lcls ii instrumentation time-resolved photoelectron ... · lcls ii instrumentation time-resolved...
TRANSCRIPT

LCLS II InstrumentationTime-Resolved
Photoelectron Spectroscopy
Albert StolowNational Research Council
Canada

Atomic & Molecular SciencesA microscopic view of NatureA microscopic view of Nature
A bl th t l f d f th 20th tArguably, the great leap forward of the 20th century
• Quantum mechanics, spectroscopy, diffraction
• The detailed structure of matter
• Shapes of molecules, solids, biomolecules
More Nobel prizes in Physics have gone to AMO than to any other discipline

Structure-Function RelationsThe dominant paradigm of the 20th centuryThe dominant paradigm of the 20th century
Th t t f l l di t t th i f ti• The structure of molecules dictates their function• A very powerful but static view of matter
STRUCTURE FUNCTION

But Nature is not StaticNew perspectives for the 21st centuryNew perspectives for the 21st century
N t i l ft ti• Nature involves many, often competing, processes
A t ti i ill t ffi• A static view will not suffice
• We will also need a dynamical view of Nature• We will also need a dynamical view of Nature

Why does “Dynamics” matter?Beyond Structure-Function Relationships
Vision (Rhodopsin)
FUNCTION
The first step in Vision occurs on the 10-13 s timescale.The overall visual response occurs on the ~10-1 stimescale.
Do the first steps in Vision need to be so fast?
Yes Otherwise very fast competing dissipativeYes. Otherwise very fast competing dissipativeprocesses due to the surrounding protein and aqueous environment will completely terminate the desired signal transduction.
Nature designed this device for speedDynamics is central to the function

Statement: Chemistry is Not Possibley
• Typical bond energy ~ 3-4 eV
• Typical collision energy at kT ~ 25 meV
• Therefore chemistry is impossible.
What is wrong with this statement?
• The old bonds do NOT need to be broken before the ne• The old bonds do NOT need to be broken before the new bonds form. It happens simultaneously, concertedly.
• It is this complex, coupled dance of both atomsand valence electrons that we call CHEMISTRY.

The Arrow of Chemistry
?
Reactant
Product
A concerted rearrangement of both the atoms and the valence electrons

General Case: Non-Born-Oppenheimer Molecular Dynamics
hConical
IntersectionIntersection
Both electronic charge and vibrational energy flow during chemical processes
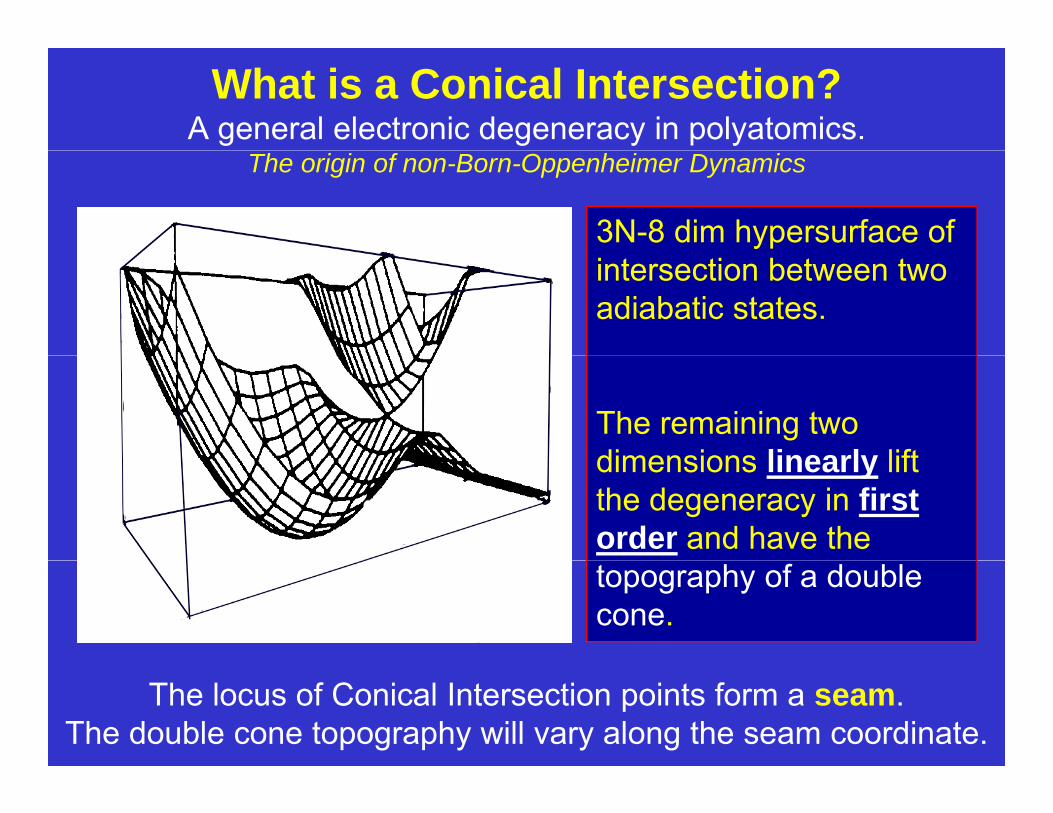
What is a Conical Intersection?A general electronic degeneracy in polyatomics.
The origin of non-Born-Oppenheimer Dynamics
3N-8 dim hypersurface of intersection between two adiabatic states.
The remaining two dimensions linearly liftdimensions linearly lift the degeneracy in first order and have the topography of a double cone.
The locus of Conical Intersection points form a seam.The double cone topography will vary along the seam coordinate.

General Features of Polyatomic Molecules 3 atoms 3 atoms
•Configuration Interaction:Electronic states composed of several Molec Orb (MOs)Electronic states composed of several Molec. Orb.(MOs)Phase and amplitude of MO coefficients internal coord.
•C i l I t ti•Conical Intersections: (charge flow)Ultrafast non-adiabatic coupling of electronic with vibrational degrees of freedomdeg ees o eedo
•Vibrational Mode Coupling: (energy flow)

General Probes of Ultrafast Molecular DynamicsDynamics of Both Valence Electrons & Molecular Vibrations
• Methods Sensitive to Atomic Motions• Ti R l d Vib ti l (IR R ) S t i• Time Resolved Vibrational (IR, Raman) Spectroscopies• Time Resolved X-ray Diffraction• Time Resolved Electron Diffraction
Th b th k• Methods Sensitive to Electronic Motions
• Attosecond Science
These both make use of the Molecular
Ionization ContinuumAttosecond Science
• Methods Sensitive to Both Atomic & Electronic Motionsincluding their Coupling.g p g
• Time Resolved Photoelectron Spectroscopy• Time Resolved High Harmonic Generation • Time Resolved X-ray AbsorptionTime Resolved X ray Absorption• Time Resolved 2D Electronic Spectroscopy• Time Resolved Stimulated Raman Scattering

General Probes of Ultrafast Molecular Dynamics
Si l/N i I f AMOSignal/Noise Issues for AMO
Time-resolved X-ray Absorption Signals: I/I measurements (where I = I - I)I/I0 measurements (where I = I0- I)
not well suited to dilute samples (gases)
Time-resolved Photoelectron Signals: I measured directly, extreme sensitivity. y y

The Molecular Ionization Continuum
e-
lmil e-iYlm() lm
(j)
lmil e-iYlm() lm
e- |j+, vj
+, Jj+>
E l d|o+, vo
+, Jo+> I.P.
Energy-resolvedPhotoelectronSpectroscopy
Angle-resolvedPhotoelectronSpectroscopy
h Sensitive to electronic
and
Sensitive to electronic
and
ION ELECTRONIC STRUCTUREION VIBRATIONAL STRUCTURE
vibrationaldynamics
rotationaldynamics
ION VIBRATIONAL STRUCTURE
ELECTRON PARTIAL WAVE COMPOSITION

Disentangling Electronic from Vibrational DynamicsMaking use of Koopmans’ Theorem
e-, 1 e-, 2++
g p
Non-adiabaticProcess
Nature 401, 52, (1999)

Disentangling Electronic from Vibrational Dynamics
Example: Internal Conversion in a Linear PolyeneThe first step in Vision
all-trans 2,4,6,8 Decatetraene
CCH3
C
CC
CC
CCH3
C
3
21A g, S1(0,0) 287 nm
11B u, S2 5762 cm -1
11A g, S0

D1 + e-
Time-resolved Valence Shell Photoelectron Spectroscopy
D0 + e-D1 + e
8.5 eV
Photoionization
I.P.7.3 eV
PhotoionizationProbe
S24 3 eV
S1Pump
4.3 eV
Non-AdiabaticCoupling 1
3.6 eVPump
S00 0 eV
Koopmans’ Correlations in Linear Polyenes
0.0 eV
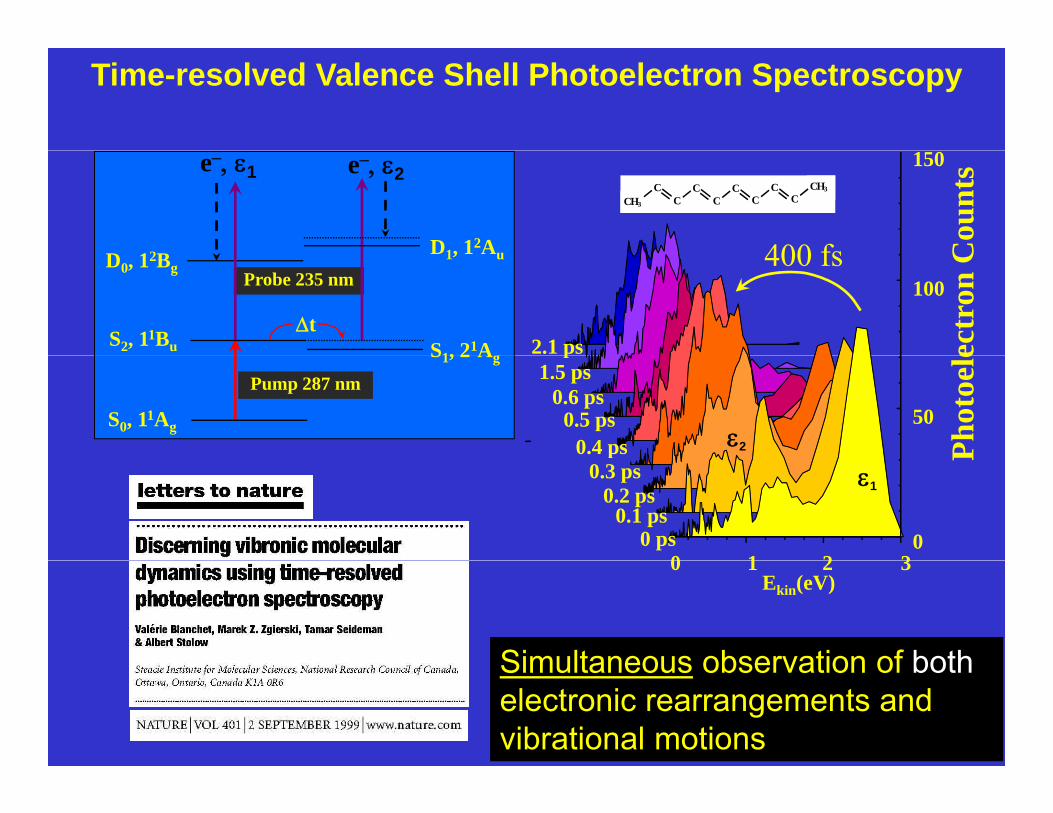
Time-resolved Valence Shell Photoelectron Spectroscopy
150
Cou
nts
400 fs
e2e1
D1, 12Au12
CCH3
CC
CC
CC
CH3
C
100
2.1 ps ectr
on C400 fs
Probe 235 nm
tS1, 21A
D1, 1 Au
S2, 11Bu
D0, 12Bg
50
p1.5 ps
0.5 ps0.4 ps
0.6 ps
2 Phot
oele
Pump 287 nm
S0, 11Ag
S1, 2 Ag
0 1 2 30
p0.3 ps
0.2 ps0.1 ps
0 ps
1
P
0 1 2 3Ekin(eV)
Simultaneous observation of bothSimultaneous observation of bothelectronic rearrangements and vibrational motions

Example: Excited State Proton Transfer
1Ion
E (e
V) 1 2
18.7 Ion
PT
hPROBE
1
3.43.9
PT
IC
S1-0.2
S0
hPUMP
0.0 0 5 1 0 0 80.6
0.40.2
0.0
me Dela
y (ps)2
0O
H
H
O O
H
OH
0.5 1.0 0.8Tim
eElectron Energy (eV)
Journal of Chemical Physics 114, 2519 (2001)ketoenol

An example: DNA Photostability
Early life developed under harsh & hostile conditions:• no significant stratospheric ozone layer• building blocks exposed to harmful UV radiation
How did the genetic material survive this period?
What is the self-protection mechanism?
El t i ll it d t t d
p
Electronically excited states are dangerous because they can lead to photochemistry.
(mutations).
To avoid this, electronic energy must be converted to less dangerous vibrational
energy which can be rapidly cooled in water.
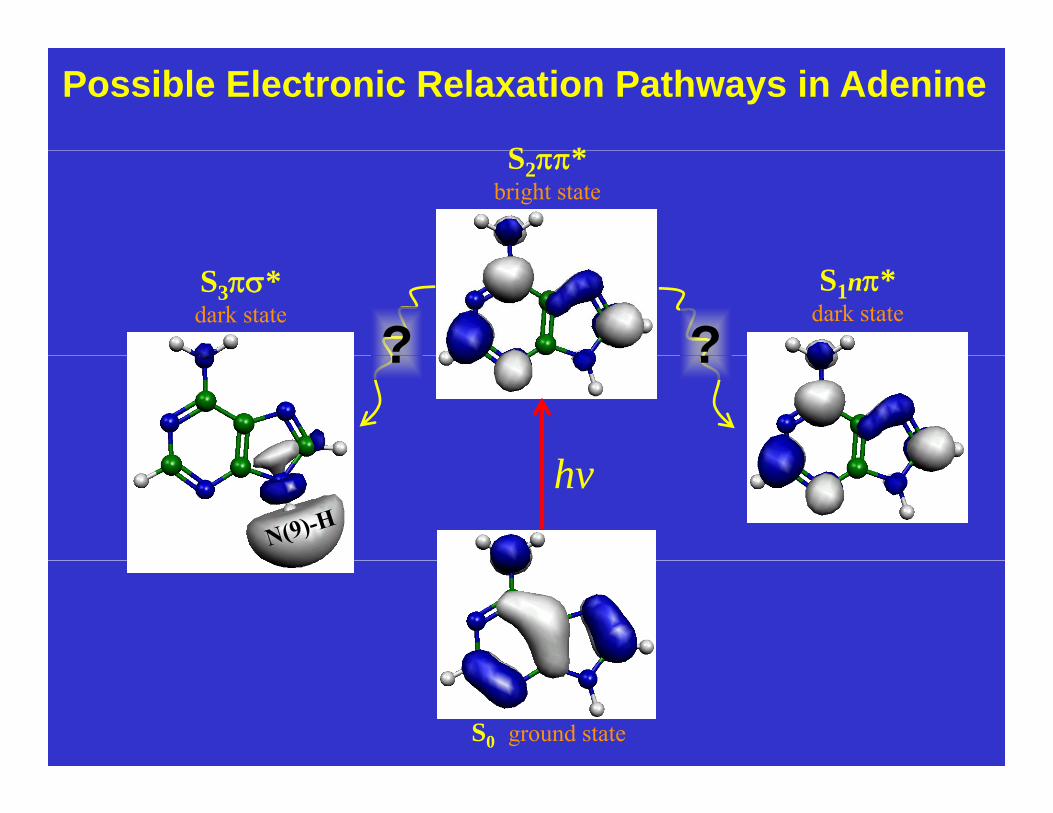
Possible Electronic Relaxation Pathways in Adenine
S *S2*bright state
S3*dark state
S1n*dark state
? ?? ?
hv
S0 ground state

Nature uses Ultrafast Processes to protect DNA bases from UV photochemical damagebases o U p otoc e ca da age

So how can we get the most information?
Femtosecond Dynamics in the Molecular Frame
• Ultrafast measurements made in the Lab Frame (LF)
• Scattering events occur in the Molecular Frame (MF) and are vectorial in natureWe need to think about the other component of
the Free Electron Continuum:• Averaging over all molecular orientations reduces the information obtained
the Free Electron Continuum:
• Can we make ultrafast measurements in the MF?Molecular Frame
Photoelectron Angular DistributionsCan we make ultrafast measurements in the MF?Photoelectron Angular Distributions

Time-Resolved Coincident 3D Momentum Vector Imaging3D Momentum Vector Imaging
e-
Time-Position Ion Detector
R+ (E )e
PROBE
|I+>R+ (Ekin, , )
RXYPumpt
PUMP
|Sn>t
-e (kin, , )Probe
PUMP
|S0>Time-Position Electron Detector
Kinematically complete (6D) time-resolved measurements

Example: (NO)2 Photodissociatione-
10 1
e
10.1
8.7(NO)2 X
~+ NO (X) + NO(X) ~+ ~
Probe6.0
t(e
V) (NO)2*
?
Probe~NO (A) + NO(X) ~*
E po
Pump
(NO)2 X~0
3s Rydberg
RN-N

Time-Resolved 6D Vector Correlations in the Molecular Frame (MF)in the Molecular Frame (MF)
y
electronk
N N
Ey
Pumpz
x
ionk
N Np
Ey
P b ionO O
Probe
via 3D momentum vectors measured in coincidence, the PAD may be transformed into the recoil frame (RF).

Dynamics from the Molecule’s Point of View
Molecular Frame Photoelectron Angular Distributions as a function of time.
(NO)2* → (NO)2
*† (3py)
NO(A) + NO(X)
N t R l d D i St k Eff t
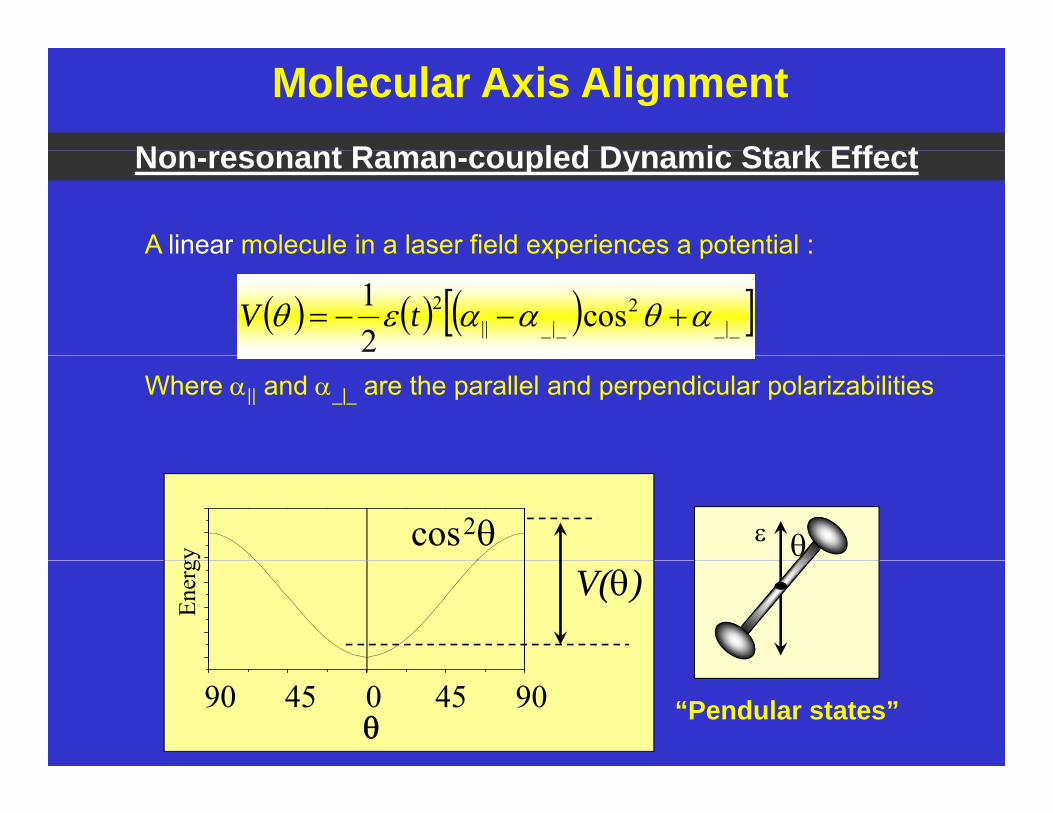
Molecular Axis Alignment
A linear molecule in a laser field experiences a potential :
Non-resonant Raman-coupled Dynamic Stark Effect
A linear molecule in a laser field experiences a potential :
_|_2
_|_||2 cos
21 tV
Where || and _|_ are the parallel and perpendicular polarizabilities
gy cos2
V()
Ener
g
904590 45 0 “Pendular states”

Why Alignment needs to be Field-Free:
• Cannot measure innate molecular properties in the presence of a strong non-resonant laser field.
• Molecular axis alignment methods must be field-free

1D Field-free Alignment of Linear Molecules
3D Field-free Alignment of General Molecules
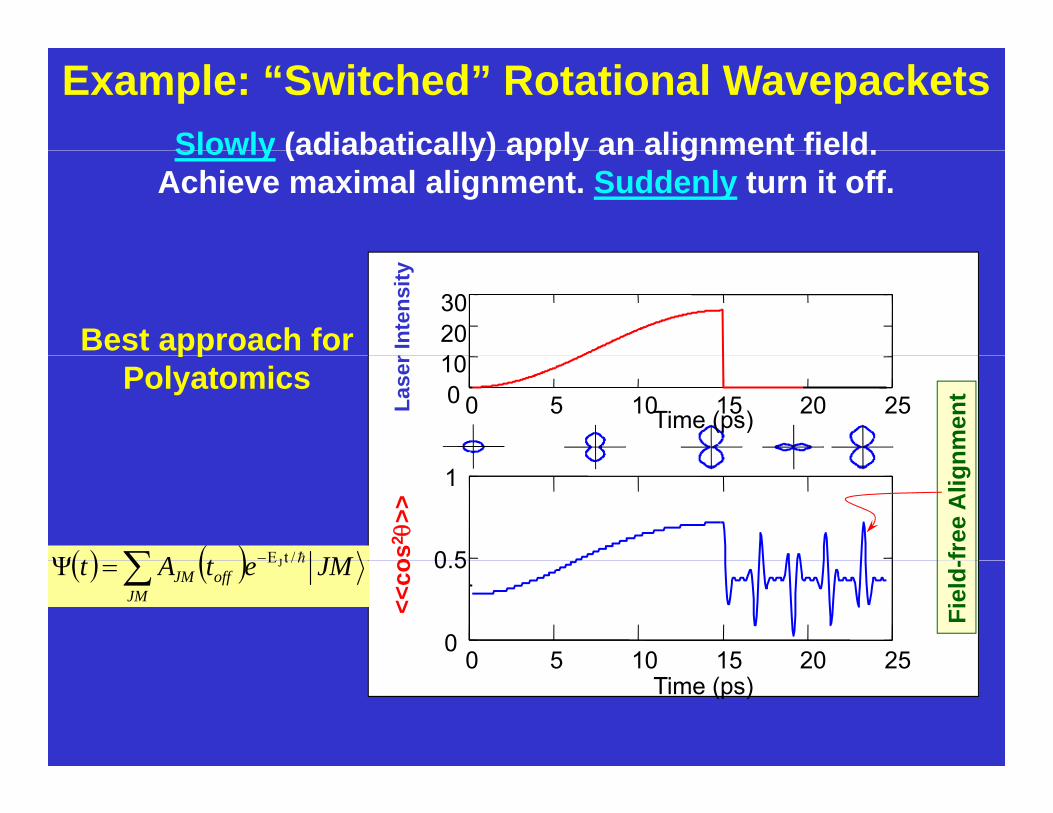
Example: “Switched” Rotational WavepacketsSlowly (adiabatically) apply an alignment field.Slowly (adiabatically) apply an alignment field.
Achieve maximal alignment. Suddenly turn it off.
102030
r Int
ensi
ty
Best approach for
nmen
t0 5 10 15 20 25010
Time (ps)La
serpp
Polyatomics
0 5
1
os2
>>
free
Alig
n
JMtAt /tEJΨ
0 5 10 15 20 250
0.5
<<co
Fiel
d- JMetAtJM
offJM /tEJΨ
0 5 10 15 20 25Time (ps)
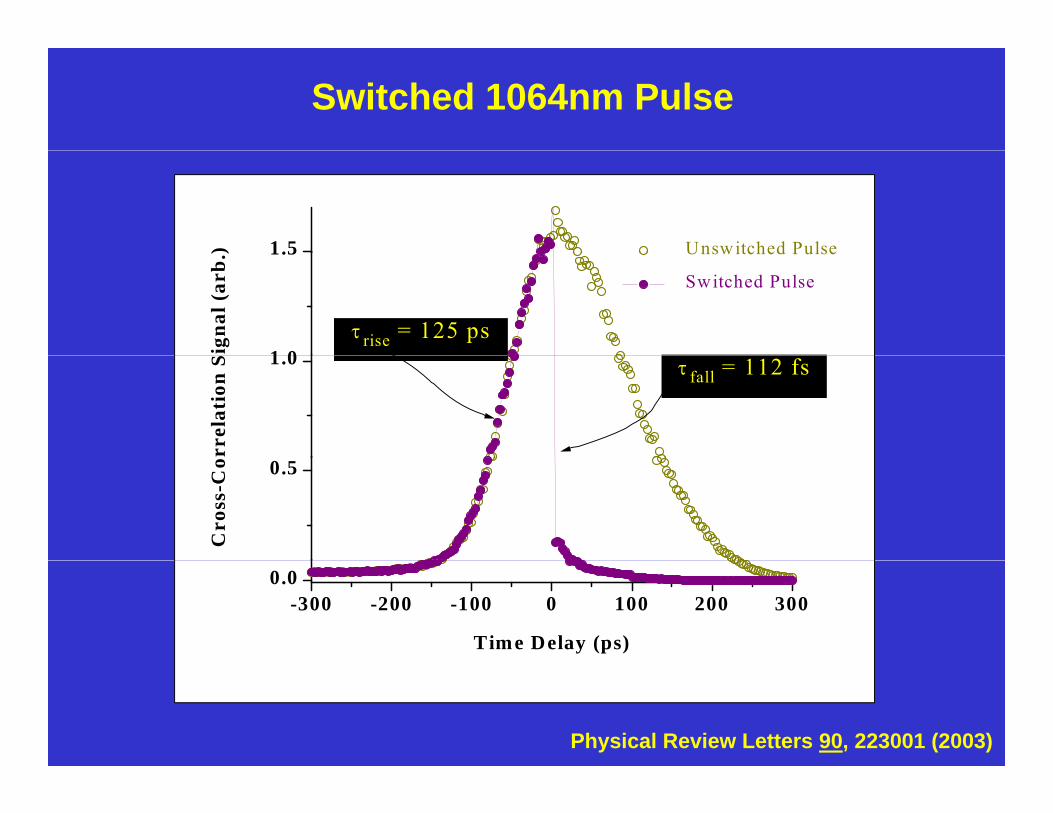
Switched 1064nm Pulse
1.5 Unswitched Pulse)
1 0
1.5 Unswitched Pulse
Switched Pulse
igna
l (ar
b.)
rise = 125 ps1.0
rrel
atio
n S fall = 112 fs
0.5
Cro
ss-C
o
-300 -200 -100 0 100 200 3000.0
Time Delay (ps)
Physical Review Letters 90, 223001 (2003)

Switched Rotational Wavepacket in CO22 . 5
2 . 0
Field-free wavepacket evolution
1 . 5
Alig
nmen
t
1 . 0
A
1 . 0
nal (
arb.
)
18 B
ent
- 2 0 0 - 1 0 0 0 1 0 0 2 0 0 3 0 00 . 5
T i D l ( )
OK
E S
ign
Alig
nme
T i m e D e l a y ( p s )
6 0 6 5 7 0 7 5 8 0 8 5 9 0 9 5 1 0 0 1 0 5 1 1 0 1 1 5 1 2 00 . 5
6 0 6 5 7 0 7 5 8 0 8 5 9 0 9 5 1 0 0 1 0 5 1 1 0 1 1 5 1 2 0
T i m e D e l a y ( p s )Time Delay (ps)

Example: CS2 Photodissociation
CS (X) + S +e-
I.P.CS2 X+
Probe6.0
(eV
)
CS2 (1B2)*
?
Probe
CS(X) + S(1D)
Epo
t CS(X) + S(3P)
Pump
CS2 X~0
p• Linear ground state• Strongly bent excited states.
RCS-S
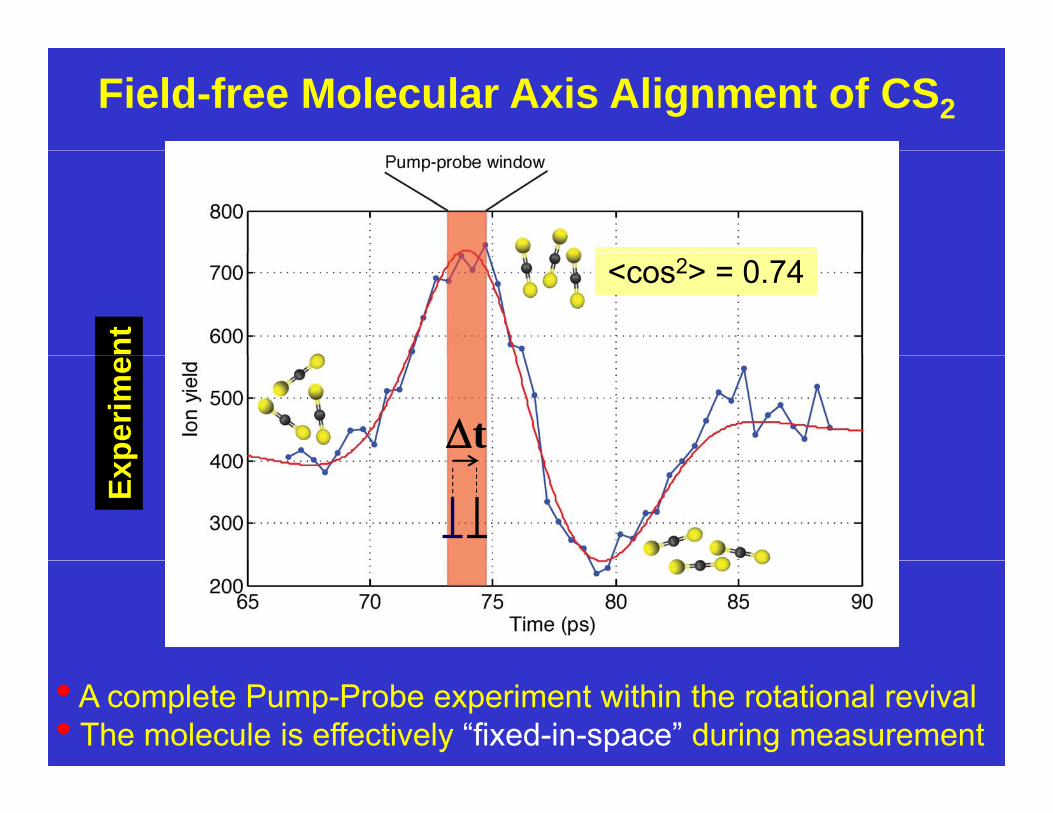
Field-free Molecular Axis Alignment of CS2
<cos2> = 0.74
nt
tperim
eEx
p
• A complete Pump-Probe experiment within the rotational revival• The molecule is effectively “fixed-in-space” during measurement

Time-resolved 3DPhotoelectron Imaging ithi R t ti l R i l
CS *(1B ) CS +(X 0) + ( k)
within a Rotational Revival
CS2 *(1B2) CS2
+(X, v=0) + e-(, k)
S
CEpump Eprobe
S

Real-time observation of valence electron motion
Attosecond time-scale valence electronic dynamics
i tin atoms

Real-time observation of valence electron motion during Chemical Reaction
CS2 *(1B2)
1 3 CS(X) + S(1D)/(3P)
purely electronic dynamicspurely electronic dynamicsMF Photoelectron Angl Dist’ns
August 2011

Other Probes of M l l W k t D i ?Molecular Wavepacket Dynamics?
Spectroscop can be sometimes anno ingSpectroscopy can be sometimes annoying….So what about other probes of wavepacket dynamics?
e.g. Time-resolved Electron or X-ray Diffraction
This promises to give directly the positions of the atoms as a function of time.

• X-Ray diffraction: Free Electron Lasers
• Ultrafast electron diffractionUltrafast electron diffraction

Hard X-ray Probes of Ultrafast DynamicsRelevance to ChemistryRelevance to Chemistry
Time-resolved X-ray Diffractiony
• Diffraction-based methods probe the spatial distribution of the total electron density whichdistribution of the total electron density, which tends to be localized (inner shell) around the atomic cores.
• Diffraction methods will not be very sensitive to the more diffuse valence electron distributions, ,the ones that are relevant to chemical dynamics.

John Arthur: Possible AMO instrumentation
• Seeded source with narrow bandwidth, high power• Branches optimized so user can choose either high• Branches optimized so user can choose either high
energy resolution or high peak power• Multiple end stations accommodating various sample
sources and spectrometers– Gas jets, cluster sources ovens, laser ablation sources,
ion sourcesion sources– Ion and electron spectrometers including magnetic
bottle, high energy resolution, and angle resolving spectrometersp
– X-ray spectrometer• Lasers for time-resolved experiments• Diagnostics for spent X-ray beam

LCLS seeded source characteristicsSoft X-ray Beam Liney
100
10(mJ)
Seeded
SXR10
per p
ulse
SASE120 Hz
or 60 Hz
1
Ener
gy p
100pC, 110m SXR undulator
60 Hz
0.10.1
1 10Photon Energy (keV)

Resonant X-ray Absorption
All elements ha e an absorption edge• All elements have an absorption edge between 250 and 1500 eV
• Ultrafast NEXAFS is emerging an important element specific probe which is sensitive to chemical environment
• Problem: transient absorption does not easily apply to dilute samples (gases)

Resonant X-ray Photoelectron Spectroscopy
SXR t neable bet een 250 and 1500 eV• SXR tuneable between 250 and 1500 eV• Photoelectron detection is extremely
sensitive, applies to gases• Ultrafast, elemental specificity, resonant
Auger decay, sensitive to chemical environment
• Problem: high SXR pulse energy means multiple targets ionized / shotmultiple targets ionized / shot

Time-resolved Auger Photoelectron SpectroscopyM. Gühr @ SXR@
Photostability of DNA Bases: Thymine
UVPUMP
NON-BOA
DYNAMICS
SXRPROBE
Eki
n
n
DYNAMICS
DE
CAY
E
nTIME
t=?
AU
GE
R
DELAY
O 1sO 1s

Time-resolved Auger Photoelectron Spectroscopy
p*Thymine Auger spectra (BL8 ALS)
p*
nN OC
pN OC
Auger Matrix element:<core cont|1/r12|valval’>
Spatial sensitivity for valence orbital at core hole.O 1s

LCLS AMO Hutch
70 fs pulse @ 565 eV photon energy47
70 fs pulse @ 565 eV photon energyTime resolution < 100 fs (jitter monitor)

Time-resolved Auger Photoelectron Spectroscopy
Thymine
48

Marcus’ Collaboration
Nora Berrah, WMUChristoph Bostedt LCLS SLAC
Melanie Mucke, Uppsala UniversityBrendan Murphy WMUChristoph Bostedt, LCLS SLAC
John Bozek, LCLS SLACPhil Bucksbaum, PULSE SLACRyan Coffee, LCLSJ C PULSE SLAC
Brendan Murphy, WMU Shungo Miyabe, PULSE SLACAdi Natan, PULSE SLACTimur Osipov, WMUVl di i P t i PULSE SLACJames Cryan, PULSE SLAC
Li Fang, WMUJoe Farrell, PULSE SLACRaimund Feifel, Uppsala University
Vladimir Petrovic, PULSE SLACSebastian Schorb, LCLS SLAC Thomas Schultz, MBI, BerlinLimor Spector, PULSE SLACa u d e e , Uppsa a U e s ty
Kelly Gaffney, PULSE SLACMike Glownia, PULSE SLACMarkus Guehr, PULSE SLAC, Spokesperson
o Specto , U S S CFrancenso Tarantelli, Univ. PerugiaIan Tenney, PULSE SLAC Song Wang, PULSE SLACBill White LCLS SLACSpokesperson
Todd Martinez, PULSE SLAC,Brian McFarland, PULSE SLAC
Bill White, LCLS SLACJames White, PULSE SLAC

X-ray Photoelectron SpectroscopyCovariance Imagingg g
• Molecular frame information obtains from l l d i id tangle-resolved coincidence measurements
• 1st Born Approx limit for photoelectron imaging means orbital reconstruction may be possible without presence of a strong laser field
• Problem: Multiple target ionization p gprecludes true coincidences
• Can we develop a covariance approach toCan we develop a covariance approach to molecular frame photoelectron imaging?

Discussion?