langerhans’ caenldl histiocytosis: case reports …€™ caenldl histiocytosis: case reports...
TRANSCRIPT

Langerhans’ Caenldl histiocytosis:case reports literature reviewRobert J. Henry, DDS, MS Edward A. Sweeney, DMD
L angerhans’ cell histiocytosis (LCH), formerlyknown as histiocytosis X, is one of a group ofpoorly understood diseases of histiocytes. The
clinical spectrum of disease ranges from the chronic,localized form to an acute leukemia-like disease with afatal outcome. Alfred Hand was the first to report a caseof histiocytosis in 1893.1 Later, in 1941, Farber describedthis condition when reporting the overlap among dis-eases that would later be termed histiocytosis X.2,3 Sincethat time, numerous case reports have appeared in thedental literature, each having a diverse focus and usinginconsistent terminology. The purpose of this reviewarticle is to enhance the understanding of LCH and toreport two cases of very young children demonstratingskull and jaw lesions.
LCH previously has been considered a reactivepolyclonal disease of immune regulation and not a trueneoplasm. More recent evidence, however, has demon-strated clonal proliferation, a key neoplastic feature.4-6
LCH afflicts young children primarily, some adoles-cents, and a few young adults. The classic presentationinvolves lytic lesions of bone, particularly of the skull.7
The pathophysiology of LCH involves histiocytes --cells derived from monocytes of the granulocyte/mac-rophage series after extravascular diapedesis. Histio-cytes function either as antigen-processing cells, phago-cytic cells, or as antigen-presenting cells,s Langerhans’cells are specialized histiocytes with immune functionssimilar to other dendritic cells and macrophages.Langerhans’ cells function immunologically by present-ing antigen to T lymphocytes.9’ 10 In LCH, these cellsundergo pathologic change and appear histologicallywithin an inflammatory background with a varied mi-croscopic appearance that necessitates additional analy-sis in order to establish a diagnosis.TM 12 The number ofLangerhans’ cells may be scarce and mixed with mac-rophages, lymphocytes, eosinophils, giant cells, neutro-phils and plasma cells when viewed under light micros-copy. 13 Pathologic Langerhans’ cells are characterizedby the presence of antigenic surface markers that reactwith a specific monoclonal antibody and by the histo-
logic presence of Langerhans’ granules (also calledBirbeck’s granules).14 Birbeck’s granules are rod-shapedultrastructural organelles that may have a vesicularportion giving it a so called "tennis racquet" appearanceunder electron microscopy.14-16 The presence ofBirbeck’s granules is pathognomonic of LCH.s,17
Although the etiology of LCH has not been estab-lished, several theories exist regarding this disease.Pathologic Langerhans’ cells are thought to be derivedfrom precursor cells or through alteration of normalhistiocytes.16, is The mechanism by which this prolifera-tion and accumulation takes place remains unknown.Viruses have been implicated as inciting agents in LCHbut their involvement remains theoretical,s, 16 Consider-able effort has been made to assess the role of the im-mune system in the etiology of LCH. The bulk of evi-dence for this theory relates to abnormal thymicbiopsies of patients with LCH, particularly those withmultisystem involvement. Findings such as thymus in-volvement and macrophage activation in the childhoodhistiocytoses seem to support an immune system eti-ology.16,19, 20 However, the fact that there seems to beno histologic difference between patients with localizeddisease and those with multisystem involvement hasprompted some to suggest that the above findings maybe secondary occurrences in the disease process21Newer x-linked polymorphic DNA probes have dem-onstrated that LCH is a highly variable monoclonalneoplastic disorder whose specific etiology remainsunestablished.6
ClassificationThe term histiocytosis X was a generic term devel-
oped as a result of similarities in the pathophysiologyamong the diseases of histiocytes as well as the sharedclinical and histologic features.B, 22 Letterer-Siwe syn-drome was an earlier term used to describe an acutedisseminated multisystem disease process of the reticu-loendothelial system that typically affected childrenyounger than 3 years of age. Hand-Sch~iller-Christiansyndrome was used to describe an intermediate,
Pediatric Dentistry- 18:1, 1996 American Academy of Pediatric Dentistry 11

chronic, disseminated form of the disease that gener-ally affected children older than 3. The classic triad as-sociated with Hand-Sch611er-Christian disease, whichoften did not occur together, included exophthalmos,diabetes insipidus, and multiple bone lesions of theskull, the second two largely due to sella turcica in-volvement. The mildest form of diseases of histiocyteswas termed eosinophilic granuloma and presented assolitary bony lesions of the ribs, pelvis, or mandible andusually afflicted older children and young adults.Hashimoto-Pritzker syndrome is yet another term thatwas used to describe a congenital form of the diseasethat presented with deep subcutaneous skin lesions,u,16 Numerous other terms also were utilized to describe
diseases of histiocytes, which often confused the diag-nosis. By 1987, it was widely established that the Langer-hans’ cell was pathognomonic of diseases of histiocytesand that classification should now be determined on thecellular basis of the disease.21 The International Histio-cyte Society in 1987 established a classification of his-tiocytoses into three groups as follows:
I. Langerhans’ cell histiocytosis
II. Histiocytoses of mononuclear phagocytes otherthan Langerhans’ cells
III. Malignant histiocytic disorders2~,17,21
Even though this classification does not reflect re-cent neoplastic evidence, it is universally accepted. Thevast majority (99%) of patients are diagnosed with thetype I or II variety.23 The diagnostic criteria for a de-finitive diagnosis for type I (LCH) is the presence Birbeck’s granules and is utilized by the InternationalHistiocyte Society for establishing a positive diagnosisof LCH27A more recent method for LCH diagnosis isthe positive immunostaining for $100 protein and theCDla antigen,s,lB,2° This review will focus on the type Ivariety, LCH.
Clinical presentationApproximately .200 new cases of LCH are diagnosed
each year in the US2~ Children from I to 15 years of ageare the most commonly afflicted. The peak incidence isfrom 2 to 4 years of age, and a predilection for blackpatients has been reported21,24 Pathologic Langerhans’cells infiltrate various organs such as bone, skin, liver,spleen, lung, and brain and may result in a variety ofclinical signs and symptoms depending on the degreeof individual organ infiltration and functional compro-mise. The clinical course of LCH varies considerablydepending on the extent and number of organs involvedas well as the age of the patient at the time of diagnosis.Bones of the skull, particularly orbital and temporalbones, the sella turcica, and mandible, as well as the ribsand pelvis are commonly involved,y, 22 The relative fre-quency of organ system involvement is as follows: bone,80%; skin, 60%; liver, spleen, lymph nodes, 33%; lungs,25%; orbit, 25%; and maxillofacial, 20%.s Initial physi-cal findings often include skin rash, otitis media, fever,
organomegaly, anemia, and diabetes insipidus.TM 22, 25Pituitary and pulmonary involvement also is common,particularly in males.2° The differential diagnosis relatesto the particular organ system involved and includessome of the following conditions: juvenile xanthogranu-loma, chronic osteomyelitis, interstitial pneumonia, scle-rosing cholangitis, dermatopathic lymphadenopathy,odontogenic cysts, and periodontal disease.11, 22
Oral involvement is a frequent finding. Ten to twentypercent of initial symptoms are nonspecific oral findingsthat include: gingival enlargement, oral ulceration,mobility of teeth with alveolar expansion, jaw pain, fa-cial swelling, as well as the classic intraosseous lesionsand scooped out radiolucencies of the alveolar process,s,22, 26, 28. Children with multiple organ and bone lesions
commonly have mandibular involvement with destruc-tive radiolucencies, alveolar bone loss, and teeth thatappear to be "floating in air". 22 Mandibular lesions arecommonly associated with maxillary involvement, butrarely is maxillary disease seen without mandibularradioluciencies
Dental considerationsLCH is included in the differential diagnosis for chil-
dren presenting with advanced periodontal diseaseand/or bone loss in the primary dentition. As men-tioned, oral involvement is common in LCH and maybe the initial chief complaint. Other entities with simi-lar presentations may include: prepubertal periodonti-tis, leukemia, neutropenia, hypophosphatasia, fibrousdysplasia, and Papillon-Lef6vre syndrome. Prepubertalperiodontitis is associated with the microorganism Ac-tinobacillus actinomycetemcomitans (Aa) and may resultin mobility and tooth loss by age 3. Differentiation fromLCH is based on the limited gingival inflammation withmarginal bone loss and demonstration of Aa from sub-gingival culture. 29 Acute myelogenous leukemia (AML)may present with gingival hypertrophy and appear asLCH. AML accounts for 15% of childhood leukemia andis distinguished from LCH by systemic symptoms thatgenerally lead to confirmation of leukemia throughbone marrow aspiration. B° Various neutropenic disor-ders also may result in significant gingival inflamma-tion and alveolar bone loss. Reduced neutrophil countsmay be the result of bone marrow production defectsor neutrophil destruction and generally are associatedwith systemic findings such as splenomegaly and infec-tion. Distinction from LCH is based on laboratory data.3~
Hypophosphatasia is characterized by low serum alka-line phosphatase levels and excessive excretion ofphosphoethanolamine in the urine. The classic oral find-ings differ from LCH in that premature tooth loss gen-erally involves the mandibular primary incisors, whichoften have abnormally large pulp spaces.32 Fibrous dys-plasia is a progressive expansile non-neoplastic bonelesion that may result in primary tooth loss. Cheekswelling produces the so-called "eyes-raised-to-heaven"or cherub look that distinguishes this condition from
12 American Academy of Pediatric Dentistry Pediatric Dentistry - 18:1, 1996

LCH.33 Papillon-Lef6vre syndrome is associated withmarked destruction of alveolar bone with prematureprimary tooth loss. This condition is differentiated fromLCH by the associated hyperkeratosis of the palms andsoles.B4 This brief list includes conditions commonlyincluded in the differential diagnosis for patients withdestructive periodontal disease and tooth loss in theprimary dentition.
The long-term dentofacial development of patientstreated for LCH has not been reported. Sequelae forpatients with childhood leukemia are known and cor-relate to LCH patients undergoing similar treatmentprotocols. Chemoradiation’s influences on craniofacialgrowth and dental development are age related andmost significant for children younger than 5 years ofage.35 The incidence of salivary gland dysfunction,enamel dysplasias, tooth / root agenesis, and alterationof mandibular growth is greater in patients receivingcombined radiation and chemotherapy.B6, 37 The long-term dental management of LCH patients begins witha review of the specific disease, location, and the thera-peutic protocol the child received. Displaced teeth and /or developing follicles with altered eruption patterns arecommon for children with a history of oral involve-ment.2s Parents should be aware of the possibility oflocalized enamel defects, agenesis of roots and/orcrowns, altered dentoalveolar growth, as well as ortho-pedic sequelae for those having undergone craniofacialradiation therapy.
Medical management and prognosis of LCHPrognostic indicators for LCH include: 1) age -- chil-
dren less than 2 years generally have disseminated dis-ease and a poorer prognosis; 2) number of sites involved-- multisystem disease carries a poorer prognosis; and3) organ dysfunction, which, if present, also results in diminished outlook21,38 Liver dysfunction at the time ofdiagnosis (indicated by hypoproteinemia or hyperbi-lirubinemia) as well as hemopoietic system involvement(indicated by thrombocytopenia, neutropenia, and ane-mia) both are associated with a poorer outlook.24’ 39 Theprognosis is particularly grave for children youngerthan 2 years old who present with multiple organ in-volvement and associated dysfunction.4° Mortality ratesfor this group exceed 60%.6, 41 Second tumors such asleukemia or thyroid carcinoma are potential complica-tions for long-term suvivors.42 Older children with mul-tisystem disease at presentation also demonstrate con-siderable morbidity. Many develop chronic problemssuch as liver dysfunction, diabetes insipidus, chronicotitis, and learning disabilities that stem from the vari-ous treatment modalities.", 24
The heterogeneity of disease in patients with LCHhas made development of specific treatment protocolsdifficult. Often the particular therapeutic approach haslittle influence on the prognosis for infants presentingwith disseminated disease, who have a relatively poorprognosis, or those presenting with the mild form of
LCH, who have an excellent prognosis,s Treatment ofsingle system disease is generally benign and mayinclude observation for spontaneous regression,radiation, surgical curettage, or intralesional infiltra-tion with steroids.41
Protocols for young children with disseminated dis-ease are similar to those for childhood leukemia andemploy chemotherapeutic agents such as vinblastinesulfate, vincristine sulfate, prednisone, methotrexate,cyclophosphamide, and 6-mercaptopurine.S, 24, 41, 43 Theuse of cyclosporine is also thought to be effective in com-bination therapy for children with milder forms ofLCH.s Although no chemotherapeutic regimen has beendemonstrated as superior, evidence suggests thatetoposide, which is known to be effective for malignan-cies of monocyte / macrophage origin, maybe useful inpatients with vital organ involvement or those not re-sponding to more conservative treatment,s, 41, 43, 44
These newer combination therapies are being recom-mended for front-line therapy only for the severe mul-tisystem form of LCH. Significant morbidity, such assecond tumor development, may be associated withaggressive therapy and should be reserved for patientswith a poor prognosis and those with disease progres-sion while undergoing conservative therapy.41, 43
Young patients with disseminated LCH are at in-creased risk for disease progression because of the con-cern over radiation effects and reluctance to employ thistreatment modality in the younger age groups.41 Radia-tion therapy in children younger than 6 years of age hasbeen associated with altered growth, adverse intellectualeffects, and arrested dental development as well as sec-ond tumor development.22 Radiation treatment of 600-1500 cG in 200-cG daily fractions is highly effective inchildren beyond the age of such concerns or in solitarylesions in very critical areas, such as sella turcica.22,45
Case reportsCase 1
A 15-month-old Hispanic female presented toMedical Center Hospital emergency room in May of1993 with chief complaints of diarrhea, diminished ap-petite, and refusal to walk because of ankle swelling.Clinical examination revealed significant hepatosple-nomegaly. The recent medical history consisted of ane-mia and recurrent otitis media. After medical evalua-tion, a provisional diagnosis of malignant histiocytosiswas made. This diagnosis was based largely on resultsof liver biopsy and bone marrow aspiration. Labvalues were as follows: WBC 8,000/mm3, Hb 8.2 g/dL,MCV 89.4 ~ g/dL, and platelets 145,000/mm3, The factthat Birbeck’s granules had not been identified from theinitial liver biopsy, although not unusual, led tothe provisional malignant histiocytosis diagnosis. Thechild was placed on a regimen consisting of methylpred-nisolone (10 kg/day) and vinblastine (0.1 mg/kg weekly intervals). She was transferred to Santa RosaChildren’s Hospital.
Pediatric Dentistry- 18:1, 1996 American Academy of Pediatric Dentistry 13
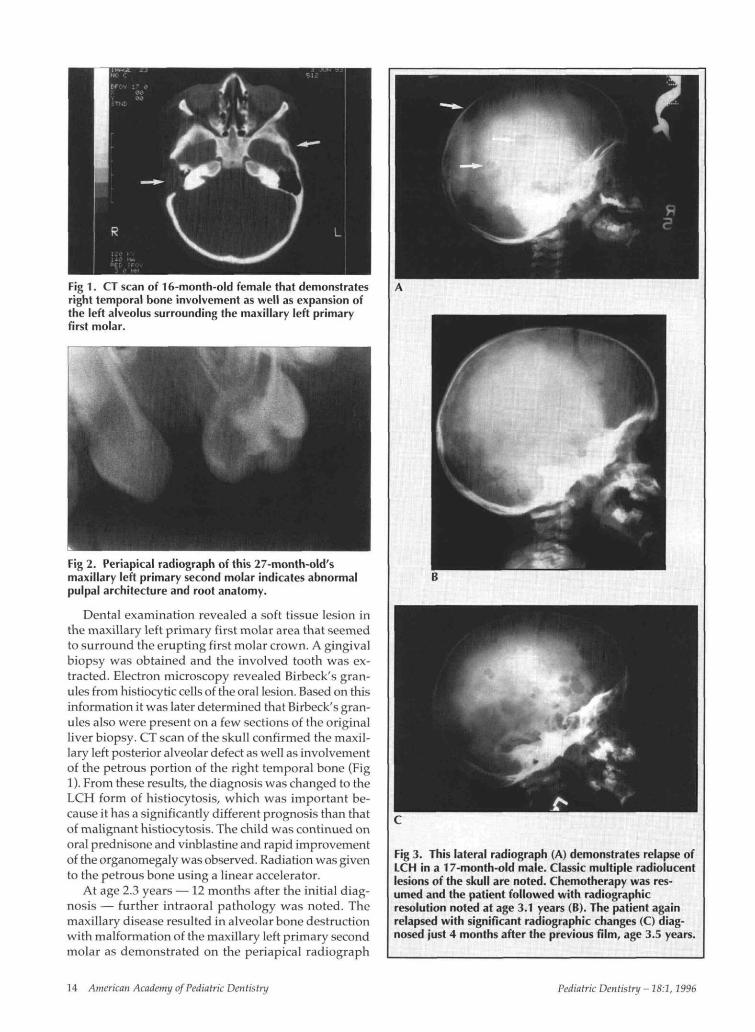
Fig 1. CT scan of 16-month-old female that demonstratesright temporal bone involvement as well as expansion ofthe left alveolus surrounding the maxillary left primaryfirst molar.
Fig 2. Periapical radiograph of this 27-month-old'smaxillary left primary second molar indicates abnormalpulpal architecture and root anatomy.
Dental examination revealed a soft tissue lesion inthe maxillary left primary first molar area that seemedto surround the erupting first molar crown. A gingivalbiopsy was obtained and the involved tooth was ex-tracted. Electron microscopy revealed Birbeck's gran-ules from histiocytic cells of the oral lesion. Based on thisinformation it was later determined that Birbeck's gran-ules also were present on a few sections of the originalliver biopsy. CT scan of the skull confirmed the maxil-lary left posterior alveolar defect as well as involvementof the petrous portion of the right temporal bone (Fig1). From these results, the diagnosis was changed to theLCH form of histiocytosis, which was important be-cause it has a significantly different prognosis than thatof malignant histiocytosis. The child was continued onoral prednisone and vinblastine and rapid improvementof the organomegaly was observed. Radiation was givento the petrous bone using a linear accelerator.
At age 2.3 years — 12 months after the initial diag-nosis — further intraoral pathology was noted. Themaxillary disease resulted in alveolar bone destructionwith malformation of the maxillary left primary secondmolar as demonstrated on the periapical radiograph
Fig 3. This lateral radiograph (A) demonstrates relapse ofLCH in a 17-month-old male. Classic multiple radiolucentlesions of the skull are noted. Chemotherapy was res-umed and the patient followed with radiographicresolution noted at age 3.1 years (B). The patient againrelapsed with significant radiographic changes (C) diag-nosed just 4 months after the previous film, age 3.5 years.
14 American Academy ofPediatric Dentistry Pediatric Dentistry - 18:1, 1996

(Fig 2). The maxillary left primary second molar extrac-tion was accomplished during conscious sedation us-ing midazolam and local anesthesia. Three months later,it became apparent radiographically and by physicalexamination that the lesion in the petrous portion of thetemporal bone had not resolved and indeed seemedactive. An excisional biopsy and mastoidectomy re-vealed significant necrosis and residual disease. Afterhealing, radiation with an additional 1000 cG was ad-ministered to the petrous bone. The patient has beenasymptomatic since that time (15 months). Mainte-nance therapy with dexamethasone and vinblastine isanticipated to continue until radiographic resolution ofthe cranial lesions has occurred.
Case 2
A 2-month-old male was referred to Santa RosaChildren's Hospital in September 1990 by his pediatri-cian because of fever, anemia, and splenomegaly. Thefamily history revealed a maternal cousin had died of"histiocytosis X" in childhood. Physical examinationidentified multiple small, flaky skin and scalp lesions.The abdomen was enlarged with no lymphadenopathynoted. Lab values were as follows: WBC 10,600/mm3,Hb 9.1 g/dL, MCV 77 pg/dL, and platelets 137,000/mm3. A diagnosis of Langerhans' cell histiocytosis wasmade based on skin biopsy of the abdomen and needlebiopsy of the liver, both suggestive of LCH. No os-teolytic lesions were seen on a skeletal survey. Therapywas begun with six courses of intravenous etoposideand methylprednisolone and continued for 12 months.In December 1991, 3 months after discontinuation oftherapy, the child presented with fever, otitis and skinrash. A skeletal survey demonstrated multiple lytic le-sion of the frontal and parietal bones (Fig 3A). Weeklychemotherapy was resumed using intravenous vinblas-tine and was continued until September of 1993. Thepatient was followed with resolution noted radio-graphically (Fig 3B).
In January 1994 the patient again relapsed and im-pressive lytic lesions were evident radiographically(Fig 3C). These as well as rib lesions were not present 4months earlier. The panoramic radiograph of this now3.6-year-old child demonstrates the multiple mandibu-lar lesions associated with LCH (Fig 4). Intravenouschemotherapy with vinblastine was reinstituted alongwith cranial radiation SOOcG to the base of the skull. Thetumor board's decision was to avoid mandibular radia-tion. The concern related to diminished intellectual de-velopment and the adverse growth effects to the max-illa and mandible associated with cranial radiation inchildren of this age.
Radiographic surveys in December 1994 revealed amarked improvement of the lytic lesions of the skulland mandible compared with those taken at the time ofrelapse in early 1994. Despite the major mandibular in-volvement, no periodontal involvement, mobility ofteeth, alveolar expansion, jaw pain or facial swelling
were found during the periodic dental evaluations fromJanuary 1994 to the time of this report. The patient con-tinues on maintenance chemotherapy with vinblastineawaiting radiographic resolution of the cranial lesions.He is currently in remission, 5.4 years after the initialdiagnosis, but with a guarded long-term prognosis.
The presence of destructive periodontal diseaseand/or tooth loss during the primary dentition shouldalert the practitioner to the possibility of serious sys-temic disease. LCH is included in the differential diag-nosis of such a condition, which may be the patients'initial chief complaint. The diagnosis for LCH may beconfirmed through immunofluorscence for the SI 00protein (Fig 5). Sufficient gingival biopsy material, withor without tooth extraction, should be obtained for thatpurpose. The two cases presented both had oral in-volvement but a quite different presentation at the timeof diagnosis. The first case involved maxillary involve-ment without mandibular association, which is rare. Inthe second case, the impressive mandibular radiolucen-cies were evident, yet the intraoral soft tissues were un-remarkable with no apparent dental involvement.
Fig 4. Panoramic radiograph of the patient at 3.6 years ofage depicts mandibular involvement often associatedwith LCH. The numerous lytic lesions are apparent.
Fig 5. This gingival biopsy exemplifies the immuno-histochemical staining of LCH cells for S-100 protein(original magnification 25x). The reaction product isdiagnostic of LCH since the cytoplasm of normalhistiocytes does not stain in this manner.
Pediatric Dentistry - 18:1,1996 American Academy of Pediatric Dentistry 15

The authors thank Dr. Clementina Geiser, Department of Hema-tology/Oncology at Santa Rosa Children’s Hospital, for makingdata available for this publication.
Dr. Henry is Associate Professor and Postdoctoral Program Di-rector, Department of Pediatric Dentistry, University of TexasHealth Science Center at San Antonio and Dr. Sweeney is Profes-sor Department of Pediatric Dentistry, University of Texas HealthScience Center at San Antonio and Chief of Dental Services, SantaRosa Children’s Hospital.
1. Hand A Jr: Polyuria and tuberculosis. Arch Pediatr 10:673-75, 1893.
2. Farber S: The nature of "solitary eosinophilic granuloma" ofbone. Am J Patho117:625-29, 1941.
3. Lichtenstein L: Histiocytosis X: integration of eosinophilicgranuloma of bone. "Letterer-Siwe disease" and "Schiiller-Christian disease" as related manifestations of a single no-sologic entity. Arch Patho156:84-102, 1953.
4. Willman C: Detection of clonal histiocytes in Langerhans’ cellhistiocytosis: biology and clinical significance. Br J Cancer70(23):$29-33,1994.
5. Yu RC, Chu C, Buluwela L, Chu AC: Clonal proliferation ofLangerhans’ cells in Langerhans’ cell histiocytosis. Lancet343:767-68, 1994.
6. Willman CL, Busque L, Griffith BB, Favara BE, McClain KL,Duncan MH, Gilliland DG: Langerhans’ cell histiocytosis°
(histiocytosis X) -- a clonal proliferative disease. N Engl Med 331:154-60, 1994. [Comment N EnglJ Med 331:191-93]
7. Moore AT, Pritchard J, Taylor DS: Histiocytosis X: an oph-thalmological review. Br J Ophthalmo169:7-11, 1985.
8. Ladisch S, Jafe E: Principals and Practice of Pediatric Oncol-ogy, 2nd Ed. Pizzo PA, Poplack DG, eds. Philadelphia: JBLippincott Co, 1993, pp 617-28,.
9. Scarpelli DG: Histiocytosis X. Arch Dermato1122:402-3, 1986.10. Lombardi T, Hauser C, Budtz-J6rgensen E: Langerhans’ cells:
structure, function and role in oral pathological conditions.J Oral Pathol Med 22:193-202, 1993.
11. Malone M: The histiocytoses of childhood. Histopathology19:105-19, 1991.
12. Ben-Ezra JM, Koo CH: Langerhans" cell histiocytosis andmalignancies of the M-PIRE system. Am J Clin Patho199:464-71, 1993.
13. Favara BE, Jaffe R: The histopathology of Langerhans’ cellhistiocytosis. Br J Cancer 70(23):$17-23,1994.
14. Mierau GW, Favara BE, Brenman JM: Electron microscopyin histiocytosis X. Ultrastruct Pathol 3:137-42,1982.
15. Birbeck MS, Breatl~nach AS, Everall JD: An electron micro-scope study of basal melanocytes and high-level clear cells(Langerhans’ cells) in vitiligo. J Invest Dermatol 37:51-64,1961.
16. JaffeR: Pathology of histiocytosis X. Perspect Pediatr Pathol9:4-47, 1987.
17. Writing Group of the Histiocyte Society: Histiocytosis syn-dromes in children. Lancet 208-9, Jan 24,1987.
18. Murphy GF, Messadi D, Fonferko E, Hancock WW: Pheno-typic transformation of macrophages to Langerhans’ cell inthe skin. Am J Patho1123:401-6, 1986.
19. Zaizov R, Stark B, Friedman I: Cell mediated immunity andactivation of macrophages in childhood histiocytoses. MedPediatr Onco114:110-15, 1986.
20. Favara BE: Langerhans’ cell histiocytosis pathobiology andpathogenesis. Semin Onco118:3-7, 1991.
21. Osband ME, Pochedly C: Histiocytosis X: an overview.Hematol Oncol Clin North Am 1:1-7, 1987.
22. Hartman KS: Histiocytosis X: areviewof 114 cases with oralinvolvement. Oral Surg 49:38-54, 1980.
23. Prichard J, Broadbent V: Histiocytosis -- an introduction.Br J Cancer 70(23):sl-3, 1994.
24. Matus-Ridley M, Raney RB Jr, Thawerani H, Meadows AT:Histiocytosis X in children: patterns of disease and results oftreatment. Med Pediatr Onco111:99-105, 1983.
25. Smith RJ, Evans JN: Head and neck manifestations of histio-cytosis X. Laryngoscope 94:395-99, 1984.
26. Jones JC, Lilly GB, Marlette RH: Histiocytosis X. J Oral Surg28:461-69,1970.
27. Loh HS, Quah TC: Histiocytosis X (Langerhans’-cell histio-cytosis) of the palate: case report. Aust Dent J 35:117-20,1990.
28. Dagenais M, Pharoah M, Sikorski P: The radiographic char-acteristics of histiocytosis X: a study of 29 cases that involvethe jaws. Oral Surg Oral Med Oral Patho174:230-36,1992.
29. Sj6din B, Matson L, Unell L, Egelberg J: Marginal bone lossin the primary dentition of patients with juvenile periodon-titis. J Clin Periodonto120:32-36, 1993.
30. Diamond CA, Matthay KK: Childhood acute lymphoblasticleukemia. Pediatr Ann 17:156-61, 164-70, 1988.
31. Abraham J: Internal Medicine for Dentistry, 2nd Ed. Rose LF,Kaye D, Eds. St Louis: CV Mosby Co, 1990, pp 310-17.
32. Macfarlane JD, Swart JG: Dental aspects ofhypophosphatasia: a case report, family study, and literaturereview. Oral Surg Oral Med Oral Patho167:521-26, 1989.
33. Faircloth WJ Jr, Edwards RC, Farhood VW: Cherubism in-volving a mother and daughter: case reports and review ofthe literature. J Oral Maxillofac Surg 49:535-42,1991.
34. Kamen S, Crespi P, Eisenbud L, Dolan T: Papillon-Lef6vresyndrome: pediatric dental management. J Pedodont 10:356-64, 1986.
35. Sonis AL, Tarbell N, Valachovic R, Gelber R, Schwenn M,Sallan S: Dentofacial development in long-term survivors ofacute lymphoblastic leukemia: a comparison of threetreetment modalities. Cancer 66:2645-52, 1990.
36. Goho C: Chemoradiation therapy: effect on dental develop-ment. Pediatr Dent 15:6-12, 1993.
37. N/isman M, BjOrk O, SOderh~ill S, Ringden O, Dahllof G: Dis-turbances in the oral cavity in pediatric long-term survivorsafter different forms of antineoplastic therapy. Pediatr Dent16:217-23, 1994.
38. Lipton JM: The pathogenesis, diagnosis and treatment of his-tiocytosis syndromes. Pediatr Derrnatol 1:112-20, 1983.
39. Lahey ME: Histiocytosis-X: an analysis of prognostic factors.J Pediatr 87:184-89, 1975.
40. Komp DM: Long-term sequelae of histiocytosis-X. Am JPediatr Hematol / Oncol 3:165-68, 1983.
41. Ladisch S, Gadner H: Treatment of Langerhans’ cell histio-cytosis -- evolution and current approaches. Br J Cancer70(23):$41-46,1994.
42. Komp DM: Langerhans" cell histiocytosis. (Editorial) N EnglJ Med 316:747-48, 1987.
43. Yu LC, Shenoy S, Ward K, Warrier RP: Successful treatmentof multisystem Langerhans’ cell histiocytosis (histiocytosisX) with etoposide. Am J Pediatr Hematol Oncol 16:275-77,1994.
44. Ishii E, Matsuzaki A, Okamura J, Inoue T, Kajiwara M,Uozumi T, Yoshida N, Miyazaki S, et al: Treatment of Langer-hans’ cell histiocytosis in children with etoposide. Am J ClinOnco115:515-17,1992.
45. Greenberger JS, Crocker AC, Vawter G, Jaffe H, Cassady R:Results of treatment of 127 patients with systemic histiocy-tosis (Letterer-Siwe syndrome, Sch~iller-Christian syndromeand multifocal eosinophilic granuloma). Medicine (Balti-more) 60:311-38, 1981.
16 American Academy of Pediatric Dentistry Pediatric Dentistry - 18:1, 1996