irb approval process 2016
TRANSCRIPT

IRB Approval ProcessJames R. Galt, PhDAssociate ProfessorPhysics and Computing Laboratory, Division of ResearchDivision of Nuclear Medicine and Molecular ImagingDepartment of Radiology and Imaging Sciences
Member IRB Committee B3 (Biomedical)
I am grateful to RK Halkar, MD (Professor, Radiology and Imaging Sciences) Shara Karlebach, WHNP-BC (IRB Research Services Consultant)for help developing these slides.
Disclosure: Royalties from the sale of Emory QuantEM software for processing nuclear renograms and Emory ExSPECT software for attenuation correction of myocardial perfusion SPECT.

The IRB Approval Process
•Why IRB?•What needs IRB approval?•The IRB Submission•Recruitment and Consent• IRB Committees and Resources

Aulus Cornelius Celsus (ca 25 BC---ca 50 AD)• Roman Physician• De Medicina
• Only surviving work• Draws upon knowledge from ancient Greek works• Considered the best surviving treatise on Alexandrian medicine• Topics include: removal of arrows, stopping bleeding, removal of
bladder stones, the amputation of limbs, setting fractures…• Covers the preparation of numerous ancient remedies, including
opoids.• Practiced human experimentation • Quote
"It is not cruel to inflict on a few criminals sufferings which may benefit multitudes of innocent people through all centuries." (De Medicina, Prooem. 26)
http://toxipedia.org/display/toxipedia/Aulus+Cornelius+Celsus

Headline: UC Davis Doctors Who Infected Brain Cancer Patients With Bowel Bacteria ResignAugust 25, 2013 4:13 PM
• SACRAMENTO (AP) – Two California neurosurgeons who infected brain-cancer patients with bowel bacteria in an effort to save their lives have resigned their positions at the University of California, Davis, after officials concluded their actions violated the school’s code of conduct.• had the permission of the three patients to try the injections, • failed to get the required prior approval from either the school or the FDA• experimental treatment that had not been tested on animals.
• The three patients, two middle-aged women and a man with glioblastoma• The doctors hoped that… live bowel bacteria would stimulate their immune systems and
prolong their lives.• The first patient died a little more than a month after the treatment, but the second lived for
more than a year, giving the doctors hope that the treatment was working.• When the third patient developed sepsis and died within two weeks, however, the university
launched an investigation…• The doctors told the Bee they weren’t trying to do unapproved research or create a
treatment they could profit from. They said they only wanted to give their patients a last-ditch chance at survival…
• “I was simply thinking that I could help patients,” Muizelaar said. “My whole medical practice is guided by actually only one principle, namely: What would I do for my mother, my son, myself?”
(Note: Enterobacter aerogenes)

Medical ResearchBiomedical Research or Experimental Medicine
• Clinical Research• Evaluation of treatments for
safety and efficacy • Includes clinical trials
• Preclinical Research• Expanding the knowledge base• Investigating the basics for the
development of new treatments• Translational Research
• Integrating the basic and clinical research domains
• Bench to bedside
99mTc Folate SPECT/CT, Pituitary Adenoma
99mTc MDP Bone Scan of a Mouse

Ethics in Medical ResearchBalancing Risks and Benefits
• Benefit• A positive value for the subject
• A greater chance at a good therapeutic outcome• Access to treatment when conventional therapy fails
• Better understanding of a disease or condition• Risk
• Harm that might come from the study• Probability of adverse reactions to treatment• Magnitude of the risk: Itchiness or death
• Social, legal, economic, or psychological risks • Will this treatment stigmatize the subject?

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
The Emory IRB• The Emory IRB is an
administrative body established to protect the rights and welfare of human subjects recruited to participate in research activities under the auspices of Emory University
• Responsible for the initial review and continuing oversight of human subjects research at Emory• Approve, disapprove, or modify• Conduct continuing review• Provide oversight and monitoring • Suspend or terminate approval• HIPAA Privacy Board

What requires IRB review?• The question:
“Is an activity research involving human subjects?”• Research
a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge.
• Human Subjecta living individual about whom an investigator (whether professional or student) conducting research obtains• Data through intervention or interaction with the individual, OR• Identifiable private information

Is It Research?• Is the intent to distribute the information beyond
the institution?• Would the same thing be done if there was not
professional recognition attached?• Activities that do not need review
• Review of clinical cases solely for quality assurance purposes
• Comparison of techniques for development or assessment of internal procedures
• No intervention, no interaction, and no identifiable subjects

Research with Human Subjects?• Study
A researcher wants to review internet sites with chat rooms and an ”Ask the Experts” feature. She will review sites commonly used by the diabetes community. She will visit 35 sites and review the posts, recording how long it takes to get a response and the quality of the response based on established criteria. Results will be published.
• The activity is:A. Not human subjects researchB. Exempt human subjects researchC. Human subjects research, eligible to EXPEDITED reviewD. Human subjects research, requiring FULL BOARD review
No:• Intervention• Interaction• Identifiable subjects

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
STUDY TYPESFull Board
All other studies must be reviewed at a convened meeting
of the IRB where quorum is present
Expedited Study poses no more than minimal risk AND all study procedures are surveys, questionnaires, focus groups, noninvasive biological samples
Exempt Informed Consent usually must be obtained; HIPAA may still apply. Many surveys and interviews of adults, educational program evaluations and secondary analysis of de-identified pre-existing data or samples. The IRB is the only unit authorized to make this determination
Not “Research” with “Human Subjects” Always check with the IRB to make this determination. Also consult PI and coordinator.
Is it “research”? Term of art defined at 45 CFR Section 46.102(d): a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to
generalizable knowledge.Does it involve “human subjects”? Term of art defined at 45 CFR Section 46.102(f): a living individual
about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information

Example: Evaluation of a Unique SPECT System in Obese Patients
• Study• Dedicated cardiac SPECT of unique design• Assemble MPI SPECT images of 80 patients in 4 categories:
No obesity; WHO obese class 1, 2, 3 (including some ≥ 45 kg/m2)• 2 physicians evaluate image quality to answer the question:
Should use of this camera be discontinued for obese patients?• IRB Status
A. IRB approval is required, this is a research study on human subjectsB. IRB approval is required, images are protected health informationC. IRB approval is required, but informed consent can be waivedD. IRB approval is not required, this is a quality assurance exercise

Example: Evaluation of a Unique SPECT System in Obese Patients
• Study Protocol• Dedicated cardiac SPECT of unique design• Assemble MPI SPECT images of 80 patients in 4 categories:
No obesity; WHO obese class 1, 2, 3 (including some ≥ 45 kg/m2)• Class 3 patients are also imaged on a conventional SPECT/CT system
for comparison• 2 physicians evaluate image quality to answer the question:
Should use of this camera be discontinued for obese patients?• IRB Status
A. IRB approval is required, this is a research study on human subjectsB. IRB approval is required, images are protected health informationC. IRB approval is required, but informed consent can be waivedD. IRB approval is not required, this is a quality assurance exercise
Protocol calls of intervention (another scan) and additional radiation (the CT portion of the SPECT/CT).

The IRB Approval Process
•Why IRB?•What needs IRB approval?•The IRB Submission•Recruitment and Consent• IRB Committees and Resources

RequiredTraining for Clinical Investigators 12
• Collaborative IRB Training Initiative (CITI)• An on-line course maintained by the University of Miami.• Recertification is required every 2 years (Emory and VA)• Website: www.citiprogram.org
• Key Concepts in Clinical Research for Investigators • Maintained by the Office of Clinical Research• Developed upon advice and approval of the Clinical Trials Executive
Committee, the Clinical Trials Task Force and other clinical trials advisory groups to provide supplementary content for clinical investigators specific to clinical trials involving human subjects.
• Website: www.ocr.emory.edu/training/courses/key-concepts.html

Other Approvals• Department (i.e. Radiology and Imaging Sciences)• Clinical Translational Research Committee
(CTRC; Winship Cancer Center, If related to oncology)• School of Medicine Conflict of Interest Committee• Environmental Health and Safety Office
(Radiation, Chemical, Biological safety) • Radiation Safety
• Does the research involve exposure to radiation?• Does the research involve radiology procedures that are
required as part of the study (even though standard-of-care)?• An Authorized User (of radioactive materials for medical use)
is required on the study team if the Nuclear Medicine scan is research driven or uses a non-standard radiotracer.

Radiation Safety: Informed Consent Dose Language
• Emory Radiation Safety• Dose calculator• Consent language generator
• Example: 1 DXA (Bone Densitometry) • 0.001 mGy• You will be exposed to radiation from x-rays.
These procedures are not necessary for your medical care and will occur because you participate in this study. The estimated radiation dose that you will receive is equal to or less than the natural environmental radiation the average person receives in the United States annually. The principal risk associated with a radiation dose is the possibility of developing a radiation-induced cancer later in life. The risk for radiation-induced cancer from this study is negligible.
http://www.ehso.emory.edu/documents/forms/radiation.html

Radiation Safety: Informed Consent Dose Language
• Emory Radiation Safety• Dose calculator• Consent language generator
• Example: 1 Myocardial Perfusion• 26 mGy• You will be exposed to radiation from
nuclear medicine. These procedures are not necessary for your medical care and will occur because you participate in this study. The estimated radiation dose that you will receive is equal to or less than the annual radiation exposure limit allowed for persons who are occupationally exposed to radiation (for example, x-ray technologist, radiologist). The principal risk associated with a radiation dose is the possibility of developing a radiation-induced cancer later in life. The risk for radiation-induced cancer from this study is minimal.
http://www.ehso.emory.edu/documents/forms/radiation.html

Radiation Safety: Informed Consent Dose Language
• Emory Radiation Safety• Dose calculator• Consent language generator
• Example: 5 Myocardial Perfusion• 130 mGy• … The radiation dose estimate that you will
receive is equal to or less than the radiation exposure allowed to be received by a radiation worker for 3 years. The principal risk associated with a radiation dose is the possibility of developing a radiation-induced cancer later in life. Although the risk from radiation is cumulative it is not expected to adversely affect your condition or treatment. The Emory University Radiation Safety Committee has reviewed and approved the use of radiation in this research study.
http://www.ehso.emory.edu/documents/forms/radiation.html

Radiology Checklist• Is a radiology procedure a standard procedure? No, if
• The imaging protocol is not standard in Emory Healthcare• The images must be modified (removal of PHI) before transferal
to a core lab.• Special image analysis must be performed• A non-standard report must be generated
• Radiology Checklist must be filled out and submitted for any study requiring Radiology Services.• Pre-study imaging (phantoms, QC scans, sample images…)• Exam (Chest x-ray, DEXA, CT, MR, Nuclear Medicine, PET…)• Human Studies Application for Radionuclide Use (if applicable)
The checklist states:ATTENTION COORDINATORS AND OCR: IF A STUDY NEEDS IMAGES SENT TO A SPONSOR/CORE LAB, THE COORDINATOR MUST REQUEST A BURNED CD FROM THE RADIOLOGY FILE ROOM. THERE IS A FEE FOR THE SERVICE. RADIOLOGY WILL NOT BE RESPONSIBLE FOR SENDING ANY IMAGES ELECTRONICALLY TO OUTSIDE ENTITIES NOR WILL THEY SHIP CDS.

Components of the IRB Submission(forms to be filled out in eIRB)
• Study Identification• Name, staff, start/stop dates
• Required Reviews• Radiology, radiation safety, etc.
• Study Sites• EUH, EUHM, CHOA, VA, Grady…
• Funding• NIH, corporate, internal
• Research Design• Lay summary, protocol
• Type of Research• Human subjects?, intervention?
• Banking• Blood samples…
• Drugs and/or Devices• FDA approved, IND, IDE…• Clinical investigator's brochures
• Radiation Safety• HIPPA / PHI
• Partial HIPPA waiver• Study Population
• # of subjects• Patients
• Population, vulnerable groups• Pregnancy
• Social• Surveys, questionnaires
• Recruitment and Payment• Advertisements
• Informed Consent• Consent, HIPPA consent, Assent (pediatrics)
• Data Safety Monitoring Plan• Miscellaneous Documents• Conflict of Interest

HIPPA Waivers• Full HIPPA Waiver
• No Authorization will be required for a covered entity to use and disclose PHI for a particular research project.
• Partial HIPPA Waiver• Authorization is not needed for all PHI uses.
Disclosure is solely for research purposes, such as disclosing PHI for research recruitment purposes.
Note: Most studies require a partial HIPPA waiver.

Lay Summary• The general purpose and method of the study
explained in non-technical language• Why is the study being done?• Who is being studied?• What is being asked of the subjects?
• Should include a brief description of procedures• How many subjects?• What interventions will be done?• If applicable, will the subjects be randomized to different
procedures?• How long will the study last?

Lay Summary• Should not be cut and paste from the consent!
• “You are being asked to participate in a research study because you have…”
• Should not be cut-and-paste from the protocol!• “Tc-99m MAA SPECT or SPECT/CT may be used for RTP of Y-90 SIRT
for HPN and MN. RDE using SPECT may be inferior to PET post SIRT…”
• A good Lay Summary makes the IRB reviewer’s task much easier• “I begin all of my IRB reviews with the Lay Summary because (if done
correctly) it helps me understand the research project.” JR Galt

Protocol Format• Background• Objective• Selection Criteria• Diagnostic Test, Rx, Pathology, End point• Statistical analysis*• References*
* IRB Reviewers are specifically asked if the protocol contains these sections. If this is a pilot study and a detailed statistical analysis does not make sense, state that explicitly.
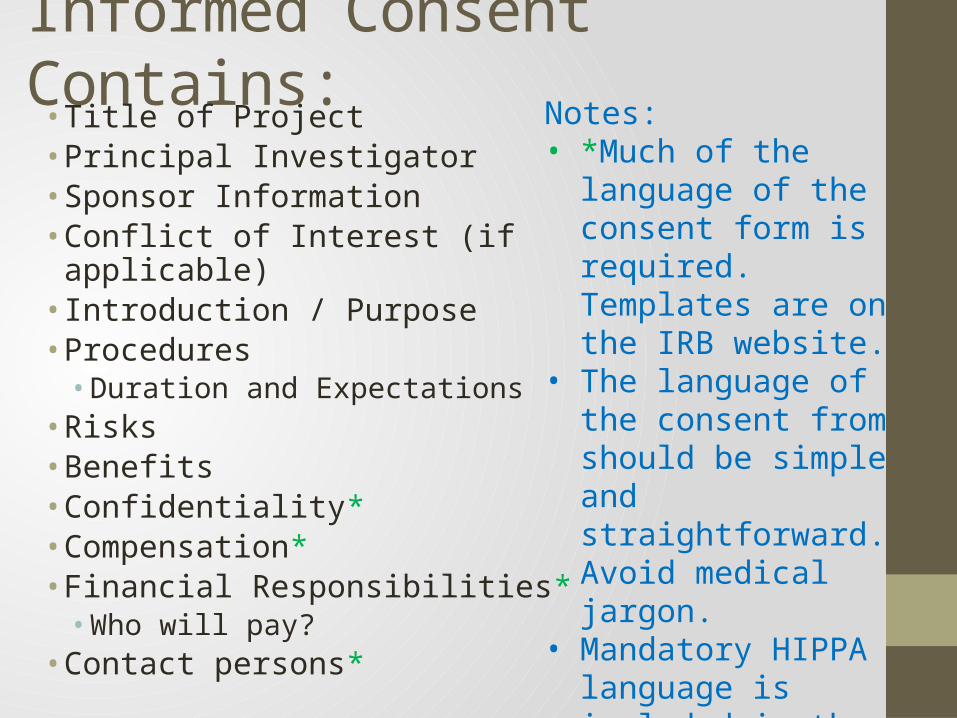
Informed Consent Contains:• Title of Project• Principal Investigator• Sponsor Information• Conflict of Interest (if applicable)• Introduction / Purpose• Procedures
• Duration and Expectations• Risks• Benefits• Confidentiality*• Compensation*• Financial Responsibilities*
• Who will pay?• Contact persons*
Notes: • *Much of the language
of the consent form is required. Templates are on the IRB website.
• The language of the consent from should be simple and straightforward. Avoid medical jargon.
• Mandatory HIPPA language is included in the main consent. In the past a separate document was used.

ClinicalTrials.gov Required Language• Effective March 7, 2012, the FDA will enforce a new requirement for
all qualifying clinical trials approved by the IRB on or after that date. The consent forms for these trials must include the following language verbatim:“A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. law. This Web site will not include information that can identify you. At most the Web site will include a summary of the results. You may search this Web site at any time.”
• The Emory IRB has included this language in Biomedical, Grady, and CHOA consent templates since 2011, study teams on FDA-regulated trials should ensure the required language is present. The VA consent template also already includes this language. Using the latest consent templates from the IRB website will ensure compliance.

Pointers onInformed Consent Documents• Avoid ambiguous language that may imply support beyond the study
• “After the study imaging, you will have standard-of-care imaging which we will track…”
• Should be “After the study imaging we will monitor other imaging procedures you have within Emory Healthcare for a period of three years.”
• Include all procedures directly related to the research• Any drugs, no matter how commonplace, if they are required for the
research procedures (example – acetaminophen)• Include any follow-up communications
• Will you contact the subjects post-procedure?• Include any surveys or questionnaires• Be specific on storage of samples or images
• Can subjects be identified?• Will genetic testing be done?• How long will samples or images be kept?

Common Problems with Informed Consent Documents• Nicely Written Consent contains
• Description of research, procedures, etc.• Extensive lists of risks and side effects of 2 (research provided)
immunosuppression study drugs. No other drugs are mentioned.• Under Costs:
You will be started on Bactrim DS 1 tablet by mouth three times a week for the 6-month treatment period to prevent pneumonia infection. You or your insurance company will be responsible for the cost of Bactrim.
• Reviewer comments.A. Subject should not be responsible for cost of Bactrim.B. Bactrim should be listed in the procedure section. Bactrim’s risks should be
listed. Cost language is ok.C. Bactrim should be listed in the procedure section Bactrim’s risks should be
listed. Subject should not be responsible for cost of Bactrim.D. Bactrim is a common, relatively inexpensive drug. There is really no
concern.Any drug or procedure required as part of the research must be included in the IFC procedures and the risks given. Costs of non-research drugs may be the responsibility of the subject.

Common Problems with Informed Consent Documents
• Incomplete Study Information – Example:• Title: PET/CT versus MRI in Toenail Cancer• PET/CT scan using a new tracer is compared to MRI• Subjects recruited from patients scheduled for MRI• Consent states that the goal of the study is to study the utility of PET/CT in
cancer of the toenail. No mention of MRI.• Reason for omission – “The MRI is not done for research so we left it out of
the consent.”• Un-necessary Language – Example:
• Study: Test of new imaging procedure. Study duration is 1 hour in the Radiology Department on 1 day
• IFC contains instructions on “preventing pregnancy during the study.”• Reason for inclusion – “IRB staff usually requires that language…”
• Risk Information• Risks should be listed for all study related activities…• Even standard-of-care if required for the study.

Common Problems with Informed Consent Documents
• Grammar, punctuation, formatting – • Incoherent text that is obviously cut and pasted from other documents• Pages with page breaks that remain from other versions
• Example – • Emory required language states:
“The researchers will review the results of certain study tests and procedures only for the research. The researchers will not be looking at the results of these tests and procedures to make decisions about your personal health or treatment. For this study, those things include “
• The last sentence should be filled in by research team: “… tests and procedures to make decisions about your personal health or treatment. For this study, those things include the research MRI scan.“
• Badly written consent forms reflect poorly on Emory, the Department, and the research team to the study’s subjects (most often Emory’s patients).

Punctuation Saves Lives
• Let’s eat, Grandma!
• Let’s eat Grandma!

Special ConsiderationsWhen enrolling• Cognitively impaired subjects• Pregnant women, • Students
• Students: considered vulnerable for potential coercion or undue influence
• Prisoners • Children• Etc.
99mTc Sestamibi Clinical Trial (~1987)Emory University Graduate Student as Healthy Control
Nuclear Medicine waiting room in Kuwait City.
James R Galt, 2009

Submit to e-IRB and Approvals

Emory eIRB• Access:
Emory University ID, Password
• To get an account: Log into ENID at http://enid.emory.edu/myaccount
• If you do not already have an eIRB account, you will see a link on the HOME tab saying “Enable access to Electronic Institutional Review Board (eIRB)”. Please click this link.
• Roles• Principal Investigator
(Faculty)• Co-investigator• Study Coordinator

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
Submit to e-IRB and Approvals

IRB CommunicationseIRB Frequently Asked Questions• Who can create and edit study applications?
Users who have been assigned as PI, Co-Iinvestigator, or Coordinator can create or edit a submission. They can also edit the submission if the IRB sends it back for changes.
• Who can "submit" applications in eIRB?
Only the person named as PI in the application can effect the initial submission in eIRB, after which the IRB can begin its review. The PI, Co-Investigator or Coordinator can make changes later requested by the IRB, however.
• Who can submit changes to the application requested by the IRB?
The PI, Co-Investigator, or Coordinator can make changes requested by the IRB.
• How does eIRB notify me when I need to do something?
eIRB will send an email to your regular email account if you need to do something. Although the email will not elaborate on the requested action, you can click the link in the email to go to the eIRB login page and then to the relevant study, where you will see what needs to be done.
http://www.irb.emory.edu/researchers/eresearch/#12

IRB CommunicationseIRB Frequently Asked Questions
• How do I ask a department approver to review and approve a study?After the PI submits a study, eIRB will email the department approver’s regular email account. Although the email will not elaborate on the requested action, the department approver can click the link in the email to go to the eIRB login page and then to the relevant study.
• How do I ask a faculty advisor to review and approve a study?After the PI submits a study, eIRB will email the faculty advisor’s regular email account. Although the email will not elaborate on the requested action, the faculty advisor can click the link in the email to go to the eIRB login page and then to the relevant study.
http://www.irb.emory.edu/researchers/eresearch/#12

The IRB Approval Process
•Why IRB?•What needs IRB approval?•The IRB Submission•Recruitment and Consent• IRB Committees and Resources

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
Recruitment• Before recruiting/enrolling a subject,
make sure you are an IRB approved study staff.
• Make sure recruitment tools are IRB approved before using (including flyers, email blasts, brochures, etc.)
• Consider privacy when recruiting a subject
• Do not mention compensation as a reason to participate

Willowbrook Hepatitis Study (1955) • Investigation
• Investigate the natural history of viral hepatitis• Test the effectiveness of gamma globulin for inoculating against
hepatitis• Laboratory
• Institution for mentally retarded children in Staten Island, NY.• Study
• Permission was obtained from parents.• Children were deliberately infected with a mild form of hepatitis.
• Justification• Most new children at Willowbrook became infected within a year.
• Concerns• Parents were not fully informed of the hazards of the study.• Parents may have been led to believe that the study was required for
admission to the school.

Informed Consent / Recruitment• Person obtaining consent must be listed in the IRB submission
• Principal investigator, study coordinator, research nurse…• Not: radiologic technologist or radiology resident that happen
to be around that day• Subject must review consent
• Many subjects will say: “Just show me where to sign.”• Subject must sign and date documents.• You must give the subject time to review the document.• You must give the subject a copy of
the consent forms.• Special Populations
• Pediatrics – consent from the parents, assent from the subject
• VA and CHOA have additional requirements

Disclosure in Informed Consent
An investigational SPECT scanner is evaluated in comparison to a standard scanner. Subjects will be scanned on both scanners and the images compared. A clinical radiopharmaceutical is used in the standard dosage (a single dose is administered). There is no radiation dose outside of the radiopharmaceutical. The study is funded by the manufacturer of the investigational SPECT system. The consent form must disclose:A. The source of funding for the studyB. The investigators relationship to the manufacturerC. The status of the investigational device
(FDA approved or investigational)D. All of the above

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
IRB reportable adverse events (UP), PD and Noncompliance
• Unanticipated Problem (UP): any adverse event considered unexpected, related to study participation, and increasing risk for participant or others
• Protocol deviations (PD): substantive deviation from the IRB approved protocol affecting rights, welfare, safety of subjects, willingness to continue with study participation, or the integrity of the research data
• Noncompliance: failure to follow regulations, IRB policies and procedures, university policies (for example, COI) and the approved protocol guidelines

Shara Karlebach, WHNP-BCIRB Education and QA Team - 2012
• Research misconduct is considered noncompliance.
• Research Misconduct (Policy 7.8) is defined as:• Fabrication – making up of Research data or results
and recording or reporting them• Falsification – manipulating Research materials,
equipment, or processes, or changing or omitting data or results such that the Research is not accurately represented in the Research Records
• Plagiarism – the appropriation of another person’s ideas, processes, results or words without giving appropriate credit
• Anonymous reports can be made to the Emory University Trust Line at: 1-888-550-8850.
IRB reportable adverse events (UP), PD and Noncompliance

IRB reportable adverse events and NoncomplianceUsual Suspects…• Adverse Events
• Unexpected reactions to drugs or device complications • May be brought to IRB Committee for re-assessment of risks
• Protocol Deviations• Failure to follow protocol procedures
• Sometimes unavoidable (e.g. equipment failure)• Subject non-compliance
• Enrollment• Too many (IRB approval is for number of subjects consented)• Protocol lapsed (Failure to submit renewal on time)
• Unauthorized person obtains consent

IRB reportable adverse events and NoncomplianceMay also include failure to Disclose Financial Interests• When noted the action plan is reviewed by Committee• Actions taken have included
• 7 acquitted• 7 death sentences• 9 prison sentences ranging from 10 year to life• Removal from project• Removal as Principal Investigator• Prohibition from research
*Nuremberg – Doctor’s Trials(1946 -1947)

The IRB Approval Process
•Why IRB?•What needs IRB approval?•The IRB Submission•Recruitment and Consent• IRB Committees and Resources

IRB Committees• Committee A – Biomedical
• 2nd, 3rd, 4th Wednesday
• Committee B – Biomedical• 1st Wednesday, 2nd Thursday, 4th Tuesday
• Committee C – Social\Humanist\Behavioral• 1st Friday
• Committee Q –Quality Assurance: Noncompliance, Unanticipated Problems• 3rd Thursday
Each sub-committee has its own membership. Meetings are run by the sub-committee’s Vice Chair.
Committee B3 – Biomedical Membership as of 11/2014
MD (B3 Chair) Pediatrics, Hematology & Oncology
PhD (IRB Co-Chair) Public Health, Nutrition
PhD Clinical Psychology, AutismMD CardiologyPhD Pediatric EndocrinologyBA or MBA or JDNon-scientist Community
MD Pediatrics, Hematology, Sickle Cell
MD Hematology, OncologyPhD Radiology
MPH, CCRC, ELS Pediatrics, Oncology
MD Infectious DiseaseMD, PhD Ophthalmology, RetinologyMD Pediatrics, NeonatologyMD EndocrinologyMD Psychiatry, Neurology, QuipsRN, PhD Orthopedics, Sociology

IRB Committee B3 MeetingOctober 2014• 7 Full Board Amendments
• Amendments are usually changes to protocol, informed consent, or study staff (study staff change usually does not go before full board)
• If prompted by new risk information or protocol changes that involve risk changes will get more scrutiny.
• 2 members review each Amendment and present their findings.• 10 Continuing Reviews
• Yearly or 6 month review of progress, enrollment, safety information.• 2 members review each Continuing Report and present their findings.
• 6 New Studies • Require the most work• 3 members review each New Study and present their findings.• Evaluate: Risk/Benefit, Subject Selection, Protocol, Funding, Vulnerable
Population, etc.

IRB Committee Decisions• Approval - All criteria are met.• Pending – Minor changes are needed.
• Investigator’s response reviewed by Vice-Chair or designee
• Deferred – Major changes needed.• Investigator’s response is returned to Full
Committee.• Disapproval – Study does not meet criteria.
• The study may not be re-submitted for review.• Tabled – Lack or quorum or expertise.
• Postponed until another meeting.

IRB Approval CriteriaHealthy subjects are recruited as controls for a study of imaging markers of Parkinson’s disease. Each subject will undergo MRI and SPECT imaging, both of which carry some risk. The IRB may approve the study if: A. The radiation dose is determined be less than the annual
natural exposure.B. There are potential benefits in the form of additional
information regarding Parkinson’s disease and the risks are properly disclosed.
C. The Radiation Safety Committee approved the study.D. The study is appropriately funded
The risk/benefit equation is often the deciding factor for the IRB. Answers c and d are required but are not sufficient for IRB approval.

Resources• Research Office –
Radiology and Imaging Sciences• IRB Website: www.emory.edu/irb
http://www.irb.emory.edu/index.html http://www.irb.emory.edu/forms/index.html

Headlines:Baby Study Spurs Ethics Debate Over Research Risksby THE ASSOCIATED PRESS August 29, 2013 1:21 PM
• WASHINGTON (AP) — When Dagen Pratt's parents enrolled their tiny premature baby in a study of oxygen treatment, they didn't understand it was to test whether one dose works better than another. No one mentioned any risks.
• Now 6, Dagen struggles with cerebral palsy, and they wonder: Is that long-ago study to blame?
• "Tell me that the Support study did not hurt Dagen in any way," her father, Shawn Pratt, challenged a government panel on Wednesday as his daughter, dressed in a bright sundress, stood quietly by.
• A major controversy has erupted over what sounds like a straightforward question: How much should patients be told about the potential risks before they're enrolled in certain kinds of medical research?
• While the experts debated how to explain research risks, two families who traveled to Washington for the unusual meeting outlined a bigger hurdle: Reeling from the stress of having a vulnerable preemie, they simply didn't understand that they were participating in an experiment.

Support Study (2005-2009)Surfactant, Positive Pressure, and Oxygenation Randomized Trial
• Designed to test level of oxygen appropriate for premature babies• Too much – retinopathy of prematurity (blindness)• Too little – neurologic damage• No definitive study showing optimal dose
• Subjects were randomized to higher or lower oxygen dose • Both within standard of care• Parents signed consent
• Findings• Lower oxygen does →less likely to get severe eye disease• Lower oxygen does →slightly increased number of deaths (unexpected)• Note: Premature babies not in the study had higher risk of death
• Office for Human Research Protections (OHRP) conclusion• Informed consent forms failed to the forms failed to describe
“the reasonably foreseeable risks of blindness, neurological damage and death”

Support Study (2005-2009)Surfactant, Positive Pressure, and Oxygenation Randomized Trial
Nature -Editorial Comment• Put yourself in the position of a parent
with an extremely premature infant. • Would you make the decision to enroll
your child in the trial if the consent form stated in simple language that babies assigned to one group were more likely to go blind, and that those in the other were at a higher risk of getting neurodevelopmental disabilities?
• Equally, would you decide to enroll if the form spelled out that, if you do not take part, your own physician and institution might keep your infant in the middle of the range, trying to avoid either outcome?
• Perhaps you might, but you would do so with full knowledge of the attendant risks. The parents in this case could not do so.
22 AUGUST 2013 | VOL 500 | NATURE | 377

The End