isolation and quantification of nucleic acids in plants chris yuen, ph.d. june 13, 2006 advances in...
TRANSCRIPT

Isolation and Quantification of Nucleic Acids in Plants
Chris Yuen, Ph.D.
June 13, 2006Advances in Bioscience Education
Leeward Community College

The Central Dogma of Genetics
ReplicationTranscription
Translation
mRNA
non-coding RNA (rRNA, tRNA, siRNA, etc.)

Deoxyribonucleotide
Ribonucleotide

Plants cells contain three distinct sets of DNA: nuclear, plastidic, mitochondrial
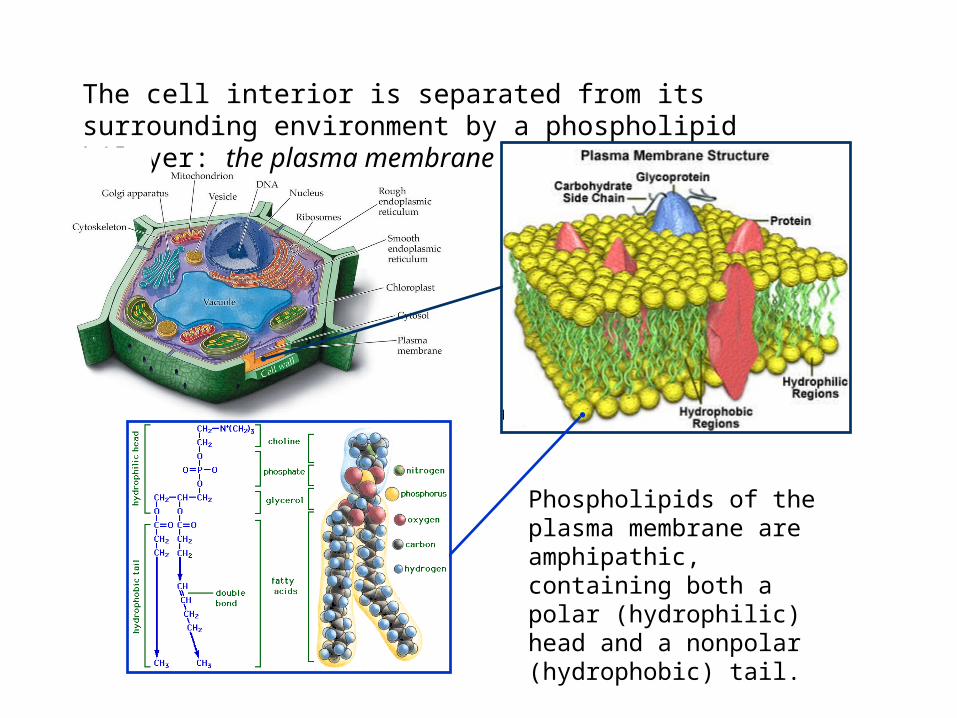
The cell interior is separated from its surrounding environment by a phospholipid bilayer: the plasma membrane
Phospholipids of the plasma membrane are amphipathic, containing both a polar (hydrophilic) head and a nonpolar (hydrophobic) tail.

Plant cells are enclosed within a rigid extracellular polysaccharide matrix: the cell wall
Cellulose microfibrils, the main constituent of plant cell walls, as viewed through an electron microscope

Nucleic Acid Extraction Requirements
1. Disruption of cell wall and membranes to liberate cellular components.
2. Inactivation of DNA- and RNA-degrading enzymes (DNases, RNases).
3. Separation of nucleic acids from other cellular components.• Extraction/Precipitation method• Adsorption Chromatography method

Getting Prepared: Creating a Nuclease-Free Environment
Living organisms produce several enzymes designed to degrade DNA and RNA molecules. There are several things you can do to minimize the risk of exposing your samples to external DNases and RNases.
• Autoclave solutions. This is usually sufficient for getting rid of DNases, and most RNases as well.
• Treat solutions with 0.1% DEPC. DEPC inactivates nucleases by covalently modifying the His residues in proteins. Generally considered unnecessary for DNA extraction. Not compatible with solutions containing Tris or HEPES.
• Have a dedicated set of pipettors or use aerosol barrier tips.
• Wear gloves. You should be doing this anyway for safety reasons, but skin cells also produce RNase7, a potent RNA-degrading enzyme.
• Bake glass, metal, or ceramic equipment at high temp.

Overview of the Extraction/Precipitation Method

Step 1: Disruption of cell walls by grinding
Step 2: Lysis of cells in extraction buffer
Step 1+2: mechanical disruption and homogenization in extraction buffer
Extraction/Precipitation Method
Grind sample into a fine powder to shear cell walls and membranes
Mix thoroughly with extraction buffer to dissolve cell membranes and inhibit nuclease activity
A homogenizer allows cells to be mechanically disrupted within the extraction buffer
Crude lysate

Purposes of the Extraction Buffer1. Dissolve cellular membranes2. Inactivation of DNase and RNase3. Assist in the removal of contaminants
DetergentsChaotropic saltsMetal chelatorsSaltsReducing agentsCTAB PVP
Extraction/Precipitation Method
+
Plasma membrane(phospholipid bilayer) Detergent molecules
Use of Detergents to Lyse Cells: Like Dissolves Like
Mixed micelle
SDS

Crude lysate containing nucleic acids and other cell constituents
Mix thoroughly with an equal volume of organic solvent
e.g. phenol, chloroform, or phenol:chloroform
Centrifuge
The aqueous phase contains water-soluble molecules, including nucleic acids. Proteins and lipids become trapped in the organic phase, and are thus separated away. Insoluble plant debris become trapped in the interphase between the two layers
Perform additional extractions for increased purity
Collect aqueous phase
Extraction/Precipitation MethodStep 3: Organic extraction
Organic
Aqueous
Interphase

• Pellet down nucleic acids. • Pellet down nucleic acids.
• Wash pellet with 70% ethanol to remove residual salts and other contaminants.
• Pellet down nucleic acids.
• Wash pellet with 70% ethanol to remove residual salts and other contaminants.
• Discard ethanol and allow pellet to dry.
After
Add alcohol and salt to precipitate nucleic acids from the aqueous fraction
Supernatant
Pellet
70% EtOH
Dissolve pellet (H2O, TE, etc.)
Step 4: Nucleic Acid Precipitation
Extraction/Precipitation Method
Before After
Centrifuge Wash Centrifuge

Overview of the Adsorption Chromatography Method
Adsorption: the binding of molecules or particles to a surface

Basic Principle
Nucleic acids within a crude lysate are bound to a silica surface
The silica surface is washed with a solution that keeps nucleic acids bound,
but removes all other substances
The silica surface is washed with a solution unfavorable to nucleic acid binding. The solution,
containing purified DNA and/or RNA, is recovered.

Step 1: Prepare crude lysate
Silica-gel membrane
Apply to column
Step 2: Adsorb to silica surface
Adsorption Chromatography Method
Centrifuge
Flow through(discard)
Nucleic acids
Surface silanol groups are weakly acidic, and will repel nucleic acids at near neutral or high pH due to their negative charge
Extraction Buffer composition favors DNA and RNA adsorption to silica: • Low pH• High ionic strength• Chaotropic salt Nucleic acids bind to the
membrane, while contaminants pass through the column.

Centrifuge
Nucleic acids
Step 3: Wash away residual contaminants
Adsorption Chromatography Method
Wash buffer
Nucleic acids
Flow through(discard)
Nucleic acids
Elution buffer
Elution Buffer composition is unfavorable to surface binding: High pHLow ionic strength
Step 4: Elute nucleic acids
Centrifuge
Nucleic acids

Using Nucleases to Remove Unwanted DNA or RNA
Add DNase
Add RNase
+ DNase (protein)
+ RNase (protein)
Depending on when nuclease treatment is performed, it may be necessary to repeat purification steps for protein removal (e.g. phenol/chloroform extraction).

Assessing the Quality and Yield of Nucleic Acids

Nucleic Acid Analysis via UV Spectrophotometry
By measuring the amount of light absorbed by your sample at specific wavelengths, it is possible to estimate the concentration of DNA and RNA. Nucleic acids have an absorption peak at ~260nm.
[dsDNA] ≈ A260 x (50 µg/mL)[ssDNA] ≈ A260 x (33 µg/mL)[ssRNA] ≈ A260 x (40 µg/mL)
DNA Absorption Spectra

How pure is your sample?
The A260/A280 ratio is ~1.8 for dsDNA, and ~2.0 for ssRNA. Ratios lower than 1.7 usually indicate significant protein contamination.
The A260/A230 ratio of DNA and RNA should be roughly equal to its A260/A280 ratio (and therefore ≥ 1.8). Lower ratios may indicate contamination by organic compounds (e.g. phenol, alcohol, or carbohydrates).
Turbidity can lead to erroneous readings due to light interference. Nucleic acids do not absorb light at the 320 nm wavelength. Thus, one can correct for the effects of turbidity by subtracting the A320 from readings at A230, A260 and A280.

Running your sample through an agarose gel is a common method for examining the extent of DNA degradation. Good quality DNA should migrate as a high molecular weight band, with little or no evidence of smearing.
genomicDNA
RNA(degraded)
Checking for Degradation: DNA

Checking for Degradation: RNA
Ribosomal RNA (rRNA) makes up more than 80% of total RNA samples. Total RNA preps should display two prominent bands after gel electrophoresis. These correspond to the 25S and 18S rRNAs, which are 3.4 kb and 1.9 kb in Arabidopsis (respectively).
Good quality RNA will have:No evidence of smearing25S/18S ratio between 1.8 - 2.3
25S
18S

Today’s Lab Objectives:Use the RNeasy Extraction Kit to isolate total RNA from Arabidopsis thaliana.
Determine RNA yield