5. Souza, W., Prokaryotic cells: structural organisation of the cytoskeleton and organelles. Mem Inst Oswaldo Cruz, 2012. 107(3): p. 283-93.
6. Shih, Y.L. and L. Rothfield, The bacterial cytoskeleton. Microbiol Mol Biol Rev, 2006. 70(3): p. 729-54.
7. Nogales, E., Structural insights into microtubule function. Annu Rev Biochem, 2000. 69: p. 277-302.
8. de Forges, H., A. Bouissou, and F. Perez, Interplay between microtubule dynamics and intracellular organization. Int J Biochem Cell Biol, 2012. 44(2): p. 266-74.
9. Etienne-Manneville, S., Microtubules in cell migration. Annu Rev Cell Dev Biol, 2013. 29: p. 471-99.
10. Luders, J. and T. Stearns, Microtubule-organizing centres: a re-evaluation. Nat Rev Mol Cell Biol, 2007. 8(2): p. 161-7.
11. Weisenberg, R.C., W.J. Deery, and P.J. Dickinson, Tubulin-nucleotide interactions during the polymerization and depolymerization of microtubules. Biochemistry, 1976. 15(19): p. 4248-54.
12. Desai, A. and T.J. Mitchison, Microtubule polymerization dynamics. Annu Rev Cell Dev Biol, 1997. 13: p. 83-117.
13. Mitchison, T. and M. Kirschner, Dynamic instability of microtubule growth. Nature, 1984. 312(5991): p. 237-42.
14. Akhmanova, A. and M.O. Steinmetz, Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol, 2008. 9(4): p. 309-22.
15. Lansbergen, G. and A. Akhmanova, Microtubule plus end: a hub of cellular activities. Traffic, 2006. 7(5): p. 499-507.

CHAPTER 8: References
The University of Sydney 2016
179
16. Etienne-Manneville, S., From signaling pathways to microtubule dynamics: the key players. Curr Opin Cell Biol, 2010. 22(1): p. 104-11.
17. Lodish, H., Berk, A., Zipursky, S.L., Matsudaira, P., Baltimore, D., and Darnell. J., Section 19.2: Microtubule Dynamics and Associated Proteins, in Molecular Cell Biology. 2000, W. H. Freeman: New York.
18. Herrmann, H., et al., Functional complexity of intermediate filament cytoskeletons: from structure to assembly to gene ablation. Int Rev Cytol, 2003. 223: p. 83-175.
19. Herrmann, H., et al., Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol, 2007. 8(7): p. 562-73.
20. Goldman, R.D., et al., The function of intermediate filaments in cell shape and cytoskeletal integrity. J Cell Biol, 1996. 134(4): p. 971-83.
21. Gruenbaum, Y., et al., The nuclear lamina comes of age. Nat Rev Mol Cell Biol, 2005. 6(1): p. 21-31.
22. Huber, F., et al., Emergent complexity of the cytoskeleton: from single filaments to tissue. Adv Phys, 2013. 62(1): p. 1-112.
23. Carlier, M.F., et al., Actin-based motility: from molecules to movement. Bioessays, 2003. 25(4): p. 336-45.
24. Plastino, J. and C. Sykes, The actin slingshot. Curr Opin Cell Biol, 2005. 17(1): p. 62-6.
25. Vandekerckhove, J. and K. Weber, At least six different actins are expressed in a higher mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J Mol Biol, 1978. 126(4): p. 783-802.
26. Perrin, B.J. and J.M. Ervasti, The actin gene family: function follows isoform. Cytoskeleton (Hoboken), 2010. 67(10): p. 630-4.
27. Rubenstein, P.A., The functional importance of multiple actin isoforms. Bioessays, 1990. 12(7): p. 309-15.
28. Khaitlina, S., Mechanisms of spatial segregation of actin isoforms. Tsitologiia, 2007. 49(5): p. 345-54.
29. Dugina, V., et al., Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J Cell Sci, 2009. 122(Pt 16): p. 2980-8.
30. Carlier, M.F., et al., Control of polarized assembly of actin filaments in cell motility. Cell Mol Life Sci, 2015. 72(16): p. 3051-67.
31. Disanza, A., et al., Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Mol Life Sci, 2005. 62(9): p. 955-70.

CHAPTER 8: References
The University of Sydney 2016
180
32. Campellone, K.G. and M.D. Welch, A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol, 2010. 11(4): p. 237-51.
33. Kirschner, M.W., Implications of treadmilling for the stability and polarity of actin and tubulin polymers in vivo. J Cell Biol, 1980. 86(1): p. 330-4.
34. Pollard, T.D. and G.G. Borisy, Cellular motility driven by assembly and disassembly of actin filaments. Cell, 2003. 112(4): p. 453-65.
35. Chesarone, M.A., A.G. DuPage, and B.L. Goode, Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol, 2010. 11(1): p. 62-74.
36. Goode, B.L. and M.J. Eck, Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem, 2007. 76: p. 593-627.
37. DeWard, A.D. and A.S. Alberts, Microtubule stabilization: formins assert their independence. Curr Biol, 2008. 18(14): p. R605-8.
38. Goley, E.D. and M.D. Welch, The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol, 2006. 7(10): p. 713-26.
39. Amann, K.J. and T.D. Pollard, Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A, 2001. 98(26): p. 15009-13.
40. Mullins, R.D., J.A. Heuser, and T.D. Pollard, The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A, 1998. 95(11): p. 6181-6.
41. Chereau, D., et al., Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc Natl Acad Sci U S A, 2005. 102(46): p. 16644-9.
42. Stradal, T.E., et al., Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol, 2004. 14(6): p. 303-11.
43. Moreau, V., et al., A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat Cell Biol, 2000. 2(7): p. 441-8.
44. Aspenstrom, P., A. Fransson, and J. Saras, Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J, 2004. 377(Pt 2): p. 327-37.
45. Humphries, A.C., S.K. Donnelly, and M. Way, Cdc42 and the Rho GEF intersectin-1 collaborate with Nck to promote N-WASP-dependent actin polymerisation. J Cell Sci, 2014. 127(Pt 3): p. 673-85.
46. Raftopoulou, M. and A. Hall, Cell migration: Rho GTPases lead the way. Dev Biol, 2004. 265(1): p. 23-32.

CHAPTER 8: References
The University of Sydney 2016
181
47. Quinlan, M.E., et al., Drosophila Spire is an actin nucleation factor. Nature, 2005. 433(7024): p. 382-8.
48. Schuldt, A., Spire: a new nucleator for actin. Nat Cell Biol, 2005. 7(2): p. 107.
49. Hartman, M.A. and J.A. Spudich, The myosin superfamily at a glance. J Cell Sci, 2012. 125(Pt 7): p. 1627-32.
50. Spudich, J.A., In pursuit of myosin function. Cell Regul, 1989. 1(1): p. 1-11.
51. Berg, J.S., B.C. Powell, and R.E. Cheney, A millennial myosin census. Mol Biol Cell, 2001. 12(4): p. 780-94.
52. Hasson, T., Myosin VI: two distinct roles in endocytosis. J Cell Sci, 2003. 116(Pt 17): p. 3453-61.
53. Vicente-Manzanares, M., et al., Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol, 2009. 10(11): p. 778-90.
54. Coue, M., et al., Inhibition of actin polymerization by latrunculin A. FEBS Lett, 1987. 213(2): p. 316-8.
55. Spector, I., et al., Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science, 1983. 219(4584): p. 493-5.
56. Braet, F., et al., Microfilament-disrupting agent latrunculin A induces and increased number of fenestrae in rat liver sinusoidal endothelial cells: comparison with cytochalasin B. Hepatology, 1996. 24(3): p. 627-35.
57. Cooper, J.A., Effects of cytochalasin and phalloidin on actin. J Cell Biol, 1987. 105(4): p. 1473-8.
58. Cramer, L.P., Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr Biol, 1999. 9(19): p. 1095-105.
59. Bear, J.E., M. Krause, and F.B. Gertler, Regulating cellular actin assembly. Curr Opin Cell Biol, 2001. 13(2): p. 158-66.
60. Favoreel, H.W., L.W. Enquist, and B. Feierbach, Actin and Rho GTPases in herpesvirus biology. Trends Microbiol, 2007. 15(9): p. 426-33.
61. Harries, P.A., et al., Differing requirements for actin and myosin by plant viruses for sustained intercellular movement. Proc Natl Acad Sci U S A, 2009. 106(41): p. 17594-9.
62. Humphries, A.C., et al., Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell host & microbe, 2012. 12(3): p. 346-59.
63. Leite, F. and M. Way, The role of signalling and the cytoskeleton during Vaccinia Virus egress. Virus Res, 2015.

CHAPTER 8: References
The University of Sydney 2016
182
64. Naghavi, M.H. and S.P. Goff, Retroviral proteins that interact with the host cell cytoskeleton. Curr Opin Immunol, 2007. 19(4): p. 402-7.
65. Iyengar, S., J.E. Hildreth, and D.H. Schwartz, Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J Virol, 1998. 72(6): p. 5251-5.
66. Liu, Y., N.V. Belkina, and S. Shaw, HIV infection of T cells: actin-in and actin-out. Sci Signal, 2009. 2(66): p. pe23.
67. Barrero-Villar, M., et al., Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J Cell Sci, 2009. 122(Pt 1): p. 103-13.
68. Jimenez-Baranda, S., et al., Filamin-A regulates actin-dependent clustering of HIV receptors. Nat Cell Biol, 2007. 9(7): p. 838-46.
69. Thomas, A., et al., Involvement of the Rac1-IRSp53-Wave2-Arp2/3 Signaling Pathway in HIV-1 Gag Particle Release in CD4 T Cells. J Virol, 2015. 89(16): p. 8162-81.
70. Arhel, N., et al., Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat Methods, 2006. 3(10): p. 817-24.
71. Fackler, O.T., et al., Association of human immunodeficiency virus Nef protein with actin is myristoylation dependent and influences its subcellular localization. Eur J Biochem, 1997. 247(3): p. 843-51.
72. Gaudin, R., et al., HIV trafficking in host cells: motors wanted! Trends Cell Biol, 2013. 23(12): p. 652-62.
73. Rey, O., J. Canon, and P. Krogstad, HIV-1 Gag protein associates with F-actin present in microfilaments. Virology, 1996. 220(2): p. 530-4.
74. Lehmann, M., D.S. Nikolic, and V. Piguet, How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses, 2011. 3(9): p. 1757-76.
75. Sherer, N.M., et al., Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol, 2007. 9(3): p. 310-5.
76. Carlson, L.A., et al., Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog, 2010. 6(11): p. e1001173.
77. Aggarwal, A., et al., Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells. PLoS Pathog, 2012. 8(6): p. e1002762.
78. Mattila, P.K. and P. Lappalainen, Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol, 2008. 9(6): p. 446-54.
79. Chhabra, E.S. and H.N. Higgs, The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol, 2007. 9(10): p. 1110-21.

CHAPTER 8: References
The University of Sydney 2016
183
80. Mitchison, T. and M. Kirschner, Cytoskeletal dynamics and nerve growth. Neuron, 1988. 1(9): p. 761-72.
81. Lehmann, M.J., et al., Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol, 2005. 170(2): p. 317-25.
82. Akhtar, J. and D. Shukla, Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J, 2009. 276(24): p. 7228-36.
83. Clement, C., et al., A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol, 2006. 174(7): p. 1009-21.
84. Dixit, R., V. Tiwari, and D. Shukla, Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci Lett, 2008. 440(2): p. 113-8.
85. Oh, M.J., et al., A role for heparan sulfate in viral surfing. Biochem Biophys Res Commun, 2010. 391(1): p. 176-81.
86. Schelhaas, M., et al., Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog, 2008. 4(9): p. e1000148.
87. Ehrlich, M., et al., Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell, 2004. 118(5): p. 591-605.
88. McMahon, H.T. and E. Boucrot, Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol, 2011. 12(8): p. 517-33.
89. Praefcke, G.J. and H.T. McMahon, The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol, 2004. 5(2): p. 133-47.
90. McNiven, M.A., et al., Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol, 2000. 151(1): p. 187-98.
91. Humphries, A.C. and M. Way, The non-canonical roles of clathrin and actin in pathogen internalization, egress and spread. Nat Rev Microbiol, 2013. 11(8): p. 551-60.
92. Merrifield, C.J., et al., Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol, 2002. 4(9): p. 691-8.
93. Taylor, M.J., M. Lampe, and C.J. Merrifield, A feedback loop between dynamin and actin recruitment during clathrin-mediated endocytosis. PLoS Biol, 2012. 10(4): p. e1001302.
94. Boucrot, E., et al., Role of lipids and actin in the formation of clathrin-coated pits. Exp Cell Res, 2006. 312(20): p. 4036-48.

CHAPTER 8: References
The University of Sydney 2016
184
95. Yarar, D., C.M. Waterman-Storer, and S.L. Schmid, A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell, 2005. 16(2): p. 964-75.
96. Cureton, D.K., et al., Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog, 2009. 5(4): p. e1000394.
97. Saffarian, S., E. Cocucci, and T. Kirchhausen, Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol, 2009. 7(9): p. e1000191.
98. Rust, M.J., et al., Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol, 2004. 11(6): p. 567-73.
99. Banerjee, I., et al., Influenza A virus uses the aggresome processing machinery for host cell entry. Science, 2014. 346(6208): p. 473-7.
100. Acosta, E.G., V. Castilla, and E.B. Damonte, Infectious dengue-1 virus entry into mosquito C6/36 cells. Virus Res, 2011. 160(1-2): p. 173-9.
101. Greene, W. and S.J. Gao, Actin dynamics regulate multiple endosomal steps during Kaposi's sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog, 2009. 5(7): p. e1000512.
102. Hernaez, B. and C. Alonso, Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J Virol, 2010. 84(4): p. 2100-9.
103. van der Schaar, H.M., et al., Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog, 2008. 4(12): p. e1000244.
104. Galindo, I., et al., African swine fever virus infects macrophages, the natural host cells, via clathrin- and cholesterol-dependent endocytosis. Virus Res, 2015. 200: p. 45-55.
105. Lim, J.P. and P.A. Gleeson, Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol, 2011. 89(8): p. 836-43.
106. Swanson, J.A. and C. Watts, Macropinocytosis. Trends Cell Biol, 1995. 5(11): p. 424-8.
107. Araki, N., M.T. Johnson, and J.A. Swanson, A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol, 1996. 135(5): p. 1249-60.
108. Amyere, M., et al., Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol Biol Cell, 2000. 11(10): p. 3453-67.
109. Koivusalo, M., et al., Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol, 2010. 188(4): p. 547-63.

CHAPTER 8: References
The University of Sydney 2016
185
110. Mercer, J. and A. Helenius, Virus entry by macropinocytosis. Nat Cell Biol, 2009. 11(5): p. 510-20.
111. Mercer, J. and A. Helenius, Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science, 2008. 320(5875): p. 531-5.
112. Schmidt, F.I., et al., Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J, 2011. 30(17): p. 3647-61.
113. Sieczkarski, S.B. and G.R. Whittaker, Dissecting virus entry via endocytosis. J Gen Virol, 2002. 83(Pt 7): p. 1535-45.
114. Wang, S., et al., Entry of a novel marine DNA virus, Singapore grouper iridovirus, into host cells occurs via clathrin-mediated endocytosis and macropinocytosis in a pH-dependent manner. J Virol, 2014. 88(22): p. 13047-63.
115. Merrifield, C.J., et al., Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nat Cell Biol, 1999. 1(1): p. 72-4.
116. Taunton, J., Actin filament nucleation by endosomes, lysosomes and secretory vesicles. Curr Opin Cell Biol, 2001. 13(1): p. 85-91.
117. Welch, M.D. and R.D. Mullins, Cellular control of actin nucleation. Annu Rev Cell Dev Biol, 2002. 18: p. 247-88.
118. Khaitlina, S.Y., Intracellular transport based on actin polymerization. Biochemistry (Mosc), 2014. 79(9): p. 917-27.
119. Gouin, E., M.D. Welch, and P. Cossart, Actin-based motility of intracellular pathogens. Curr Opin Microbiol, 2005. 8(1): p. 35-45.
120. Lakadamyali, M., et al., Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U S A, 2003. 100(16): p. 9280-5.
121. Hao, X., et al., Single-particle tracking of hepatitis B virus-like vesicle entry into cells. Small, 2011. 7(9): p. 1212-8.
122. Burke, E., et al., Profilin is required for optimal actin-dependent transcription of respiratory syncytial virus genome RNA. J Virol, 2000. 74(2): p. 669-75.
123. Koga, R., et al., Actin-Modulating Protein Cofilin Is Involved in the Formation of Measles Virus Ribonucleoprotein Complex at the Perinuclear Region. J Virol, 2015. 89(20): p. 10524-31.
124. Arber, S., et al., Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature, 1998. 393(6687): p. 805-9.
125. Bravo-Cordero, J.J., et al., Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol, 2013. 14(7): p. 405-15.
126. Xiang, Y., et al., Cofilin 1-mediated biphasic F-actin dynamics of neuronal cells affect herpes simplex virus 1 infection and replication. J Virol, 2012. 86(16): p. 8440-51.

CHAPTER 8: References
The University of Sydney 2016
186
127. Yoder, A., et al., HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell, 2008. 134(5): p. 782-92.
128. Cibulka, J., M. Fraiberk, and J. Forstova, Nuclear actin and lamins in viral infections. Viruses, 2012. 4(3): p. 325-47.
129. Ohkawa, T. and L.E. Volkman, Nuclear F-actin is required for AcMNPV nucleocapsid morphogenesis. Virology, 1999. 264(1): p. 1-4.
130. Goley, E.D., et al., Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science, 2006. 314(5798): p. 464-7.
131. Ohkawa, T., A.R. Rowe, and L.E. Volkman, Identification of six Autographa californica multicapsid nucleopolyhedrovirus early genes that mediate nuclear localization of G-actin. J Virol, 2002. 76(23): p. 12281-9.
132. Wang, Y., et al., Identification of a novel regulatory sequence of actin nucleation promoting factor encoded by Autographa californica multiple nucleopolyhedrovirus. J Biol Chem, 2015. 290(15): p. 9533-41.
133. Marek, M., et al., Baculovirus VP80 protein and the F-actin cytoskeleton interact and connect the viral replication factory with the nuclear periphery. J Virol, 2011. 85(11): p. 5350-62.
134. Hofmann, W., et al., Cofactor requirements for nuclear export of Rev response element (RRE)- and constitutive transport element (CTE)-containing retroviral RNAs. An unexpected role for actin. J Cell Biol, 2001. 152(5): p. 895-910.
135. Kimura, T., et al., Rev-dependent association of the intron-containing HIV-1 gag mRNA with the nuclear actin bundles and the inhibition of its nucleocytoplasmic transport by latrunculin-B. Genes Cells, 2000. 5(4): p. 289-307.
136. Schudt, G., et al., Live-cell imaging of Marburg virus-infected cells uncovers actin-dependent transport of nucleocapsids over long distances. Proc Natl Acad Sci U S A, 2013. 110(35): p. 14402-7.
137. Dolnik, O., et al., Interaction with Tsg101 is necessary for the efficient transport and release of nucleocapsids in marburg virus-infected cells. PLoS Pathog, 2014. 10(10): p. e1004463.
138. Feierbach, B., et al., Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog, 2006. 2(8): p. e85.
139. Bosse, J.B., et al., Nuclear herpesvirus capsid motility is not dependent on F-actin. MBio, 2014. 5(5): p. e01909-14.
140. Berghall, H., et al., Role of cytoskeleton components in measles virus replication. Arch Virol, 2004. 149(5): p. 891-901.
141. Kallewaard, N.L., A.L. Bowen, and J.E. Crowe, Jr., Cooperativity of actin and microtubule elements during replication of respiratory syncytial virus. Virology, 2005. 331(1): p. 73-81.

CHAPTER 8: References
The University of Sydney 2016
187
142. Dietzel, E., L. Kolesnikova, and A. Maisner, Actin filaments disruption and stabilization affect measles virus maturation by different mechanisms. Virol J, 2013. 10: p. 249.
143. Wakimoto, H., et al., F-actin modulates measles virus cell-cell fusion and assembly by altering the interaction between the matrix protein and the cytoplasmic tail of hemagglutinin. J Virol, 2013. 87(4): p. 1974-84.
144. Giuffre, R.M., et al., Evidence for an interaction between the membrane protein of a paramyxovirus and actin. J Virol, 1982. 42(3): p. 963-8.
145. Roberts, K.L., B. Manicassamy, and R.A. Lamb, Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol, 2015. 89(3): p. 1537-49.
146. Kadiu, I. and H.E. Gendelman, Human immunodeficiency virus type 1 endocytic trafficking through macrophage bridging conduits facilitates spread of infection. J Neuroimmune Pharmacol, 2011. 6(4): p. 658-75.
147. Gousset, K., et al., Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol, 2009. 11(3): p. 328-36.
148. Haglund, C.M. and M.D. Welch, Pathogens and polymers: microbe-host interactions illuminate the cytoskeleton. J Cell Biol, 2011. 195(1): p. 7-17.
149. Heindl, J.E., et al., Requirement for formin-induced actin polymerization during spread of Shigella flexneri. Infect Immun, 2010. 78(1): p. 193-203.
150. Quinlan, M.E. and E. Kerkhoff, Actin nucleation: bacteria get in-Spired. Nat Cell Biol, 2008. 10(1): p. 13-5.
151. Vance, R.E., R.R. Isberg, and D.A. Portnoy, Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe, 2009. 6(1): p. 10-21.
152. Welch, M.D. and M. Way, Arp2/3-mediated actin-based motility: a tail of pathogen abuse. Cell Host Microbe, 2013. 14(3): p. 242-55.
153. Welch, M.D., et al., Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science, 1998. 281(5373): p. 105-8.
154. Smith, G.A., J.A. Theriot, and D.A. Portnoy, The tandem repeat domain in the Listeria monocytogenes ActA protein controls the rate of actin-based motility, the percentage of moving bacteria, and the localization of vasodilator-stimulated phosphoprotein and profilin. J Cell Biol, 1996. 135(3): p. 647-60.
155. Loisel, T.P., et al., Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature, 1999. 401(6753): p. 613-6.
156. Welch, M.D., A. Iwamatsu, and T.J. Mitchison, Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature, 1997. 385(6613): p. 265-9.

CHAPTER 8: References
The University of Sydney 2016
188
157. Jeng, R.L., et al., A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cell Microbiol, 2004. 6(8): p. 761-9.
158. Sitthidet, C., et al., Actin-based motility of Burkholderia thailandensis requires a central acidic domain of BimA that recruits and activates the cellular Arp2/3 complex. J Bacteriol, 2010. 192(19): p. 5249-52.
159. Bernardini, M.L., et al., Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A, 1989. 86(10): p. 3867-71.
160. Suzuki, T., et al., Neural Wiskott-Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell Microbiol, 2002. 4(4): p. 223-33.
161. Egile, C., et al., Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol, 1999. 146(6): p. 1319-32.
162. Burton, E.A., T.N. Oliver, and A.M. Pendergast, Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway. Mol Cell Biol, 2005. 25(20): p. 8834-43.
163. Leung, Y., S. Ally, and M.B. Goldberg, Bacterial actin assembly requires toca-1 to relieve N-wasp autoinhibition. Cell Host Microbe, 2008. 3(1): p. 39-47.
164. Charlton, C.A. and L.E. Volkman, Penetration of Autographa californica nuclear polyhedrosis virus nucleocapsids into IPLB Sf 21 cells induces actin cable formation. Virology, 1993. 197(1): p. 245-54.
165. Ohkawa, T., L.E. Volkman, and M.D. Welch, Actin-based motility drives baculovirus transit to the nucleus and cell surface. J Cell Biol, 2010. 190(2): p. 187-95.
166. Rohrmann, G.F., M.A. Erlandson, and D.A. Theilmann, The genome of a baculovirus isolated from Hemileuca sp. encodes a serpin ortholog. Virus Genes, 2013. 47(2): p. 357-64.
167. Lanier, L.M. and L.E. Volkman, Actin binding and nucleation by Autographa california M nucleopolyhedrovirus. Virology, 1998. 243(1): p. 167-77.
168. Cudmore, S., et al., Actin-based motility of vaccinia virus. Nature, 1995. 378(6557): p. 636-8.
169. Cudmore, S., et al., Vaccinia virus: a model system for actin-membrane interactions. J Cell Sci, 1996. 109 ( Pt 7): p. 1739-47.
170. Ward, B.M. and B. Moss, Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J Virol, 2001. 75(23): p. 11651-63.
171. Paavilainen, V.O., et al., Regulation of cytoskeletal dynamics by actin-monomer-binding proteins. Trends Cell Biol, 2004. 14(7): p. 386-94.

CHAPTER 8: References
The University of Sydney 2016
189
172. Rosenblatt, J., et al., Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J Cell Biol, 1997. 136(6): p. 1323-32.
173. Carlier, M.F., et al., Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol, 1997. 136(6): p. 1307-22.
174. Abella, J.V., et al., Isoform diversity in the Arp2/3 complex determines actin filament dynamics. Nat Cell Biol, 2016. 18(1): p. 76-86.
175. Haller, S.L., et al., Poxviruses and the evolution of host range and virulence. Infect Genet Evol, 2014. 21: p. 15-40.
176. Iyer, L.M., et al., Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res, 2006. 117(1): p. 156-84.
177. Buller, R.M. and G.J. Palumbo, Poxvirus pathogenesis. Microbiol Rev, 1991. 55(1): p. 80-122.
178. Philippe, N., et al., Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science, 2013. 341(6143): p. 281-6.
179. Popgeorgiev, N., et al., Marseillevirus-like virus recovered from blood donated by asymptomatic humans. J Infect Dis, 2013. 208(7): p. 1042-50.
180. Abrahao, J.S., et al., Acanthamoeba polyphaga mimivirus and other giant viruses: an open field to outstanding discoveries. Virol J, 2014. 11: p. 120.
181. Condit, R.C., N. Moussatche, and P. Traktman, In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res, 2006. 66: p. 31-124.
182. Lefkowitz, E.J., C. Wang, and C. Upton, Poxviruses: past, present and future. Virus Res, 2006. 117(1): p. 105-18.
183. Gubser, C., et al., Poxvirus genomes: a phylogenetic analysis. J Gen Virol, 2004. 85(Pt 1): p. 105-17.
184. Upton, C., et al., Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol, 2003. 77(13): p. 7590-600.
185. McFadden, G., Poxvirus tropism. Nat Rev Microbiol, 2005. 3(3): p. 201-13.
186. Fenner, F. and F.M. Burnet, A short description of the poxvirus group (vaccinia and related viruses). Virology, 1957. 4(2): p. 305-14.
187. Fenner, F., Henderson, D.A., Arita, I., Jezek, Z., Ladnyi, I.D., Smallpox and its Eradication, G. World Health Organisation, Switzerland Editor. 1988.
188. Sanchez-Sampedro, L., et al., The evolution of poxvirus vaccines. Viruses, 2015. 7(4): p. 1726-803.

CHAPTER 8: References
The University of Sydney 2016
190
189. Studdert, M.J., Experimental vaccinia virus infection of horses. Aust Vet J, 1989. 66(5): p. 157-9.
190. Esparza, J., Has horsepox become extinct? Vet Rec, 2013. 173(11): p. 272-3.
191. Hughes, A.L., S. Irausquin, and R. Friedman, The evolutionary biology of poxviruses. Infect Genet Evol, 2010. 10(1): p. 50-9.
192. Mahy, B.W., An overview on the use of a viral pathogen as a bioterrorism agent: why smallpox? Antiviral Res, 2003. 57(1-2): p. 1-5.
193. Smith, G.L. and G. McFadden, Smallpox: anything to declare? Nat Rev Immunol, 2002. 2(7): p. 521-7.
194. Stone, R., Smallpox. WHO puts off destruction of U.S., Russian caches. Science, 2002. 295(5555): p. 598-9.
195. Artenstein, A.W. and J.D. Grabenstein, Smallpox vaccines for biodefense: need and feasibility. Expert Rev Vaccines, 2008. 7(8): p. 1225-37.
196. Moss, B., Vaccinia virus: a tool for research and vaccine development. Science, 1991. 252(5013): p. 1662-7.
197. Miner, J.N. and D.E. Hruby, Vaccinia virus: a versatile tool for molecular biologists. Trends Biotechnol, 1990. 8(1): p. 20-5.
198. Jefferson, A., V.E. Cadet, and A. Hielscher, The mechanisms of genetically modified vaccinia viruses for the treatment of cancer. Crit Rev Oncol Hematol, 2015. 95(3): p. 407-16.
199. Shen, Y. and J. Nemunaitis, Fighting cancer with vaccinia virus: teaching new tricks to an old dog. Mol Ther, 2005. 11(2): p. 180-95.
200. Abrahao, J.S., et al., Outbreak of severe zoonotic vaccinia virus infection, Southeastern Brazil. Emerg Infect Dis, 2015. 21(4): p. 695-8.
201. Pereira Oliveira, G., et al., Intrafamilial Transmission of Vaccinia virus during a Bovine Vaccinia Outbreak in Brazil: A New Insight in Viral Transmission Chain. The American Journal of Tropical Medicine and Hygiene, 2014. 90(6): p. 1021-1023.
202. Chapman, J.L., et al., Animal models of orthopoxvirus infection. Vet Pathol, 2010. 47(5): p. 852-70.
203. Peres, M.G., et al., Dogs and Opossums Positive for Vaccinia Virus during Outbreak Affecting Cattle and Humans, Sao Paulo State, Brazil. Emerg Infect Dis, 2016. 22(2): p. 271-3.
204. Goebel, S.J., et al., The complete DNA sequence of vaccinia virus. Virology, 1990. 179(1): p. 247-66, 517-63.

CHAPTER 8: References
The University of Sydney 2016
191
205. Smith, G.L., Y.S. Chan, and S.T. Howard, Nucleotide sequence of 42 kbp of vaccinia virus strain WR from near the right inverted terminal repeat. J Gen Virol, 1991. 72 ( Pt 6): p. 1349-76.
206. DeFilippes, F.M., Restriction enzyme mapping of vaccinia virus DNA. J Virol, 1982. 43(1): p. 136-49.
207. Roberts, K.L. and G.L. Smith, Vaccinia virus morphogenesis and dissemination. Trends Microbiol, 2008. 16(10): p. 472-9.
208. Moss, B., Poxvirus entry and membrane fusion. Virology, 2006. 344(1): p. 48-54.
209. Carter, G.C., et al., Vaccinia virus cores are transported on microtubules. J Gen Virol, 2003. 84(Pt 9): p. 2443-58.
210. Chung, C.S., et al., Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol, 2006. 80(5): p. 2127-40.
211. Yoder, J.D., et al., Pox proteomics: mass spectrometry analysis and identification of Vaccinia virion proteins. Virol J, 2006. 3: p. 10.
212. Resch, W., et al., Protein composition of the vaccinia virus mature virion. Virology, 2007. 358(1): p. 233-47.
213. Sanderson, C.M., M. Hollinshead, and G.L. Smith, The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J Gen Virol, 2000. 81(Pt 1): p. 47-58.
214. Ward, B.M., Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J Virol, 2005. 79(8): p. 4755-63.
215. Rietdorf, J., et al., Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat Cell Biol, 2001. 3(11): p. 992-1000.
216. Hollinshead, M., et al., Vaccinia virus utilizes microtubules for movement to the cell surface. J Cell Biol, 2001. 154(2): p. 389-402.
217. Ward, B.M. and B. Moss, Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol, 2001. 75(10): p. 4802-13.
218. Ward, B.M. and B. Moss, Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J Virol, 2004. 78(5): p. 2486-93.
219. Dodding, M.P., et al., An E2-F12 complex is required for intracellular enveloped virus morphogenesis during vaccinia infection. Cell Microbiol, 2009. 11(5): p. 808-24.
220. Morgan, G.W., et al., Vaccinia protein F12 has structural similarity to kinesin light chain and contains a motor binding motif required for virion export. PLoS Pathog, 2010. 6(2): p. e1000785.

CHAPTER 8: References
The University of Sydney 2016
192
221. Dodding, M.P., et al., A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J, 2011. 30(22): p. 4523-38.
222. Carpentier, D.C., et al., Vaccinia virus protein complex F12/E2 interacts with kinesin light chain isoform 2 to engage the kinesin-1 motor complex. PLoS Pathog, 2015. 11(3): p. e1004723.
223. Parkinson, J.E. and G.L. Smith, Vaccinia virus gene A36R encodes a M(r) 43-50 K protein on the surface of extracellular enveloped virus. Virology, 1994. 204(1): p. 376-90.
224. van Eijl, H., M. Hollinshead, and G.L. Smith, The vaccinia virus A36R protein is a type Ib membrane protein present on intracellular but not extracellular enveloped virus particles. Virology, 2000. 271(1): p. 26-36.
225. Johnston, S.C. and B.M. Ward, Vaccinia virus protein F12 associates with intracellular enveloped virions through an interaction with A36. J Virol, 2009. 83(4): p. 1708-17.
226. Arakawa, Y., et al., The release of vaccinia virus from infected cells requires RhoA-mDia modulation of cortical actin. Cell Host Microbe, 2007. 1(3): p. 227-40.
227. Handa, Y., et al., Vaccinia virus F11 promotes viral spread by acting as a PDZ-containing scaffolding protein to bind myosin-9A and inhibit RhoA signaling. Cell Host Microbe, 2013. 14(1): p. 51-62.
228. Valderrama, F., et al., Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science, 2006. 311(5759): p. 377-81.
229. Cordeiro, J.V., et al., F11-mediated inhibition of RhoA signalling enhances the spread of vaccinia virus in vitro and in vivo in an intranasal mouse model of infection. PLoS One, 2009. 4(12): p. e8506.
230. van Eijl, H., et al., The vaccinia virus F12L protein is associated with intracellular enveloped virus particles and is required for their egress to the cell surface. J Gen Virol, 2002. 83(Pt 1): p. 195-207.
231. Horsington, J., et al., Sub-viral imaging of vaccinia virus using super-resolution microscopy. J Virol Methods, 2012. 186(1-2): p. 132-6.
232. Frischknecht, F., et al., Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella. Curr Biol, 1999. 9(2): p. 89-92.
233. Frischknecht, F., et al., Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature, 1999. 401(6756): p. 926-9.
234. Newsome, T.P., N. Scaplehorn, and M. Way, SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science, 2004. 306(5693): p. 124-9.
235. Reeves, P.M., et al., Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med, 2005. 11(7): p. 731-9.

CHAPTER 8: References
The University of Sydney 2016
193
236. Newsome, T.P., et al., Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol, 2006. 8(2): p. 233-41.
237. Alvarez, D.E. and H. Agaisse, Casein kinase 2 regulates vaccinia virus actin tail formation. Virology, 2012. 423(2): p. 143-51.
238. Wolffe, E.J., A.S. Weisberg, and B. Moss, The vaccinia virus A33R protein provides a chaperone function for viral membrane localization and tyrosine phosphorylation of the A36R protein. J Virol, 2001. 75(1): p. 303-10.
239. Newsome, T.P. and N.B. Marzook, Viruses that ride on the coat-tails of actin nucleation. Semin Cell Dev Biol, 2015.
240. Snapper, S.B., et al., N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol, 2001. 3(10): p. 897-904.
241. Weisswange, I., et al., The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature, 2009. 458(7234): p. 87-91.
242. Alvarez, D.E. and H. Agaisse, The formin FHOD1 and the small GTPase Rac1 promote vaccinia virus actin-based motility. J Cell Biol, 2013. 202(7): p. 1075-90.
243. Scaplehorn, N., et al., Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr Biol, 2002. 12(9): p. 740-5.
244. Zettl, M. and M. Way, The WH1 and EVH1 domains of WASP and Ena/VASP family members bind distinct sequence motifs. Curr Biol, 2002. 12(18): p. 1617-22.
245. Donnelly, S.K., et al., WIP provides an essential link between Nck and N-WASP during Arp2/3-dependent actin polymerization. Curr Biol, 2013. 23(11): p. 999-1006.
246. Snetkov, X., et al., NPF motifs in the vaccinia virus protein A36 recruit intersectin-1 to promote Cdc42:N-WASP-mediated viral release from infected cells. Nat Microbiol, 2016. 1(10): p. 16141.
247. Arakawa, Y., J.V. Cordeiro, and M. Way, F11L-mediated inhibition of RhoA-mDia signaling stimulates microtubule dynamics during vaccinia virus infection. Cell Host Microbe, 2007. 1(3): p. 213-26.
248. Morales, I., et al., The vaccinia virus F11L gene product facilitates cell detachment and promotes migration. Traffic, 2008. 9(8): p. 1283-98.
249. Padrick, S.B. and M.K. Rosen, Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem, 2010. 79: p. 707-35.
250. Kovar, D.R., Molecular details of formin-mediated actin assembly. Curr Opin Cell Biol, 2006. 18(1): p. 11-7.

CHAPTER 8: References
The University of Sydney 2016
194
251. Alvarez, D.E. and H. Agaisse, A role for the small GTPase Rac1 in vaccinia actin-based motility. Small GTPases, 2014. 5(2): p. e29038.
252. Mueller, J., et al., Electron tomography and simulation of baculovirus actin comet tails support a tethered filament model of pathogen propulsion. PLoS Biol, 2014. 12(1): p. e1001765.
253. Rottger, S., et al., Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J Virol, 1999. 73(4): p. 2863-75.
254. Chakrabarti, S., J.R. Sisler, and B. Moss, Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques, 1997. 23(6): p. 1094-7.
255. Zhang, G., V. Gurtu, and S.R. Kain, An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem Biophys Res Commun, 1996. 227(3): p. 707-11.
256. Tabor, S., Expression using the T7 RNA polymerase/promoter system. Curr Protoc Mol Biol, 2001. Chapter 16: p. Unit16 2.
257. Ball, L.A., High-frequency homologous recombination in vaccinia virus DNA. J Virol, 1987. 61(6): p. 1788-95.
258. Ball, L.A., Fidelity of homologous recombination in vaccinia virus DNA. Virology, 1995. 209(2): p. 688-91.
259. Gammon, D.B. and D.H. Evans, The 3'-to-5' exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination. J Virol, 2009. 83(9): p. 4236-50.
260. Willer, D.O., et al., In vitro concatemer formation catalyzed by vaccinia virus DNA polymerase. Virology, 2000. 278(2): p. 562-9.
261. Cubitt, A.B., et al., Understanding, improving and using green fluorescent proteins. Trends Biochem Sci, 1995. 20(11): p. 448-55.
262. Inouye, S. and F.I. Tsuji, Aequorea green fluorescent protein. Expression of the gene and fluorescence characteristics of the recombinant protein. FEBS Lett, 1994. 341(2-3): p. 277-80.
263. Chalfie, M., et al., Green fluorescent protein as a marker for gene expression. Science, 1994. 263(5148): p. 802-5.
264. Cormack, B.P., R.H. Valdivia, and S. Falkow, FACS-optimized mutants of the green fluorescent protein (GFP). Gene, 1996. 173(1 Spec No): p. 33-8.
265. Chudakov, D.M., et al., Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev, 2010. 90(3): p. 1103-63.
266. Heim, R. and R.Y. Tsien, Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol, 1996. 6(2): p. 178-82.

CHAPTER 8: References
The University of Sydney 2016
195
267. Tomosugi, W., et al., An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat Methods, 2009. 6(5): p. 351-3.
268. Kremers, G.J., et al., Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry, 2006. 45(21): p. 6570-80.
269. Wiedenmann, J., et al., A far-red fluorescent protein with fast maturation and reduced oligomerization tendency from Entacmaea quadricolor (Anthozoa, Actinaria). Proc Natl Acad Sci U S A, 2002. 99(18): p. 11646-51.
270. Merzlyak, E.M., et al., Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods, 2007. 4(7): p. 555-7.
271. Matz, M.V., et al., Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol, 1999. 17(10): p. 969-73.
272. Shaner, N.C., et al., Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol, 2004. 22(12): p. 1567-72.
273. Shu, X., et al., Novel chromophores and buried charges control color in mFruits. Biochemistry, 2006. 45(32): p. 9639-47.
274. Campbell, R.E., et al., A monomeric red fluorescent protein. Proc Natl Acad Sci U S A, 2002. 99(12): p. 7877-82.
275. Wang, S. and T. Hazelrigg, Implications for bcd mRNA localization from spatial distribution of exu protein in Drosophila oogenesis. Nature, 1994. 369(6479): p. 400-03.
276. Kogure, T., et al., Fluorescence imaging using a fluorescent protein with a large Stokes shift. Methods, 2008. 45(3): p. 223-6.
277. Kogure, T., et al., A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat Biotechnol, 2006. 24(5): p. 577-81.
278. Shaner, N.C., P.A. Steinbach, and R.Y. Tsien, A guide to choosing fluorescent proteins. Nat Methods, 2005. 2(12): p. 905-9.
279. Moradpour, D., et al., Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol, 2004. 78(14): p. 7400-9.
280. Zordan, R.E., et al., Avoiding the ends: internal epitope tagging of proteins using transposon Tn7. Genetics, 2015. 200(1): p. 47-58.
281. Chen, X., J.L. Zaro, and W.C. Shen, Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev, 2013. 65(10): p. 1357-69.
282. Margolin, W., The price of tags in protein localization studies. J Bacteriol, 2012. 194(23): p. 6369-71.

CHAPTER 8: References
The University of Sydney 2016
196
283. Vats, P. and L. Rothfield, Duplication and segregation of the actin (MreB) cytoskeleton during the prokaryotic cell cycle. Proc Natl Acad Sci U S A, 2007. 104(45): p. 17795-800.
284. Vats, P., Y.L. Shih, and L. Rothfield, Assembly of the MreB-associated cytoskeletal ring of Escherichia coli. Mol Microbiol, 2009. 72(1): p. 170-82.
285. Swulius, M.T. and G.J. Jensen, The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-Terminal yellow fluorescent protein tag. J Bacteriol, 2012. 194(23): p. 6382-6.
286. Dunn, K.W., M.M. Kamocka, and J.H. McDonald, A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol, 2011. 300(4): p. C723-42.
287. Bolte, S. and F.P. Cordelieres, A guided tour into subcellular colocalization analysis in light microscopy. J Microsc, 2006. 224(Pt 3): p. 213-32.
288. Zinchuk, V. and O. Zinchuk, Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr Protoc Cell Biol, 2008. Chapter 4: p. Unit 4 19.
289. Stryer, L., Fluorescence energy transfer as a spectroscopic ruler. Annu Rev Biochem, 1978. 47: p. 819-46.
290. Piston, D.W. and G.J. Kremers, Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci, 2007. 32(9): p. 407-14.
291. van Rheenen, J., M. Langeslag, and K. Jalink, Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J, 2004. 86(4): p. 2517-29.
292. van Munster, E.B. and T.W. Gadella, Fluorescence lifetime imaging microscopy (FLIM). Adv Biochem Eng Biotechnol, 2005. 95: p. 143-75.
293. Oida, T., Y. Sako, and A. Kusumi, Fluorescence lifetime imaging microscopy (flimscopy). Methodology development and application to studies of endosome fusion in single cells. Biophys J, 1993. 64(3): p. 676-85.
294. Ishikawa-Ankerhold, H.C., R. Ankerhold, and G.P. Drummen, Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules, 2012. 17(4): p. 4047-132.
295. De Los Santos, C., et al., FRAP, FLIM, and FRET: Detection and analysis of cellular dynamics on a molecular scale using fluorescence microscopy. Mol Reprod Dev, 2015. 82(7-8): p. 587-604.
296. Frommer, W.B., M.W. Davidson, and R.E. Campbell, Genetically encoded biosensors based on engineered fluorescent proteins. Chem Soc Rev, 2009. 38(10): p. 2833-41.
297. Aoki, K., et al., Stable expression of FRET biosensors: a new light in cancer research. Cancer Sci, 2012. 103(4): p. 614-9.

CHAPTER 8: References
The University of Sydney 2016
197
298. Carisey, A., et al., Fluorescence recovery after photobleaching. Methods Mol Biol, 2011. 769: p. 387-402.
299. Banting, G., Photobleaching (FRAP/FLIP) and dynamic imaging. Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics. 2005, John Wiley & Sons, Ltd.
300. Patterson, G.H. and J. Lippincott-Schwartz, A photoactivatable GFP for selective photolabeling of proteins and cells. Science, 2002. 297(5588): p. 1873-7.
301. Chudakov, D.M., S. Lukyanov, and K.A. Lukyanov, Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc, 2007. 2(8): p. 2024-32.
302. Schermelleh, L., R. Heintzmann, and H. Leonhardt, A guide to super-resolution fluorescence microscopy. J Cell Biol, 2010. 190(2): p. 165-75.
303. Nienhaus, K. and G.U. Nienhaus, Fluorescent proteins for live-cell imaging with super-resolution. Chem Soc Rev, 2014. 43(4): p. 1088-106.
304. Ward, B.M., Pox, dyes, and videotape: making movies of GFP-labeled vaccinia virus. Methods Mol Biol, 2004. 269: p. 205-18.
305. Costantini, L.M. and E.L. Snapp, Going Viral with Fluorescent Proteins. J Virol, 2015. 89(19): p. 9706-8.
306. Sun, E., J. He, and X. Zhuang, Live cell imaging of viral entry. Curr Opin Virol, 2013. 3(1): p. 34-43.
307. Hogue, I.B., et al., Fluorescent Protein Approaches in Alpha Herpesvirus Research. Viruses, 2015. 7(11): p. 5933-61.
308. Francis, A.C., et al., Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog, 2016. 12(6): p. e1005709.
309. Lo, M.K., S.T. Nichol, and C.F. Spiropoulou, Evaluation of luciferase and GFP-expressing Nipah viruses for rapid quantitative antiviral screening. Antiviral Res, 2014. 106: p. 53-60.
310. Kwanten, L., B. De Clerck, and D. Roymans, A fluorescence-based high-throughput antiviral compound screening assay against respiratory syncytial virus. Methods Mol Biol, 2013. 1030: p. 337-44.
311. Smith, G.L. and B. Moss, Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene, 1983. 25(1): p. 21-8.
312. Heuser, J., Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic "honeycomb" surface coat. J Cell Biol, 2005. 169(2): p. 269-83.
313. Al Ali, S., et al., Use of Reporter Genes in the Generation of Vaccinia Virus-Derived Vectors. Viruses, 2016. 8(5).

CHAPTER 8: References
The University of Sydney 2016
198
314. Byrd, C.M. and D.E. Hruby, Construction of recombinant vaccinia virus: cloning into the thymidine kinase locus. Methods Mol Biol, 2004. 269: p. 31-40.
315. Mackett, M., G.L. Smith, and B. Moss, Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci U S A, 1982. 79(23): p. 7415-9.
316. Mackett, M., G.L. Smith, and B. Moss, General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J Virol, 1984. 49(3): p. 857-64.
317. Chakrabarti, S., K. Brechling, and B. Moss, Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol, 1985. 5(12): p. 3403-9.
318. Liu, G.Q., et al., Selection of recombinant vaccinia viruses (Tian Tan strain) expressing hepatitis B virus surface antigen by using beta-galactosidase as a marker. Sci China B, 1990. 33(2): p. 188-97.
319. Panicali, D., A. Grzelecki, and C. Huang, Vaccinia virus vectors utilizing the beta-galactosidase assay for rapid selection of recombinant viruses and measurement of gene expression. Gene, 1986. 47(2-3): p. 193-9.
320. Dominguez, J., M.M. Lorenzo, and R. Blasco, Green fluorescent protein expressed by a recombinant vaccinia virus permits early detection of infected cells by flow cytometry. J Immunol Methods, 1998. 220(1-2): p. 115-21.
321. Boyle, D.B. and B.E. Coupar, A dominant selectable marker for the construction of recombinant poxviruses. Gene, 1988. 65(1): p. 123-8.
322. Falkner, F.G. and B. Moss, Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J Virol, 1988. 62(6): p. 1849-54.
323. Sanchez-Puig, J.M. and R. Blasco, Isolation of vaccinia MVA recombinants using the viral F13L gene as the selective marker. Biotechniques, 2005. 39(5): p. 665-6, 668, 670 passim.
324. Blasco, R. and B. Moss, Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene, 1995. 158(2): p. 157-62.
325. Holzer, G.W., et al., Dominant host range selection of vaccinia recombinants by rescue of an essential gene. Virology, 1998. 249(1): p. 160-6.
326. Rodriguez, J.F. and M. Esteban, Plaque size phenotype as a selectable marker to generate vaccinia virus recombinants. J Virol, 1989. 63(2): p. 997-1001.
327. Falkner, F.G. and B. Moss, Transient dominant selection of recombinant vaccinia viruses. J Virol, 1990. 64(6): p. 3108-11.
328. Carlson, R., The changing economics of DNA synthesis. Nat Biotechnol, 2009. 27(12): p. 1091-4.

CHAPTER 8: References
The University of Sydney 2016
199
329. Kosuri, S. and G.M. Church, Large-scale de novo DNA synthesis: technologies and applications. Nat Methods, 2014. 11(5): p. 499-507.
330. Lynn, H., The role of the microtubule cytoskeleton in poxvirus replication and pathogenesis, in School of Life and Environmental Sciences. 2015, The University of Sydney: Sydney.
331. Zhang, W.H., D. Wilcock, and G.L. Smith, Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J Virol, 2000. 74(24): p. 11654-62.
332. Horsington, J., et al., A36-dependent actin filament nucleation promotes release of vaccinia virus. PLoS Pathog, 2013. 9(3): p. e1003239.
333. Doceul, V., et al., Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science, 2010. 327(5967): p. 873-6.
334. Ansarah-Sobrinho, C. and B. Moss, Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J Virol, 2004. 78(12): p. 6335-43.
335. Katz, E. and B. Moss, Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc Natl Acad Sci U S A, 1970. 66(3): p. 677-84.
336. Kato, S.E., et al., Temperature-sensitive mutants in the vaccinia virus 4b virion structural protein assemble malformed, transcriptionally inactive intracellular mature virions. Virology, 2004. 330(1): p. 127-46.
337. Jesus, D.M., et al., Vaccinia virus protein A3 is required for the production of normal immature virions and for the encapsidation of the nucleocapsid protein L4. Virology, 2015. 481: p. 1-12.
338. Pedersen, K., et al., Characterization of vaccinia virus intracellular cores: implications for viral uncoating and core structure. J Virol, 2000. 74(8): p. 3525-36.
339. Moussatche, N. and R.C. Condit, Fine structure of the vaccinia virion determined by controlled degradation and immunolocalization. Virology, 2015. 475: p. 204-18.
340. Veyer, D.L., et al., Analysis of the anti-apoptotic activity of four vaccinia virus proteins demonstrates that B13 is the most potent inhibitor in isolation and during viral infection. J Gen Virol, 2014. 95(Pt 12): p. 2757-68.
341. Wasilenko, S.T., et al., Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc Natl Acad Sci U S A, 2003. 100(24): p. 14345-50.
342. Wasilenko, S.T., et al., The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J Virol, 2005. 79(22): p. 14031-43.

CHAPTER 8: References
The University of Sydney 2016
200
343. Postigo, A., et al., Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ, 2006. 13(10): p. 1651-62.
344. Taylor, J.M., et al., The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem, 2006. 281(51): p. 39728-39.
345. Stewart, T.L., S.T. Wasilenko, and M. Barry, Vaccinia virus F1L protein is a tail-anchored protein that functions at the mitochondria to inhibit apoptosis. J Virol, 2005. 79(2): p. 1084-98.
346. Campbell, S., et al., Structural insight into BH3 domain binding of vaccinia virus antiapoptotic F1L. J Virol, 2014. 88(15): p. 8667-77.
347. Yao, X.D. and D.H. Evans, Effects of DNA structure and homology length on vaccinia virus recombination. J Virol, 2001. 75(15): p. 6923-32.
348. Rizzo, M.A., et al., An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol, 2004. 22(4): p. 445-9.
349. Shu, X., et al., A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol, 2011. 9(4): p. e1001041.
350. McKenzie, C.D., Activation of oncogenic signalling pathways by vaccinia virus, in School of Life and Environmental Science. 2016, The University of Sydney: Sydney.
351. Wong, Y.C., et al., Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection. J Virol Methods, 2011. 171(1): p. 295-8.
352. Blasco, R. and B. Moss, Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J Virol, 1991. 65(11): p. 5910-20.
353. Qin, L. and D.H. Evans, Genome scale patterns of recombination between coinfecting vaccinia viruses. J Virol, 2014. 88(10): p. 5277-86.
354. Kettle, S., et al., Vaccinia virus serpins B13R (SPI-2) and B22R (SPI-1) encode M(r) 38.5 and 40K, intracellular polypeptides that do not affect virus virulence in a murine intranasal model. Virology, 1995. 206(1): p. 136-47.
355. Hughes, S.J., et al., Vaccinia virus encodes an active thymidylate kinase that complements a cdc8 mutant of Saccharomyces cerevisiae. J Biol Chem, 1991. 266(30): p. 20103-9.
356. Unterholzner, L., et al., Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog, 2011. 7(9): p. e1002247.

CHAPTER 8: References
The University of Sydney 2016
201
357. Mulligan, R.C. and P. Berg, Selection for animal cells that express the Escherichia coli gene coding for xanthine-guanine phosphoribosyltransferase. Proc Natl Acad Sci U S A, 1981. 78(4): p. 2072-6.
358. Jackson, R.J. and H.G. Bults, A myxoma virus intergenic transient dominant selection vector. J Gen Virol, 1992. 73 ( Pt 12): p. 3241-5.
359. Szybalska, E.H. and W. Szybalski, Genetics of human cess line. IV. DNA-mediated heritable transformation of a biochemical trait. Proc Natl Acad Sci U S A, 1962. 48: p. 2026-34.
360. Mitra, K. and J. Lippincott-Schwartz, Analysis of mitochondrial dynamics and functions using imaging approaches. Curr Protoc Cell Biol, 2010. Chapter 4: p. Unit 4 25 1-21.
361. Fuerst, T.R., et al., Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A, 1986. 83(21): p. 8122-6.
362. Thiel, V., et al., Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J Gen Virol, 2001. 82(Pt 6): p. 1273-81.
363. Alexander, W.A., B. Moss, and T.R. Fuerst, Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J Virol, 1992. 66(5): p. 2934-42.
364. Wyatt, L.S., et al., Elucidating and minimizing the loss by recombinant vaccinia virus of human immunodeficiency virus gene expression resulting from spontaneous mutations and positive selection. J Virol, 2009. 83(14): p. 7176-84.
365. Tsien, R.Y., The green fluorescent protein. Annu Rev Biochem, 1998. 67: p. 509-44.
366. Waters, J.C., Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol, 2009. 185(7): p. 1135-48.
367. Zimmermann, T., J. Rietdorf, and R. Pepperkok, Spectral imaging and its applications in live cell microscopy. FEBS Lett, 2003. 546(1): p. 87-92.
368. Fereidouni, F., A.N. Bader, and H.C. Gerritsen, Spectral phasor analysis allows rapid and reliable unmixing of fluorescence microscopy spectral images. Opt Express, 2012. 20(12): p. 12729-41.
369. Nadrigny, F., et al., Detecting fluorescent protein expression and co-localisation on single secretory vesicles with linear spectral unmixing. Eur Biophys J, 2006. 35(6): p. 533-47.
370. Newsome, T.P., Marty, A. J., Lynn, H., Procter, D. J., Navigating the subcellular space: Lessons from vaccinia virus, in Viral Transport, Assembly and Egress, R.J. Diefenbach, Cunningham, A. L., Editor. 2011, Research Signpost. p. 155-177.

CHAPTER 8: References
The University of Sydney 2016
202
371. Jeshtadi, A., et al., Interaction of poxvirus intracellular mature virion proteins with the TPR domain of kinesin light chain in live infected cells revealed by two-photon-induced fluorescence resonance energy transfer fluorescence lifetime imaging microscopy. J Virol, 2010. 84(24): p. 12886-94.
372. Dower, K., et al., Development of Vaccinia reporter viruses for rapid, high content analysis of viral function at all stages of gene expression. Antiviral Res, 2011. 91(1): p. 72-80.
373. Dénes, B., Fodor, N., Obenaus, A., Fodor, I., Engineering Oncolytic Vaccinia Viruses for Non-Invasive Optical Imaging of Tumors. The Open Biotechnology Journal, 2008(2): p. 252-261.
374. Sanderson, C.M., M. Way, and G.L. Smith, Virus-induced cell motility. J Virol, 1998. 72(2): p. 1235-43.
375. Yilmaz, M. and G. Christofori, EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev, 2009. 28(1-2): p. 15-33.
376. Heerboth, S., et al., EMT and tumor metastasis. Clin Transl Med, 2015. 4: p. 6.
377. Chevalier, S.A., et al., Gem-induced cytoskeleton remodeling increases cellular migration of HTLV-1-infected cells, formation of infected-to-target T-cell conjugates and viral transmission. PLoS Pathog, 2014. 10(2): p. e1003917.
378. Stoker, A.W. and M.H. Sieweke, v-src induces clonal sarcomas and rapid metastasis following transduction with a replication-defective retrovirus. Proc Natl Acad Sci U S A, 1989. 86(24): p. 10123-7.
379. Zwilling, J., et al., Functional F11L and K1L genes in modified vaccinia virus Ankara restore virus-induced cell motility but not growth in human and murine cells. Virology, 2010. 404(2): p. 231-9.
380. Irwin, C.R. and D.H. Evans, Modulation of the myxoma virus plaque phenotype by vaccinia virus protein F11. J Virol, 2012. 86(13): p. 7167-79.
381. Irwin, C.R., et al., Myxoma virus oncolytic efficiency can be enhanced through chemical or genetic disruption of the actin cytoskeleton. PLoS One, 2013. 8(12): p. e84134.
382. Sit, S.T. and E. Manser, Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci, 2011. 124(Pt 5): p. 679-83.
383. Schepis, A., et al., Vaccinia virus-induced microtubule-dependent cellular rearrangements. Traffic, 2006. 7(3): p. 308-23.
384. Marchal, J., Infectious ectromelia. A hitherto undescribed virus disease of mice. The Journal of Pathology and Bacteriology, 1930. 33(3): p. 713-728.
385. Chen, N., et al., The genomic sequence of ectromelia virus, the causative agent of mousepox. Virology, 2003. 317(1): p. 165-86.

CHAPTER 8: References
The University of Sydney 2016
203
386. Sigal, L.J., The Pathogenesis and Immunobiology of Mousepox. Adv Immunol, 2016. 129: p. 251-76.
387. Esteban, D.J. and R.M. Buller, Ectromelia virus: the causative agent of mousepox. J Gen Virol, 2005. 86(Pt 10): p. 2645-59.
388. Fenner, F., Mousepox (infectious ectromelia): past, present, and future. Lab Anim Sci, 1981. 31(5 Pt 2): p. 553-9.
389. Panchanathan, V., G. Chaudhri, and G. Karupiah, Protective immunity against secondary poxvirus infection is dependent on antibody but not on CD4 or CD8 T-cell function. J Virol, 2006. 80(13): p. 6333-8.
390. Niemialtowski, M.G., et al., The inflammatory and immune response to mousepox (infectious ectromelia) virus. Acta Virol, 1994. 38(5): p. 299-307.
391. Smith, V.P. and A. Alcami, Expression of secreted cytokine and chemokine inhibitors by ectromelia virus. J Virol, 2000. 74(18): p. 8460-71.
392. Smith, V.P., N.A. Bryant, and A. Alcami, Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J Gen Virol, 2000. 81(Pt 5): p. 1223-30.
393. Parker, A.K., et al., Induction of natural killer cell responses by ectromelia virus controls infection. J Virol, 2007. 81(8): p. 4070-9.
394. Lynn, H., et al., Loss of cytoskeletal transport during egress critically attenuates ectromelia virus infection in vivo. J Virol, 2012. 86(13): p. 7427-43.
395. Szulc-Dabrowska, L., et al., Remodeling of the fibroblast cytoskeletal architecture during the replication cycle of Ectromelia virus: A morphological in vitro study in a murine cell line. Cytoskeleton (Hoboken), 2016. 73(8): p. 396-417.
396. Boratynska, A., et al., Contribution of rearranged actin structures to the spread of Ectromelia virus infection in vitro. Acta Virol, 2010. 54(1): p. 41-8.
397. Hand, E.S., et al., Ectopic expression of vaccinia virus E3 and K3 cannot rescue ectromelia virus replication in rabbit RK13 cells. PLoS One, 2015. 10(3): p. e0119189.
398. Briody, B.A., Response of mice to ectromelia and vaccinia viruses. Bacteriol Rev, 1959. 23(2): p. 61-95.
399. Tscharke, D.C. and G.L. Smith, A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J Gen Virol, 1999. 80 ( Pt 10): p. 2751-5.
400. Roberts, J.A., Histopathogenesis of mousepox. II. Cutaneous infection. Br J Exp Pathol, 1962. 43: p. 462-8.
401. Kurosaka, S. and A. Kashina, Cell biology of embryonic migration. Birth Defects Res C Embryo Today, 2008. 84(2): p. 102-22.

CHAPTER 8: References
The University of Sydney 2016
204
402. Riedl, J., et al., Lifeact: a versatile marker to visualize F-actin. Nat Methods, 2008. 5(7): p. 605-7.
403. Kato, S.E., et al., An alternative genetic method to test essential vaccinia virus early genes. J Virol Methods, 2004. 115(1): p. 31-40.
404. Szajner, P., A.S. Weisberg, and B. Moss, Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J Virol, 2004. 78(1): p. 257-65.
405. Lin, S. and S.S. Broyles, Vaccinia protein kinase 2: a second essential serine/threonine protein kinase encoded by vaccinia virus. Proc Natl Acad Sci U S A, 1994. 91(16): p. 7653-7.
406. Davison, A.J. and B. Moss, Structure of vaccinia virus late promoters. J Mol Biol, 1989. 210(4): p. 771-84.
407. Baldick, C.J., Jr., J.G. Keck, and B. Moss, Mutational analysis of the core, spacer, and initiator regions of vaccinia virus intermediate-class promoters. J Virol, 1992. 66(8): p. 4710-9.
408. Knutson, B.A., et al., Vaccinia virus intermediate and late promoter elements are targeted by the TATA-binding protein. J Virol, 2006. 80(14): p. 6784-93.
409. Cordeiro, J.V., Modulation of Rho GTPase signalling during vaccinia virus infection, in Cancer Research UK. 2008, University College London: London.
410. Marzook, N.B. and T.P. Newsome, Viruses That Exploit Actin-Based Motility for Their Replication and Spread, in Handbook of Experimental Pharmacology. 2016, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 1-25.
411. Wolffe, E.J., A.S. Weisberg, and B. Moss, Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology, 1998. 244(1): p. 20-6.
412. Doolittle, L.K., M.K. Rosen, and S.B. Padrick, Measurement and analysis of in vitro actin polymerization. Methods Mol Biol, 2013. 1046: p. 273-93.
413. Spudich, J.A. and S. Watt, The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem, 1971. 246(15): p. 4866-71.
414. Cooper, J.A., S.B. Walker, and T.D. Pollard, Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil, 1983. 4(2): p. 253-62.
415. Kouyama, T. and K. Mihashi, Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem, 1981. 114(1): p. 33-8.
416. Goldberg, M.B., Actin-based motility of intracellular microbial pathogens. Microbiol Mol Biol Rev, 2001. 65(4): p. 595-626, table of contents.

CHAPTER 8: References
The University of Sydney 2016
205
417. Kocks, C., et al., The unrelated surface proteins ActA of Listeria monocytogenes and IcsA of Shigella flexneri are sufficient to confer actin-based motility on Listeria innocua and Escherichia coli respectively. Mol Microbiol, 1995. 18(3): p. 413-23.
418. Wiesner, S., et al., A biomimetic motility assay provides insight into the mechanism of actin-based motility. J Cell Biol, 2003. 160(3): p. 387-98.
419. Cameron, L.A., et al., Motility of ActA protein-coated microspheres driven by actin polymerization. Proc Natl Acad Sci U S A, 1999. 96(9): p. 4908-13.
420. Bugyi, B., et al., How do in vitro reconstituted actin-based motility assays provide insight into in vivo behavior? FEBS Lett, 2008. 582(14): p. 2086-92.
421. Zuchero, J.B., et al., p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol, 2009. 11(4): p. 451-9.
422. Roy, P., et al., Local photorelease of caged thymosin beta4 in locomoting keratocytes causes cell turning. J Cell Biol, 2001. 153(5): p. 1035-48.
423. Bergeron, S.E., et al., Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms. J Biol Chem, 2010. 285(21): p. 16087-95.
424. Hofer, D., W. Ness, and D. Drenckhahn, Sorting of actin isoforms in chicken auditory hair cells. J Cell Sci, 1997. 110 ( Pt 6): p. 765-70.
425. Shmerling, D., et al., Strong and ubiquitous expression of transgenes targeted into the beta-actin locus by Cre/lox cassette replacement. Genesis, 2005. 42(4): p. 229-35.
426. Shawlot, W., et al., Restricted beta-galactosidase expression of a hygromycin-lacZ gene targeted to the beta-actin locus and embryonic lethality of beta-actin mutant mice. Transgenic Res, 1998. 7(2): p. 95-103.
427. Belyantseva, I.A., et al., Gamma-actin is required for cytoskeletal maintenance but not development. Proc Natl Acad Sci U S A, 2009. 106(24): p. 9703-8.
428. Bunnell, T.M. and J.M. Ervasti, Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton (Hoboken), 2010. 67(9): p. 564-72.
429. Hill, M.A. and P. Gunning, Beta and gamma actin mRNAs are differentially located within myoblasts. J Cell Biol, 1993. 122(4): p. 825-32.
430. Hoock, T.C., P.M. Newcomb, and I.M. Herman, Beta actin and its mRNA are localized at the plasma membrane and the regions of moving cytoplasm during the cellular response to injury. J Cell Biol, 1991. 112(4): p. 653-64.
431. Micheva, K.D., et al., beta-Actin is confined to structures having high capacity of remodelling in developing and adult rat cerebellum. Eur J Neurosci, 1998. 10(12): p. 3785-98.

CHAPTER 8: References
The University of Sydney 2016
206
432. Bassell, G.J., et al., Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci, 1998. 18(1): p. 251-65.
433. Chaponnier, C. and G. Gabbiani, Pathological situations characterized by altered actin isoform expression. J Pathol, 2004. 204(4): p. 386-95.
434. Joseph, R., O.P. Srivastava, and R.R. Pfister, Downregulation of beta-actin and its regulatory gene HuR affect cell migration of human corneal fibroblasts. Mol Vis, 2014. 20: p. 593-605.
435. Pasquier, E., et al., gamma-Actin plays a key role in endothelial cell motility and neovessel maintenance. Vasc Cell, 2015. 7: p. 2.
436. Bunnell, T.M., et al., beta-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell, 2011. 22(21): p. 4047-58.
437. Peckham, M., et al., Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci, 2001. 114(Pt 7): p. 1367-77.
438. Simiczyjew, A., et al., Effect of overexpression of beta- and gamma-actin isoforms on actin cytoskeleton organization and migration of human colon cancer cells. Histochem Cell Biol, 2014. 142(3): p. 307-22.
439. Namba, Y., et al., Human T cell L-plastin bundles actin filaments in a calcium-dependent manner. J Biochem, 1992. 112(4): p. 503-7.
440. Kashina, A.S., Differential arginylation of actin isoforms: the mystery of the actin N-terminus. Trends Cell Biol, 2006. 16(12): p. 610-5.
441. He, F., et al., Beta-actin interacts with the E2 protein and is involved in the early replication of classical swine fever virus. Virus Res, 2014. 179: p. 161-8.
442. Wang, J., et al., Interaction of the coronavirus infectious bronchitis virus membrane protein with beta-actin and its implication in virion assembly and budding. PLoS One, 2009. 4(3): p. e4908.
443. Kuhbacher, A., et al., Genome-Wide siRNA Screen Identifies Complementary Signaling Pathways Involved in Listeria Infection and Reveals Different Actin Nucleation Mechanisms during Listeria Cell Invasion and Actin Comet Tail Formation. MBio, 2015. 6(3): p. e00598-15.
444. Smith, G.L. and M. Law, The exit of vaccinia virus from infected cells. Virus Res, 2004. 106(2): p. 189-97.
445. Rokita, H., et al., Vaccinia virus-regulated acute phase cytokine production in human fibroblasts, U937 cells and endothelium. Mediators Inflamm, 1998. 7(2): p. 73-8.
446. Smith, S.A., et al., Conserved surface-exposed K/R-X-K/R motifs and net positive charge on poxvirus complement control proteins serve as putative heparin binding sites and contribute to inhibition of molecular interactions with human endothelial cells: a novel mechanism for evasion of host defense. J Virol, 2000. 74(12): p. 5659-66.

CHAPTER 8: References
The University of Sydney 2016
207
447. Kirn, D.H., et al., Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med, 2007. 4(12): p. e353.
448. Weibel, S., et al., Viral-mediated oncolysis is the most critical factor in the late-phase of the tumor regression process upon vaccinia virus infection. BMC Cancer, 2011. 11: p. 68.
449. Latham, S.L., A morphological and molecular approach to understanding fine mechanisms of endothelian vesiculation: a novel role for the actin cytoskeleton, in Department of Pathology. 2014, The University of Sydney: Sydney.
450. Duggal, R., et al., Vaccinia virus expressing bone morphogenetic protein-4 in novel glioblastoma orthotopic models facilitates enhanced tumor regression and long-term survival. J Transl Med, 2013. 11: p. 155.
451. Kober, C., et al., Microglia and astrocytes attenuate the replication of the oncolytic vaccinia virus LIVP 1.1.1 in murine GL261 gliomas by acting as vaccinia virus traps. J Transl Med, 2015. 13: p. 216.
452. Lechuga, S., et al., Loss of gamma-cytoplasmic actin triggers myofibroblast transition of human epithelial cells. Mol Biol Cell, 2014. 25(20): p. 3133-46.
453. Leung, B.O. and K.C. Chou, Review of super-resolution fluorescence microscopy for biology. Appl Spectrosc, 2011. 65(9): p. 967-80.
454. Shum, M.S., et al., gamma-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB J, 2011. 25(12): p. 4423-33.
455. Roper, R.L., et al., The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J Virol, 1998. 72(5): p. 4192-204.
456. Mathew, E., et al., The extracellular domain of vaccinia virus protein B5R affects plaque phenotype, extracellular enveloped virus release, and intracellular actin tail formation. Journal of Virology, 1998. 72(3): p. 2429-38.
457. Rodger, G. and G.L. Smith, Replacing the SCR domains of vaccinia virus protein B5R with EGFP causes a reduction in plaque size and actin tail formation but enveloped virions are still transported to the cell surface. J Gen Virol, 2002. 83(Pt 2): p. 323-32.
458. Gong, S.C., et al., A single point mutation of Ala-25 to Asp in the 14,000-Mr envelope protein of vaccinia virus induces a size change that leads to the small plaque size phenotype of the virus. J Virol, 1989. 63(11): p. 4507-14.
459. Wolffe, E.J., S.N. Isaacs, and B. Moss, Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J Virol, 1993. 67(8): p. 4732-41.
460. Wolffe, E.J., et al., The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. Journal of Virology, 1997. 71(5): p. 3904-15.

CHAPTER 8: References
The University of Sydney 2016
208
461. Sanderson, C.M., et al., Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. Journal of General Virology, 1998. 79(Pt 6): p. 1415-25.
462. Payne, L.G., Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J Gen Virol, 1980. 50(1): p. 89-100.
463. Blasco, R. and B. Moss, Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol, 1992. 66(7): p. 4170-9.
464. Payne, L.G. and K. Kristensson, Extracellular release of enveloped vaccinia virus from mouse nasal epithelial cells in vivo. J Gen Virol, 1985. 66 ( Pt 3): p. 643-6.
465. Smith, G.L. and A. Vanderplasschen, Extracellular enveloped vaccinia virus. Entry, egress, and evasion. Adv Exp Med Biol, 1998. 440: p. 395-414.
466. Marchand, J.B., et al., Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat Cell Biol, 2001. 3(1): p. 76-82.
467. Hufner, K., et al., The verprolin-like central (vc) region of Wiskott-Aldrich syndrome protein induces Arp2/3 complex-dependent actin nucleation. J Biol Chem, 2001. 276(38): p. 35761-7.
468. Katz, E., et al., Mutations in the vaccinia virus A33R and B5R envelope proteins that enhance release of extracellular virions and eliminate formation of actin-containing microvilli without preventing tyrosine phosphorylation of the A36R protein. J Virol, 2003. 77(22): p. 12266-75.
469. McIntosh, A.A. and G.L. Smith, Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J Virol, 1996. 70(1): p. 272-81.
470. Doceul, V., et al., Protein B5 is required on extracellular enveloped vaccinia virus for repulsion of superinfecting virions. J Gen Virol, 2012. 93(Pt 9): p. 1876-86.
471. Smith, G.L., A. Vanderplasschen, and M. Law, The formation and function of extracellular enveloped vaccinia virus. J Gen Virol, 2002. 83(Pt 12): p. 2915-31.
472. Co, C., et al., Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell, 2007. 128(5): p. 901-13.
473. Payne, L.G. and K. Kristensson, The effect of cytochalasin D and monensin on enveloped vaccinia virus release. Arch Virol, 1982. 74(1): p. 11-20.
474. Po'uha, S.T., et al., Partial depletion of gamma-actin suppresses microtubule dynamics. Cytoskeleton (Hoboken), 2013. 70(3): p. 148-60.
475. Takenawa, T. and S. Suetsugu, The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol, 2007. 8(1): p. 37-48.
476. Cory, G.O., et al., Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell, 2003. 11(5): p. 1229-39.

CHAPTER 8: References
The University of Sydney 2016
209
477. Lambrechts, A., et al., Listeria comet tails: the actin-based motility machinery at work. Trends Cell Biol, 2008. 18(5): p. 220-7.
478. Latham, V.M., et al., A Rho-dependent signaling pathway operating through myosin localizes beta-actin mRNA in fibroblasts. Curr Biol, 2001. 11(13): p. 1010-6.
479. Yuan, M., et al., A marker-free system for highly efficient construction of vaccinia virus vectors using CRISPR Cas9. Mol Ther Methods Clin Dev, 2015. 2: p. 15035.
480. Yuan, M., et al., Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J Virol, 2015. 89(9): p. 5176-9.
481. Progatzky, F., M.J. Dallman, and C. Lo Celso, From seeing to believing: labelling strategies for in vivo cell-tracking experiments. Interface Focus, 2013. 3(3): p. 20130001.
482. Terasaki, M. and L.A. Jaffe, Labeling of cell membranes and compartments for live cell fluorescence microscopy. Methods Cell Biol, 2004. 74: p. 469-89.
483. Schnell, U., et al., Immunolabeling artifacts and the need for live-cell imaging. Nat Methods, 2012. 9(2): p. 152-8.
484. Liu, Z., L.D. Lavis, and E. Betzig, Imaging live-cell dynamics and structure at the single-molecule level. Mol Cell, 2015. 58(4): p. 644-59.
485. Benanti, E.L., C.M. Nguyen, and M.D. Welch, Virulent Burkholderia species mimic host actin polymerases to drive actin-based motility. Cell, 2015. 161(2): p. 348-60.
486. Krause, M., et al., Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol, 2003. 19: p. 541-64.
487. Zeile, W.L., et al., Vaccinia locomotion in host cells: evidence for the universal involvement of actin-based motility sequences ABM-1 and ABM-2. Proc Natl Acad Sci U S A, 1998. 95(23): p. 13917-22.
488. Grosse, R., et al., A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J, 2003. 22(12): p. 3050-61.
489. Chakraborty, T., et al., A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanovii to the actin-based cytoskeleton of mammalian cells. EMBO J, 1995. 14(7): p. 1314-21.
490. Laurent, V., et al., Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J Cell Biol, 1999. 144(6): p. 1245-58.
491. Skoble, J., et al., Pivotal role of VASP in Arp2/3 complex-mediated actin nucleation, actin branch-formation, and Listeria monocytogenes motility. J Cell Biol, 2001. 155(1): p. 89-100.

CHAPTER 8: References
The University of Sydney 2016
210
492. Hansen, S.D. and R.D. Mullins, VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol, 2010. 191(3): p. 571-84.
493. Walders-Harbeck, B., et al., The vasodilator-stimulated phosphoprotein promotes actin polymerisation through direct binding to monomeric actin. FEBS Lett, 2002. 529(2-3): p. 275-80.
494. Barzik, M., et al., Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem, 2005. 280(31): p. 28653-62.
495. Bear, J.E., et al., Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell, 2002. 109(4): p. 509-21.
496. Samarin, S., et al., How VASP enhances actin-based motility. J Cell Biol, 2003. 163(1): p. 131-42.
497. Plastino, J., S. Olivier, and C. Sykes, Actin filaments align into hollow comets for rapid VASP-mediated propulsion. Curr Biol, 2004. 14(19): p. 1766-71.
498. Breitsprecher, D., et al., Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J, 2008. 27(22): p. 2943-54.
499. Trichet, L., C. Sykes, and J. Plastino, Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J Cell Biol, 2008. 181(1): p. 19-25.
500. Gaucher, J.F., et al., Interactions of isolated C-terminal fragments of neural Wiskott-Aldrich syndrome protein (N-WASP) with actin and Arp2/3 complex. J Biol Chem, 2012. 287(41): p. 34646-59.
501. Huttelmaier, S., et al., Characterization of the actin binding properties of the vasodilator-stimulated phosphoprotein VASP. FEBS Lett, 1999. 451(1): p. 68-74.
502. Kabsch, W., et al., Atomic structure of the actin:DNase I complex. Nature, 1990. 347(6288): p. 37-44.
503. Ferron, F., et al., Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J, 2007. 26(21): p. 4597-606.
504. Aloy, P. and R.B. Russell, Interrogating protein interaction networks through structural biology. Proc Natl Acad Sci U S A, 2002. 99(9): p. 5896-901.
505. Schreiber, G. and A.R. Fersht, Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J Mol Biol, 1995. 248(2): p. 478-86.
506. Artman, L., et al., Planning your every move: the role of beta-actin and its post-transcriptional regulation in cell motility. Semin Cell Dev Biol, 2014. 34: p. 33-43.
507. Palmer, T.D., et al., Targeting tumor cell motility to prevent metastasis. Adv Drug Deliv Rev, 2011. 63(8): p. 568-81.

CHAPTER 8: References
The University of Sydney 2016
211
508. Katsantonis, J., et al., Differences in the G/total actin ratio and microfilament stability between normal and malignant human keratinocytes. Cell Biochem Funct, 1994. 12(4): p. 267-74.
509. Stournaras, C., et al., Altered actin polymerization dynamics in various malignant cell types: evidence for differential sensitivity to cytochalasin B. Biochem Pharmacol, 1996. 52(9): p. 1339-46.
510. Popow-Wozniak, A., et al., Cofilin overexpression affects actin cytoskeleton organization and migration of human colon adenocarcinoma cells. Histochem Cell Biol, 2012. 138(5): p. 725-36.