la dystrophie myotonique de type ii
TRANSCRIPT

Dr KHELLAF
ENCADRÉ PAR: PR SIFI
SERVICE DE NEUROLOGIE
CHU BENBADIS – CONSTANTINE
FEVRIER 2015

INTRODUCTION
HISTORIQUE
EPIDEMIOLOGIE
GENETIQUE
PRESENTATION CLINIQUE
DIAGNOSTIC DIFFERENTIEL
PRISE EN CHARGE
AVANCEES SCIENTIFIQUES 2014

Le groupe des dystrophies myotoniques comporte deux
affections autosomiques dominantes génétiquement
distinctes:
1. la dystrophie myotonique de type 1 (maladie de Steinert
ou DM1)
2. la dystrophie myotonique de type 2 (PROMM ou DM2)
identifiée en 1994.
Elles ont en commun trois signes cardinaux : la faiblesse
musculaire, la myotonie et la cataracte précoce.
C’est une maladie multi systémique.
Le diagnostic clinique de DM2 est moins facile que celui de la
maladie de Steinert.
Un élément distinctif clair est l’absence de forme congénitale
dans la DM2

L’anomalie génétique en cause dans la DM2 est connue
depuis 2001.
Elle consiste en l’amplification de répétitions d’un
quadruplet CCTG situé dans l’intron 1 du gène ZFN9
localisé sur le chromosome 3q21.
Le diagnostic moléculaire est rendu difficile par la très
grande taille de l’expansion et sa grande instabilité
somatique.
les manifestations pluritissulaires semblent résulter des
effets pathogènes de l’expansion sur le métabolisme des
ARN.

l’identification de la (DM2) est récente. Ce n’est
qu’en 1994 que furent décrites les premières
observations de patients ayant une maladie de TAD ,
comportant les signes cardinaux de la maladie de
Steinert (faiblesse musculaire, myotonie et cataracte
précoce), mais chez qui la recherche d’une expansion
de triplets CTG pour le gène DMPK sur le
chromosome 19 s’est avérée négative.
En 1998, le locus est localisé sur le chromosome
3q21et ce locus est nommé DM2.

L’identification de la mutation responsable du
phénotype DM2 date de 2001 : il s’agit de
l’expansion de répétitions d’un quadruplet CCTG
dans le gène ZNF9.
En conséquence, la nomenclature actuelle distingue
la dystrophie myotonique de type 1 (maladie de
Steinert ou DM1) et la dystrophie myotonique de
type 2 (myopathie proximale myotonique ou
PROMM).

La prévalence minimale de DM2:1/100 000 hab
L’âge moyen se situe autour de 30-40 ans, mais l’affection peut se manifester après 60 ans.
Répartition géographique: nord de l’Europe le plus souvent l’Allemagne et la Pologne.
la prévalence de DM2 en Allemagne est équivalente à celle de DM1.
Quelques familles non Européennes (Maghreb, Asie)

Transmission autosomique dominante.
Anomalie : amplification des répétitions d’un
tétranucléotide CCTG situé dans l’intron 1 du gène codant
la protéine en doigt de zinc 9 (ZNF9) localisé en 3q21
La DM2 est la première maladie qui soit rapportée à
l’expansion d’un tétranucléotide.
La taille de l’expansion des allèles mutés peut être
considérable, variant de 75 à plus de 11 000 répétitions,
avec une médiane de 5 000 répétitions.
Il s’agit d’amplifications beaucoup plus larges que dans la
DM1

Le phénomène d’anticipation nettement moins marqué
que dans la maladie de Steinert
Il y’a une corrélation positive entre la taille de l’expansion
et l’âge de début
Une famille française de dystrophie myotonique AD avec
atteinte multisystémique, associée à une démence fronto
temporale, liée au locus 15q21-24, a été récemment
rapportée par Le Ber et al élargissant ainsi le groupe des
dystrophies myotoniques d’une dystrophie myotonique de
type 3 (DM3).



L’âge moyen aux premiers symptômes se situe autour de
30-40 ans, mais l’affection peut se manifester après 60
ans.
À ce jour, aucune forme congénitale de DM2 n’a été
rapportée.
Le symptôme initial le plus fréquent de la DM2 est :
la faiblesse musculaire proximale.
Les autres symptômes d’appel sont:
des douleurs ou une raideur musculaires 15%
une myotonie 40%

A. MANIFESTATIONS MUSCULAIRES
SQUELETTIQUES:
1) Les Douleurs musculaires: 60%
Elles sont épisodiques et fluctuantes.
décrites comme des brûlures ou des tiraillements.
Elles siègent préférentiellement aux membres inférieurs et
prédominent au repos.
Elles peuvent précéder la faiblesse musculaire.

2) La Faiblesse musculaire:
prédomine sur la musculature axiale et proximale des
membres.
Le déficit intéresse précocement les fléchisseurs du cou et
les abdominaux,
La faiblesse des muscles pelviens devient symptomatique
après 50 ans chez 30 %.
Au niveau des membres, la faiblesse apparaît non sélective.
Le déficit distale est au second plan et coexiste toujours
avec la faiblesse proximale
la faiblesse des muscles faciaux et bulbaires est minime ou
absente et il n’y a pas d’aspect dysmorphique de la face.

3) l’Atrophie musculaire:
L’atrophie musculaire s’observe dans moins de 10 % des
cas.
Une hypertrophie des mollets a été rapportée chez
quelques patients.
Les rétractions musculaires ne font classiquement pas
partie du tableau de DM2 mais ont été observées dans un
cas d’expression précoce par Bassez et al.

4) La Myotonie:
La myotonie clinique est inconstante et beaucoup moins
franche que dans la maladie de Steinert.
Elle est peut être asymétrique et focale, et elle est
variable dans le temps, autrement dit les patients
peuvent décrire des symptômes suggestifs de myotonie
sans que celle-ci soit décelée par l’examen.
Une exacerbation de la myotonie pendant la grossesse a
été rapportée .

B. AUTRES MANIFESTATIONS:
1) La cataracte:
a les mêmes caractéristiques que celle observée dans la
maladie de Steinert. Elle est sous-capsulaire postérieure et
donne des opacités multicolores irisées.
Sa fréquence est estimée de 61à 84 %
précoce : elle est observée à la lampe à fente chez 2/10
patients avant l’âge de 20 ans.
Elle constitue, avant l’âge de 50 ans, un précieux signe
d’appel d’une dystrophie myotonique.

2) l’atteinte cardiaque:
Elle paraît moins sévère et moins fréquente que dans la
DM1
Des troubles de la conduction auriculo-ventriculaire ou
intra ventriculaire sont observés sur l’ECG de surface chez
20 %
Les patients DM2 peuvent développer des troubles du
rythme responsables de décès.
Une cardiomyopathie progressive, sans autre cause
identifiable, a été observée chez 7 % des patients DM2 mais
3) Une hypersomnie diurne:
dont le mécanisme reste à documenter, s’observe aussi
bien dans la DM2 que dans la DM2

4) l’atteinte du système nerveux central:
il n’a été décrit jusqu’alors aucun cas avec retard mental
comme dans la maladie de Steinert.
Une atrophie cérébrale, mesurée par (IRM) en 3D, est
observée ,elle n’est corrélée à aucun paramètre clinique,
non plus qu’à la taille de l’expansion.
Des hyper signaux confluents péri ventriculaires de la
substance blanche cérébrale ont été observés sur l’IRM en
T2 chez quelques patients appartenant à des familles
distinctes.
Hypoacousie rare
Tremblements d’action dans 20% des cas

5) Une calvitie:
précoce est notée chez les hommes. Elle concerne environ
un tiers des patients entre 21 et 34 ans.
6) L’atteinte endocrinienne et métabolique:
Un diabète est présent dans environ 20 % des cas, des
anomalies du L’hyperglycémie est associée à un
hyperinsulinisme témoignant d’une résistance périphérique à
l’insuline.
Un hypogonadisme hyper gonadotrope (avec élévation de la
[FSH]) est démontré chez deux tiers des sujets masculins
L’élévation du taux de gamma-GT apparaît fréquente,
indépendamment de toute maladie hépatique
une diminution du taux sérique des immunoglobulines de
classe G et M.

1. Les CK:
le taux sérique est normal ou peu élevé, le plus souvent
inférieur à 5 fois la normale.

2. L’électromyogramme (EMG) :
Examen précieux pour le diagnostic quand il permet
d’enregistrer une myotonie cliniquement indétectable.
Il faut savoir le répéter et multiplier les muscles
explorés car les salves myotoniques sont parfois
difficiles à obtenir, rares et inconstantes.
Une myotonie est présente à l’EMG chez 90 % des
individus atteints.
Sander et al ont rapporté l’intérêt du test d’effort
bref (10 secondes) pour distinguer les dystrophies
myotoniques de types 1 et 2 : ce test est normal chez
les patients DM2 alors qu’une franche réduction de
l’amplitude du potentiel moteur est observée chez les
patients DM1.

3. La biopsie musculaire :
ne montrant aucune lésion spécifique.
une inégalité de taille des fibres musculaires, des noyaux musculaires internalisés, parfois disposés en chaînettes, des petites fibres anguleuses disséminées. Des fibres très atrophiques à noyau pycnotique, réduites à l’état de sacs nucléaires(Fig.1), sont présentes dans près de 90 % des cas.
Des fibres en nécrose sont exceptionnellement observées. Contrairement à DM1(Fig.2)

Fig1Fig2

Fig. 3 DM2, muscle deltoïde, hématéine éosine. Le calibre des fibres
musculaires est hétérogène. Les flèches indiquent les fibres atrophiques
réduites à l'état de sacs nucléaires. D'assez nombreuses fibres contiennent
des noyaux internalisés.
I. Pénisson-Besnier
Dystrophie myotonique de type 2
EMC - Neurologie, Volume 2, Issue 2, 2005, 182 - 190
http://dx.doi.org/10.1016/j.emcn.2004.10.003

Fig. 4 Muscle deltoïde. L’étude histoenzymologique (ATPase pH 9,4) montre des
fibres de type 2 (foncées) de taille normale dans la DM1 (A), fréquemment
atrophiques (indiquées par des flèches) dans la DM2 (B).
I. Pénisson-Besnier
Dystrophie myotonique de type 2
EMC - Neurologie, Volume 2, Issue 2, 2005, 182 - 190
http://dx.doi.org/10.1016/j.emcn.2004.10.003

Se pose avec Les affections génétiques comportant une
myotonie incluent
les dystrophies myotoniques : DM1
les maladies en rapport avec des mutations
affectant les gènes des canaux ioniques
musculaires:
myotonie congénitale [canal chlore],
paramyotonie et paralysie périodique
hyperkaliémique [canal sodium],
syndrome d’Andersen Tawil[canal potassium])
certains médicaments sont susceptibles d’induire une
myotonie acquise : fibrates et statines, chloroquine,
colchicine
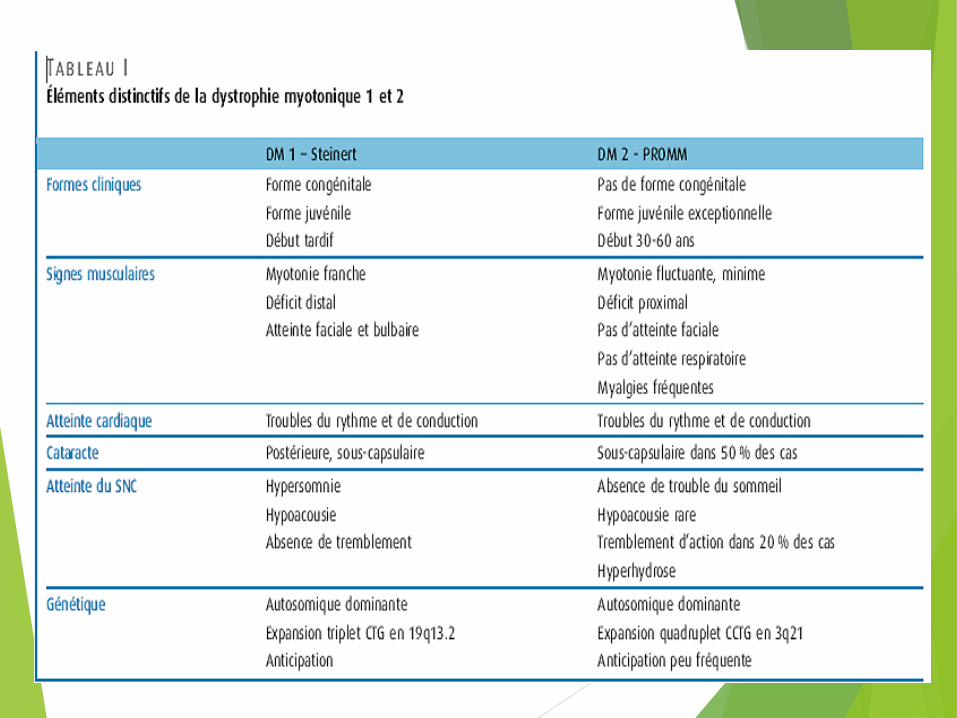

DM 1 (Steinert) DM 2 (PROMM)ÉPIDÉMIOLOGIE Quasi universelle Surtout en Europe
ÂGE DE DÉBUT Tout âge Ado à adulte tardif
ANTICIPATION +++ Faible
FORME CONGÉNITALE + Non
FAIBLESSE :
FACIALE + +/–PROXIMALE + ++DISTALE ++ +
MYALGIES Non +CATARACTE + +CALVITIE + +TROUBLES CARDIAQUES ++ +HYPOGONADISME + +HYPERGLYCÉMIE + +HYPERSOMNIE DIURNE + +MYOTONIE À L'EMG ++ +ATTEINTE DU SNC ++ +LOCUS 19q13.3 3q21
GÈNE MUTÉ DMPK ZNF9
EXPANSION Triplets CTG Quadruplets CCTG
TAILLE DE L’EXPANSION 50 à 4 000 75 à 11 000 (moyenne 5 000)


Le pronostic de DM2 paraît dans l’ensemble meilleur que
celui de DM1.
Evaluation fonctionnelle (force musculaire)
Kinésithérapie régulière, mobilisation douce, massage,
sport sans excès, balnéothérapie.
Traitement de la douleur: la carbamazépine.
Cardiopathie : anti-arythmiques.
la cataracte : traitement chirurgicale.
NB: Le diagnostic prénatal n’est pas recommandé dans la
DM2 en raison d’une anticipation plus faible, de l’absence
de formes congénitales ou infantiles sévères, et d’une
expression clinique tardive.





La dystrophie myotonique de type 2 ou DM2 (aussi
appelée PROMM pour proximal myotonic myopathy) est une
maladie neuromusculaire rare, qui fait partie du groupe des
dystrophies myotoniques. cette maladie touche les muscles,
avec faiblesse musculaire proximale et myotonie. Elle
atteint aussi d'autres organes (le cœur, l’œil (cataracte),
l’appareil respiratoire et le système endocrinien) : c'est une
maladie multisystémique.
La dystrophie myotonique de type 2 (DM2) atteint
les femmes comme les hommes et débute généralement à
l'âge adulte. Les symptômes peuvent être très variables
d'une personne à l'autre.
La prise en charge vise essentiellement à prévenir les
complications, notamment cardiaques, et à améliorer la
qualité de vie des personnes atteintes de DM2.



